

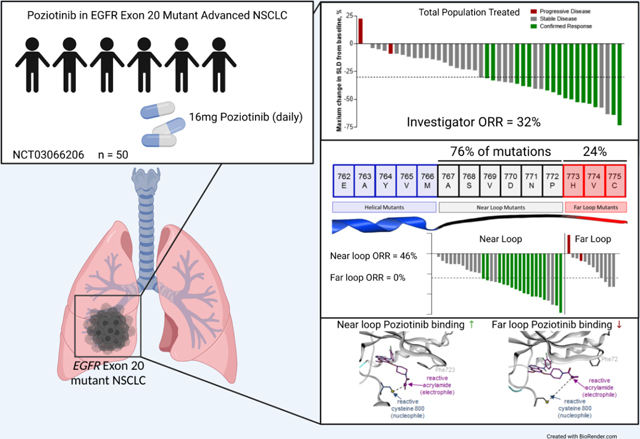
Summary
We report a phase II study of 50 advanced NSCLC patients with point mutations or insertions in EGFR exon 20 treated with poziotinib (NCT03066206). The study achieved its primary endpoint, with a confirmed objective response rate (ORR) of 32% and 31% by investigator and blinded independent review, respectively, with a median progression-free survival of 5.5 months. Using preclinical studies, in silico modelling and molecular dynamics simulations, we found that poziotinib sensitivity was highly dependent on the insertion location, with near loop insertions (amino acids A767 to P772) being more sensitive than far loop insertions, an observation confirmed clinically with an ORR of 46% and 0% observed in near vs far loop, respectively (p=0.0015). Putative mechanisms of acquired resistance included EGFR T790M, MET amplifications, and epithelial to mesenchymal transition (EMT). Our data demonstrate that poziotinib is active in EGFR exon 20 mutant NSCLC, although this activity is impacted by insertion location.
Keywords: Non-small cell lung carcinoma, epidermal growth factor receptor, exon 20 insertions
In Brief
Elamin et al. show that poziotinib is active in EGFR exon 20 mutant non-small cell lung cancer. The activity of poziotinib is impacted by insertion location in exon 20 with near loop insertion being more sensitive than far loop insertions. Poziotinib acquired resistance is mediated via EGFR-dependent and independent mechanisms.
Graphical Abstract
Introduction
Approximately 15% of patients with non-small cell lung cancer (NSCLC) harbor activating mutations in the epidermal growth factor receptor (EGFR) gene, the majority of which are ‘classical’ sensitizing EGFR mutations (exon 19 deletions and L858R point mutations). In recent studies, tyrosine kinase inhibitors (TKIs) such as osimertinib, dacomitinib, and afatinib provide dramatic clinical benefit for these patients, with an objective response rates (ORR) of 70–80% and median progression free survival (mPFS) of 11–19 months in treatment naïve patients (Mok et al., 2018; Soria et al., 2018; Wu et al., 2014). However, approximately 10% of EGFR-mutant NSCLC tumors have mutations (often in-frame insertions) within exon 20 of EGFR, and are generally resistant to EGFR TKIs approved for classical mutations, with the exception of insertions at amino acid A763(Robichaux et al., 2018; Vasconcelos et al., 2020; Vyse and Huang, 2019; Yasuda et al., 2013) and the S768I point mutation. Historical data for patients with EGFR exon 20 insertion mutations have shown that overall response rates are 0–10% to treatment with first generation TKIs erlotinib, gefitinib, or the second generation TKI, afatinib (Beau-Faller et al., 2014; Naidoo et al., 2015; Robichaux et al., 2018; Yang et al., 2020; Yang et al., 2015; Yasuda et al., 2012). Recently, mobocertinib, an irreversible EGFR TKI, received FDA approval for patients with platinum pretreated NSCLC harboring EGFR exon insertions based on an ORR of 28% and median PFS of 7.3 months (Zhou et al., 2021). The EGFR/MET bispecific monoclonal antibody amivantamab has also demonstrated activity in patients with EGFR exon 20 insertion NSCLC, with an ORR of 40%, and median PFS of 8.3 months, and recently received FDA approval for this population (Park et al., 2021). Patients with exon 20 mutant NSCLC typically receive modest benefit from other current standard of care therapies, with a mPFS of 2–6 months in patients treated with platinum doublet chemotherapy in the first line setting (Beau-Faller et al., 2014; Byeon et al., 2019; Naidoo et al., 2015; Yang et al., 2020), and less benefit from immunotherapy or docetaxel in the setting of platinum-refractory disease (Gainor et al., 2016; Garon et al., 2014; Lisberg et al., 2018; Mazieres et al., 2019; Negrao et al., 2021). Further, mechanisms of acquired resistance have yet to be explore for this subset of EGFR mutations. Therefore, there is a substantial need for new therapies for this subset of patients with NSCLC, and studies to explore acquired resistance in this patient subset.
Previously, using a multidisciplinary approach including in silico, in vitro, and in vivo testing of exon 20 mutant models, we identified the structural features of exon 20 insertions that limit TKI binding and identified that poziotinib was an effective inhibitor for these mutations owing to the small size and flexibility of the inhibitor (Robichaux et al., 2018; Robichaux et al., 2019). More recently, we classified insertions in EGFR exon 20 into insertions in αC-helix (which are broadly sensitive to approved EGFR TKIs) and insertions occurring in the C-terminal loop of the αC-helix, with the latter group further classified into near-and far-loop insertions (Robichaux et al., 2021).
Here we report the clinical efficacy, safety, potential mechanisms of resistance, and molecular determinants of response for poziotinib from an open-label, phase II study of patients with advanced EGFR exon 20 mutant NSCLC. We found that poziotinib was a clinically active and tolerable exon 20 inhibitor; and, that acquired resistance to poziotinib was associated with both EGFR-dependent and -independent mechanisms of resistance in patients and preclinical models, reminiscent of mechanisms observed in patients with classical EGFR mutations. Furthermore, we demonstrate that in preclinical models, poziotinib sensitivity is highly dependent on the location of the insertion within exon 20, and observation confirmed in our clinical analysis. This supports that insertion position may be a determinant of drug response for poziotinib and potentially other TKIs.
Results
Patients Characteristics
We investigated the clinical activity of poziotinib treatment in a phase II investigator-initiated study in patients with NSCLC harboring EGFR exon 20 mutations (NCT03066206). Initial results from the first eleven patients in this cohort were reported previously (Robichaux et al., 2018). The primary endpoint of the study was the ORR according to RECIST version 1.1 (Eisenhauer et al., 2009), with a pre-defined ORR of 30% or greater considered to be clinically meaningful. Patients received daily 16mg orally poziotinib until objective disease progression and could continue beyond progression for as long as clinical benefit was observed, as judged by the investigator and in the absence of other discontinuation criteria (patient withdrawal, adverse event). Clinical trial design including primary and secondary endpoints, timeline of follow-up scans, inclusion criteria, and dose reduction plan are included in Fig. S1 and STAR Methods. Fifty patients with EGFR exon 20 mutant metastatic NSCLC were enrolled and, baseline characteristics are shown in Table S1. The patient population was heavily pre-treated: 94% (n = 47) of patients had received at least one prior systemic therapy, and 68% (n = 34) of patients received two or more prior lines of therapy, including six patients (12%) that had received four or more prior lines of treatment. Notably, 88% (n = 44) of patients had received previous platinum-containing chemotherapy, and 34% (n = 17) of patients had received previous EGFR TKI treatment (Table S1 and Table S2). Lastly, 94% of patients (n = 47) had an EGFR exon 20 insertion mutations, and three patients had either individual or compound point mutations in exon 20. A list of patients’ mutations is provided in Table S3.
Efficacy and Safety
Among the intention-to-treat (ITT) patient population (n = 50), the investigator-assessed, confirmed ORR was 32.0% (95% CI: 20.7 to 45.8, n = 16), and the disease control rate was 84.0% (95% CI: 71.5 to 92.0, Table 1, Fig. 1A,). In a subset of 42 patients that consented for retrospective blinded independent central review (BICR), the confirmed ORR was 31.0% (95% CI: 19.1 to 46.0, n = 13, Table 1, Fig. S2). Only two of the radiologically-evaluable patients (n = 44), as assessed by the investigator, had evidence of progressive disease on the first restaging scans. One of the two patients had a germline EGFR T790M mutation and somatic EGFR H773R point mutation. The second patient had shrinkage of target lesions by 9% but also had new lesions and, therefore, was deemed to have progressive disease as their best response. Notably, both patients had concurrent TP53 mutation, an aberration reported to be associated with poor outcomes in classical EGFR mutations (Canale et al., 2020; Skoulidis and Heymach, 2019). Both patients with exon 20 point mutations other than T790M had confirmed partial responses. The median duration of investigator-assessed response was 8.6 months (95% CI: 3.7 to 19.3, Fig. 1B), and the mPFS was 5.5 months (95% CI: 5.4 to 10.4) (Fig. 1C). The PFS rate at six and twelve months was 43% (95% CI: 30 to 60) and 29% (95% CI 18 to 46), respectively (Fig 1C). In the subset of patients with prior platinum-based chemotherapy treatment (n = 44), the mPFS was 5.5 months (95% CI: 5.0 to 9.4, Fig. S3A) and the investigator-assessed confirmed ORR was 34.1% (95% CI: 21.9 to 48.9, n=15, Fig. S3B). In patients with prior platinum-based chemotherapy treatment who consented for BICR (N = 36), the confirmed ORR was 33.3% (95% CI: 20.2 to 49.7, n=12, Fig. S3C). Among 14 (28%) patients with baseline brain metastases, 12 were treated previously with local radiotherapy and two hadn’t received prior brain radiation. The intracranial ORR and DCR were 14% (n=2) and 86% (n=12), respectively. Both patients who achieved intracranial PR had received prior brain radiation at least 12 weeks prior to starting poziotinib. At data cutoff in the ITT population (April 1, 2021), all patients were off study treatment. The median duration of poziotinib treatment, irrespective of dose interruptions, was 5.8 months (IQR 3.6–12.3; range 0.1–32.5, Fig. 2). After radiological progression, 32 patients were still alive and 50.0% (n = 16) of patients continued poziotinib treatment post progression, and the median time on poziotinib after progression was 3.2 months (IQR 2.5–5.6; range 1.5–11.3, Fig. 2). At the time of data cutoff, 56% (n = 28) of patients had died. Median overall survival (mOS) was 19.2 months (95% CI: 11.8 to 24.1, Fig. S4A), and all deaths were considered related to the disease.
Table 1.
Response to Treatment by RECIST 1.1 Criteria
| Investigator Assessed (n=50) | BICR (n=42) | |
|---|---|---|
| Partial response | 16 (32%) | 13 (31%) |
| Stable disease* | 26 (52%) | 19 (45%) |
| Disease progression╬ | 6 (12%) | 8 (19%) |
| Not evaluable♯ | 2 (4%) | 2 (5%) |
| Objective response | 32% (95% CI:20.7 to 45.8) | 31% (95% CI: 19.1 to 46.0) |
| Disease control | 84% (95% CI:71.5 to 91.6) | 76.0% (95% CI:61.5 to 86.5) |
Stable disease greater than or equal to 8 weeks
Disease progression includes 4 early deaths
No evaluable follow-up assessments
BICR: Blinded independent central review
Figure 1. Tumor response to poziotinib in EGFR exon 20 mutant NSCLCA.

A. Waterfall plot of maximum percent decrease from baseline in the sum of diameters of target tumors based on investigator assessment in patients with evaluable disease. The dashed line at −30% represents the cutoff for RECIST response. Patients harboring point mutation(s) are indicated. SLD, sum of the longest diameters. B. Kaplan-Meier curve for estimated duration of response. Duration of response was defined as time from first objective response per RECIST 1.1 to objective disease progression or death from any cause. Median duration of response was 8.6 months (95% CI: 3.7 to 19.3) C. Kaplan-Meier curve for progression-free survival of patients treated with poziotinib (N=50). Progression free survival was defined as the time from the administration of the first dose of poziotinib to objective disease progression or death from any cause. Median progression-free survival was 5.5 months (95% CI: 5.4 to 10.4).
See also Figures S1-S3 and Tables S1-S3.
Figure 2. Duration of poziotinib treatment.

Duration of poziotinib treatment in all treated patients (n=50). Each bar each bar denotes a single patient.
See also Figures S4 and S5.
We also evaluated plasma circulating free DNA (cfDNA) prior to and during treatment in a subgroup of patients (n = 25). When comparing the variant allelic frequency (VAF) of EGFR exon 20 mutations in plasma prior to poziotinib initiation and at eight weeks of treatment, there was a significant drop in VAF at eight weeks overall (p=0.0016, Fig. S4B). Patients with partial response (PR) and stable disease (SD) as best response both had decreases in median VAF (−98% and - 94%, respectively), whereas the patient with progressive disease (PD) had an increase in VAF of 167%. The reduction in VAF trended towards a greater decrease in patients with PR compared to patients with SD (Fig. S4C). Taken as a whole, these data support that ctDNA VAF may serve as a useful biomarker of response for EGFR exon 20 NSCLC, as previously observed for patients with classical EGFR mutations (Lam et al., 2021; Moding et al., 2020; Tran et al., 2021).
The majority of patients experienced treatment-related adverse events (Table 2). The most common events were diarrhea (92%), skin rash (90%), oral mucositis (68%), paronychia (68%), and dry skin (60%). Most of the treatment related adverse events were grade 1 or 2. There were no grade 4 or 5 treatment related events. Adverse events leading to dose reduction occurred in 72% (n = 36) of patients, and of these patients, 22 patients (44%) had one dose reduction while 14 patients (28%) had two dose reductions. Median PFS was similar in all poziotinib-treated patients and those with dose reductions (Fig. S4D), suggesting that dose modification enabled patients to continue with poziotinib treatment without adversely impacting clinical outcomes. Therapy was discontinued because of treatment-related adverse events in only three patients (6%). Two patients discontinued due to grade 3 diarrhea, and one patient discontinued due to grade 3 skin rash. The median relative dose intensity (RDI) was 76.0% (range 46.2–100). Skin rash was the most common grade 3 toxicity seen in this study (34%). It typically presented in the form of acneiform eruption, exfoliative dermatitis, or overlapping of the two. Management of theses skin toxicities, in our experience, benefits from close collaboration with a dermatologist and patients were typically offered follow up with a dermatologist starting during the first month of treatment. For the exfoliative dermatitis, a combination of medium potency topical steroids (e.g. triamcinolone 0.1% cream BID) to intact skin and silver sulfadiazine to eroded or ulcerated skin can provide symptomatic relief, decrease potential infections, and allow patients to continue poziotinib. Management of acneiform eruptions is provided in (Fig. S5). Taken together with efficacy data above, these data demonstrate that poziotinib is a clinically active and tolerable inhibitor of EGFR exon 20 mutant NSCLC, although dose reductions were common.
Table 2.
Adverse Events*
| Adverse Event | Grade 1 | Grade 2 | Grade 3 | All Grades |
|---|---|---|---|---|
| Number of patients (percent) | ||||
| Diarrhea | 20 (40%) | 15 (30%) | 11 (22%) | 46 (92%) |
| Skin rash | 15 (30%) | 13 (26%) | 17 (34%) | 45 (90%) |
| Paronychia | 27 (54%) | 2 (4%) | 5 (10%) | 34 (68%) |
| Oral mucositis | 28 (56%) | 5 (10%) | 1 (2%) | 34 (68%) |
| Dry skin | 26 (52%) | 4 (8%) | - | 30 (60%) |
| Anorexia | 20 (20%) | - | - | 20 (40%) |
| Alopecia | 18 (36%) | 1 (2%) | - | 19 (38%) |
| Hypokalemia | 14 (28%) | - | 3 (6%) | 17 (34%) |
| Nausea | 11 (22%) | - | 3 (6%) | 14 (28%) |
| Vomiting | 13 (26%) | 3 (6%) | - | 16 (32%) |
| Fatigue | 7 (14%) | 3 (6%) | - | 10 (20%) |
| Weight loss | 6 (12%) | - | 2 (4%) | 8 (16%) |
| Pruritus | 7 (14%) | - | - | (14%) |
Listed are adverse events that were reported in at least 10% of the 50 study patients and were deemed by the investigators to be related to treatment. No grade 4 or 5 treatment-related adverse events were reported.
A higher response rate is observed in patients with near-loop vs far-loop mutations
While the majority of patients had clinical benefit from poziotinib treatment, responses were heterogenous with some patients having durable clinical benefit while other patients’ disease quickly progressed. Therefore, we sought to investigate if the heterogeneity of patient responses to poziotinib treatment were related to the location of the exon 20 insertion. To this end, we analyzed the location of patient mutations (Fig. 3A) and patient outcomes (Fig. 3B). Patients with insertions occurring in the near loop region proximal to the αC-helix (amino acids A767-P772) had a significantly higher ORR than patients with insertions in the distal far loop region (H773-C775) (46% vs 0%; P=0.0015, Fig. 3B-D). In addition, patient RECIST responses directly correlated with mutation location (R=0.32, p=0.03, Fig. 3E). While the median progression free survival did not differ significantly between patients with near- and far-loop insertions (Fig. 3F), the PFS hazard ratio (0.76), the six month PFS rate of 47% (95% CI: 33 to 67) vs 30% (95% CI: 12 to 76); and the twelve month PFS rate of 32% (95% CI: 19 to 52) vs 20% (95% CI: 6 to 68) all favored patients with near-loop insertions having better outcomes compared to patients with far-loop insertions. While the power of the PFS analysis for patients with near- and far loop mutations is limited by the sample size, the higher PFS rate, response rate, and better RECIST responses in patients with near-loop mutations suggest that poziotinib is more active in patients bearing near-loop EGFR exon 20 insertions and point mutations, although clinical activity was clearly observed in both groups.
Figure 3. Far loop mutants are lessensitive to poziotinib.

A. Schematic representation of the first 15 amino acids of exon 20 of EGFR divided by structural features corresponding to amino acids. Mutations are listed with the frequency observed in this study. Bars are representative of overall frequency of variants at the indicated amino acid. B-C. (B) Waterfall plot and (C) bar graph of evaluable patient confirmed response to poziotinib divided by mutation location. Objective response rate (ORR) and disease control rate (DCR) are shown for the intent to treat population for near and far. Statistical differences were determined by Chi-square test. D. Bar graph of RECIST responses for patients’ best objective response divided by amino acid location. Bars are representative of average RECIST response + SEM, and dots are representative of individual evaluable patients (n=44). Statistical differences were determined by two-tailed students’ t-test. E. Dot plot of patients’ best RECIST response after poziotinib treatment plotted against amino acid location of mutation (n=44). F. Kaplan-Meier plot of progression free survival of patients with near loop and far loop mutants in the intent to treat population. Long-Rank Mantel-Cox approach was used to determine p-value. B-F. Near loop: A767-P772, n=32 patients and far loop: H773-C775, n=12 patients.
EGFR exon 20 insertion location affects receptor conformation and drug binding
To determine the structural underpinnings of how the location of EGFR exon 20 insertions impacted the conformation of the drug binding pocket and drug binding, microsecond time scale molecular dynamics simulations (MDS) were carried out as previously described (Robichaux et al., 2019) to model the noncovalent enzyme–inhibitor complex of poziotinib with EGFR S768dupSVD and EGFR H773insNPH, near- and far-loop mutants, respectively. RMSD plots revealed the noncovalent complex to be structurally stable and pointed to conformational transition between states during the simulation (Fig. S6A). Principal component analysis of MD trajectories revealed sampling of conformational states not previously observed in EGFR X-ray ensembles in addition to sampling conformations previously observed in X-ray ensembles (Fig. S6B-C). Taken together, these data showed that the mutant structures remained structurally stable during the simulation, and these mutations impacted the conformational dynamics of EGFR.
Previous studies by our lab showed that HER2 mutations with a smaller drug binding pocket were more drug resistant (Robichaux et al., 2019), therefore we calculated the drug binding pocket volume of EGFR S768dupSVD and H773insNPH and found that the H773insNPH mutant had a significantly smaller drug binding pocket (p<0.0001, Fig. 4A). Next, cluster analysis was carried out on the MD trajectories and representative structures identified from clusters representing the most frequently sampled conformational sub-states of S768dupSVD and H773insNPH were investigated for structural differences in regions of the receptor known to effect drug binding interactions including the P-loop and the β3-αC loop which is N-terminal of the αC-helix (Callegari et al., 2018). Within the P-loop we found that EGFR S768dupSVD had a closed P-loop conformation compared to EGFR H773insNPH which had an extended P-loop conformation (Fig. 4B). This change in the orientation of the P-loop altered the orientation of the phenyl ring within amino acid F723, which is known to play an active role in TKI stabilization, drug binding, and selectivity (Fig. 4B, red arrow) (Callegari et al., 2018). Calculations of free energy of binding on the MDS trajectories using MM/GBSA and MM/PBSA end-point methods were completed for poziotinib in S768dupSVD and H773insNPH mutants. Poziotinib was predicted to bind to both exon 20 insertions, however, poziotinib had a lower free energy of binding (better affinity) for S768dupSVD with a delta of 2–3 kCal/mol for the near mutant (Fig. 4C).
Figure 4. Exon 20 insertion location effects the orientation of the P-loop and drug-receptor interactions.

A. Box plot of binding pocket volumes (Å3) of poziotinib bound to S768dupSVD and H773insNPH over the duration of the simulation. B. In silico model of the most conformations of S768dupSVD (blue) and H773insNPH (purple) in poziotinib bound simulations. Dashed line shows differences in orientation of the loop connecting the α-c-helix and the ß3 strand. Red arrow shows differences in orientation of P-loop highlighting F723 that interacts with most TKIs in the binding cleft. The dark blue arrow indicates the location of the SVD insertion (light blue) at S768, and the purple arrow indicates the location of the NPH insertion (light blue) at H773. C. Table of binding energy calculations in kCal/mol for poziotinib for indicated mutations. D. In silico representation of poziotinib (purple) bound to S768dupSVD (light blue) demonstrate a small distance (black dashed line) between reactive acrylamide group (purple arrow) and reactive cysteine (dark blue arrow). E. In silico representation of poziotinib (purple) bound to H773insNPH (teal) demonstrate a larger distance (black dashed line) between reactive acrylamide group (purple arrow) and reactive cysteine (dark grey arrow). F. Distance between the reactive acrylamide (electrophile) and reactive cysteine (nucleophile) during MDS with poziotinib and S768dupSVD (black) and H773insNPH (red) over time. G. Bar graph of average IC50 values of Ba/F3 cell lines expressing EGFR exon 20 point mutation or insertion mutations treated with poziotinib for 72 hours. Dots are representative of average IC50 value of each cell line determined in triplicate, and line is representative of average IC50 value of all point mutations or insertion mutations + SEM. Differences between groups was determined by two-sided students’ t-test. H. Dot plots of average IC50 values of Ba/F3 cell lines expressing various EGFR exon 20 insertion mutations (n = 24 cell lines) treated with poziotinib compared to amino acid residue of EGFR exon 20 insertion mutation. Red dashed line indicates the average IC50 value of Ba/F3 cells expressing WT EGFR + 10ng/ml EGF. Dots are representative of the average IC50 value determined in biological triplicate. Data was fit to a linear regression model, and two-tailed Pearson correlation was determined.
See also Figure S6.
Structural analysis of the MDS trajectories of poziotinib with S768dupSVD showed that the closed P-loop conformation pushed the acrylamide warhead toward the reactive cysteine residue placing the compound in in proximity of C800 (C797 in WT EGFR, Fig. 4D). The extended conformation of the P-loop in H773insNP, by contrast, resulted in a rotation of the acrylamide group away from C800 (Fig. 4E).
In addition, analysis of between the thiol group of the C800 of S768dupSVD and the C2 alkene carbon of the poziotinib acrylamide showed a peak distribution at 6.69 Å, whereas the peak distribution between C800 of H773insNPH and the reactive acrylamide of poziotinib had a bimodal distribution with peak distributions around 7Å and 9.5Å (Fig. 4F), demonstrating that the reactive carbon of poziotinib is more frequently in closer proximity of C800 when bound to the S768dupSVD mutation compared to the H773insNPH mutation. Finally, we generated a panel of Ba/F3 cell lines expressing 24 different exon 20 insertion mutations and eight exon 20 point mutations and screened these cell lines against poziotinib. Since we previously reported that helical exon 20 insertions (e.g A763insFQEA) were broadly sensitive to EGFR TKIs and had sensitivity similar to that of classical EGFR mutations (and were hence classified as “classical-like” mutations) (Robichaux et al., 2021), these mutations were excluded from the analysis. We observed that point mutations were significantly more sensitive than insertions to poziotinib inhibition (p = 0.003, Fig 4G), and poziotinib had a positive correlation between drug sensitivity and mutation location in vitro (R = 0.67, p=0.0003, Fig. 4H). Taken together these data demonstrate that location of the insertion at the C-terminal end of the αC-helix influences the orientation of distinct residues of the P-loop that stabilize EGFR TKIs and influence drug binding affinities. Moreover, these data support a structural mechanism for the observed differential sensitivity of poziotinib in near-loop mutants compared to far-loop mutants due to the structural features of the compounds.
Poziotinib Acquired Resistance Mechanisms
To identify potential mechanisms of acquired resistance to poziotinib, we developed multiple models of acquired resistance preclinically using cell lines expressing different EGFR exon 20 mutations. As a starting point, we cultured Ba/F3 cells stably expressing various EGFR exon 20 mutation in escalating doses of poziotinib until resistance clones developed and were then subjected to Sanger sequencing of EGFR. Acquired poziotinib-resistant Ba/F3 cells were found to harbor EGFR T790M and EGFR C797S mutations (Fig. S7A). Further, Ba/F3 cells engineered to co-express EGFR exon 20 mutations and T790M or C797S were resistant to poziotinib (Fig. 5A). Structural modeling of EGFR exon 20 insertion (D770_N771insNPG) and T790M with poziotinib showed distinct interference between the terminal halogenated benzene ring of poziotinib and the gatekeeper region of EGFR (Fig. 5B). The methionine gatekeeper substitution at residue 790 was predicted to cause a steric clash, displacing poziotinib away from the hinge region of the receptor (Fig. 5B), consistent with the reduced sensitivity of this mutation to the drug.
Figure 5: Poziotinib resistance may be driven by EMT but retains sensitivity to SAC inhibitors in preclinical models.

A. IC50 values of Ba/F3 cells expressing the indicated EGFR exon 20 mutations. Bars are representative of the average IC50 value ± SEM. Dotted red line indicates the average IC50 value of Ba/F3 cells expressing WT EGFR. All IC50 values were determined in at least three independent replicates. B. In silico modelling of EGFR D770insNPG (purple) with T790M (orange) and poziotinib (blue). The methionine at 790 (orange) displaces the terminal halogenated ring of poziotinib (blue) away from the hydrophobic cleft, increasing the distance between the quinazoline core of poziotinib and methionine 793 (yellow, dashed lines). C. Heatmap of protein expression from YUL-0019 (YUL) and YUL-0019 poziotinib resistant (PR) cell lines as determined by RPPA analysis of EMT associated proteins. D. Dot plots of relative fold change in protein expression determined by RPPA for indicated EMT associated markers in YUL-0019 parental (YUL) and YUL-0019 PR cell lines (PR), normalized to YUL parental controls. Dots are representative of fold change in protein expressing for biological replicates for YUL-0019 parental cells and the average of n=4 replicates for each poziotinib-resistant cell line (PR1-PR-8). Bars are representative of average ± standard deviation (SD), and p-values were determined by two-sided, unpaired students’ t-test. E. Average IC50 values of indicated inhibitors as determined by the colony formation assay after 14 days. Bars are representative of average ± SEM (n=2).
See also Figure S7.
To validate if EMT is a mechanism of EGFR-independent resistance in preclinical models of EGFR exon 20 mutant NSCLC, similar to classical EGFR mutant NSCLC (Byers et al., 2013; Nilsson et al., 2020b; Zhang et al., 2012), we generated a panel of poziotinib-resistant exon 20 mutant NSCLC cell lines as previously described (Nilsson et al., 2020a) using the YUL-0019 parental cell line which harbors an exon 20 insertion (N771del insFH) and is sensitive to poziotinib in vitro and in vivo (Robichaux et al., 2018). Protein was isolated from poziotinib sensitive and resistant cell lines analyzed by high throughput reverse phase protein array (RPPA) analysis and analyzed for markers of EMT. Differential protein expression associated with an EMT was observed in poziotinib-resistant NSCLC cell lines (Fig. 5C,D). Poziotinib-resistant cells had increased AXL expression (Byers et al., 2013; Ghiso et al., 2017; Zhang et al., 2012) (p<0.001), and down regulated E-cadherin (Byers et al., 2013; Suda et al., 2011) (p=0.002) and p-YAP S127 (Ghiso et al., 2017; Hsu et al., 2016; Kurppa et al., 2020; Lee et al., 2018; Nilsson et al., 2020b) (p<0.001); and interestingly, unlike classical EGFR mutant NSCLC, ß-catenin and ZEB1 were not upregulated in poziotinib-resistant cells (Fig. 5C,D, Fig. S7B). EMT markers were validated by Western blotting in a subset of cell lines (Fig. S7C), further supporting EMT as a mechanism of resistance to poziotinib in EGFR exon 20 mutant NSCLC. Previous reports demonstrated that EGFR TKI resistant cells are sensitive to spindle assembly complex (SAC) inhibitors (Kurppa et al., 2020; Nilsson et al., 2020b; Shah et al., 2019); therefore, we tested if poziotinib-resistant EGFR exon 20 mutant NSCLC cell lines were sensitive to SAC inhibitors, and we found that poziotinib-resistant cell lines were sensitive to multiple SAC inhibitors including CDK, PLK, KSP, and Aurora kinase inhibitors (Fig. 5E).
We next investigated whether mechanisms of resistance identified in preclinical models were also observed in patients treated with poziotinib. To do this, we analyzed matched baseline and on progression samples from 23 patients where samples were available. Possible resistance mechanisms were identified in 14 patient (60.8%) samples. Eleven patients (47.8%) had potential EGFR-independent mechanisms of resistance, and at least four patients had EGFR-dependent resistance mechanisms (17.4%), including three patients (13.0%) with acquired EGFR gatekeeper, T790M, mutations (Fig. 6A, a list of matched pre-poziotinib and on progression tumor samples analysis provided in Table S4). Interestingly, the C797S mutation, which also causes resistance to other covalent inhibitors such as osimertinib (Ercan et al., 2015; Thress et al., 2015), and to poziotinib in preclinical models, was not observed in patients. There were also acquired mutations observed within exon 20, including V774A and D770A. Notably, co-expressing EGFR V774A and D770A with exon 20 insertions in Ba/F3 cells did not result in poziotinib resistance (Fig S8A-B), additionally we observed an acquired EGFR V1128I in one patient. EGFR V1128I impact on poziotinib sensitivity was not tested in vitro In addition, a 42-year-old female with metastatic adenocarcinoma of the lung presenting with an EGFR exon 20 A767_V769dupASV insertion mutation was treated on this trial with a confirmed partial response for 13.8 months. The patient subsequently developed cancer progression on poziotinib with kidney as the only site of progression and underwent a nephrectomy and continued poziotinib treatment. Transcriptomic analysis from the resected tumor demonstrated a high epithelial to mesenchymal transition (EMT) score (Byers et al., 2013) of 1.05 (positive score indicates a mesenchymal phenotype whereas negative score indicates an epithelial phenotype). By comparison, a treatment-naïve EGFR exon20 (H773dupH) tumor from TCGA cohort showed an EMT score of 0.114 (Fig S8C), indicating that resistance may be associated with a mesenchymal phenotype in at least some cases. Further, a cell line was generated from the resected kidney tissue (MDA-004K). The resulting cell line was resistant to pharmacological inhibition of EGFR including poziotinib (IC50 values = 3.5μM – >10μM, Fig S8D-E). By both RT-PCR and western blotting, we found that the cell line had elevated expression of mesenchymal markers and reduced expression of epithelial markers, except E-cadherin, compared to the poziotinib sensitive, patient derived cell line, YUL-0019(Nilsson et al., 2020a; Robichaux et al., 2018) (Fig. 6B,C). Taken together, these data suggest that EMT may be a mechanism of resistance to poziotinib and that patients with EGFR exon 20 mutant NSCLC share at least some common mechanisms of resistance as patients with classical EGFR mutant NSCLC (e.g. EGFR T790M gatekeeper mutations, MET amplification), although novel potential mechanisms are observed as well.
Figure 6: Resistance to poziotinib is driven by both EGFR-dependent and –independent mechanisms.

A. Potential poziotinib-acquired resistance mechanisms identified in 14 out of 23 patients with matched pre-poziotinib and on disease progression samples. Each column represents a patient. The letters on top of each column denotes the primary exon 20 mutation as follows: A: H773_V774VdupHV, B: D770_N771insG, C: S768_D770dupSVD, D: H773_V774insAH, E: A767_V769dupASV, F: D770_N771dupDN, G: H773dupH, H: P772_H773dupPH, I: P772_H773insPNP. The left-side column lists the alterations acquired at resistance. Red box: mutation, blue box: amplification. Objective response to poziotinib is shown in the bottom row. Green box: partial response, orange box: stable disease. B. Relative mRNA expression of epithelial and mesenchymal markers determined by RT-PCR from indicated cell lines. Bars are representative of average ± standard deviation (SD) fold change in mRNA expression normalized to GAPDH and YUL-0019 cell line expression, and p-values were determined by two-sided unpaired t-tests. C. Western blot of epithelial and mesenchymal markers from YUL-0019 and MDA-004K cell lines.
Discussion
EGFR exon 20 insertions exhibit de novo resistance both clinically and preclinically to first-, second-, and third-generation TKIs (Arcila et al., 2013; Kosaka et al., 2017; Massarelli et al., 2013; Oxnard et al., 2013; Yasuda et al., 2012; Yasuda et al., 2013). Previously, based on 3D modeling of EGFR D770insNPG and other mutations, we observed that conformational changes induced by these insertions sterically hinder access to the drug binding pocket and contribute to this resistance. Furthermore, based on preclinical studies, we found that the smaller size, increased halogenation, and flexibility of poziotinib give it greater specificity for exon 20 mutant EGFR (Robichaux et al., 2018) than approved EGFR TKIs, prompting clinical studies of the drug. Here we report the full results of a Phase II investigator-initiated study of poziotinib for the treatment of patients with NSCLC harboring EGFR exon 20 mutations and identify structural response determinants and putative mechanisms of resistance to poziotinib in patients.
Previous studies have demonstrated that EGFR exon 20 mutations have differential sensitivity to EGFR inhibitors and that helical mutations are more similar to classical EGFR TKIs than other exon 20 insertion mutations in structure and sensitivity to first-, second-, and third generation TKIs. Here we show that near-loop and far-loop mutations are distinct in both drug sensitivity and structural dynamics with poziotinib. Using Ba/F3 cell lines expressing 24 different exon 20 loop insertions, we show that poziotinib have a direct correlation between drug activity and insertion location, and that near-loop insertions are more sensitive to poziotinib. While exon 20 insertions are located away from the drug binding pocket, MDS revealed that the location of the insertion caused marked differences in conformations of distinct regions of the receptor known to effect drug binding interactions, and that the near-loop insertions adopted a conformation that oriented the reactive acrylamide group of poziotinib into closer proximity with the C800 residue with which it forms covalent linkage, compared with the far-loop insertions. In the clinical trial, we found that the likelihood of response was highly dependent on the location of the exon 20 insertion, and that there was an inverse correlation between distance of the insertion from the αC-helix and sensitivity to the drug both preclinically and patients. Specifically, an ORR of 46% was observed in near-loop mutants compared to 0% in the far-loop mutations.
In this study we find that 32% (16/50) and 31% (13/42) of patients with EGFR exon 20 mutant NSCLC treated with poziotinib achieved a confirmed objective response as assessed by investigator and independent central review, respectively, meeting the pre-determined primary endpoint of this study (30% ORR). In addition, the median duration of response was 8.6 months, and median progression free survival was 5.5 months. In the subset of patients with platinum-refractory disease, the independent central review confirmed ORR was 33.3% (12/36) while the investigator-assessed ORR was 34.1% (15/44) with a median progression free survival of 5.5 months. In comparison, in molecularly unselected patients with platinum-refractory NSCLC, standard second-line chemotherapy, docetaxel, has an ORR of 7%–13% and median PFS of 2–4 months (Hanna et al., 2004; Shepherd et al., 2000). Similarly, retrospective studies have reported that patients with non-classical EGFR mutant NSCLC had a mPFS of 2.7–3.7 months to second-line immune checkpoint (PD-1/PDL-1) blockade (Mazieres et al., 2019; Negrao et al., 2021). Poziotinib displayed a toxicity profile that is characteristic of EGFR TKIs with the most frequent adverse events observed including diarrhea, skin rash, paronychia and oral mucositis. These adverse events were generally manageable with dose reduction and interruption. The rate of dose reduction for poziotinib treated patients was 72% compared to 66% and 53% with dacomitinib (Mok et al., 2018) and afatinib (Wu et al., 2014), respectively. Together, these data demonstrate that poziotinib is a clinically active inhibitor of EGFR exon 20 mutations and has a similar toxicity profile as other FDA-approved second-generation EGFR TKIs. Given the frequent dose reductions and the short half-life of the drug(Kim et al., 2018), twice-daily dosing regimens are now being studied; preliminary data suggests that this may improve tolerability by reducing peak drug concentrations while maintaining inhibitor trough concentrations with comparable or higher efficacy (Le X, 2021; Sacher et al., 2021).
Preliminary results have been reported recently from a multicenter phase II trial of poziotinib in NSCLC patients with EGFR exon 20 insertions (ZENITH20, NCT03318939). In the initial 115 patients treated at a starting dose of 16 mg daily, the DCR was 68.7%, the tumor shrinkage rate was 65%, and the ORR was 14.8% in the ITT population (19.3% in the evaluable population) with a median duration of response of 7.4 months (Le et al., 2020). The difference observed in ORR, between these two studies may be explained by several factors. Our study allowed patients with EGFR exon 20 point mutations while ZENITH20 excluded patients with point mutations, and exon 20 point mutations (except T790M) are more sensitive than insertion mutations. Of note, in this study, the two patients with exon 20 point mutations (V769L and G719A/S768I) both had confirmed partial responses and long duration of response (~20 months). Second, as the dose intensity of poziotinib appears to be higher in the current study, the difference may reflect more aggressive management of adverse events, particularly skin toxicities. As an example, patients in the current study were routinely scheduled to be evaluated by a dermatologist during the first cycle of treatment, a practice not commonly followed in the setting of multicenter studies. Importantly, it appears that modifying the dosing regimen may improve tolerability and efficacy; in a cohort of ZENITH20, patients were treated with poziotinib on 8-mg twice daily dose and had an ORR of 31.6% (Le X, 2021), which is comparable to our findings presented here. Notably, when administered at 8 mg and 6 mg twice daily, poziotinib resulted in grade 3 or higher TRAEs in 26% and 16% patients, respectively, which compared favorably with 50% of patients receiving it at 16 mg daily, and 44% of patients receiving 12 mg daily.
Here, we showed that acquired resistance to poziotinib in preclinical models of EGFR exon 20 was associated with both EGFR-dependent mechanisms, including acquired T790M and C797S mutations, and EGFR-independent mechanisms such as EMT. Notably, T790M, but not C797S, mutations were detected in patients resistant to poziotinib treatment. In addition, signal by-pass through Met amplifications, PI3K mutations was also observed in patients but not in preclinical models. Our analysis of a patient-derived model of poziotinib resistance indicated that the resistant cells had undergone EMT as indicated by increased expression of mesenchymal markers ZEB1, vimentin, and Snail. Moreover, poziotinib-resistant cells displayed increased expression of the glutathione peroxidase GPX4 (Viswanathan et al., 2017). Previous studies have indicated that tumor cells that have undergone EMT may have an enhanced GPX4-dependency and that GPX4 protects mesenchymal cells from ferroptotic cell death. There are a variety of potential pharmacological approaches to exploit the EMT to overcome resistance including SHP2 and AXL inhibitors (Davis et al., 2014) in combination with an EGFR exon 20 inhibitor. Similar to previous preclinical studies of classical EGFR acquired TKI resistance cells that have undergone EMT (Nilsson et al., 2020b), poziotinib-resistant EGFR exon 20 mutant NSCLC cell lines were also resistant to other all other EGFR inhibitors, but retained sensitivity to SAC inhibitors, suggesting combinations of poziotinib with drugs targeting the SAC complex such as aurora kinase inhibitors may warrant further investigation in patents with EGFR exon 20 mutant NSCLC similar to studies being conducted in patients with classical EGFR mutations (NCT04479306, NCT05017025, NCT03455829).
In conclusion, these data demonstrate that poziotinib is a clinically active and tolerable inhibitor of EGFR exon 20 insertion mutations with meaningful clinical activity in platinum-refractory patients, where limited therapeutic options are currently available, and that mechanisms of resistance to poziotinib in patients with EGFR exon 20 mutant NSCLC at least in part overlap with reported mechanisms of resistance to other EGFR inhibitors in patients with classical EGFR mutant NSCLC. Poziotinib is undergoing further clinical development in an international multicenter trial. Finally, our data demonstrate that the location of the exon 20 insertion impacts drug sensitivity in preclinical models, a finding consistent with the higher responses observed in our study in the near-loop vs far loop insertions. This study highlights that exon 20 mutations, particularly insertion mutations, are heterogeneous and more detailed location information (e.g. helical, near-loop, or far-loop) may help guide further testing and treatment selection for EGFR exon 20 -directed kinase inhibitors.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Requests for additional information, regents, and resources should be directed and will be fulfilled by the Lead Contact John V. Heymach (JHeymach@mdanderson.org).
Materials availability
Plasmids generated in this study will be made available upon request and completion of a Material Transfer Agreement.
Data and code availability
The patient-derived data reported in this study cannot be deposited in a public repository because of legal restrictions. To request access, contact John V. Heymach (JHeymach@mdanderson.org). A detailed proposal including a scientific description needs to be submitted to the lead author and internal IRB approval and the completion of a Material Transfer Agreement is required. Processed data used in this manuscript is fully provided. Clinical trial description for NCT03066206: https://clinicaltrials.gov/ct2/show/NCT03066206?term=Poziotinib&rank=6.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
Patients were eligible if they were at least 18 years of age and had histologically or cytologically confirmed, locally advanced or metastatic NSCLC (stage IIIB and IV), measurable disease (using CT, PET-CT or MRI) as defined by Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1 guidelines, Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1. The study originally had two independent cohorts; EGFR cohort (cohort 1, reported here) and HER2 cohort (cohort 2, not reported here). Eligible patients for the EGFR cohort (cohort 1) had treatment naïve or previously treated (with any number of therapy lines) NSCLC with documented EGFR exon 20 mutation by one of the following CLIA certified tests: OncoMine Comprehensive Assay (OCA), Guardant360 Assay (using plasma), or FoundationOne Assay or by an FDA approved device using cobas® EGFR mutation test v2 or therascreen EGFR RGQ PCt kit. Previously untreated patients are eligible only if the EGFR exon 20 mutation was confirmed using an FDA approved device: cobas® EGFR Mutation Test v2 or therascreen® EGFR RGQ PCR Kit prior to study enrollment. Prior treatment with an EGFR TKI was allowed including TKIs with reported specific EGFR exon 20 insertion activity. Eligible mutations included D770_N771insSVD, D770_N771insNPG, V769_D770insASV, H773_V774insNPH, or any other exon 20 in-frame insertion or point mutation excluding acquired T790M. Patients with CNS metastases were eligible if the disease was asymptomatic, stable, and did not require steroids for at least 4 weeks before the first dose of poziotinib. Patients had adequate organ function.
Key exclusion criteria included acquired EGFR T790M mutation or any other acquired EGFR exon 20 mutation (patients with coexisting primary EGFR exon 20 and germline T790M mutations were eligible); treatment with chemotherapy, investigational agent, or other anticancer drugs within 14 days of the first dose of poziotinib; uncontrolled illness including, but not limited to, uncompensated respiratory, cardiac, hepatic, or renal disease, active infection (including hepatitis B, hepatitis C, HIV, and active clinical tuberculosis), or renal transplant; ongoing or active infection, symptomatic congestive heart failure, unstable angina pectoris, cardiac arrhythmia, active peptic ulcer disease or gastritis, or psychiatric illness/social situations that would limit compliance with study requirements; history of another primary malignancy within 2 years prior to starting study treatment, except for adequately treated basal or squamous cell carcinoma of the skin or cancer of the cervix in situ; cardiac ejection fraction <50% by either echocardiogram or multi-gated acquisition (MUGA) scan.
The protocol (Text S1) was approved by MD Anderson Cancer Center Institutional Research Ethics Board (IRB protocol ID 2016–0783). All patients provided written informed consent before study procedures, sampling, and analyses, and the study was done in accordance with the Declaration of Helsinki. This study is registered with ClinicalTrials.gov, NCT03066206.
Cell lines
BA/F3 cells were cultured in RPMI containing 10% FBS, 1% penicillin/streptomycin, and 10ng/ml recombinant mIL-3. MDA-004K were cultured in RPMI + 10% FBS at 37°C. YUL-0019 and YUL-PR1–8 were cultured in RPMI containing 10% FBS and 1% penicillin/streptomycin. Phoenix 293T-amphi were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin.
METHOD DETAILS
Study design and treatment
This is an investigator-initiated, single center, phase 2, open-label study that enrolled eligible patients at the University of Texas MD Anderson Cancer Center. The primary endpoint was the proportion of patients who achieved an objective response as assessed by investigator review according to RECIST version 1.1. Secondary endpoints included progression free survival, duration of response, disease control, overall survival, and safety.
Patients received poziotinib 16 mg orally once daily, until objective disease progression, and could continue beyond progression for as long as clinical benefit was observed, as judged by the investigator and in the absence of other discontinuation criteria (patient withdrawal, adverse event). Patients who discontinued poziotinib for reasons other than disease progression continued with tumor assessments until disease progression. Dose interruption or reduction occurred if patients had a grade 3 or more non-disease related adverse event or unacceptable toxicity. If the adverse event resolved or reverted to National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 (CTCAE) grade 1, poziotinib treatment could be restarted at the same dose (16 mg) or a lower dose (12 mg). A second reduction of dose to 8 mg was allowed for reoccurrence of same toxicity. If toxicity recurs following two dose modifications, poziotinib was discontinued.
Study Assessments
Tumor response according to RECIST version 1.1 was assessed by blinded central review and by investigator using CT, PET-CT or MRI scans taken at baseline, and every eight weeks from the start of poziotinib. During the treatment period, serum chemistry, hematology, vital signs, physical examination, weight, digital electrocardiogram, and ECOG performance status were assessed every four weeks; left ventricular ejection fraction was assessed every twelve weeks; adverse events (graded according to CTCAE version 4.03) were monitored continuously throughout the treatment period.
Biomarker Analysis
Archival and/or optional fresh tumor biopsies were obtained at baseline (pre-treatment) and on disease progression. Optional serial plasma was collected at baseline, 8 weeks of poziotinib treatment and on disease progression. Tumor biopsies were analyzed using targeted next generation sequencing (MD Anderson Cancer Center Solid Tumor Assay) that covers the coding sequence of 134 genes and selected copy number variations (amplifications) in 47 genes as well as inter- and intragenic fusions involving 51 genes (list of genes provided in Table S5 and Table S6). Analysis of plasma samples was performed using a targeted next generation panel (LB-70) that covers 70-cancer related genes with analytic sensitivity of 0.1–0.3% developed in collaboration with Guardant Health (list of genes provided in Table S7). Resistance mechanisms identified in patients’ samples were validated in preclinical models using Ba/F3 cells.
Generation of patient derived cell line
MDA-004K patient derived cell line was established from a malignant kidney resection obtained from a patient with metastatic adenocarcinoma of the lung harboring an EGFR exon 20 A767_V769dupASV. The patient was treated with poziotinib with a partial response for 13.8 months. The site of progression was in the kidney, which was resected and used to generate the cell line under protocol number PA140276. Briefly, after resection, the tumor was manually minced and then incubated at 37°C overnight in a solution of RPMI containing 1mg/ml collagenase A and 0.4mg/ml hyaluronidase. Cells were then spun down and resuspended in trypsin-EDTA. Dispase and DNase I was added to the cell solution and pipetted until cells dissociated, and then cells were filtered in 40μm cell strainer. Red blood cells were removed by ammonium chloride solution. Resulting single cell suspension was then plated and maintained in RPMI + 10% FBS.
EMT Score
Fresh resected sample was obtained in RPMI and RNAseq was performed. RNA-Seq gene expression raw read counts generated were normalized into Transcripts Per Kilobase Million (TPM). TCGA lung adenocarcinoma case TCGA-50–6673 was obtained from publicly available dataset. The EMT score was calculated using a 74-gene signature published previously (Byers et al., 2013). Data for the gene expression of the 74 genes is provided in Table S8.
Ba/F3 cell generation and IC50 approximation
As previously described (Robichaux et al., 2018; Robichaux et al., 2019) Ba/F3 cells were acquired from Dr. Gordon Mills (MD Anderson Cancer Center), and cultured in RPMI (Sigma) containing 10% FBS, 1% penicillin/streptomycin, and 10ng/ml recombinant mIL-3 (R&D Biosystems). To generate stable Ba/F3 cell lines, retroviral transduction containing mutant EGFR plasmids was completed for 12–24 hours. Retroviruses were generated using Phoenix 293T-ampho cells (Orbigen) transfected with Lipofectamine 2000 (Invitrogen) and indicated retrovirus vectors. Stable lines were selected after 24–48 hours using 2μg/ml puromycin (Invitrogen). Cells were then stained with PE-EGFR (Biolegend) and sorted by FACS. After sorting, EGFR positive cells were maintained in complete RPMI media containing 1ng/ml EGF to support cell viability. Drug screening was performed as previously described (Robichaux et al., 2018; Robichaux et al., 2019). Cell Titer Glo was used to determine cell viability and raw bioluminescence values were normalized to DMSO control treated cells, and values were plotted in GraphPad Prism. Non-linear regression models were used to fit the normalized data with a variable slope, and IC5o values were determined by GraphPad prism by interpolation of concentrations at 50% inhibition. Drug screens were performed in technical triplicate on each plate and triplicate independent replicates.
In silico modeling
The molecular model of poziotinib bound to T790M D770insNPG EGFR was constructed using MOE (Boyd, 2005) (Molecular Operating Environment, 2020.09; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2020). The X-ray structure (PDB 4LRM) of D770insNPG EGFR with quinazoline irreversible inhibitor PD168383 bound was loaded into MOE and processed using the protein preparation workflow followed by tethered minimization (Yasuda et al., 2013). The ligand was manually transformed to poziotinib and the T790M mutation was incorporated using the MOE protein builder. The resulting protein-ligand complex was subjected to restrained minimization followed by full minimization of poziotinib, Cys797, and M790 while holding the rest of the protein fixed.
Tyrosine Kinase inhibitors
All inhibitors for in vitro use were purchased from Selleck Chemicals. Inhibitors were reconstituted in DMSO at a concentration of 10mM and stored at −80°C as single use aliquots.
Resistant cell line generation
YUL-0019 cells were a generous gift from the Politi lab and were generated from the pleural effusion of a patient with NSCLC harboring an exon 20 insertion as previously described (Robichaux et al., 2018). Ba/F3 cell lines expressing exon 20 insertions were generated as described above. Cell lines were made resistant to poziotinib through continuous culturing of cells with increasing doses of poziotinib until cell viability was no longer affected at 10μM poziotinib. For the YUL-0019 cell line, clones were selected and grown as single cell lines, and for Ba/F3 cells, cells were analyzed by Sanger sequencing as a heterogeneous population.
Reverse Phase Protein Array
RPPA printing was completed using lysates from YUL-0019 parental cells and YUL-0019 poziotinib resistant cell lines. RPPA studies were carried out and quantified as previously described using the SuperCurve method and R packages (version 2.10.0) (Byers et al., 2009). Data has been transformed to Z-scores and untransformed data is presented in Table S9.
Western Blotting and Antibodies
Western blotting was performed as previously described (Robichaux et al., 2018). Briefly, cells were washed and lysed in lysis buffer (Cell Signaling #9803) including protease inhibitor tablets from Roche. Protein was loaded onto pre-casted gels (BioRad) and semidry transfer was completed onto methylcellulose membranes. Antibodies E-Cadherin (#3195S), N-Cadherin (#4061), Vimentin (#5741S), and Caveolin (#3238) were purchased from Cell Signaling. ß-actin (#A5441) was purchased from Sigma. SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher) and BioRad’s ChemiDoc Touch Imaging System were used to visualize protein bands.
Clonogenic assay
Colonogeic assays was completed as previously described (Nilsson et al., 2020b). Briefly, YUL-0019 and YUL-0019 PR cells were plated into 6-well plates and incubated overnight in RPMI + 10% FBS. The next day, media was replaced with RPMI +10%FBS with or without inhibitors. After 14 days, cells were fixed with 6.0% glutaraldehyde and stained with a 0.5% crystal violet solution. Colonies were quantified and normalized to DMSO controls using Image J software, and IC50 values were determined using GraphPad Prism.
Sanger Sequencing
DNA was isolated by Qiagen DNeasy kit (Cat# 69504) according to manufacture instructions and eluted in water. EGFR was amplified in three overlapping segments by traditional PCR using the primers the following primers: EGFR #1 Forward: ATGCGACCCTCCGGGAC, EGFR #2 Reverse: TCATGCTCCAATAAATTCACTGCT, EGFR #2 Forward: ATGCGACCCTCCGGG, EGFR #2 Reverse: TCATGCTCCAATAAATTCACTGCTT, EGFR #3 Forward: CTCCGGTCAGAAAACCAAAA, and EGFR #3 Reverse: CTTCCAGACCAGGGTGTTGT. Sanger sequencing was performed using Applied Biosystems Sequence Detection System by the MD Anderson Advanced Technology Genomics Core.
Protein modeling and Molecular Dynamics Simulations (MDS)
In the absence of an experimental structure of EGFR S768dupSVD and EGFR H773insNPH mutants, homology modeling was undertaken to generate a structural model of the mutants. Prime homology modeling implemented in the Schrodinger package was used for modeling the structure of the mutants (Zhu et al., 2014). A multi-template homology modeling approach was employed to improve model coverage. Accordingly, PDB entries 2ITO,3UG2, and 5GTZ were considered for modeling the structure of EGFR H773insNPH insertion mutant bound to poziotinib. None of the template structures considered for homology modeling had a poziotinib bound to EGFR. However, a ligand similarity search identified PDB entry 2ITO, to contain a structurally similar ligand (gefitinib) complexed to EGFR. Hence, PDB entry 2ITO was used as the primary template for homology modeling, and the coordinates of gefitinib were transferred to the modeled structure. Subsequently, loop refinement was carried out to refine flexible loop regions around the binding pocket. A docked model of poziotinib covalently bound to EGFR mutants was generated using the covalent docking program CovDock (Zhu et al., 2014), available through the Schrodinger package. The covalent link was removed, and the noncovalent complex was used as the starting structure for subsequent MD simulations. PDB entries 6JWL and 3UG2 were considered for modeling the structure of EGFR H773insNPH insertion mutant bound to mobocertinib. The crystal structure of AZD9291 (osimertinib) bound to mutant EGFR (6JWL) was identified to have an inhibitor structurally similar to mobocertinib. Hence, a model of EGFR insertion mutants was generated using AZD9291 bound to EGFR the binding mode of mobocertinib was modeled by making a simple modification to AZD9291.
All-atom molecular dynamics simulations of the mutants bound to poziotinib and mobocertinib were carried out using the AMBER simulation package (Version 18). The system was prepared using the LEaP module of Within AmberTools (Version 19).
Amber force field 14SB (Maier et al., 2015), GAFF (Wang et al., 2004), and TIP3P (Jorgensen et al., 1983) parameter sets were used for parametrizing the protein, ligand, and water, respectively. AM1BCC partial charges (Jakalian et al., 2002) were assigned for the ligand. The structures were then solvated in a cubic box, keeping the boundary of the box at least 10.0 Å away from any solute atom. Additional Na+/Cl− ions were added to the simulation box to neutralize the system and to ensure a salt concentration of 0.1M concentration.
A thorough minimization and equilibration scheme was applied to relax the system. MDS was then performed under periodic boundary conditions. Bond lengths of hydrogen atoms were constrained using the SHAKE algorithm. The simulation used a time step of 2 fs. Particle Mesh-Ewald method was used to evaluate long-range electrostatic and short-range van der Waals forces interactions with a distance cut-off of 9.0 Å. The system was linearly heated from 10 K to 300 K in NVT ensemble for 200 ps and held at 300 K for over 50 ps, with harmonic restraints on the system. Subsequently, 250 ps of NVT equilibration was carried out, applying harmonic constraints on the protein and the ligand to allow the solvent to equilibrate around the solute. The system was further equilibrated for 1.5 ns in the NPT ensemble prior to the NPT production run. A Langevin thermostat and a Berendsen barostat were used for maintaining a constant temperature (300 K) and pressure (1 bar). Weak harmonic restraints were imposed on the protein backbone and were retained throughout the equilibration runs and were gradually scaled in a phased manner. Finally, the equilibrated bimolecular systems were subjected to 2 μs of NPT production simulation using the parallel CUDA version (pmemd) of AMBER 18. The first 5 ns of the production simulations were considered as equilibration and therefore discarded from all analysis.
The binding free energies of poziotinib and mobocertinib towards the mutants were calculated using two end-point-based methods, namely MM/PBSA and MM/GBSA. A single trajectory approach was employed (Biswas et al., 2017), and the binding energies were calculated using MD snapshots taken at 1ns interval from the simulation.
The binding free energy (ΔGbind) is obtained by subtracting the free energies of the unbound receptor (GProtein) and ligand (GLigand) from the free energy of the bound complex (GComplex)
| (1) |
The free energy (G) for each state is evaluated using contribution from three different terms
| (2) |
where ΔEMM, ΔGsol, and −TSSolute are the changes of the gas phase MM energy, the solvation free energy and the conformational entropy upon binding.
The EMM term includes Einternal (bond, angle and dihedral energies), electrostatic Eelectrostatic, and van der Waals energy Evdw
| (3) |
The solvation free energy (Gsol) component is the sum of polar contributions calculated using either an GB or PB model, while the non-polar contribution is estimated by a linear relationship to the change in solvent accessible surface area (SASA)
| (4) |
T ˂SSolute>, is the product of the absolute temperature T and the solute entropy Ssolute. The solute
entropy term was ignored in our calculations. Hence, the values reported herein essentially represent the binding energy component.
QUANTIFICATION AND STATISTICAL ANALYSIS
The primary objective of this study was to evaluate the efficacy of poziotinib, measured by objective response rate (ORR), in treating patients with EGFR exon 20 mutant NSCLC. We considered an ORR of 30% to be clinically meaningful. A sample size of 50 ensured that, if the trial was not terminated early, a posterior 90% credibility interval for OR will have width of 0.209 at most, under the assumption of a 30% of OR. Efficacy and toxicity were monitored in cohorts of 10 patients. The intention to treat (ITT) population was defined as all patients enrolled who received at least one dose of poziotinib. All safety summaries were produced on the ITT population. Progression-free survival and duration of response outcomes were calculated with the Kaplan-Meier method. Data were analyzed using an April 1, 2021, data cut-off. Statistical analysis was performed using GraphPad prism and student’s t-test two sided, was used where applicable. p < 0.05 was defines as threshold for significance.
Supplementary Material
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| E-Cadherin (24E10) Rabbit mAb | Cell Signaling | #3195S |
| N-Cadherin Antibody | Cell Signaling | #4061 |
| Vimentin (D21H3) XP® Rabbit mAb | Cell Signaling | #5741S |
| Caveolin-1 Antibody | Cell Signaling | #3238 |
| Monoclonal Anti-β-Actin antibody produced in mouse | Sigma | #A5441 |
| EGFR | Cell Signaling | #2232 |
| p-EGFR | Abcam | Ab32578 |
| Napsin A | Abcam | Ab129189 |
| Vinculin | Sigma | SAB4200080 |
| Zeb1 | Cell Signaling | #3396 |
| Axl | Cell Signaling | #8661 |
| GPX4 | Abcam | ab125066 |
| Col1A1 | Cell Signaling | #72026 |
| Twist | Cell Signaling | #69366 |
| Snail | Cell Signaling | #3895 |
| pS127-Yap1 | Cell Signaling | #13008 |
| Yap1 | Cell Signaling | #4912 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Tumor biopsies | This paper | N/A |
| Plasma | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | G9243 |
| 4–15% Criterion TGX Precast Midi Protein Gel | BioRad | 5671084 |
| Cell Lysis Buffer (10X) #9803 | Cell Signaling | #9803 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | 34580 |
| Erlotinib | Selleck Chem | S7786 |
| Neratinib | Selleck Chem | S2150 |
| Dacomitinib | Selleck Chem | S2727 |
| Afatinib | Selleck Chem | S1011 |
| Osimertinib | Selleck Chem | S7297 |
| Nazartinib | Selleck Chem | S7824 |
| Poziotinib (in vitro / in vivo) | Selleck Chem | S7358 |
| Poziotinib (clinical trial) | Spectrum Pharmaceuticals | |
| Dinaciclib | Selleck Chem | S2768 |
| ON-01910 | Selleck Chem | S1362 |
| SB743921 | Selleck Chem | S2182 |
| AMG900 | Selleck Chem | S2719 |
| Critical commercial assays | ||
| Deposited data | ||
| RPPA | This Paper | Table S9 |
| Gene expression | This Paper | Table S8 |
| Crystal structure of EGFR kinase domain G719S mutation in complex with Iressa | PDB | ID: 2ITO |
| Crystal structure of the mutated EGFR kinase domain (G719S/T790M) in complex with gefitinib | PDB | ID: 3UG2 |
| Crystal structure of EGFR 696–1022 T790M in complex with JTS-1–39 | PDB | ID: 5GTZ |
| Crystal structure of EGFR 696–1022 L858R in complex with AZD9291 | PDB | ID: 6JWL |
| EGFR D770_N771insNPG in complex with PD168393 | PDB | ID: 4LRM |
| Experimental models: Cell lines | ||
| Ba/F3 | Dr. Gordon Mills (MD Anderson Cancer Center) | N/A |
| Phoenix 293T-amphi | Orbigen | N/A |
| YUL-0019 | Dr. Katerina Politi (Yale) | N/A |
| YUL-PR1 | This paper | N/A |
| YUL-PR2 | This paper | N/A |
| YUL-PR3 | This paper | N/A |
| YUL-PR4 | This paper | N/A |
| YUL-PR5 | This paper | N/A |
| YUL-PR6 | This paper | N/A |
| YUL-PR7 | This paper | N/A |
| YUL-PR8 | This paper | N/A |
| MDA-004K | MDACC | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| EGFR #1 Forward: ATGCGACCCTCCGGGAC | This paper | N/A |
| EGFR #1 Reverse: TCATGCTCCAATAAATTCACTGCT | This paper | N/A |
| EGFR #2 Forward: ATGCGACCCTCCGGG | This paper | N/A |
| EGFR #2 Reverse: TCATGCTCCAATAAATTCACTGCTT | This paper | N/A |
| EGFR #3 Forward: CTCCGGTCAGAAAACCAAAA | This paper | N/A |
| EGFR #3 Reverse: CTTCCAGACCAGGGTGTTGT | This paper | N/A |
| Recombinant DNA | ||
| Software and algorithms | ||
| GraphPad Prism | Dotmatics | https://www.graphpad.com/ |
| MOLECULAR OPERATING ENVIRONMENT | Chemical Computing group | https://www.chemcomp.com/Products.htm |
| ImageJ | NIH | https://imagej.nihgov/ij/ |
| R v2.1 | R Foundation for Statistical Computing | https://www.R-project.org/ |
| Amber | UCSF | https://ambermd.org/ |
| CovDock | Schrödinger | https://www.schrodinger.com/science-articles/covdock |
| Other | ||
| Study protocol | This paper | Text S1 |
Highlights.
Poziotinib yields a 32% response rate in EGFR exon 20 mutant NSCLC
Poziotinib sensitivity is highly dependent on insertion location
Near loop exon 20 insertions are more sensitive to poziotinib than far loop insertions
Mechanisms of acquired poziotinib resistance include EGFR T790M and MET amplifications
Acknowledgements
Supported by Spectrum Pharmaceuticals, grants from funds from R01 CA190628-NIH/NCI, R01 CA234183-NIH/NCI, R01 CA247975-NIH/NCI, CPRIT IIRA RP200150, the David Bruton, Jr. Endowment, Rexanna’s Foundation for Fighting Lung Cancer, CCSG Lung Cancer Program, the Lung Cancer Moon Shot Program, the Kopelman Fund for Lung Cancer Research, the Exon 20 Group, the Lung Cancer Research Fund, the Hallman fund, Lung SPORE grant 5 P50 CA070907, and 1U54CA224065-01 (to Dr Heymach) and by grants from Lung Cancer Research Foundation and ASCO Career Development Award (57112) (to Dr Elamin).
We thank the participating patients and their families, as well as the research nurses, study coordinators and operations staff; and the Exon 20 Group, Marcia Horn, Robert Hanlon, and Kevin Hanlon.
Declaration of interests
The research being reported in this publication is research in which The University of Texas MD Anderson Cancer Center has an institutional financial conflict of interest. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson’ s conduct of this research. MD Anderson, including JPR, MBN, & JVH have filed a patent for the use of poziotinib for treating EGFR & HER2 mutant cancers and licensed the technology to Spectrum Pharmaceuticals (SP). JPR and JVH receive research support from SP and Takeda. MD Anderson, including MBN, JPR, and JVH have a pending patent submitted for treatment of EGFR TKI resistant NSCLC; and another, including JPR, SH, and JVH, for the classification of EGFR mutations. JPR and MBN have no non-financial competing interests. JPR is now fulltime employee of AstraZebeca. JVH also receives grant or research support from AstraZeneca (AZ) and GlaxoSmithKline (GSK) and has served on advisory committees for AZ, Boehringer Ingelheim (BI), Bristol-Myers Squibb, Catalyst, EMD Serono, Foundation Medicine (FMI), Hengrui Therapeutics, Genentech, GSK, Guardant Health (GH), Eli Lilly, Merck, Novartis, Pfizer, Roche, Sanofi, and Seattle Genetics. As Non-financial competing interests JVH serves as scientific advisor for Rexanna Foundation and the EGFR resisters. YYE discloses research support from Spectrum, AstraZeneca, Takeda, Eli Lilly, Xcovery, Tuning Point Therapeutics; advisory role for AstraZeneca, Eli Lilly and Turning Point; Accommodation expenses Eli Lilly. X.L. receives consultant and advisory fees from Eli Lilly, AstraZeneca and EMD Serono, and research funding from Eli Lilly, Boehringer Ingelheim and Spectrum Pharmaceuticals. MA reports research funding to the MD Anderson Cancer Center from Genentech, Nektar therapeutics, Merck, GlaxoSmithKline, Novartis, Jounce therapeutics, Bristol Myers Squibb, Eli Lilly and Adaptimmune, and receives advisory fees from GlaxoSmithKline and Shattuck labs. VKL reports advisory role fees from Takeda, Seattle Genetics, Bristol-Myers Squibb, and AstraZeneca and research funding from Bristol-Myers Squibb, AstraZeneca, and Merck. DLG reports honoraria for scientific advisory boards from AstraZeneca, Sanofi, Alethia Biotherapeutics, Lilly and Janssen, and research support from Janssen, Takeda, Ribon Therapeutics, Astellas and AstraZeneca. GB receives personal fees and research funding from Amgen, Bayer, Bristol Myers Squibb, Celgene, Daiichi Sankyo, Genentech, MedImmune, Merck, Roche and Xcovery, research funding from Adaptimmune, Exelixis, GlaxoSmithKline, Immatics, Immunocore, Incyte, Kite pharma, Macrogenics, Torque, AstraZeneca, Tmunity, Regeneron, Beigene, Novartis and Repertoire Immune Medicines, and personal fees from Abbvie, Adicet, Amgen, Araid, Clovis Oncology, AstraZeneca, Bristol Myer Squibb, Celgene, Genentech, Gilead, Merck, Novartis, Roche, Virogin Biotech, John & Johnson/Janssen and Maverick Therapeutics. AST reports advisory board/consultant fees from Bristol Myers Squibb, Eli Lilly, Genentech, Roche, Novartis, Ariad, EMD Serono, Merck, Seattle Genetics, AstraZeneca, Boehringer Ingelheim, Sellas Life Science, Takeda, Epizyme and Huron, and receives research grants from Eli Lilly, Millennium, Polaris, Genentech, Merck, Boehringer Ingelheim, Bristol Myers Squibb, Ariad, Epizyme, Seattle Genetics, Takeda and EMD Serono.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, Zakowski MF, Kris MG, and Ladanyi M. (2013). EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 12, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beau-Faller M, Prim N, Ruppert AM, Nanni-Metellus I, Lacave R, Lacroix L, Escande F, Lizard S, Pretet JL, Rouquette I, et al. (2014). Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann Oncol 25, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Shukla A, Vijayan RSK, Jeyakanthan J, and Sekar K. (2017). Crystal structures of an archaeal thymidylate kinase from Sulfolobus tokodaii provide insights into the role of a conserved active site Arginine residue. J Struct Biol 197, 236–249. [DOI] [PubMed] [Google Scholar]
- Boyd S. (2005). Molecular operating environment. Chemistry World 2, 66–66. [Google Scholar]
- Byeon S, Kim Y, Lim SW, Cho JH, Park S, Lee J, Sun JM, Choi YL, Lee SH, Ahn JS, et al. (2019). Clinical Outcomes of EGFR Exon 20 Insertion Mutations in Advanced Non-small Cell Lung Cancer in Korea. Cancer Res Treat 51, 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. (2013). An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, Cascone T, Mills GB, Heymach JV, and Johnson FM (2009). Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res 15, 6852–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callegari D, Ranaghan KE, Woods CJ, Minari R, Tiseo M, Mor M, Mulholland AJ, and Lodola A. (2018). L718Q mutant EGFR escapes covalent inhibition by stabilizing a non-reactive conformation of the lung cancer drug osimertinib. Chem Sci 9, 2740–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canale M, Petracci E, Delmonte A, Bronte G, Chiadini E, Ludovini V, Dubini A, Papi M, Baglivo S, De Luigi N, et al. (2020). Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J Clin Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Stewart TA, Thompson EW, and Monteith GR (2014). Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci 35, 479–488. [DOI] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. (2009). New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, and Janne PA (2015). EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res 21, 3913–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, et al. (2016). EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin Cancer Res 22, 4585–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, Park K, Gorbunova V, Kowalyszyn RD, Pikiel J, et al. (2014). Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet 384, 665–673. [DOI] [PubMed] [Google Scholar]
- Ghiso E, Migliore C, Ciciriello V, Morando E, Petrelli A, Corso S, De Luca E, Gatti G, Volante M, and Giordano S. (2017). YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 19, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, et al. (2004). Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol 22, 1589–1597. [DOI] [PubMed] [Google Scholar]
- Hsu PC, You B, Yang YL, Zhang WQ, Wang YC, Xu Z, Dai Y, Liu S, Yang CT, Li H, et al. (2016). YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 7, 51922–51933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakalian A, Jack DB, and Bayly CI (2002). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79, 926–935. [Google Scholar]
- Kim TM, Lee KW, Oh DY, Lee JS, Im SA, Kim DW, Han SW, Kim YJ, Kim TY, Kim JH, et al. (2018). Phase 1 Studies of Poziotinib, an Irreversible Pan-HER Tyrosine Kinase Inhibitor in Patients with Advanced Solid Tumors. Cancer Res Treat 50, 835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Tanizaki J, Paranal R, Endoh H, Lydon C, Capelletti M, Repellin CE, Choi J, Ogino A, Calles A, et al. (2017). Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. [DOI] [PMC free article] [PubMed]
- Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, et al. (2020). Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 37, 104–122 e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam VK, Zhang J, Wu CC, Tran HT, Li L, Diao L, Wang J, Rinsurongkawong W, Raymond VM, Lanman RB, et al. (2021). Genotype-Specific Differences in Circulating Tumor DNA Levels in Advanced NSCLC. J Thorac Oncol 16, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le X, Goldman JW, Clarke JM, Tchekmedyian N, Piotrowska Z, Chu D, Bhat G, Lebel FM, and Socinski MA (2020). Poziotinib shows activity and durability of responses in subgroups of previously treated EGFR exon 20 NSCLC patients. Journal of Clinical Oncology 38, 9514–9514. [Google Scholar]
- Le X SE, Suga JM, Brahmer JR, Dooms C, Mamdani H, Nechushtan H, Riess JW, Spira A, Barrett JA, Dreiling LK, Socinski M (2021). Poziotinib administered twice daily improves safety and tolerability in patients with EGFR or HER2 exon 20 mutant NSCLC(ZENITH20–5). Proceedings of the 112th Annual Meeting of the American Association for Cancer Research. [Google Scholar]
- Lee TF, Tseng YC, Nguyen PA, Li YC, Ho CC, and Wu CW (2018). Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci Rep 8, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisberg A, Cummings A, Goldman JW, Bornazyan K, Reese N, Wang T, Coluzzi P, Ledezma B, Mendenhall M, Hunt J, et al. (2018). A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naive Patients With Advanced NSCLC. J Thorac Oncol 13, 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C. (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massarelli E, Johnson FM, Erickson HS, WistubaI, and Papadimitrakopoulou V. (2013). Uncommon epidermal growth factor receptor mutations in non-small cell lung cancer and their mechanisms of EGFR tyrosine kinase inhibitors sensitivity and resistance. Lung Cancer 80, 235–241. [DOI] [PubMed] [Google Scholar]
- Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, Thai AA, Mascaux C, Couraud S, Veillon R, et al. (2019). Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol 30, 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, Bonilla RF, Ko RB, Yoo CH, Gojenola L, et al. (2020). Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nature Cancer 1, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok TS, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, Lee M, Linke R, Rosell R, Corral J, et al. (2018). Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients With Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J Clin Oncol 36, 2244–2250. [DOI] [PubMed] [Google Scholar]
- Naidoo J, Sima CS, Rodriguez K, Busby N, Nafa K, Ladanyi M, Riely GJ, Kris MG, Arcila ME, and Yu HA (2015). Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 121, 3212–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrao MV, Skoulidis F, Montesion M, Schulze K, Bara I, Shen V, Xu H, Hu S, Sui D, Elamin YY, et al. (2021). Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. Journal for ImmunoTherapy of Cancer 9, e002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson MB, Robichaux J, Herynk MH, Cascone T, Le X, Elamin Y, Patel S, Zhang F, Xu L, Hu L, et al. (2020a). Altered Regulation of HIF-1alpha in Naive- and Drug-Resistant EGFR-Mutant NSCLC: Implications for a Vascular Endothelial Growth Factor-Dependent Phenotype. J Thorac Oncol. [DOI] [PMC free article] [PubMed]
- Nilsson MB, Sun H, Robichaux J, Pfeifer M, McDermott U, Travers J, Diao L, Xi Y, Tong P, Shen L, et al. (2020b). A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, Jackman DM, Johnson BE, and Janne PA (2013). Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol 8, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, Viteri S, Han JY, Kim SW, Lee CK, et al. (2021). Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol, JCO2100662. [DOI] [PMC free article] [PubMed]
- Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, Li S, Chen T, Poteete A, Estrada-Bernal A, et al. (2018). Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 24, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux JP, Elamin YY, Vijayan RSK, Nilsson MB, Hu L, He J, Zhang F, Pisegna M, Poteete A, Sun H, et al. (2019). Pan-Cancer Landscape and Analysis of ERBB2 Mutations Identifies Poziotinib as a Clinically Active Inhibitor and Enhancer of T-DM1 Activity. Cancer Cell 36, 444–457 e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux JP, Le X, Vijayan RSK, Hicks JK, Heeke S, Elamin YY, Lin HY, Udagawa H, Skoulidis F, Tran H, et al. (2021). Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 597, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher A, Le X, Cornelissen R, Shum E, Suga J, Socinski M, Molina JR, Haura E, Clarke J, Bhat G, et al. (2021). Safety, tolerability and preliminary efficacy of poziotinib with twice daily strategy in EGFR/HER2 Exon 20 mutant non-small cell lung cancer. Annals of Oncology 32, S15–S15. [Google Scholar]
- Shah KN, Bhatt R, Rotow J, Rohrberg J, Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al. (2019). Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat Med 25, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd FA, Dancey J, Ramlau R, Mattson K, Gralla R, O’Rourke M, Levitan N, Gressot L, Vincent M, Burkes R, et al. (2000). Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol 18, 2095–2103. [DOI] [PubMed] [Google Scholar]
- Skoulidis F, and Heymach JV (2019). Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer 19, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, et al. (2018). Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 378, 113–125. [DOI] [PubMed] [Google Scholar]
- Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, and Mitsudomi T. (2011). Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol 6, 1152–1161. [DOI] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Lam VK, Elamin YY, Hong L, Colen R, Elshafeey NA, Hassan ISA, Altan M, Blumenschein GR, Rinsurongkawong W, et al. (2021). Clinical Outcomes in Non-Small-Cell Lung Cancer Patients Treated With EGFR-Tyrosine Kinase Inhibitors and Other Targeted Therapies Based on Tumor Versus Plasma Genomic Profiling. JCO Precis Oncol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos P, Gergis C, Viray H, Varkaris A, Fujii M, Rangachari D, VanderLaan PA, Kobayashi IS, Kobayashi SS, and Costa DB (2020). EGFR-A763_Y764insFQEA Is a Unique Exon 20 Insertion Mutation That Displays Sensitivity to Approved and In-Development Lung Cancer EGFR Tyrosine Kinase Inhibitors. JTO Clin Res Rep 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyse S, and Huang PH (2019). Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct Target Ther 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, and Case DA (2004). Development and testing of a general amber force field. J Comput Chem 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, et al. (2014). Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15, 213–222. [DOI] [PubMed] [Google Scholar]
- Yang G, Li J, Xu H, Yang Y, Yang L, Xu F, Xia B, Zhu VW, Nagasaka M, Yang Y, et al. (2020). EGFR exon 20 insertion mutations in Chinese advanced non-small cell lung cancer patients: Molecular heterogeneity and treatment outcome from nationwide real-world study. Lung Cancer 145, 186–194. [DOI] [PubMed] [Google Scholar]
- Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O’Byrne K, Feng J, et al. (2015). Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16, 141–151. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Kobayashi S, and Costa DB (2012). EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 13, e23–31. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, Huberman MS, Cohen DW, Nakayama S, Ishioka K, et al. (2013). Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 5, 216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al. (2012). Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Ramalingam SS, Kim TM, Kim SW, Yang JC, Riely GJ, Mekhail T, Nguyen D, Garcia Campelo MR, Felip E, et al. (2021). Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients With EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol 7, e214761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, and Harder E. (2014). Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J Chem Inf Model 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The patient-derived data reported in this study cannot be deposited in a public repository because of legal restrictions. To request access, contact John V. Heymach (JHeymach@mdanderson.org). A detailed proposal including a scientific description needs to be submitted to the lead author and internal IRB approval and the completion of a Material Transfer Agreement is required. Processed data used in this manuscript is fully provided. Clinical trial description for NCT03066206: https://clinicaltrials.gov/ct2/show/NCT03066206?term=Poziotinib&rank=6.