

Abstract
Esophageal atresia/tracheoesophageal fistula (EA/TEF) is a life-threatening birth defect that often occurs with other major birth defects (EA/TEF+). Despite advances in genetic testing, a molecular diagnosis can only be made in a minority of EA/TEF+ cases. Here, we analyzed clinical exome sequencing data and data from the DECIPHER database to determine the efficacy of exome sequencing in cases of EA/TEF+ and to identify phenotypic expansions involving EA/TEF. Among 67 individuals with EA/TEF+ referred for clinical exome sequencing, a definitive or probable diagnosis was made in 11 cases for an efficacy rate of 16% (11/67). This efficacy rate is significantly lower than that reported for other major birth defects, suggesting that polygenic, multifactorial, epigenetic and/or environmental factors may play a particularly important role in EA/TEF pathogenesis. Our cohort included individuals with pathogenic or likely pathogenic variants that affect TCF4 and its downstream target NRXN1, and FANCA, FANCB, and FANCC, which are associated with Fanconi anemia. These cases, previously published case reports, and comparisons to other EA/TEF genes made using a machine learning algorithm, provide evidence in support of a potential pathogenic role for these genes in the development of EA/TEF.
Keywords: Esophageal atresia, tracheoesophageal fistula, exome sequencing, TCF4, NRXN1, Fanconi anemia
INTRODUCTION
Esophageal atresia (EA) and tracheoesophageal fistula (TEF) are common, life-threatening birth defects seen in approximately 1:3,500 to 1:4,500 live births (Depaepe et al., 1993; Lupo et al., 2017). These defects arise from failure of the proximal foregut to properly separate into distinct respiratory (ventral) and gastrointestinal (dorsal) tubes between 28 days and 37 days post-fertilization (Ioannides & Copp, 2009). In the majority of cases, EA and TEF occur together, with isolated EA accounting for approximately 8% of cases, and isolated H-type TEF with no EA accounting for approximately 3% of cases (Scott, 1993). EA/TEF is associated with high rates of mortality, which correlate positively with the co-existence of other structural birth defects, and significant morbidity, including the need for multiple hospitalizations and surgeries, feeding intolerance, gastroesophageal reflux, failure to thrive, and recurrent infections (Little et al., 2003; Seo et al., 2010; Sulkowski et al., 2014).
Between 50 and 84% of individuals with EA/TEF have at least one additional congenital anomaly (EA/TEF+) with congenital heart defects (CHD) being particularly common (Cassina et al., 2016; Seo et al., 2010; Sulkowski et al., 2014). A subset of individuals with EA/TEF+ can be described as having VACTERL association, which describes the non-random association of vertebral (V), anal (A), cardiac (C), tracheoesophageal fistula with esophageal atresia (TE), renal (R) and limb (L) anomalies (MIM# 192350). In a subset of cases, a large chromosomal disorder, a microdeletion/microduplication, or a single gene disorder can be identified as the cause (Brosens, de Jong, et al., 2014; Brosens et al., 2016; Felix et al., 2007; Scott, 1993). Common examples include trisomy 18, DiGeorge syndrome (MIM# 188400) caused by recurrent microdeletions of chromosome 22q11.2, and CHARGE syndrome (MIM# 214800) caused by pathogenic variants in CHD7. Failure to identify an underlying cause in the majority of EA/TEF cases is likely due to failure to employ optimal genetic testing, limited knowledge about the genes that cause EA/TEF, and/or the role of multifactorial or non-genetic factors in the development of EA/TEF (Brosens, Ploeg, et al., 2014).
Here we analyze a clinical database of ~17,000 exome sequencing test results to determine the diagnostic efficacy of exome sequencing in individuals with EA/TEF+. We then use data from this clinical cohort and individuals cataloged in the DECIPHER database to identify associations between disease-causing genes and EA/TEF+.
MATERIALS AND METHODS
Ethical approval
This work was approved by the institutional review board of Baylor College of Medicine (protocol H-47546) and was conducted in accordance with the ethical standards of this institution’s committee on human research and international standards.
Database analysis and clinical review
We searched coded information from a clinical database of ~17,000 individuals who were referred to Baylor Genetics for exome sequencing to identify individuals with EA/TEF+ based on the phenotypes included in their test indications (Supplemental Table S1). Individuals for whom a molecular diagnosis was made using array-based copy number variant detection assays or other genetic tests were not included in this study. Data for this cohort did not contain information on whether exome sequencing was performed on a proband, duo, or trio basis. Among the 18 individuals in which a molecular finding was reported, inheritance data suggests that no parental samples were submitted for two subjects, at least one parental sample was submitted for five subjects, and both parental samples were submitted for 11 subjects. One individual in the cohort with TEF had a sister with TEF, and the cohort included two brothers, both of whom had TEF. None of these individuals had a molecular finding reported on their exome sequencing report.
We also searched for individuals with EA/TEF who carried sequence variants, or rare, small (< 2 Mb, and containing 1–20 protein-coding genes) copy number variants cataloged in the DECIPHER database (Firth et al., 2009). Contact was made with each of the submitting centers who then approved the publication of their patient’s clinical and molecular data (Supplemental Table S2 and S3). Subject S32 is part of the Deciphering Developmental Disorders (DDD) Study. Subjects S35 and S45 were previously published by Mattioli et al. and Umana et al, respectively (Mattioli et al., 2017; Umana et al., 2011).
Variants reported by Baylor Genetics to be potentially related to the clinical phenotypes listed in the indication for ES testing, and sequence variants reported in DECIPHER, were reanalyzed and classified as pathogenic, likely pathogenic, or variants of uncertain significance (VUS) based on American College of Medical Genetics and Genomics (ACMG) standards for variant interpretation using the most current data available (Richards et al., 2015). Each potential diagnosis was then designated as definitive, probable, or provisional based on previously published criteria set forth by Scott et al. (Tiana M Scott et al., 2021). These criteria take into account the ACMG classification of the variant(s), the inheritance pattern within the family, variant configuration (cis vs. trans), the sex of the proband, and the overlap between the phenotypes listed in the indication and phenotypes previously shown to be associated with disorders caused by the affected gene.
Calculating diagnostic efficacy
The number of EA/TEF+ cases with a definitive or probable molecular diagnosis was divided by the total number of cases to determine the efficacy rate. This process was then repeated for the subset of EA/TEF+ cases in which CHD was also included in the indication, and the subset of EA/TEF+ cases in which the criteria for VACTERL association were fulfilled (at least two defining phenotypes in addition to EA/TEF) based on the phenotypes listed in the indication.
Literature and database searches
Genes affected by putatively deleterious variants that have not been clearly associated with the development of EA/TEF, were designated as EA/TEF candidate genes. We subsequently searched the literature for reports in which each candidate gene, and/or their associated genetic disorder(s), were mentioned in conjunction with EA and/or TEF. We also searched the Mouse Genome Informatics database (MGI; http://www.informatics.jax.org/) to determine if EA and/or TEF were identified in mice bearing deleterious alleles in the mouse homologs of these candidate genes (Blake et al., 2011).
Machine learning
We have previously developed a machine learning algorithm that integrates knowledge from genome-scale data sources including Gene Ontology (GO), the Mouse Genome Database (MGI), the Protein Interaction Network Analysis (PINA) platform, the GeneAtlas expression distribution, and transcription factor binding and epigenetic histone modifications data from NIH Roadmap Epigenomics Mapping Consortium, to rank genes based on their similarity to a set of training genes known to cause a phenotype of interest (Callaway et al., 2018; Campbell et al., 2013; Tiana M Scott et al., 2021). Briefly, the method is a supervised analysis procedure that employs the input training set of known disease genes to construct a pattern in genomic feature space and ranks genes with respect to their similarity to the training set using quantitative similarity metrics. For protein-protein interactions, the method considers the number of paths from the protein encoded by a target gene to the proteins encoded by the training genes using proteome-wide protein interaction network data (Campbell et al., 2014), and then compares these path counts to a genome wide distribution. For the other knowledge sources, a centroid is created by the mean characteristics of the training set, be they annotations or tissue expression patterns. A multivariate distance metric motivated by Mahalanobis distance computes the similarity of the feature characteristics of a target gene, as annotated in the knowledge source, to this centroid. Each gene is then ranked from highest to lowest according to its similarity to the training set.
To generate EA/TEF-specific pathogenicity scores for all RefSeq genes, we trained this machine learning algorithm with a set of 42 manually curated human genes that are known to cause EA/TEF or whose mouse homologs have been shown to cause EA/TEF. This EA/TEF training gene set included AAAS, AXIN1, CHD7, CHRD, CPLANE2, CTNNB1, DYNC2H1, EFNB2, EFTUD2, FGFR3, FOXF1, FUZ, GDF3, GDF6, GLI2, GLI3, IFT172, MKKS, MYCN, NDN, NKX2-1, NKX3-2, NOG, PAX2, PCSK5, POR, RAB25, SHH, SNRPN, SOX17, SOX2, SOX4, TBX1, TBX5, TCOF1, TERC, UBE3A, UPK3A, VANGL1, WDR35, WHSC1, and WNT3 (Blake et al., 2011; Brosens, Ploeg, et al., 2014). The inclusion of genes shown to cause EA/TEF in mice was supported by the strong biological evidence of causality demonstrated in these mouse models. The addition of these genes to the training gene set also resulted in an improvement in cross validation scores (data not shown).
Cross validation studies were performed to test the performance of our machine learning procedure (Callaway et al., 2018; Campbell et al., 2013). First, the full set of training genes was randomly broken into four subsets of approximately equal size. The machine learning procedure was then trained on each respective set, and a genome wide evaluation of all genes was performed including the excluded subset of training genes. The percentiles of the excluded genes were then recorded to assess performance. The procedure was repeated, reciprocally, so that all training genes received cross-validated scores.
To visualize the performance of the procedure, we tabulated the fraction of EA/TEF training set genes with percentile scores exceeding various cutoffs. This allowed us to generate receiver operating characteristic (ROC) style curves where the effectiveness of the procedure corresponds to the area between the curve and a diagonal line which represents the result that would be generated by chance alone (Figure 1A). In this case, the ROC curves generated using data from each knowledge source, and the average of the scores across all knowledge sources, were positive. This provided evidence that our scoring procedure can identify the EA/TEF training genes more efficiently than random chance.
Figure 1. Generation and evaluation of EA/TEF-specific pathogenicity scores.
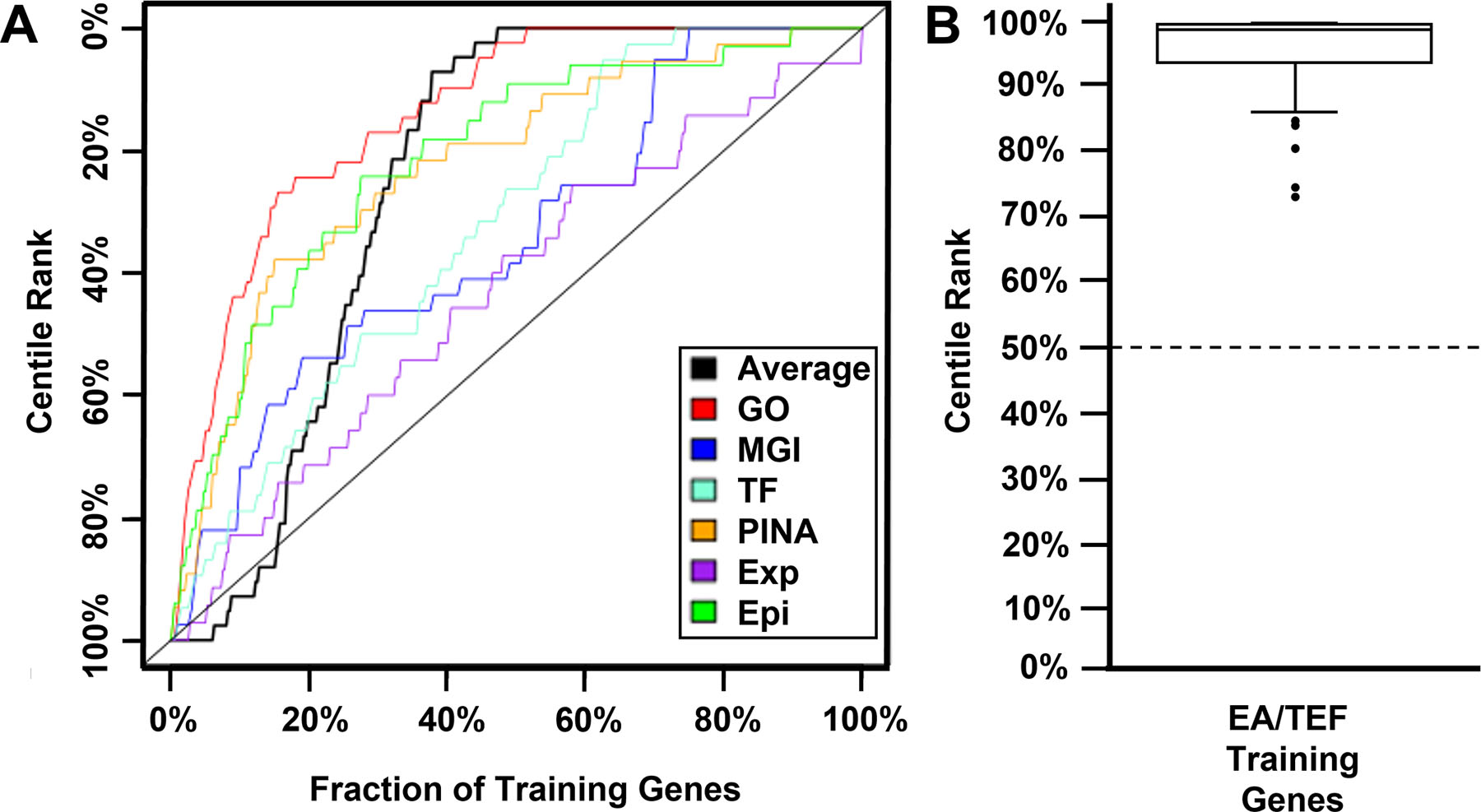
A) Receiver operating characteristic (ROC) style curves were generated in validation studies of our machine learning scoring approach using individual knowledge sources (colored) and the average score from all knowledge sources (black). The positive area underneath each curve indicates that our scoring approach identified training set genes known to cause EA/TEF more efficiently than random chance (diagonal dashed line). B) After validation, EA/TEF-specific pathogenicity scores were calculated for all RefSeq genes. The EA/TEF training gene set had a range of 72.6%−99.9% with median score of 99% with CPLANE2 (72.6%), TCOF1 (74.2%), EFTUD2 (80.2%), SNRPN (83.8%) and VANGL1 (84.6%) being outliers. This is significantly higher than the 50% median score (dashed line) for all RefSeq genes. Epi = Epigenetic histone modifications data from NIH Roadmap Epigenomics Mapping Consortium, Exp = the GeneAtlas expression distribution, GO = Gene Ontology, MGI = the Mouse Genome Database, PINA = the Protein Interaction Network Analysis platform, TF = transcription factor binding data from NIH Roadmap Epigenomics Mapping Consortium.
Having validated the algorithm, we then generated EA/TEF-specific pathogenicity scores for all RefSeq genes based on the centile rank of an omnibus score generated using fit data from all knowledge sources as previously described (Callaway et al., 2018; Campbell et al., 2013). By definition, the resulting EA/TEF-specific pathogenicity scores ranged from 0 to 100%, with a median of 50%. In contrast, the EA/TEF training gene set had a range of 72.6%−99.9% with median score of 99% (Figure 1B, Supplemental Table S4), with CPLANE2 (72.6%), TCOF1 (74.2%), EFTUD2 (80.2%), SNRPN (83.8%) and VANGL1 (84.6%) being outliers.
Statistical analyses
Two-tailed Fisher’s exact tests were performed using a 2×2 contingency table calculator available through GraphPad QuickCalcs (https://www.graphpad.com/quickcalcs/contingency1/) to compare the diagnostic yields between sub-cohorts. Box plots were generated using the Alcula.com Statistical Calculator: Box Plot program (http://www.alcula.com/calculators/statistics/box-plot/).
RESULTS
Diagnostic efficacy of clinical exome sequencing
In a clinical database of approximately 17,000 individuals who were referred for exome sequencing, we identified 67 individuals with EA/TEF+ based on phenotypes included in their test indications. Exome sequencing provided a definitive (n = 10; 15%) or probable (n = 1; 1.5%) diagnosis in 11 individuals for an efficacy rate of 16.4% (11/67). A provisional diagnosis was made in 7 additional EA/TEF+ case (7/67, 10%). If there provisional cases were included, the efficacy rate for EA/TEF+ would be 26.9% (18/67; Table S1).
In the subset of 39 individuals with EA/TEF+ who had CHD, a definitive or probable diagnosis was made in 8, for an efficacy rate of 20.5% (8/39). In the subset of 23 individuals with EA/TEF+ who met criteria for VACTERL association, a definitive or probable diagnosis was made in 3 for an efficacy rate of 13% (3/23). Although a higher efficacy rate was found for individuals with CHD, and a lower efficacy rate was found for individuals who met criteria for VACTERL association, these differences were not found to be statistically different than the subset of cases that did not have CHD (3/28, 10.7%; p = 0.3366) and the subset of cases that did not meet criteria for VACTERL association (8/44, 18.2%; p = 0.736), respectively.
Genes known to cause EA/TEF
The only gene that was recurrently found to cause EA/TEF in our clinical cohort was CHD7. Heterozygous, loss-of-function variants in this gene cause CHARGE syndrome (MIM# 214800). It has been previously reported that approximately 15%−20% of individuals with CHARGE syndrome have EA/TEF (Lalani et al., 1993). In our cohort, Subjects S3 and S4 carried frameshift variants in CHD7, and S5 carried a stop-gain variant (Supplemental Table S1). Other individuals in our clinical cohort carried pathogenic or likely pathogenic variants in other genes that have been clearly associated EA/TEF. These genes included EFTUD2 (Subject S6) that causes mandibulofacial dysostosis, Guion-Almeida type (MIM# 610536), FGFR3 (Subject S9) that causes a variety of skeletal dysplasias, and MYCN (Subject S12) that causes Feingold syndrome 1 (MIM# 164280).
Candidate genes for EA/TEF
The majority of individuals in our clinical cohort who carried a definitive or probable diagnosis had associated variants in human disease genes that have not been clearly shown to cause EA/TEF. For the remainder of our study, we considered these genes as candidate genes for EA/TEF.
To identify additional candidate genes, we searched the DECIPHER database for individuals with EA/TEF who had sequence changes or rare, small (< 2 Mb and containing 1–20 protein-coding genes) copy number variants (CNVs) (Firth et al., 2009; MacDonald et al., 2014). We obtained permission to publish the clinical and molecular data from 41 individuals with EA/TEF who met these criteria; 14 individuals with sequence variants (Subjects S19-S32; Supplemental Table S2), and 27 individuals with CNVs (Subjects S33-S59); Supplemental Table S3).
Some of the genes which were altered in individuals with EA/TEF from DECIPHER have been shown to cause EA/TEF including CHD7 (Subjects S19 and S20) and EFTUD2 (Subjects S21-S24). The remainder had changes affecting genes not clearly associated with EA/TEF. These genes were considered candidate genes for EA/TEF if they, 1) were affected by single nucleotide variants associated with a definitive or probable diagnosis, 2) were affected by a deletion and were predicted to have high loss-of-function intolerance (pLI >0.8 in gnomAD), or 3) if they may have been disrupted by a duplication based on the presence of one or more breakpoints within the gene (marked by an † in Supplemental Table S3) and were predicted to have high loss-of-function intolerance (pLI >0.8 in gnomAD) (Karczewski et al., 2020).
Associations between candidate genes and EA/TEF
Since EA/TEF is a relatively common birth defect, it is possible that it arose in some individuals in our cohort independent of the changes detected by exome sequencing or copy number variant analysis. As a means of determining which candidate genes for EA/TEF were the most likely to be contributing to the development of this disorder, we used an approach previously employed by Scott et al. (T. M. Scott et al., 2021). Briefly, we determined whether each gene associated with a definitive or probable diagnosis, had been previously reported in association with EA/TEF in humans, is known to cause a genetic syndrome previously associated with EA/TEF, and/or had a high similarity to genes known to cause EA/TEF based on a high EA/TEF-specific pathogenicity score (≥ 85% rank compared to all RefSeq genes) generated using our machine learning procedure. The results of these evaluations are summarized in Tables 1–3.
Table 1.
Genes with sufficient evidence to support an association with EA/TEF
| Gene | Disorder, MIM number | Subject ID; variant; ACMG interpretation | Number of individuals in our cohort with changes in this gene; level of diagnostic certainty | EA/TEF-specific pathogenicity score | Other cases of EA/TEF reported for this gene/disorder | References |
|---|---|---|---|---|---|---|
| TCF4 | Pitt-Hopkins syndrome, MIM# 610954 | S17; c.1486+1G>T [NM_001083962.2] p.(?); Pathogenic | 1; Definitive | 90.7% | Yes/Yes | Whalen et al., 2012 |
| NRXN1 | Pitt-Hopkins-like syndrome 2, MIM# 614325; Susceptibility to schizophrenia, MIM# 614332; Susceptibility to developmental delay, intellectual disability, abnormal behaviors, autism spectrum disorder, and seizures | S31; c.2138C>G, [NM_001135659.3] p.(Ser713*); Likely Pathogenic | 1; Probable | 90.2% | Yes/Yes | Lowther et al., 2017 |
| FANCA | Fanconi anemia complementation group A, MIM# 227650 | S25; c.[2175_2182del];[3788_3790del] [NM_000135.4] p.[(Phe726Glufs*65)];[(Phe1263del)]; Pathogenic/Pathogenic | 1; Definitive | 41.8% | Yes/Yes | Faivre et al., 2005 |
| FANCB | Fanconi anemia complementation group B, MIM# 300514 | S41†; Deletion; Pathogenic | 1; Definitive | 40.4% | Yes/Yes | Holden et al., 2006, McCauley et al., 2011 |
| FANCC | Fanconi anemia complementation group C, MIM# 227645 | S7; c.[1642C>T];[1642C>T] [NM_000136.3] p.[(R548*)];[(R548*)] Pathogenic/Pathogenic | 1; Definitive | 51.3% | Yes/Yes | Kutler & Auerbach, 2004 |
Previously reported by Umana et al. (Umana et al., 2011).
In Table 1, we list genes for which there is sufficient evidence to suggest an association with EA/TEF. In Tables 2 and 3, we list genes carrying sequence variants, or genes potentially disrupted by CNVs, respectively, for which there is currently insufficient evidence to suggest an association with EA/TEF.
Table 2.
Genes with definitive or probable diagnoses that do not currently have sufficient evidence to support an association with EA/TEF
| Gene | Disorder, MIM# number | Subject ID; variant; ACMG interpretation | Level of diagnostic certainty | EA/TEF specific pathogenicity score | Other cases EA/TEF reported for this gene/disorder in humans |
|---|---|---|---|---|---|
| NEDD4L | Periventricular nodular heterotopia 7, MIM# 617201 | S13; c.814-6T>A [NM_015277.6] p.(?); Likely Pathogenic | Probable | 41.9% | No/No |
| NSD1 | Sotos syndrome 1, MIM# 117550 | S32; c.5351T>C [NM_022455.5] p.(Ile1784Thr); Likely Pathogenic | Probable | 85.4% | No/No |
| PTPN11 | LEOPARD syndrome 1, MIM# 151100; Noonan syndrome 1, MIM# 163950; Metachondromatosis, MIM #156250 | S15; c.922A>G [NM_002834.5] p.(N308D); Pathogenic | Definitive | 71% | No/No |
| SLC20A2 | Basal ganglia calcification, idiopathic, 1, MIM# 213600 | S10; c.731-18_738del26 [NM_006749.5] p.(?); Pathogenic | Definitive | 43.6% | No/No |
| TPO | Thyroid dyshormonogenesis 2A, MIM# 274500 | S18; c.[1184_1187dupGCCG];[1472G>A] [NM_000547.6] p.[(A397Pfs*76)];[(R491H)]; Pathogenic/Pathogenic | Definitive | 77.4% | No/No |
Table 3.
Genes with high pLI scores (> 0.8) that are deleted or potentially disrupted for which there is currently insufficient evidence to support an association with EA/TEF
| Gene (pLI score) | Disorder, MIM# | Subject ID, CNV type | EA/TEF-specific pathogenicity score | Other cases of EA/TEF reported for this gene/disorder in humans |
|---|---|---|---|---|
|
BRPF1 (pLI = 1) |
Intellectual developmental disorder with dysmorphic facies and ptosis, MIM# 617333 | S35, Deletion | 47.6% | No/No |
|
FUBP1
(pLI = 1) |
None | S34, Deletion | 59.9% | No/N/A |
|
GLRA2† (pLI = 0.97) |
Intellectual developmental disorder, X-linked, syndromic, Pilorge type, MIM# 301076 | S45, Deletion | 61.5% | No/N/A |
|
LARGE1 (pLI =0.99) |
Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 6, MIM# 613154; Muscular dystrophy-dystroglycanopathy (congenital with mental retardation), type B, 6, MIM# 608840 | S44, Deletion | ND | No/No |
|
LINGO2
(pLI = 0.86) |
None | S38, Deletion | 0.7% | No/N/A |
|
MOSPD2† (pLI = 0.99) |
None | S45, Deletion | 63.3% | No/N/A |
|
NRDC (pLI = 1) |
None | S33, Deletion | ND | No/N/A |
|
USP33
(pLI = 0.82) |
None | S34, Deletion | 58.5% | No/N/A |
|
SVEP1 (pLI = 1) |
None | S39, Deletion | 91% | No/N/A |
|
TMEM108
(pLI = 1) |
None | S36, Deletion | 84.4% | No/N/A |
|
ZFYVE9 (pLI = 0.9) |
None | S33, Deletion | 89.1% | No/N/A |
|
ZNF592
(pLI = 1) |
None | S42, Deletion | 38.8% | No/N/A |
|
ZZZ3 (pLI = 1) |
None | S34, Deletion | 45.5% | No/N/A |
|
DIO2 (pLI = 0.96) |
None | S53, Breakpoint of Duplication | 40.9% | No/N/A |
|
FLNA (pLI = 1) |
FLNA-related disorders, MIM# 300321, 314400, 300048, 305620, 300049, 300048, 309350, 311300, 304120, 300244 | S59, Breakpoint of Duplication | 74.3% | No/No |
|
GMDS (pLI = 0.99) |
None | S49, Breakpoint of Duplication | 7.3% | No/N/A |
|
OTUD7A (pLI = 0.95) |
None | S55, Breakpoint of Duplication | 11.8% | No/N/A |
|
PACS1 (pLI = 1) |
Schuurs-Hoeijmakers syndrome, MIM# 615009 | S51, Breakpoint of Duplication | 79.5% | No/No |
|
PDE4D (pLI = 1) |
Acrodysostosis 2, with or without hormone resistance, MIM# 614613 | S48, Breakpoint of Duplication | 73.8% | No/No |
|
RORA (pLI = 1) |
Intellectual developmental disorder with or without epilepsy or cerebellar ataxia, MIM# 618060 | S56, Breakpoint of Duplication | 80.9% | No/No |
|
SART1 (pLI = 0.88) |
None | S51, Breakpoint of Duplication | 19.4% | No/N/A |
Found in a male previously reported by Umana et al. who carried a deletion of FANCB (Umana et al., 2011).
N/A = not applicable, ND = not determined
DISCUSSION
Isolated EA/TEF is associated with an empiric sibling recurrence risk of 1%, and an approximately 2–4% recurrence risk in the offspring of an affected individual (Shaw-Smith, 2006). These low recurrence risks, and a twin concordance rate of ~2.5%, are consistent with a polygenic/multifactorial inheritance pattern (Robert et al., 1993). The sibling recurrence risk for non-isolated EA/TEF is often described as low in most families, with a minority having a higher risk (~25 to 50%) due to a Mendelian disorder. Similarly, in the absence of a genetic diagnosis, the offspring recurrence risk for EA/TEF and/or other VACTERL-associated malformations has been estimated at ~2–4%, but the offspring recurrence risk for individuals with a genetic diagnosis can be up to 50% (Robert et al., 1993; Scott, 1993). Therefore, identifying an underlying cause for EA/TEF through genetic testing can provide more accurate, and individualized recurrence risk estimates.
Other potential benefits of obtaining a molecular diagnosis include improved prognostication and medical management that can optimize the use of medical resources and can lead to the early identification and treatment of associated medical problems (Matias et al., 2019; Niguidula et al., 2018). Despite these potential benefits, exome sequencing is not universally ordered for individuals with EA/TEF+ without a molecular diagnosis. This may be due to uncertainty about the efficacy of exomes sequencing in individuals with EA/TEF+.
Efficacy of clinical ES in cases of EA/TEF+
In our clinical cohort of 67 individuals with EA/TEF+, exome sequencing provided a definitive or probable diagnosis in 11 individuals for an efficacy rate of 16.4% (11/67). Given the potential benefits of a molecular diagnosis, this efficacy rate is sufficiently high to suggest that such testing should be considered for all individuals with EA/TEF+ who do not have a molecular diagnosis.
The efficacy rate of clinical exome sequencing in individuals with EA/TEF+ is significantly lower than the efficacy rate reported in individuals with other birth defects (Meng et al., 2017; Retterer et al., 2016) including the 37% (28/76; p = 0.0082) rate for individuals with congenital diaphragmatic hernia plus (CDH+) determined using data from the same clinical cohort (T. M. Scott et al., 2021). This difference is maintained if provisional diagnoses are included with the efficacy rate for EA/TEF+ being 26.9% (18/67) compared to 46.1% (35/76) for CDH+ (p = 0.024) (T. M. Scott et al., 2021).
To further investigate this pattern, we reviewed chromosomal microarray (CMA) data from Baylor Genetics. We found that pathogenic or likely pathogenic copy number variants were reported in 2% (1/51) of CMA cases in which EA/TEF was listed as a phenotype and 10% (22/223) of CMA cases in which CDH was listed as a phenotype. Although this is consistent with the pattern seen in our exome sequencing results, the difference in CMA efficacy rates did not reach statistically significance (p = 0.09).
One possible reason for the difference in clinical exome sequencing efficacy rates between EA/TEF+ and CDH+ is that polygenic, multifactorial, or non-genetic factors—environmental, epigenetic, or stochastic factors—may play a greater role in the development of EA/TEF than CDH. Unlike CDH, EA/TEF is a feature of VACTERL association. Several maternal risk factors, such as conception via assisted reproductive technologies, pregestational diabetes, and chronic obstructive lower pulmonary diseases, are positively correlated with the risk of having a child with VACTERL association (Lubinsky, 2018; van de Putte et al., 2020). Epigenetic factors have also been postulated to contribute to the development of VACTERL association (Lubinsky, 2018; Solomon, 2018).
In this study, the efficacy rate of exome sequencing in the subset of individuals with EA/TEF+ who met criteria for VACTERL association (13%; 3/23) was lower than that the subset of cases that did not meet criteria for VACTERL association (18.2%; 8/44), although the difference was not statistically significant (p = 0.736). The efficacy rate observed in VACTERL association cases from our cohort is comparable to the 14% (38/271) positivity rate of individuals referred to a different reference lab (GeneDx) for exome sequencing who had VACTERL association annotated as part of their clinical characteristics (Solomon, 2018). Additionally, 10% (28/271) of individuals in this GeneDx cohort had a reportable variant in a plausible candidate gene, and 34% (91/271) had variants of unknown significance in genes that correlated with the reported phenotypes. Solomon went on to report that the exome sequencing efficacy rate for this cohort was lower than the overall exome sequencing efficacy rate and, more specifically, the exome sequencing efficacy rate of individuals with multiple congenital anomalies (Retterer et al., 2016; Solomon, 2018). Further research into the genetic, epigenetic, and environmental factors that contribute to the development of EA/TEF and VACTERL association is warranted.
Phenotypic expansions involving EA/TEF
TCF4 and NRXN1
Autosomal dominant, pathogenic variants in TCF4 are associated with Pitt-Hopkins syndrome (MIM# 610954), a neurodevelopmental disorder characterized by global developmental delay, intellectual disability, autism spectrum disorder, distinctive facial features, episodic hyperventilation and/or breath-holding, seizures and severe myopia (Amiel et al., 2007; Pitt & Hopkins, 1978; Zweier et al., 2007). Whalen et al. described identical twins with Pitt-Hopkins syndrome, one of whom had congenital anomalies including esophageal atresia and a sacral mass, that were not seen in her twin (Whalen et al., 2012). Subject S17 had EA/TEF, neurodevelopmental phenotypes, dysmorphic features, congenital esotropia and myopia associated with a de novo c.1486+1G>T, p.(?) [NM_001083962.2] pathogenic variant in TCF4. This gave her a definitive diagnosis of Pitt-Hopkins syndrome and suggests that loss of TCF4 function may lead to an increased risk of developing EA/TEF.
TCF4 encodes a basic helix-loop-helix transcription factor that transactivates the NRXN1β and CNTNAP2 promoters in luciferase assays (Forrest et al., 2012). This suggests that TCF4, NRXN1 and CNTNAP2 may function in a common pathway during development and that disruption of this pathway may lead to a common set of phenotypes. Consistent with that hypothesis, autosomal recessive variants in NRXN1 are associated with Pitt-Hopkins-like syndrome 2 (MIM# 614325), and autosomal dominant NRXN1 variants are associated with susceptibility to schizophrenia (MIM# 614332), developmental delay, intellectual disability, abnormal behaviors, autism spectrum disorder, and seizures (Dabell et al., 2013; Harrison et al., 2011; Kim et al., 2008; Lowther et al., 2017; Schaaf et al., 2012; Zweier et al., 2009). Similarly, autosomal recessive and autosomal dominant variants in CNTNAP2 are associated with Pitt-Hopkins-like syndrome 1 (MIM# 610042) and autism susceptibility 15 (MIM# 612100), respectively (Alarcon et al., 2008; Zweier et al., 2009).
EA/TEF are not common features seen in NRXN1 and CNTNAP2-related disorders. However, TEF has been previously documented in one individual with a deletion overlapping the 5′ end of NRXN1, indicating a deletion of one or more of exons 1–4 (Lowther et al., 2017). Subject S31 had TEF, butterfly vertebrae, a dilated vestibule of the inner ear, and hearing abnormality and carried a likely pathogenic c.2138C>G, p.(Ser713*) [NM_001135659.3] stop-gain variant in NRXN1. In contrast, no individuals with EA/TEF harboring putatively deleterious variants in CNTNAP2 were identified in this or previous studies. However, large (>13 Mb) terminal 7q deletions including CNTNAP2 have been documented in two patients with EA/TEF (Busa et al., 2016; Speleman et al., 1992) and one patient with esophageal stenosis (Zen et al., 2010).
Taken together these data suggest that deleterious variants in genes associated with Pitt-Hopkins syndrome and related disorders—especially TCF4 and NRXN1—may predispose to the development of EA/TEF. Consistent with this possibility, we note that TCF4, NRXN1 and CNTNAP2 had high EA/TEF-specific pathogenicity scores of 90.7%, 90.2% and 81.1%, respectively.
FANCA, FANCB, FANCC, FANCL
Fanconi anemia (FA) is a genetically heterogenous disorder that can be caused by pathogenic variants in at least 23 genes and is characterized by a variety of structural birth defects, bone marrow failure, and increased risk for malignancy (Mehta & Ebens, 1993). EA and TEF are seen in approximately 1.4% and 3.5%, respectively, of individuals with FA (Giampietro et al., 1993). Here we report individuals with EA/TEF who carried variants in four FA-associated genes: FANCA, FANCB, FANCC, and FANCL.
Faive et al. described a boy with Fanconi anemia with EA/TEF who carried two pathogenic FANCA deletions, one of exons 6–8, and one of exons 11–21 (Faivre et al., 2005). More recently, Feng et al. identified four rare, heterozygous variants in FANCA in individuals with EA/TEF (Feng et al., 2018). In our study, Subject S25 is a 2 year-old male with TEF, a ventricular septal defect, aplasia/hypoplasia of the forearm bones, a short thumb, penoscrotal hypospadias, and 2–3 toe syndactyly who carried compound heterozygous, pathogenic c.[2175_2182del];[3788_3790del], p.[(Phe726Glufs*65)];[(Phe1263del)] [NM_000135.4] FANCA variants.
Holden et al. described a three-generation family with X-linked VACTERL-H syndrome caused by a c.1496+5G>A [NM_001018113.3] splice donor variant in FANCB that caused skipping of exon 7 (Holden et al., 2006). This family included a stillborn male fetus with EA/TEF, hydrocephalus associated with an Arnold-Chiari malformation, CHD, unilateral renal agenesis on the right, dysplastic left kidney, missing thumbs, bilateral absent radii, lumbar spina bifida occulta, and abnormal ears. McCauley et al. described two boys with X-linked VACTERL-H and EA/TEF (McCauley et al., 2011). The first had esophageal atresia, dilation of the lateral and third ventricles with a small 4th ventricle, right renal agenesis, left ureteric and pelvicalyceal dilation, absent thumbs and radii, micropenis, a short neck, and eye and ear anomalies associated with a maternally inherited deletion of FANCB exons 8–10. The second had a tracheoesophageal fistula, ventriculomegaly, absent thumbs and radii, and carried a c.2150T>G, p.Leu717* stop-gain variant in FANCB. In our study, Subject S45 is a previously published male infant with TEF, ventricular septal defect, renal hypoplasia, anal atresia, aplasia/hypoplasia of the thumb, and hypoplasia of the radius, who carries an chrX:14,543,045-15,074-800 deletion (hg38) that affects FANCB (Umana et al., 2011). This deletion also includes GLRA2, whose loss is associated with intellectual developmental disorder, X-linked, syndromic, Pilorge type (MIM# 301076), and MOSPD2, which has a pLI score of 0.99 in gnomAD but is currently not associated with a genetic disorder in humans (Pilorge et al., 2016).
Cox et al. reported an Ashkenazi Jewish, non-identical twin pair with VACTERL-H (Cox et al., 1997). The male fetus had a TEF, dilated lateral ventricles, right lung lobulation defects, duodenal atresia, intestinal malrotation, an ectopic right kidney, absent right radius and thumb, and small, dysplastic ears abnormal ears. The affected female fetus did not have EA/TEF. Both twins were homozygous for the pathogenic c.456+4A>T [NM_000136.3] variant in FANCC that is commonly seen in individuals of Ashkenazi Jewish ancestry (Kutler & Auerbach, 2004). In our study, Subject S7 was a female infant with EA/TEF, ventricular septal defect, horseshoe kidney, duodenal atresia, anteriorly placed anus, a flexion contracture of the left wrist, short thumb, radial defect, conductive hearing impairment, atresia of the external auditory canal, absence of clitoris, vaginal prolapse, clubbing of fingers, and a sacral dimple who was homozygous for a pathogenic c.1642C>T, p.(Arg548*) [NM_000136.3] stop variant in FANCC.
Vetro et al. reported a female and a male fetus from the same family that had EA/TEF, cardio/pulmonary, renal anomalies, genital, radial, and thumb anomalies. Genetic testing performed on the male fetus revealed a homozygous c.268del, p.(Leu90Phefs*6) [NM_018062.4] frameshift variant in FANCL (Vetro et al., 2015). In our study Subject S8 is a woman with EA/TEF, abnormal position of inferior vena cava, pyloric stenosis, inguinal hernia, polyarticular arthritis, irritable bowel syndrome, skeletal abnormalities, and upslanting palpebral fissures who carries a maternally inherited c.1096_1099dup, p.(Thr367Asnfs*13) [NM_018062.4] variant of unknown significance in the FANCL gene. This change occurs in the final exon of the gene and is not expected to trigger nonsense mediated mRNA decay. A second FANCL variant was not identified. Hence, it is unclear whether this variant contributed to the phenotypes seen in Subject S8.
Although there is sufficient evidence to suggest an association between EA/TEF and FANCA, FANCB, FANCC, and possibly FANCL, the EA/TEF-specific pathogenicity scores of these genes are relatively low—41.8%, 40.4%, 51.3% and 38.7% respectively. This suggests that they represent a distinct group of EA/TEF genes whose characteristics differ from those of the other EA/TEF genes in the training set. This difference is likely due, at least in part, to their unique role in DNA damage repair, bone marrow function, and cancer.
Other EA/TEF Candidate Genes
There is currently insufficient evidence to suggest an association between the genes listed in Tables 2 and 3. However, some have high EA/TEF-specific pathogenicity scores (>80%). These genes include SVEP1 (91%), TMEM108 (84.4%), and ZFYVE9 (89.1%) that have not been associated with a specific genetic disorder but have high pLI scores (≥ 0.9). It is possible that their haploinsufficiency may ultimately be found to cause a genetic syndrome in which EA/TEF is a phenotype.
Supplementary Material
ACKNOWLEDGEMENTS
This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. We note that those who carried out the original analysis and collection of the DECIPHER data bear no responsibility for the further analysis or interpretation of the data.
Some individuals described in this manuscript are part of the DDD study. The views expressed in this publication are those of the authors and not necessarily those of Wellcome or the Department of Health. The DDD study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
This study was conducted, in part, within the European Reference Network ITHACA.
FUNDING
This research was funded in part by NIH/NICHD grant HD098458 to D.A.S. Funding for the DECIPHER project was provided by Wellcome. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute [grant number WT098051].
Footnotes
CONFLICT OF INTEREST: Baylor College of Medicine (BCM) and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs genetic testing and derives revenue. The Department of Molecular & Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at BG.
DATA AVAILABILITY STATEMENT
The data generated during this study can be found within the published article and its supplementary files. The clinical exome sequencing variants described in this manuscript have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, Nelson SF, Cantor RM, & Geschwind DH (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet, 82(1), 150–159. 10.1016/j.ajhg.2007.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, Plouin P, Carter NP, Lyonnet S, Munnich A, & Colleaux L (2007). Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet, 80(5), 988–993. 10.1086/515582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT, & Mouse Genome Database, G. (2011). The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res, 39(Database issue), D842–848. 10.1093/nar/gkq1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosens E, de Jong EM, Barakat TS, Eussen BH, D’Haene B, De Baere E, Verdin H, Poddighe PJ, Galjaard RJ, Gribnau J, Brooks AS, Tibboel D, & de Klein A (2014). Structural and numerical changes of chromosome X in patients with esophageal atresia. Eur J Hum Genet, 22(9), 1077–1084. 10.1038/ejhg.2013.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosens E, Marsch F, de Jong EM, Zaveri HP, Hilger AC, Choinitzki VG, Holscher A, Hoffmann P, Herms S, Boemers TM, Ure BM, Lacher M, Ludwig M, Eussen BH, van der Helm RM, Douben H, Van Opstal D, Wijnen RM, Beverloo HB, … de Klein A (2016). Copy number variations in 375 patients with oesophageal atresia and/or tracheoesophageal fistula. Eur J Hum Genet, 24(12), 1715–1723. 10.1038/ejhg.2016.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosens E, Ploeg M, van Bever Y, Koopmans AE, H IJ, Rottier RJ, Wijnen R, Tibboel D, & de Klein A (2014). Clinical and etiological heterogeneity in patients with tracheo-esophageal malformations and associated anomalies. Eur J Med Genet, 57(8), 440–452. 10.1016/j.ejmg.2014.05.009 [DOI] [PubMed] [Google Scholar]
- Busa T, Panait N, Chaumoitre K, Philip N, & Missirian C (2016). Esophageal atresia with tracheoesophageal fistula in a patient with 7q35–36.3 deletion including SHH gene. Eur J Med Genet, 59(10), 546–548. 10.1016/j.ejmg.2016.09.001 [DOI] [PubMed] [Google Scholar]
- Callaway DA, Campbell IM, Stover SR, Hernandez-Garcia A, Jhangiani SN, Punetha J, Paine IS, Posey JE, Muzny D, Lally KP, Lupski JR, Shaw CA, Fernandes CJ, & Scott DA (2018). Prioritization of Candidate Genes for Congenital Diaphragmatic Hernia in a Critical Region on Chromosome 4p16 using a Machine-Learning Algorithm. J Pediatr Genet, 7(4), 164–173. 10.1055/s-0038-1655755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, James RA, Chen ES, & Shaw CA (2014). NetComm: a network analysis tool based on communicability. Bioinformatics, 30(23), 3387–3389. 10.1093/bioinformatics/btu536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Rao M, Arredondo SD, Lalani SR, Xia Z, Kang S-HL, Bi W, Breman AM, Smith JL, & Bacino CA (2013). Fusion of large-scale genomic knowledge and frequency data computationally prioritizes variants in epilepsy. PLoS Genet, 9(9), e1003797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina M, Ruol M, Pertile R, Midrio P, Piffer S, Vicenzi V, Saugo M, Stocco CF, Gamba P, & Clementi M (2016). Prevalence, characteristics, and survival of children with esophageal atresia: A 32-year population-based study including 1,417,724 consecutive newborns. Birth Defects Res A Clin Mol Teratol, 106(7), 542–548. 10.1002/bdra.23493 [DOI] [PubMed] [Google Scholar]
- Cox PM, Gibson RA, Morgan N, & Brueton LA (1997). VACTERL with hydrocephalus in twins due to Fanconi anemia (FA): mutation in the FAC gene. Am J Med Genet, 68(1), 86–90. https://www.ncbi.nlm.nih.gov/pubmed/8986283 [PubMed] [Google Scholar]
- Dabell MP, Rosenfeld JA, Bader P, Escobar LF, El-Khechen D, Vallee SE, Dinulos MB, Curry C, Fisher J, Tervo R, Hannibal MC, Siefkas K, Wyatt PR, Hughes L, Smith R, Ellingwood S, Lacassie Y, Stroud T, Farrell SA, … Shaffer LG (2013). Investigation of NRXN1 deletions: clinical and molecular characterization. Am J Med Genet A, 161A(4), 717–731. 10.1002/ajmg.a.35780 [DOI] [PubMed] [Google Scholar]
- Depaepe A, Dolk H, & Lechat MF (1993). The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch Dis Child, 68(6), 743–748. 10.1136/adc.68.6.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Portnoi MF, Pals G, Stoppa-Lyonnet D, Le Merrer M, Thauvin-Robinet C, Huet F, Mathew CG, Joenje H, Verloes A, & Baumann C (2005). Should chromosome breakage studies be performed in patients with VACTERL association? Am J Med Genet A, 137(1), 55–58. 10.1002/ajmg.a.30853 [DOI] [PubMed] [Google Scholar]
- Felix JF, Tibboel D, & de Klein A (2007). Chromosomal anomalies in the aetiology of oesophageal atresia and tracheo-oesophageal fistula [Review]. Eur J Med Genet, 50(3), 163–175. 10.1016/j.ejmg.2006.12.004 [DOI] [PubMed] [Google Scholar]
- Feng Y, Chen R, Da M, Qian B, & Mo X (2018). Identification of rare heterozygous missense mutations in FANCA in esophageal atresia patients using next-generation sequencing. Gene, 661, 182–188. DOI: 10.1016/j.gene.2018.03.097 https://www.ncbi.nlm.nih.gov/pubmed/29621589 [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, & Carter NP (2009). DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet, 84(4), 524–533. 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest M, Chapman RM, Doyle AM, Tinsley CL, Waite A, & Blake DJ (2012). Functional analysis of TCF4 missense mutations that cause Pitt-Hopkins syndrome. Hum Mutat, 33(12), 1676–1686. 10.1002/humu.22160 [DOI] [PubMed] [Google Scholar]
- Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, & Auerbach AD (1993). The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics, 91(6), 1116–1120. https://www.ncbi.nlm.nih.gov/pubmed/8502512 [PubMed] [Google Scholar]
- Harrison V, Connell L, Hayesmoore J, McParland J, Pike MG, & Blair E (2011). Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A, 155A(11), 2826–2831. 10.1002/ajmg.a.34255 [DOI] [PubMed] [Google Scholar]
- Holden ST, Cox JJ, Kesterton I, Thomas NS, Carr C, & Woods CG (2006). Fanconi anaemia complementation group B presenting as X linked VACTERL with hydrocephalus syndrome. J Med Genet, 43(9), 750–754. 10.1136/jmg.2006.041673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannides AS, & Copp AJ (2009). Embryology of oesophageal atresia. Semin Pediatr Surg, 18(1), 2–11. 10.1053/j.sempedsurg.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, Descartes M, Holt L, Braddock S, Troxell R, Kaplan L, Volkmar F, Klin A, Tsatsanis K, Harris DJ, … Gusella JF (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet, 82(1), 199–207. 10.1016/j.ajhg.2007.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutler DI, & Auerbach AD (2004). Fanconi anemia in Ashkenazi Jews. Fam Cancer, 3(3–4), 241–248. 10.1007/s10689-004-9565-8 [DOI] [PubMed] [Google Scholar]
- Lalani SR, Hefner MA, Belmont JW, & Davenport SLH (1993). CHARGE Syndrome. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews® https://www.ncbi.nlm.nih.gov/pubmed/20301296
- Little DC, Rescorla FJ, Grosfeld JL, West KW, Scherer LR, & Engum SA (2003). Long-term analysis of children with esophageal atresia and tracheoesophageal fistula. J Pediatr Surg, 38(6), 852–856. 10.1016/s0022-3468(03)00110-6 [DOI] [PubMed] [Google Scholar]
- Lowther C, Speevak M, Armour CM, Goh ES, Graham GE, Li C, Zeesman S, Nowaczyk MJ, Schultz LA, Morra A, Nicolson R, Bikangaga P, Samdup D, Zaazou M, Boyd K, Jung JH, Siu V, Rajguru M, Goobie S, … Bassett AS (2017). Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genet Med, 19(1), 53–61. 10.1038/gim.2016.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubinsky M (2018). An epigenetic association of malformations, adverse reproductive outcomes, and fetal origins hypothesis related effects. J Assist Reprod Genet, 35(6), 953–964. 10.1007/s10815-018-1197-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo PJ, Isenburg JL, Salemi JL, Mai CT, Liberman RF, Canfield MA, Copeland G, Haight S, Harpavat S, Hoyt AT, Moore CA, Nembhard WN, Nguyen HN, Rutkowski RE, Steele A, Alverson CJ, Stallings EB, Kirby RS, & and The National Birth Defects Prevention, N. (2017). Population-based birth defects data in the United States, 2010–2014: A focus on gastrointestinal defects. Birth Defects Res, 109(18), 1504–1514. 10.1002/bdr2.1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, & Scherer SW (2014). The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res, 42(Database issue), D986–992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias M, Wusik K, Neilson D, Zhang X, Valencia CA, & Collins K (2019). Comparison of medical management and genetic counseling options pre- and post-whole exome sequencing for patients with positive and negative results. J Genet Couns, 28(2), 182–193. 10.1002/jgc4.1054 [DOI] [PubMed] [Google Scholar]
- Mattioli F, Schaefer E, Magee A, Mark P, Mancini GM, Dieterich K, Von Allmen G, Alders M, Coutton C, van Slegtenhorst M, Vieville G, Engelen M, Cobben JM, Juusola J, Pujol A, Mandel JL, & Piton A (2017). Mutations in Histone Acetylase Modifier BRPF1 Cause an Autosomal-Dominant Form of Intellectual Disability with Associated Ptosis. Am J Hum Genet, 100(1), 105–116. 10.1016/j.ajhg.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley J, Masand N, McGowan R, Rajagopalan S, Hunter A, Michaud JL, Gibson K, Robertson J, Vaz F, Abbs S, & Holden ST (2011). X-linked VACTERL with hydrocephalus syndrome: further delineation of the phenotype caused by FANCB mutations. Am J Med Genet A, 155A(10), 2370–2380. 10.1002/ajmg.a.33913 [DOI] [PubMed] [Google Scholar]
- Mehta PA, & Ebens C (1993). Fanconi Anemia. In Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, & Amemiya A (Eds.), GeneReviews® https://www.ncbi.nlm.nih.gov/pubmed/20301575
- Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, Zhang J, He W, Dharmadhikari AV, Qu C, Ward P, Braxton A, Narayanan S, Ge X, Tokita MJ, Santiago-Sim T, Dai H, Chiang T, Smith H, … Lalani SR (2017). Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr, 171(12), e173438. 10.1001/jamapediatrics.2017.3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niguidula N, Alamillo C, Shahmirzadi Mowlavi L, Powis Z, Cohen JS, & Farwell Hagman KD (2018). Clinical whole-exome sequencing results impact medical management. Mol Genet Genomic Med, 6(6), 1068–1078. 10.1002/mgg3.484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilorge M, Fassier C, Le Corronc H, Potey A, Bai J, De Gois S, Delaby E, Assouline B, Guinchat V, Devillard F, Delorme R, Nygren G, Rastam M, Meier JC, Otani S, Cheval H, James VM, Topf M, Dear TN, … Betancur C (2016). Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol Psychiatry, 21(7), 936–945. 10.1038/mp.2015.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt D, & Hopkins I (1978). A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J, 14(3), 182–184. 10.1111/jpc.1978.14.3.182 [DOI] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, McKnight D, Bai R, Suchy S, Friedman B, Tahiliani J, Pineda-Alvarez D, Richard G, Brandt T, Haverfield E, … Bale S (2016). Clinical application of whole-exome sequencing across clinical indications. Genet Med, 18(7), 696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, & Committee ALQA (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert E, Mutchinick O, Mastroiacovo P, Knudsen LB, Daltveit AK, Castilla EE, Lancaster P, Kallen B, & Cocchi G (1993). An international collaborative study of the epidemiology of esophageal atresia or stenosis. Reprod Toxicol, 7(5), 405–421. http://www.ncbi.nlm.nih.gov/pubmed/8274816 [DOI] [PubMed] [Google Scholar]
- Schaaf CP, Boone PM, Sampath S, Williams C, Bader PI, Mueller JM, Shchelochkov OA, Brown CW, Crawford HP, Phalen JA, Tartaglia NR, Evans P, Campbell WM, Tsai AC, Parsley L, Grayson SW, Scheuerle A, Luzzi CD, Thomas SK, … Cheung SW (2012). Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur J Hum Genet, 20(12), 1240–1247. 10.1038/ejhg.2012.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA (1993). Esophageal Atresia / Tracheoesophageal Fistula Overview. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews® https://www.ncbi.nlm.nih.gov/pubmed/20301753 [PubMed]
- Scott TM, Campbell IM, Hernandez-Garcia A, Lalani SR, Liu P, Shaw CA, Rosenfeld JA, & Scott DA (2021). Clinical exome sequencing data reveal high diagnostic yields for congenital diaphragmatic hernia plus (CDH+) and new phenotypic expansions involving CDH. J Med Genet, 59(3), 270–278. 10.1136/jmedgenet-2020-107317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J, Kim DY, Kim AR, Kim DY, Kim SC, Kim IK, Kim KS, Yoon CH, & Pi SY (2010). An 18-year experience of tracheoesophageal fistula and esophageal atresia. Korean J Pediatr, 53(6), 705–710. 10.3345/kjp.2010.53.6.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Smith C (2006). Oesophageal atresia, tracheo-oesophageal fistula, and the VACTERL association: review of genetics and epidemiology. J Med Genet, 43(7), 545–554. 10.1136/jmg.2005.038158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD (2018). The etiology of VACTERL association: Current knowledge and hypotheses. Am J Med Genet C Semin Med Genet, 178(4), 440–446. 10.1002/ajmg.c.31664 [DOI] [PubMed] [Google Scholar]
- Speleman F, Van Roy N, Wiegant J, Verschraegen-Spae MR, Benoit Y, Govaert P, Goossens L, & Leroy JG (1992). Detection of subtle reciprocal translocations by fluorescence in situ hybridization. Clin Genet, 41(4), 169–174. 10.1111/j.1399-0004.1992.tb03657.x [DOI] [PubMed] [Google Scholar]
- Sulkowski JP, Cooper JN, Lopez JJ, Jadcherla Y, Cuenot A, Mattei P, Deans KJ, & Minneci PC (2014). Morbidity and mortality in patients with esophageal atresia. Surgery, 156(2), 483–491. 10.1016/j.surg.2014.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umana LA, Magoulas P, Bi W, & Bacino CA (2011). A male newborn with VACTERL association and Fanconi anemia with a FANCB deletion detected by array comparative genomic hybridization (aCGH). Am J Med Genet A, 155A(12), 3071–3074. 10.1002/ajmg.a.34296 [DOI] [PubMed] [Google Scholar]
- van de Putte R, van Rooij I, Haanappel CP, Marcelis CLM, Brunner HG, Addor MC, Cavero-Carbonell C, Dias CM, Draper ES, Etxebarriarteun L, Gatt M, Khoshnood B, Kinsner-Ovaskainen A, Klungsoyr K, Kurinczuk JJ, Latos-Bielenska A, Luyt K, O’Mahony MT, Miller N, … Roeleveld N (2020). Maternal risk factors for the VACTERL association: A EUROCAT case-control study. Birth Defects Res, 112(9), 688–698. 10.1002/bdr2.1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetro A, Iascone M, Limongelli I, Ameziane N, Gana S, Della Mina E, Giussani U, Ciccone R, Forlino A, Pezzoli L, Rooimans MA, van Essen AJ, Messa J, Rizzuti T, Bianchi P, Dorsman J, de Winter JP, Lalatta F, & Zuffardi O (2015). Loss-of-Function FANCL Mutations Associate with Severe Fanconi Anemia Overlapping the VACTERL Association. Hum Mutat, 36(5), 562–568. 10.1002/humu.22784 [DOI] [PubMed] [Google Scholar]
- Whalen S, Heron D, Gaillon T, Moldovan O, Rossi M, Devillard F, Giuliano F, Soares G, Mathieu-Dramard M, Afenjar A, Charles P, Mignot C, Burglen L, Van Maldergem L, Piard J, Aftimos S, Mancini G, Dias P, Philip N, … Giurgea I (2012). Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat, 33(1), 64–72. 10.1002/humu.21639 [DOI] [PubMed] [Google Scholar]
- Zen PR, Riegel M, Rosa RF, Pinto LL, Graziadio C, Schwartz IV, & Paskulin GA (2010). Esophageal stenosis in a child presenting a de novo 7q terminal deletion. Eur J Med Genet, 53(5), 333–336. 10.1016/j.ejmg.2010.06.008 [DOI] [PubMed] [Google Scholar]
- Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MA, Ekici AB, Reis A, Schenck A, & Rauch A (2009). CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet, 85(5), 655–666. 10.1016/j.ajhg.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier C, Peippo MM, Hoyer J, Sousa S, Bottani A, Clayton-Smith J, Reardon W, Saraiva J, Cabral A, Gohring I, Devriendt K, de Ravel T, Bijlsma EK, Hennekam RC, Orrico A, Cohen M, Dreweke A, Reis A, Nurnberg P, & Rauch A (2007). Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am J Hum Genet, 80(5), 994–1001. 10.1086/515583 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during this study can be found within the published article and its supplementary files. The clinical exome sequencing variants described in this manuscript have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).