

Abstract

The multifactorial nature of Alzheimer’s disease necessitates the development of agents able to interfere with different relevant targets. A series of 22 tailored chromanones was conceptualized, synthesized, and subjected to biological evaluation. We identified one representative bearing a linker-connected azepane moiety (compound 19) with balanced pharmacological properties. Compound 19 exhibited inhibitory activities against human acetyl-, butyrylcholinesterase and monoamine oxidase-B, as well as high affinity to both the σ1 and σ2 receptors. Our study provides a framework for the development of further chromanone-based multineurotarget agents.
Keywords: chromanones, Alzheimer’s disease, multineurotarget agents, σ1 and σ2 receptors, monoamine oxidases, human cholinesterases
The therapy of multifactorial, complex disorders, such as Alzheimer’s disease (AD), might be managed more efficiently by using multitarget small molecules (MSMs) with affinity for multiple biological targets.1 The corresponding design has to combine diverse pharmacophores structurally overlapping or separated by appropriate linkers.2,3 Such a paradigm shift in AD drug development from compounds addressing a single target to MSMs has been emphasized, but this strategy has not yet been translated into an approved drug for treating AD patients.4,5
AD is a neurodegenerative disorder characterized by a progressive decline of memory and learning capabilities resulting from a number of not completely identified biological processes, including synaptic loss and neuronal death, oxidative stress, deficiency of neurotransmitters, and the presence of abnormal proteinaceous deposits in neurons and in the extracellular space.6
AD is a significant unmet medical need in neurology and will become a major challenge for worldwide health care systems. Current drugs improve symptoms, but do not have profound disease-modifying effects. Strong efforts in drug discovery are based on the first line AD hypotheses, tau- and beta-amyloid. Moreover, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) or monoamine oxidases (MAOs) constitute frequently selected biological targets for the development of AD therapeutics.7−9
AD neuropathology is characterized by a loss of cholinergic neurons and synapses and a deficit of acetylcholine in certain brain regions. Cholinesterase inhibitors, such as donepezil (Figure 1), decrease the extrasynaptic metabolism of acetylcholine and increase the synaptic residence time of the neurotransmitter.10 Beyond the inhibition of the enzymatic function of AChE by selective agents, dual inhibition to both AChE and BuChE has been explored as a therapeutic tactic for AD. BuChE can compensate for AChE in hydrolyzing acetylcholine, and a shift from a supportive to a leading role in the late stages of AD has been assumed. Inhibitors of BuChE were shown to improve memory and cognitive functions in mice. Hence, BuChE is also thought to be a potential pharmacological target for the diagnosis and treatment of AD.10,11
Figure 1.

Ligands of cholinesterases, monoamine oxidases, σ receptors, and multineurotargeting compounds.
However, besides acetylcholine, the levels of other neurotransmitters are also lower throughout the stages of AD. In particular, reduced amounts of dopaminergic neurotransmitters were linked with the AD pathophysiology.12,13 MAOs catalyze the oxidative deamination of biogenic amines, such as dopamine and other specific neurotransmitters, leading to the important role of these enzymes in the central nervous system (CNS). In the course of degradation of biogenic amines, MAOs produce hydrogen peroxide and further reactive oxygen species which cause oxidative stress, neuronal damage and neurodegeneration. The neuroprotective effects of MAO inhibitors, such as L-deprenyl (selegiline) and safinamide (Figure 1), have frequently been considered to be beneficial for AD treatment.12−14 The two isoforms of monoamine oxidase, MAO-A and MAO-B, share about 70% sequence identity but differ in their substrate specificities and inhibitor sensitivities. MAO-A is ubiquitously present in human tissues, while MAO-B mainly resides in the brain, in particular in the basal ganglia, where it is predominantly located in glial cells. During aging, an increase in MAO-B activity but a constant activity of the isoenzyme A was observed in post-mortem brains, pointing to the particular role of MAO-B in age-related neurodegeneration.9,15
As a ligand-operated intracellular chaperone, the sigma-1 (σ1) receptor interferes with a large number of proteins. Located in the mitochondrial-associated membrane part of the endoplasmatic reticulum (ER), the σ1 receptor is a key player in the regulation of cellular stress-induced signaling pathways.16 NE-100 (Figure 1) acts as a σ1 receptor antagonist.17 The structure of the complex with a further selective σ1 antagonist, PD144418 (Figure 1), was solved.18 With a high expression level in cells of the CNS, the σ1 receptor fulfills important physiological tasks, such as synaptic growth and the maturation of neural stem cells into neurons. Upon σ1 receptor stimulation, signalization pathways leading to ER stress, inflammation, or oxidative damage can be modulated. Since ER stress is associated with the aging process, targeting the σ1 receptor represents an approach toward the treatment of AD.19,20 First investigated as a drug against Huntington’s disease, the σ1 receptor agonist pridopidine (Figure 1) also featured neuroprotective properties in cellular and animal models of AD due to the stabilization of mushroom-shaped memory spines.19 Antagonism at the σ1 receptor has been associated with cell death promoting effects, whereas agonism preserved cell survival.21
The sigma-2 (σ2) receptor also constitutes a therapeutic target for neurocognitive disorders including AD. Some σ2 antagonists, i.e., SAS-0132 and CT1812 (Figure 1), produced neuroprotective effects in a Caenorhabditis elegans model, improved cognitive performance in a transgenic AD mice model, displaced Aβ oligomers bound to synaptic receptors of neuronal cells and increased CSF concentration of Aβ oligomers in the cerebrospinal fluid of AD patients, respectively.20,22,23
In this study, we conceptualized new multineurotargeting compounds that are capable of interacting with the aforementioned biological targets. Our molecular design was inspired by the MSM contilisant (Figure 1), an advanced development compound for AD with antioxidant and neuroprotective properties, inhibitory potency against AChE, BuChE, MAO-B, and MAO-A, as well as affinity toward the histamine H3 and the σ1 receptor.9,24,25 Furthermore, being active in appropriate AD animal models, contilisant exceeded the efficacy of donepezil.25 The chromenone derivative VII (Figure 1) caused a concomitant inhibition of AChE and MAO-B and exhibited σ1/σ2 affinity.26,27
Results and Discussion
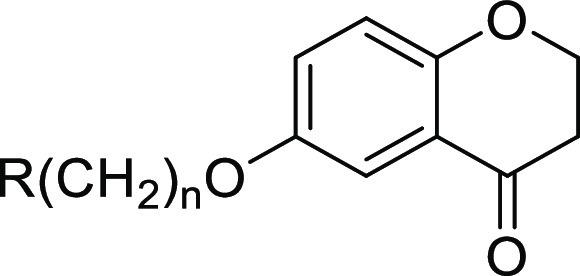
The common feature of the bioactive compounds depicted in Figure 1 is the aromatic core connected via a 1- to 6-atom linker to the basic nitrogen of a secondary or tertiary amine substructure. We conceived the chemotype of chromanones which have not yet been associated with multitarget activity. Differently sized cyclic amines were attached through alkyl chains of various lengths to position 6 of the chromanone skeleton via an ether linkage. The corresponding set of contemplated compounds, all of which with a tertiary amine substructure, are listed in Table 1.
Table 1. Inhibitory Activity of Chromanones 1–22 against Human Cholinesterases and Monoamine Oxidases (IC50 ± SE, μM) and Affinity Values (Ki, nM) at the Human σ1 (hσ1) and Rat σ2 (rσ2) Receptors24,25,27.

n.i. (no inhibition) refers to <5% inhibitory activity at 10 μM for MAO.
n.d. (not determined).
Herein, a systematic design and the straightforward assembly of such chromanone derivatives is reported. The biological evaluation enabled us to build structure–activity relationships and to explore the effect of exchanging the double bond in the heterocyclic core of chromenones with the single bond in chromanones.
The final compounds were prepared from 6-hydroxychroman-4-one (Scheme 1), whose phenolic group was converted to an ether moiety, either by Mitsunobu–Tsunoda28,29 or Williamson chemistries.30,31 The former method made use of (cyanomethylene)tributylphosphorane (CMBP), a single reagent to be employed instead of the combination of triphenylphosphine and a dialkyl azodicarboxylate. However, in most cases, we applied Williamson ether synthesis. The syntheses were carried out in a remote-controlled automated fashion and monitored in real time through LC-MS.32 The final compounds were purified using a strong cation exchange (SCX) cartridge containing benzenesulfonic acid as sorbent. The basic products were eluted from the resin with 2 N ammonia in methanol and obtained in high purity.
Scheme 1. Synthesis of Chromanones 1–22 Using a Mitsunobu–Tsunoda protocol (Method A) or a Two-Step Procedure (Method B).

The resulting chromanones were subjected to biological evaluation, giving initial priority to their cholinesterase and MAO inhibitory activities. Chromanones, functionalized at position 7 with a linker-connected dithiocarbamate group, have already been investigated as inhibitors of AChE from Electrophorus electricus and equine BuChE.33 In our study, the activity of human AChE and human BuChE was monitored photometrically in the presence of 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB) to detect thiocholine, which was released by the enzyme-catalyzed cleavage of acetylthiocholine or butyrylthiocholine. Some chromanones exhibited inhibition of human AChE and the azepane substructure turned out to be particularly favorable (compounds 19–22, Table 1). The linker length did affect AChE inhibition and a butylene tether (n = 4), as present in 3, 9, and 21, was advantageous. However, strongest AChE inhibition was achieved with azepane derivative 19 possessing an ethylene linker (IC50 = 4.88 μM, see Figure S1).
Based on our docking investigations, the accommodation of 19 in the active-site gorge of AChE was similar to those of comparable ligands, including donepezil (Figure 1) and related coumarins,34,35 homoisoflavonoids,31 and chromenones.36 The protonated azepane moiety pointed toward the catalytic anionic subsite (CAS) and made van der Waals interactions with the catalytic triad residues His447 and Ser203. The ligand formed π-alkyl interactions with Trp86 and a hydrogen bond with Tyr337. The chromanone core was oriented toward the peripheral anionic subsite (PAS) making a π–π stacking with Tyr341 and van der Waals interactions with Asp74 and Trp286 (Figure S2). However, it has been suggested that chromenone VII (Figure 1) resided in the active-site gorge in an inverse manner with the heterobicycle directed toward the CAS.26
The majority of chromanones did not affect human BuChE, but four azepanes, 19–22, had IC50 values lower than 25 μM and were, hence, identified as dual cholinesterase inhibitors. Compounds 19–22 possess the most extended aliphatic substructure among our set of chromanones, which accounted for a favorable accommodation within the active site of BuChE. Unexpectedly, we also observed BuChE inhibition for a comparable small chromanone, i.e., 1, which showed preference for BuChE (IC50 = 3.86 μM, see Figure S3) over AChE.
In order to explore the MAO-inhibitory activities of chromanones 1-22, human recombinant MAO-B and MAO-A enzymes were used and the Amplex Red monoamine oxidase assay was carried out.37 Only two chromanones, i.e., 21 and 22, exhibited modest inhibition of MAO-A (Table 1). However, MAO-B was inhibited by the majority of the chromanones at a single concentration of 10 μM (data not shown). For these compounds, IC50 values were determined (Table 1). The most potent compounds belong to the subgroup of morpholine derivatives (11–14) with 12 possessing an IC50 value of 399 nM (see Figure S4) and exhibiting a reversible binding mode at MAO-B (see Figure S5). MAO-B inhibition was also determined with pyrrolidine and azepane derivatives, whereas the introduction of the N-methyl piperazine moiety led to inactive compounds (15–18). In the case of morpholine (11–14) and azepane subseries (19–22), short linkers were favorable for MAO-B inhibition (n = 2 and 3 versus n = 4 and 5; Table 1).
Molecular docking of compound 19 into the MAO-B binding site revealed that this inhibitor spanned both cavities, its fused benzene ring was located between the “entrance” and “catalytic” cavity, separated by the residues Ile199 and Tyr326 (Figure S6). The azepane ring was oriented toward the bottom of the catalytic cavity, interacting with the FAD cofactor as well as Phe343 and Tyr398. The 2,3-dihydro-4H-pyran-4-one ring was hosted in the large entrance cavity made up by several hydrophobic amino acid residues. This binding mode was unexpected since most of the related MAO-B ligands oriented their bicyclic part toward the FAD cofactor.26,34−36,38
The basicity of analogous derivatives is ranked as follows: pyrrolidine > azepane ∼ piperidine ≫ morpholine.39 Moreover, the basicity decreases with shorter linkers, in particular with n = 2. When inspecting the influence of basicity on cholinesterases and MAO-B inhibition, there was no clear correlation, except the finding that the poor basic morpholine derivatives failed to inhibit both cholinesterases.
Brain iron accumulation is a common characteristic in neurodegenerative disorders such as AD,40 Hence, we investigated the inhibition of human matriptase-2, a transmembrane serine protease leading to enhanced plasma iron levels.41 However, none of the chromanones, at 10 μM, caused a matriptase-2 inhibition of more than 30% (data not shown).
Since both σ1 and σ2 receptors have been implicated in AD, we chose five chromanones to study their affinity to σ1 and σ2. The selection was done for the following reasons. Compounds 19 and 21 showed IC50 values lower than 15 μM at three or four targets, respectively. Compounds 1 and 12 performed best at BuChE and MAO-B, respectively, and compound 3 was a dual AChE and MAO-B inhibitor. All these ligands exhibited strong and preferred σ1 affinity in the nanomolar range (Table 1) with the azepane 19 (Ki = 42.8 nM) being the most potent σ1 ligand. The selectivity in favor of σ1 (ratio σ2/σ1) was between 4.5 (19) and 321 (12).
To visualize the interaction with the σ1 binding site, compound 19 was docked into a three-dimensional model of the σ1 receptor based on the crystal structure of the complex with PD144418 (Figure 1).18 This ligand performed an electrostatic interaction of its basic amine moiety with the side chain of receptor residue Glu172, a key feature for σ1 binding.18,42−48 Compound 19 occupied the same region of the binding site as PD144418, but displayed a reversed docking mode (Figure S7). Irrespective of the agonistic or antagonistic activity, structurally related σ1 ligands, such as the agonist PRE-084 and the antagonists PD144418 and NE-100, oriented their basic structures away from the α4 helix, which came in close contact with aromatic moieties of the ligands.49 Hence, agonistic/antagonistic behavior cannot be predicted from the binding mode, and reliable in vitro protocols to distinguish between agonist or antagonist properties have not yet been established.21 Notably, a common feature of contilisant (Table 1) and 19 is the nanomolar affinity at the σ1 and σ2 receptors. However, contilisant exhibited a better balanced multitarget profile which was not attained by our chromanones.
In this study, relevant targets for a MSM approach toward drug candidates against AD have been selected. Our approach was based on the evaluation of a systematically combined library of chromanones and led to the identification of three compounds, 3, 19, and 21, which displayed biological activity at three AD targets, i.e., AChE, MAO-B, and the σ1 receptor. The azepane subseries provided the most intriguing results with representatives 19 and 21 also inhibiting BuChE. We introduce 19 as a multineurotargeting hit compound, which additionally featured high affinity to the σ2 receptor. A comparison of 19 and 21 is illustrated in the radar chart, Figure 2.
Figure 2.

Multitarget activities of chromanones 19 and 21.
Next, the question arose to which extent the biological activities of the herein report chromanones differ from those of the analogous chromenones. Chromenones have been extensively investigated as MSMs.7,26,27,34,36 Despite the obvious similarity of both chemotypes, the structural features diverge due to the aromatic character of the γ-pyrone ring in chromenones which was confirmed by nucleus independent chemical shift values.50 Chromenone XXI, the direct structural counterpart of 19, exhibited weaker effects at the five targets (Figure 3).26,27 Our compound 19 also outperformed the chromenone VII, which has been designated as a particular valuable MSM in two previous studies (Figure 4).26,27
Figure 3.

Multitarget activities of 19 and chromenone XXI.
Figure 4.

Multitarget activities of 19 and chromenone VII.
Taken together, this investigation was attempted to pinpoint the features of chromanones as new MSMs with emphasis on neurological disorders. A systematic access to heterodimers was devised by linking a fixed chromanone core to an amine moiety. A comparison with selected analogous chromenones revealed the class of chromanones to be promising. We disclose chromanone 19 as a penta-neurotargeting agent. Our results are expected to offer opportunities for the future discovery of new molecules with optimized multitarget-directed biological properties.
Experimental Section
Chemistry
NMR spectra were recorded on a Bruker Avance III 600 (600 MHz 1H NMR, 151 MHz 13C NMR) or a Bruker Avance DRX 500 (500 MHz 1H NMR, 126 MHz 13C NMR) spectrometer. Chemical shifts δ are given in ppm. HPLC/MS chromatograms were recorded on a LCC_APL_U_LC1 instrument equipped with an HPH Phenomenex Kinetex EVO column (2.1 × 30 mm2), at a flow rate of 0.85 mL/min. Solvent A was 10 mM ammonium bicarbonate; solvent B was acetonitrile. A gradient of 5–100% B was used over 2 min. The purity of all products was determined with LC-MS using DAD detection and was higher than 95%.
General Method A for the Synthesis of Chromanones via Mitsunobu–Tsunoda Reaction51,52
A mixture of 6-hydroxychroman-4-one53 (82.1 mg, 0.5 mmol), the appropriate aminoalcohol (0.5 mmol; 2-morpholinoethan-1-ol, 65.6 mg; 2-(4-methylpiperazin-1-yl)ethan-1-ol, 72.1 mg; 2-(azepan-1-yl)ethan-1-ol, 71.6 mg), and cyanomethylene-tributylphosphorane (CMBP) (0.7 mmol, 169 mg) was heated in toluene (5 mL) at 100 °C for 16 h. The reaction mixture was diluted with methanol and loaded into a 10 g SCX cartridge. The resin was washed with methanol, and the crude product was eluted with a 2 N solution of ammonia in methanol, followed by evaporation of the solvent.
General Method B for the Synthesis of Chromanones via O-Alkylation of 6-Hydroxychroman-4-one30
A mixture of potassium carbonate (345 mg, 2.5 mmol), 6-hydroxychroman-4-one (82.1 mg, 0.5 mmol), and the corresponding α,ω-dibromoalkane (1.0 mmol; 1,2-dibromoethane, 188 mg; 1,3-dibromopropane, 202 mg; 1,4-dibromobutane, 216 mg; 1,5-dibromopentane, 230 mg) in acetonitrile (5 mL) was heated at 50 °C for 20 h. After cooling, acetonitrile (10 mL) was added. The inorganic salts were filtered off, and the solvent was evaporated. To a stirred solution of the crude intermediate in acetonitrile (5 mL) was added the appropriate cyclic amine (0.5 mmol; pyrrolidine, 35.6 mg; (R)-2-methylpyrrolidine, 42.6 mg; piperidine, 42.6 mg; morpholine, 43.6 mg; N-methylpiperazine, 50.1 mg; azepane, 49.6 mg). After stirring at 50 °C for 15 h, the reaction mixture was diluted with methanol and loaded into a 10 g SCX cartridge. The resin was washed with methanol, and the crude product was eluted with 2 N ammonia in methanol, followed by evaporation of the solvent.
6-(2-(Pyrrolidin-1-yl)ethoxy)-4H-chroman-4-one (1)
The title compound was prepared following general procedure B. Yield 58.5 mg (45%), C15H19NO3, 261.32 g/mol). 1H NMR (600 MHz, DMSO-d6) δ 1.62–1.70 (m, 4H), 2.75 (t, J = 6.0 Hz, 4H), 4.03 (t, J = 5.9 Hz, 2H), 4.46 (t, J = 6.0 Hz, 2H), 6.96 (dd, J = 8.4, 1.0 Hz, 1H), 7.15–7.18 (m, 2H); one signal at ∼2.50 (4H) is obscured by the solvent. 13C NMR (151 MHz, DMSO-d6) δ 23.30, 37.32, 54.11, 54.37, 66.94, 67.52, 108.52, 119.32, 121.09, 124.95, 152.82, 156.15, 191.60. LC-MS: m/z = 262.0 ([M + H]+).
6-(3-(Pyrrolidin-1-yl)propoxy)-4H-chroman-4-one (2)
The title compound was prepared following general procedure B. Yield 64.4 mg (47%), C16H21NO3, 275.35 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.63–1.70 (m, 4H), 1.84 (quin, J = 6.6 Hz, 2H), 2.38–2.43 (m, 4H), 2.75 (t, J = 6.5 Hz, 2H), 3.97 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.6 Hz, 2H), 6.96 (d, J = 8.7 Hz, 1H), 7.13–7.18 (m, 2H); one signal at ∼2.50 (2H) is obscured by the solvent. 13C NMR (151 MHz, DMSO-d6) δ 23.26, 28.28, 37.32, 52.30, 53.75, 66.63, 66.94, 108.37, 119.30, 121.06, 124.97, 152.98, 156.10, 191.63. LC-MS: m/z = 276.1 ([M + H]+).
6-(4-(Pyrrolidin-1-yl)butoxy)-4H-chroman-4-one (3)
The title compound was prepared following general procedure B. Yield 89.8 mg (62%), C17H23NO3, 289.38 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.43–1.81 (m, 8H), 2.32–2.42 (m, 4H), 2.75 (t, J = 6.5 Hz, 2H), 3.94 (t, J = 6.6 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H),), 6.92–7.00 (m, 1H), 7.08–7.20 (m, 2H); one signal at ∼2.50 (2H) is obscured by the solvent. 13C NMR (151 MHz, DMSO-d6) δ 23.24, 24.99, 26.84, 37.33, 53.69, 55.41, 66.94, 68.11, 108.45, 119.29, 121.08, 124.95, 152.98, 156.09, 191.63. LC-MS: m/z = 290.2 ([M + H]+).
6-((5-(Pyrrolidin-1-yl)pentyl)oxy)-4H-chroman-4-one (4)
The title compound was prepared following general procedure B. Yield 83.6 mg (55%), C18H25NO3, 303.40 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.36–1.51 (m, 4H), 1.60–1.75 (m, 6H), 2.29–2.43 (m, 6H), 2.74 (t, J = 5.9 Hz, 2H), 3.92 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.94–6.98 (m, 1H), 7.10–7.20 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 23.23, 23.70, 28.28, 28.70, 37.33, 53.75, 55.77, 66.93, 68.18, 108.43, 119.28, 121.08, 124.93, 152.99, 156.08, 191.61. LC-MS: m/z = 304.2 ([M + H]+).
(R)-6-(2-(2-Methylpyrrolidin-1-yl)ethoxy)chroman-4-one (5)
The title compound was prepared following general procedure B. Yield 54.1 mg (39%), C16H21NO3, 275.35 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 1.01 (d, 3J = 6.0 Hz, 3H), 1.21–1.32 (m, 1H), 1.56–1.69 (m, 2H), 1.80–1.88 (m, 1H), 2.17 (q, J = 8.8 Hz, 1H), 2.30–2.37 (m, 1H), 2.38–2.45 (m, 1H), 2.75 (t, J = 6.4 Hz, 2H), 3.00–3.13 (m, 2H), 4.02 (t, J = 6.0 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.8 Hz, 1H), 7.12–7.20 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 19.22, 21.74, 32.42, 37.31, 52.03, 54.10, 59.46, 66.91, 67.72, 108.65, 119.27, 121.08, 124.89, 152.80, 156.12, 191.55. LC-MS: m/z = 276.1 ([M + H]+).
(R)-6-(3-(2-Methylpyrrolidin-1-yl)propoxy)chroman-4-one (6)
The title compound was prepared following general procedure B. Yield 67.8 mg (47%), C17H23NO3, 289.38 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 0.97 (d, 3J = 6.0 Hz, 3H), 1.22–1.32 (m, 1H), 1.55–1.68 (m, 2H), 1.76–1.89 (m, 3H), 2.01 (q, J = 8.7 Hz, 1H), 2.06–2.12 (m, 1H), 2.18–2.27 (m, 1H), 2.74 (t, J = 6.4 Hz, 2H), 2.83–2.91 (m, 1H), 3.01–3.08 (m, 1H), 3.97 (t, J = 6.4 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.7 Hz, 1H), 7.12–7.18 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 19.19, 21.51, 28.11, 32.64, 37.31, 49.83, 53.44, 59.47, 66.62, 66.91, 108.46, 119.26, 121.06, 124.93, 153.01, 156.08, 191.58. LC-MS: m/z = 290.2 ([M + H]+).
(R)-6-(4-(2-Methylpyrrolidin-1-yl)butoxy)chroman-4-one (7)
The title compound was prepared following general procedure B. Yield 86.8 mg (57%), C18H25NO3, 303.40 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 0.99 (d, 3J = 6.0 Hz, 3H), 1.21–1.30 (m, 1H), 1.45–1.87 (m, 7H), 1.92–2.05 (m, 2H), 2.15–2.23 (m, 1H), 2.67–2.77 (m, 3H), 2.98–3.06 (m, 1H), 3.95 (t, J = 6.9 Hz, 2H), 4.46 (t, J = 6.3 Hz, 2H), 6.96 (dd, J = 8.6, 0.9 Hz, 1H), 7.12–7.19 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 19.21, 21.48, 24.83, 26.85, 32.65, 37.32, 53.06, 53.34, 59.47, 66.91, 68.12, 108.44, 119.25, 121.07, 124.89, 152.98, 156.06, 191.56. LC-MS: m/z = 304.2 ([M + H]+).
(R)-6-((5-(2-Methylpyrrolidin-1-yl)pentyl)oxy)chroman-4-one (8)
The title compound was prepared following general procedure B. Yield 88.8 mg (56%), C19H27NO3, 317.43 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 0.99 (d, J = 6.0 Hz, 3H), 1.22–1.29 (m, 1H), 1.36–1.51 (m, 4H), 1.54–1.65 (m, 2H), 1.66–1.76 (m, 2H), 1.80–1.87 (m, 1H), 1.93–2.00 (m, 2H), 2.15–2.22 (m, 1H), 2.67–2.73 (m, 1H), 2.73–2.77 (m, 2H), 2.99–3.04 (m, 1H), 3.94 (t, J = 6.5 Hz, 2H), 4.47 (t, J = 6.4 Hz, 2H), 6.97 (d, J = 8.5 Hz, 1H), 7.14–7.19 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 19.04, 21.29, 23.53, 27.97, 28.49, 32.48, 37.15, 53.22, 53.26, 59.35, 66.75, 68.02, 108.26, 119.11, 120.90, 124.75, 152.81, 155.90, 191.44. LC-MS: m/z = 318.0 ([M + H]+).
6-(4-(Piperidin-1-yl)butoxy)chroman-4-one (9)
The title compound was prepared following general procedure B. Yield 87.4 mg (58%), C18H25NO3, 303.40 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.32–1.37 (m, 2H), 1.41–1.49 (m, 4H), 1.49–1.56 (m, 2H), 1.64–1.72 (m, 2H), 2.20–2.34 (m, 6H), 2.74 (t, J = 6.4 Hz, 2H), 3.94 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.0 Hz, 2H), 6.96 (dd, J = 8.2, 1.1 Hz, 1H), 7.14–7.17 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 22.95, 24.37, 25.78, 26.81, 37.33, 54.19, 58.32, 66.93, 68.12, 108.43, 119.28, 121.07, 124.94, 152.97, 156.07, 191.62. LC-MS: m/z = 304.2 ([M + H]+).
6-((5-(Piperidin-1-yl)pentyl)oxy)chroman-4-one (10)
The title compound was prepared following general procedure B. Yield 95.6 mg (60%), C19H27NO3, 317.43 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.30–1.40 (m, 4H), 1.41–1.49 (m, 6H), 1.64–1.72 (m, 2H), 2.16–2.33 (m, 6H), 2.74 (t, J = 6.4 Hz, 2H), 3.92 (t, J = 6.4 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.5 Hz, 1H), 7.13–7.17 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 23.67, 24.37, 25.77, 26.28, 28.70, 37.33, 54.26, 58.74, 66.93, 68.17, 108.44, 119.29, 121.08, 124.92, 153.00, 156.09, 191.62. LC-MS: m/z = 318.2 ([M + H]+).
6-(2-Morpholinoethoxy)chroman-4-one (11)
The title compound was prepared following general procedure A. Yield 52.5 mg (38%), C15H19NO4, 277.32 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 2.40–2.48 (m, 4H), 2.65 (t, J = 5.7 Hz, 2H), 2.75 (t, J = 6.4 Hz, 2H), 3.56 (t, J = 4.6 Hz, 4H), 4.05 (t, J = 5.7 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.9 Hz, 1H), 7.15–7.21 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 37.33, 53.76, 57.11, 66.15, 66.33, 66.94, 108.69, 119.31, 121.09, 124.99, 152.80, 156.19, 191.60. LC-MS: m/z = 278.1 ([M + H]+).
6-(3-Morpholinopropoxy)chroman-4-one (12)
The title compound was prepared following general procedure B. Yield 50.8 mg (35%), C16H21NO4, 291.35 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.81–1.87 (m, 2H), 2.30–2.41 (m, 6H), 2.75 (t, J = 6.5 Hz, 2H), 3.56 (t, J = 4.7 Hz, 4H), 3.98 (t, J = 6.4 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.7 Hz, 1H), 7.14–7.18 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 25.94, 37.32, 53.52, 54.90, 66.36, 66.55, 66.94, 108.42, 119.32, 121.07, 125.00, 152.96, 156.13, 191.64. LC-MS: m/z = 292.2 ([M + H]+).
6-(4-Morpholinobutoxy)-4H-chromen-4-one (13)
The title compound was prepared following general procedure B. Yield 83.9 mg (55%), C17H23NO4, 305.37 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.51–1.57 (m, 2H), 1.67–1.73 (m, 2H), 2.24–2.35 (m, 6H), 2.75 (t, J = 6.6 Hz, 2H), 3.54 (t, J = 4.7 Hz, 4H), 3.95 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.7 Hz, 1H), 7.13–7.18 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 22.48, 26.66, 37.32, 53.47, 57.93, 66.37, 66.93, 68.05, 108.42, 119.29, 121.07, 124.94, 152.95, 156.09, 191.62. LC-MS: m/z = 306.2 ([M + H]+).
6-((5-Morpholinopentyl)oxy)chroman-4-one (14)
The title compound was prepared following general procedure B. Yield 85.9 mg (54%), C18H25NO4, 319.40 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.36–1.42 (m, 2H), 1.42–1.49 (m, 2H), 1.66–1.72 (m, 2H), 2.21–2.34 (m, 6H), 2.75 (t, J = 6.4 Hz, 2H), 3.54 (t, J = 4.7 Hz, 4H), 3.93 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.6 Hz, 1H), 7.13–7.17 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 23.52, 25.78, 28.64, 37.32, 53.53, 58.34, 66.37, 66.93, 68.14, 108.44, 119.29, 121.07, 124.92, 152.98, 156.09, 191.62. LC-MS: m/z = 320.2 ([M + H]+).
6-(2-(4-Methylpiperazin-1-yl)ethoxy)chroman-4-one (15)
The title compound was prepared following general procedure A. Yield 58.4 mg (40%), C16H22N2O3, 290.36 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 2.13 (s, 3H), 2.64 (t, J = 5.5 Hz, 2H), 2.75 (t, J = 6.4 Hz, 2H), 4.03 (t, J = 5.8 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.8 Hz, 1H), 7.15–7.20 (m, 2H); piperazine signals ∼2.20 to ∼2.50 (8H) were not resolved. 13C NMR (151 MHz, DMSO-d6) δ 37.32, 45.90, 53.15, 54.87, 56.67, 66.37, 66.93, 108.68, 119.29, 121.09, 124.97, 152.81, 156.17, 191.58. LC-MS: m/z = 291.2 ([M + H]+).
6-(3-(4-Methylpiperazin-1-yl)propoxy)chroman-4-one (16)
The title compound was prepared following general procedure B. Yield 62.1 mg (41%), C17H24N2O3, 304.39 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.80–1.85 (m, 2H), 2.13 (s, 3H), 2.38 (t, J = 7.1 Hz, 2H), 2.75 (t, J = 6.4 Hz, 2H), 3.96 (t, J = 6.4 Hz, 2H), 4.46 (t, J = 6.5 Hz, 2H), 6.96 (dd, J = 8.3, 1.1 Hz, 1H), 7.13–7.17 (m, 2H); piperazine signals ∼2.20 to ∼2.50 (8H) were not resolved. 13C NMR (151 MHz, DMSO-d6) δ 26.28, 37.32, 45.88, 52.86, 54.44, 54.91, 66.61, 66.93, 108.41, 119.31, 121.07, 124.99, 152.96, 156.11, 191.62. LC-MS: m/z = 305.2 ([M + H]+).
6-(4-(4-Methylpiperazin-1-yl)butoxy)chroman-4-one (17)
The title compound was prepared following general procedure B. Yield 81.3 mg (51%), C18H26N2O3, 318.42 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 1.49–1.56 (m, 2H), 1.64–1.72 (m, 2H), 2.12 (s, 3H), 2.28 (t, J = 7.1 Hz, 2H), 2.75 (t, J = 6.4 Hz, 2H), 3.94 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.5 Hz, 1H), 7.14–7.18 (m, 2H); piperazine signals ∼2.20 to ∼2.50 (8H) were not resolved. 13C NMR (126 MHz, DMSO-d6) δ 22.84, 26.72, 37.31, 45.88, 52.79, 54.93, 57.49, 66.91, 68.07, 108.45, 119.25, 121.06, 124.91, 152.95, 156.06, 191.57. LC-MS: m/z = 319.2 ([M + H]+).
6-((5-(4-Methylpiperazin-1-yl)pentyl)oxy)chroman-4-one (18)
The title compound was prepared following general procedure B. Yield 95.1 mg (57%), C19H28N2O3, 332.44 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 1.34–1.41 (m, 2H), 1.41–1.48 (m, 2H), 1.64–1.72 (m, 2H), 2.12 (s, 3H), 2.23 (t, J = 7.2 Hz, 2H), 2.74 (t, J = 6.5 Hz, 2H), 3.92 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.5 Hz, 1H), 7.12–7.18 (m, 2H); piperazine signals ∼2.20 to ∼2.50 (8H) were not resolved. 13C NMR (126 MHz, DMSO-d6) δ 23.56, 26.16, 28.64, 37.31, 45.89, 52.86, 54.93, 57.90, 66.91, 68.15, 108.47, 119.25, 121.07, 124.89, 152.98, 156.06, 191.57. LC-MS: m/z = 333.2 ([M + H]+).
6-(2-(Azepan-1-yl)ethoxy)chroman-4-one (19)
The title compound was prepared following general procedure A. Yield 68.0 mg (47%), C17H23NO3, 289.38 g/mol. 1H NMR (500 MHz, CDCl3) δ 1.46–1.53 (m, 4H), 1.54–1.67 (m, 4H), 2.63–2.70 (m, 4H), 2.75 (t, J = 6.4 Hz, 2H), 2.81 (t, J = 6.0 Hz, 2H), 3.99 (t, J = 6.0 Hz, 2H), 4.46 (t, J = 6.5 Hz, 2H), 6.97 (d, J = 8.9 Hz, 1H), 7.14–7.20 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 26.70, 28.10, 37.32, 55.27, 56.14, 66.92, 67.11, 108.69, 119.25, 121.08, 124.95, 152.91, 156.12, 191.57. LC-MS: m/z = 290.2 ([M + H]+).
6-(3-(Azepan-1-yl)propoxy)chroman-4-one (20)
The title compound was prepared following general procedure B. Yield 66.3 mg (44%), C18H25NO3, 303.40 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 1.37–1.62 (m, 8H), 1.70–1.88 (m, 2H), 2.50–2.70 (m, 6H), 2.75 (t, J = 6.5 Hz, 2H), 3.97 (t, J = 6.4 Hz, 2H), 4.46 (t, J = 6.6 Hz, 2H), 6.95–6.97 (m, 1H), 7.12–7.21 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 26.69, 27.28, 28.13, 37.31, 54.04, 54.99, 66.63, 66.92, 108.45, 119.28, 121.07, 124.95, 153.02, 156.08, 191.59. LC-MS: m/z = 304.2 ([M + H]+).
6-(4-(Azepan-1-yl)butoxy)chroman-4-one (21)
The title compound was prepared following general procedure B. Yield 86.8 mg (55%), C19H27NO3, 317.43 g/mol. 1H NMR (500 MHz, DMSO-d6) δ 1.39–1.62 (m, 10H), 1.64–1.73 (m, 2H), 2.40–2.47 (m, 2H), 2.51–2.55 (m, 2H), 2.74 (t, J = 6.4 Hz, 2H), 3.95 (t, J = 6.6 Hz, 2H), 4.46 (t, J = 6.5 Hz, 2H), 6.93–6.99 (m, 1H), 7.12–7.19 (m, 2H); one signal (2H) is obscured by the solvent. 13C NMR (126 MHz, DMSO-d6) δ 23.89, 26.29, 26.68, 28.18, 37.31, 54.99, 57.31, 66.91, 68.17, 108.45, 119.25, 121.06, 124.91, 152.96, 156.05, 191.57. LC-MS: m/z = 318.2 ([M + H]+).
6-((5-(Azepan-1-yl)pentyl)oxy)chroman-4-one (22)
The title compound was prepared following general procedure B. Yield 92.1 mg (56%), C20H29NO3, 331.46 g/mol. 1H NMR (600 MHz, DMSO-d6) δ 1.35–1.45 (m, 4H), 1.46–1.52 (m, 4H), 1.52–1.58 (m, 4H), 1.66–1.71 (m, 2H), 2.40 (t, J = 7.0 Hz, 2H), 2.53 (t, J = 5.5 Hz, 4H), 2.74 (t, J = 6.4 Hz, 2H), 3.92 (t, J = 6.5 Hz, 2H), 4.46 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 8.7 Hz, 1H), 7.13–7.18 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 23.49, 26.69, 27.08, 28.06, 28.66, 37.32, 55.00, 57.59, 66.93, 68.19, 108.44, 119.28, 121.07, 124.91, 152.99, 156.08, 191.61. LC-MS: m/z = 332.2 ([M + H]+).
Biochemistry and Pharmacology
All studies involving animals were carried out according to the national laws on the protection of animals and were approved by the responsible authorities (Landesdirektion Sachsen, Reference no. DD24.1–5131/446/19, TVV 18/18).
Stock solutions (10 mM) of chromanone inhibitors were prepared in acetonitrile for cholinesterase assays and in DMSO for MAO, σ receptor binding, and cell viability assays.
Cholinesterase Assays
Human erythrocyte ghosts as the source of acetylcholinesterase (AChE, activity: 110 mE/min) were prepared from commercial blood units following a literature procedure.54 Aliquots of the preparation were stored at −80 °C, thawed carefully on ice, and homogenized by sonication prior to analysis. Kinetic measurements were carried out using a Tecan Infinite M200 PRO plate reader (Crailsheim, Germany) in 24-well plates thermostatted at 37 °C. Sodium phosphate buffer (1820 μL, 100 mM, pH 7.4) followed by 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB; 100 μL, 10 mM in sodium phosphate buffer (100 mM, pH 7.4)) and the respective test solution (20 μL, chromanones, acetonitrile as blank) were transferred to the wells using a Tecan Fluent liquid handling system. Subsequently, human erythrocyte ghosts (10 μL) or butyrylcholinesterase (BChE, activity: 110 mE/min; 10 μL, Sigma-Aldrich, Germany) as well as aqueous acetylthiocholine (20 μL, 28.4 mM) or butyrylthiocholine (20 μL, 63.2 mM) were added, and the reaction was monitored for 5 min at 412 nm. Five different inhibitor concentrations were analyzed in duplicate experiments to calculate IC50 values by nonlinear regression using GraphPad Prism (version 5.04, San Diego, USA). Standard errors are given in reference to these nonlinear regression analyses.
Monoamine Oxidase Assays26,37
Recombinant human MAO-A and MAO-B enzymes expressed in baculovirus-infected insect cells were purchased from Sigma-Aldrich (Germany). The assays were carried out at room temperature in 96-well plates in a final volume of 200 μL. The test compound (2 μL) dissolved in DMSO was added, followed by the enzyme solution (88 μL) in sodium phosphate buffer (50 mM, pH 7.4), which was flushed with nitrogen for 15 min prior to use. The mixture was preincubated for 30 min at room temperature. To each well containing 2 μg of MAO-A or MAO-B was added 90 μL of freshly prepared Amplex Red reagent. For each plate, 1 mg of Amplex Red (Invitrogen, Carlsbad, CA), dissolved in 200 μL of DMSO and 100 μL of reconstituted horseradish peroxidase (200 U/mL, Sigma-Aldrich, Germany) were added to 9700 μL of sodium phosphate buffer (50 mM, pH 7.4). To each well, 20 μL of an aqueous solution of p-tyramine (Alfa Aesar, Tewksbury, MA) was added to reach a final concentration of 150 μM, starting the enzymatic reaction. The production of hydrogen peroxide and consequently of resorufin was quantified in a Fluostar BMG Labtech microplate fluorescence reader (λex = 485 nm, λem = 590 nm, gain = 1000) over 60 min. Non-MAO-B enzyme activity was determined in the presence of selegiline (Sigma-Aldrich, Germany, final concentration 1 μM) and subtracted from the total activity measured. Concentration–response curves of selegiline served as a positive control. Non-MAO-A enzyme activity was determined in the presence of clorgyline (Sigma-Aldrich, Germany, final concentration 10 μM) and subtracted from the total activity measured. Concentration–response curves of clorgyline served as a positive control. Data were analyzed using GraFit.
Matriptase-2 Assay
This assay was performed as described elsewhere.41
Assays for Determination of Affinity to the hσ1 and rσ2 Receptors
To estimate the binding potential of the test compounds toward σ1 and σ2 receptors, radioligand displacement assays were performed using suitable radioligands and target preparations.27 Inhibition constants Ki toward human σ1 (hσ1) and rat σ2 (rσ2) receptors are listed in Table 1. To assess the σ1 affinity, increasing concentrations of the respective test compound (10–11–10–5 M) were coincubated with the selective σ1 receptor ligand (+)-[3H]pentazocine (PerkinElmer; AM = 995 GBq/mmol) at a single concentration (5 nM) and cell membrane homogenates obtained from HEK293 cells stably transfected with the human σ1 receptor (provided by Olivier Soriani, Institut de Biologie Valrose, Nice, France) in binding buffer (50 mM TRIS-HCl, pH 7.4, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2) at room temperature for 2 h.
The evaluation of σ2 receptor affinity was carried out according to a slightly modified literature procedure.55 Briefly, liver freshly isolated from female SPRD rats after decapitation of the deeply anaesthetized animals was weighed and homogenized on ice, and the pellet obtained by centrifugation was frozen in aliquots at a stock concentration of 10 mg/mL and stored at −25 °C until the day of the experiment.
To assess the σ2 receptor affinity, increasing concentrations of the respective test compound (10–11–10–5 M) were coincubated with the nonselective σ1/σ2 ligand (+)-[3H]DTG (American Radiolabeled Chemicals; AM = 1850 GBq/mmol) at a single concentration (3 nM) in the presence of 10 μM dextrallorphan (Hoffmann-La Roche) to mask σ1 receptor binding sites. The incubation with rat liver membrane homogenates was performed for 2 h at room temperature in 50 mM TRIS-HCl buffer (pH 7.4) and terminated by filtration through GF/B filters.
Nonspecific binding of both radioligands was determined by coincubation with 10 μM haloperidol. The incubations were terminated by filtration via GF/B glass fiber filters (48-Sample Semi-Auto Harvester) and filter-bound radioactivity was quantified by liquid scintillation counting (Hidex 600 SL). The inhibition binding data were expressed as percentage specific binding of the radioligand versus a logarithmic molar concentration of the test compound, and the inhibition curve was generated by nonlinear regression using the “one-site competition” equation in GraphPad Prism (Prism 3.0). From the IC50 values, the inhibition constants Ki were calculated using the Cheng–Prusoff equation implemented in GraphPad Prism (KD, (+)-[3H]pentazocine, σ1 = 33 nM; KD, [3H]DTG, σ2 = 29 nM).
Molecular Docking Studies
The interaction of selected compounds with human AChE, human MAO-B, and the human σ1 receptor was investigated using AutoDock Vina56 and Discovery Studio version 2.1, software package, for simulations and visualizations. Compound 19 was included as protonated amine for AChE and σ1 simulations. The molecular geometry of the compound was energy-minimized using the adopted-based Newton–Rapson algorithm with the CHARMm force field57 until the rms gradient was below 0.01 kcal mol–1 Å–1. The ligand was set up for docking with the help of AutoDockTools (ADT; version 1.5.6), and all the rotatable bonds were allowed to rotate freely. Three-dimensional crystal structures of human AChE crystallized with fasciculine (Protein Data Bank (PDB) ID: 1B41), human MAO-B crystallized with safinamide (PDB ID: 2 V5Z), and human σ1 receptor bound to the antagonist PD144418 (PDB ID: 5HK1) were obtained from the PDB.
First, in the PDB crystallographic structures of AChE and the σ1 receptor, water molecules, any cocrystallized solvent, and the ligand were removed. For MAO-B, six water molecules located around the FAD cofactor were considered in the docking experiments because they are essential for stabilizing the complexes. Then, proper bonds, bond orders, hybridization, and charges were assigned using a protein model tool in Discovery Studio software package.
ADT was used to add hydrogens and partial charges using Gasteiger charges and to generate the docking input files. The docking approach included protein flexibility through a set of different conformations of selected side chains into the AChE and σ1 receptor macromolecules.
The accuracy of the docking procedure was evaluated by redocking the cocrystallized ligands, safinamide and PD144418, into the binding sites of MAO-B and σ1, respectively. After each inhibitor was docked, the best-ranked orientations of safinamide and PD144418 exhibited RMSD values of 0.60 and 1.06 Å, respectively, from the position of the cocrystallized ligand.
AutoDock Vina software was employed for all the protein–ligand calculations with the default settings except num_modes, which was set to 40. The docking boxes were constructed, with suitable dimensions to include the most or even the whole protein target, using ADT and were positioned at the middle of the macromolecules.
AChE Molecular Modeling
The inclusion of the receptor flexibility has been taken into account by allowing side chain flexibility to the AChE binding site. Using the AutoTors module, side chains of eight residues were allowed to move, i.e., Trp286, Tyr124, Tyr337, Tyr341, Tyr72, Asp74, Thr75, and Trp86. The following grid box parameters were employed: 60 × 60 × 72 (number of grid points), 116.546, 110.330, −134.181 (center grid box), and 1 Å (grid point spacing). The more energetically favorable conformation was selected as the best pose.
MAO-B Molecular Modeling
The following grid box parameters were employed: 46 × 40 × 40 (number of grid points), 55.743, 161.175, 19.420 (center grid box), and 1 Å (grid point spacing). The more energetically favorable conformation was selected as the best pose. Compound 19 was docked as the neutral amine.58,59
σ1 Receptor Molecular Modeling
For the σ1 receptor, side chain flexibility was allowed for 12 amino acid residues, i.e., Tyr103, Glu172, Phe107, Asp126, Val152, Phe146, Gln135, His154, Glu158, Ser117, Tyr120, and Tyr206. The following grid box parameters were employed: 28 × 22 × 34 (number of grid points), −6.978, 20.413, −27.539 (center grid box), and 1 Å (grid point spacing). The more energetically favorable conformation was selected as the best pose.
Acknowledgments
T.K. was supported by a fellowship from the Jürgen Manchot Foundation, Düsseldorf, Germany. The automated cholinesterase assays were performed by A. Wosar (Bundeswehr Institute of Pharmacology and Toxicology) which is very much appreciated. The authors acknowledge support by Dr. Christian Steinebach, Nicole Schmitt, and Ablam-Thomas Koumako.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00097.
IC50 determination of AChE inhibition by 19, docked pose of inhibitor 19 in the active site of AChE, IC50 determination of BuChE inhibition by 1, IC50 determination of MAO-B inhibition by 12, reversibility assay for MAO-B inhibition by 12, docked pose of nonprotonated inhibitor 19 in the active site of MAO-B, docked pose of compound 19 bound to the σ1 receptor (PDF)
Author Contributions
J.M.C. and M.G. conceived the study. C.L. and M.C. performed chemical analyses. C.L., P.W.E., and W.D.C. performed biochemical and pharmacological experiments. M.A.M.G., C.D.B., and T.V. synthesized compounds. I.I. and F.L.M. carried out docking analyses. All authors contributed to data analyses. T.W., U.B., W.D.C., J.M.C., and M.G. supervised the project. T.K. and M.G. wrote the manuscript with contributions from all coauthors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): M.A.M.G. and C.D.B. are former employees of Eli Lilly & Company.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- León R.; Garcia A. G.; Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. 10.1002/med.20248. [DOI] [PubMed] [Google Scholar]
- Oset-Gasque M. J.; Marco-Contelles J. Alzheimer’s disease, the ″one-molecule, one-target″ paradigm, and the multitarget directed ligand approach. ACS Chem. Neurosci. 2018, 9, 401–403. 10.1021/acschemneuro.8b00069. [DOI] [PubMed] [Google Scholar]
- Proschak E.; Stark H.; Merk D. Polypharmacology by design: A medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2019, 62, 420–444. 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. 10.3892/mmr.2019.10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C. X.; Dai C. L.; Liu F.; Iqbal K. Multi-targets: An unconventional drug development strategy for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 837649. 10.3389/fnagi.2022.837649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman D. S.; Amieva H.; Petersen R. C.; Chételat G.; Holtzman D. M.; Hyman B. T.; Nixon R. A.; Jones D. T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. Chromone scaffolds in the treatment of Alzheimer’s and Parkinson’s disease: An overview. ChemistrySelect 2022, 7, e202200540 10.1002/slct.202200540. [DOI] [Google Scholar]
- Knez D.; Sova M.; Košak U.; Gobec S. Dual inhibitors of cholinesterases and monoamine oxidases for Alzheimer’s disease. Future Med. Chem. 2017, 9, 811–832. 10.4155/fmc-2017-0036. [DOI] [PubMed] [Google Scholar]
- do Carmo Carreiras M.; Ismaili L.; Marco-Contelles J. Propargylamine-derived multi-target directed ligands for Alzheimer’s disease therapy. Bioorg. Med. Chem. Lett. 2020, 30, 126880. 10.1016/j.bmcl.2019.126880. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zhang H. Reconsideration of anticholinesterase therapeutic strategies against Alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 852–862. 10.1021/acschemneuro.8b00391. [DOI] [PubMed] [Google Scholar]
- Košak U.; Brus B.; Knez D.; Žakelj S.; Trontelj J.; Pišlar A.; Sink R.; Jukič M.; Živin M.; Podkowa A.; Nachon A.; Brazzolotto X.; Stojan J.; Kos J.; Coquelle N.; Sałat K.; Colletier J. P.; Gobec S. The magic of crystal structure-based inhibitor optimization: Development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J. Med. Chem. 2018, 61, 119–139. 10.1021/acs.jmedchem.7b01086. [DOI] [PubMed] [Google Scholar]
- Cai Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. 10.3892/mmr.2014.2040. [DOI] [PubMed] [Google Scholar]
- Pan X.; Kaminga A. C.; Wen S. W.; Wu X.; Acheampong K.; Liu A. Dopamine and dopamine receptors in Alzheimer’s disease: A systematic review and network meta-analysis. Front. Aging Neurosci. 2019, 11, 175. 10.3389/fnagi.2019.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Zhang G.; Qin Z.; Yin M.; Chen W.; Zhang Y.; Liu X. Safinamide protects against amyloid β (Aβ)-induced oxidative stress and cellular senescence in M17 neuronal cells. Bioengineered 2022, 13, 1921–1930. 10.1080/21655979.2021.2022262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaroli L.; Daina A.; Favre E.; Bravo J.; Carotti A.; Leonetti F.; Catto M.; Carrupt P. A.; Reist M. Impact of species-dependent differences on screening, design, and development of MAO B inhibitors. J. Med. Chem. 2006, 49, 6264–6272. 10.1021/jm060441e. [DOI] [PubMed] [Google Scholar]
- Wu N. H.; Ye Y.; Wan B. B.; Yu Y. D.; Liu C.; Chen Q. J. Emerging benefits: Pathophysiological functions and target drugs of the sigma-1 receptor in neurodegenerative diseases. Mol. Neurobiol. 2021, 58, 5649–5666. 10.1007/s12035-021-02524-5. [DOI] [PubMed] [Google Scholar]
- Malik M.; Rangel-Barajas C.; Sumien N.; Su C.; Singh M.; Chen Z.; Huang R. Q.; Meunier J.; Maurice T.; Mach R. H.; Luedtke R. R. The effects of sigma (σ1) receptor-selective ligands on muscarinic receptor antagonist-induced cognitive deficits in mice. Br. J. Pharmacol. 2015, 172, 2519–2531. 10.1111/bph.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. R.; Zheng S.; Gurpinar E.; Koehl A.; Manglik A.; Kruse A. C. Human sigma-1 receptor bound to PD144418. Nature 2016, 532, 527–530. 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogár F.; Fülöp L.; Penke B. Novel therapeutic target for Prevention of neurodegenerative diseases: Modulation of neuroinflammation with Sig-1R ligands. Biomolecules 2022, 12, 363. 10.3390/biom12030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena E.; Dichiara M.; Floresta G.; Parenti C.; Marrazzo A.; Pittala V.; Amata E.; Prezzavento O. Novel Sigma-1 receptor antagonists: from opioids to small molecules: what is new?. Future Med. Chem. 2018, 10, 231–256. 10.4155/fmc-2017-0164. [DOI] [PubMed] [Google Scholar]
- Fallica A. N.; Pittalà V.; Modica M. N.; Salerno L.; Romeo G.; Marrazzo A.; Helal M. A.; Intagliata S. Recent advances in the development of sigma receptor ligands as cytotoxic agents: A medicinal chemistry perspective. J. Med. Chem. 2021, 64, 7926–7962. 10.1021/acs.jmedchem.0c02265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi B.; Sahn J. J.; Ardestani P. M.; Evans A. K.; Scott L. L.; Chan J. Z.; Iyer S.; Crisp A.; Zuniga G.; Pierce J. T.; Martin S. F.; Shamloo M. Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem. 2017, 140, 561–575. 10.1111/jnc.13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo N. J.; Yuede C. M.; LaBarbera K. M.; Limegrover C. S.; Rehak C.; Yurko R.; Waybright L.; Look G.; Rishton G.; Safferstein H.; Hamby M. E.; Williams C.; Sadlek K.; Edwards H. M.; Davis C. S.; Grundman M.; Schneider L. S.; DeKosky S. T.; Chelsky D.; Pike I.; Henstridge C.; Blennow K.; Zetterberg H.; LeVine H. 3rd; Spires-Jones T. L.; Cirrito J. R.; Catalano S. M. Preclinical and clinical biomarker studies of CT1812: A novel approach to Alzheimer’s disease modification. Alzheimers Dement. 2021, 17, 1365–1382. 10.1002/alz.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista-Aguilera O. M.; Hagenow S.; Palomino-Antolín A.; Farré-Alins V.; Ismaili L.; Joffrin P.-L.; Jimeno M. L.; Soukup O.; Janockova J.; Kalinowsky L.; Proschak E.; Iriepa I.; Moraleda I.; Schwed J. S.; Romero Martínez A.; Lopez-Muñoz F.; Chioua M.; Egea J.; Ramsay R. R.; Marco-Contelles J.; Stark H. Multitarget-directed ligands combining cholinesterase and monoamine oxidase inhibition with histamine H3R antagonism for neurodegenerative diseases. Angew. Chem., Int. Ed. 2017, 56, 12765–12769. 10.1002/anie.201706072. [DOI] [PubMed] [Google Scholar]
- Bautista-Aguilera Ó. M.; Budni J.; Mina F.; Medeiros E. B.; Deuther-Conrad W.; Entrena J. M.; Moraleda I.; Iriepa I.; López-Muñoz F.; Marco-Contelles J. Contilisant, a tetratarget small molecule for Alzheimer’s disease therapy combining cholinesterase, monoamine oxidase inhibition, and H3R antagonism with S1R agonism profile. J. Med. Chem. 2018, 61, 6937–6943. 10.1021/acs.jmedchem.8b00848. [DOI] [PubMed] [Google Scholar]
- Lemke C.; Christmann J.; Yin J.; Alonso J. M.; Serrano E.; Chioua M.; Ismaili L.; Martínez-Grau M. A.; Beadle C. D.; Vetman T.; Dato F. M.; Bartz U.; Elsinghorst P. W.; Pietsch M.; Müller C. E.; Iriepa I.; Wille T.; Marco-Contelles J.; Gütschow M. Chromenones as multineurotargeting inhibitors of human enzymes. ACS Omega 2019, 4, 22161–22168. 10.1021/acsomega.9b03409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuther-Conrad W.; Diez-Iriepa D.; Iriepa I.; López-Muñoz F.; Martínez-Grau M. A.; Gütschow M.; Marco-Contelles J. Studies on the affinity of 6-[(n(cyclo)aminoalkyl)oxy]-4H-chromen-4-ones for sigma 1/2 receptors. RSC Med. Chem. 2021, 12, 1000–1004. 10.1039/D1MD00105A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda T.; Ozaki F.; Shirakata N.; Tamaoka Y.; Yamamoto H.; Itô S. Formation of heterocycles by Mitsunobu reaction. Tetrahedron Lett. 1996, 37, 2463–2466. 10.1016/0040-4039(96)00319-X. [DOI] [Google Scholar]
- Swamy K. C.; Kumar N. N.; Balaraman E.; Kumar K. V. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- Nowakowska Z.; Kedzia B.; Schroeder G. Synthesis, physicochemical properties and antimicrobial evaluation of new (E)-chalcones. Eur. J. Med. Chem. 2008, 43, 707–713. 10.1016/j.ejmech.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Sun Y.; Guo Y.; Wang Z.; Huang L.; Li X. Dual functional cholinesterase and MAO inhibitors for the treatment of Alzheimer’s disease: synthesis, pharmacological analysis and molecular modeling of homoisoflavonoid derivatives. J. Enzyme Inhib. Med. Chem. 2016, 31, 389–397. 10.3109/14756366.2015.1024675. [DOI] [PubMed] [Google Scholar]
- Godfrey A. G.; Masquelin T.; Hemmerle H. A remote-controlled adaptive medchem lab: An innovative approach to enable drug discovery in the 21st Century. Drug Discovery Today 2013, 18, 795–802. 10.1016/j.drudis.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Jiang N.; Ding J.; Liu J.; Sun X.; Zhang Z.; Mo Z.; Li X.; Yin H.; Tang W.; Xie S. S. Novel chromanone-dithiocarbamate hybrids as multifunctional AChE inhibitors with β-amyloid anti-aggregation properties for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 89, 103027. 10.1016/j.bioorg.2019.103027. [DOI] [PubMed] [Google Scholar]
- Farina R.; Pisani L.; Catto M.; Nicolotti O.; Gadaleta D.; Denora N.; Soto-Otero R.; Mendez-Alvarez E.; Passos C. S.; Muncipinto G.; Altomare C. D.; Nurisso A.; Carrupt P. A.; Carotti A. Structure-based design and optimization of multitarget-directed 2H-chromen-2-one derivatives as potent inhibitors of monoamine oxidase B and cholinesterases. J. Med. Chem. 2015, 58, 5561–5578. 10.1021/acs.jmedchem.5b00599. [DOI] [PubMed] [Google Scholar]
- Ekström F.; Gottinger A.; Forsgren N.; Catto M.; Iacovino L. G.; Pisani L.; Binda C. Dual reversible coumarin inhibitors mutually bound to monoamine oxidase B and acetylcholinesterase crystal structures. ACS Med. Chem. Lett. 2022, 13, 499–506. 10.1021/acsmedchemlett.2c00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. B.; Yin F. C.; Huang M.; Jiang N.; Lan J. S.; Kong L. Y. Chromone and donepezil hybrids as new multipotent cholinesterase and monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. RSC Med. Chem. 2020, 11, 225–233. 10.1039/C9MD00441F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens M. D.; Hinz S.; Müller C. E.; Gütschow M. Alkynyl-coumarinyl ethers as MAO-B inhibitors. Bioorg. Med. Chem. 2014, 22, 1916–1928. 10.1016/j.bmc.2014.01.046. [DOI] [PubMed] [Google Scholar]
- Fonseca A.; Reis J.; Silva T.; Matos M. J.; Bagetta D.; Ortuso F.; Alcaro S.; Uriarte E.; Borges F. Coumarin versus chromone monoamine oxidase B inhibitors: Quo vadis?. J. Med. Chem. 2017, 60, 7206–7212. 10.1021/acs.jmedchem.7b00918. [DOI] [PubMed] [Google Scholar]
- Hall H. K. Jr. Correlation of the base strengths of amines. J. Am. Chem. Soc. 1957, 79, 5441–5444. 10.1021/ja01577a030. [DOI] [Google Scholar]
- Masaldan S.; Bush A. I.; Devos D.; Rolland A. S.; Moreau C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. 10.1016/j.freeradbiomed.2018.09.033. [DOI] [PubMed] [Google Scholar]
- Gitlin-Domagalska A.; Mangold M.; Dębowski D.; Ptaszyńska N.; Łęgowska A.; Gütschow M.; Rolka K. Matriptase-2: Monitoring and inhibiting its proteolytic activity. Future Med. Chem. 2018, 10, 2745–2761. 10.4155/fmc-2018-0346. [DOI] [PubMed] [Google Scholar]
- Brune S.; Pricl S.; Wünsch B. Structure of the σ1 receptor and its ligand binding site. J. Med. Chem. 2013, 56, 9809–9819. 10.1021/jm400660u. [DOI] [PubMed] [Google Scholar]
- Greenfield D. A.; Schmidt H. R.; Skiba M. A.; Mandler M. D.; Anderson J. R.; Sliz P.; Kruse A. C. Virtual screening for ligand discovery at the σ1 receptor. ACS Med. Chem. Lett. 2020, 11, 1555–1561. 10.1021/acsmedchemlett.9b00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnier-Maréchal M.; Carato P.; Le Broc D.; Furman C.; Melnyk P. Synthesis and pharmacological evaluation of benzannulated derivatives as potent and selective sigma-1 protein ligands. Eur. J. Med. Chem. 2015, 92, 575–582. 10.1016/j.ejmech.2014.01.013. [DOI] [PubMed] [Google Scholar]
- Donnier-Maréchal M.; Carato P.; Larchanché P. E.; Ravez S.; Boulahjar R.; Barczyk A.; Oxombre B.; Vermersch P.; Melnyk P. Synthesis and pharmacological evaluation of benzamide derivatives as potent and selective sigma-1 protein ligands. Eur. J. Med. Chem. 2017, 138, 964–978. 10.1016/j.ejmech.2017.07.014. [DOI] [PubMed] [Google Scholar]
- Szczepańska K.; Podlewska S.; Dichiara M.; Gentile D.; Patamia V.; Rosier N.; Mönnich D.; Ruiz Cantero M. C.; Karcz T.; Łażewska D.; Siwek A.; Pockes S.; Cobos E. J.; Marrazzo A.; Stark H.; Rescifina A.; Bojarski A. J.; Amata E.; Kieć-Kononowicz K. Structural and molecular insight into piperazine and piperidine derivatives as histamine H3 and sigma-1 receptor antagonists with promising antinociceptive properties. ACS Chem. Neurosci. 2022, 13, 1–15. 10.1021/acschemneuro.1c00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg E.; Weber F.; Brune S.; Schepmann D.; Almansa C.; Friedland K.; Laurini E.; Pricl S.; Wünsch B. Synthesis and structure-affinity relationships of spirocyclic benzopyrans with exocyclic amino moiety. J. Med. Chem. 2019, 62, 4204–4217. 10.1021/acs.jmedchem.9b00449. [DOI] [PubMed] [Google Scholar]
- Holtschulte C.; Börgel F.; Westphälinger S.; Schepmann D.; Civenni G.; Laurini E.; Marson D.; Catapano C. V.; Pricl S.; Wünsch B. Synthesis of aminoethyl-substituted piperidine derivatives as σ1 receptor ligands with antiproliferative properties. ChemMedChem 2022, 17, e202100735 10.1002/cmdc.202100735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. R.; Betz R. M.; Dror R. O.; Kruse A. C. Structural basis for σ 1 receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. 10.1038/s41594-018-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos M. A.; Sousa C. C.; Miranda M. S.; Morais V. M.; Liebman J. F. Energetics of coumarin and chromone. J. Phys. Chem. B 2009, 113, 11216–11221. 10.1021/jp9026942. [DOI] [PubMed] [Google Scholar]
- Lukin K.; Kishore V.; Gordon T. Development of a scalable synthesis of oxadiazole based S1P1 receptor agonists. Org. Process Res. Dev. 2013, 17, 666–671. 10.1021/op300345v. [DOI] [Google Scholar]
- Koshizawa T.; Yamazaki Y.; Ohgiya T.; Shibuya K. A facile method for converting alcohol to thioether and its application in the synthesis of a novel GPR119 agonist. Tetrahedron 2015, 71, 3231–3236. 10.1016/j.tet.2015.04.002. [DOI] [Google Scholar]
- Chand K.; Tiwari R. K.; Kumar S.; Shirazi A. N.; Sharma S.; Van der Eycken E. V.; Parmar V. S.; Parang K.; Sharma S. K. Synthesis, antiproliferative, and c-Src kinase inhibitory activities of 4-oxo-4H-1-benzopyran derivatives. J. Heterocyclic Chem. 2015, 52, 562–572. 10.1002/jhet.2106. [DOI] [Google Scholar]
- Worek F.; Reiter G.; Eyer P.; Szinicz L. Reactivation kinetics of acetylcholinesterase from different species inhibited by highly toxic organophosphates. Arch. Toxicol. 2002, 76, 523–529. 10.1007/s00204-002-0375-1. [DOI] [PubMed] [Google Scholar]
- Chu U. B.; Ruoho A. E. Sigma receptor binding assays. Curr. Protoc. Pharmacol. 2015, 71, 1.34.1–1.34.21. 10.1002/0471141755.ph0134s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B.; Bruccoleri R.; Olafson B.; States D.; Swaminathan S.; Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Jones T. Z.; Balsa D.; Unzeta M.; Ramsay R. R. Variations in activity and inhibition with pH: the protonated amine is the substrate for monoamine oxidase, but uncharged inhibitors bind better. J. Neural. Transm. 2007, 114, 707–712. 10.1007/s00702-007-0675-y. [DOI] [PubMed] [Google Scholar]
- Dunn R. V.; Marshall K. R.; Munro A. W.; Scrutton N. S. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS J. 2008, 275, 3850–3858. 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.