


Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive muscle wasting disease caused by the absence of a muscle cytoskeletal protein, dystrophin. Utrophin is the autosomal homologue of dystrophin. We previously demonstrated that overexpression of utrophin in the muscles of dystrophin-null transgenic mice completely prevented the phenotype arising from dystrophin deficiency. Two independently regulated promoters control utrophin expression and the upstream promoter (promoter A) is synaptically regulated in muscle. In this study, we have investigated basal regulation and myogenic induction of promoter A. Interactions between Ap2 and Sp1 and their cognate DNA motifs are critical for basal transcription from the minimal promoter region. During differentiation of C2C12 myoblasts in vitro, a 2-fold increase in A-utrophin mRNA level was observed. Expression of a reporter gene, whose transcription was driven by a 1.3 kb promoter A fragment, paralleled expression of the endogenous transcript. Myogenic induction mapped to a conserved upstream muscle-specific E-box, which was shown to bind myogenic regulatory factors, transactivating the promoter up to 18-fold in transient assays. This study provides a basis for further understanding the regulatory mechanisms that control utrophin expression in muscle and may facilitate the development of reagents to effect therapeutic up-regulation of utrophin in DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked muscle wasting disease caused by the absence of a cytoskeletal protein, dystrophin, from the cytoplasmic surface of the sarcolemma (1). Treatment is palliative; patients die from cardiac or respiratory complications in their late teens or early twenties. Utrophin is the autosomal homologue of dystrophin and binds similar complexes of proteins in muscle (2). In adult muscle, utrophin is localised at the neuromuscular junction, where it plays an important role in the structure of the post-synaptic cytoskeleton (2). The structural similarity between the two proteins led us to speculate that utrophin may be able to replace dystrophin in DMD (3,4). We subsequently showed that enhanced expression of utrophin in skeletal muscle of dystrophin-deficient mdx mice compensated for the absence of dystrophin and completely prevented muscle pathology (4,5). One strategy for therapeutic intervention in DMD involves up-regulation of utrophin. This approach has the inherent attraction that difficulties surrounding gene delivery to muscle are circumvented. It is therefore of great importance to determine the mechanisms regulating utrophin expression in muscle.
The level and localisation of utrophin in skeletal muscle change dramatically during development and in response to disease. During early myogenic differentiation, utrophin levels increase (6,7). In embryonic muscle tissue, utrophin localises to the sarcolemma along the entire length of developing fibres (8,9). Following the establishment of synaptic contacts, utrophin levels gradually decline and the protein becomes preferentially localised to the neuromuscular and myotendinous junctions (9–11). In DMD patients, mdx mice and certain inflammatory myopathies, however, utrophin persists at the sarcolemma in extrasynaptic regions (12–14).
Two independently regulated promoters, A (15) and B (16), control expression of the utrophin transcript. Each promoter gives rise to a transcript with unique 5′ exons that splice into a common utrophin mRNA at exon 3. Promoter A lies within a CpG island at the 5′-end of the gene; promoter B lies within the large second exon of utrophin. Promoter A is similar in structure to the acetylcholinesterase gene, which is CG-rich, regulated during muscle cell differentiation and localised specifically at the neuromuscular junction (17,18). Synaptic expression of utrophin in adult muscle is partially attributable to enhanced transcription in sub-synaptic nuclei, with consequent synaptic accumulation of mRNA. A reporter gene driven by a 1.3 kb fragment of promoter A is preferentially expressed at post-synaptic nuclei in adult muscle. In common with other synaptically expressed muscle genes, this is attributable to interaction of GABP, an ets-related factor, with an upstream N-box motif (TTCCGG) activating transcription of promoter A at sub-junctional nuclei (19). Interaction of Sp1 and Sp3 with both GABP and the core promoter A sequence enhances this process (20).
In contrast to synaptic regulation, less information is available on the induction of utrophin expression observed during muscle differentiation (7) or on basal mechanisms of transcription. In this paper we show that basal transcription from utrophin promoter A is dependent on Ap2 and Sp1. Furthermore, we demonstrate that the upstream promoter (promoter A) drives myogenic induction of utrophin expression and that this effect is caused by interaction of muscle-specific regulatory factors with an evolutionarily conserved consensus E-box upstream from the minimal element.
MATERIALS AND METHODS
Bioinformatics
Sequence data were assembled and analysed using the GCG Wisconsin package. Transcription factor databases were accessed using the TESS (http://www.cbil.up enn. edu/tess) or Signalscan (http://www.bimas.dcrt.nih.gov/molbio/signal) web sites.
Electrophoretic mobility shift (EMSA) and antibody supershifting assays
Synthetic oligonucleotides (Genosys) representing consensus transcription factor binding sites and putative binding regions were diluted to 1 µg/µl. Complementary primers were annealed in 536 mM Tris–HCl, pH 7.5, 104 mM MgCl2, 10.4 mM EDTA and 53.6 mM DTT by heating for 5 min at 85°C and cooling to room temperature overnight. For the 300 bp region containing the E-box, the utrophin promoter A sequence in pBluescript K/S (Stratagene) was XhoI and PstI digested and labelled with 50 mCi [α-32P]dCTP (Amersham). EMSA analysis was performed using a Gel Shift Assay System (Promega) with human recombinant Ap2, Sp1 (2 µl) (Promega), MyoD or myogenin (2–5 µg) (Santa Cruz Biotechnology) using the instructions provided. Nuclear extracts from HeLa (15–30 µg) and C2C12 myoblast-like cells (15 µg) were supplied by Promega and Geneka Biotechnology, respectively. Supershifting assays with ap2/sp1 antibodies (1.2 µg) (ap2, AP-2α/C-18; sp1, Sp1/PEP2; Santa Cruz Biotechnology) were performed as previously described (21). Following electrophoresis (250 V for 90 min), gels were dried and exposed to film for 0.5–24 h at room temperature.
DNase I protection analysis
A 298 bp PstI–EagI:SacII fragment containing the core promoter region was excised from pBluescript (Stratagene) by restriction digestion, dephosphorylation and agarose gel extraction. The probe was end-labelled using 10 mCi [α-32P]ATP, digested with Sau3AI to obtain a 276 bp double-stranded DNA probe that was labelled at the 3′-end, extracted with phenol:chloroform:isoamyl alcohol (25:24:1 v/v) and precipitated using 500 µl of 100% ethanol. The pellet was washed with 1 ml of 70% ethanol, dried and resuspended in 50 µl of 10 mM Tris–HCl, 1 mM EDTA, pH 8.0. Footprinting reactions were performed with a DNase I core footprinting system, recombinant human protein and nuclear extract (Promega) using the instructions provided. Probes were digested with 2 µl of a 2.5 ng/ml (or 16 ng/ml for HeLa nuclear extracts) DNase I stock solution for 90 s. A 5000 c.p.m. sample was loaded on a 6% polyacrylamide sequencing gel and run for ∼2 h. Gels were dried, exposed at –70°C and visualised by autoradiography.
Construction of utrophin–luciferase vectors
Mutant sequences were introduced into the cloning vector pBluescript KS (+/–) (Stratagene) containing either the 1.3 kb (a HindIII fragment spanning positions 1–1246, previously described as HH.F; 15) or core (0.3 kb) promoter fragment (a PstI–EagI fragment spanning positions 659–944, outlined in Fig. 1) using PCR mutagenesis. Consensus sequences and mutations that abolish binding of transcription factors were ap2 (gcccgcgg→gcttcggg), sp1 (ggggcggggc→ggttcggggc) and myoD (caggtg→cacgtg) (22) and were used in mutant oligonucleotide design for utrophin sequences (changes indicated in bold) ap2-747 (ccgggg→ccattgg), sp1-755 (ggggcgg→ggttcgg) ap2-850 (cggggaggagg→cgttgaggagg) and the E-box (caggtg→cacgtg). Introduction of mutations was performed using PCR-mediated mutagenesis: conditions were 95°C for 30 s followed by 12 cycles of 95°C for 30 s, 50°C for 1 min and 58°C for 2 min per kb amplified. We were unable to mutate the sp1-780 region, presumably due the very high local CG content, a common difficulty in this type of study. Positive clones were identified by the abolition of restriction sites (with SmaI, TauI, MspAI and HphI) and confirmed through sequencing both strands of the entire promoter fragment using an automated PCR sequencing method outlined in the Taq Dye Deoxy Terminator Cycle FS Sequencing Kit (Applied Biosystems). Sequences were excised by restriction digestion and directionally cloned into pGL3 basic (Promega) for tissue culture analysis. Large-scale endotoxin-free preparations of plasmids were column purified using a commercially available kit (Qiagen), using the instructions provided.
Figure 1.

Comparison of mouse and human utrophin promoter A sequences. The regions of interest in the 1.3 kb HindIII human promoter A region (EMBL accession no. X95523; top) and sequence conservation with the mouse region (EMBL accession no. X95524; bottom) are shown. The N-box and conserved E-box motifs are boxed, with restriction sites used in footprinting and luciferase constructs underlined. Regions that bind Sp1 or Ap2 are indicated in black, with a plus symbol indicating the Sp1 region unable to be mutated in this study. Grey regions refer to sites that did not bind either factor in EMSA studies. The multiple transcription start sites of the human sequence (15) are marked by asterisks.
Tissue culture and transfection
Mammalian cell lines (HeLa, C2C12 and IN157) were maintained and transfected as previously described (16). For co-transfection of the myogenic expression vectors (EMSV, EMSV-MyoD, EMSV-myogenin, EMSV-MRF4 and EMSV-myf5; 23), 0.5 µg vector and test plasmid were used. Cells were allowed to express the fusion genes for 12–24 h and were harvested by scraping into 1× lysis buffer (Promega) after two washes in phosphate-buffered saline at room temperature. Samples were freeze–thawed on dry ice, vortexed for 10 s and centrifuged at 15 000 g for 30 s, with the supernatant assayed directly.
Luciferase reporter gene assays
Cell extracts were assayed for luciferase activity using a commercial reagent (Promega). The light output was read using a Turner TD20e luminometer (delay 5 s, integration 10 s). All assays were performed in triplicate for three separate cultures of transfected cells. In addition, the transfection/assay process was repeated in triplicate for all promoter studies.
RESULTS
Binding of Ap2 and Sp1 are essential for optimal transcription of the A-utrophin core promoter
Database analysis of this 300 bp minimal element identified putative binding sites for the ubiquitous transcription factors Ap2 and Sp1; many of these sites were conserved between the human and murine sequences, indicating elements of potential functional importance (Fig. 1). In order to determine whether the identified sites would bind to the relevant factors, double-stranded oligonucleotides containing sequences spanning the regions of interest in human promoter A were used in EMSA studies with recombinant human Ap2 and Sp1. Of eight sites studied (Fig. 1), four formed complexes with retarded electrophoretic mobility on incubation with either Ap2 or Sp1 (ap2-747, sp1-755, sp1-780 and ap2-850; Fig. 2A). All DNA–protein complexes were specifically competed by consensus oligonucleotides and abolished by point mutations targeting important residues within the transcription factor-binding motifs (data not shown). EMSA supershifting analyses using HeLa and C2C12 nuclear extracts with antibodies to Ap2 and Sp1 were used to confirm the formation of specific protein–DNA complexes in the four regions of the core promoter A region identified with recombinant factors (Fig. 2B).
Figure 2.

Specific regions of the core utrophin promoter A region bind Ap2 and Sp1. (A) Double-stranded nucleotides spanning conserved binding sites of Ap2 and Sp1 were allocated a number corresponding to their location in the human promoter sequence (Fig. 1) and assayed with the appropriate recombinant transcription factor to determine binding ability. Formation of specific promoter A DNA–protein complexes are indicated by hash/cross marks. A grey arrowhead indicates the protein–DNA complex, with the black arrowhead indicating unbound DNA probe. Lane pr., consensus oligonucleotide only; lane c, control lane containing the consensus oligonucleotide and appropriate transcription factor. For Ap2: lane 1, ap2-709; lane 2, ap2-720; lane 3, ap2-747; lane 4, ap2-810; lane 5, ap2-850. For Sp1: lane 1, sp1-709; lane 2, sp1-755; lane 3, sp1-780; lane 4, sp1-850. (B) Formation of protein–DNA complexes using nuclear extract EMSA supershift analysis. Regions of interest were incubated with nuclear extract (30 µg HeLa and 15 µg C2C12) and antibodies (1.2 µg) as indicated. Cold comp. indicates the addition of a 100 or 500 molar excess of unlabelled probe for the Ap2 and Sp1 regions, respectively. Grey arrows indicate protein–DNA complexes that are supershifted in the presence of factor antibodies (white arrow). A black arrow indicates unbound labelled probe.
To confirm regions of transcription factor binding in the wider context of the entire core promoter region, we used DNase I protection analysis. An end-labelled double-stranded DNA probe encompassing the human 276 bp core promoter A region was incubated with recombinant protein factors or total nuclear extract, then subjected to DNase I digestion. As shown in Figure 3, recombinant human Ap2 protein protected regions at positions 106–116 (ap2-850), 182–190 (human only ap2 site) and 210–219 (ap2-747). Incubation with HeLa nuclear extracts confirmed these sites and in addition footprinted the two Sp1 regions (determined using EMSA) at positions 165–172 (sp1-780) and 200–208 (sp1-755) that correspond to previously identified regions binding Sp1 (20). These footprints therefore confirm sequence-specific binding of Sp1 and Ap2 observed using EMSA. As anticipated, large regions of protection were observed at the multiple transcriptional start sites (at positions 42–46, 53, 54 and 61 with respect to the footprinting probe). Regions of protection that were not predicted by sequence and/or targeted by EMSA analysis lay in immediate proximity to the transcription start site and, in the light of recent studies, are thought to bind Sp3 and/or Sp1 (20). A titration study with recombinant human Ap1, which is not predicted to bind to this region (Fig. 3, lanes 2–4), was used as a negative control, thus confirming the specificity of the footprints shown.
Figure 3.
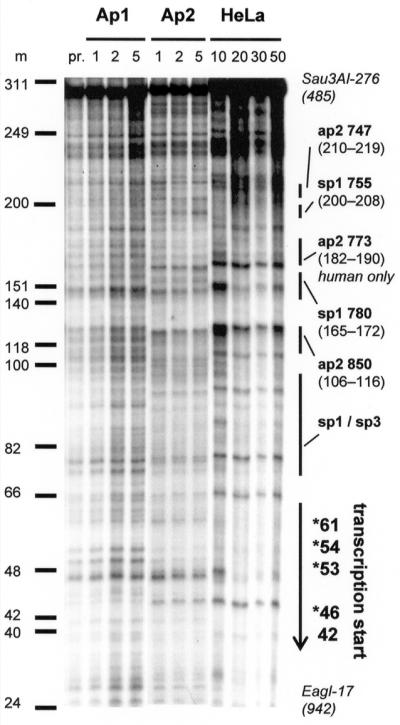
DNase I footprinting of the minimal promoter region. A radiolabelled non-coding strand of the minimal Sau3AI–EagI promoter A element was DNase I digested as outlined in Materials and Methods. Titrations of recombinant human enzyme or total nuclear extract are as indicated. For Ap1 and Ap2 numbers refer to the amount added in footprinting units (f.p.u.) with nuclear extract in µg total protein/lane. Regions of protection that are of interest are as indicated, with multiple transcription start sites marked by asterisks. Lane pr., probe only; lanes Ap1, rhap1 protein; lanes Ap2, rhap2 protein; lanes HeLa, total HeLa nuclear extract; m, molecular weight markers.
After demonstrating transcription factor binding to these sites, we next sought to delineate their functional importance in the context of the 300 bp minimal region through mutagenesis of their cognate sequences. Three regions were successfully mutated and functionally assayed by measuring their ability to drive reporter gene expression in a number of cultured cell lines (Fig. 4). All mutations decreased luciferase reporter activity, although the pattern of reduction in activity varied, reflecting the expression profiles of the individual factors within the cell lines used (as an example, the level of Sp1 expression varies at least 100-fold in different tissues; 24). We have therefore shown that Ap2 and Sp1 bind promoter A through their cognate sequences and are crucial for basal transcription in all cell lines studied.
Figure 4.

In vitro activity of the core promoter A is affected by mutagenesis of the Ap2 and Sp1 sites. Activity of the 300 bp core promoter mutant constructs as compared to the wild-type construct. For C2C12, prolif indicates proliferating and diff indicates differentiated cells. Normal minimal promoter activity is represented by white columns, with the mutants represented as a percentage activity: light grey, ap2-747; dark grey, sp1-755; black, ap2-850. All values are averages of triplicate assays from separately transfected wells.
A-utrophin mRNA is induced during myogenesis
In order to investigate the mechanisms giving rise to utrophin induction during myogenesis, we first sought to identify which utrophin promoter was responsible for the ∼2-fold increase in utrophin mRNA previously observed during myogenic differentiation in vitro (7). RNA was extracted from proliferating C2C12 cells before and 1, 4 and 7 days after changing the culture medium to induce their differentiation to myotubes. We measured isoform-specific utrophin mRNA using a quantitative RNase protection assay, as previously described (16). A 2.4-fold induction of the endogenous A-utrophin transcript was observed during differentiation (Fig. 5A), suggesting that promoter A is activated by factors that are involved in the myogenic regulatory programme.
Figure 5.
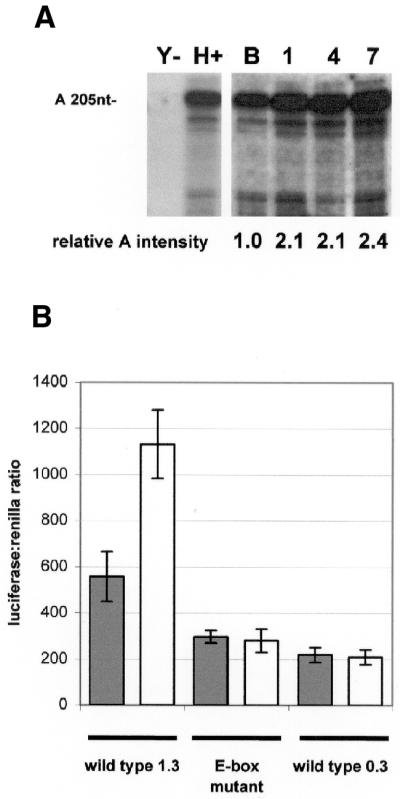
A-utrophin transcript and a promoter A construct are both up-regulated during myogenic differentiation in vitro. (A) Quantitative RNase protection analysis of the endogenous utrophin transcripts was undertaken in C2C12 myoblasts as previously described (16), before (B) and 1, 4 and 7 days after starting differentiation by changing the culture medium to DMEM with 5% horse serum. Yeast RNA (Y) and mouse heart RNA (H) were included as negative and positive controls for the assay. A specific protected band for the A-utrophin transcript (205 bp) was detected in all C2C12 RNA samples. The amount of A-utrophin mRNA relative to total RNA increases by >2-fold during differentiation. These data indicate that promoter A drives the increase in total utrophin expression seen during myogenesis. (B) Proliferating and differentiating C2C12 cells were transfected with a construct in which 1.3 kb of utrophin promoter A drives luciferase. There was a 2-fold increase in luciferase activity on differentiation of the cells. This effect was not seen when the cells were transfected with the core promoter element only (0.3 kb) or an identical 1.3 kb fragment with a single point mutation in the conserved upstream E-box. Grey columns represent proliferating and white columns represent differentiating C2C12 cell lines.
Promoter A myogenic induction maps to an upstream conserved E-box
As the activity of the 300 bp core promoter–reporter construct was similar in proliferating and differentiating C2C12 cells, it was postulated that the major myogenic response element(s) of promoter A (as indicated by RNase protection analysis) lay outside this region. Furthermore, the pattern of activity reduction of the point mutants shown in Figure 4 was identical in both proliferating and differentiating C2C12 cells, implying that Ap2 and Sp1 are not exclusively responsible for the increase in utrophin expression observed during myogenic differentiation in vitro. To determine the element within promoter A responsible for the 2-fold induction observed during myogenesis, studies were extended to include the 1 kb sequence 5′ to the core promoter region. This region contains a likely candidate region; a conserved canonical E-box motif (CANNTG) that is identical in sequence (CAGGTG) to previously characterised E-boxes responsible for myogenic regulation of a number of muscle-specific promoters (25).
To demonstrate that the upstream 1 kb region contains elements responsible for myogenic induction, a 1.3 kb promoter region (including the 300 bp core promoter region) was coupled to a luciferase reporter system. Proliferating and differentiating (1 day after medium change) C2C12 cells were transfected, and normalised reporter activity in each compared after 2 days. A 2-fold increase (2.04-fold) in luciferase activity was found in differentiating (1131.74 ± 14) compared with proliferating (557.40 ± 108, Fig. 5B) C2C12 cells. This mirrored the 2-fold increase in endogenous A-utrophin transcript levels observed under similar circumstances in vitro (2.1-fold 1–4 days after inducing differentiation), implying that elements contributing to the increase in A-utrophin mRNA production during myogenic differentiation reside within the upstream 1 kb element of promoter A, including the candidate E-box element. To test the hypothesis that the E-box was directly involved, we induced point mutations, based on published mutations that abolish MyoD binding (22), and assayed luciferase activity in both proliferating and differentiating C2C12 cells. In contrast to the wild-type promoter, the 1.3 kb E-box mutant promoter showed no evidence of increased activity on myogenic differentiation. Studies using proliferating (297.71 ± 27) and differentiating (280.25 ± 50) C2C12 cell extracts exhibited a decrease in luciferase activity towards levels observed with the smaller 0.3 kb core promoter fragment (proliferating, 219.73 ± 31; differentiating, 209.69 ± 32), which does not include the E-box. From this observation, it is therefore likely that factors present during myogenic differentiation activate utrophin promoter A via DNA–protein interactions at the upstream E-box, resulting in increased expression of A-utrophin during myogenesis. The observed decrease in promoter activity of the E-box mutant in proliferating cells (45%) compared to the wild-type element is not unexpected; unfused C2C12 cells are partially differentiated biochemically (26) and express myogenic factors in relative abundance (excepting herculin/MRF4) which bind E-boxes of transcriptionally active acetylcholine receptor subunit promoters at this stage in vivo (27).
MyoD, myogenin and MRF4 activate the utrophin promoter through interaction with the E-box motif
After the identification of the E-box as a functionally important upstream element, we sought more direct evidence of its importance in myogenic induction through the identification of interacting factors. Other previously characterized E-boxes that enhance transcription during myogenesis, such as the muscle creatine kinase promoter (28), are canonical with the promoter A motif and bind helix–loop–helix myogenic regulatory factors (MRFs) that activate a number of genes in skeletal muscle (28–32). Expression of the candidate factors MyoD, myogenin and MRF4/herculin induces myogenesis (33) and accompanies terminal differentiation of C2C12 cells (34).
In order to determine whether these factors interact with utrophin promoter A, we assayed binding of MyoD and myogenin to both (i) a 300 bp XhoI–PstI promoter A fragment containing the E-box region and (ii) a synthetic double-stranded 22mer oligonucleotide spanning the E-box (Fig. 6A). Following incubation with either MyoD or myogenin, both the 300 bp and E-box-specific probe formed a complex with retarded electrophoretic mobility. Complexes were not formed in the presence of 100-fold molar excess of unlabelled probe or with a mutant E-box oligonucleotide, demonstrating protein–DNA complex specificity.
Figure 6.

Promoter A contains a functional E-box that binds myogenic regulatory factors (MRFs). (A) EMSA studies with recombinant human protein showing both a XhoI–PstI promoter A region and the E-box motif specifically bind MyoD and myogenin. The addition of protein and excess competitor probe were as indicated. For the E-box only, the normal (n) oligonucleotide binds both myoD and myogenin, with the protein–DNA complex lost when the region is mutated (m). (B) Promoter A is transactivated by MyoD, myogenin and MRF4 in C2C12 cells. This activation is abolished by mutation of the E-box motif. Values are given as fold induction of activity expressed by the promoter A construct. Grey columns represent the 1.3 kb wild-type promoter A construct with white columns representing an identical fragment with the mutated E-box motif. Promoter, promoter A only; vector, EMSV expression vector backbone; myoD, EMSV-MyoD; myoG, EMSV-myogenin; mrf4, EMSV-MRF4; myf-5, EMSV-myf-5.
To determine the functional importance of these interactions, C2C12 cells were co-transfected with (i) the 1.3 kb promoter A–reporter construct and (ii) expression vectors for MyoD, myogenin, MRF4 and myf-5 (23; Fig. 6B). We observed transactivation of utrophin promoter A by MyoD (17.72 ± 3.9-fold increase), myogenin (10.71 ± 2.9-fold increase) and MRF4 (9.89 ± 0.9-fold increase). Identical studies on the 1.3 kb E-box mutant promoter showed no evidence of transactivation (with the highest increase observed with MyoD of 1.747 ± 0.4-fold), supporting the EMSA studies and confirming the binding site of these factors. These combined results demonstrate that these identified MRFs are able to activate utrophin expression during myogenesis by interaction with the E-box upstream of promoter A.
DISCUSSION
CpG-rich TATA-less promoters were initially considered to regulate ubiquitously expressed genes whose products performed ‘housekeeping’ functions. However, several such promoters are highly regulated (35,36). Studies of utrophin promoter A show that interactions between a GC-rich TATA-less core promoter element and upstream elements are able to confer a highly regulated expression profile to A-utrophin mRNA. Initially, we showed that the upstream N-box is responsible for synaptic induction of utrophin promoter A activity by binding to GABP, which is concentrated in the post-synaptic compartment of skeletal muscle (37). A recent study revealed that Sp1 and Sp3 bind adjacent to the utrophin promoter A core element and interact with GABP at the N-box to induce transcription (20), in a similar way to the reported activation of the neuronal nicotinic acetylcholine receptor β4 subunit gene by Sp1/Sp3 (38). We have extended these findings by (i) precisely locating nucleotides responsible for Sp1 activation and (ii) showing that binding of both Sp1 and Ap2 is essential for full activity of the core promoter region in vitro. Mutual enhancement of basal transcription from promoter A by these factors may account for the multiple tissue expression of A-utrophin (15).
The second important finding reported here is the identification of a second upstream element that binds specific regulatory factors to modulate the activity of the core promoter. We have shown that myogenic induction of utrophin is driven by promoter A, through interaction of MRFs with the upstream E-box. In this respect, utrophin promoter A appears to be regulated in a similar manner to other genes expressed in muscle (17,30,39,40). Mechanistically, this type of regulation may involve direct interactions between myogenic factors and basal factors bound to the promoter, analogous to the association of Sp1 and MyoD/myogenin factors required for activation of the human cardiac α-actin promoter in skeletal muscle cells (40).
Our goal was to identify ways of effecting therapeutic up-regulation of utrophin in DMD patients. One way to achieve this may be through identification of relevant transcriptional regulatory mechanisms operating in muscle, in order to isolate molecular targets for drugs. Characterisation of a muscle-specific regulatory mechanism and further elucidation of the events occurring at the core promoter element are important steps towards this goal. Further definition of the protein–DNA and protein–protein interactions involved in determination of the temporal and spatial pattern of utrophin A promoter activity in muscle can follow from these studies. This may enable characterisation of specific events that can be assayed for susceptibility to pharmacological modulation to yield transcriptional activation. Importantly, it may be possible to transactivate utrophin promoter A in DMD muscle by delivery of myogenic factors or by drugs that directly or indirectly alter the levels or activity of such factors. Our current work is directed towards further characterisation of these regulatory processes and ways of manipulating them.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge Dr Margaret Buckingham (Pasteur Institute, France) for the provision of myogenic regulatory expression vectors, Allyson Potter and Dr Andrew Weir for tissue culture and Dr Nanda Rodrigues for manuscript suggestions. K.J.P. is the recipient of an Oxford Overseas Bursary, ORS Award and Sir Edward Dunlop Memorial Award (Aust.). This work was supported by the Medical Research Council (UK), the Muscular Dystrophy Campaign (UK), the Muscular Dystrophy Associations of the USA and South Australia and the Association Francaise Contre les Myopathies.
REFERENCES
- 1.Koenig M., Hoffman,E.P., Bertelson,C.J., Monaco,A.P., Feener,C. and Kunkel,L.M. (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell, 50, 509–517. [DOI] [PubMed] [Google Scholar]
- 2.Love D.R., Byth,B.C., Tinsley,J.M., Blake,D.J. and Davies,K.E. (1993) Dystrophin and dystrophin-related proteins: a review of protein and RNA studies. Neuromusc. Disord., 3, 5–21. [DOI] [PubMed] [Google Scholar]
- 3.Tinsley J.M. and Davies,K.E. (1993) Utrophin: a potential replacement for dystrophin? Neuromusc. Disord., 3, 537–539. [DOI] [PubMed] [Google Scholar]
- 4.Tinsley J.M., Potter,A.C., Phelps,S.R., Fisher,R., Trickett,J.I. and Davies,K.E. (1996) Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature, 384, 349–353. [DOI] [PubMed] [Google Scholar]
- 5.Deconinck A.E., Rafael,J.A., Skinner,J.A., Brown,S.C., Potter,A.C., Metzinger,L., Watt,D.J., Dickson,J.G., Tinsley,J.M. and Davies,K.E. (1997) Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell, 90, 717–727. [DOI] [PubMed] [Google Scholar]
- 6.Schofield J., Houzelstein,D., Davies,K., Buckingham,M. and Edwards,Y.H. (1993) Expression of the dystrophin-related protein (utrophin) gene during mouse embryogenesis. Dev. Dyn., 198, 254–264. [DOI] [PubMed] [Google Scholar]
- 7.Gramolini A. and Jasmin,B. (1999) Expression of the utrophin gene during myogenic differentiation. Nucleic Acids Res., 27, 3603–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khurana T.S., Watkins,S.C., Chafey,P., Chelly,J., Tome,F.M.S., Fardeau,M., Kaplan,J.-C. and Kunkel,L.M. (1991) Immunolocalisation and developmental expression of dystrophin related protein in skeletal muscle. Neuromusc. Disord., 1, 185–194. [DOI] [PubMed] [Google Scholar]
- 9.Pons F., Robert,A., Marini,J.F. and Leger,J.J. (1994) Does utrophin expression in muscles of mdx mice during postnatal development functionally compensate for dystrophin deficiency? J. Neurol. Sci., 122, 162–170. [DOI] [PubMed] [Google Scholar]
- 10.Cartaud A., Ludosky,M.A., Tome,F.M., Collin,H., Stetzkowski-Marden,F., Khurana,T.S., Kunkel,L.M., Fardeau,M., Changeux,J.P. and Cartaud,J. (1992) Localization of dystrophin and dystrophin-related protein at the electromotor synapse and neuromuscular junction in Torpedo marmorata. Neuroscience, 48, 995–1003. [DOI] [PubMed] [Google Scholar]
- 11.Ohlendieck K., Ervasti,J.M., Matsumura,K., Kahl,S.D., Leveille,C.J. and Campbell,K.P. (1991) Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron, 7, 499–508. [DOI] [PubMed] [Google Scholar]
- 12.Helliwell T.R., Man,N.T., Morris,G.E. and Davies,K.E. (1992) The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromusc. Disord., 2, 177–184. [DOI] [PubMed] [Google Scholar]
- 13.Matsumura K., Ervasti,J.M., Ohlendieck,K., Kahl,S.D. and Campbell,K.P. (1992) Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature, 360, 588–591. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno Y., Nonaka,I., Hirai,S. and Ozawa,E. (1993) Reciprocal expression of dystrophin and utrophin in muscles of Duchenne muscular dystrophy patients, female DMD-carriers and control subjects. J. Neurol. Sci., 119, 43–52. [DOI] [PubMed] [Google Scholar]
- 15.Dennis C.L., Tinsley,J.M., Deconinck,A.E. and Davies,K.E. (1996) Molecular and functional analysis of the utrophin promoter. Nucleic Acids Res., 24, 1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burton E.A., Tinsley,J.M., Holzfeind,P.J., Rodrigues,N.R. and Davies,K.E. (1999) A second promoter provides an alternative target for therapeutic up-regulation of utrophin in Duchenne muscular dystrophy. Proc. Natl Acad. Sci. USA, 96, 14025–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y., Camp,S., Rachinsky,T.L., Bongiorno,C. and Taylor,P. (1993) Promoter elements and transcriptional control of the mouse acetylcholinesterase gene. J. Biol. Chem., 268, 3563–3572. [PubMed] [Google Scholar]
- 18.Getman D.K., Mutero,A., Inoue,K. and Taylor,P. (1995) Transcription factor repression and activation of the human acetylcholinesterase gene. J. Biol. Chem., 270, 23511–23519. [DOI] [PubMed] [Google Scholar]
- 19.Gramolini A.O., Burton,E.A., Tinsley,J.M., Ferns,M.J., Cartaud,A., Cartaud,J., Davies,K.E., Lunde,J.A. and Jasmin,B.J. (1998) Muscle and neural isoforms of agrin increase utrophin expression in cultured myotubes via a transcriptional regulatory mechanism. J. Biol. Chem., 273, 736–743. [DOI] [PubMed] [Google Scholar]
- 20.Galvagni F., Capo,S. and Oliviero,S. (2001) Sp1 and Sp3 physically interact and co-operate with GABP for the activation of the utrophin promoter. J. Mol. Biol., 306, 985–996. [DOI] [PubMed] [Google Scholar]
- 21.Kim H.S., Seo,H., Yang,C., Brunet,J.F. and Kim,K.S. (1998) Noradrenergic-specific transcription of the dopamine beta-hydroxylase gene requires synergy of multiple cis-acting elements including at least two Phox2a-binding sites. J. Neurosci., 18, 8247–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J., Blackwell,T.K., Kedes,L. and Weintraub,H. (1996) Differences between MyoD DNA binding and activation site requirements revealed by functional random sequence selection. Mol. Cell. Biol., 16, 3893–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis R.L., Weintraub,H. and Lassar,A.B. (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell, 51, 987–1000. [DOI] [PubMed] [Google Scholar]
- 24.Saffer J.D., Jackson,S.P. and Annarella,M.B. (1991) Developmental expression of Sp1 in the mouse. Mol. Cell. Biol., 11, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molkentin J.D. and Olson,E.N. (1996) Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl Acad. Sci. USA, 93, 9366–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S., Spinner,D.S., Schmidt,M.M., Danielsson,J.A., Wang,S. and Schmidt,J. (2000) Interaction of MyoD family proteins with enhancers of acetylcholine receptor subunit genes in vivo. J. Biol. Chem., 275, 41364–41368. [DOI] [PubMed] [Google Scholar]
- 27.Hauschka S.D. (1994) The scientific basis of myology. In Engel,A.G. and Franzini-Armstrong,C. (eds), Myology. McGraw Hill, New York, NY, Vol. 1, pp. 3–73.
- 28.Lassar A.B., Buskin,J.N., Lockshon,D., Davis,R.L., Apone,S., Hauschka,S.D. and Weintraub,H. (1989) MyoD is a sequence-specific DNA binding protein requiring a region of myc homology to bind to the muscle creatine kinase enhancer. Cell, 58, 823–831. [DOI] [PubMed] [Google Scholar]
- 29.Shield M.A., Haugen,H.S., Clegg,C.H. and Hauschka,S.D. (1996) E-box sites and a proximal regulatory region of the muscle creatine kinase gene differentially regulate expression in diverse skeletal muscles and cardiac muscle of transgenic mice. Mol. Cell. Biol., 16, 5058–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donoviel D.B., Shield,M.A., Buskin,J.N., Haugen,H.S., Clegg,C.H. and Hauschka,S.D. (1996) Analysis of muscle creatine kinase gene regulatory elements in skeletal and cardiac muscles of transgenic mice. Mol. Cell. Biol., 16, 1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fabre-Suver C. and Hauschka,S.D. (1996) A novel site in the muscle creatine kinase enhancer is required for expression in skeletal but not cardiac muscle. J. Biol. Chem., 271, 4646–4652. [DOI] [PubMed] [Google Scholar]
- 32.Apone S. and Hauschka,S.D. (1995) Muscle gene E-box control elements. Evidence for quantitatively different transcriptional activities and the binding of distinct regulatory factors. J. Biol. Chem., 270, 21420–21427. [DOI] [PubMed] [Google Scholar]
- 33.Miner J.H. and Wold,B.J. (1991) c-myc inhibition of MyoD and myogenin-initiated myogenic differentiation. Mol. Cell. Biol., 11, 2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montarras D., Chelly,J., Bober,E., Arnold,H., Ott,M.O., Gros,F. and Pinset,C. (1991) Developmental patterns in the expression of Myf5, MyoD, myogenin, and MRF4 during myogenesis. New Biol., 3, 592–600. [PubMed] [Google Scholar]
- 35.Miner J.H. and Wold,B. (1990) Herculin, a fourth member of the MyoD family of myogenic regulatory genes. Proc. Natl Acad. Sci. USA, 87, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luskey K.L. (1987) Conservation of promoter sequence but not complex intron splicing pattern in human and hamster genes for 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mol. Cell. Biol., 7, 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gramolini A.O., Angus,L.A., Schaeffer,L., Burton,E.A., Tinsley,J.M., Davies,K.E., Changeux,J.-P. and Jasmin,B.J. (1999) Induction of utrophin expression by heregulin in skeletal muscle cells: role of the N-box and GA binding protein. Proc. Natl Acad. Sci. USA, 96, 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bigger C.B., Melnikova,I.N. and Gardner,P.D. (1997) Sp1 and Sp3 regulate expression of the neuronal nicotinic acetylcholine receptor beta4 subunit gene. J. Biol. Chem., 272, 25976–25982. [DOI] [PubMed] [Google Scholar]
- 39.Lassar A.B., Buskin,J.N., Lockshon,D., Davis,R.L., Apone,S., Hauschka,S.D. and Weintraub,H. (1989) MyoD is a sequence-specific DNA binding protein requiring a region of myc homology to bind to the muscle creatine kinase enhancer. Cell, 58, 823–831. [DOI] [PubMed] [Google Scholar]
- 40.Biesiada E., Hamamori,Y., Kedes,L. and Sartorelli,V. (1999) Myogenic basic helix-loop-helix proteins and Sp1 interact as components of a multiprotein transcriptional complex required for activity of the human cardiac alpha-actin promoter. Mol. Cell. Biol., 19, 2577–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]