

SUMMARY
Therapeutic cancer vaccines are designed to increase tumor-specific T cell immunity. However, suppressive mechanisms within the tumor microenvironment (TME) may limit T cell function. Here we assessed how the route of vaccination alters intratumoral myeloid cells. Using a self-assembling nanoparticle vaccine that links tumor antigen peptides to a Toll-like receptor 7/8 agonist (SNP-7/8a), we treated tumor-bearing mice subcutaneously (SNP-SC) or intravenously (SNP-IV). Both routes generated antigen-specific CD8+ T cells that infiltrated tumors. However, only SNP-IV mediated tumor regression, dependent on systemic type I interferon at the time of boost. Single cell RNA-sequencing revealed that intratumoral monocytes expressing an immunoregulatory gene signature (Chil3, Anxa2, Wfdc17) were reduced after SNP-IV boost. In humans, the Chil3+ monocyte gene signature is enriched in CD16− monocytes and associated with worse outcomes. Our results show that the generation of tumor-specific CD8+ T cells combined with remodeling of the TME is a promising approach for tumor immunotherapy.
Graphical Abstract
In Brief
Intravenous administration of a cancer vaccine promotes tumor regression via antigen-specific CD8+ T cells and type I interferon-dependent modulation of the tumor microenvironment.
INTRODUCTION
A key tenet of cancer immunotherapy is to harness the patient’s own immune system to mediate tumor regression. Central to the anti-tumor immune response are T cells, which can kill tumor cells in an antigen-specific manner. Advances in our understanding of T cell biology has led to several important therapeutic strategies including checkpoint blockade, adoptive cell therapy and cancer vaccines (Waldman et al., 2020). A primary goal of therapeutic cancer vaccines is to promote tumor regression by inducing antigen-specific T cells in vivo (Saxena et al., 2021). Research efforts have focused on uncovering tumor-associated antigens, in particular targeting tumor-specific mutations termed neoantigens (neoAg). The selective expression of neoAgs on tumor cells would potentially minimize autoimmune-related toxicities. Although promising, the clinical outcomes and immune responses measured in cancer vaccine trials have been limited in late-stage patients thus far (Melero et al., 2014; Romero et al., 2016). Hence, several challenges need to be addressed to improve the efficacy of personalized cancer vaccines (Hegde and Chen, 2020). A better understanding of the tumor immune contexture is critical: knowing the key molecular and cellular drivers dampening the T cell response at the tumor site can inform the rational design of vaccine formulations and influence delivery strategies.
We previously developed a self-assembling nanoparticle vaccine platform that co-delivers long peptides containing neoAgs with a Toll-like receptor 7/8 agonist (SNP-7/8a) (Lynn et al., 2020). By modifying the route of vaccination, we can alter the quality of neoAg-specific CD8+ T cells: subcutaneous (SNP-SC) or intravenous delivery (SNP-IV) generated more terminally-differentiated or stem-like CD8+ T cells respectively (Baharom et al., 2021). It was notable that following SNP-SC vaccination, despite high magnitude CD8+ T cell repsonses, there was limited control of tumor growth in therapeutic murine tumor models. In contrast, SNP-IV was able to control the growth of established tumors; this was associated with the generation of stem-like CD8+ T cells capable of replenishing effector cells upon treatment with checkpoint inhibitors such as anti-PD-L1. In addition to providing high quality CD8+ T cells, we hypothesized that SNP-IV could also be potentially beneficial in altering the tumor microenvironment (TME) through systemic innate activation by TLR7/8a.
The TME is a highly complex and dynamic milieu populated by numerous different cell types that may play unique roles in promoting or suppressing the anti-tumor activity of T cells. An immune infiltrate continuum ranging from “inflamed” to “immune desert” is often used to describe the immune microenvironment of human tumors. Preclinical studies in mice face challenges in modeling human tumors in terms of reflecting the true immune landscape (Mosely et al., 2017). MC38 tumors, often used as a murine model of human colorectal cancer (CRC), is largely composed of immunosuppressive cells (Mariathasan et al., 2018). Using single cell RNA-sequencing (scRNA-seq), recent studies carefully characterized the immune infiltrate in tumors of CRC patients and murine models, and identified highly conserved myeloid cells, including macrophages, monocytes and conventional dendritic cells (cDCs) that are present in both human and mouse tumors (Zhang et al., 2020). cDCs can be further subdivided into type 1 (cDC1) or type 2 (cDC2) lineages: cDC1s excel at cross-presentation for priming of CD8+ T cells whereas cDC2s are specialized at priming CD4+ T cells (Guilliams et al., 2014).
The heterogeneity of myeloid cells with T cell immune-suppressive functions have been reported in various murine and human cancer types. Recent data using high dimensional single cell technologies have refined our understanding of the developmental relationships and phenotypic markers used to identify and classify these cells (Cheng et al., 2021). Myeloid-derived suppressor cells (MDSCs), often described in the context of late-stage cancers as having regulatory functions, can be thought of as a cellular state rather than a cellular identity: different myeloid cells can upregulate suppressive genes involved in inhibitory pathways such as arginine metabolism depending on the environmental stimuli (Hegde et al., 2021). Tumor-associated macrophages (TAMs) is another broad term used to refer to a heterogenous population of myeloid cells of embryonic or monocytic origin that evolved with or infiltrate into the tumors respectively (Bleriot et al., 2020; Hourani et al., 2021). An area of therapeutic interest is in modulating TAMs to polarize them towards more pro-inflammatory or anti-tumoral capacity, often referred to as an “M1 phenotype” as opposed to a more pro-tumoral “M2 phenotype” characterized by anti-inflammatory signaling (Yang et al., 2020). An important cytokine involved in modulating macrophages are type I interferons (IFNα and IFNβ), known to regulate the induction of more than 100 downstream interferon-stimulated genes (Borden, 2019; Dunn et al., 2006; U’Ren et al., 2010; Zitvogel et al., 2015). Importantly, the polarization of macrophages within the TME has been described in the context of successful or improved response to checkpoint blockade (Gubin et al., 2018; Lam et al., 2021).
In this study, we investigated how SNP-IV mediated tumor regression through two distinct innate and adaptive immune mechanisms. The results reported here show that neither vaccine-induced tumor-specific CD8+ T cells alone nor systemic innate immune activation alone were sufficient for controlling tumor growth. Rather, the optimal cancer vaccine regimen may require priming of tumor-specific CD8+ T cells followed by systemic IFN-I to alter the TME. These findings have broad implications in our understanding of cancer vaccines and immunotherapies, highlighting the need for innate immune activation to modulate the TME for optimal T cell function.
RESULTS
NeoAg-specific CD8+ T cells generated by SNP-SC controlled tumor growth when followed by SNP-IV
We previously reported that subcutaneous administration of SNP-7/8a (SNP-SC), a self-assembling nanoparticle vaccine, generated neoAg-specific CD8+ T cells that are terminally differentiated based on transcriptional profiling (Baharom et al., 2021). Although SNP-SC-generated CD8+ T cells could control tumor growth in a prophylactic setting, they were ineffective in a therapeutic setting. In contrast, SNP-7/8a administered intravenously (SNP-IV) generated stem-like CD8+ T cells that were effective in mediating tumor regression in established tumors when delivered in combination with anti-PD-L1 treatment (Baharom et al., 2021). Of note, SNP-IV also induced systemic innate activation marked by high levels of pro-inflammatory mediators such as IFNα and IL-12. Here, we delineated the role of CD8+ T cell magnitude and quality from the effects of systemic innate immune activation by SNP-IV.
Mice were implanted with MC38 tumors on the subcutaneous flank and treated on day 7 (prime) and day 14 (boost) with SNP-7/8a containing Reps1, an MC38 neoAg, together with anti-PD-L1 (Figure 1A). Consistent with our prior data, the group that received SNP-IV (prime and boost) mediated tumor regression whereas the group that received SNP-SC (prime and boost) could not control tumor growth (Figure 1B). However, mice that were first primed with SNP-SC followed by an SNP-IV boost could also control tumors and promote improved survival similar to SNP-IV given twice (Figure 1C). To confirm and extend these findings in a different tumor model, mice were implanted with TC-1 tumors and treated with SNP-7/8a containing HPV E6 antigen. Consistent with our MC38 observation, TC-1 tumor-bearing mice that were treated with SNP-SC followed by SNP-IV had significantly smaller tumors than mice boosted with SNP-SC (Figure S1A) despite the generation of similar magnitudes of antigen-specific CD8+ T cells (Figure S1B). These data suggest that SNP-IV is not required at the time of priming and terminally-differentiated CD8+ T cells generated by SNP-SC can also mediate tumor regression when boosted with SNP-IV.
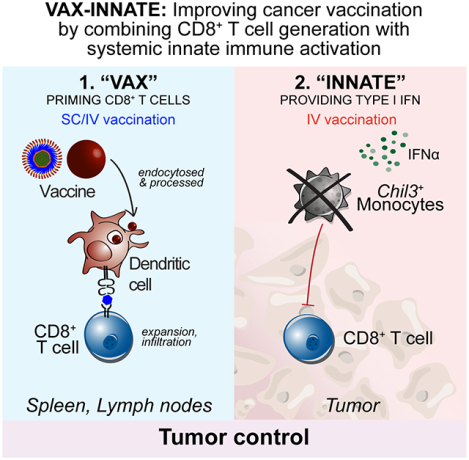
Figure 1. Tumor-specific CD8+ T cells generated by SNP-SC controlled tumor growth when followed by IV adjuvant delivery.
(A) Schematic of therapeutic study design. Mice were implanted with MC38 and treated with SNP-7/8a (Reps1) on day 7 and day 14 together with CPI.
(B) Tumor growth following treatment with SNP-IV prime and boost (red), SNP-SC prime and boost (blue) or SNP-SC prime and SNP-IV boost (green) (n=10). Statistics were assessed by two-way ANOVA.
(C) Survival curve following treatment or in untreated mice (grey) (n=30). Statistics were assessed by log-rank test.
(D) Tumor growth following treatment with SNP-SC prime with Reps1 (solid line) or an irrelevant antigen (dashed line) (n=10). Statistics were assessed by two-way ANOVA.
(E) Tumor growth following treatment SNP-SC prime (Reps1) followed by SNP-IV boost containing an irrelevant antigen (purple) or polyIC:LC (orange) (n=10). Statistics were assessed by two-way ANOVA.
(F) Survival curve following SNP-IV boost containing an irrelevant antigen (purple) or polyIC:LC (orange) or untreated (grey) (n=30). Statistics were assessed by log-rank test.
(G) Flow cytometry analysis of blood stained with tetramer and CD44 antibody (concatenated, n=10).
(H) Bar graph summarizes the frequency of tetramer+ CD8 T cells in blood after treatment (n=30). Statistics were assessed by Kruskal Wallis test.
(I) Heatmaps represent the median MFI of PD-1, Tim-3 and NKG2A on tetramer+ CD8+ T cells in blood (n=10) and spleens (n=5) on day 21.
(J) Histograms summarize the MFI PD-1, Tim-3 and NKG2A and CD39 on CD8+ T cells in tumors (n=5) on day 21.
To first show that tumor regression required neoAg-specific CD8+ T cells, mice were vaccinated with SNP-IV containing an irrelevant antigen (prime and boost). SNP-IV was ineffective in controlling tumors (Figure S1C) or extending survival (Figure S1D) in the absence of antigen-specific CD8+ T cells (Figure S1E). Mice that were primed with SNP-SC containing no tumor-specific antigen followed by SNP-IV containing the MC38 antigen was also not effective at controlling tumor growth (Figure 1D). To assess the requirement for antigen at the time of boost, mice were primed with SNP-SC containing Reps1, followed by SNP-IV containing an irrelevant antigen (Figure 1E). Remarkably, SNP-SC prime followed by SNP-IV boost with an irrelevant antigen resulted in improved control of tumor growth and a 50% survival rate (Figure 1F). These data show that neoAg+ CD8+ T cells generated by SNP-SC given once was sufficient to mediate tumor regression when followed by systemic innate stimulation, even in the absence of antigen at the time of boost. Furthermore, the anti-tumor effect by the boost is not unique to TLR7/8a signaling as IV administration of polyIC:LC, another adjuvant that induces IFN-I signaling, after SNP-SC prime could also significantly mediate tumor regression and prolong survival when given 7 days after priming with SNP-SC (Figures 1E and F).
Tumor regression induced by SNP-SC prime followed by adjuvant administered IV did not require expansion of tumor-specific CD8+ T cell responses
To assess whether there are differences in the magnitude of neoAg-specific CD8+ T cells after SNP-SC or SNP-IV boost, we sampled whole blood on day 21, a week after the boost and measured the tetramer+ population by flow cytometry (Figure 1G). SNP-SC given twice, or SNP-SC followed by SNP-IV generated similar frequencies of tetramer+ CD8+ T cells at approximately 10% of total CD8+ T cells in circulation. The magnitude of neoAg+ CD8+ T cells is also comparable in the spleen and tumor (Figures S1F and S1G). Groups that received SNP-IV containing an irrelevant antigen or polyIC:LC IV at the time of boost showed no increase in the magnitude of neoAg-specific CD8+ T cells compared to mice that only received one shot of SNP-SC containing Reps1 at approximately 1% of total CD8+ T cells in circulation (Figure 1H). In summary, SNP-SC primed neoAg-specific CD8+ T cells may promote anti-tumor efficacy if followed by IV administration of SNP-7/8a or polyIC:LC even with ten-fold lower CD8+ T cell responses compared to the other vaccinated groups.
As markers of exhaustion and antigen experience on CD8+ T cells are important for improving the therapeutic effects of immune checkpoint blockade, we assessed the expression of PD-1, TIM-3 and NKG2A post vaccination. SNP-SC boosted cells in circulation showed a higher expression of exhaustion markers PD-1, TIM-3 and NKG2A compared to the other groups that received SNP-IV (Reps1 or irrelevant antigen) or polyIC:LC IV (Figure 1I and S1H). However, the tumor-infiltrating Reps1+ CD8+ T cells showed similar expression levels of PD-1, TIM-3, NKG2A and CD39 regardless of treatment received (Figure 1J and S1I). Given the protective effect of boosting with SNP-IV containing an irrelevant antigen (Trp1), we compared the quality of tumor-infiltrating CD8+ T cells to assess whether they may be playing a bystander effect. Compared to intratumoral Reps1+ CD8+ T cells, Trp1+ CD8+ T cells expressed lower levels of exhaustion markers including CD39, reflecting a lack of tumor antigen experience (Figure S1J). Overall, there were no striking differences in the expression of exhaustion markers on CD8+ T cells, especially in the tumor, suggesting that the systemic innate stimulation provided by SNP-IV or polyIC:LC given IV may be affecting immune cells other than T cells.
SNP-IV but not SNP-SC resulted in intratumoral vaccine distribution and DC maturation
To further demonstrate differences between boosting with SNP-SC or SNP-IV that may influence anti-tumor immunity, we tracked the vaccine pharmacokinetics by performing live imaging of animals using an Alexa Fluor 647-labeled SNP-7/8a. Systemic distribution of the vaccine was detected by in vivo imaging after SNP-IV but not SNP-SC (Figure 2A). Moreover, fluorescently labeled vaccine could be localized in the tumor primarily in the first hour and remained detectable at low levels after 24 hours (Figure 2B). Assessment of explanted tumors and tumor-draining lymph nodes collected at 6 hours, 24 hours and 72 hours post vaccination confirmed the detection of fluorescently labeled vaccine after SNP-IV but not SNP-SC vaccination (Figure 2C). Additionally, SNP-IV also led to detectable vaccine in the spleens, suggesting systemic vaccine distribution (Figure S2A). Following tissue digestion, single cell suspensions were stained for flow cytometry. Consistent with the live imaging data, a population of vaccine+ cells could be detected at 6 hours post vaccination within the CD45+ leukocyte compartment (Figure 2D). In the tumor, tumor-draining LN and the spleen, multiple different myeloid cell populations including monocytes, monocyte-derived DCs (moDCs), cDC1s and cDC2s had taken up the vaccine (Figure 2E and Figure S2B).
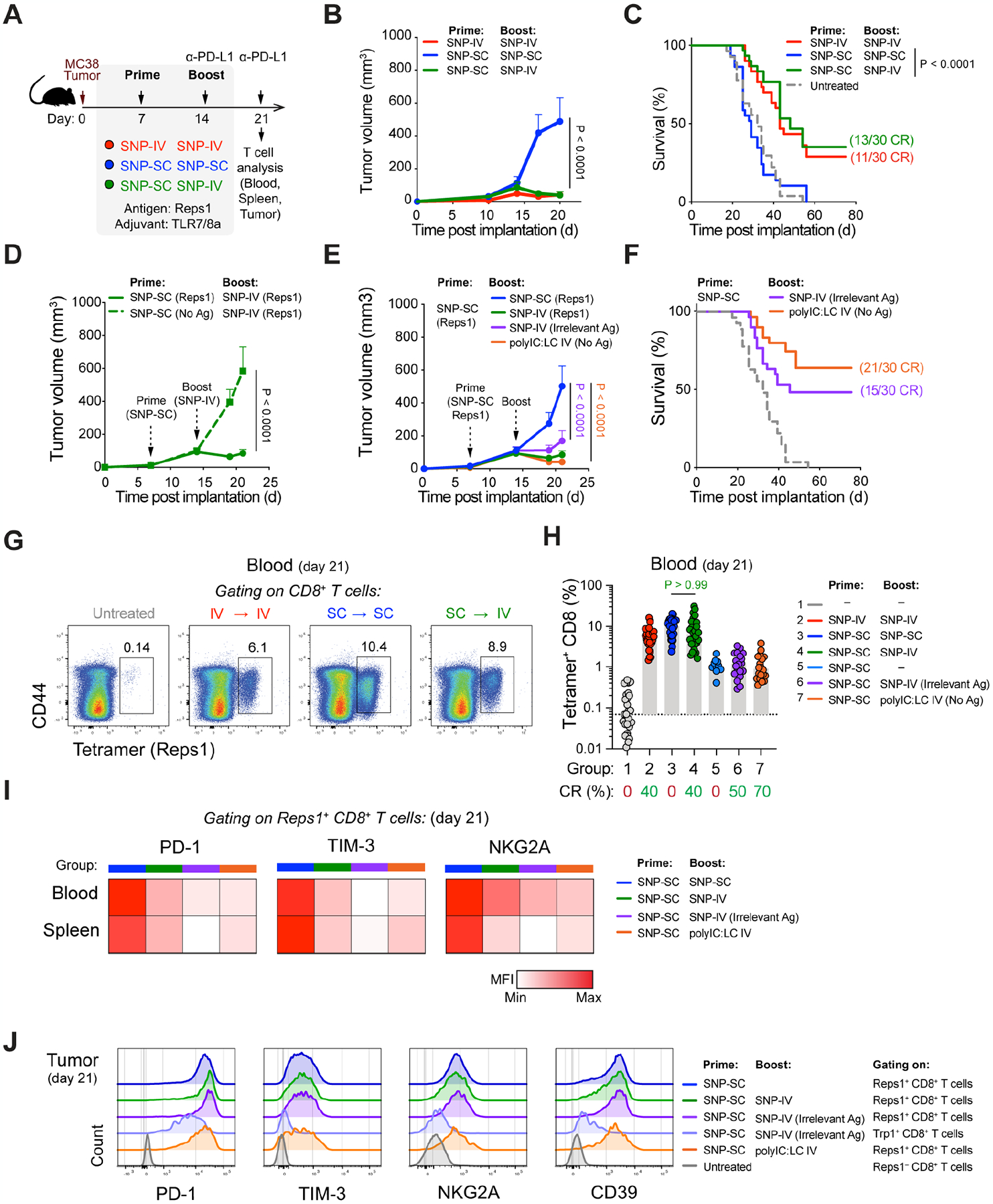
Figure 2. SNP-IV but not SNP-SC resulted in intratumoral vaccine distribution and DC maturation.
(A) In vivo imaging of mice following vaccination with fluorescently-labeled SNP-7/8a (n=4).
(B) Fluorescence radiant efficiency over time after fluorescently-labeled SNP-SC or SNP-IV gating on tumor as the region of interest (ROI) (n=4).
(C) Harvested tumor (top) and tumor-draining LNs (bottom) after SNP-IV or SNP-SC over time (n=2).
(D) Flow cytometry analysis of fluorescently-labeled SNP-7/8a and CD80 in tumor (top) and tumor-draining LN (n=4).
(E) Bar graphs summarize the frequency of myeloid cell populations of total vaccine+ cells in tumor (left) or tumor-draining LN (right) (n=4).
(F) Measurement of cytokines IFNα (left) and IL-12 (right) in sera of mice after SNP-SC or SNP-IV at 8 nmol and 32 nmol (n=3).
(G) Bar graphs summarize the numbers of cDC1 in spleen (left), tumor (middle) and tumor-draining LN (right) of mice that were untreated (white) or treated with SNP-IV prime and boost (red), SNP-SC prime and boost (blue) or SNP-SC prime followed by SNP-IV boost (green) (n=4–6).
(H) Flow cytometry analysis of cDC1s 24 h after SNP-SC or SNP-IV vaccination in the spleen (top) and tumor (bottom) (n=6).
(I) Heatmaps represent the median MFI of CD86 after SNP-IV prime and boost (red), SNP-SC prime and boost (blue) or SNP-SC prime followed by SNP-IV boost (n=6).
Given the systemic distribution of vaccine after SNP-IV but not SNP-SC, we assessed the levels of pro-inflammatory cytokines in the sera of mice 6 hours after vaccination (Figure 2F). High levels of systemic IFNα and IL-12 was measured by ELISAs after SNP-IV, especially at the higher dose of 32 nmol that is used in the therapeutic studies, compared to SNP-SC (Figure 2F). As IFNα is known to promote cross-presentation of antigens by cDC1s for CD8+ T cell priming (Le Bon et al., 2003), we quantified cDC1s in the spleen, tumor and tumor-draining LNs 24 hours after vaccination. Unsupervised uniform manifold approximation and projection (UMAP) analysis of myeloid cells revealed 6 clusters including eosinophils, neutrophils, macrophages, monocytes and cDCs (Figure S2C). Consistent with our earlier observation (Baharom et al., 2021), cDC1 numbers were reduced in the spleen and tumor after SNP-IV (Figure 2G). Compared to untreated animals, a three-fold increase in cDC1s was detected in the tumor-draining LNs after SNP-IV but not SNP-SC. To assess DC maturation, we used a combination of markers including the co-stimulatory molecules CD80 and CD86, the chemokine receptor CCR7 as well as MHCII (Figure S2D). We observed a higher proportion of mature cDC1s after SNP-IV compared to SNP-SC in the spleen and tumor (Figure 2H and Figures S2E) as well as the tumor-draining LNs (Figure 2I). Monocyte-derived cells can also be identified with varying expression levels of MHCII (Figure S2F). Interestingly, a larger proportion of moDC expressing high MHCII can be detected in the tumors after SNP-IV compared to SNP-SC or untreated mice (Figures S2G and Figures S2H). The adjuvant polyIC:LC administered by IV route was also able to induce DC maturation in the spleen and tumor-draining LN, comparable to SNP-IV (Figures S2I). Altogether, the data suggest that IV administration of SNP-7/8a results in systemic vaccine distribution including the spleen, tumor and tdLN where DCs can undergo maturation to promote licensing of neoAg-specific CD8+ T cells.
ScRNA-seq of tumors revealed that intratumoral Chil3+ monocytes were reduced after SNP-IV
To further characterize the immune responses occurring in the TME, we collected tumors 24 hours after boosting with SNP-SC or SNP-IV containing the tumor-specific antigen (Reps1) or SNP-IV containing an irrelevant antigen (Trp1). Myeloid cells were sorted by flow cytometry to obtain sufficient cell numbers for downstream analyses based on hematopoietic CD45+ cells expressing CD11b and/or CD11c (Figure 3A). Spleens were also collected to provide a baseline of major myeloid cell populations. Cells from individual mice were barcoded before scRNA-seq using the droplet-based system of 10x Genomics. Cells were clustered based on gene expression using an unsupervised inference analysis using Seurat (v4) pipeline. Focusing on monocyte, macrophages and DCs, nineteen “original clusters” were identified and visualized by UMAP dimension reduction (Figure S3A). For simplification, we organized the clusters into 9 “metaclusters” following their hierarchical ordering based on their Euclidian distance (Figure S3B) as well as formation of stable states as depicted on density plots (Figure S3C). Based on expression of canonical markers, the 9 metaclusters included 4 DC populations: cDC1 (Batf3, Clec9a, Cd24a), cDC2 (Mgl2, H2-Dmb2, Itgax), pDC (Siglech, Ly6d, Bst2), migratory/regulatory DC (mregDC, Ccr7, Fscn1, Relb), 3 macrophage subpopulations (Cd68, Apo3, Trem2) and 2 monocyte subpopulations (Lyz2, Csf1r, Ccr2) (Figure 3B and 3C). Although all 9 metaclusters were present in both spleens and tumors, the TME was primarily composed of monocytes and macrophages (Figure 3D and S3D). Based on cell cycle analysis, we identified Mac-1 as a proliferating subset of macrophages due to high expression of genes related to G2/M phase (Figure S3E). We then annotated Mac-2, Mac-3, Mono-1 and Mono-2 based on high expression of C1qb, Plin2, Ace and Chil3 (YM1) respectively (Figure 3E and S3F).
Figure 3. scRNA-seq of tumors revealed that intratumoral Chil3+ monocytes were significantly reduced after SNP-IV.
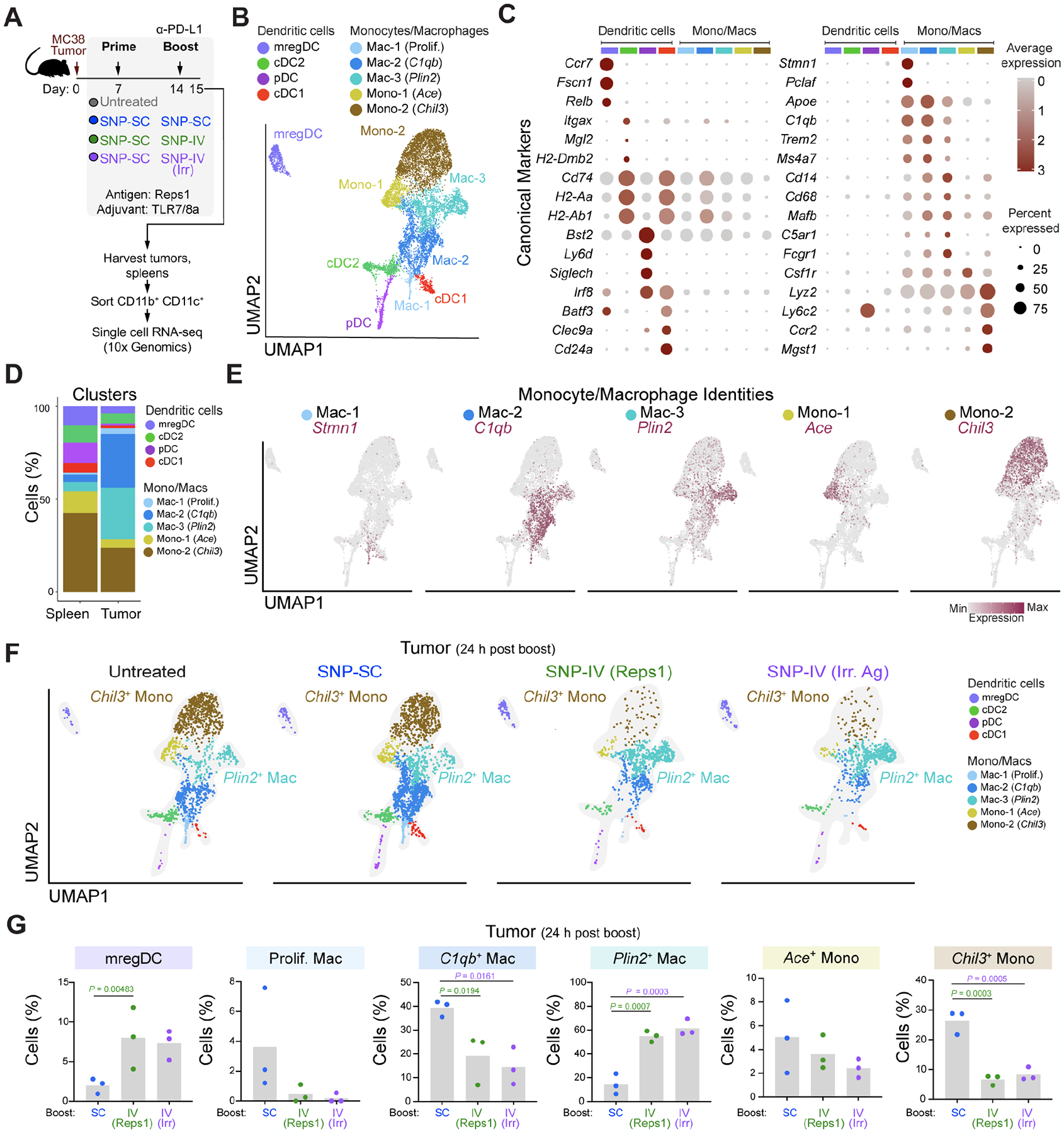
(A) Schematic of therapeutic study design. Mice (n=3) were implanted with MC38 and treated with SNP-7/8a (Reps1) on day 7 and day 14 together with CPI. Spleens and tumors were harvested on day 15. scRNA-seq was performed on flow sorted myeloid cells.
(B) UMAP of total monocytes, macrophages and DCs identified as 9 metaclusters in spleen and tumor on day 15.
(C) Dot plot of canonical markers identifying specific DC, monocyte and macrophage subsets.
(D) Bar graph shows proportion of individual metaclusters identified in spleen or tumor.
(E) Feature plots highlight individual genes C1qb, Plin2, Ace and Chil3 used to annotate monocyte/macrophage clusters.
(F) UMAPs of tumor MNP in untreated mice or mice treated with SNP-SC prime followed by SNP-SC boost (blue), SNP-IV (Reps1) boost (green) or SNP-IV (irrelevant antigen) boost (purple).
(G) Bar graphs summarize frequencies of individual metaclusters in SNP-SC (blue), SNP-IV (Reps1) (green), or SNP-IV (irrelevant antigen) boosted animals. Statistics were assessed by one-way ANOVA.
Across treatment groups, we identified a pattern corresponding to anti-tumor efficacy: untreated and SNP-SC boosted animals showed enrichment of Chil3+ monocytes in the tumors whereas groups boosted with SNP-IV (Reps1) and SNP-IV (irrelevant antigen) were enriched for Plin2+ macrophages (Figure 3F). Indeed, the frequency of intratumoral Plin2+ macrophages was significantly increased in SNP-IV boosted animals (Figure 3G and S3G). In contrast, the frequencies of Chil3+ monocytes and C1qb+ macrophages were significantly diminished in SNP-IV boosted animals, both in the tumors and spleens (Figure S3H). Consistent with our flow cytometry data, an enrichment of mregDCs (Maier et al., 2020) was observed in the spleen and tumors after SNP-IV (Figure 3G and S3I).
Chil3+ monocytes express gene signatures associated with immunoregulatory responses
Given the striking differences in the transcription profiles of monocytes and macrophages across groups, further investigation on the intratumoral monocyte/macrophage compartment was performed to understand their unique gene signatures and enriched pathways in each subpopulation (Figure 4A). More than 100 genes were upregulated following SNP-IV compared to only 40 genes upregulated after SNP-SC (Figure 4B). Additionally, about 300 genes were downregulated after SNP-IV compared to the 55 genes downregulated after SNP-SC. Each subpopulation can be distinguished by their unique top differentially expressed genes (DEGs) (Figure S4A and Table S1). Among the top DEGs, Plin2+ macrophages upregulated genes related to interferon signaling, including Irf7, Cxcl2, Ifitm1 and Isg15 whereas Chil3+ monocytes upregulated genes regulatory or suppressive activity (Figure 4C and 4D). For example, Hp encodes haptoglobin that can form complexes with HMGB1 to elicit anti-inflammatory enzymes and cytokines (Yang et al., 2016); Mgst1 encodes microsomal glutathione S-transferase, an enzyme that regulates prostaglandin E2 production, also involved in promoting anti-inflammatory cytokines such as IL-10 (Castoldi et al., 2020; MacKenzie et al., 2013); Wfdc17 encodes a WAP domain protein expressed in MDSCs (Veglia et al., 2021); Anxa2 encodes Annexin A2, a cytoskeletal protein widely implicated in promoting cancer progression (Zhang et al., 2012). Referring to the MetaScape database (Zhou et al., 2019) revealed an enrichment of pathways unique to each monocyte/macrophage population (Figure S4B). Consistent with a pro-inflammatory response, Plin2+ macrophages were enriched in pathways involved in regulation of adaptive immune responses, cell activation, TLR signaling and regulation of TNF production (Figure S4B). In contrast, Chil3+ monocytes were enriched in pathways involved in anti-inflammatory response such as wound healing.
Figure 4. Chil3+ monocytes expressed immunoregulatory gene signature while Plin2+ macrophages expressed interferon-related gene signature.
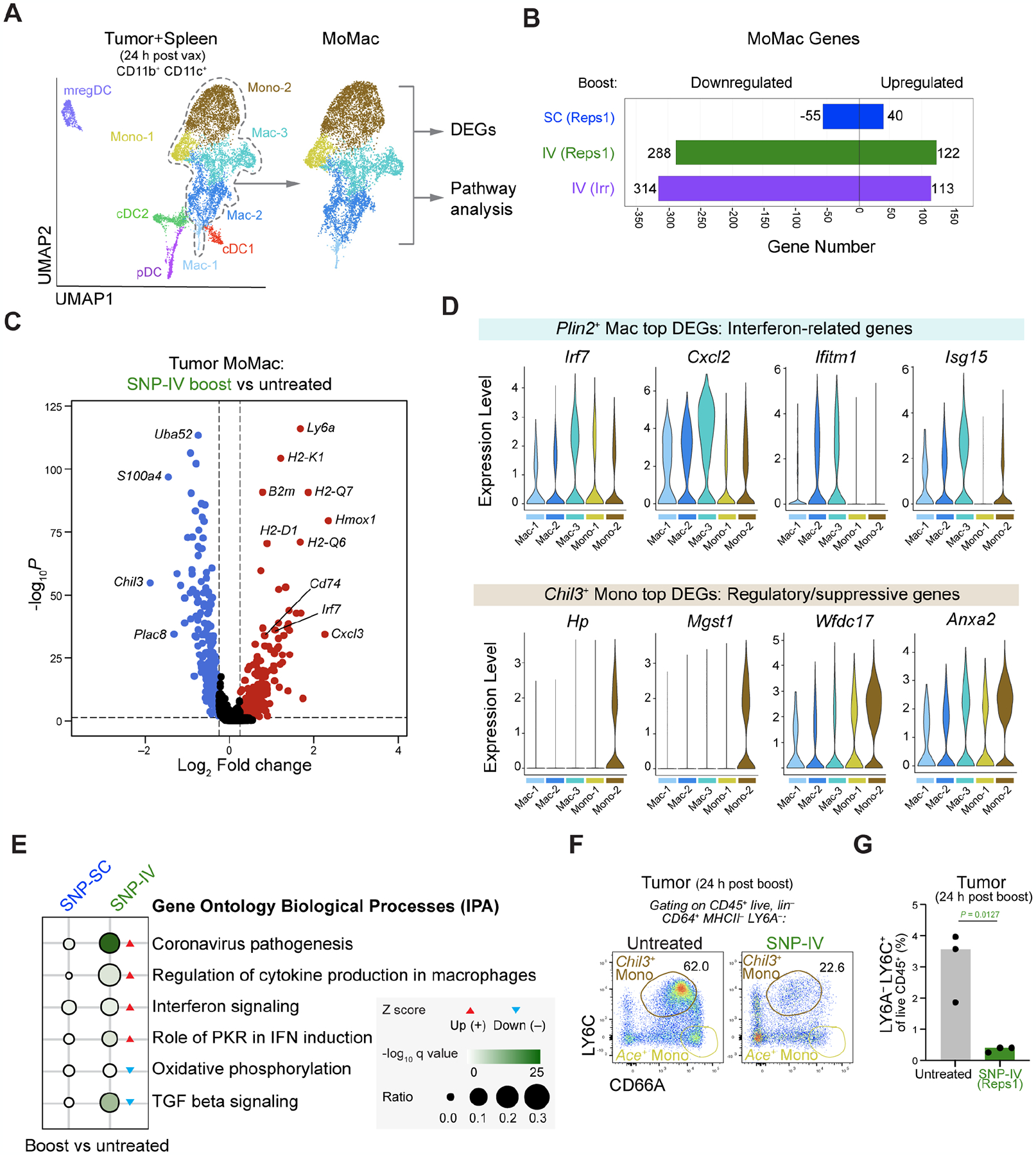
(A) Downstream analyses focused on monocyte/macrophage (MoMac) populations.
(B) Bar graph shows number of genes downregulated or upregulated by monocyte/macrophage populations following SNP-7/8a boost compared to untreated controls.
(C) Volcano plot comparing significantly (P value < 0.05) upregulated (fold change > 0.25, red) or downregulated (fold change <0.25, blue) genes within tumor macrophages in SNP-IV treated animals compared to untreated.
(D) Violin plots highlighting top DEGs related to Plin2+ macrophages (top) and Chil3+ monocytes (bottom).
(E) Dot plot highlighting top pathways upregulated (red arrow) or downregulated (blue arrow) in SNP-SC or SNP-IV treated groups compared to untreated.
(F) Flow cytometry plots show identification of Chil3+ monocytes in tumors 24 h after boosting with SNP-IV compared to untreated animals (concatenated, n=3).
(G) Bar graph summarizes the frequency of Chil3+ monocytes in tumors 24 h after boosting with SNP-IV compared to untreated animals (n=3).
To better interpret the changes in gene expression, we explored patterns of pathway changes using Ingenuity Pathway Analysis (IPA). Genes associated with coronavirus pathogenesis pathway was significantly and highly upregulated after SNP-IV compared to untreated groups in monocytes/macrophages (Figure 4E). This pathway includes genes involved in IFN-I signaling and inflammasome activation such as Irf7, Ccl5, Oas1 and Pycard, consistent with a viral gene signature. In contrast, genes involved in oxidative phosphorylation, a metabolic process favoring anti-inflammatory phenotypes, was significantly downregulated after SNP-IV. In summary, the observed differences in gene expression across groups further confirm a pattern of macrophages expressing interferon-stimulated genes following SNP-IV and monocytes expressing regulatory genes following SNP-SC.
Next, we determined whether cell surface markers could be used to identify Chil3+ monocytes by flow cytometry. We first used LY6A/Sca-1 and MHCII, markers that are highly expressed on macrophages but not monocytes (Figure S4C) as an exclusion marker. As Chil3 encodes a secreted protein, we stained with antibodies against CD66A (Ceacam1) and LY6C (Ly6c2) that are highly expressed on Ace+ monocytes and Chil3+ monocytes respectively (Figure S4C). Tumors harvested 24 hours post SNP-IV boost showed a three-fold reduction in the frequency of MHCII− LY6A− CD66dim LY6C+ monocytes compared to untreated animals (Figure 4F and 4G). This finding confirms the identification and reduction of a comparable subpopulation of monocytes in the tumor after SNP-IV that may contribute to the difference in anti-tumor efficacy following vaccination.
Interferon alpha required for mediating anti-tumor efficacy after SNP-IV treatment
Given that interferon-related genes were highly enriched after SNP-IV, we directly assessed the role of IFN-I in mediating anti-tumor control after SNP-IV. TLR7/8a and polyIC:LC are both potent inducers of IFN-I with pleiotropic effects across innate and adaptive immune responses (McNab et al., 2015; Sultan et al., 2020). IFN-I signaling plays an important role in promoting anti-tumor function (Duong et al., 2022; Fuertes et al., 2011). We had previously shown that IFN-I signaling is required for CD8+ T cell priming after SNP-7/8a vaccination using Ifnar−/− mice that lack a functional IFNαβ receptor (IFNAR). As boosting T cell responses is not required to mediate anti-tumor effect after SNP-IV given on day 14 (Figure 1F and 1H), we injected tumor-bearing mice with control or blocking antibodies against IFNAR on days 13 and 15 (Figure 5A) to assess its potential role on the innate immune response by SNP-IV. Sera collected 6 hours after SNP-IV confirmed that IFNα was depleted by three-fold after IFNAR blocking although a residual amount of IFNα was detectable (Figure 5B). Blocking IFNAR signaling had no additional impact on untreated animals but abrogated control of tumor growth following SNP-IV boost (Figure 5C) as well as decreased survival (Figure 5D) compared to SNP-IV treated animals without IFNAR blocking. As animals were boosted with SNP-IV containing an irrelevant antigen, blocking IFNAR had no impact on the frequency of neoAg-specific CD8+ T cells (Figures 5E). Other pro-inflammatory cytokines such as IL-6 and TNF as well as chemokines such as CXCL10 and CCL2 were also significantly reduced following IFNAR blocking (Figure 5F and S5A).
Figure 5. Interferon alpha required for mediating anti-tumor efficacy after SNP-IV treatment.
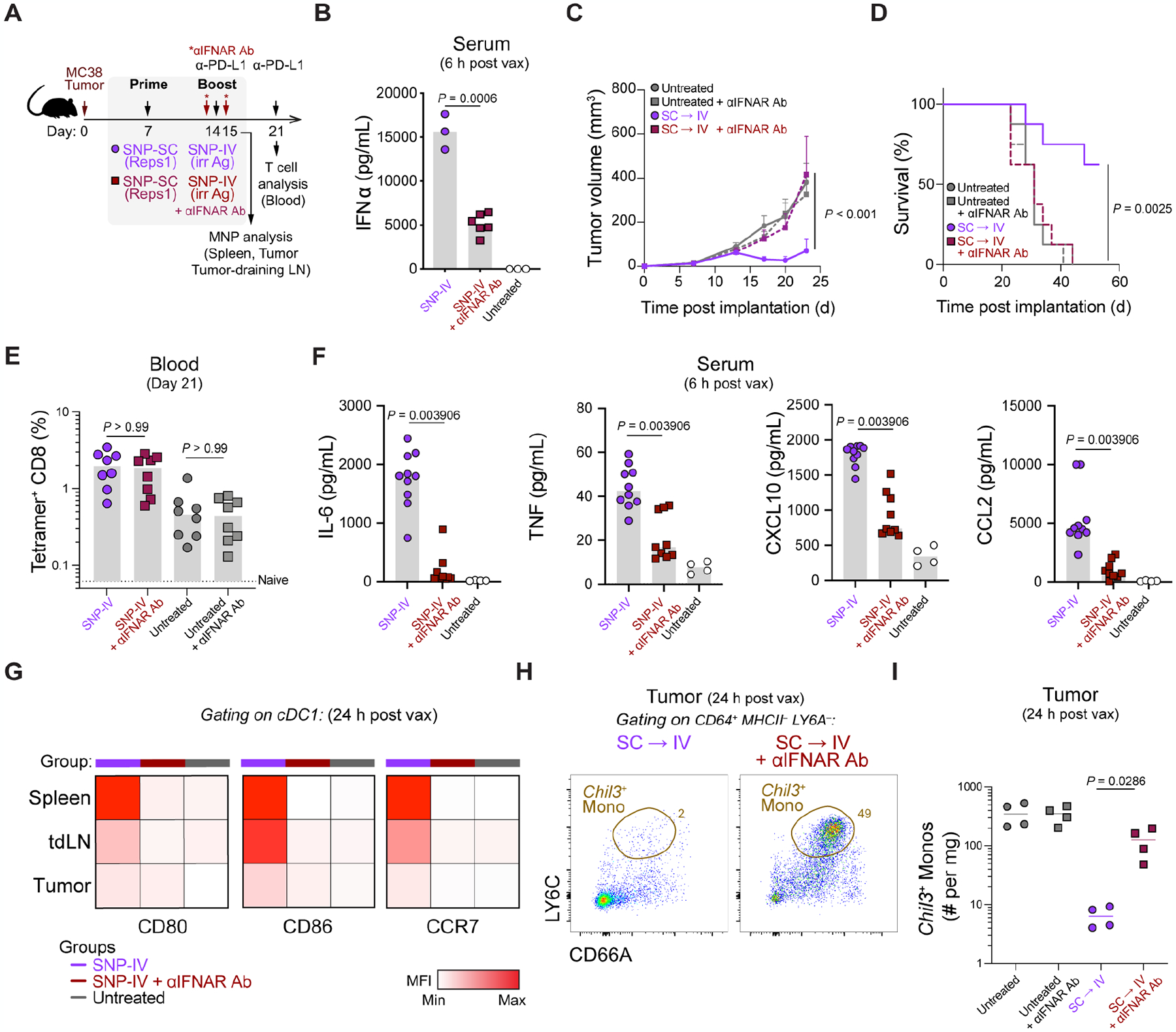
(A) Schematic of therapeutic study design. Mice were implanted with MC38 and treated with SNP-7/8a (Reps1) on day 7 and SNP-7/8a (Irrelevant antigen) on day 14 together with CPI. Blocking antibodies against IFNAR (MAR1–5A3) were given on day 13 (500 μg) and day 15 (200 μg).
(B) Measurement of IFNα in sera of mice after SNP-IV boost with isotype control or IFNAR blocking antibody (n=3–6). Statistics were assessed by Kruskal Wallis test.
(C) Tumor growth following treatment with SNP-SC prime followed by SNP-IV with isotype control (purple) or IFNAR blocking antibody (maroon) (n=8). Statistics were assessed by two-way ANOVA.
(D) Survival curve following treatment with SNP-SC prime followed by SNP-IV with isotype control (purple) or IFNAR blocking antibody (maroon) (n=8). Statistics were assessed by log-rank test.
(E) Bar graph summarizes the frequency of tetramer+ CD8 T cells in blood after treatment (n=8). Statistics were assessed by Kruskal Wallis test.
(F) Measurement of cytokines and chemokines in sera of mice after SNP-IV boost with isotype control or IFNAR blocking antibody (n=3–6). Statistics were assessed by Mann-Whitney test.
(G) Heatmaps represent the median MFI of CD80, CD86 and CCR7 on cDC1s in the spleen, tumor-draining LNs and tumors after treatment (n=5).
(H) Flow cytometry plots show identification of “Chil3+ monocytes” in tumors 24 h after boosting with SNP-IV with isotype control (purple) or IFNAR blocking antibody (maroon) (concatenated, n=4).
(I) Bar graph summarizes the frequency of “Chil3+ monocytes” in tumors of untreated animals (gray) or 24 h after boosting with SNP-IV with isotype control (purple) or IFNAR blocking antibody (maroon) (n=4). Statistics were assessed by Mann-Whitney test.
To investigate the mechanism of how IFN-I is promoting T cell-mediated tumor control, we first assessed whether blocking IFNAR would impair DC maturation. Blocking IFNAR resulted in lower expression of maturation markers such as CD80, CD86 and CCR7 on cDC1s and cDC2s in the spleen, tumor and tumor-draining LN a day after SNP-IV boost (Figures 5G and S5B). To directly test the involvement of DCs in licensing neoAg-specific CD8+ T cells, conditional knockouts of cDCs known as zDC-DTR bone marrow chimeric mice were generated (Meredith et al., 2012) (Figure S5C). Mice were injected with diphtheria toxin (DT) on day 13, day 15 and day 17 i.e. 1 day prior to SNP-IV boost as well as 1 day and 3 days after to sustain the depletion. Groups that received SNP-SC prime followed by SNP-IV boost had significantly smaller tumors compared to untreated controls, regardless of whether the mice received DT injections or not, suggesting that cDCs may be dispensable at the time of boost in promoting tumor regression (Figure S5D). Next, to test our hypothesis that monocytes and macrophages may be playing an important role in promoting anti-tumor immunity via IFN-I signaling induced by SNP-IV, tumors were harvested 24 h after vaccination with or without blocking IFNAR. Indeed, the depletion of Chil3+ monocytes following SNP-IV was reversed when blocking IFNAR in MC38 tumors (Figures 5H and 5I) or TC-1 tumors (Figures S5E–G). These data show that anti-tumor efficacy after SNP-IV at the time of boost was dependent on type I IFN leading to depletion of immunoregulatory Chil3+ monocytes in the tumor.
Chil3+ monocyte gene signature is enriched in human tumor-associated monocytes
The data presented thus far in two murine tumor models suggest that altering the TME through IFN-I-mediated modulation of monocytes may be important for T cells to control tumor growth. To determine whether the TME in human cancers may also contain similar monocyte populations, we created a list of human ortholog markers (referred to as “huChil3 geneset” based on the top 50 differentially expressed genes by Chil3+ monocytes (Table S2 and Figure S6A). Droplet-based cancer datasets from the human monocyte macrophage atlas (MoMac-VERSE) (Mulder et al., 2021) consisting of 61,353 cells were analyzed (Figure 6A). We found an enrichment of various huChil3 genes in the MoMac-VERSE with a bias towards monocytes rather than macrophages in line with our prior annotations (Figure 6B). The huChil3 geneset was found to be enriched in CD16− monocytes (cluster #8 of MoMac-VERSE) (Figure 6C) across various tumors including breast (Azizi et al., 2018; Mulder et al., 2021; Wu et al., 2021), colon (Lee et al., 2020; Zhang et al., 2020), head and neck (Cillo et al., 2020), liver (Sharma et al., 2020; Zheng et al., 2017), lung (Kim et al., 2020; Lambrechts et al., 2018; Zilionis et al., 2019), pancreas (Peng et al., 2019) and stomach cancer (Mulder et al., 2021) (Figure 6D). To confirm that this observation is not an artifact of large-scale dataset integration, we performed unbiased clustering of individual datasets of pancreatic cancer, lung cancer and liver cancer; huChil3 geneset was enriched in the CD16− monocyte cluster of each study (Figure S6B).
Figure 6. Chil3+ monocyte markers in human tumor-associated myeloid cells.
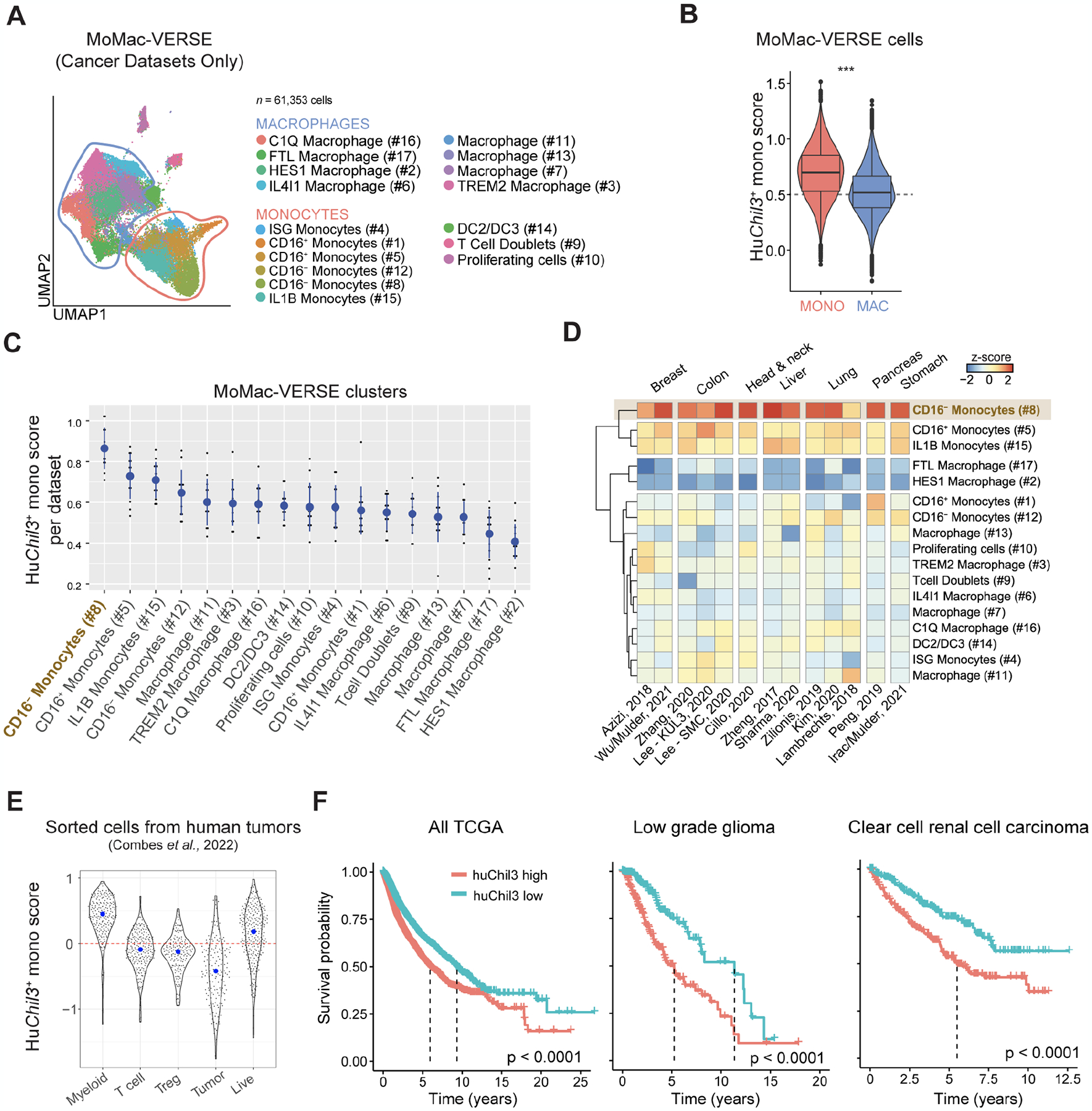
(A) UMAP representation of macrophages and monocytes in the MoMac-VERSE (Mulder et al. Immunity 2021) filtered to contain cancer studies sequenced with 10x technology.
(B) Violin plot comparing the scores for huChil3 between monocytes and macrophages from (B). Statistics were assessed by Wilcoxon Rank Sum test (***, P < 0.0001).
(C) Median score (y-axis) of huChil3 in each dataset of the MoMac-VERSE (dots) for each of the macrophage/monocyte subsets (x-axis). Mean ± SD across studies represented as blue circles and lines, respectively. Statistics were assessed by one-way Anova (P < 0.0001). Adjusted P value (Tukey’s HSD test) < 0.1 comparing #8 with any other cluster.
(D) Heatmap showing a hierarchical clustering of median scores for huChil3 in each dataset and cluster (z-scored per dataset).
(E) Scores (y-axis) for huChil3 in bulk RNA-seq samples (dots) from sorted populations (x-axis) of 364 individual tumors across 12 cancer types (Combes et al. Cell 2022). Blue dots indicate median in each group.
(F) Survival curves across all TCGA (left), low grade glioma (middle) and clear cell renal cell carcinoma (right). Patients (n = 8,911) were stratified as high- or low-expression cohorts based on median huChil3 geneset scores. Statistics were assessed by log-rank test.
Finally, we assessed how these observations may be related to disease outcome. Bulk RNA-seq samples analyzed from 364 individual tumors across 12 cancer types (Combes et al., 2022) showed that the huChil3 geneset is enriched in sorted myeloid cells but not T cells, regulatory T cells or tumor cells (Figure 6E). This enrichment suggests that this geneset allows us to infer h huChil3+ monocyte abundance via deconvolution in bulk RNA-seq data. We therefore turned to expression and associated survival data from The Cancer Genome Atlas (TCGA) and found that low huChil3 levels were associated with better survival in a pan-TCGA analysis. This was also true for general monocyte abundance as inferred via xCell (Aran et al., 2017). Notably, a subset of cancer indications including clear cell renal cell carcinoma (ccRCC) and low-grade glioma (LGG) deviated from this overall observation, where high huChil3 expression but not general monocyte expression was associated with worse outcome (Figure 6F and S6C). In summary, large-scale datasets from various human tumors support the identification of tissue monocytes that express immunosuppressive genes such as TMSB10 (Thymosin β10, a key regulator of tumorigenesis (Zhang et al., 2017) and ANXA2 (Annexin A2) shared by Chil3+ monocytes which we identify here as a negative regulator of anti-tumor immunity that may contribute to worse disease outcomes.
DISCUSSION
Efforts in developing therapeutic cancer vaccines have primarily focused on expanding the magnitude or quality of tumor-specific T cell responses in combination with checkpoint inhibitors that can enhance CD8+ T cell function (Saxena et al. 2021 Nat Rev Cancer). While generating tumor-specific T cells may be necessary for protection, tumor-induced immune suppression may be a major obstacle in achieving complete tumor regression in patients. Here, we provide direct evidence in a preclinical tumor model that systemic induction of IFN-I provided by an immunostimulant delivered intravenously can have a profound effect on remodeling the TME thus enabling improved anti-tumor efficacy of vaccine-generated tumor-specific CD8+ T cells.
Previously, we showed how modifying the route of administration of the SNP-7/8a vaccines can alter the quality of neoAg+ CD8+ T cells. SNP-IV generated more stem-like cells compared to SNP-SC that resulted in more terminally differentiated cells. A prime-boost regimen of SNP-IV given twice resulted in significant tumor regression but not SNP-SC given twice, despite high magnitude responses. One explanation for why SNP-IV was effective is that the stem-like cells responded to checkpoint inhibitors by replenishing the pool of effector cells. In this study, we provide a second explanation as to why SNP-IV may be beneficial: systemic innate immune activation. This paradigm that we refer to as “vax-innate” emphasizes two immunological events that can lead to effective tumor regression: (1) generation of tumor-specific CD8+ T cells and (2) systemic innate immune activation to reprogram the suppressive TME.
The requirement for systemic immunity in the context of cancer immunotherapies have been explored by others, especially in the context of sustaining T cell immunity (Allen et al., 2020; Spitzer et al., 2017). As a large secondary lymphoid organ, the spleen would be the primary site for positive DC-T cell interactions following SNP-IV; stem-like CD8+ T cells have been shown to form niches within the spleen (Im et al., 2016). Given that tumors are also vascularized, we show that systemic delivery of a nanoparticle peptide-based TLR7/8a vaccine led to intratumoral distribution. In translating this to humans, IV injection may be the most effective approach for treating metastatic tumors rather than direct intratumoral injections of innate stimulation to one or a few sites. An important caveat will be how IV delivery of an innate stimuli can be clinically tolerated.
Investigations into the TME and how the immune compartment influences tumor progression or regression have led to a focused effort on myeloid cells (Binnewies et al., 2018). Myeloid-targeting therapies have emerged as a promising approach for cancer immunotherapies, given the greater flexibility for tumor-antigen agnostic treatments. Various approaches include antibodies activating CD40 (a co-stimulatory receptor expressed by DCs) or blocking CSF1R (a survival receptor expressed by macrophages) or agonists to innate immune receptors such as stimulator of interferon genes (STING) or toll-like receptors (TLRs) could potentially reprogram suppressive myeloid cells to a pro-inflammatory state. These monotherapies may not be effective in patients that do not have sufficient levels of endogenous tumor-infiltrating T cells (Chaib et al., 2020).
Vaccines than induce CD8+ T cells usually require the induction of IFN-I for cross presentation. Here, we show that the vaccine-induced innate stimulation can have an added effect of altering the TME when delivered systemically. Other immune modulators that have shown superior anti-tumor efficacy when given systemically include polyIC:LC (an agonist that binds MDA5 and TLR3) (Sultan et al., 2020). Production of IFN-I leading to increased CXCL9 and CXCL10, chemokines that may be important in recruiting T cells into the tumor. Here we show that the efficacy of SNP-IV relies on the systemic production of IFN-I. However, we did not observe a significant difference in the infiltration of neoAg-specific CD8+ T cells measured in digested single cell suspensions. Ongoing efforts are focused on visualizing the TME using spatial profiling technologies to better understand the role of systemic inflammation in dictating the localization of CD8+ T cells within the tumor.
RNA-lipoplexes encoding tumor antigens have also been given systemically with the rationale that targeting the spleen offers the highest density of DCs to prime high magnitude T cells (Kranz et al., 2016). Although not directly addressed by Kranz et al., the RNA-lipoplexes themselves may act as an immunostimulant that could have modified the TME. The requirement for positive T cell-myeloid cell interactions in the TME following vaccination has been described by Thoreau et al. where the authors co-delivered E7 peptide (an HPV antigen) together with IFNα (Thoreau et al., 2015). Interestingly, administering systemic IFNα alone was ineffective, i.e. antigen-specific T cells still need to be present. This is consistent with our observations that while systemic inflammation can modify the TME, there is still a dependency on CD8+ T cells to promote tumor regression.
Although antibodies inhibiting CSF1R are often used to deplete TAMs, Sluis et al. showed that peptide-based vaccine-induced tumor regression was lost upon depletion of TAMs (van der Sluis et al., 2015). This points to the anti-tumoral role of macrophages that has not been as well described as their pro-tumoral contributions. Given the heterogeneity of monocytes and macrophages, a better characterization of subpopulations can provide a more defined approach in targeting specific cells rather than broadly depleting all monocytes and macrophages. Here, we have identified Plin2+ macrophages as upregulated in tumors after SNP-IV but not in untreated or SNP-SC treated mice. As PLIN2 is involved in lipid droplet formation, lipid metabolism may play an important role in supporting inflammatory conditions in the TME, similar to classically-activated pro-inflammatory macrophages (M1) (Rosas-Ballina et al., 2020). Lipid-associated macrophages (LAMs) have also been described as having a protective role in maintaining metabolic homeostasis, both in mice and humans (Jaitin et al., 2019). In contrast, Chil3+ monocytes and C1qb+ macrophages were downregulated in tumors after SNP-IV but highly expressed in untreated or SNP-SC treated mice. Chil3 is a gene also highly expressed in alternatively-activated anti-inflammatory macrophages (M2). Further expression of genes encoding inhibitory molecules such as ANXA2, MGST1 and WFDC17 suggest a mechanism of inhibiting T cells via soluble factors. Although we did not specifically deplete Chil3+ monocytes in this study, Shibuya et al. treated Chil3-DTR mice with DT and observed reduced tumor metastases in a systemic B16 tumor model (Shibuya et al., 2021). Their findings in a different tumor model further support the generalizability of our hypothesis that Chil3+ monocytes may play a more pro-tumoral role. C1qb+ macrophages, which were also found to be modulated by IFN-I, express high levels of Trem2 that encodes TREM2. The high expression of TREM2 on TAMs in both mice and humans (Katzenelenbogen et al., 2020) has made these cells a leading candidate for targeted therapies to overcome the immunosuppressive microenvironment in the tumor (Molgora et al., 2020). Thus, the identification of CD16− monocytes in tumors expressing the human orthologs of Chil3+ monocyte geneset offers another potential human candidate for targeted therapies.
While our TLR7/8a-based vaccine triggers a wide range of cytokines, it will be important to identify whether other stimulants of IFNα could also remodel the TME. We have achieved similar tumor regression when combining SNP-7/8a (prime) with viral vaccines (boost) that can also induce systemic innate activation including IFNα production (data not shown). IFNα alone has long been tested as a cancer therapy but dampened by the occurrence of toxic side effects (Sleijfer et al., 2005). The challenge of balancing systemic innate activation with tolerated toxicity will shape the future development of vaccines and immune interventions that can promote efficient T cell-mediated control of the tumor. Finally, the “vax-innate” paradigm can be extended to other approaches of introducing tumor-specific T cells such as adoptive cell therapy and chimeric antigen receptor T cell therapy and then coupling these T cell-based approaches with providing exogenous systemic innate immune activation.
Limitations of the study
There are several limitations to this study. First, the key observations were performed in murine tumor models that may not accurately reflect the tumor immune microenvironment in humans. Key features including tumor architecture, vascularization, growth rate and immune infiltrate may differ in patients when compared to a transplanted tumor in mice. Second, we have used two murine tumor models (MC38 and TC-1) that are both immune-inflamed tumors. As such, the findings of this study may not model immune-excluded or immune-desert tumors. Third, although we have performed experiments using IFNAR1 blocking antibodies to show that vaccine-induced IFN-I can modulate the Chil3+ monocytes in the tumor, we did not provide direct evidence that Chil3+ monocytes are responsible for suppressing the antigen-specific CD8+ T cells. Finally, the human TCGA data included in this study highlight low grade glioma and clear cell renal cell carcinoma, two cancer indications where the enrichment of HuChil3 monocyte genes is associated with worse survival compared to pan monocyte genes; this observation may not be generalizable to all tumors. Nevertheless, despite these limitations we believe these data provide a translatable approach for using innate stimulation by intravenous vaccination to potentially improve T cell function in tumors that express such inhibitory profiles.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Seder (rseder@mail.nih.gov).
Materials Availability
All unique reagents generated in this study including SNP-7/8a vaccines and cancer cell lines are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The scRNA-seq data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of publication. Accession number is listed in the key resources table.
All code used for analyses are publicly available and listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse B220 (PE-Cy7); clone RA3–6B2 | BD Biosciences | Cat# 552772 |
| Anti-mouse CCR7 (BV421); clone 4B12 | Biolegend | Cat# 120120 |
| Anti-mouse CD3 (BUV395); clone145–2C11 | BD Biosciences | Cat# 563565 |
| Anti-mouse CD3 (Alexa700); clone 17A2 | Biolegend | Cat# 100216 |
| Anti-mouse CD4 (BUV395); clone RM4–4 | BD Biosciences | Cat# 740209 |
| Anti-mouse CD8 (APCeFluor780); clone 53–6.7 | eBioscience | Cat# 47-0081-82 |
| Anti-mouse CD11b (AF700); clone MI/70 | BD Biosciences | Cat# 557960 |
| Anti-mouse CD11c (PE); clone HL3 | BD Biosciences | Cat# 553802 |
| Anti-mouse CD16/32; clone 2.4G2 | BD Biosciences | Cat# 553142 |
| Anti-mouse CD19 (BUV395); clone 1D3 | BD Biosciences | Cat# 563557 |
| Anti-mouse CD39 (PE-Dazzle594); clone Duha59 | Biolegend | Cat# 143812 |
| Anti-mouse CD40 (AF647); clone 3/23 | Biolegend | Cat# 123613 |
| Anti-mouse CD44 (BUV737); clone IM7 | BD Biosciences | Cat# 564392 |
| Anti-mouse CD45 (BUV661); clone 30-F11 | BD Biosciences | Cat# 565079 |
| Anti-mouse CD64 (BV785); clone X54–5/7.1 | BD Biosciences | Cat# 741024 |
| Anti-mouse CD66a (BV605); clone CC1 | BD Biosciences | Cat# 750882 |
| Anti-mouse CD80 (PE-Dazzle594); clone 16–10A1 | BD Biosciences | Cat# 562504 |
| Anti-mouse CD86 (BV711); clone GL1 | BD Biosciences | Cat# 740688 |
| Anti-mouse CD172α (PerCP-eF710); clone P84 | Life Technologies | Cat# 46-1721-82 |
| Anti-mouse Eomes (PerCP-eF710); clone Dan11mag | Invitrogen | Cat# 46-4875-82 |
| Anti-mouse F4/80 (PE-Cy5); clone BM8 | eBioscience | Cat# 15-4801-82 |
| Anti-mouse IA/IE (AF488); clone M5/114.15.2 | Biolegend | Cat# 107616 |
| Anti-mouse LY6A/E (PE-CF594); clone D7 | BD Biosciences | Cat# 562730 |
| Anti-mouse Ly6C (APC-eF780); clone AL-21 | BD Biosciences | Cat# 560596 |
| Anti-mouse Ly6G (BUV563); clone 1A8 | BD Biosciences | Cat# 565707 |
| Anti-mouse NK1.1 (BUV395); clone PK136 | BD Biosciences | Cat# 564144 |
| Anti-mouse NKG2A (PE-Cy7); clone 16A11 | Biolegend | Cat# 142810 |
| Anti-mouse PD-1 (BV421); clone 29F.A12 | Biolegend | Cat# 135218 |
| Anti-mouse SiglecH (BUV805); clone 440C | BD Biosciences | Cat# 748291 |
| Anti-mouse Tim-3 (BV605); clone RMT3–23 | Biolegend | Cat# 119721 |
| Anti-mouse TNFα (BV650); clone MP6-XT22 | Biolegend | Cat# 506333 |
| Anti-mouse XCR1 (BV650); clone ZET | Biolegend | Cat# 148220 |
| Anti-mouse IFNα/β receptor-1; clone MAR1–5A3 | BioXCell | Cat# BP0241 |
| Anti-mouse PD-L1; clone 10F.9G2 | BioXCell | Cat# BP0101 |
| TotalSeq-C0301 anti-mouse Hashtag 1; clones M1/42, 30-F11 | Biolegend | Cat# 155861 |
| TotalSeq-C0302 anti-mouse Hashtag 2; clones M1/42, 30-F11 | Biolegend | Cat# 155863 |
| TotalSeq-C0303 anti-mouse Hashtag 3; clones M1/42, 30-F11 | Biolegend | Cat# 155865 |
| TotalSeq-C0304 anti-mouse Hashtag 4; clones M1/42, 30-F11 | Biolegend | Cat# 155867 |
| TotalSeq-C0305 anti-mouse Hashtag 5; clones M1/42, 30-F11 | Biolegend | Cat# 155869 |
| TotalSeq-C0306 anti-mouse Hashtag 6; clones M1/42, 30-F11 | Biolegend | Cat# 155871 |
| TotalSeq-C0307 anti-mouse Hashtag 7; clones M1/42, 30-F11 | Biolegend | Cat# 155873 |
| TotalSeq-C0308 anti-mouse Hashtag 8; clones M1/42, 30-F11 | Biolegend | Cat# 155875 |
| TotalSeq-C0309 anti-mouse Hashtag 9; clones M1/42, 30-F11 | Biolegend | Cat# 155877 |
| TotalSeq-C0310 anti-mouse Hashtag 10; clones M1/42, 30-F11 | Biolegend | Cat# 155879 |
| Chemicals, peptides, and recombinant proteins | ||
| Dasatinib | Selleckchem | S1021 |
| Isoflurane, USP | Baxter Healthcare Corp. | NDC 10019-360-60 |
| Paraformaldehyde (PFA, 16%) | Electron Microscopy Sciences | 15710 |
| Tween-20 | Sigma | P-7949 |
| Heparin | Fresenius Kabi | NDC 63323-540-05 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | 276855–100mL |
| Reps1 SNP vaccine | Vaccitech North America | N/A |
| Reps1 SNP vaccine labelled with AF647 | Vaccitech North America | N/A |
| Trp1 SNP vaccine | Vaccitech North America | N/A |
| ABTS (2,2’-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) | Sigma | A3219 |
| Murine IL-12 ABTS ELISA Kit | Peprotech | 900-K97 |
| Reps1 Tetramer (H-2Db, AQLANDVVL) | Gift from J. Finnigan | N/A |
| Trp1 Tetramer (H-2Db, TAPDNLGYM) | Gift from J. Finnigan | N/A |
| Critical commercial assays | ||
| Mouse IFN Alpha All Subtype ELISA Kit, High Sensitivity | PBL Assay Science | Cat# 42115–1 |
| eBioscience™ Foxp3 / Transcription Factor Staining Set | Invitrogen | Cat# 00-5523-00 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34962 |
| ArC™ Amine Reactive Compensation Bead Kit | ThermoFisher Scientific | Cat# A10346 |
| BD Horizon Brilliant Stain Buffer | BD Biosciences | 566385 |
| Streptavidin PE (SaPE) | BD Biosciences | S866 |
| Trypsin-EDTA (0.25%) | Gibco, Life Tech | 25200–056 |
| ViaStain AOPI Staining Solution | Nexcelom Bioscience | CS2–0106 |
| Milliplex MAP mouse cytokine/chemokine Magnetic Kit | Millipore Sigma | MCYTMAG-70K-PX32 |
| Phosphate buffered saline (PBS) | Gibco | 10010–023 |
| RPMI 1640 | Cytiva, HyClone Labs | SH30027.02 |
| FBS | Gibco | 10438–026 |
| Penicillin/Streptomycin/Glutamine (100X) | Gibco, Life Tech | 10378–016 |
| Non-essential Amino Acids (100X) | Cytiva, HyClone Labs | SH30238.01 |
| Sodium Pyruvate (100mM) | Cytiva, HyClone Labs | SH30239.01 |
| Collagenase D | Roche | 11088882001 |
| DNase I | Roche | 04536282001 |
| ACK (Ammonium-Chloride-Potassium) lysing buffer | Quality Biological | 118-156-101 |
| Chromium Single Cell 50 Reagent Kit | 10X Genomics | |
| Dynabead MyOne Silane | Thermo Fisher Scientific | Cat#37002D |
| SPRIselect for Size Selection | Beckman Coulter | Cat#B23319 |
| Experimental models: Cell lines | ||
| MC38 Murine colorectal cancer cell line | Genentech, L. Delamarre | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: wild-type C57BL/6J mice | Jackson Labs | 000664 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | Jackson Labs | 002014 |
| Mouse: B6(Cg)-Zbtb46tm1(HBEGF)Mnz/J | Jackson Labs | 019506 |
| Software and algorithms | ||
| Flowjo v10 | Tree Star | N/A |
| GraphPad Prism v8 | GraphPad software | N/A |
| xPONENT software | Luminex | N/A |
| Living Image v4.5 | PerkinElmer | N/A |
| R, v4.1.2 | R Project | https://www.r-project.org/ |
| Cellranger v6.0.0 | 10X Genomics | https://support.10xgenomics.com |
| Seurat v4.1.0 | Seurat | https://cran.r-project.org/web/packages/Seurat/index.html |
| Monocle 3 v1.0.0 | Monocle | https://github.com/cole-trapnell-lab/monocle3/releases/tag/1.0.0 |
| Slingshot v2.2.0 | Slingshot | https://bioconductor.org/packages/release/bioc/html/slingshot.html |
| DoubletFinder v 2.0.3 | DoubletFinder | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| pheatmap v1.0.12 | Pheatmap | https://cran.r-project.org/web/packages/pheatmap/index.html |
| dittoSeq v1.6.0 | DittoSeq | https://bioconductor.org/packages/release/bioc/html/dittoSeq.html |
| biomaRt v2.50.3 | BiomaRt | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| EnhancedVolcano v1.12.0 | Enhanced Volcano | https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html |
| MetaScape v20220101 | MetaScape | https://metascape.org/gp/index.html#/main/ |
| QIAGEN IPA vDec. 2021 | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Deposited data | ||
| scRNA-seq | Gene Expression Omnibus | GSE214741 |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Wild-type (WT) C57BL/6J, B6.SJL-Ptprca Pepcb/BoyJ, and B6(Cg)-Zbtb46tm1(HBEGF)Mnz/J (zDC-DTR) mice were purchased from The Jackson Laboratory and housed in specific-pathogen-free conditions. Upon arrival, mice were given 1 week to adjust to the new animal facility prior to being used. Mice used in studies were between 8–10 weeks old. All mice used were females. All animal experiments were performed at the Vaccine Research Center at the National Institutes of Health (NIH) with the approval of the Institutional Animal Care and Use Committee at the NIH. Experiments complied with the ethical guidelines set by the Institutional Animal Care and Use Committee and animals were humanely killed at defined end points
Tumor cell lines
The MC38 cell line was a kind gift from L. Delamarre (Genentech). The MC38 cells were grown in media comprised of DMEM + 10% FBS + 1% penicillin/streptomycin/glutamine + 1% non-essential amino acids + 1 mM sodium pyruvate. Stocks of MC38 were generated upon receipt of the cells and used for tumor experiments. Cells were tested regularly for Mycoplasma contamination; none tested positive throughout the studies.
METHOD DETAILS
Vaccines
SNP vaccines were produced as described previously (Lynn et al., 2020). Peptide antigens modified to form nanoparticles as part of a SNP vaccine were produced by GenScript. These peptides were linked to hydrophobic blocks containing an imidazoquinoline-based TLR-7/8 agonist (Vaccitech North America, USA) using a click chemistry reaction. For the pharmacokinetics studies, SNP vaccines were produced by linking Alexa Fluor 647 to hydrophobic blocks.
Immunizations and treatments
SNP vaccines were prepared in sterile PBS (Gibco) and administered subcutaneously to each footpad (50 μl per site) or intravenously via tail vein injection (100 μl) at a dose of 8 nmol and 32 nmol respectively. 50 μg of polyIC:LC (Hiltonol) was administered intravenously via tail vein injection (100 μL). Animals were treated with 200 μg per mouse of anti-PD-L1 (10F.9G2; Bio × Cell) in 100 μl of PBS via intraperitoneal injection. For IFNα receptor (IFNAR) blockade, mice were treated 500 μg of anti-IFNAR1 antibody (MAR1–5A3; Bio × Cell) in 100 μl of PBS via intraperitoneal injection.
Tumor Implantation
For each tumor implantation, a frozen cell aliquot was thawed and cultured in MC38 media at 37 °C and 5% CO2, passaged once and collected using trypsin EDTA (Gibco). Then, 105 cells in sterile PBS per mouse were implanted subcutaneously on the right flank. Tumors were measured twice a week using digital calipers. Tumor volume was estimated using the formula: (tumor volume = short × short × long/2). Animals were killed when tumors surpassed 1,000 mm3.
Generation of zDC-DTR bone marrow chimeras
Eight-week-old recipient CD45.1 mice received 13 Gy of γ-irradiation (2 doses of 6.5 Gy each) before IV reconstitution with bone marrow from zDC-DTR mice. Eight weeks after reconstitution, successful chimerism was assessed by flow cytometry. Mice were used in studies eight weeks after reconstitution. To deplete DCs, mice were treated with 20 ng/g of diptheria toxin (DT) either intraperitoneally or intratumorally on day 13 (1 day before boosting) followed by 4 ng/g of DT on day 16.
Blood and tissue processing
Heparin-treated blood was collected and lysed with ACK lysis buffer (Quality Biological) to isolate PBMCs. Lungs, liver, kidneys and tumors were collected in digestion media containing Roswell Park Memorial Institute (RPMI) 1640, 10% FCS, 50 U ml–1 DNase I (Sigma-Aldrich) and 0.2 mg/ml collagenase D (Sigma-Aldrich). Tissues were mechanically disrupted using the respective programs on the gentleMACS dissociator (Miltenyi Biotec) and incubated at 37 °C for 30–45 min in a shaking incubator. Spleens were mechanically disrupted and lysed with ACK lysis buffer. Lymph nodes were mechanically disrupted in BioMasher tubes (Nippi). All single-cell suspensions were filtered through a 70-μm cell strainer and resuspended in PBS for flow cytometry staining.
Flow cytometry
For T cell tetramer analysis, cells were assessed for viability with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) in PBS containing 50 nM dasatinib (STEMCELL Technologies) for 30 min at room temperature. Samples were then washed and blocked with anti-CD16/CD32 (BD Biosciences). Cells were then stained with fluorescently conjugated tetramer in cell staining buffer (PBS and 2% FCS) containing 50nM dasatinib to enhance staining. Cells were simultaneously stained with the following surface antibodies to: CD8 (clone 53–6.7), PD-1 (clone 29F.A12), Tim-3 (clone RMT3–23), CD44 (clone IM7), CD39 (clone Duha59), and NKG2A (clone 16A11) purchased from BioLegend and CD4 (clone RM4–4) purchased from BD Biosciences. After a 1-h incubation at 4 °C, cells were washed twice in cell staining buffer, fixed and permeabilized using the FoxP3 transcription factor staining buffer set (eBioscience). Cells were stained overnight at 4 °C with CD3 (clone 17A2) from BD Biosciences. Stain was washed off the following morning and samples resuspended in eBioscience FoxP3 transcription factor permeabilization wash buffer after which samples were acquired on an LSRFortessa X50 (BD Biosciences) using the FACSDiva software v8.0.1 (BD Biosciences).
For the mononuclear phagocyte uptake analysis, cells were assessed for viability with the LIVE/DEAD Fixable Blue Dead Cell Stain Kit for 10 min at room temperature. After FcR blocking, cells were stained for 20 minutes at room temperature with the following surface antibodies: NK1.1 (clone PK136), CD19 (clone 1D3), CD3 (clone 145–2C11), Ly6G (clone 1A8), CD45 (clone 30-F11), Siglec-H (clone 440c), CD86 (clone GL1), CD11c (clone HL3), CD80 (clone 16–10A1), B220 (clone RA3–6B2), CD64 (clone X54–5/7.1), CD11b (clone MI/70) and Ly6C (clone AL-21) purchased from BD Biosciences, CCR7 (clone 4B12), MHC class II (I-A/I-E, clone M5/114.15.2), CD169 (clone 3D6.112) and XCR1 (clone ZET) purchased from BioLegend, and CD172a (clone P84) from Thermo Fisher Scientific. The stain was washed off and cells were then fixed in 0.5% PFA in PBS until they were acquired on an LSRFortessa X50 (BD Biosciences) using the FACSDiva software v8.0.1 (BD Biosciences).
In vivo imaging
Whole-body imaging of mice after immunization with Alexa Fluor 647-labeled vaccines was performed using the IVIS Spectrum In Vivo Imaging System (Perkin Elmer) and analyzed using the manufacturer’s software (Living Image 4.5, PerkinElmer).
ELISA and Luminex
Serum from whole blood was collected at specified time points after vaccination. Commercially available ELISA kits were used to measure IL-12 subunit p40 (PeproTech) and all subtypes of IFNα (PBL Assay Science) according to the manufacturer’s protocols. A commercially available Luminex kit (Millipore Sigma) was used according to the manufacturer’s protocols to assess multiple analytes from serum samples.
Cell sorting for scRNA-seq
Spleens and tumors from mice that had been boosted one day prior were collected and processed into single cell suspensions by mechanical dissociation. Samples were stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit for 10 min at room temperature. Then samples were washed with FACS buffer (2% FBS in PBS) and stained with Fc block (Anti-mouse CD16/32, BD Biosciences) prior to addition of a surface stain. The surface stain antibody master mix contained: CD3 BUV395, CD19 BUV395, CD45 BUV661, CD11c PE, and CD11b AF700. Each sample was also stained with a unique hashtag antibody. Samples were incubated in surface stain for 20 minutes at room temperature after which all surface stain antibodies were washed off. Samples were resuspended in FACS buffer and sorted by fluorescence activated cell sorting to isolate the live CD45+ CD11b+ CD11c+ cells. Sorted samples were pooled together by tissue prior to loading in duplicate into a Chromium single cell sorting system (10x Genomics). Expression and hashtag library construction was performed following the Chromium Single Cell VDJ Library protocol with a loading target of 1 × 104 per lane. At the conclusion, there were 4 expression and hash tag libraries from spleen samples and another 4 from tumor samples. The libraries were sequenced on a NovaSeq 6000 S2 chip.
Pre-processing of scRNA-seq data
The raw scRNA-seq data (FASTQ files, 10X Genomics) were aligned to mm10 mouse reference genome using the Cell Ranger Single Cell software v6.0.0 (10x Genomics). The output count matrices were imported into Seurat R package v4.1.0 (Hao et al., 2021), and Seurat objects were created using CreateSeuratObject function (min.cells= 3). The hashtagged data were demultiplexed using the HTODemux function, and singlet cells were predicted based on the Hashtag oligo classification for downstream analysis. Following dimension reduction and unsupervised clustering, further doublet cells were predicted and removed using DoubletFinder R package v2.0.3 (McGinnis et al., 2019), by Artificial Nearest Neighbours and assuming 12% doublet formation rate. Finally, all Seurat objects were merged as a single Seurat object and used for integration.
Integration of scRNA-seq datasets by Seurat
The scRNA-seq expression count data were log-normalized using NormalizeData function, and the variable genes were identified using FindVariableFeatures (selection.method = “vst”, nfeatures = 2000) of Seurat package. Next, the cross-dataset anchors were identified using FindIntegrationAnchors function (dims = 1:20, k.anchor= 5, k.filter= 30, reduction = “cca”) of Seurat. These anchors were then used for the integration step in which a cell-to-cell distance (batch-corrected) matrix was imputed using IntegrateData function (dims= 1:20, k.weight = 100) of Seurat.
Dimension reduction and clustering
Two rounds of dimension reduction and clustering was performed on the imputed cell-to-cell distance matrix using specific functions embedded in Seurat R package. In the first round, the linear-dimensionality reduction (PCA) was performed on the whole scaled distance matrix using RunPCA function (npcs = 50), and the data passed through FindNeighbors function and a non-linear dimensionality reduction by means of Uniform Manifold Approximation and Projection (UMAP) using RunUMAP function and umap-learn method (dims = 1:12). Next, a graph-based unsupervised clustering of the single cells was performed using FindClusters function (resolution = 0.8). The generated clusters were annotated based on their expression of canonical markers, and the clusters aligning with Mono/Mac/DC identity were subjected to the second round of the PCA and UMAP (dims = 1:10) and unsupervised clustering (resolution = 1.5). The second-round generated clusters, termed as original clusters were combined into meta-clusters following their hierarchical ordering based on their Euclidian distance calculated by mean expression of their top 50 DEGs and using pheatmap R package v1.0.12.
Comparing clusters distribution
The dittoSeq R package v1.6.0 (Bunis et al., 2020) was used to calculate and plot the distribution of each Mon/Mac/DC meta-cluster within each experimental group (condition). A meta-cluster distribution was defined as:
The statistical cross-condition comparison of the meta-cluster distribution (carried out in triplicates) was done in GraphPad Prism software v8.0 using parametric one-way ANOVA followed by Dunnett’s multiple comparison test comparing SC-IV (Reps1) and SC-IV (irr) groups versus SC-SC (Reps1). A P value < 0.05 was considered as statistically significant.
Predicting cell cycle phase
Human cell cycle gene lists (embedded in Seurat package) were converted to their mouse equivalents using biomaRt R package v2.50.3 (Durinck et al., 2009), and then were implemented by Seurat pipeline for calculating cell-cycle scores and predicting cell-cycle phase of each single cell (G1, S and G2M).
Differential expression gene (DEG) analysis
DEG analysis was performed on the merged log-normalized count matrix using Wilcoxon Rank Sum test embedded in FindMarkers function of Seurat (min.pct = 0.1, logfc.threshold = 0.25). A Bonferroni-corrected P value < 0.05 was used to describe significant DEGs among Mon/Mac meta-clusters. Cross-condition up-regulated and down-regulated genes were characterized following intra-condition combining of whole Mono/Mac meta-clusters and preforming DEG testing in SC-SC (Reps1), SC-IV (Reps1), SC-IV (irr) groups versus untreated group (min.pct = 0.1, logfc.threshold = 0.25); P value < 0.05 was considered as statistically significant.
Biological pathways analysis
To infer the biological pathways enriched in each Mono/Mac meta-cluster, their top-50 DEGs was imported into the web-based MetaScape portal (Zhou et al., 2019). In addition, to explore the cross-condition changes of the biological pathways, ingenuity pathways analysis (IPA) was performed on all statistically significant up-regulated and down-regulated genes [SC-SC (Reps1), SC-IV (Reps1), SC-IV (irr) groups versus untreated group] using QIAGEN IPA software.
Human bulk RNA-seq data analysis
Human orthologues (“huChil3”) of mouse Chil3+ monocytes were identified using the nicheNet R package (Browaeys et al., 2020). Normalized count data of sorted bulk RNA-seq populations from 12 tumor types was obtained from GEO under accession GSE184398 and log2 transformed. Subsequently, the average expression of huChil3 genes was calculated in each sample after scaling (z-score transformation) the expression of each gene across samples.
Human scRNA-seq data analysis
Human single-cell RNA-seq data: The MoMac-VERSE dataset (“2021_MoMac_VERSE.rds”) was obtained online (https://gustaveroussy.github.io/FG-Lab/) and subsequently analyzed with the Seurat package in R (Hao et al., 2021). First, we filtered the atlas to contain only datasets including cancer patients that were sequenced with 10x/Droplet sequencing technology. Next, we restricted the huChil3 geneset to only those genes robustly expressed in the dataset (average expression across clusters >5) and calculated an enrichment score for the resulting geneset in each cell (“AddModuleScore”). To compare the score across datasets and clusters, we calculated the median huChil3 geneset score in each cluster for each dataset and compared these median scores per dataset across clusters.
For individual dataset analyses the MoMac-VERSE was further filtered to only contain cells from a particular study and Variable gene selection, scaling, PCA and UMAP calculation were carried out with standard parameters (custom: Number of PCs used for nearest neighbor and UMAP calculation: 30 PCs, resolution for clustering: 0.3).
For survival analyses using TCGA datasets, Batch corrected normalized TCGA Pan-Cancer mRNA data was obtained from UCSC Xenabrowser (https://xenabrowser.net/) (n = 11,060). Samples containing NA expression values were removed. We additionally filtered the data to only contain samples from primary solid tumors (sample code 01; n = 9,702). Survival data was obtained from Table S1 in Thorsson et al. (Thorsson et al., 2018) and linked to the Pan-Cancer dataset via the unique TCGA Participant Barcode. Inferred general monocyte levels were obtained from Table S6 of Aran et al. (Aran et al., 2017) and similarly linked to the expression data via the unique TCGA Participant Barcode. Survival information and estimated monocyte levels were available for n = 8,963 samples analyzed. HuChil3 geneset scores were calculated in each of these TCGA sample as the average of the human orthologs found expressed in the MoMac-VERSE after gene-wise scaling (z-score) across samples. High and low expression groups were defined using a median split of scores and Kaplan Meier curves were calculated in R.
QUANTIFACTION AND STATISTICAL ANALYSIS
Statistical analysis of biological data
All results are presented as the median with s.d. Statistics were assessed using a Kruskal–Wallis test with Dunn’s correction for multiple comparisons [immunogenicity], two-way analysis of variance (ANOVA) with Bonferroni correction [tumor growth curves], log-rank test [survival curves], Mann–Whitney U-test [cytokines] or one-way ANOVA with Dunnett’s test for multiple comparisons [scRNA-seq [populations] (Prism; GraphPad Software v8.4.2).
Supplementary Material
Figure S1. NeoAg+ CD8+ T cells generated by SNP-SC controlled tumor growth when followed by IV adjuvant delivery, related to Figure 1 (A)TC-1 tumor growth following treatment with SNP-IV prime and boost (red), SNP-SC prime and boost (blue), SNP-SC prime and SNP-IV boost (green) or SNP-IV boost with irrelevant antigen (purple) (n=8). Statistics were assessed by two-way ANOVA.
(B)Bar graph summarizes the frequency of tetramer+ CD8 T cells in blood after treatment with SNP-7/8a containing E6 antigen (n=5–8). Statistics were assessed by Kruskal Wallis test.
(C)Tumor growth following treatment with SNP-IV prime and boost with Reps1 (solid line) or irrelevant antigen (dashed line) (n=10). Statistics were assessed by two-way Anova.
(D)Survival curve following treatment with SNP-IV containing irrelevant antigen (dashed red line) or in untreated mice (dashed grey line) (n=10). Statistics were assessed by log-rank test.
(E) Bar graphs summarize the frequency of tetramer+ CD8 T cells in blood after treatment (n=10).
(F,G) Bar graphs summarize the frequency of tetramer+ CD8 T cells in spleen (D) and tumor (E) after treatment (n=5).
(H)Bar graphs summarize the median MFI of PD-1, TIM-3 and NKG2A of tetramer+ CD8 T cells in blood (n=10) and spleen (n=5) after treatment.
(I)Flow cytometry analysis of tetramer+ CD8 T cells in the tumor after boosting with SNP-IV containing Trp1, an irrelevant antigen.
(J)Histograms show the expression of CD39, NKG2A, PD-1 and Tim-3 on Reps1+ (red) or Trp1+ (green) CD8+ T cells.
Figure S2. SNP-IV but not SNP-SC resulted in intratumoral vaccine distribution and DC maturation, related to Figure 2 (A)Harvested spleens after SNP-IV or SNP-SC over time (n=2).
(B)Bar graph summarizes the frequency of myeloid cell populations of total vaccine+ cells in the spleen (n=4).
(C)UMAP of live, non-tumor lineage− cells identified 6 clusters of myeloid cells. Histograms show the expression of phenotypic markers expressed by cDC1, cDC2, monocytes and macrophages.
(D)UMAP focused on cDCs. Histograms show the expression of CCR7, CD80, CD86 and MHCII, highly expressed by subgroup ‘B’.
(E)Pie charts summarize the proportion of cDC1-A and cDC1-B (left) or cDC2-A and cDC2-B (n=5).
(F) UMAP focused on monocytes. Histograms show the expression of CCR7, CD80, CD86 and MHCII, highly expressed by subgroup ‘C’ (monocyte-derived DC, moDC) but not subgroup ‘A’ (myeloid-derived suppressor cell, MDSC).
(G) Pie charts summarize the proportion of Mono-A (MDSC), Mono-B and Mono-C (MDSC) after treatment (n=5).
(H) Graph summarize the log fold change of moDC/MDSC after treatment with SNP-SC or SNP-IV (n=5). Statistics were assessed by one-way ANOVA.
(I) Median MFI of CD86 on cDC1s in the spleen and tumor-draining LNs after treatment (n=3). Statistics were assessed by one-way ANOVA.
Figure S3. scRNA-seq of tumors revealed that intratumoral Chil3+ monocytes were significantly reduced after SNP-IV, related to Figure 3 (A)UMAP shows original clusters identified by Seurat after filtering out contaminating lymphocytes and granulocytes.
(B)Generation of 9 metaclusters based on hierarchical ordering.
(C)Density plots identify stable states within UMAP clusters.
(D)UMAP of clusters separated by tissue: spleen (top row) or tumor (bottom row).
(E)Bar graph shows proportion of cell cycle genes in each metacluster.
(F)Violin plots visualize expression of specific genes used to annotate monocyte/macrophage clusters.
(G) Bar graphs show frequencies of pDC, cDC1 and cDC2 in tumors based on scRNA-seq data collected on day 15.
(H) UMAPs of splenic MNP in untreated mice or mice treated with SNP-SC prime followed by SNP-SC boost (blue), SNP-IV (Reps1) boost (green) or SNP-IV (irrelevant antigen) boost (purple).
(I) Bar graphs show frequencies of mregDC, pDC, cDC1, cDC2 and monocyte/macrophages in spleens based on scRNA-seq data collected on day 15.
Figure S4. Chil3+ monocytes expressed immunoregulatory gene signature while Plin2+ macrophages expressed interferon-related gene signature, related to Figure 4 (A) Heatmap of top ten differentially expressed genes (DEGs) of each monocyte/macrophage population.
(B) Dot plot highlighting top pathways identified by Metascape
(C) Violin plots highlighting genes encoding cell surface receptors to distinguish Plin2+ macrophages Chil3+ monocytes and Ace+ monocytes by flow cytometry.
Figure S5. Interferon alpha required for mediating anti-tumor efficacy after SNP-IV treatment, related to Figure 5 (A) Heatmap of cytokines and chemokines analyzed by Luminex on sera collected 24 h after SNP-IV boosting in mice that were treated with isotype control (purple) or IFNAR blocking antibodies (maroon) compared to untreated animals (gray) (n=10).
(B) Bar graphs summarize the median MFI of CD80, CD86 and CCR7 in the spleen (top row), tumor-draining lymph node (middle row) or tumor (bottom row) 24 h after SNP-IV in mice treated with isotype control (purple) or IFNAR blocking antibodies (maroon) (n=10).
(C) Schematic of therapeutic study design. zDC-DTR bone marrow chimeric mice were implanted with MC38 and treated with SNP-7/8a (Reps1) on day 7 and day 14 together with CPI. Mice were not given diphtheria toxin (solid) or given DT via IP (open square) or IT (open triangle) injection on days 13, 15 and 17.
(D) Tumor growth following treatment with SNP-SC prime followed by SNP-IV (green) (n=5) with (open symbols) or without (closed symbols) DT injection in zDC-DTR mice. Statistics were assessed by two-way ANOVA.
(E) TC-1 tumor growth following treatment with SNP-SC prime (E6 antigen) followed by SNP-IV (Irrelevant antigen) with isotype control (purple) or IFNAR blocking antibody (maroon) (n=8). Statistics were assessed by two-way ANOVA.
(F) Bar graph summarizes the frequency of tetramer+ CD8 T cells in blood (n=5–8). Statistics were assessed by Kruskal Wallis test.
(G) Bar graph summarizes the frequency of “Chil3+ monocytes” in TC-1 tumors of untreated animals (gray) or 24 h after boosting with SNP-IV with isotype control (purple) or IFNAR blocking antibody (maroon) (n=4).
Figure S6. Chil3+ monocyte markers in human tumor-associated myeloid cells, related to Figure 6 (A) Average expression of a gene set of human orthologs of Chil3+ monocyte markers expressed in the MoMac-VERSE dataset (referred to as the huChil3+ geneset).
(B) Macrophages and monocytes from Peng et al. (top) and Kim et al. (middle) and Zheng et al. (bottom). Left: UMAP reduction and unbiased cluster assignment (resolution: 0.3, cluster membership indicated by color). Middle: Score for the huChil3 geneset in cells of each cluster. Right: Distribution of logFC between the indicated cluster and all other macrophages/monocytes in the dataset for all genes (grey) and huChil3 genes (turquoise).
(C) Survival curves across all TCGA patients (left), low grade glioma (middle) and clear cell renal cell carcinoma (right). Patients were stratified as high- or low-expression cohorts based on the median xCell monocyte geneset (Aran et al. 2017).
Table S1. List of top genes expressed in monocyte/macrophage clusters from mice bearing MC38 tumors; 24 h after boosting with SNP-7/8a, related to Figures 3 and 4.
Table S2. List of human orthologs of Chil3+ monocyte markers expressed in the MoMac-VERSE dataset, related to Figure 6.
Highlights.
Tumor-specific T cells are necessary but not sufficient for therapeutic efficacy
IV vaccination promotes tumor regression by remodeling the TME
Systemic IFN-I following IV vaccination alters intratumoral Chil3+ monocytes
Enrichment of human homologs of Chil3+ monocytes is associated with worse outcomes
ACKNOWLEDGEMENTS
We thank members of the Seder lab for scientific discussions and members of the Translational Research Program (Vaccine Research Center) for their valuable support with the animal studies, especially Dr. Ruth Woodward, Dr. Diana Scorpio, Nina Callaham, Marqui Dorsey, Sienna Rush and Arelis Island. This work was supported by the Intramural Research Program of the U.S. (R.A.S.), EMBO YIP and the Singapore Immunology Network core funding (F.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A.S.I., G.M.L. and R.A.S. are listed as inventors on patents describing polymer-based vaccines. A.S.I., and G.M.L. are employees of Vaccitech North America, which is commercializing polymer-based drug delivery technologies for immunotherapeutic applications. F.B. and S.M. are employees of Genentech, a member of the Roche group, which develops and markets drugs for profit.
REFERENCES
- Allen BM, Hiam KJ, Burnett CE, Venida A, DeBarge R, Tenvooren I, Marquez DM, Cho NW, Carmi Y, and Spitzer MH (2020). Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med 26, 1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308 e1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mule MP, et al. (2021). Intravenous nanoparticle vaccination generates stem-like TCF1(+) neoantigen-specific CD8(+) T cells. Nat Immunol 22, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleriot C, Chakarov S, and Ginhoux F (2020). Determinants of Resident Tissue Macrophage Identity and Function. Immunity 52, 957–970. [DOI] [PubMed] [Google Scholar]
- Borden EC (2019). Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discov 18, 219–234. [DOI] [PubMed] [Google Scholar]
- Browaeys R, Saelens W, and Saeys Y (2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 17, 159–162. [DOI] [PubMed] [Google Scholar]
- Bunis DG, Andrews J, Fragiadakis GK, Burt TD, and Sirota M (2020). dittoSeq: Universal User-Friendly Single-Cell and Bulk RNA Sequencing Visualization Toolkit. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi A, Monteiro LB, van Teijlingen Bakker N, Sanin DE, Rana N, Corrado M, Cameron AM, Hassler F, Matsushita M, Caputa G, et al. (2020). Triacylglycerol synthesis enhances macrophage inflammatory function. Nat Commun 11, 4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaib M, Chauhan SC, and Makowski L (2020). Friend or Foe? Recent Strategies to Target Myeloid Cells in Cancer. Front Cell Dev Biol 8, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809 e723. [DOI] [PubMed] [Google Scholar]
- Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, Liu A, Duvvuri U, Kim S, Soose RJ, et al. (2020). Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 52, 183–199 e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes AJ, Samad B, Tsui J, Chew NW, Yan P, Reeder GC, Kushnoor D, Shen A, Davidson B, Barczak AJ, et al. (2022). Discovering dominant tumor immune archetypes in a pan-cancer census. Cell 185, 184–203 e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Koebel CM, and Schreiber RD (2006). Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6, 836–848. [DOI] [PubMed] [Google Scholar]
- Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, and Spranger S (2022). Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity 55, 308–323 e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, and Gajewski TF (2011). Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 208, 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. (2018). High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 175, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde PS, and Chen DS (2020). Top 10 Challenges in Cancer Immunotherapy. Immunity 52, 17–35. [DOI] [PubMed] [Google Scholar]
- Hegde S, Leader AM, and Merad M (2021). MDSC: Markers, development, states, and unaddressed complexity. Immunity 54, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourani T, Holden JA, Li W, Lenzo JC, Hadjigol S, and O’Brien-Simpson NM (2021). Tumor Associated Macrophages: Origin, Recruitment, Phenotypic Diversity, and Targeting. Front Oncol 11, 788365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, Bornstein C, Moshe A, Keren-Shaul H, Cohen M, et al. (2020). Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 182, 872–885 e819. [DOI] [PubMed] [Google Scholar]
- Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, Lee JI, Suh YL, Ku BM, Eum HH, et al. (2020). Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. (2016). Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401. [DOI] [PubMed] [Google Scholar]
- Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, Lopes A, Johnson SB, Schwarz B, Bohrnsen E, et al. (2021). Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 184, 5338–5356 e5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwe H, Pircher A, Van den Eynde K, et al. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, and Tough DF (2003). Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 4, 1009–1015. [DOI] [PubMed] [Google Scholar]
- Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, Vanhecke J, Verbandt S, Hong H, Min JW, et al. (2020). Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet 52, 594–603. [DOI] [PubMed] [Google Scholar]
- Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, Tobin K, Nichols SR, Itzkowitz Y, Zaidi N, et al. (2020). Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol 38, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie KF, Clark K, Naqvi S, McGuire VA, Noehren G, Kristariyanto Y, van den Bosch M, Mudaliar M, McCarthy PC, Pattison MJ, et al. (2013). PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol 190, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F, Mayer-Barber K, Sher A, Wack A, and O’Garra A (2015). Type I interferons in infectious disease. Nat Rev Immunol 15, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al. (2014). Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 11, 509–524. [DOI] [PubMed] [Google Scholar]
- Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, et al. (2012). Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med 209, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, et al. (2020). TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182, 886–900 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. (2017). Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol Res 5, 29–41. [DOI] [PubMed] [Google Scholar]
- Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, Khalilnezhad S, Irac SE, Dubuisson A, Chevrier M, et al. (2021). Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 54, 1883–1900 e1885. [DOI] [PubMed] [Google Scholar]
- Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, Liu L, Huang D, Jiang J, Cui GS, et al. (2019). Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, Gilboa E, Hammond SA, Hershberg R, Korman AJ, et al. (2016). The Human Vaccines Project: A roadmap for cancer vaccine development. Sci Transl Med 8, 334ps339. [DOI] [PubMed] [Google Scholar]
- Rosas-Ballina M, Guan XL, Schmidt A, and Bumann D (2020). Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Front Immunol 11, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, van der Burg SH, Melief CJM, and Bhardwaj N (2021). Therapeutic cancer vaccines. Nat Rev Cancer 21, 360–378. [DOI] [PubMed] [Google Scholar]
- Sharma A, Seow JJW, Dutertre CA, Pai R, Bleriot C, Mishra A, Wong RMM, Singh GSN, Sudhagar S, Khalilnezhad S, et al. (2020). Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 183, 377–394 e321. [DOI] [PubMed] [Google Scholar]
- Shibuya T, Kamiyama A, Sawada H, Kikuchi K, Maruyama M, Sawado R, Ikeda N, Asano K, Kurotaki D, Tamura T, et al. (2021). Immunoregulatory Monocyte Subset Promotes Metastasis Associated With Therapeutic Intervention for Primary Tumor. Front Immunol 12, 663115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleijfer S, Bannink M, Van Gool AR, Kruit WH, and Stoter G (2005). Side effects of interferon-alpha therapy. Pharm World Sci 27, 423–431. [DOI] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan H, Salazar AM, and Celis E (2020). Poly-ICLC, a multi-functional immune modulator for treating cancer. Semin Immunol 49, 101414. [DOI] [PubMed] [Google Scholar]
- Thoreau M, Penny HL, Tan K, Regnier F, Weiss JM, Lee B, Johannes L, Dransart E, Le Bon A, Abastado JP, et al. (2015). Vaccine-induced tumor regression requires a dynamic cooperation between T cells and myeloid cells at the tumor site. Oncotarget 6, 27832–27846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U’Ren L, Guth A, Kamstock D, and Dow S (2010). Type I interferons inhibit the generation of tumor-associated macrophages. Cancer Immunol Immunother 59, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluis TC, Sluijter M, van Duikeren S, West BL, Melief CJ, Arens R, van der Burg SH, and van Hall T (2015). Therapeutic Peptide Vaccine-Induced CD8 T Cells Strongly Modulate Intratumoral Macrophages Required for Tumor Regression. Cancer Immunol Res 3, 1042–1051. [DOI] [PubMed] [Google Scholar]
- Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 21, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman AD, Fritz JM, and Lenardo MJ (2020). A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N, et al. (2021). A single-cell and spatially resolved atlas of human breast cancers. Nat Genet 53, 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Levine YA, Gunasekaran MK, Wang Y, Addorisio M, Zhu S, Li W, Li J, de Kleijn DP, et al. (2016). Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Guo J, and Huang L (2020). Tackling TAMs for Cancer Immunotherapy: It’s Nano Time. Trends Pharmacol Sci 41, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459 e429. [DOI] [PubMed] [Google Scholar]
- Zhang X, Liu S, Guo C, Zong J, and Sun MZ (2012). The association of annexin A2 and cancers. Clin Transl Oncol 14, 634–640. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ren D, Guo L, Wang L, Wu S, Lin C, Ye L, Zhu J, Li J, Song L, et al. (2017). Thymosin beta 10 is a key regulator of tumorigenesis and metastasis and a novel serum marker in breast cancer. Breast Cancer Res 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356 e1316. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334 e1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, and Kroemer G (2015). Type I interferons in anticancer immunity. Nat Rev Immunol 15, 405–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NeoAg+ CD8+ T cells generated by SNP-SC controlled tumor growth when followed by IV adjuvant delivery, related to Figure 1 (A)TC-1 tumor growth following treatment with SNP-IV prime and boost (red), SNP-SC prime and boost (blue), SNP-SC prime and SNP-IV boost (green) or SNP-IV boost with irrelevant antigen (purple) (n=8). Statistics were assessed by two-way ANOVA.
(B)Bar graph summarizes the frequency of tetramer+ CD8 T cells in blood after treatment with SNP-7/8a containing E6 antigen (n=5–8). Statistics were assessed by Kruskal Wallis test.
(C)Tumor growth following treatment with SNP-IV prime and boost with Reps1 (solid line) or irrelevant antigen (dashed line) (n=10). Statistics were assessed by two-way Anova.
(D)Survival curve following treatment with SNP-IV containing irrelevant antigen (dashed red line) or in untreated mice (dashed grey line) (n=10). Statistics were assessed by log-rank test.
(E) Bar graphs summarize the frequency of tetramer+ CD8 T cells in blood after treatment (n=10).
(F,G) Bar graphs summarize the frequency of tetramer+ CD8 T cells in spleen (D) and tumor (E) after treatment (n=5).
(H)Bar graphs summarize the median MFI of PD-1, TIM-3 and NKG2A of tetramer+ CD8 T cells in blood (n=10) and spleen (n=5) after treatment.
(I)Flow cytometry analysis of tetramer+ CD8 T cells in the tumor after boosting with SNP-IV containing Trp1, an irrelevant antigen.
(J)Histograms show the expression of CD39, NKG2A, PD-1 and Tim-3 on Reps1+ (red) or Trp1+ (green) CD8+ T cells.
Figure S2. SNP-IV but not SNP-SC resulted in intratumoral vaccine distribution and DC maturation, related to Figure 2 (A)Harvested spleens after SNP-IV or SNP-SC over time (n=2).
(B)Bar graph summarizes the frequency of myeloid cell populations of total vaccine+ cells in the spleen (n=4).
(C)UMAP of live, non-tumor lineage− cells identified 6 clusters of myeloid cells. Histograms show the expression of phenotypic markers expressed by cDC1, cDC2, monocytes and macrophages.
(D)UMAP focused on cDCs. Histograms show the expression of CCR7, CD80, CD86 and MHCII, highly expressed by subgroup ‘B’.
(E)Pie charts summarize the proportion of cDC1-A and cDC1-B (left) or cDC2-A and cDC2-B (n=5).
(F) UMAP focused on monocytes. Histograms show the expression of CCR7, CD80, CD86 and MHCII, highly expressed by subgroup ‘C’ (monocyte-derived DC, moDC) but not subgroup ‘A’ (myeloid-derived suppressor cell, MDSC).
(G) Pie charts summarize the proportion of Mono-A (MDSC), Mono-B and Mono-C (MDSC) after treatment (n=5).
(H) Graph summarize the log fold change of moDC/MDSC after treatment with SNP-SC or SNP-IV (n=5). Statistics were assessed by one-way ANOVA.
(I) Median MFI of CD86 on cDC1s in the spleen and tumor-draining LNs after treatment (n=3). Statistics were assessed by one-way ANOVA.
Figure S3. scRNA-seq of tumors revealed that intratumoral Chil3+ monocytes were significantly reduced after SNP-IV, related to Figure 3 (A)UMAP shows original clusters identified by Seurat after filtering out contaminating lymphocytes and granulocytes.
(B)Generation of 9 metaclusters based on hierarchical ordering.
(C)Density plots identify stable states within UMAP clusters.
(D)UMAP of clusters separated by tissue: spleen (top row) or tumor (bottom row).
(E)Bar graph shows proportion of cell cycle genes in each metacluster.
(F)Violin plots visualize expression of specific genes used to annotate monocyte/macrophage clusters.
(G) Bar graphs show frequencies of pDC, cDC1 and cDC2 in tumors based on scRNA-seq data collected on day 15.
(H) UMAPs of splenic MNP in untreated mice or mice treated with SNP-SC prime followed by SNP-SC boost (blue), SNP-IV (Reps1) boost (green) or SNP-IV (irrelevant antigen) boost (purple).
(I) Bar graphs show frequencies of mregDC, pDC, cDC1, cDC2 and monocyte/macrophages in spleens based on scRNA-seq data collected on day 15.
Figure S4. Chil3+ monocytes expressed immunoregulatory gene signature while Plin2+ macrophages expressed interferon-related gene signature, related to Figure 4 (A) Heatmap of top ten differentially expressed genes (DEGs) of each monocyte/macrophage population.
(B) Dot plot highlighting top pathways identified by Metascape
(C) Violin plots highlighting genes encoding cell surface receptors to distinguish Plin2+ macrophages Chil3+ monocytes and Ace+ monocytes by flow cytometry.
Figure S5. Interferon alpha required for mediating anti-tumor efficacy after SNP-IV treatment, related to Figure 5 (A) Heatmap of cytokines and chemokines analyzed by Luminex on sera collected 24 h after SNP-IV boosting in mice that were treated with isotype control (purple) or IFNAR blocking antibodies (maroon) compared to untreated animals (gray) (n=10).
(B) Bar graphs summarize the median MFI of CD80, CD86 and CCR7 in the spleen (top row), tumor-draining lymph node (middle row) or tumor (bottom row) 24 h after SNP-IV in mice treated with isotype control (purple) or IFNAR blocking antibodies (maroon) (n=10).
(C) Schematic of therapeutic study design. zDC-DTR bone marrow chimeric mice were implanted with MC38 and treated with SNP-7/8a (Reps1) on day 7 and day 14 together with CPI. Mice were not given diphtheria toxin (solid) or given DT via IP (open square) or IT (open triangle) injection on days 13, 15 and 17.
(D) Tumor growth following treatment with SNP-SC prime followed by SNP-IV (green) (n=5) with (open symbols) or without (closed symbols) DT injection in zDC-DTR mice. Statistics were assessed by two-way ANOVA.
(E) TC-1 tumor growth following treatment with SNP-SC prime (E6 antigen) followed by SNP-IV (Irrelevant antigen) with isotype control (purple) or IFNAR blocking antibody (maroon) (n=8). Statistics were assessed by two-way ANOVA.
(F) Bar graph summarizes the frequency of tetramer+ CD8 T cells in blood (n=5–8). Statistics were assessed by Kruskal Wallis test.
(G) Bar graph summarizes the frequency of “Chil3+ monocytes” in TC-1 tumors of untreated animals (gray) or 24 h after boosting with SNP-IV with isotype control (purple) or IFNAR blocking antibody (maroon) (n=4).
Figure S6. Chil3+ monocyte markers in human tumor-associated myeloid cells, related to Figure 6 (A) Average expression of a gene set of human orthologs of Chil3+ monocyte markers expressed in the MoMac-VERSE dataset (referred to as the huChil3+ geneset).
(B) Macrophages and monocytes from Peng et al. (top) and Kim et al. (middle) and Zheng et al. (bottom). Left: UMAP reduction and unbiased cluster assignment (resolution: 0.3, cluster membership indicated by color). Middle: Score for the huChil3 geneset in cells of each cluster. Right: Distribution of logFC between the indicated cluster and all other macrophages/monocytes in the dataset for all genes (grey) and huChil3 genes (turquoise).
(C) Survival curves across all TCGA patients (left), low grade glioma (middle) and clear cell renal cell carcinoma (right). Patients were stratified as high- or low-expression cohorts based on the median xCell monocyte geneset (Aran et al. 2017).
Table S1. List of top genes expressed in monocyte/macrophage clusters from mice bearing MC38 tumors; 24 h after boosting with SNP-7/8a, related to Figures 3 and 4.
Table S2. List of human orthologs of Chil3+ monocyte markers expressed in the MoMac-VERSE dataset, related to Figure 6.
Data Availability Statement
The scRNA-seq data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of publication. Accession number is listed in the key resources table.
All code used for analyses are publicly available and listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse B220 (PE-Cy7); clone RA3–6B2 | BD Biosciences | Cat# 552772 |
| Anti-mouse CCR7 (BV421); clone 4B12 | Biolegend | Cat# 120120 |
| Anti-mouse CD3 (BUV395); clone145–2C11 | BD Biosciences | Cat# 563565 |
| Anti-mouse CD3 (Alexa700); clone 17A2 | Biolegend | Cat# 100216 |
| Anti-mouse CD4 (BUV395); clone RM4–4 | BD Biosciences | Cat# 740209 |
| Anti-mouse CD8 (APCeFluor780); clone 53–6.7 | eBioscience | Cat# 47-0081-82 |
| Anti-mouse CD11b (AF700); clone MI/70 | BD Biosciences | Cat# 557960 |
| Anti-mouse CD11c (PE); clone HL3 | BD Biosciences | Cat# 553802 |
| Anti-mouse CD16/32; clone 2.4G2 | BD Biosciences | Cat# 553142 |
| Anti-mouse CD19 (BUV395); clone 1D3 | BD Biosciences | Cat# 563557 |
| Anti-mouse CD39 (PE-Dazzle594); clone Duha59 | Biolegend | Cat# 143812 |
| Anti-mouse CD40 (AF647); clone 3/23 | Biolegend | Cat# 123613 |
| Anti-mouse CD44 (BUV737); clone IM7 | BD Biosciences | Cat# 564392 |
| Anti-mouse CD45 (BUV661); clone 30-F11 | BD Biosciences | Cat# 565079 |
| Anti-mouse CD64 (BV785); clone X54–5/7.1 | BD Biosciences | Cat# 741024 |
| Anti-mouse CD66a (BV605); clone CC1 | BD Biosciences | Cat# 750882 |
| Anti-mouse CD80 (PE-Dazzle594); clone 16–10A1 | BD Biosciences | Cat# 562504 |
| Anti-mouse CD86 (BV711); clone GL1 | BD Biosciences | Cat# 740688 |
| Anti-mouse CD172α (PerCP-eF710); clone P84 | Life Technologies | Cat# 46-1721-82 |
| Anti-mouse Eomes (PerCP-eF710); clone Dan11mag | Invitrogen | Cat# 46-4875-82 |
| Anti-mouse F4/80 (PE-Cy5); clone BM8 | eBioscience | Cat# 15-4801-82 |
| Anti-mouse IA/IE (AF488); clone M5/114.15.2 | Biolegend | Cat# 107616 |
| Anti-mouse LY6A/E (PE-CF594); clone D7 | BD Biosciences | Cat# 562730 |
| Anti-mouse Ly6C (APC-eF780); clone AL-21 | BD Biosciences | Cat# 560596 |
| Anti-mouse Ly6G (BUV563); clone 1A8 | BD Biosciences | Cat# 565707 |
| Anti-mouse NK1.1 (BUV395); clone PK136 | BD Biosciences | Cat# 564144 |
| Anti-mouse NKG2A (PE-Cy7); clone 16A11 | Biolegend | Cat# 142810 |
| Anti-mouse PD-1 (BV421); clone 29F.A12 | Biolegend | Cat# 135218 |
| Anti-mouse SiglecH (BUV805); clone 440C | BD Biosciences | Cat# 748291 |
| Anti-mouse Tim-3 (BV605); clone RMT3–23 | Biolegend | Cat# 119721 |
| Anti-mouse TNFα (BV650); clone MP6-XT22 | Biolegend | Cat# 506333 |
| Anti-mouse XCR1 (BV650); clone ZET | Biolegend | Cat# 148220 |
| Anti-mouse IFNα/β receptor-1; clone MAR1–5A3 | BioXCell | Cat# BP0241 |
| Anti-mouse PD-L1; clone 10F.9G2 | BioXCell | Cat# BP0101 |
| TotalSeq-C0301 anti-mouse Hashtag 1; clones M1/42, 30-F11 | Biolegend | Cat# 155861 |
| TotalSeq-C0302 anti-mouse Hashtag 2; clones M1/42, 30-F11 | Biolegend | Cat# 155863 |
| TotalSeq-C0303 anti-mouse Hashtag 3; clones M1/42, 30-F11 | Biolegend | Cat# 155865 |
| TotalSeq-C0304 anti-mouse Hashtag 4; clones M1/42, 30-F11 | Biolegend | Cat# 155867 |
| TotalSeq-C0305 anti-mouse Hashtag 5; clones M1/42, 30-F11 | Biolegend | Cat# 155869 |
| TotalSeq-C0306 anti-mouse Hashtag 6; clones M1/42, 30-F11 | Biolegend | Cat# 155871 |
| TotalSeq-C0307 anti-mouse Hashtag 7; clones M1/42, 30-F11 | Biolegend | Cat# 155873 |
| TotalSeq-C0308 anti-mouse Hashtag 8; clones M1/42, 30-F11 | Biolegend | Cat# 155875 |
| TotalSeq-C0309 anti-mouse Hashtag 9; clones M1/42, 30-F11 | Biolegend | Cat# 155877 |
| TotalSeq-C0310 anti-mouse Hashtag 10; clones M1/42, 30-F11 | Biolegend | Cat# 155879 |
| Chemicals, peptides, and recombinant proteins | ||
| Dasatinib | Selleckchem | S1021 |
| Isoflurane, USP | Baxter Healthcare Corp. | NDC 10019-360-60 |
| Paraformaldehyde (PFA, 16%) | Electron Microscopy Sciences | 15710 |
| Tween-20 | Sigma | P-7949 |
| Heparin | Fresenius Kabi | NDC 63323-540-05 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | 276855–100mL |
| Reps1 SNP vaccine | Vaccitech North America | N/A |
| Reps1 SNP vaccine labelled with AF647 | Vaccitech North America | N/A |
| Trp1 SNP vaccine | Vaccitech North America | N/A |
| ABTS (2,2’-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) | Sigma | A3219 |
| Murine IL-12 ABTS ELISA Kit | Peprotech | 900-K97 |
| Reps1 Tetramer (H-2Db, AQLANDVVL) | Gift from J. Finnigan | N/A |
| Trp1 Tetramer (H-2Db, TAPDNLGYM) | Gift from J. Finnigan | N/A |
| Critical commercial assays | ||
| Mouse IFN Alpha All Subtype ELISA Kit, High Sensitivity | PBL Assay Science | Cat# 42115–1 |
| eBioscience™ Foxp3 / Transcription Factor Staining Set | Invitrogen | Cat# 00-5523-00 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34962 |
| ArC™ Amine Reactive Compensation Bead Kit | ThermoFisher Scientific | Cat# A10346 |
| BD Horizon Brilliant Stain Buffer | BD Biosciences | 566385 |
| Streptavidin PE (SaPE) | BD Biosciences | S866 |
| Trypsin-EDTA (0.25%) | Gibco, Life Tech | 25200–056 |
| ViaStain AOPI Staining Solution | Nexcelom Bioscience | CS2–0106 |
| Milliplex MAP mouse cytokine/chemokine Magnetic Kit | Millipore Sigma | MCYTMAG-70K-PX32 |
| Phosphate buffered saline (PBS) | Gibco | 10010–023 |
| RPMI 1640 | Cytiva, HyClone Labs | SH30027.02 |
| FBS | Gibco | 10438–026 |
| Penicillin/Streptomycin/Glutamine (100X) | Gibco, Life Tech | 10378–016 |
| Non-essential Amino Acids (100X) | Cytiva, HyClone Labs | SH30238.01 |
| Sodium Pyruvate (100mM) | Cytiva, HyClone Labs | SH30239.01 |
| Collagenase D | Roche | 11088882001 |
| DNase I | Roche | 04536282001 |
| ACK (Ammonium-Chloride-Potassium) lysing buffer | Quality Biological | 118-156-101 |
| Chromium Single Cell 50 Reagent Kit | 10X Genomics | |
| Dynabead MyOne Silane | Thermo Fisher Scientific | Cat#37002D |
| SPRIselect for Size Selection | Beckman Coulter | Cat#B23319 |
| Experimental models: Cell lines | ||
| MC38 Murine colorectal cancer cell line | Genentech, L. Delamarre | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: wild-type C57BL/6J mice | Jackson Labs | 000664 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | Jackson Labs | 002014 |
| Mouse: B6(Cg)-Zbtb46tm1(HBEGF)Mnz/J | Jackson Labs | 019506 |
| Software and algorithms | ||
| Flowjo v10 | Tree Star | N/A |
| GraphPad Prism v8 | GraphPad software | N/A |
| xPONENT software | Luminex | N/A |
| Living Image v4.5 | PerkinElmer | N/A |
| R, v4.1.2 | R Project | https://www.r-project.org/ |
| Cellranger v6.0.0 | 10X Genomics | https://support.10xgenomics.com |
| Seurat v4.1.0 | Seurat | https://cran.r-project.org/web/packages/Seurat/index.html |
| Monocle 3 v1.0.0 | Monocle | https://github.com/cole-trapnell-lab/monocle3/releases/tag/1.0.0 |
| Slingshot v2.2.0 | Slingshot | https://bioconductor.org/packages/release/bioc/html/slingshot.html |
| DoubletFinder v 2.0.3 | DoubletFinder | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| pheatmap v1.0.12 | Pheatmap | https://cran.r-project.org/web/packages/pheatmap/index.html |
| dittoSeq v1.6.0 | DittoSeq | https://bioconductor.org/packages/release/bioc/html/dittoSeq.html |
| biomaRt v2.50.3 | BiomaRt | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| EnhancedVolcano v1.12.0 | Enhanced Volcano | https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html |
| MetaScape v20220101 | MetaScape | https://metascape.org/gp/index.html#/main/ |
| QIAGEN IPA vDec. 2021 | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Deposited data | ||
| scRNA-seq | Gene Expression Omnibus | GSE214741 |