

Summary
Checkpoint inhibition immunotherapy has revolutionized cancer treatment, but many patients show resistance. Here we perform integrative transcriptomic and proteomic analyses on emerging immuno-oncology targets across multiple clinical cohorts of melanoma under anti-PD-1 treatment, on both bulk and single-cell levels. We reveal a surprising role of tumor-intrinsic SIRPA in enhancing antitumor immunity, in contrast to its well-established role as a major inhibitory immune modulator in macrophages. The loss of SIRPA expression is a marker of melanoma de-differentiation, a key phenotype linked to immunotherapy efficacy. Inhibition of SIRPA in melanoma cells abrogates tumor-killing by activated CD8+ T cells in a co-culture system. Mice bearing SIRPA-deficient melanoma tumors show no response to anti-PD-L1 treatment, whereas melanoma-specific SIRPA overexpression significantly enhances immunotherapy response. Mechanistically, SIRPA is regulated by its pseudogene, SIRPAP1. Our results suggest a complicated role of SIRPA in the tumor ecosystem, highlighting cell type-dependent antagonistic effects of the same target on immunotherapy.
eTOC Blurb
Zhou et al. reveal that tumor-intrinsic SIRPA can enhance the sensitivity to anti-PD-1 treatment in melanoma patients, whereas macrophage SIRPA has a well-established role as a major inhibitory modulator in antitumor immunity. This study highlights that the same target in different cell types can have antagonistic effects on immunotherapy.
Graphical Abstract
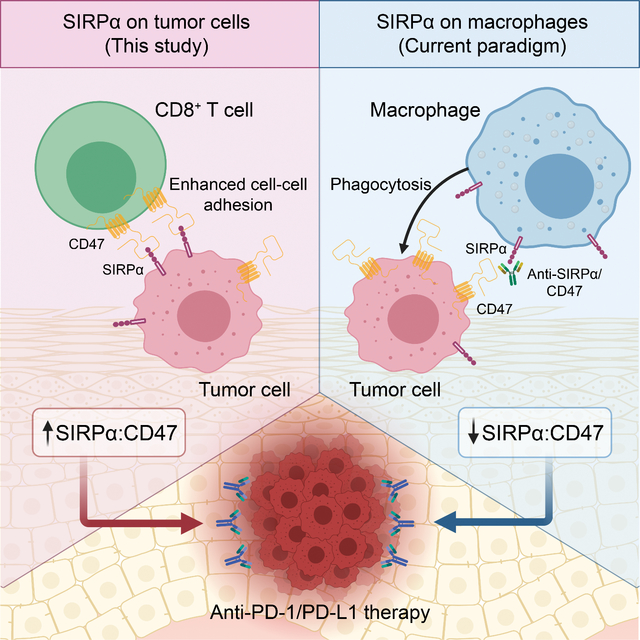
Introduction
Checkpoint inhibition immunotherapy has become one of the most successful strategies for cancer treatment and functions through the stimulation of the patient’s immune system (Hammerbacher and Snyder, 2017; Liu and Mardis, 2017). Many cancer patients, even those with advanced refractory cancers, show beneficial clinical responses, sometimes long-lasting ones, to checkpoint inhibitors targeting programmed death-1 (PD-1), programmed death-ligand 1 (PD-L1), and cytotoxic T lymphocyte antigen 4 (CTLA-4) (Hodi et al., 2010; Postow et al., 2015; Topalian et al., 2012). In particular, PD-1/PD-L1 blockades have achieved the most success in clinical development (Tan et al., 2020). However, the response rate varies by cancer type, ranging from ~10% to ~60% (Ansell et al., 2015; Yarchoan et al., 2017), and the average objective response rate is only ~26% across all cancer types (Shen et al., 2020). Combination therapy is a promising approach to overcoming PD-1/PD-L1 resistance and increasing the response rate (Zhang et al., 2020a). A recent study shows that ~80% of active trials in PD-1/PD-L1 blockades are testing combination regimens with other cancer therapies, such as immuno-oncology agents (Upadhaya et al., 2021). The daunting complexity of the human immune system and increased toxicity associated with combination therapy, however, necessitates the identification of key factors affecting the immunotherapy response and elucidating synergistic/antagonistic effects of different agents in the context of the whole tumor ecosystem (Bagaev et al., 2021; Ho et al., 2022; Newell et al., 2022).
In recent years, several studies have generated transcriptomic or proteomic profiles of clinical patient cohorts under anti-PD-1 treatment, which provide rich resources to characterize key regulators affecting immunotherapy response. However, it remains challenging to digest these data to make translational impacts for several reasons. First, due to limited sample sizes of clinical cohorts, the datasets obtained are often underpowered to detect key changes, especially given the multiple-testing burden in a genome-wide survey. To overcome this, it would be more powerful to focus on a subset of clinically actionable targets and assess the signal robustness across multiple cohorts. Second, clinical samples have highly heterogeneous cell compositions that confer additional complexity in analysis (Zaitsev et al., 2022): bulk samples reflect the average of mixed cell types, while single-cell data usually have very limited patient representativeness. Therefore, it is essential to perform complementary bulk sample and single-cell analyses and borrow the information from each other (Davis-Marcisak et al., 2021). Last but not least, pure clinical phenotype-driven association analyses often generate statistically significant but biologically trivial hits. It is also important to consider the pattern of potential targets in disease progression or lineage plasticity. The convergent hits of the above two analyses would give more creditable hypotheses for subsequent investigation. With these considerations in mind, we focused on the anti-PD-1 treated patient cohorts of melanoma, the frontier of checkpoint inhibition immunotherapy, and performed an integrative analysis of bulk and single-cell data across multiple clinical cohorts, to obtain insights that can maximize the benefits of immunotherapy.
Results
High SIRPA expression correlates with response to PD-1 blockade in bulk samples
To identify effective therapies that can potentially overcome the resistance to checkpoint inhibitors, we focused on a set of emerging immuno-oncology target genes and performed an integrative analysis across five melanoma patient cohorts under anti-PD-1 treatment (four with available transcriptomic data and one with proteomic data) (Figure 1A). We collected 60 immuno-oncology target genes that have active agents tested in ≥ 10 active clinical trials (Table S1). Given these highly actionable targets, for each of the five patient cohorts, we performed two parallel analyses: (i) differential expression analysis between responding and non-responding groups; and (ii) patient survival analysis between high expression and low expression groups of each gene (Figure 1A). Through the differential expression analysis, seven genes showed significant expression-response correlations in multiple cohorts (P ≤ 0.05, FDR ≤ 0.15, Figure 1B). Among these genes, SIRPA showed the most consistent pattern in the differential expression analysis across different patient cohorts (Figure 1C).
Figure 1. An integrative IO-target analysis across anti-PD-1 patient cohorts.

(A) The overall procedure of our integrative IO-target analysis. The bar plot indicates the number of active clinical trials per target. (B) Venn diagram showing the overlap of differentially expressed (DE) genes identified in five anti-PD-1-treated melanoma patient cohorts. (C) A summarized plot showing seven DE genes identified in at least two cohorts; red and dark-blue, strong significance with FDR ≤ 0.15; pink, regular significance with P’ ≤ 0.05; * indicates their concordance with the results of survival analysis. (D, E) Boxplots and Kaplan-Meier (KM) plots showing the associations between the mRNA- (D) and protein-level (E) expressions of the top candidate gene, SIRPA, and anti-PD-1 responses or patient survival times in the anti-PD-1 response cohorts. For box plots, the middle line in the box is the mean, the bottom and top of the box are the first and third quartiles, and the whiskers extend to the 1.5× interquartile range of the lower and the upper quartiles, respectively. To assess the differences in SIRPA mRNA expression between anti-PD-1 responding and non-responding groups robustly, three differential expression tests, DESeq2, edgeR, and limma, were used. All three tests yielded strong significant results (P ≤ 0.05 with FDR ≤ 0.15), and the most significant two-sided P value from the three is shown on the boxplot. Otherwise, one-sided P’ values are shown to indicate marginal significance. To assess the difference in SIRPα protein expression, the Mann-Whitney U test was used, and the P-value from a permutated random distribution is shown on the boxplot. For Kaplan-Meier plots, patients were split into two equal-size groups with the median as the cutoff. Log-rank tests were used to assess the difference in patient survival times between the two groups. See also Figure S1 and Table S1.
SIRPA is an emerging target in the cancer immunotherapy (Uger and Johnson, 2020; Xiang et al., 2021). The protein product of SIRPA, signal regulatory protein α1 (SIRPα), also known as CD172a or SHPS-1, is a multifunctional transmembrane glycoprotein (Barclay and Brown, 2006). SIRPα was thought to be selectively expressed on myeloid cells (e.g., macrophages, dendritic cells, and neutrophils) and neurons (Adams et al., 1998). But recently, many tumor cell lines have been shown to express SIRPα on their surface (Yamasaki et al., 2007; Yanagita et al., 2017). SIPRα exerts its effects through interaction with its ligand CD47, a transmembrane glycoprotein, ubiquitously expressed in different cell types and often overexpressed in solid and hematological tumors. Our results revealed that tumor samples with high SIRPA expression were significantly more sensitive to PD-1 blockade not only at the RNA level (P = 0.01, and FDR < 0.15 in the Hugo cohort, and P = 0.05 in the Gide and Liu cohorts, Figure 1D) but also the protein level (P = 0.01 and FDR < 0.15 in the Harel cohort, Figure 1E). Furthermore, patient survival analyses in multiple cohorts showed that the patient group with high SIRPA expression had a better prognosis than that with low SIRPA expression (log-rank test, P = 0.016 in the Hugo cohort, Figure 1D; P = 0.026 in the Harel cohort, Figure 1E). Among the five cohorts assessed, the Riaz cohort is the only one that did not show any significant patterns in tumor response or patient survival analyses, likely because (i) this cohort has a small sample size and (ii) the majority of the patients underwent complicated treatments. To further evaluate its potential as an immunotherapy biomarker, we found that SIRPA expression showed no correlation with response to other therapies such as BRAF inhibitors (Figure S1A, S1B), established ICB biomarkers such as tumor mutation burden (Figure S1C), or prognosis in the general patient population (Figure S1D). These results suggest a positive and unique role of SIRPA in tumors responding to anti-PD-1 immunotherapy.
High SIRPA expression in anti-PD-1 responding tumors comes from tumor cells
SIRPα on macrophages, interacting with CD47 on tumor cells or T cells, was recently established as the first macrophage checkpoint (Chao et al., 2019; Jalil et al., 2020; Uger and Johnson, 2020). Since the interaction between SIRPα-bearing macrophages and CD47-positive tumor cells triggers a “do not eat me” signaling cascade to inhibit the phagocytosis of tumor cells, the blockade of such an interaction would lead to efficient phagocytosis of tumor cells by macrophages. In light of this mechanism, several clinical trials were launched to target the SIRPα-CD47 interaction, alone or in combination with the anti-PD-1 treatment (Jalil et al., 2020), for a number of cancer types, including melanoma. However, this specific paradigm seemingly contradicts the SIRPA-related favorable response to the anti-PD-1 treatment we observed here.
To resolve this paradox, one key question is which cells contribute to the high expression of SIRPA in anti-PD-1-responding tumors since all the above analyses were based on bulk samples consisting of a mixture of tumor cells and various non-tumor cells. Since tumor cells constitute a substantial proportion of a bulk tissue sample, as demonstrated by tumor purity often >70% both in the surveyed melanoma dataset (Hugo et al., 2016) and a larger pan-cancer cohort (Aran et al., 2015), we hypothesized that the difference in SIRPA expression level between patients with distinct clinical benefits could mainly reflect a tumor-intrinsic SIRPA pattern. To validate this, we collected multiple single-cell RNA-seq (scRNA-seq) datasets from melanoma patients with diverse clinical backgrounds, where the expression patterns of SIRPA can be investigated in individual cell populations. First, we performed the analysis based on two published scRNA-seq datasets of melanoma patients (Jerby-Arnon et al., 2018; Smalley et al., 2021). In the Jerby-Arnon dataset, we found that SIRPA mRNA expression was comparably enriched in melanoma, macrophages, and monocyte cells while completely depleted in stromal, endothelial, and non-monocyte immune cells (Figure 2A–C). We observed the same phenomenon in another scRNA-seq dataset of distant melanoma metastases (Smalley et al., 2021), suggesting a stable and abundant expression of SIRPA in melanoma cells even after developing a secondary malignant growth, regardless of the metastatic site (Figure 2D–F). Importantly, although tumor cells were primarily clustered by patients, indicating significant inter-patient transcriptomic heterogeneity, considerable SIRPA expression was detected in most of the patients, demonstrating a ubiquitous presence of SIRPA in melanoma cells. Consistent with the findings in human patient samples, melanoma cell lines are among those with the highest SIRPA expression at both RNA and protein levels across >20 lineages (Figure S2A, B). Finally, we analyzed a single-cell proteomics dataset of a melanoma cell line (WM989) and a monocyte cell line (U937) (Leduc et al., 2022) and found that SIRPA was expressed at a ubiquitously high level in both cell types, indicating that SIRPA expression is constitutive of the melanoma cell state (Figure 2G, H).
Figure 2. SIRPA expression in tumor cells and macrophages in melanoma patient samples.

(A, B) UMAP plot visualizing cell type annotations (A) and SIRPA expression (B) in single cells of a melanoma patient cohort from Jerby-Arnon et al. (C) Top panel, a heatmap showing the proportion of cells positive for four melanoma gene markers in all melanoma cells of each patient. Middle panel, a heatmap showing the proportion of SIRPA+ cells in different cell types of each patient. Right panel, a bar plot showing the proportion of SIRPA+ cells in different cell types where cells from all patients are combined. (D-F) Same as (A-C) but for another melanoma patient cohort from Smalley et al. (G) UMAP plot visualizing protein expression levels of six melanoma markers (top panel) and six monocyte markers (bottom panel) in single cells of a melanoma cell line (top panel) and a monocyte cell line (bottom panel), respectively. (H) UMAP plot visualizing SIRPα protein expression level in the two cell lines as mentioned in (G). (B, E, G, H) The color key indicates normalized mRNA expression for a gene of interest. (C, F) The color key indicates the proportion of positive cells. (I) Left panel, the workflow of deconvoluting bulk gene expression profiles into cell type-specific gene expression profiles. Right panel, violin plots showing differential SIRPA expression by deconvolution between responding and non-responding groups in melanoma cells and macrophages, respectively. R, responding to anti-PD-1 therapy; NR, non-responding. (J, K) Violin plots showing differential SIRPA expression by scRNA-seq between anti-PD-1 treatment-naïve and post-treatment resistant groups in melanoma cells (J) or between responding and non-responding groups in macrophages (K). (I, J, K) Each violin plot shows the data distribution using a kernel density estimation. The width of the violin plot represents a probability that the data points will take on the given value, and the top and bottom lines indicate the maximal and minimal data values. The bottom and top of the inner box are the first and third quartiles, and the whiskers extend to the 1.5× interquartile range of the lower and the upper quartiles, respectively. See also Figure S2.
We next aimed to elucidate the exact contribution of tumor cells and macrophages to high SIRPA signals in tumors responding to anti-PD-1 treatment. As there is no single-cell profile of both cell types in the bulk sample cohorts surveyed, we first applied a computational deconvolution algorithm, CIBERSORTx (Newman et al., 2019) to the bulk RNA-seq dataset of Hugo et al., to infer tumor- and macrophage-specific gene expression profiles for each sample, using the Tirosh scRNA-seq dataset (Tirosh et al., 2016) as a cell signature reference (Figure 2I). We found that SIRPA was significantly up-regulated in melanoma cells of patients responding to anti-PD-1 treatment (P = 0.022, Figure 2I) but significantly down-regulated in macrophages for the responding group (P = 0.033, Figure 2I). To further confirm this pattern, we obtained another two scRNA-seq datasets that respectively surveyed tumor and macrophage transcriptomes in melanoma patients receiving anti-PD-1 treatment. With the first dataset (Jerby-Arnon et al., 2018), we showed that SIRPA was expressed less abundantly in the melanoma cells of the post-treatment resistant group compared to the treatment-naïve group (P = 8.5×10−4, Figure 2J). In the second dataset (Sade-Feldman et al., 2018), we found that SIRPA was significantly down-regulated in macrophages responding to anti-PD-1 therapy (P = 1.1×10−7, Figure 2K). Collectively, these results, from both deconvoluted bulk samples and single-cell profiling data, suggest that the high expression of SIRPA correlated to anti-PD-1 treatment response is due to melanoma cells rather than macrophages.
SIRPA is a melanocytic marker that decreases during melanoma progression
Having established the clinically relevant, high expression of SIRPA in melanoma cells, we sought to further explore its role in melanoma biology. Because cellular de-differentiation is a key axis of the melanoma phenotype (Agaimy et al., 2016; Kohler et al., 2017; Riesenberg et al., 2015), we asked whether the SIRPA expression dynamics reside in a meaningful topology along the melanoma de-differentiation trajectory. To address this question, we analyzed an RNA-seq dataset comprising a panel of human melanoma cell lines spanning four consecutive differentiation stages, namely, un-differentiated, neural crest-like, transitory, and melanocytic (Tsoi et al., 2018). Intriguingly, SIRPA expression appears to be a monotonically increasing function of the melanoma differentiation status, as readily visualized in the embedded PCA space, with the most differentiated melanoma cells showing the strongest expression of SIRPA (Figure 3A, B). We further measured the SIRPα protein expression using reverse phase protein arrays (RPPAs) in an independent collection of 48 melanoma cell lines with parallel publicly available RNA-seq and quantitative proteomics data. After scoring each cell line for differentiation status using a signature derived from a previous study (Tsoi et al., 2018), we found a very strong correlation between SIRPα protein abundance and melanoma differentiation (Figure 3C–E). Melanocytes are known to be the cell of origin of melanoma tumors (Gupta et al., 2005; Kohler et al., 2017; Moon et al., 2017). Thus, we hypothesized that SIRPA would show a similar pattern of expression on a melanocyte maturation trajectory. Indeed, based on an RNA-seq dataset derived from cultured human melanocytes and their progenitors (Mica et al., 2013), we showed that the high-expression status of SIRPA is gradually instituted as melanocytes reach maturity (Figure 3F, G).
Figure 3. SIRPA expression dynamics in melanocyte maturation and melanoma de-differentiation.

(A, B) PCA projection of human melanoma cell lines from Tsoi et al., based on gene expression profiles and colored by de-differentiation stages (A) or normalized SIRPA expression level (B). (C-E) PCA projection of human melanoma cell lines from CCLE, based on gene expression profiles and colored by differentiation score (C), SIRPα protein by RPPA (D), or SIRPα protein expression by quantitative proteomics (E). (F, G) PCA projection of in vitro differentiating human melanocytes from Mica et al., based on gene expression profiles and colored by differentiation time (F) or normalized SIRPA expression level (G). (H-J) Two-dimensional t-SNE projection of human skin single cells from Belote et al., colored by cell type (H), developmental stage (I), and normalized SIRPA expression level (J). The color key indicates the normalized SIRPA expression (A, B, F, G, J), differentiation score (C), SIRPα protein expression by RPPA (D), or SIRPα protein expression by quantitative proteomics (E). (K) Top panel, a scatterplot of TCGA melanoma (TCGA-SKCM) samples ranked by SIRPA expression level. Bottom panel, heatmap showing biological and clinical features of ordered TCGA-SKCM samples. The Kruskal-Wallis test was used to compute P values for the association of SIRPA expression with de-differentiation stages, mutational subtypes, and tumor sites. The Spearman’s rank correlation was used to evaluate the association of SIRPA expression with tumor stage and tumor purity. See also Figure S3.
Bridging the tumorigenic melanoma de-differentiation process and the physiological melanocytic differentiation process, we next aimed to monitor SIRPA expression in a setting where melanocyte-to-melanoma transformation is captured. A recent study on the stepwise introduction of oncogenic mutations into primary human melanocytes generated scRNA-seq data on genetically distinct melanoma cellular models (Hodis et al., 2022) (Figure S3A). This allowed us to query SIRPA expression along a phylogenetically related trajectory of melanoma from its normal cell of origin and associate SIRPA expression shifts with key oncogenic events. Using well-established melanocytic markers (e.g., PMEL, MLANA, MITF, and TYR) to form a reference for the differentiation program, we observed that this highly matched SIRPA expression changes between the populations, including a drop when cells obtained replicative immortality through a TERT promoter mutation and an increase when a PTEN exon mutation led to MITF duplication (Figure S3B).
Finally, to validate our findings on the in vitro SIRPA expression dynamics in melanocytes and melanoma cells in real-world physiological and pathological contexts, we further performed two analyses on data from human samples. First, we analyzed a scRNA-seq dataset of human skin samples across three developmental stages (Belote et al., 2021): fetal, neonatal, and adult. Consistent with the in vitro data, we found a ubiquitous expression pattern of SIRPA in melanocytes of all developmental stages (Figure 3H–J). Interestingly, SIRPA expression was exclusive to melanocytes, emphasizing its important role in establishing the identity of this cell type. A pseudo-time trajectory-based analysis of fetal-stage melanocytes during the establishment of their identity further characterized SIRPA as a marker gene of melanocytic maturation (Figure S3C). Second, we re-analyzed TCGA cutaneous melanoma cohort (n = 472) with a specific focus on the association between SIRPA expression and well-established pathological/clinical features. We found that the expression level of SIRPA was highly negatively correlated with melanoma tumor de-differentiation (Figure 3K), consistent with our observation in the cell line data. Together, these results support that SIRPA is a melanocytic marker whose loss is a hallmark of melanoma progression.
SIRPA loss confers anti-PD-L1 resistance through the interaction of tumor cells and CD8+ T cells
Melanoma de-differentiation is a known mechanism of resistance to T cell-mediated immunotherapy through loss of melanocytic antigens, which has been shown in both mice (Landsberg et al., 2012) and humans (Mehta et al., 2018). Based on our observations that SIRPA, a previously under-studied melanocytic antigen gene, showed strong positive correlations with both anti-PD-1 immunotherapy response and melanoma differentiation status, we hypothesized that SIRPA is directly involved in enhancing T cell-mediated immunotherapy, and its loss then serves as a mediator of immunotherapy resistance that accompanies the attenuation of melanocytic identity.
To identify a possible molecular mechanism for this phenotype, we first focused on the canonical SIRPα-CD47 interaction model. According to the current research paradigm, this interaction provides an antiphagocytic signal that modulates the crosstalk between macrophages and tumor cells (Morrissey et al., 2020; Takizawa and Manz, 2007; Willingham et al., 2012). However, given our findings on SIRPA expression prevailing in tumor cells and a widespread presence of CD47 in all major immune cell populations (Figure S4A–D), SIRPα-CD47 interaction may contribute to the communication among alternative cell-type combinations. To identify the donor and receptor cell populations that host SIRPα and CD47 in the melanoma ecosystem, we analyzed a melanoma spatial transcriptome dataset (Thrane et al., 2018), where the localization of the tumor, stromal, and immune cells was determined using H&E staining (Figure 4A) and was consistent with our gene expression-based deconvolution results (Figure 2I). The juxtaposition of slide-wide cell population distributions and the expression patterns of SIRPA and CD47 showed enrichment of SIRPA in melanoma cells and its co-localization with CD47 in T cells (Figure 4A). Given that tumor-intrinsic SIRPA overexpression in patient samples correlates with a favorable response to PD-1 blockade therapy, we hypothesized that such an effect is caused by an enhanced SIRPα-CD47 communication between melanoma cells and T cells. To test this hypothesis, we inferred a SIRPα-CD47 interaction score using CellPhoneDB (Efremova et al., 2020) based on the co-expression patterns of SIRPA in melanoma cells and CD47 in CD8+ T cells in the Tirosh scRNA-seq dataset (Tirosh et al., 2016). Indeed, we observed a significant SIRPα-CD47 interaction score between melanoma cells and CD8+ T cells (P = 0.008, permutation test, Figure 4B).
Figure 4. Effect of SIRPα inhibition on T-cell-mediated antitumor response in melanoma cells.

(A) Hematoxylin and eosin (H&E) stained tissue image of a melanoma biopsy (left panel, adopted from Fig. 3A in Thrane et al.) with the slide-wide distribution of cell type signature scores (middle panel), and the expression patterns of SIRPA and CD47 (right panel). The color key indicates signature score or normalized gene expression. (B) Histogram showing the distribution of receptor-ligand interaction scores for SIRPA in melanoma cells and CD47 in CD8+ T cells computed from the random shuffling of cell type labels. The red dotted line denotes the real score corresponding to the co-expression pattern of SIRPA in melanoma cells and CD47 in CD8+ T cells of the Tirosh cohort. (C) A co-culture system quantifying tumor cell viability upon perturbations. (D) Smoothed histograms showing cell surface SIRPα expression detected by flow cytometry after SIRPA perturbation, knockdown (KD), or overexpression (OE). The knockdown cell lines were constructed by multiple shRNAs. SIRPA KD-E, showing the most robust knockdown efficiency, was selected for downstream experiments. Isotype and B16F10 NTC are the negative and positive controls, respectively. (E, F) Line charts showing relative survival rates of tumor cells at 24 h in co-cultures of different ratios of B16F10 and CD8+ T cells (Pmel-1) without (E) and with (F) the addition of mouse anti-PD-L1 antibody (mPD-L1). (G) Bar plot showing gene expression levels of SIRPA along with six melanoma differentiation antigens in B16F10 cells with SIRPA KD, OE, or control. sig., log2 fold change > 1 and adjusted P < 0.05 by DESeq2; ns., log2 fold change < 1 or adjusted P > 0.05 by DESeq2. (H) Bar plots showing relative survival rates of tumor cells at 24h in co-cultures with CD8+ T cells (Pmel-1) pre-treated with MIAP410 or MIAP430 antibodies to block CD47-SIRPα interaction. (E, F, H) The results are based on three independent mouse experiments, each with three replicates, and the error bars indicate mean ± SEM. P values are based on Student’s t-test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S4.
To directly examine the effect of SIRPA on T cell-dependent tumor immune responses, we designed a co-culture system and performed cytotoxic T cell killing assays (Figure 4C). We established two stable cell lines with verified SIRPα knockdown (KD) and overexpression (OE) (Figure 4D) and confirmed that SIRPA perturbation in tumor cells had no impact on in vitro tumor growth during the co-culture experiment (Figure S4E). We then co-cultured these cells with activated cytotoxic CD8+ T cells for 24 h. We found that cytotoxic T cells killed tumor cells with SIRPα overexpression more efficiently compared to the non-targeting controls (NTC), whereas tumor cells with the loss of SIRPα were more resistant to T cell-mediated killing (Figure 4E). This suggests a positive role of SIRPα on the tumor cell surface in T cell-mediated tumor cytotoxicity. To further confirm the role of SIRPA in the context of anti-PD-1/PD-L1 treatment, we added an anti-mouse PD-L1 antibody into the co-culture system and observed that the difference in T cell killing effect was even more striking (Figure 4F) than in the setting without the antibody (Figure 4E). The same results were observed when adopting an additional SIRPA-knockdown cell line with an extended co-culture time of 96 h (Figure S4F, G). To rule out the possibility that SIRPA perturbation leads to expression changes of melanoma differentiation antigens (MDA), which directly mediate the immunogenicity of melanoma cells (Pitcovski et al., 2017), we profiled the transcriptomes of SIRPA-KD and SIRPA-OE B16F10 cells and found no significant expression changes of six well-established MDA-encoding genes, PMEL, TYR, TYRP1, DCT, MLANA, and MITF (Figure 4G). To provide more direct support for the proposed immunostimulatory activity of SIRPA via CD47-mediated interactions with CD8+ T cells, we utilized two antibodies capable of blocking the SIRPα-CD47 interaction, MIAP410 and MIAP430 (Han et al., 2000; Willingham et al., 2012), to pretreat the T cells before the co-culture and tested whether this blockade would affect T cell-mediated cytotoxicity. The results showed that the tumor-killing effect was hampered in T cells with CD47 blocked, and the effect was enhanced in the culture with SIRPA-overexpressing melanoma cells (Figure 4H). Taken together, these results suggest that melanoma-intrinsic SIRPA can effectively trigger T cell immunogenicity through CD47 interaction in PD-1/PD-L1 blockade immunotherapy.
Tumor-intrinsic SIRPA enhances the antitumor response of checkpoint blockade in mice
To investigate whether melanoma-intrinsic SIRPA affects the efficacy of checkpoint inhibition immunotherapy in vivo, we utilized the well-established B16F10 murine melanoma model. We subcutaneously inoculated B16F10 cells with different SIRPA perturbations-overexpression (mSIRPA-OE), knockdown (mSIRPA-KD), and non-target control (NTC)- into C57BL/6J mice. We implemented a two-step randomization strategy to ensure homogenous tumor volumes before treatment with anti-mPD-L1 or isotype control (Figure 5A). Compared to tumors treated with isotype control, tumors overexpressing SIRPA showed the most remarkable response to anti-PD-L1 treatment, both at specific time points and across time points (P < 10−3); NTC tumors showed a moderate but significant response to the treatment (P < 10−2); whereas tumors bearing SIRPA knockdown essentially exhibited no response (Figure 5B, C, Figure S5). We next examined the tumor volume changes from baseline (anti-mPD-L1 vs. isotype control) at a single mouse scale. At different time points (day 10, 12, 14, and 16), the three mouse groups showed distinct tumor-volume change rates from each other: all the mice in the mSIRPA-OE group consistently benefited from anti-mPD-L1 treatment and showed considerable tumor shrinkage, and in some cases, tumors even disappeared completely; most mice in the NTC group showed response to the treatment, but in a small proportion of mice, the tumor size increased; in sharp contrast to mSIRPA-OE, approximately half of the mice in the mSIRPA-KD group suffered from tumor expansion (Figure 5D, E). Finally, we examined the impact of different SIRPA perturbations on animal survival. The mSIRPA-OE mice treated with anti-mPD-L1 showed a much better prognosis than those treated with isotype control (log-rank test, P < 10−4), and the median survival time increased from 14 days to 20 days; NTC mice showed a marginally significant survival benefit (log-rank test, P < 0.08); mSIRPA-KD mice showed no difference at all (log-rank test, P < 0.8) (Figure 5F). Collectively, these results provide strong evidence supporting a positive role of tumor-intrinsic SIRPA in PD-1/PD-L1 induced, T-cell-mediated antitumor immunity.
Figure 5. Effects of SIRPα expression levels on anti-PD-L1 treatment response in mice.

(A) A graphic description of the mouse experiment design. Left panel, the workflow; right panel, the schedule of tumor inoculation and treatments. Three tumor cell lines, NTC, mSIRPA-OE, and mSIRPA-KD were inoculated, followed by two treatments, Isotype control, and mPD-L1. In total, six mouse groups were tested and compared. (B, C) Curves showing the tumor growth in 16 days for each mouse group. Averaged tumor volumes of the six mouse groups are shown in one plot for a universal comparison (B). The effects of the mPD-L1 antibody on tumor volumes are shown in the three tumor cell lines (C). Student’s t-tests (two-sided) were used to compare the mouse groups of different treatments at each time point. Paired Student’s t-tests (two-sided) were used to compare the two curves within each tumor cell line based on the averaged tumor volumes. The error bars indicate mean ± SEM. (D) Waterfall plots visualizing the tumor volume changes from the baseline (Isotype control) after mPD-L1 antibody treatment for every single mouse of the three tumor cell lines on days 10, 12, 14, and 16. Student’s t-tests (two-sided) were used to compare the mouse groups of different cell lines. (E) Boxplots showing averaged changes from baseline of the four time points for each tumor cell line. The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to the 1.5× interquartile range of the lower and the upper quartiles, respectively. The three groups were compared by using paired Student’s t-tests (two-sided). (F) Kaplan-Meier plots showing the mice survival rate upon anti-PD-L1 treatment for each tumor cell line. The difference between curves was tested by log-rank tests. (C-E) *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. See also Figure S5.
SIRPα expression is positively regulated by SIRPAP1 in melanoma
To identify potential regulators that affect the heterogeneity of SIRPα protein expression in melanoma patients, we quantified SIRPα protein expression in 349 TCGA melanoma samples using RPPAs and then performed an association analysis with other TCGA molecular profiling data, including DNA methylation, somatic mutation, somatic copy-number alteration, and the expression of its endogenous pseudogene, SIRPAP1. Among the three cis-regulatory features, SIRPA gene amplification showed a significant positive correlation, but the effect size was limited (Figure 6A, B). In the Hugo cohort (Hugo et al., 2016), SIRPA gene amplification showed no relation to SIRPA expression (Figure S6A). Intriguingly, SIRPAP1 RNA expression was strongly correlated with SIRPα expression (Figure 6A, C). We further confirmed this pattern in CCLE melanoma cancer cell lines (Figure S6B) based on quantitative proteomics data. These results suggest that SIRPAP1 is a key noncoding regulator for SIRPα.
Figure 6. Regulation of SIRPα protein expression in melanoma patients.

(A-C) Summarized plot showing possible regulations of SIRPα protein expression across TCGA-SKCM patients assayed by RPPA in this study, including SIRPA DNA methylation, somatic mutation, somatic copy-number alteration (SCNA), and SIRPAP1 expression. (A) Top panel, barplot of TCGA-SKCM samples ranked by SIRPα protein expression. Color keys indicate normalized levels for SIRPα protein expression, SIRPA DNA methylation, and SIPRAP1 expression, respectively. Bottom panel, heatmap showing different regulatory features. (B) Boxplots showing the association between gene amplification and SIRPα protein expression. Patient samples with somatic copy number with log2 transformation > 1 are denoted as amplified, and the others as non-amplified. The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to the 1.5× interquartile range of the lower and the upper quartiles, respectively. (C) Scatterplot showing the correlation between SIRPα and SIRPAP1 RNA expression. The correlation coefficient and P-value are based on Spearman’s rank correlation. (D) Correlation plot showing the miRNAs which are negatively correlated with SIRPA expression in TCGA-SKCM samples. The color bar represents Spearman’s rank correlation coefficient. (E, F) Scatterplots showing the correlations between let-7a-2–3p and SIRPA (E) or SIRPAP1 (F) expression. (G) Cartoon summary of SIRPAP1 overexpression by lentivirus transduction. (H, I) Western blot (H) and Flow cytometry (I) of SIRPα protein expression upon vector-based SIRPAP1 overexpression. (J) Cartoon summary of SIRPAP1 overexpression by CRISPR/Cas9 SAM system. (K, L) Western blot (K) and flow cytometry (L) of SIRPα protein expression upon CRISPR/Cas9-based SIRPAP1 overexpression. See also Figure S6.
To dissect the competitive relationship between SIRPA and SIRPAP1 and its mediators, we conducted a miRNA-centered analysis (Figure S6C). We adopted three common miRNA target prediction tools, RNAhybrid (Kruger and Rehmsmeier, 2006), miRDB (Wong and Wang, 2015), and TarPmiR (Ding et al., 2016), to identify potential miRNAs and their corresponding target sites shared by SIRPA and SIRPAP1. We then performed an association analysis on the expression levels between the miRNA candidates and SIRPA or SIRPAP1 using TCGA melanoma data. We identified three miRNAs, let-7a-2–3p, miR-149–3p, and miR-3154, that targeted the homologous regions of SIRPA and SIRPAP1 and exhibited a significantly negative correlation with both of their expression levels (Figure 6D, S6D). In particular, let-7a-2–3p showed the strongest anti-correlation with SIRPA/SIRPAP1 expression (Figure 6E, F). These results suggest that the co-regulation of SIRPA and SIRPAP1 in melanoma is mediated by a group of miRNAs.
To test whether SIRPAP1 can causally regulate SIRPα expression in melanoma cells and whether such regulation can affect the protein abundance on the cell surface, we overexpressed SIRPAP1 in A375 melanoma cells (Figure 6G) and measured the total and membrane-anchored SIRPα level by western blot and flow cytometry, respectively (Figure 6H, I). Indeed, we found that the SIRPAP1 overexpression greatly increased the total and cell surface SIRPα level. To further confirm this observation, we utilized the CRISPR/Cas9 synergistic activation mediator (SAM) system to transcriptionally activate SIRPAP1 in A375 cells (Figure 6J). We observed the same increased protein expression after induced SIRPAP1 overexpression (Figure 6K, L). Thus, we established SIRPAP1 as a positive regulator of the SIRPα expression on the surface of melanoma cells.
Taken together, we propose a model of tumor-intrinsic SIRPα-mediated immunotherapy response (Figure 7). Specifically, multiple mechanisms contribute to SIRPα expression heterogeneity, including SIRPAP1 as a ceRNA to upregulate SIRPA. The tumor cells with high SIRPα expression on the surface then interact with CD47 on CD8+ T cells. Such interaction may enhance cell-cell adhesion between tumor cells and CD8+ T cells, thereby facilitating T cell killing activity. As a result, patients whose tumors carry a high expression of SIRPA show favorable responses to anti-PD-1 immunotherapy.
Figure 7. The proposed model of SIRPα-mediated T-cell-centric immunotherapy response.

The proposed model consists of three consecutive layers, from the molecular mechanism to impacts on clinical outcomes. The left and right sides show the contrasting situations of SIRPA high expression vs. SIRPA low expression in tumor cells. On the left side, within tumor cells, SIRPAP1 upregulates SIRPA by functioning as a competing endogenous RNA; within the tumor microenvironment (TME), SIRPα on the surface of tumor cells interacts with CD47 on CD8+ T cells to enhance cell-cell adhesion between these two cell types; and consequently, the enhanced cell-cell interaction increases the T cell killing effect, leading to a better response to anti-PD-1/PD-L1 therapy. In contrast, on the right side, tumor cells with low SIRPA expression have moderate cell-cell adhesion with CD8+ T cells, thereby rendering resistance to anti-PD-1/PD-L1 therapy. The bottom panels show clinical implications of the proposed model in terms of indication for treatment, immunotherapy biomarker, and therapeutic development.
Discussion
Checkpoint inhibition immunotherapy has revolutionized cancer treatment via leveraging the cytotoxic potential of the human immune cells, especially cytotoxic T cells. Yet, we still have a very limited ability to predict patients’ responses to immunotherapy. In this study, focusing on emerging immune-oncology targets, we developed an integrative analysis strategy to prioritize actionable targets in combination with anti-PD-1/PD-L1 therapy. Combining bulk and single-cell RNA-seq datasets from melanoma patients, we showed that high SIRPA expression correlated with a favorable response to anti-PD-1 treatment and that it is melanoma cells, rather than macrophages, that contribute to the observed pattern. Through both in vivo and in vitro experiments, we further demonstrated that tumor-intrinsic SIRPA promotes T cell-mediated immunotherapy response. This is in sharp contrast to the well-established role of SIRPA as a major inhibitory immune modulator in macrophages.
Although the CD47-SIRPα signaling axis is an innate immune checkpoint in cancer, durable antitumor responses require an adaptive immune cell stimulation (Sockolosky et al., 2016). Several studies suggest the synergic effect of the anti-CD47-SIRPα signaling axis and anti-PD-1/PD-L1 therapy in syngeneic mouse models (Kuo et al., 2020; Sockolosky et al., 2016; Yanagita et al., 2017). Our results from the analysis of clinical patient cohorts and functional assays reveal a positive role of tumor-intrinsic SIRPA in the activated T cell-mediated cytotoxicity. Interestingly, a recent study identified a functional subpopulation of SIRPA+ CD8+ T cells in humans and mice during chronic immune exhaustion, and they show that these T cells kill the CD47+ target more efficiently compared to the SIRPA− CD8+ T cells both in vivo and in vitro (Myers et al., 2019). Similarly, our data showed that tumor cells with SIRPA overexpression were sensitive to cytotoxic T-cell-mediated killing. One possible mechanism is that CD47-SIRPα interaction stabilizes cell-to-cell contacts and cytolytic synapses. The strength of cell-to-cell interaction is determined not only by the affinity between the receptor and the ligand but also by their avidity. As the spanning distance of the end-to-end bound CD47-SIRPα complex (~14 nm) is very similar to TCR-MHC, CD28-CD86, and CD40-CD40L, tumor cells or CD8+ T cells bearing more SIRPα could have stronger and longer interactions with cells expressing CD47, leading to a more cytotoxicity effect (Myers et al., 2019). Furthermore, another study shows that the interaction of SIRPα on dendritic cells and CD47 on T cells is important for the T-cell activation (Seiffert et al., 2001). Thus, these studies and ours collectively suggest that any disruption to the interaction between SIRPα and CD47 may affect T cell activation and the related antitumor activity.
The CD47-SIRPα signaling axis is a hot topic in the field of immunotherapy and has been under intensive clinical investigation. Results from multiple initial clinical trials show that monotherapy with anti-CD47 exhibited varied efficacy between different cancer types (Huang et al., 2017; Zhang et al., 2020b). The ubiquitous expression of CD47 on all cell types may contribute to the low efficiency and side effect of the monotherapy. Given the notion that SIRPA is predominately expressed in neurons, dendritic cells, and macrophages, some clinical trials were initiated to evaluate the possibility of SIRPA as an alternative target. However, we found that many different types of cancer cells expressed SIRPA. Some cancer cells, for example, melanoma cells, express as much SIPRA as tumor-associated macrophages. Given the dual role of SIRPA in immunotherapy response, we would like to emphasize that it is the overall net effect that determines the clinical benefits.
Specifically, our study may have significant clinical implications for SIRPα-related therapies (Figure 7). First, for indication selection, the effect of SIRPα blockade may depend on the relative abundance of tumor-intrinsic SIRPα in a given cancer lineage. In cancer types such as melanoma, where tumor-intrinsic SIRPα expression is high, the blockade may significantly dampen tumor-killing mediated by CD8+ T cells; whereas in other cancer types such as breast cancer and lymphoma, where SIRPA expression is much lower, the efficacy of SIRPα blockade in preclinical models (Gauttier et al., 2020; Ring et al., 2017) likely reflects a dominant role of macrophage-mediated phagocytosis of tumor cells. Therefore, the relative abundance of SIRPA in tumor cells must be considered when choosing suitable cancer types for anti-SIRPα treatment. Second, to stratify patients for better immunotherapy response within a cancer type, tumor-specific SIRPA expression may be a more effective biomarker than the bulk-level SIRPA expression, as the latter would be confounded by signals from different cell components within a tumor. Currently, biomarker identification is largely based on the analysis of bulk-level expression data, and more efforts should be made to assess cell-type-specific gene expression signatures as potential biomarkers. Finally, for drug development, we propose that antibodies that specifically bind to SIRPα on the surface of macrophages would be more effective. For that purpose, it would be of particular interest to design bispecific antibodies that can simultaneously bind to two different types of antigens, one to target macrophage-specific antigens and the other to target SIRPα.
Our study highlights cell type-dependent antagonistic effects of the same target on immunotherapy, an issue largely ignored in the field. Besides SIRPA, tumor-intrinsic PD-1 and CTLA-4 have been reported (Wang et al., 2020; Zhang et al., 2019). The unexpected expression of these immune checkpoints on the surface of tumor cells may antagonize or agonize the immune cells’ antitumor activity (Kleffel et al., 2015; Zhang et al., 2019). Considering the high fraction of tumor cells in the tumor ecosystem, it would be critical to systematically elucidate the effects of immunotherapeutic targets in different cell types and take this into account in the design of clinical studies.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Han Liang (hliang1@mdanderson.org).
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
The newly generated bulk RNA-seq data of B16F10 cells with SIRPA perturbations are available at Gene Expression Omnibus (GEO) with an accession number GSE211226; and SIRPα RPPA data of TCGA-SKCM and CCLE samples are available at TCPA data portal (https://tcpaportal.org). The source and accession numbers for previously published datasets used in this study are as follows: bulk RNA-seq data from melanoma patients under anti-PD-1 treatment: GEO, GSE78220 and GSE91061, ENA, PRJEB23709, and dbGaP, phs000452.v3.p1; whole-exome sequencing data of melanoma patients under anti-PD-1 treatment, SRA: SRP090294 and SRP067938; bulk proteomics data from melanoma patients under anti-PD-1 treatment, Harel et al.; bulk RNA-seq data from melanoma patients under BRAFi treatment, GEO, GSE50509 and GSE99898; TCGA genomic data and clinical data, NCI Genome Data Commons; single-cell RNA-seq data of melanoma patients, GEO, GSE115978, GSE70630, GSE174401, GSE120575; single-cell proteomics data from human melanoma and monocyte cell lines, Leduc et al., 2022; CCLE quantitative mass spectrometry data, Nusinow et al., 2020; CCLE gene expression data, CCLE data portal (https://portals.broadinstitute.org/ccle); bulk RNA-seq data of patient-derived melanoma cell lines, GEO, GSE80829; bulk RNA-seq data of in vitro differentiating melanocytes derived from ESC/iPSC, GEO: GSE45227; single-cell RNA-seq data from human normal skin samples of different developmental stages, GEO: GSE151091; Single-cell RNA-seq data from stepwise-edited melanoma cell lines, Single Cell Portal: SCP1334; spatial transcriptomics data from treatment-naïve melanoma patients, http://www.spatialomics.org/SpatialDB; DICE immune cell type gene expression data, https://dice-database.org; bulk proteomics data from human hematopoietic cell populations sorted from peripheral blood, http://www.immprot.org. Code used for all processing and analysis is available upon request.
Experimental Model and Subject Details
Cell lines
B16F10, A375, and HEK293T cells were cultured in complete DMEM media (10% FBS and 50U/mL Penicillin-Streptomycin). B16F10 cells with SIRPA perturbation and A375 cells stably expressing SIRPAP1 were cultured in complete DMEM media supplemented with 2 ug/mL puromycin. A375 cells stably integrated with synergistic activation mediator (SAM) dCas9 and effector components were cultured as described previously (Konermann et al., 2015). CD8+ T cells isolated from Pmel-1 mice were cultured in complete RPMI-164 media supplemented with 10% FBS, 20 mM HEPES, 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, and 2mM L-glutamine. All cell lines prepared at MD Anderson Cancer Center were confirmed by short tandem repeat analysis and were periodically tested for mycobacterium contamination at the MD Anderson Characterized Cell Line Core.
Mice
We purchased 6–8 weeks old C57BL/6J female mice from the Jackson Laboratory (#000664). All animal experiments and procedures were performed according to the protocol approved by the Institutional Animal Care and Use Committee at the University of Texas MD Anderson Cancer Center. Our sample size predetermination experiments indicated that ≥15 mice were needed in each group to detect the expected effect. In the experiment, all the animals were randomized before tumor inoculation; and three days after inoculation, mice with established tumors were randomized again before the treatment.
Method Details
Expression plasmids
The full-length human SIRPAP1 and mouse Sirpa were synthesized by Epoch Life Science and cloned into pDONR221. The sequences of the two genes were verified by Sanger Sequencing. The expression clones were generated via the LR Clonase Reaction between the Entry clone and the pLenti CMV Puro DEST (w118–1). The pLenti CMV Puro DEST was a gift from Eric Campeau & Paul Kaufman (Addgene plasmid # 17452; http://n2t.net/addgene:17452; RRID: Addgene_17452) (Campeau et al., 2009).
Generation of stable cell lines
HEK293T cells were co-transfected with lentiviral vectors encoding the gene of interest or shRNAs together with packaging vectors pCMV-dR8.2 dvpr and pCMV-VSV-G using JetPRIME. At 48 h post-transfection, the supernatant was collected and filtered using a sterile syringe filter with a 0.45 μm pore size hydrophilic PVDF membrane. The filtered supernatant was applied to infect the target cells for 18 h. The infected cells were selected using the appropriate antibiotics for a week before the conduction of the experiments.
Generation of CRISPR gene activation system
Lentivectors encoding dCAS9-VP64, MS2-P65-HSF1, and lenti-sgRNA(MS2) zeo backbone were gifts from Feng Zhang (Konermann et al., 2015). Human A375 cells stably expressing dCAS9-VP64, MS2-P65-HSF1 were infected with lentivirus expressing gRNA. We designed gRNAs to target 700 bp upstream of SIRPAP1’s transcription start site.
Curation of immuno-oncology targets
We collected the immuno-oncology (IO) targets listed in Global Immuno-Ontology Drug Development Pipeline 2020, a pdf file downloaded by selecting “Specific sheets from this dashboard” and “2020”). Then, we mapped the collected 508 IO targets (504 non-redundant names) to HGNC-approved symbols by HGNC multi-symbol checker (https://www.genenames.org/tools/multi-symbol-checker/). We manually reviewed the mapped gene symbols, filled blanks if a proper gene symbol could be found in GeneCards (https://genecards.org), and then excluded IO targets without gene symbols. Next, we merged redundant target names by summing the numbers of clinical trials from each record. Lastly, we focused on those IO targets with at least 10 active agents/clinical trials and obtained 60 IO targets for the subsequent analyses.
Integrative analysis of bulk transcriptomic and proteomic data from anti-PD-1 treatment patient cohorts
To collect suitable datasets, we first reviewed all available anti-PD-1 trial studies in the literature and at the website of Tumor Immune Dysfunction and Exclusion (TIDE; http://tide.dfci.harvard.edu). We collected the anti-PD1 treated melanoma patient cohorts with available RNA-seq or proteomic data, leading to four transcriptomic datasets (Gide et al., 2019; Hugo et al., 2016; Liu et al., 2019; Riaz et al., 2017) as well as one proteomic dataset (Harel et al., 2019) (see Key Resources Table). We followed quality control of the samples from each study and then removed samples collected from sites other than skin to exclude potential confounders in survival analysis. In particular, seven non-skin samples were removed from the 41 anti-PD1 treated samples from the Gide dataset; two unqualified and 33 non-skin samples were removed from the 121 samples of the Liu dataset; 7 unqualified samples were removed from the 74 samples of the Harel dataset. Next, to analyze RNA-seq data, we used gene annotation (version 37) from GENCODE. Then, we adopted Salmon v1.4.0 to obtain read counts and TPMs for each dataset using the default parameter setting. We developed an integrative analytic procedure combining proteomic and transcriptomic data with the treatment response and survival information. Specifically, we computed expression-response and expression-survival associations in anti-PD-1-treated melanoma patients and then examined the concordance of the two associations to evaluate the combinational potential of an active IO target for anti-PD-1 therapy in melanoma. First, we performed differential expression analysis. For transcriptomic data, we adopted three well-established methods (DE-seq2/edgeR/limma) to detect differentially expressed (DE) genes between the responding and non-responding patients given anti-PD-1 treatment. To identify the most robust differentially expressed IO targets, we then constructed a consensus set of DE features by overlapping the three DE lists (P ≤ 0.05 and FDR ≤ 0.15 among all three tests with two-sided testing). To detect differential signals more sensitively, we also computed P values with one-sided testing (P’) to identify significant associations. For proteomic data, we used the Mann-Whitney U test (MW test), incorporating a permutation procedure to identify DE proteins. For each protein, we randomly shuffled the sample labels and calculated U scores of the MW test 10,000 times to obtain the background distribution and re-computed a P-value. Proteins with a P-value < 0.05 were defined as DE proteins. Second, we retained the IO targets that were differentially expressed in at least two patient cohorts. Third, we adopted a log-rank test (by ggsurvplot with the option log.rank.weights=“n” to identify early survival differences) for survival analysis. For each candidate IO target, the patients in a cohort were divided into high- and low- expression groups based on the median expression. We then computed the association between the overall survival rate and the expression groups. Lastly, we examined the concordance between DE and survival analyses to identify the IO targets showing a consistent pattern in terms of tumor response and patient survival rate.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD172a (SIRPa)-FITC, human | Miltenyi Biotec Inc | 130-099-896 |
| REA Control-FITC | Miltenyi Biotec Inc | 130-113-449 |
| Anti-SIRPα antibody | Abcam | ab8120 |
| hFAB Rhodamine anti-GAPDH Primary Antibody | Bio-Rad Laboratories | 12004168 |
| hFAB™ Rhodamine anti-Actin Primary Antibody | Bio-Rad Laboratories | 12004163 |
| Brilliant Violet 510™ anti-mouse CD172a (SIRPα) | Biolegend | 144032; RRID: AB_2810411 |
| Brilliant Violet 510™ Rat IgG1, κ Isotype Ctrl Antibody | Biolegend | 400435 |
| InVivoPlus rat IgG2b isotype control, anti-keyhole limpet hemocyanin | Biolegend | BP0090; RRID: AB_1107780 |
| InVivoPlus anti-mouse PD-L1 (B7-H1) | Bioxcell | BP0101; RRID: AB_10949073 |
| InVivoPlus anti-mouse/human/rat CD47 (IAP) | Bioxcell | MIAP410; RRID: AB_2687806 |
| InVivoMAb mouse IgG1 isotype control, | Bioxcell | MOPC-21;RRID: AB_1107784 |
| Anti-CD47 mIAP430 | Absolute Antibdody | AB00738-7.1 |
| Bacterial and virus strains | ||
| Endura™ Chemically Competent Cells | Lucigen | 60240-2 |
| Chemicals, peptides, or recombinant proteins | ||
| jetPRIME® Versatile DNA/siRNA transfection reagent(0.75ml) | Polyplus | 114-07 |
| A 33 mm diameter sterile syringe filter with a 0.45 μm pore size hydrophilic PVDF membrane | EMD Millipore Corp | SLHV033RS |
| Recombinant Mouse IL-2 Protein | R&D System,lnc | 402-ML |
| RIPA Lysis and Extraction Buffer | Thermo Fisher | 89901 |
| Western Lightning® Plus-ECL, Enhanced Chemiluminescence Substrate | PerkinElmer Inc. | NEL103001EA |
| Pierce BCA Protein Assay Kit | Thermo Fisher | 23227 |
| RPMI 1640 | Fisher Scientific | MT10040CV |
| DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Fisher Scientific | 10013CV |
| Penicillin-Streptomycin (10,000U/mL) | Thermo Fisher | 15140122 |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher | 25300120 |
| InVivoPure pH 7.0 Dilution Buffer | Bioxcell | IP0070 |
| InVivoPure pH 6.5 Dilution Buffer | Bioxcell | IP0065 |
| GP100(25-33) | ANASPEC | AS-62589 |
| Critical commercial assays | ||
| EasySep™ Mouse CD8+ T Cell Isolation Kit | Stemcell | 19853 |
| CountBright Absolute Counting Beads, for flow cytometry | Thermo Fisher | C36950 |
| Dynabeads™ Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation | Thermo Fisher | 11456D |
| Deposited data | ||
| Immuno-oncology targets (Figure 1A) | Immuno-Oncology Landscape, Cancer Research Institute (online published on Sep 18, 2020) | https://www.cancerresearch.org/scientists/immuno-oncology-landscape |
| Bulk proteomics data from anti-PD-1-treatment-responding and non-responding melanoma patients (Figure 1A–C, E) | Harel et al., 2019 | https://www.sciencedirect.com/science/article/pii/S0092867419309006 |
| Bulk RNA-seq data from anti-PD-1-treatment-responding and non-responding melanoma patients (Figure 1A–D; Figure 2I; Figure S6A) | Hugo et al., 2016 | GEO: GSE78220 |
| Bulk RNA-seq data from anti-PD-1-treatment-responding and non-responding melanoma patients (Figure 1A–D) | Gide et al., 2019 | ENA: PRJEB23709 |
| Bulk RNA-seq data from anti-PD-1-treatment-responding and non-responding melanoma patients (Figure 1A–D) | Riaz et al., 2017 | GEO: GSE91061 |
| Bulk RNA-seq data from anti-PD-1-treatment-responding and non-responding melanoma patients (Figure 1A–D) | Liu et al., 2019 | dbGaP: phs000452.v3.p1 |
| Bulk RNA-seq data from BRAFi-treatment-responding and non-responding melanoma patients (Figure S1A, B) | Rizos et al., 2014 | GEO: GSE50509 |
| Bulk RNA-seq data from BRAFi-treatment-responding and non-responding melanoma patients (Figure S1A, B) | Kakavand et al., 2017 | GEO: GSE99898 |
| Tumor mutation burden in melanoma patients (Figure S1C) | Wang et al., 2019 | https://elifesciences.org/articles/49020 |
| Survival data in TCGA melanoma patients (Figure S1D) | The Cancer Genome Atlas (TCGA) | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Single-cell RNA-seq data from pre- and post-anti-PD-1-treatment melanoma patients (Figure 2A–C, J) | Jerby-Arnon et al., 2018 | GEO: GSE115978 |
| Single-cell RNA-seq data from treatment-naïve melanoma patients (Figure 2I; Figure 4B) | Tirosh et al., 2016 | GEO: GSE70630 |
| Single-cell RNA-seq data from metastatic melanoma patients (Figure 2D–F) | Smalley et al., 2021 | GEO: GSE174401 |
| Single-cell proteomics data from human melanoma and monocyte cell lines (Figure 2G, H) | Leduc et al., 2022 | https://scp.slavovlab.net/Leduc_et_al_2022 |
| Single-cell RNA-seq data from anti-PD-1-treatment-responding and -non-responding melanoma patients (Figure 2K) | Sade-Feldman et al., 2018 | GEO: GSE120575 |
| CCLE quantitative mass spectrometry data (Figure S2A; Figure 3E) | Nusinow et al., 2020 | https://www.sciencedirect.com/science/article/pii/S0092867419313856 |
| CCLE gene expression data (Figure S2B; Figure 3C) | Cancer Cell Line Encyclopedia | https://portals.broadinstitute.org/ccle |
| SIRPα RPPA data in melanoma cell lines (Figure 3D) | This study | https://tcpaportal.org |
| Bulk RNA-seq data of patient-derived melanoma cell lines (Figure 3A–C) | Tsoi et al., 2018 | GEO: GSE80829 |
| Bulk RNA-seq data of in vitro differentiating melanocytes derived from ESC/iPSC (Figure 3F, G) | Mica et al., 2013 | GEO: GSE45227 |
| Single-cell RNA-seq data from human normal skin samples of different developmental stages (Figure 3H–J; Figure S3C) | Belote et al., 2020 | GEO: GSE151091 |
| Single-cell RNA-seq data from stepwise-edited melanoma cell lines (Figure S3A, B) | Hodis et al., 2022 | Single Cell Portal: SCP1334 |
| Spatial transcriptomics data from treatment-naïve melanoma patients (Figure 4A) | Thrane et al., 2018 | http://www.spatialomics.org/SpatialDB/download.php |
| DICE immune cell type gene expression data (Figure S4A, B) | Schmiedel et al., 2018 | https://dice-database.org |
| Bulk proteomics data from human hematopoietic cell populations sorted from peripheral blood (Figure S4C, D) | Rieckmann et al., 2017 | http://www.immprot.org |
| Bulk RNA-seq data of B16F10 cells with SIRPA perturbations | This study | GEO: GSE211226 |
| SIRPα RPPA data in TCGA-SKCM samples (Figure 6A–C; Figure S6B) | This study | https://tcpaportal.org |
| TCGA-SKCM RNA-seq data (bam files; Figure 3K; Figure 6D–F; Figure S6B) | The Cancer Genome Atlas (TCGA) | https://tcga-data.nci.nih.gov/docs/publications/tcga |
| Processed TCGA SCNA data (Figure 6A, B) | The Cancer Genome Atlas (TCGA) | Synapse: syn5049520.1 |
| Processed TCGA DNA methylation, mutation, and miRNA expression data (Figure 6A, C–G) | The Cancer Genome Atlas (TCGA) | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Whole-exome sequencing data from anti-PD-1-treatment-responding and non-responding melanoma patients (Figure S6A) | Hugo et al., 2016 | SRA: SRP090294 and SRP067938 |
| Experimental models: Cell lines | ||
| HEK293T | MD Anderson Characterized Cell Line Core Facility | HEK293T |
| A375 | MD Anderson Characterized Cell Line Core Facility | A375M |
| A375-dCas9-SAM | This study | N/A |
| B16F10 | ATCC | CRL-6475 |
| Experimental models: Organisms/strains | ||
| Mouse:B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J | The Jackson Laboratory | JAX:005023; RRID:IMSR_JAX:005023 |
| Mouse:C57BL/6J | The Jackson Laboratory | JAX:000664; RRID:IMSR_JAX:005023 |
| Oligonucleotides | ||
| Scramble gRNA-F: 5’-CACCGGTATTACTGATATTGGTGGG-3’ | This study | N/A |
| Scrambel gRNA-R: 5’-AAACCCCACCAATATCAGTAATACC-3’ | This study | N/A |
| SIRPAP1-1-F: 5’-CACCGGTAGGGTCGCGAGACGGATG -3’ | This study | N/A |
| SIRPAP1-1-R: 5’-AAACCATCCGTCTCGCGACCCTACC-3’ | This study | N/A |
| Recombinant DNA | ||
| pCMV-VSV-G | Addgene | 8454; RRID:Addgene_8454 |
| pCMV-dR8.2 dvpr | Addgene | 8455; RRID:Addgene_8455 |
| lenti sgRNA(MS2)_zeo backbone | Addgene | 61427; RRID:Addgene_61427 |
| lenti dCAS-VP64_Blast | Addgene | 61425; RRID:Addgene_61425 |
| lentiMPH v2 | Addgene | 89308; RRID:Addgene_89308 |
| plenti-CMV-Puro-Dest | Addgene | 17452; RRID:Addgene_17452 |
| pDONR221-Human SIRPAP1 | Epoch Life Science | GS65919 |
| pDONR221-Mouse SIRPA | Epoch Life Science | GS68006 |
| plenti-CMV-Puro-Human SIRPAP1 | This study | N/A |
| plenti-CMV-Puro-Mouse SIRPA | This study | N/A |
| MISSION® pLKO.1-puro Non-Target shRNA Control Plasmid DNA | Sigma-Aldrich | SHC016-1EA |
| SHCLNG MISSION shRNA-1 | Sigma-Aldrich | TRCN0000029914 |
| SHCLNG MISSION shRNA-2 | Sigma-Aldrich | TRCN0000055053 |
| SHCLNG MISSION shRNA-3 | Sigma-Aldrich | TRCN0000029915 |
| SHCLNG MISSION shRNA-4 | Sigma-Aldrich | TRCN0000029916 |
| SHCLNG MISSION shRNA-5 | Sigma-Aldrich | TRCN0000029917 |
| Software and Algorithms | ||
| FlowJo10.0.7 | FlowJo LLC | https://www.flowjo.com; RRID:SCR_008520 |
| Salmon v1.4.0 | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| Subread v2.0.1 | Liao et al., 2014 | http://subread.sourceforge.net/; RRID:SCR_009803 |
| CIBERSORTx | Newman et al., 2019 | https://cibersortx.stanford.edu/index.php |
| GSVA | Hanzelmann et al., 2013 | https://pypi.org/project/GSVA/; RRID:SCR_021058 |
| RNAhybrid | Krüger et al., 2006 | https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid; RRID:SCR_003252 |
| miRDB | Chen et al., 2020 | http://www.mirdb.org/; RRID:SCR_010848 |
| TarPmiR | Ding et al., 2016 | http://hulab.ucf.edu/research/projects/miRNA/TarPmiR/ |
| CellPhoneDB | Efremova et al., 2020 | https://www.cellphonedb.org/; RRID:SCR_017054 |
| Scanpy | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/; RRID:SCR_018139 |
| tSNE | van der Maaten and Hinton, 2008 | https://github.com/DmitryUlyanov/Multicore-TSNE |
| Python v3.6 | Python, 2015 | https://python.org; RRID:SCR_008394 |
| R v3.6 | The R Foundation | https://www.r-project.org; RRID:SCR_001905 |
| Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| BioRender | BioRender | https://app.biorender.com/; RRID:SCR_018361 |
Analysis of single-cell RNA-seq data of melanoma and normal skin samples
We downloaded gene expression profiles of single cells in raw counts (UMIs for data generated with droplet-based platforms) along with clustering annotations and clinical metadata from the Gene Expression Omnibus (GEO) and Single Cell Portal (SCP; see Key Resources Table). No quality control was further applied beyond what was already conducted by the original studies. We followed the Scanpy workflow (Wolf et al., 2018) for downstream analyses. Specifically, we 1) applied the log1pCP10K normalization to the raw counts, 2) selected highly variable genes, 3) regressed out the effects of the total count per cell and the percentage of mitochondrial gene count, 4) calculated the first 50 principal components, 5) applied Harmony (Korsunsky et al., 2019) to remove sample-level batch effects, 6) reduced the data dimension through Uniform Manifold Approximation and Projection (UMAP), 7) clustered the single cells using an unsupervised graph-based clustering algorithm (Leiden), 8) identified cluster-specific marker genes using Student’s t-test, and 9) annotated the clusters based on the expression patterns of literature-derived marker genes along with referring to the annotations from the original studies.
Pseudo-time trajectory analysis of melanocyte scRNA-seq data
We extracted the single-cell gene expression profiles of the human melanocytes at the fetal stage from the original dataset to build a pseudo-time trajectory. The data were normalized and processed following the same scRNA-seq data analysis procedure described above. Force Atlas was applied to generate a graph layout of the cells, upon which a differentiation progression was inferred by Diffusion Pseudo-time with the root set to be the cells with the lowest expression levels of canonical melanocytic lineage gene markers, including PMEL, MITF, and TYR. SIRPA expression was then visualized along the same trajectory to be compared against the differentiation progression.
Analysis of single-cell proteomics data of human melanoma and monocyte cell lines
We downloaded protein expression profiles of single cells in log-normalized and batch-corrected units from the Single-cell Proteomics Data Repository (see Key Resources Table). Data processing and analysis were conducted using the same procedure as in scRNA-seq analysis.
In-silico inference of cell-type-specific gene expression from bulk samples
To computationally enumerate seven cell types, namely, tumor cells, T cells, B cells, macrophages, natural killer cells, fibroblasts, and endothelial cells, from bulk melanoma RNA-seq samples from Hugo et al., we used CIBERSORTx to estimate their relative fractions. Following estimations by the CIBERSORTx algorithm, we constructed gene expression signature matrices for the seven cell types based on the scRNA-seq from Tirosh et al. Briefly, for each of the desired cell types, half of all the single cells were randomly selected without replacement and merged into a mega cell with average TPM values. Such random combinations were conducted 10 times to generate replicates for each cell type and were used as an input for a DE gene analysis to identify cell-type signature genes. Specifically, aggregated gene expression replicates of each cell type were compared against replicates of all other cell types using a Mann-Whitney U test, and the top 200 genes with an adjusted P-value < 0.01 and the highest log2FC were defined as signature genes. The average expression levels of these genes across all single cells were pooled into a final signature matrix for that cell type. We excluded 671 genes involved in the cell cycle (GO:0007049), 108 genes involved in ribosome biogenesis (GO:0042254), 21 genes involved in cell apoptosis (GO:0008637), and 37 genes mapped to the mitochondrial genome. With the signature matrices as a reference, we first ran the “Impute Cell Fractions” task in CIBERSORTx to estimate the relative fractions of the cell types within each bulk RNA-seq sample and then the “Impute Cell Expression” task with the “High-Resolution” mode to obtain sample-specific expression profiles of all cell types.
Analysis of melanoma spatial transcriptomic data
Spatial transcriptomic data of a stage III cutaneous malignant melanoma sample was downloaded from SpatialDB (http://www.spatialomics.org/SpatialDB/). To decompose the spatial distribution of tumor cells and immune cells from the expression data, we first queried the top 200 marker genes of each of the seven cell types (see above) surveyed in the scRNA-seq study by Tirosh et al. We then used GSVA to compute cell-type signature scores for all the dots, which quantified the enrichment or depletion of each of the cell populations at each position. Based on the concordance between transcriptome-deconvoluted and H&E staining-informed cell type distributions, the cell type-specific expression patterns of SIRPA and CD47 were visualized across the slide.
Estimation of cell-cell communication using single-cell RNA-seq data
The melanoma patients’ scRNA-seq data from Tirosh et al. were used to evaluate the confidence in SIRPα-CD47-mediated cell-cell communication between melanoma cells and CD8+ T cells based on their co-expression patterns. We ranked all T cells by the expression level of CD4 and CD8 and extracted the gene expression profiles of the top 1,044 CD8+ T cells that were completely depleted of CD4. These cells were then combined with all melanoma cells in the TPM scale as input to CellPhoneDB.
Generation and analysis of SIRPA-perturbed B16F10 RNA-seq data
For RNA-seq experiments, total RNA was extracted from mSIRPA KD, OE, or control B16F10 cells using the Qiagen RNeasy Mini kit according to the manufacturer’s instructions and were subjected to mRNA paired-end sequencing at Novogene Co., LTD. Each cell line had three biological repeats. We employed a similar processing pipeline to the integrative analysis of public datasets to analyze the B16F10 data. Briefly, we (i) performed quality control of RNA-seq raw reads using MultiQC, (ii) pseudo-aligned reads to GENCODE mm39 mouse reference genome and obtained read counts and TPM using Salmon, (iii) identified differentially expressed genes in each condition through log2 fold changes and adjusted p-values using DESeq2, and (iv) focused on the changes of six well-established MDA-encoding genes, namely PMEL, TYR, TYRP1, DCT, MLANA, and MITF.
Profiling of SIRPα protein expression in TCGA melanoma and CCLE samples
We quantified SIRPα protein expression in 349 TCGA melanoma samples and 48 CCLE melanoma samples using a reverse phase protein array at the RPPA core facility at MD Anderson. The SIRPα antibodies (Abcam, Cat # ab8120) were validated by comparison with immunoblotting, as previously described (Hennessy et al., 2010; Li et al., 2017). Briefly, lysates were manually serial-diluted in 5 two-fold dilutions with lysis buffer and printed on nitrocellulose-coated slides using an Aushon Biosystems 2470 arrayer. Slides were probed with validated primary antibodies. Signals were captured by Dako GenePoint Tyramide Signal Amplification System (Agilent, Cat. # K0620). Stained RPPA slides were first quantified using ArrayPro (Media Cybernetics) to generate signal intensities. The raw data were further normalized by SuperCurve, median polish, and replicate-based normalization (Akbani et al., 2014; Ju et al., 2015) for downstream analyses.
Analysis of potential regulators of SIRPα protein expression
To identify potential regulators of SIRPα expression, we integrated the SIRPα protein expression of TCGA-SKCM samples with other TCGA-SKCM molecular profiles (DNA methylation, mutation, somatic copy-number alteration, and pseudogene expression) that were obtained from the TCGA Pan-Cancer Atlas website (https://gdc.cancer.gov/about-data/publications/pancanatlas), except for pseudogene expression. For the pseudogene SIRPAP1, we downloaded TCGA-SKCM bam files from the GDC data portal (https://portal.gdc.cancer.gov/), extracted uniquely mapped reads, and computed RPM by using featureCounts. Then, we used multiple statistical methods to examine associations between SIRPα protein expression and other types of molecular data. For categorical variables such as wild-type versus mutant, we performed two-sample Wilcoxon tests (Mann-Whitney tests). For continuous variables, including DNA methylation and SIRPAP1, we computed Spearman’s rank correlation. To understand whether copy-number alternation of SIRPA affects the gene expression in the Hugo anti-PD1-treated patient cohort, we downloaded the whole-exome sequencing data of Hugo et al. and mapped them to human reference genome GRCh37 with the BWA-MEM algorithm (version 0.7.17). Picard (version 2.23.8) was used to mark duplications. The R package PureCN was used to estimate sample purity and ploidy. All normal samples were combined to build a reference panel, against which each tumor sample was compared to infer integer and categorical copy numbers using the cnvkit package (version 0.9.6). We categorized “Amp” for integer somatic copy number > 3, “Del” for integer somatic copy number < 1, and all others are considered somatic copy number neutral (“N”).
MicroRNA analysis
We employed three miRNA-mRNA interaction prediction tools, RNAhybrid, miRDB, and TarPmiR, which use different methodologies to infer miRNA targets, to search for miRNAs that potentially bind to the shared regions of SIRPA and SIRPAP1 transcripts. We first built a union set of miRNAs combining the results from all three tools and then used an additional filter that relied on the co-expression between SIRPA/SIRPAP1 and the miRNAs across patient samples of the TCGA melanoma cohort. With these two filters, we identified the miRNAs as potential mediators of the competitive endogenous mechanism that governed SIRPA-SIRPAP1 coordination (Figure S6C).
Flow cytometry analysis
The surface expression of SIRPα on B16F10 and A375 cells was examined by flow cytometry as described previously (Motegi et al., 2003). Briefly, cells were washed by PBS twice and then detached from the culture dishes by treatment with 0.05% Trypsin-EDTA. 1×106 cells were resuspended in 100 ul staining buffer (pH 7.2 PBS, 0.5% bovine serum albumin (BSA), and 2 mM EDTA). 10 ul FITC-conjugated SIRPα antibody was added to the cell suspension. Cells were incubated with the antibody for 10 min in the dark in the refrigerator (4°C), washed twice with PBS, and then analyzed by a BD FACSCelesta™ cytometer. Data were analyzed using the Flowjo software (version 10.7 Flowjo).
T cell killing assay based on co-cultures of B16F10 and T cells
We performed T cell killing assays as described previously (Pan et al., 2018). Briefly, CD8+ T cells were isolated from the spleen of Pmel-1 transgenic mice using the EasySep mouse CD8+ T cell isolation kit according to the manufacturer’s protocol. Freshly isolated CD8+ T cells were then activated with anti-CD3/CD28 beads according to the manufacturer’s protocol. 20 ng/ml mouse IL-2 was added to the culture medium. T cells were in vitro stimulated for at least 6 days before the co-culture with B16F10 cells. B16F10 cells (1×105) were plated into a well of 6-well plate. The next day, cells were pulsed with 1 μM gp10025−33 for 2 h. After that, the in vitro activated Pmel-1 T cells were added into co-culture with tumor cells at 0, 50%, or 100% of the number of tumor cells. We added 10ug/ml anti-mPD-L1 to the co-culture system together with T cells. For CD47 blockade treatments, the indicated antibodies (10ug/ml) were pre-incubated with T cells for 2 h. Then, the T cells were washed with PBS twice and added to the B16F10 cells. Each condition had 3 replicates. At 24 h after co-culture, the tumor cells were washed with PBS and then detached from the culture dishes by treatment with 0.05% Trypsin-EDTA. Before flow cytometry analysis, 10 ul of CountBright™ absolute counting beads were added to each sample. The samples were run through the flow cytometer and set to stop after 500 beads were acquired. The number of tumor cells in each sample was computed based on the reference beads. The average cell number in each condition was normalized to its corresponding no-T-cell control to get the relative cell viability proportion. The experiments were repeated three times using T cells from three separate mice. In each experiment, each condition was measured in triplicate. The relative cell survival in each condition was normalized to its corresponding no-T-cell control of each cell type within the experiment. The controls’ relative cell survival was set as 1. The error bar represents the standard error. For 96 h co-culture experiments, the B16F10 cells were not pulsed with 1uM gp10025–33. The in vitro activated Pmel-1 T cells were added into co-culture with tumor cells at 0, 50%, or 100% of the number of tumor cells. At 96 h after co-culture, the tumor cells were counted by flow cytometer as described previously.
Cell growth assay
On day 0, B16F10 cells (1×104) with different SIPRA perturbations were seeded into one well of 6 well plates. At different time points, cancer cells were detached from the plates by the treatment with 0.05% Trypsin-EDTA and counted using Cellometer Auto T4. Each condition has 3 repeats. The error bar represents the standard deviation.
Immunoblotting
These experiments were performed as described previously (Xu et al., 2019). The cells were lysed in RIPA buffer. Protein concentrations were measured using the Pierce BCA protein assay kit. Cell lysates were boiled and separated on a 10% SDS-PAGE gel. The proteins were transferred to a PVDF membrane, which was then incubated with specific primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies. The protein expression was detected with an ECL western blot detection kit. The membranes were imaged with the ChemiDoc MP Imaging System.
In vivo experiments using B16F10 cells
B16F10 syngeneic mouse melanoma models were performed as described previously (Overwijk and Restifo, 2001; Pan et al., 2018). Briefly, all the animals were randomized before tumor inoculation. Then 0.4×106 control (non-targeting knockdown RNA), mSIRPA-KD, or mSIRPA-OE B16F10 cells were subcutaneously injected into 7–8 weeks old female C57BL/6 mice (The Jackson Laboratory #000664). Three days after inoculation, mice in each condition with established tumors were randomized again before the treatment. For each condition of mice implanted with control, mSIRPA-KD or mSIRPA-OE B16F10 tumor cells, two treatments were performed: anti-mPD-L1 and isotype control antibody. Specifically, αPD-L1 (clone B7-H1, #BP0101, 200μg/mice) or isotype control antibodies (clone LTF-2, #BP0090, 200 ug/mouse) mAbs were administered on days 3, 7, 10, 14, and 17. Accordingly, there were six groups: control tumors treated with isotype antibody (n = 17); mSIRPA-KD tumors treated with isotype antibody (n = 15); mSIRPA-OE tumors treated with isotype antibody (n = 17); control tumors treated with anti-mPD-L1 (n = 17); mSIRPA-KD tumors treated with anti-mPD-L1 (n = 17); and mSIRPA-OE tumors treated with anti-mPD-L1 (n = 17). Tumors were measured 3–6 times a week, beginning on day 10 after inoculation, until either the survival endpoint (37 days) was reached or no palpable tumor remained. Tumor volumes were calculated using the formula for a hemiellipsoid (volume = length×width×height/2). Mice were sacrificed when tumors reached 20 mm in diameter or 1500 mm3 in volume.
Quantification and Statistical Analysis
We performed quantification and statistical analysis using GraphPad Prism 6, R (version 3.4) and python (version 3.6). Detailed descriptions of statistical tests are provided in the Method Details section and the respective figure legends.
Supplementary Material
Table S1. Immuno-oncology targets used in this study, related to Figure 1.
Highlights.
High SIRPA expression correlates with response to anti-PD-1 treatment in melanoma
The loss of SIRPA expression is a key marker of melanoma de-differentiation
Tumor-specific SIRPA overexpression enhances anti-PD-L1 response
Multiple mechanisms affect SIRPA heterogeneity, including SIRPAP1 co-regulation
Acknowledgments
This study was supported by the National Institutes of Health (U24CA209851, U24CA264128, R01CA251150, U01CA25347, U01CA217842, P50CA221703 to H.L., R50CA221675 to Y.L., and P30CA016672), Cancer Prevention and Research Institute of Texas (CPRIT) (RP140126 to H.L. and RP170593 to M.M.C. and H.C.), the Lorraine Dell Program in Bioinformatics for Personalization of Cancer Medicine (to H.L.), MD Anderson Faculty Scholar Award (to H.L.) and Barnhart Family Distinguished Professorship in Targeted Therapies (to H.L.). This work was supported by the National Cancer Institute’s Office of Cancer Genomics Cancer Target Discovery and Development (CTD2) initiative. The results published here are based in whole or in part upon data generated by the CTD2 Network (https://ocg.cancer.gov/programs/ctd2/data-portal) established by the National Cancer Institute’s Office of Cancer Genomics.
Footnotes
Declaration of Interests
H.L. is a shareholder and scientific advisor of Precision Scientific Ltd.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams S, van der Laan LJ, Vernon-Wilson E, Renardel de Lavalette C, Dopp EA, Dijkstra CD, Simmons DL, and van den Berg TK (1998). Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J Immunol 161, 1853–1859. [PubMed] [Google Scholar]
- Agaimy A, Specht K, Stoehr R, Lorey T, Markl B, Niedobitek G, Straub M, Hager T, Reis AC, Schilling B, et al. (2016). Metastatic Malignant Melanoma With Complete Loss of Differentiation Markers (Undifferentiated/Dedifferentiated Melanoma): Analysis of 14 Patients Emphasizing Phenotypic Plasticity and the Value of Molecular Testing as Surrogate Diagnostic Marker. Am J Surg Pathol 40, 181–191. [DOI] [PubMed] [Google Scholar]
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al. (2014). A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 5, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. (2015). PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Sirota M, and Butte AJ (2015). Systematic pan-cancer analysis of tumour purity. Nat Commun 6, 8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, Osokin N, Kozlov I, Frenkel F, Gancharova O, et al. (2021). Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 39, 845–865 e847. [DOI] [PubMed] [Google Scholar]
- Barclay AN, and Brown MH (2006). The SIRP family of receptors and immune regulation. Nat Rev Immunol 6, 457–464. [DOI] [PubMed] [Google Scholar]
- Belote RL, Le D, Maynard A, Lang UE, Sinclair A, Lohman BK, Planells-Palop V, Baskin L, Tward AD, Darmanis S, and Judson-Torres RL (2021). Human melanocyte development and melanoma dedifferentiation at single-cell resolution. Nat Cell Biol 23, 1035–1047. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, Volkmer JP, Weissman IL, and Majeti R (2019). Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front Oncol 9, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Marcisak EF, Deshpande A, Stein-O’Brien GL, Ho WJ, Laheru D, Jaffee EM, Fertig EJ, and Kagohara LT (2021). From bench to bedside: Single-cell analysis for cancer immunotherapy. Cancer Cell 39, 1062–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Li X, and Hu H (2016). TarPmiR: a new approach for microRNA target site prediction. Bioinformatics 32, 2768–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Gauttier V, Pengam S, Durand J, Biteau K, Mary C, Morello A, Neel M, Porto G, Teppaz G, Thepenier V, et al. (2020). Selective SIRPalpha blockade reverses tumor T cell exclusion and overcomes cancer immunotherapy resistance. J Clin Invest 130, 6109–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, Lim SY, Velickovic R, Wongchenko M, et al. (2019). Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238–255 e236. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, and Weinberg RA (2005). The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 37, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerbacher J, and Snyder A (2017). Informatics for cancer immunotherapy. Ann Oncol 28, xii56–xii73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Sterling H, Chen Y, Saginario C, Brown EJ, Frazier WA, Lindberg FP, and Vignery A (2000). CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J Biol Chem 275, 37984–37992. [DOI] [PubMed] [Google Scholar]
- Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, Baruch EN, Tripple V, Arama-Chayoth M, Greenberg E, et al. (2019). Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 179, 236–250 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K, Meric-Bernstam F, et al. (2010). A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics 6, 129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WW, Pittet MJ, Fukumura D, and Jain RK (2022). The local microenvironment matters in preclinical basic and translational studies of cancer immunology and immunotherapy. Cancer Cell 40, 701–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Torlai Triglia E, Kwon JYH, Biancalani T, Zakka LR, Parkar S, Hutter JC, Buffoni L, Delorey TM, Phillips D, et al. (2022). Stepwise-edited, human melanoma models reveal mutations’ effect on tumor and microenvironment. Science 376, eabi8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Ma Y, Gao P, and Yao Z (2017). Targeting CD47: the achievements and concerns of current studies on cancer immunotherapy. J Thorac Dis 9, E168–E174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalil AR, Andrechak JC, and Discher DE (2020). Macrophage checkpoint blockade: results from initial clinical trials, binding analyses, and CD47-SIRPalpha structure-function. Antib Ther 3, 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Z, Liu W, Roebuck PL, Siwak DR, Zhang N, Lu Y, Davies MA, Akbani R, Weinstein JN, Mills GB, and Coombes KR (2015). Development of a robust classifier for quality control of reverse-phase protein arrays. Bioinformatics 31, 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. (2015). Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 162, 1242–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C, Nittner D, Rambow F, Radaelli E, Stanchi F, Vandamme N, Baggiolini A, Sommer L, Berx G, van den Oord JJ, et al. (2017). Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell Stem Cell 21, 679–693 e676. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger J, and Rehmsmeier M (2006). RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 34, W451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo TC, Chen A, Harrabi O, Sockolosky JT, Zhang A, Sangalang E, Doyle LV, Kauder SE, Fontaine D, Bollini S, et al. (2020). Targeting the myeloid checkpoint receptor SIRPalpha potentiates innate and adaptive immune responses to promote anti-tumor activity. J Hematol Oncol 13, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, and Tuting T (2012). Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416. [DOI] [PubMed] [Google Scholar]
- Leduc A, Huffman RG, Cantlon J, Khan S, and Slavov N (2022). Exploring functional protein covariation across single cells using nPOP. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu WB, Ju ZL, Ling SY, Vellano CP, Roebuck P, Yu QH, Eterovic AK, et al. (2017). Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, et al. (2019). Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, and Mardis ER (2017). Applications of Immunogenomics to Cancer. Cell 168, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Kim YJ, Robert L, Tsoi J, Comin-Anduix B, Berent-Maoz B, Cochran AJ, Economou JS, Tumeh PC, Puig-Saus C, and Ribas A (2018). Immunotherapy Resistance by Inflammation-Induced Dedifferentiation. Cancer Discov 8, 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mica Y, Lee G, Chambers SM, Tomishima MJ, and Studer L (2013). Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep 3, 1140–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon H, Donahue LR, Choi E, Scumpia PO, Lowry WE, Grenier JK, Zhu J, and White AC (2017). Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure. Cell Stem Cell 21, 665–678 e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey MA, Kern N, and Vale RD (2020). CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity 53, 290–302 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi S, Okazawa H, Ohnishi H, Sato R, Kaneko Y, Kobayashi H, Tomizawa K, Ito T, Honma N, Buhring HJ, et al. (2003). Role of the CD47-SHPS-1 system in regulation of cell migration. EMBO J 22, 2634–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers LM, Tal MC, Torrez Dulgeroff LB, Carmody AB, Messer RJ, Gulati G, Yiu YY, Staron MM, Angel CL, Sinha R, et al. (2019). A functional subset of CD8(+) T cells during chronic exhaustion is defined by SIRPalpha expression. Nat Commun 10, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell F, Pires da Silva I, Johansson PA, Menzies AM, Wilmott JS, Addala V, Carlino MS, Rizos H, Nones K, Edwards JJ, et al. (2022). Multiomic profiling of checkpoint inhibitor-treated melanoma: Identifying predictors of response and resistance, and markers of biological discordance. Cancer Cell 40, 88–102 e107. [DOI] [PubMed] [Google Scholar]
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, and Restifo NP (2001). B16 as a mouse model for human melanoma. Curr Protoc Immunol Chapter 20, Unit 20.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcovski J, Shahar E, Aizenshtein E, and Gorodetsky R (2017). Melanoma antigens and related immunological markers. Crit Rev Oncol Hematol 115, 36–49. [DOI] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. (2015). Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenberg S, Groetchen A, Siddaway R, Bald T, Reinhardt J, Smorra D, Kohlmeyer J, Renn M, Phung B, Aymans P, et al. (2015). MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat Commun 6, 8755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer JP, George BM, Lietzenmayer M, McKenna KM, Naik TJ, McCarty A, et al. (2017). Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci U S A 114, E10578–E10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert M, Brossart P, Cant C, Cella M, Colonna M, Brugger W, Kanz L, Ullrich A, and Buhring HJ (2001). Signal-regulatory protein alpha (SIRPalpha) but not SIRPbeta is involved in T-cell activation, binds to CD47 with high affinity, and is expressed on immature CD34(+)CD38(−) hematopoietic cells. Blood 97, 2741–2749. [DOI] [PubMed] [Google Scholar]
- Shen Y, Chen Y, Wang D, and Zhu Z (2020). Treatment-related adverse events as surrogate to response rate to immune checkpoint blockade. Medicine (Baltimore) 99, e22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley I, Chen Z, Phadke M, Li J, Yu X, Wyatt C, Evernden B, Messina JL, Sarnaik A, Sondak VK, et al. (2021). Single-Cell Characterization of the Immune Microenvironment of Melanoma Brain and Leptomeningeal Metastases. Clin Cancer Res 27, 4109–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, Ploegh HL, and Garcia KC (2016). Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U S A 113, E2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H, and Manz MG (2007). Macrophage tolerance: CD47-SIRP-alpha-mediated signals matter. Nat Immunol 8, 1287–1289. [DOI] [PubMed] [Google Scholar]
- Tan S, Li D, and Zhu X (2020). Cancer immunotherapy: Pros, cons and beyond. Biomed Pharmacother 124, 109821. [DOI] [PubMed] [Google Scholar]
- Thrane K, Eriksson H, Maaskola J, Hansson J, and Lundeberg J (2018). Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res 78, 5970–5979. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uger R, and Johnson L (2020). Blockade of the CD47-SIRPalpha axis: a promising approach for cancer immunotherapy. Expert Opin Biol Ther 20, 5–8. [DOI] [PubMed] [Google Scholar]
- Upadhaya S, Neftelino ST, Hodge JP, Oliva C, Campbell JR, and Yu JX (2021). Combinations take centre stage in PD1/PDL1 inhibitor clinical trials. Nat Rev Drug Discov 20, 168–169. [DOI] [PubMed] [Google Scholar]
- Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, Tong Z, Tan S, Zhang H, Saw PE, et al. (2020). Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc Natl Acad Sci U S A 117, 6640–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. (2012). The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A 109, 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, and Wang X (2015). miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 43, D146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Wang J, Lu D, and Xu X (2021). Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther 6, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang Y, Mojumdar K, Zhou Z, Zhou Z, Jeong KJ, Mangala LS, Yu S, Tsang YH, Rodriguez-Aguayo C, et al. (2019). A-to-I-edited miRNA-379–5p inhibits cancer cell proliferation through CD97-induced apoptosis. J Clin Invest 129, 5343–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki Y, Ito S, Tsunoda N, Kokuryo T, Hara K, Senga T, Kannagi R, Yamamoto T, Oda K, Nagino M, et al. (2007). SIRPalpha1 and SIRPalpha2: their role as tumor suppressors in breast carcinoma cells. Biochem Biophys Res Commun 361, 7–13. [DOI] [PubMed] [Google Scholar]
- Yanagita T, Murata Y, Tanaka D, Motegi SI, Arai E, Daniwijaya EW, Hazama D, Washio K, Saito Y, Kotani T, et al. (2017). Anti-SIRPalpha antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2, e89140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Hopkins A, and Jaffee EM (2017). Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 377, 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev A, Chelushkin M, Dyikanov D, Cheremushkin I, Shpak B, Nomie K, Zyrin V, Nuzhdina E, Lozinsky Y, Zotova A, et al. (2022). Precise reconstruction of the TME using bulk RNA-seq and a machine learning algorithm trained on artificial transcriptomes. Cancer Cell 40, 879–894 e816. [DOI] [PubMed] [Google Scholar]
- Zhang H, Dutta P, Liu J, Sabri N, Song Y, Li WX, and Li J (2019). Tumour cell-intrinsic CTLA4 regulates PD-L1 expression in non-small cell lung cancer. J Cell Mol Med 23, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Yan YY, Li JJ, Adhikari R, and Fu LW (2020a). PD-1/PD-L1 Based Combinational Cancer Therapy: Icing on the Cake. Front Pharmacol 11, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang Q, Xiao W, Zhao Y, Pi J, Xu H, Zhao H, Xu J, Evans CE, and Jin H (2020b). Advances in Anti-Tumor Treatments Targeting the CD47/SIRPalpha Axis. Front Immunol 11, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Immuno-oncology targets used in this study, related to Figure 1.
Data Availability Statement
The newly generated bulk RNA-seq data of B16F10 cells with SIRPA perturbations are available at Gene Expression Omnibus (GEO) with an accession number GSE211226; and SIRPα RPPA data of TCGA-SKCM and CCLE samples are available at TCPA data portal (https://tcpaportal.org). The source and accession numbers for previously published datasets used in this study are as follows: bulk RNA-seq data from melanoma patients under anti-PD-1 treatment: GEO, GSE78220 and GSE91061, ENA, PRJEB23709, and dbGaP, phs000452.v3.p1; whole-exome sequencing data of melanoma patients under anti-PD-1 treatment, SRA: SRP090294 and SRP067938; bulk proteomics data from melanoma patients under anti-PD-1 treatment, Harel et al.; bulk RNA-seq data from melanoma patients under BRAFi treatment, GEO, GSE50509 and GSE99898; TCGA genomic data and clinical data, NCI Genome Data Commons; single-cell RNA-seq data of melanoma patients, GEO, GSE115978, GSE70630, GSE174401, GSE120575; single-cell proteomics data from human melanoma and monocyte cell lines, Leduc et al., 2022; CCLE quantitative mass spectrometry data, Nusinow et al., 2020; CCLE gene expression data, CCLE data portal (https://portals.broadinstitute.org/ccle); bulk RNA-seq data of patient-derived melanoma cell lines, GEO, GSE80829; bulk RNA-seq data of in vitro differentiating melanocytes derived from ESC/iPSC, GEO: GSE45227; single-cell RNA-seq data from human normal skin samples of different developmental stages, GEO: GSE151091; Single-cell RNA-seq data from stepwise-edited melanoma cell lines, Single Cell Portal: SCP1334; spatial transcriptomics data from treatment-naïve melanoma patients, http://www.spatialomics.org/SpatialDB; DICE immune cell type gene expression data, https://dice-database.org; bulk proteomics data from human hematopoietic cell populations sorted from peripheral blood, http://www.immprot.org. Code used for all processing and analysis is available upon request.