

Abstract
With the aim of expediting drug target discovery and validation for respiratory diseases, we developed an optimized method for in situ somatic gene disruption in murine lung epithelial cells via AAV6-mediated CRISPR-Cas9 delivery. Efficient gene editing was observed in lung type II alveolar epithelial cells and distal airway cells following assessment of single- or dual-guide AAV vector formats, Cas9 variants, and a sequential dosing strategy with combinatorial guide RNA expression cassettes. In particular, we were able to demonstrate population-wide gene disruption within distinct epithelial cell types for separate targets in Cas9 transgenic animals, with minimal to no associated inflammation. We also observed and characterized AAV vector integration events that occurred within directed double-stranded DNA break sites in lung cells, highlighting a complicating factor with AAV-mediated delivery of DNA nucleases. Taken together, we demonstrate a uniquely effective approach for somatic engineering of the murine lung, which will greatly facilitate the modeling of disease and therapeutic intervention.
Keywords: adeno-associated virus, AAV, viral delivery, CRISPR, Cas9, gene editing, lung epithelium, disease modeling, inflammatory response, genome integration
Graphical abstract
Chen et al. present optimized methodology for AAV-mediated delivery of CRISPR-Cas9 machinery to the murine lung, enabling population-wide gene disruption in several epithelial cell types. Separately, using long-read genome sequencing, they characterize a potential genotoxic risk via AAV integration into on-target Cas9 cut sites.
Introduction
CRISPR-Cas9 has proven to be revolutionary for basic and translational research, and it has facilitated a broad range of previously challenging applications. This is particularly true of disease modeling and drug target discovery or validation in vivo. Here, it has enabled the development of somatically engineered mouse models (SEMMs) through unprecedented gains in speed and cost-efficiency, thereby presenting a distinct complement to traditional germline-derived equivalents. In this context, adeno-associated virus (AAV) technology as a means to deliver the necessary CRISPR-Cas9 cargoes has attracted significant attention due to its potential for tissue-enriched infection, effective delivery efficiency, and relatively low immunogenicity. In addition, the tropism of naturally existing AAV capsid variants has been extensively characterized for transgene delivery into the central nervous system, retina, heart/muscle, liver, and lung, thus affording a variety of options for broad or tissue-specific genome manipulation.1,2,3,4,5,6,7,8,9,10,11,12
Several studies have described viral vector-based methods for somatic editing of oncogenic or tumor suppressor genes in situ, leading to the establishment of sophisticated SEMMs for non-small cell lung cancer,13,14,15,16,17,18,19 glioma,20 pancreatic cancer,21 breast cancer,22 and colorectal cancer.23 While the generation of cancer models can often be achieved with modest editing efficiency in the target cell population, owing to a growth advantage of the edited cells harboring CRISPR-Cas9-induced oncogenic mutations, higher editing rates could provide appropriate conditions for recreating non-transforming disease states. Indeed, in vivo gene perturbation efficiencies impacting ∼80% of target alleles, as measured by direct DNA sequence or cellular phenotype, have been observed through the use of AAV-CRISPR-Cas9 in retina,24 muscle,25,26 and brain,27 suggesting that cell population-level effects are feasible. However, the ability to achieve this degree of editing outside of select organs and across a variety of key cell types or tissues, therefore maximizing opportunities for disease modeling, remains a challenge.
To create an in vivo platform that enhances the potential for respiratory disease modeling and drug target discovery or validation, we sought to optimize AAV-mediated delivery of CRISPR-Cas9 and the related gene perturbation protocols with a specific focus on the murine lung epithelium. Achievement of a high editing efficiency in alveolar cell type II (AECII) and airway epithelium, e.g., club and ciliated cells, is of particular value given the roles these cell types play in cystic fibrosis, chronic obstructive pulmonary disease, and idiopathic pulmonary fibrosis.28,29,30,31 Through fine-tuning the AAV dosage, applying a unique repeat-dosing protocol and a multi-vector strategy to deliver the full suite of CRISPR-Cas9 machinery, we were able to disrupt ≥65% of surveyed alleles in AECII and airway epithelial cells in Ai9 mice.32 With further optimization by applying a single-guide expression vector in combination with a Cas9 transgenic model, we could induce near-complete depletion of target protein in AECII or significantly alter differentiation of airway epithelial cells following disruption of Usp30 and Notch2, respectively. Furthermore, long-read sequencing analysis and locus reconstruction of edited cells identified a substantial degree of AAV genome integration at on-target double-stranded DNA (dsDNA) break sites, revealing the potential for unintended mutagenic events or genotoxicity concerns related to gene editing methods that utilize AAV-CRISPR-Cas9.33,34,35
Results
Optimization and biodistribution profiling of AAV transduction in murine lung epithelium
Previously, AAV9 was combined with intratracheal (i.t.) delivery to express CRISPR-Cas9 modalities in the murine lung.16 Using this as a baseline to identify an optimal AAV-mediated transgene delivery method for lung epithelial cells, we compared intranasal (i.n.) versus i.t. routes of administration in combination with a reporter virus (AAV9-CAG-luciferase). Use of the strong, constitutive CAG promoter to drive reporter expression enabled us to gauge general transduction characteristics.36 i.t. delivery produced a qualitative improvement of in vivo bioluminescent signal across the whole lung compared with the i.n. method (Figure S1A). Whole-lung coverage after i.t. delivery was also examined by administering either a trypan blue dye (Figure S1B) or a fluorescent reporter virus (AAV9-CAG-GFP). Relatively homogeneous biodistribution of the GFP transgene was observed in 3D images of AAV9-reporter-infected lung lobes (Figure S1C), demonstrating the superiority of i.t. administration in achieving widespread infection of the murine lung. The i.t. method was therefore selected for experiments conducted throughout the remainder of our study.
With a goal of delivering to the lung epithelial cells with broadly attainable AAV variants, we compared transduction efficiencies after i.t. administration of AAV6,16,36 AAV8,11 and AAV916 CAG-luciferase or CAG-GFP reporter particles. All AAV serotypes provided sustained luciferase expression for at least 23 weeks after delivery (Figure S1D). Both AAV6 and AAV9 provided a stronger in vivo bioluminescent signal compared with AAV8. However, AAV9 also showed nonspecific bioluminescent signals in the abdomen of male mice (Figure S1D). These results are consistent with previous observations of improved lung delivery for AAV6 compared with AAV8 and broader tropism of the AAV9 capsid variant relative to AAV6 or AAV8.37,38 Immunofluorescent staining (IF) of lung tissue from animals intubated with either AAV6-CAG-GFP or AAV9-CAG-GFP showed high %GFP expression in AECII compared with modest expression with AAV8-CAG-GFP (Figures S1E and S1F). Relative to AAV9, AAV6 provided greater transduction efficiency in club cells (Figures S1E and S1F). Due to its higher transduction efficiency in the cell types of interest (AECII and airway epithelium), we proceeded with AAV6 for the remainder of our study.
Through the use of i.t. delivery, we sought to identify the optimal dose of AAV6-CAG-GFP necessary to achieve maximal transduction efficiency in lung epithelial cells. IF staining of GFP and cell type markers showed that transduction efficiency reached a plateau of ∼80% in AECII starting at 8 × 1011 vg (vector genome)/mouse, with similar levels of efficiency requiring higher doses in ciliated (8 × 1012 vg/mouse) and club cells (1.2 × 1013 vg/mouse) (Figure 1A). We subsequently examined the potential for co-delivery of separate AAV6 reporter viruses (CAG-GFP and CAG-mCherry). IF staining of GFP and mCherry revealed a high frequency of overlapping expression (>80%) in all three cell types after simultaneously delivered AAV6 viruses (Figures 1B and 1C). This latter strategy supports the application of multi-AAV strategies to deliver cargos that cannot fit into a single AAV vector due to packaging size limitations.
Figure 1.

Profiling AAV6 transduction in murine lung
(A) Quantification of GFP expression within cells dosed with increasing titers of AAV6-CAG-GFP. IF staining of GFP and various cell markers were performed on paraffin-embedded lung tissue harvested at week 1 after delivery of AAV6-CAG-GFP. Lung sections containing five lung lobes were quantified for each animal. n = 5. Error bars represent SEM. (B) Representative IF images showing GFP stained in green, mCherry in red, cell markers (SPC, CC10, and acetyl-alpha tubulin, respectively) in white, and nuclei (DAPI) in blue on paraffin-embedded lung sections. Animals were co-transduced with AAV6-CAG-GFP and AAV6-CAG-mCherry at 4 × 1012 vg/virus/mouse. Lung tissues were harvested at week 1 after infection. Scale bars, 500 μm. The yellow box shows the zoomed-in region. Scale bars, 50 μm (inset). (C) Quantification of the colocalization of GFP, mCherry, and cell markers. IF staining of GFP, mCherry, and various cell markers was performed on paraffin-embedded lung tissue harvested at week 1 after co-transduction of AAV reporter viruses (GFP and mCherry). Lung sections containing five lung lobes were quantified for each animal. n = 5. Error bars represent SEM. (D) The composition of lung cell types presented in GFP– versus GFP+ samples as shown in the pie charts. (E) The percentage of each lung cell type presented in GFP– versus GFP+ samples.
In a separate experiment aimed at dissecting AAV6 transduction efficiency in cell types of the lung in an unbiased manner, single-cell RNA sequencing (scRNA-seq) analysis was conducted in animals that received two sequential doses of co-delivered AAV6 vectors at 2 × 1012 vg/dose/mouse. Here, one vector expressed dCpG-Staphylococcus pyogenes (Sp)Cas9 and the other expressed a sgRNA cassette linked with a GFP marker (Figure S2A). Non-hematopoietic lineage cell populations (CD45–/GFP+ versus CD45–/GFP−) were extracted from the lungs of treated animals and subjected to scRNA profiling. Consistent with the high AAV co-transduction efficiency shown in our previous experiments (Figures 1B and 1C), scRNA-seq analysis revealed that nearly all cells that expressed Cas9 also expressed GFP (Figure S2B). Clustering analysis of cell types based on gene expression identified the major lung cell subpopulations, including AECII, alveolar type I (AT1), fibroblasts, club, ciliated, and endothelial cells (Figure S2C). The GFP transgene was enriched within AECII, club, and ciliated cells, as well as a small portion of AT1 cells (Figures 1D, 1E, and S2D). Altogether, our scRNA-seq results are consistent with the IF data demonstrating that AAV6 has skewed tropism for AECII, club, and ciliated cells relative to other cell types in the murine lung when using our i.t. delivery protocol.
Cas9 and sgRNA expression vector parameters that enable editing in the lung epithelium of Ai9 reporter mice
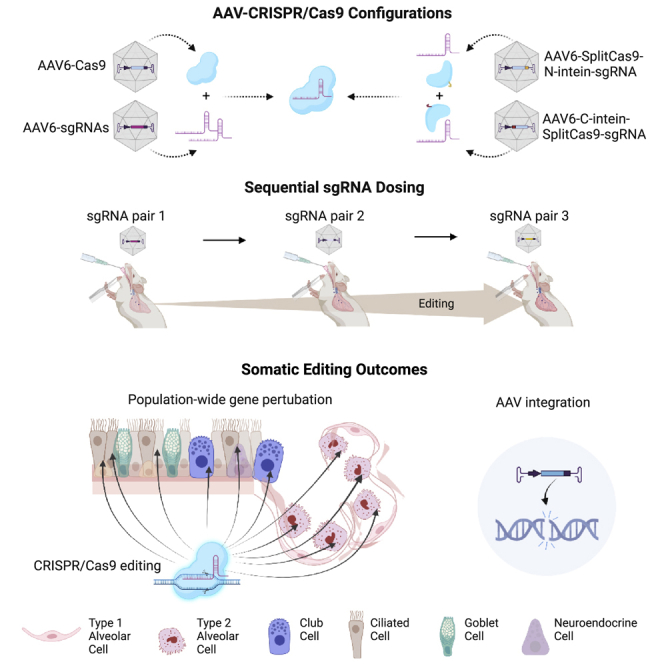
To evaluate the application of CRISPR-Cas9 in vivo, we designed Staphylococcus aureus (Sa)Cas939 all-in-one vectors carrying both Cas9 and pairs of sgRNAs targeting the genomic regions flanking a LoxP-Stop-LoxP (LSL)-tdTomato cassette in Ai9 reporter mice (Figure S3A). Here, genome editing is expected to generate indels at one or both of the paired sgRNA target sites, as well as deletion of the LSL cassette, potentially resulting in tdTomato expression (Figure 2A). As a result of the extended protospacer adjacent motif (PAM) sequence requirement for SaCas9, designs were limited to two sgRNAs with acceptable activity scores on each side of the LSL cassette. Use of the CAG promoter to drive Cas9 expression would result in an AAV genome beyond the conventional packaging size limit. Therefore, different ubiquitous promoters with a compact size, such as EF1a short and Rous sarcoma virus (RSV) long terminal repeat,36 were used for expression of SaCas9. We tested the two promoters through transient transfection of plasmids in mouse embryonic fibroblasts (MEFs) derived from Ai9 mice. We observed higher Cas9 expression and more gene editing (as measured by tdTomato-expressing cells) with the RSV promoter (Figures S3B and S3C). Hence, this promoter was selected for experiments using AAV-delivered Cas9 in vivo. Before investigations of in situ somatic editing in AECII and airway epithelial cells, we developed a FACS protocol based on cell type-specific lineage markers to sort these two cell populations for analysis (Figures S4A and S4B).40,41,42 The sorted airway epithelial cells are mainly from distal airways and are reported to contain ∼10%–15% club cells, 80% ciliated cells, and <1% lineage-negative epithelial progenitors.41,42 Upon delivery of SaCas9 and LSL-targeting sgRNAs via AAV6 in vivo, we observed relatively low (based on %tdTomato) to moderate (based on %indels) editing efficiency in AECII and distal airway epithelial cells (Figures S3D and S3E).
Figure 2.

Somatic editing in Rosa26-LSL-tdTomato mice by dual AAV delivery of Cas9 and sgRNA pair
(A) Schematic of somatic editing outcomes. Upon the delivery of AAV vectors, CRISPR-Cas9-mediated genome editing can lead to the formation of indels and deletion of the LSL cassette, which leads to tdTomato expression. (B) Two dual vector designs (configurations 1 and 2) to deliver the CRISPR/SpCas9 components. The genetic components were colored and labeled. The size of each vector was listed under the vector. (C) Indel analysis of sgRNA pair 1 in animals received one dose of AAV-SpCas9/sgRNA pair or AAV-split Cas9/sgRNA pair. %Indels formation at the two on-target sites (left panel: left sgRNA; right panel: right sgRNA) was examined in the sorted AECII and airway epithelial cells at week 3 after intubation of AAV at 2 × 1012 vg/mouse (n = 5–6). Each dot represents one animal. Error bars represent SEM. ∗p < 0.05; n.s., not significant. (D) Representative IF images of tdTomato and GFP staining of paraffin-embedded lung tissues collected at week 3 after the first round of AAV intubation. GFP stained in green, tdTomato stained in red. Cell nuclei stained by DAPI in blue. Scale bars, 200 μm. (E) Quantification of %tdTomato expression in sorted AECII and airway epithelial cells at week 3 after the intubation of the AAV-SpCas9/sgRNA pair or the AAV-SplitCas9/sgRNA pair. Data determined by two methods are shown: upper panel, FACS analysis. %tdTomato expression was quantified in AECII lineage marker-positive cells and integrin β4-positive cells, respectively. n = 4–6; lower panel, IF staining. %tdTomato expression was quantified in cells stained positive for SPC (AECII marker) and CC10 (club cell marker), respectively. n = 3. Each dot represents one animal. Error bars represent SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (F) Quantification of %GFP and %tdTomato/GFP double expression in sorted AECII and distal airway epithelial cells at week 3 after the first round of AAV intubation. Data determined by two methods are shown: upper panel, FACS analysis. %tdTomato expression was quantified in AECII lineage marker-positive cells and integrin β4-positive cells, respectively. n = 6–7; lower panel, IF staining. %tdTomato expression was quantified in cells stained positive for SPC (AECII marker) and CC10 (club cell marker), respectively. n = 3–6. Each dot represents one animal. ∗p < 0.05; n.s., not significant. (G) ddPCR analysis of the ITR region of the AAV vectors. The genome copy per cell was analyzed for AECII and distal airway epithelial cells sorted from the animals in (D). n = 8–10. Each dot represents one animal. Error bars represent SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant.
To further optimize editing efficiency, we investigated separate AAV co-delivery approaches to accommodate the larger SpCas9 (4.1 kb), which has a shorter and less stringent PAM sequence (NGG) requirement compared with SaCas9. The PAM requirement of SpCas9 enabled us to design several sgRNA pairs intended to flank and disrupt the LSL cassette, as described above. Building from our data showing efficient co-vector transduction in AECII, club, and ciliated cells (Figures 1B and 1C), we tested two dual-vector configurations to deliver SpCas9 and corresponding LSL-specific sgRNA pairs. In one configuration (configuration 1), an AAV-vector was designed to deliver full-length SpCas9 driven by the RSV promoter, while a separate vector was engineered to co-express a pair of sgRNAs (U6 promoter) and a GFP marker (CAG promoter) (Figure 2B; Table S1). A second configuration (configuration 2) used dual AAV vectors where each vector would deliver a corresponding N- or C-terminal half of an intein-mediated split-SpCas943,44 and one of two sgRNAs targeting the LSL cassette (Figure 2B). Packing size limited additional features that could be inserted within the full-length SpCas9 vector. The split SpCas9 constructs, on the other hand, allowed the inclusion of a woodchuck hepatitis virus PRE (WPRE) element, which is known to enhance expression from virus-derived transgenes (Figure 2B).45 Using %tdTomato+ cells as a measure of efficiency, we observed greater activity with FL-SpCas9 compared with split-Cas9 in Ai9-derived embryonic fibroblasts (Figure S5A). The addition of a WPRE in the split-SpCas9 constructs enhanced target disruption compared with the same constructs without a WPRE (Figure S5A). Protein analysis confirmed expression of both the FL-SpCas9 protein and intein-mediated reconstitution of split-SpCas9, which occurred as early as 4 days after delivery of AAV vectors into MEFs (Figure S5B). A small quantity of un-reconstituted split-SpCas9 protein was also detected (Figure S5B).
Following validation of the dual vector strategy in vitro, we moved to assess gene editing efficiency in vivo via the Ai9 model. Animals received one dose (1 × 1012 vg/vector/mouse) of either AAV-FL-SpCas9/sgRNA pair (configuration 1) or AAV-split- SpCas9/sgRNA pair (configuration 2). Indel formation was examined from sorted AECII and distal airway epithelial cells. The sgRNA-right showed a comparably high indel frequency (∼85% in AECII and ∼65% in distal airway epithelial cells) in animals that received either configuration 1 or configuration 2, indicating the successful reconstitution of the split-SpCas9 into functional full-length SpCas9 (Figure 2C, right panel). However, we observed the split-SpCas9 to be more sensitive to differences in sgRNA efficiency, as shown by a modest but significant reduction in indel formation with the weaker sgRNA-left versus the more efficient sgRNA-right (Figures 2C and S5C).
Despite the high proportion of indels (65%) generated with the right-side guide RNA, use of the relatively weaker left-side guide RNA (≤50%, Figure S5C) was expected to decrease the chance of a co-targeting event and, consequently, LSL deletion. This was reflected in lower %tdTomato expression (20%) as measured by immunostained lung tissues or dissociated lung cells, compared with the frequency of indels formed at either target site alone from animals that received either the AAV-FL-SpCas9/sgRNA pair (configuration 1) or the AAV-split-SpCas9/sgRNA pair (configuration 2) (Figures 2C–2E). The %tdTomato expression (∼15%–20%) as measured by IF staining of club cells in Ai9 animals that received the AAV-FL-SpCas9/sgRNA pair was consistent with, albeit slightly lower than, a recent study demonstrating ∼19%–26% editing in lung airway epithelial cells.46 While a dual vector strategy was used in each case, differences in efficiency between these methods could be related to the discrepant AAV serotypes, promoters driving the FL-SpCas9, unique right-side sgRNA sequences, and standard (our study) versus self-complementary viral genome formats (Figure 2E, bottom panel).46 Reduced %tdTomato via split- compared with FL-SpCas9 (Figure 2F) correlated with significantly lower editing efficiency at the left-sgRNA site (Figure 2E) rather than fewer genome copies within transduced cells, as animals injected with AAV-split-SpCas9 showed a trend toward higher vector copies per cell compared with AAV-FL-SpCas9 (Figure 2G). This is consistent with previous results that suggest split-SpCas9 may have comparable but often lower intrinsic DNA damaging activity relative to FL-SpCas9.44,47,48
In an effort to increase the efficacy of gene targeting, we examined the effect of two sequential doses of AAV-FL-SpCas9 (hereafter AAV-SpCas9) with separate AAV-sgRNA pairs (configuration 1) in Ai9 mice. Since we expected the original sites to be damaged, the second dose of AAV-SpCas9/sgRNA pair contained an LSL-targeting sgRNA pair distinct from that used for the first dose (Table S1, sgRNA pair 2). The second dose was provided 7 days after the initial AAV-SpCas9/sgRNA pair infection. In this context, our sgRNA vectors contained a GFP marker, which enabled simultaneous tracking of infection and LSL cassette deletion (i.e., tdTomato/GFP double-positive). While we did not observe a positive dose response in AECII after the second administration of viral particles, a significant increase of %tdTomato/GFP double expression was obtained in distal airway epithelial cells, as measured by both FACS and IF analysis (Figure 2F). Consistent with this finding, the AAV vg copy number per cell in sorted distal airway epithelial cells increased significantly after two doses, whereas no significant difference was observed in AECII (Figure 2G). This indicated that two rounds of AAV delivery at 1 × 1012 vg/vector/mouse can efficiently increase the vector transduction potential and subsequent genome editing efficiency in airway epithelial cell types. However, some cell types, such as AECII, may have maximized the potential for infection after a single dose.
Population-wide disruption of USP30 in AECII of SpCas9 transgenic mice
The potent indel formation (>65% of alleles) triggered by the right-side guide RNA in the Ai9 mice study (Figure 2C) indicated that population-wide gene disruption may be possible through further optimization of the sgRNA targeting scheme and SpCas9 expression. Accordingly, germline expression of SpCas9 permits the use of a single, compact AAV-sgRNA vector for in situ somatic editing. To evaluate whether this approach offered similar or greater efficiency relative to AAV-mediated co-delivery of SpCas9 and sgRNAs to AECII and distal airway epithelial cells, we proceeded to target an endogenous gene of interest, Usp30, a deubiquitinase that is expressed in these cell types and is suggested to play a role in mitophagy in neurons.49 Three separate sgRNA pairs were designed to perturb or delete portions of the coding sequences within exons 1–7 of the Usp30 gene (Figure 3A, upper panel; Table S1). Successful deletion would excise the C77 catalytic residue in exon 3, while indels at each of the distinctive sgRNA target sites would generate frame-shifted Usp30 alleles. AAV vectors were designed to express each pair of Usp30-specific sgRNAs as well as a mitoQC fluorescent reporter50 that served as a transduction marker in current experiments and would facilitate future studies aimed at assessing the effects of USP30 depletion on mitophagy in vivo (Figure 3A, lower panel). Before packaging into AAV particles, the gene disruption potential of each sgRNA pair was evaluated in vitro via transient plasmid transfection into MLE12 cells engineered to stably express SpCas9. Based on near-complete depletion of USP30 in these assays, we concluded that each of the sgRNA pairs were highly and similarly functional (Figure 3B).
Figure 3.

Highly efficient USP30 depletion on a populational level in murine lung epithelium
(A) Schematic of Usp30 genomic targeting sites by the sgRNA pairs (upper panel) and the AAV vector design carrying dual sgRNA cassettes and a mitoQC fluorescent reporter (lower panel). (B) The sgRNA pairs effectively downregulated USP30 protein expression in MLE-12-SpCas9 cells. Cells were infected by a single AAV at a multiplicity of infection (MOI) of 1 × 106 vg or three AAVs combined (each at an MOI of 1 × 106 vg). Proteins were extracted from the pooled cell population (without selection) at day 7 after infection and subject to western blot analysis. (C) Schematic of animal study design. Three rounds of AAV at 2 × 1012 vg/mouse/round were delivered to Rosa26-CAG-SpCas9-mKate2 transgenic mice with 7 days apart. Animals were taken down at weeks 5 and 8 after the first round of AAV delivery for analysis. (D) AAV transduction efficiency in cell types of interest as measured by %mitoQC expression. FACS analysis of %mitoQC expression at week 5 (n = 9) and week 8 (n = 8) after the first round of AAV delivery in AECII and distal airway epithelial cells. Each dot presents one animal. Error bars represent SEM. ∗∗∗p < 0.001; n.s., not significant. (E) USP30 protein downregulation in sorted AECII at week 8 after the first round of AAV delivery. GAPDH served as the housekeeping control. The digital images of the capillary electrophoresis immunoblotting of USP30 and GAPDH are shown. (F) Quantification of USP30 protein expression shown in (E). ImageJ was used to quantify the total pixel signal of USP30 and GAPDH. The pixel signal of USP30 was normalized to that of GAPDH. n = 10–15. Each dot presents one animal. Error bars represent SEM. ∗∗p < 0.01.
Building off our findings in the Ai9 model, showing that back-to-back dosing of sgRNA pairs was tolerated and improved gene editing efficiencies in select cell types, we employed a sequential dosing strategy to deliver all three of the Usp30-specific sgRNA pairs via AAV6 particles (AAV-sgRNApair-mitoQC), one pair at time, every 7 days at 2 × 1012 vg/animal/dose to Rosa26-CAG-SpCas9-mKate2 mice (Figure 3C). Animals were sacrificed at weeks 5 or 8 after injection of the first dose. AECII and distal airway epithelial cells were collected by FACS for analysis of USP30 expression or genomic deletion. FACS analysis of %mitoQC marker expression (i.e., mCherry and GFP double-positive) suggested an AAV transduction efficiency of ∼70% in AECII at week 5, which was consistent with our earlier observations (Figures 3D and 1A). However, expression of the mitoQC marker dropped significantly by week 8 (Figure 3D). Approximately 30%–35% of distal airway epithelial cells remained positive for the mitoQC marker throughout the course of this study, which may reflect longer transduction stability within this cell type relative to AECII (Figure 3D).
We then investigated CRISPR/SpCas9-mediated disruption of USP30 in AECII by protein analysis. Quantitative analysis of USP30 protein levels in AECII cells revealed an average of ∼91% depletion in the treatment group compared with the control animals (Figures 3E, 3F, and S6A), indicating potent CRISPR/SpCas9 activity in situ. Due to an insufficient number of sorted distal airway epithelial cells for protein extraction, DNA was prepared from this population and was followed by genomic PCR assessment for the presence of a truncated product indicative of a CRISPR/SpCas9-mediated deletion event within the Usp30 locus (Figure S6B; Table S2). Consistent with the efficient protein depletion in sorted AECII, robust Usp30 deletion fragments were detected in the sorted distal airway epithelial cells (Figure S6C). Taken together, we have demonstrated the potential for near-complete, population-wide gene knockout through AAV-mediated sgRNA delivery into lung epithelial cells of an SpCas9 transgenic model.
Efficient transdifferentiation of club cells to ciliated cells following AAV-mediated perturbation of Notch2
Extending our demonstration of potent USP30 depletion through a sequential sgRNA delivery approach, we proceeded to target a separate endogenous gene of interest, Notch2, which plays an important developmental role in the lung airway epithelial cells.51 It has been reported that inhibition of Notch signaling by anti-JAG1 antibodies can induce trans-differentiation of club cells into ciliated cells.52 Therefore, we reasoned that targeted disruption of Notch2 in the adult mouse lung could phenocopy antibody-mediated suppression of this signaling axis. Here, we designed a series of unique sgRNA pairs targeting Notch2, and screened them for activity via NOTCH2 depletion in MLE-12-SpCas9 cells. Following that assessment, the top three most active sgRNA pairs were cloned into AAV vectors carrying an mCherry marker (Figures 4A and 4B; Table S1) and packaged into AAV6 particles (AAV-sgRNApair-mCherry). Using Rosa26-CAG-SpCas9-eGFP transgenic animals,16 we provided three sequential doses by i.t. administration, with a unique sgRNA pair per dose at 4 × 1012 vg/mouse; a viral titer that permitted efficient targeting of club cells, the cell type of interest (Figure 1A). Animals were sacrificed at weeks 4 and 8 following the initial dose, and lung tissues were processed for downstream analysis (Figure 4C). Consistent with our previous findings (Figure 3D), FACS analysis showed ∼80% mCherry-positive AECII at week 4 with a significant decrease by week 8 (Figure 4D). Approximately 60% of distal airway epithelial cells were mCherry positive at week 4, with stable values through week 8 (Figure 4D). We then examined NOTCH2 expression in whole lung tissue of AAV-infected animals via immunohistochemistry (IHC) (Figure 4E). Pixels per area quantification of the IHC stained samples indicated a trend with lower NOTCH2 levels in the lung of treated animals compared with the AAV control group at both time points (Figure 4F). Notch2 genomic deletion fragments were identified in sgRNA-treated animals via PCR analysis of the sorted distal airway epithelial cells (Figures S7A and S7B; Table S2).
Figure 4.

CRISPR/SpCas9-mediated NOTCH2 knockdown led to efficient trans-differentiation of club cells into ciliated cells
(A) Schematic of Notch2 genomic targeting sites by sgRNA pairs (upper panel) and the AAV vector design carrying the paired sgRNA cassette and an mCherry reporter (lower panel). (B) The sgRNA pairs effectively downregulated NOTCH2 expression in MLE-12-SpCas9 cells. Cells were infected by a single AAV at an MOI of 1 × 106 or three AAVs combined (each at an MOI = 1 × 106). Proteins were extracted from the pooled cell population (without selection) on day 7 after infection and subjected to capillary protein analysis. (C) Schematic of animal study design. Three sequential doses of AAV-Notch2-sgRNA pairs at 4 × 1012 vg/mouse/round were delivered to Rosa26-CAG-SpCas9-eGFP transgenic mice at 7-day intervals. Animals were taken down for analysis at weeks 4 or 8 after the first round of infection. (D) AAV transduction efficiency in cell types of interest as measured by %mCherry expression. FACS analysis of %mCherry expression at week 4 (n = 13) and week 8 (n = 16) after the first round of sgRNA pair delivery in AECII and distal airway epithelial cells. Each dot represents one animal. Error bars represent SEM. ∗∗∗p < 0.001; n.s., not significant. (E) Representative NOTCH2 IHC staining of the paraffin-embedded lung tissue from the control group (left picture) and treatment group (right picture) at week 4 time point. Scale bars, 200 μm. (F) Quantification of NOTCH2 expression by IHC at weeks 4 and 8 time points. Data represented the NOTCH2 protein level of the whole lung tissue (not specific to AECII, club, and ciliated cells). Data were normalized to the PBS control animals. Each dot represents one animal. Error bars represent SEM. p values are indicated. (G) Representative IF images of club and ciliated cells at the week 4 time point. Club cells stained in white (CC10: a cell marker for club cells); ciliated cells stained in green (acetylated alpha-tubulin: a cell marker for ciliated cells). mCherry stained red. mCherry expression was an indicator of AAV-infected cells. Cell nuclei stained by DAPI in blue. Scale bars, 200 μm. (H) Quantification of club cells as stained by the cell marker CC10 (CC10+ area). Data were pooled from weeks 4 and 8 time points. Each dot represents one animal. Error bars represent SEM. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n.s., not significant. (I) Quantification of ciliated cells as stained by the nuclear marker FoxJ1 (FoxJ1+ area). Data was pooled from weeks 4 and 8 time points. Each dot represents one animal. Error bars represent SEM. ∗∗∗∗p < 0.0001; n.s., not significant.
To investigate the effect of NOTCH2 disruption on airway epithelia, we examined the total proportion of airway cell types through IF staining with markers of club and ciliated cells, as well as mCherry (an indicator of AAV-infected cells). We observed a significant reduction in the numbers of club cells with a corresponding increase in ciliated cells in the AAV-Notch2-sgRNApair-mCherry treatment group, compared with the AAV-mCherry control group (Figures 4G–4I and S7C). The morphology of nearby lung tissue appeared unchanged (Figure S7D), but we cannot exclude the possibility that neighboring cell types might have been altered due to Notch2 disruption. In summary, our study recapitulated the role of NOTCH signaling in regulating the differentiation of airway epithelia, and we have shown that using AAV-mediated somatic gene editing of the lung is an effective and rapid way to validate the biological mechanisms underlying a putative therapeutic agent.
Minimal host inflammatory responses in murine lung tissues after AAV6-mediated gene delivery
We recognized that, while high AAV concentrations and repeated dosing strategies provided robust gene disruption within the lung epithelia, these insults offered the potential of triggering an inflammatory response. To investigate this, we further evaluated tissues processed from various studies mentioned above. We used an ordinal grading system to assess inflammation, taking into account the overall changes in all lung lobes available for each animal. The i.t. procedure per se can cause some minor inflammation in animals, as shown by the PBS control mice (Table 1, column C). However, in the AAV6-CAG-GFP dosage study, inflammation was negligible in almost all treated animals even at the dosage of 1.2 × 1013 vg/mouse (Table 1, columns A and B). The low grades of minimal and mild (grades 1 and 2) were characterized by minimal perivascular and/or peribronchiolar mononuclear cell infiltrates and occasional histiocytes in the alveolar spaces (Figures S8A and S8B), which are common incidental findings in the lungs of mice. These lesions were not caused by or associated with treatment and their incidence is similar in all cohorts.
Table 1.
Pathological grading of inflammation severity of lung tissue after high- and re-dosing of AAVs
| Inflammation severity (grade 0–5) | Single dose of AAV6-CAG-GFP or mCherry (at week 1 after dosing) |
1–2 doses of AAV6-SpCas9 and AAV6-LSL-sgRNApair-GFP (2 × 1012 vg/mouse/dose, at week 2 after the first doing) |
3 doses of AAV6-Notch2-sgRNApair-mCherry (4 × 1012 vg/mouse/dose, at weeks 4 or 8 after the first dosing) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 8 × 1012 vg/mouse, n (%) | 1.2 × 1013 vg/mouse, n (%) | PBS control, n (%) | 1 dose, n (%) | 2 doses, n (%) | PBS, n (%) | Week 4, n (%) | Week 4 PBS, n (%) | Week 8, n (%) | Week 8 PBS, n (%) | |
| Column ID | A | B | C | D | E | F | G | H | I | J |
| Grade 0 | 3 (27.3) | 7 (63.6) | 1 (50) | 11 (91.7) | 7 (77.8) | 3 (100) | 3 (21.4) | 3 (100) | 6 (46.2) | 2 (100) |
| Grade 1 | 4 (36.4) | 2 (18.2) | 1 (50) | 1 (8.3) | 2 (22.2) | 0 | 7 (50) | 0 | 6 (46.2) | 0 |
| Grade 2 | 2 (18.2) | 2 (18.2) | 0 | 0 | 0 | 0 | 4 (28.6) | 0 | 1 (7.6) | 0 |
| Grade 3 | 2 (18.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Grade 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Grade 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
We next probed for overt signs of an inflammatory response as a result of our sequential dosing strategy. Histopathological analysis of the lung tissue from the one- versus two-dose treatment groups (AAV6-SpCas9/AAV6-LSL-gRNApair) in the Ai9 study showed 8.3% versus 22.2% of animals exhibiting minimal focal alveolar histiocytosis, respectively (grade 1) (Table 1, columns D and E). The overall inflammation grade was well within the normal range for all animals (Table 1, column D–F). Similar findings were made in SpCas9 transgenic animals that received three doses of AAV6-Notch2-sgRNApair-mCherry. In this cohort, histopathological analysis showed limited inflammation at weeks 4 and 8 after injection (Table 1, columns G and I). The proportion of animals with minimal and mild (grades 1 and 2) inflammatory responses fell within the normal expected background. The proportion of animals with grade 2 inflammation decreased between weeks 4 and 8 (28.6% versus 7.6%), suggesting that this is a temporary effect. Further analysis of the cellular immune responses as measured by FACS analysis of CD45+ and CD45+CD11b+ populations revealed no significant cell infiltrates at either week 4 or 8 time points compared with control mice (Figures S8C and S8D). Overall, our observations are consistent with a previous study, which found a relatively low immunogenic response with AAV6 compared with other evaluated serotypes.53 Taken together, we concluded that the i.t. delivery and sequential dosing strategies could be considered unlikely to influence cellular phenotypes due to procedure or AAV6-related inflammatory responses.
AAV genome integration within on-target dsDNA breaks
Although AAV remains primarily episomal following transduction, integration of viral DNA into the host genome is a viable safety concern.54 This was exemplified in a long-term follow-up study in a canine hemophilia model, which showed potential genotoxicity caused by AAV vector integration events within transduced hepatocytes.55 This phenomenon is expected to be more prominent when AAV is used to deliver nucleases intended to create double-stranded breaks in the host genome.34 To investigate whether our method can result in unintended AAV genome integration within lung epithelium, we developed a PCR-based approach to detect AAV ITR integration in one of the Usp30 genomic sites targeted by the right sgRNA in pair 14 (Figures 3A–3C). Here, ITR-host genome fusion events were amplified by primers binding to the D region in the ITR35 and to an Usp30 genomic region ∼260 bp downstream of the sgRNA target site (Figure 5A, top panel; Table S2). PCR analysis on sorted distal airway epithelial cells showed amplification of the fusion events in ∼88% of animals in the treatment group (Figure 5A, middle panel). Notably, this approach cannot be used to quantify the frequency of integration events on a per-cell basis. Sequence analysis of the PCR products confirmed the presence of genome-integrated ITR fragments (Figure 5A, lower panel). Similarly, we detected ITR integration events in the edited MLE-12-SpCas9 cell line after AAV-Usp30-sgRNApair transduction, suggesting that this effect is not specific to the in vivo method of delivery (Figure S9A).
Figure 5.

Characterization of AAV integration at the CRISPR-Cas9 on-target site
(A) PCR analysis of ITR-genomic fusion events in sorted AECII from animals treated with three doses of AAV or AAV-sgRNA pair targeting Usp30 (Figure 3C). Upper panel: schematic of primer design; middle panel: PCR amplification of ITR-genomic fusion events as shown in 2% agarose gel. Lower panel: representative individual AAV ITR integration clones by TOPO cloning of the amplified PCR products (indicated by arrows) were shown. The integrated ITR segments were underscored by red lines. The Usp30 genomic sequence was underscored by black lines. The CRISPR-Cas9 PAM site was underscored by orange lines. The Topo vector sequence was underscored by blue lines. (B) Quantification of ONT reads mapped to the tdTomato genomic locus that were also mapped to the AAV vector genome. (C) Integrated genomic view of the sequence alignments to the reference AAV vector genome. The upper track showed the position of genetic elements within each AAV vector genome; the lower track showed the coverage and read alignments. (D) Characterization of the integrated vector segments. Top panel: pie chart showed the composition of vector segments. Lower panel: violin plot showed the size distribution of the integrated vector segments. (E) Representative examples of AAV vector genome integration events. The reference sequence is composed of the genomic locus with AAV-SpCas9 (top) or AAV-LSL-sgRNA pair (bottom) vector inserted within the CRISPR-Cas9 on-target site. The genomic sequence is colored red. AAV vectors are annotated with different genetic components in distinct colors. Each AAV-genomic fusion read is plotted under the reference genome in the corresponding alignment position and orientation. The sequence of the fusion read is indicated by a line labeled with nucleotide positions. Rearrangement events were visualized in the order of appearance and connected by curved links. (F) Coverage analysis of ITR segments presented in ITR-tdTomato fusion junctions. The data were plotted based on all integration events from both AAV-SpCas9 and AAV-LSL-sgRNA pair vectors.
Our PCR-based approach for detecting integration events provided a sensitive analysis of short, select regions of the host and viral genomes. To characterize the potential for genomic integration of AAV-delivered transgenes in their entirety, we applied Oxford Nanopore long-read sequencing technology (ONT). We examined the AAV integration events in sorted AECII and distal airway epithelial cells obtained from Ai9 animals that received one dose of the co-delivered AAV6-SpCas9 and AAV6-LSL-sgRNA pair (Figure 2B, configuration 1). To circumvent the low amount of DNA obtained from the sorted AECII and distal airway epithelial cells, we took advantage of Samplix Xdrop technology,56 which permitted enrichment of DNA from a region of interest and, subsequently, ONT sequencing with limited input material. In this study, Xdrop droplet PCR primers were designed to enrich DNA molecules containing the tdTomato transgene present in Ai9 mice (Figures S9B and S9C; Tables S2 and S3).
To identify AAV vector integration events, ONT reads containing the tdTomato sequence were aligned to the AAV-SpCas9 and AAV-LSL-sgRNA pair vgs (Figures 5B, 5C, and S9D; Table S4). AAV vector-aligned reads were not observed in control animals (Table S4). In the treatment group, approximately 12% of the reads corresponded to AAV vgs, indicating the presence of integration events (Figures 5B and 5C). Furthermore, a bias toward integration was observed for the AAV-SpCas9 vector compared with the AAV-LSL-sgRNA pair counterpart, suggesting an unexpected influence of the vector design and/or size on integration frequency (Figures 5B and 5C). A small fraction of the reads aligned to mixed segments belonging to both the AAV-SpCas9 and AAV-LSL-sgRNA pair vectors (Figure 5B), revealing that vector recombination may have occurred during the process of AAV integration in animals exposed to co-delivered vectors. However, the potential for co-delivered vectors to recombine during the process of integration had not been fully explored and warrants further analysis.
Long-read sequencing of targeted loci after transduction revealed the extent to which full or partial AAV genome integration had occurred. Consistent with previous findings,33,34,35 ITR sequences were observed with a high frequency, relative to other segments of integrated AAV genomes (Figure 5C). Most AAV-SpCas9 vector integration events contained one intact fragment spanning from 48 bases to almost the full vg of ∼4.5 kb in length (Figure 5D). The remaining AAV-SpCas9 vector integration events contained separate, discontinuous fragments of a few hundred bases of the vg (Figure 5D). Conversely, we observed a distinct integration signature for the AAV-LSL-sgRNA pair vector, which was a single fragment, in the size range of a few hundred bases (Figure 5D).
We next characterized the rearrangement of the integrated AAV vector fragments in the context of the host genome by creating alignments to the reference LSL-tdTomato locus from Ai9 animals with the AAV vector inserted within the right-side target site of LSL sgRNA pair 1 (Figures S9D and S9E). The tdTomato transgenic cassette was fused with AAV fragments resulting in a heterogeneous pattern of truncation, inversion, and duplication (Figure 5E). These findings were consistent with a recent report where PacBio long-read sequencing was applied to identify the rearrangement of integrated AAV genomes in hepatocytes.57 Notably, ITR fragments dominated the fusion junctions among the reads analyzed in our experiments (Figures 5E and S9E). A closer analysis of these ITR-tdTomato fusion junctions revealed a higher frequency of the D to A′ region, compared with other regions in the ITR, indicating a potential role of this region in mediating AAV integration (Figures 5F and S9F).
Discussion
Somatic genome editing of lab animals holds incredible promise for disease and therapeutic intervention modeling. This is particularly true for diseases of the respiratory system, owing to the minimally invasive approaches that could be used for transgene delivery to lung epithelial cells, which play a role in both acute and chronic disorders.58,59 While approaches, specifically those employing viral vectors, to deliver gene editing cargo into the lungs of rodent models have proven useful for preclinical studies, these efforts have yet to achieve the population-level efficiency of gene perturbation observed in other tissues.14,15,16,17,18,19 Therefore, we sought to develop the toolsets and strategies that maximize editing outcomes in key lung epithelial cells so as to unlock the full breadth and potential of these necessary modeling approaches.
In this study, we describe an optimized protocol for somatic genome editing in murine lung epithelial cells through AAV6-mediated delivery of CRISPR-Cas9 machinery. By tuning AAV6 dosages it was possible to achieve robust transgene expression in key cell types, such as AECII cells, or a broader population, including club and ciliated cells. Moreover, we describe a well-tolerated AAV sequential dosing approach, which provided superior AAV infection efficiency as well as potent gene editing. We demonstrated this by targeting an integrated transgene (LSL-tdTomato) in Cas9-naive animals with up to ∼85% indel frequency achieved in the lung epithelium, as well as two distinct endogenous loci (Usp30 and Notch2) in SpCas9-transgenic animals, resulting in near complete gene disruption across a large fraction of the desired cell types. Thus, our method has paved the way for effective, rapid generation of SEMMs. Moreover, the versatility of our approach provides the basis for expanded CRISPR tool usage within the murine lung, including split Cas9 variants for gene activation (CRISPRa), inhibition (CRISPRi), base editing, and RNA targeting (via Cas13), while also offering a path for further vector optimization through the application of relatively small Cas enzymes60,61 or self-complementary AAV-formatted sgRNAs, which have shown superior activity to the ssAAV-sgRNAs used in our study.25 We note that AAV6 does not have unique tropism for lung epithelial cells,38 nor did the promoters used in our study have selectivity for our target cell types. Coupling our method with engineered AAV capsids and/or enhancements to the transgene regulatory elements could therefore result in yet greater tissue specificity or expressivity of the gene modifying cargo.62,63
It is widely established that immune responses can neutralize AAV or target transgene-expressing cells following virus exposure.43 Attempts at redosing may therefore require immunosuppression or a substantial waiting period after the initial exposure.10,64 Strikingly, some serotypes, such as AAV6, show a minimal neutralizing immune response against themselves following a single intranasal dose into the murine lung, therefore offering the potential for productive re-administration.53 Consistent with this finding, we did not observe a substantial or limiting inflammatory response in animals that received sequential i.t. doses with AAV6-packaged cargo, and this approach was surprisingly effective and well tolerated. Consequently, this strategy facilitated maximized transduction efficiency with repeated lower viral doses, which could potentially reduce the risk of high dose-related side effects, at least for the subsets of lung epithelial cells investigated here. In addition, a sequential dosing strategy creates the opportunity to use multiple sets of sgRNAs per target locus, thus increasing the likelihood of gene disruption. Throughout these studies, we did not find evidence of cell death, a known byproduct of excessive dsDNA breaks,65 nor unexpected cell morphology/phenotype after knockout of targeted genetic loci. However, repeated dosing with multiple sgRNAs does invite the potential for increased off-target editing events, and experiments should be designed and interpreted accordingly. Extension of our method through regulated control of CRISPR-Cas9 machinery expression,66,67,68 application of HiFi-Cas9 variants,69 or with Cas9 base editor proteins70 may enable greater precision and reduced off-targeting potential.
AAV-mediated delivery of DNA nucleases and/or homology donor sequences has become a viable approach for clinical gene editing ex vivo71 and in vivo.72,73,74,75 However, AAV vector integration, either random or at the sites of directed DNA damage, has raised safety concerns.54 Several groups have reported AAV integration using short-read sequencing methods.33,34,35,55 To fully characterize on-target editing sites, we applied a long-read sequencing approach (Samplix Xdrop/ONT), which led to the observation, for the first time in lung cells, that the entire AAV genome (up to 4.5 kb) could integrate into the intended CRISPR-Cas9 target site. While we observed a propensity for integration of ITR-proximal sequences, integration rates as a whole differed across separate vector sequences and sizes, warranting further investigation into the mechanism of AAV insertion at both on- and off-target sites of DNA damage.
We acknowledge a limitation of the Samplix Xdrop workflow, in which a multiple displacement amplification (MDA) step was carried out, leading to the generation of chimeric long reads with repetitive palindrome compartments into a single long read. Although it does not affect the mapping of these reads to the reference genome, splitting of the chimeric section by software, such as Pacusus, is recommended for de novo assembly.76 Also, we obtained only a limited number (∼hundreds) of reads containing the tdTomato sequence after Samplix Xdrop enrichment, amplification, and ONT sequencing. Further optimization of our sample and library preparation is needed to increase Xdrop enrichment efficiency and to facilitate a greater number of relevant reads for AAV integration analysis.
Taken together, our study has established an effective somatic editing platform in murine lung epithelium, and also provided insights related to in situ editing outcomes and potential genotoxicity.
Materials and methods
AAV vector construction, production, and titration
AAV vector cloning was conducted by GenScript (Piscataway, NJ). AAV constructs were cloned into the following vectors provided by GenScript: pX601, pX551, and pX552 vectors. Split-Cas9 configuration was cloned into the ssAAV vector backbone obtained from CellBiolabs (San Diego, CA). AAV6 viruses were produced and purified by ViroVek (Hayward, CA) using an Sf9 production platform. All reporter AAV viruses (GFP and mCherry) were purchased from ViroVek. AAV titers were determined using SYBR Green qPCR assay (Applied Biosciences, TX). ITR primer sequences are: ITR-F, 5′-GACCTTTGGTCGCCCGGCCT-3′; ITR-R, 5′-GAGTTGGCCACTCCCTCTCTGC-3′. An AAV expression plasmid pAV-CAG-GFP was used to generate the standard curve. AAV viruses were pretreated with proteinase K in 1:10 dilution at 37°C for 30 min. The proteinase digested samples were then serially diluted with the starting dilution of 1:100 followed by 2-fold dilutions. A qPCR reaction was composed of 12.5 μL 2× SYBR Green master mix, 1 μL primers (300 nM working concentration), 2 μL DNA, and 10.35 μL H2O. Each sample was prepared in triplicate. qPCR was carried out in a 7500 Real-Time PCR System (Applied BioSciences, TX). Cycling conditions were 95°C for 15 min, followed by 40 cycles of two-step protocol of 95°C for 30 s and 60°C for 1 min. A melting curve was performed at the end of the cycling. AAV titer was calculated based on the standard curve.
sgRNA design and in vitro assessment
Guide design was carried out using a Benchling specificity score that also calculated the relative genome-wide off-target propensity (Benchling.com, San Francisco, CA). The mm10 genome build was used to identify appropriate targeting sequences. For Ai9/Rosa26-LSL-tdTomato targeting, sgRNA pairs were designed to delete the stop cassette or perturb the LoxP elements (Table S1); the activity of each sgRNA pair was tested in MEFs derived from Ai9 animals and cultured in regular Gibco Dulbecco’s modified Eagle’s (DMEM) growth medium supplemented with 10% L-glutamine, 1% penicillin/streptomycin and 10% fetal bovine serum (FBS). Plasmids (5 μg total per nucleofection) were nucleofected into MEFs using the Amaxa 4D nucleofector (Lonza BioSciences, NC) with a pulse code of CZ-167. tdTomato expression was measured by FACS at day 4 after nucleofection. In USP30 and NOTCH2 studies, sgRNA pairs were designed to target the functional domain in Usp30 and Notch2, with the goal of ablating expression of protein by exon deletion and/or frameshift (Table S1). The efficiency of sgRNA pairs were evaluated in MLE-12-Cas9-GFP cells, a stable cell line generated from MLE-12 cells (cat. no. CRL-2110, ATCC, VA) and cultured in DMEM/F12 medium supplemented with 0.005 mg/mL insulin, 0.01 mg/mL transferrin, 30 nM sodium selenite, 10 nM hydrocortisone, 10 nM β-estradiol, 10 mM HEPES, 2 mM L-glutamine, and 2% FBS. Alt-R guide RNAs synthesized by Integrated DNA Technologies (Coralville, IA) or plasmids expressing a sgRNA pair were nucleofected into cells using a pulse code of DS-130 in SE nucleofector solution. To evaluate AAV-sgRNAs, cells were infected by AAVs at a multiplicity of infection of 1-3e6 vg. Protein and DNA were extracted from cells at 10 days after nucleofection or AAV transduction to evaluate the levels of protein expression by immunoblotting or genomic deletion by PCR.
Experimental animals and animal procedures
Rosa26-LSL-tdTomato (Ai9) mice were licensed from the Allen Institute for Brain Science, Seattle, WA. Rosa26-CAG-SpCas9-mKate2 mice were generated at Genentech by gene targeting in C57BL/6N ES cells (details, including sequence information, is available upon request). Rosa26-CAG-SpCas9-eGFP mice were licensed from the Broad Institute of MIT and Harvard, Cambridge, MA, and obtained from The Jackson Laboratory (strain no. 027891). All mice were maintained in accordance with American Association of Laboratory Animal Care guidelines. The experiments were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Genentech Institutional Animal Care and Use Committee. The i.t. intubation procedure was conducted at Genentech for Rosa26-CAG-SpCas9-mKate2 mice and at The Jackson Laboratory for Rosa26-LSL-tdTomato (Ai9) and Rosa26-CAG-SpCas9-eGFP mice. In brief, mice were anesthetized using ketamine, 75–80 mg/kg, and xylazine, 7.5–15 mg/kg, via intraperitoneal injection. The mouse was placed in dorsal recumbency on a board slanted at ∼60° and secured in position by placing the upper incisors over a rubber band affixed to the board. Fifty microliters of the liquid to be instilled was drawn up into a syringe. The tongue of the mouse was extended with a cotton tipped applicator. The mouse was intubated with the aid of an otoscope fitted with a mouse speculum (cat. no. 000A3747, HalloWell, MA). An i.v. catheter or similar device was used as an “endotracheal tube.” For mice less than 20 g body weight a 22-gauge i.v. catheter was used. For mice equal to or greater than 20 g a 20-gauge catheter was used. A dose of saline or AAV to be instilled was injected into the endotracheal tube. Animals were placed on a warm pad for recovery and then returned to the cage.
Antibodies
Antibodies used in immunoblotting and IHC include: USP30 antibody developed in-house at Genentech; anti-NOTCH2 antibody (rabbit mAb clone D76A8, cat. no. 5732S) and anti-GAPDH (cat. no. 2118) were purchased from Cell Signaling (Danvers, MA). Antibodies used in FACS analysis include: anti-mouse CD326 (EpCam)-BV421 (cat. no. 118225, BioLegend, CA); anti-mouse CD45-BV510 (cat. no. 103137, BioLegend); L/D sytox eFluor 780 (cat. no. 65-0865-14, Thermo Fisher Scientific, CA); and anti-mouse CD104 (integrin beta4) (cat. no. 553745, BD Pharmagen, CA). The anti-mouse CD104 antibody was labeled with Alexa 647 using the labeling kit (Thermo Fisher Scientific) and purified using p30 Gel columns (cat. no. 7326231, Bio-Rad, Hercules, CA). AECII lineage marker is composed of the following antibody cocktail: PE-CD11b (cat. no. 553311, BD Biosciences, CA); PE-CD31 (cat. no. 12-0311-82, Invitrogen Life Technologies, CA); PE-CD45 (cat. no. 553081, BD Biosciences); PE-Ter110 (cat. no. 553673, BD Biosciences); and PE-Cy7-CD24 (cat. no. 25-0242-82, Invitrogen Life Technologies). Control antibodies include IgG-Alexa647 (cat. no. 557690, BD Biosciences) and anti-mouse IgG-Alexa488 (cat. no. ab150073, Abcam, MA). Antibodies used in IF staining include: anti-NOTCH2 (cat. no. 5732S, Cell Signaling, MA); anti-GFP (cat. no. ab13970, Abcam); anti-tdTomato (cat. no. AB8181-200, OriGene, MD); anti-SPC (cat. no. AB3786, Millipore, MA); anti-CC10 (cat. no. 10490-1-AP, Proteintech, IL and cat. no. ab40873, Abcam); anti-acetylated alpha-tubulin (cat. no. 12152, Cell Signaling); anti-rabbit, -chicken, -mouse IgG-Alexa conjugates (Invitrogen Life Technologies); anti-CK8/18 (cat. no. M3652, Agilent Dako, CA); anti-rabbit HRP (cat. no. 760-4311, Ventana Medical Systems, Tucson, AZ); TSA-Cy5 (Discovery Cy5 Kit, cat. no. 760-238, Ventana Medical Systems); TSA-FAM (Discovery FAM Kit, cat. no. 760-243, Ventana Medical Systems); anti-FoxJ1 (cat. no. ab235445, Abcam); TSA-Rhodamine 6G (Discovery Rhodamine 6G Kit, cat. no. 760-244, Ventana Medical Systems).
IF staining and quantification
To quantify AAV transduction efficiency, we conducted IF staining of AAV reporters (GFP, mCherry) and cell markers. IF staining was performed on 4-μm-thick formalin-fixed, paraffin-embedded tissue sections mounted on glass slides. The slides were baked at 70°C for 30 min, with deparaffinization and antigen retrieval using a Cell Conditioning CC1 Standard at 95°C for 64 min (cat. no. 950-500, Ventana Medical Systems). Slides were then blocked with PBS containing 5% FBS and 1% BSA for 1 h and incubated with primary antibodies in the blocking buffer overnight at 4°C. After 3 washes with PBS, secondary antibodies with multiplexed Alexa conjugates (1:1,000) were applied to slides and incubated for 1 h. Slides were washed and incubated with DAPI (1:1,000) (cat. no. 1306, Invitrogen/Thermo Fisher Scientific, Waltham, MA) for 5 min. Washed again and mounted with coverslips using ProLong Gold antifade mounting medium (cat. no. P36930, Invitrogen/Thermo Fisher Scientific).
To quantify the transdifferentiation of club cells into ciliated cells after Notch2 genomic editing, a sequential triple IF staining of FoxJ1/CC10/Ck8_18 was performed on a Ventana Discovery Ultra platform (Ventana Medical Systems). First marker, rabbit anti-CK8/18 at 1:100 in 3% BSA/PBS for 32 min and detected with Discovery OmniMap anti-rabbit HRP for 16 min, and the signal was amplified with TSA-Cy5 for 12 min. Second marker, rabbit anti-CC10 was applied at 1:250 in 3% BSA/PBS for 32 min followed by Discovery OmniMap anti-rabbit HRP for 16 min and the signal was amplified with TSA-FAM for 12 min. Third marker, rabbit anti-FoxJ1 was applied at 1:250 in 3% BSA/PBS for 32 min and detected with Discovery OmniMap anti-rabbit HRP for 16 min and the signal was amplified with TSA-Rhodamine 6G for 12 min. To overcome the cross-reactivity, tissue sections went through a denaturing step before application of second and third markers. The denature step was achieved by incubation in Cell Conditioning CC2 (cat. no. 950-223, Ventana Medical Systems) at 100°C for 8 min. Sections were counterstained with DAPI at 0.2 μg/mL for 20 min at room temperature offline. Slides were then cover-slipped with Prolong Gold and dried overnight, in the dark, at room temperature.
For signal quantification, slides were scanned on a NanoZoomer S60 whole slide imager (Hamamatsu, Bridgewater, NJ) at 200× magnification. Individual cell nuclei were identified using an algorithm based on radial symmetry77 or Stardist78 using DAPI, and then scored independently for each fluorescent marker in MATLAB 2019a (MathWorks, Natick, MA) or Python 3.7 with scikit-image. Fluorescence intensity was measured after subtraction of autofluorescence in the CFP channel and a threshold for positive signal was applied to generate a binary mask for each marker. Nuclei were scored as positive or negative based on the percentage of positive area within the nucleus and a surrounding dilated ring. Total percentage of positive nuclei and percentage of nuclei positive for various phenotypic combinations of markers were reported. Large airways were sometimes excluded from analysis using standard morphological operations supplemented with manual curation.
Lung dissociation and flow cytometry analysis
Lungs were perfused with 20 mL PBS followed by the inflation with 2 mL sterile protease cocktail containing collagenase/dispase (cat. no. 11097113001, Roche, Switzerland), elastase (cat. no. LS002279, Worthington Biochemical, NJ), and DNaseI (cat. no. 10104159001, Roche) in DMEM/F12 medium. Lungs were disrupted with scissors and incubated with the protease cocktail in a warm room for 45 min on a shaking platform. The protease digestion was halted by adding 8 mL DMEM/F12 medium containing 10% FBS. The dissociated lung was pelleted by centrifugation. Cell pellets were washed with PBS, resuspended in 2 mL ACK red cell lysis buffer (NH4Cl, KHCO3, EDTA disodium, pH 7.4) and incubated at room temperature for 2 min. Five milliliters of DMEM/F12 containing 10% FBS was added to the cell suspension, which was then filtered through a 40-μm cell strainer to obtain a single-cell suspension. After spin down to remove the lysis buffer, cells were resuspended in a cell-staining buffer (BioLegend, cat. no. 420201) containing antibody cocktails. Cell number was counted and cell suspension was dilute as 1–2 × 107 cells/mL. Cells were then incubated with the antibody cocktail on ice for 20 min to 1 h, washed twice with FACS staining buffer and subjected to FACS analysis and sorting.
Single-cell sequencing and data analysis
Dissociated lung cells were stained and sorted for CD45−/GFP+ and CD45−/GFP− populations. Single-cell gene expression libraries were generated from the sorted cell suspensions using the Chromium Next GEM Single Cell 3ʹ Reagent Kits v.3.1 (10X Genomics, Pleasanton, CA), loading an estimated 10,000 cells per sample and following the manufacturer’s instructions. Libraries were quantified with the KAPA Library Quantification Kit (Roche) and profiled using the Bioanalyzer High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA). Each library was sequenced in one lane of a HiSeq 4000 (Illumina, Foster City, CA) to generate 300 million paired-end reads at a configuration of 28 base pairs (read 1) and 98 base pairs (read 2). Sequencing reads were processed through 10X Genomics Cell Ranger (v.3.1.0) and aligned to GRCm38 with added sequences to measure EGFP reporter gene. We used the R package Seurat for downstream analysis. Cells with fewer than 200 genes expressed or more than 8,000 genes expressed were filtered out. Cells with 15% or more mitochondrial content were also removed. The Seurat function IntegrateData was used for data integration of the two populations. Clustering analysis was performed using FindClusters with a resolution set to 0.5.
Next-generation sequencing analysis of CRISPR indel formation
Amplicons in the 200–300-bp range centered around the CRISPR targeted sites were generated with PCR primers flanking the sgRNA binding site. Each PCR reaction was purified with a DNA Clean & Concentrator-5 kit (Zymo Research, Irvine, CA) and eluted in 25 μL H2O. DNA concentrations were quantified using the Qubit dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA). Forty nanograms of amplicons were used to generate sequencing libraries with the KAPA HyperPrep Kit (Roche), which incorporated custom adapters and library amplification PCR primers from Integrated DNA Technologies. Amplicon libraries were quantified and the average library size was determined using the NGS Fragment kit on Fragment analyzer (Agilent Technologies). Libraries were pooled and sequenced on Miseq (Illumina) to generate 200,000 single-end 200-base pair reads for each sample. Analysis of the sequencing data was performed as described in Anderson et al.79
Digital droplet PCR
Levels of AAV vector genome copy number in sorted AECII and distal airway epithelial cells were analyzed using digital droplet PCR (ddPCR). DNA samples were extracted from animals in control and treatment groups using QIAGEN DNeasy and blood kit (QIAGEN, Redwood City, CA). The primers and probes used in the ddPCR assays include: the ITR-F: 5′- GGAACCCCTAGTGATGGA GTT; ITR-R: 5′-CGGCCTCAGTGAGCGA; ITR-probe: 5′ FAM, internal ZEN, and 3′ BFQ: 5′- CACTCCCTCTCTGCGCGCTCG; the house control gene mRLP19-F: 5′-ATGTATCACAGCCTGTACCTG; mRLP19-R: 5′-TTCTTGGTCTCTTCCTCCTTG; mRLP19-Probe: 5′ Hex and 3′ BFQ. 5′- AGGTCTAAGACC AAGGAAGCACGCAA. The reaction mixture was composed of 12.5 μL 2× ddPCR Supermix for probes (no dUTP) (Bio-Rad), 1 μL of primers and probe mix (final concentration of primers and probe was 900 and 250 nM, respectively), 0.5 μL sample (15 ng of DNA), and 11 μL H2O (total volume 25 μL). Each sample was tested in duplicate. Droplets were generated using a QX200 Automated Droplet Generator (Bio-Rad) and amplified in a C1000 Touch Thermal Cycler (Bio-Rad). Cycling conditions were 10 min at 95°C followed by 40 cycles of a two-step cycling protocol (95°C for 30 s and 60°C for 1 min). The ramp rate between these steps was set at 2°C/s. After cycling, the plate was transferred to a QX200 Droplet Reader (Bio-Rad). Data analysis was performed using QuantaSoft software (v.1.7.4) and quantitation of target molecules presented as copies/μL of PCR reaction. Data were normalized using the housekeeping gene mRLP19. Control group receiving PBS showed zero AAV vector copy number per cell (data not shown).
Capillary electrophoresis immunoblotting
A high-sensitivity capillary protein electrophoresis system (Peggy Sue platform) was used for protein analysis according to the manufacturer’s protocol (ProteinSimple, Santa Clara, CA). In brief, protein was extracted using the RIPA buffer from FACS-sorted AECII. Four microliters of protein lysate (0.8 μg) were mixed with 1 μL of fluorescent 5× master mix. The samples were then heat denatured at 95°C for 5 min. The samples, biotinylated ladder, primary antibody, HRP-conjugated secondary antibody, blocking reagent, chemiluminescent substrate, and separation and stacking matrixes were dispensed into a 384-well plate. After plate loading, the default electrophoresis was carried out in Peggy Sue with 12 s of sample loading time, 45 min of sample separation time, 2 h of primary antibody incubation time. Data were analyzed and digital images were visualized using Compass software (ProteinSimple v.3.1). Levels of the USP30 were quantified using ImageJ by normalizing to levels of GAPDH.
NOTCH2 IHC and quantification
IHC staining was performed on 4-μm-thick formalin-fixed, paraffin-embedded tissue sections mounted on glass slides using the Ventana Discovery XT automated platform (Ventana Medical Systems). The slides were baked at 70°C for 30 min, with deparaffinization and antigen retrieval using Cell Conditioning CC1 Standard at 95°C for 64 min (cat. no. 950-500, Ventana Medical Systems). Slides were then incubated with rabbit mAb clone D76A8 (Cell Signaling) at 8 μg/mL for 32 min and detected using Discovery OmniMap anti-rabbit HRP for 16 min. Slides were counterstained with Ventana Hematoxylin II (cat. no. 790-2208, Ventana Medical Systems). The quantification method was the same as described in the IF quantification method.
Histology analysis of inflammatory response
H&E-stained slides were examined under a light microscope by a pathologist blinded to the treatment groups. The degree of inflammation was assessed according to a semi-quantitative scale as follows: 0, no inflammatory infiltrate; 1, scattered single neutrophils or macrophages on airway epithelial surfaces, in alveoli, and/or in perivascular space; 2, small loose aggregates of extravasated neutrophils and/or macrophages in one or two airways and/or alveoli; 3, loose to compact aggregates of extravasated neutrophils and/or macrophages in multiple airways and/or alveoli with some effacement of lung architecture; 4, coalescing aggregates of neutrophils and/or macrophages in multiple airways and/or alveoli effacing most adjacent lung architecture; 5, severe inflammation.
Samplix Xdrop workflow, ONT sequencing, and analysis
High-molecular weight DNA was extracted from FACS-sorted distal airway epithelial and AECII cells from seven Ai9/Rosa26-LSL-tdTomato animals from each group (control versus treatment) using QIAGEN DNeasy and blood kit. DNA was pooled in each group to obtain enough material for Xdrop workflow and its quality was evaluated using the TapeStation 2200 System (Agilent Technologies), using Genomic DNA ScreenTape according to the manufacturer’s instructions (average sizes: control group 20 kb; treatment group: 24 kb). Xdrop enrichment, amplification, and ONT sequencing were performed by Samplix Services (Birkerød, Denmark).56 The “Detection Sequence” droplet PCR (dPCR) primers and accompanying evaluation qPCR primers were designed to bind to the tdTomato sequence close to the CRISPR-Cas9 on-target sites (Figure S8B; Table S2). This design eliminated the sequencing contamination from the episomal AAV vector genome. DNA (10 ng) was partitioned in droplets by Xdrop and subjected to dPCR using the Detection Sequence assay. The dPCR productions were then stained and sorted by FACS to select for DNA molecules containing the tdTomato sequence. The selected DNA molecules were then amplified by droplet MDA (dMDA) as described previously. After amplification, DNA was isolated and quantified, and enrichment was validated by evaluation qPCR assays (data not shown) before Oxford Nanopore Sequencing.
The GridION Oxford Nanopore Sequencing platform (Oxford Nanopore, Oxford, UK) was used to generate long-read sequencing data from the dMDA samples as described in the manufacturer’s instructions (premium whole-genome amplification protocol [SQK-LSK109] with the Native Barcoding Expansion 1–12 [EXP-NBD104]). In short, 1.5 μg of amplified DNA of each sample was treated with T7 endonuclease I, followed by size selection, end-repair, barcoding, and adaptor ligation. After library generation the samples were loaded onto a flow cell 9.4.1 (10 fmol) and run for 16–24 h on the GridION instrument under standard conditions as recommended by the manufacturer. Generated raw data (FAST5) were subjected to base calling with super high accuracy base calling and quality filtering 10, using Guppy 5.0.12. Quality filtering of 10 was applied to the data delivered in this project. Approximately 1 Gb sequencing data was obtained from each sample.
The ONT read analysis was carried out using custom in-house software written in Python and utilizing Pandas. In summary, ONT reads were filtered by matching to a 30-bp tdTomato sequence adjacent to where Samplix detection and evaluation primers were bound. These filtered reads were aligned against AAV vector sequences as the reference, using Minimap280 using -ax map-ont parameters. Coverage profile was generated and reads mapping specific to the AAV vector were extracted using SAMTools,81 Bedtools,82 and SeqKit.83 These reads containing the AAV vector were aligned to a theoretical reference constructed of the AAV vector inserted at the CRISPR-Cas9 site targeted by the right-sgRNA in the LSL-sgRNA pair 1. Observed AAV-tdTomato genomic fusion species were visualized using IGV2.3.96 and reconstructed manually using the primary and supplementary reads, their reference alignment position and read sequence alignment position. Reads in length greater than 5 kb contained the concatemers of inverted repeats of the same sequence, which were artifacts caused by the MDA step in the Samplix X drop workflow. The contigs were manually assembled from these palindromic compartments based on the sequence orientation, position, and reference alignment position.
Statistical analysis
All analysis was carried out using Prism 6 (GraphPad Software, San Diego, CA). Dot plots show mean value and error bars indicate standard error of the mean (SEM). All data points are shown. Two-tailed unpaired t test was used to test for significance of difference between the means of different groups.
Illustrations
Illustrations of vector design, animal study design, and AAV integration events were created with BioRender.com (Toronto, ON, Canada).
Data availability
Data is available upon request.
Acknowledgments
We thank several Genentech core teams, including the Micro-injection lab, Colony Resources and Genetic Analysis lab for assistance with generating the Rosa26-CAG-SpCas9-mKate2 mice; Animal care, Necropsy and Histology lab for maintaining animals, dissecting and processing animal tissues; FACS core team in helping with the sorting of the dissociated lung cells; Microscopy core team in imaging of IF and IHC stained slides; NGS core team in DNA sequencing and scRNA-seq support. We also thank Genentech colleagues in Dr. Mark Chen’s and Dr. Hans Brightbill’s labs for helping with intranasal and intratracheal procedures, Dr. Jonas Doerr for helping with lung clearing and 3D imaging of lung lobes, Mr. Ahmet Kurdoglu for helping with scRNA-seq data analysis, and Drs. Ning Ding and Daniel Lafkas for discussions regarding USP30 and NOTCH2, respectively. We also thank The Jackson Laboratory for assisting with animal handling.
Author contributions
H.C., Y.L., and B.H. conceptualized the study. H.C. conducted experiments and data collection. H.C., St.D., H.P., O.F., K.M., J.E., A.L., and Sp.D. contributed to data analysis, interpretation, and visualization. R.C., R.J.N., M.R.-G., and S.W. established the Rosa26-CAG-SpCas9-mKate2 transgenic mouse model. H.C. wrote the original manuscript draft. All authors read and approved the manuscript. H.C., Y.L., and B.H. supervised the study.
Declaration of interests
During the course of these studies, all authors were full-time employees of Genentech, Inc., and potential shareholders of Roche. No other disclosures were reported.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.10.016.
Contributor Information
Yuxin Liang, Email: yliang@gene.com.
Benjamin Haley, Email: benjamih@gene.com.
Supplemental information
References
- 1.Gao G.-P., Alvira M.R., Wang L., Calcedo R., Johnston J., Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao G., Vandenberghe L.H., Alvira M.R., Lu Y., Calcedo R., Zhou X., Wilson J.M. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/jvi.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inagaki K., Fuess S., Storm T.A., Gibson G.A., Mctiernan C.F., Kay M.A., Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacak C.A., Mah C.S., Thattaliyath B.D., Conlon T.J., Lewis M.A., Cloutier D.E., Zolotukhin I., Tarantal A.F., Byrne B.J. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 2006;99:e3–e9. doi: 10.1161/01.res.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- 5.Kurosaki F., Uchibori R., Mato N., Sehara Y., Saga Y., Urabe M., Mizukami H., Sugiyama Y., Kume A. Optimization of adeno-associated virus vector-mediated gene transfer to the respiratory tract. Gene Ther. 2017;24:290–297. doi: 10.1038/gt.2017.19. [DOI] [PubMed] [Google Scholar]
- 6.Cearley C.N., Wolfe J.H. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol. Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 7.Burger C., Gorbatyuk O.S., Velardo M.J., Peden C.S., Williams P., Zolotukhin S., Reier P.J., Mandel R.J., Muzyczka N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Vercauteren K., Hoffman B.E., Zolotukhin I., Keeler G.D., Xiao J.W., Basner-Tschakarjan E., High K.A., Ertl H.C., Rice C.M., Srivastava A., et al. Superior in vivo transduction of human hepatocytes using engineered AAV3 capsid. Mol. Ther. 2016;24:1042–1049. doi: 10.1038/mt.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Limberis M.P., Vandenberghe L.H., Zhang L., Pickles R.J., Wilson J.M. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol. Ther. 2009;17:294–301. doi: 10.1038/mt.2008.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limberis M.P., Wilson J.M. Adeno-associated virus serotype 9 vectors transduce murine alveolar and nasal epithelia and can be readministered. Proc. Natl. Acad. Sci. USA. 2006;103:12993–12998. doi: 10.1073/pnas.0601433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Payne J.G., Takahashi A., Higgins M.I., Porter E.L., Suki B., Balazs A., Wilson A.A. Multilineage transduction of resident lung cells in vivo by AAV2/8 for α1-antitrypsin gene therapy. Mol. Ther. Methods Clin. Dev. 2016;3 doi: 10.1038/mtm.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai K.X., Tse L.Y., Leung C., Tam P.K.H., Xu R., Sham M.H. Suppression of lung tumor growth and metastasis in mice by adeno-associated virus-mediated expression of vasostatin. Clin. Cancer Res. 2008;14:939–949. doi: 10.1158/1078-0432.ccr-07-1930. [DOI] [PubMed] [Google Scholar]
- 13.Lima A., Maddalo D. SEMMs: somatically engineered mouse models. A new tool for in vivo disease modeling for basic and translational research. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.667189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maddalo D., Manchado E., Concepcion C.P., Bonetti C., Vidigal J.A., Han Y.-C., Ogrodowski P., Crippa A., Rekhtman N., de Stanchina E., et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blasco R.B., Karaca E., Ambrogio C., Cheong T.-C., Karayol E., Minero V.G., Voena C., Chiarle R. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014;9:1219–1227. doi: 10.1016/j.celrep.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 16.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M., et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sánchez-Rivera F.J., Papagiannakopoulos T., Romero R., Tammela T., Bauer M.R., Bhutkar A., Joshi N.S., Subbaraj L., Bronson R.T., Xue W., Jacks T. Rapid modeling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428–431. doi: 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciampricotti M., Karakousi T., Richards A.L., Quintanal-Villalonga A., Karatza A., Caeser R., Costa E.A., Allaj V., Manoj P., Spainhower K.B., et al. Rlf-Mycl gene fusion drives tumorigenesis and metastasis in a mouse model of small cell lung cancer. Cancer Discov. 2021;11:3214–3229. doi: 10.1158/2159-8290.cd-21-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann O., Reissland M., Maier C.R., Fischer T., Prieto-Garcia C., Baluapuri A., Schwarz J., Schmitz W., Garrido-Rodriguez M., Pahor N., et al. Implementation of CRISPR/Cas9 genome editing to generate murine lung cancer models that depict the mutational landscape of human disease. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.641618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cook P.J., Thomas R., Kannan R., de Leon E.S., Drilon A., Rosenblum M.K., Scaltriti M., Benezra R., Ventura A. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat. Commun. 2017;8 doi: 10.1038/ncomms15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ideno N., Yamaguchi H., Okumura T., Huang J., Brun M.J., Ho M.L., Suh J., Gupta S., Maitra A., Ghosh B. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab. Invest. 2019;99:1233–1244. doi: 10.1038/s41374-018-0171-z. [DOI] [PubMed] [Google Scholar]
- 22.Annunziato S., Lutz C., Henneman L., Bhin J., Wong K., Siteur B., van Gerwen B., de Korte-Grimmerink R., Zafra M.P., Schatoff E.M., et al. In situ CRISPR-Cas9 base editing for the development of genetically engineered mouse models of breast cancer. EMBO J. 2020;39 doi: 10.15252/embj.2019102169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roper J., Tammela T., Akkad A., Almeqdadi M., Santos S.B., Jacks T., Yilmaz Ö.H. Colonoscopy-based colorectal cancer modeling in mice with CRISPR–Cas9 genome editing and organoid transplantation. Nat. Protoc. 2018;13:217–234. doi: 10.1038/nprot.2017.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Angeli P., Reuter P., Hauser S., Schöls L., Stingl K., Wissinger B., Kohl S. Effective splicing restoration of a deep-intronic ABCA4 variant in cone photoreceptor precursor cells by CRISPR/SpCas9 approaches. Mol. Ther. Nucleic Acids. 2022;29:511–524. doi: 10.1016/j.omtn.2022.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Li H., Min Y.-L., Sanchez-Ortiz E., Huang J., Mireault A.A., Shelton J.M., Kim J., Mammen P.P.A., Bassel-Duby R., Olson E.N. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aay6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A., et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hana S., Peterson M., McLaughlin H., Marshall E., Fabian A.J., McKissick O., Koszka K., Marsh G., Craft M., Xu S., et al. Highly efficient neuronal gene knockout in vivo by CRISPR-Cas9 via neonatal intracerebroventricular injection of AAV in mice. Gene Ther. 2021;28:646–658. doi: 10.1038/s41434-021-00224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ulrich M., Worlitzsch D., Viglio S., Siegmann N., Iadarola P., Shute J.K., Geiser M., Pier G.B., Friedel G., Barr M.L., et al. Alveolar inflammation in cystic fibrosis. J. Cyst. Fibros. 2010;9:217–227. doi: 10.1016/j.jcf.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parimon T., Yao C., Stripp B.R., Noble P.W., Chen P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020;21:2269. doi: 10.3390/ijms21072269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuo W.-L., Rostami M.R., LeBlanc M., Kaner R.J., O’Beirne S.L., Mezey J.G., Leopold P.L., Quast K., Visvanathan S., Fine J.S., et al. Dysregulation of club cell biology in idiopathic pulmonary fibrosis. PLoS One. 2020;15 doi: 10.1371/journal.pone.0237529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokoyama T., Yanagihara T., Suzuki K., Hamada N., Tsubouchi K., Ogata-Suetsugu S., Mikumo H., Ikeda-Harada C., Maeyama T., Kuwano K., Nakanishi Y. Depletion of club cells attenuates bleomycin-induced lung injury and fibrosis in mice. J. Inflamm. 2017;14:20. doi: 10.1186/s12950-017-0168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E., et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breton C., Clark P.M., Wang L., Greig J.A., Wilson J.M. ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genom. 2020;21:239. doi: 10.1186/s12864-020-6655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halbert C.L., Lam S.-L., Miller A.D. High-efficiency promoter-dependent transduction by adeno-associated virus type 6 vectors in mouse lung. Hum. Gene Ther. 2007;18:344–354. doi: 10.1089/hum.2006.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang M.-S., Park M.-J., Lee J., Oh B., Kang K.W., Kim Y., Lee S.-M., Lim J.-O., Jung T.-Y., Park J.-H., et al. Non-invasive administration of AAV to target lung parenchymal cells and develop SARS-CoV-2-susceptible mice. Mol. Ther. 2022;30:1994–2004. doi: 10.1016/j.ymthe.2022.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 39.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z., Newton K., Kummerfeld S.K., Webster J., Kirkpatrick D.S., Phu L., Eastham-Anderson J., Liu J., Lee W.P., Wu J., et al. Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc. Natl. Acad. Sci. USA. 2017;114:3903–3908. doi: 10.1073/pnas.1621177114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chapman H.A., Li X., Alexander J.P., Brumwell A., Lorizio W., Tan K., Sonnenberg A., Wei Y., Vu T.H. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Invest. 2011;121:2855–2862. doi: 10.1172/jci57673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaughan A.E., Brumwell A.N., Xi Y., Gotts J.E., Brownfield D.G., Treutlein B., Tan K., Tan V., Liu F.C., Looney M.R., et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chew W.L., Tabebordbar M., Cheng J.K.W., Mali P., Wu E.Y., Ng A.H.M., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Truong D.-J.J., Kühner K., Kühn R., Werfel S., Engelhardt S., Wurst W., Ortiz O. Development of an intein-mediated split–Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi J.-H., Yu N.-K., Baek G.-C., Bakes J., Seo D., Nam H.J., Baek S.H., Lim C.-S., Lee Y.-S., Kaang B.-K. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Mol. Brain. 2014;7:17. doi: 10.1186/1756-6606-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang S.-Q., Walkey C.J., Martinez A.E., Su Q., Dickinson M.E., Wang D., Lagor W.R., Heaney J.D., Gao G., Xue W. AAV5 delivery of CRISPR-Cas9 supports effective genome editing in mouse lung airway. Mol. Ther. 2022;30:238–243. doi: 10.1016/j.ymthe.2021.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zetsche B., Volz S.E., Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright A.V., Sternberg S.H., Taylor D.W., Staahl B.T., Bardales J.A., Kornfeld J.E., Doudna J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA. 2015;112:2984–2989. doi: 10.1073/pnas.1501698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bingol B., Tea J.S., Phu L., Reichelt M., Bakalarski C.E., Song Q., Foreman O., Kirkpatrick D.S., Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 50.McWilliams T.G., Prescott A.R., Allen G.F.G., Tamjar J., Munson M.J., Thomson C., Muqit M.M.K., Ganley I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016;214:333–345. doi: 10.1083/jcb.201603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morimoto M., Nishinakamura R., Saga Y., Kopan R. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development. 2012;139:4365–4373. doi: 10.1242/dev.083840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lafkas D., Shelton A., Chiu C., de Leon Boenig G., Chen Y., Stawicki S.S., Siltanen C., Reichelt M., Zhou M., Wu X., et al. Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature. 2015;528:127–131. doi: 10.1038/nature15715. [DOI] [PubMed] [Google Scholar]
- 53.Halbert C.L., Rutledge E.A., Allen J.M., Russell D.W., Miller A.D. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J. Virol. 2000;74:1524–1532. doi: 10.1128/jvi.74.3.1524-1532.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabatino D.E., Bushman F.D., Chandler R.J., Crystal R.G., Davidson B.L., Dolmetsch R., Eggan K.C., Gao G., Gil-Farina I., Kay M.A., et al. Evaluating the state of the science for adeno-associated virus integration: an integrated perspective. Mol. Ther. 2022;30:2646–2663. doi: 10.1016/j.ymthe.2022.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.-A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madsen E.B., Höijer I., Kvist T., Ameur A., Mikkelsen M.J. Xdrop: targeted sequencing of long DNA molecules from low input samples using droplet sorting. Hum. Mutat. 2020;41:1671–1679. doi: 10.1002/humu.24063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dalwadi D.A., Calabria A., Tiyaboonchai A., Posey J., Naugler W.E., Montini E., Grompe M. AAV integration in human hepatocytes. Mol. Ther. 2021;29:2898–2909. doi: 10.1016/j.ymthe.2021.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manicone A.M. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 2014;5:63–75. doi: 10.1586/1744666x.5.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holtzman M.J., Byers D.E., Alexander-Brett J., Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat. Rev. Immunol. 2014;14:686–698. doi: 10.1038/nri3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim E., Koo T., Park S.W., Kim D., Kim K., Cho H.-Y., Song D.W., Lee K.J., Jung M.H., Kim S., et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017;8 doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt M.J., Gupta A., Bednarski C., Gehrig-Giannini S., Richter F., Pitzler C., Gamalinda M., Galonska C., Takeuchi R., Wang K., et al. Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases. Nat. Commun. 2021;12:4219. doi: 10.1038/s41467-021-24454-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang M.H., van Lieshout L.P., Xu L., Domm J.M., Vadivel A., Renesme L., Mühlfeld C., Hurskainen M., Mižíková I., Pei Y., et al. A lung tropic AAV vector improves survival in a mouse model of surfactant B deficiency. Nat. Commun. 2020;11:3929. doi: 10.1038/s41467-020-17577-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Lieshout L.P., Domm J.M., Rindler T.N., Frost K.L., Sorensen D.L., Medina S.J., Booth S.A., Bridges J.P., Wootton S.K. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Mol. Ther. Methods Clin. Dev. 2018;9:323–329. doi: 10.1016/j.omtm.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halbert C.L., Standaert T.A., Wilson C.B., Miller A.D. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 1998;72:9795–9805. doi: 10.1128/jvi.72.12.9795-9805.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sheel A., Xue W. Genomic amplifications cause false positives in CRISPR screens. Cancer Discov. 2016;6:824–826. doi: 10.1158/2159-8290.cd-16-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dow L.E., Fisher J., O’Rourke K.P., Muley A., Kastenhuber E.R., Livshits G., Tschaharganeh D.F., Socci N.D., Lowe S.W. Inducible in vivo genome editing with CRISPR/Cas9. Nat. Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davis K.M., Pattanayak V., Thompson D.B., Zuris J.A., Liu D.R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown W., Zhou W., Deiters A. Regulating CRISPR/Cas9 function through conditional guide RNA control. Chembiochem. 2021;22:63–72. doi: 10.1002/cbic.202000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levy J.M., Yeh W.-H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13:eabf2444. doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruan G.-X., Barry E., Yu D., Lukason M., Cheng S.H., Scaria A. CRISPR/Cas9-Mediated genome editing as a therapeutic approach for leber congenital Amaurosis 10. Mol. Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCullough K.T., Boye S.L., Fajardo D., Calabro K., Peterson J.J., Strang C.E., Chakraborty D., Gloskowski S., Haskett S., Samuelsson S., et al. Somatic gene editing of GUCY2D by AAV-CRISPR/Cas9 alters retinal structure and function in mouse and macaque. Hum. Gene Ther. 2019;30:571–589. doi: 10.1089/hum.2018.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X., et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lattanzi A., Duguez S., Moiani A., Izmiryan A., Barbon E., Martin S., Mamchaoui K., Mouly V., Bernardi F., Mavilio F., Bovolenta M. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol. Ther. Nucleic Acids. 2017;7:11–19. doi: 10.1016/j.omtn.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Warris S., Schijlen E., van de Geest H., Vegesna R., Hesselink T., Te Lintel Hekkert B., Sanchez Perez G., Medvedev P., Makova K.D., de Ridder D. Correcting palindromes in long reads after whole-genome amplification. BMC Genom. 2018;19:798. doi: 10.1186/s12864-018-5164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Veta M., van Diest P.J., Kornegoor R., Huisman A., Viergever M.A., Pluim J.P.W. Automatic nuclei segmentation in H&E stained breast cancer histopathology images. PLoS One. 2013;8 doi: 10.1371/journal.pone.0070221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmidt U., Weigert M., Broaddus C., Myers G. 2018. Medical image computing and computer assisted intervention – MICCAI 2018, 21st international conference, Granada, Spain, september 16-20, 2018, proceedings, Part II; pp. 265–273. (Lecture Notes in Computer Science). [DOI] [Google Scholar]
- 79.Anderson K.R., Haeussler M., Watanabe C., Janakiraman V., Lund J., Modrusan Z., Stinson J., Bei Q., Buechler A., Yu C., et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods. 2018;15:512–514. doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shen W., Le S., Li Y., Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11 doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is available upon request.