

SUMMARY
Chimeric antigen receptor (CAR) T cell can revolutionize cancer medicine. However, overactivation, lack of tumor-specific surface markers, and antigen escape have hampered CAR T cell development. A multi-antigen targeting CAR system regulated by clinically-approved pharmaceutical agents is needed. Here, we presented VIPER CARs (VersatIle ProtEase Regulatable CARs), a collection of inducible ON and OFF switch CAR circuits engineered with a viral protease domain. We established their controllability using FDA-approved antiviral protease inhibitors in a xenograft tumor and a cytokine release syndrome mouse model. Furthermore, we benchmarked VIPER CARs against other drug-gated systems and demonstrated best-in-class performance. We showed their orthogonality in vivo using the ON VIPER CAR and OFF Lenalidomide CAR systems. Finally, we engineered several VIPER CAR circuits by combining various CAR technologies. Our multiplexed, drug-gated CAR circuits represent the next progression in CAR design capable of advanced logic and regulation for enhancing the safety of CAR T cell therapy.
Li et al. develop a collection of CARs controllable with FDA-approved antiviral drugs. They show that their systems can limit toxicity in a cytokine storm animal model and has best-in-class performance. They also develop complex CAR circuits with logic and multiplex control functions.
Graphical Abstract
INTRODUCTION
CAR T cells are exciting cancer immunotherapy, in which T cells are redirected to tumors by engineering them to express CARs (Abramson et al., 2017; Brown et al., 2016; Davila et al., 2014; Maude et al., 2014; O’Rourke et al., 2017; Qasim et al., 2017; Rapoport et al., 2015). However, despite their promise, these engineered T cells can display adverse side effects such as cytokine release syndrome (CRS) (Brudno and Kochenderfer, 2016; Morgan et al., 2010). Current options for controlling such adverse side effects involve systemic immunosuppression through drug administration (Bonifant et al., 2016; Davila et al., 2014; Kumar et al., 2001; Le et al., 2018), or eradication of the engineered T cells through activation of a kill switch installed in the T cells (Di Stasi et al., 2011). While these strategies can mitigate side effects, they adversely interfere with the therapy’s ability to fight cancer, often causing patients to succumb to tumors after the intervention (Brudno and Kochenderfer, 2019). Furthermore, re-administration of the engineered T cells is undesirable and, at times, not feasible because of the prohibitive cost and complications involved in grafting T cells. Although a kill switch is useful as a last resort safety measure, it would be highly advantageous to reversibly regulate the activity of engineered T cells in vivo without sacrificing them when preventing or minimizing the onset of severe side effects.
Similar to inducible gene switches, there are two types of controllable CARs - ON and OFF switches. Each type has its unique advantages, and therefore both are needed. ON switches are beneficial when off-target toxicity is an issue, and T cells should only activate when dictated. However, with CARs that are deemed relatively safe, an OFF switch may be preferable. This would result in CAR T cells being active by default, while allowing for temporary suppression of T cell activity or an immediate response to CRS if necessary. Furthermore, given that CRS can progress rapidly and therapy can never be too safe, both ON and OFF switch capabilities in the same CAR would be beneficial for maximum controllability and safety.
Due to their recognized importance to CAR T cell development, small-molecule or protein inducible ON switch CARs (Juillerat et al., 2016; Kudo et al., 2014; Ma et al., 2016; Urbanska et al., 2012; Wu et al., 2015) and OFF switch CARs (Giordano-Attianese et al., 2020) have been developed. While these controllable CARs have their unique advantages, they also have limitations. For example, some existing systems use small molecules or proteins as inducers with poor pharmacokinetics or tissue penetrance. Moreover, many of these inducers target human proteins, have undetermined safety profiles, and are not clinically approved. In the context of addressing CRS, current inducible systems have not demonstrated their use in CRS-specific in vivo models, including OFF switches that are often designed to stop this adverse effect. Furthermore, multiple antigen targeting CARs have been developed to improve targeting specificity, but safety switches integrated into these types of CARs are not well established. Another critical drawback to CAR-T therapy is antigen escape. Being able to orthogonally switch target antigens would prove highly valuable, but no such drug-inducible systems are available. An ideal approach to generating clinically-relevant inducible CARs is to use a system that relies on an FDA-approved drug with favorable toxicity and pharmacokinetic profiles, which can target multiple antigens.
To develop inducible CARs with safe and clinically-approved drugs, we leveraged the NS3 (Non-Structural Protein 3) system to create VersatIle ProtEase Regulatable CAR (VIPER CAR) systems. NS3 is a critical protein to the Hepatitis C Virus (HCV) life-cycle, which proteolytically cleaves the viral polyprotein at junction sites of non-structural proteins downstream of itself. Regulatory agencies have approved several drugs (e.g. grazoprevir (GZV) and danoprevir (DNV)) to inhibit the proteolytic activity of NS3 (De Clercq and Li, 2016). Notably, GZV has an exceptional safety profile and is typically administered at 100 mg/day for 12 weeks, making it an ideal small molecule for clinical applications (Bell et al., 2016). This feature of drug-gated control over proteolytic activity has been exploited in other synthetic biology applications by generating drug-controllable imaging tags or degrons using the NS3 domain (Chung et al., 2015; Jacobs et al., 2018; Lin et al., 2008; Tague et al., 2018).
The computational design of ‘chemical reader’ systems, such as protein domains that can recognize NS3 complexes bound to specific drugs, has also enabled the development of inducible dimerization and dissociation systems (Foight et al., 2019). Furthermore, using a peptide initially designed as an NS3 inhibitor (Kügler et al., 2012), along with a catalytically dead mutant of NS3, it is possible to form a heterodimerizing pair whose binding can be disrupted by GZV (Cunningham-Bryant et al., 2019). These protein engineering and chemical biology tools can be used further to increase the design and clinical potential of VIPER CAR systems.
Leveraging the unique advantages of the NS3 system and other regulatable CAR technologies, we sought to create the next generation of controllable CAR circuits with best-in-class performance and advanced multiplex features (Figure 1A). Here, we describe the efficacy of these various CAR circuits at regulating T cell activity and limiting toxicity in vitro and in multiple animal tumor models, illustrating how a drug-inhibited viral protease domain can extend and improve the versatility of CAR designs.
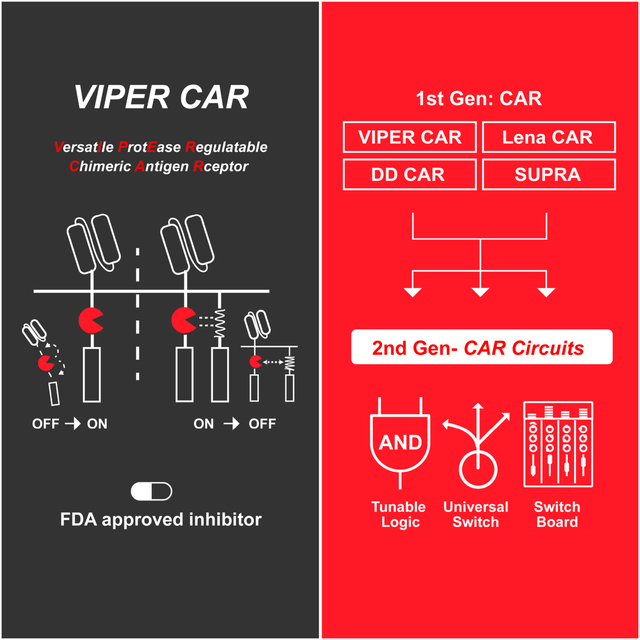
Figure 1: Design of the next-generation CAR circuits and characterization of the ON VIPER CAR.
A) Design flow of the next generation of drug-gated CAR circuits. First, drug regulatable components are identified for engineering drug-gated CAR systems. Next, the components are systematically designed and tested for the best ON and OFF switches. Subsequently, the newly designed drug-gated CAR systems are compared against other systems to determine their relative performance. Finally, the best systems are integrated together to create the next generation of CAR circuits with advanced functionalities. B) Schematic of the ON VIPER CAR mechanism. C) Primary T cell lines were treated with combinations of NS3 inhibitor and target NALM6 cells, and cytokine levels quantified (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). D) Comparison of cytotoxicity levels of the ON VIPER CAR with traditional CAR and wild-type cells (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). E) Target cell killing ability of ON VIPER CAR T cells when treated with the drug, the drug is washed out, and the drug is reintroduced after five days (mean ± s.d., n = 3). F) GZV dose-response profile of various cell lines, as measured in cytokine levels and cell killing (mean ± s.d., n = 3). E:T ratio at 1:1.
RESULTS
ON VIPER CAR
We developed an ON switch CAR by inserting the NS3 cis-protease into the CAR framework (ON VIPER CAR), producing a single fusion protein in which the linkage between the scFv and signaling domains could be precisely controlled via an NS3 inhibitor. In this design, the NS3 domain is flanked by NS5A/5B and NS4A/4B cleavage sites. In the absence of a protease inhibitor (e.g. GZV), cis-proteolysis by the inserted enzyme is anticipated to produce a fragmented CAR, which we hypothesized would be unable to transduce any activation signals in response to a target antigen. In the presence of an NS3 inhibitor, however, we anticipated that blockage of protease activity would result in the formation of a full-length and signaling-competent CAR, which would be able to mediate T cell activation following engagement with its specified antigen target (Figure 1B). Using an anti-CD19 CAR as a model system, we generated three NS3-containing ON CAR constructs, varying the positioning of the NS3 domain within the receptor to identify an optimal insertion configuration (Figure S1A).
Next, we evaluated the signaling activities of the receptors. The three ON VIPER CAR versions were introduced into human primary T cells and activated with CD19+ NALM6 cells in the presence and absence of GZV. Activation of all three led to an increase in CD69 expression, cytokine production, and cytotoxicity (Figure 1C–D, and S1B–C). In comparison, a traditional CAR (with no NS3 domain) and wild-type T cells did not respond to GZV. Of the three configurations tested, the most effective version had the NS3 domain placed between the two costimulatory domains CD28 and 4-1BB. This design was therefore selected as the final ON CAR design moving forward. Of note, our ON VIPER CAR is implemented entirely in a single chain, which should be more convenient to use than other dual chain CAR designs (Jan et al., 2021; Labanieh et al., 2022). To determine the reusability of the ON VIPER CAR, we tested receptor reactivation after it had been switched on once already. We induced ON VIPER CAR T cells with GZV, confirming the killing of CD19+ target cells, and then removed the drug, which led to a decrease in cytotoxicity. When GZV was re-introduced, we found that killing efficiency returned to similar levels as the initial activation (Figure 1E), demonstrating the ability of the ON CAR to re-activate following previous stimulation. Based on the cytotoxicity dose-response curve, the EC50 of the CAR to GZV was 23.56 nM (Figures 1F and S1D). Noticeably, the percentage of CAR-positive T cells obtained for the ON VIPER CAR following transduction was lower than that of the traditional CAR, possibly due to the larger DNA footprint of the VIPER CAR (Figure S1E). Additionally, we discovered that ON VIPER CAR T cells show slightly lower cytotoxicity than traditional CAR-T cells at a lower E: T ratio, but they offer sufficient effectiveness at the ratios between 3:1 to 1:3. (Figure S1F). The ON VIPER CAR can also be activated by three other FDA-approved NS3 inhibitors (glecaprevir, simeprevir, and boceprevir) (McCauley and Rudd, 2016), with grazoprevir and glecaprevir able to induce cell killing at lower concentrations (Figure S1G). Finally, we validated the generality of the ON VIPER CAR design by evaluating signaling and killing efficiency for a second scFv: an anti-Her2 scFv. The anti-Her2 ON VIPER CAR led to elevated production of CD69 and IL-2 in the presence of target cells and GZV (Figure S2A) and comparable killing efficiency and GZV doseresponsiveness as that of the anti-CD19 ON CAR (Figures S2B and S2C).
CARs are functional in other immune cell types and have immense therapeutic potential beyond cancer treatment. Of particular interest is the usage of CARs in regulatory T (Treg) cells, which function to suppress immune responses. Treg cells are responsible for maintaining immune homeostasis and tolerance and preventing autoimmunity. Due to their unique role in the immune system, Treg cells have been under investigation as therapeutic agents for treating autoimmune diseases, such as type 1 diabetes, or preventing organ transplant rejection (Kohm et al., 2004; Mukherjee et al., 2003; Prinz and Koenecke, 2012; Tang et al., 2012). We sought to establish that our ON VIPER CAR system is functional in Treg cells. We introduced the anti-Her2 ON VIPER CAR into Treg cells to evaluate whether GZV can regulate Treg activity. We incubated these cells with anti-CD19 CAR-expressing CD4+ T cells and Her2+/CD19+ NALM6 cells in the absence and presence of GZV (Figure S2D). CD4+ proliferation was evaluated by staining with a fluorescent cell tracer and tracking proliferation following six days of incubation. The resulting data showed that ON VIPER CAR Tregs were able to activate (via CD69 levels) in the presence of GZV (Figure S2E) and suppress CD4+ CAR T cell activation at a level comparable to that of Tregs expressing a traditional anti-Her2 CAR (Figure S2F–G).
OFF VIPER CAR
To complement the ON VIPER CAR system, we also devised strategies to inhibit CAR signaling activity in response to NS3 inhibition. An OFF VIPER CAR was constructed using a 2-component split-polypeptide configuration similar to previously described drug-controllable receptor systems (Wu et al., 2015). The first component contains the scFv, a transmembrane domain, and an NS3-binding peptide. A second membrane-tethered component contains the DAP10 ectodomain, NS3 domain, and CD3ζ signaling domain. By incorporating a catalytically-dead version of the NS3 domain (S139A), we effectively converted NS3 into an affinity domain. In the absence of GZV, the NS3-targeting peptide is expected to bind to NS3, bringing the two components together to drive CAR signaling. Upon GZV addition, the inhibitor competes with the peptide for binding to the NS3 substrate recognition site, thus facilitating the dissolution of the CAR complex (Figure 2A). As before, we tested various designs to identify optimal configurations. Specifically, we designed four permutations of each component (first component: a-d, second component: i-iv) (Figure S3A). In these variations, the NS3-binding peptide sequence was positioned C-terminally either to the hinge region or the individual costimulatory domains in the first component. The NS3 was placed at various positions in the second component.
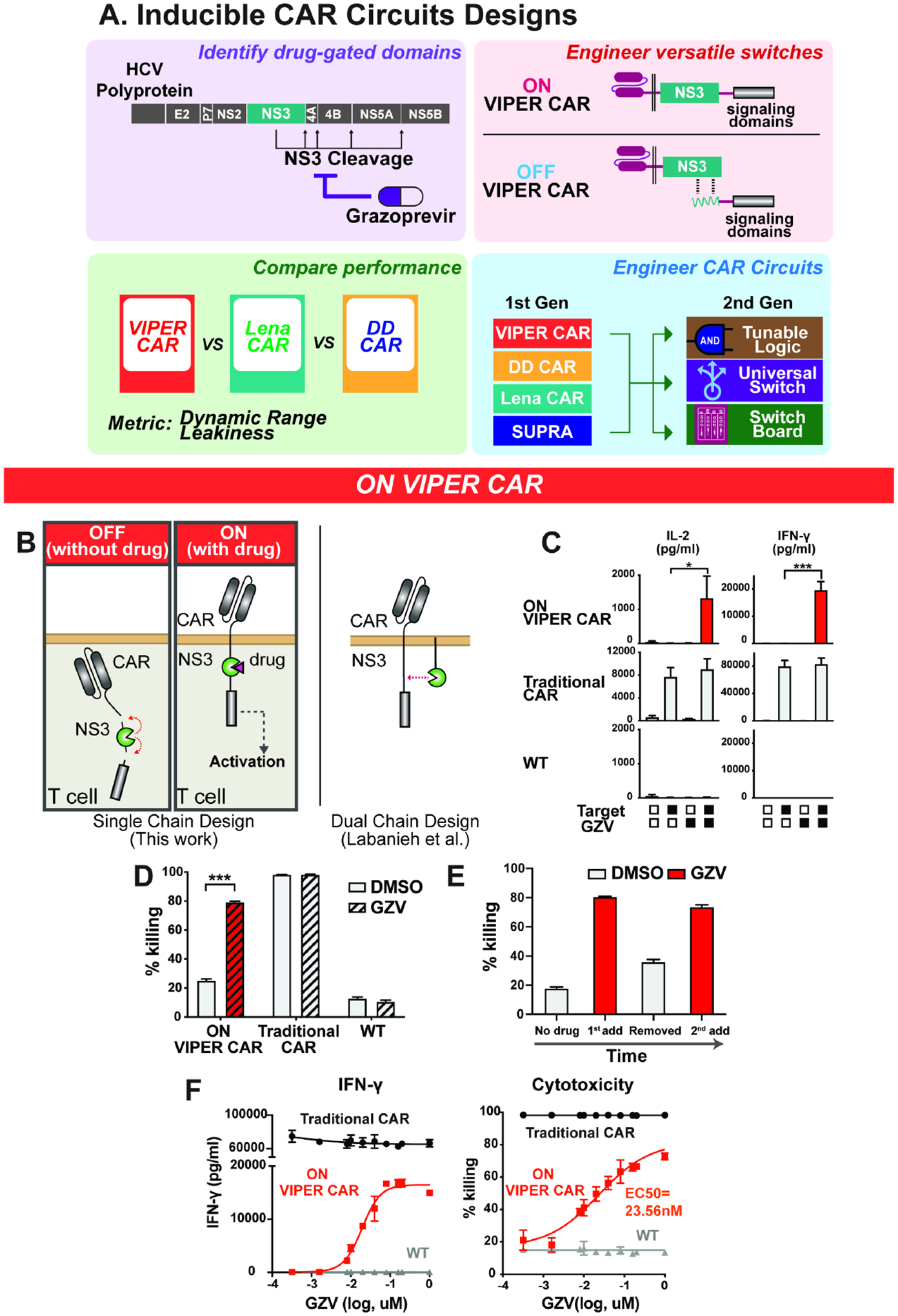
Figure 2: Characterization of the OFF VIPER CAR.
A) Schematic of the OFF VIPER CAR mechanism. B) Primary T cell lines were treated with or without GZV (all in the presence of target cells), and cytokine levels quantified (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). C) Comparison of cytotoxicity levels of the OFF VIPER CAR with traditional CAR and wild-type cells (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). D) Cytotoxicity response of OFF VIPER CAR T cells to GZV being added, washed out, and re-introduced after two days (mean ± s.d., n = 3). E) GZV dose-response profile of various cells, as measured in cytokine levels and cell killing (mean ± s.d., n = 3). E:T ratio at 1:1.
We tested the performance of each configuration in Jurkat T cells first, with CD19 used as the target for these CARs. These Jurkat T cells had been modified to express GFP upon activation of the NFAT pathway, thus allowing us to detect T cell activation through GFP expression (Lin and Weiss, 2003). CAR-expressing Jurkat T cells were incubated with CD19+ NALM6 cells in the presence or absence of GZV, and the resulting GFP levels were measured. We found that some of the first component permutations (components a and b) had weak expression and therefore did not induce the NFAT transcription reporter (data not shown). However, first component permutations that contain CD28 (components c and d) resulted in CARs that could be activated when stimulated with target cells and shut off when GZV was present. NFAT activity and CD69 expression were generally higher if both CD28 and 4–1BB costimulatory domains were in the first component (attached to the scFv), and the combination with a second component consisting of 4–1BB followed by NS3 and CD3ϛ (component i) allowed for even stronger T cell activation (Figure S3C). When paired with components c (scFv-CD28-peptide) and d (scFV-CD28–41BB-peptide), second components i (DAP10–41BB-NS3-CD3ϛ) and iii (DAP10-NS3-CD3ϛ) resulted in the largest CD69 fold change. Thus, we proceeded with component combinations c+i, c+iii, d+i and d+iii in further analysis with human primary T cells (Figure S3D).
Among the variations tested in primary T cells, component c resulted in higher T cell activation (as measured in cytokine levels) in the absence of GZV than component d. Of the two variations incorporating component c, version c+i of the OFF CAR could shut off T cell activity most effectively. This version of the OFF VIPER CAR was thus chosen as our final design (Figure S3B). The OFF VIPER CAR has similar basal activity as unmodified T cells and comparable induced activity to traditional CAR-expressing T cells (Figure 2B–C). However, it should be noted that at lower E:T ratios, the traditional CAR outperformed the OFF VIPER CAR in terms of target cell killing, which could be attributed to the lower percentage of CAR-positive cells in T cells transduced with the larger OFF VIPER CAR construct (Figures S3E and S3F). The reusability of the OFF VIPER CAR was additionally tested in a cell-killing assay by adding, removing, and re-adding GZV, with results indicating that the OFF VIPER CAR can respond a second time following previous inhibition (Figure 2D). Moreover, varying the concentration of GZV indicated that we could regulate the level of T cell activity in a dose-dependent manner, with an IC50 of around 100 pM (Figure 2E), >200-fold lower than that of the ON VIPER CAR.
VIPER CARs in a xenograft leukemia model
After confirming ON and OFF VIPER CAR activity in vitro, we proceeded to test their functionality using anti-CD19 VIPER CARs in a blood tumor model commonly used as a preclinical model for evaluating CD19 CARs (Barrett et al., 2011). For mice receiving the VIPER CARs, GZV was dosed every day at 25 mg/kg for 14 days. Tumor growth was monitored via IVIS imaging of luciferase from NALM6 cancer cells over 28 days (Figure 3A). As a control, it was verified that a daily injection of GZV of up to 25mg/kg for 14 days was not toxic to the NSG mice, nor did it reduce tumor growth (Figures S4A–D).
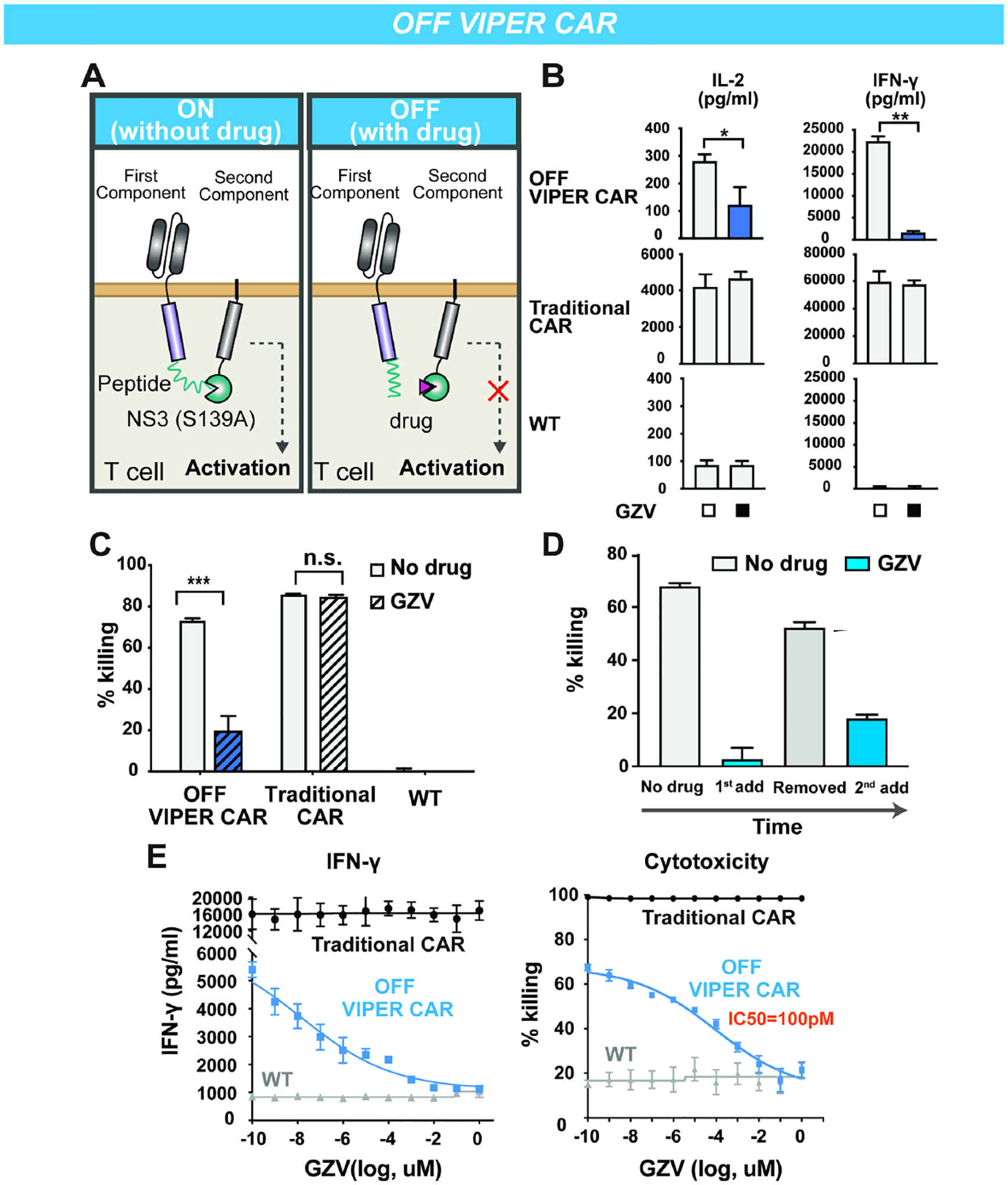
Figure 3: ON and OFF VIPER CAR are functional in a mouse xenograft tumor model.
A) Timeline of in vivo experiments. B) IVIS imaging of groups treated with (1) no T cells, (2) non-transduced T cell (NT-WT), (3) ON VIPER CAR T cells, (4) ON VIPER CAR T cells with GZV, (5) OFF VIPER CAR T cells, (6) OFF VIPER CAR T cells with GZV, or (7) Traditional CAR T cells by day 21. C) Tumor burden was quantified as total flux (photons/s) from the luciferase activity of each mouse using IVIS imaging (n = 5, *P < 0.05, **P < 0.01 and ***P < 0.001). D) Kaplan-Meier survival curves for the various mice treatment groups.
The appropriate working concentration of GZV was determined by testing the following concentrations of GZV and dosing durations on mice receiving the ON VIPER CAR: 50 mg/kg for seven days, 25 mg/kg for 14 days, and 10 mg/kg for 14 days. It was found that 25 mg/kg of GZV for 14 days was sufficient for controlling ON VIPER CAR activity and eradicating tumor cells. Conversely, tumor regrowth was observed in mice receiving 50 mg/kg GZV for seven days, possibly due to the shorter duration of drug administration (data not shown). Given the lower IC50 observed from in vitro experiments with the OFF VIPER CAR system, it was presumed that a dose of 25 mg/kg GZV should be sufficient for controlling OFF VIPER CAR activity as well.
The ON and OFF VIPER CARs were then tested in vivo for their responsiveness to GZV and ability to clear tumors. We observed that mice injected with ON VIPER CAR T cells and treated with GZV could fully clear the tumor within 28 days, while those that did not receive GZV bore high tumor burdens. Additionally, mice receiving OFF VIPER CAR T cells cleared tumors without GZV, but displayed increased tumor burden when treated with GZV (Figures 3B–C and Figures S4E). We observed 0% survival in the group of mice that had not received GZV with the ON VIPER CAR, and those that had received GZV with the OFF VIPER CAR. In contrast, of the mice receiving the ON VIPER CAR and GZV, or the OFF VIPER CAR without GZV, 100% and 80% of them survived until day 49, respectively (Figure 3D). These results demonstrate that the ON and OFF VIPER CARs are functional in an in vivo leukemia model.
Preventing cytokine release syndrome (CRS) with OFF VIPER CAR
A critical application of CAR OFF switches is to shut off CAR activity in the event of a severe adverse side effect. As such, we tested our OFF VIPER CAR system in a CRS mouse model (Mestermann et al., 2019). Here, Raji cells were I.P injected into SCID beige mice to develop an I.P. tumor; OFF VIPER CAR T cells were subsequently injected near the tumor site to simulate a surge of cytokines when the active T cells interact with the tumor. To inhibit cytokine release, the OFF VIPER CAR T cells were then switched off through daily injections of GZV (Figure 4A). As a control, dasatinib (a tyrosine kinase inhibitor that can shut down T cell activation regardless of CAR presence) was injected daily into mice that received traditional CAR-T cells. Through measurements of blood cytokine levels, we confirmed that the OFF VIPER CAR could efficiently inhibit cytokine release using GZV. In contrast, the traditional CAR alone or OFF VIPER CAR without GZV led to elevated cytokine levels in the blood. Mice treated with traditional CAR T cells and dasatinib also led to a decreased level of cytokines. However, the GZV-inhibited OFF VIPER CAR was more effective in shutting off the cytokine release (Figure 4B). Of the mice with CRS, a 100% survival rate was achieved in those receiving GZV-inhibited OFF VIPER CAR T cells and 80% of mice that received dasatinib-inhibited traditional CAR T cells. In contrast, only 40% of mice that received either solely traditional CAR or OFF VIPER CAR T cells survived (Figure 4C). These results demonstrate that the OFF VIPER CAR system can prevent the onset of CRS in an animal model.
Figure 4: OFF VIPER CAR prevents CRS in an in vivo CRS SCID-beige mouse model.
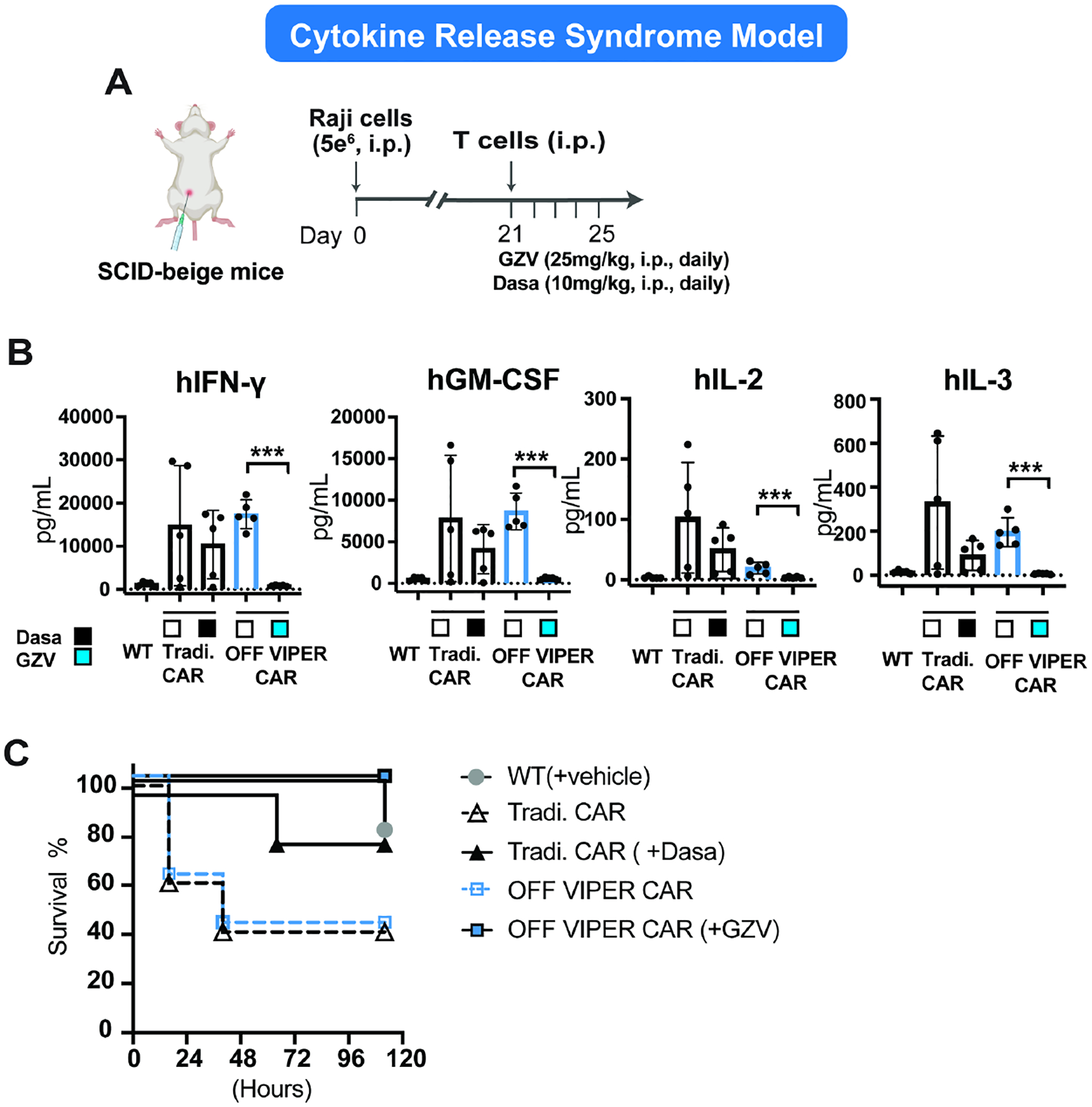
A) Timeline of the in vivo experiment. B) Cytokines (hIFN—γ, hGM-CSF, hIL-2, and hIL-3) detected in the blood 24 hours post T cell injection via Luminex assay. (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). C) Kaplan-Meier survival curves were drawn between 0–120 hours post T cell injection for the various treatment groups. (Dasa: dasatinib)
Benchmarking VIPER CAR switches
To compare the functionality of our VIPER CARs with other existing drug inducible CAR technologies, we performed parallel assays with some of the current ON and OFF CARs: ON VIPER CAR (Figure 5A, D), an ON DD-CAR (Weber et al., 2021) (Figure 5B, E), an ON Lenalidomide-CAR (Jan et al., 2021) (Figure 5C, F), OFF VIPER CAR (Figure 5G, I), and an OFF Lenalidomide-CAR (Jan et al., 2021) (Figure 5H, J). These systems were chosen based on their use of FDA-approved drug inducers and promising published results. To perform head-to-head comparisons, the same type of CAR (CD19-specific, second-generation) was used in all systems. A cytotoxicity assay was used as the readout as it has a low dynamic range (from 0–100%) and a high sensitivity to basal activity, thus providing a stringent metric to evaluate the induction performance of the systems. We found that the ON VIPER CAR, OFF VIPER CAR, and OFF Lena-CAR had a 2.54-, 3.49-, and 3.39- fold dynamic range (ratio in cytotoxicity between the ON and OFF states) respectively, and 2.72-, 1.86-, and 1.74- fold leakiness (difference between the OFF state and untransduced T cells) respectively (Figure 5K). In contrast, the ON Lena-CAR had a 1.67-fold dynamic range and 1.87-fold leakiness. The ON-DD CAR displayed the worst performance, with very high leakiness (12.05-fold), thus leading to almost no inducible activation (1.02-fold dynamic range).
Figure 5: Head-to-head comparison study between VIPER CARs and other clinically relevant drug-gated CARs.
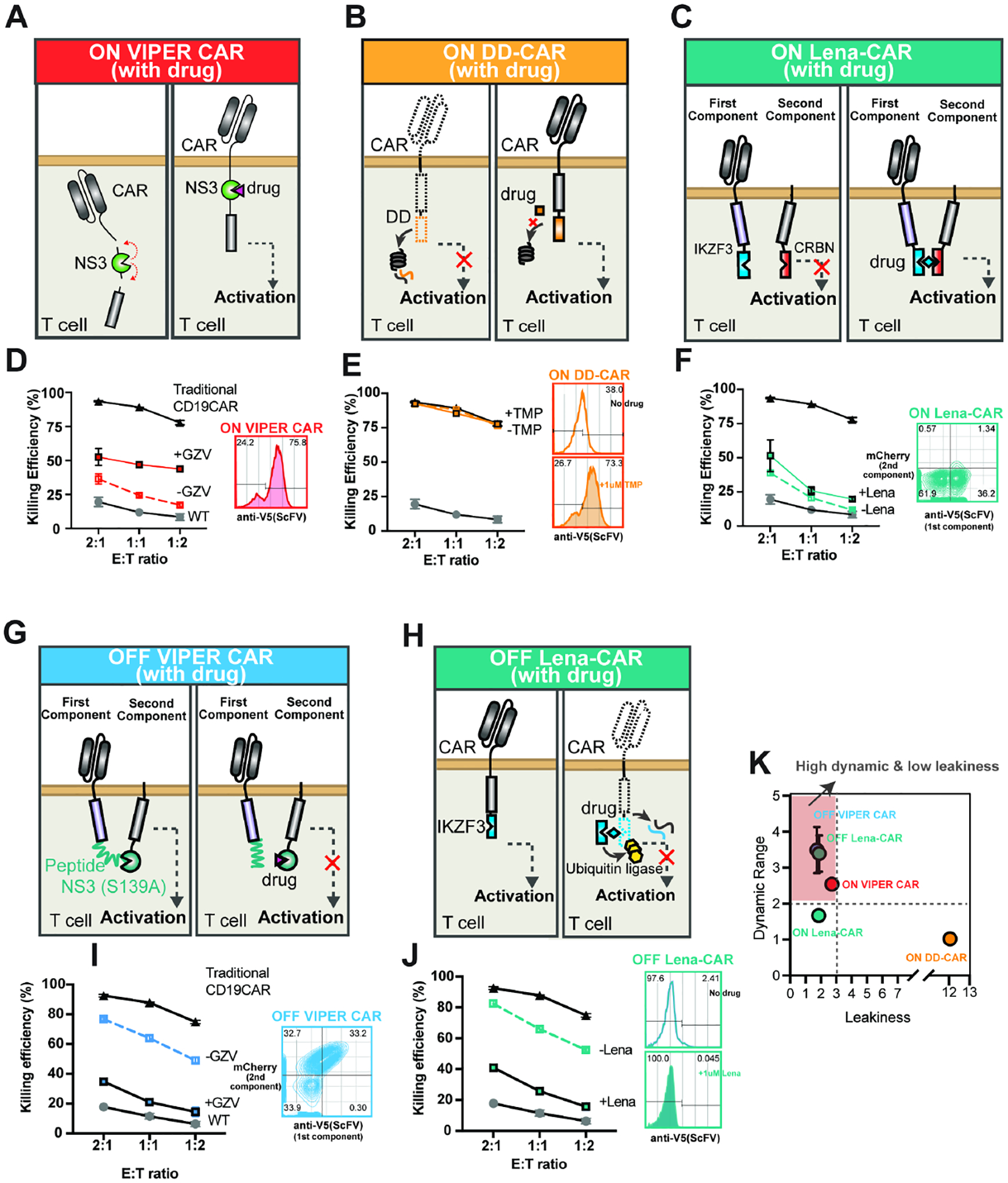
Five inducible systems, including VIPER CARs, were separately expressed on primary T cells (three ON switches (A-C) and two OFF switches (G-H)). CAR expression and killing efficacy were compared. CD19-specific scFV with a CD28 costimulatory domain were used in all ON-switch inducible systems. A) ON VIPER CAR: Schematic diagram of the system. VIPER CAR expression was determined by surface staining of the V5-tagged scFv (D, right). B) On DD-CAR: eDHFR degron domain (DD) (orange) was fused to intracellular signaling domains in the order of CD28-CD3z-DD. The DD induces rapid CAR degradation in the absence of TMP, whereas the addition of TMP stabilizes the DD, thereby preventing CAR degradation. DD CAR expression was determined by surface staining of the V5-tagged scFV after exposure to either no TMP or 1uM TMP overnight (E, right). C) ON Lena-CAR: The CAR was split into two pieces, with the IKZF3 domain (blue) in the first component (scFv-CD28-IKZF3) and the CRBN domain (dark orange) in the second component (CD28-CRBN-CD3z-mCherry). The two components are brought together using lenalidomide (or pomalidomide). ON Lena-CAR expression was determined by surface staining of the V5-tagged scFV at the first component and by a mCherry fluorescence protein fused to the second component (F, right). For comparison studies between OFF switches, we kept CAR features the same as the original design. G) OFF VIPER CAR: CAR structure was the same as shown in Figure S3B, and CAR expression was evaluated the same way as described in Figure S5A. H) OFF Lena-CAR: IKZF3 - a super degron - was fused to the intracellular signaling domain (scFV-41BB-CD3z-IKZF3). Its degradation is induced by Lenalidomide (or pomalidomide) through ubiquitin ligase recruitment. OFF Lena-CAR expression was determined by surface staining of the V5-tagged scFV. Target cell (Nalm6) killing by five different inducible CARs in the absence and presence of corresponding drugs was compared with traditional CAR and wild-type T cells at various E: T ratios (D, E, F, I, and J). K) The dynamics range and leakiness of each inducible system were calculated and compared based on the following equation at the E: T ratio of 1:2. Dynamic range= killing efficiency (%) at ON state/killing efficiency (%) at OFF state. Leakiness= killing efficiency (%) at OFF state/killing efficiency (%) of non-transduced T cell.
Orthogonally switchable dual-gated CARs
We next wanted to examine whether our VIPER CARs would be compatible with other inducible systems to create a dual-drug gated CAR T cell product. We chose to use the OFF Lena-CAR in conjunction with our ON VIPER CAR, as the OFF Lena-CAR switch performed at a comparable level with the OFF VIPER CAR. Using these two systems, we built inducible CAR-T cells that target two different tumors – the ON VIPER CAR targeted NALM6 (CD19+) cells, and the OFF Lena-CAR targeted MSTO-211H (Her2+) cells (Figure 6A). To test these CAR-T cells, we created a dual-tumor model in NSG mice by generating a systemic NALM6 (CD19+) tumor and an MSTO-211H (Her2+) subcutaneous tumor (Figure 6B). The systemic tumor should be cleared by ON VIPER CAR T cells in the presence of GZV, and the subcutaneous tumor should be cleared by OFF Lena-CAR T cells in the absence of pomalidomide (POM, an analog of lenalidomide).
Figure 6: Orthogonal regulation of dual gated-CARs in vivo.
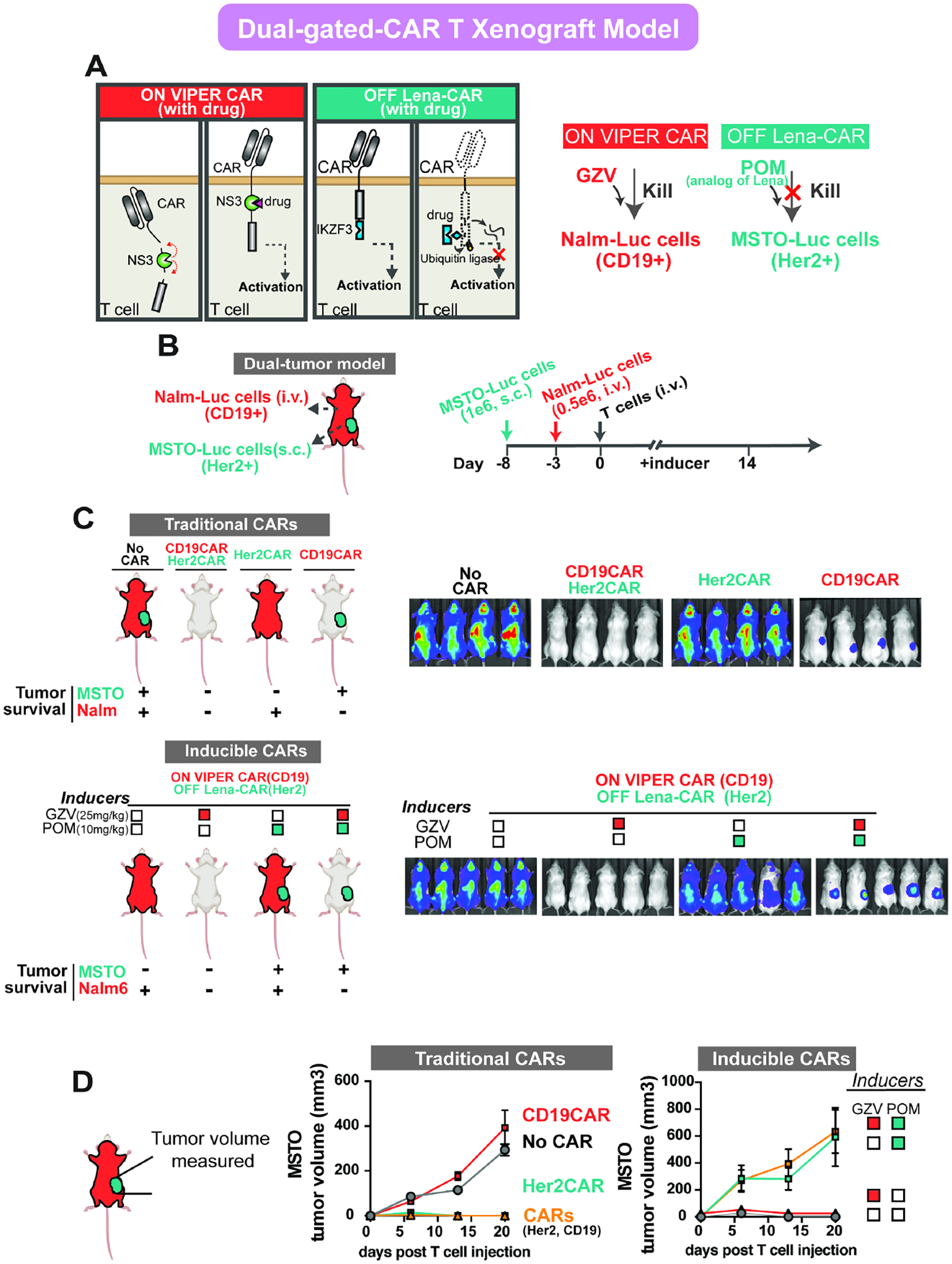
A) Schematic of the mechanism of dual gated CARs: ON VIPER CAR and OFF Lena-CAR. B) Illustration of the dual-tumor model generated by systemically growing Nalm6 cells via i.v. and regional growth of MSTO-211H cells via s.c (left). Timeline for the in vivo experiment (right). Inducers (GZV or POM) were injected daily through i.p. C) IVIS imaging of groups treated with various drugs conditions on day 13. Both Nalm6(Luc+) cells and MSTO-211H(Luc+) were imaged simultaneously. Mice treated with (1) WT, (2) CD19CAR+Her2CAR, (3) Her2CAR, or (4) CD19CAR T cells are shown at the top of the panel, and mice treated with dual gated-CARs and various conditions of drugs are shown at the bottom of the panel. D) Tumor volume of subcutaneous cells was measured by caliper every week. (WT or Traditional CARs: n=4, Inducible CARs: n=5) (POM: pomalidomide)
We tested our dual-gated CAR-T cells in these dual tumor-bearing mice, treated with different combinations of the inducers. Though both NALM6 and MSTO-211H cells express luciferase, the systemic tumor burden was imaged and quantified by introducing D-luciferin, while the subcutaneous tumor volume was measured by caliper. In the absence of POM, the anti-Her2 OFF Lena-CAR remained at an ON state and effectively cleared the subcutaneous tumor. In contrast, in the absence of GZV, the ON VIPER CAR was inactive, which allowed for the growth of the systemic CD19+ tumor. When only GZV was present, both CARs were in an active state, and it was found that both systemic and subcutaneous tumors were cleared. If only POM was present, both CARs were in an OFF state, which led to the growth of both tumors. Lastly, when both GZV and POM were present, GZV switched on the ON VIPER CAR, and POM switched off the OFF Lena-CAR, leading to the growth of the subcutaneous tumor only (Figures 6C–D, Figures S5). Using this in vivo model, we found that the two drug-inducible systems can work in an orthogonal and controlled manner to target multiple antigens.
Reconfigurable VIPER CAR-based circuits
Drug-gated AND gate:
Given the flexibility of the VIPER CAR system and its ability to integrate with existing CAR platforms, we reasoned that it could be used to rapidly generate a diverse array of multi-functional drug-gated CAR circuits toward upgrading CAR-T cell safety and efficacy. For instance, a major goal in next-generation CAR T cell designs is to improve tumor specificity, and combinatorial logic CAR systems have been successfully developed to address this challenge (Kloss et al., 2013; Lanitis et al., 2013; Roybal et al., 2016). Specifically, AND gate CARs where two or more antigens are needed to fully activate T cells have shown to enhance specificity and prevent the ON-target OFF-tumor effect (Srivastava et al., 2019). However, AND gate CARs typically require careful tuning of receptor signaling. We tested whether the VIPER CAR system could be reconfigured to create a drug controllable AND gate, which would allow for more precise control over CAR signaling.
To accomplish this, we used the orthogonal DD degron (Iwamoto et al., 2010) in combination with our NS3 domain. We generated a combinatorial logic AND gate by distributing CAR intracellular signaling domains between two distinct scFvs, with each signaling domain associated with either the NS3 or DD. Our AND gate VIPER CAR system comprises an anti-Her2 receptor with the NS3 domain positioned between the scFv and CD3ζ domains, followed by a second anti-Axl receptor with the DD positioned after the CD28 domain, thus generating a functional dual-molecule controllable CAR (Figure 7A).
Figure 7: NS3 domains enable the design of dual gated VIPER CAR circuits.
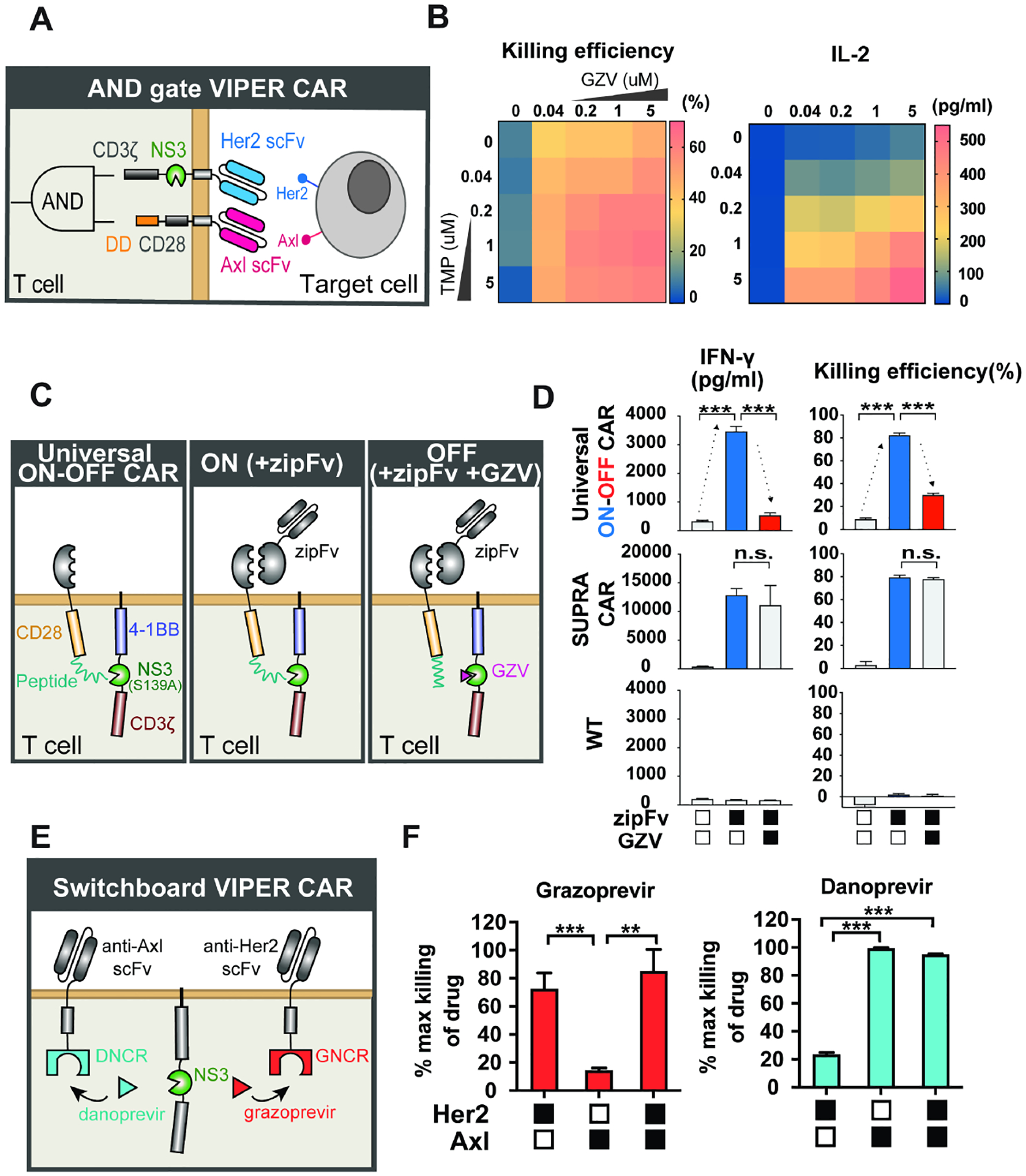
A) Schematic of the AND gate VIPER CAR that incorporates an ON VIPER CAR and a distinct DD CAR. B) Dose-response of the AND gate VIPER CAR T cell killing (CD8+ T cell) and IL-2 level (CD4+ T cell) in response to increased levels of GZV and TMP (n = 3, mean values are displayed). C) Schematic the universal ON-OFF VIPER CAR mechanism. D) Comparison of universal ON-OFF VIPER CAR T cell activity with traditional CAR and wild-type T cells, treated with combinations of zipFv and GZV, as measured by IFN-γ levels and cell killing (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). E) Schematic of switchboard VIPER CAR mechanism. F) Cytotoxicity levels of switchboard VIPER CAR in primary T cells when treated with various antigen-expressing target cells, normalized to the maximum killing observed under each drug (mean ± s.d., n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001).
The VIPER CAR/DD AND gate was expressed in primary T cells, and the drug tunability of the system was tested. To activate the VIPER CAR AND gate, we co-cultured these T cells with Her2+/Axl+ NALM6 cells and varied the amount of GZV and TMP. We found that cytotoxicity could be modulated by varying the amounts of the small molecules, although there was basal killing observed in the presence of solely GZV (Figure 7B). This expected response stems from the activation of the NS3-containing component, which contains the CD3z domain from the TCR. Without a costimulatory domain, CD3z alone can activate T cells to a weaker extent than CARs containing both CD3z and a costimulatory domain, but can still be sufficient for triggering basal levels of cytotoxicity in vitro (Brocker and Karjalainen, 1995; Hombach and Abken, 2013; Imai et al., 2004). Nonetheless, recruitment of the co-stimulatory domain is associated with improved anti-tumor efficacy. We found that TMP-induced recruitment of the CD28 domain resulted in an increase in cytotoxicity for these AND gate VIPER CAR T cells. Further studies of CD69 and cytokine levels also supported our earlier observation that the level of T cell activation could be tuned using different doses of the two drugs (Figure S6A).
Universal OFF CAR:
Another desirable CAR feature is universality where a universal CAR can target a wide array of antigens through adaptor proteins, which can prevent relapse due to antigen escape while minimizing DNA footprint. However, a multi-targeting CAR system could have a higher chance of OFF-tumor side effects. Therefore, additional safety features are required. Installing an OFF-switch to a universal CAR (Universal OFF CAR) would simultaneously improve the safety while broadening the targeting specificity.
We created a universal OFF CAR by applying the OFF VIPER CAR design to existing SUPRA CAR technology. The SUPRA CAR consists of two components: a universal zipCAR formed by the fusion of a leucine zipper and intracellular CAR signaling domains, and an adaptor protein called zipFv formed by the fusion of an scFv and the cognate leucine zipper. When the zipFv bridges the zipCAR T cells and targets cancer cells, it allows for activation through the zipCAR. To create our universal ON-OFF VIPER CAR, we divided the intracellular CAR domains into two components, similar to the design of the OFF VIPER CAR. We replaced the scFv with a leucine zipper system similar to that of the SUPRA CAR. In the absence of zipFv or GZV, the CAR remains inactive as there is no scFv targeting it to any cell. However, when zipFv is added, the zipCAR can be activated by target cells. This activation is then inhibited when GZV outcompetes the NS3-specific peptide for binding to the catalytically-dead NS3 domain (Figure 7C).
This universal ON-OFF VIPER CAR was introduced into primary CD8+ T cells, and through IFN-γ production measurements, we found that these cells were able to switch ON in the presence of anti-Her2 zipFv and target Her2+ NALM cells, and shut OFF when GZV was present. Cytotoxicity of these T cells was additionally measured in a killing assay involving Her2+ NALM6 target cells, and the results recapitulated what was observed with the cytokine data (Figure 7D). Furthermore, we found that the activation level obtained by cells expressing this ON-OFF VIPER CAR could be adjusted by varying zipFv concentration, demonstrating a high level of tunability (Figure S6B).
Switchboard:
The ability to redirect the CAR T cell to a different antigen after administering the CAR T cell represents an alternative approach to address antigen escape without the added risk of simultaneously multi-antigen targeting and the complexity of administering protein and cell therapy. However, the existing strategy is to engineer two different batches of CAR T cells with distinct antigen specificity, which is prohibitively expensive. Therefore, a drug-gated switchboard CAR system where the addition of different drugs can lead to the activation of CAR targeting distinct antigens would be very useful.
To generate the switchboard, we applied the NS3 “reader” proteins to CAR design, which allows it to switch to a different target antigen through GZV or DNV administration. These “reader” proteins bind specific inhibitor-bound states of NS3 and are expected to behave orthogonally (Foight et al., 2019). To develop the switchboard VIPER CAR circuit, we adopted the DNV/NS3 complex reader (DNCR) and GZV/NS3 complex reader (GNCR), which are regulated by DNV and GZV, respectively. Similar to the OFF VIPER CAR, the NS3 reader CARs were designed by splitting the CAR into two components. The first component consists of the scFv, CD28 costimulatory, and reader domains, while the second component encodes a catalytically-dead NS3 and the CD3ζ signaling domain. The NS3 domain binds to the GNCR in the presence of GZV, and the DNCR in the presence of DNV, allowing different scFvs to bind to the CD3ζ depending on the drug added (Figure 7E).
We first tested the function of our DNCR and GNCR CARs separately in cells using an anti-Axl and anti-Her2 CAR, respectively. Primary T cells were transduced with lentivirus encoding the reader CARs and incubated with or without inhibitors, and NALM6 target cells expressing Her2, Axl, or both antigens. Both reader CARs showed an increase in cytotoxicity and cytokine release levels when their respective inducer was present, along with the target cell line expressing their specific antigen (Figures S6C–D). With both reader CARs functional, we proceeded to express both CARs within the same cell. While the GNCR CAR indicated lower activity than the DNCR CAR, these dual CAR-expressing cells indicated increased cytokine levels and killing of Her2+ target cells when GZV was present, and increased cytokine levels and killing of Axl+ target cells when DNV was present (Figures 7F and S6E). This suggests that we can switch CAR antigen specificity using these two drugs.
DISCUSSION
Several drug-inducible CAR technologies have been developed, yet few are reliant on a clinically-approved pharmaceutical agent with a favorable safety profile. Here, we have exploited the NS3 cis-protease domain and its corresponding inhibitors to devise a chemogenetic toolset for regulating the timing and magnitude of CAR T cell activities. Features that distinguish these systems from previously reported designs are their ability to respond to clinically-approved anti-viral compounds and the versatility with which they can be used to generate on and off signaling systems. Here we demonstrated an ON switch, OFF switch, and three complex reconfigurable CAR circuits (AND-gate, universal ON-OFF, and switchboard VIPER CARs). VIPER CARs were combined with other well-characterized inducer responding domains to achieve regulable and safe CARs. These CARs have a unique design and can be regulated effectively by clinically-approved drugs.
Interestingly, an ON switch CAR based on NS3 with a different design had also been reported recently (Labanieh et al., 2022). This design incorporates two different molecules working in trans - a second-generation CAR with an NS3 cleavage site placed in between the transmembrane and signaling domains, and a membrane-tethered NS3 domain. Optimization in the transmembrane domain is needed to ensure that both chains co-localized together. Unlike our ON VIPER CAR, the single-chain design failed to perform well for them. While the exact reason for the discrepancy remains to be explored, our results highlight that subtle variation in CAR designs (e.g. the location of the NS3 domain within the CAR molecule) can have a profound effect on their function.
We demonstrated the efficacy of the ON and OFF VIPER CARs in vivo using a xenograft mouse model, with the OFF VIPER CAR being further characterized in a CRS model. The use of FDA-approved drugs as inducers in these technologies is beneficial as it can accelerate their clinical use. Furthermore, the tunability of the ON and OFF VIPER CARs using these drugs allows for the tailoring of T cell activation levels to individual patients’ needs. We envision that VIPER CAR T cell immunotherapy will be able to, in real-time, modulate T cell activity to mitigate complications like CRS and off-target activities. Additionally, as exhibited with the ON CAR, we have shown the potential of these systems to target different antigens (anti-CD19 and anti-Her2) and operate in different cell types (CD4+, CD8+, and regulatory T cell), which suggests the applicability of these VIPER CARs to a broad range of cancer and cell types.
In addition to ON and OFF switches, we have demonstrated how the combination of different technologies can create more complex CAR circuits with expanded and necessary capabilities. We have developed our dual-gated AND gate CAR by combining NS3 technology with an existing degradation domain. By carefully controlling the CD3z and CD28 components separately with different drugs, we not only regulate safety but also modulate the level of activity stemming from each component of the AND gate CAR. We have further developed a universal ON-OFF VIPER CAR with SUPRA CAR. The universal ON-OFF VIPER CAR is not only able to mitigate antigen escape by changing with zipFv of interest when desired but also incorporates a safety switch that shuts off activity immediately when needed. Lastly, the compatibility of our VIPER CARs with other existing inducible CARs is demonstrated in a dual-tumor xenograft model by combining these CAR technologies. This exemplifies how applying the technologies of the NS3 domain to other synthetic biology tools can help to further the possibilities of CAR T cell therapy.
While some of our NS3-based CAR designs have demonstrated in vitro and in vivo efficacy, others still require further optimization. For example, the basal activity of our AND gate CAR in the presence of GZV alone, and the reduced effector function of the switchboard VIPER CARs indicate that these engineered CAR-T cells can benefit from further improvement. However, we have included these VIPER CAR technologies to demonstrate the potential uses of the NS3 domain in CARs and the versatility that its associated components (e.g. inhibitor-specific reader domains) afford.
We have expanded the NS3 domain beyond HVC therapeutic target for use in CAR T cells. Its uses as a self-cleaving entity and CID make it advantageous in receptor design due to its versatility. Other viral proteases, such as the SAR-CoV2 MPro (Jin et al., 2020; Sacco et al., 2020), have been targeted for developing inhibitors to treat viral infection. The NS3 protease and its inhibitors utilized here are prime examples of how they can be integrated into CAR design, providing clinically-ready tools for enhancing the safety and efficacy of CAR-T cell therapy. Given the availability of multiple NS3-targeting drugs and the continued development of new inhibitory compounds, this research provides a streamlined framework for the translation and adaptation of antiviral strategies into effective treatment for malignancies.
STAR★METHODS
RESOURSE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wilson Wong (wilwong@bu.edu).
Materials availability
All plasmid constructs and cell lines generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and core availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Source of Primary Human T Cells
Blood of donor was obtained from the Blood Donor Center at Boston Children’s Hospital (Boston, MA) as approved by the University Institutional Review Board. Peripheral blood mononuclear cells (PBMCs) were isolated from blood using Lymphoprep (STEMCELL Technologies, 07851) and activated with Human T-activator CD3/CD28 (Thermo Fisher, 11131D) for further lentiviral transduction. CD4+ T cell and CD8+ T cell were isolated from blood using RosetteSep Human T cell enrichment cocktail (STEMCELL Technologies, 15022 and 15023), and T reg cells were isolated using the EasySep human CD4+CD127lowCD25+ regulatory T cell isolation kit (STEMCELL Technologies, 18063) according to the manufacturer’s protocol.
Animals
Female NSG mice, 6–8 weeks of age, were purchased from Jackson Laboratories (#005557) and were used for in vivo xenograft tumor mouse experiments. Female SCID-beige mice, 6–8 weeks of age, were purchased from Taconic Bioscience (CBSCBG-F) and used for simulating the CRS mouse model. For all experiment, mice of the same sex were randomly assigned to experimental groups. Animal studies were conducted at the Boston University Medical School Animal Science Center under a protocol approved by the Boston University Institutional Animal Care and Use Committee. All animal experiments were performed in accordance with the relevant institutional and national guidelines and regulations.
METHOD DETAILS
CAR Constructs Design
All CAR constructs were introduced into primary human T cells and Jurkat T cells using pHR lentivectors, and plasmids were packaged into lentiviruses using pDelta, Vsvg, and pAdv packaging and envelope plasmids(Zufferey et al., 1998). Expression of the CARs was driven by an EF-1 alpha promoter and Kozak sequence. CARs contained either a myc or V5 tag after the scFv, or a mCherry fluorescent protein at the C-terminus to verify construct expression. In OFF VIPER CAR, Universal ON-OFF VIPER CAR experiments, both components were introduced into cells using a single construct, with the components separated by either a P2A or T2A sequence. The two components of the AND gate VIPER CAR and switchable VIPER CAR were introduced on separate constructs.
Cell culture
Lentiviruses were generated using HEK293FT cells, which were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher, 10437028), penicillin/streptomycin (Corning, 30001CI), L-glutamine (Corning, 25005CI) and 1mM sodium pyruvate (Lonza, 13115E). Primary peripheral blood mononuclear cells (PBMCs), CD4+ and CD8+ T cells were isolated from whole peripheral blood obtained from healthy donors at the Blood Donor Center at Boston Children’s Hospital (Boston, MA) as approved by the University Institutional Review Board (IRB). CD4+ and CD8+ T cells were isolated using the RosetteSep Human T cell enrichment cocktail (STEMCELL Technologies, 15022 and 15023), and PBMCs were isolated using Lymphoprep (STEMCELL Technologies, 07851). Regulatory T cells were isolated using the EasySep human CD4+CD127lowCD25+ regulatory T cell isolation kit according to the manufacturer’s protocol (STEMCELL Technologies, 18063). Primary T cells were cultured in X-Vivo 15 media (Lonza, 04–418Q) supplemented with 5% human AB serum (Valley Biomedical, HP1022), 10mM N-acetyl L-Cysteine (Sigma, A9165), 55μM 2-Mercaptoethanol (Thermo Fisher, 21985023), and 30–200U/ml IL-2 (NCI BRB Preclinical Repository). N-acetyl L-Cysteine and 2-mercaptoethanol were removed during the assay of the regulatory T cell suppression experiment. Jurkat T cells and NALM6 cells were cultured in RPMI 1640 supplemented with 5% FBS, L-glutamine, and penicillin/streptomycin. CD19-positive NALM6 cells expressing Her2, mCherry, and GFP, or just Luc-BFP alone, were used as target cells in assays. Raji cells were cultured in RPMI 1640 supplemented with 10% FBS, L-glutamine, and penicillin/streptomycin. MSTO-211H (Luc-BFP) cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin/streptomycin, L-glutamine, and 1mM sodium pyruvate.
Lentivirus generation and transduction
HEK293FT cells were co-transfected with lentivirus packaging/envelope plasmids (described above) and CAR-encoding vectors using polythylenimine (PEI). After 24 hours, media was replaced with ultraculture (Lonza, 12–725F) supplemented with penicillin/streptomycin, L-glutamine, 0.5M sodium butyrate, and 1mM sodium pyruvate. Virus-containing media was collected for the following 48 hours and spun down to remove cell debris. Viral media was then concentrated either using Lenti-X Concentrator (Takara, 631232) at a 3:1 ratio and incubated at four °C overnight before centrifugation at 1500xg for 45 minutes at four °C, or using Lentivirus concentration solution (40% PEG8000 and 1.5M NaCl) at a 3:1 ratio and incubated at four °C overnight before centrifugation at 1600xg for 60minutes at 4°C. To transduce primary T cells, lentiviral spinfection was performed. Cells were thawed two days before spinfection, and one day before spinfection, cells were activated using Human T-activator CD3/CD28 Dynabeads (Thermo Fisher, 11131D) and cultured in 200U/ml IL-2. The concentrated virus was plated on non-TC treated 6-well plates coated with retronectin (Takara T100B) and spun for 90 minutes at 1200xg. The virus was then aspirated and activated T cells added to the virus-coated wells, followed by incubation at 37°C. To transduce Jurkat T cells, the concentrated virus was diluted in RPMI 1640, mixed with cells, and incubated for 72 hours at 37°C before the virus was washed out.
Antibodies and cell dyes
To stain for CD69, an APC-Cy7-conjugated mouse anti-human CD69 antibody (BD Pharmingen, 557756) was used at a dilution of 1:100. Myc-tagged and V5-tagged CARs were stained using an Alexa Fluor 488-conjugated mouse anti-myc antibody (R&D Systems, IC3696G) and an Alexa Fluor 647-conjugated mouse anti-V5 antibody (Invitrogen, 451098) respectively. For surface staining, cells were washed twice with FACS buffer (1x phosphate buffered saline (PBS), 0.1% NaN3, 1% BSA, 2mM EDTA) before incubating with antibody in the dark at room temperature for 40 minutes. Cells were then washed and resuspended with FACS buffer before analysis on an Attune NxT flow cytometer.
Cytotoxicity assay
Cytotoxicity assays were carried out by incubating primary T cells and NALM6 target cells at various effector-to-target (E:T) ratios overnight at 37°C. In the case of the switchboard VIPER CAR experiments, a 1:2 E:T ratio was used. 0.5–1μM GZV (MedChemExpress, HY-15298) was added at the time of assay, and DMSO was used as a control vehicle. When testing other NS3 inhibitors, various concentrations in the range of 0–5 μM of GZV, danoprevir, simeprevir, glecaprevir, and boceprevir were used in dose-response experiments. For dose-response experiments for ON and OFF VIPER CAR, 0–1μM of GZV were served, and for AND gate VIPER CAR, 0–5μM of GZV and (or) 0–5μM of TMP were served. For head-to-head comparison studies, 1uM of GZV, 1μM of TMP, and 1uM of Lenalidomide were used. Following a 16-hour incubation, the supernatant was saved, and cells were analyzed by flow cytometry to count the number of remaining live NALM6 cells by gating for either GFP+/mCherry+ cells or BFP+ positive cells depending on the target NALM6 cell line used. Killing efficiency was calculated as the percentage of dead cells compared to control wells containing NALM6 cells and no T cells.
Cytokine release assay
Supernatant from cells incubated with various combinations of target cells and NS3 inhibitors was saved and tested in an enzyme-linked immunosorbent assay (ELISA) to measure IFN- γ and IL-2 levels. The BD OptEIA human IFN- γ and IL-2 ELISA kits (BD Biosciences, 555142 and 555190) were used according to the manufacturer’s instructions with a 0.05% Tween-20 in PBS (Thermo Scientific, 28352) wash buffer and remaining reagents from BD OptEIA Reagent Set B (BD Biosciences, 550534). ELISAs were performed with 96-well MaxiSorp plates (Thermo Scientific, 442404).
Cell proliferation assay
Effector cells were stained with the Cell Trace Violet kit (Thermo Fisher, C34571) as per the manufacturer’s instructions. Treg and Teff cells were mixed with NALM6 target cells expressing Her2 and Axl as target cells at Treg: Teff: Target cell = 2:1:1 ratio. 1μM GZV (MedChemExpress, HY-15298) was added at the time of assay, and cells were collected for flow cytometry analysis after incubation for 5–6days.
Xenograft mouse tumor model
Female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, 6–8 weeks of age, were purchased from Jackson Laboratories (#005557) and maintained in the BUMC Animal Science Center (ASC). All protocols were approved by the Institutional Animal Care and Use Committee at BUMC.
To generate the intravenous blood tumor xenograft models, NSG (NOD Cg-Prkdcscid Il2rgtm1Wjl/ SzJ) mice were initially injected with 0.5 × 106 luciferized Nalm-6 cells intravenously. After 4 days, CAR-T (15 ×106, CAR+=55%) or VIPER CAR -T cells (15 ×106, ON VIPER CAR: CAR+=51.5%; OFF VIPER CAR: CAR+ (first component) =30%, mCherry+(second component)= 52%) were infused intravenously. Grazoprevir potassium salts (MedChemExpress, HY-15298A) were dissolved in 2.5% DMSO, 30% PEG400, and 67.5% PBS and i.p. injected every day for indicated dosage and times. For the dual-tumor xenograft model, the subcutaneous tumor was generated by injecting 1×106 MSTO-211H (Her2+, Luc+) cells subcutaneously in the right flank, and the systemic intravenous blood tumor was generated via the same method as described above. Seven days after the initial subcutaneous tumor implantation, the drug-inducible CAR-T cells (7.5×106 of each inducible CARs, ON VIPER CAR: CAR+=50–60%; OFF Lena-CAR: CAR+=50%) or traditional CAR-T cells (7.5×106 of each traditional CARs, CD19 CAR: CAR+=50–60%, Her2 CAR: CAR+=62%) were intravenously infused by tail vein injection. Grazoprevir potassium salts were injected using the same method as above, and pomalidomide was dissolved in 10% DMSO, and 90% corn oil (MedChem Express, HY-Y1888) and i.p. injected every day for indicated dosage and times. Tumor burden was measured by IVIS Spectrum (Xenogen) and was quantified as total flux (photons per sec) in the region of interest. Images were acquired within 10 minutes following intraperitoneal injection of 150mg/kg of D-luciferin (PerkinElmer #122799). For the dual-tumor animal model, systemic and subcutaneous tumor burden was quantified through luciferase signal via IVIS Spectrum, and separately the subcutaneous tumor size was measured by caliper regularly.
CRS model
SCID/beige mice (female, 6 to 8 weeks old, Taconic Bioscience) were intraperitoneally injected with 3×106 Raji tumor cells that were left to grow for 20 days. Mice were treated with either 6 ×107 traditional CAR-T cells (CAR+ = 49%) or 6 ×107 OFF VIPER CAR T cells (OFF VIPER CAR: CAR+ (first component) = 27%, mCherry+ (second component) = 32%) via intraperitoneal injection. GZV and dasatinib were administered intraperitoneally daily starting on the same day as CAR-T cell infusion. Blood was collected 24 hours post-T cell injection by submandibular blood collection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data between two groups was compared using an unpaired two-tailed t-test. All curve fitting was performed with Prism 7 (Graphpad) and p values are reported (not significant = p > 0.05, * = p < 0.05, ** = p < 0.01, *** =p < 0.001). All error bars are represented either SEM or SD.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-Cy7-anti-human CD69 | BD Pharmingen | 557756 |
| FITC Mouse Anti-Human CD4 Clone M-T477 | BD Biosciences | 556615 |
| Pacific Blue™ Mouse Anti-Human CD8 | BD Biosciences | 558207 |
| Dynabeads™ Human T-Activator CD3/CD28 | Thermo Fisher Scientific | 11132D |
| Alexa Fluor 647 anti-V5 Tag | Invitrogen | 451098 |
| Alexa Fluor 488- anti-myc Tag | R&D Systems | IC3696G |
| Cell Culture Reagents | ||
| RPMI-1640 | Lonza | 12-702Q |
| Ultraculture | Lonza | 12-725F |
| DMEM | Corning | 10-013 |
| X-VIVO 15 | Lonza | 04-418Q |
| Human AB serum | Valley Medical | HP1022 |
| FBS | Thermo Fisher Scientific | 10437-028 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | 31350010 |
| L-glutamine | Corning | 25-005-CL |
| Penicillin/Streptomycin | Caisson | PSL01 |
| Sodium Butyrate | Alfa Aesar | A11079 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Grazoprovir | MedChemExpress | HY15298 |
| Grazoprovir potasium salt | MedChemExpress | HY-15298A |
| Danoprevir | ApexBio | A4024 |
| Glecaprevir | MedChemExpress | HY-17634 |
| Simeprevir | MedChemExpress | HY-10241 |
| Boceprevir | MedChemExpress | HY-10237 |
| TMP | Sigma-Aldrich | 92131 |
| Lenalidomide | MedChemExpress | HY-A0003 |
| Pomalidomide | MedChemExpress | HY-10984 |
| XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate | Perkin Elmer | 122799 |
| N-Acetyl-L-cysteine | Sigma-Aldrich | A9165 |
| Lenti-X™ Concentrator | Clontech | 631232 |
| RetroNectin® Recombinant Human Fibronectin Fragment | Clontech | T100B |
| Phosphate-Buffered Saline (10X) pH 7.4 | Thermo Fisher Scientific | AM9625 |
| PEG8000 | Thermo Fisher Scientific | BP233-1 |
| Sodium Chloride | Thermo Fisher Scientific | AAJ2161836 |
| DMSO | CHEM-IMPEX INT’L INC | 00635 |
| PEG400 | MedChemExpress | HY-Y0873A |
| Corn oil | MedChemExpress | HY-Y1888 |
| Critical Commercial Assays | ||
| RosetteSep™ Human CD4+ T Cell Enrichment Cocktail | STEMCELL Technologies | 15062 |
| RosetteSep™ Human CD8+ T Cell Enrichment Cocktail |
STEMCELL Technologies | 15063 |
| EasySep™ Human CD4+CD127lowCD25+ Regulatory T Cell Isolation Kit | STEMCELL Technologies | 18063 |
| Human IL-2 ELISA Set | BD Biosciences | 555190 |
| Human IFN-γ ELISA Set | BD Biosciences | 555142 |
| XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate | PerkinElmer | 122799 |
| CellTrace™ Violet Cell Proliferation Kit, for flow cytometry | Thermo Fisher Scientific | C34557 |
| RosetteSep™ Human CD4+ T Cell Enrichment Cocktail | STEMCELL Technologies | 15062 |
| Experimental Models: Cell Lines | ||
| HEK293FT | N/A | N/A |
| NFAT-Jurkat | Art Weiss (UCSF) | N/A |
| NALM6 (Luc+ BFP+) | This paper | N/A |
| NALM6 (HER2+ Axl+ Mesothilin+ Luc+) | This paper | N/A |
| NALM6 (HER2+ mCherry+) | This paper | N/A |
| NALM6 (Axl+ GFP+) | This paper | N/A |
| MSTO-211H (Luc+ BFP+) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (female 6–8 weeks) | Jackson Laboratory | 005557 |
| Scid-beige(female 6–8 weeks) | Taconic Biosciences | CBSCBG-F |
| Recombinant DNA | ||
| pHR-SFFV vector | Addgene | 79121 |
| PB-pCAG-MCS-EF1a-zeocine-BFP vector | This paper | N/A |
| Software and Algorithms | ||
| Graph Pad Prism 7 | Graph Pad | N/A |
| FlowJo V10 | TreeStar | N/A |
| Living Image | Perkin Elmer | N/A |
Highlight.
Inducible ON and OFF switches using FDA-approved drugs
Best-in-class performance determined through direct comparison
Multiplex CAR circuits with advanced functionalities
ACKNOWLEDGEMENTS
W.W.W. acknowledges funding from the Boston University Ignition Award and NIH Award (DP2CA186574, U01CA265713, R56EB027729, R01EB029483). A.S.K. acknowledges funding from the NIH (R01EB029483), NIH Director’s New Innovator Award (1DP2AI131083), DARPA Young Faculty Award (D16AP00142), and DoD Vannevar Bush Faculty Fellowship (N00014-201-2825). J.T.N and E.P.T were supported by the NIH NIGMS grant (R35-GM128859-01) and the Reidy Family Career Development Award. E.P.T. was funded through a T32 NIH Training Grant awarded to Boston University (EB006359). We also thank Wong lab members for suggestions on the manuscript; Dr. Todd Blute from the BU Proteomics & Imaging Core Facility for flow cytometry assistance; BU IVIS imaging core facility for mice in vivo imaging (NIH grant 1S10RR024523-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A patent application has been filed based on this work (H.S.L., N.M.W., E.T., J.T.N., and W.W.W). W.W.W. is a scientific co-founder and shareholder of Senti Biosciences. A.S.K. is a scientific advisor and shareholder of Senti Biosciences and Chroma Medicine.
INCLUSION AND DIVERSITY
We work to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
REFERENCES
- Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen Y-B, Palmer E, Albertson T, Ferry JA, and Arrillaga-Romany IC (2017). Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. The New England journal of medicine 377, 783. [DOI] [PubMed] [Google Scholar]
- Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, and Grupp SA (2011). Treatment of advanced leukemia in mice with mRNA engineered T cells. Human gene therapy 22, 1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AM, Wagner JL, Barber KE, and Stover KR (2016). Elbasvir/Grazoprevir: A Review of the Latest Agent in the Fight against Hepatitis C. Int J Hepatol 2016, 3852126. 10.1155/2016/3852126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifant CL, Jackson HJ, Brentjens RJ, and Curran KJ (2016). Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 3, 16011. 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker T, and Karjalainen K (1995). Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. Journal of Experimental Medicine 181, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, and Simpson J (2016). Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New England Journal of Medicine 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno JN, and Kochenderfer JN (2016). Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno JN, and Kochenderfer JN (2019). Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood reviews 34, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Jacobs CL, Huo Y, Yang J, Krumm SA, Plemper RK, Tsien RY, and Lin MZ (2015). Tunable and reversible drug control of protein production via a self-excising degron. Nature chemical biology 11, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham-Bryant D, Dieter EM, Foight GW, Rose JC, Loutey DE, and Maly DJ (2019). A chemically disrupted proximity system for controlling dynamic cellular processes. Journal of the American Chemical Society 141, 3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, and Olszewska M (2014). Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine 6, 224ra225–224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E, and Li G (2016). Approved antiviral drugs over the past 50 years. Clinical microbiology reviews 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, and Grilley B (2011). Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365, 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foight GW, Wang Z, Wei CT, Warner KM, Cunningham-Bryant D, Park K, Brunette T, Sheffler W, Baker D, and Maly DJ (2019). Multi-input chemical control of protein dimerization for programming graded cellular responses. Nature biotechnology 37, 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano-Attianese G, Gainza P, Gray-Gaillard E, Cribioli E, Shui S, Kim S, Kwak MJ, Vollers S, Osorio ADJC, and Reichenbach P (2020). A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nature Biotechnology 38, 426–432. [DOI] [PubMed] [Google Scholar]
- Hombach AA, and Abken H (2013). Of chimeric antigen receptors and antibodies: OX40 and 41BB costimulation sharpen up T cell-based immunotherapy of cancer. Immunotherapy 5, 677–681. [DOI] [PubMed] [Google Scholar]
- Imai C, Mihara K, Andreansky M, Nicholson I, Pui C, Geiger T, and Campana D (2004). Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18, 676–684. [DOI] [PubMed] [Google Scholar]
- Iwamoto M, Björklund T, Lundberg C, Kirik D, and Wandless TJ (2010). A general chemical method to regulate protein stability in the mammalian central nervous system. Chemistry & biology 17, 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs CL, Badiee RK, and Lin MZ (2018). StaPLs: versatile genetically encoded modules for engineering drug-inducible proteins. Nature methods 15, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, Gasser JA, Słabicki M, Bouffard AA, and Castano AP (2021). Reversible ON-and OFF-switch chimeric antigen receptors controlled by lenalidomide. Science translational medicine 13, eabb6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, and Peng C (2020). Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature, 1–5. [DOI] [PubMed] [Google Scholar]
- Juillerat A, Marechal A, Filhol J-M, Valton J, Duclert A, Poirot L, and Duchateau P (2016). Design of chimeric antigen receptors with integrated controllable transient functions. Scientific reports 6, 18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, and Sadelain M (2013). Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nature biotechnology 31, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohm AP, Williams JS, and Miller SD (2004). Cutting edge: ligation of the glucocorticoid-induced TNF receptor enhances autoreactive CD4+ T cell activation and experimental autoimmune encephalomyelitis. The Journal of Immunology 172, 4686–4690. [DOI] [PubMed] [Google Scholar]
- Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, and Campana D (2014). T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer research 74, 93–103. [DOI] [PubMed] [Google Scholar]
- Kügler J, Schmelz S, Gentzsch J, Haid S, Pollmann E, van den Heuvel J, Franke R, Pietschmann T, Heinz DW, and Collins J (2012). High affinity peptide inhibitors of the hepatitis C virus NS3–4A protease refractory to common resistant mutants. Journal of Biological Chemistry 287, 39224–39232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Keller B, Makalou N, and Sutton RE (2001). Systematic determination of the packaging limit of lentiviral vectors. Human gene therapy 12, 1893–1905. [DOI] [PubMed] [Google Scholar]
- Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, Pacheco KZB, Malipatlolla M, Xu P, and Hui JH (2022). Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 185, 1745–1763. e1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, and Powell DJ (2013). Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer immunology research 1, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, Przepiorka D, Farrell AT, and Pazdur R (2018). FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell‐induced severe or life‐threatening cytokine release syndrome. The oncologist 23, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, and Weiss A (2003). The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. Journal of Cell Biology 162, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, Glenn JS, and Tsien RY (2008). A drug-controllable tag for visualizing newly synthesized proteins in cells and whole animals. Proceedings of the National Academy of Sciences 105, 7744–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JS, Kim JY, Kazane SA, Choi S. h., Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, and Sun SB (2016). Versatile strategy for controlling the specificity and activity of engineered T cells. Proceedings of the National Academy of Sciences 113, E450E458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, and Lacey SF (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine 371, 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley JA, and Rudd MT (2016). Hepatitis C virus NS3/4a protease inhibitors. Current opinion in pharmacology 30, 84–92. [DOI] [PubMed] [Google Scholar]
- Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, Mades A, Sadelain M, Einsele H, and Hudecek M (2019). The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Science translational medicine 11, eaau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy 18, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Chaturvedi P, Qin H-Y, and Singh B (2003). CD4+ CD25+ regulatory T cells generated in response to insulin B: 9–23 peptide prevent adoptive transfer of diabetes by diabetogenic T cells. Journal of autoimmunity 21, 221–237. [DOI] [PubMed] [Google Scholar]
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJ, Martinez-Lage M, Brem S, Maloney E, and Shen A (2017). A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science translational medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz I, and Koenecke C (2012). Therapeutic potential of induced and natural FoxP3+ regulatory T cells for the treatment of Graft-versus-host disease. Archivum immunologiae et therapiae experimentalis 60, 183–190. [DOI] [PubMed] [Google Scholar]
- Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, and Somana K (2017). Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science translational medicine 9. [DOI] [PubMed] [Google Scholar]
- Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, and Finklestein J (2015). NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nature medicine 21, 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016). Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco MD, Ma C, Lagarias P, Gao A, Townsend JA, Meng X, Dube P, Zhang X, Hu Y, and Kitamura N (2020). Structure and inhibition of the SARS-CoV-2 main protease reveals strategy for developing dual inhibitors against Mpro and cathepsin L. Science advances, eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, and Rader C (2019). Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer cell 35, 489–503. e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tague EP, Dotson HL, Tunney SN, Sloas DC, and Ngo JT (2018). Chemogenetic control of gene expression and cell signaling with antiviral drugs. Nature methods 15, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA, and Kang S-M (2012). CD4+ Foxp3+ regulatory T cell therapy in transplantation. Journal of molecular cell biology 4, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, and Powell DJ (2012). A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer research 72, 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, and Klysz D (2021). Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372, eaba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-Y, Roybal KT, Puchner EM, Onuffer J, and Lim WA (2015). Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, and Trono D (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. Journal of virology 72, 9873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.