

Abstract
Cytochrome P450 46A1 (CYP46A1) is a crucial enzyme in brain that converts cholesterol to 24 (S) hydroxy cholesterol thereby increasing its polarity to facilitate removal of excess cholesterol from the CNS. The inhibition of CYP46A1 with several synthetic molecules has been investigated extensively for treatment of Alzheimer’s disease, Huntington’s disease, glaucoma, and in hippocampal neurons from aged mice. However, phytochemicals have received far little attention in studies involving development of potential CYP46A1 inhibitors. Thus, in the present study phytoconstituents from Indian traditional medicinal plants; Bacopa monnieri, Piper longum, and Withania somnifera, were virtually screened for interaction with CYP46A1 using computational tools. Out of three plants, six molecules from P. longum and three molecules from W. somnifera were shortlisted to study interactions with CYP46A1 based on the physio-chemical parameters. Fargesin, piperolactam A and coumaperine from P. longum showed the higher binding affinity and the values were − 10.3, − 9.5, − 9.0 kcal/moles respectively, whereas, withaferin A from W. somnifera had a binding affinity of − 12.9 kcal/mol. These were selected as potential modulators as they exhibited suitable interactions with active site residues; Tyr109, Leu112, Trp368, Gly369, and Ala474. The selected molecules were further subjected to molecular dynamics simulation. Further, the pharmacological properties of molecules were also predicted using ADMET calculator and the data revealed that all the selected compounds had good absorption as well as solubility characteristics. In addition, sesamin, fargesin, piperolactam A, and coumaperine had minimal or no toxic effects. Thus, the study successfully identified compounds from Indian medicinal plants that may serve as potential inhibitors of CYP46A1 or base structures to design novel CYP46A1 inhibitors, which may be effective in treating neurological conditions involving perturbed cholesterol homeostasis.
Supplementary Information
The online version contains supplementary material available at 10.1007/s42485-022-00098-x.
Keywords: Brain, Cholesterol, Cytochrome P450, In Silico, Lipids, Phytochemicals
Introduction
Cholesterol is an important component of cell membranes that plays a vital role as a signalling molecule by acting as a precursor for numerous bioactive substances such as bile acids, steroid hormones and vitamin D (Maxfield and van Meer 2010). The cholesterol levels in brain are higher and blood–brain barrier (BBB) precludes the exchange of cholesterol from circulation (Björkhem and Meaney 2004; Zhang and Liu 2015). Thus, in situ synthesis of cholesterol occurs in neurons, astrocytes, and oligodendrocytes via a series of enzymatic steps to maintain homeostasis (Hughes et al. 2013). It has been estimated that approximately 25% of unesterified cholesterol is produced in the brain that accounts for 15–30 mg/g of brain tissue. Cholesterol acts as a crucial molecule for neuronal functions such as axonal growth, neuronal repair, synapse formation and remodelling (Petrov et al. 2016). The elimination of excess cholesterol from the brain occurs through the reverse cholesterol pathway as it cannot cross the BBB due to its hydrophobic nature. Therefore, apolipoprotein E (ApoE), ATP-binding cassette transporters (ABCA1 and ABCG1) transport apoE containing lipids to the plasma via the cerebrospinal fluid (Mahley 2016). However, this mode eliminates cholesterol at a rate of 1 mg/24 h (Chobanian et al. 1962). The major pathway for excretion of cholesterol involves the conversion of cholesterol to 24 (S) hydroxy cholesterol by brain-specific cytochrome P450 46A1 (CYP46A1) that removes 6.4 mg of cholesterol per 24 h from brain to via circulation through BBB by increasing its polarity (Lütjohann et al. 1996).
Altered cholesterol homeostasis has been observed in the aging brain and various neurodegenerative diseases such as Alzheimer’s disease (Schönknecht et al. 2002; Tian et al. 2010; Azizidoost et al. 2022) and Huntington’s disease (Boussicault et al. 2016). Altered cholesterol homeostasis has been associated with cognitive deficits in Alzheimer’s disease (Bettio et al. 2017; Martin et al. 2014). Animal studies revealed that the CYP46A1 effects are not limited to cholesterol maintenance but also involve critical cellular pathways like gene transcription, endocytosis, misfolded protein clearance, vesicular transport, and synaptic transmission (Pikuleva and Cartier 2021). It has been reported that reduction in cholesterol levels prevents N-methyl-D-aspartate (NMDA)-dependent Ca2+ influx and NMDA-induced long-term potentiation (LTP) in cultured hippocampal pyramidal neurons (Martín et al. 2014). The mechanism of LTP and long-term depression (LTD) that acts analogous to memory formation have been compromised in aged brains that are associated with the greater loss of memory (Rosenzweig and Barnes 2003). Moreover, increased activity of CYP46A1 leads to toxicity of 24 S-hydroxycholesterol, impairs various biological functions such as NMDA signalling, inflammation, oxidative stress and necroptosis (Alexandrov et al. 2005; Li et al. 2008). Shi et al. (2021) found that polymorphism of CYP46A1 (rs754203) in Chinese with Type 2 diabetes mellitus had high levels of plasma 24 (S) hydroxy cholesterol that resulted in early cognitive impairment including attention and executive deficits.
The crystal structure of CYP46A1 has been determined and reveals that the active site of enzyme is flexible and susceptible to ligand-induced conformational changes (Mast et al. 2010). In addition to cholesterol, the enzyme has been identified to act on several drugs that belong to different classes such as anti-depressants (tranylcypromine), anti-convulsants (thioperamide), anti-fungals (voriconazole and clotrimazole) (Mast et al. 2012, 2013a, 2013b) and steroids (diclofenac, testosterone and turinabol) (Mast et al. 2003). Many structurally distinct synthetic molecules have been tested for pharmacological inhibition of CYP46A1. Although, they have been shown to significantly reduce 24S-hydroxycholesterol levels in brain, but these drugs also interfere with extracerebral cholesterol metabolism (Shafaati et al. 2010). The therapeutic potential of soticlestat, a novel small molecule that inhibits CYP46A1 in associated with neural hyperexcitation has been recently studied (Nishi et al. 2020). Several adverse events such as acute psychosis and confusional state have been reported with soticlestat (Wang et al. 2022). On the other hand, the role of plant derived sterols (Pérez-Cañamás et al. 2016), polyphenol-rich oriental plums (Kuo et al. 2015), quercetin (Lu et al. 2010) were observed to be effective in inhibiting CYP46A1 along with reduction in oxidative stress, inflammatory response, LTP impairment and cognitive defects associated with age as a consequence of cholesterol reduction. Several medicinal plants including Withania somnifera (Ashwagandha) (Durg et al. 2015), Bacopa monnieri (Brahmi) (Ramasamy et al. 2015), and Piper longum (Long Pepper) which have a long history of use in the treatment of cognitive defects and various other diseases, have not yet been studied for potential CYP46A1 inhibitors. Interestingly, W. somnifera and B. monnieri primarily act as memory boosters or brain tonics in later age, whereas P. longum, used in households as a spice, possesses anti-oxidative, anti-inflammatory, immunomodulatory, anti-anxiolytic, anti-neoplastic, anti-convulsant properties (Srinivasan 2007; Go et al. 2018). Therefore, in the present study phytochemicals derived from these plants were virtually screened against CYP46A1 and the lead molecules with best binding interactions were further selected for simulations in order to evaluate the behaviour and stability of these ligands inside the active site. The phytochemicals were also evaluated for their absorption, distribution, metabolism, elimination, toxicity profiles (Cheng et al. 2012) with the aim to identify a potent, highly-specific and brain-penetrable CYP46A1 inhibitor with potential as therapeutic agents to treat neurological conditions involving altered cholesterol metabolism.
Materials and methods
Retrieval and refinement of receptor protein
Cytochrome P450 Family 46 Subfamily A Member 1 (CYP46A1) that plays key role as regulator of cholesterol clearance and turnover in brain was selected as a molecular target. The crystal structure of human CYP46A1 P450 with ligand cholesterol-3-sulphate bound (PDB: 2Q9F; X-ray diffraction 1.9 Å) and unliganded structure of the same protein (PDB: 2Q9G; X-ray diffraction 2.4 Å) were downloaded from Research Collaboratory Structural Bioinformatics- Protein Data Bank (RCSB-PDB) (Berman et al. 2002). For docking studies unliganded structure of CYP46A1 (PDB: 2Q9G) was used to screen phytochemicals. Heteroatom removal, addition of polar hydrogen atoms and Kolmann charges were done using AutoDockTools 1.5.6 and converted to PDBQT format (Trott and Olson 2009).
Mining and preparation of ligands structure
Based on the literature survey a total of 74, 153, and 89 phytochemicals were shortlisted from B. monnieri, P. longum, and W. somnifera, respectively (Supplementary Data Table S1, S2, S3). However, many phytoconstituents (40, 59, 48) from the respective plants could not be used for docking as their three-dimensional structures were not available on ZINC (Irwin and Shoichet 2005) or PubChem (Kim et al. 2016) databases. Eventually, PubChem database yielded 169 molecules from all three plants in the form of structure data file (SDF). The structure of cholesterol 3-sulphate (Fig. 1b) and CYP46A1 inhibitor clotrimazole were downloaded as control in SDF form from PubChem. Further, the molecules were prepared using AutoDockTools 1.5.6 in which hydrogen atoms, Gasteiger charges, and torsion were defined for each molecule that automatically detects a number of rotatable bonds.
Fig. 1.

a Validation of docking method by superimposition of co-crystal protein–ligand complex (ligand-red, chain A-green, heme-blue) over the docked conformation of the protein–ligand complex (ligand-aquamarine, chain A-yellow, heme-pink) b 3-dimensional structure of cholesterol 3-sulfate
Molecular docking of ligands with protein
AutoDock Vina software was used for virtual screening and molecular docking. It performs docking calculations based on the gradient optimization method (Trott and Olson 2009). The screened ligands were converted to PDBQT format by using open babel GUI (O’Boyle et al. 2011). The binding site or grid box for molecular docking was made around the previously bound ligand (Cholesterol 3-sulfate) with dimensions: centre (X, Y, Z): (26.65, 0.69, 18.48) Å and size (X, Y, Z): (36.0, 24.0, 40.0) Å respectively. The grid file was saved in text format as conf_vs with grid spacing 0.375 Å. The molecular interactions between ligand and protein was studied using a Discovery Studio Visualizer 4.0 and the ICM browser was used for structure superimposition (Raush et al. 2009).
ADME properties
The drug likeliness of all the ligands was assessed by thresholds set by ‘Lipinski’s Rule of Five’ through the online server admetSAR viz. molecular weight less than 500 Da and lipophilicity (log P, octanol–water partition coefficient) less than 3.5 for greater BBB penetration and highest uptake (Lipinski 2004). The ADMET profiling of all the compounds and clotrimazole were performed using the same software (Cheng et al. 2012).
Molecular dynamic studies of complexes
Molecular Dynamic simulation is very useful approach to investigate the behaviour and stability of the potential inhibitors inside the active site of the target (Bowers et al. 2006; Uniyal et al. 2022). Therefore, MD simulations for clotrimazole and four compounds viz. withaferin A, fargesin, piperolactam A, and coumaperine that showed good binding affinity along with clotrimazole as reference drug were performed for 100 ns using Desmond molecular dynamics tool implemented in Schrodinger Molecular Modelling Suite. The complex was solvated using TIP3P water model to accommodate the orthorhombic type box around the protein bound with ligand and neutralized with 0.15 Na+ and Cl− ions. Before the production run, the system was minimized with a maximum iteration of 2000 steps. It was then subjected to 100 ns MD simulation for equilibration and production run. The temperature and pressure were kept at 300 K and 1.01325 bar, respectively using Isothermal-isobaric (NPT) ensemble.
Results
Validation of docking approach
The structure of CYP46A1 in complex with cholesterol 3-sulfate (PDB: 2Q9F) was solved recently (Mast et al 2008). Cholesterol 3-sulfate was found to occupy the banana shape orientation and interact with the hydrophobic active site cavity over the entire length of the protein (Mast et al. 2008). The position of the docking site was confirmed by redocking cholesterol 3-sulfate to the active site of CYP46A1 using AutoDock Vina. The binding affinity for CYP46A1 complexed with cholesterol 3-sulfate was calculated to be − 13.1 kcal/mol. The docked structure of the protein–ligand complex was superimposed to the experimentally determined ligand bound structure obtained from RCSB using ICM-browser (Fig. 1a). Both the structures were found to be fully superimposed indicating that the position selected for the docking of the ligand with the receptor was correct. These observations validated the ability of the docking protocol to predict the bioactive conformation of inhibitors.
Structure based virtual screening
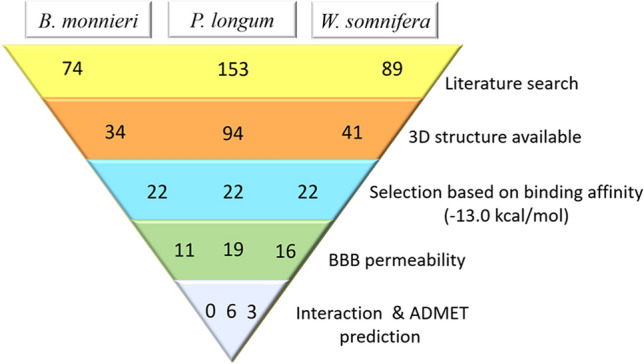
Molecular docking studies showed strong interaction of 22 (out of 34) molecules from B. monnieri, 22 (out of 94) molecules from P. longum, and 22 (out of 41) from W. somnifera with CYP46A1. However, ADMETsar profiling revealed 11, 19, 16 molecules from respective plants; B. monnieri, P. longum, W. somnifera have the ability to cross BBB (Fig. 2). It is observed that binding energy of known inhibitor clotrimazole for CYP46A1 is − 8.1 kcal/mol (Table 1), which is lower than the top hit compounds from three plants (Table S1, 2, 3). It indicates top scoring hits have stronger binding affinity for CYP46A1 as compared to reference ligand (clotrimazole).
Fig. 2.
Steps in the selection of lead molecules
Table 1.
Binding interactions of clotrimazole and phytochemicals selected from Piper longum based on docking studies
| Compounds | MW (Da) | Affinity (kcal/mol) | logP | Amino acids involved in interaction |
|---|---|---|---|---|
| Clotrimazole | 344 | − 8.1 | 5 | Gly369, Ala474, Trp368, Tyr109, Leu112, Ala111 |
| Sesamin | 354.36 | − 11.1 | 3.22 | Leu112, Ala111, Phe80, Arg226, His81 |
| Fargesin | 370.40 | − 10.3 | 3.51 | Leu112, Ala111, HOH732, Tyr109, Ala367, Hem602, Phe121, Phe80, Arg226 |
| Pisatin | 314.29 | − 9.9 | 2.14 | Gly369, Leu112, Ala111, Ala367, Ala302, HOH732, HOH765 |
| Piperolactam A | 265.27 | − 9.5 | 3.21 | Arg110, Met108, Leu112, Ala111, Tyr109, HOH770, Glu472, Ala474, Arg226 |
| Piperine | 285.34 | − 9.4 | 3.00 | Leu112, Ala111, Tyr109, Ala302, Val126 |
| Coumaperine | 257.33 | − 9.0 | 2.97 | Ala474, Ala111, Trp368, Asn227, His81, Arg226, Phe371, Phe80 |
Amino acid residues at the active site are shown in bold
Lead compounds from Bacopa monnieri
After analyzing small molecules from B. monnieri, the compounds with similar structures (cucurbitacin A, cucurbitacin B, cucurbitacin C, cucurbitacin E, and stigmastanol) were eliminated from further ligand-receptor interactions. Moreover, monnieraside and plantainoside B had a predicted log P = 0.03 and 0.13 respectively, which indicates equal partition of both the compounds in liquid and aqueous phase, thus these two were also eliminated. Finally, three molecules; ebelin lactone (binding affinity − 14.1 kcal/mol), sitosterol (− 12.0 kcal/mol), and asiatic acid (− 11.8 kcal/mol) had molecular weight < 500 Da but their logP values were more than 3.5, which were not in the acceptable range. Only cucurbitacin D had logP of 2.93 but its molecular weight was > 500 Da. Therefore, none of the molecules were selected for further ligand–protein interactions (Supplementary data Table S1).
Lead compounds from Piper longum
In P. longum akin molecules (sitosterol, sesamin, dehydropipernonaline, pipernonaline, piperdardine, piperchabamide B, piperchabamide C, brachyamide B, piperundecalidine, piperanine) and other phytochemicals that did not obey the rules of molecular weight and logP were discarded (Supplementary data Table S2). Among all, 6 phytochemicals; sesamin, fargesin, pisatin, piperolactam A, piperine, coumaperine with binding affinity − 11.1, − 10.3, − 9.9, − 9.5, − 9.4, − 9.0 kcal/mol respectively were selected to study their interactions with CYP46A1 (Table 1). The docking of all the compounds showed significant interactions with the active site amino acid residues; Tyr109, Leu112, Trp368, Gly369, and Ala474 like that of clotrimazole (as shown in bold in Table 1 and Fig. 3) (Mast et al. 2008). They also showed polar bond interactions with neighboring water molecules HOH732, HOH765, HOH770. The sesamin, pisatin, and piperine formed π-alkyl interactions with residues (His81, Ala111, Arg226), (Leu112, Ala367) and (Ala111, Tyr109) respectively (Supplementary Fig. S1 a, b, c). In addition to π-alkyl interactions in fargesin (Ala367, Phe121, Phe80, Ala111) that established conventional hydrogen bonding with residue Tyr109 (Fig. 4a). Similarly, in coumaperine (Ala111, Phe80, Arg226, Trp368) conventional hydrogen bonding with Asn227 and C-H with Ala474 were observed (Fig. 4c). Among all piperolactam A exhibited higher occupancy of the active site volume and displaced more water molecules. It formed three hydrogen bonds with Phe371, Phe80, Tyr109 and π-alkyl interactions with Ala111, HIS81, Phe80, Arg226 (Fig. 4b). It was identified that the entrance to active site channel is between the helices B′, F, and the β-1 sheet therefore His81, Phe80, Phe371 of β-1 sheet, Arg110, Leu112 of B′ helices, and Ile222, Asn227 of F helices are crucial amino acids that form the entrance to substrate access channel (Mast et al. 2008) The results identified that all the molecules interacting with these residues including clotrimazole. Moreover, all compounds had hydrophobic interactions with Phe80, Ala367, Val126 except piperolactam A.
Fig. 3.

2-dimensional and 3-dimensional interactions of clotrimazole with amino acid residues (cyan) at a particular bond distance in the pocket of protein CYP46A1 (silver). (atoms are coloured according to their type: carbon- orange, oxygen- red, nitrogen- blue; hydrogens are not shown)
Fig. 4.

2-dimensional and 3-dimensional interactions of a Fargesin b Piperolactam A c Coumaperine (orange) with amino acid residues (cyan) at a particular bond distance in the pocket of CYP46A1 (silver). (atoms are coloured according to their type: carbon- orange, oxygen- red, nitrogen- blue; hydrogens are not shown)
Lead compounds from W. somnifera
From the sixteen phytochemicals withanolide D, withaphysalin M, withaferin A with binding affinity − 13.8, − 13.7, − 12.9 kcal/mol were investigated (Table 2). Rest of the molecules were eliminated because they were either structurally similar (withaphysalin N, withanolide B, withanolide C, withanolide S, dihydrowithaferin A, withaferin, withaphysalin F, withanone, withacnistin) or had greater molecular weight and logP values (diosgenin, stigmasta-3,5-dien-7-one, sitoindoside IX, withacnistin) (Supplementary data Table S3). Ligand-receptor interactions displayed interactions of withanolide D, withaphysalin M, withaferin A with active site residues (Table 2) similar to clotrimazole (Fig. 3). Single hydrogen bond interactions with residues His81 (conventional) and Arg226 (C-H) were seen with withaphysalin M and withaferin A respectively. Further, withanolide D displayed three hydrogen bonds with HOH765, Gly369 (conventional), Thr223 (C-H). The π-alkyl interactions of withanolide D with residues Tyr109 and Trp368, withaphysalin M with Phe371, withaferin A with His81, Phe371 were observed (Supplementary Fig. S2a, b, Fig. 5a). Similarly, like clotrimazole all the three molecules interacted with residues of substrate access channel.
Table 2.
Phytochemicals selected from W. somnifera based on docking studies
| Phytochemicals | MW (Da) | Affinity (kcal/mol) | logP | Amino acids involved in interaction |
|---|---|---|---|---|
| Withanolide D | 470.61 | − 13.8 | 3.50 | Thr223, Met108, Leu112, Ala111, Tyr109, Trp368, HOH765, Gly369, Thr223, Ala302, Hem602 |
| Withaphysalin M | 482.57 | − 13.7 | 3.04 | Ile222, Met108, Ala111, Leu112, Ala474, Arg226, His81, Phe371 |
| Withaferin A | 470.61 | − 12.9 | 3.35 | Ile222, Ala111, Met108, Leu112, Ala474, Trp368, Ala367, Arg226, Phe371, His81 |
Amino acid residues at the active site are shown in bold
Fig. 5.

2-dimensional and 3-dimensional interactions of Withaferin A with amino acid residues (cyan) at a particular bond distance in the pocket of CYP46A1 (silver). (atoms are coloured according to their type: carbon- orange, oxygen- red, nitrogen- blue; hydrogens are not shown)
ADMET profiling
To predict the pharmacological properties such as absorption, distribution, metabolism, excretion and toxicity of phytochemicals, the ADMET calculations were performed for compounds from P. longum and W. somnifera and were compared with clotrimazole as reference compound (Table 3).
Table 3.
ADMET predictions of clotrimazole and phytochemicals from P. longum and W. somnifera
| ADMET Properties | Clotrimazole | Sesamin | Fargesin | Pisatin | Piperolactam A | Piperine | Coumaperine | Withanolide D | Withaphysalin M | Withaferin A |
|---|---|---|---|---|---|---|---|---|---|---|
| HIA | Positive | Positive | Positive | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| Caco-2 | Positive | Positive | Positive | Positive | Positive | Positive | Positive | Negative | Negative | Negative |
| HOB | Negative | Negative | Negative | Negative | Positive | Negative | Negative | Negative | Positive | Positive |
| Subcellular localization | Mito | Mito | Mito | Mito | Mito | Mito | Mito | Mito | Mito | Mito |
| P-GP inhibitor | N-inhibitor | Inhibitor | Inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | Inhibitor | Inhibitor |
| P-GP substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | Substrate | Substrate |
| CYP3A4 substrate | N-substrate | N-substrate | N-substrate | Substrate | Substrate | N-substrate | N-substrate | Substrate | Substrate | Substrate |
| CYP2C9 substrate | Substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | Substrate | N-substrate | N-substrate | N-substrate |
| CYP2D6 substrate | N-substrate | N-substrate | Substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate | N-substrate |
| CYP3A4 inhibition | Inhibitor | Inhibitor | Inhibitor | Inhibitor | N-inhibitor | Inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor |
| CYP2C9 inhibition | Inhibitor | Inhibitor | Inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor |
| CYP2C19 inhibition | Inhibitor | Inhibitor | Inhibitor | Inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor |
| CYP2D6 inhibition | Inhibitor | Inhibitor | Inhibitor | Inhibitor | N-inhibitor | Inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor |
| CYP1A2 inhibition | Inhibitor | Inhibitor | Inhibitor | N-inhibitor | Inhibitor | Inhibitor | N-inhibitor | N-inhibitor | N-inhibitor | N-inhibitor |
| Carcinogenicity | N-cancerous | N-cancerous | Ncancerous | N-cancerous | N-cancerous | N-cancerous | N-cancerous | N-cancerous | N-cancerous | N-cancerous |
| Ames test | Negative | Positive | Positive | Negative | Positive | Negative | Positive | Negative | Negative | Negative |
| Hepato-toxicity | Positive | Negative | Negative | Positive | Positive | Negative | Negative | Negative | Negative | Negative |
Bold indicates property that is different from rest of the molecules
N-cancerous Non-cancerous, N-inhibitor Non-inhibitor, N-substrate Non-substrate
P. longum: ADMET properties, as derived from admetSAR server, revealed that all the compounds were highly absorbed from the intestine and were also Caco-2 permeable. However, except piperolactam A all had poor oral bioavailability similar to clotrimazole. The subcellular localization of all the compounds were predicted to mitochondria. Nevertheless, pisatin, piperolactam A, piperine, and coumaperine and fargesin were not P-glycoprotein (P-GP) substrates like clotrimazole suggesting that they may be actively excluded from the cells by P-glycoprotein. Moreover, in terms of metabolism the compounds showed diverse results. As some of the cytochrome P450 isoforms could be inhibited by one or more of the test compounds and some acted as their substrates. The results of toxicity parameter predicted similar to the clotrimazole. Pisatin and piperine revealed negative for Ames test, which suggests that they are non-mutagenic. The carcinogenic profile also revealed that the ligands were non-carcinogenic in nature. The results of hepatotoxicity assessment predicted no toxicity for sesamin, fargesin, piperine and coumperine suggesting them as better molecules as compared to clotrimazole. (Table 3).
W. somnifera: The absorption parameters for W. somnifera revealed that all three compounds had greater Human intestinal absorption (HIA) but were Caco-2 impermeable. Withaphysalin M and Withaferin A were found to have poor oral bioavailability. Moreover, Withaphysalin N was found to be the only non-inhibitor and non-substrate for P-GP like clotrimazole. Furthermore, through metabolic parameters it was found that all the three tested compounds showed variable results in comparison to clotrimazole as they could act as substrate for only CYP3A4, the cytochrome P450 isoform, while non-substrate and non-inhibitor for other cytochrome P450 isoforms, which indicates that the molecules may not hamper biotransformation of drug metabolised by CYP450 enzymes. Fortunately, all compounds were suggested not to have any acute toxicity and mutagenic effects with regard to hepatoxicity and Ames test. The detailed results for the pharmacokinetic parameters and toxicity analysis are shown in Table 3.
MD simulation studies
To understand the type of interactions between the ligand and protein and their thermodynamic stability, molecular dynamic simulations for time period of 100 ns were performed. The results of MD for each complex were analyzed by calculating root mean square deviation (RMSD) and root mean square fluctuation (RMSF) for whole trajectory that are presented in the following sections:
Clotrimazole: Trajectory analysis of Clotrimazole-protein complex is shown in Fig. 6. The RMSD of the protein provides insights into its structural conformation throughout the simulation. RMSD analysis (Fig. 6a, blue line) indicates the changes in the order of 1.0–1.6 Å are perfectly acceptable for small, globular proteins while ligand RMSD (Fig. 6a, purple line) is lower than that of protein swapping in between 0.4 and 1.4 Å. The stable RMSD plot reveals that protein and ligand are well equilibrated and there is no drastic change through the simulation. The RMSF plot (Fig. 6b) for protein ranged from 0.4 to 1.8 Å, which is acceptable and the plot indicates that the residues that are involved in binding of ligand have less fluctuations. Interactions (Fig. 6c) involved in stabilisation of complex reveal that the phenyl ring, chloro-substituted phenyl ring and imidazole ring interact with Phe80, Tyr109 and Trp368 respectively. The nitrogen atom of imidazole ring is also responsible for hydrogen bond formation with Trp368. Ligand RMSF value is between 0.58 and 0.8 Å provided in the Fig. 6d where the phenyl ring atoms have higher RMSF values indicating interactions within the catalytic pocket of the enzyme.
Fig. 6.

Molecular dynamics trajectory analysis of the Clotrimazole-protein complex. a Protein–ligand RMSD; b RMSF for protein, green colored lines marked to indicate the residues which interacts with ligand; c Interaction of protein and ligand and d RMSF for ligand
Protein ligand interactions and their distribution are depicted in Fig. 7, the distribution and type of interactions are depicted in Fig. 7a along with the residues that interact with the ligand in each trajectory frame are depicted in Fig. 7b, the top panel shows the total number of specific contacts of protein with the ligand over the course of the trajectory. It is clear that most of the residues form hydrophobic interactions such as Phe80, Tyr109, Ile222, Phe371 throughout the simulation indicating that these interactions have major role in stabilisation of ligand with protein. The Trp368 and Gly369 also appear to participate in hydrogen bond formation for about half of the simulation time period.
Fig. 7.

Contact Analysis of MD trajectory of Clotrimazole complex a Protein–ligand contact distribution histogram; b protein–ligand contacts
Coumaperine: RMSD plot showed in Fig. 8a (blue line) reveals that all the protein frames aligned with RMSD values between 1.4–1.8 Å on the reference frame C alpha except for initial 11 ns having RMSD of 1.2–1.4 Å. Overall RMSD value for protein trajectory remain below 1.8 Å which is within the acceptable limit. As expected the RMSD of small molecule i.e., coumaperine (Fig. 8a, purple line) fluctuates along the whole trajectory in the range of 0.4–1.8 Å, which is likely to the protein RMSD indicating that the complex was stable during the simulation. To check the flexibility and stability of amino acid residues throughout the time period of simulation RMSF plot was generated. Figure 8b shows the RMSF for protein with the value ranging between 0.5–3.5 Å that is in the acceptable range. In addition, the ligand that interacts with the protein amino acid residues are marked with green colour in the graph. Further the interaction formed within the complex can be seen from the Fig. 8c, which reveals that carboxyl group of the ligand forms hydrogen bond with Tyr109 for about 33% of simulation. Ligand RMSF value is between 1.4 and 2.5 Å provided in the Fig. 8d where the carboxyl group have higher RMSF value indicating the flexibility of that atoms during the simulation.
Fig. 8.

Molecular dynamics trajectory analysis of the Coumaperine-protein complex. a Protein–ligand RMSD; b RMSF for protein, green colored lines marked to indicate the residues which interacts with ligand; c Interaction of protein and ligand and d RMSF for ligand
Protein ligand interactions and their distribution were also monitored throughout the simulation, represented in the Fig. 9, where the distribution and type of interaction is depicted in Fig. 9a and the residues that interact with the ligand in each trajectory frame are depicted in Fig. 9b where the top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The ligand forming hydrophobic contacts with various amino acids viz. Phe80, Ala111, Leu112, Phe121, Val126, water bridges with His81, Arg226, Ser225 and forming hydrogen bonds with Tyr109, Ser225, Arg226 throughout the simulation at different time intervals with varying percentage. According to the graphical plot, interactions in terms of hydrogen bonding, water bridges and hydrophobic interactions were essential for the linkage of ligands with the protein active site.
Fig. 9.

Contact Analysis of MD trajectory of Coumaperine complex a Protein–ligand contact distribution histogram; b protein–ligand contacts
Fargesin:RMSD plot for fargesin and protein is given in Fig. 10a which indicates that protein RMSD remained between 1.1 and 1.8 Å, while RMSD of the fargesin rises initially from 1.1 to 1.8 Å upto 19 ns then was stable between 1.2 and 1.7 Å throughout the simulation period. Overall RMSD values for protein and ligand remain below 1.8 Å, which is within the acceptable limit. Figure 10b shows the RMSF for protein with values ranging between 0.4 and 3.2 Å that is within the acceptable range. Further Fig. 10c represents the protein–ligand interaction diagram, which reveals that benzopyran moiety of the ligand is forming pi-pi stacking with Tyr109 and Phe80 for about half of the simulation time. The ligand RMSF values are between 0.5 and 1.2 Å plotted in Fig. 10d, where the oxygen and carbon atom of benzopyran ring have higher RMSF values than the other atoms indicating the flexibility of these atoms during the simulation.
Fig. 10.

Molecular dynamics trajectory analysis of the Fargesin-protein complex. a Protein–ligand RMSD; b RMSF for protein, green colored lines marked to indicate the residues that interacts with ligand; c Interactions of protein and ligand and d RMSF for ligand
The distribution and type of interactions by fargesin are depicted in Fig. 11a and the residues that interact with it in each trajectory frame are depicted in Fig. 11b where the top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The ligand forming hydrophobic contacts with various amino acids viz. Phe80, Tyr109, Leu112, Phe371, water bridges with Aag226, Thr306 throughout the simulation at different time intervals with varying percentage. According to the graphical plot interactions in terms of hydrophobic and water bridges interactions are essential for the linkage of ligands with the active site of protein.
Fig. 11.

Contact Analysis of MD trajectory of Fargesin complex a Protein–ligand contact distribution histogram; b protein–ligand contacts
Piperolactam: Piperolactam and protein RMSD plot is depicted in Fig. 12a that reveals that RMSD of protein remained stable between 1.0 and 1.7 Å throughout the simulation, while RMSD of piperolactam fluctuates from 0.2 to 1.4 Å upto 55 ns then was stable for last 45 ns between 1.3 and 1.7 Å. Overall RMSD values for protein and ligand remained below 1.7 Å which is within the acceptable range. Figure 12b shows RMSF for protein with the values ranging between 0.4 and 3.2 Å that is also within the acceptable range. Further Fig. 12c represented the protein–ligand interaction diagram, revealing that dibenzo rings of the ligand are forming pi-pi stacking with Tyr109, Phe80 and Trp368 for about half of the simulation time and oxygen atom and -NH group of the indolone ring form hydrogen bond with Trp368 and Ala474 respectively. The ligand RMSF values ranged between 0.5 and 2.0 Å plotted in Fig. 12d, where the atoms of dibenzo ring of piperolactam were found to have higher RMSF values than the other atoms indicating the flexibility of these atoms during the simulation.
Fig. 12.

Molecular dynamics trajectory analysis of the Piperolactam-protein complex. a Protein–ligand RMSD; b RMSF for protein, green colored lines marked to indicate the residues which interacts with ligand; c Interaction of protein and ligand and d RMSF for ligand
The distribution and type of interactions formed by the piperolactam are depicted in Fig. 13a and the residues that interact with it in each trajectory frame are depicted in Fig. 13b where the top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The ligand forming hydrophobic contacts with various amino acids viz. Phe80, Tyr109, Leu112, Phe371, and hydrogen bonding with Trp368, Ala474 and Gly369 throughout the simulation at different time intervals with varying percentage. According to the graphical plot interactions in terms of hydrophobic and hydrogen bond are essential for the linkage of ligands with the active site of protein.
Fig. 13.

Contact Analysis of MD trajectory of Piperolactam complex a Protein–ligand contact distribution histogram; b protein–ligand contact
Withaferin A: RMSD plot of withaferin and protein are shown in Fig. 14a which indicates that protein RMSD remained stable between 1.0 and 1.7 Å for first half of the simulation and increased to 1.8 Å in the second half of simulation and remained stable at the same value, while ligand RMSD fluctuated from 0.4 to 1.7 Å. Overall RMSD values for protein and ligand remain below 1.8 Å, which is within the acceptable range. Figure 14b shows the RMSF for protein with the values ranging between 0.4 and 3.0 Å that is also within the acceptable range. Further Fig. 14c represents the protein–ligand interaction diagram, showing that hydroxyl group of phenanthroxirenone moiety and pyran form hydrogen bond with Thr306 for 91% of trajectory and Met108 for about 40% of the simulation time respectively. The carboxyl group of phenanthroxirenone moiety participated in water bridge with Lys104 for about 36% of trajectory. The ligand RMSF values were between 0.5 and 2.0 Å and are presented in Fig. 14d, where the carboxyl oxygen atom of pyran ring was found to have higher RMSF values than the other atoms indicating the flexibility of this atom during the simulation.
Fig. 14.

Molecular dynamics trajectory analysis of the Withferin-protein complex. a Protein–ligand RMSD; b RMSF for protein, green colored lines marked to indicate the residues which interacts with ligand; c Interaction of protein and ligand and d RMSF for ligand
The distribution and type of interaction with withaferin are shown in Fig. 15a and the residues that interact with it in each trajectory frame are depicted in Fig. 15b where the top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The ligand forming hydrophobic contacts with various amino acids Phe80, Ala302, Ala367, Trp368; water bridge with His81, L104, Ile222 and hydrogen bonding with Met108 and Thr306 throughout the simulation at different time intervals with varying percentage. According to the graphical plot interactions in terms of hydrophobic, water bridge and hydrogen bond interactions are essential for the linkage of ligands with the active site of protein.
Fig. 15.

Contact Analysis of MD trajectory of Withferin complex a Protein–ligand contact distribution histogram; b protein–ligand contacts
Discussion
CYP46A1, a cholesterol-catabolic enzyme specific to the brain, has received attention as a potential drug target because of its role in neurodegenerative conditions. Many seminal studies have tested drugs such as bicalutamide, clotrimazole, fluvoxamine, thioperamide, tranylcypromine, and voriconazole (Mast et al. 2003) and non-pharmaceutical agents like soticlestat for inhibition of CYP46A1 (Nishi et al. 2020). However, long-term use of such molecules has been linked to increased risk of other co-morbidities for example interference of voriconazole with extracerebral cholesterol metabolism (Shafaati et al. 2010). Therefore, focus across the world is to identify potential plant-based natural molecules as therapeutics. Moreover, the active site of CYP46A1 is flexible and undergoes ligand-induced conformational fit (Mast et al. 2008), which explains the ability of CYP46A1 to bind wide range of structurally varied and unrelated compounds. In addition, a study has shown that that the residues far from the active site are also involved in interaction of compounds with CYP46A1 (Anderson et al. 2016). Plant derived phytochemicals because of their pleiotropic properties have been a source of potential therapeutics since ages. Thus, the current study attempts to identify phytochemicals from Indian traditional medicinal plants as CYP46A1 inhibitors using in silico approach.
The molecular docking is one of the most prominent methods for drug discovery in which binding orientation and interactions of small ligands with receptor protein is monitored. In the present study by comprehensive literature survey of three plants; B. monnieri, P. longum, and W. somnifera provided 74, 153, and 89 phytochemicals respectively which belong to different classes such as flavonoids, indoles, prenol lipids, and derivatives of alkaloids. However, only 34, 94, and 41 compounds had three-dimensional structures available in the PubChem. Therefore, these molecules were examined for their inhibitory potential using in silico approach wherein virtual screening was performed against CYP46A1 protein and ranking of different ligand conformations was based on predicted binding affinities with default scoring function in Vina. The energy comparison and selection of ligand molecules from three plants were done based on the standard values of cholesterol 3-sulfate and clotrimazole which was − 13.0 and − 8.1 kcal/mol respectively. Although, plants derived compounds are much safer than synthetic drugs still their fundamental physiological properties determine whether they can be used as a therapeutic drug. For this reason, compounds with molecular weight less than 500 Da and lipophilicity (log P) less than 3.5 for greater BBB penetration and highest uptake were selected. As a result, all the phytochemicals from B. monnieri were eliminated from further study either due to their structural similarity or insufficient molecular weight or log P values. The analysis of docking results of six phytochemicals; sesamin, fargesin, pisatin, piperolactam A, piperine, coumaperine from P. longum and three phytochemicals; withanolide D, withaphysalin M, withaferin A from W. somnifera displayed better binding affinity. The interaction analysis with protein–ligand complex suggested that all the molecules had significant binding pattern similar to clotrimazole due to their interaction with active site amino acid residues; Tyr109, Leu112, Trp368, Gly369, and Ala474, which were also in accordance with prediction by Mast et al (2008). The molecules also exhibited entrance to substrate access channel by interacting with His81, Phe80, Phe371 of β-1 sheet, Arg110, Leu112 of B′ helices, and Ile222, Asn227 of F helices residues (Mast et al. 2008). Multiple hydrogen bonds were displayed between ligand molecule and target protein, except sesamin and piperine of P. longum. For instance, fargesin, piperolactam A and coumaperine from P. longum laid their structures in a way that they can form critical hydrogen bonding (Mast et al. 2012) with residues HOH732, Tyr109; Leu112, Tyr109; Asn227, Ala474 respectively. Previously, fargesin from the Magnolia plants which are used in the treatment of headache, nasal congestion and sinusitis was shown to improve dyslipidemia and hyperglycemia in high fat-induced obese mice by phosphorylating and activating Akt, AMP-activated protein kinase (AMPK), and acetyl-CoA carboxylase in both 3T3-L1 adipocytes and white adipose tissue (Lee et al. 2012). In addition, in vitro inhibitory activities of fargesin from Flos Magnoliae on eight different cytochrome P450 enzymes were determined suggesting that fargesin may modulate drug metabolism, but more in vivo studies are required to investigate drug interactions (Kim et al. 2017). A recent study showed that fargesin inhibits atherosclerosis by promoting reverse cholesterol transport process and reduced inflammatory response via CEBPαS21/LXRα and TLR4/NF-κB pathways (Wang et al. 2020) Similarly, alteration of CYP46A1 and HMG-CoA reductase on Lipopolysaccharide stimulation through TLR4/MyD88/NF-κB signalling pathway leading to neuroinflammation has been reported (Na et al. 2021). Coumaperine and its derivatives, CP-9 and CP-38 have been shown to act as a potent inhibitors of NF-κB subunits p50 and p65 in L428 and A549 cancer cells (Nandakumar et al. 2017). Therefore, it might be possible that coumaperine and its derivatives may inhibit NF-κB signalling pathway in neurons. However, further studies to evaluate the effects are needed. Inhibitory activity of Piperolactam A was only analysed against pathogenic bacteria and SARS-CoV-2 (Kothandan et al. 2021; Nongmai et al. 2022). In W. somnifera single critical hydrogen bonding of withaferin A with His81 was observed. In silico studies revealed inhibition of NPC1 and SRB1 by withaferin A with binding energies − 5.73 and − 7.16 kcal/mol respectively in hypercholesterolemia (Ulhas and Malaviya 2022). Moreover, permeability of withaferin A through computational membrane models such as POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) revealed that interaction of oxygen at fifth position of withaferin A with phosphate group of the membranes drive high permeability across the bilayer (Wadhwa et al. 2021). Modi et al. (2022) studied the pharmacokinetics of withaferin A (500 mg/kg) in rats using UHPLC-MS/MS. The results revealed half-life of withaferin of 7.6 ± 3.3 h and bioavailability of 32.4 ± 4.8%. Thus, these studies revealed that withaferin can be further designed to test against the CYP46A1 in vivo. Further, the three lead molecules fargesin, piperolactam A and coumaperine from P. longum and withaferin A from W. somnifera were subjected to molecular dynamic studies which revealed their structural stability and less conformational changes of receptor-ligand complex during 100 ns trajectory.
In order to assess the safety and efficacy of ligand molecules, their pharmacokinetics was studied using ADMET profiling. According to Lipinski's “Rule-of-Five”, ADMET screening of all the nine molecules from P. longum, and W. somnifera showed greater HIA absorptivity whereas piperolactam A, withaphysalin M and withaferin A showed good oral bioavailability. In addition, none of the molecules displayed acute toxicity and mutagenic effects evaluated in terms of hepatoxicity and Ames test and have favorable drug-likeness features. The information available in literature also supports the fact that these plant-derived molecules have no reported toxicity (Kumar and Patnaik 2016; Mandlik and Namdeo 2020; Kaushik et al. 2021; Xia et al. 2021). Based on the data obtained it appears that many of the compounds studied may be potential inhibitors of the CYP46A1. Furthermore, all of the phytochemicals were found to be even BBB permeable, which suggests that they may be useful against neurodegenerative conditions with perturbed cholesterol homeostasis. Thus, the predicted results from both plants indicate that ADMET characteristics of most phytochemicals are similar with those of clotrimazole. Interestingly, mice treated with voriconazole, a CYP46A1 inhibitor showed decrease in 24S-hydroxycholesterol levels which is product of CYP46A1 suggesting inhibition. Furthermore, administration of CYP46A1-containing adenovirus to APP23 or APP/PS mice revealed enhanced CYP46A1 activity increased CYP46A1 expression and levels of 24S-hydroxycholesterol in the brain. This was accompanied by reduction in amyloid β load and rescuing of cognitive deficits (Hudry et al 2010), Therefore, in vitro and in vivo studies would be needed to evaluate the bioavailabity of these molecules especially BBB permeability and their ability of reverse pathology observed in various neurodegenerative conditions wherein CYP46A1 may be a potential target. A recent study has shown that treatment with distinct classes of CYP46A1 inhibitors led to central 24S-hydroxycholesterol reduction in vivo and ablation of long term depression in hippocampal slices (Popiolek et al. 2021).
In conclusion, the present study has provided lead compounds as novel CYP46A1 inhibitors based on binding interactions and MD studies. Three molecules were identified from P. longum; fargesin, piperolactam A and coumaperine that had binding affinity values of − 10.3, − 9.5, − 9.0 kcal/moles and one lead molecule from W. somnifera; withaferin A with binding affinity value of − 12.9 kcal/mol for CYP46A1. However, further improvements in these ligands may be required through structural optimization and validation through in vitro and in vivo studies to design potential phytochemical CYP46A1 inhibitors that may be effective in treating neurological conditions involving perturbed cholesterol homeostasis.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors acknowledge the facilities provided by the departments.
Declarations
Conflict of interest
Authors do not have any conflict of interest.
Ethical approval
No human or animal studies were included in study and therefore no ethical clearance was required.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Alexandrov P, Cui JG, Zhao Y, Lukiw WJ. 24S-hydroxycholesterol induces inflammatory gene expression in primary human neural cells. NeuroReport. 2005;16:909–913. doi: 10.1097/00001756-200506210-00007. [DOI] [PubMed] [Google Scholar]
- Anderson KW, Mast N, Hudgens JW, Lin JB, Turko IV, Pikuleva IA. Mapping of the allosteric site in cholesterol hydroxylase CYP46A1 for efavirenz, a drug that stimulates enzyme activity. J Biol Chem. 2016;291:11876–11886. doi: 10.1074/jbc.M116.723577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizidoost S, Babaahmadi-Rezaei H, Nazeri Z, Cheraghzadeh M, Kheirollah A. Amyloid beta increases ABCA1 and HMGCR protein expression, and cholesterol synthesis and accumulation in mice neurons and astrocytes. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867(1):159069. doi: 10.1016/j.bbalip.2021.159069. [DOI] [PubMed] [Google Scholar]
- Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The protein data bank. Acta Crystallogr Sect D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/S0907444902003451. [DOI] [PubMed] [Google Scholar]
- Bettio LEB, Rajendran L, Gil-Mohapel J. The effects of aging in the hippocampus and cognitive decline. Neurosci Biobehav Rev. 2017;79:66–86. doi: 10.1016/j.neubiorev.2017.04.030. [DOI] [PubMed] [Google Scholar]
- Björkhem I, Meaney S. Brain Cholesterol: Long Secret Life behind a Barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Boussicault L, Alves S, Lamazière A, Planques A, Heck N, Moumné L, et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain. 2016;139:953–970. doi: 10.1093/brain/awv384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen B A, et al. (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE conference on super computing (SC06) pp 86
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- Chobanian AV, Burrows BA, Hollander W. Body cholesterol metabolism in man. II. Measurement of the body cholesterol miscible pool and turnover rate. J Clin Invest. 1962;41:1738–1744. doi: 10.1172/JCI104632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durg S, Dhadde SB, Vandal R, Shivakumar BS, Charan CS. Withania somnifera (Ashwagandha) in neurobehavioural disorders induced by brain oxidative stress in rodents: A systematic review and meta-analysis. J Pharm Pharmacol. 2015;67:879–899. doi: 10.1111/jphp.12398. [DOI] [PubMed] [Google Scholar]
- Go J, Park TS, Han GH, Park HY, Ryu YK, Kim YH, et al. Piperlongumine decreases cognitive impairment and improves hippocampal function in aged mice. Int J Mol Med. 2018;42:1875–1884. doi: 10.3892/ijmm.2018.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, et al. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of alzheimer’s disease. Mol Ther. 2010;18:44–53. doi: 10.1038/mt.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TM, Rosano C, Evans RW, Kuller LH. Brain cholesterol metabolism, oxysterols, and dementia. J Alzheimer’s Dis. 2013;33:891–911. doi: 10.3233/JAD-2012-121585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. Zinc- A free database of commercially available compounds for Virtual Screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik P, Ali M, Salman M, Tabassum H, Parvez S. Harnessing the mitochondrial integrity for neuroprotection: Therapeutic role of piperine against experimental ischemic stroke. Neurochem Int. 2021;149:105138. doi: 10.1016/j.neuint.2021.105138. [DOI] [PubMed] [Google Scholar]
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kwon SS, Jeong HU, Lee HS. Inhibitory effects of dimethyllirioresinol, epimagnolin a, eudesmin, fargesin, and magnolin on cytochrome P450 enzyme activities in human liver microsomes. Int J Mol Sci. 2017 doi: 10.3390/ijms18050952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothandan R, Rajan CASG, Arjun J, Raj RRM, Syed S. Virtual screening of phytochemical compounds as potential inhibitors against SARS-CoV-2 infection. Beni-Suef Univ J Basic Appl Sci. 2021 doi: 10.1186/s43088-021-00095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G, Patnaik R. Exploring neuroprotective potential of Withania somnifera phytochemicals by inhibition of GluN2B-containing NMDA receptors: An in silico study. Med Hypotheses. 2016;92:35–43. doi: 10.1016/j.mehy.2016.04.034. [DOI] [PubMed] [Google Scholar]
- Kuo PH, Lin CI, Chen YH, Chiu WC, Lin SH. A high-cholesterol diet enriched with polyphenols from Oriental plums (Prunus salicina) improves cognitive function and lowers brain cholesterol levels and neurodegenerative-related protein expression in mice. Br J Nutr. 2015;113:1550–1557. doi: 10.1017/S0007114515000732. [DOI] [PubMed] [Google Scholar]
- Lee YS, Cha BY, Choi SS, Harada Y, Choi BK, Yonezawa T, et al. Fargesin improves lipid and glucose metabolism in 3T3-L1 adipocytes and high-fat diet-induced obese mice. BioFactors. 2012;38:300–308. doi: 10.1002/biof.1022. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang X, Ma C, Qiao J, Zhang C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat’s cultured cortical neurons. Neurosci Lett. 2008;447:120–123. doi: 10.1016/j.neulet.2008.08.037. [DOI] [PubMed] [Google Scholar]
- Lipinski CA. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, Shan Q, et al. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J Pathol. 2010;222:199–212. doi: 10.1002/path.2754. [DOI] [PubMed] [Google Scholar]
- Lütjohann D, Breuer O, Ahlborg G, Nennesmo I, Sidén Å, Diczfalusy U, et al. Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305–1315. doi: 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandlik DS, Namdeo AG. Pharmacological evaluation of Ashwagandha highlighting its healthcare claims, safety, and toxicity aspects. J Diet Suppl. 2020 doi: 10.1080/19390211.2020.1741484. [DOI] [PubMed] [Google Scholar]
- Martín MG, Pfrieger F, Dotti CG. Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep. 2014;15(10):1036–1052. doi: 10.15252/embr.201439225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Norcross R, Andersson U, Shou M, Nakayama K, Bjorkhem I, et al. Broad substrate specificity of human Cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry. 2003;42:14284–14292. doi: 10.1021/bi035512f. [DOI] [PubMed] [Google Scholar]
- Mast N, White MA, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci U S A. 2008;105:9546–9551. doi: 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Charvet C, Pikuleva IA, Stout CD. Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem. 2010;285:31783–31795. doi: 10.1074/jbc.M110.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Linger M, Clark M, Wiseman J, Stout CD, Pikuleva IA. In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol Pharmacol. 2012;82:824–834. doi: 10.1124/mol.112.080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Zheng W, Stout CD, Pikuleva IA. Antifungal azoles: Structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1S. Mol Pharmacol. 2013;84:86–94. doi: 10.1124/mol.113.085902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Zheng W, Stout CD, Pikuleva IA. Binding of a cyano-and fluoro-containing drug bicalutamide to cytochrome P45046A1 unusual features and spectral response. J Biol Chem. 2013;288:4613–4624. doi: 10.1074/jbc.M112.438754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, van Meer G. (2010) Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. 2010;22(4):422–429. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi SJ, Tiwari A, Ghule C, Pawar S, Saste G, Jagtap S, et al. Pharmacokinetic study of withanosides and withanolides from Withania somnifera using Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry (UHPLC-MS/MS) Molecules. 2022;27:1–24. doi: 10.3390/molecules27051476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S, Duan X, Wang R, Fan Y, Xue K, Tian S, et al. Chronic neuroinflammation Induced by lipopolysaccharide injection into the third ventricle induces behavioral changes. J Mol Neurosci. 2021;71:1306–1319. doi: 10.1007/s12031-020-01758-7. [DOI] [PubMed] [Google Scholar]
- Nandakumar N, Muthuraman S, Gopinath P, Nithya P, Gopas J, Kumar RS. Synthesis of coumaperine derivatives: Their NF-κB inhibitory effect, inhibition of cell migration and their cytotoxic activity. Eur J Med Chem. 2017;125:1076–1087. doi: 10.1016/j.ejmech.2016.10.047. [DOI] [PubMed] [Google Scholar]
- Nishi T, Kondo S, Miyamoto M, Watanabe S, Hasegawa S, Kondo S, et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci Rep. 2020;10:1–10. doi: 10.1038/s41598-020-74036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nongmai C, Kanokmedhakul K, Promgool T, Paluka J, Suwanphakdee C, Kanokmedhakul S. Chemical constituents and antibacterial activity from the stems and leaves of Piper wallichii. J Asian Nat Prod Res. 2022;24:344–352. doi: 10.1080/10286020.2021.1933959. [DOI] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An Open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cañamás A, Sarroca S, Melero-Jerez C, Porquet D, Sansa J, Knafo S, et al. A diet enriched with plant sterols prevents the memory impairment induced by cholesterol loss in senescence-accelerated mice. Neurobiol Aging. 2016;48:1–12. doi: 10.1016/j.neurobiolaging.2016.08.009. [DOI] [PubMed] [Google Scholar]
- Petrov AM, Kasimov MR, Zefirov AL. Brain cholesterol metabolism and its defects: Linkage to neurodegenerative diseases and synaptic dysfunction. Acta Naturae. 2016;8:58–73. doi: 10.32607/20758251-2016-8-1-58-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikuleva IA, Cartier N. Cholesterol hydroxylating Cytochrome P450 46A1: From mechanisms of action to clinical applications. Front Aging Neurosci. 2021;13:1–17. doi: 10.3389/fnagi.2021.696778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popiolek M, Izumi Y, Hopper AT, Dai J, Miller S, Shu HJ, Zorumski CF, Mennerick S. Effects of CYP46A1 inhibition on long-term-depression in hippocampal slices ex vivo and 24S-hydroxycholesterol levels in mice in vivo. Front Mol Neurosci. 2021;13:568641. doi: 10.3389/fnmol.2020.568641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy S, Chin SP, Sukumaran SD, Buckle MJC, Kiew LV, Chung LYC. In silico and in vitro analysis of bacoside A aglycones and its derivatives as the constituents responsible for the cognitive effects of Bacopa monnieri. PLoS One. 2015;10:1–19. doi: 10.1371/journal.pone.0126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raush E, Totrov M, Marsden BD, Abagyan R. A new method for publishing three-dimensional content. PLoS One. 2009;4:2–6. doi: 10.1371/journal.pone.0007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Barnes CA. Impact of aging on hippocampal function: Plasticity, network dynamics, and cognition. Prog Neurobiol. 2003;69(3):143–179. doi: 10.1016/s0301-0082(02)00126-0. [DOI] [PubMed] [Google Scholar]
- Schönknecht P, Lütjohann D, Pantel J, Bardenheuer H, Hartmann T, von Bergmann K, et al. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer’s disease compared to healthy controls. Neurosci Lett. 2002;324:83–85. doi: 10.1016/S0304-3940(02)00164-7. [DOI] [PubMed] [Google Scholar]
- Shafaati M, Mast N, Beck O, Nayef R, Heo GY, Björkhem-Bergman L, Lütjohann D, Björkhem I, Pikuleva IA. The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase in vitro and in vivo. J Lipid Res. 2010;51(2):318–323. doi: 10.1194/jlr.M900174-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Jia J, Tian S, Zhang H, An K, Zhu W, et al. Increased plasma level of 24S-hydroxycholesterol and polymorphism of CYP46A1 SNP (rs754203) are associated with mild cognitive impairment in patients with Type 2 diabetes. Front Aging Neurosci. 2021;13:1–11. doi: 10.3389/fnagi.2021.619916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K. Black pepper and its pungent principle-piperine: A review of diverse physiological effects. Crit Rev Food Sci Nutr. 2007;47:735–748. doi: 10.1080/10408390601062054. [DOI] [PubMed] [Google Scholar]
- Tian G, Kong Q, Lai L, Ray-Chaudhury A, Lin CL. Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer's disease. J Neurochem. 2010;113(4):978–989. doi: 10.1111/j.1471-4159.2010.06661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulhas RS, Malaviya A. In-silico validation of novel therapeutic activities of withaferin a using molecular docking and dynamics studies. J Biomol Struct Dyn. 2022;24:1–12. doi: 10.1080/07391102.2022.2078410. [DOI] [PubMed] [Google Scholar]
- Uniyal A, Mahapatra MK, Tiwari V, Sandhir R, Kumar R. Targeting SARS-CoV-2 main protease: structure based virtual screening, in silico ADMET studies and molecular dynamics simulation for identification of potential inhibitors. J Biomol Struct Dyn. 2022;40:3609–3625. doi: 10.1080/07391102.2020.1848636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R, Yadav NS, Katiyar SP, Yaguchi T, Lee C, Ahn H, et al. Molecular dynamics simulations and experimental studies reveal differential permeability of withaferin-A and withanone across the model cell membrane. Sci Rep. 2021;11:1–15. doi: 10.1038/s41598-021-81729-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Gao JH, He LH, Yu XH, Zhao ZW, Zou J, Wen FJ, Zhou L, Wan XJ, Tang CK. Fargesin alleviates atherosclerosis by promoting reverse cholesterol transport and reducing inflammatory response. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(5):158633. doi: 10.1016/j.bbalip.2020.158633. [DOI] [PubMed] [Google Scholar]
- Wang S, Chen G, Merlo Pich E, Affinito J, Cwik M, Faessel HM. Pharmacokinetics, pharmacodynamics and safety assessment of multiple doses of soticlestat in healthy volunteers. Br J Clin Pharmacol. 2022 doi: 10.1111/bcp.15225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Yan M, Wang P, Hamada K, Yan N, Hao H, et al. Withaferin A in the treatment of liver diseases: progress and pharmacokinetic insights. Drug Metab Dispos. 2021;50(5):685–693. doi: 10.1124/dmd.121.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6:254–264. doi: 10.1007/s13238-014-0131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.