

Abstract
Enhancer of Zeste Homolog 2 (EZH2) is the catalytic component of the Polycomb Repressive Complex 2, a chromatin modifying complex, which mediates methylation of lysine 27 on histone 3 (H3K27me3), a repressive chromatin mark. Genetic alterations in EZH2 in melanoma include amplifications and activating point mutations at tyrosine 641 (Y641) whose underlying oncogenic mechanisms remain largely unknown. Here, we found that expression of Ezh2Y641F causes upregulation of a subset of interferon-regulated genes in melanoma cells. Upregulation of these genes was not a direct effect of changes in H3K27me3, but via a non-canonical interaction between Ezh2 and Signal Transducer and Activator of Transcription 3 (Stat3). Ezh2 and Stat3 together function as transcriptional activators to mediate gene activation of numerous genes, including MHC Class 1b antigen processing genes. Furthermore, expression of Stat3 is required to maintain an anti-tumor immune response in Ezh2Y641F melanomas and to prevent melanoma progression and recurrence.
Keywords: EZH2, STAT3, PRC2, chromatin, melanoma, anti-tumor immune response
INTRODUCTION
Epigenetic modifiers are frequently mutated in melanoma, and are associated with poor survival, response to treatment, or relapse 1,2. A chromatin modifier frequently mutated in melanoma is EZH2, the catalytic component of the Polycomb Repressive Complex 2 (PRC2), which methylates lysine 27 on histone 3 (H3K27me3), a chromatin mark associated with transcriptional repression. Genetic alterations in EZH2 in melanoma include amplifications and activating point mutations at tyrosine 641(Y641) (Supp. Fig. 1a, b) 3–5. Patients with genetic alterations in PRC2 components and other chromatin modifiers that are highly connected with PRC2 exhibit lower survival rates, as do patients with high protein expression of EZH2 (Supp. Fig. 1c,d).
To investigate the role of EZH2 mutations in melanoma, we previously generated a mouse model permitting conditional expression of mutant Ezh2Y641F and showed that Ezh2Y641F expression in murine melanocytes accelerates melanomagenesis. Molecularly, Ezh2Y641F mutations also caused a global increase and redistribution in H3K27me3 across the genome 6. Ezh2Y641F in mouse models strongly co-operated with mutant BrafV600E but failed to co-operate with mutant NrasQ61R, two of the main oncogenic drivers in human melanoma 3. This result was consistent with the fact that in human patients, EZH2Y641 mutations tend to co-occur with BRAFV600E but not NRASQ61 mutations. Despite our understanding of the consequences of the Ezh2Y641F mutations on chromatin and distribution of H3K27me3, we still do not know how its downstream molecular mechanisms contribute towards melanoma initiation or progression. Similar to EZH2Y641 mutations, high EZH2 expression is also associated with disease progression and more malignant forms of melanoma 7–11 suggesting that increased Ezh2 activity in melanocytes may promote transformation and cancer initiation through epigenetic silencing of tumor suppressors or other mechanisms 12.
To understand the oncogenic mechanisms of Ezh2 in melanoma, we compared the gene expression profiles of Ezh2WT vs Ezh2Y641F mouse melanoma tumors and identified an interferon-related gene expression signature. Since we did not observe significant changes to H3K27me3 at these loci, we hypothesized that expression of these genes is driven via non-canonical interactions of Ezh2 with non-histone proteins. One such candidate protein was the immunomodulatory factor Stat3, which had previously been shown to be methylated by Ezh2 13,14. Stat3 is a member of the STAT family and is activated in response to extracellular signaling proteins, including growth factors and cytokines such as IL-6 15,16. Stat3 is activated by phosphorylation, which promotes its dimerization and nuclear transport, where it functions as a transcription factor 17, often in association with other transcription factors and co-activators 18–20. In addition to being a transcriptional activator, Stat3 can also repress its target genes 21,22; however, the mechanism of how Stat3 represses gene expression is not clear. Stat3 is implicated in many oncogenic mechanisms, contributing to both cell-intrinsic and -extrinsic hallmarks of cancer such as cell growth, proliferation, differentiation, apoptosis, inflammation, and immune cell regulation23.
In this study, we investigated the downstream molecular mechanisms of Ezh2Y641F mutations in melanoma, and show that Ezh2Y641F, in co-ordination with Stat3, causes changes in tumor antigenicity. We thus present significant insight into how Ezh2Y641F mutations favor non-canonical interactions and their biological significance during melanoma progression.
MATERIALS & METHODS
Animals
Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility and treated in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) for animal research at Washington University in St. Louis.
In vivo tumor models
C57Bl/6 mice were obtained from Taconic Biosciences. Tumor cells were injected subcutaneously in the flank at 0.5 × 10^6 cells per injection, two injections per mouse. Anti-CD8 (InVivoMAb anti-mouse CD8β Cat# BE0223 Lot# 733219O1) and Stat3 inhibitor STATTIC (MedChemExpress Cat# HY-13818 Batch# 21868) were administered intraperitoneally at 10 mg/kg. For flow cytometry analysis, tumors were harvested at Day 9 or, for mice treated with anti-CD8, 15 days after last injection. Tumors were chopped, dispersed by passing through a syringe with 18G needle, and filtered through a 0.40 µm filter. For the anti-PD-1 and JQEZ5 experiment, tumor cells were injected in immunocompetent mice, and the anti-PD-1 (200μg) and JQEZ5 (100 mg/kg) were administered every other day intraperitoneally.
Flow Cytometric analysis
Single cell suspensions from dissociated tumors were washed with PBS containing 1% FBS and stained with the following antibody cocktails. Lymphoid antibody cocktail contained: anti-CD45-PerCP/Cy5.5 (BioLegend 103132), anti-NK1.1-FITC (BioLegend 108706), anti-CD3-PB (BioLegend 100214), anti-CD4-APC (BioLegend 100412), anti-CD8-AF700 (BioLegend 100730), and anti-PD-1(CD279)-PE/Cy7 (BioLegend 135216). Myeloid antibody cocktail: anti-CD45-PerCP/Cy5.5 (BioLegend 103132), anti-CD19-FITC (BioLegend 115506), anti-B220-FITC (BioLegend 103206), anti-CD3-FITC (BioLegend 100204), anti-CD11b(Mac1)-PB (BioLegend 101224), anti-CD11c-PE/Cy7 (BioLegend 117318), and anti-Ly-6G(Gr1)-AF700 (BioLegend 127622). Propidium Iodide was used to detect dead cells. Samples were run on an Attune Nxt Flow Cytometer (ThermoFisher Scientific) at the Siteman Flow Cytometry Core Facility. Analysis was done in FlowJo V10 and statistically significant differences were identified using One-way ANOVA.
Analysis of RNA-seq data
Gene Set Enrichment Analysis (GSEA) was performed as described here 24. Enrichment of differentially expressed genes was performed using a pre-ranked gene list using log2 ratio of expression of Ezh2WT vs Ezh2Y641F. Functional annotation of RNA-seq data was performed with the Database for Annotation, Visualization, Integrated Discovery (DAVID) tools 25.
Cell culture, shRNA, cell growth assay
Six syngeneic mouse melanoma cell lines were used: Ezh2WT (234, 855, and 480; Tyr-CreERT2 BrafV600E PtenF/+ Ezh2+/+) or Ezh2Y641F (234Δ, 855Δ, and 480Δ; Tyr-CreERT2 BrafV600E PtenF/+ Ezh2Y641F/+). Cells were cultured in DMEM (Sigma D6429), 10% FBS (Corning Cat# MT35010CV) and 1% penicillin-streptomycin (Genesee Scientific Cat# 25–512). Stat3 knockdown cell lines were generated by transducing cells with lentiviral shRNA (TRCN0000071456, TRCN0000071454, TRCN0000071453, Sigma). Lentiviruses were generated using 293T cells via transfection with PEI. Stable cell lines were selected with puromycin (3 µg/ml). Knockdown was confirmed by qPCR and immunoblotting. For the growth assay, cells were plated in triplicate in 96-well plates at 500 cells/well and growth was determined using Alamar Blue assay. Statistically significant differences were detected using Student’s t-test.
Chromatin Immunoprecipitation (ChIP) sequencing and analysis
Ten million cells were used for each ChIP and were prepared using the protocol from Lee et. al. 26. Briefly, cells were lysed and sheared using a probe sonicator to achieve a size of 200–300 bp, and chromatin was isolated using phenol: chloroform for library preparation. Immunoprecipitations were performed overnight at 4°C using anti-Stat3 (Cell Signaling #9139) and anti-Ezh2 (Cell Signaling #5246) antibodies at 1:100 dilution. Libraries were prepared using TruSeq ChIP kit (Illumina) and multiplexed for sequencing using 50-bp single reads. Libraries were sequenced on a Hi-Seq 2000 at the Genome Access Center of Washington University in St. Louis. Processing of ChIP-seq data was carried out using Encode standards. Binding sites were identified using MACS2. The functional significance of Ezh2 and Stat3 binding sites/peaks was evaluated using the Genomic Regions Enrichment of Annotations Tool (GREAT) 27. Data was visualized using the Integrated Genomics viewer 28. De novo motif analysis was carried out using HOMER 29. The Ezh2 and Stat3 differentially bound peaks were annotated with the R package ChipSeeker. For ChIP-qPCR and sequential ChIP (ChIP-reChIP), forty million cells per sample were used and prepared according to Furlan-Magaril et al. 30
Protein Immunoprecipitation (IP)
Cells were lysed using RIPA buffer plus protease and phosphatase inhibitors. Lysates were incubated with Stat3 antibody (1:100, Cell Signaling #9139) for 24 hours, then incubated with protein A/G magnetic beads (Bio-Rad SureBeads Cat# 1614023) for 1 hour. Samples were eluted by incubating in 1X Laemmli buffer (Bio-Rad Cat# 161–0747) with beta-mercaptoethanol at 70°C for 10 minutes.
Immunoblotting
Samples were prepared in Laemmli buffer with beta-mercaptoethanol, run on 4–20% pre-cast gels (Bio-Rad Mini-PROTEAN TGX Gels Cat# 4561095), transferred onto nitrocellulose membranes using the Bio-Rad Trans-Blot Turbo Transfer System, blocked for 1 hour in 5% milk in TBS-T, then incubated with primary antibodies overnight at 4°C. Primary antibodies: anti-Ezh2 (Cell Signaling #5246 at 1:1000), anti-beta actin (Abcam ab213262 at 1:2000), anti-Stat3 (Cell Signaling #9139 at 1:500), and anti-phospho-Stat3 (Tyr705) (Cell Signaling #9145 at 1:2000). Membranes were washed with TBS-T before secondary antibody staining at room temperature for 1 hour. Secondary antibodies: anti-rabbit IgG (H+L) (DyLight 800 4X PEG Conjugate, Cell Signaling #5151) and anti-mouse IgG (H+L) (DyLight 680 Conjugate, Cell Signaling #5470), both at 1:15,000. Membranes were imaged using a Licor Odyssey Infrared Imager, and Licor Image Studio software or ImageJ were used for densitometry analysis.
Quantitative PCR
RNA was isolated from melanoma cells using the Aurum Total RNA Mini Kit (Bio-Rad Cat# 7326820) according to the manufacturer’s instructions. The igScript First Strand cDNA Synthesis Kit (Intact Genomics Cat# 4314) was used to synthesize the cDNA. qPCR was performed with the iTaq Universal SYBR Green Supermix (Bio-Rad Cat# 1725120) using primers listed in Supplemental Table 1.
Statistical analysis
Data was analyzed using GraphPad Prism software or Microsoft Excel. Bar graphs represent mean and standard deviation unless otherwise noted. Sample sizes were determined based on existing literature and prior pilot studies. Power analysis was also carried out, assuming normal distribution, two-tailed, to test hypotheses with significance level of 0.05 and power 0.8. Differences between two groups were compared using Student’s t-test.
RESULTS
Ezh2Y641F induces an interferon-related gene expression signature dependent on Ezh2 methyltransferase activity but not H3K27me3
To better understand the underlying oncogenic mechanisms of Ezh2Y641F mutations, we analyzed gene expression patterns from isogenic cell lines derived from Tyr-CreERT2 BrafV600E/+ PtenF/+ mouse melanomas that differ only in expression of the Ezh2 mutation (WT vs Y641F). Functional annotation 25,31 and gene set enrichment analysis of differentially expressed genes identified several enriched gene ontology terms with an immune response category, with interferon signaling being the most significant (Fig. 1a-b). A subset of these differentially expressed, IFN-regulated genes was validated on separate biological samples by qPCR (Fig. 1c).
Figure 1. EZH2Y641F melanomas exhibit an interferon-related gene expression profile independent of H3K27me3.

(a) Functional annotation of gene expression profile analysis of Ezh2WT vs Ezh2Y641F mutant melanoma cells. Upregulated genes in Ezh2Y641F cells were analyzed using the DAVID functional annotation tool to identify enriched Gene Ontology (GO) terms.
(b) GSEA plot of the Hallmark: Interferon gamma response category.
(c) Validation of differentially expressed interferon-related genes by qPCR in three separate biological controls. ** p<0.01.
(d) Log2 fold change of upregulated interferon-related genes in Ezh2Y641F melanoma cell lines treated with vehicle or the Ezh2 inhibitor JQEZ5. Expression is shown as the ratio between Ezh2Y641F and Ezh2WT melanoma cell lines (Y641F/WT).
(e) Analysis of H3K27me3 ChIP signal 5kb upstream and downstream of the transcriptional start sites (TSS) of the genes from panel (d).
Next, we investigated whether expression of these IFN-regulated genes was dependent on Ezh2’s methyltransferase activity. We analyzed gene expression after treatment with the Ezh2 inhibitor JQEZ5 6,32 and found that treatment with the inhibitor reversed the overexpression driven by the Ezh2Y641F mutation (Fig. 1d). To determine whether expression of these genes was also dependent on the repressive H3K27me3 mark mediated by Ezh2, we analyzed H3K27me3 distribution at the promoter regions of these differentially expressed genes. Surprisingly, we did not find a decrease in H3K27me3 (Fig. 1e), suggesting that even though expression of these IFN-related genes is dependent on Ezh2 activity it is not dependent on canonical H3K27 methylation, but instead occurs through an indirect or non-canonical function.
Immune response signatures in patients with increased EZH2 activity or Y641 mutations
To investigate the effect of EZH2Y641F mutations and increased EZH2 activity on gene expression in melanoma patients, we analyzed TCGA cohorts with available mutation and expression data. EZH2Y641F mutations exhibit increased enzymatic activity and, in some ways, they may be equivalent to increased EZH2 activity via overexpression. The two, however, are not necessarily identical. We previously showed that even though Ezh2Y641F mutations lead to increased methyltransferase activity resulting in a global increase of H3K27me3 as expected, they also cause redistribution of H3K27me3 away from promoters and towards gene bodies 6. We thus proposed that Ezh2Y641 mutations are not merely activating mutations, but neomorphic 6. Based on the above observations, we expect EZH2Y641F mutations and EZH2 overexpression to share some functions based on increased enzymatic activity. On the other hand, we also expect the two to exhibit significant differences stemming from the neomorphic properties of the Y641 mutant. We thus hypothesized that these similarities and differences would be reflected in the global gene expression signatures of mouse and human models with these alterations. Towards that end, we analyzed the gene expression signatures of 52 melanoma patients that exhibited at least a two-fold increase in EZH2 expression (exp>2) (Supp. Fig. 2a-c). Functional annotation of the downregulated genes in the “EZH2-high” patient group, and Ezh2Y641F mouse melanoma cells, showed strong enrichment for lipid metabolism in both groups (Supp. Fig. 2d). These changes are likely due to canonical PRC2 functions via increased deposition of the repressive H3K27me3, common to both groups. In contrast, we observed very different gene expression signatures in the upregulated genes between the “EZH2-high” human group and the Ezh2Y641F mouse model. Upregulated genes in the “EZH2-high” group were enriched for cell cycle, DNA replication and repair pathways, whereas the predominant pathway in Ezh2Y641F cells was related to immunity, response to type I interferon and the JAK/STAT pathway, among others (Supp. Fig. 2e). We were also able to identify three patients with EZH2Y641 mutations with available mutation and gene/protein expression data (TCGA Pan Cancer group). These patients exhibited enrichment for Immune pathways, IL-7 signaling, JAK-STAT pathway and PD-1 checkpoint compared to patients without EZH2 alterations. These pathways are consistent with what we observe in the Ezh2Y641F mouse model and provide further support to how relevant our model and is to human melanoma. We hypothesized, therefore, that the immune cell profile of Ezh2Y641F melanomas is a consequence of its neomorphic nature and contributes to the oncogenic activity of Ezh2.
Ezh2Y641F protein directly interacts with Stat3 in melanoma cells
Given the enrichment of the JAK-STAT pathway in the Ezh2Y641F mouse model and melanoma patients, we considered whether Ezh2 interacts with known IFN regulatory factors, such as the STAT proteins (Fig. 1a, Supp. Fig. 2). Previous studies have shown that Ezh2 directly interacts with and methylates Stat3 13,14,33, however the consequences of Stat3 methylation were not consistent between the three studies. To investigate the relevance of Stat3 in melanoma, we first determined Stat3 expression in our models and found increased protein levels in the nucleus of Ezh2Y641F than Ezh2WT cells, however, there was no difference in phosphorylated/activated Stat3 (Fig. 2a). To test whether Ezh2 interacts or forms a complex with Stat3 in melanoma cells, we performed co-affinity immunoprecipitation in Ezh2WT and Ezh2Y641F cells and successfully pulled down both Ezh2 and Stat3 (Fig. 2b). Additionally, we found that Stat3 was hypermethylated in the presence of Ezh2Y641F protein, while a much weaker interaction was observed in the presence of Ezh2WT protein in one cell line.
Figure 2. Stat3 co-immunoprecipitates with Ezh2 in Ezh2Y641F mutant cells and is hypermethylated.
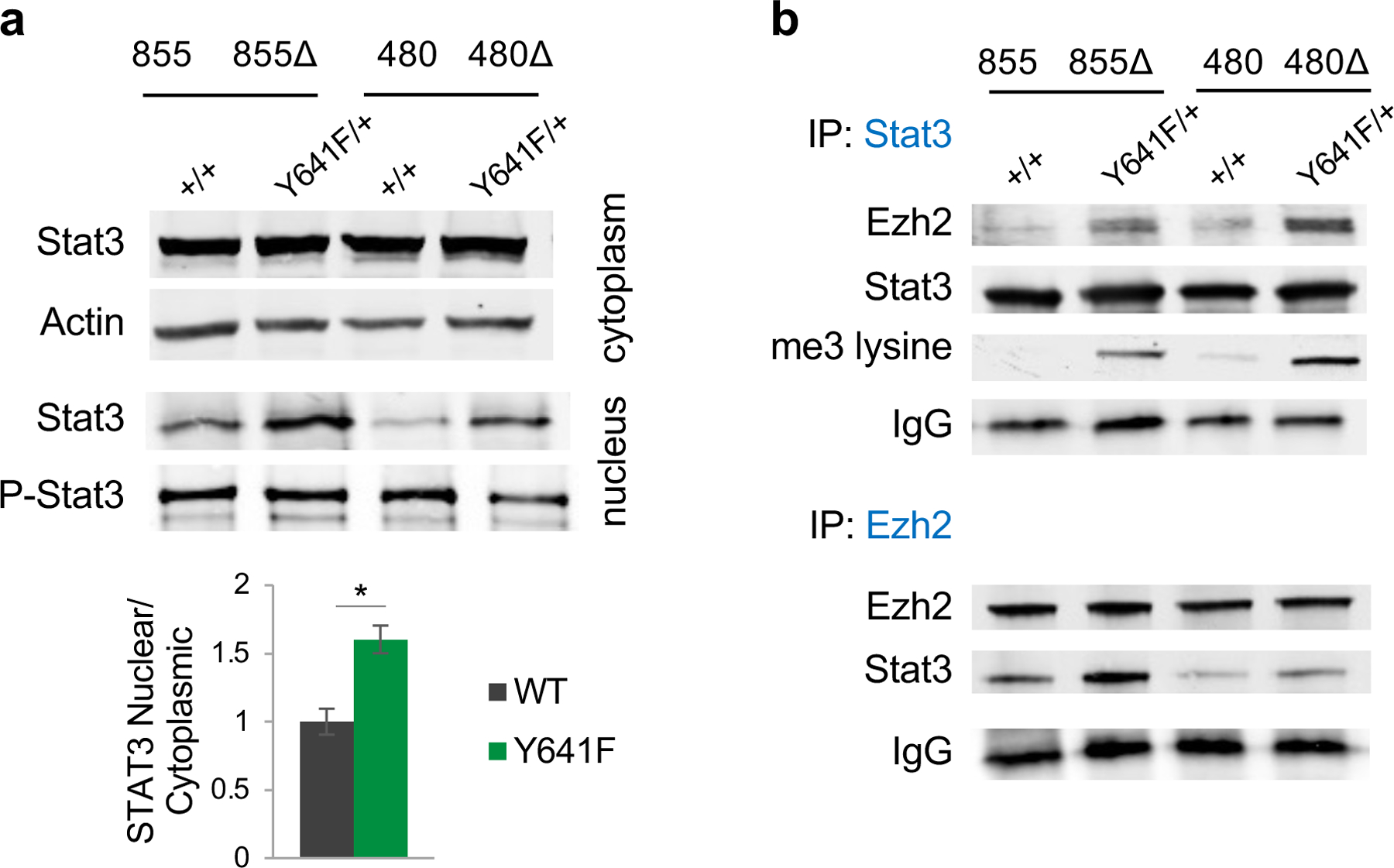
(a) Top: Representative image of immunoblotting analysis for Stat3 and actin in the cytoplasm, and Stat3 and phospho-Stat3 in the nucleus of mouse melanoma cell lines 480 and 855 (Δ = Y641F mutant). The bar graph represents quantification of the Stat3 nuclear/cytoplasmic ratio and is plotted as average ± SD (N = 4/group, p = 0.051).
(b) Immunoprecipitation for Stat3 and Ezh2, immunoblotted for Stat3, Ezh2, and tri-methylated lysine.
Stat3 knockdown in Ezh2Y641F melanoma cells restores expression of IFN-regulated genes without affecting cell growth
To investigate the potential role of Stat3 in the differential gene expression patterns observed in Ezh2Y641F melanoma cells, we used lentiviral short hairpin RNA (shRNA) to knockdown Stat3 in the syngeneic melanoma cell lines described above (Fig. 3a). Stat3 shRNA knockdown resulted in at least 90% knockdown efficiency and coincided with decreased expression of many of the upregulated IFN-regulated genes in Ezh2Y641F cells (Fig. 3b), suggesting that the changes in gene expression are at least partially mediated by Stat3. Furthermore, we found that Stat3 knockdown had no significant effect on in vitro melanoma cell growth (Fig. 3c), suggesting that if Stat3 plays a role in the pathogenesis of Ezh2Y641F melanoma, it is not through regulation of intrinsic cell growth.
Figure 3. Stat3 knockdown in melanoma cell lines partially restores IFN-related gene expression without affecting intrinsic cell growth.

(a) Lentiviral shRNA Stat3 knockdown (KD) in melanoma cell lines confirmed by qPCR (left) and by Western blot (***p<0.001). Control shRNA targets the luciferase gene.
(b) Expression of interferon-related genes in control and shRNA Stat3 knockdown. Data were averaged from triplicate experiments (*p<0.05, **p<0.01).
(c) Effect of shRNA Stat3 knockdown in Ezh2Y641F melanoma cell growth in vitro.
Ezh2Y641F changes global Ezh2 and Stat3 association with chromatin and DNA
To understand the molecular effects and the nature of the interaction between Ezh2 and Stat3 in melanoma, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) for Ezh2 and Stat3 in Ezh2WT vs Ezh2Y641F melanoma cells. Three separate syngeneic cell lines were used for these experiments as biological controls. Comparative analysis between genotypes revealed several global differences mediated by expression of Ezh2Y641F. For example, in Ezh2Y641F melanoma cells, Ezh2 protein was bound less frequently on promoter regions but more within the first intron (Supp. Fig. 3a, c), consistent with our previous observations where H3K27me3 in Ezh2Y641F is redistributed away from promoters and towards gene bodies 6. Stat3 binding across the genome was also altered in Ezh2Y641Fcells, exhibiting less binding in promoter regions compared to Ezh2WT cells (Supp. Fig. 3b, d). Systematic analysis of all differential Stat3 peaks showed binding/association of Stat3 with new genomic loci in Ezh2Y641F cells (Supp. Fig. 3e). Analysis of called peaks within proximal genes in Ezh2 and Stat3 ChIP-seq identified numerous known loci/targets bound by either protein. For example, Ezh2 was associated with known PRC2-regulated regions, such as Hoxa and Hoxc clusters, and Stat3 was associated with known Stat3 targets, such as Hif1a, Pim2, Foxo1, Il10 34 (Supp. Fig. 4).
Next, we used the Genomic Regions Enrichment of Annotations Tool (GREAT) to analyze the functional significance of cis-regulatory regions with Ezh2 and Stat3 binding 27 and identified genes known to be regulated by Ezh2 and Stat3 (Supp. Fig. 5a), suggesting that Ezh2 and Stat3 continue to interact with some of their canonical targets. Functional annotation analysis of these targets identified multiple pathways implicated in cancer cell mechanisms, such as cell migration, differentiation, IL-6 signaling and JAK/STAT3 signaling (Supp. Fig. 5b-d). In addition to their canonical targets, we found that Ezh2 and Stat3 also bind to new, non-canonical targets in Ezh2Y641F cells. Comparison of ChIP-seq peaks for Ezh2 and Stat3 in Ezh2Y641F cells identified several loci co-occupied by Ezh2 and Stat3 (Fig. 4a) where binding to these loci was not detected in Ezh2WT cells. Motif discovery analysis using HOMER tools 29 identified transcription factor binding motifs enriched in the co-occupied loci, including several transcription factors that are directly implicated in melanomagenesis such as MITF, cell proliferation (E2F1), or the immune response (IRF or STAT transcription factors, Fig. 4b). This is consistent with the initial observations in both mouse and human cells which exhibit alterations in immune regulatory pathways (Fig. 1).
Figure 4. Changes in global chromatin binding by Ezh2 and Stat3 in Ezh2Y641F melanoma cells and its effect on gene expression.

(a) ChIP-seq peaks for Ezh2 (left circle) and Stat3 (right circle) in Ezh2Y641F melanoma cells reveal new binding sites for Ezh2 and Stat3, and sites co-occupied by both proteins.
(b) Transcription factor binding motifs enriched in the co-occupied peaks of Ezh2 and Stat3 in Ezh2Y641F melanoma cells from panel (a).
(c) ChIP-seq peaks of Ezh2, Stat3 and H3K27me3 in Ezh2WT and Ezh2Y641F melanoma cells, proximal to the Ezh2 gene locus on chromosome 7.
(d) Expression of Ezh2 in Ezh2WT vs Ezh2Y641F melanoma cells after Stat3 knockdown (***p<0.001, *p<0.05).
(e) Sequential ChIP-reChIP with Ezh2 and Stat3 at the co-occupied peaks in Ezh2Y641F melanoma cells (*p<0.05, **p<0.01).
Ezh2 expression becomes dependent on the presence Stat3 in Ezh2Y641F melanoma cells
One locus co-occupied by Ezh2 and Stat3 in Ezh2Y641F melanoma cells was on chromosome 6 upstream of the Ezh2 gene itself and which was predicted to control its expression (Fig. 4c). To determine whether regulation of Ezh2 expression in Ezh2Y641F mutant melanoma cells was dependent on the presence of Stat3, we knocked down Stat3 in three mouse melanoma cell lines and found a ten-fold decrease in Ezh2 expression in Ezh2Y641F cells, while only a two-fold decrease in Ezh2WT cells (Fig. 4d). This suggests that Ezh2 and Stat3 can function as transcriptional activators in a unique interaction mediated by mutant Ezh2Y641F. We further verified co-occupancy of Ezh2 and Stat3 at this region using sequential ChIP followed by qPCR (Fig. 4e). Notably, there was no difference in H3K27me3 at this locus between Ezh2WT vs Ezh2Y641F cells, and expression of the mutant Ezh2Y641F allele does affect expression of Ezh2 itself (data not shown). However, expression of the mutant renders this locus more sensitive to regulation by Stat3.
Ezh2 and Stat3 regulate expression of MHC Class 1b antigen processing genes
One of the most significantly Ezh2/Stat3 co-occupied regions in Ezh2Y641F melanomas was the Major Histocompatibility Cluster (MHC) 1b on chromosome 17, which encodes for multiple H2-Q genes (Fig. 5a). Expression of multiple H2-Q genes was increased in Ezh2Y641F melanomas (Fig. 5b) and was dependent on Ezh2 activity, since treatment with the Ezh2 inhibitor, JQEZ5, decreased expression of these genes (Fig. 5c). The levels of the canonical PRC2-mediated repressive mark, H3K27me3, at this region were also significantly decreased in Ezh2Y641F mutant melanomas, consistent with increased expression of these genes. To determine whether expression required the presence of Stat3, we knocked down Stat3 and observed decreased expression of multiple H2-Q genes (Fig. 5d).
Figure 5. Direct regulation of MHC Class 1b H2-Q genes by Ezh2 and Stat3.
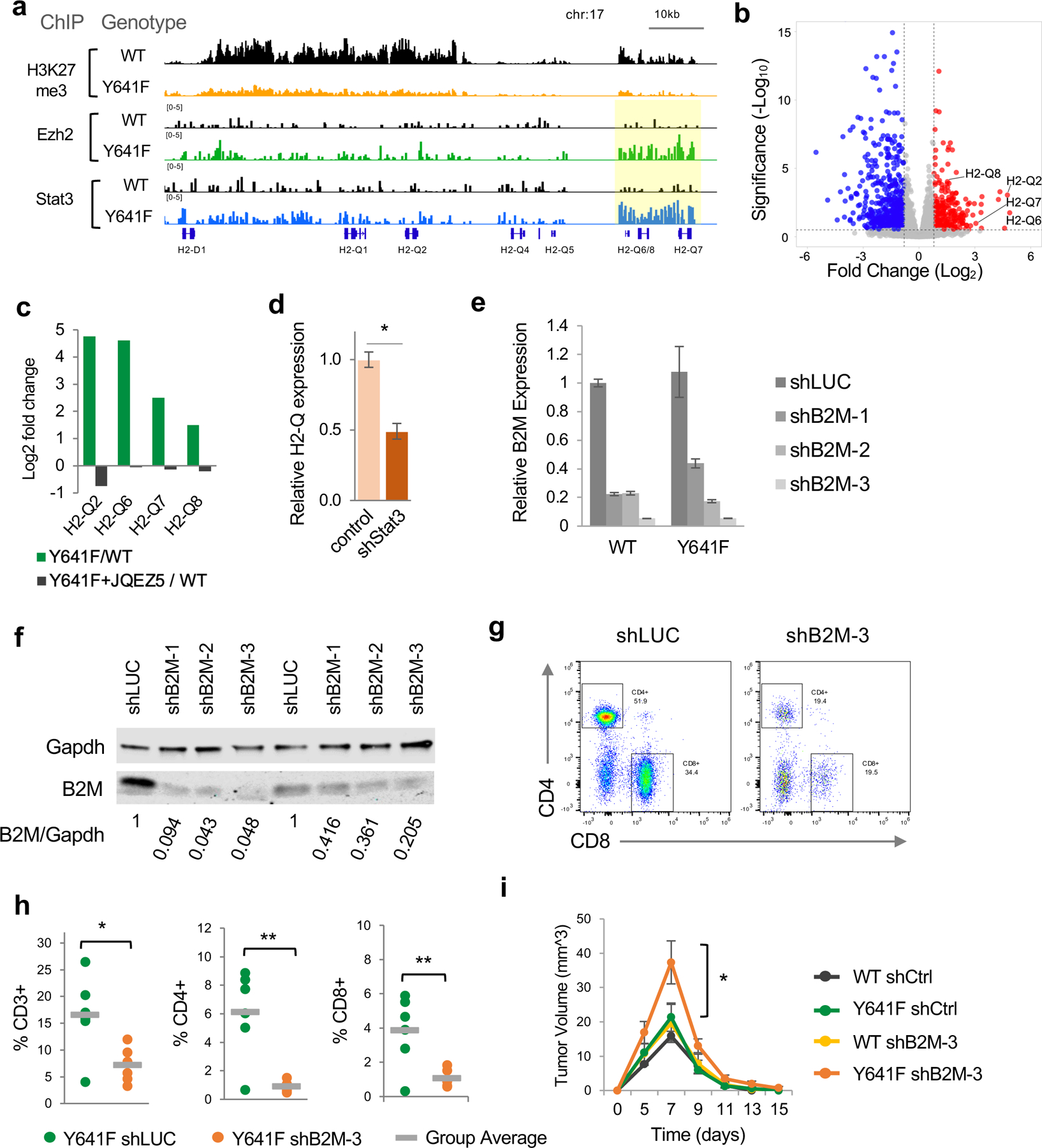
(a) ChIP-seq tracks for H3K27me3, Ezh2 and Stat3 at the H2-Q locus on chromosome 17.
(b) Volcano plot of differential gene expression in Ezh2Y641F vs Ezh2WT melanoma cells, highlighting upregulation of multiple H2-Q genes.
(c) Ratio of expression of H2-Q genes between Ezh2Y641F/Ezh2WT in vehicle vs cells treated with the Ezh2 inhibitor JQEZ5 (p adj<0.01).
(d) Expression of H2-Q genes after Stat3 knockdown in Ezh2Y641F melanoma cells (*p<0.01).
(e) Relative expression of B2M by quantitative PCR after shRNA knockdown using three different shRNAs (N = 3/group). shRNA against luciferase used as control.
(f) Knockdown efficiency confirmed by immunoblotting for B2M for WT (left) and Y641F (right) cell lines. Ratio of B2M/Gapdh shown at the bottom of the blots.
(g) Representative plots of CD4 and CD8 expression in tumor-infiltrating immune cells from Ezh2Y641F tumors. shLUC was used as a control shRNA targeting luciferase.
(h) Analysis of tumor-infiltrating lymphocytes in Ezh2Y641F tumors by flow cytometry using antibodies for CD3, CD4, and CD8 (N = 6/group, *p<0.05, **p<0.01).
(i) Tumor volume over time for Ezh2WT and Ezh2Y641F melanoma cells with B2M knockdown (N = 4–8/group, *p<0.05 for Y641F shB2M-3 vs all other groups at Day 7).
To determine whether MHC Class I complex plays a role in Ezh2Y641F mutated tumors, we knocked down expression of beta-2-microglobulin (B2M) using shRNA. MHC class Ib H2 molecules reside on the surface of the cell and require the presence of B2M to become stable and present antigens to CD8+ T cells 35. Inactivating B2M would therefore inactivate MHC class Ib function. We used three shRNAs against B2M, identified two with 80–90% knockdown, and generated two stable melanoma cell lines, which were then injected orthotopically into recipient mice and we monitored tumor growth over time. B2M knockdown tumors showed decreased infiltration of CD8+ T cells, in both Ezh2WT and Ezh2Y641F tumors (Fig. 5g-h), consistent with prior melanoma studies 36. B2M knockdown caused a significant increase in tumor growth in the presence of mutant Ezh2Y641F, but not in Ezh2WT (Fig. 5i), suggesting a specific dependency and interaction with the MHCI complex created by Ezh2Y641F mutations. B2M knockdown did not completely prevent tumor clearance, but that was not expected since the shRNA did not generate a complete B2M knockout and CD8+ T cells are still present in the tumors (Fig. 5h). Consequently, with 10–20% remaining B2M expression and CD8+ cells still present in the tumor microenvironment, the tumors were eventually rejected. Taken together, these results support that Ezh2 and Stat3 together promote expression of MHC Class Ib H2-Q genes, which may render Ezh2Y641F melanomas more antigenic.
Ezh2Y641F melanomas exhibit increased infiltration of CD8+ T cells which is dependent on Stat3 expression
Given the changes in expression of antigen-processing genes in Ezh2Y641F melanomas, we hypothesized that the immune system plays an important role in melanomas with Ezh2Y641F mutations. To investigate the tumor microenvironment of Ezh2Y641F mutant tumors, we used isogenic melanoma cell lines to generate Ezh2WT and Ezh2Y641F orthotopic tumors and analyzed the tumor microenvironment using flow cytometry. Interestingly, even though we had previously shown that expression of mutant Ezh2Y641F accelerates the development of melanoma 6, orthotopic injections of already established melanoma cell lines did not show any growth advantage for Ezh2Y641F mutant melanomas. In fact, Ezh2Y641F mutant tumors grew slightly slower than Ezh2WT, albeit the difference was not significant (Fig. 6b). This result is not surprising though, since different oncogenes drive oncogenesis at different stages, such as initiation, progression, or maintenance. These data suggest that perhaps Ezh2Y641F mutations might be more important during melanoma initiation rather than progression.
Figure 6. Assessment of tumor growth and tumor-infiltrating immune cells in Ezh2WT vs Ezh2Y641F melanoma cells.
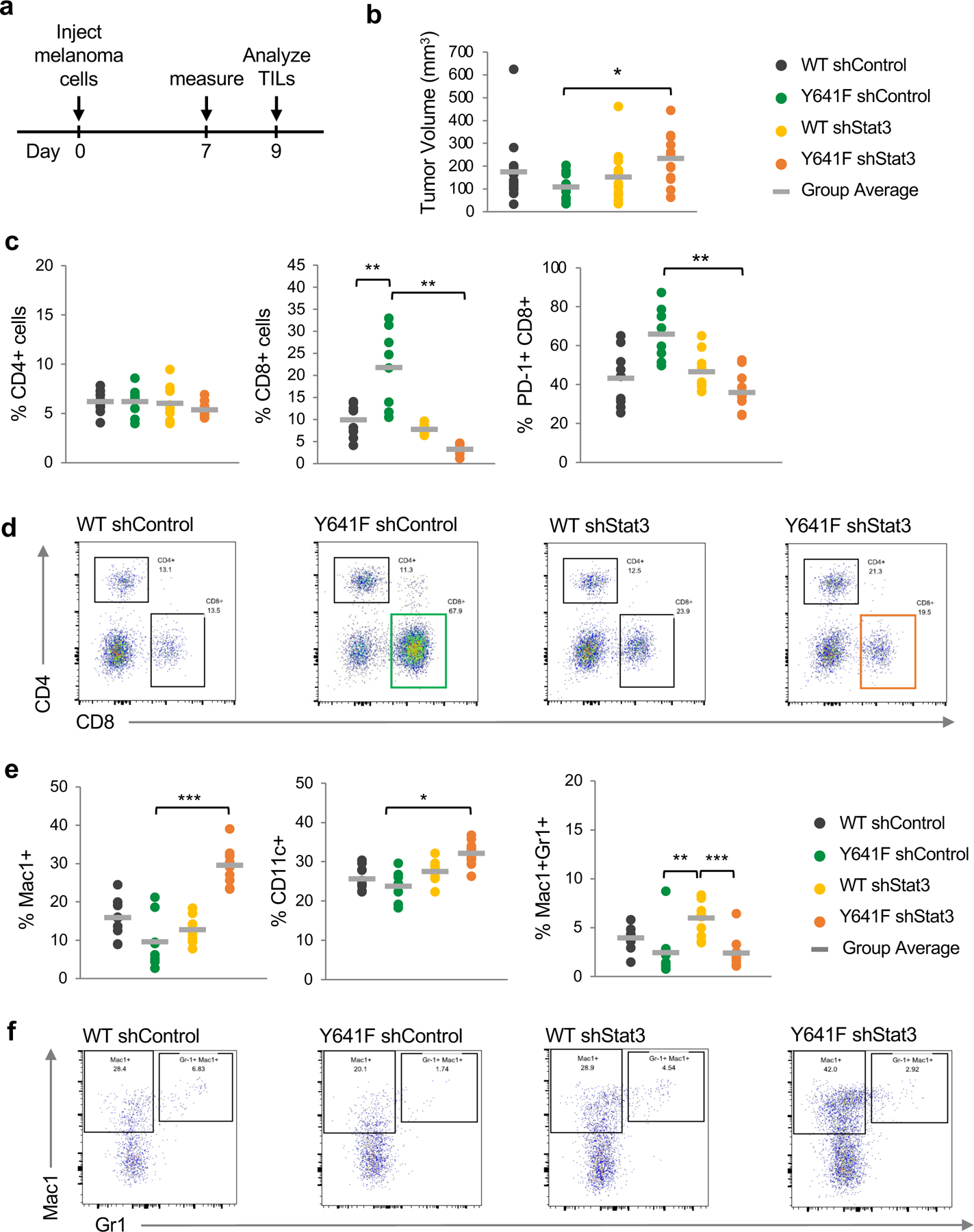
(a) Schematic of experimental design. Melanoma cells were injected subcutaneously into the flanks of C57Bl/6 mice. Tumor dimensions were measured 7 days later, and tumors were collected on day 9 to analyze tumor-infiltrating lymphocytes (TILs). N = 8–10/group.
(b) In vivo tumor volume of Ezh2WT vs Ezh2Y641F melanoma cell lines, with and without Stat3 knockdown (*p<0.05).
(c) Analysis of tumor-infiltrating lymphocytes by flow cytometry, focusing on CD4+, CD8+ and CD8+/PD-1+ cells (**p<0.01).
(d) Representative flow cytometry plots for CD4+ and CD8+ cells from panel (c).
(e) Analysis of tumor-infiltrating myeloid cells by flow cytometry, using antibodies for Mac1, CD11c, and Gr1 (*p<0.05, **p<0.01, ***p<0.001).
(f) Representative flow cytometry plots for Mac1+ and Mac1+/Gr1+ cells from panel (e).
One of the many functions of Stat3 is regulation of inflammation, a cancer hallmark, promoting a microenvironment more supportive of tumor growth. Persistent Stat3 activation is a recurring feature in many epithelial tumors 23 and Stat3 expression and activation has strong immunosuppressive properties 37. To investigate the role of Stat3 in the tumor immune response in Ezh2Y641F melanomas, we generated stable cell lines with an shRNA-mediated knock down of Stat3. Knockdown of Stat3 in Ezh2WT cell lines did not affect tumor growth, however, Stat3 knockdown in Ezh2Y641F mutant melanomas led to increased tumor growth (Fig. 6b). Notably, this is the opposite of what one would expect based on the previously well-established immunosuppressive properties of activated Stat3 in solid tumors 23.
To understand why Ezh2Y641F mutant melanoma tumors grew faster despite knocking down an immunosuppressive factor, we analyzed tumor infiltrated immune populations using flow cytometry and found increased infiltration of cytotoxic CD8+ T cells in Ezh2Y641F compared to Ezh2WT tumors (Fig. 6c). Stat3 knockdown in Ezh2WT tumors had no effect on the number of tumor infiltrated CD8+ T cells, however, in Ezh2Y641F tumors it dramatically decreased the number of CD8+ cells (Fig. 6c, d), suggesting that the role of Stat3 is dependent on expression of the mutant Ezh2Y641F. Further analysis of these tumor-infiltrating T cells showed increased expression of the PD-1 receptor, which was also dependent on Stat3 expression (Fig. 6c). These results suggest that increased infiltration of cytotoxic T cells may be limiting the growth of Ezh2Y641F mutant melanomas, but they are not sufficient to completely inhibit growth, as increased PD-1 levels indicates that these T cells are exhausted and not fully functional. With regards to myeloid populations, Stat3 knockdown induced an increase in Mac1+ myeloid and CD11c+ dendritic cells in Ezh2Y641F cells, and an increase in granulocytes (Mac1+ Gr1+ cells) in Ezh2WT cells, albeit not statistically significant (Fig. 6e, f). Taken together, these data suggest that expression of Ezh2Y641F in these tumors has created a unique dependency on Stat3 expression, with significant consequences on the tumor microenvironment.
Inhibition of Stat3 limits the anti-tumor immune response and promotes melanoma recurrence
Typically, the melanoma tumors investigated above grow rapidly for about ten days then are spontaneously rejected by the host (Fig. 7a) and shrink to undetectable size by day 25. However, melanomas with Stat3 knockdown exhibit a much higher incidence of persistent or recurrent tumors (Fig 7b, c). Pharmacologic inhibition of Stat3 using the small molecule STATTIC 38 had the same effect on tumor growth as genetic Stat3 knockdown (Fig. 7d). To determine if the tumor rejection was due to the presence of cytotoxic T cells in the tumor microenvironment, we depleted the CD8+ cells in tumor-bearing animals by administering an anti-CD8 antibody. Depletion of CD8+ cells prevented rejection of the tumors, which continued to grow rapidly compared to vehicle-treated controls (Fig. 7e, f). Additionally, we injected the same cell lines into immunodeficient NSG mice and monitored growth over time. In this setting, there was no rejection and tumors continued to grow rapidly (Fig. 7g), suggesting that the effects on tumor growth dynamics are mediated by the immune system. Finally, since tumor-infiltrated T cells exhibited increased expression of PD-1 (Fig. 6c), we tested whether anti PD-1 treatment might be beneficial in preventing tumor growth either alone or in combination with Ezh2 inhibition. Because these treatments require a longer timeframe, we used melanoma cell lines isolated from BrafV600EPtenF/+Ezh2WT or Ezh2Y641F primary tumors that were not rejected as quickly as those used in Fig. 7a. These cell lines were orthotopically injected into recipient mice and were treated with anti-PD-1 antibody, the Ezh2 inhibitor JQEZ5, or a combination or the two. We found that PD-1 inhibition was significantly more effective at inhibiting tumor growth when combined with an Ezh2 small molecule inhibitor in Ezh2Y641F melanomas (Fig. 7h).
Figure 7. Analysis of tumor growth over time of Ezh2WT vs Ezh2Y641F melanoma cells after Stat3 knockdown.
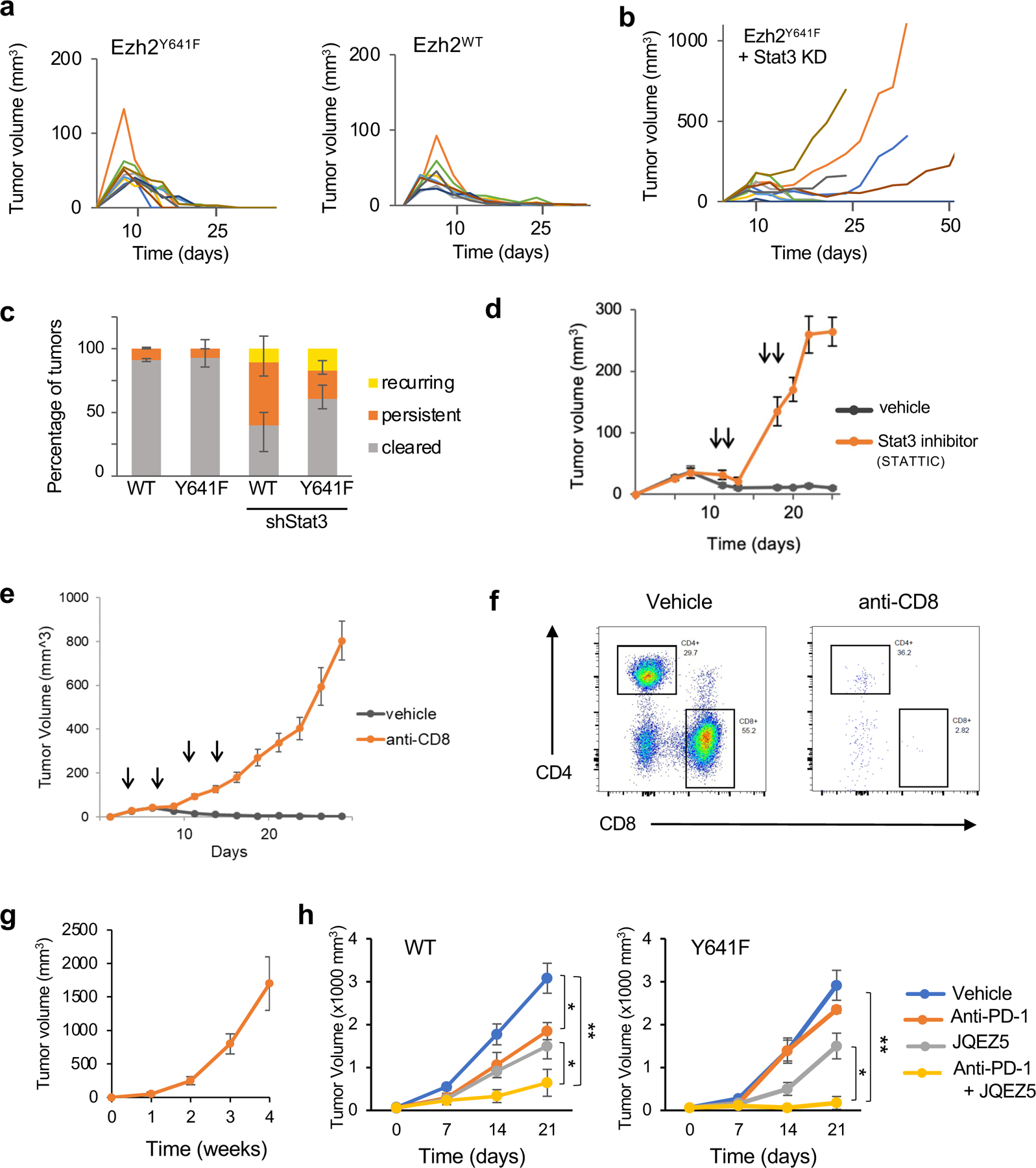
(a) Tumor volume over time for the indicated genotypes. Each line represents an individual tumor (N = 10/group). These data are from one of two replicate experiments.
(b) Tumor volume over time for Ezh2Y641F cell lines after Stat3 knockdown (N = 10/group, one of two replicate experiments).
(c) Tumor outcome of experiments in panels (a) and (b), stratified into 3 groups: cleared (rejected), persistent (tumors that grew consistently), and recurring (tumors that shrunk but then rebounded) (N = 10–14/group).
(d) Tumor volume over time after treatment with the Stat3 inhibitor, STATTIC (N = 8/group, arrows indicate the time of STATTIC treatments).
(e) Tumor volume over time with anti-CD8 treatment (N = 16/group, arrows indicate the time of anti-CD8 treatments).
(f) Representative plots of CD4 and CD8 expression in tumor-infiltrating immune cells from tumors treated with vehicle or anti-CD8 antibody.
(g) Tumor volume from an Ezh2Y641F melanoma cell line from (a), injected in immunodeficient NSG mice (N = 5).
(h) Tumor growth of Ezh2WT (left) vs Ezh2Y641F (right) melanoma cells treated with anti PD-1 antibody, Ezh2 inhibitor JQEZ5, and a combination of the two (N = 8/group). (*p<0.05, **p<0.01)
DISCUSSION
Here we investigated the consequences of a non-canonical interaction of the chromatin modifying gene Ezh2 in melanoma. We found that the Y641F mutation enhances Ezh2’s interaction with Stat3, and together they control expression of antigen processing components. This results in increased tumor infiltration of lymphocytes, however, the anti-tumor response was limited, likely due to T cell exhaustion. Molecularly, we show that expression of mutant Ezh2Y641F creates unique co-binding sites for Ezh2 and Stat3 across the genome, where they primarily function as transcriptional activators. Together, these data suggest that non-canonical interactions of Ezh2 are critical for its oncogenic activity in melanoma, and that the tumor microenvironment is a significant factor in the oncogenic properties of Ezh2 in melanoma.
Prior studies have implicated the role of Ezh2 in the anti-tumor immune response in cancer. For example, using Nras-driven melanoma models, Zingg et al. demonstrated a correlation between tumor CD8+ T cell infiltration and high PRC2 complex activity in human skin cutaneous melanoma 12. They proposed that intra-tumoral CD8+ T cell accumulation promotes upregulation of PRC2 components, which in turn silence the tumors’ own immunogenicity and antigen presentation. In several solid and blood cancers, Burr et al. suggested that canonical PRC2 silences MHC-I antigen processing, promoting T cell-mediated immunity 39. In this study, however, we suggest that Ezh2Y641F mutations lead to increased tumor immunogenicity, which does not have a significant effect on tumor growth, likely because the tumor-infiltrated T cells are exhausted/non-functional. Several variables can explain the discrepancies between these studies. First, even though Ezh2Y641F mutations exhibit increased enzymatic activity, that activity does not translate to a monotonic increase in H3K27me3, but rather to a re-distribution of the H3K27me3 mark across the genome, suggesting that these are not pure gain-of-function mutations 6 and likely do not function entirely the same way as WT Ezh2 protein in melanoma. Further supporting this neomorphic behavior, the Ezh2-Stat3 interaction, which directly drives expression of MHC-1b antigen processing genes (Fig. 5), is only weakly observed in Ezh2WT melanomas (Fig 3). This is consistent with ChIP-seq data demonstrating strong binding of Ezh2 and Stat3 at the MHC-I H2-Q region in Ezh2Y641F melanoma cells, with minimal binding in Ezh2WT cells (Fig. 5). The phenotypes we observe may therefore be specific to Ezh2Y641F and are not necessarily the same as those in melanomas that depend on canonical EZH2 activity or are driven by increased EZH2 activity through copy number alterations or amplifications. Another important variable that may determine the downstream oncogenic mechanisms mediated by Ezh2 is cellular context, such as cell physiology, genetic background, or secondary driver mutations. For example, EZH2 functions as an oncogene in some cancers (lymphoma, melanoma, ovarian, lung etc.) but as a tumor suppressor in others (AML, T-ALL). In melanoma mouse models, the Ezh2Y641F mutation cooperates with BrafV600E but not with NrasQ61R to accelerate melanomagenesis 6, consistent with the fact that in human melanoma patients, EZH2Y641F mutations are more likely to co-occur with BRAF rather than NRAS mutations4,5. The nature of the Ezh2Y641F mutations and cellular context may therefore determine how Ezh2 regulates oncogenic mechanisms.
Another interesting result in this study is the fact that Ezh2 can function as a transcriptional activator. This has previously been demonstrated in prostate cancer where Ezh2 acts as a co-activator for the androgen receptor 40, and more recently in leukemia cell lines where Ezh2 directly interacts with Myc at non-PRC2 targets to promote oncogenesis 41. Additionally, Stat3 interacts with other epigenetic regulators as well: for example, it is dependent on the presence of Brg1 (Smarca4), the ATPase subunit of the chromatin remodeling complex BAF (SWI/SNF), to activate pluripotency factors 42,43. Interactions between transcription factors and epigenetic regulators are therefore not uncommon and may explain why enzymatic inhibition of these protein complexes is not as effective as anticipated.
Despite our new findings on the non-canonical functions of Ezh2Y641F in melanoma, several questions remain unanswered. For example, we do not understand the biochemical details of how Ezh2 may be directly methylating Stat3, which lysine is methylated, whether it is it mono-, di- or tri-methylated, or whether Stat3 methylation requires the entire PRC2 complex. We also do not know if Stat3 methylation is in fact consequential, and not merely a transient interaction enhanced by the presence of a hyperactive Ezh2 protein. Our data also suggest possible downstream mechanisms of the interaction between Ezh2 and Stat3. For example, genomic loci co-occupied by Ezh2 and Stat3 are enriched for MITF binding motifs. MITF is a known regulator of melanocyte development and transformation 44,45, and has also been implicated in the anti-tumor immune response 46. MITF may therefore be a contributing factor in the oncogenic activity of Ezh2 in melanoma. E2F1 is another enriched motif in Ezh2/Stat3 binding sites. E2F1 is a known promoter of cellular proliferation so it is possible that cell intrinsic properties such as proliferation may co-operate with changes to the tumor immune response to synergistically promote melanoma progression. Future studies will address these remaining questions to fully understand the underlying oncogenic mechanisms of both Ezh2 and Stat3 in melanoma.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Siteman Flow Cytometry facility, McDonnel Genome Institute/Genome Access Center for technical assistance, and the Department of Comparative Medicine for animal expertise. We also thank all members of the Souroullas lab for critical input on the manuscript. This work was supported by the US National Cancer Institute K22-CA229612–01(GPS) and T32 CA113275–10 (SZ), the Cancer Research Foundation, Chicago IL, (GPS), The Harry J. Lloyd Charitable Trust (GPS).
Footnotes
Conflict of Interests: The authors declare no potential conflicts of interest.
CONFLIC OF INTEREST
The authors declare no relevant competing financial interests.
REFERENCES
- 1.Marine J-C, Dawson S-J, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer 2020; 20: 743–756. [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nature Publishing Group 2016; 17: 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Network TCGA, Akbani, Akdemir KC, Aksoy BA, Albert M, Ally A et al. Genomic Classification of Cutaneous Melanoma. Cell 2015; 161: 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discovery 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling 2013; 6: pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu J-Y, Paulk J et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nature Medicine 2016; 22: 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nature Communications 2015; 6: 6051. [DOI] [PubMed] [Google Scholar]
- 8.Asangani IA, Harms PW, Dodson L, Pandhi M, Kunju LP, Maher CA et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget 2012; 3: 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McHugh JB, Fullen DR, Ma L, Kleer CG, Su LD. Expression of polycomb group protein EZH2 in nevi and melanoma. J Cutan Pathol 2007; 34: 597–600. [DOI] [PubMed] [Google Scholar]
- 10.Tiffen J, Gallagher SJ, Hersey P. EZH2: an emerging role in melanoma biology and strategies for targeted therapy. Pigment Cell Melanoma Res 2014; 28: 21–30. [DOI] [PubMed] [Google Scholar]
- 11.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA et al. EZH2 Expression Is Associated With High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. JCO 2006; 24: 268–273. [DOI] [PubMed] [Google Scholar]
- 12.Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J et al. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. CellReports 2017; 20: 854–867. [DOI] [PubMed] [Google Scholar]
- 13.Dasgupta M, Dermawan JKT, Willard B, Stark GR. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci USA 2015; 112: 3985–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013; 23: 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy DE, Darnell JE. STATs: transcriptional control and biological impact. Nature Reviews Molecular Cell Biology 2002; 3: 651–662. [DOI] [PubMed] [Google Scholar]
- 16.O’Shea JJ, Gadina M, Schreiber RD. Cytokine Signaling in 2002: New Surprises in the Jak/Stat Pathway. Cell 2002; 109: S121–S131. [DOI] [PubMed] [Google Scholar]
- 17.Seidel HM, Milocco LH, Lamb P, Darnell JE, Stein RB, Rosen J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. PNAS 1995; 92: 3041–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerner L, Henriksen MA, Zhang X, Darnell JE. STAT3-dependent enhanceosome assembly and disassembly: synergy with GR for full transcriptional increase of the α2-macroglobulin gene. Genes Dev 2003; 17: 2564–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE. Interacting Regions in Stat3 and c-Jun That Participate in Cooperative Transcriptional Activation. Mol Cell Biol 1999; 19: 7138–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun W, Snyder M, Levy DE, Zhang JJ. Regulation of Stat3 transcriptional activity by the conserved LPMSP motif for OSM and IL-6 signaling. FEBS Letters 2006; 580: 5880–5884. [DOI] [PubMed] [Google Scholar]
- 21.Snyder M, Huang X-Y, Zhang JJ. Identification of Novel Direct Stat3 Target Genes for Control of Growth and Differentiation *. Journal of Biological Chemistry 2008; 283: 3791–3798. [DOI] [PubMed] [Google Scholar]
- 22.Tripathi SK, Chen Z, Larjo A, Kanduri K, Nousiainen K, Äijo T et al. Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation. CellReports 2017; 19: 1888–1901. [DOI] [PubMed] [Google Scholar]
- 23.Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer — using tissue repair as a road map. Nat Rev Cancer 2019; 19: 82–96. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 26.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nature Protocols 2006; 1: 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB et al. GREAT improves functional interpretation of cis -regulatory regions. Nature Biotechnology 2010; 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G et al. Integrative genomics viewer. Nature Biotechnology 2011; 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 2010; 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furlan-Magaril M, Rincón-Arano H, Recillas-Targa F. Sequential chromatin immunoprecipitation protocol: ChIP-reChIP. Methods Mol Biol 2009; 543: 253–266. [DOI] [PubMed] [Google Scholar]
- 31.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, Qi J, Reyes JM, Li L, Rao PK, Li F et al. Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer Discov 2016; 6: 1006–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci USA 2010; 107: 21499–21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carpenter RL, Lo H-W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014; 6: 897–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schell TD, Mylin LM, Tevethia SS, Joyce S. The assembly of functional b2-microglobulin-free MHC class I molecules that interact with peptides and CD8+ T lymphocytes 2002; : 8. [DOI] [PubMed]
- 36.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017; 8: 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nature Reviews Clinical Oncology 2018; 15: 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chemistry & Biology 2006; 13: 1235–1242. [DOI] [PubMed] [Google Scholar]
- 39.Burr ML, Sparbier CE, Chan KL, Chan Y-C, Kersbergen A, Lam EYN et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019; : 1–26. [DOI] [PMC free article] [PubMed]
- 40.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science 2012; 338: 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Yu X, Gong W, Liu X, Park K-S, Ma A et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat Cell Biol 2022. doi: 10.1038/s41556-022-00850-x. [DOI] [PMC free article] [PubMed]
- 42.Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proceedings of the National Academy of Sciences 2009; 106: 5187–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, Crabtree GR. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol 2011; 13: 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005; 436: 117–122. [DOI] [PubMed] [Google Scholar]
- 45.Kumar SM, Dai J, Li S, Yang R, Yu H, Nathanson KL et al. Human skin neural crest progenitor cells are susceptible to BRAFV600E-induced transformation. Oncogene 2014; 33: 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballotti R, Cheli Y, Bertolotto C. The complex relationship between MITF and the immune system: a Melanoma ImmunoTherapy (response) Factor? Mol Cancer 2020; 19: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.