

Abstract
Cells in complex organisms undergo frequent functional changes, but few methods allow comprehensive longitudinal profiling of living cells. Here, we introduce scission-accelerated fluorophore exchange (SAFE), a method for multiplexed temporospatial imaging of living cells with immunofluorescence. SAFE uses a rapid bioorthogonal click chemistry to remove immunofluorescent signals from the surface of labeled cells, cycling the nanomolar-concentration reagents in seconds and enabling multiple rounds of staining of the same samples. It is nontoxic and functional in both dispersed cells and intact living tissues. We demonstrate multiparameter (n≥14), nondisruptive imaging of murine peripheral blood mononuclear and bone marrow cells to profile cellular differentiation. We also show longitudinal multiplexed imaging of bone marrow progenitor cells as they develop into neutrophils over 6 days and real time multiplexed cycling of living mouse hepatic tissues. We anticipate that SAFE will find broad utility for investigating physiologic dynamics in living systems.
Keywords: Temporospatial imaging, Live cell multiplexing, Click chemistry
Introduction
Temporal processes are often studied in living cells and organisms by introducing fluorescent proteins coupled to a promoter1, protein2, or biological compartment3 of interest, making otherwise invisible physiologic events observable by fluorescence microscopy. This strategy has been intensely successful in cell biology4. However, the approach is not without shortcomings, including: i) the time required for creation and validation of cell lines; ii) the limited number of concurrent targets that can be visualized; and iii) the practical impossibility of using fluoroprotein labeling to analyze intact ensembles of primary cells, especially in complex contexts like development, differentiation, or immune function. Optimized monoclonal antibodies make immunofluorescence techniques a feasible alternative to study genetically unmodified cells both in vitro5 and in vivo6,7, but only for a handful (i.e. 1–4) of simultaneous targets. In aggregate, these liabilities sharply restrict the dimensionality of our view into living systems.
An array of analytical technologies, such as cellularly-resolved sequencing8–11, multiplexed immunostaining12–16, mass/imaging cytometry17–20, and synergistic combinations thereof21,22, can now achieve detailed snapshots of biomarker abundance, producing fine-grained but static profiles. Nevertheless, the techniques are thus far incompatible with making measurements in living cells or tissues, due to inherently lethal physicochemical processing during the analytical workflow or single-cell detection mechanisms, as in flow cytometry23, that cannot observe cells in situ (i.e. within the context of their growth environment). Of the above tools, only the methods that rely on iterative immunostaining are potentially non-destructive, as the key step of antibody-antigen recognition is intrinsically biocompatible. The competing priorities, however, of fast/complete cycling and live-cell gentleness/non-toxicity require chemical conditions that have remained orders of magnitude apart (Supplementary Fig. 1). Transient binding of antibodies (as in uPAINT24) or nucleic acid probes (as in DNA-PAINT25) have been exploited for superresolution imaging and could in principle be applied to multiplexed analyses of live cells. However, to date, the extended acquisition times and operational complexity of stochastic single molecule imaging have been limited to fixed samples. As a result, highly multiplexed temporospatial profiling of living cells has been virtually impossible.
Lacking such methods, certain biological processes have remained challenging to investigate, for example: i) the role of intercellular communication during differentiation5,26 or immunotherapy27,28, ii) functional exploration of novel cellular phenotypes, particularly in the space of immunology29–31; iii) tracing of cellular lineages, e.g., in hematopoietic development32. Experimental techniques to directly observe these events offer the possibility of interrogating biological interactions, developmental transformations, and the downstream responses. Further development of such dynamic model systems has thus far been hindered by our inability to efficiently visualize both biomolecular complexity and (multi)cellular dynamics in real time.
We envisioned that new bioorthogonal chemistries could achieve the capabilities needed for longitudinal multiplexed profiling of extracellular markers in live cells (Fig. 1). Here we describe the development and application of the scission-accelerated fluorophore exchange (SAFE) method, which leverages cooperative bioorthogonal mechanisms to first quench and then completely remove fluorescent signal from antibody-labeled cells. With a scaffold built around a cleavable C2-symmetric trans-cyclooctene (C2TCO), SAFE exploits non-covalent quencher-fluorophore interactions to accelerate already rapid tetrazine (Tz)-TCO click reactions, reaching >99% scission in seconds. Critically, this makes bioorthogonal cycling feasible at nontoxic nanomolar concentrations, which was not possible with prior methods33, all while achieving the high signal to background needed for serial profiling. We first quantify the accelerated chemical kinetics in analytical fluorescence assays and verify the scission performance in the biological context by time-resolved imaging. We then proceed to validate the accuracy and non-toxicity of 14 color imaging of living mouse peripheral blood mononuclear cells (PBMCs), perform multiplexed imaging experiments in living murine hepatic tissue, and visualize myeloid subsets in multicolor staining of living murine bone marrow. Last, we demonstrate the ability of serial bioorthogonal cycling to track the differentiation of immortalized hematopoietic progenitor cells into neutrophils in longitudinal profiling of the same cells over 6 days.
Figure 1 |. Multiplexed temporospatial profiling in living cells.
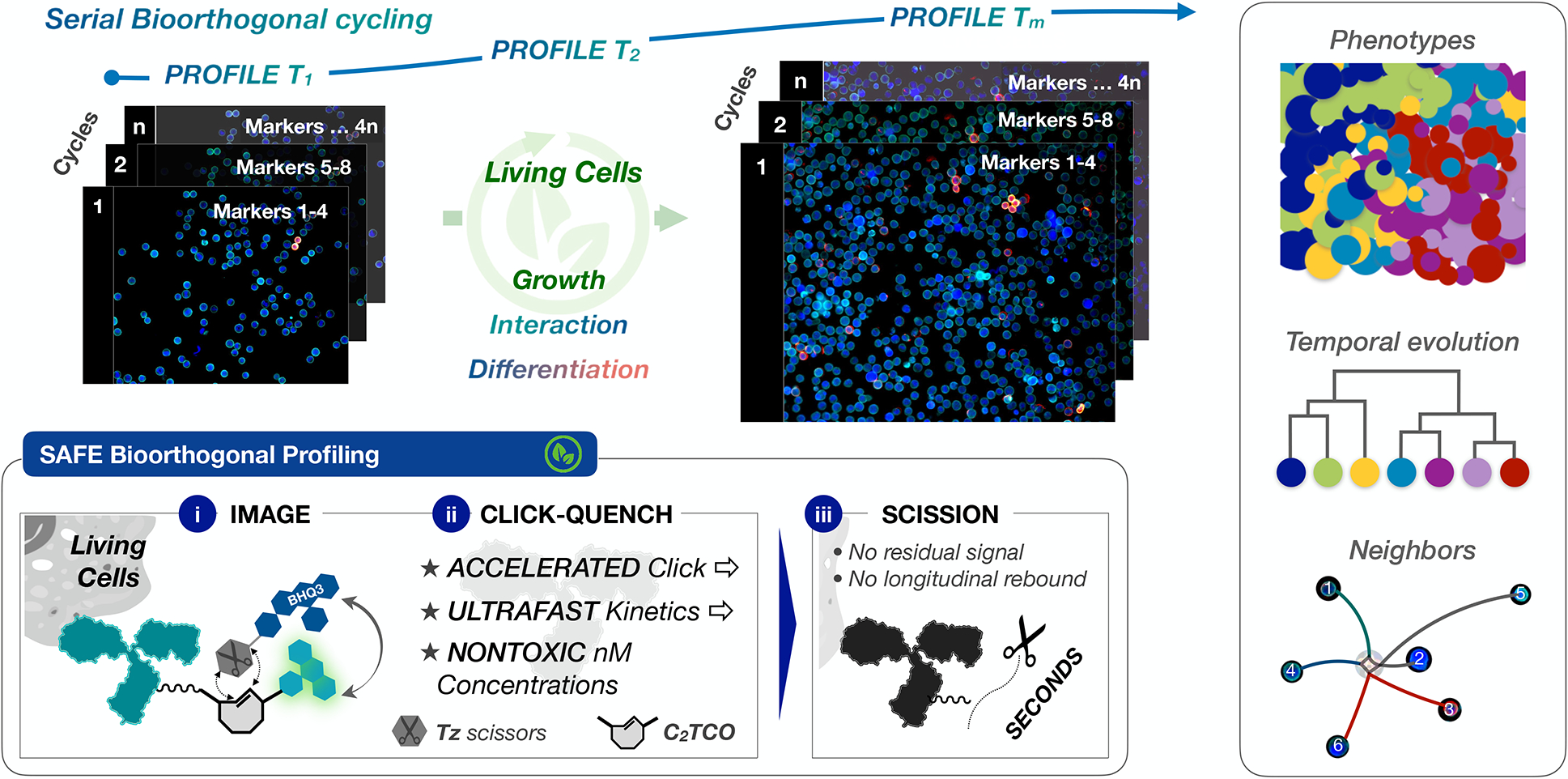
Serial bioorthogonal cycling can reveal complex cellular dynamics in living cells, enabling phenotypic characterization and longitudinal tracking of temporal evolution and/or neighbor-neighbor interactions. Inset: the design of scission-accelerated fluorophore exchange (SAFE) combines an accelerated click-qenching reaction with rapid fluorophore scission to gently, efficiently, and irreversibly erase fluorophores from the surface of immunostained living cells in seconds.
Results
Bioorthogonal cooperativity accelerates scission.
For all but the most reactive molecules, chemical processes that reach completion in the timeframe of seconds to minutes require concentrations in the high micromolar to millimolar range. While this is no obstacle for in vitro organic synthesis, it poses challenges for bioorthogonal methods: i) these same concentration ranges correspond to the level at which many organic compounds become nonspecifically cytotoxic, barring use in living cells; ii) reagents with the highest degree of reactivity are typically metastable and/or vulnerable to rapid degradation, limiting their performance in the biological environment34. To circumvent these issues, the design of SAFE exploits a cooperative interaction between the fluorescence quencher BHQ3 and Alexa Fluor dyes for rapid on/off scission of fluorophore-C2TCO-labeled antibodies (Fig. 2A) at low, nontoxic reagent concentrations. Importantly, the BHQ3-dye interaction is expected to accelerate the Tz-TCO click reaction; in this initial stage, ligation yields immediate quenching (>95%) of the dye fluorescence33. In turn, quantitative scission is driven by a functionalized tetrazine equipped with an intramolecular proton donor (NH3+ at physiologic pH) that serves to accelerate tautomerization, functioning as a ‘Tz-scissors’35–37. This click-to-cut scission reaction then releases the quenched fluorophores from antibodies, removing residual signal: i) optimizing signal to background, and ii) eliminating the downstream risk of rebounding signal from the quenched dye.
Figure 2 |. Mechanisms, synthesis and kinetics.
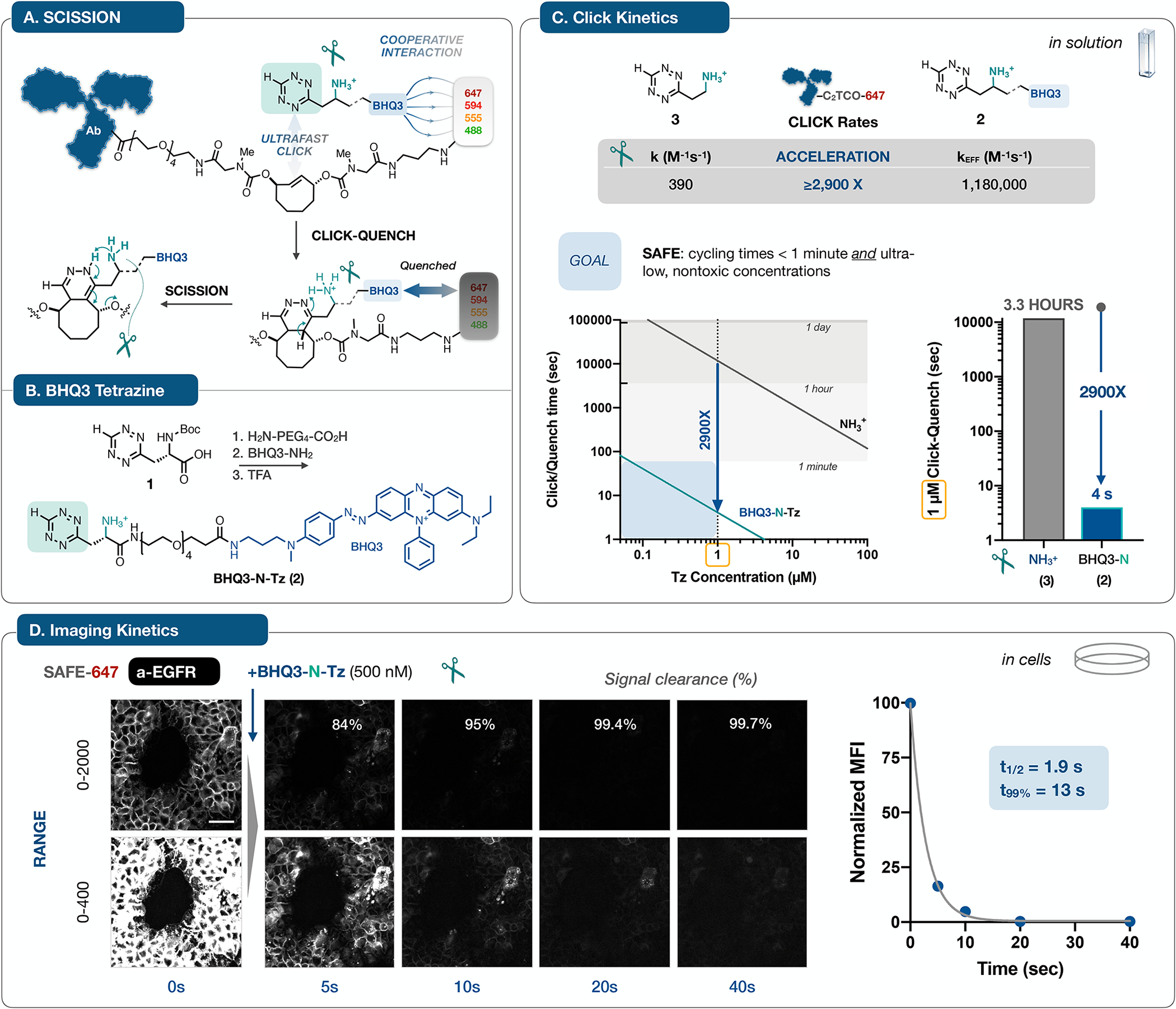
A. Antibodies labeled with C2TCO-linked dyes are rendered non-fluorescent in a concerted two step sequence: noncovalent interactions between BHQ3 and the fluorophore can accelerate the Tz/TCO click reaction33, leading to immediate quenching upon ligation; in turn, the functionalized ‘Tz-scissors’ drives instantaneous tautomerization and scission of the dye. The design of C2TCO itself ensures that the cleavage reaction proceeds irrespective of the Tz/TCO click orientation. B. Starting from the Boc-protected Tz (1), a hydrophilic PEG linker and BHQ3 are conjugated and then deprotected to yield BHQ3-N-Tz (2). C. Quantification of reaction kinetics for the click reaction of 2 with an AF647-C2TCO-labeled antibody in solution revealed an effective rate constant of >106, a marked ~3,000-fold acceleration relative to the parent aminoethyl-functionalized Tz (3)37. This acceleration shifts the concentration/time relationship into the needed range for efficient cycling at ≤ 1 μM reagent concentrations. D. To validate the accelerated kinetics in the cellular context, we collected time lapse images of A431 cells stained with a SAFE647-anti-EGFR antibody and then treated with BHQ3-N-Tz (2). Images rendered at two window levels capture the rapid (t1/2 = 1.9 s, one phase decay) and quantitative clearance of the fluorophore signal, reaching 99.7% removal within 40 seconds, concordant with the in vitro assay result. See Supplementary Fig. 3 for analytical details. Normalized mean fluorescence intensity (MFI) is plotted as mean ± SD; error bars are smaller than the symbol size). The scale bar is 50 μm
We therefore synthesized N-Boc-protected tetrazine (1) and extended it with a PEG4 amino acid linker, followed by coupling with BHQ3-amine and deprotection (Fig. 2B). The reaction of BHQ3-N-Tz (2) and an AF647-C2TCO (SAFE647) labeled antibody was followed by monitoring fluorescence intensity vs time. At an optimized concentration of 2, the reaction process could be kinetically segregated into its two component phases, resolving the quenching of the click ligation from the subsequent scission reaction. Nonlinear fitting of the biphasic fluorescence time course yielded a rate constant of 0.143 ± 0.005 s−1 (t1/2 = 5 sec) for the scission of the C2TCO linker. For the click ligation itself, we measured an effective second order rate constant of 1.18 ± 0.05 × 106 M−1s−1 (Supplementary Fig. 2), more than 2,900-fold faster than the reaction of the parent Tz (3) with C2TCO37. C2TCO itself is stable (>97% intact at t = 48h in full cell growth media) and not very reactive until accelerated by the cooperative SAFE effect, resulting in stable click reagents but also very fast kinetics. The cooperative acceleration shifts the kinetic regime from hours to seconds (Fig. 2C). To validate the solution kinetics in a biological context, we stained fixed A431 cells with a SAFE647-labeled anti-EGFR antibody and collected serial images before and after the addition of BHQ3-N-Tz (500 nM, Fig. 2D). The quenching-cleavage reaction on the surface of the cells was both rapid (t1/2 = 1.9 s) and complete, with a 99.7% reduction in signal intensity after 40 seconds (~8 scission half-lives). Control experiments confirmed that the fluorescence dynamics are not driven by photobleaching (Supplementary Fig. 3). These kinetics integrate both steps of signal removal at the extracellular surface, confirming the magnitude of the acceleration and the high dynamic range of signal elimination, critical to minimizing background accumulation and maintaining image quality across multi-cycle staining (Supplementary Fig. 1).
SAFE allows cycling in live cells.
We performed an initial short-term cycling experiment on live A549 cells stained with Hoechst and a SAFE647-conjugated anti-N-Cadherin antibody. The N-Cadherin signal was promptly eliminated with BHQ3-N-Tz (1 μM, 1 minute, Fig. 3A). A fraction of cells visually confirmed their live status by undergoing mitosis during the imaging process: in the example shown (Fig. 3A, inset) a cell is captured in metaphase before quenching and in early anaphase after quenching, 17 minutes of elapsed time later. For a longer-term assessment, we expanded the A549 experiments into serial imaging to test scission performance and visually screen for cytoxicity. Cells were stained with a SAFE647-conjugated anti-EGFR antibody, quenched/cleaved with BHQ3-N-Tz, and then returned to routine culture conditions at 37 °C, with images of the same cellular area collected at each stage. After 24 hours, the cells were re-imaged with live and dead cell staining dyes (live: calcein AM, dead: sytox red) to determine their viability (Fig. 3B, SAFE). Cell viability readouts with PrestoBlue confirmed nontoxicity across the working time and concentration range for both SAFE-conjugated antibodies and the BHQ3-N-Tz scissors (Supplementary Fig. 4).
Figure 3 |. Rapid, durable, and nontoxic multiplexing of living cells.

A. Staining and scission with SAFE647-anti-N-Cadherin confirmed efficient scission and short-term nontoxicity in living A549 cells, a subset of which progressed through mitosis during the experiment (scale bar, 50 μm). B. Longer term imaging of A549 cells was used to confirm the lack of toxicity and assess the impact of scission. Serial imaging over 24 hours demonstrated durable signal elimination and intact live cell calceinAM staining for SAFE. Control cells stained and quenched in parallel with a noncleavable anti-EGFR fluorophore-TCO conjugate and BHQ3-Tz 33 were initially dark but exhibited marked rebound of the fluorescent signal 24 hrs later, likely due to extra/intracellular degradation of the BHQ3 quencher, indicating a critical role for scission (scale bar, 100 μm). C. To quantify cell viability across multiple cycles of staining and quenching, living mouse PBMCs were isolated, aliquoted, loaded into microwells for imaging. The cells (n = 72 wells) were profiled with a SAFE panel of 12 immune markers, imaged in sets of three, followed by live/dead staining dyes to assess cell survival. Efficient scission performance produced images with high signal to background across all five cycles and no evident residual signal. The scale bar is 50 μm. D. Both cell viability after bioorthogonal profiling (96.5% SAFE vs. 96.2% FLOW) and marker abundance (% positive cells) matched well with flow cytometry controls (R2=0.99). All experiments were repeated independently three times.
Consistent with complete scission of the quenched fluorophores, cells remained non-fluorescent on day 2, with no signal rebound observed. The cells were calcein AM-bright and had increased in density, consistent with unhindered interim proliferation. As a control, we used a conventional TCO-Tz pair that can click for bioorthogonal quenching but cannot cleave fluorophores from the antibody33. Although the cells were again calcein AM-positive and proliferating, a spontaneous increase in fluorescence signal was observed in the absence of scission (Fig. 3B, CONTROL). Black hole quenchers are known to be vulnerable to azo bond cleavage, particularly in vivo38; the observed rebound is consistent with slow intracellular and/or extracellular degradation of the BHQ3, leading to loss of quenching and the reappearing signal. While both bioorthogonal methods achieve effective short-term cycling in living cells, scission is essential for serial profiling in longer timeframes where antibody internalization is expected and fluorescence rebound is a hazard.
We next sought to extend SAFE live cell cycling to mixed populations of primary immune cells. To image living mouse peripheral blood mononuclear cells (PBMCs) in suspension, we fabricated PDMS microwells (80 μm diameter, 80 μm depth) using soft lithography39. Both short-term imaging experiments and subsequent cell culture experiments (vide infra) were performed with these microwells, which showed excellent retention of the suspended cells, necessary for marker quantitation across cycles. Freshly harvested mouse PBMCs were isolated, pooled, and then loaded into microwells for imaging, reserving aliquots for parallel analysis by flow cytometry. SAFE-labeled antibodies were compared to conventionally labeled controls to validate staining specificity (Supplementary Fig. 5). We visualized 12 targets across 4 cycles of imaging and quenching in live cells over the course of <2 hours of elapsed time. At the end, we determined the cell viability (711 alive / 737 total cells in 72 microwells, 96.5% alive) in a fifth round of imaging with calcein AM/sytox red (Fig. 3C). The cells expressing each marker were counted and compared to the aliquot analyzed synchronously by flow cytometry for the same target panel. There was an excellent correlation (R2 = 0.985) between populations measured by SAFE (N = 737) and flow cytometry (N > 105) in both their marker abundance and in the measured cell viability (96.5% by SAFE vs 96.2% by flow, Fig. 3D), confirming i) the quantitative staining accuracy across multiple cycles of imaging and scission-mediated fluorophore exchange, and ii) the lack of toxicity to living PBMCs.
SAFE enables cycling in living tissue.
Whereas the in situ analysis of dispersed cells can reveal cellular interactions and differentiation in complex populations (vide infra), multiplexed imaging of living tissues would allow visualization of cellular dynamics within intact biological architectures. To explore the performance of SAFE in this context, we prepared freshly harvested liver slices from a Mer tyrosine kinase (MerTK)-GFP mouse40, in which GFP-positive macrophages provide both a positive control for immunostaining and spatial landmarks for image focus/registration during cycling. We stained the liver section with anti-CD45 (AF555) and collected a Z-stack time-lapse (Supplementary Movie 1), capturing the migration of CD45+ cells and quantifying the kinetic performance of staining/scission in living hepatic tissue. Real-time incubation of liver sections with a SAFE647-labeled anti-MHC-I antibody yielded bright cell membrane signal within minutes (t1/2 = 3 ± 0.3 min); that signal was then rapidly and selectively eliminated upon click-quench-scission with BHQ3-N-Tz (t1/2 = 2 ± 0.1 min, 98% signal clearance; Supplementary Fig. 6).
Expanding on these results, we went on to stain a fresh MerTK-GFP liver section with SAFE-modified antibodies (anti-CD8, CD4, CD11b, Ter119, CD45, MHC-I) in three cycles (Fig. 4). Bright calcein-AM staining at the end of imaging confirmed intact tissue viability after cycling, with Hoechst 33342 added as a nuclear reference marker for enumeration of all cells. Insets within the figure illustrate key marker staining patterns both within/across cycles, including positive/negative controls (e.g., GFP+, CD11b+, Ter119− macrophages) and the ability to assemble a cellular immunophenotype from successive rounds of imaging. The full multiplexed data and additional inter-cycle composite images that highlight tissue architecture are presented in Supplementary Figs. 7 and 8.
Figure 4 |. SAFE imaging of living hepatic tissue.
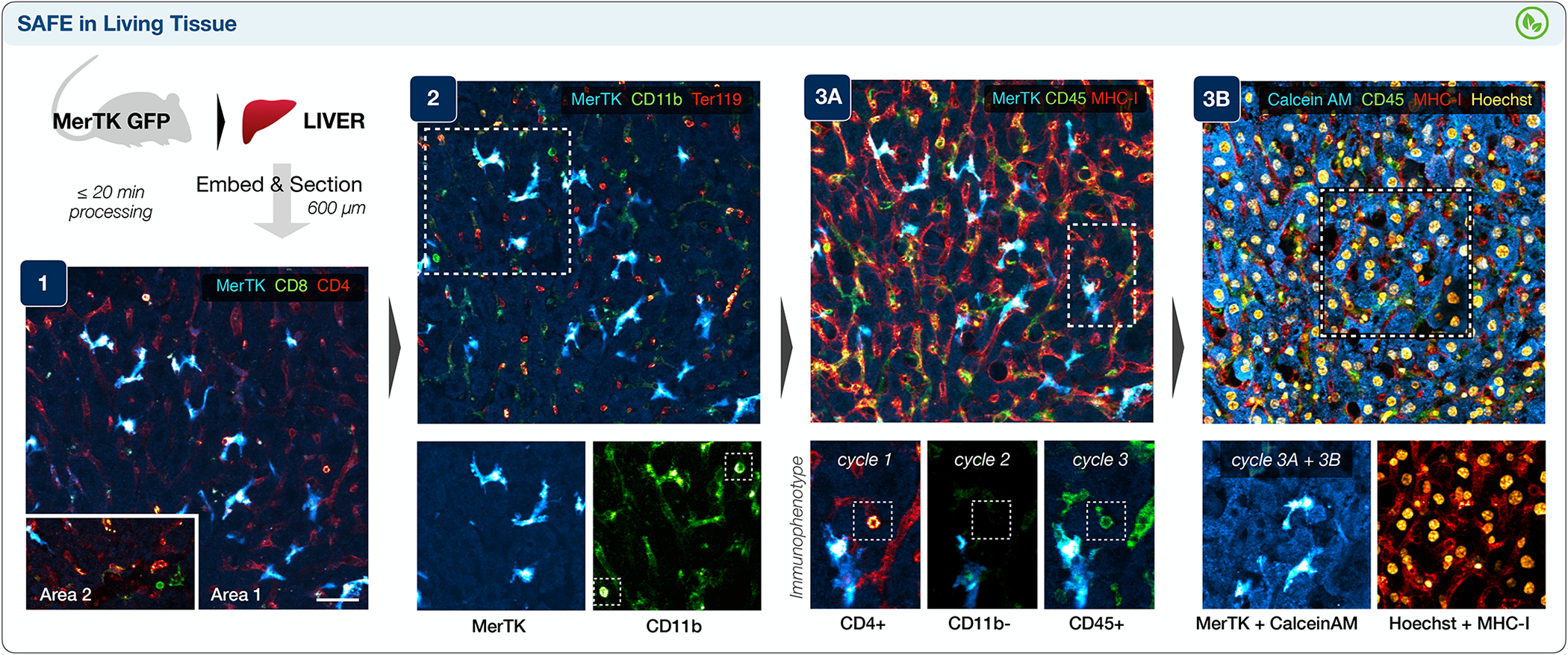
Surgically-harvested liver from a mouse bearing GFP+ macrophages (MerTK-GFP) was swiftly chilled, embedded in agarose, sectioned, mounted in live cell imaging solution at 37 °C, and profiled in three cycles. Cycle 1 highlighted CD4+ sinusoidal endothelium68, bright CD4+ T-cells, and rare CD8+ T-cells (area 2). Following scission, cycle 2 identified CD11b+ MerTK-GFP macrophages and CD11b bright immune cells (inset), as well as Ter119+ red blood cells within the sinusoids. Integration of immunophenotypic data across three cycles was also feasible with cross-registration of the MerTK channel, allowing clear identification of a (CD45+, CD4+, CD11b-) T-cell (cycle 3A). Finally, the addition of Hoechst and Calcein AM (cycle 3B) enabled nuclear identification and confirmed brisk activation of the live-cell dye across the imaged living tissue section. The experiment was repeated independently two times, with two liver sections imaged per mouse and 2 or 3 regions of interest imaged per section; the scale bar is 40 μm.
Profiling the differentiation of living bone marrow at high spatial resolution.
Hematopoietic development generates phenotypic diversity as multipotent stem/progenitor cells differentiate into their repertoire of mature progeny. High-dimensional multiplexed profiling41 and single-cell gene expression/barcoding technologies42 have traced developmental hierarchies of hematopoietic lineages. Directly visualizing the bone marrow cells and their ensemble of surface markers amidst these changes, however, has not been possible. As an initial test of the feasibility of SAFE profiling of living bone marrow, freshly harvested murine bone marrow cells were counted and put into culture overnight. We profiled 12 immune markers (3 markers/cycle, 4 cycles total), with a focus on myeloid precursors (Fig. 5A), using confocal imaging to distinguish membrane/cytoplasmic staining. Viability was confirmed by Calcein blue AM staining with the final cycle of the profiling sequence (Supplementary Fig. 9).
Figure 5 |. SAFE imaging of living bone marrow.
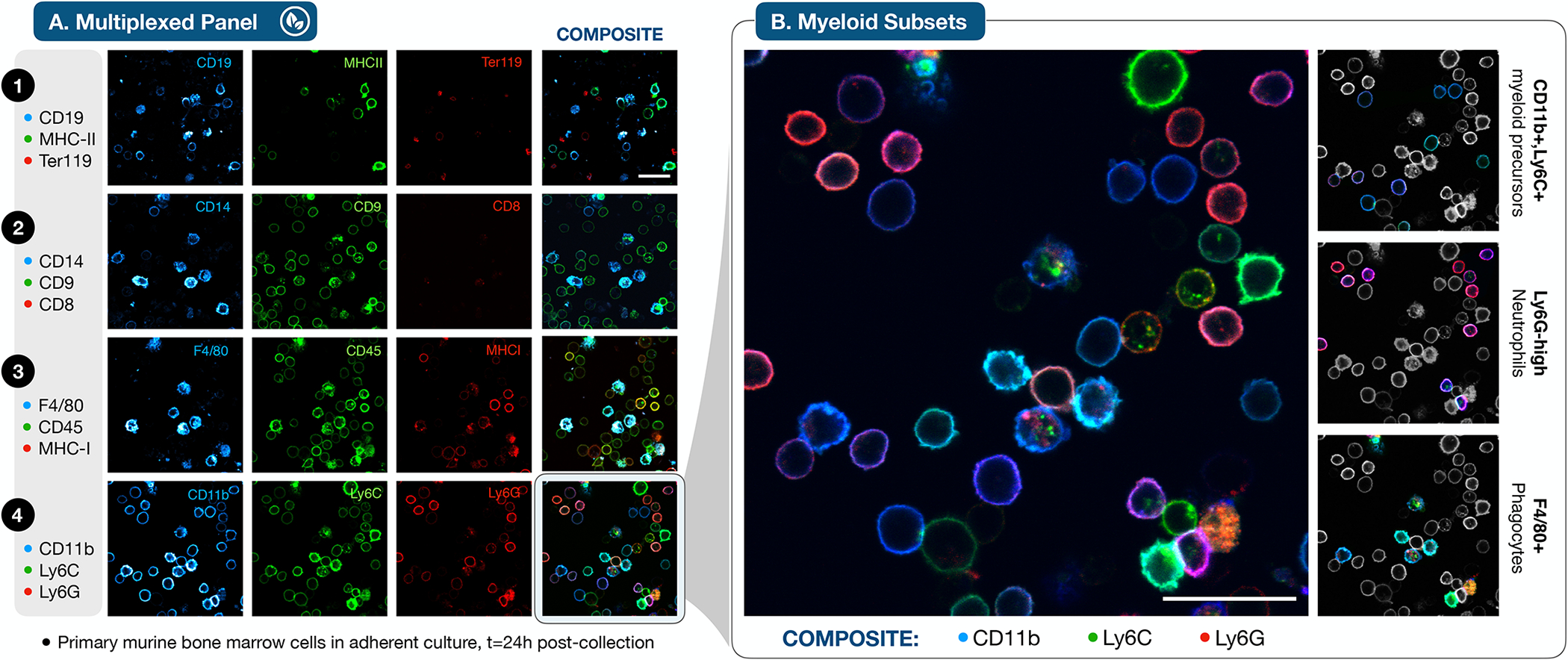
A. Primary murine bone marrow cells were isolated, plated in adherent culture and imaged after 24 hours. Confocal microscopy of 12 markers profiled in 4 cycles enabled discrimination of intracellular and extracellular fluorescent staining at high spatial resolution. A minority of cells displayed intracellular staining that was not removed by scission, suggestive of antibody internalization prior to fluorophore scission with BHQ3-N-Tz. The scale bar is 25 μm. B. A focus on the myeloid markers in cycle 4 reveals an ensemble of myeloid precursors (CD11b+, Ly6C+), Ly6G-Hi neutrophils, and F4/80+ phagocytes (F4/80+) within the developing bone marrow cell population (scale bar 25 μm), highlighted in the selectively (de)colorized individual panels at right. Experiments were repeated independently three times.
CD8 positive cells were rare, indicating that non-adherent T-cells were not well retained in this staining format. Images demonstrated the presence of erythroid (Ter119+), lymphoid (CD19+), and myeloid (CD11b+) subsets within the population, including cells across a spectrum of functional differentiation. A continuum of CD11b, Ly6C, and Ly6G expression was evident in the high-resolution composite image (Fig. 5B), with myeloid precursors (CD11b+, Ly6C+) that render as blue-cyan, and mature Ly6G+ neutrophils that appear magenta/red. A subset of phagocytically-proficient cells (CD14+ and F4/80+ in cycles 2 and 3, respectively) exhibited a pattern of multi-color intracellular staining consistent with antibody internalization prior to scission, in addition to the ON/OFF cycling of specific cell-surface markers. Apart from this subpopulation, negligible cytoplasmic signal was observed after four complete cycles, indicating the feasibility of accurate, iterative staining and scission in this cellular context.
To implement longitudinal bioorthogonal profiling on longer time scales, i.e., across not just multiple cycles but multiple days, we selected a model system with well-defined cellular dynamics. Estrogen-regulated Hoxb8 (ER-Hoxb8) hematopoietic progenitor cells are conditionally immortalized by retroviral infection of myeloid bone marrow progenitors43. Removal of ß-estradiol from culture media triggers the ER-Hoxb8 differentiation program and cells become mature neutrophils by day 4–6, a developmental sequence that has been well-characterized by flow cytometry and gene expression (Fig 6, inset)44. Loss of cKit expression and sequential upregulation of CD11b, Ly6C, and Ly6G occur in tandem with population expansion.
Figure 6 |. Longitudinal profiling of neutrophil differentiation.

A. Deep PDMS microwells retain suspension cells in culture for serial imaging (scale bar, 50μm). Bone marrow derived ER-Hoxb8 cells are conditionally immortalized when cultured with ß-estradiol; removal of ß-estradiol triggers synchronous expansion and neutrophilic differentiation. This developmental sequence induces broad changes in extracellular marker expression and cellular morphology (gray panel, inset). To track this process, CD45, cKit, CD11b, Ly6C, Ly6G, and F4/80 were serially profiled at days 0, 2, 4, and 6, before and during differentiation. CD45 (positive control) and F4/80 (negative control) expression were stable, while cKit (a progenitor cell marker) expression quickly decreased, disappearing after D0. As expected, the fraction of cells expressing CD11b, Ly6C, and Ly6G rose in sequence over the first 4–6 days. B. Quantitative expression of differentiation markers across six days and four profiles: (i) averaged across a set of n = 12 microwells (plotted as mean, SD); (ii) quantified on day 4 to track proliferation and variation across individual microwells. Ring plots track marker expression (fraction of positive cells, encoded by color) and the total number of CD45+ cells per well (in white).
We thus loaded a sparse density of ER-Hoxb8 cells into PDMS microwells (W: 60 μm, H: 80 μm, Fig. 6A), fabricated as in the PBMC studies (vide supra). The same cells were imaged longitudinally during differentiation (D0, D2, D4, D6) in two profiling cycles at each time point, staining for CD45 and F4/80 as positive/negative controls, respectively, in addition to the quartet of differentiation markers. At D0, all cells were CD45+/cKit+ and negative for the remaining panel, as expected. Rapid population growth by D2 tracked with broad upregulation of CD11b and a subset of Ly6C positive cells. The density of viable cells peaked on D4, concurrently with the appearance of bright Ly6G staining. Given the short lifespan of differentiated granulocytes45, population density had decreased slightly by D6, consistent with the fraction of dead cells normally observed by flow cytometry. F4/80 staining remained negative throughout, as expected for differentiation under neutrophil-selective culture conditions. Cell growth during temporospatial profiling was vigorous, allowing quantitative analysis of both proliferation and differentiation across the individual microwells (Fig. 6B, Supplementary Fig. 10).
Discussion
The bioorthogonal framework of SAFE creates a new method for multiplexed longitudinal profiling of living biological samples, applied here to demonstrate 9 color imaging of murine hepatic tissue and 14 color imaging of primary hematopoietic and immune populations, across a timescale spanning minutes to days. Methods that incorporate multiple cycles of fluorescent staining and quenching were originally developed for paraffin-embedded tissue sections that can withstand harsh destaining conditions12,46,47. We and others have subsequently developed gentler DNA-barcoded antibody technologies for cellular profiling14,15,48–50, but these too require long destaining times and solvent/buffer conditions incompatible with live cell analysis. Our recent method for immunostaining with Tz/TCO-driven bioorthogonal quenching33,51 achieves its speed by circumventing the need to remove fluorophores. While capable of rapid temporal cycling at nontoxic reagent concentrations, the quenching is reversed by biochemical degradation over longer timeframes in living systems, leading to signal rebound (Fig. 3B, control). In contradistinction, the SAFE method bypasses the above shortcomings and allows efficient, durable cycling (>99% signal clearance in <30 sec, Fig. 2D), while using nanomolar concentrations of reagents for gentle and irreversible fluorophore scission. By accelerating the scission sequence with cooperative BHQ3-fluorophore interactions, the SAFE reagents achieve a unique combination of intrinsic biochemical stability (C2TCO, >97% intact at 48 hrs) and high reactivity that arises as an emergent property of the system. To the best of our knowledge, SAFE is currently the only technique that allows such temporospatial profiling in living cells and tissues.
Bioorthogonal reactions have long been described in live cells as an alternative route to biomolecular tagging52, with incorporation of functionalized noncanonical amino acids into proteins53,54, modified nucleosides into DNA/RNA55, metabolic building blocks into glycans56, or clickable tags into chemical probes57,58, followed by subsequent click-mediated labeling. The impetus for developing these strategies has primarily been visualization of the different classes of biomolecules in living systems, exploiting the small footprint of compact chemical tags for tracking56 or creating a readout for replication/metabolism55. Such tags have also featured in strategies for super-resolution microscopy59–61. Here, we apply chemical tools not to deliver labels but rather to remove them via bioorthogonal bond cleavage62,63, pursuing multiplexed temporal analysis of cell-surface protein expression.
Although non-lethal methods for intracellular antibody delivery have been explored64–66, such tools are not yet compatible with routine immunostaining or the construction of multiplexed panels; these limitations currently restrict SAFE multiplexed profiling of living cells and tissues to extracellular markers. Likewise, the present iteration of the SAFE chemistry relies on diffusion for clearance of the cleaved dye, a facile process with cell-surface targets but less straightforward for hydrophilic, membrane-impermeable fluorophores released within the cytoplasm. Membrane permeability of BHQ3-Tz scissors (or other bioorthogonal scission machinery) would also need to be further defined. Differences in the speed of scission/signal clearance within tissues relative to cells (Supplementary Fig. 6) are another area for future study and suggest an opportunity for orientation-controlled cleavage as a mechanism to release only dark fluorophores (Supplementary Fig. 2, scenario i.B).
The proof-of principle experiments shown here can be further expanded to increase the number of cycles/targets within each profile, assembling broader marker panels for study of complex living tissues, especially in immunologically complex environments such as lymph node, tonsil, spleen, or tumor tissue. Observed scission efficiencies of >99.5% for cells and ~98% in tissue should permit >12 cycles of multiplexing at robust signal-background ratios (Supplementary Fig. 1b). One of the key advantages of the SAFE method is that the underlying bioorthogonal scission reaction is compatible with any conjugatable dye, including polymeric fluorochromes (e.g., the BV dye series) that are otherwise difficult to quench. Since a range of bright polymer dyes can be excited in the violet/UV, their use together with organic rhodamine/cyanine dyes could enable broader multiplexing of up to ~8 channels per cycle with minimal spectral overlap. In this mode, the potential ability of BHQ3-BVdye interactions to accelerate the click/scission reactions awaits study, but forthcoming developments in Tz/TCO scission chemistry will also provide an alternative. Finally, while our focus here was on demonstrating temporospatial analysis in well-defined model systems, the above approaches open the door to multiplexed functional profiling and longitudinal observation of a broad range of living cells and tissues, immediately following explantation or even in vivo.
Online Methods
Chemistry.
Synthesis and characterization.
Detailed synthetic methods and characterization data are presented in full in the Supplementary information, including probe activation, bioorthogonal click and scission kinetics, NMR spectra and ESI-MS data.
Bioconjugation and routine imaging methods/controls.
Antibodies.
BSA free antibodies were purchased from established vendors and stored according to manufacturer recommendations. Cetuximab (anti-EGFR antibody, Erbitux) was used to optimize staining and quenching/scission methods. Antibodies used for all SAFE imaging throughout the study are tabulated in Supplementary Table 1. All antibodies were validated with conventional immunofluorescence imaging (secondary antibody staining) on positive cell lines or mouse splenocytes before usage in SAFE imaging.
Antibody modification with SAFE probes.
For functionalization with fluorophore-C2TCO conjugates, antibodies were first exchanged from their storage solution into PBS-bicarbonate buffer (PBS supplemented with 100 mM sodium bicarbonate, pH 8.4) using a 40k Zeba Spin column (Thermo Fisher, 87765). After buffer exchange, antibodies were incubated with a 4- to 8-fold molar excess of the activated Dye-C2TCO-NHS linker for 25 mins at room temperature. The completed conjugation reaction was loaded onto another 40k Zeba column (equilibrated with PBS) for desalting and removal of unreacted dye molecules. The absorbance spectrum of the conjugated antibody was measured using a Nanodrop 1000 (Thermo Scientific) to determine the degree of labeling (DOL), applying the known extinction coefficients of the dye, IgG antibody, and standard correction factor (CF280) for the dye absorbance at 280 nm. The SAFE-conjugated antibodies were stored in the dark at 4 °C in PBS until usage.
Immunostaining and fluorophore scission.
Cells were cultured as specified (vide infra), stained with 5 μg/mL SAFE probes and incubated for 10 mins at 4 °C. Hoxb8 cells were first blocked with 0.5% BSA-PBS for 10 mins at 4 °C before staining. Cetuximab staining of A549 and A431 cells is indistinguishable with/without blocking, so no pretreatment was applied. After imaging, a 500 or 600 nM solution of BHQ3-N-Tz (2) diluted in PBS was used to wash cells for quenching and scission; while signal removal is characteristically complete in 30–60 seconds, we used a routine interval of up to 3–5 minutes to ensure complete removal of residual fluorescence. For the longitudinal imaging kinetics (Fig. 2D), cells were serially imaged during the click-scission process to quantify the aggregate quench-scission rates (Supplementary Fig 3). In routine experiments, the cells and remaining BHQ3-N-Tz were washed three times post-incubation with PBS in immediate succession. After quenching/scission, the staining, imaging, and scission cycle was repeated for multiplexed protein profiling of the same cells. Imaging was performed in Live Cell Imaging Solution (Thermo Fisher, A14291DJ). Calcein AM (Thermo Fisher, C1430) and SYTOX Red (Thermo Fisher, S34859) were used for live and dead cell staining and Hoechst 33342 (Thermo Fisher, H3570) was used for nuclear staining in a subset of experiments. Hoechst staining was associated with an increased rate of phototoxic stress in live cell microscopy, especially in longitudinal imaging, so its use was reserved for short-term experiments and for those in which nuclear morphology was particularly relevant/necessary.
Longitudinal acquisition - photobleaching control.
A431 cells were stained with 5μg/ml of SAFE647-conjugated cetuximab for 10 minutes at RT and then washed three times with PBS (5–10 sec each). Images were then acquired every 5 seconds at experimental conditions (illumination intensity, exposure time, microscope) matched to those of Fig. 2D. As illustrated by the images and intensity profiles at selected timepoints in Supplementary Fig. 3, no measurable photobleaching was observed.
SAFE specificity.
To verify that staining accuracy is not affected by the bioorthogonally cleavable linker, SAFE-modified antibodies were compared to conventionally labeled controls. A representative panel of anti-CD11b (MB488), CD45 (AF555), CD8 (MB594), and Ly6G (AF647) antibodies were modified with individual SAFE probes, as (specified). In parallel, control antibodies in a non-overlapping color/channel were prepared via conventional activated ester labeling. AZDye 555 NHS Ester (Fluoroprobes, 1166–5) was used to label anti-CD11b, while AFDye 488 TFP ester (Fluoroprobes, 1026–5) was used to label the parent anti-CD45, CD8, and Ly6G antibodies. Control antibodies with a degree of labeling (DOL) of 2–3 fluorophores/antibody were prepared to match the DOL of SAFE modified antibodies. Control and SAFE modified antibodies (each at 5 μg/mL) were then premixed and co-incubated with mouse splenocytes for 10 minutes at room temperature. After washing three times with PBS (5–10 sec each), the stained cells were imaged on μ-Slide 8 Well Glass Bottom slides (Ibidi, 80826) using a DeltaVision inverted fluorescence microscope (Supplementary Fig. 5).
Fluorescent imaging and analysis.
An Olympus BX-63 upright automated epifluorescence microscope and a DeltaVision inverted fluorescence microscope were used to acquire fluorescent images. DAPI, FITC, TxRed/Cy3, and Cy5 filter cubes were used to excite Calcein AM live cell stains, AF488, AF594, and AF647 fluorophores respectively. ImageJ was used to measure fluorescent intensities in cells.
Cell culture methods.
Cell line culture.
The A549 (CCL-185) and A431 (CRL-1555) cell lines were purchased from the American Tissue Culture Collection (ATCC) and used in method development experiments. A549 cells were passaged in F-12K (ATCC, 30–2004) with 10% FBS and 1% penicillin/streptomycin according to the specifications from ATCC. Cells were first grown in a 150 mm cell culture dish and then seeded on μ-Slide 8 Well Glass Bottom slides (Ibidi, 80826) for imaging. After 24–48 hours, confluency was assessed and cells were stained and imaged.
Cytotoxicity assays.
A431 cells were first seeded on 96 well plates at a density of ~10,000 cells/well. After 48 hr of culture (DMEM), baseline cell viability was benchmarked using the PrestoBlue Cell Viability Reagent (Thermo Fisher, A13262) to control for any well-to-well variability in post-seeding proliferation. The cells were washed with fresh DMEM after the PrestoBlue measurement and then incubated with BHQ3-N-Tz (0–10 μM) in cell culture media. DMSO control wells were prepared to match the DMSO content (0.42%) of the 10μM BHQ3-N-Tz sample. For the remaining reagent concentrations, media alone was used as the control, given minimal DMSO content (<0.13%). A continuous exposure of 1 hr at 37°C was selected, corresponding to the cumulative duration of 12 to 20 cycles of quenching/scission (3–5 minutes each). In parallel, a separate set of cells were incubated in DMEM with 5 μg/ml of Cetuximab-SAFE647 or conventionally labeled Cetuximab-AF647 as a control. Media were exchanged after incubation and the cells were returned to culture for 24 hours. Cell viability was then measured with the PrestoBlue assay, with the data normalized to the baseline values from the start of the experiment (Supplementary Fig. 4).
Microwell fabrication.
The microwells used to culture and/or image nonadherent cells were fabricated at the Soft Materials Cleanroom (SMCR), Harvard Center for Nanoscale Systems (CNS). Each microwell array consists of 1681 total wells. The microwells (W: 60 μm, H: 80 μm) were made using soft lithography with SU-8 3050. Different widths were tested (60, 80, 100 μm) and 60 μm was chosen, a dimension that demonstrated no detectable cell loss throughout multiple experimental steps (e.g., media change, incubation, washing, culture, etc.). Polydimethylsiloxane (PDMS) was spin coated on a patterned wafer, cured in 65 °C for an hour, peeled off from the wafer, and then transferred to μ-Slide 8 Well Glass Bottom slides (Ibidi) for cell culture and imaging.
Longitudinal imaging of A549 Cells.
SAFE labeling.
A549 cells were cultured in μ-Slide 8 Well Glass Bottom slides (Ibidi) for 24–48 hours before imaging. After 24–48 hours, cells were stained with 5 μg/mL of SAFE conjugated (AF647) anti-EGFR antibody for 10 mins at RT, and then rinsed with three successive washes of PBS, culminating with addition of live cell imaging solution (Thermo Fisher, A14291DJ). After image acquisition, quenching was performed by incubating the stained live cells with BHQ3-N-Tz (2) at 600 nM. Fluorescent images were acquired after staining, 1 min after quenching/scission, and 24 hours after scission. After 24 hours, cells were stained with calcein AM (Thermo Fisher, C1430) and SYTOX Red (Thermo Fisher, S34859) to check cell viability. A DeltaVision inverted fluorescence microscope was used to acquire all the fluorescent images in this study.
No scission control experiments.
A549 cells were cultured and prepared as above and then stained with a 5 μg/mL of AF647-rTCO-conjugated (FAST647, Ko et al, ACIE 2020) anti-EGFR antibody for 10 mins at RT. This antibody can click with a BHQ3-Tz quencher, but the linker/dye remain permanently attached to the antibody: i.e., no scission is possible, only quenching. After image acquisition, the stained live cells were incubated with the non-scissile BHQ3-Tz (600 nM, 1 min), as previously described. Fluorescent images were acquired after staining, 5 min after quenching, and 24 hours after quenching to quantify rebound. After imaging at 24 hours, cells were stained with Calcein AM (Thermo Fisher, C1430) and SYTOX Red (Thermo Fisher, S34859) to check cell viability.
SAFE Imaging of mouse PBMCs (Fig 3C).
Ethics and Veterinary Care.
Oversight for this and subsequent animal experiments was provided by the Institutional Animal Care and Use Committee (IACUC) for Massachusetts General Hospital and Shriner’s Hospital, approved under Animal Protocols 2013N000157 (murine tissue harvest and imaging) and 2017N000255 (murine bone marrow harvest and culture). Mice were housed at an ambient temperature of 68–73 F, with 30–70% humidity; the light/dark cycle was 12h light (7am-7pm) and 12h dark (7pm-7am).
Mouse PBMC isolation.
Whole blood was collected from C57BL/6J mice (Jackson Laboratory) by cardiac puncture. Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation. Lymphoprep (Stem Cell Technologies, 07801) was used as density gradient medium. Whole blood was diluted with an equal volume of PBS with 2 % FBS, layered with Lymphoprep on top, and centrifuged at 800 × g for 20 minutes at room temperature with break off. The upper plasma layer was discarded and the cloudy mononuclear cell layer was gently collected in a separate tube. Collected cells were washed with PBS and then cultured for imaging.
SAFE imaging in microwells.
Isolated mouse PBMC were diluted in PBS to provide an average of 10 cells per well and loaded to microwell arrays for cycling and imaging. For each cycle, cells were stained with a cocktail of three SAFE antibodies (5 μg/mL) conjugated to three different fluorophores (AF488, AF594, AF647) for 10 mins at RT. Standard scission conditions used 2 (600 nM) for one to three minutes. For the 5th cycle, cells were stained with Calcein AM (Thermo Fisher, C1430) and SYTOX Red (Thermo Fisher, S34859) per the vendor protocol to check cell viability after the cumulative profiling experiment. Staining and quenching solutions were diluted in PBS and imaging was performed with cells in live cell imaging solution (Thermo Fisher, A14291DJ). Images were acquired using a DeltaVision inverted fluorescence microscope to capture 6 fields of view (72 microwells). Cells were counted in ImageJ and results tabulated in Microsoft Excel (version 16.53) and/or Numbers (Apple, version 10.0).
Flow Cytometry Control Experiments.
Mouse PBMCs were incubated with True Stain FcX antibody (clone 93, BioLegend) in PBS with 2% FBS before staining with antibodies directly conjugated to fluorophores. Cells were divided into two tubes to stain for myeloid markers (CD11b, CD11c, Ly6C, Ly6G, MHC I, MHC II) or lymphoid markers (CD3, CD4, CD8, CD19, c-kit). Antibodies are summarized in Supplementary Table 2. DAPI was used to exclude dead cells from analysis. Cells were washed and strained through a 40 μm filter (BD Falcon, 352340) after staining and analyzed on a BD LSRII flow cytometer. AbC Total Antibody Compensation Bead Kit (Thermo Fisher, A10513) was used for single color compensation. Flow cytometry data were then analyzed using FlowJo software (version 10.8.0, Becton Dickinson) to quantify the frequency of cells positive for each marker; the gating strategy is outlined in Supplementary Fig. 6. Lymphoprep centrifugation is selective for T, B, NK, and monocyte populations and excludes granulocytes, consistent with the observed low abundance of those myeloid markers (e.g., Ly6G+) in the cells analyzed. Percentages (SAFE, FLOW) of positive cells for each marker are c-Kit (0.5%, 0.9%), CD8 (13.7%, 13.1%), CD11c (2.1%, 3.2%), CD19 (66.9%, 55.8%), CD45 (98.6%, 99.1%), CD11b (6.5%, 11.9%), CD4 (16.2%, 9.9%), CD3 (30.8%, 25%), MHCII (72.3%, 66.1%), MHCI (22.1%, 23.7%), Ly6G (0.0%, 0.2%), Ly6C (11.6%, 7.7%).
Live tissue imaging.
Specimen preparation.
Freshly harvested liver from a Mer tyrosine kinase (MerTK)-GFP mouse was embedded in 2.5% agarose and immediately sectioned with a vibrating microtome (VF-310-0Z-VM, Precisionary Instruments). The sample and the immersion bath solution were kept at 4°C during the entire slicing procedure. Slices of 500 and 700 μm thickness were obtained with optimal sectioning parameters set to 3 and 6 for the advance and oscillation speed respectively, then kept in cold media on ice until imaging. The living tissue sections were affixed to a glass slide with a small dab of cyanoacrylate adhesive. The slide was then mounted on a heated stage (37°C) on a custom-made Olympus FV1000 confocal multi-photon system and imaged using a 20x XLUMPLFLN20x water immersion objective (NA = 1.0, Olympus America).
Real-time tissue staining/scission.
A freshly cut slice was stained with anti-CD45 (AF555) at 5 μg/mL for 5 minutes at room temperature and then rinsed three times (5–10 sec each) with live cell imaging solution (LCIS, Thermo Fisher, A14291DJ). The tissue slice was then immersed in LCIS and monitored longitudinally in the MerTK-GFP, CD45-AF555, and 647 channels. After 36.5 minutes of acquisition (Z-stack, 15 planes, 4 μm spacing, frame interval 134.6 sec), the live tissue was stained with a SAFE647 modified anti-MHC-I antibody for 11 minutes (5 μg/mL in LCIS), followed by addition of BHQ3-N-Tz (1 μM, t=47 min) for real-time scission while imaging (Supplementary Fig. 6, Supplementary Movie 1). Quantitative kinetic analyses were derived from intensity profiles (pixel brightness vs coordinate) calculated in ImageJ and exported to GraphPad Prism for descriptive statistics. Mean and 95th percentile intensity data for each intensity profile were plotted as a function of time, with staining/scission kinetics synchronized to the time of antibody addition and BHQ3-N-Tz addition, respectively (Supplementary Fig. 6). The observed rates were used to guide the design of subsequent multiplexing.
Tissue multiplexing.
Experiments were conducted by staining the live MerTK-GFP liver tissue with anti-CD4 (SAFE647) and anti-CD8 (SAFE555) antibodies (cycle 1); anti-CD11b (SAFE555) and Ter119 (SAFE647) antibodies (cycle 2); and anti-CD45 (SAFE555) and anti-MHC-I (SAFE647) antibodies (cycle 3A). For cycle 3B, Calcein AM (4 μM, Thermo Fisher, C1430) and Hoechst 33342 (32.4 μM, Thermo Fisher, H3570) were added to the already-labeled tissue. For staining, the tissue was incubated with SAFE-labeled antibodies (5 μg/mL, 6 mins) in LCIS and washed 3 times (5–10 sec each) with PBS prior to imaging in fresh LCIS. For scission, the liver section was immersed in BHQ3-N-Tz (1 μM in PBS) for 15–20 minutes to allow for tissue penetration, click reaction, scission, and optimal diffusion/clearance of the cleaved fluorophores from the tissue, as derived from kinetic studies above. The tissue was then washed three times with PBS (5–10 sec each) before application of the antibody solution for the next staining cycle. See also Supplementary Figs. 7 and 8.
Mouse Bone Marrow Imaging.
Mouse bone marrow isolation and culture.
Microcentrifuge tubes (0.5 mL) were prepared by removing the lids (scissors) and piercing a hole through the bottom of the tube using a 16G sterile needle. Wild type mice (C57BL/6J females, age 10–12 weeks) were euthanized by carbon dioxide asphyxiation and the femurs removed in sterile fashion. The distal end of the femur was opened using scissors and the femur placed with the open end facing down into the bottom of the 0.5 mL microcentrifuge tube. This tube was placed inside a 1.5 mL microcentrifuge tube and the tube-within-a-tube centrifuged at 10,000 g for 15 seconds to “spin-flush” the bone marrow. The 0.5 mL microcentrifuge tube and “empty” femurs were discarded and the bone marrow pellet (~25 μL) resuspended in PBS containing 2% FBS and 1 mM EDTA. Mononuclear cells were enumerated by staining a small aliquot with acridine orange (Thermo Fisher, A3568) and counting using a fluorescence-based cell counter (Cellometer, Nexcelom).
Primary murine bone marrow cells were cultured in μ-Slide 8 Well Glass Bottom slides (Ibidi) for 24 hours in RPMI1640 media with glutamine, supplemented with 10% FBS, SCF (50 ng/mL), IL3 (10 ng/mL), and IL6 (10 ng/mL). To improve cell adhesion to the slides, various coating methods (Fibronectin, Poly-D-Lysine, Lipopolysaccharide, ibiTreat tissue culture-treated slides from Ibidi) were tested and ibiTreat slides were selected, as they showed the best cell adhesion. After 24 hours of cell culture, cells were stained with a cocktail of three SAFE antibodies (5 μg/mL, 10 mins at RT) and quenched with BHQ3-N-Tz (1 μM, 5 mins at RT) for each cycle.
Imaging.
Cells were imaged in live cell imaging solution (Thermo Fisher, A14291DJ) at room temperature. Images were collected using a custom-made Olympus FV1000 confocal multi-photon system using a 60x LUMFL N water immersion objective (NA = 1.1, Olympus America). Fluorescent channels were imaged sequentially with 405 nm, 473 nm, 559 nm and/or 635 nm lasers using a 405/473/559/635 dichroic to separate excitation light and dichroic beam splitters SDM473, SDM560, and SDM640 to separate emission light. Fluorophore signals were further separated using emission filters BA430-455, BA490-540, BA575-620 and BA655-755. Each fluorescence channel was imaged sequentially using distinct excitation and emission filter sets to ensure minimal bleed-through between channels. All lasers, beam splitters, and emission filters were purchased from Olympus. For the final cycle of the imaging sequence, cells were co-incubated with calcein blue AM (Thermo Fisher, C1429, see also Supplementary Fig. 9).
ER-Hoxb8 Differentiation Model.
ER Hoxb8 Cell culture.
ER-Hoxb8 cells were derived from the bone marrow of male ER-KO C57BL/6 CD45.1STEM mice67. Detailed protocols for construction of the retroviral vector encoding ER–Hoxb8, virus production, and transduction of bone marrow myeloid progenitor cells were described previously43 and are available online: http://sykeslab.com/reagent-request. In brief, bone marrow mononuclear cells were isolated and expanded in medium containing SCF, IL-6, and IL-3 (10 ng/mL) for 48 hrs. Cells were placed in 12-well plates pre-coated with human fibronectin (Sigma F-0895) and spin-infected with murine stem cell virus encoding ER–Hoxb8, as previously described. Infected cells were cultured in RPMI medium containing SCF (20 ng/mL) and β-estradiol (500 nM) for two days prior to addition of G418 for selection. The cells were passaged to new plates with fresh medium every 3–4 days and non-adherent immortalized cells grew out in about 3 weeks. These cells were maintained in culture no more than 4 weeks (6-well culture plate) until needed for microwell imaging experiments.
ER Hoxb8 longitudinal microwell imaging.
ER-Hoxb8 cells were imaged longitudinally on Day 0, 2, 4, and 6. On Day 0, cells were stained with a cocktail of two SAFE conjugated antibodies (anti-CD45, anti-cKit) for 10 mins at 4°C. The antibodies were diluted to 5 μg/mL in cell culture media with β-estradiol as stated above. The stained cells were washed three times with the cell culture media with β-estradiol. After washing, live cell imaging solution (Thermo Fisher, A14291DJ) containing 0.1 mM Trolox (Vector Labs, CB1000-2) was added to the cells for imaging. Images were acquired with a DeltaVision inverted fluorescence microscope, with the exposure time set to 100 ms to minimize phototoxicity to the living cells across multiple imaging sessions. Quenching was performed with BHQ3-N-Tz (2) diluted to 500 nM in cell culture media with β-estradiol and incubated with cells for 10 mins at RT. After quenching, cells were washed with cell culture media without β-estradiol for three times and placed back in the incubator for further culture. On Day 2, 4, and 6, the same procedure was conducted with two cycles of staining and quenching. The first cycle included staining with a cocktail of three SAFE conjugated antibodies (anti-CD45(MB594), anti-cKit(anti-Rat-647), anti-F4/80(AF488)) and the second cycle included staining with a cocktail of the remaining three SAFE conjugated antibodies (anti-Ly6C(MB594), anti-Ly6G(AF647), anti-CD11b(MB488)). Each image captured a set of 12 microwells, and four imaging sites on the chip were captured in each profiling session (n = 48 wells, total).
Neutrophil differentiation - flow cytometry control experiments.
ER-Hoxb8 immortalized progenitors and SCF-dependent neutrophil progenitors were kept on ice for all steps if not stated otherwise. Cells were washed and resuspended in PBS and stained with Zombie Aqua fixable viability dye (BioLegend 423101) to exclude dead cells. Cells were washed with PBS and incubated with FcBlock (clone 93, BioLegend) for 15 min at 4 °C, followed by staining with fluorescently conjugated antibodies for 25 min at 4 °C in staining buffer (0.5% BSA in PBS). The cells were washed with staining buffer and analyzed on a LSRII flow cytometer (BD). Following cell populations were identified based on cell marker expression: ER-Hoxb8 immortalized progenitors (CD45+ CD11b− cKit/CD117+), neutrophils (CD45+ CD11b+ cKit/CD117− Ly-6G+). Antibodies are reported in Supplementary Table 2.
Data Availability Statement
All data that support the observations and conclusions of the study are included in the manuscript and its Supplementary Information. Raw multichannel and/or Z-stack source data from time series images are available in TIF format at https://doi.org/10.5281/zenodo.6482316.
Supplementary Material
Acknowledgments
We are grateful to the Sykes Laboratory for preparing live bone marrow cells and to Conor Carlson-O’Fallon for assistance with imaging data analysis. This work was supported in part by grants from the CSB development fund (JCTC), R01CA257623 (RW), UH3CA202637 (RW), R01CA206890 (RW, MP), P01CA069246 (RW), U01CA206997 (RW), P01CA240239 (MP), R01CA229777 (RW); JK was supported by the Schmidt Science Fellows and K99CA256353.
Footnotes
Competing Interests Statement
The authors declare the following competing interests: Drs. Carlson, Weissleder, and Mikula declare the filing of a patent (PCT/US2021/053439, pending; Bioorthgonal linkers and reactions) which was assigned to Massachusetts General Hospital; R.W. is a consultant to ModeRNA, Tarveda Therapeutics, Lumicell, Seer, Earli, Aikili Biosystems and Accure Health, consultancies which are unrelated to the subject matter of this work.
References
- 1.Sullivan ZA et al. γδ T cells regulate the intestinal response to nutrient sensing. Science 371, eaba8310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patriarchi T et al. An expanded palette of dopamine sensors for multiplex imaging in vivo. Nat Methods 17, 1147–1155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costantini LM et al. A palette of fluorescent proteins optimized for diverse cellular environments. Nat Commun 6, 7670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez EA et al. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem Sci 42, 111–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polonsky M et al. Induction of CD4 T cell memory by local cellular collectivity. Science 360, eaaj1853 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Arlauckas SP et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zindel J et al. Primordial GATA6 macrophages function as extravascular platelets in sterile injury. Science 371, eabe0595 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Alon S et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science 371, eaax2656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho CS et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559–3572.e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng CL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stickels RR et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol 39, 313–319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin JR, Fallahi-Sichani M & Sorger PK Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun 6, 8390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goltsev Y et al. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo SM et al. Multiplexed and high-throughput neuronal fluorescence imaging with diffusible probes. Nat Commun 10, 4377 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saka SK et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat Biotechnol 37, 1080–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radtke AJ et al. IBEX: A versatile multiplex optical imaging approach for deep phenotyping and spatial analysis of cells in complex tissues. Proc Natl Acad Sci U S A 117, 33455–33465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bendall SC et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson HW et al. The single-cell pathology landscape of breast cancer. Nature 578, 615–620 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Hartmann FJ & Bendall SC Immune monitoring using mass cytometry and related high-dimensional imaging approaches. Nat Rev Rheumatol 16, 87–99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahdessian D et al. Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590, 649–654 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinnon KM Flow Cytometry: An Overview. Curr Protoc Immunol 120, 5.1.1–5.1.11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giannone G et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys J 99, 1303–1310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strauss S & Jungmann R Up to 100-fold speed-up and multiplexing in optimized DNA-PAINT. Nat Methods 17, 789–791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bechtel TJ, Reyes-Robles T, Fadeyi OO & Oslund RC Strategies for monitoring cell-cell interactions. Nat Chem Biol 17, 641–652 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Jenkins RW et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 8, 196–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voabil P et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nature Medicine 1–12 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Engblom C et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science 358, eaal5081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katzenelenbogen Y et al. Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 182, 872–885.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Pfirschke C et al. Tumor-Promoting Ly-6G+ SiglecFhigh Cells Are Mature and Long-Lived Neutrophils. Cell Rep 32, 108164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pucella JN, Upadhaya S & Reizis B The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu Rev Cell Dev Biol 36, 529–550 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Ko J, Oh J, Ahmed MS, Carlson JCT & Weissleder R Ultra-fast Cycling for Multiplexed Cellular Fluorescence Imaging. Angew Chem Int Ed Engl 59, 6839–6846 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen SS & Prescher JA Developing bioorthogonal probes to span a spectrum of reactivities. Nat Rev Chem 4, 476–489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson JCT, Mikula H & Weissleder R Unraveling Tetrazine-Triggered Bioorthogonal Elimination Enables Chemical Tools for Ultrafast Release and Universal Cleavage. J Am Chem Soc 140, 3603–3612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarris AJC et al. Fast and pH-Independent Elimination of trans-Cyclooctene by Using Aminoethyl-Functionalized Tetrazines. Chemistry 24, 18075–18081 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Wilkovitsch M et al. A Cleavable C2-Symmetric trans-Cyclooctene Enables Fast and Complete Bioorthogonal Disassembly of Molecular Probes. J Am Chem Soc 142, 19132–19141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linder KE et al. Synthesis, in vitro evaluation, and in vivo metabolism of fluor/quencher compounds containing IRDye 800CW and Black Hole Quencher-3 (BHQ-3). Bioconjug Chem 22, 1287–1297 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Zaretsky I et al. Monitoring the dynamics of primary T cell activation and differentiation using long term live cell imaging in microwell arrays. Lab on a chip 12, 5007–5015 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Mohan JF et al. Imaging the emergence and natural progression of spontaneous autoimmune diabetes. Proc Natl Acad Sci U S A 114, E7776–E7785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu P et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol 29, 886–891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loughran SJ, Haas S, Wilkinson AC, Klein AM & Brand M Lineage commitment of hematopoietic stem cells and progenitors: insights from recent single cell and lineage tracing technologies. Exp Hematol 88, 1–6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang GG et al. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3, 287–293 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Sykes DB et al. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 167, 171–186.e15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hidalgo A, Chilvers ER, Summers C & Koenderman L The Neutrophil Life Cycle. Trends Immunol 40, 584–597 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Gerdes MJ et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schubert W et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol 24, 1270–1278 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Ullal AV et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Sci Transl Med 6, 219ra9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agasti SS, Liong M, Peterson VM, Lee H & Weissleder R Photocleavable DNA barcode-antibody conjugates allow sensitive and multiplexed protein analysis in single cells. J Am Chem Soc 134, 18499–18502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giedt RJ et al. Single-cell barcode analysis provides a rapid readout of cellular signaling pathways in clinical specimens. Nat Commun 9, 4550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh J et al. Rapid Serial Immunoprofiling of the Tumor Immune Microenvironment by Fine Needle Sampling. Clin Cancer Res 27, 4781–4793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prescher JA & Bertozzi CR Chemistry in living systems. Nat Chem Biol 1, 13–21 (2005). [DOI] [PubMed] [Google Scholar]
- 53.de la Torre D & Chin JW Reprogramming the genetic code. Nat Rev Genet 22, 169–184 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Lang K & Chin JW Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev 114, 4764–4806 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Salic A & Mitchison TJ A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105, 2415–2420 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palaniappan KK & Bertozzi CR Chemical Glycoproteomics. Chem Rev 116, 14277–14306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cañeque T, Müller S & Rodriguez R Visualizing biologically active small molecules in cells using click chemistry. Nature Reviews Chemistry 2, 202–215 (2018). [Google Scholar]
- 58.Yang KS, Budin G, Tassa C, Kister O & Weissleder R Bioorthogonal approach to identify unsuspected drug targets in live cells. Angew Chem Int Ed Engl 52, 10593–10597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nikić I et al. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew Chem Int Ed Engl 53, 2245–2249 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Beliu G et al. Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy. Commun Biol 2, 261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Werther P et al. Live-Cell Localization Microscopy with a Fluorogenic and Self-Blinking Tetrazine Probe. Angew Chem Int Ed Engl 59, 804–810 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Versteegen RM, Rossin R, ten Hoeve W, Janssen HM & Robillard MS Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew Chem Int Ed Engl 52, 14112–14116 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Wang X, Fan X & Chen PR Unleashing the Power of Bond Cleavage Chemistry in Living Systems. ACS Cent Sci 7, 929–943 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh K, Ejaz W, Dutta K & Thayumanavan S Antibody Delivery for Intracellular Targets: Emergent Therapeutic Potential. Bioconjug Chem 30, 1028–1041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canton I et al. Fully synthetic polymer vesicles for intracellular delivery of antibodies in live cells. FASEB J 27, 98–108 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Röder R et al. Intracellular Delivery of Nanobodies for Imaging of Target Proteins in Live Cells. Pharm Res 34, 161–174 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Mercier FE, Sykes DB & Scadden DT Single Targeted Exon Mutation Creates a True Congenic Mouse for Competitive Hematopoietic Stem Cell Transplantation: The C57BL/6-CD45.1(STEM) Mouse. Stem Cell Reports 6, 985–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scoazec JY & Feldmann G Both macrophages and endothelial cells of the human hepatic sinusoid express the CD4 molecule, a receptor for the human immunodeficiency virus. Hepatology 12, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the observations and conclusions of the study are included in the manuscript and its Supplementary Information. Raw multichannel and/or Z-stack source data from time series images are available in TIF format at https://doi.org/10.5281/zenodo.6482316.