

Abstract
Vascular smooth muscle cells (VSMCs), a highly mosaic tissue, arise from multiple distinct embryonic origins and populate different regions of our vascular network with defined boundaries. Accumulating evidence has revealed that the heterogeneity of VSMC origins contributes to region-specific vascular diseases such as atherosclerosis and aortic aneurysm. These findings highlight the necessity of taking into account lineage-dependent responses of VSMCs to common vascular risk factors when studying vascular diseases. This chapter describes a reproducible, stepwise protocol for the generation of isogenic VSMC subtypes originated from proepicardium, second heart field, cardiac neural crest, and ventral somite using human induced pluripotent stem cells. By leveraging this robust induction protocol, patient-derived VSMC subtypes of desired embryonic origins can be generated for disease modeling as well as drug screening and development for vasculopathies with regional susceptibility.
Keywords: Induced pluripotent stem cells, Smooth muscle cell, Embryonic origin, Vascular disease, Regional susceptibility
1. Introduction
Vascular smooth muscle cells (VSMCs), the predominant cell type in medium and large blood vessels, are essential for providing vasomotor control of circulation to regulate blood pressure and for maintaining cardiovascular health through cell–cell and cell–matrix interactions [1, 2]. However, unlike other myogenic cell types, VSMCs retain a high degree of plasticity and can undergo phenotypic switching from a quiescent and contractile state to a proliferative, proinflammatory, and synthetic state in response to factors such as mechanical stretch, growth factors, and inflammatory mediators [2, 3]. Phenotypically modified VSMCs play a key role in arterial remodeling, which can consequently lead to a variety of region-specific cardiovascular complications such as atherosclerosis, myocardial infarction, stroke, in-stent restenosis, calcification, and aortic aneurysm [1].
Fate mapping studies on vertebrate embryos have identified that VSMCs with distinct embryonic origins exhibit a highly mosaic distribution along the aorta and its distal branches [4]. Specifically, VSMCs are derived from proepicardium (PE)/epicardium (EPI) in coronary arteries [5, 6]; from second heart field (SHF) in the aortic root and the outer layer of the ascending aorta [7]; from cardiac neural crest (CNC) in the inner layer of the ascending aorta, the aortic arch and branches, and the pulmonary trunk [8]; from ventral somite (VS) in the descending aorta [9, 10]; and from tissue-specific mesothelium (progenies of splanchnic mesoderm) in most coelomic organs such as gut, liver, and kidneys [11–13].
The heterogeneity of VSMC embryonic origins, in addition to other factors such as variable hemodynamics and vascular structures, contributes to a propensity for vascular diseases to exhibit regional differences. For example, a recent study showed that the development of aortic root aneurysm in a mouse model of Loeys–Dietz syndrome was the result of opposing Smad signaling activities in CNC- versus SHF-derived VSMC subtypes residing in the aortic root [14]. Likewise, single-cell transcriptomics of mouse aortas showed that the expression levels of genes associated with inflammation, adhesion, and migration are much higher in the aortic arch (the athero-prone region) SMCs than in the descending aorta (the athero-resistant region) SMCs [15].
The signaling pathways involved in diversifying embryonic origin-specific SMCs during vascular development can recur in arterial remodeling postnatally [16–18]. Therefore, a better understanding of the key molecular events that govern the diverse developmental trajectories of VSMCs under the same genetic background will help reveal the heterogeneous adaptive or pathogenic responses of anatomically distinct arteries to common risk factors. In this regard, human induced pluripotent stem cell (iPSC)-derived isogenic embryonic origin-specific VSMC subtypes represent a promising in vitro platform for understanding and modeling region-specific vascular diseases, because they can overcome the challenges of procuring arterial tissues from different segments of the same patients for comparative studies. Moreover, the scalable nature of deriving VSMCs from human iPSCs also opens up an avenue for the discovery of therapeutic targets to prevent or treat vascular diseases using high-throughput drug screening [19, 20].
Here, we detail a stepwise differentiation protocol to derive embryonic origin-specific VSMC subtypes from human iPSCs via four intermediate lineages, namely: PE/EPI, SHF, CNC, and VS. These four intermediate lineages are generated in a chemically defined medium (CDM) using different combinations of growth factors and small molecules. We temporally control differentiation of these lineages by mirroring their respective sequential signals activated at each lineage step during embryo development (Fig. 1). This stepwise differentiation approach yields highly pure, lineage-specific intermediate cell types and VSMC subtypes without the need for cell sorting to enrich target cell populations (Fig. 2). We further show that treating proliferating human iPSC-derived VSMC subtypes with a MEK inhibitor for 6 days can induce a more mature and contractile phenotype (smooth muscle myosin heavy chain 11, MYH11+) of these cells (Fig. 3). This robust and developmental trajectory-defined differentiation protocol can be deployed for the mass production of isogenic embryonic origin-specific VSMC subtypes from patient iPSCs for vascular disease modeling as well as drug screening and development.
Fig. 1.
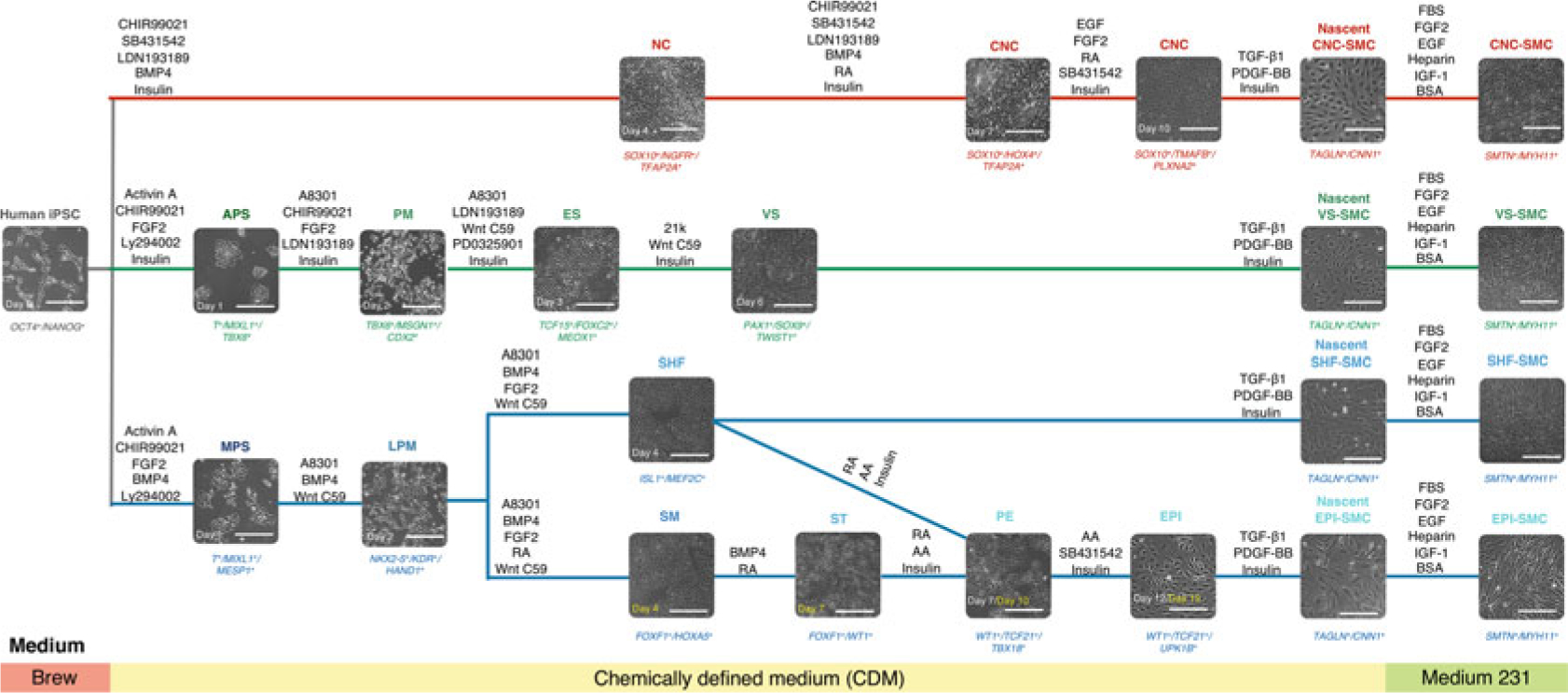
Schematic diagram of the conditions for deriving embryonic origin-specific VSMCs from human iPSCs. Lineage-specific intermediate cell types that give rise to each VSMC subtype are differentiated in a stepwise fashion. Each intermediate cell population is subjected to TGF-β1 and PDGF-BB treatment for 6 days before switching to a smooth muscle cell growth medium (Medium 231) for another 14–21 days. Phenotypic markers for specific cell types are represented in italics at the bottom of each bright-field image. NC neural crest, CNC cardiac neural crest, APS anterior primitive streak, PM paraxial mesoderm, ES early somite, VS ventral somite, MPS mid-primitive streak, LPM lateral plate mesoderm, SHF second heart field, SM splanchnic mesoderm, ST septum transversum, PE proepicardium, EPI epicardium. Scale bars represent 50 μm
Fig. 2.
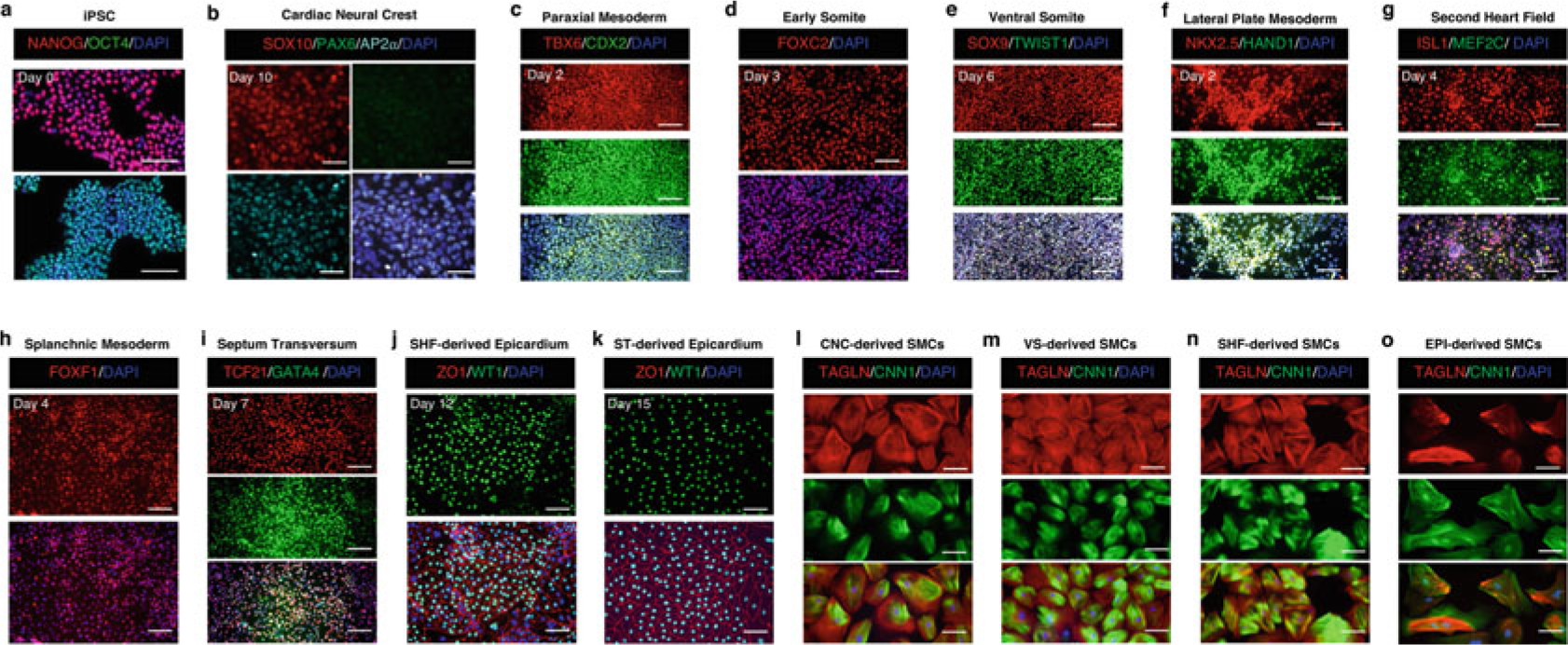
Representative immunofluorescence images showing phenotypic markers for specific cell types during embryonic origin-specific VSMC differentiation. (a–k) The time point labeled on each set of cell marker-positive fluorescence images is consistent with that shown on the bright-field image of the same cell type in Fig. 1. (l–o) Embryonic origin-specific VSMCs are positive for TAGLN and CNN1 after being cultured in the SMC growth medium for 14 days. Scale bars represent 100 μm
Fig. 3.
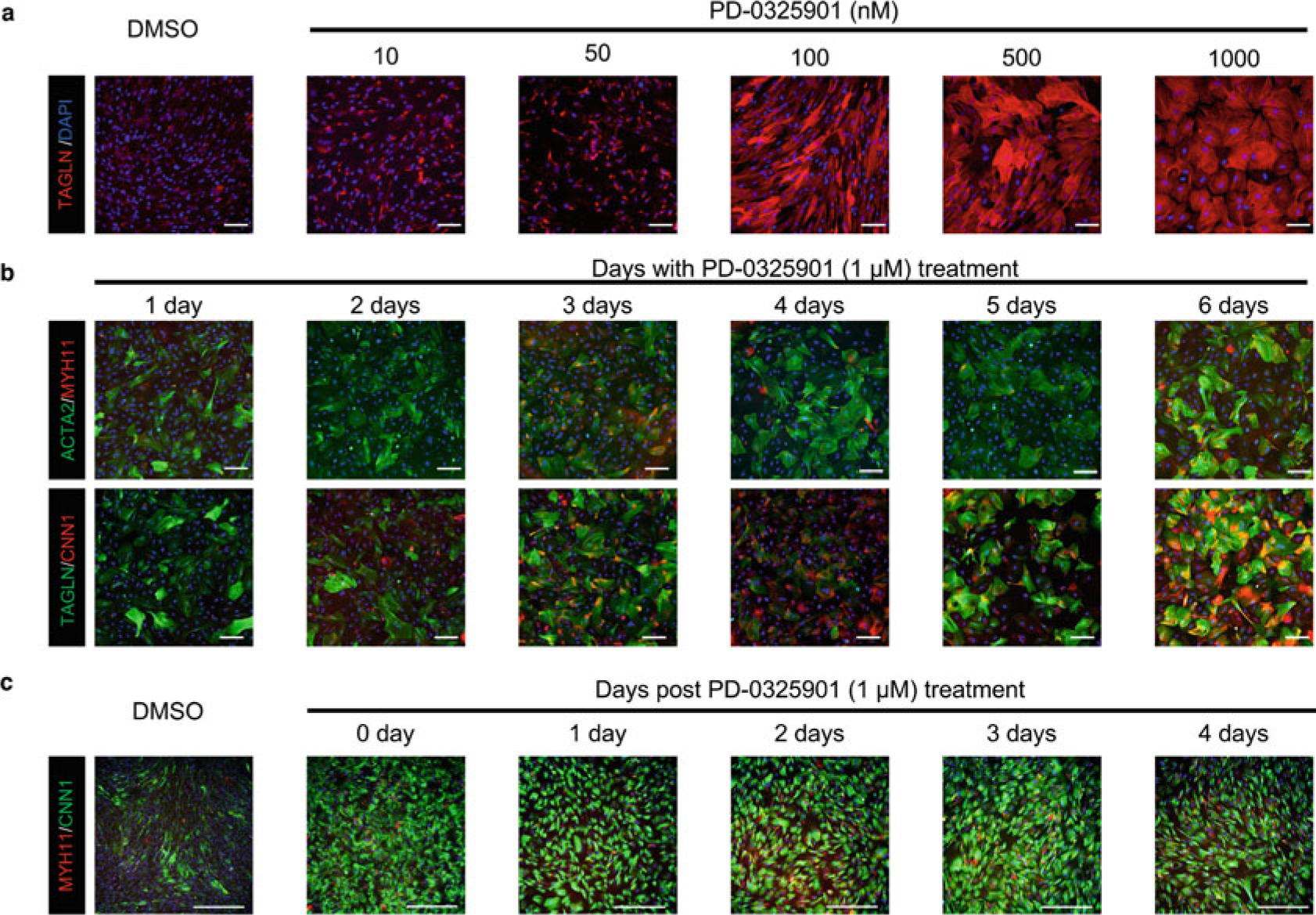
PD0325901, a MEK inhibitor, can upregulate the expression of SMC contractile proteins. (a) Human iPSC-derived VSMCs show a dose-dependent response to PD0325901-mediated upregulation of SMC contractile proteins. (b) Human iPSC-derived VSMCs show a time-dependent upregulation of SMC contractile proteins with the treatment of PD0325901 at a concentration of 1 μM. (c) Human iPSC-derived VSMCs can preserve a contractile phenotype for at least 4 days after the withdrawal of PD0325901. Because PD0325901 application demonstrates the same effect on different VSMC subtypes, only representative images generated from CNC-specific VSMCs are shown in this figure. Scale bars represent 100 μm
2. Materials
2.1. Cell Culture Reagents
Stem cell growth medium (for example, Stemmacs™ iPS-Brew XF, Miltenyi Biotech, cat # 130-104-368).
DMEM/F-12, HEPES.
Ham’s F-12 Nutrient Mix.
IMDM.
Smooth muscle cell growth basal medium (for example, Medium 231, Gibco, cat # M231500).
Smooth muscle growth supplement (for example, SMGS, Gibco, cat # S00725).
Gentamicin.
Distilled H2O.
Dimethyl sulfoxide (DMSO).
DPBS without calcium and magnesium.
Chemically defined lipid concentrate.
Glutamax.
Insulin.
Polyvinyl alcohol (PVA) (see Note 1).
Transferrin (30 mg/ml).
Bovine serum albumin (BSA, 0.1% wt/vol).
Monothioglycerol.
Gelatin solution (0.1% wt/vol).
Matrigel Growth Factor Reduced Basement Membrane Matrix, phenol red-free, LDEV-free (0.4% wt/vol, Corning, cat # 356231).
Accutase solution.
Gentle Cell Dissociation Reagent.
Bambanker (Wako Chemicals USA, cat # 30214681).
2.2. Small Molecules
ALK5 inhibitor A83-01 (1 mM).
ALK5 inhibitor SB431542 (10 mM).
L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate (25 mg/ml).
BMP inhibitor LDN193189 2HCl (1 mM).
GSK3 inhibitor CHIR99021 (10 mM).
Hedgehog activator SAG 21k (5 μM).
MEK inhibitor PD0325901 (1 mM).
PI3K-AKT inhibitor LY294002 (10 mM).
Retinoic acid (10 mM).
ROCK inhibitor Y27632 2HCl (10 mM).
Wnt inhibitor C59 (1 mM).
2.3. Growth Factors
Recombinant Human/Murine/Rat Activin A (10 μg/ml).
Recombinant Human BMP4 (100 μg/ml).
Recombinant Human EGF (100 μg/ml).
Recombinant Human FGF2 (100 μg/ml).
Recombinant Human PDGF-BB (10 μg/ml).
Recombinant Human TGF-β1 (2 μg/ml).
2.4. Preparation of Induction andMaintenanceMedia
Basal chemically defined medium (CDM), 500 ml: consisting of 240 ml of IMDM (50% vol/vol), 240 ml of Ham’s F-12 Nutrient Mix (50% vol/vol), 5 ml of chemically defined lipid concentrate (1% vol/vol); 5 ml of Glutamax (2 mM), 5 ml of PVA (1 mg/ml), 250 μl of transferrin (15 μg/ml), and 20 μl of monothioglycerol (450 μM). Basal CDM is sterile filtered after preparation and can be stored for up to 4 weeks at 4 °C (see Note 2).
Neural crest (NC) induction medium, 50 ml: consisting of 50 ml of CDM, 5 μl of CHIR99021 (1 μM), 10 μl of SB431542 (2 μM), 12.5 μl of LDN193189 (250 nM), 7.5 μl of BMP4 (15 ng/ml), and 35 μl of insulin (7 μg/ml).
Neural crest (NC) maintenance medium, 50 ml: consisting of 50 ml of CDM, 5 μl of FGF2 (10 ng/ml), 5 μl of EGF (10 ng/ml), 10 μl of SB431542 (2 μM), and 35 μl of insulin (7 μg/ml).
Anterior primitive streak (APS) induction medium, 50 ml: consisting of 50 ml of CDM, 150 μl of Activin A (30 ng/ml), 20 μl of CHIR99021 (4 μM), 10 μl of FGF2 (20 ng/ml), 10 μl of LY294002 (2 μM), and 35 μl of insulin (7 μg/ml).
Paraxial mesoderm (PM) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of A83-01 (1 μM), 15 μl of CHIR99021 (3 μM), 12.5 μl of LDN192189 (250 nM), 10 μl of FGF2 (20 ng/ml), and 35 μl of insulin (7 μg/ml).
Early somite (ES) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of A83-01 (1 μM), 12.5 μl of LDN193189 (250 nM), 50 μl of C59 (1 μM), 25 μl of PD0325901 (500 nM), and 35 μl of insulin (7 μg/ml).
Ventral somite (VS) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of SAG 21 k (5 nM), 50 μl of C59 (1 μM), and 35 μl of insulin (7 μg/ml).
Mid-primitive streak (MPS) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of Activin A (10 ng/ml), 30 μl of CHIR99021 (6 μM), 25 μl of BMP4 (50 ng/ml), 10 μl of FGF2 (20 ng/ml), and 10 μl of LY294002 (2 μM).
Lateral plate mesoderm (LPM) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of A83-01 (1 μM), 15 μl of BMP4 (30 ng/ml), and 50 μl of C59 (1 μM).
Second heart field (SHF) induction medium (see Note 3), 50 ml: consisting of 50 ml of CDM, 50 μl of A83-01 (1 μM), 15 μl of BMP4 (30 ng/ml), 50 μl of C59 (1 μM), and 10 μl of FGF2 (20 ng/ml).
Splanchnic mesoderm (SM) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of A83-01 (1 μM), 15 μl of BMP4 (30 ng/ml), 50 μl of C59 (1 μM), 10 μl of FGF2 (20 ng/ml), and 10 μl of retinoic acid (2 μM).
Septum transversum (ST) induction medium, 50 ml: consisting of 50 ml of CDM, 10 μl of retinoic acid (2 μM), and 20 μl of BMP4 (40 ng/ml).
Proepicardial cell (PE) induction medium, 50 ml: consisting of 50 ml of CDM, 200 μl of ascorbic acid (100 μg/ml), 10 μl of retinoic acid (2 μM), and 35 μl of insulin (7 μg/ml).
Epicardial cell (EPI) maintenance medium, 50 ml: consisting of 50 ml of CDM, 200 μl of ascorbic acid (100 μg/ml), 10 μl of SB431542 (2 μM), and 35 μl of insulin (7 μg/ml).
Smooth muscle cell (SMC) induction medium, 50 ml: consisting of 50 ml of CDM, 50 μl of TGF-β1 (2 ng/ml), 50 μl of PDGF-BB (10 ng/ml), and 35 μl of insulin (7 μg/ml).
Smooth muscle cell (SMC) maturation medium, 50 ml: consisting of 50 ml of Medium 231, 2.5 ml of SMGS, and 50 μl of PD0325901 (1 μM).
3. Methods
3.1. Maintenance of Human iPSCs
Human iPSCs are routinely maintained under feeder-free conditions in Stemmacs™ iPS-Brew XF (Brew) medium. Karyotype analysis should be performed periodically to ensure high quality of iPSCs. Mycoplasma contamination tests should be performed on a weekly basis, and mycoplasma positive cells should be discarded immediately. All intermediate progenitor cell types are differentiated in 6-well plates.
Incubate Matrigel-coated 6-well plates (1.5 ml per well) at 37 °C for at least 30 min before seeding iPSCs.
When iPSCs reach ~80% confluency, add 1 ml of Gentle Cell Dissociation Reagent to each well and incubate the plates in an incubator for 6 min.
Check the morphology changes of iPSCs under the microscope. Carefully aspirate the dissociation reagent when iPSCs become granular and show clear boundaries. Otherwise, put the plates back to the incubator for an additional 1–2 min.
Add 1 ml of Brew medium +10 μM Y27632 perpendicular to each well and triturate iPSCs to small clumps (~10 cells per clump).
Remove Matrigel solution from coated plates and briefly wash each well with DPBS. Add 2 ml of Brew medium +10 μM Y27632 to each well of the new plates, and then add 100 μl of iPSC clumps to each well.
Gently shake the plates in three quick back-and-forth and left-and-right motions to distribute iPSCs evenly across the wells. Cells are cultured at 37 °C in an atmosphere of 5% CO2/95% air.
On the next day, aspirate the medium from each well and add 3 ml of fresh room temperature Brew medium. Refresh the Brew medium every other day. When the cells reach ~80% confluency, passage them as described above, or proceed to differentiating lineage-specific intermediate progenitor cell types and respective VSMC subtypes.
3.2. Generation of Lineage-Specific SMC Intermediate Populations
The induction protocols for generating lineage-specific SMC precursors have been optimized based on previous studies [21–23]. Figure 1 shows a detailed schematic diagram of the differentiation steps.
3.2.1. Cardiac Neural Crest (CNC) Cell Differentiation
Day 0: Dissociate ~80% confluent iPSCs into small clumps as described in Subheading 3.1, and sparsely seed cells at a density of 5 × 103 cells/cm2 onto new Matrigel-coated 6-well plates in Brew medium +10 μM Y27632. Make sure iPSCs express high levels of pluripotency markers (Fig. 2a) before differentiation is initiated.
Day 1 ~ 4: Wash each well with DPBS and add 2 ml of NC induction medium (see Subheading 2.4, item 2). Change the NC induction medium daily.
Day 5 ~ 6: Change the NC induction medium +1 μM retinoic acid daily to facilitate the specification of cardiac NC (CNC) cells (see Note 4).
Day 7: Aspirate the spent NC induction medium and wash the wells with DPBS. Add 1 ml of Accutase solution into each well and incubate the cells at 37 °C for 5 min. Add 4 ml of CDM basal medium (see Subheading 2.4, item 1) to 15 ml conical tubes, and then collect triturated cells from each well of the 6-well plates. Centrifuge the cells at 200 × g for 5 min at room temperature. Decant the supernatant and resuspend the cell pellets at a dilution ratio of 1:6 in the NC maintenance medium (see Subheading 2.4, item 3) + 1 μM retinoic acid. Cells are seeded on Matrigel-coated 6-well plates, and 10 μM of Y27632 can be added to the medium to improve cell survival and attachment.
Day 8 ~ 9: Refresh NC maintenance medium +1 μM retinoic acid daily.
Day 10: Dissociate the cells with Accutase solution (1 ml per well) at 37 °C for 5 min. The cell pellets are resuspended in the NC maintenance medium and seeded at a split ratio of 1:3 to 1:6 on gelatin-coated 6-well plates. Replated CNC cells on Day 10 are labeled as passage 1. The majority of the cells on day 10 are positive for SOX10/AP2α and negative for PAX6 (Fig. 2b). Change the NC maintenance medium every day and passage the cells when they reach ~90% confluency. CNC cells at passage 2 can be cryopreserved for future expansion. Otherwise, proceed to Subheading 3.3 for CNC-specific (ascending and aortic arch) SMC differentiation by seeding CNCs at a density of 2 × 104 cells/cm2.
3.2.2. Ventral Somite (VS) Differentiation
Day 0: Dissociate ~80% confluent iPSCs into small clumps as described in Subheading 3.1, and sparsely seed cells at a density of 1 × 104 cells/cm2 onto new Matrigel-coated 6-well plates in Brew medium +10 μM Y27632.
Day 1: Wash each well with DPBS and add 2 ml of APS induction medium (see Subheading 2.4, item 4).
Day 2: Wash each well with DPBS and add 2 ml of PM induction medium (see Subheading 2.4, item 5). The majority of the cells on day 2 are positive for TBX6 and CDX2 (Fig. 2c).
Day 3: Wash each well with DPBS and add 2 ml of ES induction medium (see Subheading 2.4, item 6). The majority of the cells on day 3 are positive for FOXC2 (Fig. 2d).
Day 4 ~ 6: Wash each well with DPBS and add 2 ml of VS induction medium (Subheading 2.4, item 7). Change the medium daily. The majority of the cells on day 6 are positive for SOX9 and TWIST1 (Fig. 2e).
Day 7: Dissociate the cells with Accutase solution (1 ml per well) at 37 °C for 5 min. Resuspend the cell pellets in CDM + 10 μM Y27632, and seed 2 × 104 cells/cm2 on gelatin-coated 6-well plates. Proceed to Subheading 3.3 for VS-specific (descending aortic) SMC differentiation, as the maintenance conditions for VS cells have not been optimized.
3.2.3. Second Heart Field (SHF) Differentiation
Day 0: Dissociate ~80% confluent iPSCs into small clumps as described in Subheading 3.1, and sparsely seed cells at a density of 1.5 × 104 cells/cm2 onto new Matrigel-coated 6-well plates in Brew medium +10 μM Y27632.
Day 1: Wash each well with DPBS and add 2 ml of MPS induction medium (see Subheading 2.4, item 8).
Day 2: Wash each well with DPBS and add 2 ml of LPM induction medium (see Subheading 2.4, item 9). The majority of the cells on day 2 are positive for NKX2.5 and HAND1 (Fig. 2f).
Day 3 ~ 4: Wash each well with DPBS and add 2 ml of SHF cell induction medium (see Subheading 2.4, item 10). Change the medium daily.
Day 5: Dissociate the cells with Accutase solution (1 ml per well) at 37 °C for 5 min. Resuspend the cell pellets in CDM + 10 μM Y27632. The majority of cells on day 4 are positive for ISL1 and MEF2C (Fig. 2g). Seed 1 × 103 cells/cm2 (see Note 5) or 3 × 104 cells/cm2 on gelatin-coated 6-well plates, and proceed to Subheading 3.2.5 for PE/EPI differentiation or Subheading 3.3 for SHF-specific (aortic root and ascending aortic) SMC differentiation, respectively. The maintenance conditions for SHF cells have not been optimized.
3.2.4. Septum Transversum (ST) Differentiation
Day 0 ~ 2: Generate LPM cells as described in Subheading 3.2.3, steps 1–3.
Day 3 ~ 4: Wash each well with DPBS and add 2 ml of SM induction medium (see Subheading 2.4, item 11). Change the medium daily. The majority of the cells on day 4 are positive for FOXF1 (Fig. 2h).
Day 5 ~ 7: Wash each well with DPBS and add 2 ml of ST induction medium (see Subheading 2.4, item 12). Change the medium daily. The majority of the cells on day 7 are positive for GATA4 and TCF21 (Fig. 2i).
Day 8: Dissociate the cells with Accutase solution (1 ml per well) at 37 °C for 5 min. Resuspend the cell pellets in PE induction medium +10 μM Y27632. Seed 1 × 103 cells/cm2 on gelatin-coated 6-well plates and proceed to Subheading 3.2.5 for PE/EPI differentiation. The maintenance conditions for ST cells have not been optimized.
3.2.5. Proepicardium (PE)/Epicardium (EPI) Differentiation
As the PE has been shown to be derived from SHF and ST cells [24], the PE/EPI differentiation process continues after day 5 and day 8 in Subheadings 3.2.3 and 3.2.4, respectively.
Day 6 ~ 7 (for the SHF lineage, the same as below) or 9 ~ 10 (for the ST lineage, the same as below): Wash each well with DPBS and add 2 ml of PE induction medium (see Subheading 2.2, item 13). Maintain cells in the medium without change for 2 days.
Day 8 ~ 9 or 11 ~ 12: Wash each well with DPBS and 2 ml of CDM + 100 μg/ml ascorbic acid + 7 μg/ml insulin. Maintain cells in the medium without change for 2 days. Most SHF- and ST-derived EPI cells are positive for WT1 and ZO1 (Fig. 2j, k).
Day 10 ~ 12 or 12 ~ 15: Dissociate the cells with Accutase solution (1 ml per well) at 37 °C for 5 min. Resuspend the cell pellets and seed 1.5 × 104 cells/cm2 on gelatin-coated 6-well plates in EPI maintenance medium (see Subheading 2.4, item 14). Change the medium every 2 days until the cells become confluent. EPI cells can be maintained in this medium for over 15 passages without losing their cell type-specific markers. EPI cells can be cryopreserved after passage 2 for future expansion. Otherwise, proceed to Subheading 3.3 for coronary VSMC differentiation by seeding EPI cells at a density of 3 × 104 cells/cm2.
3.3. Differentiation and Maturation of Embryonic Origin-Specific VSMC Subtypes
Day 0 ~ 5: Wash lineage-specific intermediate progenitor cells from Subheadings 3.2.1, step 7 (CNC); 3.2.2, step 6 (VS); 3.2.3, step 5 (SHF); and 3.2.5, step 3 (SHF-EPI and ST-EPI) with DPBS and add 2 ml of SMC induction medium (see Subheading 2.4, item 15). Change the medium daily.
Day 6 ~ 20: Wash cells with DPBS and maintain in Medium 231 + SMGS for 2 weeks or longer as desired. Change the medium every 2 days and split the VSMCs at a 1:2 ratio (see Note 6) when the cells become confluent. Embryonic origin-specific VSMC subtypes express similar levels of VSMC contractile proteins, such as TAGLN and CNN1 (Fig. 2l–o).
Day 21 ~ 27: Wash cells with DPBS and maintain in SMC maturation medium (see Subheading 2.4, item 16) for 6 days (see Note 7). Change the medium every 2 days. We observe that treating iPSC-derived VSMC subtypes with PD0325901 at a concentration of 1 μM (Fig. 3a) for 6 days (Fig. 3b) can result in high expression levels of MYH11, a mature marker of VSMCs. This mature and contractile phenotype can be preserved for at least 4 days after the withdrawal of PD0325901 (Fig. 3c).
4. Notes
To prepare 100 mg/ml PVA solution, weigh 10 g of PVA and add the powder slowly into a sterile flask containing 100 ml of distilled H2O with constant gentle agitation until the PVA is completely dissolved. The solution can be stored at 4 °C for 2 months.
To avoid the risk of protein retention in some filters, we recommend adding growth factors (and small molecules) directly to filtered basal CDM. Make sure the stock growth factors and small molecules are prepared in sterile conditions. The complete induction and maintenance media can be stored up to 1 week at 4 °C.
Because a definitive surface marker for SHF cells is lacking, here we define SHF as ISL1+/MEF2C+/GATA4+ cells [25].
The cardiac neural crest is the most caudal of the cranial neural crest located postotically in the neural tube during development [26]. To skew the differentiation more toward a posterior axis to get CNC cells (PLXNA2+/HOX4+) [27], retinoic acid is added to the NC induction medium on day 5 as a caudalizing factor. In contrast, a cranial-dominant NC cell population (OTX2+) was reported to be successfully derived using retinoic acid-free NC induction medium [21].
We observe that it is critical to seed cardiac progenitor cells (i.e., SHF cells and ST cells) at a very low density to promote the differentiation of PE/EPI cells.
Cell–cell interaction and paracrine effects are critical to the growth of VSMCs. Therefore, we recommend using a low split ratio (<1:3) to passage human iPSC-derived VSMCs of any embryonic origins. We observe that both SHF- and EPI-VSMC subtypes are more quiescent than CNC- and VS-VSMCs in the SMC growth medium. Therefore, different seeding densities of lineage-specific progenitors are required at the VSMC induction stage to ensure adequate end-point numbers of each VSMC subtype for downstream experiments.
Although MEK inhibition can effectively upregulate the expression of VSMC contractile proteins by blocking the p38 signaling pathway, caution should be taken when highly active ERK signaling plays a pathogenic role in the development of vascular diseases such as Marfan syndrome [28]. Repsox, a TGF-β inhibitor, was reported to significantly improve the expression of MYH11 in iPSC-derived VSMCs [29]. However, this small molecule does not work even in healthy iPSC-derived VSMC subtypes in our hands. Therefore, an alternative but less effective way to promote the maturation of Marfan syndrome iPSC-derived VSMCs could be maintaining the cells in an SMC growth medium supplemented with heparin and TGF-β1 for at least 6 days [30].
Acknowledgments
This work was supported in part by research grants from NIH R01 HL1265276, R01 HL133272, R01 HL146690, R01 HL141371 (JCW), Tobacco-Related Disease Research Program (TRDRP) 30FT0852 (MS) and 27IR-0012 (JCW), and AHA 17MERIT33610009 (JCW) and AHA Career Development Award 19CDA34760019 (CL).
References
- 1.Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84(3):767–801. 10.1152/physrev.00041.2003 [DOI] [PubMed] [Google Scholar]
- 2.Berk BC (2001) Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev 81(3):999–1030. 10.1152/physrev.2001.81.3.999 [DOI] [PubMed] [Google Scholar]
- 3.Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z (2019) Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 16(12):727–744. 10.1038/s41569-019-0227-9 [DOI] [PubMed] [Google Scholar]
- 4.Majesky MW (2007) Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27(6):1248–1258. 10.1161/ATVBAHA.107.141069 [DOI] [PubMed] [Google Scholar]
- 5.Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J, Evans SM (2008) A myocardial lineage derives from Tbx18 epicardial cells. Nature 454(7200):104–108. 10.1038/nature06969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, Pu WT (2008) Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454(7200):109–113. 10.1038/nature07060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A (2017) Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta. Arterioscler Thromb Vasc Biol 37(9):1722–1726. 10.1161/ATVBAHA.117.309599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM (2000) Fate of the mammalian cardiac neural crest. Development 127(8):1607–1616 [DOI] [PubMed] [Google Scholar]
- 9.Pouget C, Gautier R, Teillet MA, Jaffredo T (2006) Somite-derived cells replace ventral aortic hemangioblasts and provide aortic smooth muscle cells of the trunk. Development 133(6):1013–1022. 10.1242/dev.02269 [DOI] [PubMed] [Google Scholar]
- 10.Wasteson P, Johansson BR, Jukkola T, Breuer S, Akyurek LM, Partanen J, Lindahl P (2008) Developmental origin of smooth muscle cells in the descending aorta in mice. Development 135(10):1823–1832. 10.1242/dev.020958 [DOI] [PubMed] [Google Scholar]
- 11.Wilm B, Ipenberg A, Hastie ND, Burch JB, Bader DM (2005) The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 132(23):5317–5328. 10.1242/dev.02141 [DOI] [PubMed] [Google Scholar]
- 12.Asahina K, Zhou B, Pu WT, Tsukamoto H (2011) Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53(3):983–995. 10.1002/hep.24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Que J, Wilm B, Hasegawa H, Wang F, Bader D, Hogan BL (2008) Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc Natl Acad Sci U S A 105(43):16626–16630. 10.1073/pnas.0808649105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacFarlane EG, Parker SJ, Shin JY, Kang BE, Ziegler SG, Creamer TJ, Bagirzadeh R, Bedja D, Chen Y, Calderon JF, Weissler K, Frischmeyer-Guerrerio PA, Lindsay ME, Habashi JP, Dietz HC (2019) Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J Clin Invest 129(2):659–675. 10.1172/JCI123547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, Jorgensen HF (2018) Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun 9(1):4567. 10.1038/s41467-018-06891-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinha S, Santoro MM (2018) New models to study vascular mural cell embryonic origin: implications in vascular diseases. Cardiovasc Res 114(4):481–491. 10.1093/cvr/cvy005 [DOI] [PubMed] [Google Scholar]
- 17.Klein D (2018) iPSCs-based generation of vascular cells: reprogramming approaches and applications. Cell Mol Life Sci 75(8):1411–1433. 10.1007/s00018-017-2730-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roostalu U, Wong JK (2018) Arterial smooth muscle dynamics in development and repair. Dev Biol 435(2):109–121. 10.1016/j.ydbio.2018.01.018 [DOI] [PubMed] [Google Scholar]
- 19.Matsa E, Ahrens JH, Wu JC (2016) Human induced pluripotent stem cells as a platform for personalized and precision cardiovascular medicine. Physiol Rev 96(3):1093–1126. 10.1152/physrev.00036.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paik DT, Chandy M, Wu JC (2020) Patient and disease-specific induced pluripotent stem cells for discovery of personalized cardiovascular drugs and therapeutics. Pharmacol Rev 72(1):320–342. 10.1124/pr.116.013003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukuta M, Nakai Y, Kirino K, Nakagawa M, Sekiguchi K, Nagata S, Matsumoto Y, Yamamoto T, Umeda K, Heike T, Okumura N, Koizumi N, Sato T, Nakahata T, Saito M, Otsuka T, Kinoshita S, Ueno M, Ikeya M, Toguchida J (2014) Derivation of mesenchymal stromal cells from pluripotent stem cells through a neural crest lineage using small molecule compounds with defined media. PLoS One 9(12):e112291. 10.1371/journal.pone.0112291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hackland JOS, Frith TJR, Thompson O, Marin Navarro A, Garcia-Castro MI, Unger C, Andrews PW (2017) Top-down inhibition of BMP signaling enables robust induction of hPSCs into neural crest in fully defined, xeno-free conditions. Stem Cell Reports 9(4):1043–1052. 10.1016/j.stemcr.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, Shyh-Chang N, Fernhoff NB, George BM, Wernig G, Salomon REA, Chen Z, Vogel H, Epstein JA, Kundaje A, Talbot WS, Beachy PA, Ang LT, Weissman IL (2016) Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166(2):451–467. 10.1016/j.cell.2016.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brade T, Pane LS, Moretti A, Chien KR, Laugwitz KL (2013) Embryonic heart progenitors and cardiogenesis. Cold Spring Harb Perspect Med 3(10):a013847. 10.1101/cshperspect.a013847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckingham M, Meilhac S, Zaffran S (2005) Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 6(11):826–835. 10.1038/nrg1710 [DOI] [PubMed] [Google Scholar]
- 26.Thattaliyath B, Hutson M (2016) Neural crest. In: Congenital heart diseases: the broken heart. Springer, Vienna, pp 41–53 [Google Scholar]
- 27.Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA, Epstein JA (2001) PlexinA2 and semaphorin signaling during cardiac neural crest development. Development 128(16):3071–3080 [DOI] [PubMed] [Google Scholar]
- 28.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC (2011) Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332(6027):358–361. 10.1126/science.1192149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, McIntosh BE, Wang B, Brown ME, Probasco MD, Webster S, Duffin B, Zhou Y, Guo LW, Burlingham WJ, Kent C, Ferris M, Thomson JA (2019) A human pluripotent stem cell-based screen for smooth muscle cell differentiation and maturation identifies inhibitors of intimal hyperplasia. Stem Cell Reports 12(6):1269–1281. 10.1016/j.stemcr.2019.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patsch C, Challet-Meylan L, Thoma EC, Urich E, Heckel T, O’Sullivan JF, Grainger SJ, Kapp FG, Sun L, Christensen K, Xia Y, Florido MH, He W, Pan W, Prummer M, Warren CR, Jakob-Roetne R, Certa U, Jagasia R, Freskgard PO, Adatto I, Kling D, Huang P, Zon LI, Chaikof EL, Gerszten RE, Graf M, Iacone R, Cowan CA (2015) Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol 17(8):994–1003. 10.1038/ncb3205 [DOI] [PMC free article] [PubMed] [Google Scholar]