

Abstract
Membrane proteins are favored drug targets and antibody therapeutics represent the fastest-growing category of pharmaceuticals. However, there remains a need for rapid and effective approaches for the discovery of antibodies that recognize membrane proteins to develop a robust clinical pipeline for targeted therapeutics. The challenges associated with recombinant expression of membrane proteins make whole cell screening techniques desirable, as these strategies allow presentation of the target membrane proteins in their native conformations. Here, we describe a workflow that employs both adherent cell-based and suspension cell-based whole cell panning methodologies to enrich for specific binders within a yeast-displayed antibody library. The first round of selection consists of an adherent cell-based approach, wherein a diverse library is panned over target-expressing mammalian cell monolayers in order to debulk the naïve library. Subsequent rounds involve the use of suspension cell-based approaches, facilitated with magnetic-activated cell sorting (MACS) or fluorescence-activated cell sorting (FACS), to achieve further library enrichment. Finally, we describe a high-throughput approach to screen target binding and specificity of individual clones isolated from selection campaigns.
Keywords: Cell panning, Molecular engineering, Membrane proteins, Directed evolution, Antibody discovery, Yeast surface display, Biopanning
1. Introduction
Membrane proteins orchestrate a wide range of biological functions such as cellular transduction, catalysis, and transfer of ions across the cell membrane. Dysregulation of these vital physiological processes can lead to the development or progression of diseased states, motivating the design of molecular interventions that target membrane proteins in order to maintain or restore homeostasis. It is therefore not surprising that over 60% of all clinically approved drugs target membrane proteins [1]. Therapeutic antibodies constitute the most rapidly growing class of pharmaceuticals because of their high affinity, specificity, and extended in vivo half-lives [2–4]. Antibodies are swiftly gaining market share, and as of 2019, antibody-based molecules accounted for 7 out of the top 10 best-selling drugs [5]. The principal methodologies employed for antibody discovery include animal immunization and in vitro directed evolution strategies using technologies such as phage display or yeast surface display [6–10]. There has been great success in discovering antibodies against relatively straightforward membrane proteins, such as single-pass targets. However, antibody discovery against more complex multipass membrane proteins such as G protein-coupled receptors (GPCRs) or ion channels has been more challenging due to poor expression, low solubility, and deficient physiological mimicry of the natural proteins with recombinantly-produced material. Biochemical methods such as detergent solubilization or nanodisk technologies may be used to address these issues [11, 12]. However, these methods require significant optimization, and they still present the risk of enriching for antibodies that recognize nonphysiological conformations of the protein or inaccessible intracellular epitopes [13].
To circumvent the complications associated with recombinant expression of membrane proteins, new approaches have emerged that use whole cells expressing the target protein, enabling presentation of the protein in its native conformation in a manner that is directly relevant for antibody binding. A common strategy used to present membrane proteins in their natural states employs an adherent cell-based platform known as biopanning. Biopanning entails presentation of a yeast-displayed library of proteins to a monolayer of target-expressing mammalian cells adhered to a surface in order to isolate binders to the membrane protein of interest [14]. We recently developed a suspension cell-based platform we call “biofloating,” which allows for the quantitative analysis of interactions between proteins displayed on the surface of yeast and membrane proteins expressed on mammalian cells [15]. We demonstrated that the sensitivity of the suspension cell-based biofloating platform we developed is superior to that of the adherent cell-based biopanning platform, with respect to assessment of both kinetic and equilibrium interaction behavior.
Building on the novel yeast/mammalian cell interaction platform we established, we have constructed a directed evolution workflow that leverages the superior binding characteristics of the suspension cell-based biofloating platform for the discovery of specific antibodies against membrane proteins. Due to the vast array of molecules present on the cell membrane, significant care must be taken to prevent enrichment of binders that are not specific for the target protein. These so-called nonspecific binders include binding proteins that recognize other molecules in the cell membrane as well as binding proteins that engage multiple different targets. We have optimized our workflow to deplete nonspecific binders to the greatest possible extent, while maximizing enrichment of target-specific clones. The general scheme for our workflow is as follows: (1) a first round of selection that debulks diverse libraries of antibody single-chain variable fragment (scFv)-expressing yeast using an adherent cell-based approach; (2) subsequent rounds of selection utilizing a suspension cell-based approach implemented via magnetic-activated cell sorting (MACS) or fluorescence-activated cell sorting (FACS); and (3) high-throughput biofloating-based screening of individual clones isolated from the enriched libraries to identify high affinity, target-specific clones.
2. Materials
2.1. Plasticware, Glassware, and Consumables
Tissue culture-treated 150 mm petri dish.
T75 tissue culture flasks.
10 mL and 25 mL serological pipette tips.
Cell scraper.
15/50 mL conical tubes.
500 mL conical disposable centrifuge bottle.
1.5 mL microcentrifuge tubes.
96-well deep-well blocks.
96-well V-bottom microplates.
Flow cytometry tubes.
70 μm cell strainers.
0.22 μm membrane bottle-top filter.
0.22 μm membrane Steriflip® filter (Millipore Sigma).
LS MACS columns (Miltenyi Biotec).
13 mL round-bottom culture tubes.
125/250 mL glass culture flasks with baffles.
2.8 L Fernbach flasks with baffles.
2.2. Buffers and Yeast Media
Prepare all solutions using Milli-Q ultrapure water. Prepare and store all buffers at 4 °C. Diligently follow all waste disposal regulations when discarding waste materials.
Phosphate-buffered saline (PBS) pH 8.
PBS pH 7.2.
PBSA: PBS pH 7.2 containing 0.1% bovine serum albumin (BSA).
PBSCMA: PBS pH 7.2 containing 0.9 mM CaCl2, 0.49 mM MgCl2, and 0.1% BSA.
SDCAA yeast media (500 mL): 10 g dextrose, 3.35 g yeast nitrogen base, 2.5 g casamino acids (-ADE, -URA, -TRP), 7.35 g sodium citrate (50 mM), 2.1 g citric acid monohydrate (20 mM), 5 mL 10,000 U/mL Penicillin Streptomycin Solution (100 U/mL). Add water to 500 mL and filter sterilize using a 0.22 μm membrane bottle-top vacuum filter.
SGCAA yeast media (500 mL):1 g dextrose, 9 g galactose, 3.35 g yeast nitrogen base, 2.5 g casamino acids (-ADE, -URA, -TRP), 7.35 g sodium citrate (50 mM), 2.1 g citric acid monohydrate (20 mM), 5 mL 10,000 U/mL Penicillin Streptomycin Solution (100 U/mL). Add water to 500 mL and filter sterilize using a 0.22 μm membrane bottle-top vacuum filter.
Poly-l-lysine solution: Dissolve poly-l-lysine to 0.1 mg/mL using sterile water and filter sterilize using a 0.22 μm membrane Steriflip® filter.
2.3. Cell Staining Reagents
CellTrace™ dye (ThermoFisher Scientific) (We have successfully used the Violet and CFSE dyes).
EZ-Link™ Sulfo-NHS-SS-Biotin (ThermoFisher Scientific).
Fluorophore-conjugated anti-cmyc tag antibody (Cell Signaling Technology, clone 9B11).
2.4. Other Reagents
Quantum™ Simply Cellular© beads (Bangs Laboratories, Inc).
Streptavidin (SA)-coated magnetic beads (Miltenyi Biotec).
2.5. Instruments
Shaking incubators at 30 °C and 37 °C.
Refrigerated centrifuge with adapters to spin 15/50 mL conical tubes, 1.5 mL microcentrifuge tubes, and 96-well plates.
Light microscope (up to 40× magnification desirable).
QuadroMACS separator™ stand (Miltenyi Biotec).
Multichannel pipettors (ideally 12-channel) are useful for sample handling.
Liquidator™ 96 (Mettler Toledo Rainin) or similar liquid handling system is useful for sample handling.
Analytical flow cytometer (minimum 2-laser instrument).
Fluorescence-activated cell sorter (FACS) (minimum 2-laser instrument).
Plate shaker.
Tube rotator.
3. Methods
The selection workflow described herein has been carried out using scFv-displaying yeast libraries with diversities ranging from 108 to 109, wherein a maximum of 1010 cells are used to oversample the library by at least tenfold (see Note 1). The first round is thus performed using the biopanning format to avoid exceeding the capacity for MACS columns (2 × 109 cells) (see Note 2). It is important to ensure high expression levels of the target membrane protein on mammalian cells, ideally over 300,000 copies/cell (see Note 3). For best results, the target-null and target-expressing cells should be genotypically and phenotypically identical except for the expression of the membrane protein of interest.
All selection steps are conducted outside of the sterile tissue culture environment, so it is critical to spray down all bench areas and pipettes with Lysol and 70% ethanol and use sterile pipette tips and tubes for all library selection procedures. All steps should be carried out either on ice or in a room maintained at 4 °C (see Note 4).
3.1. Round 1 Biopanning Selection (Debulking the Library)
Treat two 150 mm petri dishes with poly-l-lysine (see Note 5): Add 6 mL of the poly-l-lysine solution to the plate and nutate (mix the plate with a rocking or swaying motion in the plane of rotation) to enable the solution to cover the entirety of the plate. Incubate for at least 5 min. Aspirate the liquid and add 15 mL of sterile water to wash. Aspirate the liquid and add 15 mL of sterile water to wash for a second time. Aspirate the liquid and place the poly-l-lysine coated plates in a sterile environment. Allow at least 2 h to dry.
Seed both target-expressing and target-null cells into separate 150 mm petri dishes so that the cells reach 90–100% confluency on the day of the Round 1 selection.
Thaw yeast library on ice and transfer to an appropriate amount of SDCAA to achieve an optical density (OD, 600 nm) of 1, which corresponds to 1 × 107 yeast/mL. Incubate the culture overnight at 30 °C while shaking at 200 rpm.
The next day, passage yeast to an OD of 1 in 1 L fresh SDCAA. Incubate the culture overnight at 30 °C while shaking at 200 rpm.
Induce the yeast library by centrifuging 1 × 1010 cells in a 500 mL conical centrifuge bottle at 3500 × g for 5 min, discarding the supernatant, and resuspending yeast in 1 L SGCAA to an OD of 1. Incubate for 2 days at 20 °C while shaking at 200 rpm.
On the day of Round 1 selections, prepare the yeast library: Centrifuge 1 × 1010 cells in a 500 mL conical centrifuge bottle at 3500 × g for 5 min and discard the supernatant. Resuspend in 15 mL of PBSCMA. Centrifuge again 3500 × g for 5 min and discard the supernatant. Repeat this step for a total of two washes. Resuspend yeast cells in 15 mL of PBSCMA.
Prepare the target-null cells for negative selection: Tilt the 150 mm petri dish of plated target-null cells and aspirate the cell media. Use a serological pipette to slowly expel 15 mL of PBSCMA down the side wall of the petri dish. Gently rotate the plate back and forth a few times and aspirate the buffer. Repeat twice for a total of three washes.
Slowly add the 15 mL of yeast suspended in PBSCMA (from step 6) to the side of the petri dish of target-null cells from step 7 using a 25 mL serological pipette.
Rotate the petri dish very gently to distribute the yeast suspension.
Co-incubate the yeast and mammalian cells at 4 °C for 30 min without rotation.
Collect the supernatant (containing yeast that do not bind to target-null cells) while conducting five washes with cold PBSCMA as follows: Tilt the petri dish, gently pipette off the supernatant into a 25 mL serological pipette, and dispense the supernatant into a 50 mL conical tube. Slowly add 15 mL of cold PBSCMA to the side of the dish. Gently rotate the dish back and forth 20–25 times followed by gently nutating 5–10 times (see Note 6). Pipette off the supernatant and add it to the 50 mL conical tube. Repeat for a total of five washes. Use additional conical tubes as needed (see Note 7).
Spin down the collected yeast at 3500 × g for 5 min.
Discard the supernatant and resuspend yeast cells in 15 mL of cold PBSCMA.
Prepare the target-expressing mammalian cells for positive selection: Tilt the 150 mm petri dish of plated target-expressing mammalian cells and aspirate the cell media. Use a serological pipette to slowly expel 15 mL of PBSCMA down the side wall of the petri dish. Gently rotate the plate back and forth a few times and aspirate the buffer. Repeat twice for a total of three washes.
Slowly add the suspended yeast (from step 13) to the petri dish containing the target-expressing cells (from step 14) by expelling down the side of the dish.
Rotate the petri dish very gently to distribute the yeast suspension.
Co-incubate the yeast and mammalian cells at 4 °C for 120 min without rotation.
Remove unbound yeast while conducting five washes with cold PBSCMA: Tilt the petri dish, gently pipette off the supernatant into a 25 mL serological pipette, and discard the supernatant. Slowly add 15 mL of cold PBSCMA to the side of the dish. Gently rotate the dish back and forth 20–25 times followed by gently nutating 5–10 times. Pipette off and discard the supernatant. Repeat for a total of five washes (see Note 8).
Add 10 mL of SDCAA to the yeast and mammalian cells that remain in the petri dish.
Use a cell scraper to remove the bound cells from the plate (see Note 9).
Nutate the petri dish five times and collect the SDCAA-suspended yeast/mammalian cell mixture into a sterile 125 mL glass flask (see Note 10).
Add another 10 mL of SDCAA to the petri dish, nutate the dish, and add the contents of this wash to the 125 mL glass flask. The workflow for Round 1 selections has been optimized to achieve maximal enrichment of target-specific over nonspecific yeast (see Note 11, Fig. 1).
Grow the yeast culture overnight at 30 °C while shaking at 200 rpm.
The following day induce the yeast by centrifuging 3 × 107 yeast at 3500 × g for 5 min and resuspending in SGCAA (see Note 12). Grow the yeast culture at 20 °C while shaking at 200 rpm for 2 days, and proceed to Round 2 of selections.
Fig. 1.
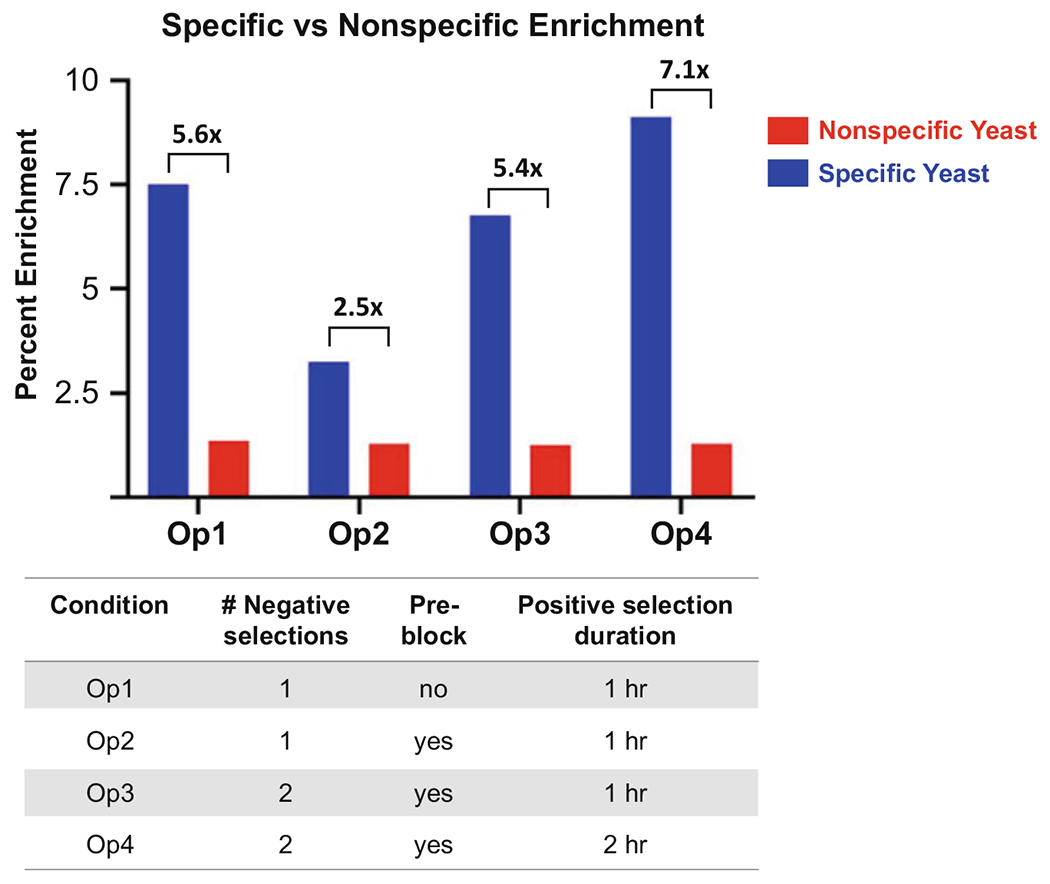
Enrichment of specific (blue) versus nonspecific (red) scFv-expressing yeast following one round of adherent cell-based selections under four different selection conditions. An anti-programmed death-ligand 1 (PD-L1) scFv-expressing yeast clone was spiked into a naïve library of 109 diversity [16], and 1010 total yeast were subjected to a single round of biopanning using the following conditions: (i) One 30 min negative selection (NS) + 60 min positive selection (PS); (ii) One 30 min NS + 30 min pre-block with soluble target-null mammalian cells at a 2:1 target-null:target-expressing cell ratio (PB) + 60 min PS; (iii) Two 30 min NS + 30 min PB + 60 min PS; and (iv) Two 30 min NS + 30 min PB + 120 min PS. Results showed that the additional NS and implementation of the PB did not significantly impact specific enrichment ratios, and in fact, implementation of PB with only a single NS actually decreased specific enrichment. However, lengthened PS incubation time (120 min) in combination with an additional NS and PB resulted in the highest specific enrichment ratio
3.2. Round 2+ MACS Selections (Enriching the Library)
Grow target-null and target-expressing mammalian cells in monolayers to 80–90% density, and detach via trypsinization. Selections will require 3 cohorts of mammalian cells: 1 × 106 biotinylated target-null cells (for negative selection), 2 × 106 non-biotinylated target-null cells (for pre-blocking), 1 × 106 biotinylated target-expressing cells (for positive selection).
Biotinylate target-null and target-expressing mammalian cells: Separately pellet 1 × 106 detached target-null and target-expressing mammalian cells in a 1.5 mL microcentrifuge tube by centrifuging at 400 × g for 5 min. Remove the supernatant. Wash cells by resuspending in 1 mL of PBS pH 8, centrifuging at 400 × g for 5 min, and removing the supernatant. Repeat this step twice for a total of three washes. Resuspend the cells in 40 μL (2.5 × 107 cells/mL) PBS pH 8 containing 2 mM of EZ-Link™ Sulfo-NHS-SS-Biotin reagent. Incubate cells at room temperature for 30 min with rotation on a tube rotator (see Note 13). To quench the reaction and remove excess reagent, conduct three washes with PBSA by resuspending cells in 1 mL PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant. Resuspend the target-null cells in 300 μL of cold PBSA and resuspend the target-expressing cells in 150 μL of cold PBSA.
Centrifuge 1 × 108 induced yeast cells from the previous round in a 1.5 mL microcentrifuge tube at 3500 × g for 5 min and aspirate supernatant.
Use of 1 × 108 yeast cells oversamples the library diversity by at least tenfold while also allowing sufficient cell throughputs during the selection.
Wash the yeast cells by adding 1 mL of cold PBSA to the microcentrifuge tube, centrifuge at 3500 × g for 5 min, and aspirate supernatant. Repeat for a total of two washes.
Resuspend the yeast cells with the 1 × 106 biotinylated target-null cells in 300 μL chilled PBSA (prepared in step 2) to perform the negative selection.
Incubate the yeast/mammalian cell suspension for 30 min at 4 °C with rotation on a tube rotator.
Bring the total volume in the microcentrifuge tube containing the cell suspension to 950 μL by adding ≈650 μL of cold PBSA.
Add 50 μL of streptavidin (SA)-coated magnetic beads to the cell mixture and incubate for 20 min at 4 °C with rotation on a tube rotator.
Centrifuge the cell suspension at 400 × g for 5 min and remove the supernatant. Wash the mixture once by resuspending in 1 mL cold PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant.
Gently resuspend the yeast/mammalian cell suspension in 5 mL of cold PBSA and add to an LS magnetic column held on a QuadroMACS separator™ stand.
Collect the flowthrough (containing the desired yeast population that does not bind to the target-null cells) in a 15 mL conical tube.
Add 3 mL of cold PBSA to wash the column. Continue to collect the flowthrough in the same 15 mL conical tube.
Repeat step 13 twice more for a total of three washes. Continue to collect the flowthrough in the same 15 mL conical tube.
Centrifuge the compiled flowthrough at 3500 × g for 5 min.
Prepare pre-blocking cells by adding 2 × 106 non-biotinylated target-null cells in a 1.5 mL microcentrifuge tube, centrifuging at 400 × g for 5 min, and removing the supernatant.
Add 1 mL of cold PBSA to the 1.5 mL centrifuge tube, centrifuge at 400 × g for 5 min, and aspirate. Repeat for a total of two washes.
Resuspend the non-biotinylated target-null cells in 150 μL of cold PBSA.
Discard supernatant of the compiled flowthrough yeast (from step 15) and resuspend with the 2 × 106 non-biotinylated target-null cells in 150 μL of chilled PBSA (from step 18) for the pre-block (see Note 14, Fig. 2).
Incubate the yeast/mammalian cell mixture for 30 min at 4 °C with rotation on a tube rotator.
Add the 1 × 106 biotinylated target-expressing cells in 150 μL of chilled PBSA (from step 2) to the tube and incubate for 60 min at 4 °C with rotation on a tube rotator to perform the positive selection.
Bring the total volume of the cell mixture in the microcentrifuge tube to 950 μL by adding ≈650 μL of cold PBSA.
Add 50 μL of streptavidin (SA)-coated magnetic beads to the cell mixture and incubate for 20 min at 4 °C with rotation on a tube rotator.
Centrifuge the cell suspension at 400 × g for 5 min and remove the supernatant. Wash the mixture once by resuspending in 1 mL of cold PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant.
Gently resuspend the yeast/mammalian cell mixture in 5 mL of cold PBSA and add to an LS magnetic column held on a QuadroMACS separator™ stand. Discard the flowthrough (containing unwanted yeast that do not bind to the target-expressing cells).
Add 3 mL of cold PBSA to wash the column. Discard the flowthrough.
Repeat two more times for a total of three washes. Discard the flowthrough from each wash.
Elute the bound cells from the magnetic column with 5 mL of cold PBSA. Collect the eluted cells in a 15 mL conical tube.
Centrifuge the eluted cells at 3500 × g for 10 min.
Carefully remove the supernatant without disrupting the cell pellet.
Grow the yeast culture in 3 mL of SDCAA overnight at 30 °C while shaking at 200 rpm.
The following day induce the yeast by spinning down 3 × 107 yeast and resuspending in 3 mL of SGCAA. Grow the yeast culture at 20 °C while shaking at 200 rpm for 2 days, and perform bulk library analysis before proceeding to the next round of selection (see Note 15).
Fig. 2.
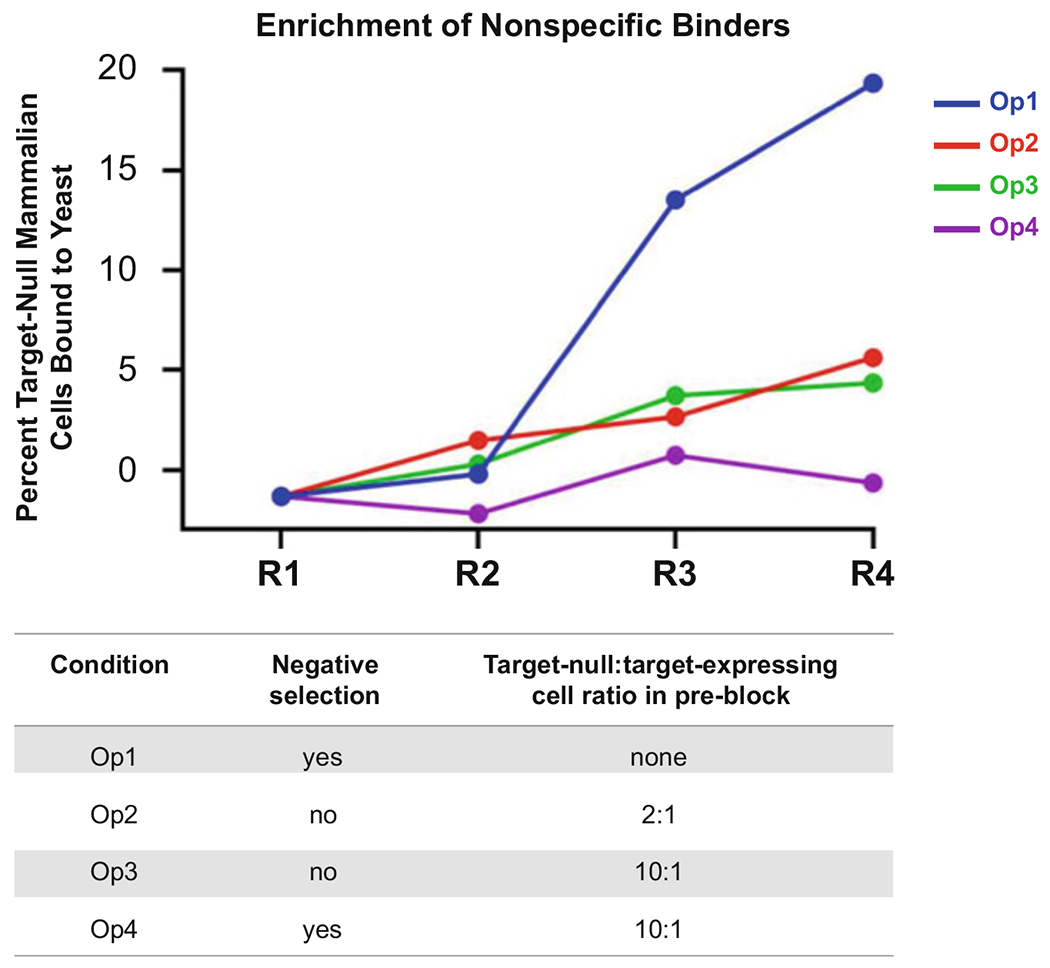
Minimizing enrichment of nonspecific yeast through rounds of suspension cell-based selections via MACS. An adherent cell-based Round 1 (R1) was conducted for a naïve yeast-displayed scFv library with diversity of 109 [16], and different suspension cell-based sorting conditions via MACS were implemented for R2-R4: (i) 30 min negative selection (NS) + 60 min positive selection (PS); (ii) 30 min pre-block (PB) with soluble target-null mammalian cells at a 2:1 target-null:target-expressing cell ratio (PB) + 60 min PS; (iii) 30 min PB with 10:1 target-null:target-expressing mammalian cell ratio + 60 min PS; and (iv) 30 min NS + 30 min PB with 10:1 target-null:target-expressing mammalian cell ratio + 60 min PS. The graph presents the percent of target-null cells that are bound to yeast (i.e., nonspecific binders). We found that employing both the NS and PB leads to reduced nonspecific clone enrichment, although the target-null:target-expressing mammalian cell ratio does not significantly affect enrichment outcome
3.3. Later Round FACS Selections (Fine-Tuning the Library)
Grow mammalian target-null and target-expressing mammalian cells in monolayers to 80–90% density, and detach via trypsinization. Selections will require three cohorts of mammalian cells: 1 × 106 biotinylated target-null cells (for negative selection), 2 × 106 non-biotinylated target-null cells (for pre-blocking), 1 × 106 CellTrace™ dye-stained target-expressing cells (for positive section).
Biotinylate the target-null cells: Pellet 1 × 106 detached target-null mammalian cells in a 1.5 mL microcentrifuge tube by centrifuging at 400 × g for 5 min. Remove the supernatant. Wash cells by resuspending in 1 mL of PBS pH 8, centrifuging at 400 × g for 5 min, and removing the supernatant. Repeat this step twice for a total of three washes. Resuspend the cells in 40 μL (2.5 × 107 cells/mL) of PBS pH 8 containing 2 mM of EZ-Link™ Sulfo-NHS-SS-Biotin reagent. Incubate cells at room temperature for 30 min with rotation on a tube rotator. To quench the reaction and remove excess reagent, conduct three washes with PBSA by resuspending cells in 1 mL PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant. Resuspend the target-null cells in 300 μL of cold PBSA.
Stain the target-expressing cells with CellTrace™ dye: Pellet 1 × 106 target-expressing mammalian cells by centrifuging at 400 × g for 5 min. Discard the supernatant. Wash cells by resuspending in 1 mL of PBS pH 7.2, centrifuging at 400 × g for 5 min, and removing the supernatant. Repeat this step for a total of three washes. Resuspend the target-expressing mammalian cells in 1 mL of PBS pH 7.2 containing 2.5 μM CellTrace™ dye. Incubate at room temperature for 20 min with rotation on a tube rotator. To quench the reaction and remove excess reagent, conduct three washes with PBSA by centrifuging at 400 × g for 5 min, removing the supernatant, and resuspending cells in 1 mL of cold PBSA. Repeat for a total of three washes. Resuspend the target-null cells in 150 μL of chilled PBSA.
Pellet 5 × 106 induced yeast cells from the previous round in a 1.5 mL microcentrifuge tube at 3500 × g for 5 min and remove the supernatant (see Note 16).
Wash the yeast cells by adding 1 mL of cold PBSA to the microcentrifuge tube, centrifuge at 3500 × g for 5 min, and aspirate supernatant. Repeat for a total of two washes.
Resuspend the yeast cells with the 1 × 106 biotinylated target-null cells in 300 μL chilled PBSA (prepared in step 2) to perform the negative selection.
Incubate the yeast/mammalian cell suspension for 30 min at 4 °C with rotation on a tube rotator.
Bring the total volume in the microcentrifuge tube containing the cell suspension to 950 μL by adding ≈650 μL of cold PBSA.
Add 50 μL of streptavidin (SA)-coated magnetic beads to the cell mixture and incubate for 20 min at 4 °C with rotation on a tube rotator.
Centrifuge the cell suspension at 400 × g for 5 min and remove the supernatant. Wash the mixture once by resuspending in 1 mL cold PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant.
Gently resuspend the yeast/mammalian cell suspension in 5 mL of cold PBSA and add to an LS magnetic column held on a QuadroMACS separator™ stand.
Collect the flowthrough (containing the desired yeast population that does not bind to the target-null cells) in a 15 mL conical tube.
Add 3 mL of cold PBSA to wash the column. Continue to collect the flowthrough in the same 15 mL conical tube.
Repeat step 13 twice more for a total of three washes. Continue to collect the flowthrough in the same 15 mL conical tube.
Centrifuge the compiled flowthrough at 3500 × g for 5 min.
Prepare pre-blocking cells by adding 2 × 106 non-biotinylated target-null cells in a 1.5 mL microcentrifuge tube, centrifuging at 400 × g for 5 min, and removing the supernatant.
Add 1 mL of cold PBSA to the 1.5 mL centrifuge tube, centrifuge at 400 × g for 5 min, and aspirate. Repeat for a total of two washes.
Resuspend the non-biotinylated target-null cells in 150 μL of cold PBSA.
Discard supernatant from step 15 and resuspend the yeast with the 150 μL of cold PBSA containing 2 × 106 non-biotinylated target-null cells for the pre-block (see Note 17, Fig. 3).
Incubate the cell mixture for 30 min at 4 °C with rotation on a tube rotator.
Add 150 μL of cold PBSA containing the 1 × 106 CellTrace™ dye-labeled target-expressing cells (from step 3) to the yeast/mammalian cell suspension.
Add fluorophore-conjugated anti-cmyc tag antibody (to a 1:100 dilution) to the mixture and incubate for 60 min at 4 °C with rotation on a tube rotator (see Note 18).
Centrifuge the cell suspension at 400 × g for 5 min and remove the supernatant. Wash the mixture once by resuspending in 1 mL of cold PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant.
Gently resuspend the cell mixture in 1 mL of cold PBSA and strain through a 70 μm strainer into a flow cytometry tube.
Sort the dual positive population on a FACS instrument, representing yeast labeled with the fluorescent anti-cmyc antibody bound to CellTrace™ dye-labeled mammalian cells (Fig. 4).
Plate approximately 1000 of the collected events from the FACS selection on an SDCAA plate (see Note 19).
Transfer the remainder of the sorted cells to a 15 mL conical tube and centrifuge at 3500 × g for 10 min (see Note 20).
Resuspend the cells in 3 mL of SDCAA and grow overnight at 30 °C while shaking at 200 rpm.
The following day induce the yeast by spinning down 3 × 107 yeast and resuspending in 3 mL of SGCAA at an OD of 1. Grow the yeast culture at 20 °C while shaking at 200 rpm for 2 days.
Perform bulk library analysis before proceeding to additional rounds of selection, if desired (see Note 21).
Fig. 3.
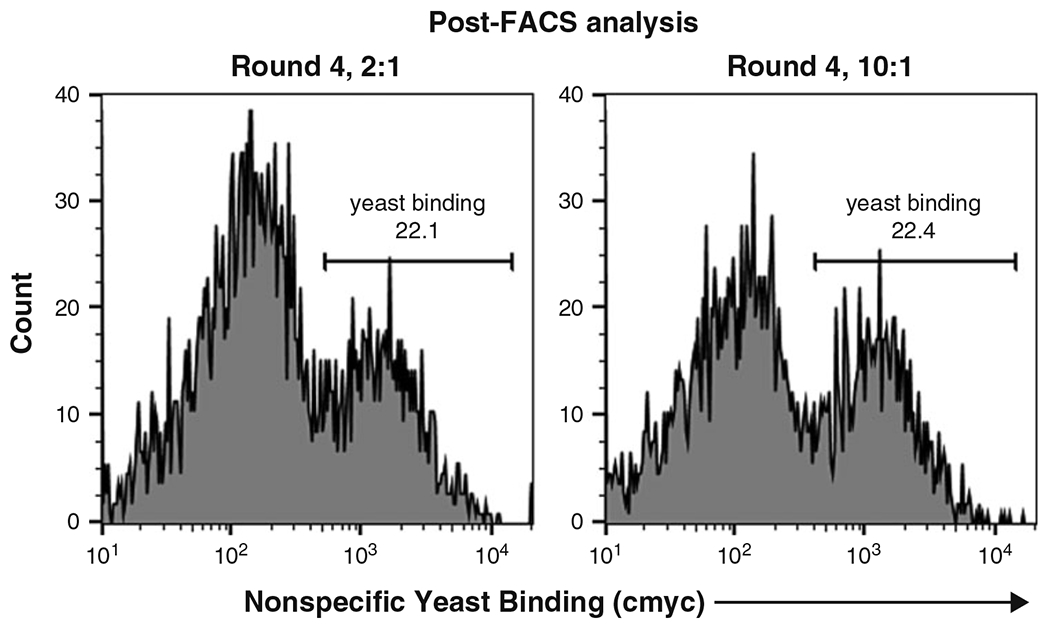
Minimizing enrichment of nonspecific yeast in suspension cell-based selections via FACS. Flow cytometry histograms depict an enriched yeast-displayed scFv library binding to target-null mammalian cells. In each plot, the population at left represents unbound mammalian cells, whereas the population at right represents mammalian cells that are bound to yeast. Suspension cell-based selections were carried out using FACS under the following conditions: (i) 2:1 ratio of target-null:target-expressing mammalian cells in the pre-block step (Round 4, 2:1); and (ii) 10:1 ratio of target-null:target-expressing mammalian cells in the pre-block step (Round 4, 10:1). We found that use of the 2:1 and 10:1 ratios led to similar enrichment of nonspecific yeast binders
Fig. 4.
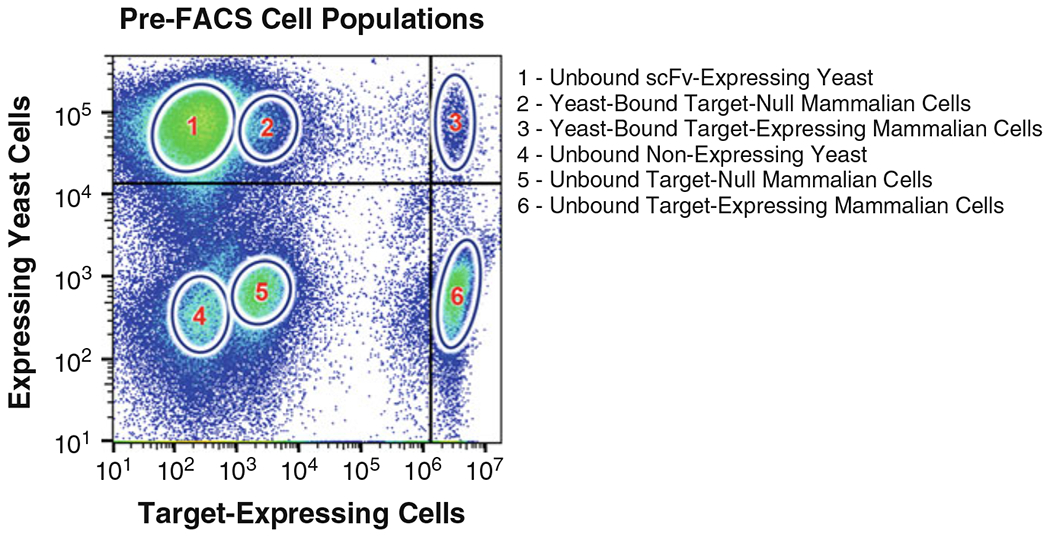
Gating scheme and cell populations for FACS selections. The sorted sample contained 5 × 106 anti-cmyc antibody-labeled yeast cells, 1 × 106 Violet Cell Trace™ dye-labeled target-expressing mammalian cells, and 2 × 106 unlabeled target-null mammalian cells. Six populations arose from this cell mixture: (1) unbound scFv-expressing yeast cells; (2) yeast-bound target-null cells; (3) yeast-bound target-expressing cells; (4) unbound non-expressing yeast cells; (5) unbound target-null cells; and (6) unbound target-expressing cells. The upper right quadrant contains the yeast-bound target-expressing cells, which represents the desired sort population
3.4. Screening Individual Yeast Clones Via Biofloating
Add 1 mL of SDCAA to each well of a 96-well deep-well block.
Pick individual yeast colonies from the SDCAA plate corresponding to the terminal round of selection, and inoculate a single clone into each of the 96 wells of the block.
Grow yeast for 1–2 days at 30 °C while shaking at 200 rpm.
Transfer 1 × 107 yeast cells from each well to a new deep-well plate.
Centrifuge the plate at 3500 × g for 5 min and discard the supernatant.
Induce the yeast cultures by resuspending each well in 1 mL of SGCAA.
Incubate for 1–2 days at 20 °C while shaking at 200 rpm (see Note 22).
The following numbers of mammalian cell and yeast cells will be needed for the clone screening process (see Note 23): 5 × 104 target-null and 5 × 104 target-expressing mammalian cells per yeast colony (total of ≈5 × 106 cells per 96 colonies) stained with a CellTrace™ dye and 5 × 105 yeast cells per well (10:1 yeast:mammalian cell ratio) to be stained with a CellTrace™ dye distinct from that used for mammalian cell labeling.
Label both target-null and target-expressing mammalian cells with a CellTrace™ dye (see Note 24): Separately pellet 5 × 106 target-null and 5 × 106 target-expressing mammalian cells in 1.5 mL microcentrifuge tubes by centrifuging at 400 × g for 5 min. Discard the supernatant. Wash twice by adding 1 mL of PBS pH 7.2, centrifuging at 400 × g for 5 min, and discarding the supernatant. Resuspend the target-null and target-expressing mammalian cells in 1 mL of PBS pH 7.2 containing 2.5 μM CellTrace™ dye. Incubate at room temperature for 20 min with rotation on a tube rotator. To quench the reaction and remove excess reagent, conduct three washes with PBSA by resuspending cells in 1 mL of cold PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant. Separately resuspend the target-null and target-expressing mammalian cells at 2.5 × 106 cells/mL (2 mL of PBSA per 5 × 106 cells). Larger capacity tubes (2 mL microcentrifuge tubes or 15 mL conical tubes as opposed to 1.5 mL microcentrifuge tubes) should be used here to accommodate the larger volume.
Label yeast cells with a CellTrace™ dye distinct from that used for mammalian cells (see Note 25, Fig. 5): Transfer 500,000 induced yeast/well from the deep-well block in step 6 to a V-bottom plate. Add the same row of yeast cells from the deep-well block to two consecutive rows on the V-bottom plate (one row will be incubated with target-null cells and the other row will be incubated with target-expressing mammalian cells). Two plates will be needed to analyze 96 clones. Centrifuge the plates at 3500 × g for 5 min and remove the supernatant. Wash the plates twice by adding 150 μL of PBS pH 7.2 to each well (we recommend use of a multichannel pipette), centrifuging at 3500 × g for 5 min, and discarding the supernatant. Resuspend the yeast cells in 20 μL of PBS pH 7.2 containing 25 μM CellTrace™ dye. Incubate yeast cells with dye at room temperature for 20 min, shaking at 530 rpm on a plate shaker. To quench the reactions and remove excess reagent, conduct three washes with PBSA by resuspending each well of yeast in 150 μL of cold PBSA, centrifuging at 400 × g for 5 min, and removing the supernatant.
Resuspend each well of yeast cells with either 20 μL of target-null cells or 20 μL of target-expressing mammalian cells from step 9 (ensure that each yeast clone receives both types of cells).
Cover the plate with aluminum foil and incubate for 60 min at 4 °C while shaking at 530 rpm on a plate shaker.
Add 80 μL of PBSA to each well and analyze on a flow cytometer.
Identify target-specific binders as the yeast clones that demonstrate binding to target-expressing cells but not to target-null cells (see Note 26, Fig. 6).
Fig. 5.
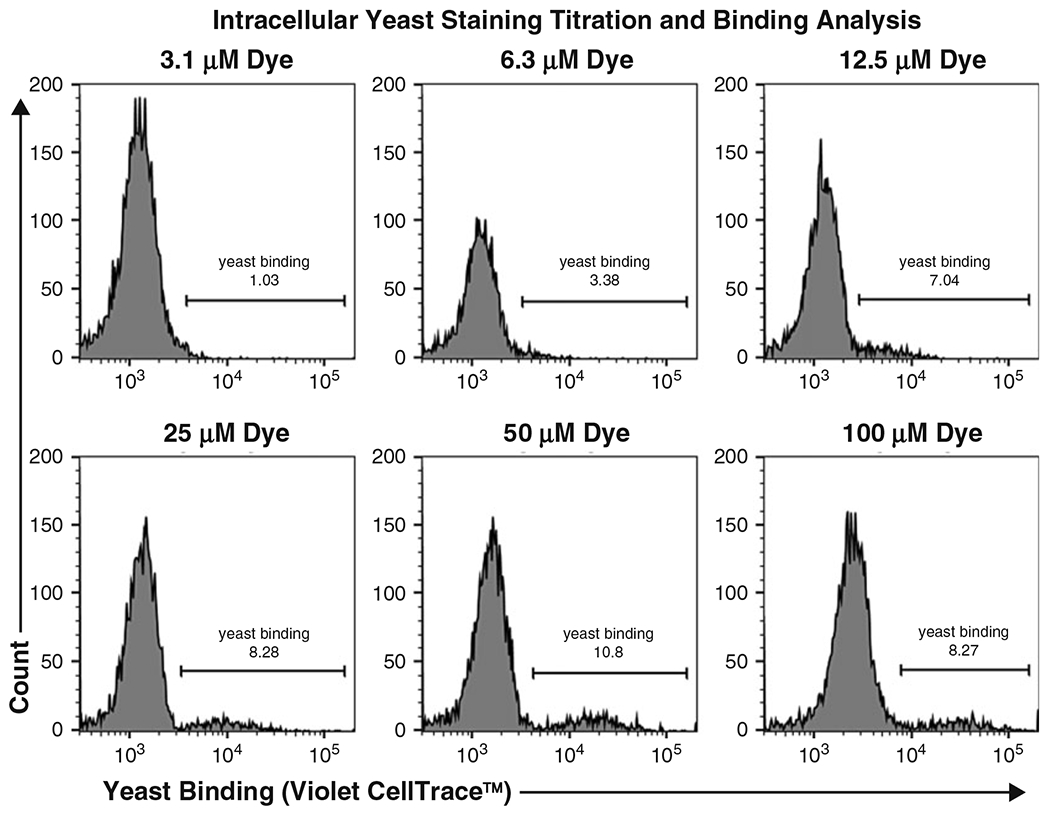
Titration of CellTrace™ dye for yeast labeling. Specific scFv-expressing yeast were labeled with various concentrations of CellTrace™ Violet dye and then incubated with unlabeled target-expressing cells. Flow cytometry analysis allows discrimination of unbound mammalian cells (left peak) from yeast-bound mammalian cells (right peak)
Fig. 6.
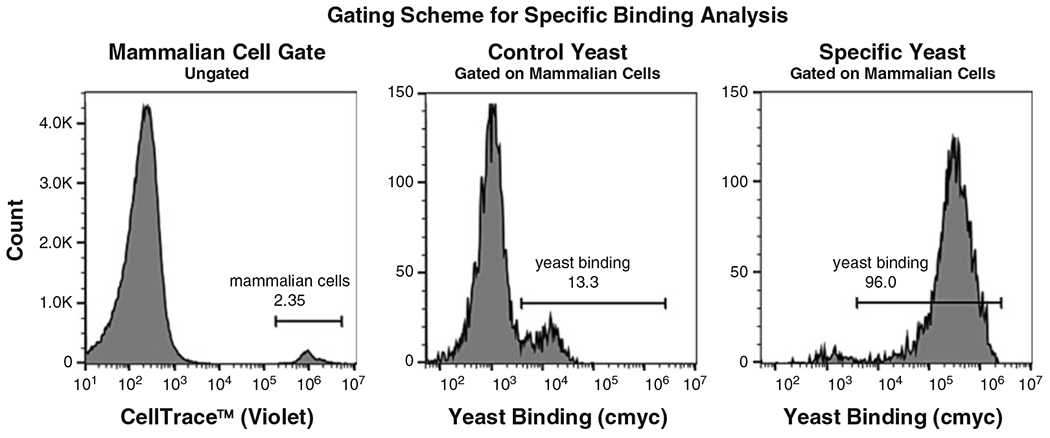
Gating scheme for yeast/mammalian cell binding analysis. CellTrace™ Violet-labeled PD-L1-expressing Chinese hamster ovary (CHO)-K1 cells were incubated with either yeast expressing an anti-programmed cell death protein 1 (PD-1) scFv (Control Yeast) or yeast expressing an anti-PD-L1 scFv (Specific Yeast). Fluorophore-conjugated anti-cmyc antibody was used for detection of scFv-expressing yeast cells. The left panel depicts the gating scheme for mammalian cells from all events (CellTrace™ Violet histogram). The middle and right panels consist of the mammalian cell population, wherein a yeast-unbound (left peak) and a yeast-bound (right peak) population exist. Distinct profiles are observed for anti-PD-1 scFv-expressing (Control) versus anti-PD-L1 scFv-expressing (Specific) yeast binding to PD-L1-expressing CHO-K1 cells, demonstrating specific binding
Acknowledgments
The authors thank Kook Bum (Dean) Kim for critical review of the manuscript. This work was supported by a Maryland Stem Cell Research Fund Discovery Award, NIH NIBIB R01EB029455, NIH NEI R01EY031097, and NIH NCI U01CA232563.
4 Notes
The three antibody fragment-displaying yeast libraries we have employed using this workflow are those constructed by Feldhaus [10], Kelly [16], and McMahon [17].
For smaller libraries, MACS or FACS selections may be feasible in the first round. It is also possible to distribute the yeast/mammalian cell suspension over multiple MACS columns.
Quantum™ Simply Cellular© beads (Bangs Laboratories, Inc) are reliable for membrane protein quantification, provided that a detection antibody of the appropriate species is available for the protein of interest.
It is important to keep yeast and mammalian cells and buffers on ice at all times throughout the selections unless otherwise specified. Not only is this important for the viability of the cells, but it will also minimize internalization of the target membrane protein.
Biopanning can be conducted successfully without coating the petri dish surface with poly-l-lysine. However, we have found certain cell lines do not adhere well to the plate, and therefore benefit from this coating. We recommend coating the plate regardless of cell type to avoid the risk of losing the mammalian cells due to surface detachment.
Rocking and nutating a plate may differ from person to person. We refer the reader to previous publications for more information regarding these wash steps [14, 18].
When conducting wash steps, it is critical to optimize handling techniques. If the dish is rocked/nutated too hard, bound yeast (and possibly even adhered mammalian cells) may fall off. Conversely, if the wash is carried out too gently, then a significant amount of settled nonbinding yeast will remain on the dish. We recommend using a light microscope to visualize the quantity of yeast bound/settled on the surface. If there are few detectable yeast cells bound, then the washing steps were likely carried out too vigorously. On the other hand, if complete saturation of settled yeast cells over the mammalian cells is observed, then more vigorous washes should be carried out.
Round 1 serves to debulk the library, meaning it is particularly important to avoid excessive stringency in implementing the washes for this round. It is advisable to wash more gently during the positive compared to the negative selection in this round. Leaving excess settled yeast on the petri dish may decrease the enrichment ratio for the first round, but will prevent loss of specific binders. Subsequent rounds utilizing MACS or FACS will enrich more stringently, so reduced stringency in Round 1 should not significantly impact selection outcomes.
Cell scraping serves to remove the adhered mammalian cells (and thus the bound yeast from the positive selection). After scraping and collection, removal of all cells from the plate should be confirmed using a light microscope.
Collecting the scraped yeast/mammalian cell mixtures in higher volumes of yeast media (such as the suggested 20 mL) allows for appreciable dilution of the mammalian cell debris. Only yeast cells will grow in SDCAA, and it is important that yeast growth not be hindered by significant amounts of mammalian cell debris collected from the dish.
To maximize the specific:nonspecific enrichment ratio in Round 1 biopanning, we carried out an experiment in which target-specific yeast were spiked into a naïve yeast-displayed library of diversity 109 [16], and selections were performed according to four different schemes. These schemes varied the number of negative selections, the use of a pre-block step (with competing unlabeled target-null mammalian cells to quench nonspecific yeast binders), and the length of positive selection incubation (Fig. 1). We determined that the optimal workflow for specific yeast enrichment involved iteration of a single negative selection and use of a 120 min incubation time for the positive selection.
After the first round of selections, the library diversity typically drops ≈1000-fold which is why we generally induce 3 × 107 yeast post-Round 1 to ensure that the library is being oversampled at least ten-fold and prevent loss of clones. Diversity can be approximated by quantifying the total amount of yeast cells remaining from the positive selection. To do this, one can take a representative image of the petri dish post-selection, count the number of yeast cells in the image, and extrapolate the total number of selected yeast from the known area of the petri dish. Alternatively, one can analyze a small sample (≈50 μL) of the cell scraped mammalian/yeast cell mixture via flow cytometry to acquire a yeast cell count (gating on the smaller yeast population based on forward scatter [FSC] and side scatter [SSC]), which can be extrapolated to determine the total number of yeast cells collected.
Cell biotinylation can be carried out at 4 °C if the target-null and target-expressing mammalian cells exhibit reduced viability when maintained in buffer at room temperature.
We found that the optimal strategy for depleting nonspecific binders in MACS selection rounds was to conduct a 30 min negative selection, followed by a pre-block with non-biotinylated target-null mammalian cells for 30 min (to quench nonspecific yeast binders) prior to adding target-expressing mammalian cells (Fig. 2). We did not observe significant differences when using a 2:1 versus 10:1 target-null: target-expressing cell ratio; thus, we proceeded using the 2:1 ratio.
After the second round of selection and following each round thereafter, we recommend characterizing yeast/mammalian cell interaction in the bulk libraries from all rounds (including the naïve library) simultaneously using the biofloating workflow, as detailed in Subheading 3.4. Biofloating analysis will enable monitoring of library enrichment over the course of selections. Ideally, yeast libraries should exhibit increased binding to target-expressing but not target-null cells as the selections progress. These analyses will inform on whether additional rounds of MACS and/or FACS are appropriate. After specific enrichment is observed following a MACS or FACS selection, we recommend conducting one final round of FACS to further deplete the nonbinding yeast, thus enabling a more efficient screening process of the individual yeast clones, described in Subheading 3.4.
There is a tradeoff between the quantity of cells subjected to FACS and the sort length. We chose to use 5 × 106 yeast cells for FACS to allow for sufficient library sampling while also not increasing the sort length too extensively. Because mammalian cells are also in the mixture, the sorting time can become problematic if the total cell numbers are too high.
Similar to the MACS optimization experiments, we tried two different target-null:target-expressing mammalian cell ratios for a FACS selection (Fig. 3). We found that using a 2:1 versus a 10:1 ratio did not lead to a significant difference in reducing nonspecific yeast binding. We therefore proceeded with the 2:1 ratio.
The libraries we use are tagged with a detection epitope, so we may fluorescently label the scFv-expressing yeast cells via fluorophore-conjugated antibodies against the epitope tag that are visualized using flow cytometry. Epitope tags including cmyc, HA, FLAG, and V5 can be used for this procedure.
We advise plating immediately after FACS selections to avoid bias when regrowing the sorted population. The number of yeast cells plated after the sort can be estimated by assuming that each yeast-bound mammalian cell binds to a single yeast cell.
The cell pellet is typically not visible after centrifuging the sorted population from FACS. It is best to leave some liquid remaining while aspirating, so that the pellet is not disturbed. If needed, an additional wash step can be performed to thoroughly exchange the buffer with yeast media.
Before picking clones and proceeding with individual clone screening procedures (Subheading 3.4), we advise using biofloating techniques to stain all library rounds against both target-null and target-expressing mammalian cells, in order to assess enrichment and specificity. Individual clones should be analyzed from the round of selection in which there is the highest percentage of yeast bound to the target-expressing mammalian cells but minimal binding to the target-null mammalian cells.
We encountered difficulties in screening individual yeast clones using biopanning methodologies. Screening 96 clones using 6-well plates is not practical, so we conducted experiments investigating potential use of 48-well and 96-well plates. We found that scaling down the surface area resulted in poor washing, most likely attributed to the higher circumference: surface area ratio (edge effects). These findings highlight an important advantage of using biofloating versus biopanning methodologies in terms of throughput and scalability.
In the FACS and biofloating workflows, yeast are typically stained with a fluorophore-labeled anti-cmyc tag antibody to visualize only the scFv-expressing yeast using flow cytometry. However, when analyzing 96 clones, this mandates the use of significant antibody quantities, which is costly. Therefore, we instead stained the yeast cells with a second CellTrace™ dye to allow for a more cost-efficient screen.
When CellTrace™ CFSE dye was used to label yeast cells, we observed heavy bleed-over of the fluorophore into the mammalian cells, making it impossible to distinguish yeast-bound mammalian cells from unbound mammalian cells. Interestingly, this problem did not arise when yeast cells were stained with CellTrace™ Violet dye. We therefore proceeded with staining the mammalian cells with CellTrace™ CFSE and the yeast cells with CellTrace™ Violet dye. Dye usage may need to be optimized for each mammalian cell line.
We titrated yeast expressing a target-specific scFv against target-expressing mammalian cells labeled with varying amounts of CellTrace™ Violet dye (Fig. 5). We found that staining the yeast cells with 25 μM of dye resulted in a clear separation of yeast-bound versus unbound mammalian cells. This was the lowest concentration tested that resulted in effective separation and thus reflected the most cost-effective approach for screening. We recommend conducting a similar titration to determine the optimal dye concentration for yeast cell staining when using multiple dyes or in cases wherein mammalian cells exhibit high amounts of autofluorescence.
It is important to implement a proper gating scheme when analyzing the yeast/mammalian cell interactions via flow cytometry. We typically gate on the CellTrace™ dye associated with mammalian cells so that the analysis will exclude any unbound yeast. The gated population is then plotted as a histogram in the channel associated with the CellTrace™ dye associated with yeast cells in order to visualize the populations of unbound mammalian cells and yeast-bound mammalian cells [15]. A general gating scheme and a demonstration of specific binding can be seen in Fig. 6.
References
- 1.Yin H, Flynn AD (2016) Drugging membrane protein interactions. Annu Rev Biomed Eng 18:51–76. 10.1146/annurev-bioeng-092115-025322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu JKH (2014) The history of monoclonal antibody development—progress, remaining challenges and future innovations. Ann Med Surg 3:113–116. 10.1016/j.amsu.2014.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu R-M, Hwang Y-C, Liu I-J et al. (2020) Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 27:1. 10.1186/s12929-019-0592-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH (2010) Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 49:493–507. 10.2165/11531280-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 5.Urquhart L (2020) Top companies and drugs by sales in 2019. Nat Rev Drug Discov 19:228–228. 10.1038/d41573-020-00047-7 [DOI] [PubMed] [Google Scholar]
- 6.Smith G (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228:1315. 10.1126/science.4001944 [DOI] [PubMed] [Google Scholar]
- 7.Scott J, Smith G (1990) Searching for peptide ligands with an epitope library. Science 249:386. 10.1126/science.1696028 [DOI] [PubMed] [Google Scholar]
- 8.Marks J, Hoogenboom H, Griffiths A, Winter G (1991) By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 222:581–597 [DOI] [PubMed] [Google Scholar]
- 9.Boder ET, Wittrup KD (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15:553–557. 10.1038/nbt0697-553 [DOI] [PubMed] [Google Scholar]
- 10.Feldhaus MJ, Siegel RW, Opresko LK et al. (2003) Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol 21:163–170. 10.1038/nbt785 [DOI] [PubMed] [Google Scholar]
- 11.Hutchings CJ, Koglin M, Marshall FH (2010) Therapeutic antibodies directed at G protein-coupled receptors. MAbs 2:594–606. 10.4161/mabs.2.6.13420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rouck JE, Krapf JE, Roy J et al. (2017) Recent advances in nanodisc technology for membrane protein studies (2012-2017). FEBS Lett 591:2057–2088. 10.1002/1873-3468.12706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo E, Kelil A, Bayliss PE et al. (2020) In situ antibody phage display yields optimal inhibitors of integrin α11/β1. MAbs 12:1717265. 10.1080/19420862.2020.1717265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tillotson BJ, Cho YK, Shusta EV (2013) Cells and cell lysates: a direct approach for engineering antibodies against membrane proteins using yeast surface display. Methods 60: 27–37. 10.1016/j.ymeth.2012.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krohl PJ, Kim KB, Lew L et al. (2020) A suspension cell-based interaction platform for interrogation of membrane proteins. AICHE J 66:e16995. 10.1002/aic.16995 [DOI] [Google Scholar]
- 16.Kelly RL, Zhao J, Le D, Wittrup KD (2017) Nonspecificity in a nonimmune human scFv repertoire. MAbs 9:1029–1035. 10.1080/19420862.2017.1356528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMahon C, Baier AS, Pascolutti R et al. (2018) Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat Struct Mol Biol 25:289–296. 10.1038/s41594-018-0028-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stern LA, Schrack IA, Johnson SM et al. (2016) Geometry and expression enhance enrichment of functional yeast-displayed ligands via cell panning: geometry and expression drive yeast biopanning. Biotechnol Bioeng 113:2328–2341. 10.1002/bit.26001 [DOI] [PMC free article] [PubMed] [Google Scholar]