

Abstract
Background:
Cerebral Cavernous Malformations (CCMs) are neurovascular lesions caused by loss-of-function mutations in one of three genes, including KRIT1 (CCM1), CCM2, and PDCD10 (CCM3). CCMs affect ~1/200 children and adults, and no pharmacologic therapy is available. CCM lesion count, size, and aggressiveness vary widely among patients of similar ages with the same mutation or even within members of the same family. However, what determines the transition from quiescent lesions into mature and active (aggressive) CCM lesions is unknown.
Methods:
We use genetic, RNA-seq, histology, flow cytometry and imaging techniques to report the interaction between CCM-endothelium, astrocytes, leukocytes, microglia/macrophages, neutrophils (CALMN interaction) during the pathogenesis of CCMs in the brain tissue.
Results:
Expression profile of astrocytes in adult mouse brains using translated mRNAs obtained from the purification of EGFP-tagged ribosomes (Aldh1l1-EGFP/Rpl10a) in the presence or absence of CCM lesions (Slco1c1-iCreERT2;Pdcd10fl/fl; Pdcd10BECKO) identifies a novel gene signature for neuroinflammatory astrocytes. CCM-induced reactive astrocytes have a neuroinflammatory capacity by expressing genes involved in angiogenesis, chemotaxis, hypoxia signaling, and inflammation. RNA-seq analysis on RNA isolated from brain endothelial cells (BECs) in chronic Pdcd10BECKO mice (CCM-endothelium), identified crucial genes involved in recruiting inflammatory cells and thrombus formation through chemotaxis and coagulation pathways. In addition, CCM-endothelium was associated with increased expression of Nlrp3 and Il1b. Pharmacological inhibition of NLRP3 significantly decreased inflammasome activity as assessed by quantification of a fluorescent indicator of caspase-1 activity (FAM-FLICA caspase-1) in BECs from Pdcd10BECKO in chronic stage. Importantly, our results support the hypothesis of the crosstalk between astrocytes and CCM endothelium that can trigger recruitment of inflammatory cells arising from brain parenchyma (microglia) and the peripheral immune system (leukocytes) into mature active CCM lesions that propagate lesion growth, immunothrombosis, and bleedings. Unexpectedly, partial or total loss of brain endothelial NF-kB activity (using Ikkbfl/fl mice) in chronic Pdcd10BECKO mice does not prevent lesion genesis or neuroinflammation. Instead, this resulted in a trend increase in the number of lesions and immunothrombosis, suggesting that therapeutic approaches designed to target inflammation through endothelial NF-kB inhibition may contribute to detrimental side effects.
Conclusions:
Our study reveals previously unknown links between neuroinflammatory astrocytes and inflamed CCM endothelium as contributors that trigger leukocyte recruitment and precipitate immunothrombosis in CCM lesions. However, therapeutic approaches targeting brain endothelial NF-kB activity may contribute to detrimental side effects.
Keywords: Blood-Brain Barrier, Cerebrovascular Malformations, Cerebrovascular Disease/Stroke, Thrombosis, Vascular Disease
Graphical Abstract
Introduction
Cerebral cavernous malformations (CCMs) are common neurovascular lesions causing a lifelong risk of brain hemorrhage, seizures, and neurological sequelae for which there is no current effective pharmacologic therapy1, 2. CCMs affect approximately 0.5% of the general population, in which children represent ~25% of diagnosed individuals. Inherited germline (~20%) and somatic (~80%) loss of function mutations in the genes KRIT1 (Krev1 interaction trapped gene 1, CCM1), CCM2 (Malcavernin), PDCD10 (programmed cell death protein 10, CCM3) propel brain vascular changes marked by the disruption of intercellular junctions, increase in reactive oxygen species (ROS), angiogenesis, altered basement membrane composition, and increased vascular permeability2–4. CCMs are dynamic lesions that can form, enlarge, regress, or behave aggressively, producing repetitive hemorrhage that contributes to the clinical manifestation of CCM disease in familial and sporadic cases5. Moreover, histological analysis in human CCM lesions has suggested that CCM bleeding in brain tissue may lead to inflammation associated with adaptive immune response and thrombosis in varying degrees6–8.
Importantly, histopathological hallmarks identified in human CCM brain tissue are closely recapitulated in the central nervous system (CNS) of genetically sensitized CCM mouse models9 and, more recently observed, in chronic CCM mouse models using inducible brain endothelial-specific genetic inactivation of CCM genes10–14. These CCM animal models have been crucial to unveiling transition phases in CCM lesion genesis that range from early-stage isolated caverns (stage 1) during CCM lesion formation to late-stage multi-cavernous lesions (stage 2) containing hemosiderin deposits and immune cell infiltration (characteristics of human active CCM lesions)9. However, the molecular and cellular mechanisms contributing to CCM lesion initiation and progression as well as the transition between quiescence to mature active lesions in the CNS remain elusive. Recent studies indicate that GFAP+ astrocytes contribute to CCM pathogenesis by integrating a circuit of neurovascular dysfunction during CCM lesion formation13. These findings suggest hypoxia programs are activated during CCM formation that affects endothelial-astrocyte crosstalk associated with changes in angiogenesis, inflammation, and endothelial-cell metabolism, involved in CCM formation13.
The present study shows that while the hypoxia and angiogenesis pathways are important in CCM lesion formation13, the inflammation and NLRP3 inflammasome activity play a more critical role in transitioning into mature and active CCM lesions. We report that neuroinflammation conferred by reactive astrocytes and CCM endothelium attracts innate and adaptive immunity to CCM lesions in a chronic and inducible CCM mouse models. Unexpectedly, partial or total loss of brain endothelial NF-kB activity in a chronic CCM animal model does not prevent lesion genesis or neuroinflammation. Instead, this loss results in elevated immunothrombosis, suggesting that therapeutic approaches designed to target inflammation through NF-kB inhibition may contribute to detrimental side effects. Here, we propose that the interaction between CCM-endothelium, astrocytes, leukocytes, microglia/macrophages, and neutrophils (CALMN interaction) play a critical role in the pathogenesis of CCMs by inducing active lesions and propensity for immunothrombosis. Our study reveals a new role for astrocytes and CCM endothelium in neuroinflammation and identifies that “CALMN interaction”, may lead to new therapeutic strategies to prevent active CCM lesions.
Methods
Data Availability
Detailed methods and major resources table can be found in the Supplemental Material. All material, data, and detailed protocols are available upon request, and the RNAseq data used in this study is available in the Gene Expression Omnibus database under GSE204979 (http://www.ncbi.nlm.nih.gov/geo).
Results
Increase of hypoxia, inflammation and inflammasome signaling pathways in CCM disease.
Histological analysis of CCM lesions in chronic animal models has found that lesions vary from early-stage isolated caverns (stage 1) that range of size to late-stage multi-cavernous lesions (stage 2) more resembling of human CCM lesions9. However, the molecular and cellular mechanism contributing to CCM lesion initiation and activation is unknown15. Therefore, to monitor changes in CCM lesion genesis, conferred exclusively to the CNS tissue, at different developmental stages, we created a brain microvascular endothelial cell-specific Pdcd10 knock out animal model (Pdcd10BECKO) by crossing a brain endothelial tamoxifen-regulated Cre (Slco1c1-CreERT2) with Pdcd10fl/fl mice (Slco1c1-CreERT2; Pdcd10fl/fl)10, 11, 13 (Fig. 1A). We have chosen to study the CCM lesions in the cerebrum because it offers a temporal pattern of lesion genesis representative to the CNS. Upon tamoxifen injection, the brains of Pdcd10BECKO animals develop CCM lesions progressively that are subdivided into acute (P15), progression (P50), and chronic (P80) stages (Fig. 1A)9, 10, 13. Moreover, differences in cerebrum histological changes correlated with changes in gene expression pattern in cerebral tissue (Fig 1A, 1B) that coincided with the induction of acute phase CCM marker genes including the HIF-target genes Loxl2, and Vegfa13(Fig. 1B, Fig S1). We observed a marked elevation in the expression of Loxl2 and Angpl4 in the progression and chronic stages. However, the levels of Vegfa mRNA were attenuated in chronic phase CCM brain tissue (Fig. 1B), suggesting that angiogenesis triggered by VEGF is critical in CCM lesion formation. Furthermore, we observed a significant increase of neuroinflammatory genes in the CCM cerebral tissue, including, Mcp1 and Cd74, as well as inflammasome genes, such as Il1b and Nlrp3, during the progression and chronic stages (Fig. 1B, Fig S1). We also observed that among genes associated with coagulation pathways in CCM, Procr (EPCR)14, 16, 17 is the best candidate gene to differentiate between disease severities (Fig S1). These results suggest that while hypoxia and angiogenesis signaling are important in the CCM lesion formation13, neuroinflammation signaling may play a more important role in the progression and chronic stages. In agreement with previous results, prominent GFAP-immunoreactivity was observed in astrocytes flanking acute and chronic cerebral CCM lesions13 (Fig. 1C, 1D). Collectively, these results suggest that hypoxia, neuroinflammation, and inflammasome signaling are present in mature CCM lesions and that reactive astrocytes are a critical feature in the disease.
Figure 1. Increase of hypoxia and neuroinflammation signaling pathways in CCM disease.
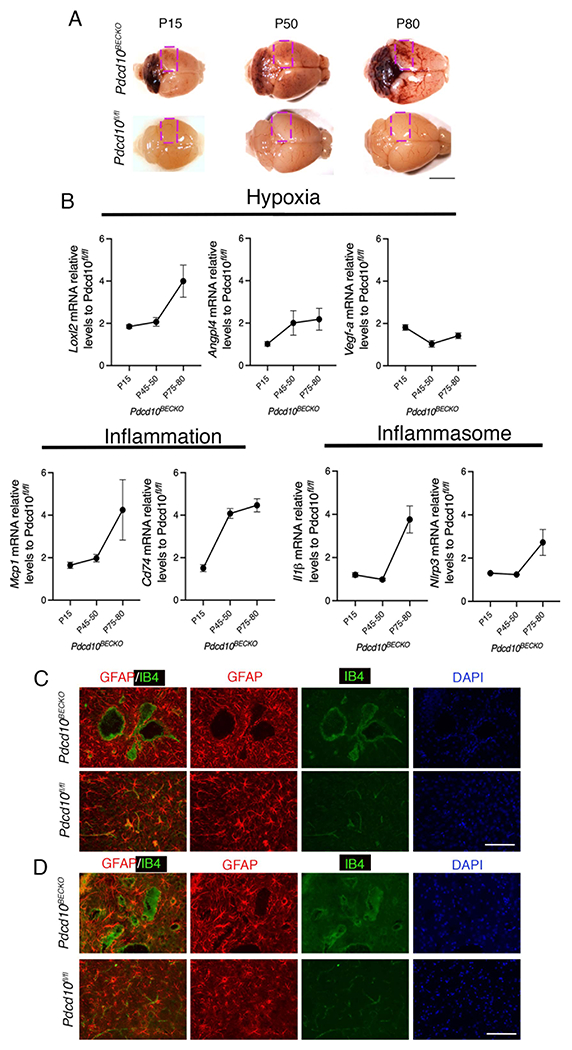
A, CCM lesions are present in the cerebellum and cerebrum of Pdcd10BECKO mice at acute (P15), progression (P50), and chronic stage (P80). A square shows the region used in B for RNA analysis. Scale bar, 5 mm. B, Analysis of Loxl2, Angpl4, Vegfa, Mcp1, Cd74, Il1β, and Nlrp3 mRNA levels by RT-qPCR at acute, progression, and chronic stage as indicated. Data are mean ±SEM, Pdcd10BECKO mice, n=3; Pdcd10fl/fl mice, n=6. C, Immunofluorescence staining of GFAP+ astrocytes (red), and endothelial marker isolectin B4 (IB4; green) of cerebral sections from P15 Pdcd10BECKO and littermate control Pdcd10flfl. DAPI staining (blue) was used to reveal nuclei. n = 4 mice in each group. Scale bar, 100 μm. D, Immunofluorescence staining of GFAP+ astrocytes (red), and endothelial marker isolectin B4 (IB4; green) of cerebral sections from P80 Pdcd10BECKO and littermate control Pdcd10flfl. DAPI staining (blue) was used to reveal nuclei. n = 5 mice in each group. Scale bar, 100 μm.
CCM endothelium induces neuroinflammatory astrocytes.
To better understand how GFAP+ astrocytes contribute to CCM pathogenesis, the astrocyte ribosome-bound mRNA levels were analyzed using Translational Ribosome Affinity Purification (TRAP) as a measurement of active translation18. For these experiments, Pdcd10BECKO and Pdcd10fl/fl control mice were crossed with a ribosome tagged mouse line (Aldh1l1-EGFP/Rpl10a). Following CCM induction with tamoxifen administration, the level of ribosome-bound and global mRNA abundance was quantified (Fig. 2). The presence of GFAP+ astrocytes was observed along with EGFP-RpL10a co-localization in Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a brain sections. We also observed that CCM lesions in Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a develop surrounded by GFAP+ astrocytes that colocalized with EGFP-RpL10a expressing cells (Fig. 2A, Fig. S2). Ribosome-bound mRNAs were highly enriched for astrocyte-specific genes and almost absent of other brain resident cell markers (Fig. S2). We identified 153 upregulated and 168 downregulated genes (FDR < 0.03) between astrocyte TRAP in Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a and astrocyte TRAP in Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a (Fig. 2B, Fig. S3). Gene functional classification analysis of differentially expressed genes (DEGs) revealed significant enrichment for terms related to antigen presentation, platelet activation/aggregation, VEGF signaling, and glucose metabolism (Fig. 2C). Genes demonstrating the most significant fold change include genes associated with neurodegenerative diseases (e.g., Ndufa4l2, App, S1pr3) (Fig. 2D). Moreover, many relevant genes with a differential expression trend were also associated with hypoxia (e.g., Adm, Igfbp3, Loxl2), chemokines (e.g., Cx3cr1, Cx3cl1, Ccl2, Ackr1), and inflammation (e.g., Cd74, Adora2a, C1qa) (Fig. 2e,2f,2g). Of particular interest are Cx3cl1 and Ccl2, which enable the capacity of reactive astrocytes to recruit microglia and leukocytes to CCM lesions19. Indeed, inflammation, neurodegeneration, and ischemia are CNS injuries that induce reactive astrocytes20. We observed that astrocytes activated during CCM disease acquire a particular transcriptional signature that we denominated CCM reactive astrocyte with neuroinflammatory capacity (Fig. 2H, 2I, 2J, Fig. S3). CCM reactive astrocytes are characterized by an increase in CX3CR1 expression in areas surrounding CCM lesions in Pdcd10BECKO brain tissue (Fig. 2h, 2J, Fig. S3). These results indicate a crucial role for a subset of neuroinflammatory reactive astrocytes in recruiting inflammatory cells to CCM mature lesions.
Figure 2. Neuroinflammatory astrocytes in CCM disease.
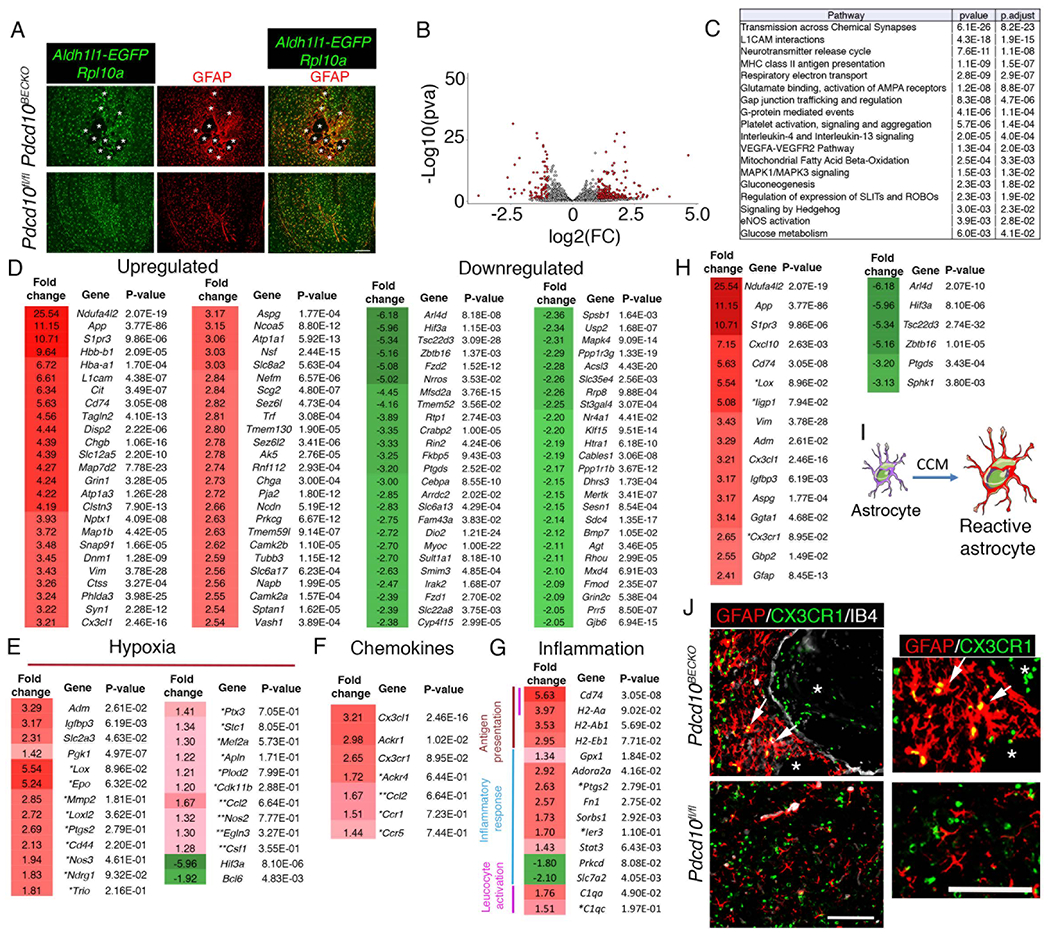
A, Immunofluorescence analysis show colocalization of GFAP+ astrocytes with cells expressing EGFP-RpL10a in Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a brains sections. Asterisks denote the vascular lumen of CCM lesions. Scale bar is 200 μm. n = 3 mice in each group. B, Volcano plot of differentially expressed transcripts in P75 Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a versus littermate control Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a. Transcripts are represented on a log2 scale. The significantly down- and up-regulated genes are labeled in red. n = 3 mice in each group. C, Top pathway enrichment analysis for Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a brain tissue. Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a littermates were used as controls. (320 DEG, fold change ≥ 1.2 and FDR < 0.03). D, List of the top 50 up- or down-regulated genes from translated mRNAs obtained from ribosomes in astrocytes from Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a brains. Fold change and P values are shown for each gene. E, Analysis of hypoxia-regulated genes. F, Analysis of chemokine transcripts. G, Analysis of inflammation-related transcripts. H, Transcriptional signature of CCM reactive astrocytes. I, CCM endothelium instructs the astrocytes to acquire a reactive astrocyte phenotype. J, CCM reactive astrocytes are positive to CX3CR1 staining. Fold change and P values are shown for each gene. n = 3 mice in each group. * and ** indicate genes that do not reach statistical significance because one of the biological replicates the fold change was too large but in the same direction (*) or when one of the biological replicates does not change (**).
Increase of hypoxia, inflammation and inflammasome signaling pathways in CCM endothelium.
CCM endothelium induces HIF-1α protein stabilization in astrocytes under normoxic conditions through elevation of nitric oxide, resulting in the synthesis of astrocyte-derived VEGF and enhancement of CCM lesion formation13. To better understand the interaction between CCM endothelium and astrocytes and potentially other neurovascular cells during CCM disease, we performed an RNA-seq profiling of brain endothelial cells (BECs) isolated from P75 Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a brains and Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a control brains (Fig. 3A). We identified 827 upregulated and 526 downregulated genes between BECs from Pdcd10BECKO and Pdcd10fl/fl controls (Fig. 3A, Fig. S4). The mRNAs obtained from the purification of BECs were highly enriched for brain endothelial-specific genes, but a small increase in leukocyte and myelin gene contaminants was also detected (Fig. 3B). Gene set enrichment analysis (GSEA) of DEGs in BECs isolated from Pdcd10BECKO brains revealed significant enrichment for terms related to neutrophil degranulation, platelet activation/aggregation, VEGF, ROS, ROCK, and EndMT signaling (Fig. 3C). The transcriptomic analysis also revealed significant upregulation of additional pathways associated with aneurysms, neurodegenerative diseases, and senescence genes (e.g., Col10a1, Serpina1e/b/d, Cyp3a13, Gkn3, Serpine1). Moreover, hypoxia genes (e.g., Apln, Egln3, Vegfa, Loxl2, Cd44) displayed an upregulation trend in BECs during chronic CCM disease (Fig. 3E). Additionally, genes implicated in neuroinflammatory response (e.g., Il6, Ccl2, Cd74, Cx3cl1, Ackr2) and inflammasome activity (e.g., Il1b, Nlrp3) suggested a potential role of CCM endothelium-induced neuroinflammation (Fig. 3F through 3H). Of note, CCM endothelium is associated with locally increased expression of anticoagulant endothelial receptors that create a local bleeding diathesis in CCMs16. We observed a significant upregulation of genes implicated in the balance between the anti- and pro-coagulation system (e.g., Procr (EPCR), Thbd, Serpine1 (PAI-1), Tfpi (TFPI), Vwf) (Fig. 3H). These results suggest that anti- and pro-thrombotic factors co-express on mature CCM lesions. Moreover, as previously reported, we observe a significant increase in genes associated with ROS in the CCM endothelium (e.g., Cybb, Slc7a2, Ncf4) (Fig. 3h)21. These transcriptomic analyses from CCM endothelium and astrocytes are consistent with our immunohistology, and bulk RNA gene expression analyses initially observed in the chronic CCM brains (Fig. 1). In addition, ligand-receptor analysis on RNA transcripts (using membrane-bound and soluble proteins) from CCM endothelium and CCM reactive astrocytes identified ligand-receptor pair considered “active”22 during CCM disease where leukocyte recruitment and neuroinflammation signaling pathways from both CCM endothelium and astrocytes are critical (Fig. 3I).
Figure 3. Inflammation and inflammasome pathways are increased in the CCM endothelium.
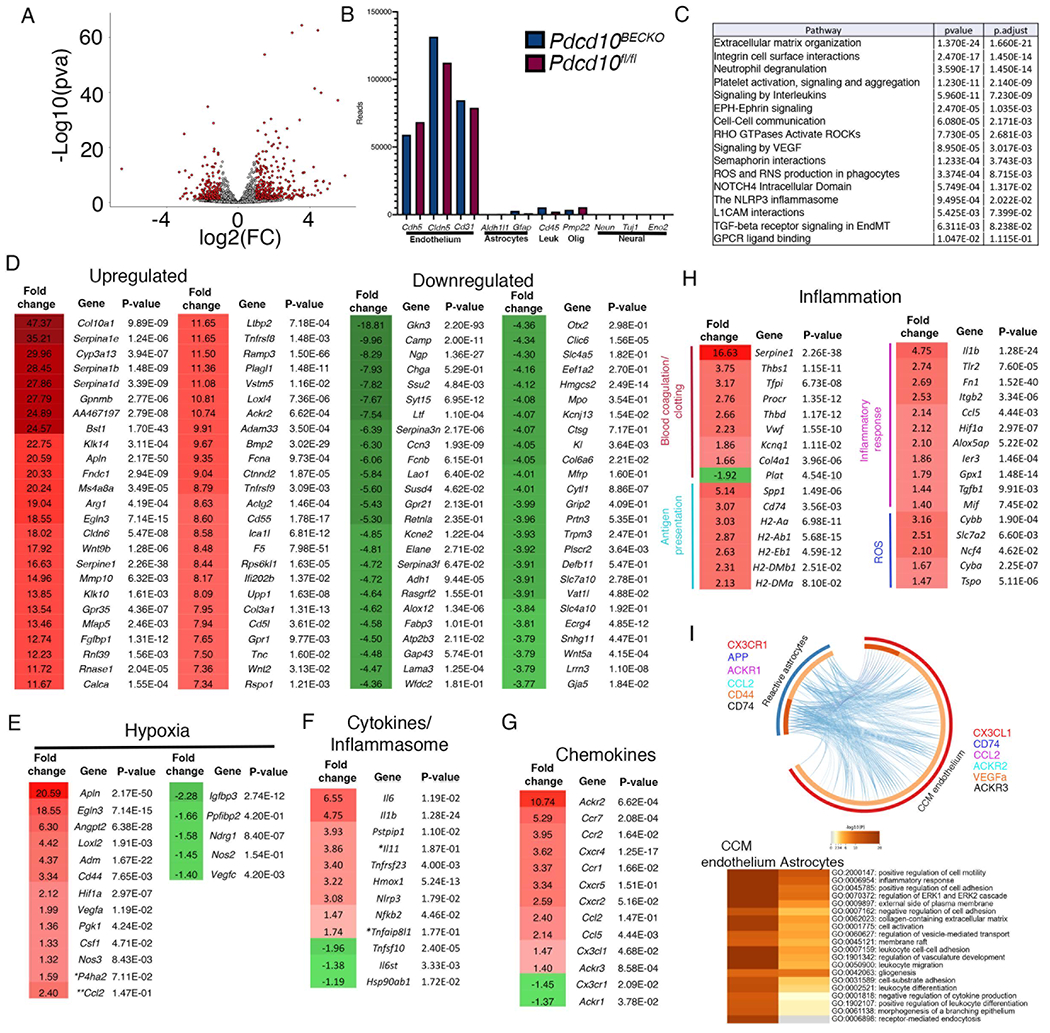
A, Volcano plot of differentially expressed transcripts in fresh isolated brain endothelial cells from P75 Pdcd10BECKO;Aldh1l1-EGFP/Rpl10a versus isolated brain endothelial cells from littermate control Pdcd10fl/fl;Aldh1l1-EGFP/Rpl10a. Transcripts are represented on a log2 scale. The significantly down- and up-regulated genes are labeled in red. Pdcd10BECKO mice, n=4; Pdcd10fl/fl mice, n=3 mice in each group. B, Analysis of known brain cell-specific gene markers. C, Top pathway enrichment analysis. D, List of the top 50 up- or down-regulated genes. Fold change and P values are shown for each gene. E, Analysis of hypoxia-regulated genes. F, Analysis of cytokines and inflammasome transcripts. G, Analysis of chemokine transcripts. H, Analysis of inflammation-related transcripts. I, Ligand-receptor analysis in reactive astrocytes (blue semicircle) and CCM endothelium (red semicircle) from Pdcd10BECKO RNAseq. Upregulated membrane bound and soluble proteins were used for analysis. Heatmap of statistical enrichment terms in I. Dark orange color represent the genes that are share by CCM endothelium and reactive astrocytes (e.g., CD74). Lines link ligand-receptor between cell types and examples are presented color coded (e.g., CX3CR1-CX3CL1). Fold change and P values are shown for each gene. Pdcd10BECKO; Aldh1l1-EGFP/Rpl10a mice, n=4; Pdcd10fl/fl; Aldh1l1-EGFP/Rpl10a mice, n=4 mice in each group.
Augmented inflammasome activity in CCM endothelium.
During inflammation, the transcription factor nuclear factor kB (NF–kb) is critical to upregulate NLRP3 gene expression23. NLRP3 inflammasome assembly and activation in endothelial cells is critical in developing and progressing chronic and neurodegenerative diseases23. Therefore, because inflammasome activation in the CCM endothelium may be an important component of the neuroinflammation observed in CCM disease, we aimed to investigate inflammasome activity in the chronic CCM mouse animal model. Caspase-1 activity is controlled by the NLRP3 inflammasome and a fluorescent indicator of caspase-1 activity (FAM-FLICA caspase-1) allows us to quantitatively assess the inflammasome activity in isolated BECs from P75 Pdcd10BECKO brains (Fig. 4A through 4C). We observed that caspase-1 activation was significantly increased in subsets of isolated BECs from Pdcd10BECKO brains when compared to BECs from Pdcd10fl/fl brains (Fig. 4A, 4B). We confirmed the specificity of BECs to elevate FAM-FLICA staining in Pdcd10BECKO brains by in situ treatment with MCC950, an inhibitor of the NLRP3 inflammasome activation24 that significantly decreases the FAM-FLICA staining in BECs from Pdcd10BECKO when compared with same BECs treated with vehicle (Fig. 4A ,4C). Moreover, the use of CCM brain tissue confirmed caspase1 activation in the brain endothelium with high levels of VCAM1 staining forming multi-cavernous lesions (stage 2) and containing leukocytes located within the multi- cavernous (Fig. 4D, Fig. S4) as well as a trend to increase levels of IL1b in Pdcd10BECKO brains. However, we did not observe an obvious caspase1 activation in single CCM lesions. Consistent with histological analysis, flow cytometry analysis of caspase-1 in CD45+ cells identifies a significant increase in caspase-1 activity in both microglia and peripheral inflammatory cells from Pdcd10BECKO brains (Fig. S6). These results suggest that CCM chronicity and inflammatory leukocytes contribute to inflammasome activation in CCM lesions, and its activation may be particular to some lesions, such as CCM lesions stage 2. However, our current data cannot discern whether inflammasome activation in CCM endothelium or other cells plays the biggest role in CCM disease. Since NF–kb activity is essential for NLRP3 expression, inflammasome priming, and assembly25, we next investigated whether RelA (p65) levels were altered in CCM brains. Levels of phospho p65 protein were increased in Pdcd10BECKO brains in comparison with littermate control Pdcd10fl/fl brains (Fig. 4E). This data revealed that mature CCM lesions are poised to innate immune response and subsequent immunothrombosis by elevation of brain endothelial and leukocyte inflammasome activity.
Figure 4. Increase in inflammasome activity in CCM endothelium.
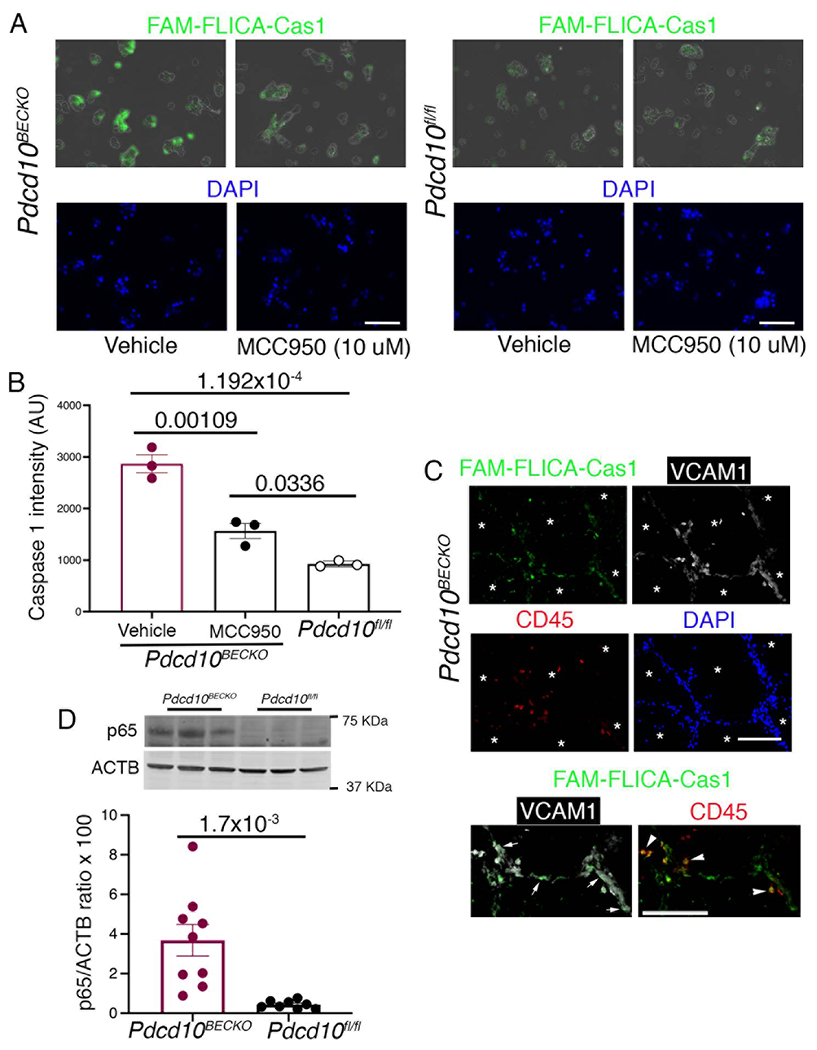
A, Visualization of inflammasome activation in P80 Pdcd10BECKO isolated brain endothelial cells in the presence or absence of MCC950 (10 μM, 1 h at 37 C). Pdcd10fl/fl isolated brain endothelial cells were used as controls in the presence or absence of MCC950. FAM-FLICA Caspase-1 assay kit was used to visualize caspase-1 signal as an indicator of inflammasome activity. Scale bars, 50 μm. B, Quantification of FAM-FLICA Caspase-1 intensity from a in Pdcd10BECKO compared to Pdcd10fl/fl or Pdcd10BECKO isolated cells exposed to MCC950. Data are mean ±SEM, n = 3 mice in each group (One-Way ANOVA followed by the Tukey post hoc test). C, FAM-FLICA fluorescence analysis (green) in multi-cavernous CCM lesions in combination with immunofluorescence for VCAM-1 (white) and CD45 (red), and DAPI labelling nuclei (blue). Asterisks denote vascular lumen of lesions. FAM-FLICA Caspase-1 signal is observed in both VCAM-1 (endothelium) and CD45 (leukocytes) positive cells as marked by white arrows and arrowheads, respectively. Scale bars, 100 μm. D, Quantification of phospho p65 protein in P80 Pdcd10BECKO brains compared to Pdcd10fl/fl brain controls by western blot. Data are mean ±SEM Pdcd10BECKO mice, n=9; Pdcd10fl/fl mice, n=8 (2-tailed unpaired t test).
Increase of immune cells in CCM brain tissue.
We next evaluated whether CCM endothelial-reactive astrocyte signaling during CCM disease prompts the recruitment of immune cells into the CCM brain tissue. As anticipated, we observed significant recruitment of CD45+ cells as well as early vascular thrombosis (staining for CD41+ platelet aggregation) in the vascular lumen of lesions in P75 Pdcd10BECKO brains (Fig. 5A). In agreement with reports from human CCM lesions8, histological analysis of P75 Pdcd10BECKO brains also showed large mature vascular thrombosis associated with CD45+ leukocyte infiltration (Fig. 5B, 5C), and in a serial section, show the accumulation of neutrophils lining the vessel wall and within the thrombus (Fig. S5, Fig. S6). Histological analysis of CCM brain lesions also demonstrated that CX3CR1+ and CD16/32+ cells constitute the larger number of cells forming the vascular thrombosis (Fig. S6). We used flow cytometry to further determine the identity and quantification of brain-infiltrating leukocytes during CCM disease. Consistent with histological analysis, we first observed a significant increase in CD45+ leukocytes isolated from P75 Pdcd10BECKO brains compared to leukocytes isolated from Pdcd10fl/fl control brains (Fig. 5D). Flow cytometry results show a significant increase in infiltrating myeloid cells in Pdcd10BECKO brains (Fig. 5E). We identified classical monocytes as CX3CR1−CD11b+Ly6C+, and microglia as CX3CR1+CD11b+F4/80+. The phenotype Ly6G+CD11b+, CX3CR1+CD11b+F4/80−Ly6C−, and CD11b+CX3CR1−Ly6C−CD11c+ identified neutrophils, non-classical monocytes, and dendritic cells, respectively (Fig 5E). Moreover, we further observed that lymphoid cells were elevated in Pdcd10BECKO brains (Fig. 5F). The phenotype CD45+TCRb+CD4+, CD45+TCRb+CD8+, and CD45+CD19+, identified CD4, CD8, T cells and B cells respectively, as the population of lymphocytes infiltrating Pdcd10BECKO brains (Fig 5F). Histological analysis of Pdcd10BECKO brains showed a significant elevation of IBA1+ microglia cells in the vicinity of CCM lesions (Fig 5G, 5H). In addition, lesion rupture and thrombosis were associated with an increase in IBA1+ microglia cells and IBA1+IB4+ leukocyte infiltration in Pdcd10BECKO brains and in Krit1BECKO;PTENBECKO/wt brains and spinal cords (Fig. 5G, 5H, Fig. S5, Fig. S7). The IBA1+ microglia cells located nearby the thrombus were hypertrophied and ameboid in morphology and enclosed by GFAP+ astrogliosis (Fig. 5G, 5I). These results confirm that neuroinflammation is a hallmark of mature CCM lesions and that inflammatory chemokines produced by CCM endothelium and CCM reactive astrocytes may contribute to attracting innate and adaptive immunity and the propensity to immunothrombosis.
Figure 5. Increased presence of immune cells in CCM brain tissue.
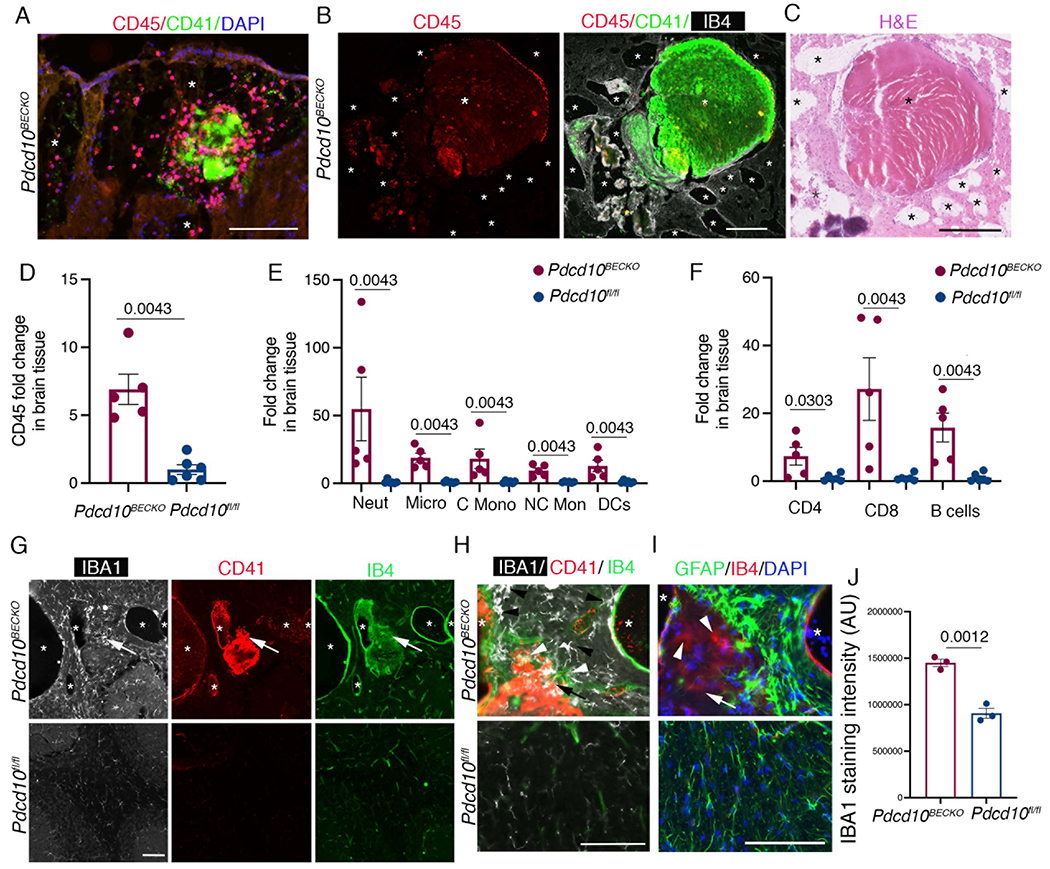
A, Immunofluorescence analysis shows leukocyte recruitment CD45+ (red) and early platelet aggregation by CD41+ (green) in the vascular lumen of lesions in P75 Pdcd10BECKO brains. Nuclei are labelled by DAPI (blue). Asterisks denote vascular lumen of CCM lesions. Scale bar, 100 μm. B, Large mature vascular thrombosis associated with CD45+ leukocyte infiltration in P75 Pdcd10BECKO brains. Scale bar, 200 μm. C, Hematoxylin and eosin staining of a serial section in b. Scale bar, 250 μm. Asterisks denote vascular lumen of CCM lesions. D, Flow cytometry of CD45+ cells isolated from P80 Pdcd10BECKO brains compared to Pdcd10fl/fl brain controls. Data are mean ±SEM, Pdcd10BECKO mice, n=5; Pdcd10fl/fl mice, n=6 (2-tailed Mann-Whitey test). E, Flow cytometry of myeloid cells. Data are mean ±SEM, Pdcd10BECKO mice, n=5; Pdcd10fl/fl mice, n=6 (2-tailed Mann-Whitey test). No multiple testing was performed. F, Flow cytometry of lymphoid cells. Data are mean ±SEM, Pdcd10BECKO mice, n=5; Pdcd10fl/fl mice, n=6 (2-tailed Mann-Whitey test). No multiple testing were performed. G, Immunofluorescence analysis for microglia IBA1+ (white), platelets CD41+ (red) cells and labeling of the brain vasculature, using isolectin B4 (green). Asterisks denote vascular lumen of CCM lesions. Scale bar, 100 μm. Arrows indicate region at high magnification observed in h. H, Increase of IBA1+ microglial cells (black arrowheads) and infiltrated IBA1+IB4+ leukocytes (white arrow heads) can be observed near thrombus (CD41+, red) of Pdcd10BECKO brains. I, Immunofluorescence analysis for GFAP (green), and IB4 (red) from a serial section in g and h, Nuclei are labelled by DAPI (blue). GFAP+ astrocytes enclosed IB4+ cells (white arrowheads) present in the thrombus in h. Asterisks denote vascular lumen of CCM lesions. J, microglia IBA1+ staining quantification. Scale bar, 100 μm. Data are mean ±SEM, n = 3 mice in each group (2-tailed student’s t test).
IkkbBECKO mice show increased thrombosis during CCM disease.
The IKK(IkB Kinase)/NF-kB signaling pathway regulates several components of the immune response and inflammation in endothelial cells, in which endothelial IKKB is the primary kinase mediating NF-kB activation (NF-kB1/p105, NF-kBp65, A20, NEMO)26, 27. Since NF-kB inhibition has anti-inflammatory effects in vivo, we wanted to investigate whether specific NF-kB inhibition in brain endothelium during CCM disease prevented the neuroinflammation observed in Pdcd10BECKO brains. Therefore, we produced mice with partial or complete brain endothelial-specific deletion of Ikkb (IkkbBECKO/wt or IkkbBECKO, respectively) by crossing mice with the Ikkb gene flanked with LoxP sites28 with Slco1c1-CreERT2;Pdcd10fl/fl mice (Pdcd10BECKO). Unexpectedly, we noticed that Pdcd10BECKO;IkkbBECKO mice showed increased mortality approximately at P70 (data not shown). Detailed histological analysis of CCM lesions in stage 1 (single cavern, ~0.02 mm2) in P15 Pdcd10BECKO brains showed a small trend increase in lesion size between brains in Pdcd10BECKO mice when compared to littermates Pdcd10BECKO;IkkbBECKO/wt (that show larger lesions) (Fig. S9). Notably, CCM lesions in stage 2 (multi-cavernous ~0.12 mm2) showed no significant difference in lesion size and number between brains in P15 Pdcd10BECKO mice when compared to littermates Pdcd10BECKO;IkkbBECKO/wt and Pdcd10BECKO;IkkbBECKO mice (Fig. S9). In contrast, CCM lesions in stage 1 and stage 2 in P50 Pdcd10BECKO brains showed a trend increase in the number of lesions in Pdcd10BECKO;IkkbBECKO/wt, and in the number of thrombi in Pdcd10BECKO;IkkbBECKO mice compared to littermates Pdcd10BECKO mice (Fig. S9). Histological analysis of CCM lesions in stage 1 and stage 2 showed no significant difference in lesion size between brains in P80 Pdcd10BECKO mice when compared to littermates Pdcd10BECKO;IkkbBECKO/wt and Pdcd10BECKO;IkkbBECKO mice (Fig 6A, 6C, 6F, 6H). However, there was a significant increase in the number of stage 1 and a trend increase in stage 2 lesions in P80 Pdcd10BECKO;IkkbBECKO mice compared to littermates Pdcd10BECKO mice (Fig 6B, 6D). Moreover, the histological analysis showed a trend of an increased presence of thrombosis in stage 2 lesions, but not stage 1 lesions, in Pdcd10BECKO;IkkbBECKO/wt mice compared to littermate Pdcd10BECKO mice (Fig. 6E–6H). Importantly, while the lesion number of CCM lesions in stage1 (Lesion number per mm2 of brain tissue) does not changes between acute (P15), progression (P50), and chronic (P80) Pdcd10BECKO brains, there was a trend increase in the number of CCM lesions in stage 2 in P50 and P80 Pdcd10BECKO brains. In addition, there was also a trend increase in the presence of thrombus formation in stage 1 and 2 lesions in P50 and P80 Pdcd10BECKO brains but not in P15 Pdcd10BECKO brains. The increase in the number of multi-cavernous and single lesions with thrombosis in P50 and P80 Pdcd10BECKO brains are consistent with the elevated inflammatory microenvironment shown at these two stages, but not in P15 Pdcd10BECKO brains (Fig. 1). Moreover, brains from Pdcd10BECKO;IkkbBECKO mice manifest increased microglia activation and multifaceted CCM lesion formation, which may contribute to the high mortality observed at P70 (Fig. 6I, 6J). These results are consistent with previous findings that indicate that NF-kB inhibition might result in severe proinflammatory effects in particular tissues29, potentially by loss of NF-kB-dependent expression of genes that participate in the resolution of neuroinflammation30, 31, enhance of NLRP3 inflammasome activity25, or by inducing apoptosis26. Further studies will be needed to evaluate pharmacological strategies targeting different nodes of neuroinflammation. Overall, our study suggests that CCM lesions transit through different stages that are histologically and molecularly different, and that neuroinflammation plays a critical role in CCM pathogenesis during the chronic stage (Fig. 7). However, therapeutic approaches designed to target brain endothelial NF-kB activity may lead to detrimental side effects.
Figure 6. Loss of brain endothelial IKKb increases lesion number and thrombosis during CCM disease.
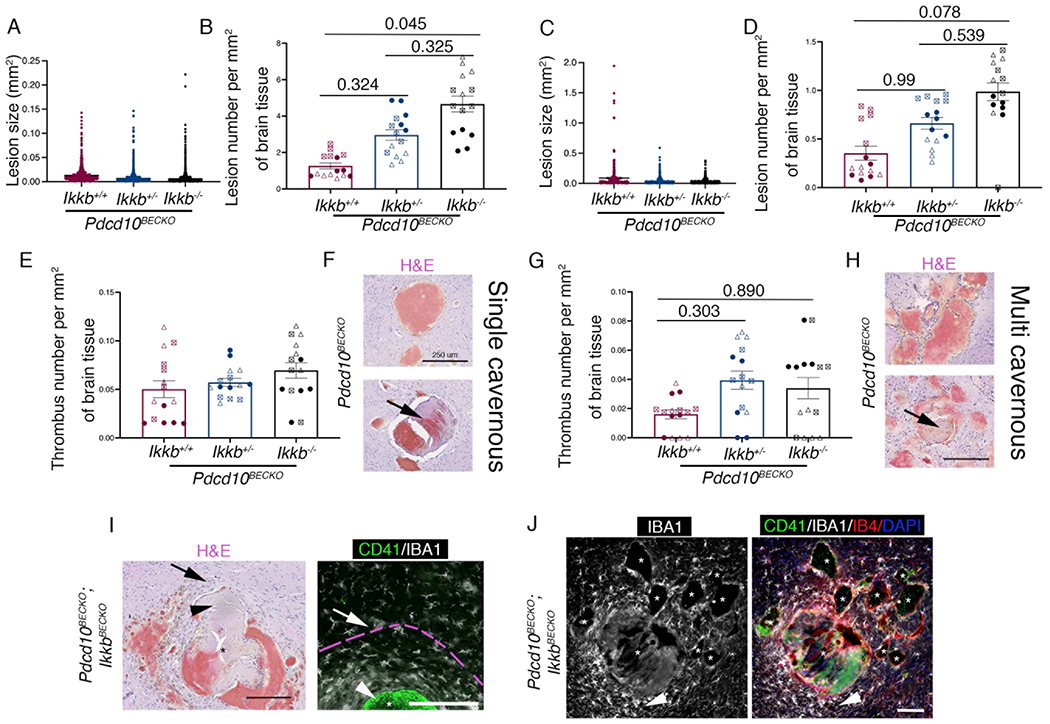
A, Analysis and quantification of stage 1 (single cavernous) lesion size in Pdcd10BECKO;Ikkbwt/wt (Pdcd10BECKO;Ikkb+/+) , Pdcd10BECKO;IkkbBECKO/wt (Pdcd10BECKO;Ikkb+/−) and Pdcd10BECKO;IkkbBECKO (Pdcd10BECKO;Ikkb−/−) brains. B, Analysis and quantification of the number of stage 1 lesions per mm2. C, Analysis and quantification of stage 2 (multi-cavernous). D, Analysis and quantification of the number of stage 2 lesions per mm2. Animals were injected at P5 with 4-hydroxi-tamoxifen to differentially assess lesion burden10. E, Analysis and quantification of the number of thrombi in stage 1 lesions per mm2. F, Hematoxylin and eosin staining of stage 1 lesions in P80 Pdcd10BECKO mice. Arrow indicates thrombus present in stage 1 lesion. G, Analysis and quantification of the number of thrombi in stage 2 lesions per mm2. H, Hematoxylin and eosin staining of stage 2 lesions in P80 Pdcd10BECKO mice. The arrow indicates thrombus present in stage 2 lesion. I, Hematoxylin and eosin staining show a multifaceted CCM lesion in Pdcd10BECKO;IkkbBECKO brains, a serial section was used for immunofluorescence analysis for CD41 (green) and IBA1 (white). Arrows indicate similar regions. Increase in IBA1+ microglial cells with hypertrophied and ameboid in morphology are observed to form a ring near the thrombus. Pink broken line indicates area of microglia activation. Arrowheads indicate thrombus. Scale bar, 200 μm. J, Immunofluorescence analysis of CD41+ (green), IBA1+ (white) and labeling of the brain vasculature, using isolectin B4 (red), of cerebral sections from P80 Pdcd10BECKO;IkkbBECKO. Exacerbated increased of IBA1+ microglial cells near a lesion with thrombosis (arrowhead). Lesions with not thrombosis show a moderate recruitment of microglial cells. Nuclei are labelled by DAPI (blue). Asterisks denote vascular lumen of CCM lesions. Animals were injected at P1 with 4-hydroxi-tamoxifen. Scale bar, 100 μm. Statistical analysis is based on the average of 5 sections per animal. Data regarding lesion number per area and thrombosis from each individual section is represented by three (B, D, E, G) different symbol shapes in the graphs, each set of shapes represents one animal (per group: filled circle, animal 1; triangle, animal 2; square, animal 3). All data are mean ±SEM, n=3 (Kruskal-Wallis post hoc Dunn’s test).
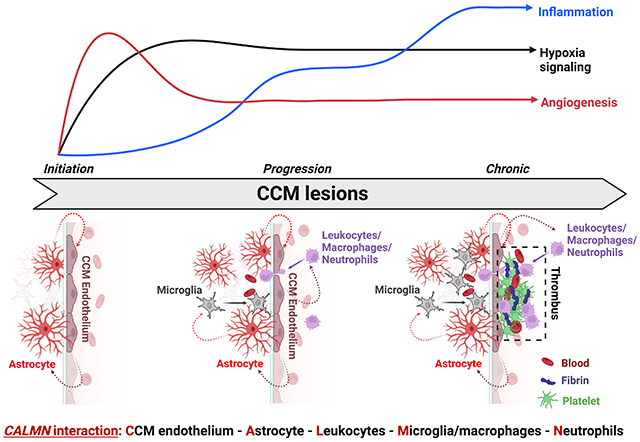
Figure 7. CALMN interaction plays a critical role in the pathogenesis of CCM disease.
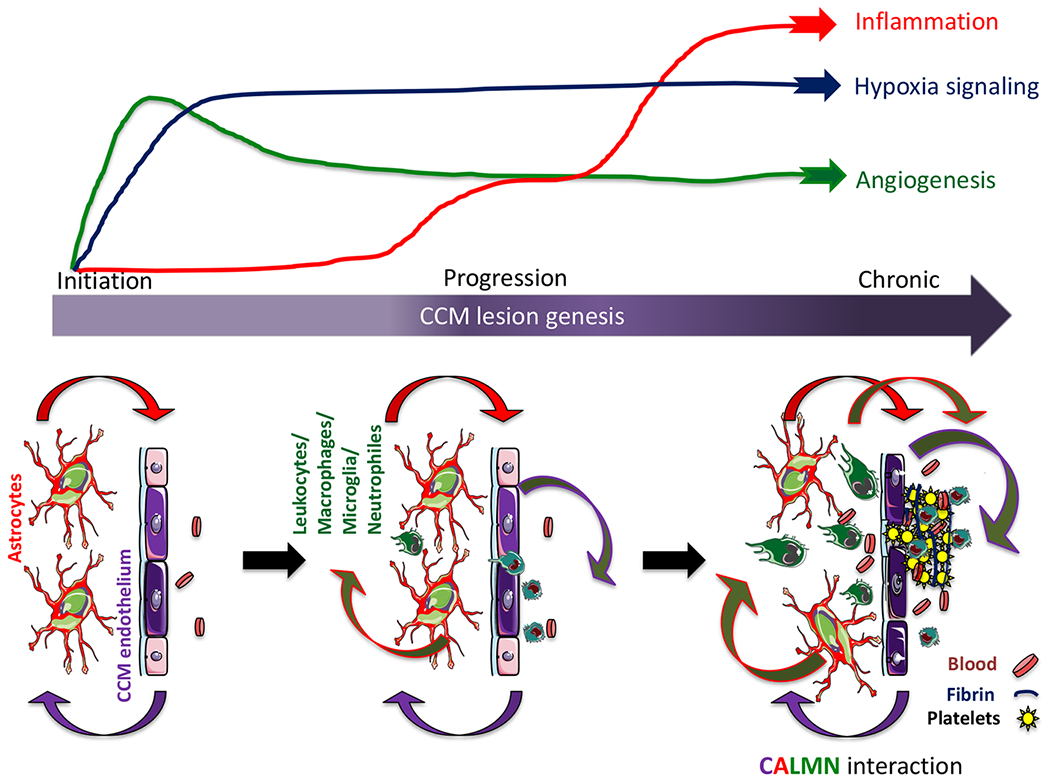
The illustration shows that angiogenesis and hypoxia signaling are essential in CCM lesion formation (initiation)13, while the inflammation pathway initiates in the progression phase and may contribute to mature active CCM lesions, lesion growth, immunothrombosis, and bleedings. Our model proposes that CCM endothelium and astrocytes synergize to recruit inflammatory cells to CCM lesions. Moreover, a reciprocal interaction between CCM endothelium, astrocytes, leukocyte, and macrophage/microglia, neutrophils, that we termed CALMN interaction, is critical for the transition of lesions into aggravating active lesions.
Discussion
We show here that neuroinflammatory signaling pathways from reactive astrocytes and CCM endothelium contribute to immune cell recruitment and thrombus formation in vascular lesions of a mouse model of CCM disease (Fig. 7). Through RNA-seq analysis from astrocyte-translated mRNA, we identified that astrocytes in the presence of CCM lesions acquire a reactive phenotype with neuroinflammatory capacity involved in the production of leukocyte chemotactic factors, antigen presentation, and inflammatory response. We show that CCM endothelium plays a crucial role in recruiting inflammatory cells and promoting thrombus formation potentially through chemotaxis and NLRP3 inflammasome activity. Additionally, our analysis reveals that neuroinflammation conferred by neuroinflammatory astrocytes and CCM endothelium attracts innate and adaptive immunity during CCM disease. Our study proposes that while hypoxia and angiogenesis pathways are essential in CCM lesion formation, inflammation and NLRP3 inflammasome activities play a more critical role in transitioning into mature and active CCM lesions. Our findings introduce the concept that “CALMN (CCM endothelium, astrocytes, leukocytes, monocytes/microglia, neutrophils) interaction” contribute to the pathogenesis of CCMs and a better understanding of the CALMN interaction may lead to new therapeutic strategies for CCMs disease (Fig. 7).
Our study reveals previously unknown links between neuroinflammatory astrocytes and inflamed CCM endothelium as contributors that trigger leukocyte recruitment and facilitates immunothrombosis in CCM lesions. Our work and others have shown that angiogenesis signaling pathways are crucial in CCM lesion formation characterized by isolated single caverns that range in size (stage 1)10, 11, 13, 32. Astrocytes, the most abundant cell type in the CNS, respond to the CCM endothelium and propel CCM formation in a non-cell-autonomous manner, mediated by hypoxia and angiogenesis programs13. However, the mechanistic linkage by which CCM lesions acquire the multi-cavernous stage (stage 2), characterized by hemosiderin deposits and immune cell infiltration in CCM brains of both humans and mice9 remains unclear. Studies have shown that polymorphisms in inflammatory pathways (e.g., Tlr4 signaling) contribute to variability in CCM disease severity33, 34 and the depletion of B lymphocytes in a genetically sensitized CCM mouse model reduces the maturation and propensity for chronic bleedings35. Recent studies in human and mouse models show that inflammation contributes to brain vascular malformations13, 14, 35–37. These observations provide direct evidence that inflammation plays a critical role in CCM lesion maturation into the clinical manifestation of CCM disease38. Our findings have important implications for explaining the mechanism behind CCM lesions maturation, by demonstrating that, in addition to CCM hemorrhage, the neuroinflammatory signaling pathway triggered by both astrocytes and CCM endothelial cells significantly contributes to lesion progression, during which inflammatory cell infiltration and thrombosis are observed. Indeed, an increase in astrocyte CX3CL1 signaling may play an important role in recruiting both microglia and peripheral inflammatory cells into CCM lesions via the CX3CR1 receptor. Moreover, the increase of astrocyte MCP1 (Ccl2), a chemoattractant for monocytes and activated T cells, may synergize with CX3CL1 to recruit the innate and adaptive immune response36 as well as to induce disassembly of tight junctions in brain endothelial cells during CCM disease3. We observed that reactive astrocytes induced in CCMs expressed high mRNA levels of CD74, a cell-surface form of the MHC class-II invariant chain. CD74 expression in astrocytes regulates the expression of COX-2 (Ptgs2)39, an angiogenic and pro-inflammatory enzyme, which inhibition ameliorates CCM disease13. Astrocyte reactivity in diseases that affect the CNS is well documented20. However, the extrinsic signals that lead to human and mouse reactive astrocytes to adopt diverse molecular states and functions in response to injury and neuroinflammation are not fully elucidated20, 40. Our data showed that, during CCM disease, CCM endothelium instructs astrocytes to acquire a particular transcriptional signature that we denominated CCM reactive astrocytes with neuroinflammatory capacity. This work proposes that future studies should aim to identify a selective mechanism to inhibit reactive astrocytes in the context of CCM, which may constitute a novel therapeutic target for the disease13. In addition, we identified CCM reactive astrocytes to express high levels of genes directly associated with neurodegenerative diseases with cerebral angiopathies, such as Cx3cr1, Ndufa4l2, App, S1pr341. The manipulation and further characterization of the role of reactive astrocytes in CCMs and other cerebrovascular malformations as well as the further understanding of the reactive astrocyte and brain endothelial interactions will be an important area for future research.
In CCMs, BECs show gross endothelial changes (CCM endothelium)3, 4, 42, leading to blood-brain barrier dysfunction that contributes to neurological deficits and, in some cases, death43, 44. Moreover, previous work has demonstrated that, during lesion formation, the CCM endothelium is hypersensitive to angiogenesis and can induce a hypoxic program associated with changes in angiogenesis, inflammation, and endothelial-cell metabolism under normoxic conditions13. Interestingly, our data regarding CCM endothelium at the chronic stage of the disease shows enrichment in genes relevant to mature and active CCM lesions. These changes included the elevation of pathways associated to neutrophil degranulation, platelet activation, ROS production45, and NLRP3 inflammasome. In addition, CCM endothelium in the chronic stage shows significant upregulation of transcripts coding for chemokines such as MCP1, CCL5, CX3CL1, known for recruiting peripheral leukocytes into inflamed tissue46. We also found that genes implicated in hypoxia signaling such as APLN, EGLN3, VEGFa, LOXL2, CD44 were highly increased in chronic CCM endothelium and may participate in disease severity. The data establish that the potential mechanistic crosstalk between astrocytes and CCM endothelium can trigger recruitment of inflammatory cells, arising from both brain parenchyma (microglia) and periphery immune system (leukocytes) into mature CCM lesions, that could potentially propagate lesion growth, thrombosis, and bleeding. Accordingly, altered levels of several genes associated with blood coagulation, TSP1 (Thbs1), EPCR (Procr), TM (Thbd), VWF, tPA (Plat), indicate that in the chronic model, some CCM lesions may have a propensity to bleeding (high levels of EPCR and TM and TFPI) while others may have a propensity to become thrombotic (high levels of PAI-1, TSP1, VWF, low in tPA, and inflammation)14, 47. It is also possible that pro-thrombotic signals in chronic CCM lesions overcome the local anti-coagulant vascular domain16 and promote immunothrombosis in largely inflamed lesions. Notably, these findings in our CCM mouse model are consistent with human studies in which inflammatory, anti-coagulant, and angiogenic plasma soluble proteins were shown to be promising predictive and prognostic biomarkers during CCM disease progression1, 16, 48.
In our study, transcriptomic data from CCM endothelium in the chronic stage of the disease suggest a critical role for genes associated with vascular homeostasis, aneurysms, neurodegenerative diseases, and senescence genes, including IL6, CD74, COL10A1, SERPINA1e/b/d, CYP3A13, GKN3, SERPINE1. Future studies could assess the impact of these genes in CCM lesion genesis and their contribution to neurological manifestation of the disease in animal models. In addition, our histological and gene expression analysis suggests a critical role for the endothelial NLRP3 inflammasome activity, which may significantly contribute to both the inflammation and thrombosis49 observed in CCM chronic lesions. Moreover, persistent inflammasome activation in endothelium and immune cells contributes to neurological and neurodegenerative conditions50 that could be relevant to the clinical symptomatology of CCM patients. Therefore, future research should focus on further identifying the molecular mechanisms that contribute to the increase of NLRP3 during CCM disease and investigate the therapeutic effect of drugs targeting NLRP3 inflammasome signaling51 in CCM disease. Indeed statins, aspirin and inhibitors of ROS activity, negatively regulate NLRP3 inflammasome activity51, 52 and have been identified as a potential therapy for CCM disease in pre-clinical and clinical studies53, 54. It is possible that different mutations in CCM genes (CCM1, CCM2 or CCM3) may result in different biological mechanisms that lead to lesion aggressiveness. However, the current evidence suggests that mutation of any CCM genes operates on similar pathways during disease initiation1, 9, 13, 35, 55. Consistently, CCM in human and murine studies has shown that neuroinflammation contributes to pathogenesis in CCM1 (most common) and CCM3 (most aggressive) diseases9, 33, 35, 56. Our findings reveal that the CCM1 mouse model sensitized with loss of one copy of the PTEN gene results in focal lesions with extensive reactive astrocytes and neuroinflammation in the brain and spinal cords. Future research should be aimed to further characterize CALMN interaction in CCM disease with PTEN/PI3K signaling because PIK3CA mutations have been recently associated with the manifestation of the disease in sporadic CCMs patients57–59.
Our findings also reveal that during the initiation, progression, and chronic phase of the CCM disease, the NF-kB inhibition through IKKB-deleted brain endothelium does not prevent CCM disease associated with an increased number of CCM lesions in stage 1 and stage 2, and immunothrombosis. These findings are consistent with studies that indicate that pharmacological therapies that target NF-kB have been proved to be impractical for treating inflammation-associated disorders in pre-clinical and clinical trials and led to the termination of several drug-development programs60. Because NF-kB signaling controls both the pro- and anti-inflammatory programs during inflammatory processes. For instance, NF-kB inhibition might result in severe proinflammatory effects in particular tissues29, potentially by loss of NF-kB dependent gene expression of genes that regulate the resolution of neuroinflammation30, 31, 61, enhancement of NLRP3 inflammasome activity25, kinase-independent effects of IKKB on endothelium or by inducing apoptosis26. Therefore, future investigations should identify potential therapeutic strategies to ameliorate CALMN interaction and inflammatory responses during the chronic stage of CCM disease without targeting brain endothelial NF-kB activity to prevent detrimental side effects.
Supplementary Material
Novelty & Significance.
What is Known?
Cerebral cavernous malformations (CCMs) are common brain and spinal cord lesions causing a lifelong risk of bleedings, thrombosis, and focal neurological deficits for which there is no current effective pharmacologic therapy.
Heterogeneity in CCM lesion count and size varies widely among patients of similar ages with the same mutation or even within members of the same family (particularly familial cases). Moreover, the majority of CCM cases (particularly sporadic cases) are often asymptomatic, suggesting that CCM lesions can remain quiescent for long periods.
CCM lesions transition through early-stage isolated caverns (stage 1) during CCM lesion formation to late-stage multi-cavernous lesions (stage 2) containing hemosiderin deposits and leukocyte infiltration. However, what determines the transition from quiescent lesions into mature and active (aggressive) CCM lesions is unknown.
What New Information Does This Article Contribute?
Using RNA-sequencing, histology, flow cytometry, and imaging techniques, we characterize CCM lesion transitions between acute, progressive, and chronic stages in the brain tissue of a murine CCM mode. We identify that astrocytes in CCM lesions acquire a reactive phenotype with neuroinflammatory capacity involved in the production of leukocyte chemotactic factors, hypoxia signaling, antigen presentation, and inflammatory response.
Our analysis identifies that CCM endothelium in chronic stage increased genes involved in recruiting inflammatory cells, hypoxia signaling, and thrombus formation through chemotaxis and coagulation pathways. We demonstrate that multi-cavernous lesions are related to enhanced NLRP3 inflammasome activity.
We show previously unknown links between neuroinflammatory reactive astrocytes and inflamed CCM endothelium as contributors that trigger leukocyte recruitment and precipitate immunothrombosis in CCM lesions. We propose that neuroinflammation and inflammasome activity play a critical role in transitioning from initial CCM formation into mature and active lesions through CALMN (CCM endothelium, astrocytes, leukocytes, microglia/macrophages, neutrophils) interactions.
Unexpectedly, partial or total loss of brain endothelial NF-kB activity in a CCM animal model results in disease worsening, suggesting that therapeutic approaches designed to target inflammation through brain endothelial NF-kB inhibition may contribute to detrimental side effects.
CCMs are neurovascular lesions that affect brain and spinal cord. An estimated 0.2% of the US population present CCM lesions, and around 20% of CCM cases are familial. Hemorrhagic stroke, thrombosis, and seizures are the most severe symptoms and significant causes of disability in patients living with cavernous angiomas or CCMs. Our study proposes that hypoxia and angiogenesis pathways are essential in CCM lesion formation, while inflammation and NLRP3 inflammasome activities play a more critical role in transitioning into mature CCM lesions. Our study reveals previously unknown links between neuroinflammatory reactive astrocytes and inflamed CCM endothelium as contributors that trigger immune cell recruitment and thrombus formation in vascular lesions of a mouse model of CCM disease. RNA-seq analysis from astrocyte-translated mRNA, shows that astrocytes in the presence of CCMs acquire a reactive phenotype with neuroinflammatory capacity involved in the production of leukocyte chemotactic factors, antigen presentation, and inflammatory response. We show that CCM endothelium plays a crucial role in recruiting inflammatory cells and promoting thrombus formation, potentially through chemotaxis and NLRP3 inflammasome activity. Our findings introduce the concept that “CALMN interaction” contributes to CCM pathogenesis, and a better understanding of the CALMN interaction may lead to new therapeutic strategies for CCM disease.
Acknowledgments
The authors thank Mark H. Ginsberg, Connie Lee, Jianbo Hu and Amy Akers from Alliance to Cure Cavernous Malformation for helpful discussion; Yin Shi (Shelly), Cassandra Bui, and Wilma McLaughlin for technical assistance; Jennifer Santini and Marcy Erb for microscopy technical assistance; and Kristen Jepsen for RNA-seq technical assistance; Ikkbfl/fl mice were the generous gift of Michael Karin (UCSD);Slco1c1-CreERT2 mice were the generous gift of Markus Schwaninger (University of Lübeck); Pdcd10fl/fl mice were the generous gift of Wang Min (Yale University).
Sources of Funding
This work was supported by the National Institute of Health, National Institute of Neurological Disorder and Stroke grant R01NS121070 (M.A.L.-R.), NS092521 (M.H.G.), and National Institute of Health, National Heart, Lung, and Blood Institute grants K01HL133530 (M.A.L.-R.), P01HL151433 (K.L., M.H.G., M.A.L.-R.), the American Heart Association AHA18POST34060251 (M.O.), the Conrad Prebys Fundation (M.O.), Crohn’s & Colitis Foundation 902590 (H.S.), UCSD School of Medicine RS295R (O.M.), Microscopy Core P30 NS047101 and as well as the UC San Diego IGM Genomics Center funding from a National Institutes of Health SIG grant S10 OD026929.
Nonstandard Abbreviations and Acronyms
- CCMs
Cerebral Cavernous Malformations
- KRIT1
Krev1 interaction trapped gene 1
- CCM2
Malcavernin
- PDCD10
Programmed cell death protein 10
- RNA-seq
RNA sequencing
- CALMN
CCM endothelium, Astrocytes, Leukocytes, Microglia/macrophages, Neuthrophils
- EGFP
Enhanced Green Fluorescent Protein
- ALDH1L1
Aldehyde Dehydrogenase 1 Family Member L1
- RPL10A
Ribosomal protein L10a
- Slco1c1
Solute Carrier Organic Anion Transporter Family Member 1C1
- iCreERT2
Tamoxifen-induced Cre Estrogen receptor
- BECKO
Brain endothelial cell knock out
- BECs
Brain endothelial cells
- NLRP3
NOD-, LRR- and pyrin domain-containing protein 3
- NF-kB
Nuclear factor kappa B
- IKKb
IkappaB kinase b
- CNS
Central nervous system
- GFAP
Glial Fibrillary Acidic Protein
- TRAP
Translational Ribosome Affinity Purification
- ROS
Reactive Oxygen Species
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists
BixRviv Preprint- https://doi.org/10.1101/2022.05.09.491214
Disclosure
There are no conflicts of interest related to this work.
Reference
- 1.Snellings DA, Hong CC, Ren AA, Lopez-Ramirez MA, Girard R, Srinath A, Marchuk DA, Ginsberg MH, Awad IA and Kahn ML. Cerebral Cavernous Malformation: From Mechanism to Therapy. Circ Res. 2021;129:195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perrelli A and Retta SF. Polymorphisms in genes related to oxidative stress and inflammation: Emerging links with the pathogenesis and severity of Cerebral Cavernous Malformation disease. Free Radic Biol Med. 2021;172:403–417. [DOI] [PubMed] [Google Scholar]
- 3.Stamatovic SM, Sladojevic N, Keep RF and Andjelkovic AV. PDCD10 (CCM3) regulates brain endothelial barrier integrity in cerebral cavernous malformation type 3: role of CCM3-ERK1/2-cortactin cross-talk. Acta Neuropathol. 2015;130:731–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Ramirez MA, Fonseca G, Zeineddine HA, Girard R, Moore T, Pham A, Cao Y, Shenkar R, de Kreuk BJ, Lagarrigue F, et al. ,. Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J Exp Med. 2017;214:3331–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clatterbuck RE, Moriarity JL, Elmaci I, Lee RR, Breiter SN and Rigamonti D. Dynamic nature of cavernous malformations: a prospective magnetic resonance imaging study with volumetric analysis. J Neurosurg. 2000;93:981–6. [DOI] [PubMed] [Google Scholar]
- 6.Shi C, Shenkar R, Kinloch A, Henderson SG, Shaaya M, Chong AS, Clark MR and Awad IA. Immune complex formation and in situ B-cell clonal expansion in human cerebral cavernous malformations. J Neuroimmunol. 2014;272:67–75. [DOI] [PubMed] [Google Scholar]
- 7.Shenkar R, Venkatasubramanian PN, Zhao JC, Batjer HH, Wyrwicz AM and Awad IA. Advanced magnetic resonance imaging of cerebral cavernous malformations: part I. High-field imaging of excised human lesions. Neurosurgery. 2008;63:782–9; discussion 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuurbier SM, Hickman CR, Tolias CS, Rinkel LA, Leyrer R, Flemming KD, Bervini D, Lanzino G, Wityk RJ, Schneble HM, et al. , Long-term antithrombotic therapy and risk of intracranial haemorrhage from cerebral cavernous malformations: a population-based cohort study, systematic review, and meta-analysis. Lancet Neurol. 2019;18:935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald DA, Shenkar R, Shi C, Stockton RA, Akers AL, Kucherlapati MH, Kucherlapati R, Brainer J, Ginsberg MH, Awad IA and Marchuk DA. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum Mol Genet. 2011;20:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Detter MR, Shenkar R, Benavides CR, Neilson CA, Moore T, Lightle R, Hobson N, Shen L, Cao Y, Girard R, et al. , Novel Murine Models of Cerebral Cavernous Malformations. Angiogenesis. 2020;23:651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardoso C, Arnould M, De Luca C, Otten C, Abdelilah-Seyfried S, Heredia A, Leutenegger AL, Schwaninger M, Tournier-Lasserve E and Boulday G. Novel Chronic Mouse Model of Cerebral Cavernous Malformations. Stroke. 2020;51:1272–1278. [DOI] [PubMed] [Google Scholar]
- 12.Zhou HJ, Qin L, Jiang Q, Murray KN, Zhang H, Li B, Lin Q, Graham M, Liu X, et al. , Caveolae-mediated Tie2 signaling contributes to CCM pathogenesis in a brain endothelial cell-specific Pdcd10-deficient mouse model. Nat Commun. 2021;12:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Ramirez MA, Lai CC, Soliman SI, Hale P, Pham A, Estrada EJ, McCurdy S, Girard R, Verma R, Moore T, et al. ,. Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maderna C, Pisati F, Tripodo C, Dejana E and Malinverno M. A murine model of cerebral cavernous malformations with acute hemorrhage. iScience. 2022;25:103943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koskimaki J, Zhang D, Li Y, Saadat L, Moore T, Lightle R, Polster SP, Carrion-Penagos J, Lyne SB, Zeineddine HA, et al. ,. Transcriptome clarifies mechanisms of lesion genesis versus progression in models of Ccm3 cerebral cavernous malformations. Acta Neuropathol Commun. 2019;7:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Ramirez MA, Pham A, Girard R, Wyseure T, Hale P, Yamashita A, Koskimaki J, Polster S, Saadat L, Romero IA, et al. , Cerebral cavernous malformations form an anticoagulant vascular domain in humans and mice. Blood. 2019;133:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malinverno M, Maderna C, Abu Taha A, Corada M, Orsenigo F, Valentino M, Pisati F, Fusco C, Graziano P, Giannotta M, et al. , Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun. 2019;10:2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. , A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim RY, Hoffman AS, Itoh N, Ao Y, Spence R, Sofroniew MV and Voskuhl RR. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;274:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhauser C, Volterra A, Carmignoto G, Agarwal A, et al. , Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goitre L, De Luca E, Braggion S, Trapani E, Guglielmotto M, Biasi F, Forni M, Moglia A, Trabalzini L and Retta SF. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic Biol Med. 2014;68:134–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armingol E, Officer A, Harismendy O and Lewis NE. Deciphering cell-cell interactions and communication from gene expression. Nat Rev Genet. 2021;22:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Afonina IS, Zhong Z, Karin M and Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol. 2017;18:861–869. [DOI] [PubMed] [Google Scholar]
- 24.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S and Brown JH. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018;138:2530–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. , NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, et al. ,. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–6. [DOI] [PubMed] [Google Scholar]
- 27.Hinz M and Scheidereit C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014;15:46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–88. [DOI] [PubMed] [Google Scholar]
- 30.Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, Lu LF, Rudensky AY and Baltimore D. Author Correction: An NF-kappaB-microRNA regulatory network tunes macrophage inflammatory responses. Nat Commun. 2018;9:3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez-Ramirez MA, Reijerkerk A, de Vries HE and Romero IA. Regulation of brain endothelial barrier function by microRNAs in health and neuroinflammation. FASEB J. 2016;30:2662–72. [DOI] [PubMed] [Google Scholar]
- 32.Orsenigo F, Conze LL, Jauhiainen S, Corada M, Lazzaroni F, Malinverno M, Sundell V, Cunha SI, Brannstrom J, Globisch MA, et al. , Mapping endothelial-cell diversity in cerebral cavernous malformations at single-cell resolution. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choquet H, Pawlikowska L, Nelson J, McCulloch CE, Akers A, Baca B, Khan Y, Hart B, Morrison L, Kim H and Brain Vascular Malformation Consortium S. Polymorphisms in inflammatory and immune response genes associated with cerebral cavernous malformation type 1 severity. Cerebrovasc Dis. 2014;38:433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polster SP, Sharma A, Tanes C, Tang AT, Mericko P, Cao Y, Carrion-Penagos J, Girard R, Koskimaki J, Zhang D, et al. , Permissive microbiome characterizes human subjects with a neurovascular disease cavernous angioma. Nat Commun. 2020;11:2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi C, Shenkar R, Zeineddine HA, Girard R, Fam MD, Austin C, Moore T, Lightle R, Zhang L, Wu M, et al. , B-Cell Depletion Reduces the Maturation of Cerebral Cavernous Malformations in Murine Models. J Neuroimmune Pharmacol. 2016;11:369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yau ACY, Globisch MA, Onyeogaziri FC, Conze LL, Smith R, Jauhiainen S, Corada M, Orsenigo F, Huang H, Herre M, et al. , Inflammation and neutrophil extracellular traps in cerebral cavernous malformation. Cell Mol Life Sci. 2022;79:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkler EA, Kim CN, Ross JM, Garcia JH, Gil E, Oh I, Chen LQ, Wu D, Catapano JS, Raygor K, et al. , A single-cell atlas of the normal and malformed human brain vasculature. Science. 2022;375:eabi7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Retta SF and Glading AJ. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: Two sides of the same coin. Int J Biochem Cell Biol. 2016;81:254–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Zhou Y, Chen S, Hu Y, Zhu Z, Wang Y, Du N, Song T, Yang Y, Guo A and Wang Y. Macrophage migration inhibitory factor facilitates prostaglandin E2 production of astrocytes to tune inflammatory milieu following spinal cord injury. J Neuroinflammation. 2019;16:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, Couturier CP, Watson BR, Scalisi G, Alkwai S, Rothhammer V, et al. , MAFG-driven astrocytes promote CNS inflammation. Nature. 2020;578:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zlokovic BV, Gottesman RF, Bernstein KE, Seshadri S, McKee A, Snyder H, Greenberg SM, Yaffe K, Schaffer CB, Yuan C, et al. , Vascular contributions to cognitive impairment and dementia (VCID): A report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 2020;16:1714–1733. [DOI] [PubMed] [Google Scholar]
- 42.Boulday G, Rudini N, Maddaluno L, Blecon A, Arnould M, Gaudric A, Chapon F, Adams RH, Dejana E and Tournier-Lasserve E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J Exp Med. 2011;208:1835–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gore AV, Lampugnani MG, Dye L, Dejana E and Weinstein BM. Combinatorial interaction between CCM pathway genes precipitates hemorrhagic stroke. Dis Model Mech. 2008;1:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choquet H, Pawlikowska L, Lawton MT and Kim H. Genetics of cerebral cavernous malformations: current status and future prospects. J Neurosurg Sci. 2015;59:211–20. [PMC free article] [PubMed] [Google Scholar]
- 45.Goitre L, Balzac F, Degani S, Degan P, Marchi S, Pinton P and Retta SF. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS One. 2010;5:e11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiss JM, Downie SA, Lyman WD and Berman JW. Astrocyte-derived monocyte-chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J Immunol. 1998;161:6896–903. [PubMed] [Google Scholar]
- 47.Much CD, Sendtner BS, Schwefel K, Freund E, Bekeschus S, Otto O, Pagenstecher A, Felbor U, Rath M and Spiegler S. Inactivation of Cerebral Cavernous Malformation Genes Results in Accumulation of von Willebrand Factor and Redistribution of Weibel-Palade Bodies in Endothelial Cells. Front Mol Biosci. 2021;8:622547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Girard R, Li Y, Stadnik A, Shenkar R, Hobson N, Romanos S, Srinath A, Moore T, Lightle R, Shkoukani A, et al. , A Roadmap for Developing Plasma Diagnostic and Prognostic Biomarkers of Cerebral Cavernous Angioma With Symptomatic Hemorrhage (CASH). Neurosurgery. 2021;88:686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, Khan N, Nair V, Bajaj N, Sharma M and Ashraf MZ. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc Natl Acad Sci U S A. 2017;114:4763–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heneka MT, McManus RM and Latz E. Author Correction: Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2019;20:187. [DOI] [PubMed] [Google Scholar]
- 51.Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, Aung LHH, Li PF, Yu T and Chu XM. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11:776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhuang T, Liu J, Chen X, Zhang L, Pi J, Sun H, Li L, Bauer R, Wang H, Yu Z, et al. , Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ Res. 2019;125:590–605. [DOI] [PubMed] [Google Scholar]
- 53.Gibson CC, Zhu W, Davis CT, Bowman-Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF, et al. , Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation. 2015;131:289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shenkar R, Shi C, Austin C, Moore T, Lightle R, Cao Y, Zhang L, Wu M, Zeineddine HA, Girard R, et al. , RhoA Kinase Inhibition With Fasudil Versus Simvastatin in Murine Models of Cerebral Cavernous Malformations. Stroke. 2017;48:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, et al. , Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;536:488. [DOI] [PubMed] [Google Scholar]
- 56.Girard R, Zeineddine HA, Koskimaki J, Fam MD, Cao Y, Shi C, Moore T, Lightle R, Stadnik A, Chaudagar K, et al. , Circ Res. 2018;122:1716–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hong T, Xiao X, Ren J, Cui B, Zong Y, Zou J, Kou Z, Jiang N, Meng G, Zeng G, et al. , Somatic MAP3K3 and PIK3CA mutations in sporadic cerebral and spinal cord cavernous malformations. Brain. 2021;144:2648–2658. [DOI] [PubMed] [Google Scholar]
- 58.Peyre M, Miyagishima D, Bielle F, Chapon F, Sierant M, Venot Q, Lerond J, Marijon P, Abi-Jaoude S, Le Van T, et al. , Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations. N Engl J Med. 2021;385:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren AA, Snellings DA, Su YS, Hong CC, Castro M, Tang AT, Detter MR, Hobson N, Girard R, Romanos S, et al. , PIK3CA and CCM mutations fuel cavernomas through a cancer-like mechanism. Nature. 2021;594:271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu D, Zhong Z and Karin M. NF-kappaB: A Double-Edged Sword Controlling Inflammation Biomedicines. 2022;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sugimoto MA, Sousa LP, Pinho V, Perretti M and Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol. 2016;7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galili T, O’Callaghan A, Sidi J, Sievert C. heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Bioinformatics. 2018;34:1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed methods and major resources table can be found in the Supplemental Material. All material, data, and detailed protocols are available upon request, and the RNAseq data used in this study is available in the Gene Expression Omnibus database under GSE204979 (http://www.ncbi.nlm.nih.gov/geo).