

Abstract

5-Fluorouracil (5-FU) is one of the most widely used chemotherapeutics for the treatment of cancers associated with the aerodigestive tract, breast, and colorectal system. The efficacy of 5-FU is majorly affected by dihydropyrimidine dehydrogenase (DPD) as it degrades more than 80% of administered 5-FU into an inactive metabolite, dihydrofluorouracil. Herein we discuss the molecular mechanism of this inactivation by analyzing the interaction pattern and electrostatic complementarity of the DPD–5-FU complex. The basis of DPD overexpression in cancer cell lines due to significantly distinct levels of the miRNAs (miR-134, miR-27b, and miR-27a) compared to normal cells has also been outlined. Additionally, some kinases including sphingosine kinase 2 (SphK2) have been reported to correlate with DPD expression. Currently, to address this problem various strategies are reported in the literature, including 5-FU analogues (bypass the DPD-mediated inactivation), DPD downregulators (regulate the DPD expression levels in tumors), inhibitors (as promising adjuvants), and formulation development loaded with 5-FU (liposomes, nanoparticles, nanogels, etc.), which are briefly discussed in this Review.
Keywords: 5-FU resistance, 5-FU analogues, DPD inhibitors, DPD downregulators, formulation development
Introduction
Since the development of 5-FU by Heidelberger and co-workers in 1957, it has been widely used as a chemotherapeutic agent, especially in the treatment of a variety of solid malignant tumors including gastrointestinal and breast cancers.1−3 It is one of the first rationally designed antimetabolites that undergo activation via physiological pyrimidine salvage pathways to exert their anticancer effects. The detailed mechanism of the action of 5-FU as an anticancer drug is displayed in Figure 1. The interruption of the normal physiological functioning of thymidylate synthase (TS) results in reduced levels of pyrimidine thymidylate (dTMP), which is a nucleotide required for deoxyribonucleic acid (DNA) replication.4 From this figure it is clear that the anticancer effect of 5-FU is not only by inhibition of the TS but also due to misincorporation of its activated form, i.e., fluorodeoxyuridine triphosphate (fdUTP) into ribonucleic acid (RNA) and DNA. However, the resistance observed after a prolonged and repeated exposure to 5-FU has become a most challenging task for most researchers.
Figure 1.

Mechanism of the action of 5-FU as an anticancer drug.
5-FU is reported to have t1/2 of about 5–20 min in plasma.5 About 60–90% of its administered dose gets metabolized in liver and extrahepatic tissues, while 10–20% get excreted unchanged in urine; thus, only a small amount of 5-FU remains to exert its anticancer activity by inhibiting TS. In addition to this, 5-FU has to withstand one more challenge, i.e., the metabolism of 5-FU to inactive metabolite due to DPD overexpression in tumor tissues before cytotoxic nucleotides can be formed.6,7 This type of inactivation belongs to one of the pharmacokinetic resistance mechanisms.8 DPD catalyzes the first step of pyrimidine degradation, i.e., nicotinamide adenine dinucleotide phosphate (NADPH) dependent reduction of thymine and uracil moieties of drug molecules. Cancer cells like TE-5R (human esophageal squamous carcinoma cells (ESCC) with overexpressed DPD) are 15.6 times more resistant to 5-FU chemotherapy as compared to wild type cells.9−11 This 15-fold reduction in anticancer activity is due to the overexpression of DPD. A similar kind of inactivation is also observed in the case of anticancer drugs including tegafur and capecitabine.12 The detailed mechanism of 5-FU inactivation via DPD is displayed in Figure 2. It can be observed that 5-FU undergoes NADPH-dependent reduction to form inactive 5,6-dihydro-5-fluorouracil (DHFU), which is further subjected to hydrolysis with the help of the enzyme dihydropyrimidinase. This enzyme of the hydrolase category results in the formation of fluoro-ureidopropionate (FUPA) that further degrades to fluoro-β-alanine (FBAL).12,13
Figure 2.

Mechanism of 5-FU inactivation via DPD.
To overcome the problem of 5-FU inactivation via DPD, various research scientists have proposed DPD inhibitors and analogues that may be given either as adjuvants or as a single new regimen to bypass the DPD-mediated inactivation, respectively.10,14−16 Additionally, some attempts have also been made to address the 5-FU resistance issue by monitoring the DPD expression via its downregulators or developing a formulation that may attenuate the 5-FU degradation. Among these strategies, designing DPD inhibitors is very tricky as its inhibition in liver or extrahepatic tissues may result in life-threatening toxicity owing to high levels of 5-FU. Therefore, its scheduling has always been strict for cancer patients. Considering this, some analogues of 5-FU derivatives were proposed which have the ability to target specific cancer cell lines and bypass DPD-mediated inactivation. Understanding the cause of DPD overexpression in tumors encouraged researchers to look for DPD downregulators to reverse the 5-FU resistance. Some efforts have been made in the development of formulations (nanogels, nanoparticles, liposomes, etc.) loaded with 5-FU to ameliorate the resistance issue and enhance its bioavailability. The molecular cause of DPD overexpression, its subsequent participation in 5-FU inactivation, and the strategies adopted to improve the treatment quality with 5-FU have been briefly discussed in this Review.
Molecular Cause of DPD Overexpression in Cancer
miRNAs Associated with DPD Overexpression in Cancer Cells
Understanding the molecular cause of 5-FU resistance due to DPD overexpression is very crucial, especially at a genetic level. It is reported that expression levels of DPD mRNA are significantly different in the cases of normal lung tissues and lung tumor tissues. The basis of this variation was explained by Hirota and co-workers in 2012. This research group proposed that some microRNAs (miRNA) in human lung tissues negatively regulated DPD expression by considering blood samples of lung tumor and normal lung tissue from 16 Japanese donors. Later they quantified DPD protein and mRNA expression by performing luciferase assays, real-time RT-PCR, and Western blotting in both tissues. miRNA regulates post-transcription by silencing the mRNAs. Likewise, their group identified that the reduced levels of miRNA (miR-134, miR-27b, and miR-27a) are responsible for the overexpression of DPD protein in tumor tissues. Upon comparing miRNA levels between tumors and normal tissue, they found that expression levels of miR-134, miR-27b, and miR-27a tended to be lower in tumor tissue than in normal tissue (3.64 ± 4.02 versus 9.75 ± 6.58 and 0.64 ± 0.75 versus 1.48 ± 1.39). DPD protein levels were significantly higher in the tumors. Thus, the decreased expression of miR-27b would be responsible for the high levels of DPD protein.17 However, contributions of this DPD overexpression toward 5-FU sensitivity and resistance were not investigated by Hirota and co-workers but were further explained by Offer and associates in 2014. The research group observed that cell lines with miR-27a or miR-27b overexpression showed decreased IC50 of 5-FU, i.e., 4.4 mmol/L (both) for HCT-116 cell lines, whereas normal cells expressing a nontargeting (scramble) control miRNA showed IC50 of 14.3 mmol/L. This clearly indicates that DPD enzyme activity was inversely correlated with expression levels of these miRNAs.18
Kinases Associated with DPD Overexpression in Cancer Cells
Recently, Zhang et al. disclosed the mechanism of 5-FU resistance via DPD-mediated inactivation in colorectal cancer.19 The research carried out by this group indicated that high SphK2 confers 5-FU resistance by promoting DPD-mediated 5-FU degradation into FBAL. In Figure 3, it can be observed that SphK2 has a direct association with sphingosine-1-phosphate (S1P) and an indirect association with histone deacetylase 1 (HDAC1). This type of observed association in the network could be explained on the basis of some reported experimental evidence. SphK2 generally generates S1P that upregulates the transcription of the DPD coding gene, i.e., DPYD, by inhibiting HDAC1. As a result of HDAC1 inhibition, the acylation of histone H3 lysine 56 acetylation (H3K56) in the nucleus is enhanced, and subsequently the expression of DPD is enhanced.19 Additionally, a study by Deng and associates reported elevated miR-21 levels in colorectal cancer which indirectly regulate thymidine phosphorylase (TP) and DPD expression levels. The results demonstrate that miR-21 may play an important role in the 5-FU resistance of colon cancer cells as the knockdown of miR-21 increased the sensitivity to 5-FU chemotherapy toward HT-29/5-FU.20
Figure 3.
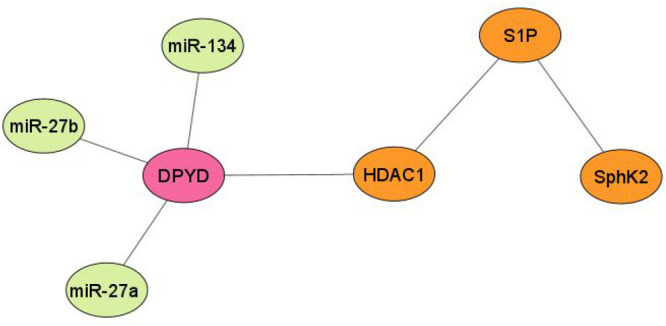
Network analysis of miRNAs associated with DPYD gene expression.
Tsuchiya and co-workers tried to identify the correlation between DPD and epidermal growth factor receptor (EGFR) mutation status based on a clinical hint that among patients with non-small cell lung cancer (NSCLC), sensitivity to 5-FU is mainly associated with the EGFR mutation. The in vitro activity carried out by this research group observed that EGFR-mutated cell lines (PC9, HCC827; exon19 E746-A750 and H1975; exon21 L858R, T790M, gefitinib resistant) showed higher levels of DPD mRNA and protein expression than wild types (H1437, H1299). This indicated that there might be a crosstalk between the EGFR signal cascade (mutated EGFR cell lines) and DPD protein expression. They identified that upon EGF treatment, the cell line with a EGFR mutation in comparison to EGFR wild type manifested the upregulation of Sp-1. Because DPYD gene expression is regulated by transcription factor Sp1, the upregulation of this factor results in high DPD expression among EGFR-mutated cell lines. This observation was justified when inhibition of Sp-1 by gefitinib, an EGFR-tyrosine kinase inhibitor (TKI), and mithramycin A suppressed the DPD expression. This type of observation was only observed in EGFR-mutated cell lines but not observed in wild types and EGFR-TKI resistant cell lines. These results suggest that DPD is regulated mainly in EGFR signal cascade enhanced lung cancers such as EGFR-mutated adenocarcinomas. 5-FU and its derivatives such as uracil-tegafur (UFT) or S-1 might be effective for EGFR wild type adenocarcinomas because of the low DPD expression.21
Interaction Pattern Analysis for the DPD–5-FU Complex
5-FU majorly binds with DPD as a substrate by interacting with active site amino acids. To identify the important amino acid residues involved in 5-FU binding, Tozer et al. developed a homology model and performed molecular docking studies of 5-FU. Molecular docking results suggested that 5-FU majorly interacts with key residues Asn668, Asn736, Thr737, and Asn609 of DPD with a binding affinity of 26.95 kcal/mol; the details of the interaction pattern are shown in Figure 4A. From the figure, it can be observed that the carbonyl and N–H group in 5-FU formed five strong H-bonds with Asn609, Asn736, and Thr737.22 The electrostatic complementarity (EC) analysis explains the detailed map of the electrostatic character of the protein active site and ligand. To gain a deep insight into the fundamental processes that underlie 5-FU–DPD binding, EC has been studied between 5-FU and the DPD complex and suggested that this drug maximally manifests EC within the active binding pocket of DPD with little electrostatic clashes. The EC map is shown in Figure 4B.
Figure 4.

(A) 2D interaction diagram; (B) 3D electrostatic complementarity map for a complex of DPD–5-FU.
Strategies to Counter DPD-Mediated Resistance to 5-FU
Many strategies have been proposed to counteract DPD-mediated 5-FU resistance, which are discussed in Table 1. Moreover, timelines have also been displayed for all these proposed strategies (Figure 5 and Figure 6), and many of them are being implemented in developed nations. Although some prevention efforts have been executed in developing countries, slight progress has been made, the gap is still wide, and there is a paucity of data on the DPD-mediated resistance issue.
Table 1. Strategies Adopted to Counter DPD-Mediated 5-FU Inactivation.
| sample no. | strategy | compound/formulation | research group | toxicity | ref |
|---|---|---|---|---|---|
| 1 | 5-FU analogues/prodrugs | RO0094889 | Hattori et al., 2002 | (14) | |
| Nippon Roche Research Center | |||||
| 2 | 5-FU analogues/prodrugs | NUC-3373 + leucovorin | Ghazaly et al., 2015 | favorable safety profile among cancer patients with no FBAL- or FUTP-associated toxicities, such as hand–foot syndrome, diarrhea, or neutropenia | (23, 24) |
| Centre for Haemato-Oncology, Barts Cancer Institute, Queen Mary University of London | |||||
| 3 | 5-FU analogues/prodrugs | novel palladium-activated 5-FU prodrug | Adam et al., 2022; Cancer Research UK Edinburgh Centre | no mortality or abnormal clinical signs | (25) |
| 4 | 5-FU analogues/prodrugs | DFP-11207 | Fukushima et al., 2017; Delta-Fly Pharma Inc., Kawauchi-cho, Tokushima, Japan | low gastrointestinal and myelosuppressive toxicities compared to 5-FU | (26, 27) |
| 5 | DPD downregulator | JTE-013 | Zhang et al., 2020; Ocean University of China, Qingdao and Capital Medical University, Beijing, China | the derivatives of JTE-013 derivatives have been reported with no observable toxicity | (28, 29) |
| 6 | DPD downregulator | wogonin | Réti et al., 2009; National Institute of Oncology, Budapest, Hungary | in experimentation animals, this component did not show any gross sign of toxicity as there was no considerable change in body weight gain and diet intake profiles | (30, 31) |
| long period of treatment with a high wogonin dose, i.e., 120 mg/kg, may induce heart injury in experimental animals | |||||
| 7 | DPD downregulator | kaempferol | Chebbi et al., 2019; Université de Tunis El Manar, 1068, Tunis, Tunisia | no or minor toxicity against normal epithelial, myeloid cells, and peripheral blood | (32, 33) |
| 8 | DPD inhibitor | eniluracil (EU) | 776C85; formulated by Glaxo Wellcome Inc., Research Triangle Park, North Carolina | diarrhea, neutropenia, with a notable absence of plantar–palmar erythrodysesthesia | (34, 35) |
| dose: tablets of EU (10 mg) and 5-FU (5 and 25 mg) administered p.o. | |||||
| 9 | DPD inhibitor | TAS-114 + S-1 | TAS-114 and S-1, FT (tegafur):CDHP:OXO = 1:0.4:1 have been developed by Taiho Pharmaceutical Co., Ltd., Japan | anemia, diarrhea, and maculopapular rash in combination with S-1 | (36−38) |
| 10 | DPD inhibitor | UFT + leucovorin | tegafur:uracil = 1:4, Osaka University, Japan | thrombocytopenia, neutropenia, infection, neurotoxicity, and cutaneous toxicity | (39, 40) |
| 11 | DPD inhibitor | BOF-A2 (emitefur) | Biwako Research Institute, Otsuka Pharmaceutical Co., Ltd. | leukopenia, thrombocytopenia, and anemia | (41, 42) |
| 12 | formulation designed to improve the bioavailabilty of 5-FU | targeted delivery of 5-FU with EGF-grafted hollow mesoporous silica nanoparticles | Chen et al., 2013; Deakin University, Australia | (43) | |
| 13 | formulation designed to improve the bioavailabilty of 5-FU | covalently mucoadhesive amphiphilic prodrug of 5-FU | Liu et al., 2018; Shenyang Pharmaceutical University, China | (44) | |
| 14 | formulation designed to improve the bioavailabilty of 5-FU | folic acid-decorated and PEGylated PLGA nanoparticles for 5-FU | Hammadi et al., 2016; University of Granada, Spain | (45) | |
| 15 | formulation designed to improve the bioavailabilty of 5-FU | methacrylic-based nanogels | Ashwanikumar et al., 2012; Chemical Biology, Rajiv Gandhi Center for Biotechnology, Poojappura, Thiruvananthapuram, Kerala, India | (46) | |
| 16 | formulation designed to improve the bioavailabilty of 5-FU | octadecylamine–retinoic acid conjugate | Varshosaz et al., 2012; Isfahan University of Medical Sciences, Isfahan, Iran | (47) | |
| 17 | formulation designed to improve the bioavailabilty of 5-FU | 5-FU-loaded pH-sensitive PEGylated liposomal nanoparticles | Udofot et al., 2016; University of Texas MD Anderson Cancer Center, Houston, TX | (48) | |
| 18 | formulation designed to improve the bioavailabilty of 5-FU | lipofufol (triple stealth liposomal formulation) | Fanciullino et al., 2013, Aix Marseille University, France | less toxic than 5-FU due to limited liver uptake, thereby makes DPD activity less important for disposition; prolonged circulation time | (49) |
| 19 | formulation designed to improve the bioavailabilty of 5-FU | pluronic-coated biogenic gold nanoparticles | Mahdi et al., 2021; King Saud University, Riyadh, South Arabia | low 5-FU-related toxicity due to its entrapment in nanoparticle | (50) |
| 20 | formulation designed to improve the bioavailabilty of 5-FU | exosomes for targeted codelivery of miR-21 inhibitor and 5-FU | Liang et al., 2020; Henan University of Science and Technology, Luoyang | - | (51) |
Figure 5.

5-FU/prodrugs and DPD inhibitors designed and evaluated to counter DPD-mediated 5-FU resistance.
Figure 6.

DPD-downregulators and formulation developed to counter DPD-mediated 5-FU resistance.
5-FU Analogues/Prodrugs
Capecitabine is a prodrug that generates 5-FU from the intermediate 5′-deoxy-5-fluorouridine upon activation via thymidine phosphorylase (dThdPase). The resulting 5-FU undergoes inactivation via DPD (Figure 7A). Hattori et al., 2003 designed a series of 5′-deoxy-5-(substituted)-cytidine derivatives as tumor-activated prodrugs of the DPD inhibitor. The designed prodrug undergoes a sequence of biotransformations by esterase, cytidine deaminase, and thymidine phosphorylase selectively in tumor tissues to form active 5(substituted)-uracil derivatives (Figure 7B). This research group reported that these designed prodrugs, i.e., RO0094889, when given as adjuvant therapy with capecitabine (5-FU analogue) significantly raised the levels of 5-FU in plasma with a Cmax value of 1 nmol/mL or g as compared with capecitabine alone (5-FU Cmax = 0. 62 nmol/mL or g). In tumor tissue, the capecitabine in combination with RO0094889 shows 13 nmol/mL as the 5-FU Cmax value, where capecitabine manifested Cmax = 3.5 nmol/mL or g. The concentration of 5-FU in tumor tissue is significantly raised by RO0094889. The study is of great utility as the efficacy of capecitabine was enhanced with minimal toxicity by proposing these designed prodrugs as adjuvants with capecitabine.14,52
Figure 7.

(A). Capecitabine biotransformation to 5-FU and its subsequent inactivation via DPD. (B) RO0094889 activation to 5-VU, an active metabolite and DPD inhibitor.
Ghazaly et al., 2015 reported the first nucleotide analogue, NUC-3373 (Figure 8), a preactivated form of the active anticancer agent FdUMP that exerts an effect similar to that of 5-FU. Interestingly, NUC-3373 is devoid of inactivation via DPD, unlike 5-FU. Moreover, this drug is more cytotoxic than 5-FU as it achieved about 2- to -333-fold lower EC50 values in the majority of the tested cancer cell lines. Upon thymidine kinase (TK) inhibition, the cytotoxicity of this analogue and FUDR was reduced by 4- to 136-fold, respectively, which clearly indicates that NUC-3373 is more independent of TK inhibition. Additionally, this research group assessed the sensitivity of NUC-3373 and 5-FU to DPD degradation by absorption spectroscopy and UPLC-MS/MS and observed that the NUC-3373 concentration remained unaffected in the cell lysate irrespective of gimeracil (DPD inhibitor). NUC-3373 demonstrated 47% tumor growth inhibition, which is greater than 5-FU alone (25%) in colorectal cancer xenografts.23
Figure 8.

NUC-3373, a pyrimidine nucleotide analogue that can bypass DPD-mediated inactivation.
Recently, Adam and associates from Cancer Research UK Edinburgh Centre proposed a novel palladium-activated prodrug (displayed in Figure 9) which is designed to evade the DPD-mediated 5-FU inactivation. In Figure 9 it is clearly observed that 5-FU is vulnerable to undergo inactivation at a site near to the carbonyl centers. To circumvent its inactivation, the region vulnerable to DPD-mediated inactivation was masked. They locked the novel prodrug in a lactim-type structure by alkylating the O atoms of 5-FU, which can also be seen in Figure 9. The in vivo pharmacokinetic analysis reported by this group suggests that the prodrug is rapidly and completely absorbed after oral administration and exhibits a longer half-life than 5-FU. The basis of the enhanced t1/2 was the blocking of the DPD recognition site. It is reported that Pd has the ability to quickly uncage N- and O-alkylated compounds in biological systems. Considering this unique ability of Pd, Adam et al., 2022 have developed a Pd-loaded microdevice that can be readily implanted in growing mouse tumors to facilitate ex vivo prodrug activation. This prodrug in the presence of a Pd-resin implant in the tumor rapidly releases 5-FU. In vivo efficacy studies in a xenograft colon cancer model served to prove such a prodrug’s ability to locally convert into active drugs by intratumorally inserted Pd implants. The developed compound 4 (modified 5-FU) manifested excellent bioavailability with a Cmax value of 83.6 mg/mL that is observed within 5 h (Tmax value).25 The Cmax value of unmodified 5-FU is 0.017 mg/mL at 0 h (Tmax value).53
Figure 9.

Novel prodrug of 5-FU (compound 4) in a lactim-type structure.
Fukushima and associates26 developed a novel antimetabolite, DFP-11207 (Figure 10), which constitutes 1-ethoxymethyl-5-fluorouracil (EM-FU; a precursor form of 5-FU), 5-chloro-2,4-dihydroxypyridine (CDHP; an inhibitor of 5-FU degrading enzyme), and citrazinic acid (CTA; an inhibitor of 5-FU phosphorylation). This prodrug rapidly hydrolyzes to produce EM-FU, CDHP, and CTA, where CDHP and CTA strongly inhibit DPD and orotate phosphoribosyl transferase activities, respectively, in a cell-free system using rat plasma and 20% homogenates of the rat liver and small intestine. The former metabolite EM-FU specifically hydrolyzes to 5-FU by various CYP subtypes, CYP1A2, 2A6, 2E1, and 3A4, to exert its anticancer activity. The inhibitory activity of DFP-11207 toward DPD and orotate phosphoribosyl transferase (OPRT) was comparable to that of CDHP and CTA alone. The pharmacokinetic study suggested that DFP-11207 resulted in lower Cmax and AUC values but longer Tmax and T1/2 values of 5-FU, respectively, than S-1, which suggested that DFP-11207 is superior to avoid the 5-FU-induced severe hematological toxicity including neutropenia, in particular, thrombocytopenia, on top of the protection of 5-FU-induced GI toxicities.26,27
Figure 10.
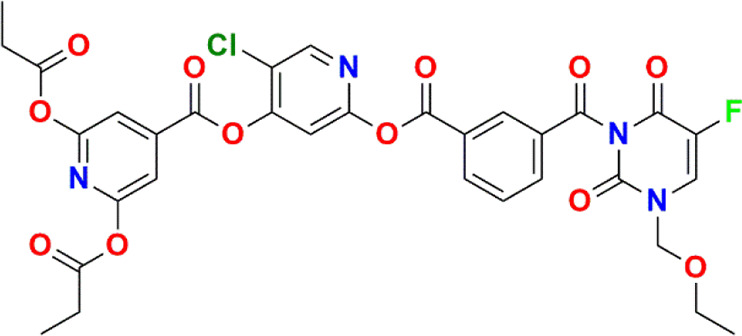
Novel prodrug of 5-FU in a lactim-type structure.
DPD Downregulators
Because sphingosine-1-phosphate receptor 2 (S1PR2) is closely associated with the expression of DPD in various cancer cells, inhibiting it may downregulate the DPD levels and finally reverse the 5-FU resistance. Considering the significance of inhibiting S1PR2, Zhang et al., 2020 synthesized a series of novel pyrrolidine–pyrazole derivatives as S1PR2 inhibitors by using JTE-013 as the reference drug and evaluated their tendency to overcome 5-FU resistance in colorectal cancer. As a resultant of this downregulation, a suppression in the degradation of intracellular 5-FU into an inactive FBAL was observed. To evaluate the potential of these derivatives, in vitro anti-5-FU resistance activities against human colorectal cell lines, including HCT116, HT-29, and NCM460, were analyzed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The structure activity relationship (SAR) of the designed series revealed that among all compounds, the molecule JTE-013 with an isopropyl group at the R1 position, a methyl group at the R2 position, and a chloro group at the R3 position conferred the most potent IC50(HCT116, 57.13 ± 0.25 μM; HT29, 59.66 ± 1.01 μM; NCM460, 68.56 ± 0.78 μM) and EC50 (HCT116, 13.28 ± 1.46 μM; HT29, 48.21 ± 2.24 μM; NCM460, 58.43 ± 0.65 μM) values. The favorable and unfavorable substituents that may improve or diminish the S1PR2 inhibitory activity are shown in Figure 11.29
Figure 11.

SAR of pyrrolidine–pyrazole-based derivatives as S1PR2 inhibitors to potently reverse 5-FU resistance by downregulating DPD expression in colorectal cancer.
In 2021, to overcome 5-FU resistance in HCT116DPD and SW620/5-FU cells, Luo and associates reported inhibitors of S1PR2 that can possibly downregulate the DPD overexpression. The designed compound selectively inhibits DPD expression by interfering with the JMJD3-H3K27me3-DPD pathway to reverse the 5-FU resistance in the aforementioned colorectal cancer cells. The combination of this compound and 5-FU significantly inhibited tumor growth and decreased tumor weight by 66.16%, which is greater than that of the control group that was given the combination of JTE-013 and 5-FU. This compound interacted with S1PR2 with a KD value of 1.32 × 10–8 M. The antiproliferative activity of 5-FU was found to be >1000 μM in SW620/5-FU and HCT116DPD cells, while it was observed to be 44 μM against NCM460 cell lines. Interestingly, the antiproliferative activity of this compound along with 5-FU was observed to be 0.21 ± 0.03, 0.18 ± 0.04, and 9.44 ± 0.13 μM in the case of SW620/5-FU, HCT116DPD, and NCM460, respectively. It is reported that possessing four chloro functional groups favored the activity in both resistance cells at a nanomolar level. There is a 95-fold improvement in potency against HCT116DPD cells (0.18 mM) and a 279-fold improvement against SW620/5-FU cells (0.21 mM) compared with JTE-013. More importantly, it showed obvious selectivity toward normal NCM460 cells (9.44 mM). Therefore, it is a potential molecule to reverse 5-FU resistance.28
In the recent past, Luo et al. have optimized the structure of the well-known S1PR2 inhibitor JTE-013 (displayed in Figure 12) to develop its analogues as DPD downregulators. The developed analogue displayed in Figure 12 is the most potent S1PR2 antagonist from the rest of the compounds with KD = 34.8 nM. This modified structure significantly inhibited the DPD expression by interfering with the transcription pattern which was evident from the Western blotting and immunohistochemistry analysis in both liver and tumor tissues. This ultimately reversed the 5-FU resistance in SW620/5-FU and HCT116DPD cells as the inhibition rates of 5-FU in both cell lines were observed to be increased from 23.97% to 65.29% and 27.23% to 60.81%. Thereby, substituting pyrazolo[3,4-b]pyridine with a 4-fluorophenyl group at the fourth position was observed to be favorable. The reported results by this research group suggest that the JTE-013 derivative could effectively reverse 5-FU resistance in both HCT116DPD and SW620/5-FU mouse xenograft models.54
Figure 12.

New JTE-013 derivative as S1PR2 inhibitors.
Zhao and associates in 2010 reported a natural product component wogonin (WOG) that can be given in combination to have a stronger antigastric cancer effect than WOG or 5-FU alone. This type of synergism was observed on MGC-803 cells due to decreased mRNA levels of DPD (5-FU inactivating enzyme) and inhibition of NF-κB nuclear translocation without altering the mRNA levels of the TS and OPRT. The preclinical study revealed that wogonin not only enhanced the anticancer effect of 5-FU at a low dose (i.p. 10 mg/kg/day) but also showed no toxicity nor had it influenced diet consumption in experimental animals.55
Réti et al., 2009 reported nonsteroidal antiinflammatory drugs (NSAIDs) which enhanced the cytotoxicity of 5-FU in high cyclooxygenase-2 (COX-2) expressing colorectal cancer cells, i.e., HCA-7 and xenografts (HT-29-C), through the modulation of DPD mRNA expression and/or enzyme activity. This research group evaluated the combination of NSAIDs and 5-FU in HT-29 cells treated with collagen IV (HT-29-C) that significantly upregulated COX-2 and DPD mRNA and also their enzyme activities. The study suggested that the NSAIDs and 5-FU in combination significantly downregulated DPD mRNA expression. They reported that the DPD enzymatic activities in HT-29 and HT-29-C are 9 ± 2.7 and 17.44 ± 2.87 pmol/min/mg protein, respectively, whereas the relative DPYD mRNA expression level in both cells is 8.09 ± 0.4 × 10–3 and 2328 ± 464 × 10–3, respectively. Upon treating the cell line HT-29-C with 5-FU alone, the mRNA expression level of DPD was found to be 0.9 ± 0.08, while the DPD activity was observed to be 14.6 ± 3.6 pmol/min/mg. Upon treating these cell lines with a combination of FU and indomethacin, the values for DPD mRNA and its enzymatic activity were observed to be 0.06 ± 0.04 and 5.1 ± 1.6 pmol/min/mg, respectively. This clearly suggests the utility of prescribing both drugs as an effective combination therapy. One of the selective COX-2 inhibitors, i.e., NS-398, in combination with 5-FU resulted in a DPD mRNA expression level of 0.005 ± 0.004, and its enzymatic activity was reported to be 5.9 ± 1.1 pmol/min/mg. Among indomethacin and NS-398, NS-398 can be suggested as a promising adjuvant therapy with 5-FU.31
As discussed in the section Molecular Cause of DPD Overexpression in Cancer, high SphK2 in colorectal tumors enhances S1P levels, which upregulates DPYD by inhibiting HDAC1 and enhancing the acylation of H3K56 in the nucleus. Under such conditions, exploring 5-FU in combination with SphK2-related inhibitors might be a potential therapeutic strategy to overcome 5-FU resistance in patients with colorectal tumors. In this context, a potential inhibitor of SphK2, i.e., SLR080811 (Figure 13), was examined to reverse 5-FU resistance by DPD inhibition. The results suggested that this amidine-based SphK1 inhibitor conferred 11-fold higher selectivity toward SphK2 by reducing the levels of S1P available for inhibiting HDAC1. This resulted in a decrease in the acylation of H3K56 and DPYD expression and favorably enhanced the 5-FU sensitivity.19,56
Figure 13.
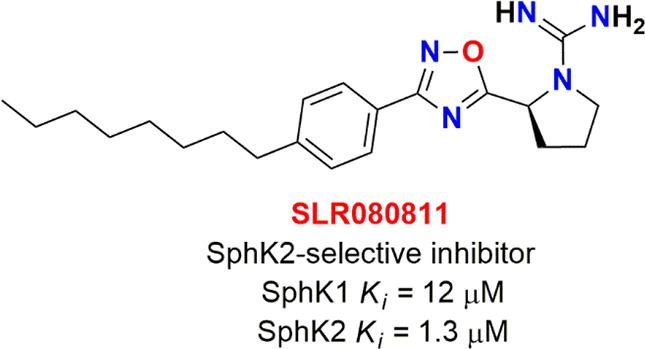
SLR080811; SphK2 selective inhibitor.
Riahi-Chebbi and associates reported the phenolic compound kaempferol to reverse 5-FU resistance in human resistant LS174 colon cancer cells. This phenolic compound was observed to significantly lower the DPD gene expression. Moreover, the cytotoxicity of 5-FU in combination with kaempferol was enhanced in the aforementioned resistant cancer cell lines. The IC50 value of 5-FU in resistant LS174-R cells is 706 μM, which is 26-fold higher than that of sensitive LS174 cells as this value was observed to be 26.9 μM. The results of this research group suggested that the inhibitory effect was enhanced to about 65% in combination with 60 μM of 5-FU.32
DPD Inhibitors as Adjuvants
Eniluracil
Eniluracil (compound 1; Figure 14; ethynyluracil, EU) is an irreversible inhibitor of DPD with an ethynyl substitution at the fifth position that has the capacity to increase the bioavailability of 5-FU to 100%. It inactivates DPD through covalent modification of an amino acid residue with 500-fold higher efficiency than any other tested 5-alkynyluracils. According to Porter et al., the inactivation mechanism of 5-FU follows a two-step process: (1) reversible binding with Km of 1.6 ± 0.2 μM and (2) irreversible inactivation of DPD following a first-order rate constant of 20 min–1 with t1/2 of 4 h.57 Within minutes of dosing, it can inhibit more than 99% of DPD activity where new DPD is resynthesized with a t1/2 of 63 h. Liver extracts prepared up to 6 h after an EU dose (2 mg/kg) inhibited >96% of DPD. It improves the elimination t1/2 and the area under the plasma concentration–time curve of 5-FU as evident from the in vivo pharmacokinetic study reported by Baccanari et al., 1993.58 Though eniluracil is itself not cytotoxic, in combination therapy it enhances the 5-FU cytotoxicity by fivefold in cancer cells with DPD overexpression. It also inhibits the formation of 5-FU catabolites that either may exert toxicity or may interfere with the antitumor activity of 5-FU. It is reported that more than 80% of 5-FU is converted to FBAL, a neurotoxic catabolite.59−61 The developed DPD inhibitor eniluracil has shown potent inhibitory activity with no significant neurotoxicity and cardiotoxicity. Thereby, this DPD inhibitor is a protective agent that may not only counter the 5-FU resistance but also reduce the toxicity induced by 5-FU.62,63 In humans, neutropenia is the main side effect caused by eniluracil as observed with a 5-day schedule and diarrhea with a 28-day schedule.64 On the basis of the negative feedback from colorectal tumor patients, the further development of eniluracil was discontinued. However, eniluracil has received Orphan Drug status from the FDA for the treatment of hepatocellular cancer in combination with fluoropyrimidines.5
Figure 14.

Reported DPD inhibitors.
TAS-114
TAS-114 (compound 2; Figure 14) is a first-in-class dual deoxyuridine 5′-triphosphate nucleotidohydrolase (dUTPase)/DPD inhibitor that has the potential to improve the therapeutic efficacy of 5-FU , as reported by Yano et al. It can moderately target capecitabine/5-FU inactivating enzyme DPD by a competitive inhibition mechanism. From the pharmacokinetic study, it was observed that TAS-114 elevated the plasma concentration of 5-FU in a dose-dependent manner. In vitro studies revealed moderate activity of TAS-114 compared with that of the potent DPD inhibitor gimeracil. In mice administered capecitabine alone, the plasma level of free 5-FU was observed to be 109 ng·h/mL at a dose of 240 mg/kg, whereas in mice administered a combination of capecitabine/TAS-114 (240/38 mg/kg) and capecitabine/gimeracil (240/0.18 mg/kg), the free 5-FU was observed to be 450 and 2247 ng·h/mL, respectively. Moreover, it dramatically decreased the free FdUMP and dUMP accumulation within the tumor in a dose-dependent manner despite the increase in 5-FU exposure. The levels of both of these metabolites in the tumor were observed to be significantly decreased by 28% and 35%, respectively.16
3-Cyano-2,6-dihydroxypyridine
3-Cyano-2,6-dihydroxypyridine (compound 3; Figure 14; CNDP) is a potent DPD inhibitor with an IC50 value of 4.4 nM which was first identified by Tatsumi and co-workers65 in 1993. This compound is reported to inhibit DPD with an inhibitory activity about 2000 times that of uracil. As per kinetic analyses with partially purified DPD from rat liver, the mechanism of inhibition is a mixed type with a Ki value of 1.51 nM. Coadministration of CNDP with 1-ethoxymethyl-5-fluorouracil (EM-FU) to rats with Yoshida sarcoma cell lines enhanced the free 5-FU levels in blood and tumor along with its antitumor activity.65 Emitefur (BOF-A2), a 5-FU derivative, was developed in 1989 by Fujii et al.42 BOF-A2, a prodrug, was first preregistered in Japan for colorectal cancer, and upon in vivo activation it releases EM-FU and CNDP. The antineoplastic effect of this prodrug was observed because of 5-FU. Following this, phase 1 of a clinical study was carried out in 2001, adding more to the cancer adverse events and pharmacokinetic section.12 The phase II trial conducted by Sugimachi and Maehara on patients with advanced gastric cancer revealed a promising response rate of 15.0%.66 Preclinical investigations of emitefur confirmed antitumor activity in several animal models xenografted with human gastric (H-81), colorectal (H-143), and breast cancers (H-31) and observed sustained 5-FU levels.67
UFT
UFT (compound 4; Figure 14) contains uracil and tegafur in a 4:1 ratio in which uracil directly competes with 5-FU for the uracil binding site on the DPD protein, allowing more 5-FU to be available for the activation pathways. The inhibitory effect of uracil on DPD is rapidly reversible. Therefore, more frequent dosing is required as the short-lived nature of the inhibition may allow more flexibility in the use of a standard dose of 5-FU.40,68 Some prospective randomized studies report the clinical effect of UFT administration for patients with NSCLC for instance: Patients with p-stage I–IIIB NSCLC who were previously administered UFT (400 mg/body/day) for 1 year after surgery reported a significantly better prognosis than those patients who had received surgical treatment alone.69 A subsequent study conducted by the same group confirmed the efficacy of adjuvant therapy, including UFT for p-stage I–II NSCLC.70 Both the postoperative disease-free interval and the 5-year survival rate of patients were significantly better for those who received UFT (8 mg/kg/day for 6 months) after adjuvant intravenous cisplatin (CDDP) and adriamycin (ADM) than those who had undergone surgery alone.71 These results suggest that administering UFT can ameliorate the postoperative survival of patients with NSCLC. Nakagawa and associates reported that patients bearing tumors with low DPD expression when administered UFT had a significantly better prognosis than those who did not receive adjuvant treatment.72 Yen and co-workers reported comparable long-term outcomes among patients receiving adjuvant UFT monotherapy after a D2 gastrectomy (pathological stage II–IIIB (except T1) gastric cancer).73 A clinical trial (UMIN000005594) was conducted on two groups, i.e., A and B, where group A received three doses of UFT (300 mg/m2 per day)/LV (75 mg per day) and showed comparable outcomes to group B who received two doses of UFT (300 mg/m2 per day)/LV (50 mg per day) with little change in adverse effects.74 All these reports support the UFT potential combination therapy for the quality treatment of resistance to cancer.
Teysuno (S-1)
Teysuno (compound 5; Figure 14, S-1) is approved in Japan and EU for the treatment of gastrointestinal cancers.75 S-1 contains tegafur, a prodrug of 5-FU, and two of the modulators of 5-FU and DPD inhibitors, i.e., gimeracil and oteracil, at a molar ratio 1:0.4:1. This combination is available as a formulated capsule with tegafur 15 mg, gimeracil 4.35 mg, and oteracil 11.8 mg. Each component of this formulation has an important utility, where tegafur is a prodrug which undergoes hydroxylation via hepatic cytochrome P450 (CYP) 2A6 enzymes to form 5-FU and later repetitively undergoes phosphorylation in the liver to its inactive and active metabolites. 5-FU is degraded mainly in the liver and in the bloodstream by DPD; to prevent this degradation or improve efficacy, gimeracil (a well-known DPD inhibitor) and oteracil (an OPRT inhibitor) are included. S-1 induces a low incidence of hand–foot syndrome (HFS) and GI toxicity and therefore marks the basis of switching from capecitabine to S-1 in patients being troubled by HFS or diarrhea, but S-1 is also used as an initial therapy.76,77 Various clinical studies have been carried out considering S-1 among Danish, Japanese, Koreans, and Asians. In phase I–II studies, the response rate to S-1 plus cisplatin was observed to be in the range of 53–74% among Japanese patients with advanced gastric cancer.78 Similar results in favor of S-1 were observed in Korean patients. Apart from these, S-1 also improved the response rate in patients with HER2-positive advanced gastric cancer about 68%. Moreover, the toxicities associated with this regimen were considered manageable.79 However, in a study in Europe, an S-1 dosage of 80 mg/m2/day was poorly tolerated because of nonhematological adverse events, particularly diarrhea.76,80 A phase III trial (NCT00150670) sponsored by Taiho Pharmaceutical Co., Ltd. considered 305 patients with advanced gastric cancer to examine if the overall survival was better in patients with advanced gastric cancer treated with S-1 plus cisplatin than with S-1 alone. It was observed that the overall survival was observed to be 13 months among patients treated with S-1 plus cisplatin and 11 months among patients treated with S-1 alone. Upon comparing the response rate, about 54% of the patients responded completely/partially to S-1 plus cisplatin in comparison to S-1 alone (only 31% of the patients could respond completely/partially). Other combinations are also being trialed, for instance, S-1 in combination with docetaxel among patients with stage III gastric cancer and S-1 with oxaliplatin in locally advanced adenocarcinoma of the gastric cancer.81,82 Recently, a rare case of reversible cardiac dysfunction associated with S-1 therapy has been studied by Oyakawa and associates in a 65-year old patient with gastric cancer. The clinicians reported S-1-related myocardial dysfunction in the patient which they found to be reversible with the administration of furosemide, human atrial natriuretic peptide, dobutamine, enalapril, spironolactone, and bisoprolol.83 S-1 is still a combination of interest for most of the researchers around the globe as evident from the frequency of published reports associated with it.
Formulation to Counter the DPD-Mediated Inactivation of 5-FU
Various efforts have been made by the research community to counter the 5-FU resistance not only by proposing new chemical entities (as DPD inhibitors, analogues for bypassing this resistance mechanism, or DPD downregulators) but also by developing formulations to improve the 5-FU bioavailability. These efforts made up to now are briefly discussed in the sections below.
EGF-Grafted Hollow Mesoporous Silica Nanoparticles
Drug-loaded nanocarriers (nanoparticles, liposomes, and dendrimers) with proper particle sizes can bypass the drug efflux pumps and counter multidrug resistance especially by ATP-binding cassette (ABC) transporters. In addition to these strategies, numerous research reports have also been published reporting these nanocarriers as effectively reversing the DME-mediated resistance/bioavailability issue. Chen et al., 2015 proposed hollow mesoporous silica nanoparticles (HMSNs) grafted with epidermal growth factor (EGF) (Figure 15A) as nanocarriers to deliver 5-FU to colorectal cancer cells to protect it from premature release and subsequent DPD-mediated inactivation. One more interesting fact about HMSNs is that they provide large surface areas which can be modified with various functional groups for target molecule grafting for exerting cancer cell specific targeting. The novelty in this reported study was that it countered 5-FU resistance via DPD-mediated inactivation by specifically internalizing 5-FU in the SW480/ADR cell lines with DPD and EGFR overexpression via receptor-mediated endocytosis, and it could escape from endolysosomes. This ultimately enhances the intracellular 5-FU accumulation for causing cell arrest at the S phase. Their research reports suggested that EGFR-HMSNs-5-FU exhibited much higher cytotoxicity on SW480/ADR cells, i.e., 1.05 ± 0.22 μg/mL, than HMSNs-5-FU and free 5-FU while the plain HMSNs did not show significant cytotoxicity (>50 and >5 μg/mL, respectively).43
Figure 15.

Formulations developed to enhance the 5-FU bioavailability.
Mucoadhesive Drug Delivery System (MDDS)
MDDS has gained a lot of attention among the pharmaceutical technologists as it has numerous advantages: (1) localization at specific sites, (2) elongation of the residence time at drug absorption sites, (3) escalation of contact with the mucosa, and (4) creation of a higher drug concentration gradient for facilitating the absorption by enterocytes. Mucoadhesive is facilitated mainly by thiolated polymers (thiomers) which generally form disulfide bridges with cysteine-rich subdomains of mucin via sulfhydryl/disulfide exchange reaction and/or an ordinary oxidation process. This drug delivery system displayed in Figure 15B has also been explored to enhance the bioavailability of 5-FU, which is mainly hampered due to DPD. Liu et al., 2018 reported a mucoadhesive amphiphilic 5- FU prodrug developed by conjugation with a maleimide group, linked by long lipophilic chains to lessen its degradation by DPD and to foster the transportation with enhanced lipophilicity and penetrability through the biomembrane. The proposed mucoadhesive possesses an maleimide end group of the ester linkage 5-FU prodrug named 1-alkylcarbonyloxymethyl 5-FU (EMC-5-FU) that chemically attaches to the sulfhydryl group of mucin glycoproteins. The EMC-5-FU unbound with mucin due to higher lipophilicity (40 times) diffuses into the intestinal tissue. The absorbed prodrug molecules will conjugate with in vivo circulating albumin to release 5-FU slowly and further ameliorate its stability in blood.44
Folic Acid-Decorated and PEGylated PLGA Nanoparticles
In 2016, Hammadi and co-workers developed the folic acid (FOL)-decorated and PEGylated poly(d,l-lactide-co-glycolide; PLGA) nanoparticles (NPs) as shown in Figure 15C to specifically deliver 5-FU to colon and breast cancers. In vitro cytotoxicity studies in folate-overexpressed HT-29 colon cancer cells and MCF-7 breast cancer cells demonstrated that the IC50 of 5-FU-loaded FOL–PEG–PLGA NPs was approximately fourfold better than that of 5-FU-loaded PLGA NPs. The formulated FOL–PEG–PLGA NPs have manifested various advantages including optimized anticancer efficiency, minimal toxicity, increased biological half-life, and countered drug resistance issues.45
Methacrylic-Based Nanogels
Ashwanikumar and co-workers implemented a microemulsion polymerization technique to prepare copolymers of the methacrylic acid and 2-ethylhexyl acrylate (MAEHA; Figure 15D) nanogel system. This nanogel system is pH sensitive and has the ability to specifically target the colon with 5-FU. These biocompatible nanogels completely accumulate in the tumor microenvironment and bring about cell death by an apoptosis mechanism. This drug delivery system can bypass the high acidity of the stomach (pH 1.5–2.5) and slightly basic pH of the duodenum (pH 6) to reach the colon at pH 7.6–7.8. The unique pH-sensitive swelling behavior of the utilized polymer in basic pH is of great interest for preparing nanogels. MTT assay suggested that 5-FU-entrapped nanogels displayed better cytotoxicity (44.1% inhibition at 100 μM concentration) than that of free 5-FU against HCT-116 cell lines upon 24 h exposure where 48 h MTT data indicated that the inhibition was 56.27% for nanogels with 5-FU, twice the value of the free drug. After 72 h of incubation, the nanogels showed the highest inhibition of 64.75% for 100 μM. The research group suggested that nanogels could be successfully used as an efficient vector for pH-sensitive and controlled delivery of drugs specifically targeted to the colon.46
Octadecylamine–Retinoic Acid Conjugate
Varshosaz et al., 2012 developed nanostructured lipid carriers (NLCs; Figure 15E) in colorectal carcinoma to enhance the cytotoxicity of 5-FU and reduce the side effects by targeting low-density lipoprotein (LDL) receptors. A fatty acyl amide derivative of retinoic acid was synthesized by conjugating 5-FU with octadecyl amine to improve the intracellular accumulation and loading capacity of 5-FU in NLCs. The results of their research work suggested that free 5-FU conferred an IC50 value of 7.6 mM. At a similar concentration, the cytotoxicity of 5-FU loaded in NLCs constituting the retinoic acid conjugate was nearly twofold that of NLCs that were loaded only with 5-FU and greater than fivefold that of free 5-FU. The loaded retinoic acid conjugate of cholesterol-releasing NLCs can target LDL receptors for HT29 cells and appears to be promising in minimizing the 5-FU dose in colorectal cancer.47
5-FU-Loaded pH-Sensitive PEGylated Liposomal Nanoparticles
Udofot and co-workers formulated 5-FU-loaded pH-sensitive PEGylated liposomal NPs as displayed in Figure 15F to improve the pharmacokinetic and biodistribution profile of 5-FU in HCT-116 tumor-bearing mice. This improved profile of pHLNP-5-FU is indicated from prolonged blood circulation time, i.e., increased t1/2, higher AUC, and increased 5-FU tumor accumulation. In addition to the improved PK and biodistribution profiles, the pHLNP-5-FU manifested a better therapeutic index of 5-FU and also showed stability at pH 7.4. Overall, their study suggested that the pHLNp delivery system for 5-FU could not only improve the therapeutic index of 5-FU but also reduce the adverse side effects of 5-FU.48
Lipofufol (Triple Stealth Liposomal Formulation)
Fanciullino et al., 2013 attempted to address the drug resistance and toxicity-related issues with 5-FU by developing a triple liposomal formulation (5-FU in combination with 2′-deoxyinosine and folinic acid; Figure 15G). This triple stealth liposomal formulation limited liver uptake to consequently improve the toxicity issues, making DPD activity less crucial for 5-FU disposition. Moreover, it restored the in vitro sensitivity of 5-FU to highly resistant human colorectal cell lines such as SW620 and LS174t models. In LS174t xenografted mice, the survival time was observed to be 55%. Thereby, this is a safer and powerful alternative to standard 5-FU in the experimental treatment of colorectal cancer, both in vitro and in vivo. This formulation also confers improved pharmacokinetic and biodistribution profiles.49
Pluronic-Coated Biogenic Gold Nanoparticles
Mahdi and co-workers prepared 5-FU-loaded biogenic gold NPs with pluronic-based coating (Figure 15H) to improve the high drug-loading capacity, sustained delivery, hemocompatibility, improved efficacy, and enhanced permeation profiles. This formulation demonstrated a 4.6-fold higher permeability across the intestine over 24 min owing to impregnated hydrophilic 5-FU within the pluronic coating over the gold core. These merits make them a promising formulation for effective colon cancer treatment with minimal side effects and improved bioavailability.50
Exosomes for Targeted Codelivery of miR-21 Inhibitor and 5-FU
As discussed in section Molecular Cause of DPD Overexpression in Cancer , the knockdown of miR-21 increased the sensitivity to 5-FU chemotherapy toward HT-29/5-FU. Downregulation of miR-21 can moreover be advantageous not only in improving the sensitivity of 5-FU but also in inducing cell cycle arrest, increased apoptosis, reduced tumor proliferation, and rescued hMSH2 and PTEN expressions, regulatory targets of miR-21. Thereby, considering the role of miR-21 in resistance to 5-FU, Liang et al. have engineered exosomes to simultaneously deliver 5-FU and miR-21 inhibitor oligonucleotide (miR-21i) to the 5-FU-resistant colorectal cancer cell line HCT-1165FR (Figure 16). This codelivery system efficiently facilitated cellular uptake and significantly downregulated miR-21 expression in the aforementioned cell lines. More excited, this combinational delivery of miR-21i and 5-FU with the engineered exosomes significantly reversed the drug resistance and also enhanced its cytotoxicity in 5-FU-resistant colon cancer cells, compared with the single treatment with either miR-21i or 5-FU.51
Figure 16.
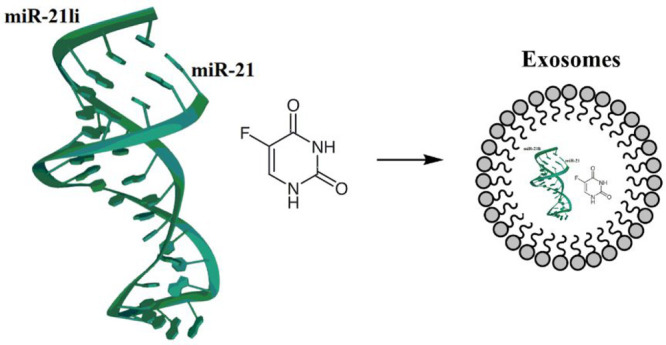
Exosomes to codeliver miR-21 inhibitor and 5-FU.
Conclusion
5-FU, a first line chemotherapeutic agent, requires prior activation via the physiologic pyrimidine and salvage pathways to exert anticancer effects by misincorporating into DNA/RNA or inhibiting TS. However, overexpression of DPD in some cancer cell lines is a major cause of inactivation of 5-FU resistance, which ultimately compromises its anticancer activity. Inactivation of 5-FU is carried out by DPD by forming a reduced inactive metabolite, i.e., 5,6-dihydro-5-FU, leaving only a scant amount of it bioavailable for cancer treatment. The molecular cause at genetic and proteomic levels has been briefly compiled in this Review, which can help molecular biologists and medicinal chemists to propose better chemotherapeutics or agents that may ameliorate the resistance issue. This Review sheds light on various strategies adopted by research communities for overcoming this issue, including 5-FU analogues (bypass the DPD-mediated inactivation), DPD downregulators (regulate the DPD expression levels in tumors), inhibitors (as promising adjuvants), formulation development loaded with 5-FU (liposomes, nanoparticles, nanogels etc.). Overall, the information provided in this Review will assist a number of researchers in developing basic knowledge on DPD-mediated resistance mechanisms at the molecular level. Moreover, the discussed strategies provide an avenue to address the resistance issue.
Glossary
Abbreviations
- 5-FU
5-fluorouracil
- DPD
dihydropyrimidine dehydrogenase
- miRNAs
microRNAs
- SphK2
sphingosine kinase 2
- TS
thymidylate synthase
- dTMP
pyrimidine thymidylate
- DNA
deoxyribonucleic acid
- fdUTP
fluorodeoxyuridine triphosphate
- RNA
ribonucleic acid
- NADPH
nicotinamide adenine dinucleotide phosphate
- ESCC
esophageal squamous carcinoma cells
- DHFU
5,6-dihydro-5-fluorouracil
- FUPA
fluoro-ureidopropionate
- FBAL
fluoro-β-alanine
- S1P
sphingosine-1-phosphate
- HDAC1
histone deacetylase 1
- H3K56
histone H3 lysine 56 acetylation
- TP
thymidine phosphorylase
- EGFR
epidermal growth factor receptor
- NSCLC
non-small cell lung cancer
- dThdPase
thymidine phosphorylase
- TK
thymidine kinase
- TKI
tyrosine kinase inhibitor
- UFT
uracil-tegafur
- EC
electrostatic complementarity
- OPRT
orotate phosphoribosyl transferase
- EM-FU
1-ethoxymethyl-5-fluorouracil
- CDHP
5-chloro-2,4-dihydroxypyridine
- CTA
citrazinic acid
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SAR
structure activity relationship
- S1PR2
sphingosine-1-phosphate receptor 2
- WOG
wogonin
- NSAIDs
nonsteroidal anti-inflammatory drugs
- COX-2
cyclooxygenase-2
- EU
ethynyluracil
- dUTPase
deoxyuridine 5′-triphosphate nucleotidohydrolase
- CNDP
3-cyano-2,6-dihydroxypyridine
- ABC
ATP-binding cassette
- HMSNs
hollow mesoporous silica nanoparticles
- MDDS
mucoadhesive drug delivery system
- FOL
folic acid
- MAEHA
methacrylic acid and 2-ethylhexyl acrylate
- NLCs
nanostructured lipid carriers
- LDL
low-density lipoprotein
This work was financially supported by the Indian Council of Medical Research (ICMR), New Delhi, under sanction no. ISRM/11(09)/2019 by providing a Senior Research Fellowship (SRF) to H.V.
The authors declare no competing financial interest.
References
- Heidelberger C.; Chaudhuri N.; Danneberg P.; Mooren D.; Griesbach L.; Duschinsky R.; Schnitzer R.; Pleven E.; Scheiner J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179 (4561), 663–666. 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- Duschinsky R.; Pleven E.; Heidelberger C. The synthesis of 5-fluoropyrimidines. J. Am. Chem. Soc. 1957, 79 (16), 4559–4560. 10.1021/ja01573a087. [DOI] [Google Scholar]
- Chamorey E.; Francois E.; Etienne M.-C.; Ferrero J.-M.; Peyrade F.; Barranger E.; Bozec A.; Largillier R.; Cassuto O.; Viotti J.; et al. DPD status and fluoropyrimidines-based treatment: high activity matters too. BMC Cancer 2020, 20 (1), 1–9. 10.1186/s12885-020-06907-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley D. B.; Harkin D. P.; Johnston P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003, 3 (5), 330–338. 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Kindler H. L.; Schilsky R. L. Eniluracil: an irreversible inhibitor of dihydropyrimidine dehydrogenase. Expert Opin. Investig. Drugs 2000, 9 (7), 1635–1649. 10.1517/13543784.9.7.1635. [DOI] [PubMed] [Google Scholar]
- Iyer L.; Ratain M. J. 5-Fluorouracil pharmacokinetics: causes for variability and strategies for modulation in cancer chemotherapy. Cancer Investig. 1999, 17 (7), 494–506. 10.3109/07357909909032859. [DOI] [PubMed] [Google Scholar]
- Raju B.; Choudhary S.; Narendra G.; Verma H.; Silakari O. Molecular modeling approaches to address drug-metabolizing enzymes (DMEs) mediated chemoresistance: a review. Drug Metab. Rev. 2021, 53 (1), 45–75. 10.1080/03602532.2021.1874406. [DOI] [PubMed] [Google Scholar]
- Karthika C.; Sureshkumar R.; Zehravi M.; Akter R.; Ali F.; Ramproshad S.; Mondal B.; Tagde P.; Ahmed Z.; Khan F. S.; et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life 2022, 12 (6), 897. 10.3390/life12060897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi O.; Ohashi S.; Nakai Y.; Nakagawa S.; Matsuoka K.; Kobunai T.; Takechi T.; Amanuma Y.; Yoshioka M.; Ida T. Novel 5-fluorouracil-resistant human esophageal squamous cell carcinoma cells with dihydropyrimidine dehydrogenase overexpression. Am. J. Cancer Res. 2015, 5 (8), 2431. [PMC free article] [PubMed] [Google Scholar]
- Takechi T.; Fujioka A.; Matsushima E.; Fukushima M. Enhancement of the antitumour activity of 5-fluorouracil (5-FU) by inhibiting dihydropyrimidine dehydrogenase activity (DPD) using 5-chloro-2, 4-dihydroxypyridine (CDHP) in human tumour cells. Eur. J. Cancer 2002, 38 (9), 1271–1277. 10.1016/S0959-8049(02)00048-5. [DOI] [PubMed] [Google Scholar]
- Van Kuilenburg A. B. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer 2004, 40 (7), 939–950. 10.1016/j.ejca.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Verma H.; Singh Bahia M.; Choudhary S.; Kumar Singh P.; Silakari O. Drug metabolizing enzymes-associated chemo resistance and strategies to overcome it. Drug Metab. Rev. 2019, 51 (2), 196–223. 10.1080/03602532.2019.1632886. [DOI] [PubMed] [Google Scholar]
- Elander N.; Aughton K.; Ghaneh P.; Neoptolemos J.; Palmer D.; Cox T.; Campbell F.; Costello E.; Halloran C.; Mackey J.; et al. Expression of dihydropyrimidine dehydrogenase (DPD) and hENT1 predicts survival in pancreatic cancer. Br. J. Cancer 2018, 118 (7), 947–954. 10.1038/s41416-018-0004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori K.; Kohchi Y.; Oikawa N.; Suda H.; Ura M.; Ishikawa T.; Miwa M.; Endoh M.; Eda H.; Tanimura H.; et al. Design and synthesis of the tumor-activated prodrug of dihydropyrimidine dehydrogenase (DPD) inhibitor, RO0094889 for combination therapy with capecitabine. Bioorg. Med. Chem. Lett. 2003, 13 (5), 867–872. 10.1016/S0960-894X(02)01082-X. [DOI] [PubMed] [Google Scholar]
- Vodenkova S.; Buchler T.; Cervena K.; Veskrnova V.; Vodicka P.; Vymetalkova V. 5-Fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. 10.1016/j.pharmthera.2019.107447. [DOI] [PubMed] [Google Scholar]
- Yano W.; Yokogawa T.; Wakasa T.; Yamamura K.; Fujioka A.; Yoshisue K.; Matsushima E.; Miyahara S.; Miyakoshi H.; Taguchi J.; et al. TAS-114, a first-in-class dual dUTPase/DPD inhibitor, demonstrates potential to improve therapeutic efficacy of fluoropyrimidine-based chemotherapy. Mol. Cancer Ther. 2018, 17 (8), 1683–1693. 10.1158/1535-7163.MCT-17-0911. [DOI] [PubMed] [Google Scholar]
- Hirota T.; Date Y.; Nishibatake Y.; Takane H.; Fukuoka Y.; Taniguchi Y.; Burioka N.; Shimizu E.; Nakamura H.; Otsubo K.; et al. Dihydropyrimidine dehydrogenase (DPD) expression is negatively regulated by certain microRNAs in human lung tissues. Lung cancer 2012, 77 (1), 16–23. 10.1016/j.lungcan.2011.12.018. [DOI] [PubMed] [Google Scholar]
- Offer S. M.; Butterfield G. L.; Jerde C. R.; Fossum C. C.; Wegner N. J.; Diasio R. B. microRNAs miR-27a and miR-27b Directly Regulate Liver Dihydropyrimidine Dehydrogenase Expression through Two Conserved Binding SitesRegulation of DPD by miR-27a and miR-27b. Mol. Cancer Ther. 2014, 13 (3), 742–751. 10.1158/1535-7163.MCT-13-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-H.; Shi W.-N.; Wu S.-H.; Miao R.-R.; Sun S.-Y.; Luo D.-D.; Wan S.-B.; Guo Z.-K.; Wang W.-Y.; Yu X.-F.; et al. SphK2 confers 5-fluorouracil resistance to colorectal cancer via upregulating H3K56ac-mediated DPD expression. Oncogene 2020, 39 (29), 5214–5227. 10.1038/s41388-020-1352-y. [DOI] [PubMed] [Google Scholar]
- Deng J.; Lei W.; Fu J.-C.; Zhang L.; Li J.-H.; Xiong J.-P. Targeting miR-21 enhances the sensitivity of human colon cancer HT-29 cells to chemoradiotherapy in vitro. Biochem. Bioph. Res. Co. 2014, 443 (3), 789–795. 10.1016/j.bbrc.2013.11.064. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T.; Tominaga T.; Mochinaga K.; Yamasaki N.; Matsumoto K.; Miyazaki T.; Hatachi G.; Kitajima Y.; Nagayasu T. Reverse translational research for identifying the correlation between EGFR mutation status and DPD protein expression in lung adenocarcinoma. Cancer Res. 2016, 76 (14_Supplement), 5028–5028. 10.1158/1538-7445.AM2016-5028. [DOI] [Google Scholar]
- Tozer T.; Heale K.; Manto Chagas C.; de Barros A. L. B.; Alisaraie L. Interdomain twists of human thymidine phosphorylase and its active–inactive conformations: Binding of 5-FU and its analogues to human thymidine phosphorylase versus dihydropyrimidine dehydrogenase. Chem. Biol. Drug Des. 2019, 94 (5), 1956–1972. 10.1111/cbdd.13596. [DOI] [PubMed] [Google Scholar]
- Ghazaly E. A.; Slusarczyk M.; McGuigan C.; Harrison D.; Blagden S. P. Abstract B46: NUC-3373: A novel pyrimidine nucleotide analogue that overcomes key cancer drug resistance limiting patient survival. Mol. Cancer Ther. 2015, 14 (12_Supplement_2), B46–B46. 10.1158/1535-7163.TARG-15-B46. [DOI] [Google Scholar]
- Kazmi F.; Ciombor K.; Graham J.; Coveler A.; Myers M.; Berlin J.; Harrison D.; Blagden S.; Evans T. J. Abstract CT140: NUC-3373, a targeted inhibitor of thymidylate synthase, in patients with advanced colorectal cancer. Cancer Res. 2021, 81 (13_Supplement), CT140–CT140. 10.1158/1538-7445.AM2021-CT140. [DOI] [Google Scholar]
- Adam C.; Bray T. L.; Pérez-López A. M.; Tan E. H.; Rubio-Ruiz B.; Baillache D. J.; Houston D. R.; Salji M. J.; Leung H. Y.; Unciti-Broceta A. A 5-FU precursor designed to evade anabolic and catabolic drug pathways and activated by Pd chemistry in vitro and in vivo. J. Med. Chem. 2022, 65 (1), 552–561. 10.1021/acs.jmedchem.1c01733. [DOI] [PubMed] [Google Scholar]
- Fukushima M.; Iizuka K.; Jin C.; Zhang C.; Hong M.; Eshima K. Development of new promising antimetabolite, DFP-11207 with self-controlled toxicity in rodents. Drug Des. Devel. Ther. 2017, 11, 1693. 10.2147/DDDT.S128420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajani J. A.; Javle M.; Eng C.; Fogelman D.; Smith J.; Anderson B.; Zhang C.; Iizuka K. Phase I study of DFP-11207, a novel oral fluoropyrimidine with reasonable AUC and low Cmax and improved tolerability, in patients with solid tumors. Invest. New Drugs 2020, 38 (6), 1763–1773. 10.1007/s10637-020-00939-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D.; Zhang Y.; Yang S.; Tian X.; Lv Y.; Guo Z.; Liu X.; Han G.; Liu S.; Wang W.; et al. Design, synthesis and biological evaluation of sphingosine-1-phosphate receptor 2 antagonists as potent 5-FU-resistance reversal agents for the treatment of colorectal cancer. Eur. J. Med. Chem. 2021, 225, 113775. 10.1016/j.ejmech.2021.113775. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-H.; Luo D.-D.; Wan S.-B.; Qu X.-J. S1PR2 inhibitors potently reverse 5-FU resistance by downregulating DPD expression in colorectal cancer. Pharmacol. Res. 2020, 155, 104717. 10.1016/j.phrs.2020.104717. [DOI] [PubMed] [Google Scholar]
- Qi Q.; Peng J.; Liu W.; You Q.; Yang Y.; Lu N.; Wang G.; Guo Q. Toxicological studies of wogonin in experimental animals. Phytother. Res: An International Journal Devoted to Pharmacological and Toxicological Evaluation of Natural Product Derivatives 2009, 23 (3), 417–422. 10.1002/ptr.2645. [DOI] [PubMed] [Google Scholar]
- Réti A.; Pap É.; Adleff V.; Jeney A.; Kralovánszky J.; Budai B. Enhanced 5-fluorouracil cytotoxicity in high cyclooxygenase-2 expressing colorectal cancer cells and xenografts induced by non-steroidal anti-inflammatory drugs via downregulation of dihydropyrimidine dehydrogenase. Cancer Chemother. Pharmacol. 2010, 66 (2), 219–227. 10.1007/s00280-009-1149-8. [DOI] [PubMed] [Google Scholar]
- Riahi-Chebbi I.; Souid S.; Othman H.; Haoues M.; Karoui H.; Morel A.; Srairi-Abid N.; Essafi M.; Essafi-Benkhadir K. The Phenolic compound Kaempferol overcomes 5-fluorouracil resistance in human resistant LS174 colon cancer cells. Sci. Rep. 2019, 9 (1), 1–20. 10.1038/s41598-018-36808-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Wei L.; Lin S.; Chen Y.; Lin J.; Peng J. Synergistic effect of kaempferol and 5-fluorouracil on the growth of colorectal cancer cells by regulating the PI3K/Akt signaling pathway. Mol. Med. Rep. 2019, 20 (1), 728–734. 10.3892/m.mr.2019.10296. [DOI] [PubMed] [Google Scholar]
- Meropol N. J.; Niedzwiecki D.; Hollis D.; Schilsky R. L.; Mayer R. J. Phase II study of oral eniluracil, 5-fluorouracil, and leucovorin in patients with advanced colorectal carcinoma. Cancer 2001, 91 (7), 1256–1263. . [DOI] [PubMed] [Google Scholar]
- Schilsky R. L.; Hohneker J.; Ratain M. J.; Janisch L.; Smetzer L.; Lucas V. S.; Khor S. P.; Diasio R.; Von Hoff D. D.; Burris H. 3rd Phase I clinical and pharmacologic study of eniluracil plus fluorouracil in patients with advanced cancer. J. Clin. Oncol. 1998, 16 (4), 1450–1457. 10.1200/JCO.1998.16.4.1450. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Hayashi H.; Planchard D.; Morán T.; Gregorc V.; Dowell J.; Sakai H.; Yoh K.; Nishio M.; Cortot A. B.; et al. A randomized, phase 2 study of deoxyuridine triphosphatase inhibitor, TAS-114, in combination with S-1 versus S-1 alone in patients with advanced non-small-cell lung cancer. Invest. New Drugs 2020, 38 (5), 1588–1597. 10.1007/s10637-020-00930-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T.; Yoh K.; Shitara K.; Takahashi H.; Ueno M.; Kobayashi S.; Morimoto M.; Okusaka T.; Ueno H.; Morizane C.; et al. First-in-human phase 1 study of novel dUTPase inhibitor TAS-114 in combination with S-1 in Japanese patients with advanced solid tumors. Invest. New Drugs 2019, 37 (3), 507–518. 10.1007/s10637-018-0697-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaka T.; Shimamato Y.; Ohshimo H.; Yamaguchi M.; Kato T.; Yonekura K.; Fukushima M. Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anti-cancer drugs 1996, 7 (5), 548–557. 10.1097/00001813-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Ribas A.; Gallardo E.; Vera R.; Casado S.; Vidal R.; Bellmunt J. Life-threatening toxicity of oral tegafur-uracil (UFT) plus leucovorin. Clin. Oncol. 1997, 9 (4), 269–270. 10.1016/S0936-6555(97)80017-9. [DOI] [PubMed] [Google Scholar]
- Fujii S.; Ikenaka K.; Fukushima M.; Shirasaka T. Effect of uracil and its derivatives on antitumor activity of 5-fluorouracil and 1-(2-tetrahydrofuryl)-5-fluorouracil. GANN Jpn. J. Cancer Res. 1978, 69 (6), 763–772. 10.20772/cancersci1959.69.6_763. [DOI] [PubMed] [Google Scholar]
- Nakai Y.; Furuse K.; Ohta M.; Yamaguchi Y.; Fujii M.; Asakawa M.; Fukuoka M.; Yoshida K.; Niitani H. Efficacy of a New 5-Fluorouracil Derivative, Bof-A2, in Advanced Non-Small Cell Lung Cancer a Multi-Center Phase II Study. Acta Oncol. 1994, 33 (5), 523–526. 10.3109/02841869409083929. [DOI] [PubMed] [Google Scholar]
- Fujii S.; Fukushima M.; Shimamoto Y.; Ohshimo H.; Imaoka T.; Shirasaka T. Antitumor activity of BOF-A2, a new 5-fluorouracil derivative. Jpn. J. Cancer Res. 1989, 80 (2), 173–181. 10.1111/j.1349-7006.1989.tb02286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; She X.; Wang T.; He L.; Shigdar S.; Duan W.; Kong L. Overcoming acquired drug resistance in colorectal cancer cells by targeted delivery of 5-FU with EGF grafted hollow mesoporous silica nanoparticles. Nanoscale 2015, 7 (33), 14080–14092. 10.1039/C5NR03527A. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhao D.; Sun M.; Wei W.; Wang Y.; Zhou J.; Zhang R.; Wang J.; Zhang H.; He Z.; et al. Covalently mucoadhesive amphiphilic prodrug of 5-fluorouracil for enhanced permeation and improved oral absorption. Drug Delivery Transl. Res. 2018, 8 (3), 645–656. 10.1007/s13346-018-0502-z. [DOI] [PubMed] [Google Scholar]
- El-Hammadi M. M.; Delgado Á. V.; Melguizo C.; Prados J. C.; Arias J. L. Folic acid-decorated and PEGylated PLGA nanoparticles for improving the antitumour activity of 5-fluorouracil. Int. J. Pharm. 2017, 516 (1–2), 61–70. 10.1016/j.ijpharm.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Ashwanikumar N.; Kumar N. A.; Nair S. A.; Kumar G. V. Methacrylic-based nanogels for the pH-sensitive delivery of 5-fluorouracil in the colon. Int. J. Nanomed. 2012, 7, 5769. 10.2147/IJN.S31201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshosaz J.; Hassanzadeh F.; Sadeghi H.; Andalib S. Synthesis of octadecylamine-retinoic acid conjugate for enhanced cytotoxic effects of 5-FU using LDL targeted nanostructured lipid carriers. Eur. J. Med. Chem. 2012, 54, 429–438. 10.1016/j.ejmech.2012.05.024. [DOI] [PubMed] [Google Scholar]
- Udofot O.; Affram K.; Smith T.; Tshabe B.; Krishnan S.; Sachdeva M.; Agyare E.. Pharmacokinetic, biodistribution and therapeutic efficacy of 5-fluorouracil-loaded pH-sensitive PEGylated liposomal nanoparticles in HCT-116 tumor bearing mouse. J. Nat. Sci. 2016, 2 ( (1), ).e171. [PMC free article] [PubMed] [Google Scholar]
- Fanciullino R.; Mollard S.; Giacometti S.; Berda-Haddad Y.; Chefrour M.; Aubert C.; Iliadis A.; Ciccolini J. In vitro and in vivo evaluation of lipofufol, a new triple stealth liposomal formulation of modulated 5-fu: impact on efficacy and toxicity. Pharm. Res. 2013, 30 (5), 1281–1290. 10.1007/s11095-012-0967-2. [DOI] [PubMed] [Google Scholar]
- Mahdi W. A.; Hussain A.; Ramzan M.; Faruk A.; Bukhari S. I.; Dev A. Pluronic-coated biogenic gold nanoparticles for colon delivery of 5-fluorouracil: In vitro and ex vivo studies. AAPS Pharm.Sci.Tech 2021, 22 (2), 1–18. 10.1208/s12249-021-01954-7. [DOI] [PubMed] [Google Scholar]
- Liang G.; Zhu Y.; Ali D. J.; Tian T.; Xu H.; Si K.; Sun B.; Chen B.; Xiao Z. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J. Nanobiotechnol. 2020, 18 (1), 1–15. 10.1186/s12951-019-0563-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M.; Miwa M.; Eda H.; Ura M.; Tanimura H.; Ishikawa T.; Miyazaki-Nose T.; Hattori K.; Shimma N.; Yamada-Okabe H.; et al. Augmentation of the antitumor activity of capecitabine by a tumor selective dihydropyrimidine dehydrogenase inhibitor, RO0094889. Int. J. Cancer. 2003, 106 (5), 799–805. 10.1002/ijc.11276. [DOI] [PubMed] [Google Scholar]
- Li S.; Wang A.; Jiang W.; Guan Z. Pharmacokinetic characteristics and anticancer effects of 5-fluorouracil loaded nanoparticles. BMC cancer 2008, 8 (1), 1–9. 10.1186/1471-2407-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D.; Guo Z.; Zhao X.; Wu L.; Liu X.; Zhang Y.; Zhang Y.; Deng Z.; Qu X.; Cui S.; et al. Novel 5-fluorouracil sensitizers for colorectal cancer therapy: Design and synthesis of S1P receptor 2 (S1PR2) antagonists. Eur. J. Med. Chem. 2022, 227, 113923. 10.1016/j.ejmech.2021.113923. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Wang J.; Zou M.-J.; Hu R.; Zhao L.; Qiang L.; Rong J.-J.; You Q.-D.; Guo Q.-L. Wogonin potentiates the antitumor effects of low dose 5-fluorouracil against gastric cancer through induction of apoptosis by down-regulation of NF-kappaB and regulation of its metabolism. Toxicol. Lett. 2010, 197 (3), 201–210. 10.1016/j.toxlet.2010.05.019. [DOI] [PubMed] [Google Scholar]
- Ding T.; Zhi Y.; Xie W.; Yao Q.; Liu B. Rational design of SphK inhibitors using crystal structures aided by computer. Eur. J. Med. Chem. 2021, 213, 113164. 10.1016/j.ejmech.2021.113164. [DOI] [PubMed] [Google Scholar]
- Porter D.; Chestnut W.; Merrill B.; Spector T. Mechanism-based inactivation of dihydropyrimidine dehydrogenase by 5-ethynyluracil. J. Biol. Chem. 1992, 267 (8), 5236–5242. 10.1016/S0021-9258(18)42757-3. [DOI] [PubMed] [Google Scholar]
- Baccanari D. P.; Davis S. T.; Knick V. C.; Spector T. 5-Ethynyluracil (776C85): a potent modulator of the pharmacokinetics and antitumor efficacy of 5-fluorouracil. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (23), 11064–11068. 10.1073/pnas.90.23.11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa Y.; Funakoshi T.; Horimatsu T.; Miyamoto S. i.; Matsubara T.; Yanagita M.; Nakagawa S.; Yonezawa A.; Matsubara K.; Muto M. Accumulation of alpha-fluoro-beta-alanine and fluoro mono acetate in a patient with 5-fluorouracil-associated hyperammonemia. Cancer Chemother. Pharmacol. 2017, 79 (3), 629–633. 10.1007/s00280-017-3249-1. [DOI] [PubMed] [Google Scholar]
- Omura K. Clinical implications of dihydropyrimidine dehydrogenase (DPD) activity in 5-FU-based chemotherapy: mutations in the DPD gene, and DPD inhibitory fluoropyrimidines. Int. J. Clin. Oncol. 2003, 8 (3), 132–138. 10.1007/s10147-003-0330-z. [DOI] [PubMed] [Google Scholar]
- Kang J.; Kim A. H.; Jeon I.; Oh J.; Jang I. J.; Lee S.; Cho J. Y. Endogenous metabolic markers for predicting the activity of dihydropyrimidine dehydrogenase. Clin. Transl. Sci. 2022, 15, 1104. 10.1111/cts.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S. T.; Joyner S. S.; Baccanari D. P.; Spector T. 5-Ethynyluracil (776C85): protection from 5-fluorouracil-induced neurotoxicity in dogs. Biochem. Pharmacol. 1994, 48 (2), 233–236. 10.1016/0006-2952(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Arellano M.; Malet-Martino M.; Martino R.; Spector T. 5-Ethynyluracil (GW776): effects on the formation of the toxic catabolites of 5-fluorouracil, fluoroacetate and fluorohydroxypropionic acid in the isolated perfused rat liver model. Br. J. Cancer 1997, 76 (9), 1170–1180. 10.1038/bjc.1997.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont E. B.; Schilsky R. L. The oral fluoropyrimidines in cancer chemotherapy. Clin. Cancer Res. 1999, 5 (9), 2289–2296. [PubMed] [Google Scholar]
- Tatsumi K.; Yamauchi T.; Kiyono K.; Kishi K.; Yanagihara Y.; Imaoka T.; Kawaguchi T.; Kubo M. 3-Cyano-2, 6-dihydroxypyridine (CNDP), a new potent inhibitor of dihydrouracil dehydrogenase. J. Biochem. 1993, 114 (6), 912–918. 10.1093/oxfordjournals.jbchem.a124276. [DOI] [PubMed] [Google Scholar]
- Sugimachi K.; Maehara Y. A phase II trial of a new 5-fluorouracil derivative, BOF-A2 (Emitefur), for patients with advanced gastric cancer. Surg. Today 2000, 30 (12), 1067–1072. 10.1007/s005950070003. [DOI] [PubMed] [Google Scholar]
- Shirasaka T.; Fujita F.; Fujita M.; Fukushima M.; Taguchi T.; Fujii S. Antitumor activity and metabolism of BOF-A2, a new 5-fluorouracil derivative, with human cancers xenografted in nude mice. Gan To Kagaku Ryoho 1990, 17 (9), 1871–1876. [PubMed] [Google Scholar]
- Milano G.; McLeod H. Can dihydropyrimidine dehydrogenase impact 5-fluorouracil-based treatment?. Eur. J. Cancer 2000, 36 (1), 37–42. 10.1016/S0959-8049(99)00211-7. [DOI] [PubMed] [Google Scholar]
- Wada H.; Hitomi S.; Teramatsu T. Adjuvant chemotherapy after complete resection in non-small-cell lung cancer. West Japan Study Group for Lung Cancer Surgery. J. Clin. Oncol. 1996, 14 (4), 1048–1054. 10.1200/JCO.1996.14.4.1048. [DOI] [PubMed] [Google Scholar]
- Wada H.; Miyahara R.; Tanaka F.; Hitomi S. Postoperative adjuvant chemotherapy with PVM (cisplatin+ vindesine+ mitomycin C) and UFT (uracil+ tegaful) in resected stage I–II NSCLC (non-small cell lung cancer): a randomized clinical trial. Eur. J. Cardiothorac. Surg. 1999, 15 (4), 438–443. 10.1016/S1010-7940(99)00031-7. [DOI] [PubMed] [Google Scholar]
- Imaizumi M. Postoperative adjuvant cisplatin, vindesine, plus uracil-tegafur chemotherapy increased survival of patients with completely resected p-stage I non-small cell lung cancer. Lung Cancer 2005, 49 (1), 85–94. 10.1016/j.lungcan.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Nakagawa T.; Tanaka F.; Takata T.; Matsuoka K.; Miyahara R.; Otake Y.; Yanagihara K.; Fukushimab M.; Wada H. Predictive value of dihydropyrimidine dehydrogenase expression in tumor tissue, regarding the efficacy of postoperatively administered UFT (Tegafur+ Uracil) in patients with p-stage I nonsmall-cell lung cancer. J. Surg. Oncol. 2002, 81 (2), 87–92. 10.1002/jso.10137. [DOI] [PubMed] [Google Scholar]
- Yen H.-H.; Chen C.-N.; Yeh C.-C.; Lai I.-R. Adjuvant tegafur-uracil (UFT) or S-1 monotherapy for advanced gastric cancer: a single center experience. World J. Surg. Oncol. 2021, 19 (1), 1–9. 10.1186/s12957-021-02233-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata T.; Hagihara K.; Tsutsui A.; Akamatsu H.; Ohue M.; Shingai T.; Tei M.; Ikenaga M.; Kim H. M.; Osawa H.; et al. Administration Method of Adjuvant Tegafur-Uracil and Leucovorin Calcium in Patients with Resected Colorectal Cancer: A Phase III Study. Oncologist 2021, 26 (5), e735–e741. 10.1002/onco.13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler M.; Johnston P. G.. The Approval of Teysuno/S-1 by the European Medicines Agency: A Potentially Important Advance for Gastric Cancer Patients; Oxford University Press: Oxford, U.K., 2011. 10.1634/theoncologist.2011-0333. [DOI] [Google Scholar]
- Pfeiffer P.; Thomseth-Lerström P.; Jensen H.; Vestermark L.; Krogh M.; Schønnemann K. Single institution experience with S-1 (TEYSUNO) in Danish patients with gastro-intestinal cancer. Ann. Oncol. 2013, 24, iv74. 10.1093/annonc/mdt203.136. [DOI] [Google Scholar]
- Sanford M. S-1 (Teysuno®): a review of its use in advanced gastric cancer in non-Asian populations. Drugs 2013, 73 (8), 845–855. 10.1007/s40265-013-0062-y. [DOI] [PubMed] [Google Scholar]
- Kang Y.-K. Phase I/II studies of combination chemotherapy with S-1 and platinum in patients with previously untreated metastatic or recurrent gastric cancer. Gastric Cancer 2009, 12 (1), 38–42. 10.1007/s10120-008-0472-9. [DOI] [Google Scholar]
- Sugimoto N.; Tanaka J.; Tsuda M.; Okamoto W.; Okuda H.; Imamura H.; Shimokawa T.; Sakai D.; Kurokawa Y.; Komatsu Y.; et al. A phase ll study of trastuzumab in combination with triweekly S-1 plus CDDP in HER2-positive advanced gastric cancer (HERBIS-1). Am. Soc. Clin. Oncol 2013, 31, 70. 10.1200/jco.2013.31.4_suppl.70. [DOI] [Google Scholar]
- Chollet P.; Schöffski P.; Weigang-Köhler K.; Schellens J.; Cure H.; Pavlidis N.; Grünwald V.; De Boer R.; Wanders J.; Fumoleau P. Phase II trial with S-1 in chemotherapy-naıve patients with gastric cancer. A trial performed by the EORTC early clinical studies group (ECSG). Eur. J. Cancer 2003, 39 (9), 1264–1270. 10.1016/S0959-8049(03)00237-5. [DOI] [PubMed] [Google Scholar]
- Kakeji Y.; Yoshida K.; Kodera Y.; Kochi M.; Sano T.; Ichikawa W.; Lee S.-W.; Shibahara K.; Shikano T.; Kataoka M.; et al. Three-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 plus docetaxel versus S-1 alone in stage III gastric cancer: JACCRO GC-07. Gastric Cancer 2022, 25 (1), 188–196. 10.1007/s10120-021-01224-2. [DOI] [PubMed] [Google Scholar]
- Iwatsuki M.; Orita H.; Kobayashi K.; Hidaka S.; Arigami T.; Kusumoto T.; Satake H.; Oki E.; Tsutsumi S.; Tobimatsu K.; et al. Phase II study of S-1 and oxaliplatin as neoadjuvant chemotherapy for locally advanced adenocarcinoma of the gastric or esophagogastric junction: KSCC1601. Gastric Cancer 2022, 25 (1), 180–187. 10.1007/s10120-021-01218-0. [DOI] [PubMed] [Google Scholar]
- Oyakawa T.; Hua Z.; Ebihara A.; Shiga T. A Rare Case of Reversible Cardiac Dysfunction Associated with Tegafur/Gimeracil/Oteracil (S-1) Therapy. Int. Heart J. 2021, 62 (3), 700–705. 10.1536/ihj.20-651. [DOI] [PubMed] [Google Scholar]