

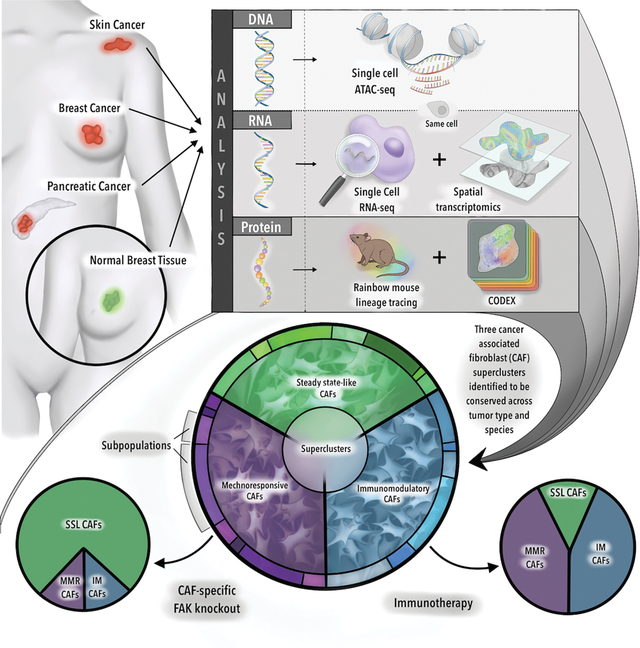
SUMMARY
Cancer associated fibroblasts (CAFs) are integral to the solid tumor microenvironment. Once thought to be a relatively uniform population of matrix-producing cells, single-cell RNA-sequencing has revealed diverse CAF phenotypes. Here, we further probed CAF heterogeneity with a comprehensive multi-omics approach. Using paired, same-cell chromatin accessibility and transcriptome analysis, we provided an integrated analysis of CAF subpopulations over a complex spatial transcriptomic and proteomic landscape to identify three superclusters – steady state-like (SSL), mechanoresponsive (MR) and immunomodulatory (IM) CAFs. These superclusters were recapitulated across multiple tissue types and species. Selective disruption of underlying mechanical force or immune checkpoint inhibition therapy resulted in shifts in CAF subpopulation distributions and impacted tumor growth. As such, the balance among CAF superclusters may have considerable translational implications. Collectively, this research expands our understanding of CAF biology, identifying regulatory pathways in CAF differentiation and elucidating therapeutic targets in a species- and tumor-agnostic manner.
eTOC:
Foster et al. integrate epigenomic, transcriptomic and proteomic changes in cancer associated fibroblasts (CAFs) both spatially and temporally through tumor development. CAF superclusters – steady state-like (SSL), mechanoresponsive (MR), and immunomodulatory (IM) – are recapitulated across tissue types and species. Immunotherapy or disruption of mechanotransduction shifts CAF distributions, suggesting a “push-pull” dynamic.
Graphical Abstract
INTRODUCTION
Solid tumors contribute to over 80% of cancer deaths. For most solid tumor types, treatment involves a combination of chemotherapy, radiation, and surgery. Approaches that target multiple cell types in the tumor microenvironment (TME) improve therapeutic efficacy. The TME is composed of transformed cancer cells along with non-transformed stromal cells including immune cells, endothelial cells, pericytes, and cancer associated fibroblasts (CAFs) (Foster et al., 2018; Ligorio et al., 2019; Norton et al., 2020). Clinically, solid tumors are often identified by their firm texture compared with benign parenchyma. This is attributable to CAFs, which secrete extracellular matrix (ECM) components (e.g., collagens), generating a dense fibrotic network (desmoplasia) in the tumor (Drifka et al., 2016).
Despite their abundance in solid tumors, CAFs are not targeted in mainstream cancer therapy. This is primarily because CAF heterogeneity and functions remain incompletely understood (Helms et al., 2021). CAFs can facilitate tumor proliferation, invasion, and metastasis, and are often associated with a poor prognosis (Park et al., 2020). However, indiscriminate CAF depletion can accelerate tumor progression (Ozdemir et al., 2014). Such paradoxical observations of CAF function support the need for further investigation of this complex cell population.
CAF heterogeneity is perhaps best characterized in terms of cell surface marker expression. They express canonical fibroblast-associated markers; however, there is no single marker that is shared by all CAFs (Sahai et al., 2020). CAF heterogeneity at the transcriptomic level is less well characterized. Further, it has yet to be determined whether individual CAF subpopulations are preserved across tumor types/sites and species, although previous analyses suggest shared phenotypes (Buechler et al., 2021).
Here, we investigate CAF single-cell gene expression and chromatin accessibility in conjunction with spatial transcriptomics across multiple solid tumor types and species, with the aim of identifying commonalities that may represent therapeutic targets. We show that CAF subpopulations fall into three broad categories – steady state-like (SSL), mechanoresponsive (MR), and immunomodulatory (IM). Each of these categories is comprised of distinct subtypes, governed by specific changes in chromatin accessibility, and spatially distinct. We validate these results functionally: fibroblast-specific disruption of mechanotransduction leads to increased SSL CAFs and more aggressive tumor biology. Alternatively, exposure to immune checkpoint inhibition results in a reduction in SSL CAFs and a proportional increase in IM CAFs in patients with treatment responses. These results suggest a “push-pull” dynamic among CAF superclusters, the balance of which suggests translational implications. Taken together, our data help to define the CAF biology spatial dynamics, identify regulatory pathways in CAF differentiation, delineate CAF subpopulation functions, elucidate novel CAF-specific therapeutic targets in a species- and tumor-agnostic manner, and provide a multimodal -omics framework for future studies in cancer biology.
RESULTS
Unlike primary tumor cells, which show distinct transcriptional and epigenomic programs in accordance with tumor type and tissue of origin (e.g., breast versus colon) (Figure 1A – left panel), the extent to which CAF signatures are recapitulated across solid tumor types and species has yet to be elucidated (Figure 1A – right panel).
Figure 1. Derivation of three CAF superclusters.
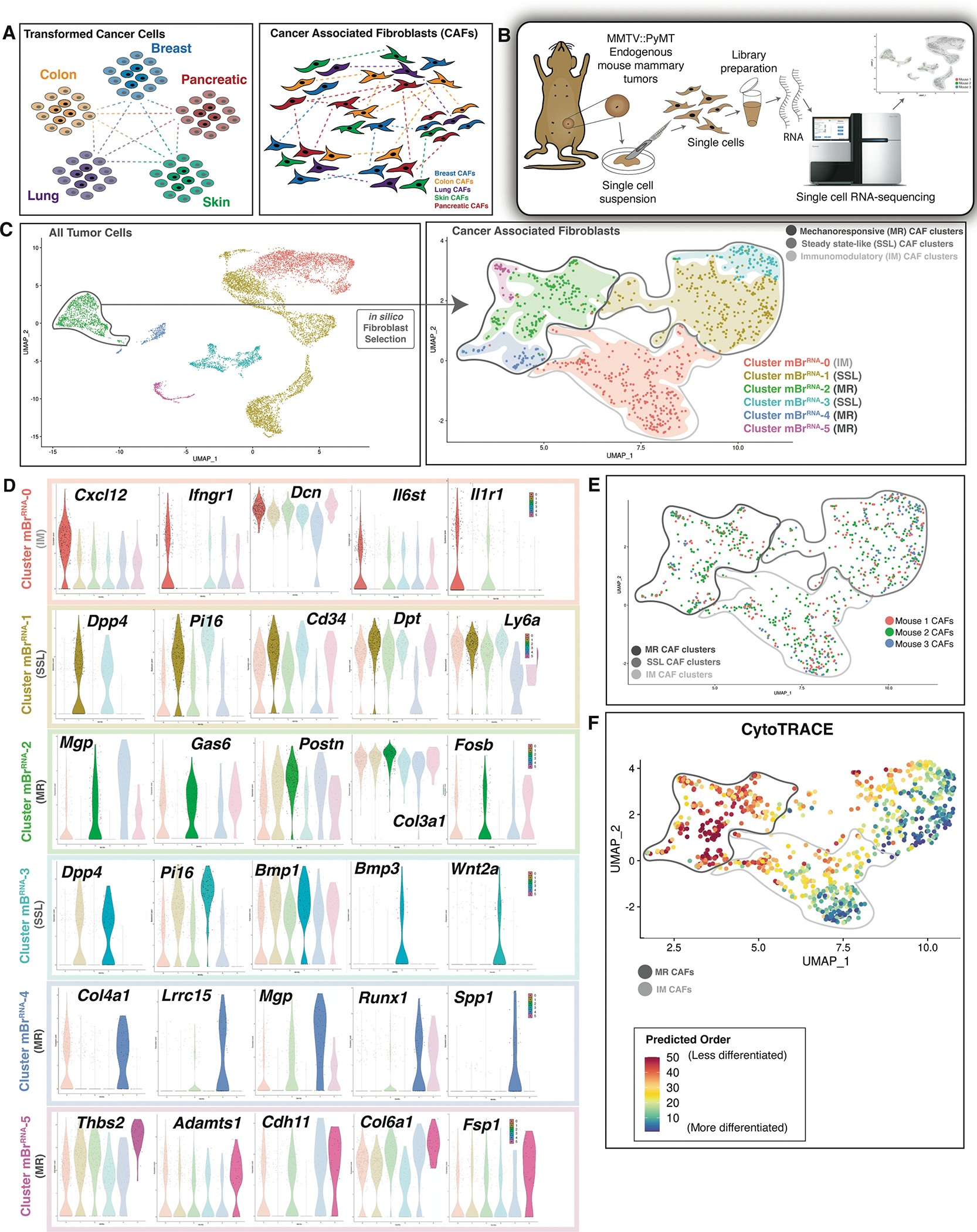
A. Schematics illustrating that transformed cancer cells show distinct transcriptional and epigenomic programs in accordance with tumor type and tissue of origin (left panel), whereas CAF subtypes may be conserved between tumor types (right panel).
B. Schematic showing the procedure of scRNA-seq applied to endogenous mouse breast tumors. MMTV::PyMt endogenous mouse tumors (n=3 biological replicates per timepoint per condition).
C. UMAP plot showing all tumor cells sequenced (left panel) → in silico CAF selection → UMAP plot demonstrating 6 distinct CAF clusters: dark grey circle indicates MR CAF clusters, medium grey circle indicates SSL CAF clusters and light grey circle indicates IM CAF clusters (right panel).
D. Violin plots showing differentially expressed genes characteristic of each mouse breast cancer CAF cluster. Colors correspond to the indicated CAF cluster in the UMAP above.
E. UMAP plot showing the sample distribution by hash-oligo data for the endogenous mouse breast tumors sequenced. MR, SSL, and IM CAF superclusters indicated as labelled in figure panel.
F. UMAP plot showing CytoTRACE analysis of mBrRNA CAF data. Mechanoresponsive and immunomodulatory CAF cluster groups are circled as labelled in figure panel.
Mouse endogenous breast CAFs are heterogeneous
CAFs are a heterogeneous cell population with a multitude of functions in the tumor microenvironment. To comprehensively delineate CAF heterogeneity by examining individual fibroblast transcriptional programs, we conducted single-cell RNA-seq (scRNA-seq) of tumor samples from syngeneic tumor-bearing mice using an endogenous mouse breast cancer model (MMTV::PyMT). (Figure 1B). Modularity-optimized clustering of the resulting dataset identified seven transcriptionally-defined populations, which we ascribed as B-cells, T-cells, Macrophages, Granulocytes, Epithelial Cells, Endothelial Cells, and CAFs (Figure 1C – left panel).
Based on in silico fibroblast selection, CAFs were re-clustered to delineate six mouse breast (MBr) CAF subgroups (Clusters 0–5) (Figure 1C – right panel, Table S1) (Januszyk et al., 2020). Cluster mBrRNA-1 demonstrated expression of Pi16, Dpp4, Dpt, and Cd34, closely aligning with recent descriptions of a stemlike phenotype in steady-state tissue (Buechler et al., 2021). mBrRNA-3 exhibited similarly high expression of Pi16 and Dpp4 with lower expression of extracellular matrix (ECM) constituents, particularly collagen, compared to mBrRNA-1 (Figure 1D). However, mBrRNA-3 was associated with higher cytokine and growth factor expression, such as Bmp1, Bmp3, and Wnt2 (Figure 1D). Given the overlap in Pi16 and Dpp4 expression, we termed these groups steady state-like (SSL) fibroblasts.
Clusters mBrRNA-2, 4, and 5 demonstrated markedly elevated expression of mechanosensitive signaling mediators and ECM components. mBrRNA-2 CAFs selectively expressed genes associated with mechanotransduction and focal adhesion kinase (FAK) pathway signaling, including Mgp, Gas6, Postn, and Fosb (Figure 1D). MBr-4 was associated with Lrrc15 and Spp1 expression, implicated in CAF pathophysiology, as well as high expression of ECM components (Dominguez et al., 2020; Nallasamy et al., 2021) (Figure 1D). mBrRNA-5 also demonstrated expression of fibrosis-associated factors, including Thbs2, Fsp1, Col6a1, and Cdh11 (Figure 1D). Supporting the functional association of mBrRNA-4 and mBrRNA-5 with mBrRNA-2, the former cells also showed upregulation of “Focal Adhesion”, “Integrin Binding”, “Protein Binding Involved in Cell-Matrix Adhesion”, and “Focal Adhesion-PI3K-akt-mTOR-signaling” pathways (Figure S1A). Taken together, we named these groups mechanoresponsive (MR) fibroblasts.
mBrRNA-0 CAFs were characterized by contributions to multiple inflammatory pathways. Interestingly, this included elements of both type II interferon and IL1 signaling. IL1-associated genes, such as Il1r1, Myd88, Il6st, and Cxcl1, were consistent with other descriptions of inflammatory CAFs (Biffi et al., 2019; Dominguez et al., 2020; Elyada et al., 2019). However, mBrRNA-0 was also enriched for IFNg-associated genes, particularly those relevant to antigen presentation, including Ifngr1, B2m, Cd74, H2-D1, and H2-K1. Finally, mBrRNA-0 cells strongly expressed Cxcl12, a regulator of cancer cell growth and local immune response (Figure 1D) (Biasci et al., 2020). This cluster likely modulates a diverse array of inflammatory elements in the microenvironment, and we therefore refer to this group as immunomodulatory (IM) fibroblasts. This is supported by pathway analysis showing upregulation of “Cytokine-mediated signaling pathway”, “Regulation of Acute Inflammatory Response”, and “Complement Activation” pathways (Figure S1A).
Importantly, the distribution of these subgroups was highly similar among tumors analyzed from different mice (Figure 1E) (Zhao et al., 2021). Given that our transcriptomic results supported functionally-significant CAF heterogeneity, we assessed the relative differentiation states of these CAF subgroups using CytoTRACE, a computational tool that leverages transcriptional diversity to order cells based on developmental potential (Figure 1F) (Gulati et al., 2020). Lineage progression among CAFs has yet to be well-defined (Sahai et al., 2020). This analysis suggested that MR CAFs may represent a less differentiated state relative to IM CAFs.
Next, we applied CellChat, a tool to understand cell-cell interactions in a given niche using single-cell gene expression data (Jin et al., 2021). This analysis suggested two “master” patterns of CAF behavior – Pattern 1 highlights the MR fibroblast subgroups (mBrRNA-2, 4, and 5) and Pattern 2 was associated with the IM and SSL subgroups (mBrRNA-0, 1, and 3) (Figure S1B – left panel). Mechanotransduction-related pathways, including GAS, PERIOSTIN, THBS, and SPP1, drive Pattern 1 CAFs, whereas cytokine signaling such as CCL, CXCL, and IL1 drive Pattern 2 (Figure S1B – right panel). Pattern 1 CAF subgroup cells interact more with each other and with ECM proteins (Figure S1C), while Pattern 2 CAF subgroup cell interactions favor immune cells (Figure S1D).
Changes in chromatin accessibility complement transcriptional phenotypes of CAF subpopulations
To evaluate the epigenomic changes associated with fibroblast activation in the context of solid tumors, we conducted scATAC-seq in parallel with our scRNA-seq assays for endogenous mBr tumors (mBrATAC). We identified considerable heterogeneity in chromatin accessibility profiles among individual CAFs, which were clustered into four epigenomically distinct subgroups using a Louvain-based modularity optimization in the ArchR platform (Granja et al., 2021) (Figure 2A). We then performed cross-platform integration to link mBr scATAC-seq data with the mBr scRNA-seq data using ArchR’s unconstrained implementation of Seurat’s label transfer algorithm (Granja et al., 2021). This resulted in four of our initial six scRNA-seq clusters mapping strongly to our scATAC-profiled cell groups (Figure 2B). To minimize nomenclature confusion, mBr scATAC-seq clusters were named based on the corresponding mBr scRNA cluster. The clusters with fewer cells (mBrRNA- 3 and 5) did not map clearly to the scATAC-seq data, potentially due to the presence of fewer transcription factors at a gene expression level compared to the other clusters. Alternatively, these groups may be underrepresented in our scATAC-seq dataset.
Figure 2. Chromatin accessibility of CAF subsets.

A. UMAP plot displaying mouse breast cancer mBrATAC clusters (bottom panel) → in silico fibroblast selection → integration with CAF mBrRNA data (top panel). (Data represent n=3 biological replicates per timepoint per condition).
B. Anchor based label transfer of scRNA clusters into scATAC data (left panel) results in integrated mBr CAF scRNA-ATAC clusters (right panel).
C. Heatmap showing characteristic differential paired chromatin accessibility-gene expression for integrated MBr CAF scRNA-ATAC clusters.
D. UMAP plots showing integrated MBr CAF scRNA-ATAC data for key genes of interest characteristic of immunomodulatory and steady state-like CAF clusters (top row) and mechanoresponsive clusters (bottom row).
E. Chromatin accessibility peaks for key genes of interest characteristic of immunomodulatory CAF clusters (Cxcl12 and Il6) and mechanoresponsive (Gas6 and Thbs1).
F. Schematic showing procedure for multiome sequencing of endogenous mouse breast tumor tissue. Data represent samples from nonmalignant breast tissue, early breast tumors and late breast tumors. Each time point included three biological replicates.
G. UMAP plot includes all cells that underwent multiome sequencing (left panel) → in silico CAF selection → UMAP plot demonstrating distinct CAF clusters: MR, IM, and SSL superclusters as indicated (right panel). Characteristic genes for each cluster are provided in figure labels. (Data represent n=3 biological replicates per timepoint per condition).
H. Violin plots indicate differentially expressed genes characteristic of each mBrMulti CAF cluster.
I. Transcription factor (TF) motif analysis was performed on mBrMulti data using the Signac and chromVAR packages. Feature plots indicate cells with highly accessible motifs for the indicated TF.
We first examined the epigenomic landscape of mBrATAC-1, which showed elevated chromatin accessibility proximal to SSL genes, including Dpp4, Ly6a, and Cd34 (Figure 2C–D). mBrATAC-0 demonstrated chromatin accessibility proximal to IM genes, such as Cxcl12, Il6, and Ccl19 (Figure 2C–E). While mBrATAC-1 demonstrated specific accessibility peaks and transcription factor footprinting in association with the Gata1 locus, mBrATAC-0 exhibited a comparable pattern at the Stat1 locus (Figure S2). These transcription factors are associated with stemness and inflammation, respectively, among other known functions. In the MR clusters, mBrATAC-4 showed significantly elevated accessibility proximal to Gas6, Yap, and Acta2 supporting a myofibroblast phenotype (Figure 2C–E). mBrATAC-2 was associated with accessibility proximal to FAK (Ptk2) as well as integrins and MR matrix factors such as Postn, Thbs1, Timp2, Fsp1, and Pdgfra. mBrATAC-2 lineage-specific transcription factors included Fos, Fosb, Junb, and Jund, all of which are related to mechanotransduction, as well as Runx1 and Runx2. These patterns of chromatin accessibility support the aforementioned SSL, IM, and MR CAF super-clusters, suggesting CAF heterogeneity may be determined, in part, at the chromatin level.
Multiome analysis follows parallel scRNA-seq/scATAC-seq derived fibroblast clusters throughout tumor development
To further correlate gene expression and chromatin accessibility changes in CAFs through the course of tumor development, simultaneous measurements were obtained using the Chromium Single Cell Multiome platform (10x Genomics) for three conditions: non-tumor mammary parenchyma, early mouse breast tumors, and late breast tumors (Figure 2F). Initial clustering revealed six transcriptionally and epigenomically distinct cell types: T-cells, B-cells, myeloid cells, epithelial cells, endothelial cells, and CAFs (Figure 2G – left panel). Following in silico fibroblast selection, eight CAF clusters were identified (Figure 2G – right panel). Fibroblasts from nonmalignant mammary tissue were largely confined to mouse breast multiome (mBrMulti-) clusters 0, 2, and 4 (Figure S3A–B). The majority of cells in the remaining clusters were obtained from early breast tumors, while late tumors showed comparatively more epithelial cells with proportionately fewer CAFs, although these CAFs were similarly distributed across the 8 clusters (Figure S3A–B). Of note, we obtained relatively fewer cell numbers overall from the late tumor timepoint, likely due to loss of blood supply centrally as tumors progress.
Cells in mBrMulti-2 demonstrated a striking resemblance to the stemlike Pi16 signature, consistent with its presence in steady-state, nonmalignant breast tissue (Figure 2G–H, S3A–E) (Buechler et al., 2021). mBrMulti-0 included both CAFs and steady-state breast tissue fibroblasts enriched for the production of basement membrane components such as laminin and type IV collagens, as well as uniquely high expression of the disintegrin and metalloprotease domain (ADAM) family, suggesting a prominent role in migration and remodeling.
Progression to malignancy introduced mBrMulti-3, which was associated with mechanotransduction, ECM production, and less diverse transcriptional programming (Figure S3F). mBrMulti-3 also aligned with the Lrrc15 signature, previously found to be associated with malignancy (Buechler et al., 2021). mBrMulti-6 demonstrated the highest expression of Trps1, recently implicated in benign fibroblast regenerative behavior (Mascharak et al., 2022). mBrMulti-1 demonstrated overlapping gene expression patterns that placed these cells on a spectrum between mBrMulti-2 and mBrMulti-3, particularly in terms of ECM production. However, these cells demonstrated several unique attributes that distinguished them from either group. Specifically, mBrMulti-1 cells expressed genes associated with hyaluronic acid synthesis (Has1, Has2), as well as the homing receptor for hyaluronic acid, Cd44. mBrMulti-1 was also unique in the expression of IL1 responsive genes, such as Cxcl1, Il6, Ccl2, and Il1r1, often associated with traditional inflammatory CAFs (Biffi et al., 2019). However, high expression of ECM genes clearly separated these fibroblasts from the more immunomodulatory clusters.
The remaining clusters expressed fewer genes associated with ECM production and remodeling. mBrMulti-4 expressed genes traditionally associated with lymphocyte signaling, such as Lef1, Dock2, Il7r, and Ptpn22. These genes have also been described in fibroblasts during wound healing and tissue repair (Guo et al., 2022; Huang et al., 2002; Phan et al., 2020; Spalinger et al., 2017). Remarkably, transcriptomic profiles of mBrMulti-5 strongly suggested a role in antigen presentation, demonstrating expression of Cd74, H2-Aa, Cd83, and Cd86. The capacity of CAFs to function as effective antigen presenting cells remains fiercely debated (Dominguez et al., 2020; Elyada et al., 2019).
We then compared our initial mouse breast scRNA-seq (mBrRNA) clusters to the mBrMulti timecourse using an anchor-based label transfer approach. The SSL clusters (mBrRNA-1 and mBrRNA-3) associated with cells from steady-state, nonmalignant tissue in the multiome analysis. Specifically, mBrMulti- 0 and mBrMulti-2 clusters were similar to the SSL mBrRNA clusters, supporting an association with non-perturbed tissue (Figure 2G, S3C–D) (Buechler et al., 2021). Clusters mBrMulti-1, 3, and 6 corresponded to the MR clusters (mBrRNA-2 and mBrRNA-4), indicative of a mechanoresponsive state. By contrast, the IM cluster mBrRNA-0 appeared to incorporate subpopulations of mBrMulti-4, 5, and 7. This may be due to the larger size of mBrRNA-0 cluster compared to the other clusters, with additional subpopulations captured by the multiome analysis. This phenomenon is exemplified by comparable IL1-associated signaling from a subpopulation of cluster mBrMulti-1 and IFNg-associated signaling from cluster mBrMulti-5, both of which were characterized within mBrRNA-0.
We then incorporated the ATAC portion (paired intra-nuclear scATAC-seq) of the multiome sequencing to evaluate patterns of chromatin accessibility in these clusters. Analysis of chromatin accessibility at transcription factor (TF) binding sites using chromVAR (Schep et al., 2017) revealed highly accessible motifs for proinflammatory TFs, such as Stat2, in IM fibroblasts. Accessibility of canonical fibrosis-related TF motifs such as TGFB-related Smad2::Smad3 was observed in MR fibroblasts (Figure 2I). Interestingly, accessibility of Runx1 motifs was found in both IM and MR CAF clusters, suggesting that these clusters may represent more differentiated CAFs relative to steady state-like fibroblasts, consistent with our prior results. Finally, specific accessibility of Sox6 motifs was found in mBrMulti-6 (mechanoresponsive). Sox6 is a TF known to regulate differentiation in a variety of tissue types, and recent knockdown studies have suggested potential profibrotic and proinflammatory roles (Li et al., 2020). Cluster identities were further supported through analysis of Lrrc15, Dpp4, Col1a1, and Cd86 peaks (Figure S3E). CellChat analysis again demonstrated prominent signaling networks between MR clusters, while the IM clusters mBrMulti-4 and mBrMulti-5 tended to be involved in immune cell interactions (Figure S4A). Overall, we observed excellent correlation between our parallel scRNA-seq/scATAC-seq and multiome timecourse datasets, supporting the conservation of fibroblast phenotypes associated with cancer.
In order to evaluate our results within the framework of the field, we again incorporated a label transfer approach using previously defined fibroblast clusters from an atlas of mouse perturbed states (Buechler et al., 2021). We observed a high degree of similarity between the stemlike Pi16 state and our SSL cluster mBrMulti-2 (Figure S4B). The well-described Lrrc15 signature was also observed in our highly mechanoresponsive cluster mBrMulti-3, consistent with previous findings evaluating myofibroblastic CAFs (Dominguez et al., 2020). The Col15a1 signature appeared to localize to both the MR cluster mBrMulti-1 and SSL cluster mBrMulti-0, and the Cxcl5 signature was particularly strong in the IM cluster mBrMulti-7. Interestingly, we did not observe an analogous group to our IFNg-associated IM cluster mBrMulti-4. Overall, however, our findings are in line with previously published fibroblast scRNA-seq series in cancer.
Breast CAF subpopulations are spatially distinct
To further explore the significance of CAF heterogeneity in solid tumor biology, we applied a spatial transcriptomic platform (Visium, 10x Genomics) to whole tumor endogenous mBr samples (Figure 3A). A unique challenge inherent to spatial transcriptomic platforms is that each “spot” can capture gene expression information from more than one cell (1–10 cells characteristically). Tumors feature a complex microenvironment composed of a variety of cell types. As such, to accurately interpret our spatial transcriptomics results in the context of our scRNA-seq CAF data, we needed to account for the contributions of non-fibroblast cells from each Visium spot (Figure 3B). In parallel with the multiome analysis, mouse breast tissue for spatial transcriptomics were obtained from the same mice used for multiome and at the same time points (Figure 3C). Using the transcriptional profiles from the multiome evaluation, we were able to predict the spatial contributions of each of our fibroblast clusters in conjunction with non-fibroblast cell types (Figure 3D–F, S5A). We found that the predicted spatial distributions for our scRNA-seq clusters were largely congruent with the transcriptional differences observed earlier for SSL, MR, and IM fibroblasts (Figure 3G), which we validated using a nearest-neighbor analytic approach applied over the timecourse of tumorigenesis. Comparing early tumors with non-tumor parenchyma, the mBrMulti-5 antigen-presenting CAFs (IM) co-localized with lymphoid immune cells, whereas mBrMulti-3 myofibroblasts (MR) co-localized with epithelial cells. Comparing late tumors with early tumors, proximity interactions between mBrMulti-4, 5, and 7 and myeloid-lineage cells (including macrophage phenotypes) became more prominent (Figure S5B). Importantly, similar results were obtained upon applying our mBr scRNA-seq (mBrRNA) clusters to the same spatial transcriptomic profiles – high expression of both MR and IM genes of interest was observed in a localized manner (Figure S5C–D). In summary, spatial transcriptomic analysis localized our fibroblast clusters within the complex tissue architecture of endogenous mBr tumors.
Figure 3. CAF cluster localization and plasticity.
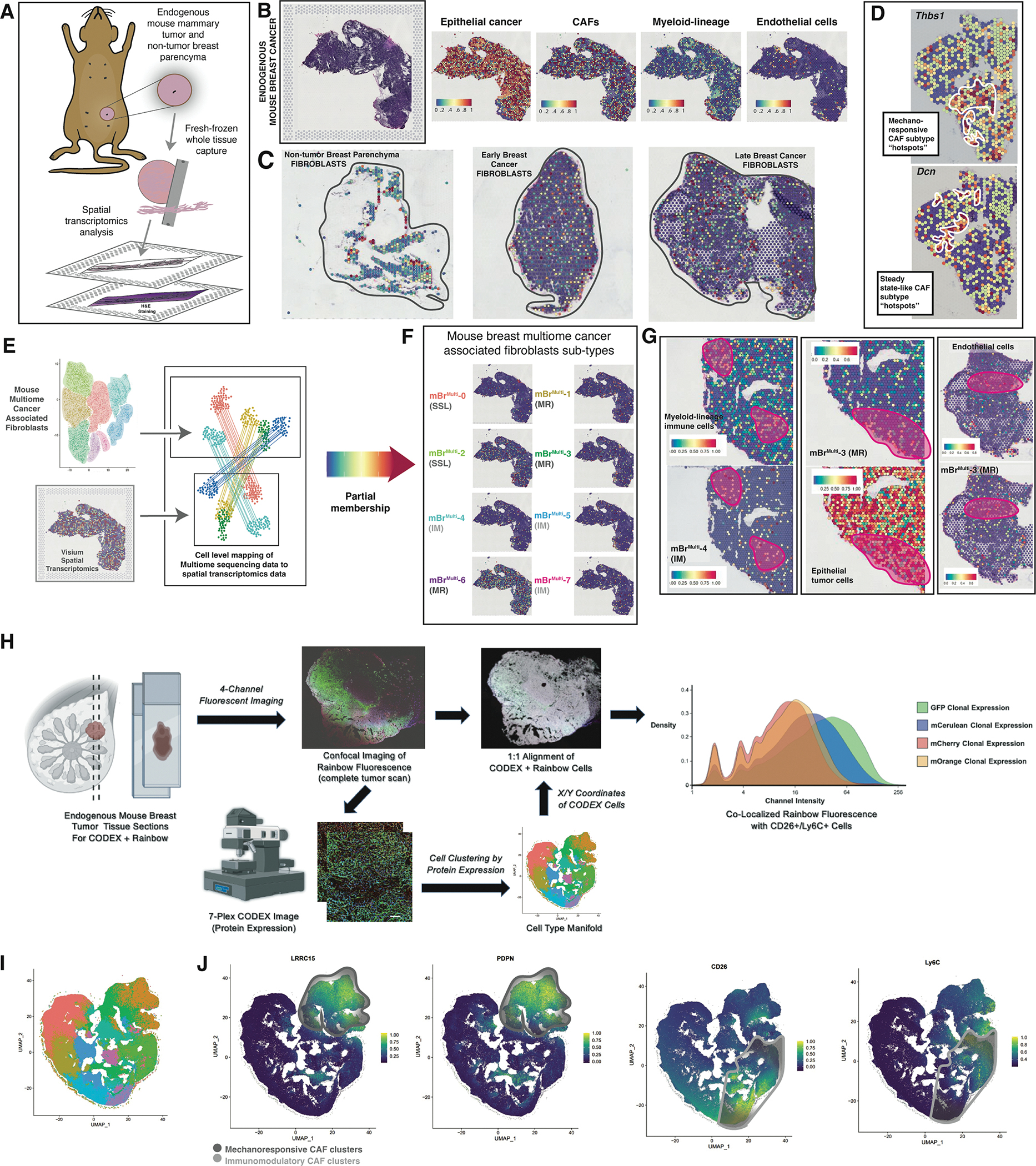
A. Schematic showing 10X Genomics Visium spatial transcriptomic analysis of endogenous mouse breast tumors.
B. H&E staining of a representative section of an endogenous mouse breast tumor used for Visium analysis (left panel). Visium spatial transcriptomic sequencing panels showing cell type represented representation in the representative tumor section (right panels).
C. Representative Visium sections showing CAF representation within normal breast parenchyma, early and late endogenous breast tumors.
D. Spatial transcriptomics plots showing “hot spots” containing CAFs with high expression of key genes characteristic of mechanoresponsive (Thbs1) and steady state-like CAF clusters (Dcn).
E. Cell level mapping of multiome sequencing data to spatial transcriptomics data → Partial membership of mBrRNA CAF clusters represented in mBrVisium data.
F. Visium spatial transcriptomics plots showing representation of each of the mBrMulti CAF clusters on a representative endogenous mouse breast tumor section.
G. Example co-localization of specific mBrMulti CAF clusters with relevant cell types from representative Visium sections, clusters and cell types as labeled in figure, pink circles highlight areas of co-localization.
H. Schematic describing the Rainbow-CODEX workflow. Tumor sections are first imaged for Rainbow fluorescence followed by CODEX staining and analysis, which included spatial analysis at the single cell level. Staining patterns for each cell are represented in two-dimensional UMAP plots, identifying populations of CD26+Ly6C+ SSL/IM CAFs. Cellular location is then identified on the corresponding confocal image and matched to Rainbow fluorophore expression, ultimately confirming the presence of poly-clonal SSL/IM CAFs that once expressed aSMA.
I. UMAP of cell populations derived from CODEX staining in Rainbow mouse breast tumors.
J. CODEX data feature plots demonstrating distinct localization of MR CAFs (LRRC15+, PDPN+) in the upper-right of manifold and SSL/IM CAFs (CD26+, Ly6C+) in the bottom-right of manifold.
Rainbow-CODEX analysis supports plasticity of CAF subpopulations
Based on CytoTRACE analysis, both our mBr scRNA-seq (Figure 1F) and multiome (Figure S3F) MR CAF subtypes appeared to be less differentiated relative to IM CAFs. To further explore the lineage of these mouse breast CAF subgroups at the tissue level, we employed the Rainbow mouse model, a multi-colored lineage tracing model that elucidates cell activation and proliferation, using an aSMA Cre driver (aSMACreERT2::Rosa26VT2/GK3) (Ransom et al., 2018; Ueno and Weissman, 2006; Yanai et al., 2013). aSMA is a well-established marker of myofibroblasts and is strongly expressed by this subgroup in our spatial transcriptomic analysis (Figure S6A – left panel). The Rainbow background was applied to the endogenous breast cancer mouse transgenic (MMTV-PyMT::aSMACreERT2::Rosa26VT2/GK3) (Figure S6A – right panel). These mice received systemic Tamoxifen induction between 4–5 weeks of age, which marks a time prior to the onset of significant tumorigenesis. On confocal analysis of tumor tissue, CAFs proliferate in the tumor stroma in a poly-clonal manner. During early tumor development, small clones of CAFs are found throughout the tumor tissue (Figure S6B – left panel), which increase in size as the tumor develops (Figure S6B – middle panel). This pattern can be further appreciated using Imaris analysis (Figure S6B – right panel). The distribution of Rainbow CAF colors was observed to be relatively equal across the tissue sections, consistent with the Rainbow model (Figure S6C). We identified similar CAF behaviors using allograft tumor models for breast cancer for pancreatic cancer (with injection of syngeneic cancer cells into the mammary fat pad or pancreatic parenchyma, respectively) (Figure S6D).
We then assessed the plasticity of myofibroblastic aSMA-expressing populations by incorporating CODEX spatial proteomic analysis into the Rainbow mouse model (Figure 3H, S7A). CODEX analysis of Rainbow mouse breast tumors identified protein expression-based cell populations. The entire dataset of cell-segmented protein expression was projected to a unified UMAP manifold. LRRC15 and PDPN strongly co-localized at the upper-right of the manifold (Figure 3H–J). CD26 and Ly6C, on the other hand, localized to the bottom right of the manifold, likely representing SSL and IM CAFs. This region of the protein UMAP was also predominated by markers such as CD8 and Ly6G, supporting an immune-like phenotype (Figure S7B–C). Additionally, Ly6C marked cell populations in this region of the manifold that most likely correspond to macrophages, with evident co-expression of CD68 (Figure S7C).
CODEX data were subsequently mapped to Rainbow fluorescence imaging using a per-specimen alignment mask, in which each specimen’s CODEX expression was directly overlaid on its corresponding Rainbow image (Figure 3H). Following alignment, individual phenotypic filters were applied for SSL and IM CAFs versus MR CAFs to restrict fluorescent GFP, membrane (m)Cerulean, mCherry, and mOrange channels to the areas co-localized with the respective CAF population. Distributions of clonal expression for IM and SSL versus MR CAFs were then analyzed using kernel density plots. Overall, both CAF subpopulations demonstrated strong poly-clonality, with heterogeneous clonal expression across all rainbow colors. Most interestingly, SSL and IM fibroblasts exhibited Rainbow fluorescence associated with aSMA lineage-induced mCerulean, mCherry, and mOrange clonal populations, confirming lineage plasticity among aSMA-positive fibroblasts. Collectively, these data support the potential capacity for MR CAFs to differentiate into SSL or IM CAFs.
Single-cell transcriptomic analysis of CAFs in human breast cancer
We next sought to evaluate the extent to which local fibroblast responses to tumors are evolutionarily conserved between mice and humans. We examined cells isolated from three breast cancer patients (Table S2). Single cells from these samples were tagged with unique oligonucleotides, followed by scRNA-seq evaluation (Figure 4A). We identified populations of cells corresponding to CAFs, epithelial cells, endothelial cells, and immune cells (Figure 4B – left panel).
Figure 4. Cross-tumor integration of CAF phenotypes.
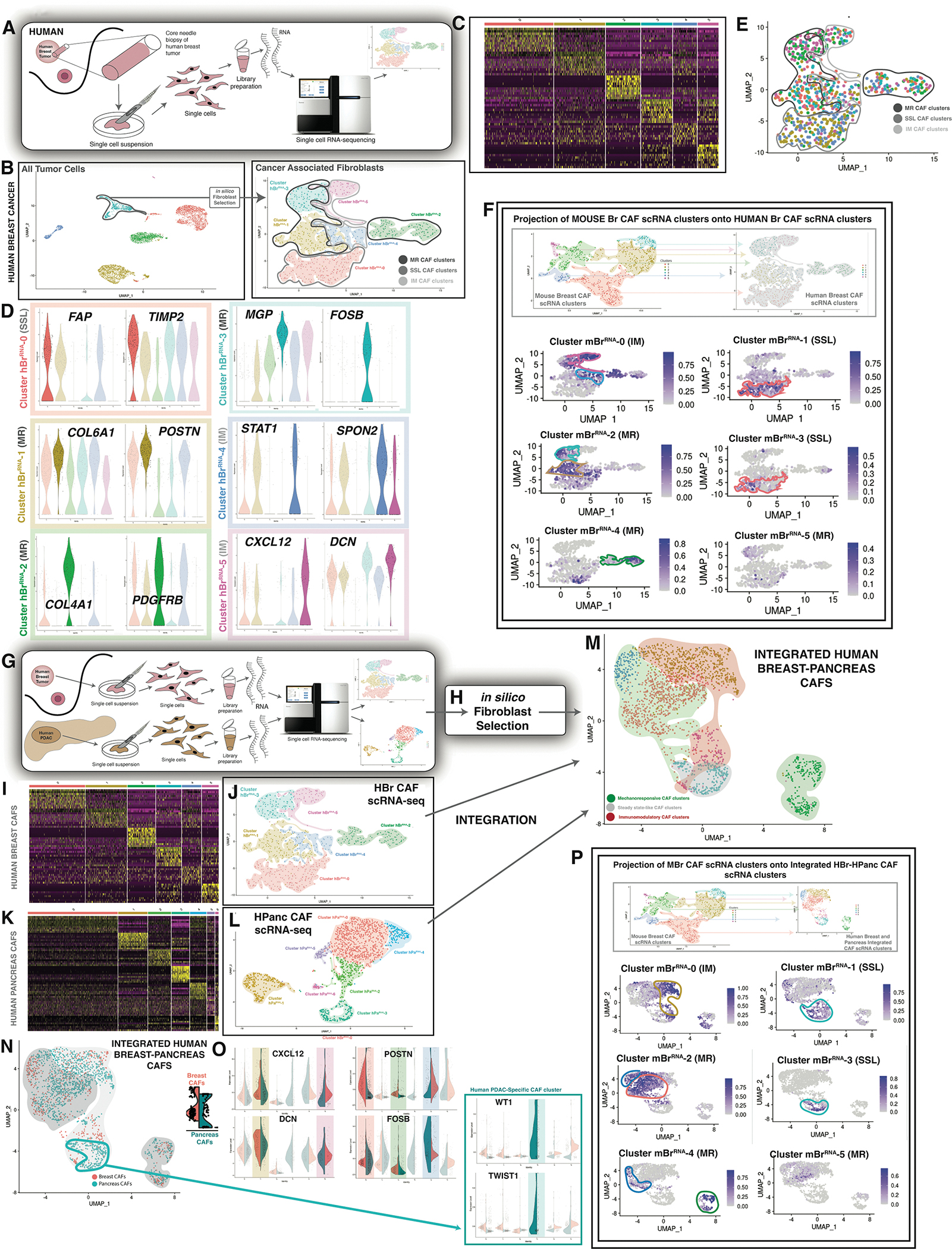
A. Schematic showing scRNA-seq of human breast tumor tissue.
B. UMAP plot showing all tumor cells sequenced (top panel) → in silico CAF selection → UMAP plot showing human breast cancer scRNA-seq (hBrRNA) CAF clusters. Clusters as labelled in figure panel (bottom panel).
C. Heatmap showing characteristic differential gene expression for hBrRNA CAF clusters.
D. Violin plots showing differentially expressed genes characteristic of each hBrRNA CAF cluster. Colors represent clusters visualized in B.
E. UMAP plot showing hash-oligo data for the human breast tumors sequenced. Each human sample was stained with two hash-oligos for validation.
F. Label transfer projection of mBrRNA clusters onto hBrRNA CAF clusters. Clusters as labeled in figure panel. CAF clusters indicated with colors corresponding to mBrRNA CAF data.
G. Schematic showing scRNA-seq of human breast and pancreas tumor tissue.
H. In silico CAF selection.
I. Heatmap showing characteristic differential gene expression for hBrRNA CAF clusters.
J. UMAP plot showing hBrRNA CAF clusters as in B (right panel).
J. Heatmap showing characteristic differential gene expression for hPaRNA CAF clusters. (Data represent n=3 unique patient tissue samples as noted).
K. UMAP plot showing hPaRNA CAF clusters.
L. UMAP plot showing integrated human breast-pancreas CAF data (7 clusters). Green shading indicates mechano-responsive CAF clusters whereas gold shading indicates immuno-modulatory CAF clusters.
M. UMAP plot showing integrated human breast-pancreas CAF data in terms of organ of origin.
N. Violin plots showing selected highest-differentially expressed genes characteristic of integrated human breast-pancreas CAF clusters. One cluster is only represented by HPanc CAFs, as indicated (blue box).
O. Label transfer projection of mBrRNA CAF clusters onto integrated human breast-pancreas CAF scRNA-seq clusters. Clusters as labeled in figure panel. CAF clusters indicated with colors corresponding to mBrRNA CAF data.
We again focused our analysis on CAFs, which were isolated in silico and re-clustered to delineate fibroblast subsets (Figure 4B – right panel). Human breast cancers demonstrated six transcriptionally-defined CAF clusters (hBrRNA-0 - hBrRNA-5) (Figure 4C). Similar to mBrRNA-2, hBrRNA-3 CAFs exhibited significantly elevated expression of genes associated with mechanotransduction and fibrosis, including MGP and FOSB (Figure 4D), as well as “Focal Adhesion” pathways, suggesting that these represent an MR subpopulation. CAFs from hBrRNA-1 and 2 were also associated with elevated expression of fibrosis-related genes such as PDGFRB and vitamin-K-dependent matrix proteins such as POSTN. By contrast, hBrRNA-0 cells were characterized by elevated expression of SSL genes such as FAP (which shares significant homology with DPP4 expressed in the SSL MBr clusters) and TIMP2, and CAFs from hBrRNA-4 and 5 were associated with expression of cytokines and other signaling elements such as CXCL12 and STAT1 consistent with IM identity (Figure 4D). The distribution of the clusters was not significantly different among tumors from three different patients despite their differences in age, tumor type, and stage (Figure 4E, Table S2).
Cross-species integration of cancer associated fibroblasts
To further evaluate whether the human breast (hBrRNA) CAF clusters were analogous to those identified in mouse breast (mBrRNA) CAFs, we performed an integrated cross-species analysis using ortholog mapping in conjunction with Seurat’s label transfer approach, as previously described (Stuart et al., 2019). This allowed us to project our six mBrRNA CAF clusters onto the hBrRNA embedding in a nearest neighbor fashion. These projections supported strong similarities between specific hBrRNA and mBrRNA clusters, such as hBrRNA-2 with mBrRNA-4 (MR), as well as hBrRNA-0 with mBrRNA-1 and mBrRNA-3 (SSL). We also observed broad overlap of transcriptional programs for hBrRNA-1 and hBrRNA-3 with mBrRNA-2 and mBrRNA-5 (MR), as well as hBrRNA-4 and hBrRNA-5 with mBrRNA-0 (IM) (Figure 4F).
To further explore these findings, we performed multiome analysis on three human breast tumors (Figure S8A–B, Table S2) and compared these findings with our mouse multiome (mBrMulti) clusters. Fibroblasts were again isolated in silico, generating six human multiome (hBrMulti) CAF clusters (Figure S8C–D). Applying CytoTRACE, MR CAFs were the least differentiated of the superclusters (Figure S8E), consistent with the aforementioned CytoTRACE analyses of other datasets. In terms of the ATAC-seq results, accessibility was seen near genes congruent with the RNA-seq data (Figure S8F). We then mapped hBrMulti clusters to the aforementioned mBrMulti UMAP using an anchor-based label transfer approach. Again, we found striking similarities: hBrMulti-0 mapped to the MR mBrMulti-3, indicative of a highly mechanoresponsive state (Figure S8G), hBrMulti-3 mapped to the SSL mBrMulti-1, and hBrMulti-5 mapped to the IM mBrMulti-4 and mBrMulti-5 (Figure S8G). Thus, each supercluster was consistently represented in both mouse and human CAF datasets. These findings suggest strong evolutionary conservation of the fibroblast response to breast tumors in the form of three transcriptionally-delineated superclusters.
Cross-tumor integration of cancer associated fibroblasts
We next sought to evaluate whether the local fibroblast response to epithelial neoplasia was conserved across different tumor types. Having characterized CAFs in both mouse and human breast cancer, we next examined another epithelial cancer type, pancreatic ductal adenocarcinoma (PDAC).
We examined three resected pancreaticoduodenectomy tumor specimens from patients who had residual pancreatic cancer following neoadjuvant chemotherapy for locally advanced PDAC (Table S2). CAFs isolated from these tumors showed strong protein expression of ASMA and COL1, consistent with pro-fibrotic fibroblast activation (Figure S9A–B). Single cells from these samples were isolated followed by scRNA-seq evaluation (Figure 4G). We again focused our analysis on the CAFs within the resulting data (Figure 4H). Relatively similar to our human breast cancer findings (Figure 4I–J), we identified seven transcriptionally-defined subgroups among human pancreatic (hPaRNA) CAFs (Figure 4K–L).
In order to compare these seven subgroups to the six subgroups previously identified in our human breast (hBrRNA) CAF dataset, we performed a label-transfer-based integration using Seurat as previously described (Stuart et al., 2019). The resulting integrated dataset containing both human breast (hBrRNA) and human pancreatic cancer (hPaRNA) CAFs was further processed to determine variable features spanning the combined data object (Stuart et al., 2019). Partitional analysis, blinded to tissue of origin, resulted in the identification of six transcriptionally-defined hBrRNA-hPaRNA CAF subgroups (Figure 4M). Five of these six clusters were present in each of the six patients (three breast and three pancreas) (Figure 4N–O). In contrast, one cluster (hBrRNA-hPaRNA-3) was comprised of cells entirely from pancreatic CAFs.
We found that CAFs within hBrRNA-hPaRNA-0, 2, and 4 demonstrated increased expression of matrix proteins such POSTN and mechanotransduction features such as FOSB (Figure 4O – top right panels). Whereas CAFs from hBrRNA-hPaRNA-1 and 5 exhibited elevated expression of immune-related genes, including CXCL12 (Figure 4O – top left panels). hBrRNA-hPaRNA-3 CAFs, unique to pancreatic cancer, were characterized by expression of WT1 and TWIST1 (Figure 4O – bottom panels). Based on studies of pancreatic fibroblasts during development, this subpopulation may represent a lineage of mesothelium-derived CAFs (Garcia et al., 2020). However, TWIST1 expression is also associated with epithelial mesenchymal transition (EMT) and CAF transdifferentiation (Lee et al., 2015). On immunofluorescent staining of the PDAC tissue specimens, FAP+ IM CAFs appeared separate from MR CAFs (aSMA/COL1+) (Figure S9A–B).
To evaluate whether these six human integrated hBrRNA-hPaRNA CAF clusters were analogous to the mBrRNA CAFs, we performed a similar cross-species analysis using ortholog mapping in conjunction with label-based transform, as above (Stuart et al., 2019). This allowed us to project our original six mBr CAF clusters onto the integrated hBrRNA-hPaRNA CAF embedding (Figure 4P). The resulting overlay demonstrated strong similarities between the putative MR mBrRNA- 2, 4, 5 and MR hBrRNA-hPaRNA-0, 2, and 4 clusters. Furthermore, we observed similarities between the putative IM mBrRNA- 0 and IM hBrRNA-hPaRNA-1 and 5 clusters. Finally, SSL mBrRNA-1 and 3 demonstrated similarity with a comparably smaller population in hBrRNA-hPaRNA-3. These results support evolutionary conservation of the fibroblast response to epithelial tumors across disparate tissue types.
Functional modulation validates MR and IM CAF subgroups
Next, we established allograft breast cancer in FAK-knockout (FAK−/−) and FAK-wildtype (FAK+/+) Rainbow mice, using a ubiquitous Cre driver (ActinCreERT2::Rosa26VT2/GK3) to capture all CAFs irrespective of cell surface marker expression. We used florescence-activated cell sorting (FACS) with lineage-negative gating to isolate rainbow-colored CAFs, representing fibroblasts that expand in the tumor milieu during cancer growth. Phospho-flow cytometry was then applied to delineate MR CAF subgroups versus SSL and IM CAFs based on protein expression. When FAK signaling was knocked out, MGP, which was highly expressed at the gene expression level by MR CAF clusters, CAF protein expression was significantly decreased (Figure S10A–C), and, conversely, CAF DPP4 (CD26) protein expression, expressed by SSL and IM CAF clusters, was increased (Figure S10C). Furthermore, we saw a significant difference in tumor aggressiveness between our FAK−/− and FAK+/+ models: with FAK-knockout among cells in the stromal compartment, tumors grew significantly larger (Figure 5A, Figure S10D – bottom panel) compared with FAK-wildtype. Of note, FAK expression was unaffected in the transformed cancer cells using the allograft model, so the effect of FAK modulation was felt primarily by CAFs in this circumstance.
Figure 5. Functional modulation affects the balance of CAF subtypes.
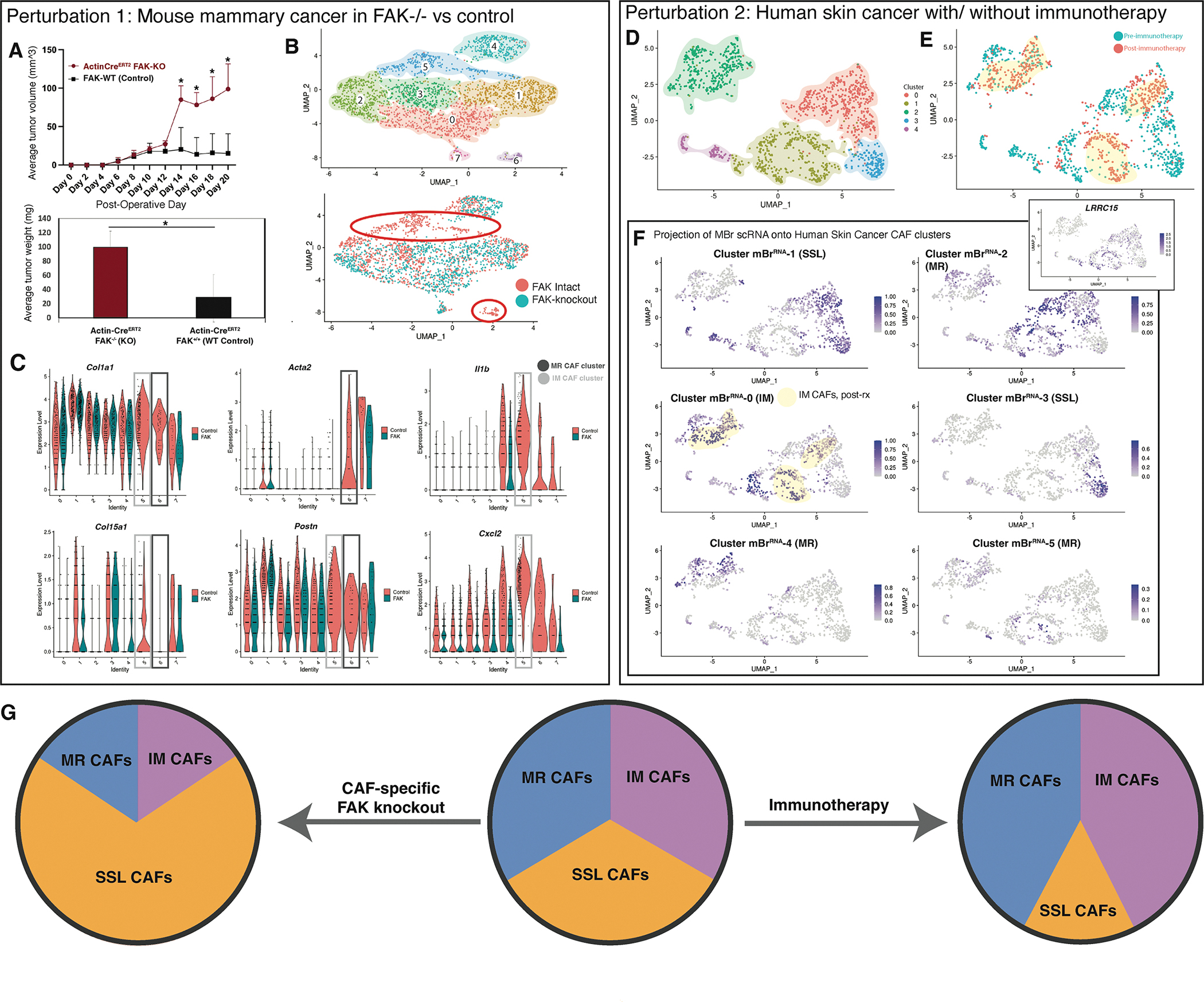
A. Average tumor size comparing allograft mouse breast tumors with global FAK knockout versus control (ActinCreERT2::FAKKO mice) (top panel). Average tumor weight comparing allograft mouse breast tumors with global FAK knockout versus control on day 20 of harvest (ActinCreERT2::FAKKO mice) (bottom panel). POD = post-operative day, KO = knockout, WT = Wildtype, * = p<0.05 (t-test). (Data represent n=3 biological replicates per timepoint per condition unless otherwise noted).
B. UMAP plot showing eight transcriptionally-defined CAF clusters for mouse allograft breast cancer specimens from FAK-intact and Col1a2CreERT2::FAKfl/fl mice (data represent n=3 biological replicates per group, hash-oligos incorporated to distinguish biological replicates) (Top panel). UMAP plot grouped by CAF origin (FAK-intact in red vs Col1a2CreERT2::FAKfl/fl in green). Primary clusters (5 and 6) lost with fibroblast-specific FAK knockout are highlighted with orange circles (bottom panel).
C. Violin plots illustrating expression of genes of interest between CAFs (FAK-intact in red vs Col1a2CreERT2::FAKfl/fl in green). MR and IM CAF clusters of interest highlighted with grey boxes as labelled in the figure panel.
D. UMAP plot showing transcriptionally-defined clusters for human BCC scRNA-seq CAF.
E. UMAP plot from CAF-specific scRNA-seq data colored according to pre- vs post- immune checkpoint blockade for human BCC.
F. Label transfer projection of mouse breast CAF scRNA-seq clusters on human BCC CAF scRNA-seq clusters. Immunomodulatory CAF cluster 0 is highly represented in post-therapy CAFs, while SSL cluster 3 is almost entirely found in pre-therapy samples. These patterns are highlighted with yellow shading. Feature plot of LRRC15 expression shown in inset corresponds closely with MBr cluster 2 (MR1) correlation as anticipated.
G. Schematic summarizing CAF subpopulation perturbations observed with FAK knockout in the context of mouse breast cancer compared with immunotherapy in the context of human BCC.
To further validate these results, we applied the aforementioned model to FAK−/− and FAK+/+ mice using CAF-specific Cre drivers (aSMACreERT2 and Col1CreERT2). In these mice, FAK expression is only modulated in cells expressing Acta2 or Col1, respectively. scRNA-seq of these tumors 10 days after tumor establishment demonstrated a loss of two prominent CAF clusters (Figure 5B–C). Cluster 6 expressed MR genes, such as Acta2, Lrrc15, Col12a1, Col7a1, and Col5a2 (Figure 5B–C). Interestingly, Cluster 5 was also lost with FAK knockout and appeared to be composed of primarily IM CAFs, expressing Il1b, Tnf, Cxcl1, and Cxcl2 (Figure 5B–C). Thus, we see a loss of both MR and IM CAFs associated with conditional FAK knockout early after tumor establishment. Similar to our ubiquitous Cre driver data noted previously, when FAK signaling is eliminated, tumors grow significantly larger using either of these CAF-specific Cre drivers (Figure S10D) and showed less dense fibrosis in the ECM (Figure S10E). Taken together, these data suggest that the ratio of CAF subpopulations can direct tumor growth.
Finally, we asked whether perturbations in our master CAF subgroups occurred clinically in the setting of an inflammatory stimulus. To do this, we analyzed our group’s published scRNA-seq data from human basal cell carcinoma (hBcRNA) from a cohort of patients before and after they received treatment with immune checkpoint blockade (Yost et al., 2019). Pooling fibroblast transcriptional profiles from these two data subsets, we identified 5 CAF clusters (Figure 5D), two of which (hBcRNA-3 and 4) were represented by CAFs only seen in the pre-treatment context (Figure 5E). Next we projected our six mBrRNA CAF clusters onto the hBcRNA CAF embedding in a nearest neighbor fashion (Figure 5F). We identified that the clusters only seen in the pre-treatment context (hBcRNA-3 and 4) were overwhelmingly ascribed to the steady state-like mBrRNA-1 and 3. By contrast, hBcRNA- 0, 1, and 2 remained present at largely stable quantities before and after immune checkpoint blockade and shared the most similarity with the MR clusters mBrRNA-2 and 4 (exemplified by Lrrc15 expression as in our other datasets, Figure 5F pop-out) as well as the IM cluster mBrRNA-0. Taken together, these results suggest that immunotherapy may stimulate more functional roles for CAFs in the TME, namely MR or IM programming associated with perturbed states. These data further support a “push-pull” relationship among the shared CAF phenotypes identified across tumor datasets (Figure 5G).
DISCUSSION
CAFs are often the most numerous cell type in solid tumors, representing an understudied therapeutic target. In order to effectively target CAFs, a more comprehensive understanding of the heterogeneity and function of these cells is warranted.
We identified six transcriptionally distinct clusters of CAFs in endogenous mouse breast tumors. Based on gene expression profiles, we characterized three of these clusters as “mechanoresponsive” (MR), two of these clusters as “steady state-like” (SSL) and the largest single cluster as “immunomodulatory” (IM). Prior work has suggested two subgroups of CAFs in pancreatic cancer (so called “myCAFs”, myofibroblast, and “iCAFs”, inflammatory) (Steele et al., 2021; Yang et al., 2016). Importantly, recent work from Buechler et al definitively identified steady state phenotypes for fibroblasts across multiple tissue types. The Pi16 signature in this investigation mapped to our steady state-like clusters in both the mBrRNA and the mBrMulti data sets, supporting this separate supercluster (Buechler et al., 2021). Our data validate the preservation of these clusters across tumor types and species.
Using Visium spatial transcriptomics applied over the timecourse of tumorigenesis, we further analyzed endogenous mouse breast tumor tissue and identified that MR, SSL, and IM CAF subtypes are located in spatially distinct regions of the tumors. Integration of the mBrRNA CAF data with mBrATAC data from the same tumors suggested that these phenotypes emerge as the result of changes at the chromatin accessibility level, which are then carried downstream.
Functionally, we found that genetic ablation of FAK function in the stromal cell compartment leads to accelerated tumor growth. Previous studies have demonstrated mixed functional results from CAF-specific FAK suppression (Demircioglu et al., 2020) (Wu et al., 2020). Interestingly, we observed a loss of both MR and IM CAF subpopulations as a result of CAF-specific FAK loss, suggesting a role for FAK in inflammatory signaling. Taken together, FAK signaling has a complex role in non-transformed stromal cells, which may have clinical implications for the continued pursuit of FAK-centered therapies.
We further investigated the CAF subtypes identified in human basal cell carcinoma (BCC) using an independently processed, published dataset and found that our proposed CAF subtypes were once again largely recapitulated (Yost et al., 2019). When these patients were treated with immune checkpoint blockade, the proportion of SSL CAF subtypes nearly disappeared, whereas the proportions of MR and IM CAF subtypes were largely unaffected in terms of numbers by this therapy. These findings may be due to increased inflammatory and/or mechanical stimuli in responding tumors, leading to functional differentiation of steady state-like CAFs into the other groups.
Given the expanding body of publicly available scRNA-seq cancer datasets, and the trend toward “in silico” isolation of cell subtypes (as opposed to prospective FACS isolation using marker-based strategies), it has become increasingly feasible to compare CAFs and other fibroblast types over diverse studies. Buechler et al. examined fibroblasts across many tissue types (most from benign disease samples) as well as from three cancer sets (Buechler et al., 2021). As the authors addressed, there are inherent decision points in any such broad survey, ranging from inclusion-exclusion criteria to algorithm selection and parameterization. While our work highlights only a handful of CAF transcriptional programs conserved across humans and mice, this integrated approach has the potential to simplify how the field of CAF heterogeneity is interpreted. Moreover, our fibroblast findings from both internal and publicly available datasets in this study are broadly congruent with data reported by other groups examining CAF subgroups in the context of pancreatic cancer (Biffi et al., 2019; Elyada et al., 2019; Ohlund et al., 2017). The identification of shared CAF subtypes across tumor types and species expands the potential of effective CAF-targeting therapy.
In terms of limitations of the study, the Visium approach is incorporated without a higher resolution confirmatory analysis, such as MERFISH. Rather, we incorporated spatial proteomic methodology in the form of CODEX to further examine architectural features of the tissue. Although aSMA is a widely accepted marker for myofibroblastic CAFs, there is a possibility for co-labeling of myoepithelial cells, a relatively rare cell type but inherent limitation of the aSMA Cre driver. To help with validation in this regard, we applied CODEX to cells that co-express CAF markers (LRRC15, PDPN, CD26, Ly6c etc.), whereas myoepithelial cells would not be expected to demonstrate such strong expression of these markers. Additionally, we used a label transfer-based method for data integration, rather than techniques more grounded in information theory such as Harmony or LIGER (Korsunsky et al., 2019; Lee et al., 2017). Label transfer is comparatively limited in its ability to reject combinations of dissimilar data, but was selected due to its significantly elevated stability relative to parametric variation. As increasing numbers of tools become available for the integration of heterogeneous data (Osorio et al., 2021; Zou et al., 2021), cross-study endeavors of this type may provide a conceptual framework for the study of both benign and malignant fibrotic diseases (Phan et al., 2021).
In summary, we present a comprehensive investigation of CAF single-cell gene expression and paired chromatin accessibility in conjunction with spatial transcriptomics across multiple solid tumor types and species. Our data identify that CAF subpopulations fall into three broad categories, which are maintained over the course of tumorigeneses and are spatially distinct. These are remarkably conserved across tumor type and tissue of origin. Each of these categories also shows functional relevance with a clear “push-pull” dynamic between superclusters. Taken together, we define the temporal and spatial dynamics of CAF biology, delineate key regulatory pathways determining CAF differentiation, identify CAF subpopulation functions with potential for further translational and clinical exploration, and introduce a multimodal -omics framework with application to the next steps in CAF investigation.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Additional information regarding resources and reagents will be fulfilled by the Lead Contact, Michael T. Longaker (longaker@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The gene expression and chromatin accessibility datasets generated during this study are publicly available and can be found through Gene Expression Omnibus (GEO) web portal using accession listing GSE212482 and its subseries (GSE212461, GSE212481, GSE212706, GSE212707, GSE212708), which includes our scRNA-seq, scATAC-seq, single cell multiome, and also spatial transcriptomics data. The CODEX spatial proteomics data will be made available by the authors upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Cell lines used for allograft tumor experiments included Py8119 and Pan02. All cells were maintained under sterile conditions in a humidified incubator under 5% CO2 at 37°C. A phase-contrast microscope (Leica) was used to image cells. Cells were grown to 70–80% confluence in DMEM-Glutamax after minimal passage prior to implantation in the mammary fat pad or pancreas of mice. 10,000 cells were implanted per site under sterile conditions.
Animal Models
The following mouse strains were purchased from Jackson Laboratories: Black/6 (C57BL/6J), Actin-CreERT2 (Tg(CAG-cre/Esr1)5Amc/J), MMTV-PyMT (FVB/N-Tg(MMTV-PyVT)634Mul/J), Col1-CreERT2 (Tg(Col1a2-cre/ERT,-ALPP)7Cpd/J), and FAKfl/fl (B6.129P2(FVB)-Ptk2tm1.1Guan/J) mice. αSMA-CreERT2 were courtesy of Dr. Ivo Kalajzic, University of Connecticut. Rainbow mice (ROSA26VT2/GK3) were courtesy of the Weissman Laboratory, Stanford University School of Medicine. All of the mice were genotyped as per manufacturer’s recommendations. Mice were housed at the Stanford University Comparative Medicine Pavilion (CMP) and Research Animal Facility (RAF). The facilities provided light- & temperature-regulated housing. Mice were given rodent chow and water ad libitum. A minimum sample size of three animals (three biological replicates) was used for all experiments (additional details noted in figure legends). Animals with appropriate genotypes for a given experiment were randomly allocated to the various experimental conditions. All experiments were completed according to the Stanford University Animal Care and Use Committee standards of care.
METHOD DETAILS
Tissue Processing and Histology
4% paraformaldehyde (Electron Microscopy Sciences) was used to fix human and mouse tumor samples for 20 h at 4 °C. Standard protocol was used to embed the tissues into paraffin. For cryopreservation, specimens were protected from cryofreeze in 30% sucrose (Sigma) then until saturation at 4 °C following fixation, followed by OCT until saturation at 4 °C, and then embedded in OCT. Representative tissue specimens were stained with hematoxylin and eosin (H&E, Sigma–Aldrich), Picrosirius Red Stain (Abcam), or Masson’s trichrome (Sigma–Aldrich) per manufacturer’s protocols.
Immunocytochemistry (ICC)
Fibroblasts were seeded onto coverslips coated with 1% Embryomax gelatin (EMD Millipore). The following day, cells were fixed with 4% with paraformaldehyde for 10 minutes, permeabilized with 0.5% Triton-X-100 (Sigma) for 15 minutes, and then incubated with 1X Powerblock (Biogenex) for 1 hr. Cells were then stained with primary antibody at 4 °C overnight. Cells were then washed with 0.1% Tween-20 (PBST; Sigma–Aldrich), stained with secondary antibody for 1 h at room temperature, and then mounted using Prolong Gold Antifade Mountant with DAPI (Life Technologies).
Immunofluorescence (IF)
Cryopreserved specimens were cryosectioned at 8um onto Superfrost Plus microscope slides (FisherSci). Samples were permeabilized with 0.5% Triton-X-100 (Sigma) for 15 minutes, and then incubated with 1X Powerblock (Biogenex) for 1 hr. Primary antibodies were applied to tissue specimens for 1 h at room temperature, and then rinsed three times with 0.1% Tween-20 (Sigma). Secondary antibodies were applied for one hour at room temperature. The antibody incubation and washing steps were repeated if multiple proteins were stained for in one specimen section. Slides were then mounted using Prolong Gold Antifade Mountant with DAPI (Life Technologies). Antibodies used for ICC and IF included: anti-aSMA (Abcam, ab32575, lot: GR282976–32, used at 1:100), anti-COL3 (Abcam, ab7778, lot: GR3234897–1, used at 1:100), anti-COL1 (Abcam, ab34710, lot: GR3244041–2, used at 1:100), anti-CD26 (Abcam, ab222716, lot: GR3220836–1, used at 1:100), anti-phospho-FAK (Thermo Fisher, 799255, lot: RG240925A, used at 1:100), IgG Alexa-Fluor 488 (Invitrogen, A32731, lot: SH251139, used at 1:1000), IgG Alexa-Fluor 555 (Invitrogen, A32732, lot: SH251140, used at 1:1000), IgG Alexa-Fluor 647 (Invitrogen, A32733, lot: SI231745, used at 1:1000).
Tissue Clearing
Tissue clearing optimized to preserve expression of endogenous fluorophores as previously described by our laboratory (Foster et al., 2019) was pursued on selected sectioned Rainbow specimens. In brief, for dehydration, tert-butanol (FisherSci) was buffered to a pH 9.5 with triethylamine (FisherSci). Fixed tissue specimens were placed into increasing gradients of tert-butanol (33%, 66%, and 100%) at room temperature for 30 minutes each and then left in 100% tert-butanol overnight. Tert-butanol and benzoic acid:benzyl benzoate (Sigma Aldrich) at a 1:2 ratio were buffered to pH 9.5 with triethylamine (FisherSci). Cleared sectioned tissue specimens were stored in BABB solution at 4°C.
Confocal Imaging and Analysis
Tissues were fixed and prepared in the dark to minimize bleaching of endogenous fluorophore expression. Laser scanning confocal microscopy was performed using a Leica WLL TCS SP8 Confocal Laser Scanning Microscope (Leica Microsystems) located in the Cell Sciences Imaging Facility (Stanford University, Stanford, CA). The ×10, ×20, and ×40 objectives were used (×10 HC PL APO, air, N.A. 0.40; ×20 and ×40 HC PL APO IMM CORR CS2, H2O/Glycerol/oil, N.A. 0.75). Precise excitation and hybrid detection of the Rainbow fluorophores (mCerulean, eGFP, mOrange, and mCherry) was captured when applicable. Raw image stacks were imported into Fiji (Image-J, NIH) or Imaris (Bitplane) software for analysis. Fiji was used to make two-dimensional micrographs of the confocal data and to quantify fluorophore expression intensity. For analysis of clonality from Rainbow mouse tissue, surfaces were created for each color of the Rainbow construct expressed using the volume surface and thresholding tools in using Imaris software.
Mouse Induction and Tumor Growth
Animal models as previously described received 5 days of 200ml (20mg/ml in corn oil) tamoxifen induction for activation of Cre recombinase (per the protocol provided by Jackson Labs). MMTV-PyMT model mice received systemic tamoxifen induction between 4–5 weeks of age, which marks a time prior to the onset of significant tumorigenesis. Mouse tumors were monitored and measured several times per week. Prior to tumor harvest, mice were sacrificed.
Sample Preparation
Mouse tumor tissue specimens (n=3 biological replicates per timepoint per condition) were minced on ice and digested for 3× 30 min in a 37 °C water dry agitator in 0.5 mg/mL collagenase (collagenase type IV, ThermoFisher) digest buffer in Medium 199 (HyClone, GE Healthcare) consisting of 5% fetal bovine serum (Gibco FBS, ThermoFisher), DNase I (Worthington), Poloxamer 188 (Cat. P5556–100ML, Sigma), HEPES, and CaCl2. For human tissue, the MACS human tumor dissociation kit was used according to the manufacturer’s specifications. The digest was quenched with quench media (DMEM (Gibco DMEM, ThermoFisher) with 15% FBS), then centrifuged at 500 × g for 5 min at 4 °C, resuspended in quench media, and filtered through 100, 70, and 40 μm cell strainers (Falcon cell strainer, ThermoFisher). Red blood cell lysis was performed using Hybri-Max (Sigma) per the manufacturer’s protocol. Histopaque was performed using Histopaque-1119 (Sigma–Aldrich), per the manufacturer’s protocol. Cells were counted and re-suspended for further processing.
Florescence Activated Cell Sorting (FACS) Analysis
Cells were counted and resuspended in FACS buffer. Primary antibodies were applied, and cells were stained in the dark with gentle agitation for 30 min. Cells were then washed thoroughly. Staining with secondary antibodies was conducted in the same manner. Propidium iodine (PI, Thermofisher, Cat. P3566, lot: 1755970, 3 μg/mL) or DAPI (Thermofisher, Cat. 3571) were used as a viability marker. Fibroblasts were isolated using the FACS Aria II system. Flow-cytometry plots shown are representative of at least three independent experiments.
Antibodies against the following cell surface markers primarily or secondarily conjugated to the same fluorophore were used for exclusion of “lineage” cells in mouse and human specimens in order to isolate fibroblasts in an unbiased manner: CD45, CD31, Ter119, Tie2, CD324, and CD326. This approach has been previously validated by our laboratory in other fibrotic pathologies.
For phospho-specific flow-cytometry analysis, a single-cell suspension was prepared using manual tissue dispersion rather than enzymatic digestion to preserve phosphorylated signal, and then prepared using the BD Biosciences Cytofix/Cytoperm™ kit according to manufacturer’s instructions. Phosphorylated protein analysis was conducted using the FACS Aria II system.
Recruitment of Human Subjects
Human tumor tissue specimens were obtained from patient’s undergoing surgery at the Stanford Hospital under Stanford University’s IRB approval. The patients were approached in the pre-operative area by one of the manuscript authors. The aims of the study were discussed with the patient. Participation was entirely voluntary. Written, informed consent was obtained from all the patients included in this study prior to surgery. Tissue specimens were collected by one of the authors on this manuscript from the pathologist once the tumor specimen had been inked (from a non-critical part of the tumor), placed directly into sterile saline, and kept on ice for transport. Tissue specimens were processed immediately to form a single cell suspension (details discussed above). Cells were either immediately processed or frozen in Bambanker. This study complies with all relevant ethical regulations for research with human participants.
Single Cell Barcoding, Library Preparation, and Sequencing
Cell suspensions from individual mouse or human samples were labeled with TotalSeq Series B hashtag oligonucleotide-labeled antibodies (BioLegend). Single-cell RNA-seq (scRNA-seq) was then performed at the Stanford Genomics Facility (SGF) for droplet-based microfluidics using the 10x Chromium Single Cell platform (Single Cell 3’ v3, 10x Genomics, USA). Droplets of the cellular suspensions, reverse transcription master mix, and partitioning oil were mixed, loaded onto a single cell chip, and processed on the Chromium Controller. Reverse transcription was performed, and cDNA was amplified using a BioRad C1000 Touch thermocycler, with cDNA size selected using SpriSelect beads (Beckman Coulter, USA). An Agilent Bioanalyzer High Sensitivity DNA chip was used to analyze cDNA for qualitative control purposes; cDNA was then fragmented using the proprietary fragmentation enzyme blend for 5min at 32°C, followed by end repair and A-tailing at 65°C for 30 min. DNA was double-sided size selected using SpriSelect beats. Sequencing adaptors were ligated to the cDNA at 20°C for 15min. cDNA was amplified using a sample-specific index oligo as primer, followed by another round of double-sided size selection using SpriSelect beads. Final libraries were analyzed on an Agilent Bioanalyzer High Sensitivity DNA chip for qualitative control purposes. Libraries were sequenced on a HiSeq 4000 Illumina platform targeting 50,000 reads per cell.
Base calls were converted to reads using the Cell Ranger (10X Genomics; version 3.1) implementation mkfastq and then aligned against either the Cell Ranger mm10 reference genome or GRCh38 v3.0.0 human reference genome, available at: http://cf.10xgenomics.com/supp/cell-exp/, or using Cell Ranger’s count function with SC3Pv3 chemistry and 5,000 expected cells per sample. Hashtag oligos (HTOs) for human and mouse samples were demultiplexed using Seurat’s implementation HTODemux. Briefly, k-medoid clustering is performed on the normalized HTO values, after which a ‘negative’ HTO distribution is calculated. For each HTO, the cluster with the lowest average value is treated as the negative group and a negative binomial distribution is fit to this cluster. Using the 0.99 quantile of this distribution as a threshold, each cell is classified as positive or negative for each HTO. Since each sample for this dataset contained exactly three pooled biological replicates (i.e., tumors from different mice or tumors from different humans), cells for which ambiguous HTO identity could not be clearly established were preserved and assigned to the hashtag for which it was most highly expressed.
Cell barcodes representative of quality cells were differentiated from apoptotic cell barcodes or background RNA based on a threshold of having at least 200 unique transcripts profiled, less than 100,000 total transcripts, and less than 10% of their transcriptome of mitochondrial origin. Unique molecular identifiers (UMIs) from each cell barcode were retained for all downstream analysis, normalized with a scale factor of 10,000 UMIs per cell, and subsequently natural log transformed with a pseudocount of 1 using the R package Seurat (version 3.1.1) (Chen et al., 2013). The first 15 principal components of the aggregated data were then used for uniform manifold approximation and projection (UMAP) analysis (Gulati et al., 2020). Cell annotations were ascribed using SingleR (version 3.11) against either the Mouse-RNAseq reference dataset, available at https://rdrr.io/github/dviraran/SingleR/man/mouse.rnaseq.html, or human primary cell atlas dataset, available at https://rdrr.io/github/dviraran/SingleR/man/hpca.html, and further refined based on inspection. Cell-type marker lists were generated using Seurat’s native FindMarkers function with a log fold change threshold of 0.25 using the ROC test to assign predictive power to each gene.
scATAC-seq and Multiome Data Preparation
Single-cell ATAC sequencing (scATAC-seq) and multiome sequencing were performed at the Stanford Genomics Facility (SGF) using the 10x Genomics platform. The relevant protocols were followed per manufacturer’s guidelines. Nuclei isolation was pursued as for scATAC-seq and multiome sequencing using 0.1x lysis buffer with 3-minute incubation.
CellChat Receptor-Ligand Analysis
To evaluate the potential for cell-cell interactions between our fibroblast populations and other cell types, including immune cells, we applied the recently developed CellChat platform (Jin et al., 2021). This was implemented using our scRNA-seq Seurat object in R, in conjunction with the standalone CellChat Shiny App for its Cell-Cell Communication Atlas Explorer. Cells were binned according to the SingleR-defined cell type classifications, with fibroblast cells subsetted based on their location within either our scarring or regenerative arms. Default parameterizations were used throughout, and Secreted Signaling, ECM-Receptor, and Cell-Cell Contact relationships were considered.
Analysis of Published scRNA-seq Datasets
Published scRNA-seq datasets from the Buechler et al. study were downloaded from their FibroXplorer website (https://www.fibroxplorer.com) as .RDS files containing Seurat objects. These were re-processed from the level of raw UMI counts, while maintaining cluster label and UMAP coordinate information for each cell.
Prediction of Differentiation States
We utilized the recently developed bioinformatics tool CytoTRACE to compare differentiation states among cells in our dataset (https://cytotrace.stanford.edu/) (Gulati et al., 2020). This tool analyzes the number of uniquely expressed genes per cell, as well as other factors like distribution of mRNA content and number of RNA copies per gene, to calculate a score assessing the differentiation and developmental potential of each cell. This algorithm then place the cells along a trajectory corresponding to cell differentiation by taking advantage of each cell’s asynchronous progression under the platform’s unsupervised framework. This analysis was performed using default parameters for each cell in our dataset.
scATAC-seq Data Processing and Analysis
Raw base call (BCL) files were demultiplexed to fastq files using the 10x Genomics Cell Ranger tool cellranger-atac mkfastq. These files were then aligned to the mouse genome (mm10) using cellranger-atac count with default parameters. Downstream analysis of scATAC-seq data were performed using ArchR, a tool developed by our collaborators (Granja et al., 2020) (Granja et al., 2021). Single cell ATAC-seq data were integrated with scRNA-seq data using ArchR’s unconstrained implementation of Seurat’s label transfer algorithm, as previously described (Foster et al., 2021).
Multiome Data Analysis
Data generated using the 10X Multiome platform were processed using 10X’s cellranger-arc toolkit in Linux per the manufacturer’s instructions. This included (1) an ATAC matrix computer step of barcode processing, read trimming, read alignment, duplicate marking, peak calling, and peak-barcode matrix generation using either the mm10 mouse or GRCh38 human reference genome; (2) a gene expression (GEX) Matrix Computer step of read trimming, genome alignment, transcriptome alignment, UMI correction, and UMI counting; and joint cell calling. Downstream secondary analysis for ATAC and GEX included dimensionality reduction, clustering, peak annotation, transcription factor analysis, differential expression analysis, differential accessibility analysis, and feature linkage as described above using the Seurat, Signac, and ArchR toolkits.
The three human breast multiome samples were initially evaluated in R using a modified implementation of Signac’s “Joint RNA and ATAC analysis: 10x multiomic” vignette (compiled May 31, 2022) in conjunction with the EnsDb.Hsapiens.v86 and BSgenome.Hsapiens.UCSC.hg38 genomic libraries. ATAC quality control (QC) thresholds of nucleosome_signal < 2 & TSS.enrichment > 1 were used for all three samples. RNA QC thresholds of nFeature_RNA > 200 were used for all three samples. For sample 1, additional RNA thresholds of nFeature_RNA < 3000 & nCount_RNA < 15.0e3 & nCount_ATAC < 4500 were employed. For sample 2, additional cutoffs of nFeature_RNA < 5000 & nCount_RNA < 15.0e3 & nCount_ATAC < 10000 were used. For sample 3, thresholds of nFeature_RNA < 6000 & nCount_RNA < 25.0e3 & nCount_ATAC < 50000 were employed. Following completion of QC, each sample underwent peak calling with MACS2 using the Signac CallPeaks() function, followed by keepStandardChromosomes() with “coarse” pruning, and subsetByOverlaps() using the blacklist_hg38_unified, after which a “peaks” assay was created the Signac CreateChromatinAssay() function. The RNA portions of each sample were then log-normalized with a scale factor of 10,000, followed by variable feature identification, and PCA identification using default Seurat parameters. FindNeighbors(dims = 1:15), FindClusters (resolution = 0.2), and FindUmap() were then applied in a standard fashion, followed by SingleR (version 3.11) against the human primary cell atlas dataset, available at https://rdrr.io/github/dviraran/SingleR/man/hpca.html, as above, and further refined based on inspection to focus on cancer associated fibroblasts, which were subsequently re-clustered using a resolution parameter of 0.6.
The nine mouse breast multiome samples were initially evaluated in R using a modified implementation of Signac’s “Joint RNA and ATAC analysis: 10x multiomic”, using the EnsDb.Mmusculus.v79 annotation. ATAC quality control (QC) thresholds of nucleosome_signal < 2 & TSS.enrichment > 1 were used for all nine mouse samples. RNA QC thresholds of nFeature_RNA > 200 were also used for all nine samples. The following additional thresholds were used for specific samples:
C1: nFeature_RNA < 2500 & nCount_RNA < 5.0e3 & nCount_ATAC < 1.5e4
C2: nFeature_RNA < 2500 & nCount_RNA < 5.0e3 & nCount_ATAC < 1.5e4
C3: nFeature_RNA < 1500 & nCount_RNA < 5.0e3 & nCount_ATAC < 1.5e4
L1: nFeature_RNA < 2600 & nCount_RNA < 5.0e3 & nCount_ATAC < 1.5e4
L2: nFeature_RNA < 2000 & nCount_RNA < 5.0e3 & nCount_ATAC < 1.5e4
L4: nFeature_RNA < 3000 & nCount_RNA < 5.0e3 & nCount_ATAC < 1.5e4
S1: nFeature_RNA < 6000 & nCount_RNA < 5.0e5 & nCount_ATAC < 2.5e6
S2: nFeature_RNA < 3000 & nCount_RNA < 5.0e4 & nCount_ATAC < 2.5e4
S3: nFeature_RNA < 2500 & nCount_RNA < 5.0e4 & nCount_ATAC <1.0e5
Following completion of QC, each sample underwent peak calling with MACS2 using the Signac CallPeaks() function, followed by keepStandardChromosomes() with “coarse” pruning, and subsetByOverlaps() using the blacklist_mm10, after which a “peaks” assay was created the Signac CreateChromatinAssay() function. The RNA portions of each sample were then log-normalized with a scale factor of 10,000, followed by variable feature identification, and PCA identification using default Seurat parameters. FindNeighbors(dims = 1:15), FindClusters (resolution = 0.2), and FindUmap() were then applied in a standard fashion, followed by SingleR (version 3.11) against the Mouse-RNAseq reference dataset, available at https://rdrr.io/github/dviraran/SingleR/man/mouse.rnaseq.html, as above, and further refined based on inspection to focus on cancer associated fibroblasts, which were subsequently re-clustered using a resolution parameter of 0.3 to arrive at the resulting 8 CAF clusters.
Spatial Transcriptomics
Mouse tissue was processed according to the Visual Spatial Protocol-Tissue Preparation Guide by 10X Genomics. In brief, we flash froze tissue samples in OCT using a Thermo Scientific Thermo-Flask benchtop container filled with liquid nitrogen and a small, round, metal bowl with 1-inch height worth of isopentane. A pair of sturdy needle-drivers was used to maneuver the isopentane bowl in and out of the Thermo-Flask. The isopentane bowl was incubated in liquid nitrogen for about 10 minutes. Tissue was completely frozen in approximately 30 seconds, wrapped in aluminum foil, stored immediately in a box containing dry ice, and transferred to a −80°C freezer. Tumor tissues were cryosectioned at −20 degrees onto gene expression slides. The Gene Expression Slide & Reagent kit was followed per the 10X Genomics protocol and used to produce sequencing libraries. The libraries were then sequenced using NextSeq (Illumina), and Bcl files were demultiplexed. Raw FASTQ files and histology images were processed by sample with the Space Ranger software, which uses STAR v.2.5.1b (Dobin et al., 2013) for genome alignment, against the Cell Ranger mm10 reference genome, available at: http://cf.10xgenomics.com/supp/cell-exp/ (Dobin et al., 2013).
To analyze the spatial distribution of cell types in Visium data, pairwise cell-cell neighbor interactions were quantified by extracting a feature matrix, f, of the probability of cell type representation in each Visium spot. An adjacency matrix, j, was then used to identify neighboring Visium spots within k=5 nearest neighbors. For each possible cell-cell pairing, a pairing score was calculated as the dot product of individual cell type vectors in f with the adjacency matrix (i.e. f1·j·f2) using AdjacencyScore in R (Govek et al., 2021). Scores were averaged at the group-level, then subtracted between groups to quantify the magnitude of differential cell pairings in early tumor vs. native breast tissue and late tumor vs. early tumor.
Rainbow-CODEX Analysis
Breast tumor specimens from Rainbow mice were sectioned at 6 μm and spatially phenotyped for fibroblast subtypes using Co-Detection by indEXing (CODEX), a spatial sequencing and quantification technique for protein expression (Black et al., 2021). Prior to CODEX processing, Rainbow fluorescence was imaged as previously mentioned, after which native fluorescence was photobleached to enable CODEX staining and phenotyping. A 7-plex CODEX antibody panel was used to define specific fibroblast markers of interest, including CD26, Ly6C, Ly6G, LRRC15, and PDPN. Antibody conjugation and cyclic imaging of n=3 Rainbow breast tumor specimens were performed by Stanford’s Cell Sciences Imaging Facility, using a Keyence BZ-X (Akoya Biosciences). Raw imaging data were processed for graphical rendering and cell segmentation using the standardized Akoya Biosciences pipeline, then exported to RStudio for computational analysis. CODEX expression data were pre-processed in STvEA with default settings (Govek et al., 2021), and the resulting single-cell protein expression matrices were batch corrected between n=3 specimens using Batchelor (Haghverdi et al., 2018). After batch correction, protein expression matrices were projected to a UMAP manifold in Seurat to visualize protein-defined cell populations.
Rainbow clonal expression was correlated with CODEX-defined fibroblast subpopulations by direct spatial alignment of Rainbow and CODEX images on a per-specimen basis. For each breast tumor specimen, a transparent overlay of CODEX-defined cell spatialization (i.e. an alignment mask) was oriented onto the corresponding Rainbow image. A cell phenotypic filter was then applied to the alignment mask to block Rainbow visualization in all spaces outside of CODEX cell populations of interest, eliminating Rainbow fluorescence not co-localized with the specified cell population. The remaining clonal expression within each channel of GFP, mCerulean, mCherry, and mOrange was then analyzed for its intensity distribution using kernel density plots in FIJI.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the software GraphPad Prism v.6 (unless otherwise noted). Results are expressed as absolute numbers, percentages, fractions, or mean +/− standard deviation (unless otherwise noted). Unpaired t-test assuming two-tailed distribution or one-way analysis of variance (ANOVA) and post hoc Tukey correction were used to compare groups where relevant. Comparisons of scRNA-seq cluster distribution among samples were performed using the DA-seq toolkit in R, with “regions” defined as cell clusters and unique “labels” assigned to each tissue sample (Zhao et al., 2021). An adjusted p-value of < 0.05 was considered statistically significant for all comparisons.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mAb to alpha Smooth Muscle Actin | Abcam | Cat# 32575 |
| mAb to FAK | Abcam | Cat# 40794 |
| pAb to Collagen I | Abcam | Cat# Ab34710 |
| pAb to Collagen III | Abcam | Cat# Ab7778 |
| mAb to FAP | Cell Signaling | Cat# 66562S |
| mAb to MGP | ThermoFisher | Cat# 60055-1 |
| Ab to Phospho-FAK (pTyr397) | Invitrogen | Cat# 700255 |
| Ab to DPP4 (CD26) | Abcam | Cat# Ab28340 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat# A-11008 |
| Goat anti-Mouse IgG2a heavy chain, PE/Cy7 ®preadsorbed | Abcam | Cat# Ab130787 |
| Donkey anti-Rat IgG H&L, Alexa Fluor 647 preadsorbed | Abcam | Cat# Ab150155 |
| Anti-mouse CD45, Alexa Fluor 647 | Biolegend | Cat# 103124 |
| Anti-mouse CD31, Alexa Fluor 647 | Biolegend | Cat# 102516 |
| Anti-mouse TER-119, Alexa Fluor 647 | Biolegend | Cat# 116218 |
| Anti-mouse/human CD324, Alexa Fluor 647 | Biolegend | Cat# 147308 |
| Anti-mouse CD326, Alexa Fluor 647 | Biolegend | Cat# 118212 |
| Anti-mouse CD202b (TIE2), Biotin | Invitrogen | Cat# 124006 |
| Streptavidin, Alexa Flour 647 conjugate | Invitrogen | Cat# S32357 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (Z)-4-Hydroxyta moxifen | Millipore Sigma | Cat# H7904 |
| Tamoxifen | Sigma | |
| Corn oil | Sigma | |
| Hematoxylin | Sigma | Cat# H3136 |
| Acetic Acid EMD | Millipore | Cat# AX0073 |
| Phosphotungstic Acid | Sigma | Cat# P4006 |
| Eosin Y solution, alcoholic | Sigma | Cat# HT1101128 |
| Ethanol Gold Shield | Sigma | Cat# 64-17-5 |
| BABB (Benzoic acid:benzyl Benzoate) | Sigma | Cat# B6630 |
| tert-butanol | Sigma | Cat# 360538 |
| Triethylamine | FisherSci | Cat# 04885 |
| Permount | Fisher Chemicals | Cat# SP15 |
| Triton X-100 | Sigma | Cat# X100 |
| Fluoromount-G | SouthernBiotech | Cat# 0100-01 |
| Prolong Gold Antifade Mountant with DAPI | ThermoFisher | Cat# P36935 |
| DAPI (4’, 6-Diamido-2-Phenylindole, Dihydrochloride) | ThermoFisher | Cat# D1306 |
| Collagenase (Collagenase Type IV) | ThermoFisher | Cat# 17104019 |
| Liberase | Sigma Aldrich | Cat# 05401020001 |
| Medium 199 | Sigma Aldrich | Cat# M2520 |
| Fetal Bovine Serum | Thermo Fisher | Cat# 10438026 |
| DNase I | Worthington | Cat# LS006330 |
| Poloxamer 188 | Sigma | Cat# P5556 |
| HEPES | Sigma Aldrich | Cat# H3375 |
| Calcium Chloride | Sigma Aldrich | Cat# 499609 |
| Dulbecco’s Modified Eagle Medium | ThermoFisher | Cat# 11320082 |
| SYTOX ADvanced Ready Flow Reagent | ThermoFisher | Cat# R37173 |
| Digitonin | Promega | Cat# G9441 |
| Tween-20 | Sigma/Roche | Cat# 11332465001 |
| 1X Power Block | BioGeneX | Cat# HK085-5K |
| NP40 | Sigma/Roche | Cat# 11332473001 |
| RNAse Inhibitor | Protector | Cat# 3335402001 |
| Critical Commercial Assays | ||
| Trichrome Stain Kit | Abcam | Cat# Ab150686 |
| Picro Sirius Red Stain Kit | Abcam | Cat# Ab150681 |
| Chromium Single Cell Chip B Kit | 10X Genomics | Cat# 1000154 |
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10X Genomics | Cat# 1000092 |
| Chromium Next GEM Chip H Single Cell Kit | 10X Genomics | Cat# 1000161 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10X Genomics | Cat#1000175 |
| Single Index Kit N Set A | 10X Genomics | Cat# 100021289 |
| Visium Spatial Gene Expression Solution v1 | 10X Genomics | Cat#PN-1000184 |
| Visium Spatial Tissue Optimization Slide & Reagent Kit | 10X Genomics | Cat#PN-1000193 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10X Genomics | Cat#PN-1000285 |
| Deposited Data | ||
| The gene expression and chromatin accessibility datasets generated during this study are publicly available and can be found through the GEO web portal using the accession listing at right. | Gene Expression Omnibus (GEO) | GSE212482 and its subseries (GSE212461, GSE212481, GSE212706, GSE212707, GSE212708) |
| Experimental Models: Organisms/Strains | ||
| Black6 (C57BL/6J) mice | The Jackson Laboratory | Stock#000664 |
| Actin-Cre-ERT2 (Tg(CAG-cre/Esr1)5Amc/J) mice | The Jackson Laboratory | Stock#004682 |
| Col1-CreERT2 (Tg(Col1a2-cre/ERT,-ALPP)7Cpd/J) mice | The Jackson Laboratory | Stock#029235 |
| MMTV-PyMT (FVB/N-Tg(MMTV-PyVT)634Mul/J) mice | The Jackson Laboratory | Stock#002374 |
| FAKfl/fl (B6.129P2(FVB)-Ptk2tm1.1Guan/J) mice | The Jackson Laboratory | Stock#031956 |
| αSMA-CreERT2 mice | Courtesy of Dr. Ivo Kalajzic, University of Connecticut | |
| Rainbow (ROSA26VT2/GK3) mice | Courtesy of the Weissman Laboratory, Stanford University School of Medicine | |
| Software and Algorithms | ||
| Adobe Photoshop CC | Adobe | RRID:SCR 014199 |
| Adobe Illustrator CC | Adobe | RRID:SCR 010279 |
| Prism 5 | GraphPad | RRID:SC R 002798 |
| FIJI/ImageJ | National Institutes of Health | RRID:SCR 002285 |
| Imaris | Oxford Instruments | RRID:SCR 007370 |
Highlights:
CAF subpopulations comprise three functional categories (superclusters)
Subpopulations are conserved across solid tumor types and species
Disruption of mechanotransduction or ICI yields shifts between CAF subpopulations
Targeting the balance of CAF subpopulations holds therapeutic potential
ACKNOWLEDGEMENTS
We would like to acknowledge the Stanford Genomics Facility, Stanford University Cell Sciences Imaging Core Facility (RRID:SCR_017787; with instruments supported by Award Number 1S10OD010580–01A1 from the National Center for Research Resources), Lokey Stem Cell FACS Core, and 10x Genomics for their assistance with experiments. We would like to thank Drs. Shannon J Turley, Akshay T Krishnamurty, and their group at Genentech for their kind gift of the LRRC15 antibody. We would also like to thank Drs. George Poultsides, Monica Dua, and Brendan Visser for their assistance acquiring patient specimens. We would like to acknowledge Art and Elaine Taylor, the Rantz Foundation, and Warren and Judy Kaplan for their generous support of our research efforts. Funding sources include NIH 1F32CA239312–01A1 (D.S.F), 1R01GM116892 (M.T.L), 1R01GM136659 (M.T.L), 5U01DK119094 (M.J., G.C.G.), 1R01DE027346 (D.C.W.), RM1-HG007735 (H.Y.C.), American College of Surgeons (D.S.F.), the Advanced Residency Training at Stanford program (D.S.F.), Goldman Sachs Foundation (J.A.N., D.S.F., M.T.L.), the Child Health Research Institute at Stanford (D.C.W.), the Scleroderma Research Foundation (H.Y.C., M.T.L.), the Damon Runyon Cancer Research Foundation (D.D., M.T.L.), the Gunn/Olivier Fund, the California Institute for Regenerative Medicine, the Hagey Laboratory for Pediatric Regenerative Medicine, and the Stinehart/Reed Foundation. Sequencing and imaging data were obtained using machines purchased with NIH funds (S10OD025212, S10OD018220, 1S10OD01058001A1). H.Y.C. is an Investigator of the Howard Hughes Medical Institute. H.Y.C. is supported by NIH R35-CA209919.
Footnotes
DECLARATION OF INTERESTS
D.S.F., M.J., D.D., H.Y.C., J.A.N., and M.T.L. have applied for a provisional patent through Stanford University related to the research presented in this manuscript. H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, Cartography Biosciences, and an advisor of 10x Genomics, Arsenal Biosciences, and Spring Discovery. K.E.Y. is a consultant for Cartography Biosciences.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found in a separate document included with this manuscript submission.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Biasci D, Smoragiewicz M, Connell CM, Wang Z, Gao Y, Thaventhiran JED, Basu B, Magiera L, Johnson TI, Bax L, et al. (2020). CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc Natl Acad Sci U S A 117, 28960–28970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson DA (2019). IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 9, 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black S, Phillips D, Hickey JW, Kennedy-Darling J, Venkataraaman VG, Samusik N, Goltsev Y, Schurch CM, and Nolan GP (2021). CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc 16, 3802–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, Yang YA, Tam L, Caothien R, Roose-Girma M, et al. (2021). Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579. [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demircioglu F, Wang J, Candido J, Costa ASH, Casado P, de Luxan Delgado B, Reynolds LE, Gomez-Escudero J, Newport E, Rajeeve V, et al. (2020). Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat Commun 11, 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 10, 232–253. [DOI] [PubMed] [Google Scholar]
- Drifka CR, Loeffler AG, Mathewson K, Keikhosravi A, Eickhoff JC, Liu Y, Weber SM, Kao WJ, and Eliceiri KW (2016). Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection. Oncotarget 7, 76197–76213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, Burcham AR, Salhotra A, Ransom RC, Henn D, et al. (2021). Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Jones RE, Ransom RC, Longaker MT, and Norton JA (2018). The evolving relationship of wound healing and tumor stroma. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Nguyen AT, Chinta M, Salhotra A, Jones RE, Mascharak S, Titan AL, Ransom RC, da Silva OL, Foley E, et al. (2019). A Clearing Technique to Enhance Endogenous Fluorophores in Skin and Soft Tissue. Sci Rep 9, 15791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia PE, Scales MK, Allen BL, and Pasca di Magliano M (2020). Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govek KW, Troisi EC, Miao Z, Aubin RG, Woodhouse S, and Camara PG (2021). Single-cell transcriptomic analysis of mIHC images via antigen mapping. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Adeyanju O, Sunil C, Mandlem V, Olajuyin A, Huang S, Chen SY, Idell S, Tucker TA, and Qian G (2022). DOCK2 contributes to pulmonary fibrosis by promoting lung fibroblast to myofibroblast transition. Am J Physiol Cell Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, and Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, Oon C, Bhattacharyya S, Sanford-Crane H, Horton W, et al. (2021). Mesenchymal Lineage Heterogeneity Underlies Non-Redundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Sharma S, Zhu LX, Keane MP, Luo J, Zhang L, Burdick MD, Lin YQ, Dohadwala M, Gardner B, et al. (2002). IL-7 inhibits fibroblast TGF-beta production and signaling in pulmonary fibrosis. J Clin Invest 109, 931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januszyk M, Chen K, Henn D, Foster DS, Borrelli MR, Bonham CA, Sivaraj D, Wagh D, Longaker MT, Wan DC, and Gurtner GC (2020). Characterization of Diabetic and Non-Diabetic Foot Ulcers Using Single-Cell RNA-Sequencing. Micromachines (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat Commun 12, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Wang J, Sa JK, Ladewig E, Lee HO, Lee IH, Kang HJ, Rosenbloom DS, Camara PG, Liu Z, et al. (2017). Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet 49, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Yeo SY, Sung CO, and Kim SH (2015). Twist1 is a key regulator of cancer-associated fibroblasts. Cancer Res 75, 73–85. [DOI] [PubMed] [Google Scholar]
- Li G, Qin Y, Qin S, Zhou X, Zhao W, and Zhang D (2020). Circ_WBSCR17 aggravates inflammatory responses and fibrosis by targeting miR-185–5p/SOX6 regulatory axis in high glucose-induced human kidney tubular cells. Life Sci 259, 118269. [DOI] [PubMed] [Google Scholar]
- Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, Ebright RY, Karabacak MN, Kulkarni AS, Liu A, et al. (2019). Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 178, 160–175 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascharak S, Talbott HE, Januszyk M, Griffin M, Chen K, Davitt MF, Demeter J, Henn D, Bonham CA, Foster DS, et al. (2022). Multi-omic analysis reveals divergent molecular events in scarring and regenerative wound healing. Cell Stem Cell 29, 315–327 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallasamy P, Nimmakayala RK, Karmakar S, Leon F, Seshacharyulu P, Lakshmanan I, Rachagani S, Mallya K, Zhang C, Ly QP, et al. (2021). Pancreatic Tumor Microenvironment Factor Promotes Cancer Stemness via SPP1-CD44 Axis. Gastroenterology 161, 1998–2013 e1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton J, Foster D, Chinta M, Titan A, and Longaker M (2020). Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214, 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio D, Kuijjer ML, and Cai JJ (2021). rPanglaoDB: an R package to download and merge labeled single-cell RNA-seq data from the PanglaoDB database. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Sahai E, and Rullan A (2020). SnapShot: Cancer-Associated Fibroblasts . Cell 181, 486–486 e481. [DOI] [PubMed] [Google Scholar]
- Phan QM, Driskell IM, and Driskell RR (2021). The Three Rs of Single-Cell RNA Sequencing: Reuse, Refine, and Resource. J Invest Dermatol 141, 1627–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, and Driskell RR (2020). Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom RC, Foster DS, Salhotra A, Jones RE, Marshall CD, Leavitt T, Murphy MP, Moore AL, Blackshear CP, Brett EA, et al. (2018). Genetic dissection of clonal lineage relationships with hydroxytamoxifen liposomes. Nat Commun 9, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalinger MR, Lang S, Gottier C, Dai X, Rawlings DJ, Chan AC, Rogler G, and Scharl M (2017). PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 13, 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, Hao Y, Oni TE, Brosnan E, Elyada E, et al. (2021). Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin Cancer Res 27, 2023–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, and Weissman IL (2006). Clonal analysis of mouse development reveals a polyclonal origin for yolk sac blood islands. Dev Cell 11, 519–533. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Hao M, Yeo SK, and Guan JL (2020). FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene 39, 2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Tanaka T, and Ueno H (2013). Multicolor lineage tracing methods and intestinal tumors. J Gastroenterol 48, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, Dang Y, Chu Y, Fan J, and He R (2016). FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res 76, 4124–4135. [DOI] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jaffe A, Li H, Lindenbaum O, Sefik E, Jackson R, Cheng X, Flavell RA, and Kluger Y (2021). Detection of differentially abundant cell subpopulations in scRNA-seq data. Proceedings of the National Academy of Sciences 118, e2100293118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou B, Zhang T, Zhou R, Jiang X, Yang H, Jin X, and Bai Y (2021). deepMNN: Deep Learning-Based Single-Cell RNA Sequencing Data Batch Correction Using Mutual Nearest Neighbors. Front Genet 12, 708981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression and chromatin accessibility datasets generated during this study are publicly available and can be found through Gene Expression Omnibus (GEO) web portal using accession listing GSE212482 and its subseries (GSE212461, GSE212481, GSE212706, GSE212707, GSE212708), which includes our scRNA-seq, scATAC-seq, single cell multiome, and also spatial transcriptomics data. The CODEX spatial proteomics data will be made available by the authors upon request.