

Abstract
Dynamic protein complexes function in all cellular processes, from signaling to transcription, using distinct conformations that regulate their activity. Conformational switching of proteins can turn on or off their activity through protein–protein interactions, catalytic function, cellular localization, or membrane interaction. Recent advances in structural, computational, and chemical methodologies have enabled the discovery of small-molecule activators and inhibitors of conformationally dynamic proteins by using a more rational design than a serendipitous screening approach. Here, we discuss such recent examples, focusing on the mechanism of protein conformational switching and its regulation by small molecules. We emphasize the rational approaches to control protein oligomerization with small molecules that offer exciting opportunities for investigation of novel biological mechanisms and drug discovery.
Conformational plasticity regulates protein activity
Proteins are characterized by conformational plasticity (see Glossary), which can allow from minor to major alterations in the topology of the protein structure and include changes in the global way that a protein folds, in the ordered/disordered regions as well as in the position of certain domains or motifs [1,2]. Minor structural rearrangements rarely regulate oligomerization of structurally dynamic proteins and are often associated with rigid body oligomerization of proteins with reduced conformational plasticity. By contrast, major alterations of the topology of a protein structure can create new binding sites or expose buried domains/motifs that can interact with other biomolecules. Such alterations allow proteins with increased conformational plasticity to switch from oligomerization-nonpermissive to oligomerization-permissive conformations, collectively known as conformational ensemble.
Typically, oligomerization-permissive and -nonpermissive conformations of a protein have different free energies and may exist in a dynamic equilibrium [3]. Conformational switching is not always energetically favorable but it is often induced by other proteins, membrane binding, metabolites, or changes in the pH, light, and temperature. Those perturbations often result in redistribution of the protein molecules among different conformational states. Thus, several proteins often have multiple binding partners and use their conformational plasticity to sense changes in their intracellular or extracellular environment to elicit a biological response [2]. For instance, Hsp70, a dynamic protein involved in diverse cellular processes, is known to interact with multiple proteins, including p53, C-myc, and RAF kinase [4]. Therefore, small molecules that can induce or stabilize a specific conformation can act as activators or inhibitors of a protein and result in gain or loss of a specific protein function [2,5]. Such small molecules are tremendously useful probes to dissect the structure–function relationships of proteins and their associated biological mechanisms, and can also expand the repertoire of druggable proteins in drug discovery.
Small molecules that control protein conformations have been commonly discovered in a serendipitous manner through phenotypic screening in cells or in a conformation and pocket agnostic screening approaches in vitro [2,5,6]. Hence, the small-molecule effect on the conformational dynamics of a protein and its mechanism of action was elucidated in a retrospective manner. However, recent advances in structural biology and computational methods, along with chemical targeting strategies, now enable a rational approach for the discovery of small molecules that promote/regulate a particular conformational state (Boxes 1 and 2). Notably, small molecules have been identified for several proteins that were considered challenging and not suitable targets for structure-based ligand discovery [6]. Here, we discuss recent examples of small-molecule activators and inhibitors that were identified to modulate a specific conformational state of a protein, and provide details about how these molecules induce structural alterations and modulate protein–protein complexes, as well as their functional consequences. Examples of allosteric small molecule modulators that allosterically modulate catalytic activity of enzymes without significant perturbation of the protein structure are not covered here but have been previously reviewed elsewhere [7,8].
Box 1. Experimental approaches for the discovery of small molecules that target specific conformations.
Advances in structural biology methods, such as X-ray crystallography and cryo-electron microscopy (cryo-EM), have enabled the high-resolution elucidation of stable conformations of proteins, while NMR spectroscopy enables structural elucidation of low-populated (‘invisible’) conformational states and conformational changes [69–73]. Other biophysical approaches, such hydrogen/deuterium exchange mass spectroscopy (HDXMS), fluorescence resonance energy transfer (FRET), and double electron–electron resonance (DEER), are suitable to sample conformational switching, distinct conformations, and the distribution of different conformations [74–76]. These approaches require the expression and purification of the recombinant protein of interest, although FRET, DEER, and NMR methods can be used to study proteins in a cellular environment.
Advances in computational power and simulation methodologies also enable simulations of larger protein systems and longer timescales that can provide insights into structural conformations and evaluation of conformational dynamics by small molecules [72,77,78]. Importantly, it has become evident that integration of experimental data from simulations, biochemical, and cellular experiments with specific controls that evaluate loss of conformational switching and function provides a better understanding and elucidation of physiologically relevant mechanisms. A common challenge for the discovery of small-molecule modulators of conformational plasticity is the identification of their binding site.
Advancements in computational biology have also facilitated the identification of allosteric or cryptic sites (see Box 2 in the main text). Such a binding site is required to satisfy the following criteria: (i) being involved in the molecular mechanism that underlies conformational switching of the protein; and (ii) having favorable physicochemical properties that would facilitate binding of small molecules. To address the latter, several chemical strategies have been used to rationally target a less favorable binding site or facilitate small-molecule binding. For example, libraries of fragments or small molecules with reactive warheads can enable covalent targeting to cysteine, lysine, and tyrosine residues close to the binding site [60,79–81], and the ‘bump and hole’ approach uses mutagenesis of the protein to generate a binding site or facilitate a specific interaction with the small molecule [82]. Several examples of small-molecule modulators show that high potency in binding is not a requirement, but rather binding to a hotspot region that controls the equilibrium between protein conformations is essential to alter the protein conformation.
Box 2. Computational strategies to rationally target specific conformations.
Resolution of a protein structure through X-ray crystallography or cryo-EM often traps low-energy conformations, which represent an end state in a conformational ensemble between an active and inactive conformations or are already part of a protein complex. They often fail to capture short-lived and less stable intermediate conformations that may be important for the activation of a protein. Thus, several in silico strategies have recently been developed to identify such short-lived conformations that could be drugged to alter the equilibrium in the conformational ensemble.
Elastic network models have been used extensively to predict intermediate conformations starting from a given conformation [67,68]. For example, elastic network models in conjunction with molecular dynamic simulations revealed a cryptic epitope that exists in an intermediate conformation of the epidermal growth factor (EGFR) and was not previously identified from known crystal structures. This finding rationalized the targeting of the therapeutic antibody, mAb806, toward EGFR [83,84]. Moreover, Markov state models (MSMs) are commonly used for the analysis of molecular dynamics simulations and can predict the probability of different conformations as well as the rate of conversion from one conformation to another [85]. A similar approach was recently used to uncover a cryptic site that is conserved among all bromodomains and that can allosterically regulate the interaction of bromodomains with DNA [86]. Such a discovery paves the way for the identification of new-generation inhibitors that would allosterically modulate bromodomain activity by targeting a particular conformation. There is an increasing body of evidence that highlights the importance of cryptic sites in small-molecule discovery, but their identification remains a significant challenge. Hence, as a solution, combinations of conformational sampling with elastic models have already been applied to uncover cryptic sites in a diverse set of proteins [87,88].
Finally, in silico drug design methods (e.g., docking, pharmacophore based, and similarity screening) are used for the identification of small molecules for a specific protein surface/cryptic site. Notably, recent advances in computational chemistry methods have enabled the screening of millions to billions of compounds, while improved scoring functions and machine learning methods have improved ranking and selection of hit molecules from such small-molecule libraries [89–93].
Modulating protein–protein interactions through conformational switching
Conformational switching allows proteins to adopt different conformational states [1–3]. This can turn on or off the activity of a protein by exposing or burying domains or motifs that regulate the capacity of a protein to interact with itself (protein homo-oligomerization), or other proteins (protein hetero-oligomerization). Small-molecule modulators may control the activity of a protein by stabilizing or destabilizing a particular conformational state that participates in protein–protein interactions (Figure 1). Several such small molecules have been rationally identified to promote the pro- or anti-oligomerization conformation of a protein and we discuss some notable examples here (Table 1).
Figure 1. Conformational switching and protein oligomerization controlled by small molecules.

(A) Alterations in the conformational ensemble of a protein induced by other biomolecules (pink oval), small-molecule ligands (green triangle), and physical factors (yellow thunder) can induce protein oligomerization by promoting the pro-oligomerization conformation of a protein (right) or inhibit protein oligomerization by promoting the anti-oligomerization conformation of a protein (left). (B) Small molecule-based modulators of protein–protein interactions can alter the dynamic equilibrium toward stabilizing an anti-oligomerization conformation (left) to prevent oligomerization or induce a pro-oligomerization conformation (right) of the protein to enable oligomerization.
Table 1.
Characteristics of small-molecule activators and inhibitors that control conformational plasticity of proteinsa
| Small molecule | Target | Effect on protein oligomerization | Effect on protein conformation | Activity in cellulo/in vivo | References |
|---|---|---|---|---|---|

|
STING | Promotes protein homo-oligomerization | Locks STING in an active conformation and induces polymerization | Reduces tumor growth in melanoma allografts | [14] |

|
BAX | Promotes protein homo-oligomerization | Triggers conformational activation and exposure of 6A7 epitope of inactive BAX | Induces apoptosis in various cancer cell lines and suppresses tumor growth in leukemia xenografts | [26,27] |

|
BAX | Inhibits protein homo-oligomerization | Inhibits allosterically the conformational activation and BH3 motif exposure |
Inhibits apoptosis induced by TNF-α+cycloheximide and staurosporine in MEFs, doxorubicin in primary cardiomyocytes; prevents doxorubicin induced cardiomyopathy in vivo | [29,30] |

|
BAX | Inhibits protein homo-oligomerization | Inhibits conformational activation and stabilizes BAX structure | Inhibits apoptosis induced by staurosporine in BAK KO MEFs, but not BAX KO MEFs, and by BH3 mimetics in NIH-3T3 cells | [31] |
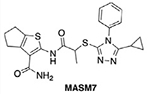
|
MFN2 | Promotes protein homo-oligomerization | Promotes pro tethering conformation | Promotes mitochondrial fusion and increases mitochondrial functionality | [39] |

|
MFN2 | Inhibits protein homo-oligomerization | Promotes anti-tethering conformation | Promotes mitochondrial fission and reduces mitochondrial functionality; Induces mitochondrial outer membrane permeabilization and potentiates cell death when combined with SMAC mimetics | [39] |

|
BCL6 | Promotes protein homo-oligomerization | N/A | Reduces proliferation of lymphoma cells | [94,95] |
| Zn+2 | Ube2T | Promotes protein homo-oligomerization | Promotes domain-swapped architecture that induces formation of two cyclic trimers | N/A | [96] |

|
β2AR | Inhibits protein hetero-oligomerization | Locks β2AR in inactive conformational state | Inhibits G-protein-mediated cAMP production and β-arrestin recruitment to activated receptor | [44,45] |

|
Axin | Promotes protein hetero-oligomerization | Releases auto inhibited conformation of Axin | Increases expression of hematopoietic stem cells markers in zebrafish and protects blood-brain barrier integrity in rat model of stroke | [64,65] |

|
KRAS G12C | Inhibits protein hetero-oligomerization | Promotes the inactive GDP-bound conformation | Inhibits binding of effectors (e.g., RAF and PI3Kα) and reduces pERK and pAKT, levels leading to antiproliferative effect in NSCLC cells. | [58,60] |

|
PKM2 | Promotes protein homo-oligomerization | Stabilizes R-state of PKM2 tetramer | Reduces tumor growth in lung xenografts | [97] |

|
RORγt | Inhibits protein hetero-oligomerization | Conformational change in ligand-binding site that blocks cofactor binding | Reduces expression of IL17a in murine lymphoblast cells | [98] |

|
TLR8 | Inhibits protein hetero-oligomerization | Locks inactive homo-dimer state | Reduces secretion of proinflammatory cytokines in human specimens from patients with rheumatoid and osteoarthritis | [99] |
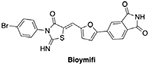
|
DR5 | Promotes protein homo-oligomerization | Not determined | Reduces survival of human glioblastoma cells | [100] |
Abbreviations: β2AR, β2 adrenergic receptor; BAX, BCL-2-associated X; CMT2A, Charcot–Marie–Tooth disease Type 2A; KO, knockout; MEF, mouse embryonic fibroblast; MFN, mitofusin; NSCLC, non-small-cell lung cancer.
Small-molecule modulators of protein homo-oligomerization
STING
Stimulator of interferon genes (STING) is an important component of the innate immune system and its activation induces type I interferon response to pathogen infection (e.g., viruses and bacteria) [9]. STING is a part of the cyclic GMP-AMP synthase (cGAS)-STING-TANK binding kinase 1 (TBK1)-interferon regulatory factor 3 (IRF3) signaling axis, which is upregulated upon detection of cytosolic DNA derived from pathogen infection or intracellular damage [9]. In particular, binding of cytosolic DNA to inactive cGAS triggers a conformational change that activates cGAS, which produces the second messenger cyclic GMP-AMP (cGAMP) by utilizing ATP and GTP as building blocks [9,10]. Next, c-GAMP binds to the cyclic nucleotide protein sensor, STING, and induces a conformational change that allows STING to polymerize and act as a docking scaffold for TBK1 and IRF3 [9–13]. Activation of TBK1 results in the phosphorylation of STING and IRF3, where the latter translocates to the nucleus and upregulates the expression of type I interferon genes [11–13].
STING resides on the endoplasmic reticulum (ER) membrane and comprises four transmembrane (TM) helices (TM1–4), a cytosolic cyclic nucleotide-binding domain (CNBD), to which c-GAMP binds, and a connector helix that links TM4 with the CNBD [11–13]. In the resting state, STING forms a symmetrical dimer in which the TM helices are organized so that they form an integrated domain-swapped architecture, while the CNBDs from each subunit form a V-shaped pocket that can accommodate cGAMP (Figure 2A) [9–13]. Moreover, the connector helices form a right-handed coiled-coil through several hydrophobic interactions. cGAMP binding to the dimer activates STING by inducing a conformational change, which promotes the formation of four stranded β-sheets, which act as a lid and seal the nucleotide-binding pocket. In addition to the lid closure, a remarkable structural rearrangement is the rotation by 180° of the CNBDs of each subunit in relation to the TM helices, which is followed by disruption of the coiled-coil formed by the connector helices [9–13]. Such disruption allows C148, located on the connector helices, to form interdimer disulfide bonds and drive polymerization in a side-by-side manner [11].
Figure 2. Analyses of pro-oligomerization and anti-oligomerization conformations for stimulator of interferon genes (STING), BCL-2-associated X (BAX), and Mitofusin 1 and 2 (MFN1/2).

(A) Structural overview of STING conformational switching [Protein Data Bank (PDB) ID: 6NT6, 6NT7] [13]. (B) Superposition of the SR-717-induced with the cyclic GMP-AMP (cGAMP)-induced STING conformation illustrates that SR-717 induces a near-identical conformation with c-GAMP [top; PDB ID: 4KSY, 6XNP; root mean square deviation (RMSD): 0.85 Å]. Superposition of the SR-717-induced STING conformation with the apo-structure of STING highlights differences in the distance of the cyclic nucleotide-binding domains (CNBDs) (bottom; PDB ID: 6NT5, 6XNP; RMSD: 4.01 Å) [13,14]. (C) Cartoon of conformational activation and oligomerization of BAX in cells. (D) NMR structure of BIM BH3 bound to BAX (left; PDB ID: 2K7W) [19]. Superposition of the BIM BH3-induced BAX conformation with the inactive BAX conformation shows α1-α2 loop opening (right; PDB ID: 1F16, 2K7W; RMSD: 1.14 Å) [18,19]. (E) Structural model of BTSA1 bound to BAX (left) [26]. Superposition of the BTSA1-induced BAX conformation with the inactive BAX conformation shows α1-α2 loop opening (right; PDB ID: 1F16; RMSD: 1.0 Å). (F) Structural model of eltrombopag bound to BAX (left) [31]. Superposition of the eltrombopag-induced BAX conformation with the inactive BAX conformation indicates that eltrombopag induces a similar conformation with inactive BAX (right; PDB ID: 1F16; RMSD: 0.50 Å). (G) Structural models of the pro- and anti-tethering conformations of MFN2 [37]. (H) Disruption of the intramolecular HR1-HR2 interactions triggers exposure of the HR2 domain. (I) MASM7 disrupts the intramolecular HR1-HR2 interactions and promotes the protethering conformation of MFN2, which allows MFN oligomerization and fusion [39]. (J) MFI8 inhibits MFN oligomerization and fusion by inhibiting intermolecular HR2-HR2 interactions [39]. Abbreviation: TM, transmembrane.
Activation of STING has been shown to augment the immune response and elicit antitumor activity in preclinical models [14]. Thus, the development of STING agonists has recently emerged as a promising strategy to combat cancer. SR-717 is a potent STING agonist that was discovered from a cGAS-STING phenotypic cell-based screening [14]. Determination of the crystal structure of the STING-SR-717 complex [Protein Data Bank (PDB) ID: 6XNP] revealed that binding of SR-717 mimics the binding of cGAMP to the STING dimer and induces a conformation that is almost identical to the active conformation induced by cGAMP (Figure 2B) [14]. Two SR-717 molecules bind at the base of the STING dimer intersubunit cleft and induce residue interactions from opposing β strands of each monomer to promote the formation of the β-sheet cap element. Furthermore, SR-717 phenocopied cGAMP treatment and induced phosphorylation of STING and IRF3, two key signaling events that follow STING activation and require STING polymerization [11–14]. Thus, SR-717 has been classified as cGAMP mimetic. Notably, SR-717 increased antitumor immunity and inhibited tumor growth in a murine melanoma model as a single treatment [14].
BAX
Proapoptotic BCL-2-associated X (BAX) has a central role in the execution of the mitochondrial apoptosis pathway through its lethal activity in inducing mitochondrial outer membrane permeabilization (MOMP) [15]. BAX is found predominantly in the cytosol in an inactive conformation. Upon stress, BAX is primarily activated by selected BH3-only activator proteins, such as BIM and BID, and undergoes conformational changes that enable its mitochondrial translocation, oligomerization, and MOMP induction [15] (Figure 2C). BAX is highly regulated by interactions with other BCL-2 family proteins, which directly activate or inhibit its cytosolic and membrane-associated conformations to promote or block BAX-mediated cell death [16]. Several studies have established its critical role in mediating apoptosis in cancer cells upon chemotherapy treatments and in normal cells (e.g., cardiomyocytes and neurons) upon stress or injury [16,17].
The inactive BAX structure (PDB ID: 1F16) comprises a typical BCL-2-fold of nine α helices linked with variable loops that shield the core hydrophobic helix α5 and provide a binding surface for the C-terminal α9 helix, which is the anchoring helix for the mitochondrial membrane (Figure 2C) [18]. A hydrocarbon-stapled BH3 helix from BIM bound to monomeric BAX revealed an activation site at the N-terminal surface of BAX formed by α1, α6, and the α1-α2 loop (Figure 2C,D) [19]. Binding of the BH3 helix induces opening of the α1-α2 loop, exposure of the 6A7 antibody epitope, and subsequent conformational changes leading to the release of the hydrophobic α9 helix and exposure of the hydrophobic α2 helix (BH3 domain) [19,20]. Once anchored to the mitochondrial membrane via helix α9, BAX can undergo further conformational changes that enable the BAX structure to separate into the latch domain (helix α6–α8) and the core domain (helix α3–α5), and dislodgement of its BH3 domain (helix α2) [21]. Symmetrical dimers can be formed by two core domains binding primarily through their BAX-BH3 domains and these are proposed to be the building blocks of the BAX oligomers [21–23] (Figure 2C). Despite incomplete understanding of the possible BAX conformations during BAX activation, several small-molecule modulators have been discovered to control conformational changes of BAX [24].
BAM7 was identified as a small-molecule activator of BAX by using an in silico docking screen for small molecules that mimic the BIM BH3 helix interactions with the BAX activation site [25]. Subsequently, a pharmacophore screening approach with the BAM7 predicted interactions with the BAX site that led to the discovery of a more potent activator of BAX, BTSA1 [26]. NMR and biochemical studies demonstrated the interaction of BTSA1 with the BAX activation site and induction of key conformational changes expected along the BAX activation pathway, leading to BAX oligomerization and MOMP [26] (Figure 2E). BTSA1 and BTSA1.2 promoted BAX-mediated apoptosis in various cancer cell lines and suppressed tumor growth in xenograft mouse models [26,27].
An inhibitor of BAX activation, BAI1, was initially identified from a mitochondrial screen for MOMP inhibitors followed by structural studies that confirmed its direct binding to soluble and monomeric BAX [28,29]. Using NMR and computational studies, BAI1 was found to bind an allosteric pocket, distant from the N-terminal activation site, and centered where helices α3, α4, α5 and α6 converge [29]. BAI1 blocked the conformational activation of BAX, particularly the exposure of the hydrophobic α2 helix and the release of the hydrophobic α9 helix, preventing BAX mitochondrial translocation and oligomerization. BAI1 exhibited promising therapeutic potential by inhibiting cardiomyocyte cell death in in vivo models of doxorubicin-induced cardiotoxicity [30].
Interestingly, using computational screening, the US Food and Drug Administration (FDA)-approved thrombopoietin receptor agonist, eltrombopag, was identified to have high similarity to BAM7 [31]. Surprisingly, eltrombopag showed a high-affinity interaction with BAX but, instead of promoting BAX conformational activation as seen with BAM7, it inhibited BAX activation [31]. NMR studies, molecular dynamics simulations, and biochemical validation showed that eltrombopag binds the N-terminal activation site with a distinct orientation compared with activators BAM7 and BTSA1 (Figure 2F). The binding mode of eltrombopag also supported stabilization of the inactive BAX structure with the loop α1-α2 closed, consistent with its inhibitory mechanism. Cellular studies supported the engagement with, and inhibition of, cellular BAX by eltrombopag upon cell death induction [31]. These findings illustrate how the same protein-binding surface of a conformational dynamic protein can be used for activation or inhibition using specific interactions with small molecules.
Mitofusins 1 and 2
Mitofusin 1 and 2 (MFN1/2) reside on the outer mitochondrial membrane (OMM) and regulate the tethering of OMMs from adjacent mitochondria through mitochondrial fusion [32]. Several studies placed mitochondrial fusion and fission (mitochondrial dynamics) at the heart of a homeostatic mechanism that regulates several diverse cellular processes, such as metabolic adaptation in response to stress, cell death, and innate immunity [33,34]. Imbalances in mitochondrial dynamics that derive from deregulated MFN2 activity have been associated with different pathologies, such as Charcot–Marie–Tooth disease Type 2A (CMT2A), neurological disease, neurodegeneration, and abnormal adipose tissue overgrowth [35,36].
MFN1/2 share high sequence homology and comprise an N-terminal GTPase domain, two coiled-coil heptad repeat domains (HR1 and HR2), and a short TM domain responsible for anchoring MFN1/2 on the OMM (Figure 2G) [37]. MFN1/2 from adjacent mitochondria tether the OMMs by forming homotypic (MFN1-MFN1 or MFN2-MFN2) or heterotypic (MFN1-MFN2) complexes. MFN1/2 require large conformational changes before the homotypic or heterotypic complex formation. Conformational switching allows MFNs to adopt an anti- or a pro-tethering conformation [37,38] (Figure 2G,H). In the anti-tethering conformation, the HR1 domain interacts intramolecularly with the HR2 domain, while in the protethering conformation, intermolecular HR2-HR2 interactions are formed that mediate homotypic or heterotypic complex formation.
A peptide 367-384 Gly derived from the HR1 domain was found to bind the HR2 domain and promoted mitochondrial fusion by inducing the protethering conformation of MFN1/2 [37,38]. Consistent with the activity of this peptide, a pharmacophore model was generated using the HR1 region 367–384. In silico screening of a small-molecule library to fit the pharmacophore model identified a mitofusin-activating small molecule 7 (MASM7) that could mimic interactions of the HR1 region 367–384 and induced the protethering conformation of MFN2 [39] (Figure 2I). MASM7 was found to bind the HR2 domain, and induced MFN2 oligomerization and, subsequently, mitochondrial fusion in cells [39].
A peptide 398–418 Gly derived from the HR1 domain was identified to bind specifically to the HR2 domain and inhibit stabilization of the protethering conformation of MFN2 [37]. Specifically, 398–418 Gly reduced the protethering conformation of MFN2 and subsequently induced mitochondrial fission in cells. A similar pharmacophore model-based screening approach that was used for the discovery of MASM7 was used for the identification of small molecules that would mimic interactions of the HR1 region 398–418 with the HR2 domain [39]. Indeed, in silico screening led to the identification of the mitochondrial fusion inhibitor 8 (MFI8), which was found to bind to the HR2 domain (Figure 2J). MFI8 also reduced the capacity of MFN2 to oligomerize, which resulted in inhibition of mitochondrial fusion in cells and, subsequently, the fragmentation of mitochondria [39]. Interestingly, MASM7 increased whereas MFI8 decreased mitochondrial functionality in cells (i.e., respiration, ATP production, and membrane potential). Furthermore, MASM7-derivative compounds reversed mitochondrial fusion and motility defects in cellular and in vivo models of CMT2A [38], while MFI8 activity promoted mitofusin-dependent OMM permeabilization, caspase-3/7 induction, and synergy with SMAC mimetics to promote apoptosis in cells [39].
Small molecule modulators of protein hetero-oligomerization
β2AR
The β2 adrenergic receptor (β2AR) belongs to the family of G-protein-coupled receptors (GPCRs) and modulates the signaling that regulates cardiac function and the vascular system [40]. Chronic overactivation of β2AR by its endogenous ligand, norepinephrine, has been associated with heart failure and aging; thus, the development of β2AR antagonists has emerged as a promising therapeutic approach to combat cardiovascular and pulmonary diseases [40]. β2AR comprises seven TM helices, three extracellular loops (ECLs), and three intracellular loops (ICLs) [41,42]. The N terminus of the protein is located in the extracellular environment, while the C terminus is located in the intracellular environment. GPCRs undergo conformational changes to transmit signals from the extracellular to the intracellular environment upon agonist binding (Figure 3A). β2AR can adopt an active conformation, which allows binding of G-protein in the intracellular environment, or an inactive conformation, which does not allow binding of G-protein in the intracellular environment [43]. Resolution of the β2AR crystal structure in complex with an orthosteric agonist (BI-167107) revealed major conformational changes that occur in the trihelical bundle formed by TM5–7 helices, where TM6 rotates 14 Å away from the helical bundle and TM5/7 move slightly toward the intracellular environment (Figure 3B) [41]. Such structural rearrangement generates a new intracellular binding site for the trimeric G-protein. Upon β2AR–G-protein complex formation, G-protein is activated by the exchange of GDP with GTP in the Gα subunit. Then, the trimeric G-protein decomposes into two smaller subunits, the dimeric Gβγ and the GTP-bound Gα, where the latter activates adenylyl cyclases that produce cAMP. Finally, increased levels of cAMP activate protein kinase A (PKA), which induces smooth muscle relaxation [40,43].
Figure 3. Analyses of pro-oligomerization and anti-oligomerization conformations for β2 adrenergic receptor (β2AR), KRAS, and Axin.

(A) Structural overview of β2AR conformational switching and signaling mechanism. Binding of an orthosteric agonist (BI-167107) to the receptor stabilizes the active conformation of β2AR, which allows binding of the G-protein [Protein Data Bank (PDB): 3SN6, 2RH1] [41,42]. (B) Superposition of β2AR active and inactive conformation highlights the differences in the topology of the transmembraITM) 5, 6, and 7 helices (PDB ID: 3SN6, 2RH1; root mean square deviation (RMSD): 1.26 Å). (C) Structural overview of the β2AR-cmpd-15PA complex uncovers an intracellular allosteric pocket that Cmpd-15PA binds to (PDB ID: 5X7D) [45]. (D) Superposition of Cmpd-15PA induced with active β2AR conformation (PDB ID: 5X7D, 3SN6; RMSD: 1.18 Å) (left) and Cmpd-15PA induced with inactive β2AR conformation (PDB ID: 5X7D, 2RH1; RMSD: 0.29 Å) (right) illustrates that cmpd-15PA locks β2AR in an inactive conformation. (E) Structural overview of RAS conformational switching and signaling mechanism (PDB ID: 4OBE, 5VQ2) [52,55]. (F) Structural overview of the RAS-AMG 510 complex (PDB ID: 6OIM, 4OBE) [52,58]. (G) Superposition of AMG 510 induced with RAS inactive conformation illustrates that AMG 510 locks RAS in an inactive conformation (PDB ID: 6OIM, 4OBE; RMSD: 1.66 Å). (H) Cartoon of Axin conformational activation. Interaction of the N terminus with the C terminus in the inactive conformation of Axin prohibits low-density lipoprotein receptor-related protein 6 (LRP6) binding, while disruption of such interaction allows Axin to adopt an active conformation, permissive to LRP6 binding. (I) HLY78 promotes the active conformation of Axin by disrupting the interaction between the N and C termini of Axin.
An in vitro screening assay was performed to discover a small molecule that negatively modulates β2AR activity [44]. Purified β2AR was mixed with a DNA-encoded small molecule library and compound 15 (Cmpd-15) emerged as a potent inhibitor of β2AR activity. A polyethylene glycol-functionalized analog of compound 15 (Cmpd-15PA) was used as a surrogate of Cmpd-15 in structural studies to shed light on the mechanism whereby Cmpd-15 modulates β2AR activity [45]. Resolution of the crystal structure of the β2AR–Cmpd-15PA complex (PDB ID: 5X7D) revealed that Cmpd-15 binds in a pocket that is formed by TM1, 2, 6, 7, Helix 8 (H8), and ICL1 in the intracellular environment, suggesting that Cmpd-15 allosterically inhibits β2AR activity (Figure 3C,D) [45]. Structural changes associated with the activation of β2AR at the junction of TM7 and H8 are incompatible with the binding interactions of Cmpd-15 and, therefore, Cmpd-15 binding would sterically clash with Ga subunit coupling. Importantly, Cmpd-15 ‘locks’ β2AR in an inactive conformational state by directly stabilizing the inactive conformation of TM6, explaining why Cmpd-15 has positive cooperativity for orthosteric inverse agonists and negative cooperativity for agonists [45]. Interestingly, at a similar location with the allosteric pocket identified for Cmpd-15 on β2AR, two other GPCR structures, CCR2 and CCR9 (PDB ID: 5T1A and 5LWE, respectively), were also reported to bind intracellular modulators, suggesting that this allosteric pocket is broadly conserved to regulate activity of diverse GPCR structures [46,47].
KRAS
The KRAS protein is a critical switch that belongs to the Ras protein family (K-RAS, H-RAS, and N-RAS) and regulates cellular processes, such as proliferation, differentiation, growth, and survival signaling [48]. The KRAS protein comprises a GTPase domain (residues 1–166) and a flexible C-terminal structural element of 20 residues that has a critical role in anchoring RAS to the membrane. Inactive RAS protein in the GDP-bound conformation is induced to the active GTP-bound conformation by the binding of guanine nucleotide exchange proteins (GEFs), such as Son of Sevenless (SOS), which catalyze the exchange of GDP to GTP (Figure 3E) [49,50]. In the active conformation, RAS interacts with multiple effector proteins, such as RAF, P13K, and RALGDS, which lead to activation of RAF/MEK/ERK, PI3K/AKT, and RALGDS/RAL signaling pathways, respectively [48]. The RAS GDP/GTP cycle also includes the conversion of active RAS back to the inactive GDP-bound conformation by the binding of GTPase-activating proteins (GAPs), such as NF1, which increases the rate of GTP hydrolysis (Figure 3E) [49,51]. Structures of the RAS GTPase domain have been determined in the inactive GDP-bound conformation or the active conformation bound to a nonhydrolyzable analog of GTP [52,53]. The GTPase domain encompasses six β-strands that form the protein core and are surrounded by five α-helices. The key differences between the inactive and active conformations are observed in the functional two regions, termed switch-I region (residues 30–38) and switch-II region (residues 60–76). The switch I/II regions have critical roles in interactions of KRAS with GAPs, GEFs, and effector proteins, such as RAF, as highlighted in several complex structures of KRAS [48].
The Ras gene is mutated in ~30% of all human cancers and KRAS mutations are the most prevalent. Mutations in the G12 and Q61 residues are most common in oncogenic KRAS, reduce the GTPase activity and induce a GTP-bound state [54]. Efforts to target the KRAS G12C mutant, which is predominant in non-small-cell lung cancer (NSCLC), with a screening strategy of thiol-reactive warhead-containing compounds, led to the identification of compounds that covalently bind to the C12 residue [55]. A covalent bond of thiol-reactive warhead with G12C allowed binding of the remaining molecule in a pocket under the switch-II region that is accessible only in the GDP-bound conformation of KRAS G12C. Similar efforts led to the development of additional G12C inhibitors [56–59]. For example, AMG 510 exploits a cryptic pocket under the switch-II region of KRAS G12C formed by residues H95, Y96, and Q99 (Figure 3F,G) [58,60]. Binding of AMG 510 covalently to G12C locks KRAS to the inactive GDP-bound conformation, which is incompatible with binding interactions of effector proteins. Therefore, AMG 510 inhibits selectively activation of KRAS and downstream ERK signaling and induced potent tumor regression in vivo in KRAS G12C NSCLC tumors. Notably, AMG 510 was the first KRAS inhibitor to gain FDA approval for the treatment of KRAS G12C NSCLC tumors [58,61].
Axin
The canonical Wnt signaling pathway has a seminal role in embryogenesis because it regulates cell differentiation, proliferation, and survival [62]. Aberrant activation of Wnt signaling is associated with cancer, while attenuation of Wnt signaling is observed in osteoporosis [62]. Activation of the canonical Wnt signaling pathway relies on the stabilization of β-catenin, which then translocates to the nucleus and activates transcription factors, such as T cell factors, which are lymphoid-enhancing factors that regulate transcription of Wnt-targeted genes [63,64]. In the presence of the Wnt ligand, the β-catenin destruction complex is sequestered on the plasma membrane and binds to low-density lipoprotein receptor-related protein 5/6 (LRP5/6) through Axin, a scaffold protein of the β-catenin destruction complex [63]. This interaction inhibits both the ubiquitination of β-catenin by the β-catenin destruction complex and, subsequently, its degradation [63].
Axin comprises a regulation of a G-protein signaling (RGS) domain in the N terminus, a glycogen synthase kinase (GSK) 3 interacting motif, and a DIX domain in the C terminus [63,64]. Axin can adopt a ‘closed’ conformation, in which its N terminus interacts with its C terminus to autoinhibit Axin-LRP6 complex formation (Figure 3H). By contrast, in the ‘open’ conformation of Axin, the interactions between its C terminus and N terminus are disrupted; thus, the C terminus of Axin is accessible to interact with LRP6. A small-molecule modulator of the Wnt signaling pathway, HLY78, promoted Axin-LRP6 complex formation [64,65]. HLY78 binds to the DIX domain of Axin and promotes the ‘open’ conformation of Axin by preventing the intramolecular interactions between the C and N termini that occur in the ‘closed’ conformation (Figure 3I). Such structural transformation enhances Axin-LRP6 complex formation, which in turn promotes the activation of the Wnt signaling [64,65]. In line with previous reports showing that Wnt signaling regulates the hematopoietic stem cell (HSC) population, HLY78 treatment in zebrafish increased the expression levels of HSC markers runx1 and cmyb, suggesting that HLY78 could find a therapeutic application for the treatment of bone marrow failure [64].
Concluding remarks
Over recent years, there has been significant progress in the discovery of small molecules that modulate the activity of a protein by directly stabilizing or promoting a particular conformation. Remarkably, such small molecules were found to control diverse protein classes, which can be either membrane bound or cytoplasmic and function as homo-oligomers or heterocomplexes. Despite the significant promise to develop both gain- and loss-of-function small molecules to control signaling pathways by manipulating specific conformations of proteins, a rational approach for the discovery of such small molecules has not been widely adopted by researchers in either academia or pharmaceutical industry. Although current computational methods have accelerated the identification of short-lived conformations, as well as the prediction of protein structures (e.g., AlphaFold) and interactions (Box 2) [3,66–68], there are still challenges to predict dynamic protein–protein interactions (see Outstanding questions).
Outstanding questions.
How can we use computational methods to predict major structural rearrangements and their interacting partners?
Can we develop more robust experimental methods suitable for high-throughput screening purposes to evaluate conformational switching in cells?
How can we use machine learning to identify druggable cryptic sites in a given conformational ensemble?
How can we predict the effect of a small molecule on the stability of a conformation?
Can we rationally control the interactions of a protein with DNA or RNA using small molecules that modulate the conformational plasticity of the protein?
Furthermore, several limitations arise from the experimental methods that can be used for the screening of small molecules because the conformational plasticity of proteins may be different in cells and in vitro. Box 1 provides a description of experimental methods that are suitable for monitoring conformational switching of proteins. Such methods, in conjunction with site-directed mutagenesis, can assess the regulatory effect of the protein or a binding site in connection with the desired conformation. In regard to small molecules, caution should be exercised over structures that are defined as pan-assay interference compounds (PAINS) or Brenk structural alerts, because they can either interfere with assay conditions or have toxic and unstable chemical moieties, giving rise to false positives. Such small molecules should be removed from both in silico and commercial compound libraries.
Targeting specific protein conformations to modulate interactions of protein complexes can lead to small molecules that can promote or inhibit a protein–protein interaction and to both activator and inhibitor chemical probes that can provide a deeper understanding of the function of a protein. Moreover, gain-of-function and temporal modulation of the activity of a protein with small molecules are desirable properties for small molecules because they may confer specificity without necessarily high affinity. Based on successful cases from our lab and others, we reason that several components are established to accelerate the discovery of chemical probes to modulate diverse protein complexes, including complexes between proteins and other biomolecules (see Outstanding questions). Ultimately, these will provide the foundation for the development of novel therapeutics that modulate dynamic protein complexes by harnessing conformational plasticity.
Highlights.
Small molecules can control protein oligomerization by stabilizing or promoting a particular conformational state.
Small molecules that turn on and off conformational changes provided important insights into protein function and biological mechanisms.
Advances in several methodologies enabled the discovery of small-molecule activators and inhibitors of protein conformational changes.
A structure-guided drug discovery platform for targeting conformational plasticity of proteins with small-molecule modulators will accelerate the discovery of novel chemical probes and drug candidates in diverse biological mechanisms.
Acknowledgments
This work is supported by R01CA178394, R01CA238229, PR191593P1, P30CA013330, P30AG038072, P01AG031782, the Irma T. Hirschl Trust Career Award, and a Pershing Square Sohn Cancer Research Alliance Award to E.G.
Glossary
- Allosteric
typically refers to a binding site that is spatially and topologically distinct from orthosteric binding sites. Chemical perturbations by ligands known as allosteric modulators or mutations at allosteric sites can modulate the activity of orthosteric binding sites.
- Bump and hole
combination of chemical and genetic methods commonly used to enhance selectivity and affinity of already existing ligands towards a specific isoform or conformation of the protein target. Typically, this approach relies on protein engineering for the generation of a ‘hole’ in the binding site of ligand, usually by substituting bulky residues with less bulky residues. The ligand is also chemically modified by generating a ‘bump’, which confers steric complementarity for the engineered protein.
- Conformation
3D shape of a protein that is defined by the position of its constituent atoms in space, which arises from the bonding and interactions within the protein structure.
- Conformational ensemble
all the distinct conformations that a protein can have. Protein molecules are partitioned in several distinct conformations, which are in dynamic equilibrium with each other.
- Conformational plasticity
ability of a protein to adopt several distinct conformations.
- Cooperativity
phenomenon whereby the affinity of a ligand or a protein subunit toward a protein is increased (positive) or decreased (negative) upon binding of another ligand or protein subunit to the protein, albeit at a spatially distinct binding site.
- DNA-encoded small molecule library
library of structurally distinct small molecules conjugated with a DNA sequence that serves as an identification barcode using high-throughput sequencing. Such libraries commonly find applications in experimental high-throughput screening drug discovery campaigns.
- Inverse agonist
ligand or small molecule that binds to the same binding site with an agonist in a constitutively active receptor protein and decreases its activity.
- Machine learning method
computational approach that utilizes algorithms to facilitate pattern recognition and classification based on already existing data with the aim of predicting the likelihood of a particular outcome in a new set of data. It is used in computational biology to generate structural models and drug design.
- Orthosteric
typically refers to a binding site where endogenous ligands and substrates bind.
- Pharmacophore model
defined spatial orientation of steric and electronic features of a ligand required for the molecular recognition and specific interaction of the ligand with its protein target. Such steric and electronic features include hydrophobic centroids, aromatic rings, hydrogen bond donor/acceptor, anions, and cations.
- Reactive warhead
chemical moiety of a small-molecule ligand that can react with amino acids (e.g., cysteine) and form covalent bonds between the protein and the ligand.
- Scoring function
mathematical function used in protein–ligand docking studies that aims to predict the likelihood of binding for a ligand toward its protein target by evaluating the favorable intermolecular interactions between the ligand and the protein. Scoring functions provide a numerical indicator that is commonly used in in silico screening campaigns to rank different ligands based on their likelihood of binding to the protein target.
- Structure-based ligand discovery
drug discovery approach that uses the structure of a protein as a starting point and aims to identify ligands that have complementary molecular properties with a specific region of the protein target. Structure-based ligand discovery utilizes in silico methods to virtually screen libraries of compounds or biochemical methods that allow the physical screening of compounds using physicochemical readouts in vitro.
Footnotes
Declaration of interests
None declared by authors.
References
- 1.Grant BJ et al. (2010) Large conformational changes in proteins: signaling and other functions. Curr. Opin. Struct. Biol 20, 142–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garlick JM and Mapp AK (2020) Selective modulation of dynamic protein complexes. Cell Chem. Biol 27, 986–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atilgan AR and Atilgan C (2022) Computational strategies for protein conformational ensemble detection. Curr. Opin. Struct. Biol. 72, 79–87 [DOI] [PubMed] [Google Scholar]
- 4.Ryu SW et al. (2020) Proteome-wide identification of HSP70/HSC70 chaperone clients in human cells. PLoS Biol. 18, e3000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kojetin DJ and Burris TP (2013) Small molecule modulation of nuclear receptor conformational dynamics: implications for function and drug discovery. Mol. Pharmacol 83, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott DE et al. (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov 15, 533–550 [DOI] [PubMed] [Google Scholar]
- 7.Astl L. et al. (2019) Interrogating regulatory mechanisms in signaling proteins by allosteric inhibitors and activators: a dynamic view through the lens of residue interaction networks. Adv. Exp. Med. Biol 1163, 187–223 [DOI] [PubMed] [Google Scholar]
- 8.Zorn JA and Wells JA (2010) Turning enzymes ON with small molecules. Nat. Chem. Biol 6, 179–188 [DOI] [PubMed] [Google Scholar]
- 9.Ablasser A. et al. (2013) cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X. et al. (2013) Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ergun SL et al. (2019) STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 178, 290–301 [DOI] [PubMed] [Google Scholar]
- 12.Zhao B. et al. (2019) A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569, 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shang G. et al. (2019) Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567, 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin EN et al. (2020) Antitumor activity of a systematic STING-activating non-nucleotide cGAMP mimetic. Science 369, 993–999 [DOI] [PubMed] [Google Scholar]
- 15.Walensky LD and Gavathiotis E (2011) BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem. Sci 36, 642–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh R. et al. (2019) Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol 20, 175–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li K. et al. (2021) Too much death can kill you: inhibiting intrinsic apoptosis to treat disease. EMBO J. 40, e107341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki M. et al. (2000) Structure of BAX: coregulation of dimer formation and intracellular localization. Cell 103, 645–654 [DOI] [PubMed] [Google Scholar]
- 19.Gavathiotis E. et al. (2008) BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavathiotis E. et al. (2010) BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell 40, 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czabotar PE et al. (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531 [DOI] [PubMed] [Google Scholar]
- 22.Bleicken S. et al. (2014) Structural model of active Bax at the membrane. Mol. Cell 56, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z. et al. (2016) BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 35, 208–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spitz AZ and Gavathiotis E (2022) Physiological and pharmacological modulation of BAX. Trends Pharmacol. Sci 43, 206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavathiotis E. et al. (2012) Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol 8, 639–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reyna DE et al. (2017) Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 32, 490–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez A. et al. (2022) Co-targeting of BAX and BCL-XL proteins broadly overcomes resistance to apoptosis in cancer. Nat. Commun 13, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bombrun A. et al. (2003) 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem 46, 4365–4368 [DOI] [PubMed] [Google Scholar]
- 29.Garner TP et al. (2019) Small-molecule allosteric inhibitors of BAX. Nat. Chem. Biol. 15, 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amgalan D. et al. (2020) A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat. Cancer 1, 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spitz AZ et al. (2021) Eltrombopag directly inhibits BAX and prevents cell death. Nat. Commun 12, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H. et al. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol 160, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bach D. et al. (2003) Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem 278, 17190–17197 [DOI] [PubMed] [Google Scholar]
- 34.Silwal P. (2021) Mitofusin-2 boosts innate immunity through the maintenance of aerobic glycolysis and activation of xenophagy in mice. Commun. Biol 4, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishikawa K. et al. (2019) Acquired expression of mutant mitofusin 2 causes progressive neurodegeneration and abnormal behavior. J. Neurosci 39, 1588–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rocha N. et al. (2017) Human biallelic MFN2 mutations mitochondrial dysfunction, upper body hyperplasia, and suppression of leptin expression. eLife 6, e23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franco A. et al. (2016) Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540, 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rocha AG et al. (2018) MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360, 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zacharioudakis E. et al. (2022) Modulating mitofusins to control mitochondrial function and signalling. Nat. Commun 13, 3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J. et al. (2018) G-protein-coupled receptors in heart disease. Circ. Res. 123, 716–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen GF Soren et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherezov V. et al. (2007) High-resolution crystal structure of an engineered human β2-Adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bai C. et al. (2021) Exploring the activation process of the β2AR-Gs complex. J. Am. Chem. Soc 143, 11044–11051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahn S. et al. (2017) Allosteric ‘beta-blocker’ isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. U. S. A 114, 1708–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X. et al. (2017) Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Y. et al. (2016) Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540, 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oswald C. et al. (2016) Intracellular allosteric antagonism of the CCR9 receptor. Nature 540, 462–465 [DOI] [PubMed] [Google Scholar]
- 48.Simanshu DK et al. (2017) RAS proteins and their regulators in human disease. Cell 170, 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bos JL et al. (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 50.Boriack-Sjodin P. et al. (1998) The structural basis of the activation of Ras by Sos. Nature 394, 337–343 [DOI] [PubMed] [Google Scholar]
- 51.Rabara D. et al. (2019) KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. U.S.A 116, 22122–22131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hunter JC et al. (2014) In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. U. S. A 111, 8895–8900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu S. et al. (2017) Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallogr. D Struct. Biol 73, 970–984 [DOI] [PubMed] [Google Scholar]
- 54.Hunter JC et al. (2015) Biochemical and structural analysis of common cancer associated KRAS mutations. Mol. Cancer Res 13, 1325–1335 [DOI] [PubMed] [Google Scholar]
- 55.Ostrem JM et al. (2015) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patricelli MP et al. (2016) Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive State. Cancer Discov. 6, 316–329 [DOI] [PubMed] [Google Scholar]
- 57.Janes MR et al. (2018) Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589 [DOI] [PubMed] [Google Scholar]
- 58.Canon J. et al. (2019) The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 [DOI] [PubMed] [Google Scholar]
- 59.Hallin J. et al. (2020) The KRAS(G12C) Inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lanman BA et al. (2020) Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J. Med. Chem 63, 52–65 [DOI] [PubMed] [Google Scholar]
- 61.Skoulidis F. et al. (2021) Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med 384, 2371–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacDonald BT et al. (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim SE et al. (2013) Wnt stabilization of β-catenin reveals principles for morphogen receptor-scaffold assemblies. Science 340, 867–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang S. et al. (2013) Small molecule modulation of Wnt signaling via modulating the Axin-LRP5/6 interaction. Nat. Chem. Biol 9, 579–585 [DOI] [PubMed] [Google Scholar]
- 65.Chen D-Z et al. (2016) Design, synthesis and structural optimization of lycorine-derived phenanthridine derivatives as Wnt/β-catenin signaling pathway agonists. J. Nat. Prod 79, 180–188 [DOI] [PubMed] [Google Scholar]
- 66.Jumper J. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orellana L. et al. (2016) Prediction and validation of protein intermediate states from structurally rich ensembles and coarse grained simulations. Nat. Commun 7,12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mahajan S and Sanejouand YH (2015) On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys 567, 59–65 [DOI] [PubMed] [Google Scholar]
- 69.Spreitzer E. et al. (2020) Probing surfaces in dynamic protein interactions. J. Mol. Biol 432, 2949–2972 [DOI] [PubMed] [Google Scholar]
- 70.Alderson TR and Kay LE (2021) NMR spectroscopy captures the essential role of dynamics in regulating biomolecular function. Cell 184, 577–595 [DOI] [PubMed] [Google Scholar]
- 71.Branden G and Neutze R (2021) Advances and challenges in time-resolved macromolecular crystallography. Science 373, 6558. [DOI] [PubMed] [Google Scholar]
- 72.Ferre G. et al. (2019) Structure and dynamics of G protein-coupled receptor-bound ghrelin reveal the critical role of the octanoyl chain. Proc. Natl. Acad. Sci. U. S. A 116, 17525–17530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wen T. et al. (2021) Structural basis for activation and allosteric modulation of full-length calcium-sensing receptor. Sci. Adv 7, eabg1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaczmarski JA et al. (2020) Altered conformational sampling along an evolutionary trajectory changes the catalytic activity of an enzyme. Nat. Commun 11, 5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hodge EA et al. (2020) Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 29, 843–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Floser A. et al. (2021) Disentangling bias between Gq, GRK2, and arrestin3 recruitment to the M3 muscarinic acetylcholine receptor. eLife 10, e58442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dhusia K. et al. (2020) Understanding the impacts of conformational dynamics on the regulation of protein-protein association by a multiscale simulation method. J. Chem. Theory Comput 16, 5323–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Orellana L. (2019) Large-scale conformational changes and protein function: breaking the in silico barrier. Front. Mol. Biosci 6, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu W. et al. (2021) Fragment-based covalent ligand discovery. RSC Chem. Biol 2, 354–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Erlanson DA et al. (2020) Fragment-to-lead medicinal chemistry publications in 2018. J. Med. Chem 63, 4430–4444 [DOI] [PubMed] [Google Scholar]
- 81.Reja RM et al. (2022) Lysine-targeting reversible covalent inhibitors with long residence time. J. Am. Chem. Soc 144, 1152–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baud MGJ et al. (2014) Chemical biology. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 346, 638–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Binder ZA et al. (2018) Epidermal growth factor receptor extracellular domain mutations in glioblastoma present opportunities for clinical imaging and therapeutic development. Cancer Cell 34, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orellana L. et al. (2019) Oncogenic mutations at the EGFR ectodomain structurally converge to remove steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. U.S.A 116, 10009–10018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pande VS et al. (2010) Everything you wanted to know about Markov State Models but you were afraid to ask. Methods 52, 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raich L. et al. (2021) Discovery of a hidden transient state in all bromodomain families. Proc. Natl. Acad. Sci. U.S.A 118, e2017427118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng W. (2021) Predicting cryptic ligand binding sites based on normal modes guided conformational sampling. Protein- Struct. Funct. Bioinf 89, 416–426 [DOI] [PubMed] [Google Scholar]
- 88.Macari G. et al. (2019) Computational methods and tools for binding site recognition between proteins and small molecules: from classical geometrical approaches to modern machine learning strategies. J. Comput. Aided Mol. Des 33, 887–903 [DOI] [PubMed] [Google Scholar]
- 89.Johnson DK and Karanicolas J (2016) Ultra-high-throughput structure-based virtual screening for small-molecule inhibitors of protein-protein interactions. J. Chem. Inf. Model 56, 399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lyu J. et al. (2019) Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sadybekov AA et al. (2022) Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Narendra G. et al. (2021) Multiple machine learning models combined with virtual screening and molecular docking to identify selective human ALDH1A1 inhibitors. J. Mol. Graph. Model. 107, 107950. [DOI] [PubMed] [Google Scholar]
- 93.Bao J. et al. (2021) DeepBSP-a machine learning method for accurate prediction of protein-ligand docking structures. J. Chem. Inf. Model 61, 2231–2240 [DOI] [PubMed] [Google Scholar]
- 94.Stabicki M. et al. (2020) Small molecule induced polymerization triggers degradation of BCL6. Nature 588, 164–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kerres N. et al. (2017) Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Rep. 20, 2860–2875 [DOI] [PubMed] [Google Scholar]
- 96.Morreale FE et al. (2017) Mind the metal: a fragment library-derived zinc impurity binds the E2 ubiquitin-conjugating enzyme Ube2T and induces structural rearrangements. J. Med. Chem 60, 8183–8191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anastasiou D. et al. (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol 8, 839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scheepstra M. et al. (2015) Identification of an allosteric binding site for RORγt inhibition. Nat. Commun. 6, 8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang S. et al. (2018) Small-molecule inhibition of TLR8 through stabilization of its resting state. Nat. Chem. Biol 14, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang G. et al. (2013) Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat. Chem. Biol 9, 84–89 [DOI] [PubMed] [Google Scholar]