

SUMMARY
Successful pancreatic ductal adenocarcinoma (PDAC) immunotherapy necessitates optimization and maintenance of activated effector T cells (Teff). We prospectively collected and applied multi-omic analyses to paired pre- and post-treatment PDAC specimens collected in a platform neoadjuvant study of granulocyte-macrophage colony-stimulating factor-secreting allogeneic PDAC vaccine (GVAX) vaccine ± nivolumab (anti-programmed cell death protein 1 [PD-1]) to uncover sensitivity and resistance mechanisms. We show that GVAX-induced tertiary lymphoid aggregates become immune-regulatory sites in response to GVAX + nivolumab. Higher densities of tumor-associated neutrophils (TANs) following GVAX + nivolumab portend poorer overall survival (OS). Increased T cells expressing CD137 associated with cytotoxic Teff signatures and correlated with increased OS. Bulk and single-cell RNA sequencing found that nivolumab alters CD4+ T cell chemotaxis signaling in association with CD11b+ neutrophil degranulation, and CD8+ T cell expression of CD137 was required for optimal T cell activation. These findings provide insights into PD-1-regulated immune pathways in PDAC that should inform more effective therapeutic combinations that include TAN regulators and T cell activators.
In brief
Li et al. perform multi-omic analyses on pre- and post-treatment specimens from a pancreatic cancer neoadjuvant platform trial, and identify sensitivity and resistance mechanisms associated with anti-PD-1 combination therapy. Results associate tumor-associated neutrophils with poor outcomes but CD137+CD8+ T cells with better outcomes, suggesting treatment strategies for future interventions.
Graphical Abstract
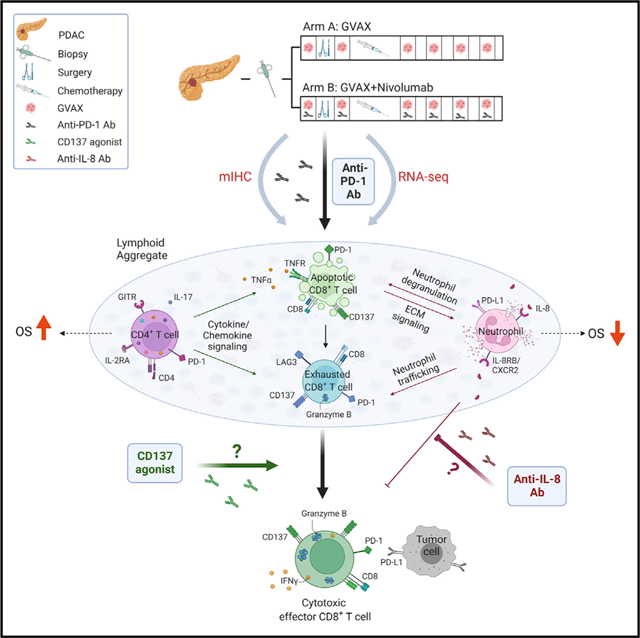
INTRODUCTION
Immune checkpoint inhibitors (ICIs) are a major breakthrough in cancer therapeutics; however, less than 20% of all cancer patients respond to ICIs as single agents (Osipov et al., 2019; Sharma and Allison, 2015). Pancreatic ductal adenocarcinomas (PDACs) are among the prototypic, immunogenically “cold” tumors because they lack natural infiltration of effector T cells (Teffs), the cells that respond to ICIs (Ho et al., 2020). To convert PDACs into ICI-responsive tumors, effective immunotherapy strategies are required to (1) increase antigenicity, (2) enhance Teff function, and (3) overcome T cell excluding and immunosuppressive signals in the tumor microenvironment (TME) (Johnson et al., 2017; Popovic et al., 2018). We have been testing the hypothesis that cancer vaccines are effective in inducing Teffs that can infiltrate PDACs. We reported that our granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting allogeneic PDAC vaccine (GVAX) can induce the formation of tertiary lymphoid aggregates (LAs) in PDACs just 2 weeks following one vaccination (Lutz et al., 2014a, 2014b). Furthermore, programmed death-ligand 1 (PD-L1) expression is induced on both the tumor epithelial cells and myeloid cells within these LAs, suggesting that vaccine therapy may prime PDACs to respond to ICIs (Lutz et al., 2014a, 2014b; Tsujikawa et al., 2017). This concept was subsequently tested in several preclinical and clinical studies (Soares et al., 2015; Tsujikawa et al., 2020). These studies showed that it is possible to turn PDACs into immune- responsive tumors. However, clinical response rates remain low. Thus, a current challenge is to identify additional immune-regulatory signals within the complex PDAC TME that require further modification to effectively enhance Teff function and to optimize and maintain activation of the most potent Teffs.
Analyses of the PDAC TME mostly come from banked bio-specimen collections instead of prospective immunotherapy clinical trials (Danilova et al., 2019; Stromnes et al., 2017). Here, we report the initial data analysis of a neoadjuvant platform study where we continue to add immune agents to those tested in the previous treatment arm guided by the data generated. Prospectively banked paired pre- and post-immunotherapy tumor biospecimens collected from the study allowed for the rigorous, multi-omic analyses of changes in the TME with treatment and the correlation of these TME changes with clinical outcomes.
RESULTS
Nivolumab enhances vaccine-induced CD4+ and CD8+ T lymphocytes and reduces CD4+PD-1+ and CD8+PD-1+ T lymphocyte infiltration into post-treatment PDAC tumors
In this platform clinical trial (NCT02451982), patients with resectable PDAC were randomized to receive GVAX only (arm A) or GVAX plus nivolumab (arm B) as neoadjuvant therapy and subsequently underwent surgical resection 2 weeks after one treatment (Figure S1A). Patients with successful PDAC resection continued to receive the same immunotherapy in addition to standard-of-care adjuvant chemotherapy. A biological endpoint, namely the increase in intratumoral T helper (Th) 17 signals in arm B versus arm A, was chosen as the primary endpoint, as suggested by our prior study (Lutz et al., 2014a, 2014b). Secondary endpoints included safety, disease-free survival (DFS), and overall survival (OS). There was a trend toward improved DFS and OS in arm B versus arm A, although the sample size in this clinical trial was small and only powered to compare intratumoral Th17 signals, not clinical outcomes (Heumann et al., 2022). To investigate changes in tumor-infiltrating immune cells in PDACs after neoadjuvant immunotherapy with GVAX or GVAX + nivolumab, we employed a sequential staining and stripping multiplex immunohistochemistry (mIHC) technique (Tsujikawa et al., 2017). Specimens were collected from 34 consecutive patients (Figure S1B; Tables S1 and S2). Among them, 19 patients whose post-treatment surgically resected tumor specimens met the criteria for mIHC staining and analysis and who were followed for OS for at least 2 years (Table S1) were included in this study. Pre-treatment tumor biopsy cores (Figures S2A and S2B) and post-treatment resected tumors were collected (Figures S2C–S2E). Tumor-infiltrating tertiary LAs, which were not present in pre-treatment tumors and were induced by vaccine therapy as previously described (Lutz et al., 2014a, 2014b; Tsujikawa et al., 2017), were identified by peripheral node addressin (PNAd)-marked high endothelial venules (HEVs) on all 19 post-treatment tumors (representative images in Figure S2C). Slides were stained by mIHC with a panel of myeloid and lymphoid cell markers (Table S3). The regions of interest (ROIs) were selected within the post-treatment surgically resected tumor areas that contain LAs and divided into the areas of LAs and the tumor areas outside LAs (designated non-LA tumor areas) (Figures 1A–1C). A minimum of three ROIs in the tumor areas that each contained at least one LA were selected from each post-treatment tumor (Figures S2D and S2E). The ROIs were also selected to cover minimally three different tumor cores from the same pre-treatment biopsy (Figures 1D, 1E, S2A, and S2B). A combination of markers was used to identify each immune subtype (Table S4). Epithelial cellular adhesion molecule (EpCAM) staining was used to help identify tumor epithelia. Interestingly, we found that EpCAM is expressed on the extracellular matrix (ECM) surrounding LAs. Notably, EpCAM can be detected on the surface of extracellular vesicles derived from PDACs (Amrollahi et al., 2019). The density of each immune cell subtype was calculated as the percentage of all cells within ROIs. The comparison of immune cell densities in pre-treatment biopsies between the two treatment arms (Figure S2F) did not show a significant difference, although this comparison may be limited by small sample sizes.
Figure 1. Multiplex immunohistochemistry of PDACs before and after immunotherapy.
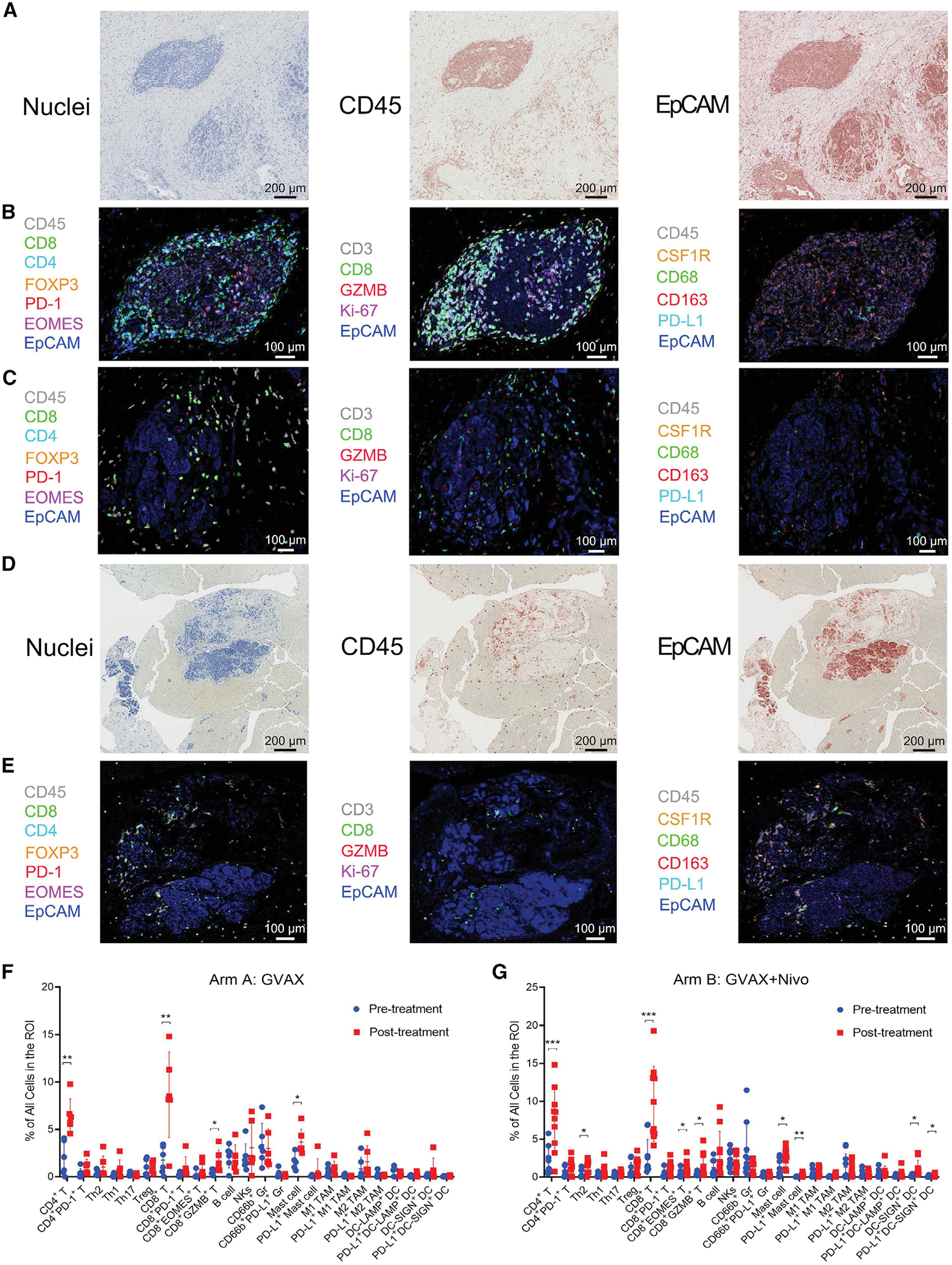
(A) ROIs were selected within the post-treatment surgical resected tumor areas. CD45 and EpCAM staining were used to identify the lymphoid cells and tumor epithelia, respectively. All scale bars, 200 μm.
(B and C) Overlaid images of representative markers assigned with pseudocolors in one representative LA (B) and one representative non-LA tumor area (C) in post-treatment tumors. All scale bars, 100 μm.
(D) CD45 and EpCAM staining of one representative ROI from a pre-treatment biopsy tumor area. All scale bars, 200 μm.
(E) Overlaid images of one representative ROI of pre-treatment biopsy. All scale bars, 100 μm.
(F and G) Summary of the density of all immune cell subtypes analyzed as indicated in the paired pre-treatment versus post-treatment non-LA tumor areas from the same arm A patients (F, n = 6) and arm B patients (G, n = 10). Data shown as the mean ± SD; comparison by paired t test; *p < 0.05, **p < 0.01, ***p < 0.001; all others, not significant. See also Figures S1, S2, Tables S1, S2, S3, and S4.
Because pre-treatment biopsy specimens did not contain LAs, we compared non-LA tumor areas between matched pre- and post-treatment tumor specimens to assess treatment-induced changes. In summary, we observed an increase in many immune cell subsets infiltrating the non-LA tumor areas outside the organized LAs in post-treatment tumors when compared with each patient’s pre-treatment biopsy in both treatment arms (Figures 1F and 1G). These results demonstrate that vaccine therapy not only induces the formation of LAs but also induces intratumoral infiltration of immune cells. Moreover, we observed a significantly higher density of CD4+ and CD8+ T cells in the post-treatment LAs in arm B versus arm A (Figure 2A). This result suggested that ICI treatment enhances Teff infiltration if T cells have been induced by a coadministered vaccine. In addition, we observed a significantly lower density of post-treatment CD4+programmed cell death protein 1 (PD-1)+ and CD8+PD-1+ T cells in LAs from tumors in arm B compared with arm A (Figure 2A), likely due to an immune response to nivolumab. There was a significant increase in Th17 cells in non-LA tumor areas from patients treated in arm B compared with arm A, supporting the primary endpoint of this trial (Figure 2B).
Figure 2. Multiplex IHC of CD8+ or CD4+ T subtypes and their correlation with OS.
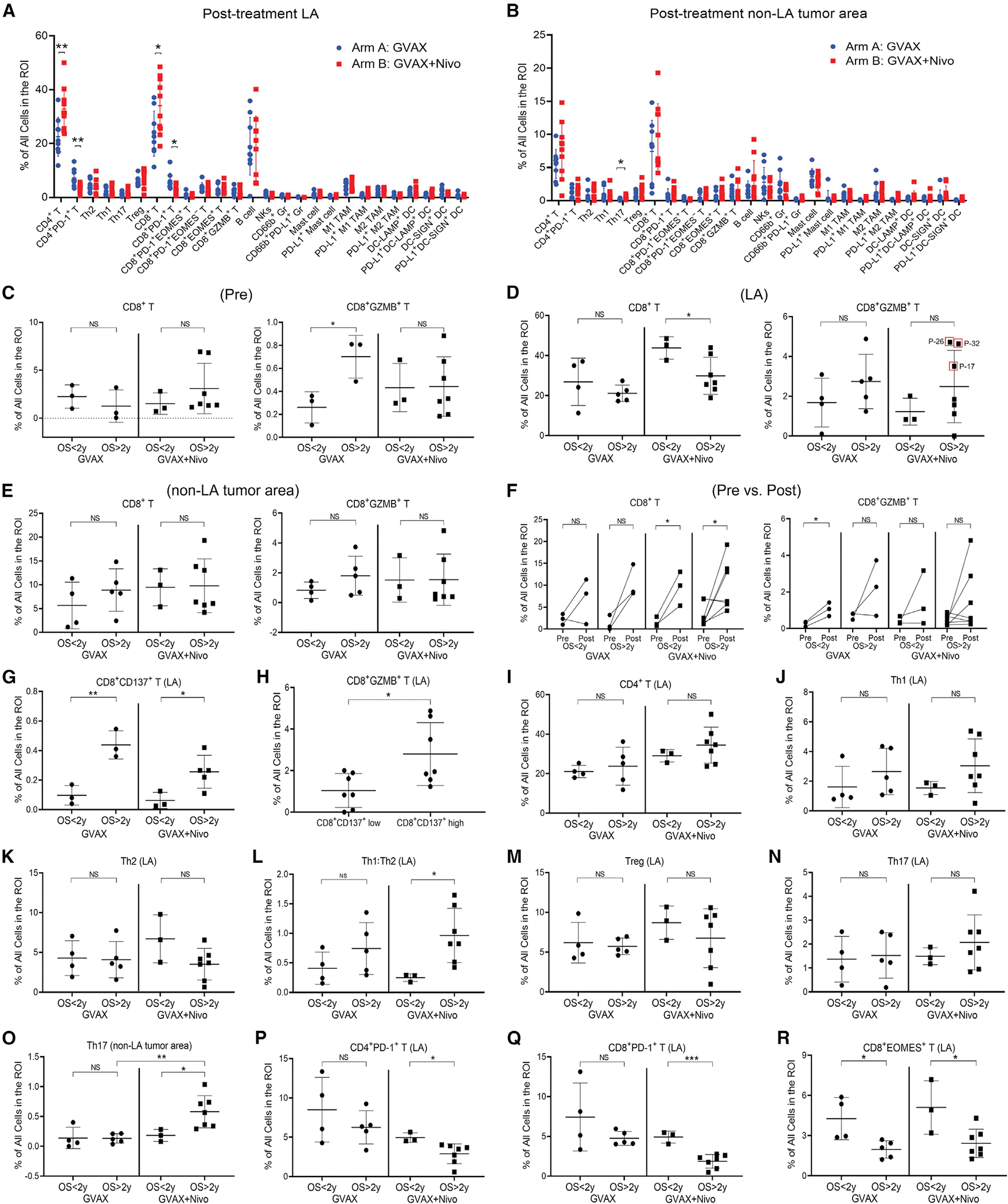
(A and B) Summary of the density of all immune cell subtypes analyzed as indicated in LA (A) and non-LA tumor areas (B) in arm A (n =9) versus arm B (n=10) patients.
(C–E) Correlation between OS and CD8+ and CD8+GZMB+ T cells in pre-treatment tumor biopsies (C, pre, n = 6 for GVAX and n = 10 for GVAX + Nivo) and in LA (D) and non-LA tumor areas (E) from post-treatment tumors (see sample numbers in A and B). Three cases with the highest densities of CD8+GZMB+ T cells and the longest OS are circled.
(F) Changes in the density of CD8+ or CD8+GZMB+ T cells between pre-treatment (pre) and matched post-treatment non-LA tumor areas (post) and their correlation with OS.
(G) Correlation between OS and CD8+CD137+ T cells in LA.
(H) Correlation between CD8+GZMB+ T cells and CD8+CD137+ T cells in LA. Tumors are subgrouped by higher versus lower density of CD8+CD137+ T cells in LA.
(I–R) Correlation between OS and CD4+ T cells (I), Th1 (J), Th2 (K), Th1:Th2 ratio (L), Treg (M), and Th17 (N) in LA, Th17 in non-LA tumor area (O), and CD4+PD-1+ T cells (P), CD8+PD-1+ T cells (Q), and CD8+EOMES+ T cells (R) in LA. All data shown as the mean ± SD; all comparisons by t test; *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant; all others, not significant. See also Figures S3 and S4.
An increase in a diverse intratumoral CD8+ T cell population occurs with combination vaccine + nivolumab compared with vaccine alone
To understand the response and resistance mechanisms elicited by ICIs in PDACs, we examined each subtype of T cells and myeloid cells and their correlation with OS. OS > 2 years is considered a standard clinical benefit outcome for PDAC patients following surgical resection in published correlative studies (Tsujikawa et al., 2017). A higher infiltration of CD8+GZMB+ T cells in the pre-treatment biopsies in arm A, but not arm B, was associated with improved survival (Figure 2C). However, quantification of low numbers of CD8+GZMB+ T cells in pre-treatment biopsies could be prone to large variations (Figure S3A). Therefore, it is also possible that this finding could be due to the small sample size of available pre-treatment biopsy specimens in arm A. Interestingly, a lower density of general CD8+ T cells, possibly due to a decrease in the CD8+PD-1+ T cell subtype, in the post-treatment LAs (Figure 2D), but not in non-LA tumor areas (Figure 2E), was associated with a significantly greater OS in arm B. The densities of CD8+ and CD8+GZMB+ T cells calculated as a percentage of CD45+ cells showed a similar trend (Figures S3B and S3C). However, anti-PD-1 therapy did not appear to alter other immune-effector cells, such as B cells and natural killer (NK) cells, in vaccine-treated PDAC patients (Figures S3D–S3G).
We next compared changes in CD8+ and CD8+GZMB+ T cell infiltration in available pre-treatment biopsies and matched post-treatment non-LA tumor areas and correlated these data with OS (Figure 2F). There was a statistically significant increase in the infiltration of CD8+GZMB+ T cells in three arm A patients with OS < 2 years (Figure 2F). However, it should be noted that they had the fewest CD8+GZMB+ T cells overall. Although the combination of GVAX + nivolumab did not result in a significant increase in CD8+GZMB+ T cells in LAs in association with improved OS, the three patients who had the highest numbers of CD8+GZMB+ T cells in their LAs had the longest OS among all the patients (Figure 2D).
We recently reported that the anti-CD137T cell agonist antibody given in combination with GVAX and anti-PD-1 antibody significantly enhanced survival in a murine PDAC orthotopic model (Muth et al., 2020). To determine the clinical relevance of these findings, we examined the density of CD3+CD8+CD137+ T cells and observed that a higher density of CD3+CD8+CD137+ T cells in post-treatment LAs was associated with OS > 2 years in both treatment arms (all p < 0.05) (Figure 2G). We further examined whether this CD137+ T cell subset in LAs was associated with the cytotoxic Teff signature and found that a higher density of CD3+CD8+CD137+ T cells was significantly (p = 0.019) associated with a higher density of CD8+GZMB+ T cells in post-treatment LAs (Figure 2H). Notably, CD3+CD8+CD137+ T cells were usually of low density and resided almost exclusively in LAs; therefore, we did not analyze their density in non-LA tumor areas. This result also suggested that this Teff subtype may need to be further expanded and mobilized by CD137 agonist treatment to generate a stronger antitumor response, as anti-CD137 agonist antibodies were shown to enhance the proliferation, memory, and activation status of Teffs (Sanchez-Paulete et al., 2016).
An increase in the Th1:Th2 CD4+ T cell ratio and in the Th17 cell density correlates with improved OS in patients treated with vaccine + nivolumab
We next evaluated the infiltration of CD4+ T cells in the post-treatment LAs and non-LA tumor areas and correlated them with OS. The overall density of CD4+ T cells and densities of the specific CD4+ T cell subtypes including Th1, Th2, Th17, and Tregs failed to significantly correlate with OS in either arm (Figures 2I–2N). However, a higher Th1 to Th2 (Th1:Th2) ratio was significantly associated with greater OS in arm B but not in arm A (Figure 2L), suggesting a role for anti-PD-1 therapy in promoting antitumor Th cell activity. We also observed a trend toward increased Th17 cells in LAs in patients with OS > 2 years compared with those with OS < 2 years (Figure 2N) and a statistically significant increase in Th17 cells in post-treatment non-LA tumor areas in patients with OS > 2 years versus OS < 2 years, suggesting that anti-PD-1 therapy expanded Th17 cells in patients with improved OS and that this expansion was not limited to the LAs (Figure 2O). We did not observe any other anti-PD-1 therapy-specific correlations between OS and CD4+ T cells and their subtypes in the pre-treatment tumors or in the treatment-induced changes (Figures S3H–S3V). Collectively, our results suggested that the Th1:Th2 ratio and Th17 density in the post-treatment tumors may predict longer OS following combination treatment with GVAX and anti-PD-1 antibody.
Anti-PD-1 therapy reduces LA-residing PD-1+CD4+ and CD8+ T cells in association with the improved OS but does not have a differential effect on EOMES+ T cell populations compared with vaccine therapy alone
We next examined whether the anti-PD-1 therapy-associated decreases in CD8+PD-1+ and CD4+PD-1+ T cells in the LAs correlated with patient survival. We found that CD8+PD-1+ and CD4+PD-1+ T cells in LAs were significantly decreased in patients with OS > 2 years compared with those with OS < 2 years only in arm B, but not in arm A, indicating that CD8+PD-1+ and CD4+PD-1+ T cells are potential outcome predictors for anti-PD1 therapy (Figures 2P and 2Q). In contrast, the Eomesodermin (EOMES)+ subgroup of CD8+ T cells was significantly decreased in patients with OS > 2 years in both arm A and arm B (Figure 2R). EOMES expression in T cells has a bimodal expression pattern, elevated in early activated T cells and exhausted T cells but absent in effector memory T cells (Li et al., 2018). Our results support an exhausted phenotype of CD8+EOMES+ T cells in post-treatment LAs as a potential resistance mechanism following treatment with either GVAX or GVAX + nivolumab.
Next, we examined additional subgroups of CD8+PD-1+ T cells, including CD8+PD-1+EOMES+ and CD8+PD-1+EOMES− T cells, to further understand which vaccine-induced T cell subsets respond to anti-PD-1 therapy. Comparing arm B with arm A, within the total CD8+ T cell population, only the percentage of CD8+PD-1+EOMES− T cells decreased significantly (p < 0.01; Figures S4A and S4B), suggesting that EOMES is not the major T cell exhaustion mechanism responding to anti-PD-1 therapy and that other T cell exhaustion mechanisms, including LAG3 and TIM3 signaling (Goldberg and Drake, 2011; Wolf et al., 2020), warrant further investigation. Importantly, analysis of the baseline densities and changes in densities between pre- and post-treatment of the CD8+PD-1+, CD4+PD-1+, and CD8+EOMES+ T cell populations infiltrating tumor areas did not correlate with survival in either treatment arms(Figures S4C–S4I). These data further suggest that anti-PD-1 therapy mainly targets PD-1+ T cells in the LAs, which are sites of T cell activation and regulation.
Changes in both TAM and TAN populations correlate with OS following vaccine + anti-PD-1 therapy
We also examined changes in myeloid cell populations following vaccine plus anti-PD-1 therapy, since they are known cellular regulators of T cell responses in PDACs. We did not observe any association between OS and the density of M1-like tumor-associated macrophages (TAMs), M2-like TAMs, or PD-L1+ TAMs in the pre-treatment tumor biopsies (Figures S5A–S5D). However, a higher density of M2-like TAMs in the post-treatment LAs was associated with poor OS in both arms A and B. In addition, a higher ratio of M1-like to M2-like TAMs was associated with improved OS, but this ratio only reached statistical significance for arm B (Figure 3A). In contrast, changes in the density of TAMs in non-LA tumor areas were not associated with OS (Figure 3B). The selective changes in TAMs residing in LAs relative to non-LA tumor areas provide additional evidence that LAs are important sites of immune modulation in PDAC tumors.
Figure 3. Multiplex IHC analysis of TAM and TAN.
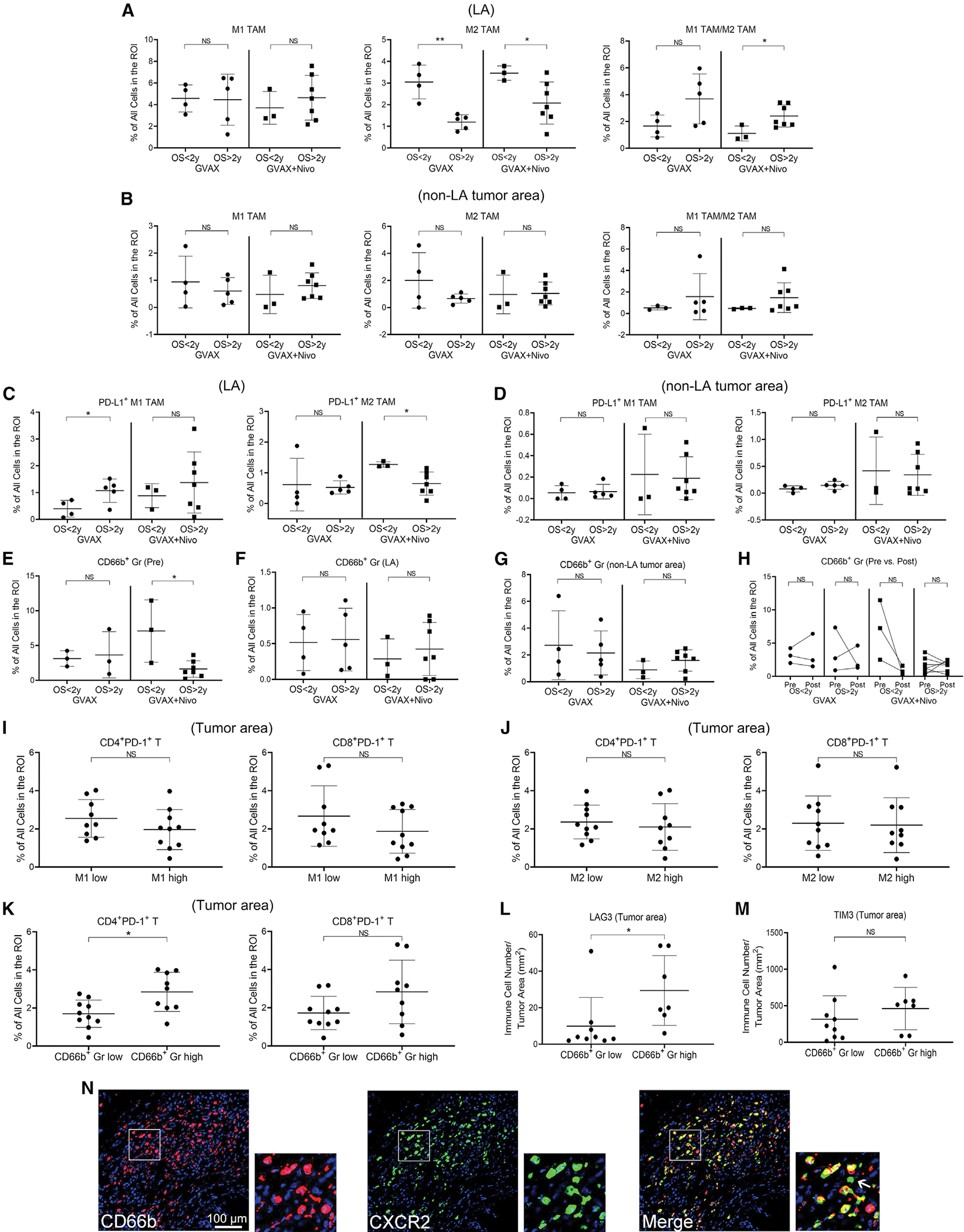
(A–G) Correlation between OS and M1-like TAM, M2-like TAM and the ratio of M1- and M2-like TAM in LA (A) and non-LA tumor area (B), PD-L1+ M1-like and M2-like TAM in LA (C) and non-LA tumor area (D), TAN (CD66b+ Gr) in pre-treatment biopsy (E, pre) and in post-treatment LA (F), and non-LA tumor area (G).
(H) Changes in TANs between pre-treatment (pre) and matched post-treatment non-LA tumor areas (post) and their correlation with OS. Sample numbers (A–H) are the same as in Figure 2.
(I–K) Correlation between CD8+PD-1+ T cells and CD4+PD-1+ T cells, as indicated, and myeloid cell densities in post-treatment tumor areas. Tumors (n = 19) subgrouped by higher versus lower density of M1-like TAM (I), M2-like TAM (J), or CD66b+ TAN (K), respectively, in LA.
(L and M) Correlation between LAG3+ cells (L) and TIM3+ cells (M), respectively, and TANs in post-treatment tumor areas. Tumors subgrouped by higher versus lower density of TANs in LA.
(N) mIHC images of CD66b and CXCR2, as indicated, and their overlaid image. All scale bars, 100 μm. Pseudocolors assigned by the Halo software. A representative LA shown; one square region enlarged and also shown. Arrow indicates the only CXCR2+ cell that was not CD66b+ within this region. All data shown as the mean ± SD; all comparisons made by t test; *p < 0.05; **p < 0.01; NS, not significant. See also Figure S5.
Next, we examined PD-L1+ TAMs. A higher density of LA-residing PD-L1+ M1-like TAMs was associated with longer OS in arm A (Figure 3C), confirming our previous reports (Tsujikawa et al., 2017). Nevertheless, vaccine-induced PD-L1 expression in TAMs may counteract the antitumor activity of Teff and, thus, limit the efficacy of vaccine therapy. Notably, a lower density of LA-residing PD-L1+ M2-like TAMs was associated with improved OS in arm B (Figure 3C). However, this correlation was not observed in non-LA tumor areas (Figure 3D). We also investigated mast cells, which are anticipated to respond to GM-CSF expression from the GVAX vaccine and did not observe any strong association between the density of mast cells or PD-L1+ mast cells and OS (Figures S5E–S5L).
We then examined tumor-associated neutrophils (TANs) marked by CD3−CD66b+ and found that the density of TANs was higher than that of TAMs in pre-treatment biopsies (Figure S2F), suggesting that neutrophils may play a more prominent role than TAMs in regulating immunity in PDAC TMEs. This possibility was supported by the finding that a higher density of TANs seen in pre-treatment biopsies was associated with a significant decrease in OS (Figures 3E–3H) in patients treated with GVAX + nivolumab. In contrast, TAN densities in post-treatment LAs or non-LA tumor areas did not appear to have an impact on OS, likely because the treatment may have changed the densities, distribution, and/or function of TANs. We also did not observe a correlation between PD-L1+ TANs and OS in either treatment arm (Figures S5M–S5P), suggesting that TANs themselves were not targets of anti-PD-1 therapy but potentially a modulatory mechanism of response to anti-PD-1 therapy. The densities of myeloid cell subtypes, including TANs, calculated as a percentage of CD45+ cells showed a similar trend (Figures S5Q–S5V). These results suggest that TANs in pre-treatment PDACs may influence the survival outcome following treatment with GVAX and anti-PD-1 antibody.
We next tested the hypothesis that a change in TANs in the post-treatment LAs, an immunoregulatory site, would be associated with different T cell functions. We found that higher density of TANs in post-treatment LAs was significantly (p = 0.011) associated with higher densities of tumor-infiltrating CD4+PD-1+ T cells and, in a near-significant trend (p = 0.081), with higher densities of CD8+PD-1+ T cells (Figures 3I–3K) in the whole tumor areas. Here, we combined cases from both treatment arms together to increase the sample size for this analysis. We performed additional immunohistochemistry (IHC) staining of T cell exhaustion markers (Figure S5W) and found that a higher density of TANs in post-treatment LAs was associated with a higher density of tumor-infiltrating LAG3+ T cells (p = 0.041) (Figure 3L). TIM3+ and EOMES+ T cell changes (Figures 3M and S5X, respectively) did not correlate with TAN density; thus, modulation of TANs may promote reinvigoration of exhausted T cell populations through LAG3.
To identify a potential therapeutic target on TANs, we examined the expression of interleukin (IL)-8RB/CXC chemokine receptor 2 (CXCR2) on TANs by IHC because IL-8RB/CXCR2 and its ligand, IL-8, are therapeutic targets being studied in cancer immunotherapy trials (Teijeira et al., 2021). The results showed that the majority of CD66b+ TANs expressed CXCR2 and that CD66b+ TANs are responsible for most of the CXCR2 expression in post-treatment tumors (Figure 3N), supporting the future development of IL-8/CXCR2-based immunotherapy for targeting TANs in PDAC.
Tumor-immune cell spatial relationships are influenced by CD8+GZMB+ T cells and TANs residing in non-LA tumor areas following GVAX + nivolumab therapy
To exert immune-effector or immunosuppressive functions, immune cells need spatial proximity to their target cells. We and others have shown that spatial distances between specific TME cell populations can inform therapeutic outcomes (Berry et al., 2021; Davis-Marcisak et al., 2021; Obradovic et al., 2021). We therefore measured the spatial distance between several key immune cells and their target cells. The ROIs from the above mIHC images were reprocessed by Halo software (Figure 4A), and the distances between epithelial tumor cells marked by EpCAM, CD8+ T cells, and myeloid cells marked by colony stimulating factor 1 receptor (CSF1R) were measured (Figure 4B). There was no statistically significant difference in distance measurements between these populations in different treatment arms or when correlated with OS (Figure S6A). The density of CD8+ T cells in LAs also did not significantly alter the distances between tumor cells, CD8+ T cells, and CSF1R+ myeloid cells (Figure 4C). Interestingly, a lower density of CD8+PD-1+ T cells in LAs, which appeared to be associated with anti-PD-1 treatment and with longer OS (Figures 2A and 2Q), was associated with longer distances between PD-1−CD8+ T cells and tumor cells (Figure 4D). Consistently, in tumors from patients treated with GVAX + nivolumab, the distance between CD8+ T cells, regardless of their PD-1 expression, and tumor cells was significantly increased in tumors with a higher versus lower density of CD8+GZMB+ T cells in LAs (Figure 4E). This result should not be attributed to the density of CD8+GZMB+ T cells themselves, which was a small subset of CD8+ T cells in LAs. Therefore, CD8+GZMB+ T cells in LAs likely influence the immune-regulating role of LAs but are not effectors that directly kill tumors. It is possible that, despite anti-PD-1 treatment, LAs containing a higher density of CD8+GZMB+ T cells and/or lower density of CD8+PD-1+ T cells are still not sufficient to fully activate CD8+ T cells with optimal effector function to kill tumor cells. Nevertheless, the distance from PD-1+CD8+ T cells to PD-L1+CSF1R+ myeloid cells in tumors with higher densities of CD8+GZMB+ T cells (p = 0.041) (Figure 4E) in LAs became significantly longer, likely due to nivolumab blockade of the PD-1/PD-L1 pathway. It will be interesting in the future to examine whether the CD8+ T cells that remained distant from tumor cells with higher densities of CD8+GZMB+ T cells and/or lower density of CD8+PD-1+ T cells in LAs have a higher potential to be activated when treated with additional T cell-activating agents such as CD137 agonists.
Figure 4. Spatial relationship between tumor cells and immune cells.
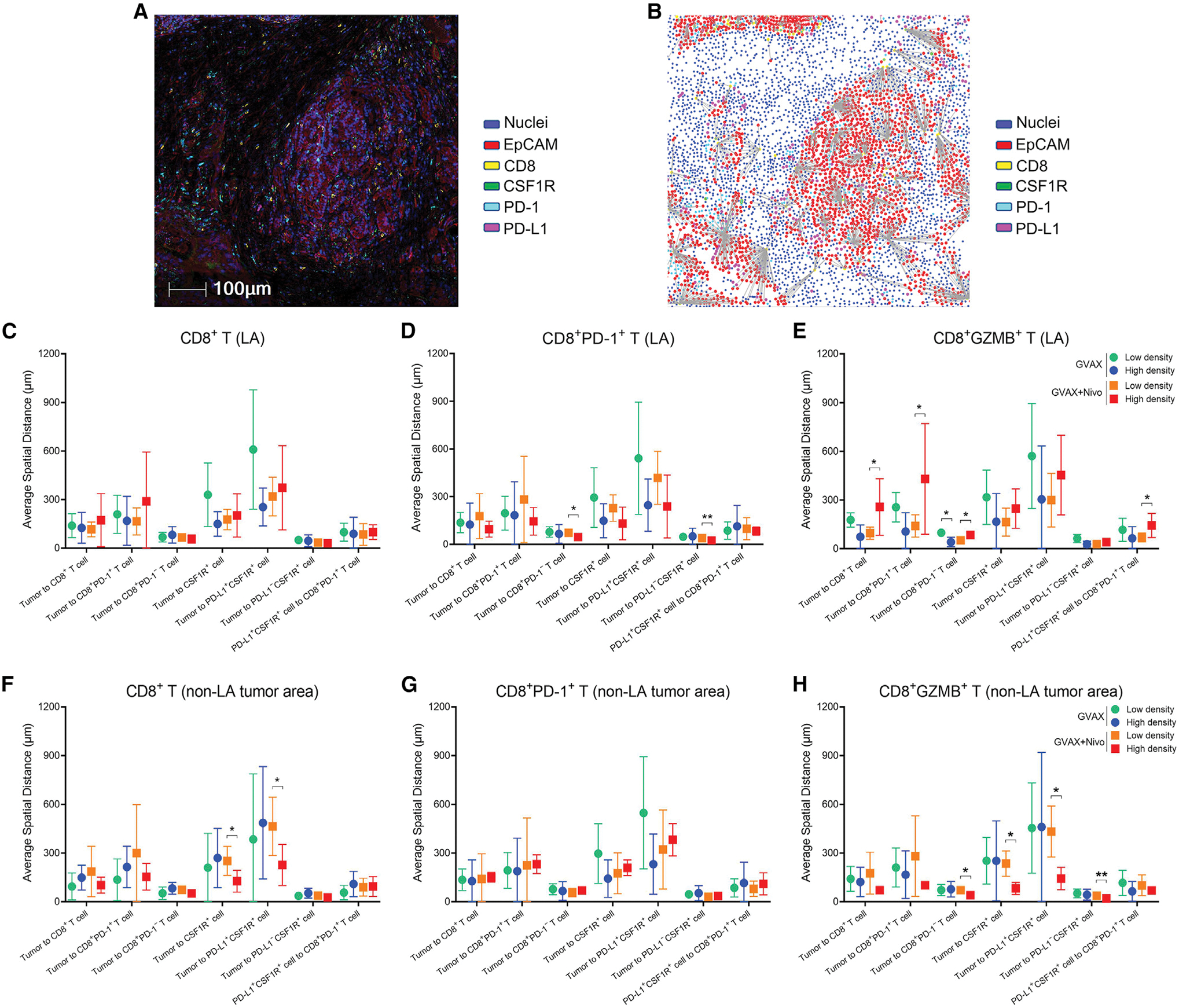
(A) A representative region from six-marker mIHC images integrated by Halo software. Scale bar, 100 μm.
(B) Distance measurement schema of a representative region. Positive signals (exemplified by the EpCAM staining signals) and the nearest neighbor cells (exemplified by the nearest CD8+ cells) are connected by lines whose lengths are measured as the distances.
(C–H) Tumors were subgrouped by higher versus lower density of CD8+ T cells (C and F), CD8+PD-1+ T cells (D and G), and CD8+GZMB+ T cells (E and H) in LA (C–E) and non-LA tumor areas (F–H). Distances from one cell type to another cell type were compared by t test between subgroups (high versus low density) in two cohorts of tumors treated with GVAX and GVAX + nivolumab, respectively. Tumor cells marked by EpCAM staining and identified by pathologists. mIHC images qualified for distance measurement: n = 7 for GVAX and n = 10 for GVAX + Nivo. All data shown as the mean ± SD; *p < 0.05; **p < 0.01; all others, not significant. See also Figure S6.
The density of general CD8+ T cells, but not that of CD8+PD-1+ T cells, in non-LA tumor areas also demonstrated a correlation with the distance between CSF1R+ myeloid cells and tumor cells (Figures 4F and 4G). Interestingly, in tumors from patients treated with GVAX + nivolumab, the distance between CD8+ T cells, particularly PD-1−CD8+ T cells, and tumor cells became significantly shorter in those with higher versus lower densities of CD8+GZMB+ T cells (p = 0.024) in non-LA tumor areas (Figure 4H). This result provides additional evidence that anti-PD-1-treated CD8+GZMB+ T cells in non-LA tumor areas facilitate access of Teffs to tumor cells.
Although the analysis was limited by the unavailability of pre-treatment biopsy specimens from some patients, the densities of CD8+ T cells, their subsets, and myeloid cell subtypes in the pre-treatment tumor areas did not appear to influence spatial relationships in post-treatment tumors (Figures S6B and S6C). However, a noteworthy finding from the remaining distance measurements (Figures S6D–S6G) was the trend of the association of higher densities of TANs in LAs and lower densities of TANs in non-LA tumor areas with shorter distances from CD8+ T cells or myeloid cells to tumor cells in post-treatment tumors. This result supports further exploration of TANs as a surrogate predictor of response and a therapeutic target to improve treatment outcomes with anti-PD-1 therapy-based combinations in PDAC patients.
The densities of TANs and CD8+GZMB+ T cells are outcome predictors of anti-PD-1 therapeutic responses in PDAC patients
We next examined the strength of immune markers in predicting outcomes using the Bayesian information criterion (BIC) as described (Claeskens and Hjort, 2008). We included two types of outcomes: survival measured by OS and DFS and immune cell distances (Table 1). Immune cell distances were considered biological outcomes that were influenced by the densities of infiltrating immune cells (Figure 4). To select a myeloid cell marker that best predicts the response to anti-PD-1 therapy and serves as a therapeutic target for further enhancing immune responses, we used BIC to compare baseline CD66b+ TANs, M1-like TAMs, and M2-like TAMs in pre-treatment tumors. We found that TANs had a small and potentially relevant BIC score to support its prediction of OS in patients treated with GVAX + nivolumab.
Table 1.
Comparison of models based on the BIC method
| Pre-treatment tumor area |
Post-treatment LA |
|||||
|---|---|---|---|---|---|---|
| CD66b+Gr (TAN) | M1 TAM | M2 TAM | CD8+ T cell | CD8+GZMB+ T cell | ||
|
| ||||||
| GVAX + Nivo Arm | survival (day) | |||||
| OS | 10.16b | 16.51 | 16.19 | 11.71a | 15.21 | |
| DFS | 12.40 | 12.93 | 14.10 | 14.53 | 14.61 | |
| distance (μm) | ||||||
| tumor to CD8+ cell | 129.34 | 129.17 | 129.36 | 128.87 | 123.73b | |
| tumor to CD8+PD-1+ cell | 141.94 | 141.87 | 142.02 | 141.22 | 136.96b | |
| tumor to CD8+PD-1− cell | 95.43 | 93.73 | 94.15 | 94.94 | 87.93b | |
| tumor to CSF1R+ cell | 125.93 | 124.72 | 125.97 | 126.04 | 124.43 | |
| tumor to PD-L1+CSF1R+ cell | 139.21 | 137.86 | 139.20 | 139.25 | 137.85 | |
| tumor to PD-L1−CSF1R+ cell | 79.02 | 79.50 | 79.47 | 79.12 | 75.49a | |
| PD-L1+CSF1R+ cell to CD8+PD-1+ cell | 114.26 | 113.34 | 112.57 | 114.11 | 108.80b | |
| GVAX Arm | survival (day) | |||||
| OS | 6.55 | 6.91 | 6.64 | 8.74a | 12.99 | |
| DFS | 7.12 | 6.43 | 6.65 | 11.04 | 9.23 | |
| distance (μm) | ||||||
| tumor to CD8+ cell | 49.21 | 49.21 | 49.21 | 85.23 | 80.03b | |
| tumor to CD8+PD-1+ cell | 52.88 | 52.88 | 52.88 | 91.72 | 88.03a | |
| tumor to CD8+PD-1− cell | 44.14 | 44.14 | 44.14 | 74.82 | 66.83b | |
| tumor to CSF1R+ cell | 47.38 | 47.38 | 47.38 | 94.36 | 95.24 | |
| tumor to PD-L1+CSF1R+ cell | 50.85 | 50.85 | 50.85 | 102.98 | 104.36 | |
| tumor to PD-L1−CSF1R+ cell | 41.56 | 41.56 | 41.56 | 70.91 | 66.81a | |
| PD-L1+CSF1R+ cell to CD8+PD-1+ cell | 49.77 | 49.77 | 49.77 | 84.28 | 83.14 | |
LA, lymphoid aggregate; OS, overall survival; DFS, disease-free survival; TAN, tumor-associated neutrophil; M1 TAM, M1-like tumor-associated macrophage; M2 TAM, M2-like tumor-associated macrophage.
Considered to have a meaningful difference.
Considered to have a strong difference.
We next selected a Teff marker in LAs that may best predict outcomes to anti-PD-1 immunotherapy. As a result, the density of CD8+GZMB+ T cells had a lower BIC score than the total CD8+ T cells and is therefore more likely to predict distances between tumor cells and multiple immune cell subtypes in both treatment arms (Table 1). In summary, this model selection analysis supports further investigation of TANs and Teffs as regulators of the immune response in PDACs.
RNA-seq analysis of TILs demonstrated reduced naïve CD4+ T cells following nivolumab treatment, and CD8+ TCR clonal expansion was associated with longer OS
The above mIHC analyses showed that cytotoxic Teffs expressing GZMB in some PDACs following GVAX or GVAX + nivolumab treatment were associated with longer OS. We leveraged sequencing data to further delineate the regulatory mechanisms underlying these T cell responses. Whole-exome sequencing (WES) of biopsied tumors from the 19 patients showed a common mutation profile (Figures S7A–S7C; Table S5) and generally low tumor mutation burden (TMB) (Figures S7D and S7E). There were no statistically significant differences observed in TMB or predicted neoantigen load (Figures S7D–S7G, S8A, and S8B) between treatment arms, between tumors with different survival outcomes, or between tumors with different densities of immune infiltrates in LAs, confirming that genetics does not explain the observed differential therapeutic responses in either treatment arm.
Next, we performed whole-transcriptome RNA sequencing (RNA-seq) on tumor-infiltrating leukocytes (TILs) isolated from the post-treatment, surgically resected PDACs of nine patients treated with GVAX and eight patients treated with GVAX and nivolumab. These cells were sorted by flow cytometry into four immune cell subtypes: CD3+CD4+, CD3+CD8+, CD3−CD11b+, and CD3−CD11b−CD19+ cells prior to RNA-seq analysis. Of the 68 samples sequenced, 46 passed quality control metrics as described in STAR Methods and were retained for analysis (Figure S1). To characterize cellular pathways, we first used RNA-seq data to predict cell type composition based upon gene expression signatures with CIBERSORTx in the samples from all cell populations (Newman et al., 2019). This analysis estimated the proportion of 22 immune cell subtypes in each of the samples (Figure 5A). Generally, the cell type composition estimated from the CIBERSORTx analysis of RNA-seq data was consistent with the flow cytometry-based sorting. Nonetheless, we observed the presence of small subpopulations of additional cell types that we attributed to imperfect flow cytometry sorting. Therefore, we compared changes in cellular abundances between treatment arms and OS for the cellular populations that were consistent with subtypes of the sorted cells for each marker protein.
Figure 5. Transcriptomic profiling of tumor-infiltrating immune cells in PDACs treated with GVAX or GVAX + nivolumab.

(A) Cibersort heatmap of immune cell subtype composition profiling sorted CD19+ (n = 10), CD4+ (n = 13), CD8+ (n = 13), and CD11b+ (n = 10) TILs. Treatment arms, OS, and sorted cell types indicated by various colors.
(B–D) The proportion of immune cell subtypes profiled by Cibersort was compared between treatment arms by t test. Boxplots display minimum and maximum values (whiskers), interquantile range (box) with median, and outliers.
(E–J) TCR and BCR clonality predicted from RNA-seq of sorted CD4+ (E and H), CD8+ (F and I), and CD19+ cells (G and J) were compared between treatment arms (E–G) and between OS > 2 years and OS < 2 years cohorts (H–J) by t test. Data shown as the mean ± SD. Cases with follow-up < 2 years shown separately. See also Figures S7, S8, and Table S5.
Comparing CIBERSORTx-estimated cellular abundances of immune cell subtypes between trial arms, we observed a nonsignificant decrease in naive CD4+ T cells (p = 0.061; Figure 5B) but a statistically significant decrease in CD8+ T cells (p = 0.047; Figure 5C) in arm B. Additionally, in arm B, we observed increased monocytes (p = 0.041; Figure 5D) but no significant changes between treatment arms in CD19+ cells (Figure 5A). Unlike the comparison between treatment arms, comparisons of the abundances of the cell subtypes associated with the sorted cell populations failed to associate with OS.
Prediction methods for T cell receptor (TCR) and B cell receptor (BCR) sequences from bulk RNA-seq data can enable further analysis of cancer-specific functional activity in each cell type. We compared changes in TCR and BCR clonality by treatment and survival from predictions of repertoires in the CD4+, CD8+, and CD19+ RNA-seq data using MiXCR (Bolotin et al., 2015). In this analysis, no significant changes in clonality were observed in intratumoral CD4+ T cells, CD8+ T cells, or B cells following each treatment when comparing the two arms (Figures 5E–5G). However, we observed a statistically significant increase in CD8+ T cell clonality (p = 0.027), but not CD4+ T cell or B cell clonality, in tumors associated with OS > 2 years (Figures 5H–5J). These results suggested that adding anti-PD-1 therapy to GVAX increased the immunogenic activity in CD4+ T cells but not in CD8+ T cells, whereas CD8+ T cell clonal expansion and cytotoxic activity were required for improved survival outcomes. These findings are consistent with the insufficient activation of Teffs through anti-PD-1 therapy in PDAC (Figures 2D and 2G) and support the need to combine additional immune-modulating agents to improve patient outcomes to immunotherapy.
Transcriptional changes from nivolumab therapy in CD4+ T cell chemokine signaling that link neutrophil degranulation and subsequent ECM remodeling to the inhibition of activated CD8+ T cell motility and function
To further characterize molecular pathways that may inform the mechanisms that underlie the observed cellular changes that are associated with longer OS, we performed differential expression analysis on each of the TIL subtypes (Figures 6A–6F; Table S6) and identified significantly differentially expressed genes (log2 fold change >0.5 or < −0.5 and false discovery rate [FDR] adjusted p value <0.05) that correlated with OS and treatment arms. To refine the molecular pathways associated with these gene expression changes, we performed additional enrichment analyses on the hallmark, Kyoto Encyclopedia of Genes and Genomes (KEGG), REACTOME, and Gene Ontology (GO) biological process (GOBP) and molecular function (GOMF) gene sets from MSigDb (Table S7). Although we did not perform the differential expression analysis by OS in CD11b+ and CD19+ cells due to too few samples with sufficient RNA quality, our analysis supported an association between longer OS and upregulation of the ubiquitin-dependent proteolysis pathway, tumor necrosis factor (TNF) signaling in both CD4+ and CD8+ T cells, and enhanced functional signaling in CD8+ T cells (Figures 6A and 6B; Table S7).
Figure 6. RNA-seq analysis of differentially expressed genes between different subcohorts from this study cohort of PDACs.
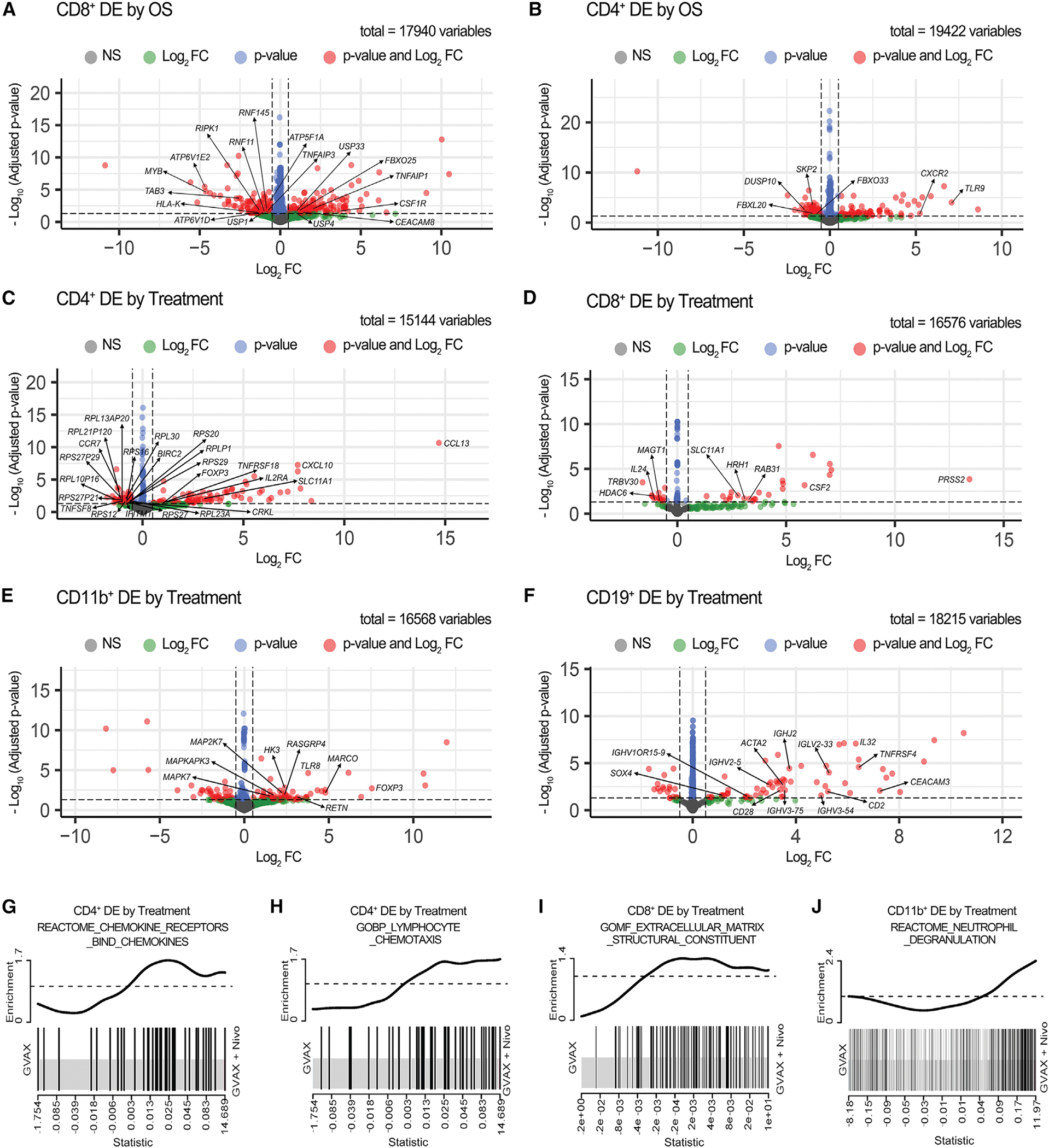
(A–B) Volcano plots showing differentially expressed (DE) genes compared between the OS > 2 years and OS < 2 years cohorts in sorted CD8+ (A) and CD4+ (B) T cells. (C-F) Volcano plots showing DE genes compared between treatment arms in sorted CD4+ T cells (C), CD8+ T cells (D), CD11b+ cells (E), and CD19+ B cells (F). The sample numbers are the same as in Figure 5. Vertical dashed lines indicate Log2 fold change (FC) at −0.5 and 0.5; horizontal dashed line indicates adjusted p value at 0.05. Genes that met either, both, or neither (NS) of the following two criteria: (1) Log2 FC > 0.5 or < −0.5 and (2) adjusted p value <0.05 are represented by color-coded dots. Total gene counts are indicated. Genes of interest are annotated.
(G–J) Enrichment plots showed upregulation of the REACTOM chemokine receptor bind chemokines pathway in sorted CD4+ T cells (G), the GOBP myeloid cells and lymphocyte chemotaxis pathway in sorted CD4+ T cells (H), the GOMF extracellular matrix structural constituent pathway in sorted CD8+ T cells (I), and the REACTOM neutrophil degranulation pathway in CD11b+ cells (J) in the GVAX + Nivo versus GVAX treatment arm. DE by treatment, differentially expressed between treatment arms. See also Tables S6 and S7.
Additional changes were attributed to anti-PD-1 therapy in arm B. These include a statistically significant downregulation of CCR7 in CD4+ T cells (Figure 6C); BIRC2, which has been shown to reduce intratumoral CD8+ T and NK cell infiltration associated with ICI therapy (Samanta et al., 2020); CRKL, which controls the generation of Tregs induced by foreign antigens (Blaize et al., 2020); IFITM1, which is an interferon (IFN)-induced antiviral protein; and TNFSF8, which is a cytokine in the TNF ligand family. Anti-PD-1 therapy was also associated with a significant increase in the expression of genes associated with immune activation. These included CXCL10, which is a Teff trafficking chemokine; IL2RA, which is associated with the Th1 response; FOXP3, which is upregulated in Tregs during CD4+ T cell activation; SLC11A1, which drives Th17 differentiation (Jiang et al., 2009); and TNFRSF18 (GITR), which is a T cell activation signal. Notably, we observed enhanced immune responses in the upregulation of multiple cytokine/chemokine signaling pathways (Table S7), such as the REACTOME pathways, including chemokine receptors bind chemokines (Figure 6G). Moreover, we observed an upregulation of the cytokine CCL13 (Figure 6C), which attracts proinflammatory myeloid cells (Mendez-Enriquez and Garcia-Zepeda, 2013), leading us to hypothesize that changes in cytokine/chemokine signaling due to anti-PD-1 therapy may further affect the function of other immune cells in the TME. This was supported by the significant upregulation of multiple GOBP pathways associated with myeloid cells and lymphocyte chemotaxis in arm B (Figure 6H). Altogether, these results suggest the activation of CD4+ T cells by anti-PD-1 therapy through regulating chemokine/cytokine signaling pathways.
Compared with CD4+ T cells, we observed fewer changes in immune response genes associated with anti-PD-1 therapy in CD8+ cells (Figure 6D). However, we did observe a statistically significant increase in the hallmark epithelial to mesenchymal transition (EMT) pathway; the REACTOME pathways, including ECM organization, collagen degradation, and ECM proteoglycans; the GOBP pathways, including collagen metabolic processes and complement activation; and the GOMF pathways for the ECM matrix structural constituent (Figure 6I). Upregulation of these pathways pointed to anti-PD-1 therapy-induced ECM regulation of CD8+ T cell trafficking rather than direct CD8+ T cell activation. These data also pointed to anti-PD-1 therapy-induced neutrophil degranulation signals in the ECM as several genes in this process were altered, including downregulation of MAGT1 (Li et al., 2011a, 2011b), upregulation SLC11A1 (Hedges et al., 2013; Jiang et al., 2009), and upregulation of PRSS2 (Sui et al., 2021) in arm B. It should be noted that an induction of neutrophil degranulation signaling is anticipated to hinder CD8+ T cell activation, likely through ECM regulation. This hypothesis was further supported by significant differential gene expression changes in the REACTOME neutrophil degranulation pathway in CD11b+ cells from arm B (Figure 6J). In this myeloid cell population, we observed significant increases in the innate immune response following anti-PD-1 therapy at the individual gene level (MARCO, RETN, and TLR8) (Figure 6E) and the pathway level (REACTOME innate immune system), which are anticipated to induce myeloid cell infiltration and may further induce neutrophil degranulation. We observed a similar trend for B cell activation signatures following anti-PD-1 therapy in the CD19+ RNA-seq data (Figure 6F), including overexpression of immunoglobulin genes and the chemokine IL32, which induces the expression of IL-8 (Khawar et al., 2015). We also observed differential gene expression in CD19+ cells from arm B, consistent with changes in B cell motility reflected at the pathway level through EMT (hallmark EMT). Altogether, these analyses suggest that anti-PD-1 therapy induced changes in multiple cytokine and chemokine signals that activated CD4+ T cells and induced myeloid cell trafficking and neutrophil degranulation, possibly through altering the ECM, to hinder the motility and activation of immune-effector cells, including CD8+ T cells and B cells.
Single-cell analysis of untreated PDACs revealed that CD137(TNFRSF9)hi CD8+ T cells signal to neutrophils via IFN-γ receptor ligands, and the CD137(TNFRSF9)hi effector CD8+ T cell subset exhibits an increased TNF-α response
The integrated analyses of immune cell protein markers by mIHC and TCR/BCR by RNA-seq from matched tumors provided the opportunity to relate T cell and B cell functions to the cellular landscape of LAs. We then examined tumor-specific T and B cell functionality by comparing the clonality of CD4+ and CD8+ T cells and B cells between the higher- and lower-density immune cell cohorts grouped according to mIHC results. We found a statistically significant increase in CD8+ T cell clonality in the cohort with a higher versus lower density of CD8+CD137+ T cells (p = 0.0089; Figure S8C). CD8+ T cell clonal expansion was also associated with longer OS (Figure 5I), consistent with the above data showing that higher CD8+CD137+ T cell density was also associated with longer OS (Figure 2G).
Given this positive association, we hoped to further mine the RNA-seq data for additional molecular changes associated with these T cell responses. However, the six samples with higher CD8+CD137+ T cell density and also with adequate quality RNA to conduct these additional studies were all associated with longer OS. Thus, we leveraged a single-cell atlas of 25,903 cells from 17 treatment-naive primary PDAC tumors previously reported (Steele et al., 2020) to evaluate the distribution of CD137 (encoded by TNFRSF9) expression across T cell subtypes and the TME changes directly associated with infiltration of CD8+CD137+ T cells. Standardized cell subtyping (Bell et al., 2022; Kinny-Koster et al., 2022) identified multiple immune cell subtypes in this atlas (Figure 7A). Expression values for TNFRSF9 were used to classify the T cells as TNFRSF9hi or TNFRSF9lo (Figure 7B; STAR Methods) and to calculate the mean proportions of CD137 status in each cell type across PDAC samples that had at least one cell of the corresponding cell type. Within the T cell populations, Tregs (13% ± 3.3%, n = 16) had the greatest average proportion of TNFRSF9-expressing cells in PDACs in the atlas, followed by effector CD8+ T cells (2.2% ± 1%, n = 15) and CD8+ T cells (1.3% ± 0.6%, n = 16) (Figures 7C and S9A).
Figure 7. Single-cell analysis of CD137 (TNFRSF9)-expressing T cells.
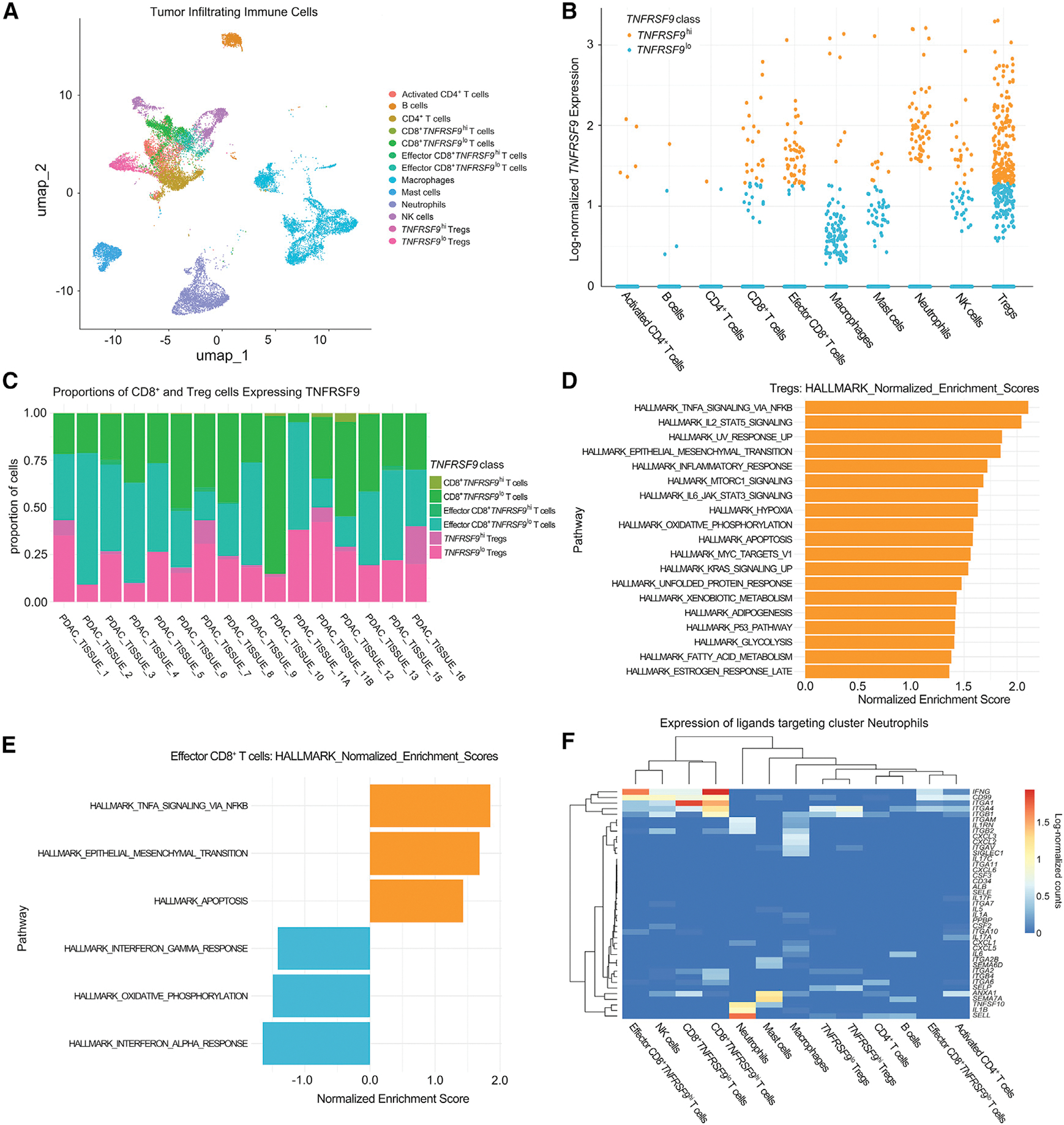
(A) Uniform Manifold Approximation and Projection (UMAP) embedding of color-coded immune cell subtypes from the PDAC atlas.
(B) Immune cells from the PDAC atlas were annotated according to high (hi) or low (lo) TNFRSF9 expression and annotated for different immune cell subtypes.
(C) Stacked bar plots of the proportions of CD8+, effector CD8+, and Treg cells classified as TNFRSF9hi or TNFRSF9lo.
(D and E) MsigDB hallmark gene sets significantly enriched in TNFRSF9hi (orange) or TNFRSF9lo (blue) Tregs (D) and effector CD8+ cells (E) ordered by normalized enrichment score.
(F) Expression of ligands by immune cells that interact with receptors expressed by neutrophils is presented in the heatmap as scaled, log-normalized counts. See also Figure S9.
To profile phenotypic differences in TNFRSF9hi and TNFRSF9lo cells in PDACs, differential expression analysis by MAST test (Finak et al., 2015) between TNFRSF9hi and TNFRSF9lo cells was carried out among CD8+ T cells, effector CD8+ T cells, and Tregs followed by gene set enrichment analysis of hallmark gene sets (Liberzon et al., 2015; Subramanian et al., 2005). No gene sets were found to be significantly enriched in either group of CD8+ T cells. TNFRSF9hi Tregs were significantly enriched for 19 gene sets, with inflammatory pathways such as TNF-α signaling via nuclear factor κB (NF-κB), IL-2, STAT5 signaling, inflammatory response, and apoptosis being among the most enriched gene sets in these cells (Figure 7D). Effector CD8+TNFRSF9hi T cells were significantly enriched for TNF-α signaling via the NF-κB, EMT, and apoptosis pathways, whereas effector CD8+TNFRSF9lo T cells were enriched for pathways including IFN-γ response, oxidative phosphorylation, and IFN-α response relative to the TNFRSF9hi cells (Figure 7E). These results suggested that increased TNFRSF9 expression in effector CD8+ T cells may lead to a shift in the inflammatory response program, where low expression of TNFRSF9 is associated with an IFN response that is important for cytotoxicity in Teffs (Cattolico et al., 2022), to a TNF-responsive state associated with high expression of TNFRSF9. Although TNF-α is involved in the CD8+ T cell antitumor activity (Calzascia et al., 2007), it can also promote apoptosis of CD8+ T cells (Zheng et al., 1995). This may explain the observed enrichment in apoptosis pathways and represent the mechanism underlying T cell exhaustion that was observed among the effector CD8+TNFRSF9hi T cells.
Our bulk RNA-seq analysis led to the hypothesis that interactions between CD4+ T cell cytokine production, CD8+ T cell activity, and neutrophil degranulation occur in PDACs following anti-PD-1 therapy. The ability to model interactions between immune cell types with ligand-receptor networks enabled further evaluation of the impact of CD137 expression on both CD8+ T cells and Tregs in modulating their interactions with neutrophils. For neutrophils, CD8+ T cells, and Tregs, incoming signals were assessed by quantification of ligands expressed by other cells that interact with receptors expressed by each cell type using Domino (Cherry et al., 2021) (Figures 7F and S9B). We found that neutrophils signaled to T cells through the expression of ITGAM, ITGB2, and ITGA5, regardless of T cell subtype or TNFRSF9 expression levels (Figure S9B), in accordance with the ECM interactions observed in our bulk RNA-seq analysis. Analysis of incoming signals directed to effector CD8+TNFRSF9hi T cells showed elevated expression of CD70 by TNFRSF9hi Tregs compared with TNFRSF9lo Tregs (Figure S9B). Expression of CD70 in expanded Tregs has been associated with loss of regulatory function exerted on other immune cells (Arroyo Hornero et al., 2020). Such dysfunction in Tregs may be a means by which the communication between CD4+ T cells and CD8+ T cells was modulated by nivolumab, as seen in the bulk RNA-seq analysis. Among signals targeting neutrophils (Figure 7F), important distinctions were found between ligand expression in TNFRSF9hi and TNFRSF9lo cells. CD8+TNFRSF9hi T cells expressed ligands that target the IFN-γ receptor (INFRG1, INFRG2) at a higher level than CD8+TNFRSF9lo T cells, including IFNG, ITGA1, ITGA6, ITGB1, ITGB2, and ITGB4. Effector CD8+TNFRSF9hi T cells also expressed substantially more IFNG directed to neutrophils than effector CD8+TNFRSF9lo T cells and did not cluster with effector CD8+TNFRSF9lo T cells on the heatmap of ligand expression. In contrast, CD8+TNFRSF9lo T cells expressed more ANXA1, which encodes Annexin 1, a ligand of formyl peptide receptor 2, and controls the resolution of inflammation and the return to homeostasis in recipient cells (Alessi et al., 2017). Thus, the TNFRSF9hi cells exhibited inflammatory signals directed to neutrophils that could induce degranulation, as similarly shown above in the CD8+ T cell RNA-seq data originating from this study, and had diminished expression of molecules such as ANXA1 that mediate the resolution of inflammation.
DISCUSSION
This prospectively collected cohort of PDAC paired pre- and post-treatment tumor biospecimens was analyzed to elucidate the mechanisms of anti-PD-1 treatment sensitivity and resistance in the neoadjuvant setting. This study also integrates multi-omic analyses to comprehensively define changes in immune subsets within PDAC TMEs following anti-PD-1 ICI therapy. Although the sample size is still small, our multi-omics approach revealed immunomodulatory signaling pathways that may serve as new therapeutic targets for testing in PD-1-blocking antibody-containing combination immunotherapies in our ongoing platform neoadjuvant study. Analysis of the single-cell RNA-seq atlas confirmed the existence of these immune subset signaling pathways in an independent cohort, further supporting the need to further assess these specific pathways in future trials. Thus, this platform study provides the opportunity to rapidly understand mechanisms of improved immunity and compensatory mechanisms of resistance with the goal of identifying the most active combinations to rapidly move forward in clinical development for treating a deadly cancer.
The combination of mIHC and RNA-seq analyses confirmed the overexpression of neutrophil trafficking factors and neutrophil degranulation signals following treatment with vaccine and anti-PD-1 therapy as well as TAN expression of IL8RB/CXCR2, differentiating TANs from other immune suppressive monocyte populations previously identified as barriers to T cell infiltration (Veglia et al., 2018). The immune activation and suppressive changes that occur either at baseline or early after initial priming provide strong support that there are compensatory mechanisms that compete in PDACs. These competing signals may serve as new targets for testing with initial administration of anti-PD-1 combination immunotherapy in an attempt to bypass compensatory mechanisms prohibiting full activation of Teff. IL-8, which is known to be secreted by PDAC cells in response to KRAS activation, is the main ligand for the IL8RB/CXCR2 receptor on TANs (Gonzalez-Aparicio and Alfaro, 2020; Teijeira et al., 2021). Our recent preclinical study showed that an anti-IL-8 antibody can potentiate the antitumor activity of anti-PD-1 antibody, possibly through the activation of an innate immune and type I cytokine response in TANs (Li et al., 2022). Taken together, these data further support testing an IL-8 pathway inhibitor with anti-PD-1 therapy currently in the same platform trial (NCT02451982) as a next step in improving Teff responses.
We show that a shorter distance between CD8+GZMB+ T cells and tumor cells is associated with an anti-PD-1-induced response and resistance. Therefore, vaccine priming of the PDAC TME can recruit functional antitumor T cells, but not all patients achieve a clinical benefit. We also identified a CD137-expressing CD8+ T cell population in both treatment arms that was associated with increased CD8+GZMB+ T cells and improved OS. Our preclinical study previously demonstrated that this Teff subtype can be further enhanced when mice with orthotopically implanted PDACs are treated with the triple combination of GVAX, anti-PD-1 antibody, and anti-CD137 agonist antibody (Muth et al., 2020). This triple combination is already being tested as a new arm in our neoadjuvant platform study and was shown in a preliminary analysis to be safe and meaningfully enhance DFS in resectable PDAC patients (Heumann et al., 2022).
Our integrated analysis approach suggested previously unrealized mechanisms promoting T cell exhaustion following PD-1 antibody therapy. Testing the hypotheses generated from the RNA-seq data that did not have available accompanying mIHC results requires further validation, particularly with functional assays. These additional analyses will provide an in silico framework to directly evaluate these predicted immunomodulatory effects of neutrophil degranulation and changes in ECM structure in human PDACs. These additional studies will be conducted on biospecimens from our ongoing platform clinical trial inhibiting TAN function with an IL-8 blockade antibody and enhancing T cell function with a CD137 agonist antibody.
This study has a few limitations. The sample size was limited by the inability to obtain pre-treatment biopsies from some patients and by the exclusion of tumor specimens with insufficient clinical follow-up time. The spatial relationship analysis of immune cells in PDAC in response to immunotherapy was limited by the number of markers and the size of ROIs that can be handled by the current software. This study relied on bulk RNA-seq of the individual major subtypes of immune cells due to technical limitations at the time when the clinical trial began. Thus, we were limited to inferences of cellular function from computational estimates of cellular composition through deconvolution methods (Korotkevich et al., 2021) and to inferences of immune cell function in single-cell RNA-seq data from treatment-naive PDAC atlases (Kinny-Koster et al., 2022; Steele et al., 2020). Nevertheless, single-cell technology is anticipated to be incorporated into the analysis of specimens from new treatment arms.
In summary, multi-omic analyses in small cohorts of surgically resectable PDAC patients comparing different immune modulatory agents may be a rapid and efficient way to identify the immunomodulatory effects that can inform combinations for testing in larger studies with primary clinical endpoints.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources or data should be directed to and will be fulfilled by the contact: Lei Zheng (lzheng6@jhmi.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The transcriptomic and clinical data used in this study are available in NCBI’s Gene Expression Omnibus (GEO) SuperSeries GSE197613 (GEO database: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197613) and are publicly available. Whole-exome sequencing data are deposited in the database of Genotypes and Phenotypes (dbGaP): phs003002.v1.p1. Details of the trial, data, contact information, proposal forms, and review and approval process are available at the following website: https://clinicaltrials.gov/ct2/show/NCT02451982. The trial protocol is provided in Data S1. The microscopy data reported in this paper will be shared by the lead contact upon request.
Code for genomics analysis available at: github.com/FertigLab/J1568_Bulk
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and specimens
A total of 34 patients were enrolled and randomized into two treatment arms to receive either GVAX (Arm A) or GVAX in combination with nivolumab (Arm B) as neoadjuvant and adjuvant therapy (Figure S2A) in PDAC from the clinical trial (NCT02451982) at Johns Hopkins Hospital under a Johns Hopkins Medical Institution Institutional Review Board-approved protocol (ID: IRB00050517). Written informed consent was obtained from all patients. All available pre-treatment and post-treatment tumor specimens from 19 patients with overall survival greater than 2 years since the start of immunotherapy or those who died within 2 years after the start of immunotherapy (OS < 2 years) by the date of data cutoff on February 17, 2020, were included in this study (Figure S1). Patients underwent pancreatectomy between March 2016 and February 2018 (Table S1). Patients who did not have a diagnosis of PDAC on final surgical pathology, who were found intraoperatively to have metastasis, and whose specimens were not adequate for mIHC or did not meet the criteria for ROI selection for analysis were excluded from this study (Table S2). Primary PDAC tumor samples were obtained from endoscopic ultrasound-guided fine needle core biopsies (EUS-FNB) or surgically resected tumors. Both fresh tissue and formalin-fixed paraffin-embedded (FFPE) tissue blocks were obtained. Pre-treatment tumor specimens for research purposes were obtained from 6 patients in Arm A and 10 patients in Arm B after the EUS-FNB specimens had been prioritized for diagnostic purposes. Post-treatment surgically resected tumor specimens and blood for peripheral blood mononuclear cells were obtained from all 19 patients.
METHOD DETAILS
Sequential IHC and image acquisition
The sequential staining-stripping multiplex immunohistochemistry (mIHC) protocol has been described previously (Tsujikawa et al., 2017). Briefly, deparaffinized 5-μm FFPE tissue sections were first stained with hematoxylin (Dako, S3301) followed by whole-slide brightfield scanning using NanoZoomer (Hamamatsu). Then, following endogenous peroxidase blocking and heat-induced antigen retrieval with citrate buffer (pH 6.0) (BioGenex, HK080–9K), sequential multiple iterative cycles of the IHC process that included staining, scanning, and chromogen stripping, were performed as described previously (Tsujikawa et al., 2017). Information on the concentration of primary antibodies, the incubation time of primary antibodies and the horseradish peroxidase (HRP)-conjugated polymer (Nichirei Biosciences Inc.), and the aminoethyl carbazole (AEC) (Vector Laboratories, SK-4200) reaction time for chromogenic detection are summarized in Table S3. Negative control images were obtained after the last antibody and chromogen stripping. In this study, two separate staining panels were used, including one panel of 15 markers primarily for lymphoid cells and another panel of 15 markers primarily for myeloid cells (Table S3). Every immune marker examined represented a specific aspect of the immune response of interest, which was determined based on previously published studies (Tsujikawa et al., 2017); therefore, we did not control for multiplicity in the analysis.
Multiplex image processing and analysis
The digitized image processing and analyzing workflow encompasses image coregistration, visualization, and quantitative image analysis. Digitized images collected from NanoZoomer were first coregistered via the specific CellProfiler v.2.1.1 pipeline designed as previously described (Tsujikawa et al., 2017). Tumor areas for subsequent analyses were circled by pathologists on hematoxylin-stained slides. A minimum of three rectangular ROIs (approximately 3000*3000 pixels per ROI) in the vicinity of tumor epithelia, which were known to have an adequate representation of the whole tumor area in a prior study (Tsujikawa et al., 2017), were chosen. For biopsy cores, quantifying any three ROIs that cover at least three biopsy cores yielded consistent immune cell density results. Tumor epithelia identified by EpCAM staining, large intratumoral blood vessels, and areas with tissue detachment were maximally excluded. Visualization was performed by converting coregistered images into individually pseudocolored single-marker staining images through ImageJ v1.48 software (National Institutes of Health) (Schneider et al., 2012) and Aperio ImageScope v.12.3.2.8013 software (Leica Biosystems). Up to five ROIs per slide were quantified. During the quantitative image analysis step, signals of single-cell segmentation and quantification were obtained using the specific CellProfiler v.2.1.1 pipeline as previously described (Tsujikawa et al., 2017), followed by image cytometry analysis via FCS Express 7 Image Cytometry software v7.10.0007 (De Novo Software). The calculation of immune cell densities as percentages of all cells took into consideration PDAC desmoplasia, although they were generally consistent with those calculated as the percentage of total CD45+ cells. Immune cell subtypes were defined by multiple markers, as listed in Table S4.
For the quantification of CXCR2+ CD66b+ cells, image analysis was conducted with Halo Image Analysis Platform software v3.4.2986 (Indica Labs). First, three sequential IHC images, including hematoxylin, CD66b, and CXCR2 staining by the above-described multiplex IHC protocol, were deconvolved, registered and fused into a pseudomultiplex immunofluorescent image. Then, tumor areas on the whole slide section except for the necrotic region were annotated by the pathologist and included in the analysis. Slides with significant tissue damage were also excluded. Cells were identified by nuclear staining with hematoxylin. Positive staining signals were detected by adjusting the threshold of the signal intensity of each marker on hematoxylin-marked cells via the HighPlex FL module. CD66b+, CXCR2+, and CXCR2+CD66b+ cells were selected and analyzed for their quantities and average intensities of positive signals on each cell type.
Singleplex immunohistochemistry
Following heat-induced antigen retrieval for 30 minutes at 95°C or 30 seconds at 125°C in citrate (BioGenex, HK080–9K) or EDTA buffer (Sigma‒Aldrich E1161), slides were incubated with rat anti-mouse/human PNAd IgM (BioLegend, clone MECA-79) at a 1:100 dilution for 1 hour at room temperature, mouse anti-human LAG3 IgG (Novus Biologicals, clone 17B4) at a 1:200 dilution overnight at 4°C, or rabbit anti-human TIM-3 IgG (Cell Signaling Technology, clone D5DR5) at a 1:50 dilution overnight at 4°C, as previously described (Avram et al., 2013; Gorris et al., 2018; Wojcik et al., 2022). After the washing steps, the slides were incubated with either biotinylated anti-rat IgM secondary antibody (BioLegend, 408903) at a 1:200 dilution followed by the VECTASTAIN ABC-HRP kit (Vector Laboratories, PK-4000), HRP anti-mouse (Vector Laboratories, MP-7452) or HRP anti-rabbit (Vector Laboratories, MP-7451) polymer detection kits. The ImmPRESS DAB peroxidase substrate (Vector Laboratories, SK-4105) was used prior to counterstaining in hematoxylin, dehydration, and mounting with coverslips. Digital images were obtained at 20X magnification via whole-slide brightfield scanning using the NanoZoomer slide scanner (Hamamatsu).
LAG3 and TIM3 IHC quantification
Image analysis was performed using HALO Image Analysis Platform software v3.0.311.402 (Indica Labs). Tumor tissue areas were defined and annotated by the pathologist. Sections without tumor representation were omitted, and tissue artifacts, including folds, detachments, and tears, were maximally excluded within the annotation layer used for analysis. Immune cells positively stained for LAG3 and TIM3 were quantified using the Immune Cell v1.3 module. The immune cell stain intensity and color were manually identified for each marker. The algorithm settings for identifying staining-positive cells, including minimum tissue optical density (0.037), tissue edge thickness (0), minimum immune stain optical density (0.13), membrane detection tolerance (0.45), and immune cell size (5,250), were held constant across all cases. The density of stained positive cells was calculated as the immune cell count divided by the tumor tissue area (mm2) on the whole slide section.
Spatial relationship assessment
All steps of the image analysis were performed using Halo Image Analysis Platform software v3.4.2986 (Indica Labs). Briefly, 1) six different sequentially stained images were deconvolved, registered and fused into a pseudomultiplex immunofluorescent image; 2) the entire tumor region except for the necrotic region was annotated by a pathologist and included in the analysis; 3) positive staining signals were detected by adjusting the threshold of the signal intensity of each marker via the HighPlex FL module; and 4) distance and proximity measurements were performed based on the positive signal detected with coordinates and by the spatial module included in the software.
TIL isolation and sorting
TILs were obtained from a nonnecrotic tumor piece approximately 2 cm in diameter that could be spared for research purposes after the clinical specimens were prioritized and dissected by pathologists. RPMI-1640 media (Life Technologies) were used to resuspend all the cells after disruption and homogenization. The cell suspension was filtered with a 70-μm cell strainer (Corning), and a layer of mononuclear cells that contained TILs was then isolated using Ficoll density gradient centrifugation with Ficoll-Paque PLUS (Cytiva) as described (Tan and Lei, 2019). TILs were washed three times using 40 mL of complete RPMI media each time. After the final wash with RPMI media, isolated TILs were cryopreserved in a liquid nitrogen freezer or subjected to cell sorting. Peripheral blood mononuclear cells were obtained from the patient’s whole blood using Ficoll-Paque™ PLUS (Cytiva) gradient centrifugation to serve as the FACS staining control. TILs and peripheral blood mononuclear cells were stained with Zombie Green viability dye (Biolegend, 423111), anti-CD4-PE-CF594 (Biolegend, 562281), anti-CD8-APC (Biolegend, 344722), anti-CD19-PE (Biolegend, 555413), and anti-CD11b-BV421 (Biolegend, 393114) antibodies. After sorting with a BD FACSAria IIu Cell Sorter (Biosciences), the sorted immune cell subtype pellets were sent to a commercial vendor (Eurofins) for RNA extraction and sequencing. Note that they were sequenced and preprocessed together with other specimens under JHMI IRB protocol numbers IRB00083132 and IRB00092443.
RNA extraction, sequencing, and transcriptomic analysis
Bulk tumor RNA was extracted from TILs and quantified by using the All Prep DNA/RNA Micro Kit (Qiagen, 80284) and the Qubit RNA HS Assay Kit (Invitrogen, Q32852) according to the manufacturer’s instructions. The isolated RNA for each of the sorted CD3+CD4+, CD3+CD8+, CD3−CD11b+, and CD3−CD11b−CD19+ cells was sent to iTeos, Inc., for bulk RNA-seq library preparation with a SMART-Seq v4 ultra low input mRNA sample prep kit and 100-bp paired end sequencing by Eurofins. Library preparation and sequencing were performed in three batches, which were analyzed for technical artifacts as described below. First, quantification of transcript abundances was performed from RNA-seq reads with pseudoalignment to the hg38 genome using Kallisto v0.45.0 (Bray et al., 2016). The hg38 cDNA reference (release 87) was obtained from Ensemble (Bray et al., 2016). Gene counts were quantified from kallisto estimates of transcript abundances using tximport (v.1.22.0) (Soneson et al., 2015), with the t lengthScaledTPM computed for samples across both batches. We evaluated sample quality from the distribution of reads as visualized in a boxplot of log counts. We observed 10 samples with zero median expression, reflective of a low read count, to be filtered from subsequent analysis as low quality. We used principal component analysis (PCA) of the variance stabilization transform (vst) RNA-seq data to evaluate sample clustering by cell type. To further evaluate sample quality, we examined marker gene expression and its relationship to the sample groups observed in the PCA. In completing this analysis, we identified 7 samples mislabeled for cell type, which were additionally filtered out of the sample cohort. To evaluate the batch effects present due to sequencing, we repeated PCA and noticed homogenized clustering of samples from both batches. For each cell type, we completed differential expression analysis with DESeq2 (Love et al., 2014), including batch as a covariate in our model along with our comparison of interest. Estimated fold changes are shrunk with apeglm using lfcShrink to account for the variation in the samples in this dataset. Genes were statistically significant if the absolute log2-fold changes after shrinkage were greater than 0.5 and the FDR-adjusted p value was below 0.05. Gene set statistics were run with fgsea using MSigDb (Zhu et al., 2019) v7.5.1 pathways annotated in the HALLMARK, KEGG, REACTOME, and GO databases. Gene sets were considered to be significantly enriched for FDR-adjusted p values below 0.05, and the results were visualized with LIMMA barcode plots (Ritchie et al., 2015). Raw counts were uploaded to CIBERSORTx (Korotkevich et al., 2021) for further deconvolution of cell types. Imputation of cell fractions utilizing the LM22 CIBERSORT DEFAULT reference (Kanehisa et al., 2017) (signature matrix) to estimate T-cell presence based on gene expression was completed using B-mode batch correction and 1000 permutations for statistical significance of estimates of cellular proportions in each sample. Comparisons of cellular abundances were calculated with Student’s t test comparing CIBERSORTx estimated proportions of each cellular population between the treatment or survival groups.
DNA extraction, sequencing, and the estimation of tumor mutation burden and neoantigen load
Genomic DNA was extracted by using the AllPrep DNA/RNA Kit (Qiagen) according to the manufacturer’s instructions and sent to a commercial vendor (BGI Genomics) for whole-exome sequencing (WES). According to the vendor, to construct whole-exome capture libraries, genomic DNA was randomly fragmented into 200–250-bp fragments, and the fragments were purified and ligated by specific adaptors according to the instructions of the MGIEasy Universal DNA Library Prep Set and then captured with the MGIEasy Exome Capture V4 Probe Set (~59 Mb). All constructed libraries were sequenced on a DIPSEQ platform at an average coverage of 268X (90–420) in normal samples and 665X (83–1251) in tumor samples. The raw sequencing data were processed by SOAPnuke software v2.0.7 with default parameters to filter out low-quality reads and adaptor contamination. The clean reads were processed by using the UCSC human reference genome (hg19) and the Sentieon pipeline that follows the GATK best practices, including read alignment, mark duplication reads, indel realignment, base quality score recalibration (BQSR), and variant calling (Table S5). Variant calls were converted to mutation annotations using the default VCF2MAF (Kandoth and Qwangmsk, 2018) pipeline and the same UCSC human reference genome (hg19). Mutation annotations were analyzed in R utilizing the Maftools (Mayakonda et al., 2018) package (v2.8.05). Tumor mutational burden was extracted using a capture size of 35.8 (Ellrott et al., 2018). Class I HLA typing was performed at the clinical laboratories of Johns Hopkins Hospital. Neoepitopes (9 to 11-mers) containing nonsynonymous SNVs or INDELs were predicted by NetMHCpan software v4.1b (Jurtz et al., 2017; Wells et al., 2020) for class I HLA-binding affinity <500 nM according to at least one method. Neoantigen load in plots was completed utilizing the “netMHCpan_total_mutationsite_valid” annotation. All statistical comparisons were performed using Student’s t test.
BCR/TCR prediction and clonality metrics
BCR and TCR sequences were predicted from the bulk RNA-seq data for CD3+CD4+, CD3+CD8+, and CD3CD19+ cells utilizing the MiXCR pipeline (Bolotin et al., 2015). FASTQ files of the same sample and direction were appended prior to running MiXCR under the following commands: ‘mixcr analyze shotgun –starting-material rna -s hsa –assemble-partial-rounds 2’. MiXCR results were then analyzed in R using IMMUNARCH (ImmunoMindTeam, 2019) v0.6.7. Clonality was measured utilizing the normalized Shannon’s Entropy (Shannon’s equitability-measure of evenness) (Hopkins et al., 2018). Clonality is calculated as (1-Shannon’s equitability), computed by:
where pi is the proportion of the ith clone from a repertoire of n clones. Clonality values range from 0–1, with 0 indicating equal representation of clones within a sample (lower clonality) and 1 indicating a monoclonal population. All statistical comparisons were performed using Student’s t test.
PDAC atlas single cell RNA-seq analysis
Single-cell RNA-seq analysis used published data from an atlas of cells from pancreatic ductal adenocarcinoma resected primary tumors and biopsies (Kinny-Koster et al., 2022; also includes methods for data preprocessing and quality control). Immune cells from the atlas were selected from 61 tumor samples by subsetting on clusters expressing PTPRC. These subset cells were clustered again at a resolution of 104 using the R package monocle3 (Cao et al., 2019; Qiu et al., 2017; Trapnell et al., 2014) (version 1.0.0). Clusters were annotated based on canonical marker genes detected by monocle3. Thirty-three clusters were detected in 52,765 cells and annotated as Macrophages (AIF1), Neutrophils (S100A8, S100A9), NK cells (NKG7, CD3E), B cells (CD79A, CD79B, CD52), Mast cells (KIT, ENPP3), CD4+ T cells (CD3E, CD4), Activated CD4+ T cells (CD3E, CD4, EOMES, GZMK), CD8+ T cells (CD3E, CD8A), Effector CD8+ T cells (CD3E, CD8A, EOMES, GZMK), or Tregs (CD3E, FOXP3, CTLA4). Clusters that could not be confidently annotated as a single cell type due to doublets were removed.
After cell type annotation, cells were further subset to include only the immune cells in 17 tumor samples from the 16 patients in (Steele et al., 2020) (25,903 cells) due to the larger proportion of immune cells represented specifically in the samples from this study. The sparse matrix of UMI counts (15,219 genes × 25,903 cells) was converted to a Seurat object with the R package Seurat (Butler et al., 2018; Hao et al., 2021; Stuart et al., 2019; Zeng et al., 2018) (version 1.4.1). Using functions from the Seurat package, counts were log-normalized and scaled. The top 50 principal components (PCs) were computed using the 2,000 most variable features. A dimensionally reduced representation of the data was calculated with UMAP using the top 25 PCs. Cells were classified as having high TNFRSF9 expression (TNFRSF9hi) or low expression (TNFRSF9lo) based on the presence of a log-normalized expression value of TNFRSF9 above or below the median expression value in all cells with nonzero expression of TNFRSF9, respectively. Cells annotated as CD8+ T cells, Effector CD8+ T cells, and Tregs were subdivided into CD8+TNFRSF9hi T cells, CD8+TNFRSF9lo T cells, Effector CD8+TNFRSF9hi T cells, Effector CD8+TNFRSF9lo T cells, TNFRSF9hi Tregs, and TNFRSF9lo Tregs. The proportions of CD8+ and Treg cells expressing TNFRSF9 were calculated as the number of cells of each type with the TNFRSF9hi classifier divided by the number of cells annotated as that type and as cells of each type with the TNFRSF9hi or TNFRSF9lo classifier divided by the total number of cells typed as CD8+, Effector CD8+, or Tregs.
Differential expression between TNFRSF9hi cells and TNFRSF9lo cells of the same cell type was carried out with MAST tests in Seurat. The results for all assayed genes were ordered by the average log fold change as rankings for gene set enrichment analysis with the R package fgsea (Korotkevich et al., 2021) using the H: HALLMARK gene set from the Molecular Signatures Database (MSigDB, version 7.5.1) (Liberzon et al., 2015; Subramanian et al., 2005).
Intercellular interactions were assessed among annotated cell types and TNFRSF9-classified CD8+ cells, Effector CD8+ cells, and Tregs based on the expression of interacting expressed transcription factor targets, receptors, and ligands using the R package Domino (Cherry et al., 2021) (version 0.1.1). In the Domino analysis, receptor–ligand networks were computed with pySCENIC (Aibar et al., 2017; Van de Sande et al., 2020) (version 0.11.0) using lists of transcription factors, motif annotations, and cisTarget motifs from the GRCh38 (hg38) reference genome (Van de Sande et al., 2020) and curated ligand and receptor interactions obtained from CellPhoneDB (Efremova et al., 2020; Garcia-Alonso et al., 2021) (version 2.0.0).
QUANTIFICATION AND STATISTICAL ANALYSIS
The density of each immune cell subtype was calculated by its percentage among all cells or CD45+ cells in ROIs. The tumors whose density of certain immune cell subtypes was higher than the average were grouped into the “high” density group for that immune cell subtype, while tumors whose density of certain immune cell subtypes was lower than the average were grouped into the “low” density group for that immune cell subtype. For the comparison between pre- and post-treatment biospecimens, a paired t test was performed, while an unpaired t test was used to compare two independent groups. Statistical analyses and graphing were performed using GraphPad Prism software v9.3.1 (GraphPad Software) or the Immunarch package (ImmunoMindTeam, 2019). All tests were two-sided, and a p value of <0.05 was considered statistically significant. Statistical analyses of the genomics data were performed as described in detail in the methods for each data modality above with code available from github.com/FertigLab/J1568_Bulk.
The Bayesian information criterion (BIC) was used to evaluate the performance of each model proposed and to determine which marker better predicts the outcomes. A linear regression model was used to evaluate the effect of each marker, dichotomized by the mean, as a predictor of each distance measure. For each binary survival outcome, logistic regression was employed, with each marker treated as continuous. A meaningful difference in BIC between the two models is 2 at a minimum, and a difference between 5–10 and above 10 is considered to be strong and very strong, respectively (Raftery, 1995).
ADDITIONAL RESOURCES
More information about the platform trial (NCT02451982) and associated resources can be found at https://clinicaltrials.gov/ct2/show/NCT02451982.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Multiplex immunohistochemistry antibody panel | See Table S3 | |
| Rat monoclonal anti-mouse/human PNAd, clone MECA-79 | BioLegend | Cat#120802; RRID: AB_493555 |
| Mouse monoclonal anti-Human LAG-3, clone 17B4 | Novus Biologicals | Cat#NBP1-97657SS; RRID: AB_11162489 |
| Rabbit monoclonal anti-Human TIM-3, clone DRDR5 | Cell Signaling Technology | Cat#45208S; RRID: AB_2716862 |
| Monoclonal Biotin anti-rat IgM, clone MRM-47 | BioLegend | Cat#408903; RRID: AB_10571208 |
| Mouse monoclonal anti-Human CD4 PE-CF594, clone RPA-T4 | BioLegend | Cat#562281; RRID: AB_11154597 |
| Mouse monoclonal anti-Human CD8 APC, clone SK1 | BioLegend | Cat#344722; RRID: AB_2075388 |
| Mouse monoclonal anti-Human CD19 PE, clone HIB19 | BioLegend | Cat#555413; RRID: AB_395813 |
| Mouse monoclonal anti-Human CD11b BV421, clone LM2 | BioLegend | Cat#393114; RRID: AB_2750258 |
|
| ||
| Biological samples | ||
|
| ||
| Tumor biopsy before treatment | This paper | N/A |
| Tumor samples after surgery | This paper | N/A |
| Peripheral blood mononuclear cells | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Hematoxylin | Dako | Cat#S3301 |
| Citra Plus Solutions | BioGenex | Cat# HK086-9K |
| EDTA buffer | Sigma-Aldrich | Cat# E1161 |
| Hydrogen peroxide solution | Sigma-Aldrich | Cat#H1009-500ML |
| HistoFine Simple Stain MAX PO (Mouse) | Nichirei Bioscience | Cat#414132F |
| HistoFine Simple Stain MAX PO (Rabbit) | Nichirei Bioscience | Cat#414142F |
| HistoFine Simple Stain MAX PO (Rat) | Nichirei Bioscience | Cat#414311F |
| VectaMount AQ Mounting Medium | Vector Laboratories | Cat#H-5501-60 |
| Ficoll-Paque Plus | Sigma-Aldrich | Cat#GE17-1440-02 |
|
| ||
| Critical commercial assays | ||
|
| ||
| AEC Substrate Kit | Vector Laboratories | Cat#SK-4200 |
| ImmPRESS HRP Goat Anti-Rabbit IgG Polymer Detection Kit | Vector Laboratories | Cat#MP-7451 |
| ImmPRESS HRP Goat Anti-Mouse IgG Polymer Detection Kit | Vector Laboratories | Cat#MP-7452 |
| ImmPRESS DAB peroxidase substrate Kit | Vector Laboratories | Cat#SK-4105 |
| AllPrep DNA/RNA Micro Kit | Qiagen | Cat#80284 |
| Qubit RNA HS Assay Kit | Invitrogen | Cat#Q32852 |
| SMART-Seq v4 Ultra Low Input RNA Kit | Takara | Cat#634894 |
| Zombie Green™ Fixable Viability Kit | BioLegend | Cat#423111 |
|
| ||
| Deposited data | ||
|
| ||
| Transcriptomic Data (raw) | This paper | GEO database: GSE197613 |
| Whole exome sequencing data | This paper | dbGAP: phs003002.v1.p1 |
| Code for genomics analysis | This paper | github.com/FertigLab/J1568_Bulk |
| scRNA-seq database | Kinny-Koster et al., 2022 | https://doi.org/10.1101/2022.07.14.500096 |
|
| ||
| Software and algorithms | ||
|
| ||
| Aperio ImageScope v.12.3.2.8013 | Leica Biosystems | https://www.leicabiosystems.com/us/digital-pathology/manage/aperio-imagescope/ |
| CellProfiler pipeline v.2.1.1 | Tsujikawa et al., 2017 | https://github.com/multiplexIHC/cppipe |
| ImageJ v1.48 | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| FCS Express 7 Image Cytometry v7.10.0007 | De Novo Software | https://denovosoftware.com/full-access/download-landing/ |
| Halo Image Analysis Platform v3.4.2986 | Indica Labs | https://indicalab.com/halo/ |
| Kallisto v0.45.0 | Bray et al., 2016 | http://pachterlab.github.io/kallisto/ |
| Tximport v1.22.0 | Soneson et al., 2015 | http://bioconductor.org/packages/tximport |
| DESeq2 v1.36.0 | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| apeglm v1.18.0 | Zhu et al., 2019 | https://bioconductor.org/packages/release/bioc/html/apeglm.html |
| MSigDb v7.5.1 | Zhu et al., 2019 | http://www.gsea-msigdb.org/gsea/msigdb |
| LIMMA v3.52.2 | Ritchie et al., 2015 | http://www.bioconductor.org/packages/release/bioc/html/limma.html |
| CIBERSORTx | Korotkevich et al., 2021 | https://cibersortx.stanford.edu/ |
| MGIEasy Universal DNA Library Prep Set | MGI Tech Co., Ltd. | https://en.mgi-tech.com/products/reagents_info/id/8 |
| MGIEasy Exome Capture V4 Probe Set | MGI Tech Co., Ltd. | https://en.mgi-tech.com/products/reagents_info/9/ |
| SOAPnuke v2.0.7 | BGI Genomics | https://github.com/BGI-flexlab/SOAPnuke |
| VCF2MAF v1.6.16 | Kandoth and Qwangmsk, 2018 | https://zenodo.org/record/1185418#.YugPFHZBxD8 |
| Maftools package v2.8.05 | Mayakonda et al., 2018 | https://github.com/PoisonAlien/Maftools |
| NetMHCpan v4.1b | Jurtz et al., 2017 | https://services.healthtech.dtu.dk/service.php?NetMHCpan-4.1 |
| MiXCR v4.0 | Bolotin et al., 2015 | https://github.com/milaboratory/mixcr/releases |
| IMMUNARCH v0.6.7 | ImmunoMindTeam, 2019 | https://immunarch.com/index.html |
| Normalized Shannon’s Entropy algorithm | Hopkins et al., 2018 | https://doi.org/10.1172/jci.insight.122092 |
| monocle3 v1.0.0 | Cao et al., 2019; Qiu et al., 2017; Trapnell et al., 2014 | http://cole-trapnell-lab.github.io/monoclerelease/monocle3/#installing-monocle-3 |
| Seurat v4.1.1 | Butler et al., 2018; Hao et al., 2021; Stuart et al., 2019; Zeng et al., 2018 | https://cran.r-project.org/web/packages/Seurat/index.html |
| fgsea v1.22.0 | Korotkevich et al., 2021 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Domino v0.1.1 | Cherry et al., 2021 | https://github.com/Shamir-Lab/DOMINO/releases |
| pySCENIC v0.11.0 | Aibar et al., 2017; Van de Sande et al., 2020 | https://github.com/aertslab/pySCENIC |
| CellPhoneDB v2.0.0 | Efremova et al., 2020; Garcia-Alonso et al., 2021 | https://github.com/Teichlab/cellphonedb |
| GraphPad Prism v9.3.1 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
|
| ||
| Other | ||
|
| ||
| Clinical trial protocol | This paper | Data S1 |
Highlights.
Prospectively collected PDAC specimens from a neoadjuvant platform clinical trial
Identified sensitivity and resistance mechanisms to anti-PD-1 therapy in PDAC
Informed studies of additional immune-modulating agents in the ongoing platform trial
Generated hypotheses of reprogramed TME signals for combination immunotherapy strategies
ACKNOWLEDGMENTS
This study was supported by NIH grants R01 CA169702 (L.Z.), R01 CA197296 (L.Z., E.M.J.), P50 CA062924 (E.M.J., L.Z.), a Bristol-Myers Squibb grant (E.M.J., L.Z.), Viragh Foundation and Skip Viragh Pancreatic Cancer Center at Johns Hopkins (E.M.J., L.Z.), and NCI P30 CA006973. RNA-seq was supported by a grant from iTeos (L.Z.), which is independent from the collaboration with Bristol-Myers Squibb. DNA sequencing and analysis was supported by a collaborative project at Johns Hopkins University inHealth Program and Precision Medicine Application Platform. We thank Drs. Dongbing Liu, Fanfan Xie, and Mengyue Lei at BGI Genomics, Inc. for their technical advice. B.K. is supported by the German Research Foundation (KI 2437/2–1).
Footnotes
DECLARATION OF INTERESTS
L.Z. receives grant support from Bristol-Meyer Squibb, Merck, AstraZeneca, iTeos, Amgen, NovaRock, Inxmed, Halozyme, and Abmeta. L.Z. is a paid consultant/Advisory Board member at Biosion, Alphamab, NovaRock, Ambrx, Akrevia/Xilio, QED, Natera, Novagenesis, Snow Lake Capitals, BioArdis, Tempus, Amberstone, Pfizer, Tavotek, and Mingruizhiyao. L.Z. holds shares at Alphamab, Amberstone, and Mingruizhiyao. E.J.F. is on the scientific advisory board of Resistance Bio and is a paid consultant for Merck and Mestag Therapeutics. E.J. receives grant support from Lustgarten Foundation, Bristol-Meyer Squibb, Genentech, and AstroZeneca; is a paid consultant for NextCure, Genocea, DragonFly, Stimit, CSTONE, Achilles, and Candel; is on the advisory board of Parker Institute and Break Through Cancer; is a founder of Abmeta Biotech; and is the Chief Medical Advisor for the Lustgarten Foundation.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2022.10.001.
REFERENCES
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi M-C, Cenac N, Si-Tahar M, and Riteau B (2017). FPR2: a novel promising target for the treatment of influenza. Front. Microbiol. 8, 1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrollahi P, Rodrigues M, Lyon CJ, Goel A, Han H, and Hu TY (2019). Ultra-sensitive automated profiling of EpCAM expression on tumor-derived extracellular vesicles. Front. Genet. 10, 1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo Hornero R, Georgiadis C, Hua P, Trzupek D, He L-Z, Qasim W, Todd JA, Ferreira RC, Wood KJ, Issa F, et al. (2020). CD70 expression determines the therapeutic efficacy of expanded human regulatory T cells. Commun. Biol. 3, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avram G, Sánchez-Sendra B, Martín JM, Terrádez L, Ramos D, and Monteagudo C (2013). The density and type of MECA-79-positive high endothelial venules correlate with lymphocytic infiltration and tumour regression in primary cutaneous melanoma. Histopathology 63, 852–861. [DOI] [PubMed] [Google Scholar]
- Bell AT, Mitchell JT, Kiemen AL, Fujikura K, Fedor H, Gambichler B, Deshpande A, Wu P-H, Sidiropoulos DN, Erbe R, et al. (2022). Spatial transcriptomics of FFPE pancreatic intraepithelial neoplasias reveals cellular and molecular alterations of progression to pancreatic ductal carcinoma. Preprint at bioRxiv. 10.1101/2022.07.16.500312. [DOI] [Google Scholar]
- Berry S, Giraldo NA, Green BF, Cottrell TR, Stein JE, Engle EL, Xu H, Ogurtsova A, Roberts C, Wang D, et al. (2021). Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockade. Science 372, eaba2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaize G, Daniels-Treffandier H, Aloulou M, Rouquié N, Yang C, Marcellin M, Gador M, Benamar M, Ducatez M, Song K. d., et al. (2020). CD5 signalosome coordinates antagonist TCR signals to control the generation of Treg cells induced by foreign antigens. Proc. Natl. Acad. Sci. USA 117, 12969–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, Mak TW, and Ohashi PS (2007). TNF-α is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Invest. 117, 3833–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattolico C, Bailey P, and Barry ST (2022). Modulation of type I interferon responses to influence tumor-immune cross talk in PDAC. Front. Cell Dev. Biol. 10, 816517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry C, Maestas DR, Han J, Andorko JI, Cahan P, Fertig EJ, Garmire LX, and Elisseeff JH (2021). Computational reconstruction of the signalling networks surrounding implanted biomaterials from single-cell transcriptomics. Nat. Biomed. Eng. 5, 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeskens G, and Hjort NL (2008). Model Selection and Model Averaging (Cambridge Books; ). [Google Scholar]
- Danilova L, Ho WJ, Zhu Q, Vithayathil T, De Jesus-Acosta A, Azad NS, Laheru DA, Fertig EJ, Anders R, Jaffee EM, et al. (2019). Programmed cell death ligand-1 (PD-L1) and CD8 expression profiling identify an immunologic subtype of pancreatic ductal adenocarcinomas with favorable survival. Cancer Immunol. Res. 7, 886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Marcisak EF, Deshpande A, Stein-O’Brien GL, Ho WJ, Laheru D, Jaffee EM, Fertig EJ, and Kagohara LT (2021). From bench to bedside: single-cell analysis for cancer immunotherapy. Cancer Cell 39, 1062–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alonso L, Handfield L-F, Roberts K, Nikolakopoulou K, Fernando RC, Gardner L, Woodhams B, Arutyunyan A, Polanski K, Hoo R, et al. (2021). Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 53, 1698–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MV, and Drake CG (2011). LAG-3 in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Aparicio M, and Alfaro C (2020). Significance of the IL-8 pathway for immunotherapy. Hum. Vaccin. Immunother. 16, 2312–2317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gorris MAJ, Halilovic A, Rabold K, van Duffelen A, Wickramasinghe IN, Verweij D, Wortel IMN, Textor JC, de Vries IJM, and Figdor CG (2018). Eight-color multiplex immunohistochemistry for simultaneous detection of multiple immune checkpoint molecules within the tumor microenvironment. J. Immunol. 200, 347–354. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM III, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges JF, Kimmel E, Snyder DT, Jerome M, and Jutila MA (2013). Solute carrier 11A1 is expressed by innate lymphocytes and augments their activation. J. Immunol. 190, 4263–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumann TR, Judkins C, Lim SJ, Wang H, Parkinson R, Gai J, Celiker B, Durham JN, Laheru DA, De Jesus-Acosta A, et al. (2022). Neoadjuvant and adjuvant antitumor vaccination alone or combination with PD1 blockade and CD137 agonism in patients with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 40, 558. [Google Scholar]
- Ho WJ, Jaffee EM, and Zheng L (2020). The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 17, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AC, Yarchoan M, Durham JN, Yusko EC, Rytlewski JA, Robins HS, Laheru DA, Le DT, Lutz ER, and Jaffee EM (2018). T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 3, e122092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ImmunoMindTeam (2019). Immunarch: An R Package for Painless Bioinformatics Analysis of T-Cell and B-Cell Immune Repertoires (Zenodo; ). 10.5281/zenodo.3367200. [DOI] [Google Scholar]
- Jiang HR, Gilchrist DS, Popoff JF, Jamieson SE, Truscott M, White JK, and Blackwell JM (2009). Influence of Slc11a1 (formerly Nramp1) on DSS-induced colitis in mice. J. Leukoc. Biol. 85, 703–710. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Yarchoan M, Lee V, Laheru DA, and Jaffee EM (2017). Strategies for increasing pancreatic tumor immunogenicity. Clin. Cancer Res. 23, 1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, and Nielsen M (2017). NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, and Qwangmsk GJ (2018). Mskcc/Vcf2Maf: Vcf2Maf V1. 6.16 (Zenodo; ). 10.5281/zenodo.1185418. [DOI] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawar B, Abbasi MH, and Sheikh N (2015). A panoramic spectrum of complex interplay between the immune system and IL-32 during pathogenesis of various systemic infections and inflammation. Eur. J. Med. Res. 20, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinny-Koster B, Guinn S, Tandurella JA, Mitchell JT, Sidiropoulos DN, Loth M, Lyman MR, Pucsek AB, Seppala TT, Cherry C, et al. (2022). Inflammatory signaling and fibroblast-cancer cell interactions transfer from a harmonized human single-cell RNA sequencing atlas of pancreatic ductal adenocarcinoma to organoid Co-culture. Preprint at bioRxiv. 10.1101/2022.07.14.500096. [DOI] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. Preprint at bioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- Li F-Y, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, Cohen JI, Uzel G, Su HC, and Lenardo MJ (2011a). Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F-Y, Lenardo MJ, and Chaigne-Delalande B (2011b). Loss of MAGT1 abrogates a Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes. Res. 24, S109–S114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, He Y, Hao J, Ni L, and Dong C (2018). High levels of Eomes promote exhaustion of anti-tumor CD8+ T cells. Front. Immunol. 9, 2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Rozich N, Wang J, Wang J, Xu Y, Herbst B, Yu R, Muth S, Niu N, Li K, et al. (2022). Anti-IL-8 antibody activates myeloid cells and potentiates the anti-tumor activity of anti-PD-1 antibody in the humanized pancreatic cancer murine model. Cancer Lett. 539, 215722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdó ttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The molecular signatures database hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz ER, Kinkead H, Jaffee EM, and Zheng L (2014a). Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. Oncoimmunology 3, e962401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al. (2014b). Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2, 616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayakonda A, Lin D-C, Assenov Y, Plass C, and Koeffler HP (2018). Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Enriquez E, and Garcia-Zepeda EA (2013). The multiple faces of CCL13 in immunity and inflammation. Inflammopharmacology 21, 397–406. [DOI] [PubMed] [Google Scholar]
- Muth ST, Saung MT, Blair AB, Henderson MG, Thomas DL 2nd, and Zheng L (2020). CD137 agonist-based combination immunotherapy enhances activated, effector memory T cells and prolongs survival in pancreatic adenocarcinoma. Cancer Lett. 499, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, Guo XV, Aggen DH, Rathmell WK, Jonasch E, et al. (2021). Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988–3005.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipov A, Murphy A, and Zheng L (2019). From immune checkpoints to vaccines: the past, present and future of cancer immunotherapy. Adv. Cancer Res. 143, 63–144. [DOI] [PubMed] [Google Scholar]
- Popovic A, Jaffee EM, and Zaidi N (2018). Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J. Clin. Invest. 128, 3209–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery AE (1995). Bayesian model selection in social research. Socio. Methodol. 25, 111–163. [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D, Huang TY-T, Shah R, Yang Y, Pan F, and Semenza GL (2020). BIRC2 expression impairs anti-cancer immunity and immunotherapy efficacy. Cell Rep. 32, 108073. [DOI] [PubMed] [Google Scholar]
- Sanchez-Paulete AR, Labiano S, Rodriguez-Ruiz ME, Azpilikueta A, Etxeberria I, Bolaños E, Lang V, Rodriguez M, Aznar MA, Jure-Kunkel M, et al. (2016). Deciphering CD137 (4–1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 46, 513–522. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, et al. (2015). PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T cell infiltration into pancreatic tumors. J. Immunother. 38, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, Lazarus J, Amir E.-a. D., Gunchick V, Espinoza C, et al. (2020). Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat. Cancer 1, 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, and Hingorani SR (2017). T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 5, 978–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, et al. (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui L, Wang S, Ganguly D, El Rayes T, Askeland C, Børretzen A, Sim D, Halvorsen OJ, Knutsvik G, Aziz S, et al. (2021). PRSS2 stimulates tumor growth by remodeling the TME via repression of Tsp1. Preprint at bioRxiv. 10.1101/2021.03.23.436667. [DOI] [Google Scholar]
- Tan YS, and Lei YL (2019). Isolation of tumor-infiltrating lymphocytes by Ficoll-Paque density gradient centrifugation. Methods Mol. Biol. 1960, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijeira A, Garasa S, Ochoa MC, Villalba M, Olivera I, Cirella A, Eguren-Santamaria I, Berraondo P, Schalper KA, de Andrea CE, et al. (2021). IL8, neutrophils, and NETs in a collusion against cancer immunity and immunotherapy. Clin. Cancer Res. 27, 2383–2393. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa T, Crocenzi T, Durham JN, Sugar EA, Wu AA, Onners B, Nauroth JM, Anders RA, Fertig EJ, Laheru DA, et al. (2020). Evaluation of cyclophosphamide/GVAX pancreas followed by listeria-mesothelin (CRS-207) with or without nivolumab in patients with pancreatic cancer. Clin. Cancer Res. 26, 3578–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, et al. (2017). Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 19, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, et al. (2020). A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276. [DOI] [PubMed] [Google Scholar]
- Veglia F, Perego M, and Gabrilovich D (2018). Myeloid-derived suppressor cells coming of age. Nat. Immunol. 19, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. (2020). Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183, 818–834.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik JB, Desai K, Avraam K, Vandebroek A, Dillon LM, Giacomazzi G, Rypens C, and Benci JL (2022). Consistent measurement of LAG-3 expression across multiple staining platforms with the 17B4 antibody clone. Preprint at bioRxiv. 10.1101/2022.02.21.481075. [DOI] [PubMed] [Google Scholar]
- Wolf Y, Anderson AC, and Kuchroo VK (2020). TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 20, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng XP, Zhu XY, Li BR, Pan CS, Hao L, Pan J, Wang D, Bi YW, Ji JT, Xin L, et al. (2018). Spatial distribution of pancreatic stones in chronic pancreatitis. Pancreas 47, 864–870. [DOI] [PubMed] [Google Scholar]
- Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, and Lenardo MJ (1995). Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 377, 348–351. [DOI] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The transcriptomic and clinical data used in this study are available in NCBI’s Gene Expression Omnibus (GEO) SuperSeries GSE197613 (GEO database: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197613) and are publicly available. Whole-exome sequencing data are deposited in the database of Genotypes and Phenotypes (dbGaP): phs003002.v1.p1. Details of the trial, data, contact information, proposal forms, and review and approval process are available at the following website: https://clinicaltrials.gov/ct2/show/NCT02451982. The trial protocol is provided in Data S1. The microscopy data reported in this paper will be shared by the lead contact upon request.
Code for genomics analysis available at: github.com/FertigLab/J1568_Bulk
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.