

Summary
The small GTPase Ras homolog enriched in brain (Rheb) plays a critical role in activating the mechanistic target of rapamycin complex 1 (mTORC1), a signaling hub that regulates various cellular functions. We recently observed nuclear mTORC1 activity, raising an intriguing question as to how Rheb, which is known to be farnesylated and localized to intracellular membranes, regulates nuclear mTORC1. In this study, we found that active Rheb is present in the nucleus and required for nuclear mTORC1 activity. We showed that inhibition of farnesyltransferase reduced cytosolic but not nuclear mTORC1 activity. Furthermore, a farnesylation-deficient Rheb mutant, with preferential nuclear localization and specific lysosome tethering, enables nuclear and cytosolic mTORC1 activities, respectively. These data suggest that non-farnesylated Rheb is capable of interacting with and activating mTORC1, providing mechanistic insights into the molecular functioning of Rheb as well as regulation of the recently observed, active pool of nuclear mTORC1.
eTOC Blurb:
Although Rheb is essential for mTORC1 activation at the lysosome, its functional role in regulating the recently observed nuclear mTORC1 signaling is unclear. Here, Zhong et al observed that active Rheb is present in the nucleus and its farnesylation is not required for nuclear mTORC1 activity.
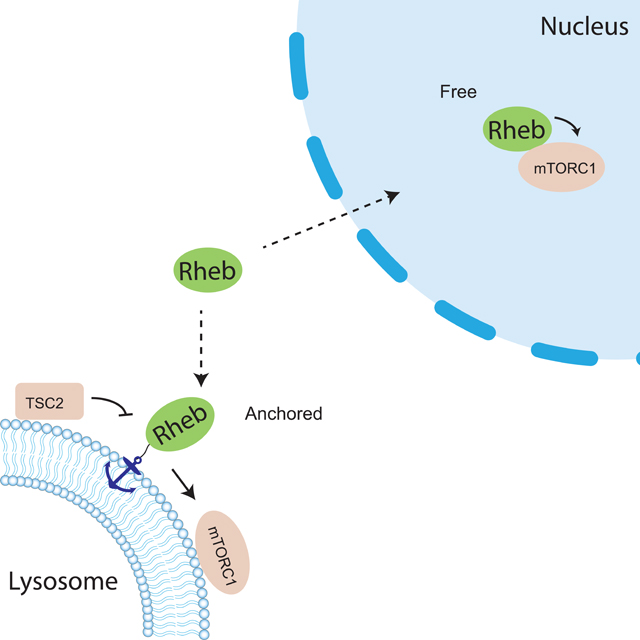
Introduction
The mechanistic target of rapamycin complex 1 (mTORC1) is a key signaling complex that consists of the catalytic kinase mTOR and numerous associated components, including the mTORC1 defining component Raptor. mTORC1 plays a central role in regulating many fundamental cellular processes, such as nutrient sensing, metabolism, macromolecule synthesis, proliferation, and autophagy. mTORC1 signaling pathway is widely implicated in pathological conditions such as cancer and type 2 diabetes (Laplante and Sabatini, 2012). In a canonical model, mTORC1 is activated by the small GTPase Rheb at the lysosomal surface in response to either amino acid or growth factor stimulation (Kim and Guan, 2019; Liu and Sabatini, 2020).
Rheb is a small GTPase and its activity is determined by nucleotide binding state (Tabancay et al., 2003; Long et al., 2005). The active GTP-bound form of Rheb interacts with mTORC1 and allosterically turns on mTOR kinase activity (H. Yang et al., 2017). It is well established that Rheb is essential for canonical mTORC1 activation. The nucleotide binding state of Rheb is regulated by the Tuberous Sclerosis Complex 2 (TSC2), a tumor suppressor and a GTPase activating protein (GAP) towards Rheb. TSC2 facilitates hydrolysis of the bound GTP to GDP, thus inactivating Rheb (Inoki et al., 2003; Tee et al., 2003). Like many other small GTPases, Rheb undergoes post-translational modifications at its C-terminus where a CAAX motif marks it for modification by a farnesyl group, which has been shown to regulate the membrane association of Rheb (Clark et al., 1997; Takahashi et al., 2005; Hanker et al., 2010). Rheb has been reported to be present on the lysosomal surface, where mTORC1 and TSC also reside (Demetriades et al., 2014; Menon et al., 2014). In response to growth factor stimulation, the active upstream kinase Akt phosphorylates TSC2 (Inoki et al., 2002; Manning et al., 2002) and triggers its disassociation from the lysosomal surface, thus relieving the inhibition of Rheb and leading to eventual mTORC1 activation on the lysosome by active Rheb (Menon et al., 2014) (Figure S1A).
In addition to the canonical lysosomal pool of mTORC1, mTORC1 components were also found to be present in the nucleus although the presence of mTORC1 activity was under debate (Kim and Chen, 2000; Bernardi et al., 2006; Havel et al., 2015; Kantidakis et al., 2010; Wan et al., 2017; Zhang et al., 2002; Rosner and Hengstschläger, 2012). We recently observed nuclear mTORC1 activity in living cells, which can be induced by platelet-derived growth factor (PDGF) or leucine-O-methyl ester, an amino acid surrogate, in NIH3T3 cells or by insulin in 3T3L1 adipocytes (Zhou et al., 2020, 2015). This observation was enabled by the development of a genetically encoded mTORC1 activity reporter (TORCAR). Briefly, the mTORC1 substrate 4EBP1 is sandwiched between a pair of cyan and yellow fluorescent proteins that can undergo fluorescence resonance energy transfer (FRET). Phosphorylation of 4EBP1 within TORCAR by mTORC1 induces a conformational change, leading to an increase in the cyan/yellow emission ratio, which serves as a readout for increased mTORC1 kinase activity. Using TORCAR variant targeted to the nucleus, we detected nuclear mTORC1 activity in response to growth factor stimulation (Zhou et al., 2015). This activity was shown to be dependent on nuclear Akt activity, which promotes Raptor translocation into the nucleus and phosphorylates PRAS40 to relieve its inhibition on mTORC1, leading to an increase in nuclear mTORC1 activity (Zhou et al., 2020) (Figure S1A). While nuclear mTORC1 is under this non-canonical regulation by nuclear Akt, whether other factors in the canonical mTORC1 pathway play a role in modulating nuclear mTORC1 activity is unclear. In particular, does Rheb regulate nuclear mTORC1 activity, given that it is a direct and essential activator of mTORC1? How does a lysosomally localized GTPase regulate a nuclear complex? Related to the latter question, emerging evidence suggests that Rheb also localizes to other cellular compartments in addition to the lysosome (Hao et al., 2018; Angarola and Ferguson, 2020, 2019). However, the functional roles of these subcellular pools of Rheb in mTORC1 signaling have not been defined.
In this study, we focused on examining whether the canonical TSC/Rheb axis is conserved in the nucleus to regulate nuclear mTORC1 activity. We found that although there is no evidence for the presence of nuclear TSC2, active Rheb is present in the nucleus and required for nuclear mTORC1 activity. We further showed that lipid modification of Rheb is not required for regulating nuclear mTORC1 activity and lysosomal tethering of farnesylation-deficient Rheb enables mTORC1 signaling. In summary, the present study reveals the mechanisms that specifically regulate nuclear mTORC1 activity and provides further insights into both the canonical and non-canonical regulation of mTORC1 by an important small GTPase, Rheb.
Results
Rheb is present in the nucleus and required for nuclear mTORC1 activity
To determine whether Rheb is required for nuclear mTORC1 activity, we generated a Rheb knockdown cell line using NIH3T3 cells infected with lentivirus carrying shRNA targeting Rheb mRNA. The protein level of Rheb was efficiently reduced, as evaluated by western blotting (Figure S1B). As expected, Rheb knockdown significantly reduced PDGF-induced phosphorylation of mTORC1 substrate, S6K1 at Thr389 (Saitoh et al., 2002; Hay and Sonenberg, 2004) (Figure S1B). Consistent with these data, the mTORC1 activity reporter TORCAR showed a diminished cyan/yellow (C/Y) emission ratio change in response to PDGF stimulation in Rheb knockdown cells (2.2 ± 0.6 %, n = 34) compared with wild type cells (10.0 ± 0.5 %, n = 32), on par with the response in Raptor knockdown cells (3.7 ± 0.5 %, n = 12), suggesting that Rheb is required for cellular mTORC1 activity (Figures S1C-S1D). To examine the impact of knocking down Rheb on nuclear mTORC1 activity, we expressed the mTORC1 biosensor targeted to the nucleus via a nuclear localization sequence (NLS) (TORCAR-NLS) (Zhou et al., 2015) in both wild type and Rheb knockdown cells and stimulated the cells with PDGF. Rheb knockdown cells showed a minimal emission ratio change upon PDGF stimulation (0.5 ± 0.5 %, n = 25) (Figures 1A–1B). As a control, wild type cells responded to PDGF stimulation with a 6.9 ± 0.4 % increase in C/Y emission ratio (n = 29), which was abolished by pretreatment with rapamycin, an mTORC1 specific inhibitor (Figures 1A–1B). These results suggest an essential role of Rheb for nuclear mTORC1 activity.
Figure 1. Rheb is present in the nucleus and required for nuclear mTORC1 activation.

(A-B) Representative averaged time course traces (A) and summary (B) of PDGF-induced maximum responses of TORCAR-NLS in double starved NIH3T3 cells. Blue, wild type cells (WT, N = 29); yellow, Rheb knockdown cells (shRheb, N = 25); green, wild type cells with rapamycin pretreatment (100 nM for 30 min) (Rapamycin, N = 20). N indicates the cell number quantified from 3 independent experiments. (C) Representative confocal immunofluorescence images of wild type (WT) NIH3T3 cells and Rheb knockdown (shRheb) cells from two independent experiments. Staining for Rheb is shown in green and nucleus is shown in blue. (D) Quantification of the nuclear signals in (C). Intensities were normalized to the maximum intensity. Control experiments were done by leaving out the primary antibody ((−) anti-Rheb). N indicates the number of cells quantified. Data are representative from 2 independent experiments. (E) Line scan shown in (C). A line was drawn across the nuclear region of the cell. Intensities along the line in the green channel were measured and shown as relative arbitrary units (AU). Shaded areas indicate standard error of the mean (SEM). Box plots show the upper and lower adjacent values, interquartile range and the median. Scale bar = 10 µm. See also Figure S1.
Rheb activates lysosomal mTORC1 via co-localization and interaction with mTORC1 (Kim and Guan, 2019; Liu and Sabatini, 2020). We hypothesized that Rheb is also present in the nucleus for activation of nuclear mTORC1. To examine whether endogenous Rheb is present in the nucleus, we probed the distribution of Rheb with an antibody previously validated for immunofluorescence (Menon et al., 2014). As shown in Figure 1C, although the primary localization of Rheb was found to be outside of the nucleus with the majority concentrated in the perinuclear region, a small amount of Rheb was clearly present in the nucleus (Figures 1C and S1E), which was consistent with previous findings (Menon et al., 2014). The signal was specific to Rheb because knockdown of Rheb significantly reduced the immunofluorescence signal throughout the cell (Figures S1E and S1F) and essentially abolished the nuclear signal, as shown by the quantifications and line scans across the nuclear region (Figures 1D–1E). To further examine the presence of nuclear Rheb, we performed nuclear fractionation experiments using a published protocol (Suzuki et al., 2010) and found that a small portion of Rheb was present in the nuclear fraction (Figure S1G). These data show that Rheb is present in the nucleus in NIH3T3 cells. To test the presence of nuclear Rheb in other cell types, we performed immunofluorescence analysis to probe Rheb distribution in Cal27, a human oral squamous carcinoma cell line (Wang et al., 2019). Similar to what we observed in NIH3T3 cells, significant signals were found in the nuclei of Cal27 cells and were sensitive to Rheb knockdown (Figure S1H-S1J). In addition, Rheb signals were also observed in the nuclear regions of other cell lines, including HeLa, Cos7, PLC/PRF/5, a human hepatoma cell line (Huo et al., 2019), and 3T3L1 adipocytes (Figure S1K), suggesting Rheb is present in the nucleus of a variety of cell types.
Nuclear mTORC1 activity is suppressed by exogenously expressed nuclear TSC2
In the canonical activation of lysosomal mTORC1, TSC2 plays an important role in growth factor-induced activation of Rheb, which in turn activates mTORC1. As a Rheb GAP, TSC2 is localized to the lysosomal surface at the basal state (Demetriades et al., 2016), keeping Rheb in the inactive, GDP-bound form by stimulating GTP hydrolysis to GDP. Growth factor stimulated Akt phosphorylates TSC2 on two key residues, Ser939 and Thr1462, leading to its dissociation from the lysosome, and thereby relieving Rheb inhibition and leading to lysosomal mTORC1 activation (Inoki et al., 2002; Manning et al., 2002; Menon et al., 2014). However, numerous studies have shown TSC2 is not present in the nucleus (Menon et al., 2014; Demetriades et al., 2016, 2014; Ferlazzo et al., 2017; Prentzell et al., 2021), suggesting that nuclear Rheb, unlike its lysosomal counterpart, is not under the typical Akt-TSC2-mediated control.
To further examine the distribution of endogenous TSC2, we performed immunofluorescence experiments in NIH3T3 cells and found that the specific signal that was sensitive to TSC2 knockdown was only present in the cytosolic regions (Figures S2A-S2C). To further support these results, we also performed nuclear fractionation and found that TSC2 was absent in the nuclear fraction in contrast to Rheb (Figure S1G). Taken together, these data support that although mTORC1 activator Rheb is present in the nucleus, the negative regulator TSC2 is unlikely to be present in the nucleus, suggesting that nuclear Rheb is not under Akt-TSC2-mediated control.
No guanine nucleotide exchange factor (GEF) has been identified for Rheb. In the absence of a GEF, it is thought that a relatively low nucleotide binding affinity allows Rheb to spontaneously exchange GDP for GTP to become active (Li et al., 2004). Given that the critical negative regulator TSC2 is absent from the nucleus, we hypothesized that spontaneous activity allows nuclear Rheb to regulate nuclear mTORC1 activity. Because of the small amount of Rheb in the nucleus and the challenges associated with pulldown assays using subcellular fractions, we tested this idea by using a synthetic biology approach. We reasoned that if active GTP bound Rheb is involved in regulating nuclear mTORC1, exogenously expressing nuclearly targeted TSC2 would inhibit Rheb through its GAP activity, thus inhibiting nuclear mTORC1 activity. To test this idea, we focused on several variants of TSC2, including the wide type TSC2, a TSC2-AA variant in which the two critical Akt phosphorylation sites, Ser939 and Thr1462, are mutated to Alanine to eliminate Akt-mediated negative control but retains GAP activity to Rheb (Manning et al., 2002; Menon et al., 2014), and a TSC2-AA-3Q mutant that has no GAP activity (Li et al., 2004) (Figure S2D). Because TSC1 forms a stable complex with TSC2 and prevents ubiquitin-mediated TSC2 degradation (Chong-Kopera et al., 2006), we first asked how the overexpression and nuclear targeting of TSC2 affect TSC1. By using immunofluorescence, we found that overexpressing nuclear TSC2 (TSC2-NLS) not only increased the level of TSC1 in the whole cell, but also significantly increased TSC1 level in the nucleus (Figures S2E-S2G), suggesting that exogenously overexpressed nuclear TSC2 may recruit TSC1 to preserve the integrity of the TSC complex in the nucleus.
Next, we tested the capability of these TSC2 variants to suppress mTORC1 activity. When co-expressed with TORCAR, wild type TSC2 showed exclusively cytoplasmic localization, whereas TSC2-AA mutant showed partial lysosomal localization (Figures 2A and S2H), which is in line with previous reports (Menon et al., 2014). We stimulated these NIH3T3 cells with PDGF and quantified PDGF-induced response of TORCAR in the cytosolic regions. As shown in Figure 2B and 2C, co-expression of wild type TSC2 did not affect the cytosolic response of TORCAR (7.8 ± 0.5 %, n = 35) in comparison with the mCherry control (8.3 ± 0.7 %, n = 27), while co-expression of TSC2-AA significantly suppressed the cytosolic response of TORCAR (4.2 ± 0.4 %, n = 32). Notably, this suppression was dependent on the GAP activity of TSC2, as TSC2-AA-3Q failed to inhibit cytosolic mTORC1 activity (Figures S2I-S2K). These data suggest that GAP activity towards Rheb could inhibit mTORC1 activity (in the case of TSC2-AA), whereas Akt-mediated phosphorylation of TSC2 could abolish the inhibitory effect of overexpressed TSC2 (in the case of TSC2 wild type). As unphosphorylatable TSC2-AA is resistant to Akt-mediated phosphorylation and lysosomal dissociation, it remains on the lysosome as a strong dominant negative regulator to inhibit lysosomal Rheb and mTORC1 activity, regardless of active Akt (Figure 2D).
Figure 2. Growth factor induced nuclear mTORC1 activity is suppressed by overexpressing TSC2 in the nucleus.

(A) Representative fluorescence images from three independent experiments showing the localization of overexpressed constructs in NIH3T3 cells. Fluorescence of mCherry, mCherry-TSC2 and mCherry-TSC2-AA mutant are shown as “RFP”. “CFP” shows fluorescence of TORCAR. (B-C) Representative averaged time course traces (B) and summary (C) of PDGF-induced maximum responses of TORCAR in the cytosolic regions of double starved NIH3T3 cells. mCherry (blue, N = 27); mCherry-TSC2 (yellow, N = 35); mCherry-TSC2-AA (green, N = 32). (D) Model depicting lysosomal TSC2-dependent spatial regulation of mTORC1 activation. (E) Representative fluorescence images from three independent experiments showing the localization of overexpressed nuclearly targeted constructs in NIH3T3 cells. Fluorescence of mCherry-NLS, mCherry-TSC2-NLS and mCherry-TSC2-AA-NLS mutant are shown as “RFP”. “CFP” shows the fluorescence of TORCAR-NLS. (F-G) Representative averaged time course traces (F) and summary (G) of PDGF-induced maximum responses of TORCAR-NLS in double starved NIH3T3 cells. Co-expressed constructs: mCherry-NLS (blue, N = 20); mCherry-TSC2-NLS (yellow, N = 23); mCherry-TSC2-AA-NLS (green, N = 26). (H) Model depicting the inhibition of nuclear mTORC1 activity by overexpressed nuclear TSC2. N indicates the cell number quantified from at least 3 independent experiments. Shaded areas indicate standard error of the mean (SEM). Box plots show the upper and lower adjacent values, interquartile range and the median. Scale bar = 10 µm. See also Figure S2.
To test our hypothesis that active Rheb in the nucleus is involved in regulating nuclear mTORC1 activity, we expressed the nuclear targeted version of TSC2 and its variants and examined nuclear mTORC1 activity by TORCAR-NLS. Both TSC2-NLS and the reporter showed appropriate nuclear localization (Figures 2E and S2L). In cells stimulated with PDGF, co-expression of both TSC2-NLS and TSC2-AA-NLS significantly reduced TORCAR-NLS response compared to the control group expressing mCherry-NLS (Figures 2F–2G; mCherry-NLS: 8.9 ± 0.5 %, n = 20; TSC2-NLS: 5.7 ± 0.5 %, n = 23; TSC2-AA-NLS: 7.0 ± 0.6 %, n = 26). The GAP deficient mutant TSC2-AA-3Q-NLS was not able to inhibit nuclear mTORC1 activity (Figures S2L-S7N), suggesting that inhibition of nuclear mTORC1 by nuclear targeted TSC2 is dependent on its GAP activity towards Rheb. Notably, wild type TSC2 showed distinct behaviors in the cytosol and in the nucleus (Figures 2C and 2G). Indeed, given that the regulation of TSC2 by Akt is achieved via phosphorylation-induced dissociation from the lysosomal surface and that this spatially controlled negative regulation of TSC2 is unlikely to occur in the nucleus, both wild type and unphosphorylatable TSC2 can exert their GAP activity towards Rheb to suppress nuclear mTORC1 activity (Figure 2H). These data suggest that active Rheb is involved in stimulating nuclear mTORC1 activity.
Farnesylation of Rheb is not required for nuclear mTORC1 activity
Like other Ras family proteins, Rheb possesses a hypervariable region at its C-terminus, ending with the sequence CSVM, which conforms to the CAAX motif that undergoes farnesylation at the cysteine residue (Clark et al., 1997). The weak and transient membrane interaction enabled by C-terminal farnesylation is thought to be important for both subcellular localization of Rheb and Rheb-mediated lysosomal mTORC1 activation (Hanker et al., 2010; Angarola and Ferguson, 2020). We asked whether this farnesylation is also critical for nuclear Rheb to activate nuclear mTORC1. To test this, we used the selective farnesyltransferase inhibitor FTI-277 to inhibit Rheb farnesylation (Hanker et al., 2010). Using NIH3T3 cells stably expressing mCherry-tagged Rheb, we showed that while DMSO treated cells showed perinuclear localization of Rheb (Figure S3A), similar to what was observed for endogenous Rheb (Figure 1C), cells treated with FTI-277 showed diffusive Rheb localization (Figure S3A), suggesting that FTI-277 treatment efficiently disrupted Rheb localization. We then evaluated the effect of the farnesyltransferase inhibitor on PDGF-induced phosphorylation of mTORC1 substrate in wild type NIH3T3 cells. Consistent with previous reports (Basso et al., 2005; Hanker et al., 2010), FTI-277 treatment significantly reduced S6K1 phosphorylation (Figure S3B). To examine the effect of the farnesyltransferase inhibitor on compartmentalized mTORC1 activity, we expressed TORCAR in the cytosol and nucleus by tagging a nuclear export signal (NES) and an NLS, respectively, and quantified PDGF-induced mTORC1 activity at subcellular locations in the presence or absence of the farnesyltransferase inhibitor. As expected, FTI-277 significantly reduced the cytosolic TORCAR (TORCAR-NES) response to PDGF (Figures 3A–3B, 7.4 ± 0.5 %, n = 32) compared with the DMSO group (Figures 3A–3B, 9.9 ± 0.6 %, n = 23), indicating that farnesylation is essential for canonical mTORC1 activation, as shown in previous studies (Hanker et al., 2010). In stark contrast, FTI-277 treatment did not affect the response of nuclear TORCAR (Figure 3C–3D; DMSO: 5.5 ± 0.6 %, n = 32; FTI-277: 5.2 ± 0.8 %, n = 17), suggesting that nuclear mTORC1 activity does not require farnesylation of Rheb.
Figure 3. Farnesyltransferase inhibitor FTI-277 did not affect nuclear mTOR1 activity.

(A-B) Representative averaged time course traces (A) and summary (B) of PDGF-induced maximum responses of TORCAR-NES in double starved NIH3T3 cells treated with DMSO (blue, N = 23) or farnesyltransferase inhibitor FTI-277 at 5 µM overnight (yellow, N = 32). (C-D) Representative averaged time course traces (C) and summary (D) of PDGF-induced maximum responses of TORCAR-NLS in double starved NIH3T3 cells treated with DMSO (blue, N = 32) or farnesyltransferase inhibitor FTI-277 at 5 µM overnight (yellow, N = 17). N indicates the cell number quantified from at least 3 independent experiments. Shaded areas indicate standard error of the mean (SEM). Box plots show the upper and lower adjacent values, interquartile range and the median. See also Figure S3.
Farnesylation-deficient Rheb differentially rescued mTORC1 activity within subcellular compartments
To further investigate the role of Rheb farnesylation in regulating mTORC1 signaling, we took advantage of a farnesylation-deficient Rheb mutant, C181S, in which the cysteine in the CAAX motif is mutated to a serine (Jiang and Vogt, 2008) and reconstituted Rheb knockdown cells with wild type Rheb, Rheb-C181S, and Rheb-Δ5A, an inactive form of Rheb which lacks five amino acid residues (amino acids 38–42) and is unable to bind nucleotide (Inoki et al., 2003) (Figure 4A). We first examined the localization of these Rheb mutants by tagging them with mCherry fluorescent protein (Figure 4B). Wild type Rheb and Rheb-Δ5A showed similar localization to endogenously or stably expressed Rheb (Figures 1C and S3A). The farnesylation-deficient Rheb-C181S showed no indication of membrane association, reminiscent of the effect caused by farnesyltransferase inhibitor treatment (Figure S3A). In fact, Rheb-C181S showed accumulation in the nucleus (Figure 4B), consistent with the previous reports (Jiang and Vogt, 2008; Hanker et al., 2010). We then examined cytosolic mTORC1 activity by cytosol-localized TORCAR-NES (Figure 4C) in these cells. As a control, Rheb knockdown abolished the response of the cytosolic TORCAR (1.0 ± 0.4 %, n = 47), which can be partially rescued by expressing wild type Rheb (7.0 ± 0.6 %, n = 44). On the other hand, Rheb-C181S (1.1 ± 0.4 %, n = 44) and Rheb-Δ5A (1.9 ± 0.7 %, n = 28) failed to rescue cytosolic TORCAR responses in Rheb knockdown cells, indicating that both farnesylation and nucleotide binding are important for Rheb to positively regulate cytosolic mTORC1 activity.
Figure 4. Farnesylation-deficient Rheb had different effects on rescuing subcellular mTORC1 activity.

(A) Schematics of different Rheb constructs tagged with mCherry. (B) Representative images of overexpressed mCherry-Rheb constructs in NIH3T3 cells from 3 independent experiments. (C-D) Representative averaged time course traces (C) and summary (D) of PDGF-induced maximum responses of TORCAR-NES in double starved NIH3T3 cells. Blue, wild type cells (WT, N = 25); yellow, Rheb knockdown cells (shRheb + None, N = 47 cells); green, Rheb knockdown cells transfected with wild type mCherry-Rheb-WT (shRheb + Rheb-WT, N = 44); orange, Rheb knockdown cells transfected with mCherry-Rheb-C181S (shRheb + Rheb-C181S, N = 44); pink, Rheb knockdown cells transfected with mCherry-Rheb-Δ5A (shRheb + Rheb-Δ5A, N = 28 cells). (E-F) Representative averaged time course traces (E) and summary (F) of PDGF-induced maximum responses of TORCAR-NLS in double starved NIH3T3 cells. Blue, wild type cells (WT, N = 29); yellow, Rheb knockdown cells (shRheb + None, N = 54); green, Rheb knockdown cells transfected with wild type mCherry-Rheb-WT (shRheb + Rheb-WT, N = 42); orange, Rheb knockdown cells transfected with mCherry-Rheb-C181S (shRheb + Rheb-C181S, N = 84); pink, Rheb knockdown cells transfected with mCherry-Rheb-Δ5A (shRheb + Rheb-Δ5A, N = 43). (G) Model showing that active Rheb in the nucleus is critical for activating nuclear mTORC1, in addition to the previously reported Akt-mediated nuclear translocation of Raptor and phosphorylation of PRAS40. N indicates the cell number quantified from at least 3 independent experiments. Shaded areas indicate standard error of the mean (SEM). Box plots show the upper and lower adjacent values, interquartile range and the median. Scale bar = 10 µm. See also Figure S3.
Although farnesylation of Rheb is known to be important for mTORC1 activity, it is not clear whether the farnesyl group itself or farnesylation-enabled membrane association of Rheb is critical for mTORC1 activation. To distinguish these mechanisms, we used the lysosomal-associated membrane protein 1 (LAMP1)-derived sequence (Zhou et al., 2015) to target Rheb-C181S to the lysosomal membrane and test if lysosomal targeting without farnesylation can rescue the cytosolic TORCAR response in Rheb knockdown cells (Figures S3C). Interestingly, while the Rheb knockdown cells did not respond to PDGF stimulation (0.0 ± 1.1 %, n = 16), localizing Rheb-C181S mutant to the lysosomal membrane can partially rescue the cytosolic TORCAR response (Lyso-Rheb-C181S: 4.6 ± 0.7 %, n = 44). Lysosomal targeting is critical for this activity as the untargeted Rheb-C181S was not able to rescue the cytosolic (Figures S3C) TORCAR response (1.7 ± 0.7 %, n = 41). Similar results were also observed in western blotting experiments examining S6K1 phosphorylation using Rheb knockdown cells stably reconstituted with Rheb mutants (Figure S3D-S3E). To further validate this finding and examine the role of farnesylation, we tested whether Lyso-Rheb-C181S could rescue cytosolic TORCAR response in cells treated with farnesyltransferase inhibitor. Indeed, in cells overexpressing Lyso-Rheb-C181S, FTI-277 was not able to inhibit cytosolic TORCAR response (Figures S3F-S3G. DMSO: 8.9 ± 0.6 %, n = 43; FTI-277: 9.2 ± 0.7 %, n = 43). Furthermore, in cells stably expressing Lyso-Rheb-C181S, FTI-277 treatment did not lead to a reduction of PDGF-induced phosphorylation of S6K1 (Figure S3H-S3I). These data support that farnesylation itself is not required for Rheb to activate mTORC1 but the lysosomal membrane association enabled by farnesylation is essential for Rheb to activate the canonical mTORC1 pathway. Thus, non-farnesylated Rheb should be capable of interacting with and allosterically activating mTORC1, which can occur on the lysosomal surface or in the nucleus.
To further test this hypothesis, we examined if farnesylation-deficient Rheb could rescue nuclear mTORC1 activity. Consistent with previous results, knockdown of Rheb abolished nuclear TORCAR response (0.3 ± 0.4 %, n = 54) compared to wild type cells (6.9 ± 0.5 %, n = 29) (Figures 4E-4F). Overexpressing wild type Rheb rescued nuclear TORCAR response to a level that is comparable to wild type cells (4.3 ± 0.7 %, n = 42) (Figures 4E-4F). Strikingly, Rheb-C181S rescued a significant amount nuclear mTORC1 activity (3.7 ± 0.5 %, n = 84). As a negative control, Rheb-Δ5A failed to rescue nuclear TORCAR response (1.2 ± 0.4 %, n = 43). These data support that Rheb regulates mTORC1 in the nucleus independent of its farnesylation and that farnesylation regulates the localization of Rheb, not its capability to stimulate mTORC1 activity.
Discussion
As a small GTPase, the activity of Rheb is determined by its nucleotide binding status. Wild type Rheb was shown to have high basal GTP binding, and yet no GEF has been identified for Rheb. Thus, a relatively low nucleotide affinity may allow Rheb to spontaneously exchange GDP for GTP to become active, given that the cellular concentration of GTP is 10-fold higher than GDP (Traut, 1994). TSC2 acts as a GAP for Rheb and colocalizes with Rheb on the lysosomal surface to keep Rheb in its GDP form to suppress the canonical mTORC1 activity, but how TSC2 is targeted to the lysosomal surface is less clear. It was reported that TSC2 directly interacts with lysosomally localized Rheb. However, only a small pool of Rheb is localized to lysosomes (Angarola and Ferguson, 2020), suggesting that there might be other factors mediating lysosomal localization of TSC2. Recent work showed that G3BPs play an important role in tethering TSC2 to lysosomes (Prentzell et al., 2021). Whether the phosphorylation of TSC2 affects its interaction with G3BPs and thus its lysosomal association requires further investigation.
Upon growth factor stimulation, active Akt phosphorylates TSC2, promoting its disassociation from the lysosomal surface and allowing Rheb to activate mTORC1 on the lysosomal surface (Figure S1A). Using live cell imaging, we showed that overexpression of the unphosphorylatable TSC2, but not wild type TSC2, suppressed the cytosolic response of TORCAR (Figures 2B–2C), supporting the lysosome-dependent spatial regulation of TSC2 as a mechanism to regulate lysosomal mTORC1 signaling. In contrast, wild type TSC2 strongly inhibited the nuclear targeted TORCAR response (Figures 2F–2G), despite the presence of nuclear Akt activity (Zhou et al., 2020). These data suggest that colocalization of TSC2 and Rheb is a prerequisite for the inhibitory effect of TSC2 on Rheb and imply that Akt-mediated phosphorylation of TSC2 disrupts its colocalization with Rheb on the lysosomal surface. Molecularly, it has been reported that phosphorylated TSC2 binds to 14–3-3 in the cytosol (Cai et al., 2006), which may prevent its binding to the lysosomal surface. On the other hand, in the nucleus, phosphorylated exogenous TSC2 cannot be spatially re-positioned or restricted by 14–3-3, and can therefore freely bind to Rheb to inhibit nuclear mTORC1 activity. In fact, the absence of endogenous nuclear TSC2 allows nuclear Rheb to be in an active “standby” status, which could allow for faster nuclear mTORC1 response to external signals (Figure 4G). It is also possible that nuclear Rheb is controlled by other mechanisms of regulation, such as other nuclear GAPs or E3 ligases (Martin et al., 2014; Ge et al., 2020), which requires future investigation.
Rheb undergoes farnesylation to achieve weak membrane association. In line with previous studies (Hanker et al., 2010), we also found that farnesylation is essential for Rheb to localize to membrane compartments and important for mTORC1 signaling since the farnesyltransferase inhibitor disrupted Rheb membrane localization as well as S6K1 phosphorylation (Figures S3A-S3B). Significantly, our genetically encoded biosensor (TORCAR) allowed us to reveal the differential effects of abolishing farnesylation on subcellular mTORC1 activities. In contrast to canonical mTORC1 regulation, we found that the farnesyltransferase inhibitor did not affect nuclear mTORC1 activity (Figures 3C–3D) and the farnesylation-deficient Rheb-C181S could stimulate nuclear mTORC1 (Figures 4E-4F). Farnesyltransferase and other enzymes responsible for Rheb farnesylation have been proposed as drug targets for cancers, but the effects of farnesyltransferase inhibitors were less satisfying than anticipated (Wang et al., 2017; W. S. Yang et al., 2017; Mullard, 2021). Given that nuclear mTORC1 does not require farnesylated Rheb, it is possible that nuclear mTORC1 activity is spared from effects of farnesyltransferase inhibitors. It would be very interesting to test the roles of subcellular pools of mTORC1 in mediating drug toxicity and resistance in the context of various diseases.
As a signaling hub, mTORC1 senses a variety of cues to regulate a broad range of biological processes, yet with high specificity and efficacy. This is likely to be achieved through the spatial compartmentalization of the signaling pathway (Mehta and Zhang, 2021; Zhang et al., 2021). Nuclear mTORC1 has been suggested to directly regulate transcription, such as those mediated by Pol III (Michels et al., 2010; Shor et al., 2010). Further investigation is needed to identify the substrates and the functions of nuclear mTORC1, which could benefit from the targeted TSC2 mutants developed in this study, given that TSC2-NLS specifically inhibits nuclear mTORC1 activity (Figure 2).
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jin Zhang, jzhang32@health.ucsd.edu .
Materials Availability
Plasmids generated in this study will be made available upon reasonable requests.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request. All original code has been deposited at https://github.com/jinzhanglab-ucsd/MatScopeSuite. And is publicly available as of the date of publication. DOIs are listed in the key resource table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-phospho-p70 S6K1(Thr389) | Cell Signaling Technology | Cat#9205 |
| Rabbit polyclonal anti-p70 S6K1 | Cell Signaling Technology | Cat#9202 |
| Rabbit polyclonal anti-Tuberin/TSC2 | Cell Signaling Technology | Cat#3612 |
| Rabbit monoclonal anti-Tuberin/TSC2 | Cell Signaling Technology | Cat#4308 |
| Rabbit monoclonal anti-Hamartin/TSC1 | Cell Signaling Technology | Cat#6935 |
| Rabbit polyclonal anti-β-tubulin | Cell Signaling Technology | Cat#2146 |
| Mouse monoclonal anti-Rheb | Abnova | Cat#H00006009-M01 |
| Mouse monoclonal anti-p84 | GeneTex | Cat#GTX70220 |
| Goat anti-Rabbit HRP 2nd antibody | Pierce | Cat#PI31460 |
| Goat anti-Mouse HRP 2nd antibody | Pierce | Cat#PI31430 |
| Goat anti-Rabbit Alexa 488 2nd antibody | Life Technologies | Cat#A11034 |
| Goat anti-Mouse Alexa 488 2nd antibody | Life Technologies | Cat#A11001 |
| Bacterial and virus strains | ||
| pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene #21339 |
| pLenti CMVTRE3G Puro DEST (w811–1) (gateway) | N/A | Addgene #6141 (discontinued) |
| pLenti CMV rtTA3G Blast | N/A | Addgene #6136 (discontinued) |
| pLenti-TetOn-mCherry-Rheb | This paper | N/A |
| pLenti-TetOn-Lyso-mCherry-Rheb-C181S | This paper | N/A |
| pLenti-TetOn-Lyso-mCherry-Rheb-𝛥5A | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PDGF | Sigma | Cat#P3201 |
| Rapamycin | LC Laboratories | Cat#R-5000 |
| FTI-277 | ApexBio | Cat#B5842 |
| DMSO | Sigma | Cat#D2650 |
| NP-40 | Millipore Sigma | Cat#492016 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15710S |
| Doxycycline | Clontech | Cat#631311 |
| Critical commercial assays | ||
| LysoTracker Green DND-26 | Invitrogen | Cat#L7526 |
| Experimental models: Cell lines | ||
| NIH3T3 cells | ATCC | Cat#CRL-1658 |
| Cal27 | Gift from Silvio Gutkind Lab, UC San Diego | N/A |
| HeLa | ATCC | Cat#CCL-2 |
| Cos7 | ATCC | Cat#CRL-1651 |
| 3T3L1 | Gift from Alan Saltiel Lab, UC San Diego | N/A |
| PLC/PRF/5 | Gift from Gen-Sheng Feng Lab, UC San Diego | N/A |
| HEK293T | ATCC | Cat#CRL-3216 |
| Oligonucleotides | ||
| shRNA targeting sequence: mouse Rheb: CAGACATACTCCATAGATATT |
The RNAi Consortium | TRCN0000075605 |
| shRNA targeting sequence: mouse TSC2: TCGCAAGACTCCGCCACATTA |
The RNAi Consortium | TRCN0000306245 |
| Nonsense mutations in reconstituted Rheb: ACtTAtTCaATcGAc |
This paper | N/A |
| Recombinant DNA | ||
| pcDNA3-TORCAR | Zhou et al., 2015 | Addgene #64927 |
| pcDNA3-TORCAR-NLS | Zhou et al., 2015 | Addgene #64931 |
| pcDNA3-TORCAR-NES | Zhou et al., 2020 | Addgene #175015 |
| pcDNA3-Flag-TSC2 | Manning et al., 2002 | Addgene #14129 |
| pcDNA3-Flag-TSC2-AA | Manning et al., 2002 | Addgene #14131 |
| pcDNA3-mCherry-TSC2 | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA-3Q | This paper | N/A |
| pcDNA3-mCherry-TSC2-NLS | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA-NLS | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA-3Q-NLS | This paper | N/A |
| myc-Rheb | Provided by Kun-Liang Guan Lab, UC San Diego | N/A |
| pcDNA3-mCherry-Rheb | This paper | N/A |
| pcDNA3-mCherry-Rheb-C181S | This paper | N/A |
| pcDNA3-mCherry-Rheb-𝛥5A | This paper | N/A |
| pcDNA3-Lyso-mCh-Rheb-C181S | This paper | N/A |
| Software and algorithms | ||
| MATscope imaging suite | Open source | DOI: 10.5281/zenodo.5908008 |
| Prism 9 | GraphPad | https://www.graphpad.com |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture, Transfection and Starvation
NIH3T3 cells (CRL-1658, ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, 11885, Gibco) supplemented with 10% calf serum (30–2030, ATCC) and 1% penicillin-streptomycin (Sigma-Aldrich). Cells were routinely tested for mycoplasma contamination and found negative. For transfection, cells were transfected with PolyJet Transfection Reagent (SL100688, SignaGen Laboratories) according to the manufacturer’s instructions and incubated in serum-free DMEM overnight (serum starvation). The next day, cells were incubated for 2 hours in modified Hank’s balanced salt solution (1×HBSS with 2 g/l glucose, pH 7.4, made from 10× HBSS (14065, GIBCO)) at 37°C (referred to as “double starvation”) before live cell imaging or immunoblotting. Cal27, HeLa and Cos7 cells were cultured in DMEM supplemented with 10% fetal bovine serum and penicillin-streptomycin. PLC/PRF/5 cells and 3T3L1 adipocytes were obtained from the Feng lab and the Saltiel lab at UC San Diego respectively to perform experiments.
METHOD DETAILS
Reagents and Drug Treatment
PDGF (P3201) was purchased from Sigma and was used at 50 ng/mL to stimulate starved cells. Rapamycin (R-5000) was purchased from LC Labs and was used at 100 nM for 30 min pretreatment before imaging experiments were performed. FTI-277 was purchased from ApexBio (B5842) and used at 5 µM for overnight treatment. DMSO was purchased from Sigma (D2650) and used as vehicle. Doxycycline was purchased from Clontech (631311) and supplemented in medium at a final concentration of 100 ng/mL to induce gene expression.
Constructs and Cloning
mTORC1 activity reporter (TORCAR) and its targeted versions (TORCAR-NES and TORCAR-NLS) were previously described (Zhou et al., 2020). The shRNA sequence targeting mouse Rheb (TRCN0000075605) and TSC2 (TRCN0000306245) were obtained from The RNAi Consortium shRNA Library. Primers containing the sense and anti-sense shRNA sequences were designed (Eton Bioscience, Inc.) to amplify a lentivirus shRNA transfer plasmid obtained from Addgene (#21339). The resulting PCR products were purified and Gibson assembled into pLKO.shRheb. Similarly, the pcDNA3-mCherry expressing vector was assembled by amplifying fragments from pcDNA3-Akt-STOPS (Zhou et al., 2020). To construct the pcDNA3-mCherry-TSC2 plasmid, pcDNA3-mCherry construct was used to amplify fragments to serve as backbone portions, and pcDNA3-Flag-TSC2 (Addgene #14129) was used to amplify TSC2. The resulting PCR fragments were purified and Gibson assembled to generate pcDNA3-mCherry-TSC2. The pcDNA3-mCherry-TSC2-AA was constructed using a strategy similar to the one used to construct pcDNA3-mCherry-TSC2 using fragments amplified from pcDNA-Flag-TSC2-AA (Addgene #14131). The pcDNA3-mCherry-TSC2-AA-3Q was generated by designing primers covering TSC2 1595–1597 (KRR) to change the sequence to QQQ and fragments with mutations were Gibson assembled. The nuclearly targeted version of these constructs were generated similarly. mCherry-tagged Rheb constructs were Gibson assembled from a human myc-Rheb plasmid (provided by the Kun-Liang Guan Lab), and pcDNA3-mCherry-Rheb was used as the backbone for Gibson assembly to generate pcDNA3-mCherry-Rheb-C181S and pcDNA3-mCherry-Rheb-Δ5A mutants using primers with mutation or deletion. pcDNA3-mCherry-Rheb-C181S was cut with BamHI/EcoRI to get mCherry-Rheb-C181S and Lyso-TORCAR (Zhou et al., 2015) was cut with BamHI/EcoRI to get the backbone with the lysosome targeting sequence. The fragments were ligated using T4 ligase to generate pcDNA3-Lyso-mCh-Rheb-C181S. pcDNA-mCherry-Rheb, pcDNA-mCherry-Rheb-C181S and pcDNA-Lyso-mCherry-Rheb-Δ5A were subcloned into a doxycycline inducible lentiviral expressing vector (Addgene #6141) using Gibson assembly, yielding pLenti-TetOn-mCherry-Rheb, pLenti-TetOn-mCherry-Rheb-C181S and pLenti-TetOn-Lyso-mCherry-Rheb-Δ5A. In these pLenti-TetOn constructs, mutations (ACtTAtTCaATcGAc) were made from Rheb 217–231 (ACATACTCCATAGAT) within the shRNA targeting sequences for Rheb to be resistant to shRNA silencing when Rheb knockdown cells were reconstituted with these exogenously expressed Rheb mutants.
Lentivirus Production and Stable Cell Lines Generation
Lentivirus was packaged in HEK293T cells. Specifically, HEK293T cells were co-transfected with corresponding lentiviral transfer plasmids + psPAX2 + pMD2.G using PolyJet. After 48hr the supernatant was collected, filtered with a 0.45 µm syringe filter and added to NIH3T3 cells in 35 mm dishes. Polybrene Transfection Reagent (TR-1003-G, Millipore) was supplemented to improve the transduction. After 48 hours, cells were passed in fresh growth medium containing puromycin (2 µg/mL) to select transduced cells for at least one week. Cells were maintained in selection medium and passed for related experiments. For doxycycline inducible cell lines, wild type or Rheb knockdown NIH3T3 cells were co-transduced with lentivirus expressing rtTA transcription factor and doxycycline inducible TetOn expressing vectors. Cells were then maintained in growth medium containing puromycin (2 µg/mL) and blasticidin (2.2 µM) and passaged for related experiments.
Nuclear Fractionation
Nuclear fractionation was done as previously reported (Suzuki et al., 2010). Briefly, two 10cm dishes of NIH3T3 fibroblasts were cultured to 80% confluence. Cells were then washed with cold PBS, collected and resuspended in 0.1% NP-40 (492016, Millipore Sigma) followed by short centrifugation. The supernatant was collected as cytosolic fraction and the pellet was washed and collected as nuclear fraction. The same proportion of samples from each fraction was used for immunoblotting analysis.
Immunoblotting
Cells were washed with ice-cold PBS and then lysed in RIPA lysis buffer containing protease inhibitor cocktail, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, and 25 nM calyculin A. Total cell lysates were incubated on ice for 30 min and then centrifuged at 15,000g at 4°C for 20 min. Equal amounts of total protein were separated via 4–15% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with TBS containing 0.1% Tween-20 and 5% bovine serum albumin and then incubated with primary antibodies overnight at 4°C. The next day, membranes were washed, incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies, and were developed using horseradish peroxidase-based chemiluminescent substrate (34579 and 34076, Thermo Fisher Scientific). The following primary antibodies were used for immunoblotting: p-S6K1 (T389) (#9205), S6K1 (#9202), TSC2 (#3612) and tubulin (#2146) antibodies from Cell Signaling Technology, Rheb antibody from Abnova (H00006009-M01), p84 (GTX70220) antibody from GeneTex. Horseradish peroxidase-labeled goat anti-rabbit (PI31460) or anti-mouse (PI31430) secondary antibodies were purchased from Pierce. Uncropped blots are shown in Figure S4.
Immunofluorescence Imaging
For immunofluorescence, upon reaching ~80% confluency in 6-well plates (353046, Corning) containing glass coverslips (12–541A, Fisher Brand), cells were washed 3 times with PBS and fixed with 4% paraformaldehyde in PBS (15710S, Electron Microscopy Sciences) for 25 min at room temperature. Cells were then washed 3 times with PBS and permeabilized with PBS containing 0.1% Triton X-100 for 25 min at room temperature. Following 1-hour incubation in blocking buffer (PBS containing 0.1% Triton X-100 and 5% BSA) at room temperature, coverslips were incubated overnight at 4ºC in primary antibody diluted in blocking buffer. Following three 5-min washes in PBS, coverslips were incubated for 1 hour at room temperature in the dark in secondary antibody (anti-rabbit Alexa Fluor 488, A11034 or anti-mouse Alexa Fluor 488, A11001, Life Technologies) at 1:1000 dilution in blocking buffer. Following three 5-min washes with PBS, coverslips were mounted in Prolong Glass antifade Mountant with NucBlue (P36981, Invitrogen). Coverslips were dried and kept in −20℃ before images were taken. Primary antibodies used were anti-Rheb (H00006009-M01) from Abnova, and anti-TSC1 (#6935) and anti-TSC2 (#4308) from Cell Signaling Technology. Confocal images for both green (488 nm ex) and blue (405 nm ex) channels were acquired by a Leica SP8 microscope with 60x oil objective (1.40 NA) or 40x oil objective (1.30 NA) in Lightning Deconvolution mode. For each coverslip, the middle slice was obtained and multiple images were taken for each experimental condition from different biological samples. Images were analyzed using CellProfiler software (Broad Institute). Briefly, the green channel was used to segment the cell and the mean intensity was measured for each cell as the whole cell signal. The blue channel was used as nuclear marker to segment the nuclear region, and the nuclear mean intensity in the green channel was measured as nuclear signal. Line scan was drawn in ImageJ software (NIH) and the intensity along the line was measured.
Live-cell Imaging
For live cell imaging, cells were plated onto sterile glass-bottomed 35-mm dishes (D35–14-1.5-N, CellVis) and grown to 40% confluency at 37°C with 5% CO2. Cells were transfected, double starved, and washed once with HBSS and imaged in the dark at room temperature. Images were acquired on a Zeiss Axio Observer Z1 microscope equipped with a 40x/1.3NA objective (Carl Zeiss), Prime95B sCMOS camera (Photometrics) and a motorized stage (Carl Zeiss). The microscope was controlled by MATLAB (Mathworks) and µmanager (Micro-Manager, an open-source microscope imaging software) -based MATScope imaging suite (Github: https://github.com/jinzhanglab-ucsd/MatScopeSuite). CFP images were acquired using ET420/20x excitation filter with a T4551pxt dichroic and AT470/40m emission filter. YFP images were acquired using ET495/10x excitation filter with a T5151p dichroic and ET535/25m emission filter. The C/Y FRET images were acquired using ET420/20x excitation filter with T4551pxt dichroic and ET535/25m emission filter. mCherry imaging was performed using HQ568/55x excitation filter with a Q600LPxr dichroic mirror and HQ653/95m emission filter. Exposure times were 50–500 ms, and images were taken every 30 s or 1 min. Imaging data were analyzed using MATLAB scripts developed within the lab. Specifically, fluorescence images were background-corrected and regions of interest (ROI) were manually selected. For TORCAR-NES and TORCAR-NLS, ROIs were within the cytosolic or nuclear regions, respectively. For TORCAR, when whole cell responses were measured (Figures S1C-S1D), individual cells were selected as the ROIs; and when the cytosolic response of TORCAR was examined (Figures 2B–2C), ROIs were within the cytosolic regions and data was labeled as “Cytosolic regions”. The cyan/yellow FRET emission ratio was calculated for each ROI on each frame of the image series. The ratio was normalized to the time point before addition of PDGF (t = 0 min). For lysosomal colocalization experiments, lysosomes were visualized in live cells using LysoTracker Green DND-26 (L7526, Invitrogen). Briefly, before images were taken under the microscope, LysoTracker Green was added to the medium and incubated with cells for 30 min, then cells were washed twice in HBSS imaging buffer. LysoTracker Green were visualized using 488 nm excitation under the microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
The data were analyzed using GraphPad Prism 9. Immunofluorescence data were obtained from two biological replicates, and all other data were obtained from at least three independent biological replicates. Unless otherwise noted, unpaired two-tailed t-test was used for two-group comparison and one-way ANOVA with Dunnett’s multiple comparisons test were used for multi-group comparisons. **** indicates a p-value<0.0001; *** indicates a p-value between 0.0001 to 0.001; ** indicates a p-value between 0.001 to 0.01; * indicates a p-value between 0.01 to 0.05; ns, p>0.05, not significant. N numbers, as indicated in figures, legends and the main text, represent the number of cells. Shaded areas in average curves indicate standard error of the mean (SEM). Violin plots and box plots show the upper and lower adjacent value, interquartile range and the median.
Supplementary Material
Significance
In this study, by combining imaging and synthetic biology approaches, we found a small pool of Rheb is present in the nucleus and required for nuclear mTORC1 activity. We clarified the role of Rheb farnesylation in subcellular mTORC1 signaling, provided new insights into the regulation of nuclear mTORC1 and developed tools for selectively perturbing subcellular pools of mTORC1 activity for functional dissection. Understanding the precise mechanisms and functions of subcellular mTORC1 signaling should provide us with the opportunity to target subcellular mTORC1 activity as a potential therapeutic strategy.
Highlights:
A small pool of Rheb is present in the nucleus but not its negative regulator TSC2.
Active Rheb exists in the nucleus and is required for nuclear mTORC1 activity.
Properly localized non-farnesylatable Rheb can turn on nuclear or cytosolic mTORC1.
Rheb farnesylation is required for proper localization but not mTORC1 activation.
Acknowledgements
We thank Olivia Molinar in the Trejo lab for providing suggestions about nuclear fractionation. We thank Ayse Z Sahan and Mingyuan Chen in the Zhang lab for critical reading of the manuscript. This work was supported by NIH grants, R01 DE030497 (J.Z.) and NS047101 to UCSD Microscopy Core.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angarola B, Ferguson SM, 2019. Weak membrane interactions allow Rheb to activate mTORC1 signaling without major lysosome enrichment. Mol. Biol. Cell 30, 2750–2760. doi: 10.1091/mbc.E19-03-0146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angarola B, Ferguson SM, 2020. Coordination of Rheb lysosomal membrane interactions with mTORC1 activation. F1000Res. 9. doi: 10.12688/f1000research.22367.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso AD, Mirza A, Liu G, Long BJ, Bishop WR, Kirschmeier P, 2005. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J. Biol. Chem. 280, 31101–31108. doi: 10.1074/jbc.M503763200 [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP, 2006. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 442, 779–785. doi: 10.1038/nature05029 [DOI] [PubMed] [Google Scholar]
- Cai S-L, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL, 2006. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 173, 279–289. doi: 10.1083/jcb.200507119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, Guan K-L, 2006. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J. Biol. Chem. 281, 8313–8316. doi: 10.1074/jbc.C500451200 [DOI] [PubMed] [Google Scholar]
- Clark GJ, Kinch MS, Rogers-Graham K, Sebti SM, Hamilton AD, Der CJ, 1997. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J. Biol. Chem. 272, 10608–10615. doi: 10.1074/jbc.272.16.10608 [DOI] [PubMed] [Google Scholar]
- Demetriades C, Doumpas N, Teleman AA, 2014. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156, 786–799. doi: 10.1016/j.cell.2014.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetriades C., Plescher M., Teleman AA., 2016. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 7, 10662. doi: 10.1038/ncomms10662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlazzo ML, Bach-Tobdji MKE, Djerad A, Sonzogni L, Devic C, Granzotto A, Bodgi L, Bachelet J-T, Djefal-Kerrar A, Hennequin C, Foray N, 2017. Radiobiological Characterization of Tuberous Sclerosis: a Delay in the Nucleo-Shuttling of ATM May Be Responsible for Radiosensitivity. Mol. Neurobiol. 55, 1–11. doi: 10.1007/s12035-017-0648-6 [DOI] [PubMed] [Google Scholar]
- Ge M-K, Zhang N, Xia L, Zhang C, Dong S-S, Li Z-M, Ji Y, Zheng M-H, Sun J, Chen G-Q, Shen S-M, 2020. FBXO22 degrades nuclear PTEN to promote tumorigenesis. Nat. Commun. 11, 1720. doi: 10.1038/s41467-020-15578-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker AB, Mitin N, Wilder RS, Henske EP, Tamanoi F, Cox AD, Der CJ, 2010. Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene 29, 380–391. doi: 10.1038/onc.2009.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao F, Kondo K, Itoh T, Ikari S, Nada S, Okada M, Noda T, 2018. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 131. doi: 10.1242/jcs.208017 [DOI] [PubMed] [Google Scholar]
- Havel JJ, Li Z, Cheng D, Peng J, Fu H, 2015. Nuclear PRAS40 couples the Akt/mTORC1 signaling axis to the RPL11-HDM2-p53 nucleolar stress response pathway. Oncogene 34, 1487–1498. doi: 10.1038/onc.2014.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N, 2004. Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704 [DOI] [PubMed] [Google Scholar]
- Huo Y, Chen WS, Lee J, Feng G-S, Newton IG, 2019. Stress conditions induced by locoregional therapies stimulate enrichment and proliferation of liver cancer stem cells. J Vasc Interv Radiol. doi: 10.1016/j.jvir.2019.02.026 [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan K-L, 2003. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834. doi: 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan K-L, 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657. doi: 10.1038/ncb839 [DOI] [PubMed] [Google Scholar]
- Jiang H, Vogt PK, 2008. Constitutively active Rheb induces oncogenic transformation. Oncogene 27, 5729–5740. doi: 10.1038/onc.2008.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ, 2010. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc. Natl. Acad. Sci. USA 107, 11823–11828. doi: 10.1073/pnas.1005188107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Guan K-L, 2019. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 21, 63–71. doi: 10.1038/s41556-018-0205-1 [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, 2000. Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc. Natl. Acad. Sci. USA 97, 14340–14345. doi: 10.1073/pnas.011511898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM, 2012. mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Inoki K, Guan K-L, 2004. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol. Cell. Biol. 24, 7965–7975. doi: 10.1128/MCB.24.18.7965-7975.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, Sabatini DM, 2020. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203. doi: 10.1038/s41580-019-0199-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J, 2005. Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713. doi: 10.1016/j.cub.2005.02.053 [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC, 2002. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162. doi: 10.1016/s1097-2765(02)00568-3 [DOI] [PubMed] [Google Scholar]
- Martin TD., Chen X-W., Kaplan REW., Saltiel AR., Walker CL., Reiner DJ., Der CJ., 2014. Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 53, 209–220. doi: 10.1016/j.molcel.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Zhang J, 2021. Biochemical Activity Architectures Visualized-Using Genetically Encoded Fluorescent Biosensors to Map the Spatial Boundaries of Signaling Compartments. Acc. Chem. Res. 54, 2409–2420. doi: 10.1021/acs.accounts.1c00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD, 2014. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785. doi: 10.1016/j.cell.2013.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, Hernandez N, 2010. mTORC1 directly phosphorylates and regulates human MAF1. Mol. Cell. Biol. 30, 3749–3757. doi: 10.1128/MCB.00319-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A, 2021. The FDA approves a first farnesyltransferase inhibitor. Nat. Rev. Drug Discov. 20, 8. doi: 10.1038/d41573-020-00213-x [DOI] [PubMed] [Google Scholar]
- Prentzell MT, Rehbein U, Cadena Sandoval M, De Meulemeester A-S, Baumeister R, Brohée L, Berdel B, Bockwoldt M, Carroll B, Chowdhury SR, von Deimling A, Demetriades C, Figlia G, Genomics England Research Consortium, de Araujo MEG, Heberle AM, Heiland I, Holzwarth B, Huber LA, Jaworski J, Kedra M, Kern K, Kopach A, Korolchuk VI, van ‘t Land-Kuper I, Macias M, Nellist M, Palm W, Pusch S, Ramos Pittol JM, Reil M, Reintjes A, Reuter F, Sampson JR, Scheldeman C, Siekierska A, Stefan E, Teleman AA, Thomas LE, Torres-Quesada O, Trump S, West HD, de Witte P, Woltering S, Yordanov TE, Zmorzynska J, Opitz CA, Thedieck K, 2021. G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell 184, 655–674.e27. doi: 10.1016/j.cell.2020.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner M, Hengstschläger M, 2012. Detection of cytoplasmic and nuclear functions of mTOR by fractionation. Methods Mol. Biol. 821, 105–124. doi: 10.1007/978-1-61779-430-8_8 [DOI] [PubMed] [Google Scholar]
- Saitoh M, Pullen N, Brennan P, Cantrell D, Dennis PB, Thomas G, 2002. Regulation of an activated S6 kinase 1 variant reveals a novel mammalian target of rapamycin phosphorylation site. J. Biol. Chem. 277, 20104–20112. doi: 10.1074/jbc.M201745200 [DOI] [PubMed] [Google Scholar]
- Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, Yu K, 2010. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J. Biol. Chem. 285, 15380–15392. doi: 10.1074/jbc.M109.071639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K, 2010. REAP: A two minute cell fractionation method. BMC Res. Notes 3, 294. doi: 10.1186/1756-0500-3-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabancay AP, Gau C-L, Machado IMP, Uhlmann EJ, Gutmann DH, Guo L, Tamanoi F, 2003. Identification of dominant negative mutants of Rheb GTPase and their use to implicate the involvement of human Rheb in the activation of p70S6K. J. Biol. Chem. 278, 39921–39930. doi: 10.1074/jbc.M306553200 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Nakagawa M., Young SG., Yamanaka S., 2005. Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J. Biol. Chem. 280, 32768–32774. doi: 10.1074/jbc.M506280200 [DOI] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J, 2003. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268. doi: 10.1016/s0960-9822(03)00506-2 [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. J Biol Chem. 2009. Mar 20. 284(12):8023–32. 10.1074/jbc.M900301200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW, 1994. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22. doi: 10.1007/BF00928361 [DOI] [PubMed] [Google Scholar]
- Wan W, You Z, Xu Y, Zhou L, Guan Z, Peng C, Wong CCL, Su H, Zhou T, Xia H, Liu W, 2017. mTORC1 Phosphorylates Acetyltransferase p300 to Regulate Autophagy and Lipogenesis. Mol. Cell 68, 323–335.e6. doi: 10.1016/j.molcel.2017.09.020 [DOI] [PubMed] [Google Scholar]
- Wang J, Yao X, Huang J, 2017. New tricks for human farnesyltransferase inhibitor: cancer and beyond. Medchemcomm 8, 841–854. doi: 10.1039/c7md00030h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Feng X, Molinolo AA, Martin D, Vitale-Cross L, Nohata N, Ando M, Wahba A, Amornphimoltham P, Wu X, Gilardi M, Allevato M, Wu V, Steffen DJ, Tofilon P, Sonenberg N, Califano J, Chen Q, Lippman SM, Gutkind JS, 2019. 4E-BP1 Is a Tumor Suppressor Protein Reactivated by mTOR Inhibition in Head and Neck Cancer. Cancer Res. 79, 1438–1450. doi: 10.1158/0008-5472.CAN-18-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, Pavletich NP, 2017. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373. doi: 10.1038/nature25023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Yeo S-G, Yang S, Kim K-H, Yoo BC, Cho JY, 2017. Isoprenyl carboxyl methyltransferase inhibitors: a brief review including recent patents. Amino Acids 49, 1469–1485. doi: 10.1007/s00726-017-2454-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J-F, Mehta S, Zhang J, 2021. Signaling microdomains in the spotlight: visualizing compartmentalized signaling using genetically encoded fluorescent biosensors. Annu. Rev. Pharmacol. Toxicol. 61, 587–608. doi: 10.1146/annurev-pharmtox-010617-053137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shu L, Hosoi H, Murti KG, Houghton PJ, 2002. Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J. Biol. Chem. 277, 28127–28134. doi: 10.1074/jbc.M202625200 [DOI] [PubMed] [Google Scholar]
- Zhou X, Clister TL, Lowry PR, Seldin MM, Wong GW, Zhang J, 2015. Dynamic Visualization of mTORC1 Activity in Living Cells. Cell Rep. 10, 1767–1777. doi: 10.1016/j.celrep.2015.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zhong Y, Molinar-Inglis O, Kunkel MT, Chen M, Sun T, Zhang Jiao, Shyy JY-J, Trejo J, Newton AC, Zhang Jin, 2020. Location-specific inhibition of Akt reveals regulation of mTORC1 activity in the nucleus. Nat. Commun. 11, 6088. doi: 10.1038/s41467-020-19937-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. All original code has been deposited at https://github.com/jinzhanglab-ucsd/MatScopeSuite. And is publicly available as of the date of publication. DOIs are listed in the key resource table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-phospho-p70 S6K1(Thr389) | Cell Signaling Technology | Cat#9205 |
| Rabbit polyclonal anti-p70 S6K1 | Cell Signaling Technology | Cat#9202 |
| Rabbit polyclonal anti-Tuberin/TSC2 | Cell Signaling Technology | Cat#3612 |
| Rabbit monoclonal anti-Tuberin/TSC2 | Cell Signaling Technology | Cat#4308 |
| Rabbit monoclonal anti-Hamartin/TSC1 | Cell Signaling Technology | Cat#6935 |
| Rabbit polyclonal anti-β-tubulin | Cell Signaling Technology | Cat#2146 |
| Mouse monoclonal anti-Rheb | Abnova | Cat#H00006009-M01 |
| Mouse monoclonal anti-p84 | GeneTex | Cat#GTX70220 |
| Goat anti-Rabbit HRP 2nd antibody | Pierce | Cat#PI31460 |
| Goat anti-Mouse HRP 2nd antibody | Pierce | Cat#PI31430 |
| Goat anti-Rabbit Alexa 488 2nd antibody | Life Technologies | Cat#A11034 |
| Goat anti-Mouse Alexa 488 2nd antibody | Life Technologies | Cat#A11001 |
| Bacterial and virus strains | ||
| pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene #21339 |
| pLenti CMVTRE3G Puro DEST (w811–1) (gateway) | N/A | Addgene #6141 (discontinued) |
| pLenti CMV rtTA3G Blast | N/A | Addgene #6136 (discontinued) |
| pLenti-TetOn-mCherry-Rheb | This paper | N/A |
| pLenti-TetOn-Lyso-mCherry-Rheb-C181S | This paper | N/A |
| pLenti-TetOn-Lyso-mCherry-Rheb-𝛥5A | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PDGF | Sigma | Cat#P3201 |
| Rapamycin | LC Laboratories | Cat#R-5000 |
| FTI-277 | ApexBio | Cat#B5842 |
| DMSO | Sigma | Cat#D2650 |
| NP-40 | Millipore Sigma | Cat#492016 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15710S |
| Doxycycline | Clontech | Cat#631311 |
| Critical commercial assays | ||
| LysoTracker Green DND-26 | Invitrogen | Cat#L7526 |
| Experimental models: Cell lines | ||
| NIH3T3 cells | ATCC | Cat#CRL-1658 |
| Cal27 | Gift from Silvio Gutkind Lab, UC San Diego | N/A |
| HeLa | ATCC | Cat#CCL-2 |
| Cos7 | ATCC | Cat#CRL-1651 |
| 3T3L1 | Gift from Alan Saltiel Lab, UC San Diego | N/A |
| PLC/PRF/5 | Gift from Gen-Sheng Feng Lab, UC San Diego | N/A |
| HEK293T | ATCC | Cat#CRL-3216 |
| Oligonucleotides | ||
| shRNA targeting sequence: mouse Rheb: CAGACATACTCCATAGATATT |
The RNAi Consortium | TRCN0000075605 |
| shRNA targeting sequence: mouse TSC2: TCGCAAGACTCCGCCACATTA |
The RNAi Consortium | TRCN0000306245 |
| Nonsense mutations in reconstituted Rheb: ACtTAtTCaATcGAc |
This paper | N/A |
| Recombinant DNA | ||
| pcDNA3-TORCAR | Zhou et al., 2015 | Addgene #64927 |
| pcDNA3-TORCAR-NLS | Zhou et al., 2015 | Addgene #64931 |
| pcDNA3-TORCAR-NES | Zhou et al., 2020 | Addgene #175015 |
| pcDNA3-Flag-TSC2 | Manning et al., 2002 | Addgene #14129 |
| pcDNA3-Flag-TSC2-AA | Manning et al., 2002 | Addgene #14131 |
| pcDNA3-mCherry-TSC2 | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA-3Q | This paper | N/A |
| pcDNA3-mCherry-TSC2-NLS | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA-NLS | This paper | N/A |
| pcDNA3-mCherry-TSC2-AA-3Q-NLS | This paper | N/A |
| myc-Rheb | Provided by Kun-Liang Guan Lab, UC San Diego | N/A |
| pcDNA3-mCherry-Rheb | This paper | N/A |
| pcDNA3-mCherry-Rheb-C181S | This paper | N/A |
| pcDNA3-mCherry-Rheb-𝛥5A | This paper | N/A |
| pcDNA3-Lyso-mCh-Rheb-C181S | This paper | N/A |
| Software and algorithms | ||
| MATscope imaging suite | Open source | DOI: 10.5281/zenodo.5908008 |
| Prism 9 | GraphPad | https://www.graphpad.com |