

Abstract
Numerous studies have linked Parkinson disease (PD) with low levels of uric acid (UA). Low UA has been associated with the risk of developing PD, and its progression and severity. The biological mechanisms underlying these relationships have never been firmly established. The most frequently proposed mechanism is that UA is an antioxidant. Low UA is thought to predispose to oxidative stress, which contributes to dopamine neuron degeneration which leads to initial appearance of symptoms of PD and its worsening over time. Several recent studies have questioned this explanation. In this review, we describe the biology of UA, its many links with PD, evidence regarding UA as an antioxidant, and questions whether UA causes PD or contributes to its progression. We also address the possibility that something about PD causes low UA (reverse causation), or that low UA is a biomarker of some other more relevant mechanism in PD. We hope the evidence provided here will stimulate additional studies to better understand the links between UA and PD. Elucidating these mechanisms remains important, because they may provide new insights into the pathogenesis of PD or novel approaches to treatments.
Keywords: urate, uric acid, Parkinson’s disease, reactive oxidant species, anti-oxidant
Introduction
The main pathological hallmark of Parkinson disease (PD) is progressive loss of midbrain dopamine neurons with accumulation of Lewy bodies.1 The biological processes causing these changes are multi-faceted and include accumulation of toxic α-synuclein species, mitochondrial dysfunction, energy failure, genetic predispositions, exposure to pesticides or other toxins, neuro-inflammation, oxidative stress, and others. The initial mechanism triggering PD may differ among individuals, multiple mechanisms may be involved simultaneously, and the most relevant mechanism may change as the disease evolves.
Whatever the mechanisms may be, numerous studies have consistently described links between PD and uric acid (UA), the end product of purine metabolism in humans.2–6 Low serum UA has been linked with a greater risk of developing PD, the severity of motor features, and faster progression of both motor and non-motor features. The links are very strong and highly reproducible. Dissecting the nature of these links is important, because they may provide clues to the pathogenesis or treatment of PD.
Serum UA is influenced by numerous factors such as age, sex, diet, body mass, physical activity, and medications.7–11 Abnormalities in serum UA have been linked with many other disorders (Table 1).7–11 Serum UA even varies according to the time of day the sample is taken,12 with significant variations within the same subject.13 These many variables affecting UA have made it challenging to dissect how UA might relate to PD. The most common viewpoint is that low UA contributes to oxidative stress that predisposes to dopamine neuron degeneration in PD. This degeneration increases the risk of developing PD and contributes to more rapid progression.
Table 1.
Disorders Associated with Changes in Uric Acid
| Condition | Serum UA |
|---|---|
|
| |
| Neurodegenerative diseases | |
| Parkinson’s disease | − |
| Alzheimer’s disease | − |
| Motor neuron disease | − |
|
| |
| Inflammatory disorders | |
| Gouty arthritis | + |
| Asthma | + |
| Autoimmune disease | − |
| Multiple sclerosis | − |
|
| |
| Vascular disease | |
| Coronary artery disease | + |
| Stroke | + |
| Vascular dementia | + |
|
| |
| Other disorders | |
| Hypertension | + |
| Diabetes mellitus | + |
| Metabolic syndrome | + |
| Hemolytic anemia | + |
| Leukemia | + |
| Renal insufficiency | − |
| Fanconi disease | − |
|
| |
| Inherited diseases | |
| Lesch-Nyhan disease | + |
| Down syndrome | + |
| Fructosemia | + |
| Glycogen storage diseases | + |
| APRT deficiency | + |
| PRPPS overexpression | + |
| Familial hyperuricemic nephropathy | − |
| Hereditary xanthinurias | − |
| Renal hypouricemia | − |
|
| |
| Medications/supplements | |
| Diuretics | + |
| Cyclosporin | + |
| Androgens | + |
| Aspirin | + |
| Fructose | + |
This table shows the direction of serum uric acid (UA) found in the conditions listed, with (+) higher than healthy controls and (−) lower than healthy controls.
If this view is correct, then treatments that elevate UA may have neuroprotective value. However, some studies have questioned the hypothesis that low UA is causally related to the onset of symptoms in PD or contributes to PD progression, and a recent neuroprotection trial that involved raising UA in PD failed to show any benefit.14 These observations suggest that alternative viewpoints regarding the relationship between UA and PD should be considered. These include the possibility that something about PD causes lower UA, or that UA is a biomarker of some other process relevant to PD.
PD and UA: Cause or Effect?
Several reviews summarizing the many studies linking PD and UA have been published.2–6 One meta-analysis summarized 13 prior case-control studies of serum UA from a combined total 2379 PD cases and 2267 controls from different parts of the world.4 Serum UA was significantly and consistently lower in PD compared to controls. In addition, serum UA was significantly lower in advanced PD compared to early PD, implying that low serum UA may contribute to disease severity or progression. A more recent meta-analysis spanning 7 studies evaluated the relationship between serum UA and non-motor features, and suggested that low serum UA may also contribute to neurocognitive dysfunction, fatigue and sleep disorders.6
However, these epidemiological case-control studies have a number of limitations. Since UA is measured in individuals who already have PD, UA levels may be confounded by the disease itself, its treatment, lifestyle changes related to PD, or one of many other factors linked to UA. Although the anti-oxidant hypothesis posits that low UA contributes to enhanced oxidative stress that predisposes to neurodegeneration in PD (Figure 1), case-control studies cannot exclude the possibility that something about PD causes low UA, a phenomenon known as “reverse causation” (Figure 1).
Figure 1.
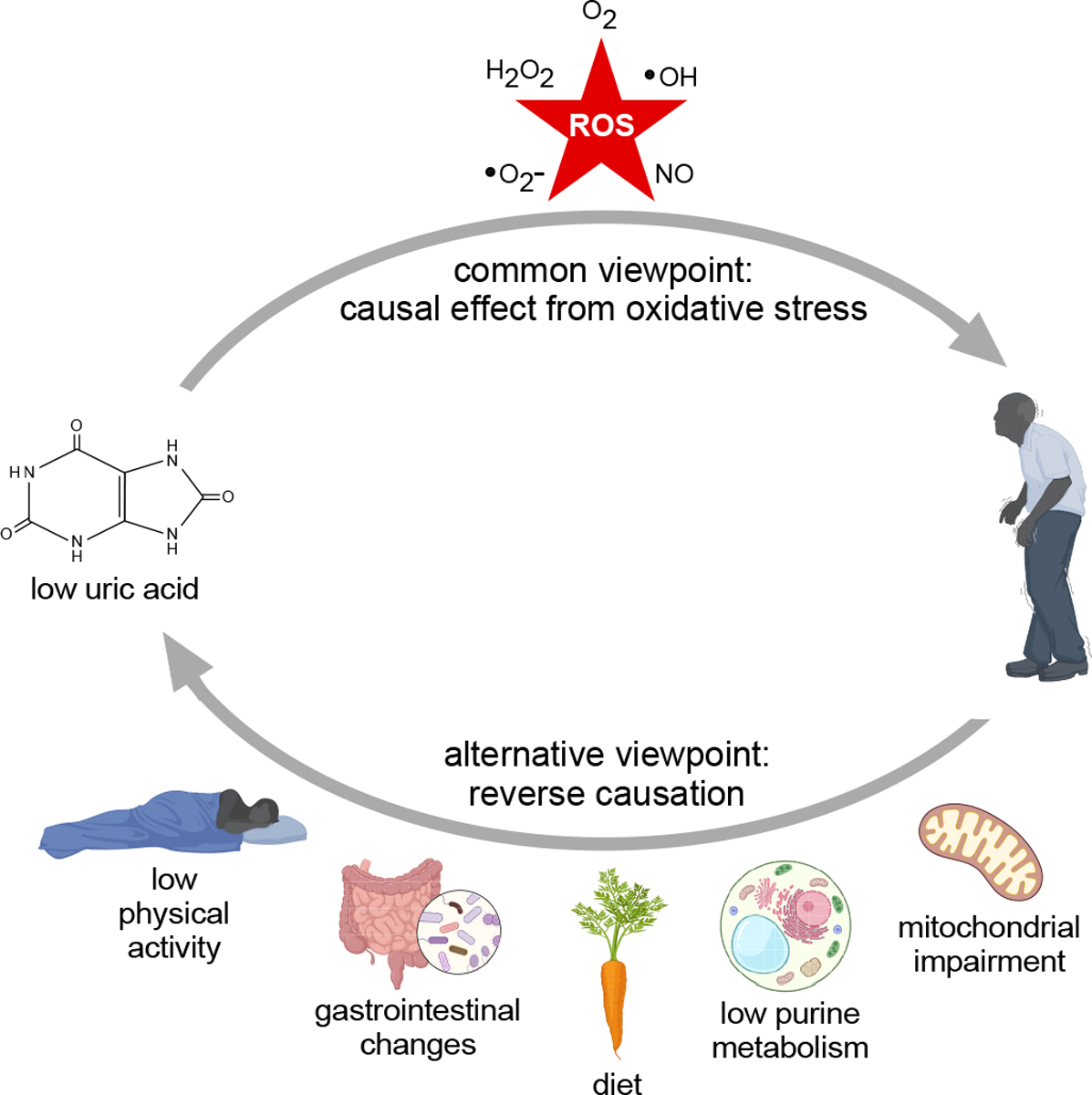
Potential causal relationships between Parkinson disease and uric acid. The commonly held viewpoint is that low uric acid leads to increased oxidative stress, which in turn contributes to dopamine neuron degeneration that triggers initial appearance of symptoms and contributes to progression. However, the direction of causation may be the opposite; Parkinson disease may cause low uric acid. Some potential mechanisms for reverse causation may include mitochondrial dysfunction, inherently low purine metabolism, dietary habits, changes in gastrointestinal motility or microbiome, or low physical activity.
The possibility of reverse causation is sometimes addressed using prospective studies, in which the risk of developing PD is evaluated. These studies depend on the argument that PD cannot be the cause of low UA, if low UA is found before PD develops. Numerous prospective studies of PD and UA have been reported, with strengths varying according to the number of cases included and the duration of the follow-up period. An early meta-analysis summarizing the incidence of new PD diagnoses from 6 studies encompassing 33,186 participants suggested a 33% reduction in PD risk among individuals with higher serum UA, a potential dose-response relationship, and an impact on progression of disease among those who received a diagnosis.3 Subsequent studies confirmed these findings. For example, another prospective study with a combined total of 90,214 participants revealed higher serum UA was associated with a 37% reduction in incident PD.15 These prospective studies provide further evidence for a link between UA and PD, but the argument that they can exclude reverse causation is undermined by evidence that the PD disease process may start decades before a clinical diagnosis is made.1, 16, 17 Therefore, prospective studies cannot exclude the possibility of reverse causation.
Other studies have examined a possible causal link between UA and PD using the method of Mendelian randomization.18–21 This method relies on evaluating relationships between PD and a biological proxy that has an established causal link with UA transport, not UA itself (Figure 2). These studies all explored links between PD and UA transporters, which are known to regulate serum UA levels (Table 2). In principle, this method can exclude reverse causation, as well as many other confounding factors in epidemiological studies. Three of the most recent studies failed to find evidence for a causal link between the PD and the UA transporters. The findings have questioned the commonly accepted viewpoint that low UA contributes to PD. However, these studies rely on certain assumptions.22, 23 One key assumption is that the study had adequate statistical power to detect a link, an assumption that is particularly important when a link is not found. The power of these studies ranged from 20–80%, so it is possible they missed detecting the causal link (Table 2). Another assumption of Mendelian randomization studies is that the biological proxy chosen (UA transporters) does not have independent effects (pleiotropy) that may impact the disorder under study. In fact, these transporters are not specific to UA; all of them transport other organic molecules,24 and it is feasible that some of these other molecules mediate any links with PD. Perhaps the most important assumption is that biological proxy chosen has a strong relationship with the putative causal variable (serum UA). UA transporters account for no more than 3–7% of the variation in serum UA levels.23 While this degree of variation is considered more than adequate for the statistical needs of Mendelian randomization studies, it is not clear that it has biological significance because it is small in relation to other common factors that influence UA levels (Figure 2). In addition to the limitations associated with these assumptions, the Mendelian randomization studies have other limitations. For example, etiological heterogeneity is likely in PD, and it is feasible that UA applies only to a specific subgroup. In view of these many limitations, it is not likely that Mendelian randomization studies can ever exclude or establish a causal role for UA in PD.
Figure 2.
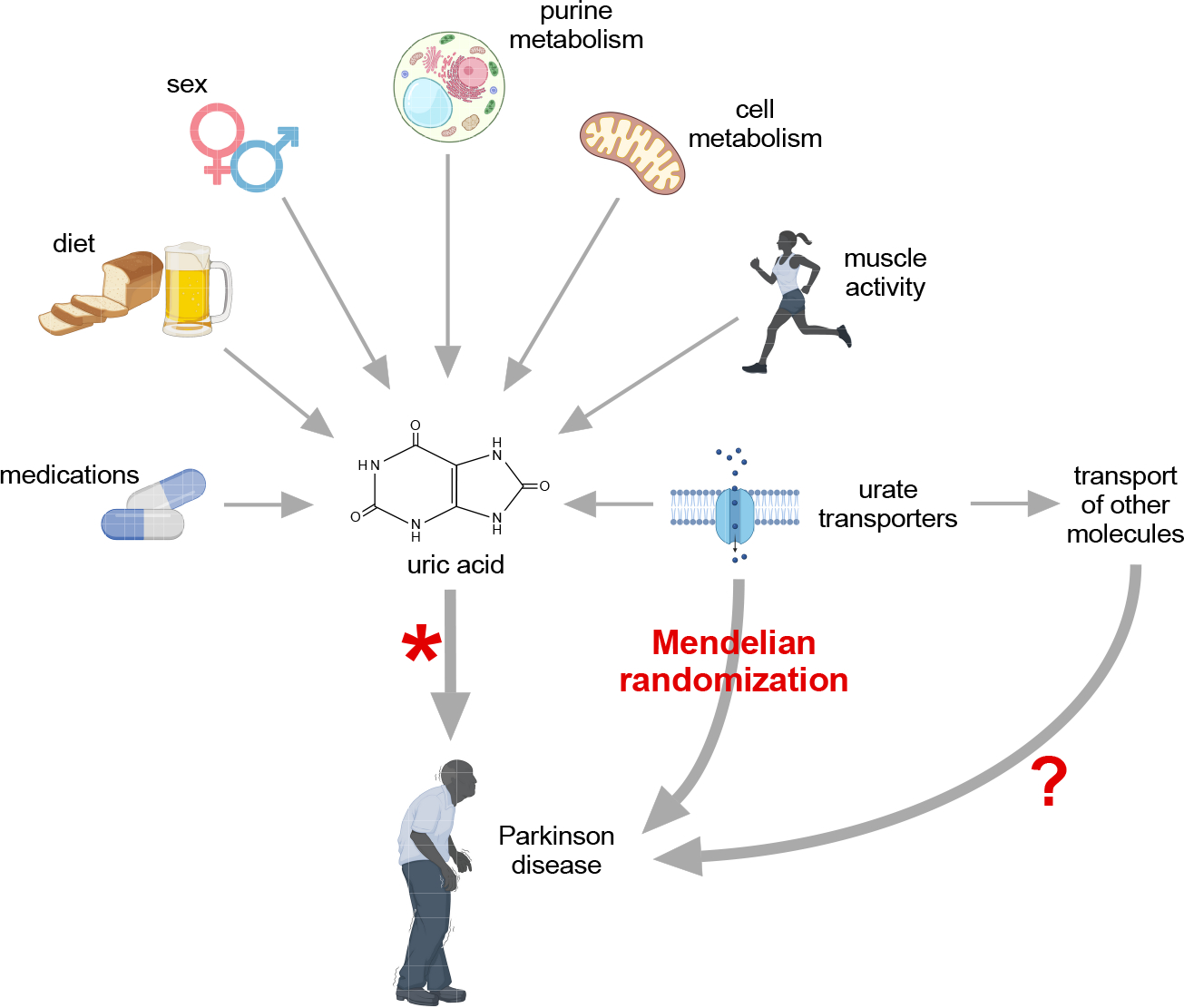
The relevance of Mendelian randomization studies in Parkinson disease. These studies did not find any association between transporters of uric acid and Parkinson disease, questioning the idea that low UA causes PD or contributes to its progression. However, the available studies cannot definitively confirm nor refute these causal associations between uric acid and PD, because they cannot exclude an impact of other possible molecules on PD (pleiotropy) and the impact of these transporters is biologically relatively small relative to other factors that may have a bigger impact on uric acid levels.
Table 2.
Summary of Mendelian Randomization Studies
| Study | PD subjects | Control subjects | Total SNPs | Statistical power | Methods to address pleiotropy | Outcome |
|---|---|---|---|---|---|---|
| Coneys, et al21 | 33,674 | 449,056 | 31 | 80% | MR-Egger method; Funnel plots |
No causal effect of UA on progression |
| Kobylecki, et al20 | 279 | 102,534 | 2 | 19% | None; pleiotropy limited by assessing only 2 SNPs | No causal effect of UA on progression |
| Kia, et al19 | 13,708 | 95,282 | 31 | 80% | MR-Egger method; penalized weighted median | No causal effect of UA on incidence |
| Simon, et al18 | 735 | NR | 3 | NR | None | UA protects against progression |
Abbreviations: NR, not reported; SNP, single nucleotide polymorphisms
In summary, there is a strong and indisputable epidemiological link between PD and UA, wherein low serum UA is associated with the risk of developing PD, faster progression, and greater severity. The nature of this association remains unclear. It is plausible that low UA contributes to neurodegeneration associated with PD, but it is equally plausible that something about PD causes low UA. The available epidemiological studies, including the Mendelian randomization studies, do not reveal the direction of causation. Nor do they address the proposed biological mechanism of exaggerated oxidative stress leading to neuronal degeneration. The following sections summarize some more biologically-oriented observations that may help to guide next steps in addressing these associations between UA and PD.
Is UA a Biologically Relevant Anti-Oxidant?
Hundreds of publications describe UA as a “powerful anti-oxidant”. These claims are an example of a hypothesis that became a “fact” in the absence of conclusive evidence. The idea that UA is a powerful oxidant came from Bruce Ames, a famous biochemist and Nobel laureate.25 Using a well-established in vitro assay, Ames demonstrated that UA had anti-oxidant properties similar to Vitamin C (ascorbic acid). Because UA is 4-fold higher than ascorbic acid in human plasma, he hypothesized that UA might be quantitatively the most important anti-oxidant in humans. Using similar in vitro assays before and after depletion of plasma UA, numerous subsequent studies endorsed his hypothesis,26–28 and it became a routinely cited fact.
Very few studies critically questioned this fact, perhaps because it originated from such a famous biochemist. However, Ames himself tested his own hypothesis.29 He recognized that in vitro assays sometimes provide results that are not relevant in vivo. For example, erythrocytes have enormous anti-oxidant capacity, because they express large amounts of enzymes such as catalase (which eliminates H2O2) and superoxide dismutase (which eliminates superoxide anions). This enormous source of natural anti-oxidants in blood is lost when studying isolated plasma or serum. In a second study, therefore, Ames compared all of the major anti-oxidants in humans using an assay that more closely resembled the situation in vivo. This study revealed the following rank order for quantitative importance: ascorbic acid = protein thiols > bilirubin > UA > alpha-tocopherol (Vitamin E).29 These findings imply that protein thiols are better anti-oxidants than UA. Protein thiols are nearly ubiquitous in plasma proteins, at amounts far greater than UA. Even bilirubin was a better anti-oxidant than UA. In a third study, Ames reported that ascorbic acid was the most potent antioxidant in human plasma.29 His follow-up studies are rarely cited, but they imply that UA is a weak anti-oxidant under natural conditions. Following these studies, Ames no longer promoted the hypothesis that UA was the most important anti-oxidant in humans.
Other investigators also cautioned against over-interpretations from common in vitro “anti-oxidant” assays, because in vivo relevance is not clear.30 The potential relevance of UA as an anti-oxidant in vivo was directly tested in one study of 26 individuals who received an intravenous infusion of recombinant uricase to reduce serum UA as an experimental treatment for gout.31 The treatment drastically reduced mean serum UA more than 90% from 10.8±1.3 to 0.9±0.5 md/dL for 15 weeks. Despite this profound and prolonged reduction of serum UA, there was no evidence for any significant increase in any of the standard in vivo biomarkers of oxidative stress in the blood.31 These results imply that UA is not a biologically relevant anti-oxidant in vivo.
How Much UA in Brain?
Regardless of whether UA may be a biologically relevant antioxidant or not, an important issue to consider in relation to PD is whether or not neurons are exposed to UA. In normal adults, serum UA is slightly higher in men than women.32 Adult males average 4.9 ±1.4 mg/dL while pre-menopausal females average 4.2±1.2 mg/dL.33 Curiously, if UA is protective against PD, men should have a lower incidence of PD than women, although the opposite is observed.34
Levels of UA in brain are much lower than blood. UA is produced from xanthine or hypoxanthine by the enzyme xanthine-oxidoreductase (XOR), which is expressed at high levels in the gastrointestinal system, and especially the liver.35, 36 Lower levels of XOR are expressed in some other tissues, but levels in brain are very low or absent (Figure 3). As a result, very little UA is produced in the brain. Further, UA does not penetrate the blood-brain barrier, either passively or through UA transporters, suggesting that the brain may not be exposed to UA from peripheral sources either.
Figure 3.
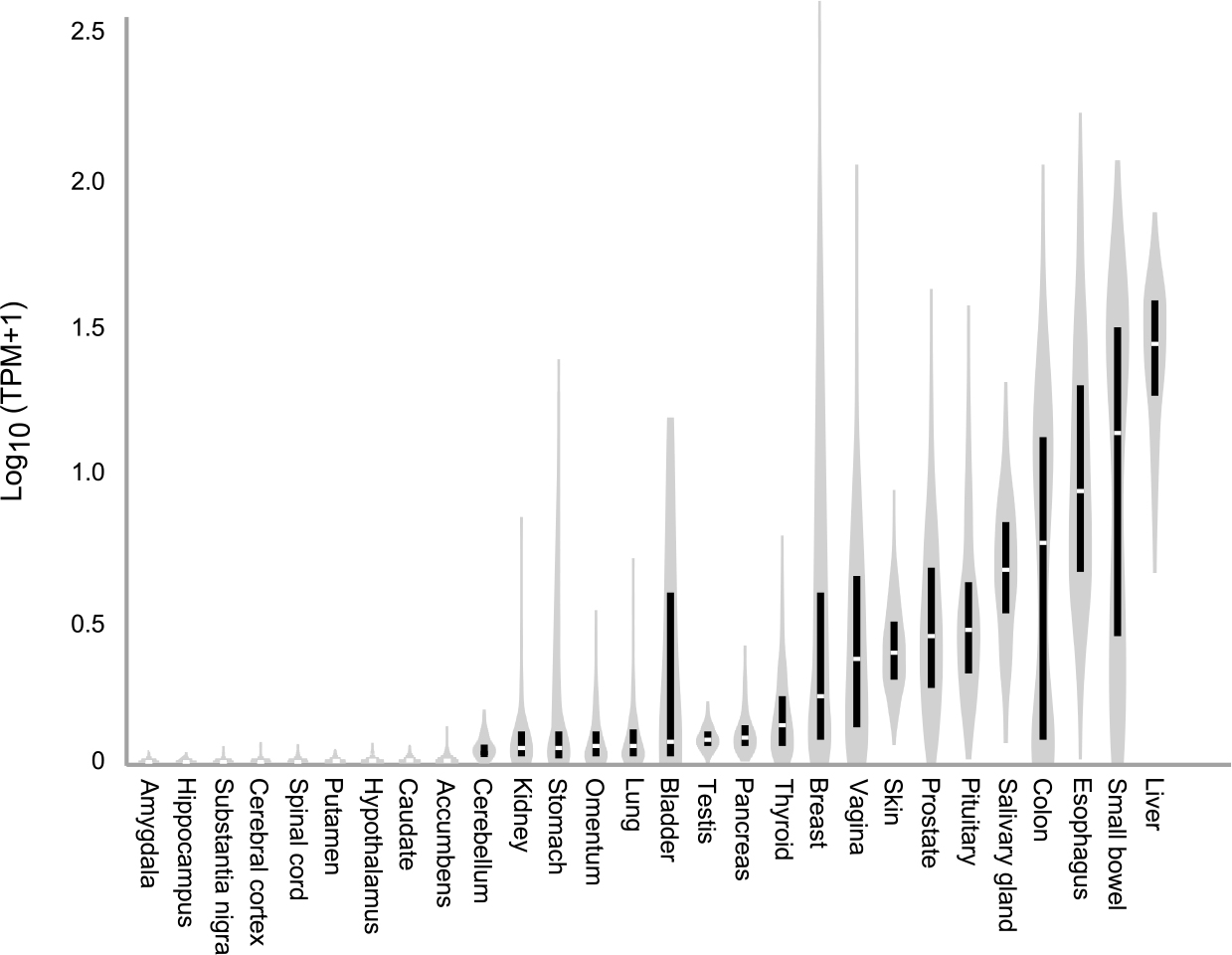
Distribution of xanthine oxidoreductase in the human body. Data for this figure were obtained from GTEx (V8), a public database containing information from ~1000 normal people regarding the relative expression of all known genes across 54 tissues (www.GTEXportal.org). The vertical black bars show inter-quartile ranges of mRNA levels, with median noted by the break in the bar. The vertical gray shapes provide a visual representation of clustering and spread of all available data. Multiple brain regions tested are shown to the left of the figure, with representative values for other tissues to the right. The highest levels of expression are in the liver, with low or absent levels in almost all areas of the brain. According to the Human Protein Atlas, another public database that provides protein expression levels across 44 human tissues, human brain also does not express detectable XOR protein (www.proteinatlas.com). Abbreviation TPM refers to transcripts per million.
In keeping with these observations, cerebrospinal (CSF) levels of UA in normal individuals are only 5–6% of blood levels.37, 38 CSF UA levels may reach 10% of blood levels in disorders that disrupt the blood-brain barrier, such as meningitis.39 There is a strong correlation between UA in blood and CSF, and the majority of UA in the CSF is believed to come from passive leak from the blood or from contamination of the CSF sample with small amounts of blood during lumbar puncture.38 Together, these observations question whether UA can have any meaningful effect on brain neurons.
To address the limited UA in brain, recent studies tested the potential value of oral inosine, a purine nucleoside that can pass the blood-brain barrier.40, 41 Once in the brain, inosine can be converted into UA, but only if the relevant enzymes (purine nucleoside phosphorylase and XOR) are present (Figure 4). In one study, a total of 75 individuals with PD received different oral doses of inosine daily for 6 months.41 The overall baseline serum UA of 4.5 mg/dL increased by an average of +3.0 mg/dL in the group treated with the highest dose. This result confirms that oral inosine can substantially raise serum UA, a result confirmed in an independent smaller study of only 10 PD subjects.42
Figure 4.
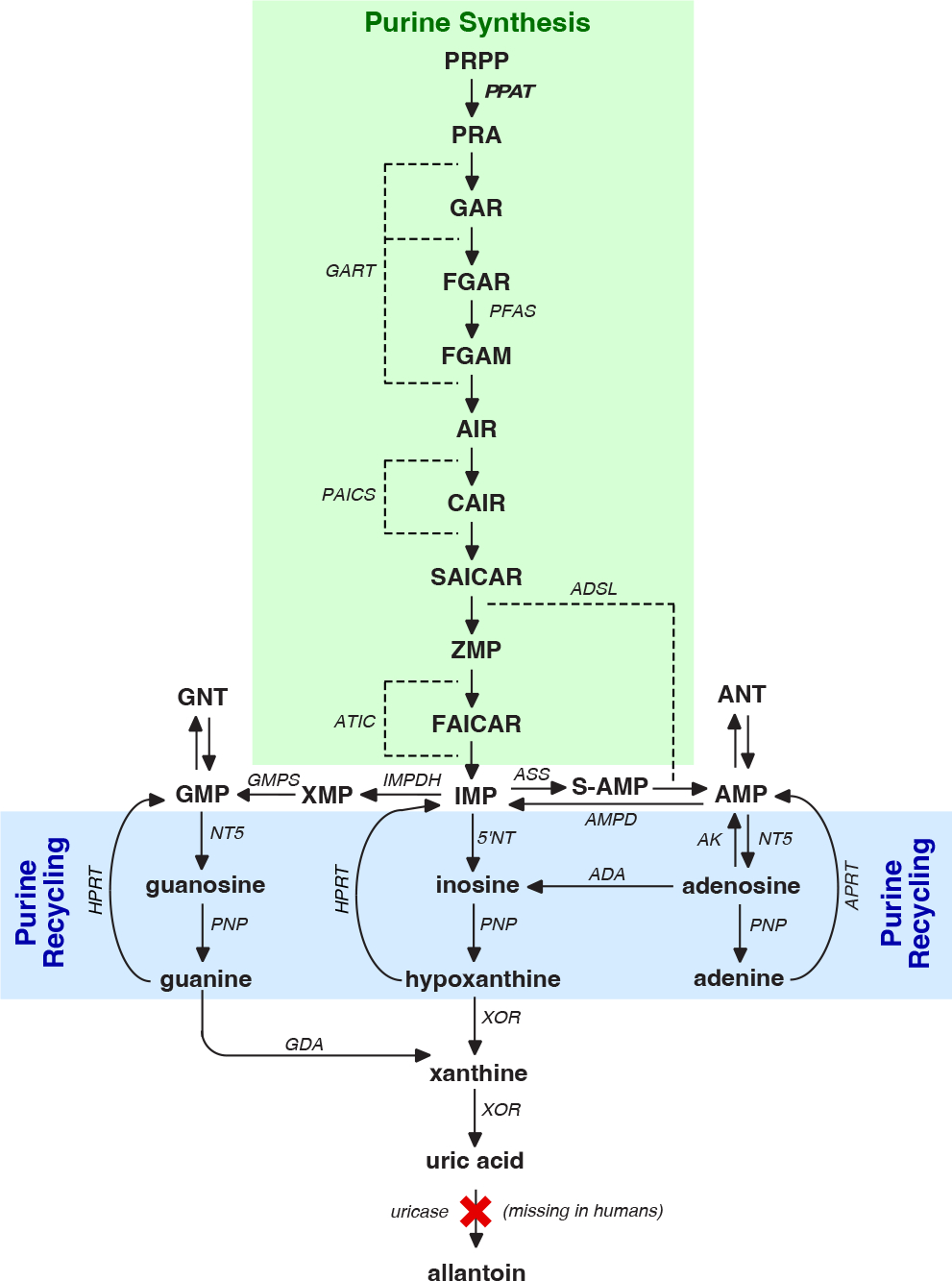
The 6 enzymes and 10 reactions of purine synthesis are shown in green. The 2 enzymes and 3 reactions of purine recycling are shown in blue. Enzymes are shown in bold font according to their gene symbols; metabolites are shown in regular font. Abbreviations: ADA, adenosine deaminase; ADSL, adenylosuccinate lyase; ASS, adenylosuccinate synthase; AMPD, adenylate deaminase; AMP, adenosine monophosphate; ANT, adenine-based nucleotides; APRT, adenine phosphoribosyltransferase; ATIC, AICAR transformylase/IMP cyclohydrolase; AK, adenylate kinase; CAIR, 5’-Phosphoribosyl-4-carboxy-5-aminoimidazole; FAICAR, formylaminoimidazole carboxamide ribotide; GART, phosphoribosylglycinamide formyltransferase/phosphoribosylglycinamide synthetase/phosphoribosylaminoimidazole synthetase; GDA, guanine deaminase; GMP, guanosine monophosphate; GMPS, GMP-synthase; HPRT, hypoxanthine-guanine phosphoribosyltransferase; IMP, inosine monophosphate or inosinate; IMPDH, IMP-dehydrogenase; NT5, 5’-nucleotidase; PNP, purine nucleoside phosphorylase; PAICS, phosphoribosylaminoimidazole carboxylase; PFAS, phosphoribosyl formylglycinamide synthase; PPAT, bifunctional phosphoribosylaminoimidazole carboxylase; PRA, 5-phosphoribosylamine; PRPP, phosphoribosylpyrophosphate; S-AMP, adenylosuccinate; SAICAR, succinylaminoimidazole carboxamide ribotide; XOR, xanthine oxidoreductase; XMP, xanthine monophosphate; ZMP, 5-aminoimidazole-4-carboxamide ribonucleotide.
In the larger study of 75 individuals, CSF UA was also measured following inosine treatment.41 The mean CSF UA was 0.43 mg/dL in the placebo group versus 0.59 mg/dL in the group receiving the highest dose of inosine. These results confirm prior studies that CSF UA is much lower than serum UA. They also demonstrate that oral inosine can raise CSF UA, but the magnitude of this increase in CSF is small.
In the same 75 individuals, the ability of the oral inosine to impact oxidative stress was evaluated.40 First, standard in vitro assays were used to measure anti-oxidant capacity in serum and CSF. Serum antioxidant capacity increased by 43% in the group treated with the highest dose of inosine, but there was no significant change in CSF anti-oxidant capacity, perhaps because the UA increase in CSF was too small. Because of the known limitations of these in vitro assays, a second assay involved the use of two sensitive and validated biomarkers of in vivo oxidant stress (oxidation of ferric tripyridyltriazine by serum and urinary 8-hydroxy deoxyguanosine).40 Neither biomarker showed any significant impact of oral inosine on serum or CSF markers of oxidant stress. These results reinforce other observations described above indicating that UA is not a biologically relevant antioxidant in human blood or brain.
Some investigators have attempted to measure UA directly in human brain tissue collected at autopsy, showing small but non-significant trends towards lower levels in PD.43 Such studies must be interpreted with caution, because the large gradient between blood and brain means that even small amounts of blood retained in the vasculature will significantly impact measures from autopsied brain tissue. In addition, purines begin to degrade within seconds following disruption of blood flow to the brain, so they cannot be reliably measured in tissue where the post-mortem interval is measured in hours. All together, these observations suggest the brain does not produce UA, and it is not exposed to significant UA from peripheral sources. These observations therefore question whether UA can have any meaningful protective effect on brain neurons.
Can Disorders of UA Provide Some Insights?
It could be argued that studies that transiently increase UA with inosine40–42 or decrease UA with uricase31 do not provide conclusive evidence regarding UA as a biologically relevant anti-oxidant because the treatment durations (weeks or even months) may be too short in comparison to the potentially life-long exposure to low UA that may occur in PD. To address this concern, some insights may be taken from inborn errors of metabolism, which can be considered “experiments of nature.” Most notable is hereditary xanthinuria caused by deficiency of XOR.44 Because these individuals lack the enzyme that makes UA, they have non-detectable serum UA from birth. Many individuals have no symptoms at all, although a third develop xanthine kidney stones. Lifespan is normal, and there is no clinical evidence for an increased risk of PD, although there are no rigorous studies specifically addressing this risk.44
Another instructive inborn error is renal hypouricemia, caused by deficiency of URAT1, a transporter critical for reabsorption of UA by the kidneys.45 Individuals with this disorder exhibit renal wasting of UA, with marked and lifelong reductions in serum UA (usually <2 μg/dL). Lifespan is normal, and many remain asymptomatic for life, although some develop UA kidney stones.45 PD is not typically described as a common clinical comorbidity in renal hypouricemia, but a recent study of 1106 subjects found the risk of PD was 1.3% among individuals with serum UA <2 μg/dL compared to a risk of only 0.2% among individuals with normal UA.46 This study implies an increased risk of PD associated with hypouricemia, but the overall incidence of PD in both groups was low, and the increased risk of PD was observed only for men.
Although these inborn errors affecting UA are rare, they seem to imply that lifelong low (or even absent) UA does not have any obvious impact on risk for PD. Of course, it could be argued that these disorders are too rare to be able to detect a clinical association with PD. While this may be the case, a more obvious increase in PD might be expected for disorders with lifelong low (or even absent) UA. Furthermore, if UA is a powerful antioxidant, we might also expect an increase in other disorders attributed to oxidative stress, most notably atherosclerosis causing cardiovascular or cerebrovascular disease. However, there does not appear to be any obvious risk for these common diseases among individuals with low UA. These rare disorders therefore further question the biological relevance of UA as an important anti-oxidant. Further studies of these rare disorders looking more specifically for evidence of oxidative stress (or evidence of potentially compensatory changes in other endogenous anti-oxidants) could be valuable for assessing the biological relevance of UA as an antioxidant in humans.
Can Experimental Models Address the Biological Relevance of UA?
Because UA is difficult to study directly in human brain, surrogate experimental models may provide useful information on its potential relevance for PD. For UA, animal models present serious species-related barriers. UA is the end-product of purine metabolism only in humans and higher apes (Figure 4). Almost all other species have an enzyme (uricase) that metabolizes UA and maintains very low serum UA levels of <1.0 mg/dL.35 In addition, the metabolism of purines is approximately 10-fold faster in rodents compared to humans. Such profound species differences in UA metabolism mean that effects of manipulating UA in rodents may be difficult to interpret in relation to humans. Studies in non-human primates may be useful, but they would have to be conducted in the few species that do not express uricase.
Cultured cells provide another potential experimental model to address the putative protective role of UA in PD. These models have been very valuable for studying purine metabolism in human cells, but their value for studying mechanisms related to oxidative stress is clouded by the relatively high oxidant stress of the in vitro environment. High levels of oxidative stress are inherent to the culture environment, because they are conducted at atmospheric levels of O2 (18–21%) while O2 levels in vivo are much lower (2–9%).47 As a result, the addition of almost any anti-oxidant to tissue culture medium has beneficial effects. In fact, virtually all tissue culture media already contain substantial amounts of added antioxidants, to combat the known problem with its oxidative environment. Because of the complex oxidant and anti-oxidant in vitro environment, results obtained with cultured cells may not translate to in vivo.
These limitations of animal and in vitro models do not mean they cannot be used to study purine metabolism or the potential role of UA in PD. However, they indicate that results obtained with these experimental models must be interpreted with caution, and appropriate controls included.
What Can We Learn From Recent Interventional Trials?
Several reports have described outcomes following attempts to increase UA in PD with oral inosine. In the largest study, 298 individuals with early PD not yet requiring treatment were randomized to receive placebo or oral inosine.14 Subjects with PD had to have baseline serum UA below the population median, to enrich for cases most likely to benefit. Clinical progression was monitored with a standard clinical rating scale (MDS-UPDRS) and dopamine transporter (DAT) imaging as a biomarker. This study was halted early when an interim analysis disclosed no benefit in clinical outcome or imaging. These results do not support the concept that UA is a biologically relevant neuroprotectant in PD,48 but several investigators have pointed to features of the trial that may have led to a false negative outcome, leading to suggestions for improved future study designs.14, 49, 50 For example, it is possible the study should be limited to men, where links with UA are strongest. It is also possible that raising UA may need to occur earlier, before the degenerative process is so advanced that it produces symptoms.
Another trial involved providing inosine in PD, but with a very different rationale.51 In this trial, inosine was given to augment ATP, rather than to reduce oxidative stress. The main rationale was that brain ATP may be limiting in PD, because of mitochondrial dysfunction. To augment brain ATP, inosine was combined with an XOR inhibitor, to prevent conversion to UA. In this situation, inosine is converted to hypoxanthine, which is ultimately recycled into nucleotides including ATP (Figure 4).52 The trial was unblinded and relatively small, with only 26 PD subjects. After 2 months of treatment, there was a 12% reduction in PD motor symptoms using the MDS-UPDRS.51 This study provides provocative results, but it needs to be repeated in a larger and blinded design.
While interventional trials in subjects with PD may provide the most compelling evidence for clinical relevance, they are also expensive and time-consuming. As a result, it is imperative that clinical trials are supported by strong preclinical evidence, to avoid unnecessarily wasting valuable and often limiting resources that might be needed elsewhere.48
Does PD Cause low UA?
The information reviewed here regarding the biology of UA raises important questions regarding whether low UA causes or contributes to the neurodegenerative process underlying PD. It may be valuable to consider the alternative perspective that something about PD causes low serum UA. This perspective is not usually considered, but there are several plausible mechanisms (Figures 1–2).
Mitochondria and energy.
Many studies have pointed to mitochondrial dysfunction in PD.1 One of the major functions of mitochondria is the production of energy in the form of ATP, which is quantitatively the most abundant of all purines in the body.53 Because serum UA is closely linked with endogenous purine metabolism,54 any process that impacts purine metabolism is likely to alter serum UA. Thus, mitochondrial dysfunction and energy failure in PD may lead to low serum UA levels. In this situation, low UA in PD could represent a biomarker for energy failure, rather than a cause of PD. If this is the explanation for low UA in PD, then efforts to raise brain ATP may be more valuable than efforts to raise UA.51, 52
Physical activity and muscle mass.
Another biological mechanism by which PD could cause low UA relates to overall levels of physical activity and muscle mass. Reduced levels of activity are well documented for all stages of PD, with some evidence that reduced activity precedes clinical diagnosis.55 As PD progresses, activity levels reduce further. These changes in physical activity are associated with progressively reduced muscle mass and strength, and sometimes frank sarcopenia.56, 57 A related phenomenon is weight loss, which is common in PD, and may precede onset of PD symptoms.
Serum UA is linked with physical activity, muscle mass and muscle strength, and body-mass index.58–61 The mechanism linking UA with muscle activity is well-established. The normal contraction of myofibrils consumes ATP, and the associated ATP turnover causes some purine degradation. Strenuous exercise also results in tearing of some myofibers, leading to leakage of ATP into the extracellular space, where it is degraded into UA. Thus the reduced levels of physical activity and/or muscle mass in PD may be responsible for low serum UA. In this situation, low serum UA in PD could reflect low muscle activity and/or weight loss in PD.
Diet.
Numerous epidemiological studies have linked PD with specific dietary factors, such as dairy, alcohol, and coffee.62 Although these links are well established, the biological mechanism underlying these links are largely unknown.
The diet also has a significant impact on serum UA.63 Almost all foods contain purines in the form of DNA, RNA, and related metabolites.64 These are partly digested, and absorbed through the gut. A substantial proportion is metabolized to UA by the gut and liver. Some foods (e. g. organ meats) raise UA much more than others (e. g. fruits, vegetables). Beverages containing alcohol also have a strong influence on UA. Although alcohol is a carbohydrate rather than a purine, it interacts with metabolic pathways that accelerate endogenous purine turnover.65 Beer also contains very high levels of guanine and guanosine, which is metabolized into UA. Fructose is another carbohydrate that is very commonly used as a sweetener. Fructose also interacts with metabolic pathways that accelerate purine synthesis, and increases serum UA.66, 67 On the other hand, coffee and milk been linked with lower serum UA, although the mechanisms are not known.68 In view of these observations, it is feasible that dietary habits in PD contribute to low UA. Although dietary influences on serum UA or PD are well-known, they can be difficult to dissect as confounding variables in epidemiological studies.69, 70
Gastrointestinal motility and microbiome.
Changes in gastrointestinal motility, particularly constipation, are common in PD. Such changes are linked with marked changes in the PD gut microbiome.71 Changes in the gut microbiome may have a significant impact on serum UA, because there are numerous organisms that metabolize or produce UA in the gut, and approximately a third of the body’s UA is eliminated by the gut.72 Therefore, changes in gastrointestinal motility or the gut microbiome may be responsible for low serum UA in PD.
An innocent bystander?
Because UA can be spontaneously oxidized, it is feasible that oxidative stress in PD leads to oxidative reductions in UA, but this biochemical reaction confers no biologically relevant protection. In this situation, low UA in PD may reflect an innocent bystander effect. This possibility was tested in 20 PD subjects and 20 controls by measuring the 24hr urinary accumulation of allantoin, the oxidative product of UA.73 This study showed no evidence for low urinary UA in PD, and no evidence for increased oxidation of UA in PD. However, this study was relatively small, and it may have been underpowered to detect any significant change in urinary UA or allantoin.
Variations in purine metabolism.
Variations in purine synthesis could also be responsible for low serum UA. One study showed that PD was associated with low serum levels of the UA precursor, hypoxanthine.74 This finding implies that purine synthesis “upstream” of UA may be abnormal in PD. Another study noted significant changes in the expression of some enzymes of purine metabolism in the PD brain, providing further evidence for variability in purine metabolism.75 There are common variants for many enzymes of purine synthesis (Figure 4).76 The functional significance of most of these variants has not been studied, but they could have a profound impact on purine metabolism and on UA production. In this situation, low serum UA in PD could reflect a biomarker of abnormal purine metabolism. Further studies of this possibility seem warranted, because they imply that interventions “upstream” of UA may be more valuable than interventions involving UA itself.
Summary and Future Directions.
Numerous independent studies from all over the world have described links between PD and UA. This link appears to be highly reproducible and irrefutable. However, the biological mechanisms responsible for this link have never been established. The traditional theory has been that the low UA is associated with reduced anti-oxidant potential, contributing to increased oxidative stress that triggers or accelerates degeneration of dopaminergic neurons in PD.
This viewpoint has been so strong that it has led to numerous studies, including clinical trials aiming to raise UA levels and to slow progression of PD. Now that these trials have failed, we are left wondering if the trial designs were adequate to demonstrate the expected neuroprotective qualities of UA, and if better trials should be designed.14, 49, 50 Others have called for a re-evaluation of the scientific premise behind these trials.48
In this review, we summarize evidence that question the long-held view that UA is a biologically relevant anti-oxidant. Even if it is an important anti-oxidant, there is evidence to suggest that UA is largely excluded from the brain where it would be needed to protect against neurodegeneration. Observations summarized here suggests we should begin to consider reverse causation. In other words, it may be time to consider the possibility that something about PD causes low serum UA. We summarize several such mechanisms. We also describe the possibility that there is no causal relationship between UA and PD, and that UA may instead be a biomarker of disease processes in PD. We hope these ideas will stimulate further studies to better understand the mechanistic links between UA and PD.
Acknowledgements
We thank Stewart Factor and Nao Kamatani for their comments on this manuscript. This work was supported in part by a pilot grant from the Emory Udall center (P50 NS123103) and NIH R01 grants NS109242, NS116025, NS119758, and NS119831.
Financial Disclosures in past 12 months
Fatemeh Seifar and Ashok Dinasarapu report no disclosures. H. A. Jinnah has grant support (recent, active, or pending) from the US government (National Institutes of Health), and industry (Abbvie, Addex, Aeon, Revance, Jazz). Dr. Jinnah has also served on advisory boards or as a consultant for Addex, Allergan, Apello, CoA Therapeutics, Cavion, Daiichi Sankyo, Ipsen, PureTech, Retrophin, Revance and Takaha/Ene. He has received honoraria or stipends for lectures or administrative work from the International Parkinson’s Disease and Movement Disorders Society. Dr. Jinnah has also served on the Scientific Advisory Boards for several private foundations including the Benign Essential Blepharospasm Research Foundation, Cure Dystonia Now, the Dystonia Medical Research Foundation, and the Tourette Association of America. He also is principle investigator for the Dystonia Coalition, which has received the majority of its support through the NIH (grants NS116025, NS065701 from the National Institutes of Neurological Disorders and Stroke TR001456 from the Office of Rare Diseases Research at the National Center for Advancing Translational Sciences). The Dystonia Coalition has received additional material or administrative support from industry sponsors (Allergan Inc. and Merz Pharmaceuticals) as well as private foundations (The Benign Essential Blepharospasm Foundation, Cure Dystonia Now, The Dystonia Medical Research Foundation, The National Spasmodic Dysphonia Association, and the National Spasmodic Torticollis Association).
Author Roles
F.S., and H.A.J contributed to conception, design, and review of literature of the manuscript. A.R.D contributed to review of literature, analysis and interpretation of data. F.S and H.A.J wrote the manuscript and all authors contributed to its editing.
Funding:
The authors declare no financial conflict of interest. The work was supported in part by a Emory-Udall Parkinson’s Disease Center Pilot Grant (P50 NS123103) along with grants to the Dystonia Coalition, a consortium of the Rare Diseases Clinical Research Network (RDCRN) that is supported by the Office of Rare Diseases Research (ORDR) at the National Center for Advancing Clinical and Translational Sciences (NCATS; U54 TR001456) in collaboration with the National Institute for Neurological Diseases and Stroke (NINDS; U54 NS065701 and U54 NS116025).
Bibliography
- 1.Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- 2.Schlesinger I, Schlesinger N. Uric acid in Parkinson’s disease. Mov Disord 2008;23:1653–1657. [DOI] [PubMed] [Google Scholar]
- 3.Shen C, Guo Y, Luo W, Lin C, Ding M. Serum urate and the risk of Parkinson’s disease: results from a meta-analysis. Can J Neurol Sci 2013;40:73–79. [DOI] [PubMed] [Google Scholar]
- 4.Wen M, Zhou B, Chen YH, et al. Serum uric acid levels in patients with Parkinson’s disease: A meta-analysis. PLoS One 2017;12:e0173731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Z, Zhang S, Wang D, et al. The significance of uric acid in the diagnosis and treatment of Parkinson disease: An updated systemic review. Medicine (Baltimore) 2017;96(45):e8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grazynska A, Adamczewska K, Antoniuk S, et al. The influence of serum uric acid level on non-motor symptoms sccurrence and severity in patients with idiopathic Parkinson’s disease and atypical parkinsonisms: A systematic review. Medicina 2021;57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neogi T Gout. N Engl J Med 2011;36(9):443–452. [DOI] [PubMed] [Google Scholar]
- 8.Saito Y, Tanaka A, Node K, Kobayashi Y. Uric acid and cardiovascular disease: A clinical review. J Cardiol 2021;78:51–57. [DOI] [PubMed] [Google Scholar]
- 9.Borghi C, Agabiti-Rosei E, Johnson RJ, et al. Hyperuricaemia and gout in cardiovascular, metabolic and kidney disease. Eur J Intern Med 2020;80:1–11. [DOI] [PubMed] [Google Scholar]
- 10.Adomako E, Moe WW. Uric acid and urate in urolithiasis: the innocent bystander, instigator, and perpetrator. Sem Nephrol 2020;40:564–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joosten LAB, Crisan TO, Bjornstad P, Johnson RJ. Asymptomatic hyperuricaemia: a silent activator of the innate immune system. Nat Rev Rheumatol 2020;16:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devgun MS, Dhillon HS. Importance of diurnal variations on clinical value and interpretation of serum urate measurements. J Clin Pathol 1992;45:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu KH, Luo SF, Tsai WP, Huang YY. Intermittent elevation of serum urate and 24-hour urinary uric acid excretion. Rheumatology (Oxford) 2004;43:1541–1545. [DOI] [PubMed] [Google Scholar]
- 14.Parkinson Study Group S-PDI, Schwarzschild MA, Ascherio A, et al. Effect of urate-elevating inosine on early Parkinson disease progression: The SURE-PD3 randomized clinical trial. JAMA 2021;326:926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao X, O’Reilly EJ, Schwarzschild MA, Ascherio A. Prospective study of plasma urate and risk of Parkinson disease in men and women. Neurology 2016;86:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savica R, Rocca WA, Ahlskog JE. When does Parkinson disease start? Arch Neurol 2010;67:798–801. [DOI] [PubMed] [Google Scholar]
- 17.Breen DP, Lang AE. Tracking the course of prodromal Parkinson’s disease. Brain 2017;140:259–262. [DOI] [PubMed] [Google Scholar]
- 18.Simon KC, Eberly S, Gao X, et al. Mendelian randomization of serum urate and parkinson disease progression. Ann Neurol 2014;76:862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kia DA, Noyce AJ, White J, et al. Mendelian randomization study shows no causal relationship between circulating urate levels and Parkinson’s disease. Ann Neurol 2018;84:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobylecki CJ, Nordestgaard BG, Afzal S. Plasma urate and risk of Parkinson’s disease: A mendelian randomization study. Ann Neurol 2018;84:178–190. [DOI] [PubMed] [Google Scholar]
- 21.Coneys R, Storm CS, Kia DA, Almramhi M, Wood NW. Mendelian randomisation finds no causal association between urate and Parkinson’s disease progression. Mov Disord 2021;36:2182–2187. [DOI] [PubMed] [Google Scholar]
- 22.Konig IR, Greco FMD. Mendelian randomization: Progressing towards understanding causality. Ann Neurol 2018;84:176–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown EG, Goldman SM, Tanner CM. Mendel and urate: Acid test or random noise? Parkinsonism Relat Disord 2018;53:1–3. [DOI] [PubMed] [Google Scholar]
- 24.Mandal AK, Mount DB. The molecular physiology of uric acid homeostasis. Annu Rev Physiol 2015;77:323–345. [DOI] [PubMed] [Google Scholar]
- 25.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci (USA) 1981;78:6858–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan M, Grayson L, Clarke DJ. The total antioxidant capacity of human serum measured using enhanced chemiluminescence is almost completely accounted for by urate. Ann Clin Biochem 1997;34 ( Pt 6):688–689. [DOI] [PubMed] [Google Scholar]
- 27.Wayner DD, Burton GW, Ingold KU, Barclay LR, Locke SJ. The relative contributions of vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity of human blood plasma. Biochim Biophys Acta 1987;924:408–419. [DOI] [PubMed] [Google Scholar]
- 28.Nalsen C, Ohrvall M, Kamal-Eldin A, Vessby B. Plasma antioxidant capacity among middle-aged men: the contribution of uric acid. Scand J Clin Lab Invest 2006;66(3):239–248. [DOI] [PubMed] [Google Scholar]
- 29.Frei B, England L, Ames BN. Ascorbate is an outstanding antioxidant in human blood plasma. Proc Natl Acad Sci U S A 1989;86:6377–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao G, Prior RL. Comparison of different analytical methods for assessing total antioxidant capacity of human serum. Clin Chem 1998;44(6 Pt 1):1309–1315. [PubMed] [Google Scholar]
- 31.Hershfield MS, Roberts LJ 2nd, Ganson NJ, et al. Treating gout with pegloticase, a PEGylated urate oxidase, provides insight into the importance of uric acid as an antioxidant in vivo. Proc Natl Acad Sci U S A 2010;107:14351–14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halperin Kuhns VL, Woodward OM. Sex differences in urate handling. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikkelsen WM, Dodge HJ, Valkenburg H. The distribution of serum uric acid values in a population unselected as to gout or hyperuricemia. Am J Med 1965;39:242–251. [DOI] [PubMed] [Google Scholar]
- 34.Meoni S, Macerollo A, Moro E. Sex differences in movement disorders. Nat Rev Neurol 2020;16:84–96. [DOI] [PubMed] [Google Scholar]
- 35.Parks DA, Granger DN. Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl 1986;548:87–99. [PubMed] [Google Scholar]
- 36.Linder N, Rapola J, Raivio KO. Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab Invest 1999;79:967–974. [PubMed] [Google Scholar]
- 37.Praetorius E, Poulsen H, Dupont H. Uric acid, xanthine and hypoxanthine in the cerebrospinal fluid. Scand J Clin Lab Invest 1957;9:133–137. [DOI] [PubMed] [Google Scholar]
- 38.Farstad M, Haug JO, Lindbak H, Skaug OE. Uric acid in the cerebrospinal fluid in cerebral atrophy. Acta Neurol Scand 1965;41:52–58. [DOI] [PubMed] [Google Scholar]
- 39.Crone C Uric acid in the cerebrospinal fluid; the relative concentration in meningitis and other neurological disorders. Dan Med Bull 1957;4:22–25. [PubMed] [Google Scholar]
- 40.Bhattacharyya S, Bakshi R, Logan R, Ascherio A, Macklin EA, Schwarzschild MA. Oral inosine persistently elevates plasma antioxidant capacity in Parkinson’s disease. Mov Disord 2016;31:417–421. [DOI] [PubMed] [Google Scholar]
- 41.Schwarzschild MA, Ascherio A, Beal MF, et al. Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: a randomized clinical trial. JAMA Neurol 2014;71(2):141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwaki H, Ando R, Miyaue N, et al. One year safety and efficacy of inosine to increase the serum urate level for patients with Parkinson’s disease in Japan. J Neurol Sci 2017;383:75–78. [DOI] [PubMed] [Google Scholar]
- 43.McFarland NR, Burdett T, Desjardins CA, Frosch MP, Schwarzschild MA. Postmortem brain levels of urate and precursors in Parkinson’s disease and related disorders. Neurodegener Dis 2013;12:189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sebesta I, Stiburkova B, Krijt J. Hereditary xanthinuria is not so rare disorder of purine metabolism. Nucleosides Nucleotides Nucleic Acids 2018;37:324–328. [DOI] [PubMed] [Google Scholar]
- 45.Nakayama A, Matsuo H, Ohtahara A, et al. Clinical practice guideline for renal hypouricemia (1st edition). Hum Cell 2019;32:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koto R, Sato I, Kuwabara M, Seki T, Kawakami K. Temporal trends in the prevalence and characteristics of hypouricaemia: a descriptive study of medical check-up and administrative claims data. Clin Rheumatol 2022;41:2113–2119. [DOI] [PubMed] [Google Scholar]
- 47.Jagannathan L SC, Costa M Oxidative stress under ambient and physiological oxygen tension in tissue culture. Curr Pharmacol Rep 2016;2:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frucht SJ. Effect of urate-elevating inosine on progression of early parkinson disease. JAMA 2022;327:85. [DOI] [PubMed] [Google Scholar]
- 49.Schwarzschild MA, Macklin EA, Bakshi R, et al. Sex differences by design and outcome in the Safety of Urate Elevation in PD (SURE-PD) trial. Neurology 2019;93:e1328–e1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macklin EA, Ascherio A, Schwarzschild MA. Effect of urate-elevating inosine on progression of early Parkinson disease: Reply. JAMA 2022;327:85–86. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe H, Hattori T, Kume A, et al. Improved Parkinsons disease motor score in a single-arm open-label trial of febuxostat and inosine. Medicine (Baltimore) 2020;99:e21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson TA, Jinnah HA, Kamatani N. Shortage of cellular ATP as a cause of diseases and strategies to enhance ATP. Front Pharmacol 2019;10:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 1994;140:1–22. [DOI] [PubMed] [Google Scholar]
- 54.Albrecht E, Waldenberger M, Krumsiek J, et al. Metabolite profiling reveals new insights into the regulation of serum urate in humans. Metabolomics 2014;10:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lord S, Godfrey A, Galna B, Mhiripiri D, Burn D, Rochester L. Ambulatory activity in incident Parkinson’s: more than meets the eye? J Neurol 2013;260:2964–2972. [DOI] [PubMed] [Google Scholar]
- 56.Yazar T, Yazar HO, Zayimoglu E, Cankaya S. Incidence of sarcopenia and dynapenia according to stage in patients with idiopathic Parkinson’s disease. Neurol Sci 2018;39:1415–1421. [DOI] [PubMed] [Google Scholar]
- 57.Drey M, Hasmann SE, Krenovsky JP, et al. Associations between early markers of Parkinson’s disease and sarcopenia. Front Aging Neurosci 2017;9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alvim RO, Siqueira JH, Zaniqueli D, Dutra DM, Oliosa PR, Mill JG. Influence of muscle mass on the serum uric acid levels in children and adolescents. Nutr Metab Cardiovasc Dis 2020;30:300–305. [DOI] [PubMed] [Google Scholar]
- 59.Molino-Lova R, Sofi F, Pasquini G, et al. Higher uric acid serum levels are associated with better muscle function in the oldest old: Results from the Mugello Study. Eur J Intern Med 2017;41:39–43. [DOI] [PubMed] [Google Scholar]
- 60.Kawamoto R, Ninomiya D, Kasai Y, et al. Serum uric acid Is positively associated with handgrip strength among Japanese community-dwelling elderly women. PLoS One 2016;11:e0151044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong XW, Tian HY, He J, Wang C, Qiu R, Chen YM. Elevated serum uric acid is associated with greater bone mineral density and skeletal muscle mass in middle-aged and older adults. PLoS One 2016;11:e0154692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurology 2016;15:1257–1272. [DOI] [PubMed] [Google Scholar]
- 63.Towiwat P, Li ZG. The association of vitamin C, alcohol, coffee, tea, milk and yogurt with uric acid and gout. Int J Rheum Dis 2015;18:495–501. [DOI] [PubMed] [Google Scholar]
- 64.Kaneko K, Aoyagi Y, Fukuuchi T, Inazawa K, Yamaoka N. Total purine and purine base content of common foodstuffs for facilitating nutritional therapy for gout and hyperuricemia. Biol Pharm Bull 2014;37:709–721. [DOI] [PubMed] [Google Scholar]
- 65.Faller J, Fox IH. Ethanol-induced hyperuricemia. N Engl J Med 1982;307:1598–1602. [DOI] [PubMed] [Google Scholar]
- 66.Choi JW, Ford ES, Gao X, Choi HK. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: the Third National Health and Nutrition Examination Survey. Arthritis Rheum 2008;59:109–116. [DOI] [PubMed] [Google Scholar]
- 67.Hallfrisch J Metabolic effects of dietary fructose. FASEB J 1990;4:2652–2660. [DOI] [PubMed] [Google Scholar]
- 68.Park KY, Kim HJ, Ahn HS, et al. Effects of coffee consumption on serum uric acid: systematic review and meta-analysis. Semin Arthritis Rheum 2016;45:580–586. [DOI] [PubMed] [Google Scholar]
- 69.Gao X, Chen H, Choi HK, Curhan G, Schwarzschild MA, Ascherio A. Diet, urate, and Parkinson’s disease risk in men. Am J Epidemiol 2008;167:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Annanmaki T, Muuronen A, Murros K. Low plasma uric acid level in Parkinson’s disease. Mov Disord 2007;22:1133–1137. [DOI] [PubMed] [Google Scholar]
- 71.Heinzel S, Aho VTE, Suenkel U, et al. Gut microbiome signatures of risk and prodromal markers of Parkinson disease. Ann Neurol 2020;88:320–331. [DOI] [PubMed] [Google Scholar]
- 72.Liu X, Tong X, Zou Y, et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat Genet 2022;54:52–61. [DOI] [PubMed] [Google Scholar]
- 73.Sampat R, Young S, Rosen A, et al. Potential mechanisms for low uric acid in Parkinson disease. J Neural Transm 2016;123:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johansen KK, Wang L, Aasly JO, et al. Metabolomic profiling in LRRK2-related Parkinson’s disease. PLOS One 2009;4:e7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garcia-Esparcia P, Hernandez-Ortega K, Ansoleaga B, Carmona M, Ferrer I. Purine metabolism gene deregulation in Parkinson’s disease. Neuropathol Appl Neurobiol 2015;41:926–940. [DOI] [PubMed] [Google Scholar]
- 76.Norman B, Glenmark B, Jansson E. Muscle AMP deaminase deficiency in 2% of a healthy population. Muscle Nerve 1995;18:239–241. [DOI] [PubMed] [Google Scholar]