

Abstract
Pyrazole is a five-membered heterocycle bearing two adjacent nitrogen atoms. Both pharmaceutical agents and natural products with pyrazole as a nucleus have exhibited a broad spectrum of biological activities. In the last few decades, more than 40 pyrazole-containing drugs have been approved by the FDA for the treatment of a broad range of clinical conditions including celecoxib (anti-inflammatory), CDPPB (antipsychotic), difenamizole (analgesic), etc. Owing to the unique physicochemical properties of the pyrazole core, pyrazole-containing drugs may exert better pharmacokinetics and pharmacological effects compared with drugs containing similar heterocyclic rings. The purpose of this paper is to provide an overview of all the existing drugs bearing a pyrazole nucleus that have been approved or in clinical trials, involving their pharmacological activities and SAR studies.
This review paper provides an overview of all the existing drugs bearing a pyrazole nucleus that have been approved or in clinical trials, involving their pharmacological activities and SAR studies.
1. Introduction
Pyrazole is an important heterocyclic motif in medicinal chemistry and organic synthesis. Although not many pyrazole-containing compounds exist in nature, recent trends have shown that the pyrazole ring plays an increasingly important role in drug pipelines. There are over 50 pyrazole-containing synthetic medicines on the market globally. Specifically, the United States Food and Drug Administration (US FDA) has approved more than 30 pyrazole-containing drugs since 2011, with a maximum number of six drugs approved solely in 2019 (Fig. 1). These marketed drugs with a pyrazole moiety target a wide range of clinical disorders, including hereditary angioedema, non-small cell lung cancer (NSCLC), sickle cell disease, cystic fibrosis, rheumatoid arthritis, etc.
Fig. 1. The number of pyrazole-containing drugs approved by the FDA (as of April 2022). For the drugs that have been approved by the FDA for the treatment of different indications many times, only one time is counted.

Pyrazole is a five-membered aromatic heterocycle with two adjacent N heteroatoms (Fig. 2).1,2 Its N-1 atom has similar properties to the NH of pyrrole which can serve as a hydrogen bond donor, and its N-2 atom behaves similarly to the nitrogen atom of pyridine which can serve as a hydrogen bond acceptor. Due to different chemical environments, all five bonds have different bond lengths. The aromaticity of pyrazoles lies at an intermediate level among the other aromatic heterocycles (Fig. 3).3,4
Fig. 2. The structure of pyrazole.
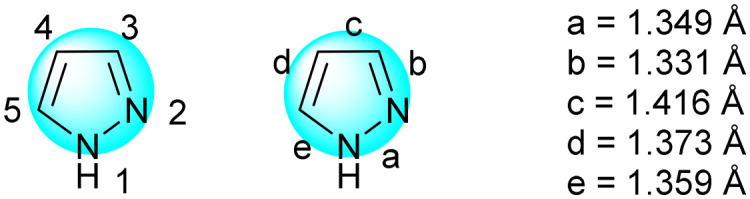
Fig. 3. The aromaticity rank of aromatic heterocycles.

Pyrazole has a pKa value of 2.5, but it is significantly less basic than imidazole with a pKa value of 7.1. With an adjacent heteroatom connected to the N atom, the basicity of the nitrogen being connected is reduced because of the inductive effect exerted by the adjacent heteroatom. Nevertheless, pyrazoles are basic enough to be protonated by most strong inorganic acids. When pyrazole is substituted with an unsymmetric substituent, it may exist as a mixture of two tautomers. For example, 5-methylpyrazole and 3-methylpyrazole often coexist in a solution, and there is an equilibrium relationship between them. The result of tautomerism is that the alkylation of unsymmetrically substituted pyrazoles produces a mixture of two isomers (Fig. 4), one is the N-1 alkylation and the other is the N-2 alkylation. The ratio depends on the nature of the substituent and the solvent. The pyrazole-containing pharmaceuticals discussed in this article include their tautomers. In drug design applications, the unique properties of the pyrazole ring compared to other aromatic rings are often considered to be critical in improving the biological activity and physicochemical properties.
Fig. 4. Tautomerization of pyrazole.

This review summarized versatile pyrazole-containing drugs on the market or under clinical development in the following aspects: target, pharmacological activity, and their structure–activity relationship. However, some withdrawn or veterinary drugs, as well as investigational and experimental drugs, are not discussed here. These pyrazole-containing drugs were categorized based on their therapeutic areas. We expect that this review could provide a useful reference to both medicinal chemists and organic chemists in the aspects of rational drug design and chemical synthesis.
2. General drug design strategies for introducing the pyrazole skeleton
Generally, pyrazole can serve as a bioisostere to replace an arene, leading to enhanced potency and improved physicochemical properties, such as lipophilicity and water solubility. Considering the aromaticity, pyrazole (61) and benzene (100) are quite different, but the lipophilicity of pyrazole (Clog P = 0.24) is significantly lower than that of benzene (Clog P = 2.14). At the same time, containing an H-bond donor, pyrazole can also be considered as a bioisostere for more lipophilic and metabolically incompetent arenes, such as phenol and other heterocycles.
For example, losartan (1) was the first selective non-peptide angiotensin II receptor antagonist on the market for treating hypertension. It was soon discovered that its carboxylic acid metabolite, imidazole acid 2, was significantly more potent than losartan with similar or longer duration of action. Ashton's group switched to pyrazole as the bioisostere of losartan's imidazole moiety. To this end, they discovered a series of pyrazole compounds as represented by pyrazole 3, which had similar potency to the imidazole derivatives (Fig. 5).5
Fig. 5. Pyrazole as a bioisostere for the arene core.

On the other hand, pyrazole can serve as a hydrogen bond donor and an acceptor. Its N-1 can be employed as a hydrogen bond donor, and its N-2 can be employed as a hydrogen bond acceptor to form a hydrogen bond interaction with the amino acid in the active site of the enzyme. The first D1/D5 antagonist with high affinity and selectivity, SCH-23390 (4), was reported in the 1980s.6,7 However, benzazepine 4 has a short duration of action and obvious off-target effects, which indicates that there may be metabolic problems in vivo. Pharmacokinetic evaluations suggested that extensive O-glucuronidation of the phenol and N-dealkylation of the N–Me group in vivo may contribute to the poor PK profile. In general, the heterocyclic bioisosteres of phenol are more lipophilic and less sensitive to phase I and II metabolisms than phenol itself. Therefore, an attempt has been made to replace the metabolically problematic phenol with a pyrazole that can provide a hydrogen bond function. Indazole 5 was tested to be approximately 10 times less potent than SCH-23390 (4), but began to display an improved PK profile. As a demonstration of the importance of the hydrogen bond donor, the affinity of methylated indazole 6 for the D1 receptor was significantly reduced (Fig. 6).8
Fig. 6. Improving the PK profile by introducing the pyrazole motif.

3. Pyrazole-containing drugs targeting cardiovascular system diseases
Berotralstat (BCX-7353) (7) was approved by the FDA in 2020, and it is a selective inhibitor of the plasma kinase releasing enzyme for the prevention of episodes of hereditary angioedema (HAE), a rare genetic disorder associated with severe swelling of the skin and upper respiratory tract (Fig. 7a).9 It is caused by mutations in the regulatory or coding region of the gene encoding the C1 inhibitor, resulting in C1 inhibitor deficiency (type I) or dysfunction (type II). The C1 inhibitor is a serine protease inhibitor that regulates the production of bradykinin, usually by covalently binding to and inactivating plasma kininase. The plasma kinin releasing enzyme is a protease that cleaves high molecular weight kininogen (HMWK) to produce cleaved HMWK. During the HAE onset, the plasma kinin releasing enzyme levels decrease, leading to cleavage of high molecular weight kininogen and release of bradykinin, a potent vasodilator that increases vascular permeability. Bradykinin plays a major role in promoting edema and pain associated with HAE. Patients with HAE are unable to properly regulate plasma kininase activity due to the lack or dysfunction of serum inhibitors of C1 inhibitors, resulting in uncontrolled increases in plasma kininase activity and recurrent angioedema episodes.10 Berotralstat (7) works by binding to plasma kininase and blocking its proteolytic activity, thereby controlling excess bradykinin production, which is used strictly to prevent rather than treat these attacks. In the drug structure, the pyrazole–benzylamine part is a factor Xa (FXa) serine protease inhibitor that has an integral role in the overall drug structure. The protonated benzylamine forms hydrogen bonds with Asp572, Gly601, and Ala573 in the S1 pocket and forms a trifluoro substitution in the pyrazole carbon stacking at the top of the disulfide bond (Cys609). The pyrazole ring forms a π–π interaction with Trp598, which is essential for drug binding (Fig. 7b).11
Fig. 7. (a) The structure of berotralstat (7); (b) the key target interactions.

Apixaban (BMS-562247) (8) was approved by the FDA in 2012, and it is an oral, direct, and highly selective FXa inhibitor12 that inhibits free and bound FXa and prothrombin,13 independent of antithrombin III, for the prevention and treatment of thromboembolic disease (Fig. 8a).14 Concerning the structure of apixaban, the pyrazolo-piperidone is critical in that it allows the carbonyl group of the piperidone to form hydrogen bonds with Gly216 and structural water molecules, enhancing the affinity of the drug to the target site.15 It also enables the adjacent amino group to form hydrogen bonds with Glu146, which further enables the drug molecule to occupy the target pocket (Fig. 8b).
Fig. 8. (a) The structure of apixaban (8) and razaxaban (9); (b) the key target interactions.

Razaxaban (BMS-561389) (9) is also an orally active, 1,2-benzisoxazole-containing inhibitor15 of FXa with anticoagulant activity. It can form an isoxazole ring-opening stable metabolite of benzamidine, which is subsequently excreted through the bile (Fig. 8a).16 The most important core of the drug structure is the C-3 methylamino pyrazole part,17 which allows the carbonyl group on the amide bond to form H-bond interactions with structural water and Gly216, while allowing the imidazole moiety to form π–π interactions with Trp215 and Phe174, which greatly enhances the affinity of the drug molecule to the target. The pyrazole 3-trifluoromethyl part fits into a small lipophilic pocket above the S1 pocket near the Cys191–220 disulfide bond, which provides a favorable protein–ligand binding interaction (Fig. 8b).18
Idiopathic thrombocytopenia (ITP) is a condition that may cause abnormal bruising or bleeding due to an abnormally low number of platelets in the blood.19 Eltrombopag (SB-497115) (10) is a thrombopoietin receptor agonist approved by the FDA for the treatment of ITP or aplastic anemia associated with various etiologies.20 It was also approved (late 2012) for the treatment of thrombocytopenia (low platelet count) in patients with chronic hepatitis C to allow them to initiate and maintain interferon-based therapy. Eltrombopag (10) is an orally bioavailable, small molecule TPO receptor agonist that interacts with the transmembrane structural domain of the human TPO receptor. The molecule docking of thrombopoietin and eltrombopag reveals their combination action mode and the pyrazole moiety forms a π–π interaction with Phe261 (Fig. 9).21
Fig. 9. The structure of eltrombopag (10).

Riociguat (BAY-412272) (11) is a soluble guanylate cyclase (sGC) agonist22 for the treatment of patients with PAH (pulmonary arterial hypertension)23 and inoperable patients with CTEPH (chronic thromboembolic pulmonary hypertension)24 or persistent PAH after pulmonary endarterectomy (Fig. 10a). Riociguat (11) is a stimulator of sGC, an enzyme and receptor for nitric oxide (NO) in the cardiopulmonary system. When NO binds to sGC, the enzyme catalyzes the synthesis of the signaling molecule cyclic guanosine monophosphate (cGMP). Intracellular cGMP plays an important role in the regulation of processes affecting vascular tone, proliferation, fibrosis, and inflammation. Pulmonary hypertension is associated with endothelial dysfunction, impaired nitric oxide synthesis, and inadequate stimulation of the NO–sGC–cGMP pathway.25 Riociguat (11) stimulates the NO–sGC–cGMP pathway and leads to an increase in cGMP production and subsequent vasodilation. This mechanism of action is also used by another drug, vericiguat (BAY-1021189) (12), which alleviates the need for a functional NO–sGC–cGMP axis26 and thus contributes to the prevention of myocardial and vascular dysfunction27 associated with reduced sGC activity in heart failure (Fig. 10b).28 Thus, vericiguat (12) is used to reduce cardiovascular death in patients with chronic systolic heart failure, and the drug was approved by the FDA in 2021. The molecular docking results suggested that riociguat (11) and vericiguat (12) exhibited a similar binding fashion with guanylate cyclase via reversible noncovalent interactions involving hydrogen bonding and π–π interaction.29 Pyrazole mainly acted as a bioisostere of the aryl group to improve lipophilicity.
Fig. 10. The structures of riociguat (11) and vericiguat (12).

Deoxygenated sickle hemoglobin (HbS) aggregation is the causative factor in sickle cell disease.30 Mutations in genes associated with this disease result in the formation of abnormal sickle-shaped red blood cells that accumulate and block blood vessels throughout the body, leading to a vaso-occlusive crisis.31 To treat sickle cell disease, voxelotor (GBT-440) (13) received accelerated approval from the FDA in 2019 because it may be a promising treatment for the 20 million people worldwide who have this disease (Fig. 11). For the mechanism of action of voxelotor (13), it irreversibly binds to the N-terminal valine of the hemoglobin alpha chain, leading to a metamorphic modification of Hb20, which increases the affinity for oxygen. Because oxygen-containing HbS does not polymerize, by directly blocking HbS polymerization, voxelotor (13) can successfully treat sickle cell disease by preventing the formation of abnormally shaped cells that eventually lead to hypoxia and blood flow to organs.32 The pyrazole ring mainly plays the role of localization, causing the N of the pyridine ring to form hydrogen bond interaction with Phe175.
Fig. 11. The structure of voxelotor (13).

Sildenafil (HIP-0908) (14) is a phosphodiesterase 5 (PDE5) inhibitor that is used for two main indications.33 One is the treatment of erectile dysfunction (ED)34 and the other is the treatment of PAH (Fig. 12a).35 The physiological mechanism of penile erection involves the release of NO from the corpus cavernosum during sexual stimulation. Nitric oxide then activates guanylate cyclase, leading to increased levels of cGMP, which produces smooth muscle relaxation in the corpus cavernosum and allows blood to flow in.36 Sildenafil (14) has no direct relaxing effect on isolated human cavernous bodies, but effectively enhances the relaxing effect of NO on this tissue. In addition, PDE5 is also present in the pulmonary vascular system, which is the reason for its second indication.37 It increases cGMP in pulmonary vascular smooth muscle cells, which leads to relaxation. In the structure of sildenafil (14), pyrazole and pyrimidine are crucial.38 The pyrazole ring can form π–π interactions with Try612, the pyrimidine ring forms π–π interactions with Phe820, and the NH and carbon groups also form H-bonding interactions with Gln817 (Fig. 12b). This makes the drug molecule well bound to the hydrophilic pocket. It is also reported that potent and selective PDE5 inhibitors could be achieved using basic alkyl or heteroaryl N-2 pyrazole substituents. However, if the pyrazole ring is substituted, the selective inhibition of PDE5 is lost, suggesting an important role of the pyrazole ring in drug interactions with PDE5.39 Udenafil (DA-8159) (15) is also a new PDE5 inhibitor used to treat ED.40 It has been approved in South Korea but not yet approved for use in the U.S. The inhibition of PDE5 by udenafil (15) enhances erectile function by increasing the amount of cGMP, which is similar to sildenafil.41 Due to the different positioning effects of other groups, udenafil (15) forms π–π interactions with Tyr612 and Phe820 on different sides at the same time.
Fig. 12. (a) The structures of sildenafil (14) and udenafil (15); (b) the key target interactions.

4. Pyrazole-containing drugs targeting endocrine system diseases
4.1. Drugs to treat endocrine organs
Niraparib (MK-4827) (16) was approved by the FDA in 2017 for the treatment of recurrent epithelial ovarian,42 fallopian tube,43 or primary peritoneal cancer that responded to platinum-based chemotherapy (Fig. 13a). For the mechanism of action, niraparib (16) is an inhibitor of the poly ADP-ribose polymerase (PARP) enzymes PARP-1 and PARP-2, which play a role in DNA repair. In vitro studies suggest that the niraparib-induced cytotoxicity may involve inhibition of PARP enzyme activity and increased PARP–DNA complex formation, leading to DNA damage, apoptosis, and cell death.44 Several [6,5] dense aromatic azabicycles were designed in which a nitrogen atom in the five-membered ring is placed to interact with the amide's anti-NH to form a critical intramolecular hydrogen bond.45 This demonstrated that the drug had the highest PARP-1 inhibitory activity when the five-membered ring was a pyrazole ring and Y = N (Fig. 13b).
Fig. 13. (a) The structure of niraparib (16); (b) [6,5] fused aromatic azabicycles.

Marginal zone lymphoma (MZL) is a rare, slowly progressive form of non-Hodgkin's lymphoma that is initially treated with rituximab (an anti-CD20 drug) alone or in combination with chemotherapy.46 Unfortunately, many patients relapse or develop resistance to these drugs. Treatment options then become limited, and alternative treatments for lymphoma are needed to control disease progression.47 In 2021, the FDA accelerated the approval of umbralisib (TGR-1202) (21), a kinase inhibitor of PI3K-delta and casein kinase CK1-epsilon, for the treatment of relapsed and refractory marginal cell lymphoma and follicular lymphoma in adults (Fig. 14a).48 The PI3K pathway is dysregulated in malignant tumors, leading to overexpression of p110 isoforms (p110α, p110β, p110δ, p110γ), which induces malignant transformation of cells.49 Umbralisib mainly inhibits PI3Kδ and the casein kinase CK1ε, the former is expressed in both healthy and malignant B cells, and CK1ε is thought to be involved in the pathogenesis of malignant cells, including lymphoma. Pyrazolopyrimidines play a crucial role in conformational studies, where the N of the pyrimidine can form H-bond interactions with Leu85 and the amino group can form H-bond interactions with Glu83, which allows the drug molecule to be well bound in the target pocket (Fig. 14b).
Fig. 14. (a) The structure of umbralisib (21); (b) the key target interactions.

Darolutamide (BAY-1841788) (22) is a nonsteroidal androgen receptor (AR) antagonist that received FDA approval in 2019 for the treatment of desmoid-resistant nonmetastatic prostate cancer (nmCRPC) (Fig. 15a).50 The AR is a key target for rational drug design for the treatment of prostate cancer, and androgens can enhance the growth and survival of prostate cancer cells by binding to the AR.51 Darolutamide (22) competitively inhibits the binding of androgens to their receptors and suppresses AR nuclear translocation as well as AR-mediated transcription. The result of these processes is a reduction in prostate cancer cell proliferation and tumor size. The androgen receptor target is a narrow pocket, and the two pyrazole structures before and after can better bind to the target. One of them can form a π–π interaction with Arg840, while N can form an H-bond interaction with Phe673 (Fig. 15b). For the mechanism of action, the face-to-face stacking interaction of the eastern pyrazole part with the indole group of Trp742 was identified as the key interaction,52 which allows darolutamide (22) to bind more tightly to the AR receptor than other AR antagonists, such as apalutamide and enzalutamide.53
Fig. 15. (a) The structure of darolutamide (22); (b) the key target interactions.

4.2. Drugs to regulate hormone levels
Since 2012, several new drugs have been approved by the FDA to treat type 2 diabetes mellitus (T2DM), including anagliptin (23) (2012), teneligliptin (24) (2013) and omarigliptin (25) (2015) (Fig. 16a). They are orally effective and selective inhibitors of dipeptidyl peptidase 4 (DPP-4) that improve glycemic control in patients with T2DM by prolonging the half-life of glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). Anagliptin (23) produces a longer-lasting inhibition of DPP-4 activity than earlier approved DPP-4 drugs like vildagliptin. In the DPP-4 structure complexed with anagliptin (23),54 the pyrazolopyrimidine ring interacts with the benzene ring of Phe357 in a π–π stacking interaction, which not only enhances the potency of the drug but also increases its selectivity (Fig. 16b). Teneligliptin (24) occupies the active site in a linear conformation, and its binding leads to a conformational change involving Arg358, which adopts a unique side-chain orientation that opens the hydrophobic pocket known as the extended S2 pocket.55 Teneligliptin (24) induces the same conformational change involving Arg358 and occupies the extended S2 pocket with the phenyl and trifluoromethyl parts in a similar manner. The structure optimization process of omarigliptin (25) is shown in Fig. 16c, where the trans-2,5-difluorophenyl-3-amino group is essential for binding to the active site of the DPP-4 protein.56 The substitution of 2-pyrazole hydrogen with a methylsulfonyl group resulted in omarigliptin, which had a shorter half-life compared to the corresponding methylsulfonyl analog in preclinical pharmacokinetic studies.57 Based on these three drugs, the importance of pyrazole for the development of DPP-4 inhibitors is demonstrated, including improvements in affinity and druggability.
Fig. 16. (a) The structure of anagliptin (23), teneligliptin (24) and omarigliptin (25); (b) the key target interactions; (c) the structure optimization process of omarigliptin (25).

5. Pyrazole-containing drugs targeting lymphatic system diseases
5.1. Drugs to treat lymphoid cells
Bruton's tyrosine kinase (BTK) is a non-receptor kinase that is a signaling molecule for B-cell receptors expressed on the surface of peripheral B cells.58 The BCR signaling pathway plays a critical role in normal B cell development as well as in the proliferation and survival of malignant B cells in many B cell malignancies, including mantle cell lymphoma (MCL).59 Once activated by upstream Src family kinases, BTK phosphorylates phospholipase-Cγ (PLCγ), leading to Ca2+ mobilization and activation of NF-κB and MAP kinase pathways. These downstream cascades promote the expression of genes involved in B cell proliferation and survival.60 Ibrutinib (PCI-32765) (29) is an irreversible, potent BTK inhibitor (Fig. 17a) that has received FDA approvals (2012, 2013, and 2017) for the treatment of MCL,61 chronic lymphocytic leukemia (CLL) and Waldenstrom's macroglobulinemia (WM), and chronic graft-versus-host disease (GVHD). Ibrutinib (29) forms a covalent bond with the cysteine residue in the BTK active site (Cys481), leading to irreversible inhibition of the enzymatic activity with sub-nanomolar IC50. It also forms four H-bonds with Thr474, Glu475, and Cys481 and several water bridges with adjacent residues in the pyrazolopyrimidine core (Fig. 17b).
Fig. 17. (a) The structure of ibrutinib (29); (b) the key target interactions.

Zanubrutinib (BGB-3111) (30) is a BTK inhibitor that received accelerated approval from the FDA in 2019 for the treatment of adult patients with MCL who have received at least one treatment62 (Fig. 18a). Compared to ibrutinib, the first-in-class BTK inhibitor, zanubrutinib (30) is more potent and selective for BTK, with fewer off-target effects.63 In the active site of BTK (Fig. 18b), the α,β-unsaturated carbonyl group in zanubrutinib forms a covalent bond with Cys481. Three critical hydrogen bonds with hinge residues Glu475 and Met477 were observed in the pyrazole group.64 Compared to the cocrystal structure of Ibrutinib with BTK, one more hydrogen bond is formed between the backbone carbonyl moiety of Met477 and zanubrutinib. In addition, a T-shape π–π stacking between the terminal phenyl group and Phe540 and a water bridge between the nitrogen atom of the pyrazolyl ring and Lys430 were observed.
Fig. 18. (a) The structure of zanubrutinib (30); (b) the key target interactions.

Asciminib (ABL-001) (31), which received FDA approval in 2021, is a tyrosine kinase inhibitor (TKI) indicated for the treatment of chronic phase Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML). More specifically, it is an allosteric inhibitor of the BCR-ABL1 tyrosine kinase, which binds to the myristyl pocket of the ABL1 part of the fusion protein and locks it into an inactive conformation, preventing its oncogenic activity. Therefore, asciminib (31) has also been shown to benefit the T315I mutation in Ph+ CML, which produces a BCR-ABL1 mutant that is generally treatment-resistant compared to wild-type BCR-ABL1. The N–H of the pyrazole ring can form a hydrogen bond interaction with Glu481, which also plays a positioning role of the π–π interaction between the pyridine ring and Tyr454 (Fig. 19).
Fig. 19. (a) The structure of asciminib (31); (b) the key target interactions.

5.2. Drugs to treat lymphatic organs
The Janus kinase (JAK) family of protein tyrosine kinases includes JAK1, JAK2, JAK3, and non-receptor tyrosine kinase 2 (TYK2).65 JAKs play a key role in intracellular signaling pathways for various cytokines and growth factors essential for hematopoiesis, such as interleukins, erythropoietin, and thrombopoietin.66 JAKs have multiple functions: JAK1 and JAK3 promote lymphocyte differentiation, survival, and function, while JAK2 promotes signaling of erythropoietin and thrombopoietin.
Ruxolitinib (INCB-018424) (32), a JAK inhibitor, was first approved by the FDA in 2011 for the treatment of adult myelofibrosis (MF) and subsequently by the EMA in 2012 (Fig. 20a). It can selectively inhibit JAK2 and JAK1, with some affinity for JAK3 and TYK2. The anticancer effect of ruxolitinib (32) was attributed to its inhibition of JAK and JAK-mediated STAT3 phosphorylation. By downregulating the JAK–STAT pathway, it inhibited myeloproliferation and suppressed plasma levels of pro-inflammatory cytokines such as IL-6 and TNF-α. Activated JAKs stimulate T-responsive cells, leading to increased proliferation of effector T cells and increased production of pro-inflammatory cytokines.67 By blocking JAK1 and JAK2, ruxolitinib (32) inhibits donor T cell expansion and suppresses pro-inflammatory responses.
Fig. 20. (a) The structure of ruxolitinib (32); (b) the key target interactions.

In 2017, Ohlson's group disclosed that maintaining pyrrolizidine nitrogen is critical as it provides a donor–acceptor interaction with the JAK2 kinase hinge.68 To obtain optimal JAK potency in this family, an alkyl or cycloalkyl group may need to be attached near the pyrazole moiety (Fig. 20b). Alkyl chains attached to pyrazoles are flexible and can adopt a conformation that allows polar isohydroxamic acids to occupy a tolerant position near the solvent (Fig. 20b). In addition, the hydrophobic interactions of pyrrolopyrimidines and pyrazoles with surface residues His500, Pro501, and Phe620 are also an important part of the drug–target interactions.
Belumosudil (SLX-2119) (33) is an inhibitor of Rho-associated convoluted helix kinase 2 (ROCK2),69 a protein that plays an important role in the pathogenesis of immune and fibrotic diseases (Fig. 21a). Inhibition of ROCK2 has been shown to resolve immune dysregulation by downregulating pro-inflammatory Th17 cells and upregulating regulatory T cells by manipulating phosphorylation of STAT3 and STAT5.70 In the structure–activity relationship, benzopyrazoles formed H-bond interactions with Glu170, while also allowing better binding of the entire molecule to the drug pocket (Fig. 21b).
Fig. 21. (a) The structure of belumosudil (33); (b) the key target interactions.

5.3. Drugs to treat autoimmune conditions
Baricitinib (INCB-028050) (34) is also a selective and reversible JAK1 and JAK2 inhibitor approved by the FDA in 2017 for the treatment of moderate to severe rheumatoid arthritis that has responded poorly to at least one TNF antagonist (Fig. 22a). Rheumatoid arthritis is a progressive autoimmune disease. While there are several disease-modifying antirheumatic drugs available for treatment, patients often experience inadequate therapeutic responses to these drugs. Baricitinib (34) selectively and reversibly inhibits JAK1 and JAK2 to modulate their signaling pathways, thereby reducing the phosphorylation and activation of STATs. In isolated enzyme assays, baricitinib (34) also exhibited an inhibitory effect on other types of JAK enzymes, tyrosine kinase 2 and JAK3, at higher concentrations needed for JAK1/2 inhibition.
Fig. 22. (a) The structure of baricitinib (34); (b) the key target interactions.

5.4. Anti-inflammatory drugs
Cyclooxygenase is a member of the animal-type heme peroxidase family, also known as prostaglandin G/H synthase.71 Cyclooxygenases (including COX-1 and COX-2) are essential for the production of beneficial prostaglandins, especially those produced in the stomach, platelets, and kidneys.72 Inhibition of COX-1 leads to decreased production of beneficial prostaglandins and is thought to be associated with the development of side effects such as gastric ulcers,73 decreased renal blood flow,74 and primary hemostasis disorders.75 COX-2 production is induced during active inflammation and leads to the production of prostaglandins that amplify pain,76 produce edema,77 and promote the production of other inflammatory mediators.78 Since the last century, the FDA has approved a variety of non-steroidal anti-inflammatory drugs (NSAIDs) that inhibit the COX, including antipyrine (1884) (35), bendazac (AF-1934) (36), benzydamine (AF-864) (37), celecoxib (SC-58635, 1998) (38), metamizole (1977) (39), phenylbutazone (1994) (40), etc. (Fig. 23).
Fig. 23. The structures of antipyrine (35), bendazac (36), benzydamine (37), celecoxib (38), metamizole (39), and phenylbutazone (40).

Antipyrine (35) inhibits both COX-1 and COX-2 isoforms.79 Bendazac (36) is an oxyacetic acid that has nonsteroidal anti-inflammatory effects, as well as analgesic, antipyretic, and platelet inhibitory effects. These effects may be accounted for in part by the substance's capability to inhibit prostaglandin synthesis by inhibiting cyclooxygenase activity in converting arachidonic acid to cyclic endoperoxides.80 Despite its anti-inflammatory, antinecrotic, anticholinergic, and antilipidemic properties, most research revolves around investigating and demonstrating the agent's primary role in inhibiting protein denaturation.81 Benzedamine (37), available as the hydrochloride salt, is a locally acting NSAID with local anesthetic and analgesic properties.82 It has a variety of physicochemical properties and pharmacological activities different from traditional aspirin-like NSAIDs, but which are beneficial to the mechanism of action of benzydamine (37) as an effective topical NSAID with local anesthetic and analgesic effects.83
Celecoxib (38) is a selective inhibitor of COX-2,84 so it causes fewer stomach-related side effects. In 2016, Dong's group demonstrated that the orientation of pyrazole provides favourable interaction between COX-2 and celecoxib (38).85 Metamizole (39) is an antipyretic and analgesic used for the relief of severe and persistent fever and pain. In an aqueous solution, metamizole (39) is immediately hydrolyzed to 4-formylaminoantipyrine (MAA), which can be further metabolized to 4-aminoantipyrine (AA), 4-formylaminoantipyrine (FAA), or 4-acetylaminoantipyrine (AAA). The mechanism of action of metamizole (39) may involve increasing the availability of arachidonic acid as a substrate for the synthesis of endogenous cannabinoids or other related molecules.86 Phenylbutazone (40) binds to and inactivates prostaglandin H synthase and prostacyclin synthase through peroxide (H2O2)-mediated inactivation.87 The pyrazole ring of all these anti-inflammatory drugs has some common effects. On the one hand, the pyrazole ring itself acts as an aryl bioisostere, improving the lipophilicity and solubility of the drug. On the other hand, although the pyrazole ring does not form direct interaction with the target protein, it facilitates the better binding of the drug to the receptor binding pocket. For some pyrazolinone drugs, carbonyl groups can often form hydrogen bonds with nearby amino acid residues to further strengthen the binding force between the drug and the receptor.
5.5. Antibacterial drugs
Ceftolozane (CXA-101) (41) is a semisynthetic broad-spectrum fifth-generation cephalosporin that was approved by the FDA in 2014 in combination with tazobactam for the treatment of serious infections such as intra-abdominal infections88 and complicated urinary tract infections (Fig. 23a). Tazobactam and ceftolozane (41) broaden their spectrum by making them effective against organisms that express β-lactamases and would normally degrade them. This occurs through irreversible inhibition of β-lactamases. In addition, tazobactam may covalently bind to plasmid-mediated and chromosome-mediated β-lactamases. Tazobactam is primarily effective against the OHIO-1, SHV-1, and TEM moieties of β-lactamases, but may also inhibit other β-lactamases. Ceftolozane (41) exerts antimicrobial effects, prevents cell wall formation, protects bacteria from injury, and confers resistance to certain antibiotics. For the structure–activity relationship, the pyrazole ring provides stability against AmpC β-lactamase-overproducing Pseudomonas aeruginosa and oxime confers β-lactamase stability.89 The 2-methylpyrazole group was found to have the best activity against Pseudomonas aeruginosa and the 2-aminoethylureido group has the optimal balance of activity (Fig. 24b).
Fig. 24. The structure–activity relationship of ceftolozane (41).

Sulfaphenazole (42) is a sulfonamide antibacterial (Fig. 25).90 In bacteria, antibacterial sulfa drugs act as competitive inhibitors of dihydropterophanate synthase (DHPS), an enzyme involved in folic acid synthesis.91 In this way, the microbes die due to the lack of folic acid. The role of the pyrazole ring in sulfaphenazole (42) is to improve the lipophilicity of the drug, making it easier to cross the bacterial cell membrane.92
Fig. 25. The structure of sulfaphenazole (42).

6. Pyrazole-containing drugs targeting central nervous system diseases
Genetic rearrangements of the mesenchymal lymphoma kinase (ALK) are genetic alterations that drive the progression of NSCLC in many patients.77 Normally, ALK is a naturally occurring endogenous tyrosine kinase receptor that plays an important role in brain development and triggers the activity of various specific neurons in the nervous system.
Lorlatinib (PF-06463922) (43) was approved by the FDA in 2018 for the treatment of ALK-positive and ROS1-positive NSCLC (Fig. 26), and demonstrated in vitro activity against multiple mutant forms of ALK enzymes, including some mutations detected in tumors upon disease progression with crizotinib (PF-2341066) (44) and other ALK inhibitors. In addition, lorlatinib (43) can cross the blood–brain barrier (BBB), allowing it to reach and treat progressive or worsening brain metastases. Lorlatinib (43) contains an N-methylamide that crizotinib (44) lacks. This methyl group points upward to the P-ring and the carbonyl group of Leu1951.93 The difference in the stability of the ALK Leu1196 P-ring may be related to specific protein–ligand interactions. During optimization of lorlatinib (43) against ALK and activating ALK mutants, cis-N-methyl amides are used as macrocyclic connectors to provide efficient interactions for the G-ring and stabilize structural water. The pyrazole ring makes the methyl reach the contact distance to TrkB Tyr635 in the ALK structure. What's more, it also brings the cyanide group closer to Tyr635, creating a dipole interaction.93
Fig. 26. The structures of lorlatinib (43) and crizotinib (44).

Crizotinib (44) was approved by the FDA in 2011 for the treatment of ALK-positive and ROS1-positive NSCLC (Fig. 26). In addition to ALK, it also inhibits the hepatocyte growth factor receptor (HGFR, c-MET) and Recepteur d'Origine Nantais (RON).94 Abnormalities in the ALK gene caused by mutations or translocations may lead to the expression of oncogenic fusion proteins. In patients with non-small cell lung cancer, they have the EML4-ALK gene.95 Crizotinib (44) inhibits the ALK tyrosine kinase, which ultimately leads to reduced proliferation and tumor survival in cells carrying the gene mutation.96 ALK gene site mutations and insertions are fully characterized mechanisms of resistance, with Leu1196M, Glu1269A and Glu1202R in direct contact with crizotinib (44).97 Ser1206Y is in close proximity to the crizotinib (44) binding site (Fig. 26). As for the role of its pyrazole scaffold, molecular docking analysis disclosed that the Gly2032 residue sits at the solvent front in the distal end of the kinase hinge and creates a turn, putting the Gly2032 alpha carbon in position to engage in a van der Waals interaction with the pyrazole ring of crizotinib.97
In addition to the two ALK inhibitors mentioned above, for the treatment of NSCLC, the FDA has approved kinase inhibitors including entrectinib (RXDX-101, 2019, TRK inhibitor) (45), larotrectinib (46) (ARRY-470, 2018, TRK inhibitor), pralsetinib (47) (BLU-667, 2020, RET inhibitor) and selpercatinib (48) (LOXO-292, 2020, RET inhibitor) over the past few years.
Entrectinib (45) is a tropomyosin receptor tyrosine kinase (TRK) TRKA, TRKB, TRKC, proto-oncogene tyrosine protein kinase ROS1 (ref. 98) and anaplastic lymphoma kinase (ALK) inhibitor for the treatment of ROS1-positive metastatic non-small cell lung cancer with NTRK gene fusion-positive solid tumors (Fig. 27a).99 This therapy is superior to similar ALK inhibitors such as erlotinib, ceritinib, and lorlatinib due to the broader range of targets. TRK receptors generate cell proliferation through downstream signaling by mitogen-activated protein kinase,100 phosphatidylinositol 3-kinase,101 and phospholipase C-γ.102 Inhibition of these pathways inhibits cancer cell proliferation and alters the balance in favor of apoptosis, leading to tumor size reduction. In 2021, Wang's group disclosed that the nitrogen atom on the pyrazole ring can form hydrogen bonds with Met592 in the hinge region (Fig. 27b). The fluorophenyl part, which is well integrated into the hydrophobic pocket, maintains a vertical conformation with the whole backbone and has hydrophobic interactions with the surrounding Phe521, Leu657, Gly667 and Asp668.
Fig. 27. (a) The structures of entrectinib (45), selitrectinib (49) and repotrectinib (50); (b) the structure–activity relationship of selitrectinib (49) and repotrectinib (50).

Selitrectinib (LOXO-195) (49) and repotrectinib (TPX-0005) (50) are two multi-kinase inhibitors designed from entrectinib (45) that are in clinical trials (Fig. 27a). Selitrectinib (49) shows strong binding to the structural domains of wild-type TRKA, TRKB, and TRKC kinases and has potent inhibitory activity in kinase assays.103 Repotrectinib (50) is a multi-kinase inhibitor with potent effects on ROS1, TRK, and JAK2.104 Both of them have shown significant efficacy in clinical trials in patients resistant to first-generation TRK inhibitors. In 2021, Wang's group also noted that the oxygen atom on the amide forms a hydrogen bond with Met592 in the hinge region via a water molecule.102 If the amide group in repotrectinib is substituted by the sulfonamide group, resulting in the loss of its activity. There is a hydrophobic interaction of the tetrahydropyrrole between the gatekeepers Phe589 and Phe521 (Fig. 27b). The methyl group at the same position of repotrectinib also has a hydrophobic effect on Phe521.105 The remaining methyl groups of selitrectinib and repotrectinib have the same hydrophobic interaction with P-loop Val524 and Gly517.
Larotrectinib (LOXO-101) (46) is an oral TRK inhibitor that was approved by the FDA in 2013 (Fig. 28). In vitro and in vivo tumor models, larotrectinib (46) has shown antitumor activity in cells with constitutive activation of the TRK protein due to gene fusions,106 deletion of the protein regulatory domain,107 or in cells with TRK protein overexpression.108 The docking results illustrate that the “linker” moiety of pyrazolo[1,5-a]pyrimidine is anchored to the hinge region through a key H-bond with the NH of Met592 while forming a CH−π interaction with Val524.109
Fig. 28. The structure of larotrectinib (46).

Rearranged during transfection (RET) is a transmembrane receptor tyrosine kinase (RTK) containing extracellular, transmembrane, and intracellular structural domains whose activity is required for normal kidney110 and nervous system development.111 Constitutive RET activation is achieved by chromosomal rearrangements that produce a fusion of the 5′ dimerizable structural domain with the 3′ RET tyrosine kinase structural domain,112 leading to constitutive dimerization and subsequent autophosphorylation.
Selpercatinib (LOXO-292) (47) is a RET receptor tyrosine kinase inhibitor approved for the treatment of RET-driven NSCLC and medullary thyroid carcinoma by the FDA in 2020 (Fig. 29a). It has enhanced specificity for the RET kinase compared to other RTK classes. Enhanced RET oncogene expression is a hallmark of many cancers.113 Pralsetinib (48), similar to the previously approved selpercatinib (BLU-667) (47), is a kinase inhibitor with greater specificity for the RET kinase than other RTK classes.114 Selpercatinib (47) and pralsetinib (48) represent the first generation of specific RET RTK inhibitors for the treatment of RET-driven cancers. Information based on natural and induced resistance mutations and molecular models suggests that selpercatinib (47) directly inhibits RET autophosphorylation by competitively binding to ATP.115 It binds to RET by forming an H-bond with Ala807 and three water-mediated H-bonds with Glu734, Asp771, and Gly810, with the pyrazole part forming a hydrogen bond with Ala807. Pralsetinib (48) was developed by screening over 10 000 agnostic-designed kinase inhibitors followed by extensive chemical modifications to improve their properties.116 For the structure–activity relationship study, both pyrazoles play a crucial role in target interaction, with one of them forming hydrogen bonds with Glu805 and Ala807 (Fig. 29b).
Fig. 29. (a) The structures of selpercatinib (47) and pralsetinib (48); (b) the key target interaction.

Rimonabant (SR-141716) (51) was the first drug to target the endocannabinoid (CB) pathway by inhibiting the action of anandamide and 2-archidonyl-glycerol on the CB1 receptor for the treatment of anorexic obesity (Fig. 30a).117 It is an inverse agonist of the cannabinoid receptor CB1 and its primary pathway of action is appetite reduction. Rimonabant (51) blocks the central effects of this neurotransmitter pathway involved in obesity and weight control, and also blocks the direct effects of CB on adipocyte and hepatocyte metabolism, thereby improving insulin resistance, triglycerides, and high-density lipoprotein cholesterol (HDL-C). The spatial overlap of the H atom of the chain amide and the methyl group on the pyrazole core and the electrostatic force between the lone pair of electrons of the pyrazole-N2 atom and the oxygen atom repel the carboxamide part,118 but can be further stabilized by weak hydrogen bonding interactions between the NH part of the formamide and the nitrogen atom in the pyrazole core (Fig. 30b). Surinabant (SR-147778) (52) is also a novel cannabinoid receptor 1 (CB1) antagonist that inhibits various tests119 such as the heart rate, body sway, and sensory highs (Fig. 30a). Surinabant (52) inhibits all THC responses, including almost complete inhibition of body sway and VAS alertness, but its effects on the heart rate and high sensation are submaximal.120 The pyrazole ring acts to improve the pharmacological activity in both drugs, serving as a key “hub” in the center of the scaffold, which allows for better positioning of the drug pocket and improved lipophilicity.
Fig. 30. (a) The structures of rimonabant (51) and surinabant (52); (b) the key target interactions.

Zaleplon (CL-284846) (53) is a sedative that is primarily used in insomnia and is known as a non-benzodiazepine hypnotic (Fig. 30), which exerts its effects through subunit modulation of the γ-aminobutyric acid–benzodiazepine (GABABZ) receptor chloride channel macromolecular complex.121 Zaleplon (53) also selectively binds to brain omega-1 receptors located on the alpha subunit of the GABAA/chlorine channel receptor complex122 and enhances the binding of tert-butyl-dicyclophosphorothioate (TBPS).123 Indiplon (NBI-34060) (54) is also a non-benzodiazepine hypnotic sedative that was developed in two formulations, i.e., an immediate-release formulation for falling asleep and an extended-release formulation for maintaining sleep (Fig. 30).124 It acts as a high-affinity orthosteric modulator of the GABAA receptor, enhancing GABA-activated chloride currents in a dose-dependent and reversible manner.125 As the molecular docking results suggested, zaleplon (53) and indiplon (54) exhibited a similar binding fashion with GABABZvia reversible noncovalent interactions involving hydrogen bonding and π–π interaction (Fig. 31). The pyrazole ring mainly forms a π–π interaction with nearby amino acid residues.
Fig. 31. The structures of zaleplon (53) and indiplon (54).

Edaravone (55) is a free radical scavenger that was approved in 2017 for the treatment of amyotrophic lateral sclerosis (ALS) (Fig. 32a). Clinical studies have shown that the treatment reduced disease progression compared to a placebo.126 As a low molecular weight molecule with good water and lipid solubility, it has therapeutic advantages in crossing the blood–brain barrier to mediate the pro-intellectual and neuroprotective effects. The pro-intellectual and neuroprotective effects are mediated through inhibition of lipid peroxidation and scavenging of free radicals.127 Edaravone (55) works by increasing prostacyclin production, reducing lipoxygenase metabolism of arachidonic acid by capturing hydroxyl radicals, and inhibiting tetraoxapyrimidine-induced lipid peroxidation and quenching reactive oxygen species. It targets a variety of cells, including neurons, endothelial cells, and cardiomyocytes. There is also evidence that neuronal nitric oxide synthase (nNOS) levels are reduced and SOD1 levels are enhanced after transient ischemia in rabbits, thereby preventing spinal cord injury. For this mechanism, edaravone (55) and its derivatives can exist in three reciprocal isomeric forms a, b, and c (Fig. 32b).128 It is now well established that the anionic form is most relevant for scavenging free radicals in polar media via a single electron transfer mechanism (Fig. 32b, pathway A). The most successful derivatives in such media are those with a good balance between the amount of anionic form and oxidation potential. In contrast, it has been hypothesized that the mechanism of H-atom extraction is dominant in the lipid phase (Fig. 32b, pathway B).129 For the structure–activity relationship, the pyrazole forms a π–π interaction with Tyr202 and the hydroxyl group forms an H-bond interaction with Glu200, allowing the whole molecule to be well bound within the target pocket.
Fig. 32. (a) The structure of edaravone (55); (b) the action mechanism of edaravone (55).

Granisetron (APF-530) (56) is a 5-HT3 antagonist used in cancer therapy and postoperative treatment of nausea and vomiting (Fig. 33a).130 A serotonin receptor (5-HT3 selective) antagonist for antiemetic and drug resistance in cancer chemotherapy patients. Granisetron (56) is a potent and selective 5-HT3 receptor antagonist.131 The antiemetic activity of the drug is achieved by inhibiting the central and peripheral 5-HT3 receptors. The serotonin receptor protein is a pentamer that can bind to five molecules, where the benzopyrazole ring can form a π–π interaction with Arg65 of the D chain, and the amino group on the bridged ring can form a hydrogen bond interaction with different amino acids on the D chain and E chain, respectively, strengthening the affinity between the drug and the receptor (Fig. 33b).132
Fig. 33. (a) The structure of granisetron (56); (b) the key target interactions.

Fibrodysplasia ossificans progressiva (FOP) is an unusually rare genetic disorder, which is caused by gain-of-function mutations in the ACVR1/ALK2 gene that lead to progressive heterotopic ossification.133 Palovarotene (RG-667) (57) is a selective retinoic acid receptor gamma (RARγ) agonist belonging to a class of drugs known as retinoic acids, similar in the mechanism to trefoil, which is a derivative of vitamin A (Fig. 34a).134 In a structure–activity relationship study, in addition to the carboxyl group required by such RARγ agonists, the pyrazole ring also plays a crucial role, which forms the π–π interaction with Phe304 (Fig. 34b).
Fig. 34. (a) The structure of palovarotene (57); (b) the key target interactions.

7. Pyrazole-containing drugs targeting gastrointestinal tract diseases
Avapritinib (BLU-285) (58), a selective tyrosine kinase inhibitor of KIT and platelet-derived growth factor receptor alpha (PDGFRα),135 was approved by the FDA in 2020 for the treatment of unresectable metastatic gastrointestinal mesenchymal tumors (GIST) carrying 18 mutations in PDGFRα activating fragment exons (Fig. 35).136 These activation fragment mutations are resistant to imatinib and represent the initial small molecule targeted therapies. Avapritinib (58) has a negative regulatory effect on the transporter proteins ABCB1 (ref. 137) and ABCG2,138 which may be due to the interaction of avapritinib (58) with the drug-binding pocket of these transporter proteins. As the molecular docking results suggested, avapritinib occupies a longer, thinner region within the active site, and is predicted to form a hydrogen bond with residue Cys673.139
Fig. 35. The structure of avapritinib (58).

Betazole (59) is a histamine H2 agonist used as a diagnostic agent to measure gastric acidity or maximum gastric acidity production (Fig. 36).140 Thus, this agonist action increases the amount of gastric acid produced. This measurement can be used to diagnose diseases such as the Zollinger–Ellison syndrome.141 Alternatively, betazole (59) can be used as a gastric secretory stimulant instead of histamine, with the advantage that it does not cause side effects and therefore it does not require the use of antihistamine compounds.
Fig. 36. The structure of betazole (59).

Cystic fibrosis (CF) is the result of a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.142 The CFTR protein produced by this gene is a transmembrane ion channel that carries sodium and chloride ions across the cell membrane – water follows chloride ions to the cell surface, thus helping to hydrate the cell surface and dilute pericellular secretions. Mutations in the CFTR gene produce insufficient amounts and functions of CFTR proteins, resulting in defective ion transport and accumulation of mucus throughout the body, leading to multi-organ diseases involving the lungs, gastrointestinal tract, and pancreatic system.143 In 2019, the small molecule CFTR corrector elexacaftor (VX-445) (60) was approved by the FDA for the treatment of CF patients with one F508del-CFTR mutation (Fig. 37). Unlike first-generation correctors such as tezacaftor and ivacaftor, elexacaftor (60) is considered a next-generation CFTR corrector because it has a different structure and mechanism. Elexacaftor (60) promotes transport to the cell surface for incorporation into the cell membrane, which increases the amount of mature CFTR proteins present on the cell surface, thus improving ion transport and CF symptoms. SAR studies have implicated that the 2,2,4-trimethylpyrrolidinyl group acts as the critical anchor in the pocket characterizing this site, in particular, π–π interacting with the side chain of Phe354.144
Fig. 37. The structure of elexacaftor (60).

Telotristat ethyl (LX-1032) (61) is a tryptophan hydroxylase inhibitor that was approved by the FDA in 2017 for carcinoid syndrome diarrhea (Fig. 38).145 Telotristat ethyl is an ethyl ester prodrug that can be hydrolyzed in vivo and in vitro to its active part, LP-778902 (62). The systemic exposure of telotristat ethyl is relatively low because of the rapid rate of hydrolysis to the active fraction. LP-778902 (62) is a potent inhibitor of TPH, which belongs to the drug under investigation. While existing treatments for carcinoid syndrome reduce the release of serotonin outside tumor cells, telotristat ethyl reduces the production of serotonin inside tumor cells at its source.146 By specifically inhibiting serotonin production, telotristat ethyl attempts to control this important driver of carcinoid syndrome, thereby providing patients with more control over their disease.
Fig. 38. The structures of telotristat ethyl (61) and LP-778902 (62).

8. Pyrazole-containing drugs targeting urinary system diseases
Uroepithelial cancer is statistically the fourth most common cancer in the world. Genetic mutations or changes, such as dysregulation of the fibroblast growth factor receptor (FGFR) pathway and FGFR aberrations, are associated with the pathogenesis of uroepithelial carcinoma, including all four FGFR genes. Thus, it is believed that changes in FGFR genes promote cell proliferation, migration, angiogenesis, and resistance to apoptosis in many cancers, including uroepithelial carcinoma. Erdafitinib (63), an oral selective pan-FGFR kinase inhibitor that inhibits the enzymatic activity of expressed FGFR1, FGFR2, FGFR3, and FGFR4, was approved by the FDA in 2019 for the treatment of locally advanced or metastatic uroepithelial cancer (Fig. 39). For the mechanism of action, erdafitinib (63) exhibited inhibition of FGFR phosphorylation and signaling and reduced cell viability in cell lines expressing altered FGFR genes, including point mutations, amplifications, and fusions (Fig. 39). Erdafitinib (63) showed antitumor activity in FGFR-expressing cell lines and in xenograft models derived from tumor types, including bladder cancer.
Fig. 39. The structures of erdafitinib (63), axitinib (64) and pazopanib (65).

In addition to erdafitinib (63), axitinib (64) is a second-generation tyrosine kinase inhibitor (Fig. 39) that treats advanced renal cell cancer after failure of prior systemic therapy by selectively inhibiting vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2, VEGFR-3).147 In 2019, Liu's group noted that axitinib (64) could fit well into the DFG-in conformation,148 with the indazole part forming two hydrogen bonds with Cys673 and Glu671 in the hinge-binding region, allowing for tighter binding to the VEGFR receptor. Pazopanib (65), also a small molecule inhibitor of multiple protein tyrosine kinases, was approved by the FDA in 2009 for the treatment of advanced renal cell carcinoma and advanced soft tissue sarcoma in patients on prior chemotherapy (Fig. 39). Pazopanib (65) is a second-generation multi-target tyrosine kinase inhibitor targeting VEGFR-1, -2, and -3, platelet-derived growth factor receptor-alpha, platelet-derived growth factor receptor-beta, and c-kit.149 These receptor targets are part of the angiogenic pathway that promotes tumor angiogenesis to facilitate tumor survival and growth (Fig. 39).
9. Pyrazole-containing drugs for other indications
Encorafenib (LGX-818) (66) is a serine/threonine protein kinase inhibitor.150 It inhibits the BRAF gene that encodes the B-raf protein, a proto-oncogene involved in mutations in multiple genes (Fig. 40). This protein functions in the regulation of the MAP kinase and ERK signaling pathway,151 affecting cell division, differentiation and secretion. Mutations in this gene, most commonly the V600E mutation, are the most common oncogenic mutations in melanoma.152 On June 27, 2018, the FDA approved encorafenib (66) and binimetinib in combination for patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations. Molecular docking studies disclosed that the drug binds tightly to the cavity of BRAF mainly through the hydrophobic stacking contact π–π interaction between the pyrazole group and Phe583.
Fig. 40. The structure of encorafenib (66).

Fomepizole (67) is a competitive inhibitor of alcohol dehydrogenase,153 which catalyzes the initial steps in the metabolism of ethylene glycol and methanol to their toxic metabolites (Fig. 41).154 Fomepizole (67) was approved as an antidote to confirmed or suspected methanol or ethylene glycol poisoning.155 For the structure–activity relationship, pyrazoles form π–π interactions and H-bond interactions with nearby amino acid residues (Fig. 41).
Fig. 41. The structure of fomepizole (67).

Conclusions
In summary, among the drugs approved by the FDA each year, the number of drugs containing pyrazole rings is increasing, and the introduction of pyrazole rings into drug molecules has become a common strategy in medicinal chemistry and drug design. On the one hand, due to its good physicochemical properties, the pyrazole ring can be used as a biological isostere for other aromatic rings to improve the druggability and lipophilicity of drugs. On the other hand, the pyrazole ring is both a hydrogen bond acceptor and donor, which allows it to better bind to drug targets, changing its affinity for different receptor pockets for optimal therapeutic effects. However, there are also some challenges to introducing pyrazole rings into drug molecules. Unlike 1,2,3-triazole, which can be synthesized by click reaction, the introduction of pyrazole rings is often required through ready-made blocks. In addition, the in vivo metabolic toxicity of pyrazoles is also a concern for medicinal chemists when introducing this group.
With the in-depth understanding of the interaction between pyrazole rings and receptor proteins, it is expected to develop more pyrazole ring-containing drug molecules substituted by bioisosteres. In the future, the pyrazole ring replacement strategy could also be widely applied to develop next-generation drugs with less resistance. We hope that this review will shed some light on the drug discovery of pyrazole ring-containing chemical entities for organic chemists and medicinal chemists.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We acknowledge the National Natural Science Foundation of China (#22071175 to Y. D.) and the Beijing Municipal Science & Technology Commission (Z211100003321007 to N. H.) for financial support.
References
- Rue K. Raptis R. G. Acta Crystallogr., Sect. E: Crystallogr. Commun. 2021;77:955–957. doi: 10.1107/S2056989021008604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krygowski T. M. Anulewicz R. Cyrański M. K. Puchala A. Rasala D. Tetrahedron. 1998;54:12295–12300. doi: 10.1016/S0040-4020(98)00749-2. [DOI] [Google Scholar]
- Blanco F. Alkorta I. Zborowski K. Elguero J. Struct. Chem. 2007;18:965–975. doi: 10.1007/s11224-007-9237-z. [DOI] [Google Scholar]
- Alkorta I. Blanco F. Elguero J. J. Mol. Struct.: THEOCHEM. 2008;851:75–83. doi: 10.1016/j.theochem.2007.10.037. [DOI] [Google Scholar]
- Ashton W. T. Hutchins S. M. Greenlee W. J. Doss G. A. Chang R. S. Lotti V. J. Faust K. A. Chen T. B. Zingaro G. J. Kivlighn S. D. et al. . J. Med. Chem. 1993;36:3595–3605. doi: 10.1021/jm00075a014. [DOI] [PubMed] [Google Scholar]
- Neisewander J. L. Fuchs R. A. O'Dell L. E. Khroyan T. V. Synapse. 1998;30:194–204. doi: 10.1002/(SICI)1098-2396(199810)30:2<194::AID-SYN9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Hyttel J. Eur. J. Pharmacol. 1983;91:153–154. doi: 10.1016/0014-2999(83)90381-3. [DOI] [PubMed] [Google Scholar]
- Wu W. L. Burnett D. A. Spring R. Greenlee W. J. Smith M. Favreau L. Fawzi A. Zhang H. Lachowicz J. E. J. Med. Chem. 2005;48:680–693. doi: 10.1021/jm030614p. [DOI] [PubMed] [Google Scholar]
- Lee A. Drugs. 2021;81:405–409. doi: 10.1007/s40265-021-01475-4. [DOI] [PubMed] [Google Scholar]
- Mathis A. Sale M. Cornpropst M. Sheridan W. P. Ma S. C. Clin. Transl. Sci. 2022;15:1027–1035. doi: 10.1111/cts.13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotian P. L. Wu M. Vadlakonda S. Chintareddy V. Lu P. Juarez L. Kellogg-Yelder D. Chen X. Muppa S. Chambers-Wilson R. Davis Parker C. Williams J. Polach K. J. Zhang W. Raman K. Babu Y. S. J. Med. Chem. 2021;64:12453–12468. doi: 10.1021/acs.jmedchem.1c00511. [DOI] [PubMed] [Google Scholar]
- Granger C. B. Alexander J. H. McMurray J. J. Lopes R. D. Hylek E. M. Hanna M. Al-Khalidi H. R. Ansell J. Atar D. Avezum A. Bahit M. C. Diaz R. Easton J. D. Ezekowitz J. A. Flaker G. Garcia D. Geraldes M. Gersh B. J. Golitsyn S. Goto S. Hermosillo A. G. Hohnloser S. H. Horowitz J. Mohan P. Jansky P. Lewis B. S. Lopez-Sendon J. L. Pais P. Parkhomenko A. Verheugt F. W. Zhu J. Wallentin L. Committees A. Investigators N. Engl. J. Med. 2011;365:981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- Wong P. C. Pinto D. J. Zhang D. J. Thromb. Thrombolysis. 2011;31:478–492. doi: 10.1007/s11239-011-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harenberg J. Wehling M. Semin. Thromb. Hemostasis. 2008;34:39–57. doi: 10.1055/s-2008-1066023. [DOI] [PubMed] [Google Scholar]
- Pinto D. J. Orwat M. J. Koch S. Rossi K. A. Alexander R. S. Smallwood A. Wong P. C. Rendina A. R. Luettgen J. M. Knabb R. M. He K. Xin B. Wexler R. R. Lam P. Y. J. Med. Chem. 2007;50:5339–5356. doi: 10.1021/jm070245n. [DOI] [PubMed] [Google Scholar]
- Badawy S. I. Gray D. B. Zhao F. Sun D. Schuster A. E. Hussain M. A. Pharm. Res. 2006;23:989–996. doi: 10.1007/s11095-006-9899-z. [DOI] [PubMed] [Google Scholar]
- Hilden L. R. Pommier C. J. Badawy S. I. Friedman E. M. Int. J. Pharm. 2008;353:283–290. doi: 10.1016/j.ijpharm.2007.11.032. [DOI] [PubMed] [Google Scholar]
- Quan M. L. Lam P. Y. Han Q. Pinto D. J. He M. Y. Li R. Ellis C. D. Clark C. G. Teleha C. A. Sun J. H. Alexander R. S. Bai S. Luettgen J. M. Knabb R. M. Wong P. C. Wexler R. R. J. Med. Chem. 2005;48:1729–1744. doi: 10.1021/jm0497949. [DOI] [PubMed] [Google Scholar]
- Erickson-Miller C. L. Delorme E. Tian S. S. Hopson C. B. Landis A. J. Valoret E. I. Sellers T. S. Rosen J. Miller S. G. Luengo J. I. Duffy K. J. Jenkins J. M. Stem Cells. 2009;27:424–430. doi: 10.1634/stemcells.2008-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feese M. D. Tamada T. Kato Y. Maeda Y. Hirose M. Matsukura Y. Shigematsu H. Muto T. Matsumoto A. Watarai H. Ogami K. Tahara T. Kato T. Miyazaki H. Kuroki R. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1816–1821. doi: 10.1073/pnas.0308530100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H. Choi N. Kim S. Jin M. S. Shen H. Kim Y. C. Pharmaceuticals. 2022;15:440. doi: 10.3390/ph15040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro-Pereira P. V. Falcai A. Kubo C. A. Oliveira-Junior E. B. Marques O. C. Antunes E. Condino-Neto A. Br. J. Pharmacol. 2012;166:1617–1630. doi: 10.1111/j.1476-5381.2011.01764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M. Ghofrani H. A. Thorax. 2016;71:73–83. doi: 10.1136/thoraxjnl-2015-207170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambly N. Granton J. Expert Rev. Respir. Med. 2015;9:679–695. doi: 10.1586/17476348.2015.1106316. [DOI] [PubMed] [Google Scholar]
- Liu R. Kang Y. Chen L. Nat. Commun. 2021;12:5492. doi: 10.1038/s41467-021-25617-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A. Sandner P. Schmidtko A. Naunyn-Schmiedeberg's Arch. Pharmacol. 2020;393:287–302. doi: 10.1007/s00210-019-01779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follmann M. Ackerstaff J. Redlich G. Wunder F. Lang D. Kern A. Fey P. Griebenow N. Kroh W. Becker-Pelster E. M. Kretschmer A. Geiss V. Li V. Straub A. Mittendorf J. Jautelat R. Schirok H. Schlemmer K. H. Lustig K. Gerisch M. Knorr A. Tinel H. Mondritzki T. Trubel H. Sandner P. Stasch J. P. J. Med. Chem. 2017;60:5146–5161. doi: 10.1021/acs.jmedchem.7b00449. [DOI] [PubMed] [Google Scholar]
- Tomasoni D. Adamo M. Anker M. S. von Haehling S. Coats A. J. S. Metra M. ESC Heart Fail. 2020;7:3505–3530. doi: 10.1002/ehf2.13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R. Li Z. Di X. Guo D. Ji J. Wang S. BioSci. Trends. 2018;12:369–374. doi: 10.5582/bst.2018.01081. [DOI] [PubMed] [Google Scholar]
- Howard J. Hemmaway C. J. Telfer P. Layton D. M. Porter J. Awogbade M. Mant T. Gretler D. D. Dufu K. Hutchaleelaha A. Patel M. Siu V. Dixon S. Landsman N. Tonda M. Lehrer-Graiwer J. Blood. 2019;133:1865–1875. doi: 10.1182/blood-2018-08-868893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchaleelaha A. Patel M. Washington C. Siu V. Allen E. Oksenberg D. Gretler D. D. Mant T. Lehrer-Graiwer J. Br. J. Clin. Pharmacol. 2019;85:1290–1302. doi: 10.1111/bcp.13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo M. K. Abraham D. J. Biochemistry. 2005;44:8347–8359. doi: 10.1021/bi050412q. [DOI] [PubMed] [Google Scholar]
- Fries R. Shariat K. von Wilmowsky H. Bohm M. Circulation. 2005;112:2980–2985. doi: 10.1161/CIRCULATIONAHA.104.523324. [DOI] [PubMed] [Google Scholar]
- Ozbeyli D. Gokalp A. G. Koral T. Ocal O. Y. Dogan B. Akakin D. Yuksel M. Kasimay O. Physiol. Behav. 2015;151:230–237. doi: 10.1016/j.physbeh.2015.07.030. [DOI] [PubMed] [Google Scholar]
- Rotella D. P. Sun Z. Zhu Y. Krupinski J. Pongrac R. Seliger L. Normandin D. Macor J. E. J. Med. Chem. 2000;43:1257–1263. doi: 10.1021/jm000081+. [DOI] [PubMed] [Google Scholar]
- Simiele M. Pensi D. Pasero D. Ivaldi F. Rinaldi M. Di Perri G. Ranieri V. M. D'Avolio A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015;1001:35–40. doi: 10.1016/j.jchromb.2015.07.023. [DOI] [PubMed] [Google Scholar]
- Veenstra J. A. Peptides. 2021;146:170667. doi: 10.1016/j.peptides.2021.170667. [DOI] [PubMed] [Google Scholar]
- Ludwig M. G. Vanek M. Guerini D. Gasser J. A. Jones C. E. Junker U. Hofstetter H. Wolf R. M. Seuwen K. Nature. 2003;425:93–98. doi: 10.1038/nature01905. [DOI] [PubMed] [Google Scholar]
- Allerton C. M. Barber C. G. Beaumont K. C. Brown D. G. Cole S. M. Ellis D. Lane C. A. Maw G. N. Mount N. M. Rawson D. J. Robinson C. M. Street S. D. Summerhill N. W. J. Med. Chem. 2006;49:3581–3594. doi: 10.1021/jm060113e. [DOI] [PubMed] [Google Scholar]
- Ku H. Y. Ahn H. J. Seo K. A. Kim H. Oh M. Bae S. K. Shin J. G. Shon J. H. Liu K. H. Drug Metab. Dispos. 2008;36:986–990. doi: 10.1124/dmd.107.020099. [DOI] [PubMed] [Google Scholar]
- Zhao C. Kim S. H. Lee S. W. Jeon J. H. Kang K. K. Choi S. B. Park J. K. BJU Int. 2011;107:1943–1947. doi: 10.1111/j.1464-410X.2010.09759.x. [DOI] [PubMed] [Google Scholar]
- Chen A. Chin. J. Cancer. 2011;30:463–471. doi: 10.5732/cjc.011.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K. Bolanos B. Smith M. Palde P. B. Cuenca P. D. VanArsdale T. L. Niessen S. Zhang L. Behenna D. Ornelas M. A. Tran K. T. Kaiser S. Lum L. Stewart A. Gajiwala K. S. J. Biol. Chem. 2021;296:100251. doi: 10.1074/jbc.RA120.016573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A. G. Ekblad T. Karlberg T. Low M. Pinto A. F. Tresaugues L. Moche M. Cohen M. S. Schuler H. J. Med. Chem. 2017;60:1262–1271. doi: 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. Wilcoxen K. Rowley M. Toniatti C. J. Med. Chem. 2015;58:3302–3314. doi: 10.1021/jm5018237. [DOI] [PubMed] [Google Scholar]
- Maharaj K. Powers J. J. Achille A. Mediavilla-Varela M. Gamal W. Burger K. L. Fonseca R. Jiang K. Miskin H. P. Maryanski D. Monastyrskyi A. Duckett D. R. Roush W. R. Cleveland J. L. Sahakian E. Pinilla-Ibarz J. Blood Adv. 2020;4:3072–3084. doi: 10.1182/bloodadvances.2020001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran E. Smith S. M. Curr. Opin. Oncol. 2014;26:469–475. doi: 10.1097/CCO.0000000000000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris H. A. Flinn I. W. Patel M. R. Fenske T. S. Deng C. Brander D. M. Gutierrez M. Essell J. H. Kuhn J. G. Miskin H. P. Sportelli P. Weiss M. S. Vakkalanka S. Savona M. R. O'Connor O. A. Lancet Oncol. 2018;19:486–496. doi: 10.1016/S1470-2045(18)30082-2. [DOI] [PubMed] [Google Scholar]
- Long A. M. Zhao H. Huang X. J. Med. Chem. 2012;55:10307–10311. doi: 10.1021/jm301336n. [DOI] [PubMed] [Google Scholar]
- Bastos D. A. Antonarakis E. S. OncoTargets Ther. 2019;12:8769–8777. doi: 10.2147/OTT.S197244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford E. D. Stanton W. Mandair D. Cancer Manage. Res. 2020;12:5667–5676. doi: 10.2147/CMAR.S227583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukovala M. Spetsieris N. Efstathiou E. Expert Opin. Pharmacother. 2020;21:1537–1546. doi: 10.1080/14656566.2020.1770726. [DOI] [PubMed] [Google Scholar]
- Yu J. Zhou P. Hu M. Yang L. Yan G. Xu R. Deng Y. Li X. Chen Y. Eur. J. Med. Chem. 2019;182:111608. doi: 10.1016/j.ejmech.2019.111608. [DOI] [PubMed] [Google Scholar]
- Watanabe Y. S. Yasuda Y. Kojima Y. Okada S. Motoyama T. Takahashi R. Oka M. J. Enzyme Inhib. Med. Chem. 2015;30:981–988. doi: 10.3109/14756366.2014.1002402. [DOI] [PubMed] [Google Scholar]
- Yoshida T. Akahoshi F. Sakashita H. Kitajima H. Nakamura M. Sonda S. Takeuchi M. Tanaka Y. Ueda N. Sekiguchi S. Ishige T. Shima K. Nabeno M. Abe Y. Anabuki J. Soejima A. Yoshida K. Takashina Y. Ishii S. Kiuchi S. Fukuda S. Tsutsumiuchi R. Kosaka K. Murozono T. Nakamaru Y. Utsumi H. Masutomi N. Kishida H. Miyaguchi I. Hayashi Y. Bioorg. Med. Chem. 2012;20:5705–5719. doi: 10.1016/j.bmc.2012.08.012. [DOI] [PubMed] [Google Scholar]
- Biftu T. Sinha-Roy R. Chen P. Qian X. Feng D. Kuethe J. T. Scapin G. Gao Y. D. Yan Y. Krueger D. Bak A. Eiermann G. He J. Cox J. Hicks J. Lyons K. He H. Salituro G. Tong S. Patel S. Doss G. Petrov A. Wu J. Xu S. S. Sewall C. Zhang X. Zhang B. Thornberry N. A. Weber A. E. J. Med. Chem. 2014;57:3205–3212. doi: 10.1021/jm401992e. [DOI] [PubMed] [Google Scholar]
- Chen P. Feng D. Qian X. Apgar J. Wilkening R. Kuethe J. T. Gao Y. D. Scapin G. Cox J. Doss G. Eiermann G. He H. Li X. Lyons K. A. Metzger J. Petrov A. Wu J. K. Xu S. Weber A. E. Yan Y. Roy R. S. Biftu T. Bioorg. Med. Chem. Lett. 2015;25:5767–5771. doi: 10.1016/j.bmcl.2015.10.070. [DOI] [PubMed] [Google Scholar]
- Ponader S. Chen S. S. Buggy J. J. Balakrishnan K. Gandhi V. Wierda W. G. Keating M. J. O'Brien S. Chiorazzi N. Burger J. A. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y. X. Zhu H. Y. Li X. T. Xia Y. Miao K. R. Zhao S. S. Wu Y. J. Wang L. Xu W. Li J. Y. Hematol. Oncol. 2019;37:392–400. doi: 10.1002/hon.2667. [DOI] [PubMed] [Google Scholar]
- Chang B. Y. Huang M. M. Francesco M. Chen J. Sokolove J. Magadala P. Robinson W. H. Buggy J. J. Arthritis Res. Ther. 2011;13:R115. doi: 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. T. Gardberg A. Pereira A. Johnson T. Wu Y. Grenningloh R. Head J. Morandi F. Haselmayer P. Liu-Bujalski L. Mol. Pharmacol. 2017;91:208–219. doi: 10.1124/mol.116.107037. [DOI] [PubMed] [Google Scholar]
- Tam C. S. Trotman J. Opat S. Burger J. A. Cull G. Gottlieb D. Harrup R. Johnston P. B. Marlton P. Munoz J. Seymour J. F. Simpson D. Tedeschi A. Elstrom R. Yu Y. Tang Z. Han L. Huang J. Novotny W. Wang L. Roberts A. W. Blood. 2019;134:851–859. doi: 10.1182/blood.2019001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal Singh S. Dammeijer F. Hendriks R. W. Mol. Cancer. 2018;17:57. doi: 10.1186/s12943-018-0779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y. Liu Y. Hu N. Yu D. Zhou C. Shi G. Zhang B. Wei M. Liu J. Luo L. Tang Z. Song H. Guo Y. Liu X. Su D. Zhang S. Song X. Zhou X. Hong Y. Chen S. Cheng Z. Young S. Wei Q. Wang H. Wang Q. Lv L. Wang F. Xu H. Sun H. Xing H. Li N. Zhang W. Wang Z. Liu G. Sun Z. Zhou D. Li W. Liu L. Wang L. Wang Z. J. Med. Chem. 2019;62:7923–7940. doi: 10.1021/acs.jmedchem.9b00687. [DOI] [PubMed] [Google Scholar]
- Delen E. Doganlar O. J. Korean Neurosurg. Soc. 2020;63:444–454. doi: 10.3340/jkns.2019.0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B. S. Howell M. D. Sun K. Papp K. Nasir A. Kuligowski M. E. Investigators I. S. J. Allergy Clin. Immunol. 2020;145:572–582. doi: 10.1016/j.jaci.2019.08.042. [DOI] [PubMed] [Google Scholar]
- Ostojic A. Vrhovac R. Verstovsek S. Future Oncol. 2011;7:1035–1043. doi: 10.2217/fon.11.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L. Mustafa N. Tan E. C. Poulsen A. Singh P. Duong-Thi M. D. Lee J. X. T. Ramanujulu P. M. Chng W. J. Yen J. J. Y. Ohlson S. Dymock B. W. J. Med. Chem. 2017;60:8336–8357. doi: 10.1021/acs.jmedchem.7b00678. [DOI] [PubMed] [Google Scholar]
- Braun L. M. Zeiser R. HemaSphere. 2021;5:e581. doi: 10.1097/HS9.0000000000000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H. Miwa Y. Kasa M. Kitano K. Amano M. Kaibuchi K. Hakoshima T. J. Biochem. 2006;140:305–311. doi: 10.1093/jb/mvj172. [DOI] [PubMed] [Google Scholar]
- Sun H. Y. Ji F. Q. Biochem. Biophys. Res. Commun. 2012;423:319–324. doi: 10.1016/j.bbrc.2012.05.120. [DOI] [PubMed] [Google Scholar]
- Kern M. A. Schubert D. Sahi D. Schoneweiss M. M. Moll I. Haugg A. M. Dienes H. P. Breuhahn K. Schirmacher P. Hepatology. 2002;36:885–894. doi: 10.1053/jhep.2002.36125. [DOI] [PubMed] [Google Scholar]
- Cox S. K. Roark J. Gassel A. Tobias K. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2005;819:181–184. doi: 10.1016/j.jchromb.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Sennello K. A. Leib M. S. J. Vet. Intern. Med. 2006;20:1291–1296. doi: 10.1111/j.1939-1676.2006.tb00741.x. [DOI] [PubMed] [Google Scholar]
- García-Lozano J. Server-Carrió J. Escrivà E. Folgado J.-V. Molla C. Lezama L. Polyhedron. 1997;16:939–944. doi: 10.1016/S0277-5387(96)00346-4. [DOI] [Google Scholar]
- Shaw A. T. Friboulet L. Leshchiner I. Gainor J. F. Bergqvist S. Brooun A. Burke B. J. Deng Y. L. Liu W. Dardaei L. Frias R. L. Schultz K. R. Logan J. James L. P. Smeal T. Timofeevski S. Katayama R. Iafrate A. J. Le L. McTigue M. Getz G. Johnson T. W. Engelman J. A. N. Engl. J. Med. 2016;374:54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper K. Cancer Discovery. 2017;7:1360. doi: 10.1158/2159-8290.CD-NB2017-151. [DOI] [PubMed] [Google Scholar]
- Grover J. Kumar V. Sobhia M. E. Jachak S. M. Bioorg. Med. Chem. Lett. 2014;24:4638–4642. doi: 10.1016/j.bmcl.2014.08.050. [DOI] [PubMed] [Google Scholar]
- El Sayed M. T. El-Sharief M. Zarie E. S. Morsy N. M. Elsheakh A. R. Voronkov A. Berishvili V. Hassan G. S. Bioorg. Med. Chem. Lett. 2018;28:952–957. doi: 10.1016/j.bmcl.2018.01.043. [DOI] [PubMed] [Google Scholar]
- Balfour J. A. Clissold S. P. Drugs. 1990;39:575–596. doi: 10.2165/00003495-199039040-00007. [DOI] [PubMed] [Google Scholar]
- Soldo L. Ruggieri A. Milanese C. Pinza M. Guglielmotti A. Ophthalmic Res. 2004;36:145–150. doi: 10.1159/000077327. [DOI] [PubMed] [Google Scholar]
- Quane P. A. Graham G. G. Ziegler J. B. Inflammopharmacology. 1998;6:95–107. doi: 10.1007/s10787-998-0026-0. [DOI] [PubMed] [Google Scholar]
- Stefania C. Andrea M. Alessio M. Mauro P. Amira G. Martin C. J. Giovanni M. Di Giannantonio M. Fabrizio S. Curr. Neuropharmacol. 2021;19:1728–1737. doi: 10.2174/1570159X19666210113151136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A. Casini A. Heine A. Kuhn D. Supuran C. T. Scozzafava A. Klebe G. J. Med. Chem. 2004;47:550–557. doi: 10.1021/jm030912m. [DOI] [PubMed] [Google Scholar]
- Dong L. Yuan C. Orlando B. J. Malkowski M. G. Smith W. L. J. Biol. Chem. 2016;291:25641–25655. doi: 10.1074/jbc.M116.757310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H. Nakagawa M. Takeuchi K. Okabe S. Dig. Dis. Sci. 1989;34:238–245. doi: 10.1007/BF01536058. [DOI] [PubMed] [Google Scholar]
- de Grauw J. C. van Loon J. P. van de Lest C. H. Brunott A. van Weeren P. R. Vet. J. 2014;201:51–56. doi: 10.1016/j.tvjl.2014.03.030. [DOI] [PubMed] [Google Scholar]
- Hong M. C. Hsu D. I. Bounthavong M. Infect. Drug Resist. 2013;6:215–223. doi: 10.2147/IDR.S36140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G. Chung P. Adam H. Zelenitsky S. Denisuik A. Schweizer F. Lagace-Wiens P. R. Rubinstein E. Gin A. S. Walkty A. Hoban D. J. Lynch, 3rd J. P. Karlowsky J. A. Drugs. 2014;74:31–51. doi: 10.1007/s40265-013-0168-2. [DOI] [PubMed] [Google Scholar]
- Hong Y. L. Hossler P. A. Calhoun D. H. Meshnick S. R. Antimicrob. Agents Chemother. 1995;39:1756–1763. doi: 10.1128/AAC.39.8.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha-Duong N. T. Dijols S. Marques-Soares C. Minoletti C. Dansette P. M. Mansuy D. J. Med. Chem. 2001;44:3622–3631. doi: 10.1021/jm010861y. [DOI] [PubMed] [Google Scholar]
- Zamora I. Afzelius L. Cruciani G. J. Med. Chem. 2003;46:2313–2324. doi: 10.1021/jm021104i. [DOI] [PubMed] [Google Scholar]
- Johnson T. W. Richardson P. F. Bailey S. Brooun A. Burke B. J. Collins M. R. Cui J. J. Deal J. G. Deng Y. L. Dinh D. Engstrom L. D. He M. Hoffman J. Hoffman R. L. Huang Q. Kania R. S. Kath J. C. Lam H. Lam J. L. Le P. T. Lingardo L. Liu W. McTigue M. Palmer C. L. Sach N. W. Smeal T. Smith G. L. Stewart A. E. Timofeevski S. Zhu H. Zhu J. Zou H. Y. Edwards M. P. J. Med. Chem. 2014;57:4720–4744. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Zou H. Y. Li Q. Lee J. H. Arango M. E. McDonnell S. R. Yamazaki S. Koudriakova T. B. Alton G. Cui J. J. Kung P. P. Nambu M. D. Los G. Bender S. L. Mroczkowski B. Christensen J. G. Cancer Res. 2007;67:4408–4417. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- Christensen J. G. Zou H. Y. Arango M. E. Li Q. Lee J. H. McDonnell S. R. Yamazaki S. Alton G. R. Mroczkowski B. Los G. Mol. Cancer Ther. 2007;6:3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- Cui J. J. Tran-Dube M. Shen H. Nambu M. Kung P. P. Pairish M. Jia L. Meng J. Funk L. Botrous I. McTigue M. Grodsky N. Ryan K. Padrique E. Alton G. Timofeevski S. Yamazaki S. Li Q. Zou H. Christensen J. Mroczkowski B. Bender S. Kania R. S. Edwards M. P. J. Med. Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Awad M. M. Katayama R. McTigue M. Liu W. Deng Y. L. Brooun A. Friboulet L. Huang D. Falk M. D. Timofeevski S. Wilner K. D. Lockerman E. L. Khan T. M. Mahmood S. Gainor J. F. Digumarthy S. R. Stone J. R. Mino-Kenudson M. Christensen J. G. Iafrate A. J. Engelman J. A. Shaw A. T. N. Engl. J. Med. 2013;368:2395–2401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton J. E. Drugs. 2021;81:697–708. doi: 10.1007/s40265-021-01503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichincheri M. Ardini E. Magnaghi P. Avanzi N. Banfi P. Bossi R. Buffa L. Canevari G. Ceriani L. Colombo M. Corti L. Donati D. Fasolini M. Felder E. Fiorelli C. Fiorentini F. Galvani A. Isacchi A. Borgia A. L. Marchionni C. Nesi M. Orrenius C. Panzeri A. Pesenti E. Rusconi L. Saccardo M. B. Vanotti E. Perrone E. Orsini P. J. Med. Chem. 2016;59:3392–3408. doi: 10.1021/acs.jmedchem.6b00064. [DOI] [PubMed] [Google Scholar]
- Murray B. W. Rogers E. Zhai D. Deng W. Chen X. Sprengeler P. A. Zhang X. Graber A. Reich S. H. Stopatschinskaja S. Solomon B. Besse B. Drilon A. Mol. Cancer Ther. 2021;20:2446–2456. doi: 10.1158/1535-7163.MCT-21-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Madrid D. Szydzik J. Lind D. E. Borenas M. Bemark M. Cui J. Palmer R. H. Hallberg B. Sci. Rep. 2019;9:19353. doi: 10.1038/s41598-019-55060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Yu P. Dong L. Wang W. Duan S. Wang B. Gong X. Ye L. Wang H. Tian J. J. Med. Chem. 2021;64:10286–10296. doi: 10.1021/acs.jmedchem.1c00712. [DOI] [PubMed] [Google Scholar]
- O'Reilly E. M. Hechtman J. F. Ann. Oncol. 2019;30:viii36–viii40. doi: 10.1093/annonc/mdz385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A. Ou S. I. Cho B. C. Kim D. W. Lee J. Lin J. J. Zhu V. W. Ahn M. J. Camidge D. R. Nguyen J. Zhai D. Deng W. Huang Z. Rogers E. Liu J. Whitten J. Lim J. K. Stopatschinskaja S. Hyman D. M. Doebele R. C. Cui J. J. Shaw A. T. Cancer Discovery. 2018;8:1227–1236. doi: 10.1158/2159-8290.CD-18-0484. [DOI] [PubMed] [Google Scholar]
- Fan Y. Zhang Y. Liu Y. Jiang H. Zhou Y. Tang C. Fan W. Bioorg. Med. Chem. Lett. 2022;63:128646. doi: 10.1016/j.bmcl.2022.128646. [DOI] [PubMed] [Google Scholar]
- Ghilardi J. R. Freeman K. T. Jimenez-Andrade J. M. Mantyh W. G. Bloom A. P. Kuskowski M. A. Mantyh P. W. Mol. Pain. 2010;6:87. doi: 10.1186/1744-8069-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khotskaya Y. B. Holla V. R. Farago A. F. Mills Shaw K. R. Meric-Bernstam F. Hong D. S. Pharmacol. Ther. 2017;173:58–66. doi: 10.1016/j.pharmthera.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Doebele R. C. Davis L. E. Vaishnavi A. Le A. T. Estrada-Bernal A. Keysar S. Jimeno A. Varella-Garcia M. Aisner D. L. Li Y. Stephens P. J. Morosini D. Tuch B. B. Fernandes M. Nanda N. Low J. A. Cancer Discovery. 2015;5:1049–1057. doi: 10.1158/2159-8290.CD-15-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S. Zhang L. Luo X. Nan J. Yang W. Bin H. Li Y. Huang Q. Wang T. Pan Z. Mu B. Wang F. Tian C. Liu Y. Li L. Yang S. J. Med. Chem. 2022;65:2035–2058. doi: 10.1021/acs.jmedchem.1c01597. [DOI] [PubMed] [Google Scholar]
- Solomon B. J. Tan L. Lin J. J. Wong S. Q. Hollizeck S. Ebata K. Tuch B. B. Yoda S. Gainor J. F. Sequist L. V. Oxnard G. R. Gautschi O. Drilon A. Subbiah V. Khoo C. Zhu E. Y. Nguyen M. Henry D. Condroski K. R. Kolakowski G. R. Gomez E. Ballard J. Metcalf A. T. Blake J. F. Dawson S. J. Blosser W. Stancato L. F. Brandhuber B. J. Andrews S. Robinson B. G. Rothenberg S. M. J. Thorac. Oncol. 2020;15:541–549. doi: 10.1016/j.jtho.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A. Lopes A. R. McCusker M. G. Garrigues S. G. Ricciardi G. R. Arensmeyer K. E. Scilla K. A. Mehra R. Rolfo C. Curr. Oncol. Rep. 2020;22:48. doi: 10.1007/s11912-020-00909-8. [DOI] [PubMed] [Google Scholar]
- Qian Y. Chai S. Liang Z. Wang Y. Zhou Y. Xu X. Zhang C. Zhang M. Si J. Huang F. Huang Z. Hong W. Wang K. Mol. Cancer. 2014;13:176. doi: 10.1186/1476-4598-13-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V. Gainor J. F. Rahal R. Brubaker J. D. Kim J. L. Maynard M. Hu W. Cao Q. Sheets M. P. Wilson D. Wilson K. J. DiPietro L. Fleming P. Palmer M. Hu M. I. Wirth L. Brose M. S. Ou S. I. Taylor M. Garralda E. Miller S. Wolf B. Lengauer C. Guzi T. Evans E. K. Cancer Discovery. 2018;8:836–849. doi: 10.1158/2159-8290.CD-18-0338. [DOI] [PubMed] [Google Scholar]
- Choudhury N. J. Drilon A. Transl. Lung Cancer Res. 2020;9:2571–2580. doi: 10.21037/tlcr-20-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles P. P. Murray-Rust J. Kjaer S. Scott R. P. Hanrahan S. Santoro M. Ibanez C. F. McDonald N. Q. J. Biol. Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- Luo Z. Wang L. Fu Z. Shuai B. Luo M. Hu G. Chen J. Sun J. Wang J. Li J. Chen S. Zhang Y. Bioorg. Med. Chem. Lett. 2021;47:128149. doi: 10.1016/j.bmcl.2021.128149. [DOI] [PubMed] [Google Scholar]
- Pamplona F. A. Prediger R. D. Pandolfo P. Takahashi R. N. Psychopharmacology. 2006;188:641–649. doi: 10.1007/s00213-006-0514-0. [DOI] [PubMed] [Google Scholar]
- Zhang B. Li J. Yang X. Wu L. Zhang J. Yang Y. Zhao Y. Zhang L. Yang X. Yang X. Cheng X. Liu Z. Jiang B. Jiang H. Guddat L. W. Yang H. Rao Z. Cell. 2019;176:636–648.e13. doi: 10.1016/j.cell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Klumpers L. E. Roy C. Ferron G. Turpault S. Poitiers F. Pinquier J. L. van Hasselt J. G. Zuurman L. Erwich F. A. van Gerven J. M. Br. J. Clin. Pharmacol. 2013;76:65–77. doi: 10.1111/bcp.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T. Vemuri K. Pu M. Qu L. Han G. W. Wu Y. Zhao S. Shui W. Li S. Korde A. Laprairie R. B. Stahl E. L. Ho J. H. Zvonok N. Zhou H. Kufareva I. Wu B. Zhao Q. Hanson M. A. Bohn L. M. Makriyannis A. Stevens R. C. Liu Z. J. Cell. 2016;167:750–762.e14. doi: 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharathi C. Prabahar K. J. Prasad Ch S. Kumar M. S. Magesh S. Handa V. K. Dandala R. Naidu A. J. Pharm. Biomed. Anal. 2007;44:101–109. doi: 10.1016/j.jpba.2007.01.051. [DOI] [PubMed] [Google Scholar]
- Darwish M. Martin P. T. Cevallos W. H. Tse S. Wheeler S. Troy S. M. J. Clin. Pharmacol. 1999;39:670–674. doi: 10.1177/00912709922008308. [DOI] [PubMed] [Google Scholar]
- Albaugh P. A. Marshall L. Gregory J. White G. Hutchison A. Ross P. C. Gallagher D. W. Tallman J. F. Crago M. Cassella J. V. J. Med. Chem. 2002;45:5043–5051. doi: 10.1021/jm0202019. [DOI] [PubMed] [Google Scholar]
- Xu L. Wang J. Xiao B. Yang J. Liu Y. Huang R. Bioorg. Med. Chem. Lett. 2012;22:963–968. doi: 10.1016/j.bmcl.2011.12.013. [DOI] [PubMed] [Google Scholar]
- Lankford A. Ancoli-Israel S. Int. J. Clin. Pract. 2007;61:1037–1045. doi: 10.1111/j.1742-1241.2007.01322.x. [DOI] [PubMed] [Google Scholar]
- Ikeda K. Iwasaki Y. PLoS One. 2015;10:e0140316. doi: 10.1371/journal.pone.0140316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K. Uchikado H. Miyagi N. Morimoto Y. Ito T. Tancharoen S. Miura N. Miyata K. Sakamoto R. Kikuchi C. Iida N. Shiomi N. Kuramoto T. Kawahara K. Int. J. Mol. Med. 2011;28:899–906. doi: 10.3892/ijmm.2011.795. [DOI] [PubMed] [Google Scholar]
- Chegaev K. Cena C. Giorgis M. Rolando B. Tosco P. Bertinaria M. Fruttero R. Carrupt P. A. Gasco A. J. Med. Chem. 2009;52:574–578. doi: 10.1021/jm8007008. [DOI] [PubMed] [Google Scholar]
- Muhlethaler T. Gioia D. Prota A. E. Sharpe M. E. Cavalli A. Steinmetz M. O. Angew. Chem., Int. Ed. 2021;60:13331–13342. doi: 10.1002/anie.202100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. Expert Opin. Pharmacother. 2003;4:1563–1571. doi: 10.1517/14656566.4.9.1563. [DOI] [PubMed] [Google Scholar]
- Feyer P. Seegenschmiedt M. H. Steingraeber M. Support Care Cancer. 2005;13:671–678. doi: 10.1007/s00520-004-0766-3. [DOI] [PubMed] [Google Scholar]
- Argenti F. Bianchi T. Alparone L. IEEE Trans. Image Process. 2006;15:3385–3399. doi: 10.1109/tip.2006.881970. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Drugs. 2022;82:711–716. doi: 10.1007/s40265-022-01709-z. [DOI] [PubMed] [Google Scholar]
- Kitoh H. Biomedicines. 2020;8:325. doi: 10.3390/biomedicines8090325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques-Abreu M. Serrano C. Expert Rev. Anticancer Ther. 2021;21:1081–1088. doi: 10.1080/14737140.2021.1963235. [DOI] [PubMed] [Google Scholar]
- Scherber R. M. Clin. Lymphoma, Myeloma Leuk. 2018;18:S141–S178. doi: 10.1016/j.clml.2018.07.281. [DOI] [Google Scholar]
- Wu C. P. Lusvarghi S. Wang J. C. Hsiao S. H. Huang Y. H. Hung T. H. Ambudkar S. V. Mol. Pharmaceutics. 2019;16:3040–3052. doi: 10.1021/acs.molpharmaceut.9b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardino A. K. Evans E. K. Kim J. L. Brooijmans N. Hodous B. L. Wolf B. Lengauer C. Mol. Cell. Oncol. 2018;5:e1435183. doi: 10.1080/23723556.2018.1435183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apsel Winger B. Cortopassi W. A. Garrido Ruiz D. Ding L. Jang K. Leyte-Vidal A. Zhang N. Esteve-Puig R. Jacobson M. P. Shah N. P. Cancer Res. 2019;79:4283–4292. doi: 10.1158/0008-5472.CAN-18-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayman C. B. Kirsner J. B. Ford H. JAMA. 1961;175:908–909. doi: 10.1001/jama.1961.63040100023020b. [DOI] [PubMed] [Google Scholar]
- Gheorghiu T. Klein H. J. Frotz H. Jirmann V. W. Ed M. Klin. Wochenschr. 1969;47:1206–1213. doi: 10.1007/BF01484885. [DOI] [PubMed] [Google Scholar]
- Heijerman H. G. M. McKone E. F. Downey D. G. Van Braeckel E. Rowe S. M. Tullis E. Mall M. A. Welter J. J. Ramsey B. W. McKee C. M. Marigowda G. Moskowitz S. M. Waltz D. Sosnay P. R. Simard C. Ahluwalia N. Xuan F. Zhang Y. Taylor-Cousar J. L. McCoy K. S. McCoy K. Donaldson S. Walker S. Chmiel J. Rubenstein R. Froh D. K. Neuringer I. Jain M. Moffett K. Taylor-Cousar J. L. Barnett B. Mueller G. Flume P. Livingston F. Mehdi N. Teneback C. Welter J. Jain R. Kissner D. Patel K. Calimano F. J. Johannes J. Daines C. Keens T. Scher H. Chittivelu S. Reddivalam S. Klingsberg R. C. Johnson L. G. Verhulst S. Macedo P. Downey D. Connett G. Nash E. Withers N. Lee T. Bakker M. Heijerman H. Vermeulen F. Van Braeckel E. Knoop C. De Wachter E. van der Meer R. Merkus P. Majoor C. Lancet. 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G. Roldan A. Hancock M. A. Da Fonte D. F. Xu H. Hussein M. Frenkiel S. Matouk E. Velkov T. Lukacs G. L. JCI Insight. 2020;5:e139983. doi: 10.1172/jci.insight.139983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baatallah N. Elbahnsi A. Mornon J. P. Chevalier B. Pranke I. Servel N. Zelli R. Decout J. L. Edelman A. Sermet-Gaudelus I. Callebaut I. Hinzpeter A. Cell. Mol. Life Sci. 2021;78:7813–7829. doi: 10.1007/s00018-021-03994-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulke M. H. O'Dorisio T. Phan A. Bergsland E. Law L. Banks P. Freiman J. Frazier K. Jackson J. Yao J. C. Kvols L. Lapuerta P. Zambrowicz B. Fleming D. Sands A. Endocr.-Relat. Cancer. 2014;21:705–714. doi: 10.1530/ERC-14-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarca A. Barriuso J. McNamara M. G. Hubner R. A. Valle J. W. Expert Opin. Pharmacother. 2016;17:2487–2498. doi: 10.1080/14656566.2016.1254191. [DOI] [PubMed] [Google Scholar]
- Beinse G. Hulin A. Rousseau B. Invest. New Drugs. 2019;37:1289–1291. doi: 10.1007/s10637-019-00743-1. [DOI] [PubMed] [Google Scholar]
- Liu X. Wang B. Chen C. Qi Z. Zou F. Wang J. Hu C. Wang A. Ge J. Liu Q. Yu K. Hu Z. Jiang Z. Wang W. Wang L. Wang W. Ren T. Bai M. Liu Q. Liu J. J. Med. Chem. 2019;62:5006–5024. doi: 10.1021/acs.jmedchem.9b00176. [DOI] [PubMed] [Google Scholar]
- Wang X. Kim J. J. Med. Chem. 2012;55:7332–7341. doi: 10.1021/jm300613w. [DOI] [PubMed] [Google Scholar]
- Li Z. Jiang K. Zhu X. Lin G. Song F. Zhao Y. Piao Y. Liu J. Cheng W. Bi X. Gong P. Song Z. Meng S. Cancer Lett. 2016;370:332–344. doi: 10.1016/j.canlet.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Pan J. H. Zhou H. Zhu S. B. Huang J. L. Zhao X. X. Ding H. Pan Y. L. Cancer Manage. Res. 2018;10:2289–2301. doi: 10.2147/CMAR.S170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbar H. S. El-Gamal M. I. Tarazi H. Lee B. S. Jeon H. R. Kwon D. Oh C. H. J. Enzyme Inhib. Med. Chem. 2020;35:1712–1726. doi: 10.1080/14756366.2020.1819260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepik K. J. Levy A. R. Sobolev B. G. Purssell R. A. DeWitt C. R. Erhardt G. D. Kennedy J. R. Daws D. E. Brignall J. L. Ann. Emerg. Med. 2009;53:439–450.e10. doi: 10.1016/j.annemergmed.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Sande M. Thompson D. Monte A. A. Am. J. Emerg. Med. 2012;30:262.e3–262.e5. doi: 10.1016/j.ajem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Bestic M. Blackford M. Reed M. J. Clin. Pharmacol. 2009;49:130–137. doi: 10.1177/0091270008327142. [DOI] [PubMed] [Google Scholar]