

Abstract
In the present study, the MCRs of hydroxylamine hydrochloride and ethyl acetoacetate with various substituted aromatic and heteroaromatic aldehydes reacted to produce 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones derivatives catalyzed by an agro-waste-based solvent medium in an oil bath at 60 °C with stirring. The developed protocol features several advantages, such as being benign and eco-friendly, efficient, avoiding the use of hazardous solvents, and inexpensive, while giving isoxazole derivatives in an 86–92% yield. The homogeneity of the product was confirmed by various spectroscopic analyses. Further, in vitro anticancer evaluation of the synthesized compounds (4h–4o) against lung cancer cells was performed, and among them, 4j, 4k, 4m, and 4o exhibited excellent anticancer activity and compounds 4i and 4n moderate inhibitory activity against lung cancer A549 cells to the reference drug doxorubicin. Furthermore, the derivatives were subjected to electrochemical behaviour studies using cyclic voltammetry and they showed intensive oxidation and reduction potential and also showed excellent anti-oxidant agents. Based on this research study, more and more novel structures of isoxazoles are being designed and synthesized, and their electrochemical behaviour and anticancer activities are studied for the development of novel drug-like candidates.
We demonstrate the MCRs of hydroxylamine hydrochloride and ethyl acetoacetate with various substituted aromatic and heteroaromatic aldehydes using an agro-waste-based solvent medium as a catalyst. The products were then screened for their anticancer activity.
1. Introduction
One of the major challenges in current organic synthesis is the development of an inexpensive, eco-friendly and efficient method for the synthesis of bioactive molecules and libraries. The discovery of high-throughput screening has tremendously increased the potential to meet the demand for the novel bioactive molecules testing. Subsequently, multicomponent reactions (MCRs) have become an increasingly in-demand tool for the synthesis of libraries of compounds to meet the demand for various substituents.1 Heterocyclic compounds have attracted considerable scrutiny in the pharmaceutical, agro-chemicals and food industries.2–4 Heterocycle compounds, which include a huge number of families, have been widely explored, and have shown interesting pharmacological profiles for a wide range of applications. Among them, nitrogen-containing heterocyclic compounds are common in nature and their application in pharmaceuticals, agrochemicals and functional materials is promising.5–9 Compounds containing both oxygen and nitrogen in a ring are referred to as isoxazolines and isoxazoles, and are known to possess anti-inflammatory, antihypertensive, bactericidal and other types of properties that are ideal for pharmacological applications. Sulfa drugs, such as sulfaisoxazole and sulfamethoxazole, and some penicillin derivatives (cloxacillin and oxacillin) having an isoxazole skeleton are known to exhibit antimicrobial properties.10–13 Isoxazole (1a) is a five-membered heterocyclic compound containing oxygen and nitrogen atoms in the 1,2-positions, and its partially saturated analogues are called isoxazolines (1b–d) and the completely saturated analogue is called isoxazolidine (1e) (Fig. 1).14
Fig. 1. Structures of different isoxazole skeletons.

Both isoxazole and isoxazolone derivatives have emerged as important heterocyclic moieties and have shown wide range of pharmacological activities in fungicidal,15 analgesic,16 antimicrobial,17 anti-oxidant,18 nematicidal,19 anti-inflammatory,20 hypoglycaemic,21 and many more interesting applications (Fig. 2).
Fig. 2. Some biologically prominent drug molecules containing the isoxazole skeleton.

Nowadays, multi-component reactions (MCRs) are the most preferred route by chemists, because of their high utility in the synthesis of complex heterocyclic compounds from simple and readily accessible starting materials. MCRs are considered a very strong technique for the synthesis of a variety of bioactive molecules in a one-pot process by three or more reactants.22 This type of reaction technique is important in the precursor construction of many natural products and in biologically active derivative synthesis.23 Since the first MCR was described by Strecker in 1850,24 now it is demonstrated to be a highly valuable tool for the expedient creation of numerous complex chemical skeleton compounds, including natural products and bioactive compounds.25 Recently, considerable attention has been focused on MCRs owing to their high efficiency, mild reaction conditions, facileness and eco-friendliness, and as they can give a high yield isolation of the product.
A literature survey showed that the synthesis of aryl-3-methylisoxazol-5(4H)-one derivatives was achieved through the coupling of an aromatic aldehyde with ethyl acetoacetate and hydroxylamine hydrochloride (Scheme 1). This reaction has been performed using different reagents26 and catalysts in basic media, such as sodium silicate,27 sodium benzoate,28 sodium azide,29 sodium saccharin,30 sodium citrate,31 sodium sulfide,32 sodium ascorbate,33 sodium tetraborate,34 tartaric acid,35 potassium phthalimide,36 mesolite,37 DOWEX(R)50WX4,38 citric acid,39 choline chloride/urea,40 nano-MMT-Sn.41
Scheme 1. Reported catalysts for the synthesis of 3-methyl-4-(hetero)arylmethylene isoxazole-5(4H)-one.

Even though, the above-mentioned catalytic approaches have their own advantages, most of them suffer from limitations, such as the use of hazardous materials (or solvents), expensive catalysts, environmental problems, production of undesirable waste, long reaction times, harsh reaction conditions and low yield product isolation.42,43 Recently, organic chemists have been striving to develop greener synthesis methods for the synthesis of various bioactive molecules. The solvent-free synthesis is one such type of reaction that has gained attention in the last few decades. This approach can offer an enhanced reaction rate by reducing the reaction time, a high product yield and easier work-up, making it a facile method for organic transformations. In conjunction, solvent-free reactions carried out under microwave irradiation have also recorded significant advantages in recent years, such as a faster reaction rate, clean process and good atom economy. Nowadays, chemists are more focused on the development of green chemistry protocols to replace the use of harmful organic solvents that could lead to waste production and/or emission to the environment. Reagents like solid-supported organic or inorganic catalysts have also gained more attention in organic transformations, as such approaches can not only avoid the use of toxic solvents for the reaction but also avoid the need for expensive catalysts and tedious work-up.
In this work, we focused on a greener protocol using the agro-waste WEOFPA (Water Extract of Orange Fruit Peel Ash), previously reported by our group,44 as an efficient catalyst for the synthesis of isoxazole from the condensation of hydroxylamine hydrochloride and ethyl acetoacetate with various substituted aromatic and heteroaromatic aldehydes, in which compounds 4i, 4j, 4l, 4m and 4o were not reported previously. Overall, the developed protocol proceeded under solvent-free conditions and so the method falls in the domain of green chemistry synthesis.
2. Result and discussion
2.1. Preparation and characterization of WEOFPA
The preparation and characterization of the agro-waste extract media required for this work were previously reported by our group,44 and the detailed preparation procedure of WEOFPA is mentioned in the experimental section (Fig. 3). The obtained ash powder was examined for elemental analysis by XRD, SEM-EDX and flame photometry.44 In this work, we report for the first time ash powder XRF analysis, which revealed the presence of a high concentration of K, P, Ca and Si, and minor elements of oxides of Ti, Fe, Cu, Mg, Br, Rb2, Nb2 and Sr (Table 1). The data were very much comparable with our reported characterization data.44 The final extract media pH was examined and found to be 11.70. This pH revealed that the extract solution was basic, and it contained oxide and carbonate forms of the K and Ca major contents, and it could serve as a superior basic catalyst alternative to the toxic chemicals presently used in various organic transformations.
Fig. 3. Schematic representation of the preparation of WEOFPA.

XRF data of the water extract of orange fruit peel ash.
| Entry | Oxide elements | Quantity (%) |
|---|---|---|
| 1 | K2O | 70.7 |
| 2 | P2O | 12.3 |
| 3 | CaO | 10.1 |
| 4 | SiO2 | 4.0 |
| 5 | MgO | 1.17 |
| 6 | Fe2O3 | 0.21 |
| 7 | CuO | 0.22 |
| 8 | TiO2 | 0.18 |
| 9 | ZnO | 0.15 |
| 10 | MnO | 0.10 |
| 11 | Br2O | 0.05 |
| 12 | Rb2O | 0.04 |
| 13 | Nb2O5 | 0.02 |
| 14 | SrO | 0.02 |
The main objective of our research work here was on the development of a greener protocol for the synthesis of 3-methyl-4-(hetero)arylmethylene isoxazole-5(4H)-ones via the reaction of aromatic or heteroaromatic aldehyde, ethyl acetoacetate, and hydroxylamine hydrochloride in the presence of the agro-waste catalyst WEOFPA and glycerol (Scheme 2). In a model reaction, benzaldehyde, ethyl acetoacetate and hydroxylamine hydrochloride were chosen for the optimization of the reaction, which was performed in a 50 mL round-bottomed flask at 60 °C on an oil bath with stirring. After completion of the reaction (as monitored by thin layer chromatography, TLC), the reaction mixture was triturated with ice-cold water for the immediate separation of the solid product in a good to excellent yield isolation.
Scheme 2. General synthetic route of 3-methyl-4-(hetero)arylmethylene isoxazole-5(4H)-one.

2.2. Optimization of the agro-waste-derived catalyst and the reaction method
Optimization of the WEOFPA and the minimal glycerol amount required for the 1 mmol scale in a model reaction were examined by selecting different volumes of WEOFPA and glycerol combinations starting with 0, 1, 2, 3, 4, 5 and 6 mL of WEOFPA and 0.4 mL of glycerol (Table 2). The studies revealed that the reaction carried out in the absence of WEOFPA did not show product formation by TLC, while with the gradual increase in volume of WEOFPA from 1 mL to 5 mL with 0.4 mL of glycerol led to an exponential increase in the product isolated at 60 °C in an oil bath under stirring. Further, with the increase in WEOFPA to 6 mL, there was no increased in the product yield isolated or decrease in the reaction time (Table 2). This optimization reaction experiment confirmed that a eutectic mixture combination volume of 5 mL of WEOFPA and 0.4 mL of glycerol gave a suitable combination medium for the reaction and gave a high yield of 3-methyl-4-(hetero) aryl methylene isoxazole-5(4H)-one. We also examined in a model reaction the use only of WEOFPA (optimized) or glycerol separately, and a low yield was obtained in the no glycerol case (entry 8, Table 2) while no product formation was noticed in the only-glycerol-mediated reaction (entry 9, Table 2).
Optimization of WEOFPA.
| Entry | WEOFPA (mL) | Glycerol (mL) | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | 0.0 | 0.0 | 70 | ND |
| 2 | 1 | 0.4 | 65 | 29 |
| 3 | 2 | 0.4 | 62 | 38 |
| 4 | 3 | 0.4 | 60 | 50 |
| 5 | 4 | 0.4 | 60 | 65 |
| 6 | 5 | 0.4 | 55 | 95 |
| 7 | 6 | 0.4 | 55 | 95 |
| 8 | 5 | 0.0 | 80 | 70 |
| 9 | 0.0 | 0.4 | 80 | ND |
To check the importance of the agro-waste solvent media role in the present reaction, we performed the reaction in the absence of WEOFPA. Surprisingly, no product was formed even after 3 h of heating on the oil bath with magnetic stirring. These studies confirmed that the reaction required the catalyst, and that adding a small amount of glycerol for the heterocyclization influenced the reaction rate and made it faster and led to a high yield of product isolated (Table 3). Before getting into the suitable method required for the reaction, we exercised different reaction conditions on a model reaction, like “on and off” mechanochemical grinding for about 50 min. The progress of the reaction was monitored by TLC, and it was found that this method gave a low product yield isolated. Then, we extended the model reaction to the ultrasonication, compared to the grinding method, and this method gave a 65% product yield after 80 min sonication. To make the synthetic method much faster, we performed the model reaction in a custom-made microwave oven having a condenser fixing facility, but no product formation was observed by TLC. Further, we tested an oil bath heating method at different temperatures (entries 5–9, Table 3) under stirring, in which 60 °C gave the product isolated in a high yield in 55–60 min (entry 7, Table 3).
Optimization of the methods and temperature in a model reaction.
| Entry | Methods | WEOFPA + glycerol (mL) | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | Mechanochemical | 5 + 0.4 | 50 | 44 |
| 2 | Stirring at rt | 5 + 0.4 | 60 | 39 |
| 3 | Ultrasonication | 5 + 0.4 | 80 | 65 |
| 4 | MW 300 W | 5 + 0.4 | 20 | ND |
| 5 | Heating at 40 °C in an oil bath | 5 + 0.4 | 85 | 65 |
| 6 | Heating at 50 °C in an oil bath | 5 + 0.4 | 70 | 75 |
| 7 | Heating at 60 °C in an oil bath | 5 + 0.4 | 55 | 95 |
| 8 | Heating at 70 °C in an oil bath | 5 + 0.4 | 55 | 88 |
| 9 | Heating at 80 °C in an oil bath | 5 + 0.4 | 55 | 85 |
Thus, the optimized synthetic approach was demonstrated to be a simple and efficient synthesis of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-one using a natural-feedstock-extracted solvent media derived from agro-waste orange peels. To explore the scope and generality of the developed method for the compatibility of various substituted aldehydes, the reaction was extended to a variety of aromatic-substituted and heteroaromatic-substituted aldehyde-containing electron-withdrawing group (EWGs) and electron-donating groups (EDGs) (Table 4). The isolated products reported in Table 4 revealed that the EWG and EDG present on the aromatic ring of the aldehyde functionality did not have any significant measurable effect on the rate of the reaction.
Synthesis and physical data of the prepared 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones.
| Entry | Aldehyde | Producta | Time (min) | Yield (%)b | M.p. (°C) |
|---|---|---|---|---|---|
| 1 |
 1a 1a |
 4a 4a |
55 | 90 | 140–142 [37] |
| 2 |
 1b 1b |
 4b 4b |
57 | 89 | 160–162[37] |
| 3 |
 1c 1c |
 4c 4c |
55 | 89 | 136–138[37] |
| 4 |
 1d 1d |
 4d 4d |
59 | 83 | 173–175[36] |
| 5 |
 1e 1e |
 4e 4e |
59 | 87 | 165–167[46] |
| 6 |
 1f 1f |
 4f 4f |
56 | 90 | 224–226[40] |
| 7 |
 1g 1g |
 4g 4g |
57 | 91 | 239–241[36] |
| 8 |
 1h 1h |
 4h 4h |
55 | 95 | 244–246 [new] |
| 9 |
 1i 1i |
 4i 4i |
58 | 88 | 220–222 [new] |
| 10 |
 1j 1j |
 4j 4j |
55 | 89 | 250–252 [new] |
| 11 |
 1k 1k |
 4k 4k |
60 | 93 | 185–187 [new] |
| 12 |
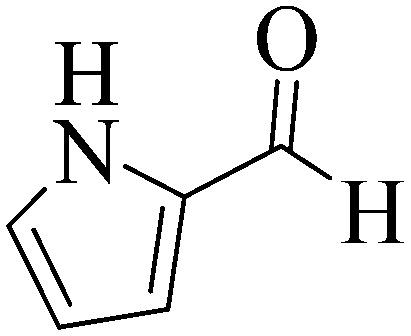 1l 1l |
 4l 4l |
59 | 90 | 196–198 [new] |
| 13 |
 1m 1m |
 4m 4m |
55 | 89 | 185–187 [new] |
| 14 |
 1n 1n |
 4n 4n |
55 | 91 | 217–219 [new] |
| 15 |
 1o 1o |
 4o 4o |
58 | 89 | 201–203 [new] |
The products were characterized by FT-IR, 1H- and 13C-NMR, and LC/HR-MS.
Yields refer to the isolated yields.
Further, we also compared the present developed method with previous reported methods, as summarized in Table 5. The reaction with the catalyst mesolite under 40 °C temperature with stirring for about 1 h afforded an 89% yield (entry 1, Table 5), but used an expensive catalyst with ethanol solvent. In entry 2, the synthesis was performed in the presence of the ion-exchange resin DOWEX 1-x8OH, H2O under room temperature stirring conditions for about 6 h to afford a 95% yield. The citric acid-catalyzed synthesis at room temperature with stirring for about 8 h gave a 90% yield (entry 3). Both entries 2 and 3 as catalyzed reactions required a long reaction time and used direct chemicals for the reaction. The potassium phthalimide (10 mol%)-catalyzed reaction gave a 96% isolation of the product (entry 4), while the use of sodium saccharin (10 mol%) under room temperature required 2 h to afford a 96% yield (entry 5). The nano-MMT functional-catalyzed reaction under neat ultrasonication required 45 min and afforded a 96% yield (entry 6); however the catalyst preparation and work-up were tedious. The reaction carried out in the presence of choline chloride : urea (1 : 2) with heating at 70 °C for about 25 min gave a 90% yield (entry 7). N,N-Diethyl-N-sulfoethanaminium methane sulfonate under room temperature required 4 h reaction to afford a 96% yield (entry 5), while the use of magnetic sulfonated polysaccharides in the presence of EtOH under room temperature for 2 h duration gave an 88% yield. The present method used inexpensive and highly abundant agro-waste after the valuable fruit juice was extracted and falls under a green chemistry protocol, with a benign nature and good atom economy compared to the various reported catalytic methods for the synthesis of 3-methyl-4-(hetero)arylmethylene isoxazole-5(4H)-one's (entry 10, Table 5).
Comparative study of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-one synthesis in the present study with the reported methods in the literature.
| S. No. | Catalyst | Method | Time | Yield (%) | Reference |
|---|---|---|---|---|---|
| 1 | Mesolite, EtOH:H2O | rt, 40 °C | 1 h | 89 | 37 |
| 2 | DOWEX 1-x8OH, H2O | rt | 6 h | 95 | 38 |
| 3 | Citric acid | rt | 8 h | 90 | 39 |
| 4 | Potassium phthalimide | rt | 2 h | 96 | 36 |
| 5 | Sodium saccharin | rt | 2 h | 96 | 30 |
| 6 | Nano-MMT-Sn | US, 60 °C, | 45 min | 96 | 41 |
| 7 | ChCl:urea | Heating 70 °C | 25 min | 90 | 40 |
| 8 | [Et3N-SO3H][MeSO3] | rt | 4 h | 85 | 45 |
| 9 | Magnetic sulfonated polysaccharides, EtOH | rt | 2 h | 88 | 46 |
| 10 | WEOFPA, glycerol | Heating 60 °C | 60 min | 94 | Present work |
2.3. Plausible mechanism
The plausible mechanistic pathway for the product formation involved the reaction between ethyl acetoacetate (2) and hydroxylamine hydrochloride (3), with the amine group nucleophilic attack on the carbonyl centre of the compound (2) to form the oxime intermediate A (Scheme 3). The catalyst then activates the carbonyl group of the oxime intermediate (A) and the nucleophilic attack occurs at the carbonyl carbon of the aromatic/heteroaromatic aldehyde (1). This is also activated by the WEOFPA and glycerol combination at 60 °C in an oil bath to form intermediate B with the removal of a water molecule. Finally, the desired product (4) is obtained by intramolecular cyclization and by the elimination of ethyl alcohol.
Scheme 3. Plausible mechanism for the formation of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-one.

2.4. Interpretation of the spectral data
The homogeneity of the isolated target product was characterized by several spectroscopic techniques. The FT-IR spectrum of compound 4l showed a prominent band at 3010 cm−1, due to the CH stretching; a band at 1773 cm−1, due to C O stretching; a band at 1655 cm−1, due to C N stretching; and a band at 1529 cm−1, due to N–O stretching (Fig. S21†). The 1H-NMR spectrum of the product 4l was obtained and showed the following results: δ at 2.16 singlet due to 3H of CH3, 6.85 triplet 1H due to CH, 7.02 and 7.51 doublet due to 2H of CH, 7.98 doublet due to 1H of the aromatic and 8.24 singlet 1H due to NH (Fig. S22†). The 13C-NMR spectrum gave different carbon environment peaks at 170.24, 166.27, 137.56, 129.78, 127.56, 120.12, 114.47, 106.51, 22.07 ppm (Fig. S23†). LC-MS: m/z (calcd.): 176.44 Da; m/z (obs.): 177.25 Da (Fig. S25†).
2.5. Electrochemical behaviour studies of selected derivatives
We studied the electrochemical behaviour of the synthesized 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones by cyclic voltammetry experiments47 (Fig. 3) using a CHI6005E Electrochemical Analyzer with general-purpose electrochemical system software. Cyclic voltammetry is generally used for the detection of the reversibility, irreversibility and quasi-reversibility of a redox process. The anodic peak potential, anodic peak current, cathodic peak potential and cathodic peak currents are the important parameters obtained from the voltammogram. Here, a three-electrode cell system was employed with the silver electrode (Ag/AgCl) as a reference cell, GC (glassy carbon) as a working electrode and platinum wire as a counter electrode. The experiments were carried out in an inert atmosphere by purging nitrogen gas (99.99% pure) for about 25 min before every experiment started. The electrochemical characterization was performed using 0.1 mM of complex TBAP (tetrabutylammonium perchlorate (https://www.sigmaaldrich.com/IN/en/product/aldrich/86885)) with supporting electrolytes in DMSO. An inlaid GC disc with a surface area 0.071 cm2 was employed as an electrode in the electrochemical characterization. The GCEs were polished with polishing cloth (Buehler, Micro Cloth) using sequentially 1.0 μm and 0.5 μm alumina in a water slurry. They were then repeatedly washed with 1 : 1 HNO3, acetone and water, respectively and subjected to ultrasonication for about 5 min.48 Herein, for the first time, we report the electrochemical behaviour studies of the bare electrode and the derivative dissolved in 0.1 mM of TBAB in DMSO of selected 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones under sonication.
The voltammograms were recorded at a scan rate of 50 mV s−1, the reduction waves (anodic current) in a range of potential observed from 0.27 to 0.72 V (Fig. 4) and the oxidation peaks (cathodic current) from −0.65 to −0.98 (Fig. 4) due to the oxazolone and amino groups present on these derivatives, which showed both oxidation and reduction potential reactions. This study revealed that compounds 4h, 4i, 4j, 4l and 4m were electrochemically more active and exhibited intensive oxidation and reduction potential peaks, and displayed good oxidation and reducing properties, The derivatives 4k, 4n and 4o showed moderate oxidation and reduction potential values, while the redox peaks were not observed in the spectra of bare GCE, which confirmed the peaks were due to the sample only.
Fig. 4. Electrochemical behaviours of selected 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones.

2.6. In vitro anticancer studies
The in vitro anticancer properties of the selected synthesized compounds were screened for their in vitro anticancer activity against lung cancer A549 cells. The anticancer efficacy of the derivatives showed comparable activities to the reference doxorubicin used, and the results are summarized in Table 6. Doxorubicin was utilized as a clinically antitumor positive control agent and also DMSO was used as a negative control. This experimental section is presented in the ESI† (S1–1.6). The cytotoxicity efficacy of the synthesized molecules was proved by the IC50 value, which is the concentration that causes a 50% reduction of the cell growth values attained. The ratio of inhibition of cell propagation of the prepared molecules was studied at six different concentrations (100, 50, 25, 12.5, 6.25 and 3.125 μg mL−1) and the IC50 values were determined and are provided in Tables 6 and S1.† It can be seen from Table 6 that most of the derivatives tested showed good to excellent anticancer activity against A549 cell lines.49–51 Compounds 4j (IC50 = 8.99), 4m (IC50 = 14.22) and 4o (IC50 = 12.31) showed almost equivalent activity to the reference drug, while compounds 4i (IC50 = 37.43), 4k (IC50 = 22.75) and 4n (IC50 = 59.16) showed activity near to that of the reference anticancer drug doxorubicin (IC50 = 8.73) in μM.
In vitro anticancer activity of compounds 4h–4o.
| S. No. | Compounds | IC50 ± SD (μM) |
|---|---|---|
| 1 | 4h | 107.43 ± 3.53 |
| 2 | 4i | 37.43 ± 0.71 |
| 3 | 4j | 8.99 ± 0. |
| 4 | 4k | 22.75 ± 0.89 |
| 5 | 4l | 182.63 ± 3.35 |
| 6 | 4m | 14.22 ± 0.51 |
| 7 | 4n | 59.16 ± 1.33 |
| 8 | 4o | 12.31 ± 0.35 |
| 9 | Doxorubicin | 8.73 ± 0.12 |
The principle relationship between the molecular structure and its biological activity is referred to as the structure–activity relationship (SAR). Based on the above outcomes, several key facts about the SAR (Fig. 5) were revealed in this framework of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones derivatives (4h–o), suggesting that the biological effects of a new chemical compound can often be predicted from its molecular structure. This is because similar compounds may have similar physical and biological properties. The SAR depends on the recognition of which structural characteristics correlate with which chemical and biological reactivity. In compounds 4i, 4j, 4k and 4o, two or more substituted rings are attached to isoxazoles. Compounds 4j, 4m and 4o demonstrated comparable in vitro anticancer activities compared to the standard doxorubicin. Upon comparing compounds 4h and 4j, we found that a substitution of Cl in 4j drastically reduced the IC50 value by 12 times. The presence of an enone/chalcone moiety in 4o and 4m gave them important pharmacophore attributes, while the “redox chameleon” sulfur in 4o clearly indicated the anticancer activities associated with their structures.
Fig. 5. SAR of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-ones.

In summary, compounds 4h, 4i, 4j, 4k, 4l, 4m, 4n and 4o were evaluated for their anticancer activity against lung cancer A549 cells. Among them, 4-((2-chloro-1H-indol-3-yl)methylene)-3-methylisoxazol-5(4H)-one (4j), 4-((2-hydroxynaphthalen-1-yl)methylene)-3-methylisoxazol-5(4H)-one (4k), 3-methyl-4-(thiophen-2-ylmethyl)isoxazol-5(4H)-one (4m) and 4-((1,4-diphenyl-1H-pyrrol-3-yl)methyl)-3-methyl isoxazol-5(4H)-one (4o) showed excellent IC50 values in μM compared with the reference drug and can be regarded as lead drug candidates for their anticancer activity against lung cancer A549 cells. Compounds 3-methyl-4-((2-phenyl-1H-indol-3-yl)methylene)isoxazol-5(4H)-one (4i) and 4-(3,4-dimethoxybenzylidene)-3-methylisoxazol-5(4H)-one (4n) showed a moderate inhibitory activity against all the tested cell lines with IC50 values of 37.43 and 59.16 μM, respectively, while the derivatives 4-((1H-indol-3-yl)methylene)-3-methylisoxazol-5(4H)-one (4h) and 4-((1H-pyrrol-2-yl)methylene)-3-methylisoxazol-5(4H)-one (4l) exhibited less inhibitory activity against lung cancer A549 cells (Table S1†).45
3. Conclusion
In this work, we demonstrated an agro-waste-extracted WEOFPA combined with glycerol combination to form a eutectic mixture as an green catalyst that was also low-cost and environmentally friendly and proved to be efficient in the synthesis of 3-methyl-4-(hetero)aryl methylene isoxazole-5(4H)-one derivatives (4h–4o). Various substituted aromatic and heteroaromatic aldehydes, ethyl acetoacetate and hydroxylamine hydrochloride heated in an oil bath at 60 °C were used to synthesize isoxazole derivatives, and presented the new derivatives 4i, 4j, 4l, 4m and 4o. The developed method offers several advantages, like being non-toxic and non-polluting, external base-free and inexpensive. For the first time, we performed electrochemical behaviour studies for the compounds 4h, 4i, 4j, 4k, 4l, 4m, 4n and 4o, which revealed that compounds 4h, 4i, 4j, 4l and 4m were electrochemically more active and exhibited intensive oxidation and reduction properties, while the derivatives 4k, 4n and 4o showed moderate oxidation and reduction characters. Further, we performed an in vitro anticancer evaluation of the same compounds (4h–4o) against lung cancer cells, and found that 4j, 4k, 4m and 4o exhibited excellent anticancer activity, while compounds 4i and 4n showed moderate inhibitory activity against lung cancer A549 cells. Based on these research findings, more and more novel structures of isoxazoles are being designed and synthesized, and their electrochemical behaviour and anticancer activities are being studied for the development of pharmacological drugs.
Conflicts of interest
The authors declare no conflict of interest, financial or otherwise.
Supplementary Material
Acknowledgments
The corresponding author thankful to the UGC for the award of Major Research Project {(UGC-MRP: F.43-181/2014 (SR)}, Interdisciplinary Research Project, RCUB and VGST, Govt. of Karnataka for SMYSR award to Dr. KK. Department of Chemistry, RCUB.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00191h
References
- Elena A. M. Volodymyr V. T. Sergey M. D. Yulia V. S. Thomas J. J. M. Elena V. V. Valentin A. C. ARKIVOC. 2013;(iii):338–371. [Google Scholar]
- Abdel Rehman M. M. Mangouma S. A. EL-Bitar H. L. Bull. Pharm. Sci. 1990;13:137–144. [Google Scholar]
- He F. Snider B. B. J. Org. Chem. 1999;64:1397–1399. doi: 10.1021/jo9820465. [DOI] [Google Scholar]
- Dabholkar V. Badhe K. Kurade S. IJAR. 2017;6:8416–8420. [Google Scholar]
- Domling A. Chem. Rev. 2006;106(1):17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- Burke M. D. Schreiber S. L. Angew. Chem., Int. Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- Spring D. R. OBM. 2003;1:3867–3870. doi: 10.1039/b310752n. [DOI] [PubMed] [Google Scholar]
- Strausberg R. L. Schreiber S. L. Science. 2003;300:294–295. doi: 10.1126/science.1083395. [DOI] [PubMed] [Google Scholar]
- Horton D. A. Bourne G. T. Smythe M. L. Chem. Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- Whest H. M. and Hoffer M., US 2430094, 1947
- Wornser G. P. Keusch G. T. Hell R. C. Drugs. 1982;24:459–518. doi: 10.2165/00003495-198224060-00002. [DOI] [PubMed] [Google Scholar]
- Doyle F. P. Long A. A. W. Nayler J. H. C. Stove E. R. J. Chem. Soc. 1963:5838. doi: 10.1039/JR9630005838. [DOI] [Google Scholar]
- Doyle F. P. Long A. A. W. Nayler J. H. C. Stove E. R. Nature. 1961;192:1183–1184. doi: 10.1038/1921183a0. [DOI] [PubMed] [Google Scholar]
- Yogesh W. Pravin M. Pramod K. Mini-Rev. Org. Chem. 2021;18:1–23. doi: 10.2174/1570193X1801210210115256. [DOI] [Google Scholar]
- Santos M. M. M. Faria N. Iley J. Coles S. J. Hursthouse M. B. Martins M. L. Moreira R. Bioorg. Med. Chem. Lett. 2010;20:193–195. doi: 10.1016/j.bmcl.2009.10.137. [DOI] [PubMed] [Google Scholar]
- Kano H. Adachi I. Kido R. Hirose K. J. Med. Chem. 1967;10:411–418. doi: 10.1021/jm00315a028. [DOI] [PubMed] [Google Scholar]
- Saikh F. Das J. Ghosh S. Tetrahedron Lett. 2013;54:4679–4682. doi: 10.1016/j.tetlet.2013.06.086. [DOI] [Google Scholar]
- Padmaja A. Rajasekhar C. Muranikrishna A. Padmavathi V. Eur. J. Med. Chem. 2011;46:5034–5038. doi: 10.1016/j.ejmech.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Srinivas A. Nagaraj A. Reddy C. S. Eur. J. Med. Chem. 2010;45:2353–2358. doi: 10.1016/j.ejmech.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Karabasanagouda T. Adhikari A. V. Girisha M. IJC. 2009;48B:430–437. [Google Scholar]
- Kang Y. Y. Shin K. J. Yo K. H. Seo K. J. Hong C. Y. Lee C. S. Park S. Y. Kim D. J. Park S. W. Bioorg. Med. Chem. Lett. 2000;10:95–99. doi: 10.1016/S0960-894X(99)00646-0. [DOI] [PubMed] [Google Scholar]
- Keshavarzipour F. Tavakol H. Appl. Organomet. Chem. 2017;31:e3682. doi: 10.1002/aoc.3682. [DOI] [Google Scholar]
- Singh M. S. Chowdhry S. RSC Adv. 2012;2:4547–4592. doi: 10.1039/C2RA01056A. [DOI] [Google Scholar]
- Strecker D. Justus Liebigs Ann. Chem. 1850;75:27–45. doi: 10.1002/jlac.18500750103. [DOI] [Google Scholar]
- Ramn D. J. Yus M. Angew. Chem., Int. Ed. 2005;44:1602–1634. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]
- Safari J. Ahmadzadeh M. Zarnegar Z. J. Org. Chem. Res. 2016;2:134–139. [Google Scholar]
- Liu Q. Wu R. T. J. J. Chem. Res. 2011;10:598–599. doi: 10.3184/174751911X13176501108975. [DOI] [Google Scholar]
- Liu Q. Zhang Y. N. Bull. Korean Chem. Soc. 2011;32:3559–3560. doi: 10.5012/bkcs.2011.32.10.3559. [DOI] [Google Scholar]
- Kiyani H. Ghorbani F. Elixir Org. Chem. 2013;58A:14948–14950. [Google Scholar]
- Kiyani H. Ghorbani F. Heterocycl. Lett. 2013;3:359–369. [Google Scholar]
- Kiyani H. Ghorbani F. Heterocycl. Lett. 2013;3:145–153. [Google Scholar]
- Liu Q. Hou X. Phosphorus, Sulfur Silicon Relat. Elem. 2012;187:448–453. doi: 10.1080/10426507.2011.621003. [DOI] [Google Scholar]
- Kiyani H. Org. Chem.: Indian J. 2013;9:97–101. [Google Scholar]
- Kiyani H. Ghorbani F. Open J. Org. Chem. 2013;1:5–9. [Google Scholar]
- Khandebharad A. U. Sarda S. R. Gill C. H. Agrawal B. R. Res. J. Chem. Sci. 2015;5:27–32. [Google Scholar]
- Hamzeh K. J. J. Saudi Chem. Soc. 2017;21:S112–S119. doi: 10.1016/j.jscs.2013.11.002. [DOI] [Google Scholar]
- Ganesh T. P. Sachin P. G. Balasaheb R. A. Machhindra K. L. Bull. Chem. React. Eng. Catal. 2017;12:32–40. doi: 10.9767/bcrec.12.1.655.32-40. [DOI] [Google Scholar]
- Davood S. J. Mex. Chem. Soc. 2015;59:191–197. [Google Scholar]
- Ashkan B. R. Davood S. Orient. J. Chem. 2016;32:1433–1437. doi: 10.13005/ojc/320317. [DOI] [Google Scholar]
- Abdul A. Maqdoom F. Int. J. ChemTech Res. 2017;10:269–273. [Google Scholar]
- Majid A. Zohre Z. Javad S. Green Chem. Lett. Rev. 2018;11:78–85. doi: 10.1080/17518253.2018.1434564. [DOI] [Google Scholar]
- Keshavarzipour F. Tavakol H. J. Iran. Chem. Soc. 2016;13:149–153. doi: 10.1007/s13738-015-0722-9. [DOI] [Google Scholar]
- Shahabi D. Tavakol H. J. Iran. Chem. Soc. 2017;14:135–142. doi: 10.1007/s13738-016-0965-0. [DOI] [Google Scholar]
- Khatavi S. Y. Kamanna K. J. Mol. Struct. 2022;1250:131708. doi: 10.1016/j.molstruc.2021.131708. [DOI] [Google Scholar]
- Sadeghi-Takallo M. Zare A. Bull. Chem. Soc. Ethiop. 2019;33:69–76. doi: 10.4314/bcse.v33i1.7. [DOI] [Google Scholar]
- Ghasemi Z. Afsaneh H. A. Sajjad A. Sepideh V. Jafar S. RSC Adv. 2021;11:36958–36964. doi: 10.1039/D1RA06472J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P. S. Sutrave D. S. Int. J. ChemTech Res. 2018;11:77–88. doi: 10.20902/IJCTR.2018.110911. [DOI] [Google Scholar]
- Elgrishi N. Rountree K. J. McCarthy B. D. Rountree E. S. Eisenhart T. T. Dempsey J. L. J. Chem. Educ. 2018;95:197–206. doi: 10.1021/acs.jchemed.7b00361. [DOI] [Google Scholar]
- Bakhotmah D. A. Tarik E. A. Mohammed A. A. Ibrahim S. Y. Polycyclic Aromat. Compd. 2020:2136–2150. [Google Scholar]
- Olha M. Roman L. Nataliya F. Rostyslav S. Tetyana Y. Anna O. Valentina V. Volodymyr M. Victoriya G. J. Appl. Pharm. 2020;10:59–63. [Google Scholar]
- Xuanrong S. Longchao Z. Mengshi G. Xiangjie Q. Chenfeng Z. Dabu Z. Yue C. Molecules. 2019;24:624–635. doi: 10.3390/molecules24030624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.