

Abstract
Sperm chromatin is typically transformed by protamines into a compact and transcriptionally inactive state1,2. Sperm cells of flowering plants lack protamines, yet they have small, transcriptionally active nuclei with chromatin condensed through an unknown mechanism3,4. Here we show that a histone variant, H2B.8, mediates sperm chromatin and nuclear condensation in Arabidopsis thaliana. Loss of H2B.8 causes enlarged sperm nuclei with dispersed chromatin, whereas ectopic expression in somatic cells produces smaller nuclei with aggregated chromatin. This result demonstrates that H2B.8 is sufficient for chromatin condensation. H2B.8 aggregates transcriptionally inactive AT-rich chromatin into phase-separated condensates, which facilitates nuclear compaction without reducing transcription. Reciprocal crosses show that mutation of h2b.8 reduces male transmission, which suggests that H2B.8-mediated sperm compaction is important for fertility. Altogether, our results reveal a new mechanism of nuclear compaction through global aggregation of unexpressed chromatin. We propose that H2B.8 is an evolutionary innovation of flowering plants that achieves nuclear condensation compatible with active transcription.
Subject terms: Plant reproduction, Nuclear organization, Spermatogenesis, Epigenetics, Histone variants
H2B.8 is identified as a histone variant that mediates a newly described mechanism of transcription-compatible chromatin condensation in flowering plant sperm cells.
Main
Sperm chromatin undergoes extensive condensation that is essential for male fertility in most animals. During animal sperm maturation, nearly all histones are replaced by small, arginine-rich protamines that facilitate dense packaging of the DNA1,2. In mammalian sperm for example, histones are retained at only 5–15% of the genome, whereas the majority of regulatory information carried by histones is lost1,5. Unwinding DNA from histones requires DNA strand breaks, which are observed in great quantities during this process6,7. Furthermore, protamines preclude transcription8. The extreme chromatin compaction enabled by protamines protects genome integrity against genotoxic factors and achieves a small and hydrodynamic sperm head that enhances swimming ability2,8. The adoption of such a process illustrates the high evolutionary pressure on sperm fitness9.
Sperm condensation also occurs in another large group of multicellular eukaryotes: plants. Similar to animals, green algae and non-seed plants (such as liverworts, mosses and ferns) produce motile sperm, which swim through water to reach the egg cell10. Consistent with the theory that protamine-mediated sperm condensation evolved to facilitate swimming2, sperm nuclei in these species are highly condensed by protamines and protamine-like proteins and transcribe very little, if any, RNA11–14.
Diverged from other land plant species approximately 150 million years ago, flowering plants no longer rely on water for fertilization10. Flowering plants produce immotile, transcriptionally active sperm3,4. The sperm cells are encapsulated in pollen grains, which are produced by mitotic divisions of the haploid meiotic product called the microspore10. The microspore divides once to produce a vegetative cell and a generative cell, the latter of which subsequently divides to generate two sperm cells10. During fertilization, the vegetative cell develops into a pollen tube that delivers the sperm to the egg apparatus15. Consistent with the high metabolic activity required for powering pollen tube growth, chromatin in vegetative cells is highly decondensed and transcriptionally active16–18. By contrast, sperm has highly condensed, histone-based chromatin and small nuclei19,20. In the absence of protamines, the mechanism of sperm chromatin condensation in flowering plants is unknown.
To understand the mechanism that underlies sperm condensation in flowering plants, we performed super-resolution imaging and comparative proteomics analyses of sperm cells, vegetative cells and somatic cells from A. thaliana. Through these experiments, we identified a specifically expressed histone variant, H2B.8, that colocalizes with chromatin aggregates in the sperm nucleoplasm. Sperm nuclei with h2b.8 mutations are enlarged and have decondensed chromatin, whereas ectopic expression of H2B.8 in somatic cells causes the opposite phenotype. This result demonstrates that H2B.8 is necessary and sufficient for nuclear and chromatin condensation. H2B.8 aggregates chromatin through a phase-separation mechanism that depends on a conserved intrinsically disordered region (IDR). H2B.8 specifically concentrates unexpressed AT-rich euchromatin, which reduces the nuclear volume while maintaining transcription. H2B.8-induced nuclear compaction is important for fertility, as h2b.8 mutations reduce male transmission. Collectively, our results explain flowering plant sperm condensation and reveal a new mechanism of transcription-compatible chromatin condensation.
Aggregation of sperm chromatin
Previous studies of Arabidopsis using DAPI staining showed that sperm chromatin is highly condensed and had small nuclear size compared to somatic and vegetative cells19. To characterize sperm chromatin in more detail, we examined Arabidopsis sperm nuclei using super-resolution 3D structured illumination microscopy (3D-SIM). Distinct chromatin aggregates were observed throughout the nucleoplasm in sperm cells (Fig. 1a). By contrast, chromatin in vegetative and leaf cells was more homogenous, although leaf cells had condensed heterochromatin foci at the nuclear periphery (Fig. 1a).
Fig. 1. Sperm chromatin is aggregated and contains a specific histone variant: H2B.8.
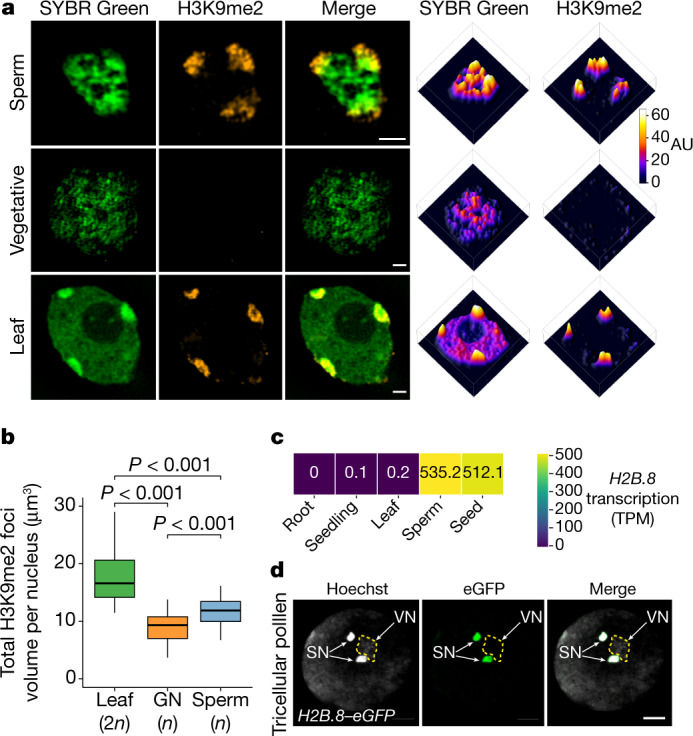
a, Super-resolution 3D-SIM images (left) and associated intensity profiles (right) of wild-type (WT) sperm and vegetative nuclei from pollen and a diploid leaf nucleus. DNA was stained with SYBR Green (green) and H3K9me2 was immunolocalized (orange). Data shown represent three independent experiments. AU, arbitrary units. b, Total volume of H3K9me2-enriched heterochromatin foci in a diploid leaf nucleus, generative nucleus (GN) and sperm nucleus. P values calculated using one-sided analysis of variance (ANOVA) followed by individual two-sample Tukey tests; n = 30 nuclei each examined over two independent experiments. All the boxplots in this work show median (thick black bar) and first and third quartiles, with lower and upper whiskers extending to 1.5-times the interquartile range of the first and third quartiles or the highest and lowest values, respectively. c, H2B.8 transcription levels in indicated tissues and cells. TPM, transcripts per million. d, Confocal images of pH2B.8::H2B.8–eGFP pollen, in which the eGFP signal is specific to the sperm nuclei (SN). VN, vegetative nucleus (outlined in a dashed line). Data shown represent three independent experiments. Scale bars, 1 μm (a) 5 μm (d).
To understand the composition of chromatin within sperm aggregates, we performed immunostaining for histone H3 lysine 9 dimethylation (H3K9me2), a modification associated with silenced heterochromatin21. Larger aggregates situated at the sperm nuclear periphery colocalized with H3K9me2 signals (Fig. 1a). This result shows that heterochromatin domains persist in the sperm, as previously reported16. However, these heterochromatin foci appeared more enlarged in sperm cells than in leaf cells (Fig. 1a). To further examine this aspect, we quantified the total volumes of H3K9me2-enriched heterochromatin foci in sperm, in diploid leaf nuclei and in the nuclei of generative cells (which are haploid mother cells that divide into sperm cells). As expected, the volume of heterochromatin foci was reduced by half in haploid generative nuclei compared to diploid leaf nuclei (Fig. 1b). Conversely, sperm heterochromatin foci were 27.5% larger than those of generative cell nuclei (Fig. 1b), which suggested that there was a reduced level of heterochromatin condensation or an increased proportion of the genome incorporated into heterochromatin. The latter scenario is less likely, as less H3K9me2 (compared with total H3) was detected in sperm cells than in leaf cells (Extended Data Fig. 1a). Apart from heterochromatin foci, other chromatin aggregates in sperm were depleted of H3K9me2 (Fig. 1a). This observation indicates that a new mechanism is used to compact the less heterochromatic part of sperm chromatin.
Extended Data Fig. 1. H2B.8 is specifically expressed in sperm and mature seeds.
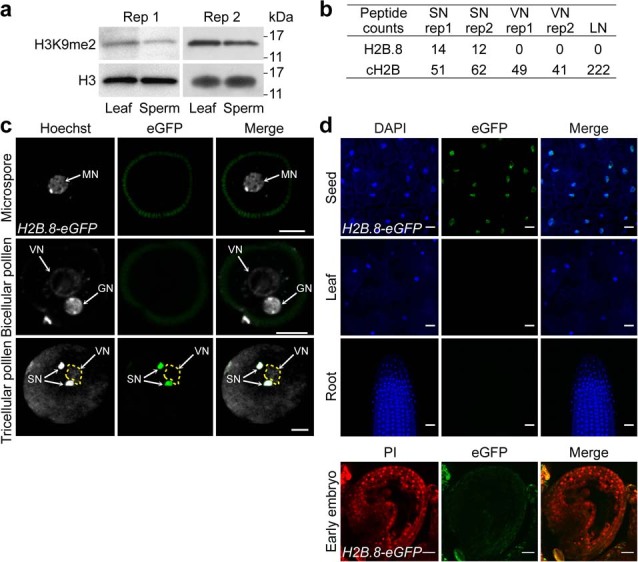
a, Western blot showing H3K9me2 levels in leaf and sperm nuclei, with histone H3 as a control. Data shown represent two independent experiments. Rep, biological replicate. For source data, see Supplementary Fig. 1. b, Peptide counts from sperm (SN), vegetative (VN) and leaf nuclei (LN) mass spectrometry. cH2B, canonical H2B. c, Confocal images of H2B.8 (pH2B.8::H2B.8-eGFP) incorporation during male gametogenesis. Lowest panel is a duplicate of Fig. 1d. MN, VN, GN and SN, respectively, microspore, vegetative, generative and sperm nucleus. Data shown represent three independent experiments. Scale bars, 5 μm. d, Confocal images of various tissues from pH2B.8::H2B.8-eGFP plants. Data shown represent three independent experiments. Scale bars, 20 μm (early embryo, leaf, and root) and 5 μm (seed).
H2B.8 marks sperm chromatin
To investigate the mechanism of sperm chromatin compaction, we searched for sperm-specific chromatin factors. To that end, we performed mass spectrometry on leaf nuclei and on sperm and vegetative nuclei isolated by fluorescence-activated cell sorting (FACS). We identified a variant of histone H2B, H2B.8 (encoded by AT1G08170), which constituted 12.6% of sperm H2B but was absent in nuclei from vegetative and leaf cells (Extended Data Fig. 1b). Consistent with this result, RNA sequencing (RNA-seq) experiments detected abundant H2B.8 transcript levels in sperm cells, but none from somatic tissues such as leaves, roots and whole seedlings (Fig. 1c). To further examine the protein expression pattern during development, we generated reporter lines by expressing a H2B.8–eGFP fusion protein with the native H2B.8 promoter in Arabidopsis (pH2B.8::H2B.8–eGFP). Confocal imaging showed that H2B.8 is incorporated into sperm following the second pollen mitotic division step, when nuclei are compacted and chromatin aggregates (Fig. 1d and Extended Data Fig. 1c). H2B.8 was rapidly lost after fertilization, but reappeared during seed maturation (Extended Data Fig. 1d). No H2B.8–eGFP was observed in any other cell or tissue except sperm and seeds (Extended Data Fig. 1c,d), which is consistent with recently published analyses of H2B expression22.
H2B.8 mediates chromatin condensation
Because the presence of H2B.8 correlated with nuclear and chromatin condensation, we proposed that H2B.8-induced chromatin condensation is responsible for sperm compaction. To test this hypothesis, we generated two independent h2b.8 CRISPR knockout mutants (h2b.8-1 and h2b.8-2) (Extended Data Fig. 2a) and examined their sperm phenotype by confocal and 3D-SIM imaging. Sperm nuclei of the two h2b.8 mutants were about 40% larger than those of the wild type (Fig. 2a), which suggested that H2B.8 contributes to sperm nuclear compaction. Notably, sperm nuclei of h2b.8 mutants were smaller than their progenitor, the generative nucleus, which is also haploid (Extended Data Fig. 2b). This result indicated that H2B.8 is not the sole mechanism involved in sperm nuclear compaction. In further support of our hypothesis, chromatin was more homogenous and had reduced aggregation in h2b.8 mutants (Fig. 2b and Extended Data Fig. 2c–e). To confirm the role of h2b.8 mutations in these phenotypes, we expressed pH2B.8::H2B.8–Myc and pH2B.8::H2B.8–eGFP transgenes in the h2b.8 mutant (h2b.8-1; unless otherwise specified, all h2b.8 mutants hereafter refer to this allele). Both transgenes successfully rescued the sizes of h2b.8 sperm nuclei to wild-type levels (Fig. 2a), which provided evidence that H2B.8 drives nuclear condensation. Chromatin aggregates were also restored in pH2B.8::H2B.8–eGFP h2b.8 sperm (Fig. 2c). Furthermore, these restored aggregates colocalized with H2B.8–eGFP (Fig. 2c and Extended Data Fig. 2f), which indicated that H2B.8 is directly involved in forming the aggregates.
Extended Data Fig. 2. H2B.8 is required for sperm chromatin aggregation.

a, Alignment of h2b.8 CRISPR lines. h2b.8-1 has a single base deletion at 76 bp as indicated by an arrow, leading to a premature stop codon after 33 amino acids. h2b.8-2 has a 12 bp deletion after 67 bp and another of 5 bp after 92 bp, producing a truncated 54 amino acid protein. b, Nuclear sizes of the generative nucleus and sperm nuclei of indicated genotypes. P-values, one-sided ANOVA followed by individual two-sample Tukey tests, which adjust for multiple comparisons. Boxplots marked as A, B and C are significantly different between groups (P < 0.001) but not within the group (P > 0.1). n = 30 (generative nucleus), 80 (WT, h2b.8-1), and 77 (h2b.8-2) nuclei examined over two independent experiments. c, Density plot of individual SYBR Green-stained voxel intensities from wild-type (WT, blue) and h2b.8 (red) sperm nuclei. n = 30 nuclei for each. d, e, Boxplots indicating the volumes (d) and intensity (e) of SYBR Green foci in WT (blue) and h2b.8 (red) sperm. P-value, independent two-sample t-test. n = 30 nuclei for each. AU, arbitrary unit. f, g, Colocalization of DAPI and H2B.8-eGFP signals in sperm (f, pH2B.8::H2B.8-eGFP) and in H2B.8 ectopically expressed seedlings (g, p35S::H2B.8-eGFP). n = 37 and 31 nuclei for sperm and seedlings, respectively. h, Confocal images and quantification of H2B.8 condensates in p35S::H2B.8-eGFP seedlings. P-value, independent two-sample t-test. n = 30 and 71 for nuclei with small and large H2B.8 condensates, respectively, examined over two independent experiments. Scale bars, 2 μm.
Fig. 2. H2B.8 is required and sufficient to drive chromatin and nuclear condensation.

a, Sperm nuclear sizes in WT and other indicated genotypes. P values calculated using one-sided ANOVA followed by individual two-sample Tukey tests. Boxplots marked as A and B are significantly different between groups (P < 0.001) but not within the group (P > 0.1). n = 80 (WT, h2b.8-1, pH2B.8::H2B.8–Myc h2b.8-1, pH2B.8::H2B.8-N234K–Myc h2b.8-1), 79 (pH2B.8::H2B.8–eGFP h2b.8-1, h2b.8-2) or 39 (pH2B.8::H2B.8ΔIDR–eGFP h2b.8-1) nuclei examined over two independent experiments. b,c, Super-resolution 3D-SIM images (left) and associated intensity profiles (right) of sperm and seedling nuclei of indicated genotypes (h2b.8 refers to h2b.8-1 unless specified otherwise). Data shown represent three independent experiments. Scale bars, 1 μm (b), 1 μm (c, upper two panels) and 2 μm (c, lower three panels). d, Root nuclear sizes in indicated genotypes (numbers after the genotype indicate independent transgenic lines). P values calculated using one-sided ANOVA followed by individual two-sample Tukey tests. Boxplots marked as A and B are significantly different between groups (P < 0.05) but not within the group (P > 0.1). n = 100 (WT), 101 (p35S::H2B.2–YFP-9), 103 (p35S::H2B.8–eGFP-3, p35S::H2B.8ΔIDR–YFP-5), 104 (p35S::H2B.8–eGFP-5), 107 (p35S::H2B.8ΔIDR–YFP-2) or 109 (p35S::H2B.2–YFP-8) nuclei examined over two independent experiments.
To test whether H2B.8 is sufficient to drive chromatin aggregation and nuclear compaction, we ectopically expressed H2B.8 using a strong constitutive promoter (p35S). Distinctive chromatin aggregates that colocalized with H2B.8–eGFP were induced in p35S::H2B.8–eGFP somatic nuclei (Fig. 2c and Extended Data Fig. 2g). Approximately 30% of nuclei contained numerous small H2B.8 aggregates; the remaining 70% had fewer and larger aggregates (Fig. 2c and Extended Data Fig. 2h). The total volume of H2B.8 aggregates was comparable between the two types of nuclei (Extended Data Fig. 2h), which suggested that H2B.8-containing chromatin aggregates were fusing over time. p35S::H2B.8–eGFP expression also reduced the nuclear size in root cells by 22% (Fig. 2d). These results demonstrate that H2B.8 is sufficient for chromatin and nuclear condensation.
H2B.8 drives chromatin phase separation
H2B.8 is distinguished from other Arabidopsis H2B variants by a much longer amino-terminal tail that contains a 93 amino acid IDR (Extended Data Fig. 3a,b). Phylogenetic analysis revealed that H2B.8 is specific to flowering plants and is present in all flowering plant species with published genomes, except the most basal Amborella trichopoda (Fig. 3a and Supplementary Table 1). Notably, all identified H2B.8 homologues shared the insertion of an IDR in the histone tail (Fig. 3a and Supplementary Table 1), which indicated its functional importance.
Extended Data Fig. 3. Phase separation property of H2B.8.

a, Alignment of Arabidopsis H2B variants (N-terminal tail – upper panel, C-terminal body – lower panel). The N-terminal tail IDR of H2B.8 is highlighted with a yellow dashed box. Amino acids are colored according to the RasMol scheme. b, Intrinsic disorder prediction of H2B.8 (red) and H2B.2, a canonical H2B (blue), by PONDR. Histone profiles are aligned at the interchange between tail and body domains (dashed line). H2B.8 N-terminal tail IDR is highlighted in yellow. c, SDS-PAGE of reconstituted histone octamers containing H2B.2, H2B.8, H2B.8ΔIDR or H2B.8-scrambledIDR. For gel source data, see Supplementary Fig. 1. d, Fluorescence recovery after photobleaching (FRAP) of H2B.2- or H2B.8-containing chromatin droplets. Average relative fluorescence intensity of the photobleached area across 6 individual chromatin droplets was calculated and displayed at each time point. Error bars, ±SD. Scale bars, 1 μm. e, Fusion of H2B.2- or H2B.8-containing chromatin droplets (histone H3 and dsDNA were labeled with AF594 and YOYO-1, respectively) over time. Data shown represent one representative experiment, which has been performed twice with similar results. Scale bars, 1 μm. f, g, In vitro phase separation of nucleosomal arrays (NA) containing H2B.2, H2B.8 or H2B.8ΔIDR at indicated salt and NA concentrations (in g, NA concentration for all panels was 50 nM). Histone H3 and dsDNA were labeled with AF594 and YOYO-1, respectively, and merged images of red and green channels are shown. Data shown represent one representative experiment, which has been performed three times with similar results. Scale bars, 2 μm. h, Nuclear size quantification of Nicotiana benthamiana epidermis cell nuclei transiently expressing native and chimeric H2B.8 as indicated. Boxplots marked as A and B are significantly different between groups (P < 0.001) but not within the group (P > 0.1). n = 30 (WT, p35S::H2B.8-EWSR1-IDR-eGFP, and p35S::H2B.8-TAF15-IDR-eGFP), 32 (p35S::H2B.8-eGFP), and 33 (p35S::H2B.8-scrambledIDR-eGFP) nuclei from two independent experiments.
Fig. 3. H2B.8 condenses chromatin through IDR-dependent phase separation.

a, Phylogenetic tree illustrating H2B.8 evolution (marked by a star). Sperm chromatin compaction state, sperm motility and the presence of protamines or protamine-like proteins are denoted for represented eukaryote lineages. Ma, million years ago. b, In vitro phase-separation assays of nucleosomal arrays (NA) bearing H2B.2, H2B.8, H2B.8ΔIDR and H2B.8 with a scrambled IDR sequence (H2B.8-scrambledIDR) under indicated salt conditions and 100 nM NA concentration. Histone H3 was labelled by Alexa Fluor 594 (AF594, red), and double-stranded DNA (dsDNA) was stained with YOYO-1 (green). BF, bright field. Data shown represent one representative experiment, which has been performed three times with similar results. Scale bars, 2 μm.
IDRs can drive the formation of biomolecular condensates through phase separation23,24. Moreover, phase separation of IDR-containing proteins can mediate the formation and condensation of heterochromatin foci25,26. On the basis of this knowledge and the distinctive H2B.8 foci pattern in sperm and somatic cells that ectopically express H2B.8 (Fig. 2c), we proposed that H2B.8 aggregates chromatin through IDR-mediated phase separation.
To test this hypothesis, we assembled nucleosomal arrays using recombinant fluorophore-labelled histone octamers and a DNA template containing 12 repeats of the 601 nucleosome positioning sequence. We then tested the phase-separation properties of these nucleosomal arrays (Fig. 3b and Extended Data Fig. 3c). The addition of cation (K+ and/or Mg2+) at physiologically relevant concentrations to chromatin reconstituted with either H2B.8 or a canonical H2B (H2B.2) that naturally lacks an IDR induced the formation of phase-separated droplets (Fig. 3b). This result is in line with a previous finding that chromatin undergoes phase separation in vitro at physiological salt conditions27,28. Consistent with the reported liquid-like property of chromatin condensates27, chromatin droplets that contained H2B.8 or H2B.2 showed fluorescence recovery after photobleaching (FRAP; Extended Data Fig. 3d) and droplet fusion following contact (Extended Data Fig. 3e).
FRAP of H2B.8 chromatin droplets was slower than that of H2B.2 droplets (Extended Data Fig. 3d), which reflected the reduced internal droplet dynamics and a more gel-like behaviour. To further test whether H2B.8 confers different phase-separation properties to chromatin, we examined H2B.8-containing and H2B.2-containing nucleosomal arrays under different salt and array concentrations. Under the same physiological salt concentration, H2B.8-containing nucleosome arrays formed phase-separated condensates at lower chromatin concentrations (Extended Data Fig. 3f). Furthermore, unlike typical chromatin phase separation that requires the assistance of physiological cations27,28, H2B.8-containing nucleosome arrays formed phase-separated droplets under low or no cation conditions (or in the presence of high concentration of the chelating agent EDTA; Fig. 3b and Extended Data Fig. 3g). To test whether this phase-separation property relies on the IDR of H2B.8, we reconstituted nucleosomal arrays using H2B.8 without the IDR (H2B.8ΔIDR) (Extended Data Fig. 3c). Similar to H2B.2-containing chromatin, H2B.8ΔIDR-containing nucleosome arrays did not phase separate at lower array concentrations (<50 nM; Extended Data Fig. 3f) and failed to undergo phase separation in the absence of salt (Fig. 3b and Extended Data Fig. 3g). Consistent with the idea that IDRs promote phase separation through the disordered state instead of specific sequence motifs23,29, chromatin that contained H2B.8 in which the IDR sequence was randomly scrambled (H2B.8-scrambledIDR) phase separated in a salt-independent manner (Fig. 3b). Taken together, our results demonstrate that the IDR of H2B.8 mediates a new form of chromatin phase separation.
To test whether H2B.8 phase separation is required for chromatin condensation in vivo, we ectopically expressed H2B.8ΔIDR (p35S::H2B.8ΔIDR–YFP) or the canonical H2B.2 in cells. In contrast to the effect of full-length H2B.8, H2B.8ΔIDR or H2B.2 expression did not induce chromatin aggregation and had no effect on nuclear size in root cells (Fig. 2c,d). This result confirmed that chromatin and nuclear condensation are dependent on the IDR of H2B.8. We next expressed H2B.8ΔIDR in h2b.8 mutant plants (pH2B.8::H2B.8ΔIDR–eGFP h2b.8) and examined whether sperm nuclear condensation is also dependent on the IDR. Unlike the full-length H2B.8, H2B.8ΔIDR failed to rescue the sperm nuclear size phenotype of h2b.8 (Fig. 2a,c). This result demonstrates the importance of the IDR for sperm nuclear condensation.
Beyond the IDR, H2B.8 has several amino acid differences to that of canonical H2Bs in the globular domain, including asparagine 234 (N234), which is canonically a lysine residue that is subject to monoubiquitylation22 (Extended Data Fig. 3a). The inability of H2B.8ΔIDR to rescue the h2b.8 sperm nuclear phenotype (Fig. 2a) shows that without the IDR, the H2B.8 globular domain cannot mediate nuclear compaction. Nonetheless, as H2B monoubiquitylation is an important modification30 that would be precluded by N234, we examined whether N234 is important for H2B.8 function by expressing a mutated H2B.8 with the 234th asparagine replaced by lysine (H2B.8-N234K). The expression of H2B.8-N234K fully complemented the h2b.8 sperm nuclear size phenotype (Fig. 2a), which shows that N234 is not essential for H2B.8 function.
The dependence of chromatin and nuclear condensation on the IDR of H2B.8 might result from the disordered state of the IDR and associated phase-separation ability or from IDR-mediated recruitment of unknown chromatin-condensing factors. To test these hypotheses, we expressed H2B.8 with a randomly scrambled IDR sequence (H2B.8-scrambledIDR) or with the native IDR replaced by animal IDR sequences of similar negative charge (H2B.8-EWSR-IDR and H2B.8-TAF15-IDR)31 in tobacco leaves and measured their effects on nuclear size. Similar to the effect of native H2B.8, the expression of H2B.8-scrambledIDR, H2B.8-EWSR-IDR and H2B.8-TAF15-IDR all effectively condensed the nuclei of tobacco epidermis (Extended Data Fig. 3h). This result indicates that the function of H2B.8 relies on phase-separation ability instead of specific sequence motifs within the IDR. In line with this, although the presence of the IDR is conserved among flowering plants, the sequences of H2B.8 IDRs are diverse (Supplementary Table 1).
H2B.8 is located in silent euchromatin
To further understand H2B.8 activity, we determined the genomic localization of H2B.8 by performing native chromatin immunoprecipitation assay with sequencing (ChIP-seq) on pH2B.8::H2B.8–eGFP h2b.8 pollen using GFP-specific antibodies. This analysis identified H2B.8 peaks that occupied about 17% of the sperm genome. This value was comparable with our mass spectrometry results, which showed that approximately 13% of canonical H2B is replaced by H2B.8 in sperm (Extended Data Fig. 1b). H2B.8 was most enriched within so-called euchromatic transposable elements (TEs) (Fig. 4a,b), which are AT-rich and depleted of H3K9me2 and other heterochromatic marks32. Heterochromatic TEs, which are typically GC-rich and H3K9me2-rich32, had comparatively little H2B.8 (Fig. 4a,b). H2B.8 was excluded from the bodies of transcribed genes compared with inactive genes and intergenic regions, and H2B.8 enrichment and gene transcription were anticorrelated (Fig. 4a–c). H2B.8 distribution along chromosomes followed that of euchromatic TEs, with H2B.8 most abundant at the edges of pericentromeric regions (Fig. 4d and Extended Data Fig. 4a).
Fig. 4. H2B.8 is localized in AT-rich TEs and transcriptionally inactive intergenic regions.

a, Genome snapshots of H2B.8 abundance in sperm (log2(IP/input)), sperm cell transcription (log2(RPKM)) and gene and TE annotations (orange, euchromatic TE; magenta, heterochromatic TE) at representative regions. RPKM, reads per kilobase of transcript per million mapped reads. b, Profiles of sperm H2B.8 enrichment over genes (grouped by sperm cell expression), TEs (grouped by chromatin state) and intergenic regions. c,f, Scatterplots showing anticorrelation of sperm H2B.8 enrichment with sperm transcription (c) or GC content (f) among indicated genomic features. ρ, Spearman’s rank. Colours are as d. d, Coverage of genes, euchromatic TEs and heterochromatic TEs (left y axis, 500-kb windows) and H2B.8 enrichment in sperm (right y axis; 1-kb windows) along chromosome 1. Chromosomes 2–5 are shown in Extended Data Fig. 4a. e, PCA of H2B.8 abundance in p35S::H2B.8–eGFP seedlings with other chromatin marks. The green and pink shaded areas represent euchromatic and heterochromatic marks, respectively.
Extended Data Fig. 4. Ectopically expressed H2B.8 in seedlings exhibits a similar deposition profile as native H2B.8 in sperm.

a, Coverage of genes, euchromatic TEs and heterochromatic TEs (left Y axis; 500 kb windows) and H2B.8 enrichment in sperm (right Y axis; 1 kb windows) along Chromosomes 2 to 5. Chromosome 1 is shown in Fig. 4d. b, Genome snapshots of H2B.8 abundance (log2(IP/input)) and transcription (log2(RPKM)) in p35S::H2B.8-eGFP seedlings, and gene and TE annotations (orange, euchromatic TE; magenta, heterochromatic TE) over representative regions. RPKM, Reads Per Kilobase of transcript per Million mapped reads. c, Profiles and associated heatmaps of H2B.8 enrichment in p35S::H2B.8-eGFP seedlings over genes (grouped by seedling expression), TEs (grouped by chromatin state) and intergenic regions. d, Proportions (%) of the genome covered by genes, TEs and intergenic regions in wild-type sperm and seedling, and by respective H2B.8 peaks in pH2B.8::H2B.8-eGFP h2b.8 sperm and p35S::H2B.8-eGFP seedling. Same color coding is used as in (c) and Fig. 4b. e, Upper panel, scatterplot showing anticorrelation of H2B.8 enrichment with GC content (%) in p35S::H2B.8-eGFP seedlings over denoted genomic features. Lower panel, scatterplot showing the WT seedling anti-GFP enrichment (log2(IP/input)) control against GC content (%). ρ, Spearman’s Rank. f, Prediction of H2B.8 using indicated chromatin features, transcription (log10(RPKM+1)) and GC content as predictors. The y-axis shows the adjusted R2 value between the predicted and observed values. g, Profiles of H2B.8 enrichment in p35S::H2B.8-eGFP seedlings over non-genic (TE and intergenic) regions. The test regions were grouped into three equal parts by GC content levels (low GC < 0.29; 0.29 < middle GC < 0.34; high GC > 0.34).
To further understand the chromatin preference of H2B.8 and whether its localization pattern is intrinsically determined by H2B.8 or other sperm-specific components, we performed native ChIP-seq with seedlings of the ectopic H2B.8 expression line (p35S::H2B.8-eGFP). This revealed an analogous H2B.8 localization pattern to that in sperm, with enrichment in euchromatic TEs and intergenic regions (Extended Data Fig. 4b–d). Together with the ability of ectopic H2B.8 to condense somatic cell chromatin and nuclei (Fig. 2c,d), these results suggest that H2B.8 deposition does not rely on sperm-specific factors. Next, utilizing available seedling epigenomic data, we explored H2B.8 associations with other chromatin features. Principal component analysis (PCA) revealed that H2B.8 clustered with neither permissive nor repressive chromatin modifications but associated with GC content (Fig. 4e). Multivariate linear regression modelling of H2B.8 further showed that transcription and GC content were the best predictors of H2B.8 localization, with which strong anticorrelations exist (Fig. 4c,f, Extended Data Fig. 4e,f and Supplementary Table 2). This may explain why H2B.8 is strongly depleted from transcribed genes (Fig. 4b and Extended Data Fig. 4c). In the remainder of the genome, in which there is little transcription, H2B.8 accumulated at GC-poor elements, mostly euchromatic TEs and intergenic regions (Extended Data Fig. 4e,g). In summary, our results suggest that H2B.8 localization is mostly driven by transcription and GC content rather than sperm-specific factors.
It is yet unclear why H2B.8 localization is associated with transcription and GC content; however, our cytological observations provided indications. 3D-SIM showed that unlike the full-length H2B.8, which is largely devoid in heterochromatin, H2B.8ΔIDR preferentially located to heterochromatin when expressed in sperm or seedling cells (Fig. 2c). To validate this observation and to examine the genomic localization of H2B.8ΔIDR, we performed ChIP-seq on H2B.8ΔIDR ectopically expressed (p35S::H2B.8ΔIDR–eGFP) seedlings and compared results with p35S::H2B.8–eGFP (and p35S::H2B.2-eGFP as a control) seedlings. Consistent with the cytology results, H2B.8ΔIDR preferentially located to pericentromeric heterochromatin, and its abundance along chromosomal arms was generally reduced compared with H2B.8 (Extended Data Fig. 5a). Notably, although the deletion of IDR markedly reduced the enrichment of H2B.8 in euchromatic TEs and increased H2B.8 deposition in heterochromatic TEs, it did not disrupt the preferential depletion of H2B.8 from transcribed genes (Extended Data Fig. 5b). This finding indicates that the IDR of H2B.8 is required for the preferential localization of H2B.8 in euchromatin, but not for its exclusion from transcribed regions.
Extended Data Fig. 5. The IDR is required for the exclusion of H2B.8 from heterochromatin.
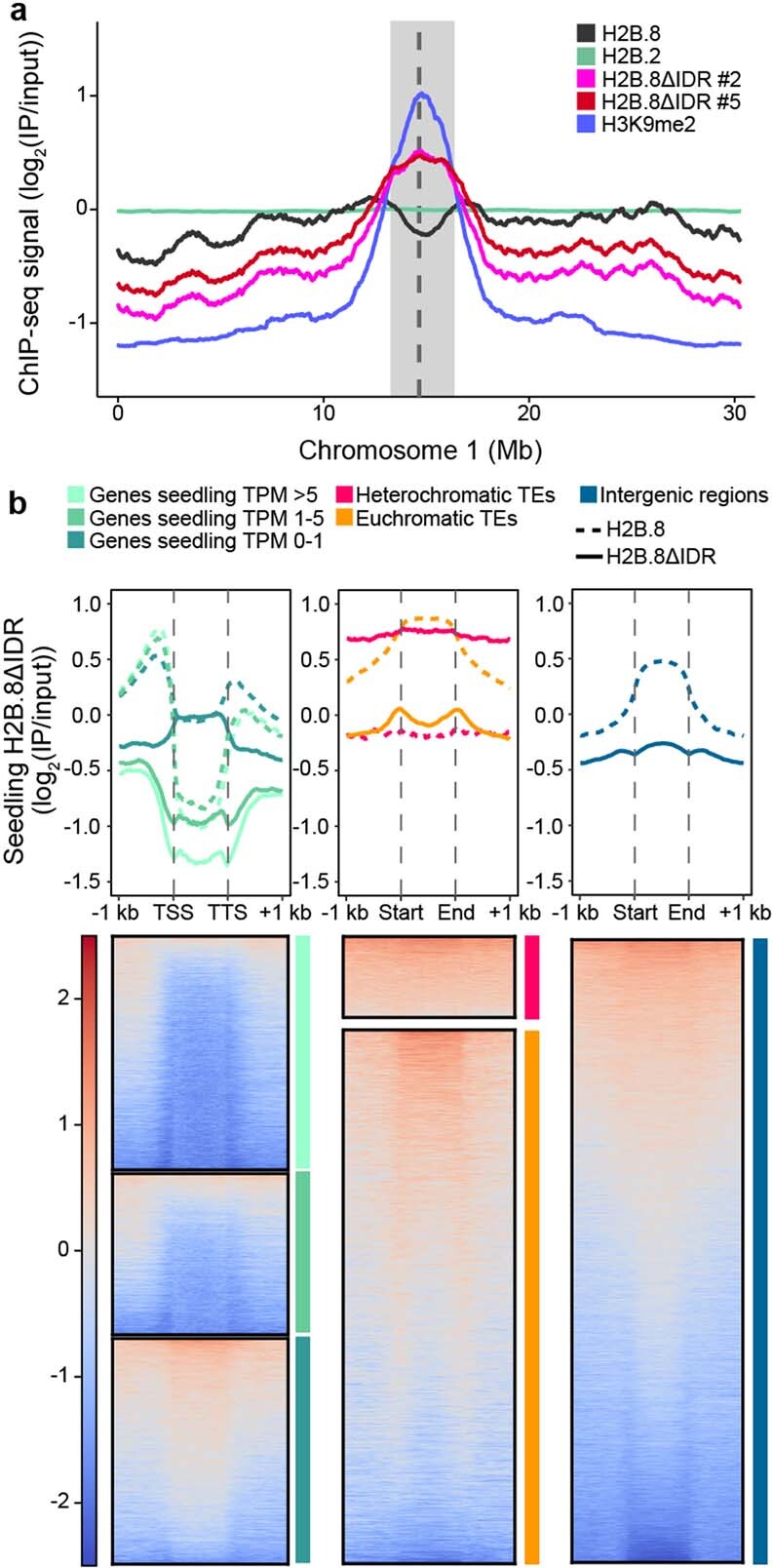
a, Enrichment of H2B.8 (p35S::H2B.8-eGFP), H2B.2 (p35S::H2B.2-eGFP), H2B.8ΔIDR (p35S::H2B.8ΔIDR-eGFP lines #2 and #5) and H3K9me2 in seedlings (log2(IP/input); 1 kb windows). Grey dashed line localizes the centromere and grey shaded area represents pericentromeric heterochromatin. b, Profiles and associated heatmaps of H2B.8ΔIDR enrichment in p35S::H2B.8ΔIDR-eGFP seedlings over genes (grouped by seedling expression), TEs (grouped by chromatin state) and intergenic regions. For comparison, p35S::H2B.8-eGFP ChIP-seq data are shown as dashed lines (as in Extended Data Fig. 4c).
H2B.8 does not suppress transcription
Chromatin condensation is frequently associated with transcriptional repression21. Therefore, the localization of H2B.8 in the non-transcribing parts of the genome could arise through two mechanisms. Either H2B.8 suppresses transcription or it is excluded from transcribed regions. To test these hypotheses, we isolated wild-type and h2b.8 mutant sperm cells and performed RNA-seq. Among the 12,198 genes expressed in either wild-type or h2b.8 sperm, none had significantly altered expression in h2b.8 cells (Fig. 5a). Similarly, we did not find any TEs that were significantly misregulated in h2b.8 sperm (Extended Data Fig. 6a). RNA-seq of wild-type and p35S::H2B.8–eGFP seedlings provided further support of the negligible effect of H2B.8 on transcription (Extended Data Fig. 6b,c). Therefore, our results indicate that unlike protamines, which condense animal sperm at the expense of transcriptional potential8, H2B.8 condenses plant sperm without suppressing transcription.
Fig. 5. H2B.8-induced chromatin aggregation does not suppress transcription but decondenses heterochromatin foci.
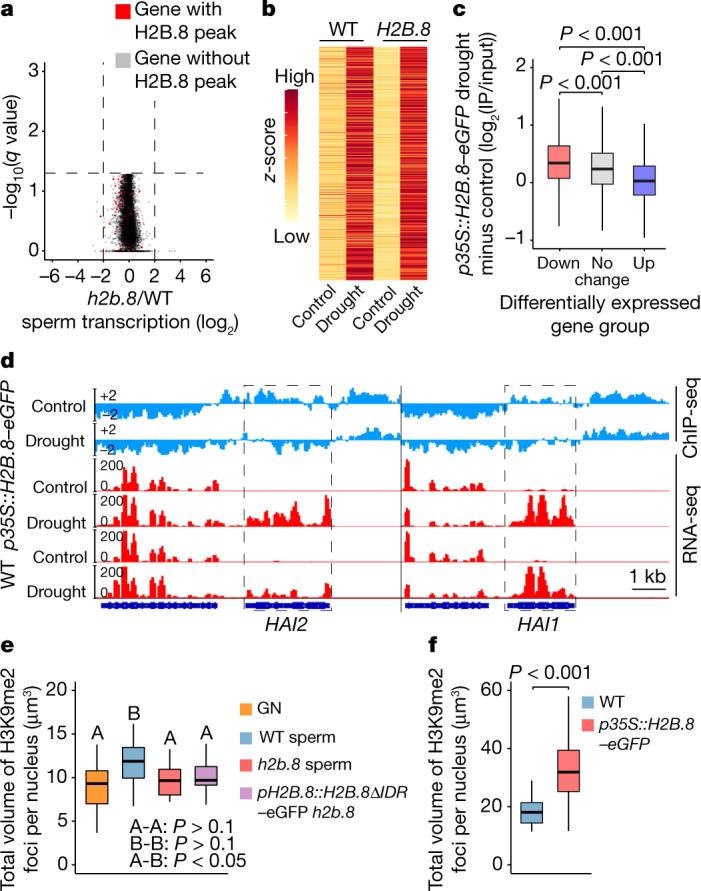
a, Volcano plot showing differential gene expression between h2b.8 mutant and WT sperm cells. n = 12,198 for genes with TPM > 1 in either WT or h2b.8 mutant. b, Heatmap depicting the transcription (z-score) of upregulated genes in response to drought stress in WT and p35S::H2B.8–eGFP seedlings subject to mock (control) and drought treatment. n = 1,605. c, Differences in H2B.8 abundance between drought and control conditions over differentially expressed gene groups in p35S::H2B.8–eGFP seedlings. P values calculated using one-sided ANOVA followed by individual two-sample Tukey tests. n = 711 (Down), 26,180 (No change) and 1,605 (Up) genes. d, Genome snapshots of example drought-response genes showing H2B.8 abundance (log2(IP/input)) and transcription (log2(RPKM)) in seedlings of indicated genotypes under control and drought conditions. e, Quantification of H3K9me2-enriched heterochromatin foci in the generative nucleus and sperm nuclei of indicated genotypes. P values calculated using one-sided ANOVA followed by individual two-sample Tukey tests. Boxplots marked as A and B are significantly different between groups (P < 0.05) but not within the group (P > 0.1). n = 30 nuclei each from two independent experiments. f, Quantification of H3K9me2-enriched heterochromatin foci in WT and p35S::H2B.8–eGFP seedling nuclei. P value calculated using independent two-sample t-test. n = 138 and 101 nuclei for WT and p35S::H2B.8–eGFP, respectively, examined from two independent experiments.
Extended Data Fig. 6. Ectopically expressed H2B.8 has a negligible effect on transcription.
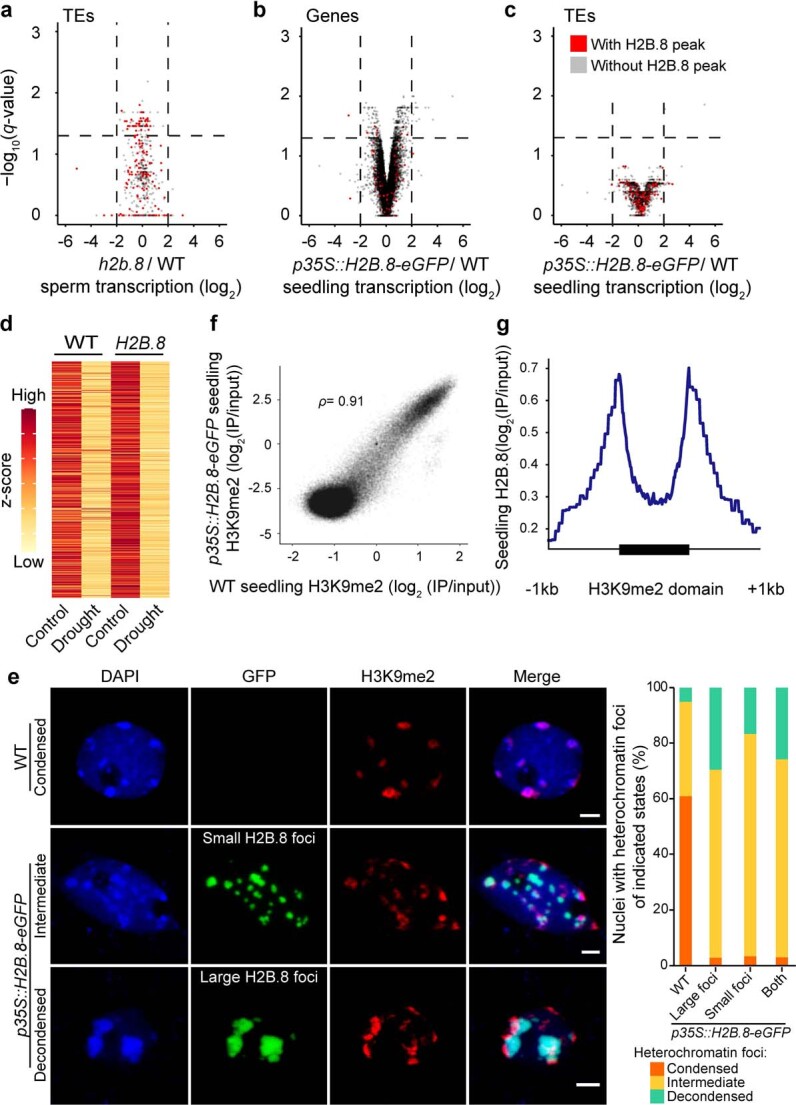
a-c, Volcano plots showing differential TE (a, c) or gene (b) expression between h2b.8 mutant and wild-type (WT) sperm cells (a), or p35S::H2B.8-eGFP and WT seedlings (b, c). TPM, Transcripts Per Million. n = 480 for TEs with TPM > 1 in either WT or h2b.8 mutant sperm, and n = 20285 (b) and 1472 (c) for genes or TEs with TPM > 1 in either p35S::H2B.8-eGFP or WT seedlings. d, Heatmap depicting the transcription of genes (z-score) downregulated by drought stress in WT and p35S::H2B.8-eGFP seedlings subject to mock (control) and drought treatment. n = 771. e, Confocal images and quantification of seedling nuclei of indicated genotypes with condensed, intermediately condensed or decondensed heterochromatin foci (measured by H3K9me2 signal). n = 138 (WT), and 71, 30 and 101 (p35S::H2B.8-eGFP nuclei with large, small, and both large and small H2B.8 foci, respectively). H2B.8 condensates are largely distinct from heterochromatin foci, overlapping only 19.0% ± 10.7% (standard deviation calculated from 53 nuclei) of the volume of heterochromatin foci. f, Scatterplot showing correlation of H3K9me2 levels (log2(IP/input)) over 1 kb windows between WT and p35S::H2B.8-eGFP seedlings. ρ, Spearman’s Rank. g, Profile of H2B.8 enrichment in p35S::H2B.8-eGFP seedlings over H3K9me2-enriched domains.
To directly test whether H2B.8 inhibits transcriptional activation, we exposed p35S::H2B.8–eGFP and wild-type seedlings to drought stress and examined their transcriptional response. Comparable transcriptomic changes between p35S::H2B.8–eGFP and wild-type seedlings following drought treatment were observed (Fig. 5b and Extended Data Fig. 6d). Moreover, ChIP-seq of drought-treated and control p35S::H2B.8–eGFP seedlings showed that H2B.8 is evicted from genes induced by drought, such as Highly ABA-Induced 1 (HAI1) and HAI2 (ref. 33), whereas genes repressed by drought gain H2B.8 (Fig. 5c,d). These results demonstrate that H2B.8 does not inhibit gene transcription but is instead removed by transcription. This process may underlie the localization of H2B.8 in unexpressed euchromatin.
H2B.8 promotes male fertility
To investigate the biological significance of H2B.8-mediated sperm condensation, we performed reciprocal crosses between the h2b.8 heterozygous mutant and wild type. When the h2b.8 heterozygous mutant was used as the male, F1 progeny were around 30% less likely to carry the h2b.8 allele than the wild-type allele (P < 0.01, Fisher’s exact test; Supplementary Table 3). By contrast, the transmission of h2b.8 was not significantly different from the wild-type allele when passed through the female (P = 0.64, Fisher’s exact test; Supplementary Table 3), which demonstrated that H2B.8 is important for male fertility. The fertility defect of h2b.8 is not caused by disrupted pollen germination, as h2b.8 pollen grains germinated at comparable rates to wild type in vitro (Supplementary Table 4). This result is consistent with the null effect of H2B.8 on transcription (Fig. 5a,b and Extended Data Fig. 6a–d), and suggests that the fertility defect is most probably caused by enlarged sperm nuclei.
Manual crossing is a stressful process, with pistils (the female organs) slightly desiccated and pollinated at an earlier developmental stage than normal. To investigate whether h2b.8 affects male fertility in a less stressful mating environment, we examined the segregation ratio of progeny generated from self-pollinated h2b.8 heterozygous plants. We observed the expected Mendelian segregation ratio (N = 1,462; Supplementary Table 5), which shows that the h2b.8 mutation does not affect fertility when plants are allowed to self-fertilize under laboratory conditions. As H2B.8 is transiently expressed in mature seeds (Extended Data Fig. 1d), this result also demonstrated that h2b.8 does not affect seed development under laboratory conditions. Collectively, our observations suggest that H2B.8 is important for sperm fertility in challenging or stressful situations, such as those created by manual crossing. This finding might be relevant to reproduction under natural environmental conditions, which are usually less favourable than standard laboratory conditions.
H2B.8 decondenses heterochromatin
In addition to the overall chromatin condensation in sperm observed by 3D-SIM, there was slight decondensation of heterochromatin foci (Fig. 1a,b). To understand whether this is caused by H2B.8, we performed immunostaining using H3K9me2-specific antibodies and measured the volume of H3K9me2 foci in h2b.8 mutant nuclei. The h2b.8 mutation significantly reduced the volume of heterochromatin foci in sperm to a level resembling that in the generative nucleus (Fig. 5e). Furthermore, H3K9me2 heterochromatin foci were more enlarged and decondensed in p35S::H2B.8–eGFP seedling nuclei than in wild type nuclei (Fig. 5f and Extended Data Fig. 6e). Most wild-type nuclei showed highly condensed heterochromatin foci, but these were rarely found in p35S::H2B.8–eGFP seedling nuclei (Extended Data Fig. 6e). The majority of p35S::H2B.8–eGFP seedling nuclei exhibited moderately dispersed heterochromatin foci (Fig. 5f and Extended Data Fig. 6e), reminiscent of those in sperm (Fig. 1a,b). This result suggests that H2B.8 causes heterochromatin foci to decondense.
Our data also suggested that decondensation of heterochromatin foci depends on H2B.8 phase separation, as the expression of H2B.8ΔIDR did not affect heterochromatin (Fig. 5e). As heterochromatin foci are also phase-separated condensates25,26,34, this suggested that there are interactions between the two types of condensates. Indeed, although H2B.8 and heterochromatic condensates were mostly distinct, some physical associations were observed (Fig. 2c and Extended Data Fig. 6e). Collectively, our results suggest that condensation of chromatin through H2B.8 phase separation affects heterochromatin condensation, probably because H2B.8-associated AT-rich euchromatic TEs are interspersed with heterochromatic TEs in pericentromeric regions (Fig. 4d and Extended Data Figs. 4a and 6f,g). Alternative hypotheses that are independent of H2B.8 phase separation are also plausible; for example, H2B.8 might directly recruit factors that interfere with heterochromatin condensation.
H2B.8 increases chromosomal arm contacts
To understand how H2B.8 mediates chromatin condensation, we performed genome-wide chromosome conformation capture analysis35 (Hi-C) on seedlings that ectopically express H2B.8 (p35S::H2B.8–eGFP) and wild-type controls. The Hi-C libraries were sequenced to single kilobase resolution (Extended Data Fig. 7a), and our wild-type contact matrices were comparable to previously published experiments36,37 (Extended Data Fig. 7b). As previously shown37–40, topologically associating domains were absent in Arabidopsis, but telomeres were frequently associated, as were centromeres (Extended Data Fig. 7c).
Extended Data Fig. 7. H2B.8 affects intra- and inter-chromosomal interactions.

a, Sequencing read counts of wild-type (WT) and p35S::H2B.8-eGFP seedling Hi-C libraries. Rep, biological replicate. b, Correlations between the Hi-C data generated in this study and previously published36,37. R, Pearson’s correlation coefficient. c, Hi-C interaction frequency heatmaps at 500 kb resolution for WT (upper; merged replicates) and p35S::H2B.8-eGFP (lower; merged replicates) seedlings. d, Genome-wide interaction frequency fold change heatmap between WT (Rep 2) and p35S::H2B.8-eGFP (Rep 2) seedlings at 500 kb resolution, as shown in Fig. 6a for Rep 1. e, Short-range intrachromosomal interaction frequency difference between p35S::H2B.8-eGFP (Rep 2) and WT (Rep 2) over quantiles of seedling H2B.8 enrichment (log2(IP/input)), as shown in Fig. 6b for Rep 1. Spearman’s ρ = 0.930. f, Long-range interaction frequency difference between p35S::H2B.8-eGFP (Rep 2) and WT (Rep 2) between chromosome arms and pericentromeric regions over quantiles of seedling H2B.8 enrichment (log2(IP/input)), as shown in Fig. 6c for Rep 1. Spearman’s ρ = 0.870.
The comparison of p35S::H2B.8–eGFP and wild-type Hi-C data revealed alterations in higher-order chromatin architecture. Within chromosomes, ectopic H2B.8 caused increased short-range contacts (200 kb–1.1 Mb) (Fig. 6a and Extended Data Fig. 7d). Short-range intrachromosomal interactions are indicative of chromatin condensation, which suggested that H2B.8 principally forms aggregates by concentrating linearly proximal regions (Fig. 6a and Extended Data Fig. 7d). In support of this idea, the increase in short-range contacts strongly correlated with local H2B.8 abundance (Spearman’s ρ = 0.97) (Fig. 6b and Extended Data Fig. 7e).
Fig. 6. Increased chromosomal interactions between H2B.8-containing loci.

a, Genome-wide interaction frequency fold-change heatmap between WT and p35S::H2B.8–eGFP seedlings at 500-kb resolution. b, Short-range intrachromosomal interaction frequency difference between p35S::H2B.8–eGFP and WT over quantiles of seedling H2B.8 enrichment (log2(IP/input)). Spearman’s ρ = 0.974. c, Long-range interaction frequency difference between p35S::H2B.8–eGFP and WT between chromosome arms and pericentromeric regions over quantiles of seedling H2B.8 enrichment (log2(IP/input)). Spearman’s ρ = 0.890.
Contacts between pericentromeric regions (heterochromatin) and distal chromosomal arms (euchromatin) were also increased in p35S::H2B.8–eGFP seedlings (Fig. 6a and Extended Data Fig. 7d). Concomitantly, interchromosomal interactions between pericentromeres were reduced (Fig. 6a and Extended Data Fig. 7d). These alterations are consistent with the cytologically observed heterochromatin decondensation and association of heterochromatin foci and H2B.8 condensates (Fig. 2c and Extended Data Fig. 6e). The effect of H2B.8 was local, as the interactions between pericentromeric regions and chromosomal arms increased at regions with abundant H2B.8 (Fig. 6c and Extended Data Fig. 7f). These observations support the hypothesis that dispersal of heterochromatin foci is caused by H2B.8-mediated aggregation of euchromatic TEs that are abundant in and near pericentromeric regions (Fig. 4d and Extended Data Figs. 4a and 6g). Taken together, our Hi-C and ChIP-seq data demonstrate that H2B.8 achieves a form of global chromatin condensation through the binding and aggregation of transcriptionally inactive AT-rich sequences dispersed throughout the genome.
Discussion
Our results revealed a mechanism of chromatin condensation driven by H2B.8-induced phase separation (Extended Data Fig. 8). Chromatin aggregates in the sperm nucleus were reduced in h2b.8 mutants (Fig. 2b and Extended Data Fig. 2c), whereas ectopically expressed H2B.8 in somatic cells was sufficient to induce chromatin aggregates in an IDR-dependent manner (Fig. 2c). Hi-C and cytological observations revealed that H2B.8 forms chromatin condensates by increasing interactions between H2B.8-enriched chromosomal regions (Figs. 2c and 6b,c and Extended Data Fig. 2h). Owing to H2B.8 deposition within AT-rich sequences in both pericentromeric regions and chromosomal arms (Fig. 4d and Extended Data Figs. 4a and 6g), broad chromosomal regions were concentrated by phase separation. In interphase somatic cells, euchromatin takes up most of the nuclear volume41. Because H2B.8 is abundant in euchromatin, H2B.8-induced chromatin condensation is highly effective at condensing nuclei (Fig. 2a,d).
Extended Data Fig. 8. Mechanism of H2B.8-mediated sperm condensation.
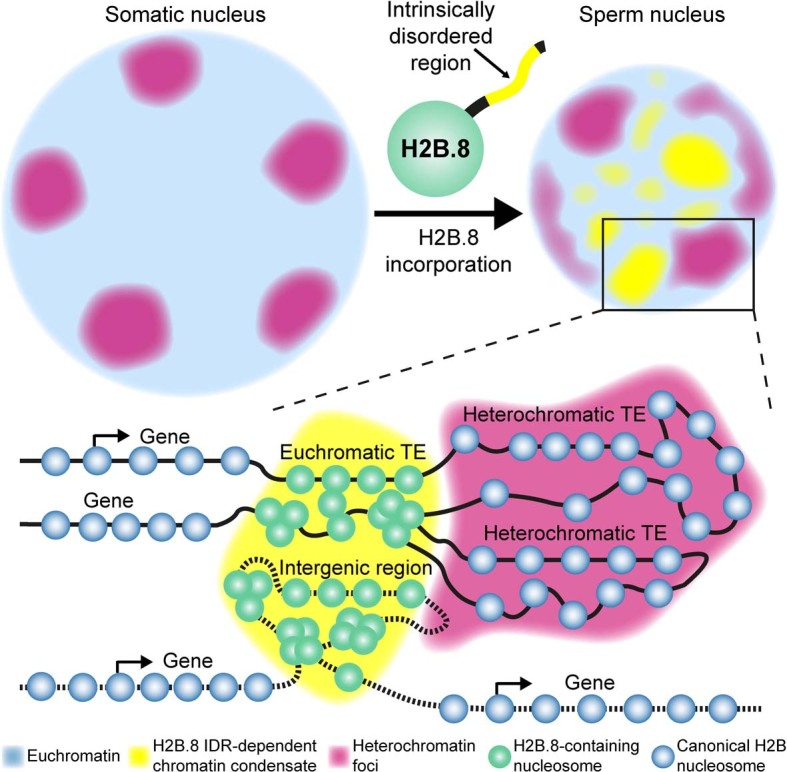
H2B.8 drives sperm nuclear condensation via the formation of chromatin condensates (yellow), which is dependent on an intrinsically disordered region (IDR) of H2B.8 conserved among flowering plants. Unlike typical chromatin condensation mechanisms, H2B.8-induced condensation does not inhibit transcription. Condensation is achieved by the specific deposition of H2B.8 into inactive AT-rich chromatin, which alters higher-order chromatin architecture to effectively compact the nucleus without sacrificing transcription. H2B.8-mediated chromatin aggregation disperses heterochromatin foci (pink), suggesting interactions between the euchromatic (yellow) and heterochromatic (pink) chromatin condensates in the nucleus.
Despite its effectiveness, H2B.8 compacts nuclei without compromising transcription (Fig. 5a,b). For species with swimming sperm, in which the size of the sperm head is crucial1,2,9, DNA condensation may be paramount and transcription dispensable. Protamines can greatly condense DNA and have been identified in the sperm of most all multicellular eukaryotic lineages except angiosperms (flowering plants) and gymnosperms (Fig. 3a). This includes bryophytes and pteridophytes, both of which have motile sperm and use protamines for sperm condensation10–14,19 (Fig. 3a). By contrast, H2B.8 is specific to flowering plants. Flowering plants have immotile sperm and may benefit from a less extreme approach that condenses nuclei without limiting transcription. This moderate level of condensation is still important for fertility, as h2b.8 mutations can significantly reduce male transmission (Supplementary Table 3). As sperm transcription (Fig. 5a and Extended Data Fig. 6a) and in vitro pollen germination (Supplementary Table 4) are not affected by the h2b.8 mutation, the reduced fertility is probably caused by the enlarged sperm nuclei. We speculate that fertilization, which takes place at ovules deeply embedded in angiosperm maternal tissues, might favour smaller sperm nuclei. Consistent with this idea, gymnosperms, which have exposed ovules, produce sperm with uncondensed nuclei11 and lack H2B.8 (Fig. 3a). Therefore, H2B.8 is an evolutionary innovation of angiosperms that achieves a moderate level of chromatin condensation that is compatible with active transcription.
Methods
Plant growth conditions
A. thaliana plants (Col-0 ecotype) used in this study were grown under long day (16 h light, 8 h dark) conditions at 22 °C and 70% humidity. Seedlings were grown on germination medium plates without glucose under the same conditions.
Generation of CRISPR–Cas9 mutants
Mutant alleles of H2B.8 were generated using the CRISPR–Cas9 technique42. Four single guide RNAs (sgRNAs; Supplementary Table 6) were designed using CHOPCHOP (v.3)43 and cloned using the Golden Gate system42. Constructs were transformed into the Agrobacterium tumefaciens strain GV3101 using floral dip to wild-type (WT) Col-0 A. thaliana44. Transformants were screened by Sanger sequencing. Selected lines were taken to the next generation to produce homozygous mutants without Cas9. Line h2b.8-1 was genotyped by dCAPS with EcoNI, whereas line h2b.8-2 was genotyped by PCR (Supplementary Table 6).
Vector cloning
For the H2B.8 reporter constructs (pH2B.8::H2B.8–eGFP and pH2B.8::H2B.8–Myc), approximately 2 kb upstream of H2B.8 was cloned as the promoter and the H2B.8 gDNA sequence was amplified (Supplementary Table 6). Using MultiSite Gateway Technology (Thermo Fisher Scientific), the PCR products were ligated to P4P1r and pDONR207, respectively. Sequences were assembled to the expression vector pK7m34GW with a carboxy-terminal eGFP or 3× Myc tag in P2rP3. The ectopic H2B.8 expression vector (p35S::H2B.8–eGFP) was generated in the same way, but using the 35S promoter.
The native and ectopic H2B.8ΔIDR expression constructs (pH2B.8::H2B.8ΔIDR–YFP and p35S::H2B.8ΔIDR–YFP, respectively) were generated by overlapping PCR to remove the IDR sequence, whereas ectopic H2B.2 (p35S::H2B.2–YFP) was cloned from gDNA (Supplementary Table 6). Products were ligated to the pCAMBIA1300 vector backbone containing the 35S promoter and a C-terminal eGFP/YFP using the In-Fusion cloning system (Takara Bio).
The pH2B.8::H2B.8-N234K–Myc construct was generated by overlapping PCR (Supplementary Table 6) and ligated to pDONR207 and subsequently conjugated to the H2B.8 promoter and a C-terminal 3× Myc tag in the pK7m34GW expression vector using MultiSite Gateway Technology (Thermo Fisher Scientific).
For the p35S::H2B.8-scrambledIDR–eGFP, p35S::H2B.8-EWSR1-IDR–eGFP and p35S::H2B.8-TAF15-IDR–eGFP constructs, the scrambledIDR (randomly shuffled amino acid sequence of the IDR by Python 3.9: DEVIQDISANPPVLENEPVTPSEPTVQEDTRECIETPEETPISVPEGEATPETKVQGDNSDFSSQTRTVDLKEVPSVPPREGTPPTPVVDDVE); EWSR1-IDR31 (ASTDYSTYSQAAAQQGYSAYTAQPTQGYAQTTQAYGQQSYGTYGQPTDVSYTQAQTTATYGQTAYATSYGQPPTGYTTPTAPQAYSQPVQGYGTGAYDTTTATVTTTQASYAAQSAYGTQPAYPAYGQQPAATAPTRPQDGNKPTETSQPQSSTGGYNQPSLGYGQSNYSYPQVPGSYPMQPVTAPPSYPPTSYSSTQPTSYDQSSYSQQNTYGQPSSYGQQSSYGQQSSYGQQPPTSYPPQTGSYSQAPSQYSQQSSSYGQQSSFRQDHPSSMGVYGQ), and TAF15-IDR31 (SDSGSYGQSGGEQQSYSTYGNPGSQGYGQASQSYSGYGQTTDSSYGQNYSGYSSYGQSQSGYSQSYGGYENQKQSSYSQQPYNNQGQQQNMESSGSQGGRAPSYDQPDYGQQDSYDQQSGYDQHQGSYDEQSNYDQQHDSYSQNQQSYHSQ) sequences, respectively, were optimized by Arabidopsis codon usage and synthesized. The synthesized fragments were then fused with H2B.8 by overlapping PCR (Supplementary Table 6). Inserts were cloned to the pCAMBIA1300 vector containing the 35S promoter and C-terminal eGFP.
The above-mentioned vectors were transformed into h2b.8-1 mutant plants of T3 generation or WT plants. Single insertion transgenic lines in T3 or T4 generations were used in this study, and at least two independent transgenic lines were included in each assay.
Sperm, vegetative and leaf nuclei total protein extraction
Sperm and vegetative nuclei were isolated by FACS (two replicates were isolated for each cell type) using a BD FACSMelody cell sorter (BD Biosciences), as previously described45 (Supplementary Fig. 2a). A total of 2 million sorted nuclei (isolated from roughly 40 ml open flowers) were pooled for each replicate, and 0.45 volume of 3.2× lysis buffer (10% SDS, 100 mM TEAB, pH 7.55) was added. Nuclei were lysed at 95 °C for 5 min, then centrifuged at 13,000g for 8 min at room temperature. The lysate was placed into a new tube. One-tenth volume of 12% phosphoric acid was added and mixed by pipetting. Then, six times the volume of S-Trap buffer (90% aqueous MeOH, 100 mM TEAB, pH 7.1) was added and mixed by pipetting. Protein was loaded onto a S-Trap Micro column (Protifi) by centrifugation at 4,000g for 30 s. The column was washed three times with 150 µl S-Trap buffer. Protein was digested on the column with 4 µg trypsin in 50 mM TEAB at 47 °C for 1 h. Peptides were eluted sequentially by centrifugation (4,000g for 30 s) with 40 µl 50 mM TEAB, 40 µl 0.2% formic acid and 35 µl 50% acetonitrile and 0.2% formic acid.
Liquid chromatography–mass spectrometry
The eluted peptide solutions were dried down and the peptides dissolved in 0.1% trifluoroacetic acid and 3% acetonitrile for liquid chromatography with tandem mass spectrometry (LC–MS/MS). Sperm and vegetative nuclei samples were analysed by nanoLC–MS/MS on an Orbitrap Fusion Tribrid mass spectrometer coupled to an UltiMate 3000 RSLCnano LC system (Thermo Fisher Scientific). The samples were loaded and trapped using a pre-column with 0.1% trifluoroacetic acid at 20 µl min–1 for 3 min. The trap column was then switched in-line with the analytical column (nanoEase M/Z column, HSS C18 T3, 100 Å, 1.8 µm, Waters) for separation using the following long gradient of solvent A (water, 0.05% formic acid) and solvent B (80% acetonitrile, 0.05% formic acid) at a flow rate of 0.3 µl min–1: 0–3 min 3% B (trap only); 3–14 min linear increase of B to 13%; 14–113 min increase of B to 39%; 113–123 min increase of B to 55%; followed by a ramp to 99% B and re-equilibration to 3% B.
Data were acquired with the following mass spectrometer settings in positive ion mode. MS1/OT: resolution of 120 K, profile mode, mass range m/z of 300–1,800, automatic grain control of 4e5 and fill time of 50 ms. MS2/IT, data-dependent analysis was performed using higher-energy collision dissociation fragmentation with the following parameters: top30 in IT rapid, centroid mode, isolation window of 1.6 Da, charge states of 2–5, threshold of 1.9e4, collision energy of 30, automatic gain control target of 1.9e4, maximum inject time of 35 ms, dynamic exclusion of 1 count, 15 s of exclusion and exclusion mass window of ±5 ppm.
For sperm and vegetative nuclear proteomes, recalibrated peak lists were generated with MaxQuant (v.1.6.1.0)46 in label-free quantitation mode using the TAIR10_pep_20101214 Arabidopsis protein sequence database (TAIR, 35386 entries) plus the MaxQuant contaminants database (245 entries). The label-free quantitation results from MaxQuant with default parameters were used together with search results from an in-house Mascot Server 2.4.1 (Matrix Science) on the same databases. For all searches, a precursor tolerance of 6 ppm and a fragment tolerance of 0.6 Da were used. The enzyme was set to trypsin/P with a maximum of 2 allowed missed cleavages, oxidation (M) and acetylation (protein N-term) were set as variable modifications and carbamido-methylation (C) was set as a fixed modification. The search results were imported into Scaffold 4 (Proteome Software) using identification probabilities of 99% for proteins and 95% for peptides.
Histone alignments and disorder predictions
Alignments of histone DNA and protein sequences were performed using CLC Main Workbench software (v.8.1; Qiagen). Predictions of intrinsic disorder were undertaken using PONDR47 with the VL-XT algorithm. Raw data were plotted using ggplot2 in R (v.3.6.0)48,49.
Histone H2B phylogenetic analysis
Plant H2B protein sequences were downloaded from Phytozome50, Congenie51, Waterlily Pond52, Magnoliid genomes53,54 and UniProt55. Human and yeast H2B sequences were obtained from UniProt and used as out-groups for phylogenetics.
Sequences were imported to MEGA-X56 and aligned using MUSCLE with default parameters. The phylogeny was generated using neighbour-joining testing, applying the Poisson model and allowing for uniform substitution rates. H2B.8 homologues were identified owing to the distinct branch formed, separate from canonical H2B variants. Several representative H2B.8 homologues were searched using BLAST57 to query whether such homologues are specific to flowering plants. H2B.8 homologues are presented in Supplementary Table 1.
Confocal microscopy and analysis
Microspores and pollen were isolated as previously described45, stained with Hoechst 33342 and examined using a Leica SP8X confocal microscope. Young embryos were dissected58 and stained with propidium iodide for imaging. Mature embryos were isolated from dry seeds using a stereomicroscope. Mature embryos and seedlings (including roots) were stained in PBS with 0.1% Triton X-100 and 0.5 µg ml–1 DAPI for 5–10 min before microscopy examination (Zeiss 880, Airyscan mode). Immunofluorescence was performed with 2-week-old seedlings as previously described18.
Sperm nuclear size was quantified from DAPI-stained whole pollen confocal images using a semi-automated pipeline in ImageJ adapted from ref. 59. In brief, autothreshold was used to obtain nuclei and then processed using Gaussian blur to smooth edges. The autothreshold was repeated and then nuclei were selected using the wand tool. Measurements were then obtained for nuclear area (μm2). Somatic nuclei selected for analysis were vascular cylinder cells in the elongation zone of the root tip. Such nuclei were selected owing to the ability to accurately identify the cell type within the tissue. Using ImageJ, Z-stacks were divided into substacks of different cell layers within the root tip. Maximum intensity projections were then obtained to account for slight differences in the depth of nuclei. Images were analysed in the same semi-automated way as per sperm nuclei. Statistical analysis was undertaken in R; we used analysis of variance followed by Tukey’s post hoc test for pairwise comparisons.
Foci were identified in WT and h2b.8 sperm using the ImageJ plug-in FociPicker3D60.
For DAPI and H2B.8 colocalization (pH2B.8::H2B.8–eGFP and p35S::H2B.8–eGFP), nuclei were segmented using Otsu thresholding with 3D ImageJ Suite tools61. Fluorescence intensity measurements were extracted for DAPI and GFP channels within each segmented nucleus. Mean values were calculated for 20 × 20 voxel areas. Each volume value was normalized by dividing with the sum of DAPI or GFP intensity for the entire corresponding nucleus. Regions were grouped into eight quantiles by GFP value and plotted against DAPI intensity.
Classification of H3K9me2 foci was undertaken as previously described18. H2B.8-mediated chromatin aggregates were classified in the same way.
To quantify the size and number of H2B.8 aggregates and H3K9me2 domains, we used the 3D ImageJ Suite tools61. In each fluorescence channel, we undertook 3D Nuclei Segmentation using Otsu thresholding61. The number and volumes of segments were plotted in R using ggplot2.
3D-SIM and analysis
Sperm and vegetative nuclei were isolated from pollen (collected from about 1 ml of flowers) as previously described45 and resuspended in 200 μl Galbraith buffer (45 mM MgCl2, 30 mM sodium citrate, 20 mm MOPS and 0.1% Triton X-100, pH 7.0). For generative nuclei, bicellular pollen were manually dissected from flower buds and individually staged by DAPI staining and fluorescence microscopy. Nuclei were extracted from seedlings by finely chopping with a razor blade in lysis buffer (15 mM Tris-HCl pH 7.5, 2 mM EDTA, 0.5 mM spermine, 80 mM KCl, 20 mM NaCl and 0.1% Triton X-100). The suspension was filtered through a 35-μm filter (Corning) into a 1.7 ml tube. Nuclei were pelleted at 500g for 3 min and resuspended in 200 μl lysis buffer.
Nuclei were fixed in solution with 4% MeOH-free formaldehyde (Thermo Scientific) for 5 min. HiQA number 1.5H coverslips (CellPath) were washed with 10% HCl for 30 min and then washed three times in H2O for 5 min to remove impurities. Fixed nuclei were spun onto coverslips at 500g for 3 min using a Shandon Cytospin 2. Fixation was repeated by blotting nuclei with 4% MeOH-free formaldehyde for 5 min. Fixative was removed and coverslips were washed three times in PBS, 5 min per wash. Nuclei were blocked with 3% BSA in PBS with 0.1% Tween-20 (PBST) for 30 min in a humidified chamber. If performing immunostaining, antibodies were diluted 200-fold in 3% BSA in PBST and then blotted to nuclei on coverslips. Antibody incubation occurred overnight at 4 °C. Primary antibodies were removed by washing three times with PBST for 5 min. Secondary antibodies were diluted similarly to primary antibodies and then added to nuclei. Incubation occurred for 1 h at room temperature in a humidified chamber. If not performing immunostaining, the protocol resumed at this point. PBST washes were repeated as before. Nuclei were stained in the dark with either DAPI or SYBR Green (Invitrogen) at 2 mg μl–1 or 100× dilution, respectively for 5 min. DNA stain was removed by washing in H2O for 5 min. Coverslips were adhered to slides in 13 μl Vectashield H-1000 mounting medium. Nuclei were imaged using a ×63 oil-immersion lens on a Zeiss Elyra PS.1 super-resolution microscope.
3D reconstructions for SIM were undertaken using Zeiss Zen Black software. Intensity profiles associated with images were acquired using the Interactive 3D Surface Plot plugin for ImageJ62,63.
To acquire voxel intensities from WT and h2b.8 sperm, nuclei were segmented using Otsu thresholding and individual voxels were extracted. Fluorescence intensity was normalized by dividing by total nuclear intensity. The density of binned voxel intensities was plotted with ggplot2 in R.
Histone purification from Escherichia coli
Sequences for A. thaliana histone H2B.8, H2B.8ΔIDR, H2B.8-scrambledIDR, H2B.2, H2A, H3, H3 mutant K9CC110A and H4 were codon optimized for protein expression in E. coli and synthesized (Genewiz) into the pET30a+ vector with a non-cleavable C-terminal 8× His-tag then transformed into the E. coli strain BL21 and purified as previously described64. In brief, cells were grown to an optical density of 0.5–0.6 at 37 °C in LB medium with 30 μg ml–1 kanamycin. Histone expression was induced by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside and incubated for 4 h at 37 °C. Cells were collected by centrifugation at 4,000 r.p.m. for 30 min and resuspended in 1× PBS. Centrifugation was repeated and cells were resuspended in wash buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA and 5 mM 2-mercaptoethanol). Cells were lysed by ultrasonication and debris was pelleted at 18,000 r.p.m. for 20 min. The supernatant was discarded and the pellet was resuspended with wash buffer and 1% Triton X-100. The sample was sonicated and then washed a further two times with wash buffer. The pellet was then resuspended with unfolding buffer (20 mM Tris-HCl pH 7.5, 7 M guanidinium HCl and 5 mM 2-mercaptoethanol) and mixed at room temperature for 1.5 h to fully dissolve the pellet. The sample was centrifuged at 18,000 r.p.m. for 30 min. The supernatant was flash-frozen in liquid nitrogen and then stored at −80 °C.
Histone octamer and nucleosome reconstitution
DNA templates of 12× 177 bp of the Widom 601 sequence were cloned and purified as previously described65. The sequence for the 177 bp DNA sequence is listed as follows:
5’-GAGCATCCGGATCCCCTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCACATATATACATCCTGTTCCAGTGCCGGACCC-3’
Respective histone octamers were reconstituted as previously described64. In brief, equimolar amounts of four individual histones were added into unfolding buffer (20 mM Tris-HCl pH 7.5, 7 M guanidinium HCl and 5 mM B-ME) and were then dialysed into refolding buffer (2 M NaCl, 10 mM Tris-HCl pH 7.5, 1 mM EDTA and 5 mM 2-mercaptoethanol) before purification using Superdex 200 columns (Cytiva).
Nucleosomes were assembled using the salt-dialysis method as previously described64. Histone octamers and DNA templates were mixed in TEN buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA and 2 M NaCl) and dialysed for 16–18 h at 4 °C in TEN buffer, which was continuously diluted by slowly pumping in TE buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA) to lower the concentration of NaCl from 2 to 0.6 M. Samples were collected after final dialysis in HE buffer (10 mM HEPES pH 7.5, 0.1 mM EDTA) for 4 h. Nucleosomal array assemblies were assessed for quality by assembling under low concentration (2.5 μg DNA and the corresponding octamer in 50 μl reconstitution system), and the stoichiometry of histone octamers to DNA template was examined using an FEI Tecnai G2 Spirit 120 kV transmission electron microscope.
Alexa Fluor 594 H3 K9CC110A labelling and labelled nucleosome reconstitution
Fluorophore labelling of histone H3 K9CC110A was performed through the addition of 1.5 M excess Alexa Fluor 594 (AF594)-C5-maleimide (Invitrogen) to histone in unfolding buffer followed by incubation in the dark for 4 h at room temperature. Conjugation reactions were quenched through the addition of 10 mM DTT, and free fluorophore was removed by flowing AF594-labelled histone H3 through a Superdex 200 Increase 10/300 column (Cytiva) in unfolding buffer. The fluorescently tagged H3 was then combined with corresponding histones in unfolding buffer for histone octamer reconstitution as described above. Histone H3 mutant C110A does not affect nucleosome structure or positioning66, and H3 mutant K9CC110A is commonly used to install analogues of methyl lysine on cysteine with similar function to their natural counterparts67,68. Fluorescently labelled and unlabelled histone octamers were combined at a ratio of 1:100 before reconstitution to reduce the potential impact on samples during fluorescent labelling27.
In vitro chromatin condensate formation
In vitro phase-separation experiments were performed as previously described27. In brief, experiments were recorded on 384-well glass bottom plates (Cellvis) and sealed with optically clear adhesive film. Before use, 384-well plates were PEGylated with 5K mPEG-silane and passivated with BSA. Nucleosomal array samples were first equilibrated in chromatin dilution buffer (25 mM Tris⋅OAc, pH 7.5, 5 mM DTT, 0.1 mM EDTA and 0.1 mg ml–1 BSA, 5% (w/v) glycerol) and incubated for 5 min at room temperature. Nucleosomal array samples were then added to 1 volume of chromatin dilution buffer in the presence of salt (KOAc and/or Mg(OAc)2) and transferred to the PEGylated and BSA-passivated 384-well plates to incubate for 30 min at room temperature. Double-strand DNA within nucleosomal arrays were stained with YOYO-1 iodide (491/509) (Invitrogen) following the addition of chromatin dilution buffer.
In vitro FRAP and droplet fusion
In vitro FRAP and droplet fusion experiments were carried out after chromatin condensates were formed using the conditions detailed above, using a NIKON A1 confocal microscope equipped with a ×100 oil-immersion objective. Droplets were bleached with a 561-nm laser pulse (2 repeats, 20% intensity, dwell time 1 s). Post-bleach intensity was normalized to pre-bleach levels to obtain a measure of recovery. NIS-Elements AR Analysis software was used for image analysis.
Transient expression in tobacco
Vectors (p35S::H2B.8–eGFP, p35S::H2B.8-scrambledIDR–eGFP, p35S::H2B.8-EWSR1-IDR–eGFP and p35S::H2B.8-TAF15-IDR–eGFP) were transformed into Nicotiana benthamiana by Agrobacterium infection for transient expression. Two days after injection, the leaf epidermis was isolated and stained by DAPI solution containing 0.2% (v/v) Triton X-100. Images were obtained using a Leica SP8X confocal microscope.
Drought stress treatment
Ten-day-old WT and p35S::H2B.8–eGFP seedlings (two replicates for each treatment) were collected and incubated for 3 h in 50% (w/v) PEG6000 1× PBS for drought treatment or 1× PBS for controls.
Histone extraction for western blotting
Leaf tissue (0.5 g) was ground in liquid nitrogen and suspended in 4 ml histone extraction buffer (10 mM Tris-HCl pH 7.5, 2 mM EDTA, 0.25 M HCl, 5 mM DTT and 1× protease inhibitor cocktail (Roche)). The suspension was filtered through one layer of miracloth (Merck Millipore) and centrifuged at 4,000 r.p.m. for 10 min at 4 °C. The supernatant was transferred to a fresh tube and mixed with 1.75 ml trichloroacetic acid solution (T0699, Sigma). The precipitate was collected by centrifugation at 4,000 r.p.m. for 20 min at 4 °C and then washed twice with 1 ml acetone. The pellet was air dried before incubating with 1× LDS sample buffer (NP0007, Invitrogen) overnight. The sample was denatured at 95 °C for 10 min. For sperm, 5 million sperm nuclei were sorted by FACS (Supplementary Fig. 2a). The sperm nuclei were centrifuged at 1,200g for 5 min at 4 °C. The nuclei pellet was resuspended with 1 ml histone extraction buffer and then 0.25 ml trichloroacetic acid solution was added and mixed. Histones were precipitated and eluted in the same way as for leaf. Anti-H3K9me2 antibody (Abcam, ab1220) was diluted 1:5,000 and used for western blotting.
Pollen and seedling native ChIP-seq library preparation, sequencing and analysis
About 0.5 g of 10-day-old seedlings (two replicates for each genotype) were ground with a pestle and mortar in liquid nitrogen and homogenized in nuclei isolation buffer (0.25 M sucrose, 15 mM PIPES pH 6.8, 5 mM MgCl2, 60 mM KCl, 15 mM NaCl, 1 mM CaCl2, 0.9% Triton X-100, 1 mM PMSF and 1× protease inhibitor cocktail (Roche)) for 15 min. Nuclei were separated from debris by filtering through two layers of miracloth (Merck Millipore). For pollen nuclei, we collected approximately 20 ml of open flowers and isolated pollen in Galbraith buffer (two replicates were isolated for each genotype). Nuclei were released by vertexing pollen with glass beads in 200 µl of nuclei isolation buffer at 2,000 r.p.m. for 3 min. The homogenate was filtered through 40-µm and 10-µm cell strainers successively to obtain nuclei. To maximize nuclei recovery, unbroken pollen grains that remained on the filters were recycled to the glass beads and vortexed with nuclei isolation buffer before proceeding to filter steps again.
Nuclei suspension from seedlings or pollen was centrifuged at 4,000g for 10 min and pellets were resuspended in TM2 (50 mM Tris-HCl, 2 mM MgCl2, 0.25 M sucrose, 1 mM PMSF and 1× protease inhibitor cocktail). After cold centrifugation at 4,000g for 5 min, nuclei were resuspended in MNase digestion buffer (50 mM Tris-HCl pH 7.5, 5 mM CaCl2, 0.25 M sucrose, 1 mM PMSF and 1× protease inhibitor cocktail) with an appropriate amount of MNase (New England Biolabs) and incubated at 37 °C for 10 min. Digestion was stopped by adding EDTA to a final concentration of 25 mM. One-tenth volume of 1% Triton X-100 and 1% sodium deoxycholate was added and the sample was left on ice for 15 min. Then, the reaction was diluted by adding low-salt buffer (50 mM Tris-HCl pH 7.5, 10 mM EDTA, 150 mM NaCl, 0.1% Triton X-100, 1 mM PMSF and 1× protease inhibitor cocktail) and rotated for 1 h at 4 °C. After centrifugation, the supernatant was used for immunoprecipitation with pre-washed GFP-Trap beads (Chromotek) overnight at 4 °C. Beads were washed twice each with low-salt buffer and high-salt buffer (50 mM Tris-HCl pH 7.5, 10 mM EDTA, 300 mM NaCl, 0.1% Triton X-100 and 1 mM PMSF) and eluted in elution buffer (0.1 M NaHCO3 and 1% SDS) by shaking at 65 °C for 15 min. The eluates were digested with proteinase K and RNase A before phenol–chloroform DNA extraction. Libraries were prepared using Ovation Ultralow System V2 and sequenced on NextSeq 500 (Illumina) with 2× 38 bp paired-end reads.
Sequencing reads were mapped to TAIR10 with Bowtie 2 (v.2.3.4.1)69, retaining mononucleosomal fragments. Duplicated reads were removed using Samtools-1.7 rmdup. Bigwig files were generated by normalizing IP bam files to respective inputs using deepTools (v.3.1.1)70. Two replicates for each experiment were confirmed to be highly correlated; a single replicate was used for downstream analyses. Profiles were visualized using IGV (v.2.6.2)71.
Data underlying metaplots and heatmaps were generated with deepTools and plotted with a custom script in R.
TE classes were defined by seedling H3K9me2 enrichment (log2(Immunoprecipitation/input)). Considering the bimodal distribution of H3K9me2 enrichment at TEs, 0.7 was selected as the cut-off for heterochromatic (>0.7) and euchromatic (<0.7) classes.
To generate peaks, H2B.8 or H2B.8ΔIDR enrichment was calculated over 50-bp windows and those with >1.2 log2(IP/input) were retained. Windows within 150 bp were merged using BEDtools (v.2.28.0)72. Regions were filtered by size, with those <200 bp removed from analysis. H2B.8 enrichment was then calculated over the new regions, and those with <1.2 log2(IP/input) were discarded. The remaining regions were defined as H2B.8 peaks.
For genome coverage, peaks were divided into 50-bp windows and partitioned into gene, TE or intergenic groups depending on overlaps. Overlaps with genes and TEs for volcano plots were determined using BEDtools; 25% of the feature was required to be covered by a peak to be defined as an overlap.
Downloaded data73–80 were mapped and processed in the same way. PCA, and Spearman and Pearson correlations were calculated with deepTools using categorial genomic regions and plotted in R with ggplot2.
Bisulfite-seq analysis
Downloaded sequencing reads81 were processed using TrimGalore (v.0.4.1) (https://github.com/FelixKrueger/TrimGalore) with default parameters. Reads were mapped to TAIR10 using Bismark (v.0.22.2)82, and methylation was called using MethylDackel (v.0.5.2) (https://github.com/dpryan79/MethylDackel), selecting --CHG and --CHH options. CG methylation data were used in PCA.
Sperm cell and seedling RNA-seq library preparation, sequencing and analysis
Sperm cells were isolated by FACS as previously described83, with two replicates isolated for each genotype (Supplementary Fig. 2b). RNA was extracted from 1 million sperm cells (isolated from approximately 100 ml of open flowers) per replicate using a Direct-zol RNA Microprep kit. A Plant RNeasy Mini kit was used to extract RNA from 10-day-old seedlings (three or two replicates of seedlings were used for each genotype in normal or drought/mock treatment conditions, respectively). Libraries were prepared using a Universal RNA-Seq library preparation kit and sequenced on NextSeq 500 (Illumina) with single end (76 bp) or paired end (2× 38 bp) reads.
Sequencing reads were mapped to TAIR10 with TopHat (v.2.0.10)84. Kallisto (v.0.43.0)85 and Sleuth-(v.0.30.0)86 were used to obtain transcript per million (TPM) values and q values, respectively. Differentially expressed genes and TEs were identified by | log2(TPM fold-change) | ≥ 2 (≥1 was used for calling drought-responsive genes) and q < 0.05. Volcano plots were generated with a custom ggplot2 R script. Heatmaps were generated using ggplot2 in R, with the transcriptional changes normalized using the som package.
Downloaded data87–89 were mapped, and TPM values were obtained in the same way.
Multivariate linear regression modelling
The genome was divided into 200-bp windows for calculating the abundance of each variable. For ChIP-seq data, log2(IP/input) was calculated for each window using multiBigwigSummary in deeptools. For bisulfite-seq data, CG methylation was calculated using the number of sequenced Cs in the CG context within the window divided by the number of (C+T) in the CG context (windows without CG sites or aligned sequencing reads were removed). for RNA-seq data, log10(reads per kilobase of transcript per million mapped reads (RPKM)) was calculated for each window.
Multivariate linear regression models were generated in R using the lm function. For each variable, a linear model was fitted to predict H2B.8 enrichment and the adjusted R2 values are shown in Supplementary Table 2. Variables were gradually added to the model, and the predictive power of the adjusted model was examined by calculating adjusted R2 and Akaike information criterion scores in R (Extended Data Fig. 4f). A separate model was built using categorial variables of genomic regions to assess the association between H2B.8 and genomic features (adjusted R2 of the model is shown in Supplementary Table 2).
Hi-C library preparation, sequencing and analysis
Two replicates of 10-day-old seedlings (1–3 g) were collected and fixed with 20 ml 2% formaldehyde solution for 15 min in vacuum conditions at room temperature and then quenched by adding 2.162 ml of 2.5 M glycine. Fixed seedling tissue was rinsed with water three times and dried with tissue paper.
The nuclei were released by grinding in liquid nitrogen and then resuspended with 25 ml of extraction buffer I (0.4 M sucrose, 10 mM Tris-HCl pH 8, 10 mM MgCl2, 5 mM β-mercaptoethanol, 0.1 mM PMSF and 13 μl protease inhibitor). Nuclei were filtered through a miracloth (Calbiochem) and then centrifuged at 4,000 r.p.m. for 20 min at 4 °C. The supernatant was discarded and the pellet was resuspended with 1 ml of extraction buffer II (0.25 M sucrose, 10 mM Tris-HCl pH 8, 10 mM MgCl2, 1% Triton X-100, 5 mM β-mercaptoethanol, 0.1 mM PMSF and 13 μl protease inhibitor). Then the mixture was centrifuged at 14,000 r.p.m. for 10 min at 4 °C and the pellet was resuspended with 300 μl of extraction buffer III (1.7 M sucrose, 10 mM Tris-HCl pH 8, 0.15% Triton X-100, 2 mM MgCl2, 5 mM β-mercaptoethanol, 0.1 mM PMSF and 1 μl protease inhibitor). The mixture was loaded onto an equal amount of clean extraction buffer III and centrifuged at 14,000 r.p.m. for 10 min. The pelleted nuclei were washed twice with 1× ice cold CutSmart buffer and finally resuspended in 0.5 ml volume. SDS was applied to permeabilize nuclei at 65 °C for 10 min, Triton X-100 was added to quench SDS. Thereafter, chromatin was digested with 400 units of MboI overnight at 37 °C with gentle rocking. MboI was then denatured to cease activity.
Digested chromatin underwent DNA end-repair with biotin-14-dCTP insertion followed by blunt-end ligation. After decrosslinking with proteinase K at 65 °C, DNA was purified using a phenol–chloroform extraction method. Biotin-14-dCTP was removed from non-ligated DNA fragment ends using T4 DNA polymerase. DNA was sheared to a range of 200–600 bp by sonication. Next, the fragments underwent end-repair and were pulled down by streptavidin C1 magnetic beads to enrich for fragments containing contact information. Fragment ends were then A-tailed, sequencing adapters were ligated and libraries were amplified by PCR for 12–14 cycles. Following purification, libraries were sequenced using an Illumina HiSeq X Ten platform with 2× 150 bp length reads. The Hi-C library construction and sequencing were conducted by Annoroad Gene Technology.
Sequencing reads were mapped to the TAIR10 reference genome using the HiC-Pro (v.2.11.1) pipeline90. The bam files (bwt2merged.bam) generated by HiC-Pro containing mapped reads were used as input files for FAN-C (v.0.9.8)91. The module ‘fanc auto’ was applied to generate 500, 100, 50, 10 and 1 kb contact matrices (hic files). The resultant hic files with 100-kb resolution were directed to the ‘fanc expected’ module to calculate the expected interaction probability against genomic distance for intrachromosomal interaction. For matrix and score comparisons, the default comparison method of fold-change was used with the ‘fanc compare’ command. The outputs (hic object) were transferred to text files by ‘fanc dump’ and were visualized as heatmaps in R using ggplot2. To explore whether higher contacts observed in p35S::H2B.8–eGFP depend on H2B.8 incorporation, the genome was binned into 1-kb windows. H2B.8 signals (log2(IP/input)) of each window were generated and sorted into 20 quantiles by strength. The interaction frequency differences (values generated by FAN-C at 1-kb resolution) of each quantile pair for either short-range interactions or interactions between pericentromeric regions and chromosome arms were averaged and plotted as heatmaps. The resolution of our Hi-C data was estimated as previously reported92. Our Hi-C data was deemed to achieve 1-kb resolution as 80% of genomic bins (1 kb) had >1,000 contacts. Our WT data were compared to published contact matrices36,37.
Male transmission assay
Heterozygous h2b.8-1 mutant and WT plants were crossed in two separate experiments by two different researchers. The inheritance of mutant versus WT alleles was determined by dCAPS genotyping. In one experiment, WT plants were used as females and crossed with heterozygous h2b.8-1 mutant plants as males; a total of 743 F1 progeny were genotyped, and the result is listed in Supplementary Table 3. In a second experiment, reciprocal crosses between heterozygous h2b.8-1 mutant and WT plants were performed; a total of 574 and 575 F1 progeny were genotyped (Supplementary Table 3). Statistical significance was tested using Fisher’s exact test in R.
In vitro pollen germination
In vitro pollen germination of WT and h2b.8 was undertaken as previously described18.
Statistics and reproducibility
Statistical tests performed on experimental data and sample sizes are noted in figure legends. All data points are derived from biological replicates. Boxplots show median (thick black bar) and first and third quartiles, with lower and upper whiskers extending to 1.5-times the interquartile range of the first and third quartiles or the highest and lowest values, respectively. Exact P values for each pairwise comparison in all figures are listed in Supplementary Table 7. Micrographs throughout are representative of at least three independent experiments.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-022-05386-6.
Supplementary information
This file contains Supplementary Figs. 1 and 2 and Supplementary Tables 2–7 (see separate file for Supplementary Table 1).
List of H2B.8 homologues identified in flowering plant species.
Acknowledgements
We thank C. Dean, S. Penfield and D. Zilberman for critical reading of the manuscript; S. Lopez and E. Wegel at the John Innes Centre Bioimaging Facility for assistance with microscopy; staff at the Norwich BioScience Institute Partnership Computing infrastructure for Science Group for High Performance Computing resources; and staff at Annoroad Gene Technology (Beijing, China) for Hi-C library preparation and sequencing. This work was funded by a UKRI-BBSRC Doctoral Training Partnerships studentship (BBM0112161 to T.B.), a Marie Skłodowska-Curie Action grant (101033109 to S.Z.), Biotechnology and Biological Sciences Research Council grants (BBS0096201 and BBP0135111 to S.H., M.V. and X.F.), a Centre of Excellence for Plant and Microbial Sciences grant (to S.H. and X.F.), grants from the National Key Research and Development Program (2019YFA0508403 to P.L.; 2017YFA0504202 to G.L.), grants from the National Natural Science Foundation of China (32100417 to L.W.; 31991161 to G.L.; 32150023, 32125010, 31871443 to P.L.), a Beijing Municipal Science and Technology Commission grant (Z201100005320013 to G.L.), a European Research Council Starting Grant (‘SexMeth’ 804981 to S.Z. and X.F.), and a EMBO Young Investigator Award (to X.F.).
Extended data figures and tables
Author contributions
T.B., S.H. and X.F. conceived the study. T.B., S.H., L.W., S.Z., P.L. and X.F. designed the experiments. L.W. performed in vitro phase separation, FRAP and droplet fusion assays, and G.L. and P.L. supervised these experiments. T.B. (proteomics, 3D-SIM, transgenics, ChIP-seq, RNA-seq and plant phenotyping), S.H. (confocal, transgenics and ChIP-seq) and S.Z. (confocal, stress treatment, ChIP-seq, RNA-seq, western blotting, Hi-C analysis and plant phenotyping) performed all other experiments and analyses (which were supervised by X.F.). G.S. and M.V. assisted T.B. with mass spectrometry and genomic analysis, respectively. T.B., S.H. and X.F. wrote the manuscript, and all authors commented on the manuscript.
Peer review
Peer review information
Nature thanks Steven Jacobsen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Sequencing data generated in this study (ChIP-seq, RNA-seq and Hi-C) have been deposited in the Gene Expression Omnibus under accession number GSE161366. All remaining data are in the main paper or the supplementary materials.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Toby Buttress, Shengbo He, Liang Wang, Shaoli Zhou
Contributor Information
Pilong Li, Email: pilongli@mail.tsinghua.edu.cn.
Xiaoqi Feng, Email: xiaoqi.feng@jic.ac.uk.
Extended data
is available for this paper at 10.1038/s41586-022-05386-6.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-022-05386-6.
References
- 1.Steger K, Balhorn R. Sperm nuclear protamines: a checkpoint to control sperm chromatin quality. Anat. Histol. Embryol. 2018;47:273–279. doi: 10.1111/ahe.12361. [DOI] [PubMed] [Google Scholar]
- 2.Rathke C, Baarends WM, Awe S, Renkawitz-Pohl R. Chromatin dynamics during spermiogenesis. Biochim. Biophys. Acta. 2014;1839:155–168. doi: 10.1016/j.bbagrm.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Twell D. Male gametogenesis and germline specification in flowering plants. Sex Plant Reprod. 2011;24:149–160. doi: 10.1007/s00497-010-0157-5. [DOI] [PubMed] [Google Scholar]
- 4.Russell SD, Jones DS. The male germline of angiosperms: repertoire of an inconspicuous but important cell lineage. Front. Plant Sci. 2015;6:173. doi: 10.3389/fpls.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammoud SS, et al. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–478. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gouraud A, et al. “Breaking news” from spermatids. Basic Clin. Androl. 2013;23:11. doi: 10.1186/2051-4190-23-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcon L, Boissonneault G. Transient DNA strand breaks during mouse and human spermiogenesis new insights in stage specificity and link to chromatin remodeling. Biol. Reprod. 2004;70:910–918. doi: 10.1095/biolreprod.103.022541. [DOI] [PubMed] [Google Scholar]
- 8.Miller D, Brinkworth M, Iles D. Paternal DNA packaging in spermatozoa: more than the sum of its parts? DNA, histones, protamines and epigenetics. Reproduction. 2010;139:287–301. doi: 10.1530/REP-09-0281. [DOI] [PubMed] [Google Scholar]
- 9.Ausio J, Eirin-Lopez JM, Frehlick LJ. Evolution of vertebrate chromosomal sperm proteins: implications for fertility and sperm competition. Soc. Reprod. Fertil. Suppl. 2007;65:63–79. [PubMed] [Google Scholar]
- 10.Hackenberg D, Twell D. The evolution and patterning of male gametophyte development. Curr. Top. Dev. Biol. 2019;131:257–298. doi: 10.1016/bs.ctdb.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Southworth D, Cresti M. Comparison of flagellated and nonflagellated sperm in plants. Am. J. Bot. 1997;84:1301–1311. [PubMed] [Google Scholar]
- 12.Reynolds WF, Wolfe SL. Changes in basic proteins during sperm maturation in a plant, Marchantia polymorpha. Exp. Cell Res. 1978;116:269–273. doi: 10.1016/0014-4827(78)90448-2. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds WF, Wolfe SL. Protamines in plant sperm. Exp. Cell Res. 1984;152:443–448. doi: 10.1016/0014-4827(84)90645-1. [DOI] [PubMed] [Google Scholar]
- 14.Renzaglia KS, Garbary DJ. Motile gametes of land plants: diversity, development, and evolution. Crit. Rev. Plant Sci. 2010;20:107–213. [Google Scholar]
- 15.Johnson MA, Harper JF, Palanivelu R. A fruitful journey: pollen tube navigation from germination to fertilization. Annu. Rev. Plant Biol. 2019;70:809–837. doi: 10.1146/annurev-arplant-050718-100133. [DOI] [PubMed] [Google Scholar]
- 16.Schoft VK, et al. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 2009;10:1015–1021. doi: 10.1038/embor.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merai Z, et al. The AAA-ATPase molecular chaperone Cdc48/p97 disassembles sumoylated centromeres, decondenses heterochromatin, and activates ribosomal RNA genes. Proc. Natl Acad. Sci. USA. 2014;111:16166–16171. doi: 10.1073/pnas.1418564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He S, Vickers M, Zhang J, Feng X. Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. eLife. 2019;8:e42530. doi: 10.7554/eLife.42530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borg M, Berger F. Chromatin remodelling during male gametophyte development. Plant J. 2015;83:177–188. doi: 10.1111/tpj.12856. [DOI] [PubMed] [Google Scholar]
- 20.Baroux C, Raissig MT, Grossniklaus U. Epigenetic regulation and reprogramming during gamete formation in plants. Curr. Opin. Genet. Dev. 2011;21:124–133. doi: 10.1016/j.gde.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Feng W, Michaels SD. Accessing the inaccessible: the organization, transcription, replication, and repair of heterochromatin in plants. Annu. Rev. Genet. 2015;49:439–459. doi: 10.1146/annurev-genet-112414-055048. [DOI] [PubMed] [Google Scholar]
- 22.Jiang D, et al. The evolution and functional divergence of the histone H2B family in plants. PLoS Genet. 2020;16:e1008964. doi: 10.1371/journal.pgen.1008964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uversky VN. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017;44:18–30. doi: 10.1016/j.sbi.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 25.Larson AG, et al. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017;547:236–240. doi: 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strom AR, et al. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241–245. doi: 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson BA, et al. Organization of chromatin by intrinsic and regulated phase separation. Cell. 2019;179:470–484. doi: 10.1016/j.cell.2019.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strickfaden H, et al. Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cells. Cell. 2020;183:1772–1784. doi: 10.1016/j.cell.2020.11.027. [DOI] [PubMed] [Google Scholar]
- 29.Borcherds W, Bremer A, Borgia MB, Mittag T. How do intrinsically disordered protein regions encode a driving force for liquid–liquid phase separation? Curr. Opin. Struct. Biol. 2021;67:41–50. doi: 10.1016/j.sbi.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuchs G, Oren M. Writing and reading H2B monoubiquitylation. Biochim. Biophys. Acta. 2014;1839:694–701. doi: 10.1016/j.bbagrm.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell. 2018;174:688–699. doi: 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sequeira-Mendes J, et al. The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell. 2014;26:2351–2366. doi: 10.1105/tpc.114.124578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhaskara GB, Nguyen TT, Verslues PE. Unique drought resistance functions of the highly ABA-induced clade A protein phosphatase 2Cs. Plant Physiol. 2012;160:379–395. doi: 10.1104/pp.112.202408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, et al. Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol. Cell. 2019;76:646–659. doi: 10.1016/j.molcel.2019.08.019. [DOI] [PubMed] [Google Scholar]
- 35.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun L, et al. Heat stress-induced transposon activation correlates with 3D chromatin organization rearrangement in Arabidopsis. Nat. Commun. 2020;11:1886. doi: 10.1038/s41467-020-15809-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grob S, Schmid MW, Grossniklaus U. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol. Cell. 2014;55:678–693. doi: 10.1016/j.molcel.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Moissiard G, et al. MORC family ATPases required for heterochromatin condensation and gene silencing. Science. 2012;336:1448–1451. doi: 10.1126/science.1221472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng SH, et al. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol. Cell. 2014;55:694–707. doi: 10.1016/j.molcel.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang CM, et al. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res. 2015;25:246–256. doi: 10.1101/gr.170332.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grewal SI, Jia S. Heterochromatin revisited. Nat. Rev. Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 42.Castel B, Tomlinson L, Locci F, Yang Y, Jones JDG. Optimization of T-DNA architecture for Cas9-mediated mutagenesis in Arabidopsis. PLoS ONE. 2019;14:e0204778. doi: 10.1371/journal.pone.0204778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Labun K, et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:171–174. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 45.Borges F, et al. FACS-based purification of Arabidopsis microspores, sperm cells and vegetative nuclei. Plant Methods. 2012;8:44. doi: 10.1186/1746-4811-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 47.Romero P, et al. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 48.The R Development Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2013).
- 49.Wickham H, et al. Welcome to the Tidyverse. J. Open Source Softw. 2019;4:1686. [Google Scholar]
- 50.Goodstein DM, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:1178–1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sundell D, et al. The plant genome integrative explorer resource: PlantGenIE.org. New Phytol. 2015;208:1149–1156. doi: 10.1111/nph.13557. [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, et al. The water lily genome and the early evolution of flowering plants. Nature. 2020;577:79–84. doi: 10.1038/s41586-019-1852-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, et al. Liriodendron genome sheds light on angiosperm phylogeny and species-pair differentiation. Nat. Plants. 2019;5:18–25. doi: 10.1038/s41477-018-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaw SM, et al. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants. 2019;5:63–73. doi: 10.1038/s41477-018-0337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:506–515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson M, et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:5–9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pillot M, et al. Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell. 2010;22:307–320. doi: 10.1105/tpc.109.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalyanikrishna, Mikulski P, Schubert D. Measurement of Arabidopsis thaliana nuclear size and shape. Methods Mol. Biol. 2020;2093:107–113. doi: 10.1007/978-1-0716-0179-2_8. [DOI] [PubMed] [Google Scholar]
- 60.Du G, et al. Spatial dynamics of DNA damage response protein foci along the ion trajectory of high-LET particles. Radiat. Res. 2011;176:706–715. doi: 10.1667/rr2592.1. [DOI] [PubMed] [Google Scholar]
- 61.Ollion J, Cochennec J, Loll F, Escude C, Boudier T. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics. 2013;29:1840–1841. doi: 10.1093/bioinformatics/btt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen P, et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013;27:2109–2124. doi: 10.1101/gad.222174.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 66.Shimko JC, Howard CJ, Poirier MG, Ottesen JJ. Preparing semisynthetic and fully synthetic histones h3 and h4 to modify the nucleosome core. Methods Mol. Biol. 2013;981:177–192. doi: 10.1007/978-1-62703-305-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanulli S, et al. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature. 2019;575:390–394. doi: 10.1038/s41586-019-1669-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramirez F, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:160–165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stroud H, et al. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma Z, et al. Arabidopsis serrate coordinates histone methyltransferases ATXR5/6 and RNA processing factor RDR6 to regulate transposon expression. Dev. Cell. 2018;45:769–784. doi: 10.1016/j.devcel.2018.05.023. [DOI] [PubMed] [Google Scholar]
- 75.Li C, et al. Concerted genomic targeting of H3K27 demethylase REF6 and chromatin-remodeling ATPase BRM in Arabidopsis. Nat. Genet. 2016;48:687–693. doi: 10.1038/ng.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen C, et al. Cytosolic acetyl-CoA promotes histone acetylation predominantly at H3K27 in Arabidopsis. Nat. Plants. 2017;3:814–824. doi: 10.1038/s41477-017-0023-7. [DOI] [PubMed] [Google Scholar]
- 77.Nassrallah A, et al. DET1-mediated degradation of a SAGA-like deubiquitination module controls H2Bub homeostasis. eLife. 2018;7:e37892. doi: 10.7554/eLife.37892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiorucci AS, et al. Arabidopsis S2Lb links AtCOMPASS-like and SDG2 activity in H3K4me3 independently from histone H2B monoubiquitination. Genome Biol. 2019;20:100. doi: 10.1186/s13059-019-1705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stroud H, et al. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA. 2012;109:5370–5375. doi: 10.1073/pnas.1203145109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choi J, Lyons DB, Kim MY, Moore JD, Zilberman D. DNA methylation and histone H1 jointly repress transposable elements and aberrant intragenic transcripts. Mol. Cell. 2020;77:310–323. doi: 10.1016/j.molcel.2019.10.011. [DOI] [PubMed] [Google Scholar]
- 81.Zemach A, et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Santos MR, Bispo C, Becker JD. Isolation of Arabidopsis pollen, sperm cells, and vegetative nuclei by fluorescence-activated cell sorting (FACS) Methods Mol. Biol. 2017;1669:193–210. doi: 10.1007/978-1-4939-7286-9_16. [DOI] [PubMed] [Google Scholar]
- 84.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2014;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 86.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017;14:687–690. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 87.Narsai R, et al. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017;18:172. doi: 10.1186/s13059-017-1302-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walker J, et al. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018;50:130–137. doi: 10.1038/s41588-017-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tannenbaum M, et al. Regulatory chromatin landscape in Arabidopsis thaliana roots uncovered by coupling INTACT and ATAC-seq. Plant Methods. 2018;14:113. doi: 10.1186/s13007-018-0381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kruse K, Hug CB, Vaquerizas JM. FAN-C: a feature-rich framework for the analysis and visualisation of chromosome conformation capture data. Genome Biol. 2020;21:303. doi: 10.1186/s13059-020-02215-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Supplementary Figs. 1 and 2 and Supplementary Tables 2–7 (see separate file for Supplementary Table 1).
List of H2B.8 homologues identified in flowering plant species.
Data Availability Statement
Sequencing data generated in this study (ChIP-seq, RNA-seq and Hi-C) have been deposited in the Gene Expression Omnibus under accession number GSE161366. All remaining data are in the main paper or the supplementary materials.