

Abstract
Background and Objectives
Variants of the apolipoprotein E (APOE) gene are the greatest known risk factors for sporadic Alzheimer disease (AD). Three major APOE isoform alleles, ε2, ε3, and ε4, encode and produce proteins that differ by only 1–2 amino acids but have different binding partner interactions. Whereas APOE ε2 is protective against AD relative to ε3, ε4 is associated with an increased risk for AD development. However, the role of APOE in gene regulation in AD pathogenesis has remained largely undetermined. Extracellular vesicles (EVs) are lipid bilayer–delimited particles released by cells to dispose of unwanted materials and mediate intercellular communication, and they are implicated in AD pathophysiology. Brain-derived EVs (bdEVs) could act locally in the tissue and reflect cellular changes. To reveal whether APOE genotype affects EV components in AD brains, bdEVs were separated from patients with AD with different APOE genotypes for parallel small RNA and protein profile.
Methods
bdEVs from late-stage AD brains (BRAAK stages 5–6) from patients with APOE genotypes ε2/3 (n = 5), ε3/3 (n = 5), ε3/4 (n = 6), and ε4/4 (n = 6) were separated using our published protocol into a 10,000g pelleted extracellular fraction (10K) and a further purified EV fraction. Counting, sizing, and multiomic characterization by small RNA sequencing and proteomic analysis were performed for 10K, EVs, and source tissue.
Results
Comparing APOE genotypes, no significant differences in bdEV total particle concentration or morphology were observed. Overall small RNA and protein profiles of 10K, EVs, and source tissue also did not differ substantially between different APOE genotypes. However, several differences in individual RNAs (including miRNAs and tRNAs) and proteins in 10K and EVs were observed when comparing the highest and lowest risk groups (ε4/4 and ε2/3). Bioinformatic analysis and previous publications indicate a potential regulatory role of these molecules in AD.
Discussion
For patients with late-stage AD in this study, only a few moderate differences were observed for small RNA and protein profiles between APOE genotypes. Among these, several newly identified 10K and EV-associated molecules may play roles in AD progression. Possibly, larger genotype-related differences exist and are more apparent in or before earlier disease stages.
Alzheimer disease (AD) is a progressive neurodegenerative disease that represents a public health crisis as the global population ages.1 AD brain pathology features extracellular neuritic plaques (NPs) formed by amyloid precursor protein (β-amyloid or Aβ) aggregation and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau.2 Variants of the apolipoprotein E (APOE) gene are the greatest known genetic risk factors for sporadic AD.3 Three major APOE isoforms exist, encoded by alleles ε2, ε3, and ε4, and produce proteins that differ by only 1–2 amino acids but have different binding partner interactions. Whereas APOE2 is protective against AD relative to APOE3, APOE4 is associated with an increased risk for the development of AD.4 In White individuals, the risk of late-onset AD is higher for APOE4 homozygosity (15-fold) and heterozygosity (2- to 3-fold), with some indication of greater risk for early-onset disease.4 APOE protein may be involved in AD development by serving as a central player in lipid homeostasis. APOE assists in intercellular lipid transfer, binding both cholesterol and low-density lipoprotein receptors.3 In addition, APOE also regulates AD signature protein Aβ deposition, aggregation, and clearance.5 The association of APOE genotypes and the clinical risk of AD has been well established, while the exact mechanism of the relationship is still a largely undermined area. Understanding the relationship between APOE genotypes and AD could shed a light on the manifold mechanisms of AD pathogenesis.
Extracellular vesicles (EVs) are lipid bilayer–delimited particles released by cells. EVs contain lipids, proteins, and RNA from the parent cell.6,7 EVs are implicated in AD pathophysiology by spreading misfolded proteins throughout the brain, including Aβ and tau.8-10 EVs are also found “at the scene of the crime”: in amyloid plaques.8 However, depending on the source cell, EVs might also block AD pathology by assisting with amyloid clearance.11,12 Dysregulation of various microRNAs was observed in serum of patients with AD,13 suggesting that the RNA cargo of EVs may also be related to AD pathogenesis.
The study of EVs and APOE is just beginning,14-16 but a rapidly growing literature suggests multiple mechanisms whereby EVs and APOE genotype might contribute to AD. APOE is prominently sorted into EVs, particularly from neurons and astrocytes.14 APOE shuttling may increase on exposure to Aβ peptides.14 In the pigment cell amyloid model system,17 APOE was associated with EVs in the multivesicular body, where it nucleated the formation of cytotoxic amyloid aggregates15 before release from the cell. APOE + EVs have recently been characterized in CSF and cell culture, with changes in the fluids of patients with AD and mild cognitive impairment.18 Studies in the periphery have also touched on the role of EVs in the context of APOE and AD.19,20 Changes in neurotropic, inflammatory, and antioxidant markers in APOE ε4 carrier plasma EVs have been proposed because they serve as predictors as early as 5 years before AD onset. Nevertheless, no systematic profile study has yet been reported on the connection between APOE genotype and EV cargoes.
We therefore obtained brain tissue of cases with late-stage AD with different APOE genotypes (ε2/3, ε3/3, ε3/4, and ε4/4) to explore changes in the molecular composition of brain-derived EVs (bdEVs) associated with APOE genotypes. We used our modification of a rigorous method for the separation of tissue EVs21,22 to obtain and characterize bdEVs in accordance with the recommendations of the Minimal Information for Studies of EVs and related initiatives of the International Society for EVs.6,23 To assess possible small RNA and protein composition changes related to APOE genotypes in the brain, we compared brain homogenate (BH), a brain-derived 10,000g pelleted extracellular fraction (10K), and further purified EV fractions. RNA sequencing and protein profiling revealed moderate differences in bdEVs (both 10K and EV)-associated small noncoding RNAs and proteins correlated with APOE genotype. The most prominent differences were observed between patients with the lowest risk (ε2/3) and highest risk (ε4/4) genotypes, but not between those with other genotypes. Several 10K and EV-associated proteins and RNAs identified in our study are implicated in pathways related to CNS function.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
Human brain tissues were obtained from the Johns Hopkins AD Research Center. All collections were approved by the Johns Hopkins University Institutional Review Board. Written informed consent was obtained from all participants (or guardians of participants) in the study.
Tissue Collection, Processing, and Approvals
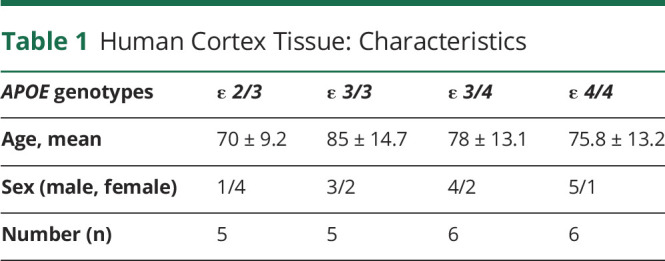
Patients with AD were diagnosed according to BRAAK24 and Consortium to Establish a Registry for Alzheimer's Disease (CERAD)25,26 criteria. At autopsy, the brain was externally examined, weighed and cut into coronal slabs, frozen on prechilled metal plates, and stored at −80°C. A mixture of temporal cortex (Brodmann area 42 and 21) and inferior parietal cortex (Brodmann area 40) tissues were used. A total of 22 AD patients with different APOE genotypes (APOE ε 2/3, ε 3/3, ε 3/4, and ε 4/4) were included (Table 1, additional information is listed in eTable 1, links.lww.com/NXG/A549). For APOE genotyping of tissue, genomic DNA was obtained using the DNeasy kit (Qiagen), and genotyping was performed using standard procedures.27
Table 1.
Human Cortex Tissue: Characteristics
Separation of EVs From Brain Tissue
bdEVs were separated from brain tissues using our published protocol.22 As stated, “before extraction, a small (∼50 mg) piece of tissue was stored at −80°C for later protein and RNA extraction. The remaining frozen tissue was weighed, briefly sliced on dry ice, and digested in 75 U/mL collagenase type 3 (Worthington #CLS-3, S8P18814) in Hibernate-E solution for 20 minutes at 37°C. The digestion was stopped by adding PhosSTOP and Complete Protease Inhibitor (SigmaAldrich PS/PI 4906837001/11697498001) solutions. The digested tissue was spun at 300g for 10 minutes at 4°C. Supernatant was spun at 2,000g for 15 minutes at 4°C. Supernatant was filtered through a 0.22 μm filter (Millipore Sigma, SLGS033SS) to remove tissue debris and was spun at 10,000g for 30 minutes at 4°C using 5 mL Ultra-Clear tubes in an AH-650 swinging-bucket rotor (Thermo Fisher; k-factor 53, acceleration and deceleration settings 9). The pellet was resuspended in 100 μL phosphate buffered saline (PBS), which was termed the “10,000g pellet” (10K). The 10,000g supernatant was then concentrated with a 100 kDa molecular weight cutoff protein concentrator (Thermo Fisher 88524) from 5 to 0.5 mL and processed by size-exclusion chromatography (SEC) (qEV original, IZON Science SP1-USD, Christchurch, New Zealand), followed by concentration by ultracentrifugation (UC) (70 minutes at 110,000g (average) at 4°C [TH-641 rotor Thermo Fisher, thinwall polypropylene tube with 13.2 mL capacity]). Supernatant was removed, and the pellet was resuspended in 100 μL PBS as purified EV fractions.22 Fractions were stored at −80°C until further use.”22
Brain Homogenate Preparation for Protein and RNA
Proteins were extracted from BHs by grinding tissue for 10 seconds using a handheld homogenizer (Kontes Pellet Pestle Motor) in cold PBS with protease inhibitor/phosphatase inhibitors. After addition of radioimmunoprecipitation assay lysis buffer (Cell Signaling Technology 9806), the mixture was sonicated at 20 kHz in an ultrasonic ice bath for 4 × 20 seconds with 10-second intervals in between. Homogenates were rotated for 2 hours at 4°C and spun for 15 minutes at 14,000g at 4°C, after which supernatants were transferred to fresh tubes and stored at −80°C.
RNA from frozen brain tissues was extracted by adding Trizol (Thermo Fisher 15596018) and homogenizing tissues with Lysing Matrix D (MP Biomedicals 116913100) in a benchtop homogenizer (FastPrep-24, MP Biomedicals) at 4.0 m/s for 20 seconds. After homogenization, the supernatant was collected and RNA was isolated by miRNeasy Mini Kit (Qiagen 217004) following the manufacturer's instructions.
Transmission Electron Microscopy
bdEV preparations (10 μL) were adsorbed to glow-discharged 400 mesh ultrathin carbon-coated grids (EMS CF400-CU-UL) for 2 minutes following our published protocol.22 Grids were rinsed 3 times with tris‐buffered saline and stained with 1% uranyl acetate with 0.05% Tylose. Grids were aspirated, dried, and immediately imaged with a Philips CM120 instrument set at 80 kV. Images were captured with an 8 megapixel AMT XR80 charge‐coupled device.
Nano-Flow Analysis
bdEV concentration and size distribution were estimated using the nanoFCM flow nanoAnalyzer (NanoFCM Co.) per manufacturer's instructions. To calibrate the instrument for concentration and size, 200 nm polystyrene beads and a cocktail of silica nanospheres (diameters of 68, 91, 113, and 151 nm, provided by NanoFCM) were used, respectively. bdEV preparations were diluted as needed (typically 1:50 dilution for 10K fractions and 1:200 dilution for EVs), and particle events were recorded for 1 minute. Particle numbers were calculated based on a calibration curve, flow rate, and side scatter intensity.
EV Surface Marker Profiling
EV surface markers were assayed using prototype S-PLEX ultrasensitive assays (Meso Scale Discovery). Each S-PLEX plates was coated with CD9, CD81, CD63 capture antibodies and one isotype IgG1 antibody. bdEV samples were diluted 20-fold and incubated with the plates at RT with continuous shaking. bdEVs captured by each antibody spot were detected by a cocktail of antibodies targeting CD9, CD81, and CD63. Assay plates were then read with MSD GOLD Read buffer B on an MSD SECTOR instrument. Signal from IgG1 isotype control and Dulbecco's phosphate-buffered saline were subsequently subtracted from signals on each detection antibody capture spot before further analysis.
RNA Extraction and Quality Control
RNA extraction and quality control were conducted following our previously published protocol22: “RNA was extracted by miRNeasy Mini Kit (Qiagen 217004) following the manufacturer's instructions. Size profiles of 10K and EV RNA were analyzed by capillary electrophoresis using a RNA 6000 Pico Kit (Agilent Technologies 5067-1513) on a Fragment Analyzer (Advanced Analytical). Total RNA and small RNA from BH were analyzed by capillary electrophoresis using a RNA 6000 Nano Kit (Agilent Technologies 5067-1511) and RNA 6000 Pico Kit.”22
Small RNA Sequencing
bdEV RNA was concentrated to 6 µL using the Savant SpeedVac Vacuum concentrator. Small RNA libraries were prepared from 50 ng of BH RNA and 5 µL of RNA from 10K and EVs using the Ion Total RNA-Seq Kit V2 (Life Technologies 4475936). Libraries were barcoded using the Ion Xpress™ RNA-Seq Barcode 1–16 Kit (Life Technologies 4471250) following the manufacturer's instructions and as previously published.28 The Agilent 2100 Bioanalyzer™ instrument and DNA 1000 chip (Agilent Technologies 5067-1504) were used to assess the yield and size distribution of the small RNA libraries (96 nt to 250 nt). Multiplexed libraries were equally pooled based on mass to a final concentration of 45 pm, prepared for sequencing using the Ion Chef system (Life Technologies 4484177), and sequenced on the Ion Torrent S5™ by Ion™ 540 chips (Life Technologies A27765).
RNA Sequencing Data Analysis
RNA sequencing data were analyzed using a pipeline as previously published22: “Original BAM files were converted into FASTQ format using picard tools (SamToFastq command). Reads shorter than 15 nt were removed from the raw FASTQ data using cutadapt software v1.18. The size-selected reads were aligned to human reference transcriptomes using bowtie software (1 mismatch tolerance) in a sequential manner. Specifically, reads were first mapped to rRNA, tRNA, RN7S, snRNA, snoRNA, scaRNA, VT-RNA, Y-RNA, as well as the mitochondrial genome. All reads that did not map to the abovementioned RNA species were aligned to human miRNA references (miRBase 22 release). The remaining reads were further aligned to protein-coding mRNAs and long noncoding RNA (lncRNA) references (GENCODE Release 29). The numbers of reads mapped to each RNA type were extracted using eXpress software based on a previous publication.”22,29 Differential gene expression between APOE4/4 and APOE2/3 was quantified using R/Bioconductor package DESeq2 as described (with significance defined as unadjusted p value <0.05).30,31
Mass Spectrometry
The mass spectrometry (MS) workflow was as previously published22: “Samples were resuspended in 1 X RIPA buffer (20 mM Tris-HCl pH7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% SDS, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin) and protease inhibitors and incubated on ice for 5 minutes. The samples were sonicated for 15 minutes in an ice water bath and centrifuged at 14,000g at 4°C for 10 minutes. The supernatant was collected and assessed for protein concentration using the microBCA assay (Thermo Fisher Scientific 23235). Three micrograms of BH and 1.5 µg of bdEV samples were buffer-exchanged prior to mass spectrometry to remove detergent. Proteins were resuspended in 8 M urea, 50 mM Tris pH = 8.3. Next, 1 µL of TCEP (tris [2-carboxyethyl] phosphine hydrochloride, 200 mM solution in water) was added to the samples and incubated for 4 hours at 21°C in a ThermoMixer (Eppendorf AG). Four microliters of 1 M IAA (iodoacetamide in water) was then added and samples were incubated in the dark at 21°C. Eight hundred microliters of 50 mM Tris (pH 8.3) and 1 μg of trypsin were then added to samples prior to overnight incubation at 37°C. Ten microliters of 10% trifluoroacetic acid was added to each sample to acidify. Samples were cleaned using stage-tips preparations using 3 plugs of Empore polystyrenedivinylbenzene (SBD-XC) copolymer disks (Sigma Aldrich, MO) for solid phase extraction. Peptides were reconstituted in 0.1% formic acid and 2% acetonitrile and loaded onto a trap column (C18 PepMap 100 μm i.d. × 2 cm trapping column, Thermo Fisher Scientific) at 5 µL/minute for 6 minutes using a Thermo Scientific UltiMate 3000 RSLCnano system and washed for 6 minutes before switching the precolumn in line with the analytical column (BEH C18, 1.7 μm, 130 Å and 75 μm ID × 25 cm, Waters). Separation of peptides was performed at 45°C, 250 nL/minute using a linear ACN gradient of buffer A (water with 0.1% formic acid, 2% ACN) and buffer B (water with 0.1% formic acid, 80% ACN), starting from 2% buffer B to 13% B in 6 minutes and then to 33% B over 70 minutes, followed by 50% B at 80 minutes. The gradient was then increased from 50% B to 95% B for 5 minutes and maintained at 95% B for 1 minute. The column was then equilibrated for 4 minutes in water with 0.1% formic acid, 2% ACN. Data were collected on a Q Exactive HF (Thermo Fisher Scientific) in Data Dependent Acquisition mode using m/z 350–1500 because MS scan range at 60,000 resolution. HCD MS/MS spectra were collected for the 7 most intense ions per MS scan at 60,000 resolution with a normalized collision energy of 28% and an isolation window of 1.4 m/z. Dynamic exclusion parameters were set as follows: exclude isotope on, duration 30 seconds, and peptide match preferred. Other instrument parameters for the Orbitrap were MS maximum injection time 30 ms with AGC target 3 × 106 and MSMS for a maximum injection time of 110 ms with AGC target of 1 × 105.”22
Proteomics Data Analysis
Proteomics data analysis was performed as previously published22: “Human protein sequences were downloaded from the Uniprot database (uniprot.org/proteomes/UP000005640) and used as the database for the search engine. Common Repository of Adventitious Proteins was used as the potential laboratory contaminant database. Protein identification was performed using the proteomics search engine Andromeda built in to Maxquant V 1.16.0. Trypsin with a maximum of 2 missed cleavages was used as the cleavage enzyme. Carbamidomethyl of cysteine was set as fixed modification, and oxidation of methionine was set as variable modification. The false discovery rate (FDR) was set to 1%. The label-free quantification was performed with match between runs using a match window of 0.7 minutes. Large label-free quantification (LFQ) ratios were stabilized to reduce the sensitivity for outliers. For human data sets, data scaling was performed using the cyclic loess method, and scaled data were visualized with a principal component analysis (PCA) plot. For differential abundance analysis, nested factorial design was set up for the analysis, where each subtype of the disease was nested within the main disease category, and contrasts for the main categories were computed by averaging the subtypes (targets were considered significant for p value <0.05).”22
Identification of statistically significantly enriched protein pathways was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG).32 Cellular component annotations of identified proteins were enriched by STRING.33 Only those categories with FDR < 0.05 were included for analysis.
Statistical Analysis
Statistical significance of particle count and size distribution differences between APOE genotypes was determined by the 2-tailed Welch t test.
Data Availability
Nucleic acid sequencing data were deposited with the Gene Expression Omnibus, accession GSE159541. Proteomics data files are available on request. We have submitted all relevant details of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV200126).23
Results
Separation of bdEVs From AD and Control Brain Tissue
Following the protocol illustrated in Figure 1A, we separated bdEVs (10K and EVs) from the brain tissue of patients with late-stage AD with different APOE genotypes (Table 1). A small amount (∼50 mg) of each tissue was set aside to produce BH to assess protein and RNA profiles of the source material. After enzymatic digestion and initial filtering of the remaining tissue, 10,000g ultracentrifuged pellets were collected and termed “10K” as an intermediate product of EV separation. The 10K supernatant was then separated by SEC22 and concentrated into a more pure EV preparation. Fractions were processed for characterization, including small RNA profiling and proteomics.
Figure 1. bdEV Enrichment and Characterization From AD Patients With Different APOE Genotypes.

(A) Workflow for bdEV enrichment, small RNA sequencing, and proteomics. After digestion, centrifugation, and filtration steps, 10,000g pellets were collected and defined as the 10K fraction. SEC was applied to 10,000g supernatants to enrich bdEVs. RNA and proteins from BH, 10K, and bdEVs were then isolated and subjected to small RNA sequencing and mass spectrometry. (B) 10K and bdEVs from AD brain tissues with different APOE genotypes (ε2/3, ε3/3, ε3/4, and ε4/4) were visualized by negative staining TEM (scale bar = 100 nm). TEM is representative of 10 images taken of each fraction from 5 independent human tissue samples. (C) Particle concentrations of 10K and EV fractions of AD patients with different APOE genotypes were measured by nanoFCM flow nanoAnalyzer. Particle concentration for each group was normalized by tissue mass (per 100 mg). (D) Size distributions of 10K and EV fractions of AD patients with different APOE genotypes were measured by nanoFCM flow nanoAnalyzer and calculated as particles in a specific size bin vs total detected particles in each sample (percentage). Data are presented as mean ± SD. ns: no significant difference (p > 0.05) between the 2 APOE genotype groups by the 2-tailed Welch t test. AD = Alzheimer disease; bdEVs = brain-derived EVs; SEC = size-exclusion chromatography; TEM = transmission electron microscopy.
Basic bdEV Characterization
Transmission electron microscopy revealed oval and round particles in brain-derived 10K and EV fractions from patients with different APOE genotypes (ε2/3, ε3/3, ε3/4, and ε4/4) that were consistent with EV morphology (Figure 1B). Particle concentration per 100 mg tissue input and particle size distribution were determined by nano-flow cytometry measurement. No significant particle count differences (Figure 1C) or size distribution (Figure 1D) shifts were observed for 10K and EV samples from patients with different APOE genotypes. GO ontology by STRING was used to determine the cellular component enrichment of proteins recovered from 10K and EV fractions. The top 10 cellular component terms (ranked by adjusted p value) (eFigure 1A, links.lww.com/NXG/A549) enriched in both 10K and EVs were EV-related terms such as EV, extracellular exosome, vesicle, extracellular space, and extracellular region. The protein number enriched for these terms was higher in EVs than 10K, as expected, and consistent with greater purity of the EV fraction. Different terms were shown between 10K and EVs: only 10K was enriched for intracellular terms, including cytosol and cytoplasm, while the cytoplasmic vesicle, plasma membrane, and cell projection terms were uniquely shown in EVs. No obvious differences were observed in 10K and EVs from brains with different genotypes. EV membrane proteins CD9, CD81, and CD63 were all detected on the EV surface (eFigure 1B, links.lww.com/NXG/A549). No significant differences were observed for 10K, but ε3/4 EVs showed higher CD63 signal compared with ε2/3 (eFigure 1B, links.lww.com/NXG/A549).
Moderate Effects of APOE Genotype on Small RNA and Protein Contents of bdEVs and BH
PCA was conducted on small RNA profiles (eFigure 2A, links.lww.com/NXG/A549) and proteins (eFigure 2B, links.lww.com/NXG/A549) of 10K, EV, and BH from patients with different APOE genotypes (ε2/3, ε3/3, ε3/4, and ε4/4). No obvious separation was observed between genotypes, which indicates that APOE genotype did not cause major changes in the molecular composition of bdEVs or the brain tissue of late-stage AD brains. In addition, differential expression analysis for individual molecules revealed that most differentially expressed molecules were found only in the comparison of the lowest risk (ε2/3) and the highest risk (ε4/4) patients. Any miRNAs that differed between other genotypes were also different between ε2/3 and ε4/4. Protein differences were found only between ε2/3 and ε4/4. We therefore focused on the comparison between ε2/3 and ε4/4.
Differential Expression of miRNAs in the Brain Tissue and bdEVs Related to APOE Genotypes
Examining individual miRNAs, 30 miRNAs (10K) and 6 miRNAs (EVs) differed significantly (p value <0.05) between ε4/4 and ε2/3 (Figure 2A). Of these, 9 miRNAs in 10K and 2 miRNAs in EVs (miR-379-5p and miR-199a-5p) differed by more than 2-fold (Figure 2A). A number of these miRNAs were also differentially expressed between other APOE genotypes (eFigure 3A–B, links.lww.com/NXG/A549). For example, miR-483-5p was more abundant in 10K vesicles from ε4 carriers (ε3/4 and ε4/4 groups) compared with non-ε4 carriers (ε2/3 and ε3/3 groups) (eFigure 3A, links.lww.com/NXG/A549). In EVs, miR-199a-5p was more abundant in ε4/4 compared with both ε2/3 and ε3/3 carriers (eFigure 3B, links.lww.com/NXG/A549). Previous studies have identified potential involvement of these miRNAs in AD pathogenesis, and their potential target genes are listed in eTable 2, links.lww.com/NXG/A549. To assess whether AD-associated miRNA differences in the extracellular space reflect overall changes in the brain tissue, we compared miRNA fold changes of 10K and EVs with BH. We highlighted miRNAs that were differentially abundant only in BH (blue dots), only in 10K or EVs (red dots), or in both bdEVs and BH (purple dots) (Figure 2B). More than 90 miRNAs were differentially abundant in BH (blue dots), while relatively few miRNAs differed in EVs or 10K (Figure 2B). In 10K, we found 14 differentially expressed miRNAs that reflected changes in BH, but only 3 reflective miRNAs were identified in EVs (Figure 2C). This suggests that many miRNAs may be differentially expressed in brain tissues, but that these differences are not well reflected in 10K or EVs.
Figure 2. bdEV miRNAs With Differential Expression Between ε4/4 and ε2/3.

(A) Volcano plots showing 10K (left) and EV (right) miRNA log2FC and p value for APOE ε4/4 vs APOE ε2/3 carriers. Thresholds for 2-fold change and p value <0.05 are indicated by dashed lines. Significant changes are indicated with different colors. Gray: nonsignificant (Not Sig), black: p value < 0.05. (B) miRNA log2FC between APOE ε4/4 and APOE ε2/3 in BH were plotted against 10K (upper left) and EVs (upper right). Dashed lines indicate log2FC of one (up or down). Colored dots indicate miRNAs with p values < 0.05 in BH (blue), in 10K/EV (red), or in both BH and 10K/EVs (purple), or unchanged transcripts (grey). Dot size represents the mean normalized abundance of individual miRNAs. (C) Venn diagrams of miRNAs differentially expressed between APOE ε4/4 and APOE ε2/3 in 10K (bottom left) vs BH, and EVs vs BH (bottom right). bdEVs = brain-derived EVs; BH = brain homogenate; EVs = Extracellular vesicles; Log2FC = log2 fold changes.
Differential Expression of tRNAs in the Brain Tissue and bdEVs Related to APOE Genotypes
Since incorporation of non-miRNA noncoding RNAs into EVs can also be modulated by external stimuli imposed on cells,34,35 we examined whether individual snoRNAs, snRNAs, Y-RNAs, and tRNAs were differentially expressed between APOE ε2/3 and ε4/4. There were no differences in the snoRNA, snRNA and Y-RNA content of EV or 10K fractions (data not shown), but 2 tRNA-Gly isodecoders (10K) and tRNA-Met, tRNA-Lys, tRNA-Thr, and tRNA-Leu (EVs) were significantly differentially abundant by 2-fold or more (p value <0.05) (Figure 3A). These apparent differences did not reflect changes in BH (Figure 3B).
Figure 3. bdEV tRNAs With Differential Expression Between ε4/4 and ε2/3.

(A) Volcano plots showing 10K (left) and EV (right) tRNA Log2FC and p values for APOE ε4/4 vs APOE ε2/3. Thresholds for 2-fold change and p value < 0.05 are indicated by dashed lines. Significant changes are indicated with different colors. Grey: nonsignificant (Not Sig), black: p value < 0.05. (B) tRNA Log2FC between APOE ε4/4 and APOE ε2/3 carriers for BH were plotted against 10K (left) and EVs (right). Dashed lines indicate log2FC of one (up or down). Colored dots indicate tRNAs with p values < 0.05 in BH (blue), in 10K/EV (red), or in both BH and 10K/EVs (purple), or unchanged transcripts (gray). Dot size represents the mean normalized abundance of individual tRNAs. bdEVs = brain-derived EVs; BH = brain homogenate; EVs = Extracellular vesicles; Log2FC = log2 fold changes.
Proteomics and Regulatory Pathways in bdEVs: APOE ε4/4 and APOE ε2/3
Mass spectrometry identified 685, 311, and 426 proteins in BH, 10K, and EVs, separately (eTable 3, links.lww.com/NXG/A549). Comparing ε4/4 and ε2/3, 71.4% (10K, Figure 4A left), 70.3% (EVs, Figure 4A right), and 81.5% (BH, eFigure 4A, links.lww.com/NXG/A549) of proteins were detected for both genotypes. KEGG pathway analyses of APOE ε4/4-“unique” proteins in 10K and EVs corresponded with metabolism-related pathways (carbon, glutathione, and galactose metabolism) (Figure 4B). KEGG pathway analysis of APOE ε2/3-“unique” proteins in 10K and EVs did not return any enriched pathways (data not shown), although numerous pathways were enriched in BH of APOE ε2/3 carriers (eFigure 4B, links.lww.com/NXG/A549). Moreover, both ε4/4 and ε2/3 contained enriched proteins in BH that have been reported to be involved in AD, Huntington disease, and oxidative phosphorylation (eFigure 4B, links.lww.com/NXG/A549). The Log2 (LFQ intensity) of individual proteins differentially expressed between ε4/4 vs ε2/3 in 10K and EVs (Figure 4C) and BH (eFigure 4C, links.lww.com/NXG/A549) is shown in Figure 4D. There were no differences when comparisons were made between other genotype groups (eFigure 5A–B, links.lww.com/NXG/A549). In contrast to miRNAs and tRNAs, there were more dysregulated proteins in the 10K and EV fractions than in BH. The potential functions of these proteins in neurodegenerative disease progression are summarized in eTable 4, links.lww.com/NXG/A549. Most of these proteins have known involvement in metabolism and mitochondrial function regulation.
Figure 4. bdEV Proteins With Differential Expression in APOE ε4/4 and APOE ε2/3 Carriers.

(A) Venn diagrams of proteins identified in 10K and EVs from APOE ε4/4 and APOE ε2/3 carriers. (B) Significant pathways of APOE ε4/4 10K and EV proteins according to the Kyoto Encyclopedia of Genes and Genomes (KEGG). (C) Volcano plots showing 10K (left) and EV (right) protein Log2FC and p values between APOE ε4/4 and APOE ε2/3 carriers. Thresholds for 2-fold change and p value <0.05 are indicated by dashed lines. Significant changes are indicated with different colors. Gray: non-significant (Not Sig), black: p value < 0.05. (D) Expression level of proteins differentially expressed between APOE ε4/4 and APOE ε2/3 in 10K (left) and EVs (right). Data are presented as mean log2 (LFQ intensity) ± SD. *p values < 0.05 between APOE ε4/4 and APOE ε2/3 from proteomics analysis. bdEVs = brain-derived EVs; EVs = extracellular vesicles; LFQ = label-free quantification; Log2FC = log2 fold changes.
Discussion
Although APOE genotype is one of the greatest known genetic risk factors for sporadic AD, it is not known whether APOE variants are associated with bdEV contents. We thus performed small RNA sequencing and proteomics of BHs, “10K” pelleted extracellular fraction, and a purified EV fraction from patients with AD with different APOE genotypes (ε2/3, ε3/3, ε3/4, and ε4/4). We observed no differences in total particle concentration or morphology in 10K or EVs, and overall small RNA and protein profiles of 10K, EVs, and tissue also did not associate with distinct APOE genotypes. A small number of apparently differentially abundant miRNAs, tRNAs, and proteins were identified between the most “distant” genotypes: ε2/3 and ε4/4 based on p value. These results indicate, at most, a modest association of APOE genotype with small RNA and protein content of late-stage AD brains and bdEVs.
Does APOE Genotype Associate With bdEV Production?
bdEVs are released as a mixture of various subtypes and from diverse cells. Both the biogenesis and cargo loading of specific EV subtypes could be affected by AD or APOE genotypes because the APOE ε4 allele is reported to be involved in neuronal, endosomal, and lysosomal system dysfunction.36 Previously, reduced EV counts were reported to be associated with the ε4 allele in nondiseased brain tissues of human and mouse (BRAAK level 0–2).16 Although no differences in particle count were observed in our study, we examined only late-stage AD samples (BRAAK stage 5–6, CERAD B-C).3,37 Direct comparisons of young, unaffected brain with brains from early and late disease stages may be needed.
Noncoding RNAs and APOE Genotype
We found several miRNAs and tRNAs, but not other noncoding RNAs, which seemed to differ with APOE genotype. For microRNAs, differences in the 10K and EV fractions were not as pronounced as in brain tissue. Nevertheless, miRNAs we identified from 10K and EVs were previously shown to be involved in regulating Aβ aggregation,38 anxiety behavior,39 neuronal cell loss,40 and neuronal development.41 As for tRNAs, the number of APOE genotype–associated differences in 10K and EV fraction were comparable with those in brain tissues, but the individual RNAs were inconsistent between the different sources. However, our findings are limited because only a small number of genotyped samples were examined and they were all from late-stage patients.
bdEV Proteins and APOE Genotype
Comparing ε4/4 and ε2/3 genotypes, and in contrast with RNA results, more proteins were differentially abundant in the 10K and EV fractions than in source brain tissue. Proteins that were detected only in ε4/4 samples, as well as those differentially abundant between ε4/4 and ε2/3, are in mitochondrial and metabolic pathways. For example, higher levels of tricarboxylic acid cycle protein mitochondrial aconitate hydratase (ACO2) were previously found in multiple brain regions of patients with AD42 and in this study appeared to be enriched in ε4/4 brain compared with that in ε2/3. While mounting evidence suggests that mitochondrial and metabolic dysfunction contributes to AD by compromising the energy supply,43-46 further exploration of protein modification and enzymatic activity of these proteins in bdEVs could unveil mechanisms of APOE-regulated energy metabolism deficits in AD. Antioxidant and neurogenesis pathway proteins also differed between ε4/4 and ε2/3 patients. Antioxidant defense protein deglucase DJ1 (PARK7) was enriched in ε4/4 compared with that in ε2/3, and neuroprotective proteins such as γ-enolase (ENO2)47,48 and brevican core protein49,50 were downregulated in ε4/4 vs ε2/3. Because oxidative damage and neurogenesis/differentiation pathway impairment are often implicated in the progression of neurodegenerative diseases, our study indicates that APOE genotypes may also affect AD progression through these pathways and extracellular proteins such as those in bdEVs may help to indicate these processes.
In summary, bdEV small RNA and protein content from patients with late-stage clinical AD with different APOE genotypes have been compared with the content of matched source tissue. Even when comparing the most extreme genotype groups in our cohort (ε4/4 and ε2/3), only modest molecular differences were observed in the brain tissue or bdEVs. However, several differences in miRNAs, tRNAs, and proteins were identified, and several of these have plausible roles in AD. Our findings have several limitations: First, only patients with late-stage AD and only a small number of samples were included in this study. Second, because APOE may affect the contents of EVs before AD disease onset, future work should examine non-AD controls with different APOE genotypes, along with patients with AD. In addition, given that female individuals have a higher risk of AD, biological sex should be examined more carefully. Last, because of the small sample size, we used raw p value and fold change to cast a relatively wide net for targets, and this may have introduced false positives. Our results should thus be further explored using larger cohorts and expanded to address questions of biological sex, AD stage, and preonset differences between genotypes.
Acknowledgment
V. Mahairaki and K.W. Witwer gratefully acknowledge support from the Richman Family Precision Medicine Center of Excellence in Alzheimer Disease including helpful comments and advice from founder and director Constantine Lyketsos. Thanks to: Kenneth Pienta, Johns Hopkins University School of Medicine, for access to the nanoFCM flow nanoAnalyzer platform; Mitchell Science Writing for manuscript editing and citation formatting; and the La Trobe University Comprehensive Proteomics Platform. The authors also thank members of the Witwer and Retrovirus Laboratories, Johns Hopkins University School of Medicine, and various members of the International Society for Extracellular Vesicles for valuable discussions and support.
Glossary
- AD
Alzheimer disease
- APOE
apolipoprotein E
- bdEVs
brain-derived EVs
- BH
brain homogenate
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- EVs
Extracellular vesicles
- FDR
false discovery rate
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LFQ
label‐free quantification
- MS
mass spectrometry
- NFTs
neurofibrillary tangles
- NPs
neuritic plaques
- PBS
phosphate buffered saline
- PCA
principal component analysis
- SEC
size-exclusion chromatography
Appendix. Authors
Study Funding
This work was supported in part by grants from the US NIH: AI144997 (to K.W. Witwer, with support for T.A.P. Driedonks), MH118164 and AG057430 (to V. Mahairaki and K.W. Witwer), by UG3CA241694 (to K.W. Witwer), supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director, by the National Health and Medical Research Council of Australia (GNT1132604 to A.F. Hill); and by JHU Alzheimer's Disease Research Centers NIH P30 AG 066507 and BIOCARD NIH U19AG033655.
Disclosure
The authors report no disclosures relevant to the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Alzheimer's Association. Alzheimer's disease facts and figures. Alzheimers Dement. 2016;12(4):459-509. [DOI] [PubMed] [Google Scholar]
- 2.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430(7000):631-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C-C, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349-1356. [PubMed] [Google Scholar]
- 5.Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81(4):740-754. Accessed February 23, 2022. /pmc/articles/PMC3983361/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. Accessed May 6, 2021. pubmed.ncbi.nlm.nih.gov/30637094/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci. 2016;113(8):E968-E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajendran L, Honsho M, Zahn TR, et al. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103(30):11172-11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez-González R, Kim Y, Miller C, Pacheco-Quinto J, Eckman EA, Levy E. Extracellular vesicles: where the amyloid precursor protein carboxyl-terminal fragments accumulate and amyloid-β oligomerizes. FASEB J. 2020;34(9):12922-12931. Accessed February 23, 2022. pubmed.ncbi.nlm.nih.gov/32772431/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker S, Polanco JC, Götz JJ. Extracellular vesicles containing P301L mutant tau accelerate pathological tau phosphorylation and oligomer formation but do not seed mature neurofibrillary tangles in ALZ17 mice. J Alzheimers Dis. 2016;54(3):1207-1217. [DOI] [PubMed] [Google Scholar]
- 11.Yuyama K, Sun H, Sakai S, et al. Decreased amyloid- pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J Biol Chem. 2014;289(35):24488-24498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An K, Klyubin I, Kim Y, et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol Brain. 2013;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng L, Doecke JD, Sharples RA, et al. , Australian Imaging, Biomarkers and Lifestyle AIBL Research Group. Prognostic serum miRNA biomarkers associated with Alzheimer's disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry. 2015;20(10):1188-1196. Accessed February 23, 2022. pubmed.ncbi.nlm.nih.gov/25349172/. [DOI] [PubMed] [Google Scholar]
- 14.Nikitidou E, Khoonsari PE, Shevchenko G, Ingelsson M, Kultima K, Erlandsson A. Increased release of apolipoprotein E in extracellular vesicles following amyloid-β protofibril exposure of neuroglial co-cultures. J Alzheimers Dis. 2017;60(1):305-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Niel G, Bergam P, Di Cicco A, et al. Apolipoprotein E regulates amyloid formation within endosomes of pigment cells. Cell Rep. 2015;13(1):43-51. [DOI] [PubMed] [Google Scholar]
- 16.Peng KY, Pérez-González R, Alldred MJ, et al. Apolipoprotein E4 genotype compromises brain exosome production. Brain. 2019;142(1):163-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watt B, van Niel G, Raposo G, Marks MS. PMEL: a pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013;26(3):300-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Keene CD, Peskind ER, et al. Cerebrospinal fluid particles in Alzheimer disease and Parkinson disease. J Neuropathol Exp Neurol. 2015;74(7):672-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khedher MRBen, Haddad M, Laurin D, Ramassamy C. Effect of APOE ε4 allele on levels of apolipoproteins E, J, and D, and redox signature in circulating extracellular vesicles from cognitively impaired with no dementia participants converted to Alzheimer's disease. Alzheimes Dement (Amst). 2021;13(1):e12231. Accessed October 21, 2021. onlinelibrary.wiley.com/doi/full/10.1002/dad2.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khedher MRBen, Haddad M, Laurin D, Ramassamy C. Apolipoprotein E4–driven effects on inflammatory and neurotrophic factors in peripheral extracellular vesicles from cognitively impaired, no dementia participants who converted to Alzheimer's disease. Alzheimers Dement Transl Res Clin Interv. 2021;7(1):e12124. Accessed October 21, 2021. onlinelibrary.wiley.com/doi/full/10.1002/trc2.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vella LJ, Scicluna BJ, Cheng L, et al. A rigorous method to enrich for exosomes from brain tissue. J Extracell Vesicles. 2017;6(1):1348885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Cheng L, Turchinovich A, et al. Influence of species and processing parameters on recovery and content of brain tissue-derived extracellular vesicles. J Extracell Vesicles. 2020;9(1):1785746. Accessed May 6, 2021. tandfonline.com/action/journalInformation?journalCode=zjev20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Deun J, Mestdagh P, Agostinis P, et al. EV-Track: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14(3):228-232. [DOI] [PubMed] [Google Scholar]
- 24.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-259. [DOI] [PubMed] [Google Scholar]
- 25.Mirra SS, Heyman A, McKeel D, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurol Neurol. 1991;41(4):479-486. [DOI] [PubMed] [Google Scholar]
- 26.Morris JC, Heyman A, Mohs RC, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurol Neurol. 1989;39(9):1159-1165. [DOI] [PubMed] [Google Scholar]
- 27.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31(3):545-548. [PubMed] [Google Scholar]
- 28.Cheng L, Hill AF. Small RNA library construction for exosomal RNA from biological samples for the ion torrent PGMTM and ion S5TM system. Methods Mol Biol. 2017;1545:71-80. [DOI] [PubMed] [Google Scholar]
- 29.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10(1):71-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiou NT, Kageyama R, Ansel KM. Selective export into extracellular vesicles and function of tRNA fragments during T cell activation. Cell Rep. 2018;25(12):3356-3370.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Driedonks TAP, van der Grein SG, Ariyurek Y, et al. Immune stimuli shape the small non-coding transcriptome of extracellular vesicles released by dendritic cells. Cell Mol Life Sci. 2018;75(20):3857-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's Disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157(1):277-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caselli RJ, Reiman EM, Locke DEC, et al. Cognitive domain decline in healthy apolipoprotein E ε4 homozygotes before the diagnosis of mild cognitive impairment. Arch Neurol. 2007;64(9):1306-1311. [DOI] [PubMed] [Google Scholar]
- 38.Hébert SS, Horré K, Nicolaï L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(7):6415-6420. Accessed June 17, 2021. pnas.org/content/105/17/6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marty V, Labialle S, Bortolin-Cavaillé ML, et al. Deletion of the miR-379/miR-410 gene cluster at the imprinted Dlk1-Dio3 locus enhances anxiety-related behaviour. Hum Mol Genet. 2016;25(4):728-739. [DOI] [PubMed] [Google Scholar]
- 40.Sabirzhanov B, Zhao Z, Stoica BA, et al. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34(30):10055-10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han K, Gennarino VA, Lee Y, et al. Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev. 2013;27(5):485-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zahid S, Oellerich M, Asif AR, Ahmed N. Differential expression of proteins in brain regions of Alzheimer's disease patients. Neurochem Res. 2014;39(1):208-215. [DOI] [PubMed] [Google Scholar]
- 43.Hauptmann S, Scherping I, Dröse S, et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30(10):1574-1586. [DOI] [PubMed] [Google Scholar]
- 44.Leuner K, Hauptmann S, Abdel-Kader R, et al. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer's disease? Antioxidants Redox Signal. 2007;9(10):1659-1675. [DOI] [PubMed] [Google Scholar]
- 45.Müller WE, Eckert A, Kurz C, Eckert GP, Leuner K. Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer's disease-therapeutic aspects. Mol Neurobiol. 2010;41(2-3):159-171. [DOI] [PubMed] [Google Scholar]
- 46.Cadonic C, Sabbir MG, Albensi BC. Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol Neurobiol. 2016;53(9):6078-6090. [DOI] [PubMed] [Google Scholar]
- 47.Hattori T, Takei N, Mizuno Y, Kato K, Kohsaka S. Neurotrophic and neuroprotective effects of neuron-specific enolase on cultured neurons from embryonic rat brain. Neurosci Res. 1995;21(3):191-198. [DOI] [PubMed] [Google Scholar]
- 48.Hafner A, Obermajer N, Kos J. γ-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem J. 2012;443(2):439-450. [DOI] [PubMed] [Google Scholar]
- 49.Hockfield S, Kalb RG, Zaremba S, Fryer H. Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harb. 1990;55:505-514. [DOI] [PubMed] [Google Scholar]
- 50.Bandtlow CE, Zimmermann DR. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. 2000;80(0):1267-1290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Nucleic acid sequencing data were deposited with the Gene Expression Omnibus, accession GSE159541. Proteomics data files are available on request. We have submitted all relevant details of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV200126).23