

Abstract

We report here the use of commercially available ZnEt2 as an efficient precatalyst for the addition of alcohols to carbodiimides to obtain a wide range of isoureas under mild conditions. In an initial screening using methanol and commercial carbodiimides as substrates, the bulky isourea (OMe)(NHDipp)C(NDipp) (Dipp = 2,6-iPr2C6H3) was prepared for the first time using a catalytic method, and its structure confirmed by an X-ray diffraction analysis. Then, the efficiency of the precatalyst was tested with two carbodiimides, C(NiPr)2 and C(Np-tol)2, toward a series of alkylic and arylic alcohols and diols, with different steric and electronic properties, including the presence of other functional groups, usually with excellent conversions, especially for the more reactive aromatic carbodiimide. Some of the new isoureas thus prepared have also been isolated and characterized. Kinetic and stoichiometric experiments allowed us to propose a plausible mechanism for these transformations.
Introduction
Isoureas, urea isomers of general formula (RN)C(OR1)(NR2R3) (R1 ≠ H), are very important compounds typically used in organic synthesis as alkylating reagents.1 Additionally, they have found applications in agriculture (stimulating plant growth),2 medicinal chemistry (enzyme inhibitors),3 physical chemistry (surfactants),4 and biochemistry.5 The starting materials traditionally used to obtain isoureas are ureas, carbodiimides, chloroformamidines, cyanamides, or organic cyanates1c,6 and more recently protocols, based on isonitriles have also been described,7 usually requiring a metal-based catalyst.7a−7c From all these methods, the most straightforward and efficient in terms of atom economy is the intermolecular addition of alcohols to carbodiimides, which is also the most widely used.1a−1c,8 Due to the low electrophilicity of carbodiimides, these reactions often require the use of a catalyst, the most typical ones being copper(I) and copper(II) salts1c,1e,8,9 but also zinc(II) salts,1c,8,9g in which the metal ion is believed to act as a Lewis acid interacting with the carbodiimide via the imine N atom to enhance the electrophilicity of the central carbon atom. More recently, improved state-of-the-art homogeneous catalysts have been reported for these transformations, significantly broadening the substrate scope for this reaction and adding valuable mechanistic studies as well. Thus, in 2016, Eisen and co-workers reported the first examples of actinide-based catalysts with amide ligands for the hydroalkoxylation of carbodiimides (A in Figure 1).10 In the following years, subsequent reports by the same group would expand this family of catalysts with iminato-supported actinide11 and hafnium12 complexes. In 2018, Cantat and co-workers reported 1,5,7-triazabicyclo[4.4.0]dec-5-ene and its alkali salts (Li+, Na+, K+) as examples of transition-metal-free catalysts for the hydroalkoxylation of carbodiimides (B in Figure 1), with excellent results in terms of activity for the K+ salt.13 In the same year, Zhao, Yao, and co-workers reported the use of rare-earth-metal catalysts for the first time for this transformation (C in Figure 1), more specifically, lanthanide and yttrium amides.14 Also in 2018, Panda and co-workers reported a binuclear Ti(IV) complex that turned out to be efficient catalysts for the addition of a variety of E–H bonds (E = O, C, N, P) to carbodiimides and other heterocumulenes (D in Figure 1).15 In 2022, Kozlowski, Zacuto, and co-workers used CuCl, a catalyst traditionally used for this reaction, to find an acceleration in the reaction rate in the presence of atmospheric O2 due to in situ formation of ClCuII(OH) as the active species (E in Figure 1).16
Figure 1.

Recent examples of catalysts for the addition of alcohols to carbodiimides.
Since 2010, our group has successfully used simple, commercially available ZnEt2 as a precatalyst in other hydroelementation reactions involving carbodiimides and (i) amines to give guanidines (N–H addition),17 or (ii) terminal alkynes to give the corresponding propiolamidines (C–H addition).18 Additionally, in 2021, Ma, Roesky, and co-workers reported the use of this compound for the hydrophosphination (P–H addition) of carbodiimides and related heterocumulenes.19 In an attempt to expand the catalytic hydroelementation repertoire of ZnEt2, we focus now our attention on the hydroalkoxylation of carbodiimides. As mentioned above, there are scarce reports in the literature employing Zn(II) salts as Lewis-acid-type catalysts for these reactions8a,9g and to the best of our knowledge, no in-depth mechanistic studies have been carried out. However, zinc alkyls have not been reported and different reaction mechanisms should be expected as well. Thus, here we report the addition of a wide range of alcohols to carbodiimides catalyzed by ZnEt2 (F in Figure 1), together with stoichiometric and kinetics experiments, allowing us to propose a plausible mechanism for this catalytic reaction.
Results and Discussion
Catalytic Addition of Alcohols to Carbodiimides
We start, as a model process to study, with the reaction between 1,3-diisopropylcarbodiimide (DIC) with methanol, or tBuOH, in order to find optimal conditions for the synthesis of isoureas (See Supporting Information, Table S1). Having established these conditions using a 5 mol % precatalyst ratio and a temperature of 60 °C, the study was extended to the reaction of different commercial carbodiimides toward methanol in the presence of ZnEt2, with the corresponding blank experiments (Table 1). As can be seen, DIC reacted with methanol to give essentially full conversion to isourea 1a in 1 h at 60 °C (entry 1), whereas the control experiment only shows low conversion after protracted heating (16 h) at the same temperature (entry 2). Similar results were obtained for 1,3-dicyclohexylcarbodiimide (DCC), achieving full conversion to 2a in 2 h at 60 °C (entry 3) with no conversion observed in the control experiment (entry 4). The bulkier 1,3-di-tert-butylcarbodiimide reacted sluggishly with MeOH in the presence of ZnEt2 and only 27% conversion to isourea 3a was obtained after 30 h at 60 °C (entry 5). It is worth mentioning the latter isourea has been reported just once before and prepared via a stoichiometric method.20 Unsurprisingly, the more electrophilic 1,3-di-p-tolylcarbodiimide (DTC) reacted under milder conditions to give full conversion to 4a in 1.5 h at 25 °C (entry 7), whereas in the absence of ZnEt2 only 64% conversion was achieved after 4 h at 60 °C (entry 8). Finally, a challenging substrate, such as the bulky 1,3-bis-(2,6-diisopropylphenyl)carbodiimide, was chosen to test the activity of ZnEt2 as a precatalyst. Pleasingly, under somewhat more demanding conditions (80 °C, 120 h), 97% conversion to isourea 5a was reached (entry 9), as opposed to the blank experiment in which virtually no conversion was obtained under the same conditions (entry 10). We should note here that the latter isourea had not been prepared by catalytic methods before and, to our knowledge, it has only been reported once and prepared by a stoichiometric method (using carbodiimide and NaOMe as starting materials).21 Moreover, an attempted synthesis of 5a by Eisen and co-workers using actinide-based catalysts showed no conversion after 24 h.11e For that reason, we decided to isolate and fully characterize compound 5a, which was obtained in excellent yields (89%) as a white crystalline solid.
Table 1. Reactivity of Different Carbodiimides toward Methanol with and without the Catalysta.
| entry | C(NR)2 | precatalyst | T (°C) | t (h) | conversion (%)b |
|---|---|---|---|---|---|
| 1 | R = iPr | ZnEt2 | 60 | 1 | >99 |
| 2 | 60 | 21 | 16 | ||
| 3 | R = Cy | ZnEt2 | 60 | 2 | >99 |
| 4 | 60 | 2 | |||
| 5 | R = tBu | ZnEt2 | 60 | 30 | 27 |
| 6 | 60 | 48 | |||
| 7 | R = p-tol | ZnEt2 | 25 | 1.5 | >99 |
| 8 | 60 | 4 | 64 | ||
| 9 | R = Dipp | ZnEt2 | 80 | 120 | 97 [89]c |
| 10 | 80 | 120 | <1 |
Reaction conditions: 0.50 mmol carbodiimide, 0.50 mmol MeOH, 0.025 mmol precatalyst (25 μL, 1.0 M solution of ZnEt2 in hexanes), 500 μL of C6D6, and Si(SiMe3)4 (0.01 mmol) as an internal standard.
Based on 1H NMR of the reaction crude versus internal standard.
Isolated yield from the preparative–scale reaction.
An X-ray diffraction analysis of crystals of 5a confirmed its structure unambiguously (Figure 2), displaying a planar, Y-shaped “N2CO” core, and an anti-conformation about the C=N double bond. The structural features are akin to those of the related isourea (OEt)(NHPh)C=NPh, also with an anti-conformation,22 as opposed to the syn isomer found in the solid state for (OPh){NH(p-tol)}C=N(p-tol), reported by Cantat and co-workers,13 most likely favored due to two N–H hydrogen bonds found between two molecules of isourea in the lattice. In fact, a density functional theory calculation for the syn and anti-isomers of 5a demonstrated that the anti-isomer is significantly more stable (ca. 8 kcal·mol–1) than the syn counterpart, hence pointing to the retention in solution of the anti-conformation found in the solid-state structure. We must highlight that this trend was also observed for other isoureas derived from DIC (1) and DTC (4) with alkylic groups on the O atom, for which the anti-arrangement remains as the most stable one (thermodynamically), although with reduced energy gaps between syn/anti isomers. However, the opposite situation (most stable syn isomers) appears to be the case for most of the isoureas with aryl groups on the O atom (see the Supporting Information). Additionally, for some of the isoureas, low energy gaps between these isomers (<2 kcal·mol–1) are consistent with the coexistence of both isomers in solution as experimentally detected by NMR (i.e., isoureas 1d,g,i, vide infra).
Figure 2.
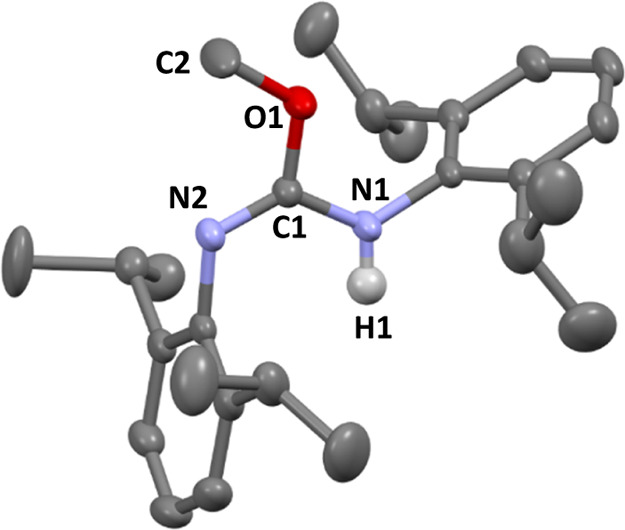
Molecular structure of 5a with ellipsoids plotted at 30% probability and H atoms omitted for clarity (except H1, attached to N1). Selected distances (Å) and angles (°): C1–O1 1.342(2), C1–N1 1.359(2), C1–N2 1.270(2), O1–C2 1.434(2), N1–C1–N2 127.1(1), O1–C1–N1 111.0(1), N2–C1–O1 122.0(1), and C1–O1–C2 116.2(1).
Having performed the initial screening with different carbodiimides toward methanol, we then decided to focus on the addition of a wide range of alcohols (and diols) to DIC (Table 2) and DTC (Table 3) using 5 mol % of ZnEt2 as a precatalyst to expand the scope of this reaction. DIC, the less electrophilic of the two carbodiimides chosen for this study, usually required temperatures of 60 °C to reach high conversions in reasonable times. Thus, reactions with EtOH and iPrOH proceeded to full conversion in 3 h to give isoureas 1b,c, respectively. Addition of the more sterically encumbered tBuOH gave 88% conversion of an isomer mixture (syn/anti)23 of 1d after 5 h. Because prolonged heating did not increase conversion, we assume that a thermodynamic equilibrium was reached at this temperature. We must highlight that ZnEt2 outperformed other catalysts reported in the literature in terms of activity and conversion for the synthesis of 1d, such as an actinide-based catalyst reported by Eisen (17%, 24 h, 75 °C),10 Na-TBD reported by Cantat (58%, 5 h, 75 °C),13 or the rare-earth-based catalyst [La{N(SiMe3)2}3] (trace amounts, 24 h, 60 °C).14 Benzyl alcohol and 1-phenylethanol were fully consumed in 1 h to give the corresponding isoureas 1e,f. The synthesis of 1f is another example in which ZnEt2 overcame the limitations of the lanthanum catalyst [La{N(SiMe3)2}3],14 only able to produce trace amounts of 1f after 24 h at 60 °C. Bulky 1-adamantanol reacted with DIC to give the novel isourea 1g with 60% conversion after 6 h. Like in the reaction with tBuOH, further heating did not increase conversion, and NMR analysis of the reaction crude also suggested that 1g was obtained as an isomer mixture. The reaction of phenol with DIC led to 58% conversion to 1h. This is in line with results obtained by Cantat and co-workers with TBD-based catalysts13 because the synthesis of 1h usually requires harsh reaction conditions due to strong product inhibition, as deduced from kinetic studies.24 Similarly, the reaction with 2,6-dimethylphenol led to 68% conversion to the novel isourea 1i. Increasing steric bulk in phenols led to lower conversions, as in the reaction of DIC with 2,6-tertbutylphenol, with 14% conversion to the corresponding isourea 1j. Functional group tolerance was also tested, as in the addition of 2-(methylthio)ethanol, which rendered full conversion to 1k in 48 h at 60 °C. This new isourea was also isolated in excellent yields as colorless oil. A pyridine-based diol was found to be also active in a double O–H addition toward 2 equiv of DIC, providing the new bisisourea 1l quantitatively after 4 h, which was also isolated in very good yields as a white solid.
Table 2. Addition of Alcohols to DIC Using ZnEt2 as a Precatalysta.


Reaction conditions: the corresponding alcohol (0.50 mmol) or diol (0.25 mmol), 79.0 μL of DIC (0.50 mmol), 25.0 μL of a 1 M solution of ZnEt2 in hexane (0.025 mmol) in C6D6 (500 μL) at 60 °C, using Si(SiMe3)4 as an internal standard.
Isomer ratio.
Isolated yield from a preparative scale reaction.
Table 3. Addition of Alcohols to DTC Using ZnEt2 as a Precatalysta.


Reaction conditions: the corresponding alcohol (0.50 mmol) or diol (0.25 mmol), 112 mg of DTC (0.50 mmol), 25.0 μL of a 1 M solution of ZnEt2 in hexane (0.025 mmol) in C6D6 (500 μL), using Si(SiMe3)4 as internal standard.
Isolated yield from preparative-scale reaction.
Isomer ratio.
Alcohol additions to the more electrophilic DTC were also carried out (Table 3). As expected, milder reaction conditions were generally required for this carbodiimide as compared to DIC. Indeed, addition of methanol, ethanol, or isopropanol proceeded in 2 h or less to give full conversion to isoureas 4a–c at 25 °C. The reaction with bulkier tBuOH required somewhat harsher conditions, and full conversion was achieved after heating at 60 °C for 1.5 h. Formation of isoureas 4e,f from benzyl alcohol and 1-phenylethanol, respectively, proceeded remarkably fast, almost upon mixing time. Moreover, isourea 4f, reported here for the first time, was also isolated as a white solid in excellent yields. The more sterically demanding 1-adamantol required heating to 60 °C to reach total conversion to 4g in 7 h. Phenols were also reactive toward DTC: while unsubstituted phenol needed heating at 60 °C to give 4h, 2,6-dimethylphenol reacted essentially upon mixing with DTC to give 4i. Likewise, bulkier 2,6-ditertbutylphenol gave 88% conversion to isourea 4j right after mixing all substrates, as an isomer mixture (syn/anti). Again, longer reaction times did not increase the conversion, suggesting a thermodynamic equilibrium between O–H addition and elimination. The presence of heteroatoms was well tolerated, as proven by the reaction with 2-(methylthio)ethanol, which gave 96% conversion to the isourea 4k in 3 h at 25 °C. Finally, glycol was chosen to test the double O–H addition toward 2 equivalents of DTC with excellent results, as denoted by the full conversion to bisisourea 4l under mild conditions (25 °C, 7 h). The latter two isoureas, 4k and 4l, have also been prepared for the first time and were isolated as white solids in excellent yields.
Mechanistic Studies
To shed some light upon the plausible mechanism for the addition of alcohols to carbodiimides using ZnEt2 as a precatalyst, we decided to carry out kinetic studies. For that purpose, we chose to study the reaction between DTC and tBuOH to give 4d, which proceeded slow enough at 25 °C allowing us to use the initial reaction rate method. Thus, after a series of experiments following product formation at the early stages of the reaction, by varying the concentration of each one of the substrates or precatalyst, while keeping the others constant we found a first-order dependence both on the concentration of ZnEt2 and the concentration of DTC and an inverse first-order dependence on the concentration of the alcohol (Figure 3).
Figure 3.

Plots of initial rate versus: (a) [DTC], (b) [tBuOH]−1, (c) [ZnEt2], for the formation of 4d from tBuOH and DTC using ZnEt2 as a precatalyst.
Experiments at different temperatures allowed us to obtain the activation parameters. Thus, an activation energy (Ea) value of 83(9) KJ mol–1 was obtained from the Arrhenius plot (Figure S33, Supporting Information), and enthalpy (ΔH‡) and entropy (ΔS‡) of activation of 80(9) KJ mol–1 and -72(28) e. u. were obtained from the Eyring plot (Figure 4), the latter suggesting a highly ordered transition state in the rate-determining step of the reaction. To complete our kinetic studies, experiments employing tBuOD instead of tBuOH allowed us to obtain KIE values of 2.6(5), indicating that a step involving direct protonolysis should be the rate determining step (vide infra).
Figure 4.

Eyring plot for the formation of 4d from tBuOH and DTC using ZnEt2 as a precatalyst.
To gain further understanding of the reaction mechanism, we also performed a series of stoichiometric experiments (Supporting Information, Figures S36–S43). In the first place, we noticed that, while monitoring the catalytic additions of tBuOH toward DIC and DTC to give isoureas 1d and 4d by 1H NMR, a new set of signals appeared that could be assigned to the ethyl [ca. 1.5 (triplet) and ca. 0.6 (quadruplet) ppm] and the tert-butoxide [ca. 1.3 ppm (singlet)] groups of the tetramer [ZnEt(OtBu)]4 (6),25 the expected mono-protonolysis product in the reaction of ZnEt2 with tBuOH, suggesting that the excess tBuOH is unable to react with the second alkyl group. To confirm our proposal, a stoichiometric reaction in a preparative scale between ZnEt2 and tert-butanol was carried out, affording compound 6 in excellent yields. We also tested that the reaction product was independent of the stoichiometry employed, and even an excess tBuOH did not give the bisalkoxide derivative, confirming our prior observations during the catalysis (Scheme 1).25
Scheme 1. Reaction of ZnEt2 with tBuOH to Form 6.

These results strongly suggest that compound 6 is one of the active species in the catalytic addition of tBuOH to DTC, which was further confirmed after running a catalytic test with 6 as a catalyst under the same reaction conditions and obtaining >99% conversions in 1.5 h at 60 °C as well. Once established the plausible role of alkoxide derivative 6 in the reaction, we also confirmed that ZnEt2 did not react with equimolar amounts of DTC or DIC under the latter conditions (>1 h, 60 °C), ruling out reaction pathways involving the insertion of carbodiimides in Zn–C(alkyl) bonds. Neither did compound 6, hinting that the insertion of carbodiimides into Zn-OR bonds is thermodynamically unfavorable. However, the addition of 1 equiv of tBuOH to the latter solutions at 60 °C (for solutions with DIC) or room temperature (for solutions with DTC) gave variable amounts of the isoureas 1d (isomer mixture) and 4d, respectively, together with the formation of the expected compound 6 in the reactions with ZnEt2. In all these reactions, zinc isoureato intermediates were not detected either, again evidencing the instability of these compounds. Further attempts were made to prepare these intermediates by mixing isourea 4d with ZnEt2 in equimolar amounts and monitoring the reaction by 1H NMR. We observed, already upon mixing at room temperature, that the signals of isourea are no longer present and instead a new set of broad signals were clearly observed in the regions 7.2–6.0 and 2.2–2.1 ppm, attributed most likely to aromatic and methyl protons from p-tolyl groups of novel isoureato derivatives. Unfortunately, attempts to isolate and/or purify these species were unsuccessful, but we must point out that keeping these solutions at room temperature for prolonged times resulted in progressive disappearance of these intermediates to give mixtures containing 6 and DTC as the major products after 3 days, which evidences the presence of unstable zinc isoureato products that de-insert carbodiimide to give back the zinc alkoxide 6.
Combining the kinetics with the stoichiometric experiments we were able to propose a plausible reaction mechanism for the addition of alcohols to carbodiimides catalyzed by ZnEt2 (Scheme 2). Initially, the diethylzinc would react with one equivalent of alcohol releasing ethane to give a zinc monoalkoxide species (A in Scheme 2), which, based on the literature of zinc(II) alkyl-alkoxides, would most likely be a tetramer.25,26 A 1H NMR study of 6 at 60 °C (the highest temperature used in catalytic experiments), in C6D6, reveals only a slight shift of the signals of the ethyl and tert-butyl groups of the compound. This could indicate that the tetrameric structure is essentially maintained at this temperature, although it cannot be ruled out that the presence of an excess of alcohol and carbodiimide during the catalytic process could alter this structure. Then, [2 + 2] cycloaddition of a molecule of carbodiimide and a Zn-OR moiety of A would give rise to a N,O-isoureato intermediate (C in Scheme 1), which may isomerize or be in equilibrium with a more stable N,N′-isoureato species (C′ in Scheme 1). Although these intermediates could not be isolated, previously described N,N′-monoguanidinato,17h propiolamidinato,18 and phosphaguanidinato19 zinc compounds structurally related to C′ displayed a dimeric structure in the solid state, as evidenced by X-ray diffraction studies. Moreover, these compounds were all proposed as intermediates in the addition of amines, terminal alkynes and phosphines to carbodiimides catalyzed by ZnEt2. As already mentioned, the insertion of carbodiimide in A to give the isoureato derivatives is an equilibrium because the inverse reaction (de-insertion of carbodiimide) slowly takes place in the absence of alcohol to give back the alkoxide A. However, in the presence of excess tBuOH, protonolysis of intermediates C and/or C′ would take place to generate free isourea and regenerate alkoxide A completing the cycle. According to the KIE analysis, this would be the rate-determining step of the reaction, which is also in good agreement with the negative value of ΔS‡ obtained in the Eyring plot, as the protonolysis of the isoureato C/C′ would involve a more ordered transition state at this step. Finally, to account for the inverse first-order on the alcohol experimentally obtained, we must propose an off–cycle equilibrium involving one or more molecules of alcohol. This equilibrium might involve the coordination of one or several molecules of alcohol to Zn centers of the alkoxide derivative A to form adducts of the type B. Thus, coordination of alcohol molecules to form adducts of the type B would compete with the coordination and insertion of carbodiimide to give isoureatos C/C′. To experimentally prove this theory, we carried out the reaction of compound 6 toward 1 and 5 equiv of tBuOH and checked for significant changes in the 1H NMR spectra (Supporting Information, Figure S44). An upfield shift was especially noticeable in the signals due to the Et moiety (ca. 0.04 ppm) after adding 5 equiv of alcohol, which can be attributed to weak interactions between molecules of tBuOH and compound 6. Similar equilibria were already proposed by Eisen and co-workers with actinide alkoxides to account for inverse first-order kinetics in alcohol in the same type of catalysis.10,11b,11d,11e
Scheme 2. Proposed Mechanism for the Addition of Alcohols to Carbodiimides Catalyzed by ZnEt2. In the Scheme, [Zn] Represents One of the Reactive Centers for the Possible Aggregates of A, B, C, and C′.

Conclusions
In summary, we have expanded the catalytic repertoire of commercially available ZnEt2 for hydroelementation reactions proving this time its effectiveness for the addition of alcohols to carbodiimides. Using this precatalyst, we have prepared a wide range of isoureas under mild conditions, some of them synthesized for the first time. An initial screening showed that ZnEt2 is a very active precatalyst for the addition of MeOH to electrophilic carbodiimides such as DTC even at 25 °C and to less reactive alkylic carbodiimides, such as DIC and DCC, at 60 °C. The catalytic hydroalkoxylation of the more sterically demanding carbodiimide C(NDipp)2, not reported until now, was even possible under slightly harsher conditions (80 °C, 5 days). Afterward, a set of different alkylic and arylic alcohols, with different steric demands and electronic properties, were tested toward DIC and DTC. Again, ZnEt2 proved to be more active toward DTC, reaching very high conversions under mild conditions (25 °C) even with the most challenging and sterically demanding substrates, whereas the less electrophilic DIC required higher temperatures (60 °C) and the conversion only dropped substantially with phenols and very bulky alcohols. The presence of other functionalities on the alcohols, such as thiolates (−SR), was well tolerated by the catalyst for both carbodiimides.
Kinetic and stoichiometric experiments combined allowed us to propose a plausible mechanism for these catalytic reactions, involving an alkyl-alkoxide zinc intermediate (detected and isolated) and unstable alkyl-isoureato zinc compounds (detected by NMR). The protonolysis of the latter species turned out to be the rate-determining step of the reaction, as determined by KIE experiments. Additionally, an inverse first-order dependence was found in the alcohol concentration, suggesting an off–cycle equilibrium probably involving weak coordination of alcohol molecules to the alkoxide compound.
Acknowledgments
We thank MCIN/AEI/10.13039/501100011033 (project numbers PID2020-117353GB-I00, PGC2018-097366-B-I00, and RED2018-102387-T), the Junta de Comunidades de Castilla-La Mancha and EU for financial support through the European Regional Development Fund (ERDF, project SBPLY/19/180501/000137), the Universidad de Castilla-La Mancha (2021-AYUDA-32015) for financial support, and the SCBI of the Universidad de Málaga, Spain, for access to computing facilities.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.2c00372.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
CCDC 2172359 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via ww.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Supplementary Material
References
- a Mathias L. J. Esterification and Alkylation Reactions Employing Isoureas. Synthesis 1979, 1979, 561–576. 10.1055/s-1979-28761. [DOI] [Google Scholar]; b Mathias L. J. Synthetic Applications of Isoureas. A Review. Org. Prep. Proced. Int. 1980, 12, 309–326. 10.1080/00304948009356484. [DOI] [Google Scholar]; c Bakibaev A. A.; Shtrykova V. V. Isoureas: Synthesis, Properties, and Applications. Russ. Chem. Rev. 1995, 64, 929–938. 10.1070/rc1995v064n10abeh000185. [DOI] [Google Scholar]; d Crosignani S.; White P. D.; Linclau B. Microwave-Accelerated O-Alkylation of Carboxylic Acids with O-Alkylisoureas. Org. Lett. 2002, 4, 2961–2963. 10.1021/ol0263705. [DOI] [PubMed] [Google Scholar]; e Crosignani S.; White P. D.; Steinauer R.; Linclau B. Polymer-Supported O-Benzyl and O-Allylisoureas: Convenient Preparation and Use in Ester Synthesis from Carboxylic Acids. Org. Lett. 2003, 5, 853–856. 10.1021/ol0275062. [DOI] [PubMed] [Google Scholar]; f Crosignani S.; White P. D.; Linclau B. Polymer-Supported O-Alkylisoureas: Useful Reagents for the O-Alkylation of Carboxylic Acids. J. Org. Chem. 2004, 69, 5897–5905. 10.1021/jo049239e. [DOI] [PubMed] [Google Scholar]; g Zohrabi-Kalantari V.; Heidler P.; Larsen T.; Link A. O,N,N’-Trialkylisoureas as Mild Activating Reagents for N-Acylsulfonamide Anchors. Org. Lett. 2005, 7, 5665–5667. 10.1021/ol052351u. [DOI] [PubMed] [Google Scholar]; h Chighine A.; Crosignani S.; Arnal M.-C.; Bradley M.; Linclau B. Microwave-Assisted Ester Formation Using O-Alkylisoureas: A Convenient Method for the Synthesis of Esters with Inversion of Configuration. J. Org. Chem. 2009, 74, 4753–4762. 10.1021/jo900476y. [DOI] [PubMed] [Google Scholar]; i Liu Y. Isoureas: Versatile Alkylation Reagents in Organic Chemistry. Synlett 2009, 2009, 1353–1354. 10.1055/s-0029-1216653. [DOI] [Google Scholar]
- Ogawa M.; Matsui T.; Oyamada K.; Tobitsuka J. Isourea and Triazinone Derivatives as Related to their Ability to Atimulate Rice Seedling Growth. Plant Cell Physiol. 1977, 18, 841–848. 10.1093/oxfordjournals.pcp.a075499. [DOI] [Google Scholar]
- a García-Moreno M. I.; Díaz-Pérez P.; Ortiz Mellet C.; García Fernández J. M. Synthesis and Evaluation of Isourea-Type Glycomimetics Related to the Indolizidine and Trehazolin Glycosidase Inhibitor Families. J. Org. Chem. 2003, 68, 8890–8901. 10.1021/jo034673m. [DOI] [PubMed] [Google Scholar]; b Trapero A.; Alfonso I.; Butters T. D.; Llebaria A. Polyhydroxylated bicyclic isoureas and guanidines are potent glucocerebrosidase inhibitors and nanomolar enzyme activity enhancers in Gaucher cells. J. Am. Chem. Soc. 2011, 133, 5474–5484. 10.1021/ja111480z. [DOI] [PubMed] [Google Scholar]; c Sevšek A.; Celan M.; Erjavec B.; Quarles van Ufford L.; Sastre Torano J.; Moret E. E.; Pieters R. J.; Martin N. I. Bicyclic isoureas derived from 1-deoxynojirimycin are potent inhibitors of beta-glucocerebrosidase. Org. Biomol. Chem. 2016, 14, 8670–8673. 10.1039/C6OB01735E. [DOI] [PubMed] [Google Scholar]
- Badache L.; Boschet F.; Lehanine Z.; Boutevin B.; Ameduri B. Synthesis and surface properties of a series of surfactants based on O-alkyl and O-perfluoro-N,N′-diisopropylisoureas. J. Fluorine Chem. 2011, 132, 382–388. 10.1016/j.jfluchem.2011.03.016. [DOI] [Google Scholar]
- Werber M. M. Nucleophilicity of isourea linkages in substituted agaroses. Anal. Biochem. 1976, 76, 177–183. 10.1016/0003-2697(76)90276-1. [DOI] [PubMed] [Google Scholar]
- Forman S. E.; Erickson C. A.; Adelman H. Chemistry of Pseudoureas1. J. Org. Chem. 1963, 28, 2653–2658. 10.1021/jo01045a041. [DOI] [Google Scholar]
- a Jiang T.; Gu Z. Y.; Yin L.; Wang S. Y.; Ji S. J. Cobalt(II)-Catalyzed Isocyanide Insertion Reaction with Sulfonyl Azides in Alcohols: Synthesis of Sulfonyl Isoureas. J. Org. Chem. 2017, 82, 7913–7919. 10.1021/acs.joc.7b01127. [DOI] [PubMed] [Google Scholar]; b Bu X. B.; Zhang Z. X.; Peng Q. Q.; Xu X.; Zhao Y. L. Rhodium-Catalyzed Tandem Reaction of Isocyanides with Azides and Oxygen Nucleophiles: Synthesis of Isoureas. J. Org. Chem. 2019, 84, 53–59. 10.1021/acs.joc.8b01842. [DOI] [PubMed] [Google Scholar]; c Cao M.; Liu L.; Tang S.; Peng Z.; Wang Y. Palladium-Catalyzed Solvent-Controlled Selective Synthesis of Acyl Isoureas and Imides from Amides, Isocyanides, Alcohols and Carboxylates. Adv. Synth. Catal. 2019, 361, 1887–1895. 10.1002/adsc.201801245. [DOI] [Google Scholar]; d Mishra D.; Phukan P. A Unified Approach for the Synthesis of Isourea and Isothiourea from Isonitrile and N,N-Dibromoarylsulfonamides. J. Org. Chem. 2021, 86, 17581–17593. 10.1021/acs.joc.1c01578. [DOI] [PubMed] [Google Scholar]
- a Däbritz E. Snytheses and Reactions of O,N,N′-Trisubstituted Isoureas. Angew. Chem., Int. Ed. Engl. 1966, 5, 470–477. 10.1002/anie.196604701. [DOI] [Google Scholar]; b Williams A.; Ibrahim I. T. Carbodiimide chemistry: recent advances. Chem. Rev. 1981, 81, 589–636. 10.1021/cr00046a004. [DOI] [Google Scholar]
- a Schmidt E.; Moosmüller F. Zur Kenntnis aliphatischer Carbodiimide, IX. Mitt. Liebigs Ann. Chem. 1955, 597, 235–240. 10.1002/jlac.19555970307. [DOI] [Google Scholar]; b Schmidt E.; Carl W. Zur Kenntnis aliphatischer Carbodiimide, XII. Liebigs Ann. Chem. 1961, 639, 24–31. 10.1002/jlac.19616390104. [DOI] [Google Scholar]; c Schmidt E.; Däbritz E.; Thulke K.; Grassmann E. Zur Kenntnis aliphatischer Carbodiimide, XIV. Liebigs Ann. Chem. 1965, 685, 161–166. 10.1002/jlac.19656850120. [DOI] [Google Scholar]; d Vowinkel E.; Büthe I. Eine einfache Methode zur Reduktion von Alkoholen zu Kohlenwasserstoffen. Chem. Ber. 1974, 107, 1353–1359. 10.1002/cber.19741070432. [DOI] [Google Scholar]; e Heinisch G.; Matuszczak B.; Rakowitz D.; Mereiter K. Diazine-derived guanidines, isothioureas, and isoureas: Synthesis and attempts of configurational assignment. J. Heterocycl. Chem. 2002, 39, 695–702. 10.1002/jhet.5570390414. [DOI] [Google Scholar]; f Crosignani S.; Young A. C.; Linclau B. Synthesis of 2-oxazolines mediated by N,N′-diisopropylcarbodiimide. Tetrahedron Lett. 2004, 45, 9611–9615. 10.1016/j.tetlet.2004.10.143. [DOI] [Google Scholar]; g Aresta M.; Dibenedetto A.; Fracchiolla E.; Giannoccaro P.; Pastore C.; Pápai I.; Schubert G. Mechanism of Formation of Organic Carbonates from Aliphatic Alcohols and Carbon Dioxide under Mild Conditions Promoted by Carbodiimides. DFT Calculation and Experimental Study. J. Org. Chem. 2005, 70, 6177–6186. 10.1021/jo050392y. [DOI] [PubMed] [Google Scholar]
- Batrice R. J.; Kefalidis C. E.; Maron L.; Eisen M. S. Actinide-Catalyzed Intermolecular Addition of Alcohols to Carbodiimides. J. Am. Chem. Soc. 2016, 138, 2114–2117. 10.1021/jacs.5b12731. [DOI] [PubMed] [Google Scholar]
- a Ghatak T.; Fridman N.; Eisen M. S. Actinide Complexes Possessing Six-Membered N-Heterocyclic Iminato Moieties: Synthesis and Reactivity. Organometallics 2017, 36, 1296–1302. 10.1021/acs.organomet.7b00037. [DOI] [Google Scholar]; b Liu H.; Fridman N.; Tamm M.; Eisen M. S. Catalytic Addition of Alcohols to Carbodiimides Mediated by Benzimidazolin-2-iminato Actinide Complexes. Organometallics 2017, 36, 4600–4610. 10.1021/acs.organomet.7b00432. [DOI] [Google Scholar]; c Liu H.; Fridman N.; Tamm M.; Eisen M. S. Addition of E-H (E = N, P, C, O, S) Bonds to Heterocumulenes Catalyzed by Benzimidazolin-2-iminato Actinide Complexes. Organometallics 2017, 36, 3896–3903. 10.1021/acs.organomet.7b00502. [DOI] [Google Scholar]; d Liu H.; Khononov M.; Fridman N.; Tamm M.; Eisen M. S. Catalytic Addition of Alcohols into Carbodiimides Promoted by Organoactinide Complexes. Inorg. Chem. 2017, 56, 3153–3157. 10.1021/acs.inorgchem.7b00357. [DOI] [PubMed] [Google Scholar]; e Makarov K.; Saha S.; Ghatak T.; Fridman N.; Eisen M. S. Remodeling of N-Heterocyclic Iminato Ligand Frameworks for the Facile Synthesis of Isoureas from Alcohols and Carbodiimides Promoted by Organoactinide (Th, U) Complexes. ACS Omega 2021, 6, 14692–14700. 10.1021/acsomega.1c01836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khononov M.; Liu H.; Fridman N.; Tamm M.; Eisen M. S. Benzimidazolin-2-iminato Hafnium Complexes: Synthesis, Characterization, and Catalytic Addition of Alcohols to Carbodiimides. Organometallics 2020, 39, 3021–3033. 10.1021/acs.organomet.0c00384. [DOI] [Google Scholar]
- Imberdis A.; Lefèvre G.; Thuéry P.; Cantat T. Metal-Free and Alkali-Metal-Catalyzed Synthesis of Isoureas from Alcohols and Carbodiimides. Angew. Chem., Int. Ed. 2018, 57, 3084–3088. 10.1002/anie.201711737. [DOI] [PubMed] [Google Scholar]
- Hu L.; Lu C.; Zhao B.; Yao Y. Intermolecular addition of alcohols to carbodiimides catalyzed by rare-earth metal amides. Org. Chem. Front. 2018, 5, 905–908. 10.1039/c7qo00991g. [DOI] [Google Scholar]
- Bhattacharjee J.; Harinath A.; Banerjee I.; Nayek H. P.; Panda T. K. Highly Active Dinuclear Titanium(IV) Complexes for the Catalytic Formation of a Carbon-Heteroatom Bond. Inorg. Chem. 2018, 57, 12610–12623. 10.1021/acs.inorgchem.8b01766. [DOI] [PubMed] [Google Scholar]
- Wells L. A.; Gau M. R.; Peddicord M. B.; Bostwick K. F.; Kozlowski M. C.; Zacuto M. J. Copper-Catalyzed Addition of Alcohols to Carbodiimides: Oxygen as an Accelerant. Org. Process Res. Dev. 2022, 26, 1803–1811. 10.1021/acs.oprd.1c00465. [DOI] [Google Scholar]
- a Pacheco-Liñán P. J.; Alonso-Moreno C.; Carrillo-Hermosilla F.; Garzón-Ruiz A.; Martín C.; Sáez C.; Albaladejo J.; Bravo I. Novel Fluorescence Guanidine Molecules for Selective Sulfate Anion Detection in Water Complex Samples over a Wide pH Range. ACS Sens. 2021, 6, 3224–3233. 10.1021/acssensors.1c00876. [DOI] [PubMed] [Google Scholar]; b Mesias-Salazar A.; Trofymchuk O. S.; Daniliuc C. G.; Antiñolo A.; Carrillo-Hermosilla F.; Nachtigall F. M.; Santos L. S.; Rojas R. S. Copper (II) as catalyst for intramolecular cyclization and oxidation of (1,4-phenylene)bisguanidines to benzodiimidazole-diylidenes. J. Catal. 2020, 382, 150–154. 10.1016/j.jcat.2019.12.002. [DOI] [Google Scholar]; c Mesías-Salazar Á.; Martínez J.; Rojas R. S.; Carrillo-Hermosilla F.; Ramos A.; Fernández-Galán R.; Antiñolo A. Aromatic guanidines as highly active binary catalytic systems for the fixation of CO2 into cyclic carbonates under mild conditions. Catal. Sci. Technol. 2019, 9, 3879–3886. 10.1039/C9CY00667B. [DOI] [Google Scholar]; d Pacheco-Liñán P. J.; Fernández-Sainz J.; Bravo I.; Garzón-Ruiz A.; Alonso-Moreno C.; Carrillo-Hermosilla F.; Antiñolo A.; Albaladejo J. Guanidine Substitutions in Naphthyl Systems to Allow a Controlled Excited-State Intermolecular Proton Transfer: Tuning Photophysical Properties in Aqueous Solution. J. Phys. Chem. C 2018, 122, 9363–9373. 10.1021/acs.jpcc.8b02576. [DOI] [Google Scholar]; e Nieto D.; Bruña S.; González-Vadillo A. M.; Perles J.; Carrillo-Hermosilla F.; Antiñolo A.; Padrón J. M.; Plata G. B.; Cuadrado I. Catalytically Generated Ferrocene-Containing Guanidines as Efficient Precursors for New Redox-Active Heterometallic Platinum(II) Complexes with Anticancer Activity. Organometallics 2015, 34, 5407–5417. 10.1021/acs.organomet.5b00751. [DOI] [Google Scholar]; f Elorriaga D.; Carrillo-Hermosilla F.; Antiñolo A.; López-Solera I.; Fernández-Galán R.; Villaseñor E. Unexpected mild C-N bond cleavage mediated by guanidine coordination to a niobium iminocarbamoyl complex. Chem. Commun. 2013, 49, 8701–8703. 10.1039/c3cc44952a. [DOI] [PubMed] [Google Scholar]; g Elorriaga D.; Carrillo-Hermosilla F.; Antiñolo A.; López-Solera I.; Menot B.; Fernández-Galán R.; Villaseñor E.; Otero A. New Alkylimido Niobium Complexes Supported by Guanidinate Ligands: Synthesis, Characterization, and Migratory Insertion Reactions. Organometallics 2012, 31, 1840–1848. 10.1021/om201192w. [DOI] [Google Scholar]; h Alonso-Moreno C.; Carrillo-Hermosilla F.; Garcés A.; Otero A.; López-Solera I.; Rodríguez A. M.; Antiñolo A. Simple, Versatile, and Efficient Catalysts for Guanylation of Amines. Organometallics 2010, 29, 2789–2795. 10.1021/om1003122. [DOI] [Google Scholar]
- Martínez A.; Moreno-Blázquez S.; Rodríguez-Diéguez A.; Ramos A.; Fernández-Galán R.; Antiñolo A.; Carrillo-Hermosilla F. Simple ZnEt2 as a catalyst in carbodiimide hydroalkynylation: structural and mechanistic studies. Dalton Trans. 2017, 46, 12923–12934. 10.1039/C7DT02700A. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Ma X.; Yan B.; Ni C.; Yu H.; Yang Z.; Roesky H. W. An efficient catalytic method for hydrophosphination of heterocumulenes with diethylzinc as precatalyst without a solvent. Dalton Trans. 2021, 50, 15488–15492. 10.1039/d1dt02706a. [DOI] [PubMed] [Google Scholar]
- Hartke K.; Radau M. Zur Addition von Alkoholen an Carbodiimide unter Tetrafluorborsäure-Katalyse. 10. Mitt. über Carbodiimide. Arch. Pharm. 1972, 305, 654–664. 10.1002/ardp.19723050905. [DOI] [PubMed] [Google Scholar]
- Minamisono K.; Suzuki M.; Yoshimura M.; Kuroda T.. Isourea derivatives for reducing terminal carboxyl concentration in polyesters. Japan JP 07145139 A, 1995.
- Ghosh R.; Samuelson A. G. Catalytic metathesis of carbon dioxide with heterocumulenes mediated by titanium isopropoxide. Chem. Commun. 2005, 2017–2019. 10.1039/b417713d. [DOI] [PubMed] [Google Scholar]
- West K. R.; Bake K. D.; Otto S. Dynamic Combinatorial Libraries of Disulfide Cages in Water. Org. Lett. 2005, 7, 2615–2618. 10.1021/ol0507524. [DOI] [PubMed] [Google Scholar]
- Tate J. A.; Hodges G.; Lloyd-Jones G. C. O-Phenylisourea Synthesis and Deprotonation: Carbodiimide Elimination Precludes the Reported Chapman Rearrangement. Eur. J. Org. Chem. 2016, 2016, 2821–2827. 10.1002/ejoc.201600386. [DOI] [Google Scholar]
- a Coates G. E.; Ridley D. 341. Alkoxy-, thio-, and amino-derivatives of methylzinc. J. Chem. Soc. 1965, 341, 1870–1877. 10.1039/jr9650001870. [DOI] [Google Scholar]; b Matsui Y.; Kamiya K.; Nishikawa M.; Tomiie Y. The Crystal Structure of Ethylzinc t-Butoxide. Bull. Chem. Soc. Jpn. 1966, 39, 1828. [Google Scholar]; c Moratti S. C.; Simpson J. Tetra-μ3-tert-butoxo-tetrakis[ethylzinc(II)]. Acta Crystallogr., Sect. E: Struct. Rep. Online 2007, 63, m2444. 10.1107/s1600536807042018. [DOI] [Google Scholar]
- a Noltes J. G.; Boersma J. Investigations on organozinc compounds IX. Coordination chemistry of organozinc compounds RZnX: organozinc derivatives of tert-butanol, some phenols, diethylhydroxylamine and some oximes. J. Organomet. Chem. 1968, 12, 425–431. 10.1016/s0022-328x(00)88695-7. [DOI] [Google Scholar]; b Geerts R. L.; Huffman J. C.; Caulton K. G. Soluble zinc bis(aryloxides). Inorg. Chem. 1986, 25, 1803–1805. 10.1021/ic00231a018. [DOI] [Google Scholar]; c Boyle T. J.; Bunge S. D.; Andrews N. L.; Matzen L. E.; Sieg K.; Rodriguez M. A.; Headley T. J. Precursor Structural Influences on the Final ZnO Nanoparticle Morphology from a Novel Family of Structurally Characterized Zinc Alkoxy Alkyl Precursors. Chem. Mater. 2004, 16, 3279–3288. 10.1021/cm0493997. [DOI] [Google Scholar]; d Lewinski J.; Dutkiewicz M.; Lesiuk M.; Sliwinski W.; Zelga K.; Justyniak I.; Lipkowski J. Solid-state conversion of the solvated dimer [{tBuZn(mu-OtBu)(thf)}2] into a long overlooked trimeric [{tBuZnOtBu}3] species. Angew. Chem., Int. Ed. 2010, 122, 8442–8445. 10.1002/ange.201004504. [DOI] [PubMed] [Google Scholar]; e Mąkolski Ł.; Szejko V.; Zelga K.; Tulewicz A.; Bernatowicz P.; Justyniak I.; Lewiński J. Unravelling Structural Mysteries of Simple Organozinc Alkoxides. Chem.—Eur. J. 2021, 27, 5666–5674. 10.1002/chem.202004222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.