

Abstract
Poly (ADP-ribose) polymerase inhibitors (PARPis) represent a major advance in ovarian cancer, now as a treatment and as a maintenance therapy in the upfront and recurrent settings. However, patients often develop resistance to PARPis, underlining the importance of dissecting resistance mechanisms. Here, we report different dosing/timing schemes of PARPi treatment in BRCA2-mutant PEO1 cells, resulting in the simultaneous development of distinct resistance mechanisms. PARPi-resistant variants PEO1/OlaJR, established by higher initial doses and short-term PARPi treatment, develops PARPi resistance by rapidly restoring functional BRCA2 and promoting drug efflux activity. In contrast, PEO1/OlaR, developed by lower initial doses with long-term PARPi exposure, shows no regained BRCA2 function but a mesenchymal-like phenotype with greater invasion ability, and exhibits activated ATR/CHK1 and suppressed EZH2/MUS81 signaling cascades to regain HR repair and fork stabilization, respectively. Our study suggests that PARPi resistance mechanisms can be governed by treatment strategies and have a molecular basis on BRCA2 functionality. Further, we define different mechanisms that may serve as useful biomarkers to assess subsequent treatment strategies in PARPi-resistant ovarian cancer.
Introduction
Ovarian cancer is a lethal gynecological malignancy responsible for nearly 5% of cancer deaths, despite accounting for only 2.6% of all gynecological malignancies in the United States [1]. Approximately 20–25% of high-grade serous ovarian cancer (HGSOC) patients carry BRCA1 or 2 (BRCA1/2) mutations [2, 3], essential for homologous recombination (HR) repair, making them sensitive to poly (ADP-ribose) polymerase inhibitors (PARPis) [4]. However, PARPi treatment is often complicated by the inevitable development of either intrinsic or acquired resistance in patients [5–9]. Thus, there is an urgent need to broaden our understanding of PARPi resistance to better utilize PARPis in the clinic.
The major resistance mechanisms of PARPis include restoration of HR repair, replication fork stabilization, and increased drug efflux [10, 11]. BRCA reversion mutations that re-establish HR functionality and replication fork stabilization are well-known PARPi resistance mechanisms both through clinical and preclinical studies [12, 13]. However, only approximately 20% of BRCA-mutant HGSOC patients develop BRCA reversion mutations against PARPi treatment, indicating a majority of PARPi resistance mechanisms in the clinic remain elusive [14]. In addition, both HR restoration and fork stabilization mechanisms are closely associated with RAD51 formation, which is thought to depend on BRCA2 [15].
Despite multiple studies describing the role of BRCA2, there is an unmet need for further understanding of the role of treatment pressures and BRCA2 in the preferential selection of PARPi resistance mechanisms. For instance, Yamamoto et al. reported that infrared radiation induces RAD51 formation and HR restoration in a BRCA2-mutant PEO1 HGSOC cell line [16] without evaluating the causes of BRCA2-independent HR restoration. Other studies demonstrated the critical roles of replication fork remodelers, e.g., EZH2, SMARCAL1, HTLF, and ZRANB3, in fork stabilization and development of PARPi resistance [17–20], but whether BRCA2-deficient cancer cells may naturally develop PARPi resistance by mitigating these fork remodelers is unclear.
In many preclinical studies on PARPi resistance, a stepwise approach has been used to develop PARPi-resistant cell lines, in which cells were initially exposed to relatively low concentrations of PARPi, and resistant clones were propagated and exposed to gradually higher doses of PARPi over an extended period [21, 22]. Dosing regimens in the clinical setting, however, might be more closely modeled by exposing cells to higher clinically relevant concentrations at the onset of treatment for a shorter period, which may lead to the development of distinct PARPi resistance mechanisms. Therefore, it is necessary to further characterize PARPi-resistant cells to understand how treatment schedule would dominate their preference in developing resistance.
In this study, we hypothesized that in a BRCA1-proficient background, treatment selection pressure-induced variations in BRCA2 status might influence the prevalence of specific resistance mechanisms over others, thus explaining the failure of some interventional treatments in such scenarios. We subjected BRCA2-mutant PEO1 cells to varying treatment and selection pressures to generate acquired olaparib-resistant (OlapR) variants that were either BRCA2-revertant or -mutant and then explored the prevalence of the different resistance mechanisms in these cells as well as in another de novo PARPi-resistant cell line, PEO4. We identified a novel BRCA2-independent HR pathway and preferential selection of some resistance mechanisms over others in a BRCA2-dependent manner. Our results help better elucidate the influences of treatment and selection pressures on PARPi resistance in a heterogenous clinical setting that might ultimately have implications on how we manage ovarian cancer.
Results
PARPi-resistant BRCA2-mutant PEO1 cell lines show distinct phenotypes
To test our hypothesis, we developed OlapR cells from the PARPi-sensitive BRCA2-deficient PEO1 cells, exposing them to higher initial doses of olaparib over 15 weeks to generate PEO1/OlaJR (Fig. 1a, upper). A second BRCA2-mutant OlapR variant (PEO1/OlaR) was obtained from Dr. Bitler’s group [16], which was developed using lower initial concentrations of olaparib over 24 weeks (Fig. 1a, lower). Growth assays demonstrated that IC50 values against olaparib were at least 59-fold and 4-fold higher in OlapR cells than in parental PEO1 and de novo PARPi-resistant PEO4, respectively (Fig. 1b, upper). These OlapR variants also presented 5-fold more resistance to another PARPi rucaparib relative to PEO1 cells (Fig. 1b, lower), indicating cross-resistance. Importantly, both OlapR cell lines maintained PARPi resistance up to 8 weeks of olaparib withdrawal which were confirmed by cell growth and colony-forming assays (Supplementary Fig. 1a–b). PARPi resistance is often associated with resistance to platinum-based therapy [23]. Here, we found that both OlapR cell lines also exhibited > 2.8-fold resistance to cisplatin compared to PEO1, affirming expected cisplatin resistance (Supplementary Fig. 1c).
Fig. 1. Different treatment schemes cause distinct phenotypes of PARPi-resistant BRCA2-mutant PEO1 cell lines.
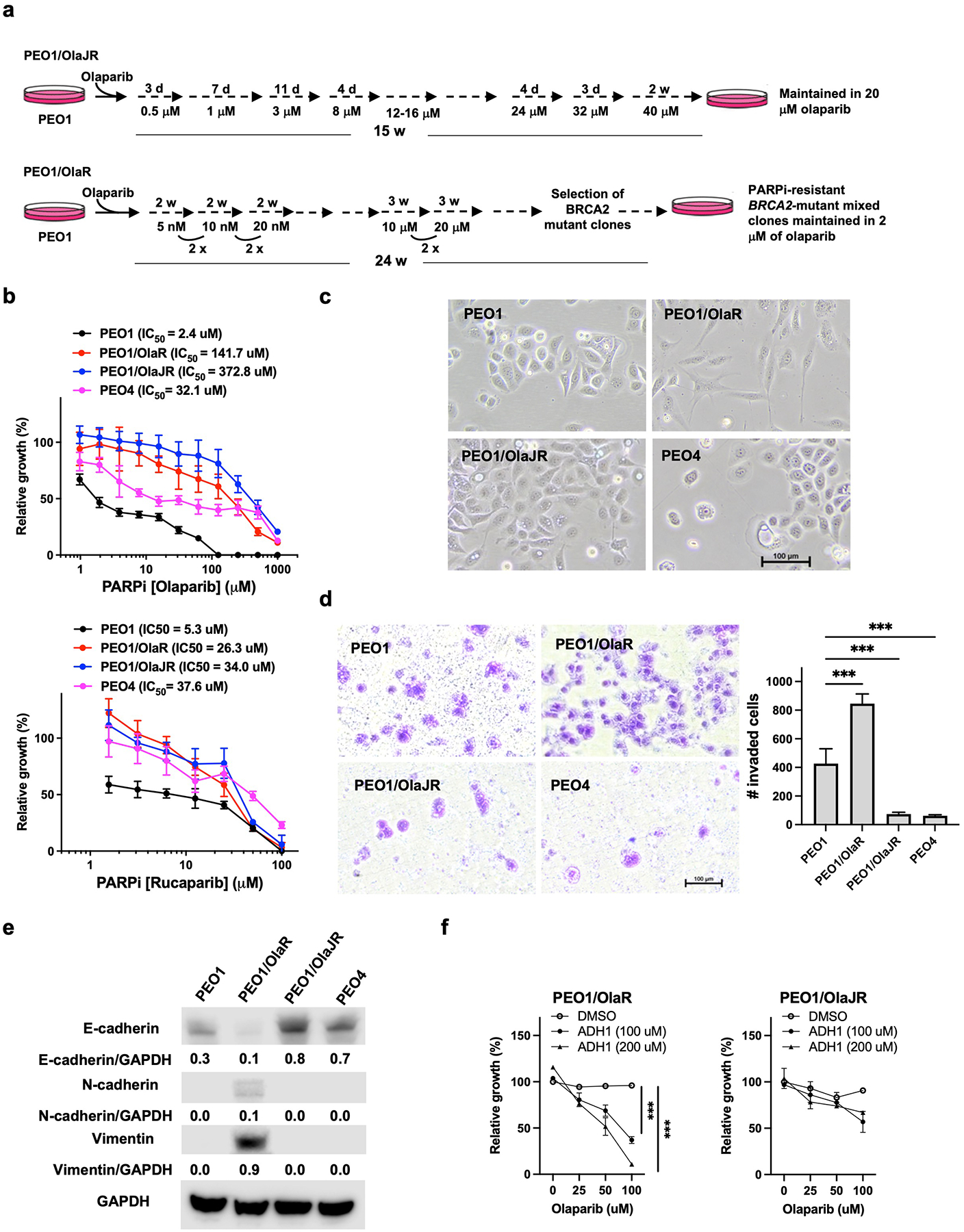
(a) Olaparib treatment schemes of PEO1/OlaJR and PEO1/OlaR. d, days; wk, weeks. (b) Cell growth was examined by XTT assay. Cells were treated with olaparib or rucaparib at indicated doses for 72 hours. IC50 values were calculated using GraphPad Prism v.7.1. (c) Cell morphology of parental PEO1, OlapR variants (PEO1/OlaR and PEO1/OlaJR), and de novo PARPi-resistant PEO4. (d) The invasion abilities were assessed by transwell invasion assay. (e) Western blotting of E-cadherin, N-cadherin, vimentin, and GAPDH. Densitometric values of these proteins relative to GAPDH are shown. (f) Cell growth was examined by XTT assay. Cells were treated with olaparib and/or N-cadherin inhibitor (ADH-1) at indicated doses for 72 hours. ***, p < 0.001.
Resistance to PARPi is characterized by epithelial-mesenchymal transition (EMT) in PEO1/OlaR
First, we noted that OlapR variants displayed distinct cell morphologies (Fig. 1c). PEO1/OlaR demonstrated a mesenchymal-like phenotype whereas PEO1/OlaJR maintained epithelial cell morphology like parental PEO1 and de novo PARPi-resistant PEO4 cells (Fig. 1c). Invasion assays exhibited a greater invasiveness in PEO1/OlaR but decreased invasive abilities in PEO1/OlaJR and PEO4 relative to PEO1 (Fig. 1d). We next performed an immunoblotting analysis of EMT-related markers considering the unique mesenchymal-like morphology of PEO1/OlaR. Immunoblotting results showed increased mesenchymal markers N-cadherin and vimentin in PEO1/OlaR, while the epithelial marker E-cadherin was overexpressed in PEO1/OIaJR and PEO4 compared to PEO1 (Fig. 1e) in agreement with their phenotypes.
As such, we used an N-cadherin inhibitor ADH-1 as data suggest N-cadherin may modulate PARPi resistance, at least in part, via activating pro-survival protein Bcl-2 [24, 25]. We found ADH-1 re-sensitized PEO1/OlaR but not PEO1/OlaJR to olaparib (Fig. 1f), suggesting increased N-cadherin expression may be involved in the resistance to PARPi in PEO1/OlaR.
A heterozygous BRCA2 reversion mutation contributes to PARPi resistance in PEO1/OlaJR
Given that PEO1/OlaJR showed similar morphology to PEO4, which has de novo PARPi resistance due to a BRCA2 reversion mutation [26], we next performed immunoblotting of BRCA2 in OlapR cells to confirm their BRCA2 status. We found the expression of BRCA2 protein in PEO1/OlaJR and the natural revertant control PEO4, but not in PEO1 and PEO1/OlaR (Fig. 2a). These data indicate that PEO1/OlaJR might have a reversion mutation that restored the open reading frame of BRCA2. Moreover, immunoblotting of the immunoprecipitates from PEO1/OlaJR with RAD51-specific antibodies showed co-immunoprecipitation of RAD51 with BRCA2 (Fig. 2a), suggesting the restored functionality of BRCA2. In addition, only PEO1/OlaJR and PEO4 showed BRCA2 foci when treated with PARPi (Supplementary Fig. 3), which is consistent with their BRCA2 status (Fig. 2a). However, the addition (Fig. 2a, left) or increasing the concentrations (Fig. 2a, right) of a replication inhibitor HU, did not necessarily increase RAD51 association with BRCA2 in PEO1/OlaJR, unlike that observed in PEO4 (Fig. 2a, left).
Fig. 2. PEO1/OlaJR restores its BRCA2 functionality by a heterozygous BRCA2 reversion mutation.
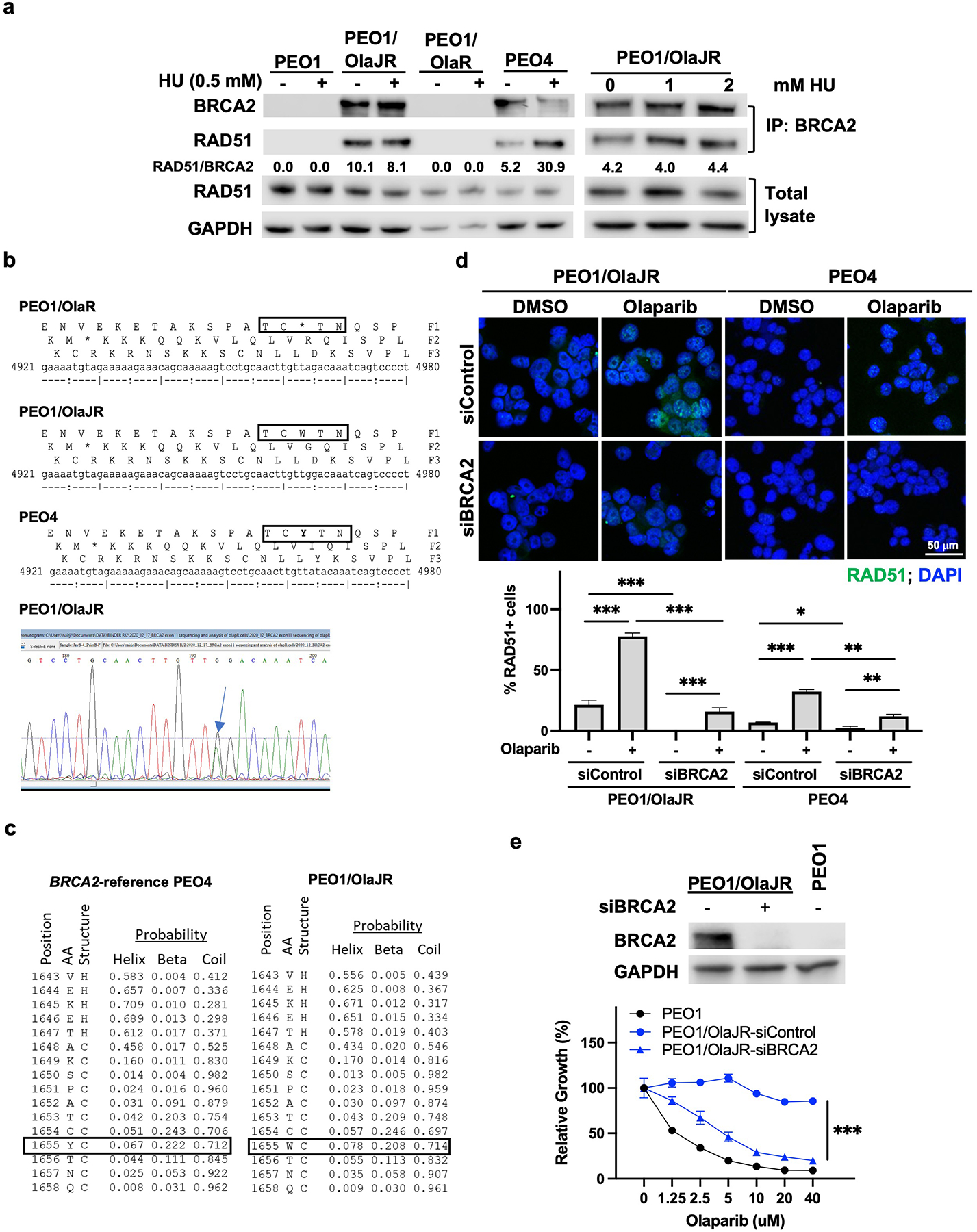
(a) Immunoblotting of co-immunoprecipitated BRCA2 with RAD51 from cells with or without HU treatment (left) and specifically from PEO1/OlaJR (right) with increasing concentrations of HU (0–2 mM). Densitometric analysis of co-immunoprecipitated RAD51 normalized to total RAD51 and GAPDH relative to BRCA2 is shown. (b) Sanger sequencing of amplicons of exon11 of BRCA2 indicates reverse mutation that restores the full-length BRCA2 transcripts in PEO1/OlaJR. The chromatogram of the target region of exon 11 of BRCA2 shows a reverse mutation that reverses the stop codon (TAG) to tryptophan (TGG). (c) The secondary protein structure prediction of the BRCA2 target region (p.1655) using RaptorX software shows similar secondary structure probability values for tryptophan (W) as opposed to tyrosine (Y) in the wildtype BRCA2 reference (GenBank refseq #NM_000059.4). (d) Immunofluorescence staining of RAD51 foci (green) was conducted to examine the HR repair activity. Representative images were taken at 63x magnification. Cells with > 5 RAD51 foci were counted as RAD51-positive (RAD51+) cells. The percentage of RAD51+ cells is plotted below. (e) Immunoblotting of cells transfected with or without siRNAs against BRCA2 (upper). Cell growth of cells with or without BRCA2 silencing was examined by XTT assay (bottom).
To further characterize BRCA2 mutations in both OlapR variants, specific primers targeting the nonsense mutation Y1655X in exon 11 of BRCA2 were used for generating and sequencing the amplicons in PEO1 and its OlapR variants. PEO1/OlaJR has a heterozygous reversion mutation (GTT[A/G]GACA, Y1665W), while parental PEO1 and PEO1/OlaR both share the predicted homozygous nonsense mutation (Y1655X) at the same location (Fig. 2b). Also, secondary protein structure predictions by RaptorX [27] showed similar probabilities for secondary structures on that location when compared to wildtype BRCA2 reference (Fig. 2c), indicating that the substituted amino acid (tryptophan, W) belongs to the same class of neutral polar amino acids as tyrosine (Y), thus unlikely changing the structure of BRCA2. This was further confirmed by whole exome sequencing (WES), which clearly showed the same heterozygous reversion mutation in one of the BRCA2 alleles in PEO1/OlaJR that was not shown in either PEO1 or PEO1/OlaR (Supplementary Table 1). These findings corroborate the immunoblotting analysis above and validate BRCA2 functionality in binding to RAD51 in PEO1/OlaJR.
Next, we measured the functionality of HR in OlapR variants by assessing RAD51 foci formation [28, 29]. Immunofluorescence staining analysis exhibited persistent RAD51 foci in PEO1/OlaJR cells even in the absence of PARPi treatment, while it was not detected in parental cells (Fig. 2d and Fig. 3a). Treatment with olaparib further increased RAD51 foci in PEO1/OlaJR (Fig. 2d), which was substantially reversed upon BRCA2 knockdown by siRNAs (Fig. 2d). Silencing BRCA2 also re-sensitized PEO1/OlaJR to olaparib (Fig. 2e), suggesting that at least in this cell line, resistance to PARPi seems to be BRCA2-dependent, further proving the functional nature of its BRCA2.
Fig. 3. Both PARPi-resistant PEO1 cell lines show intact HR repair.
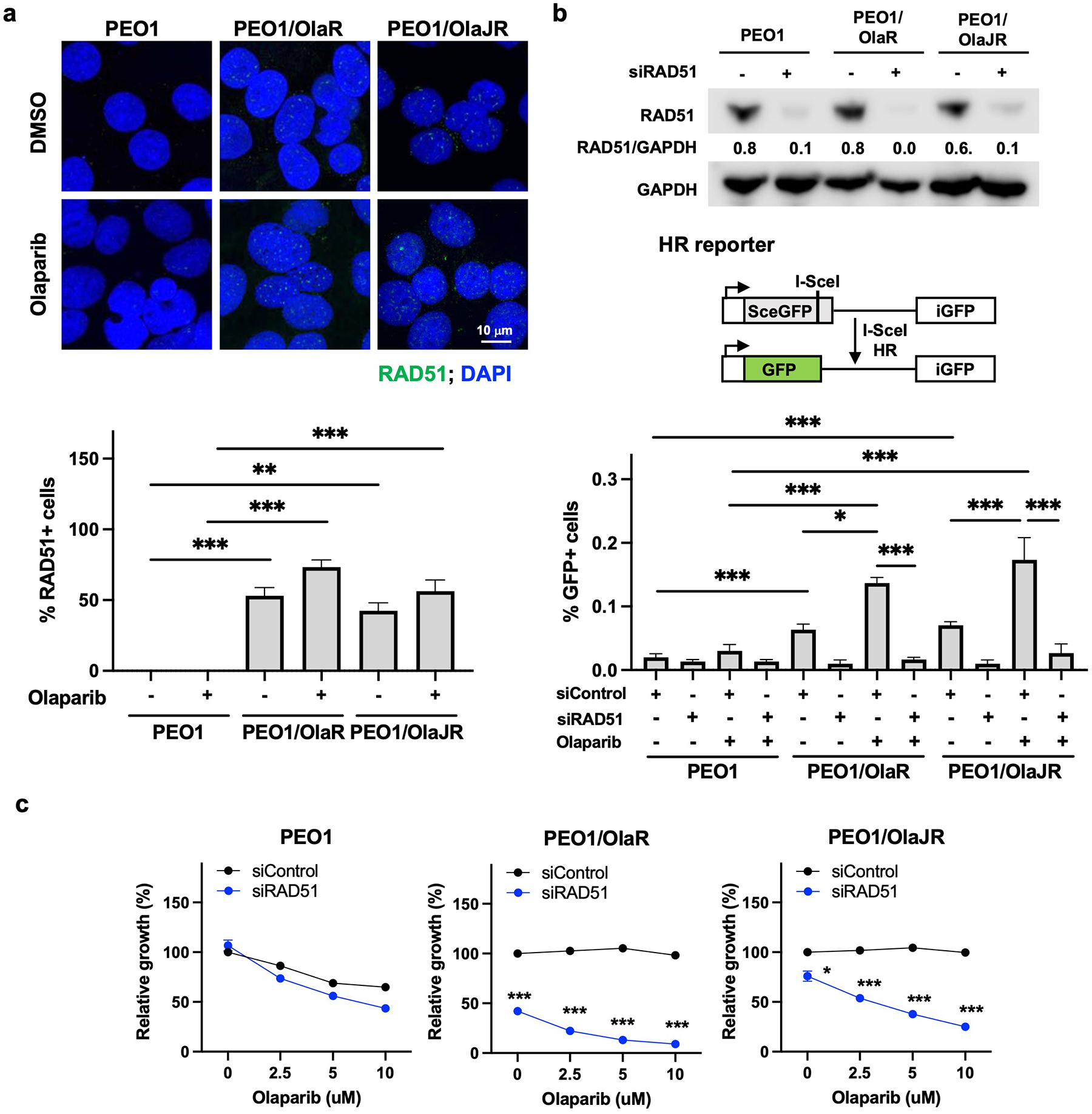
(a) Cells were treated with olaparib (5 μM) treatment for 48 hours. Immunofluorescence staining of RAD51 foci (green) was conducted for HR repair activity. Representative images were taken at 63x magnification (upper). Cells with > 5 RAD51 foci were counted as RAD51-positive (RAD51+) cells. The percentage of RAD51+ cells is plotted (bottom). (b) Immunoblotting of RAD51 in cells transfected with or without siRNAs against RAD51 (siRAD51, upper). DRGFP reporter assay was performed to measure the activity of HR repair as detailed in Materials and Methods (middle). The GFP-positive (GFP+) cells indicating HR-proficient cells were analyzed by flow cytometry (bottom). (c) Cell growth assays (XTT) were performed on cells transiently transfected with control siRNA (siControl) or RAD51-specific siRNAs (siRAD51) for 48 hours and treated with olaparib at indicated doses for a further 48 hours. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To further examine the effect of different treatment strategies on the genetic and mutational landscapes of OlapR cells, we conducted both WES and spectral karyotyping (SKY). While both OlapR lines showed a substantial number of new somatic mutations by WES analysis, it was ~3-fold higher in PEO1/OlaR (n=242) compared to PEO1/OlaJR (n=82) (Supplementary Table 2). Furthermore, translocations were the major type of chromosomal aberrations observed by SKY, and it was at least 2-fold more in PEO1/OlaR than in PEO1/OlaJR (Supplementary Fig. 2 and Supplementary Table 3), further affirming the higher degree of genomic instability in PEO1/OlaR (Supplementary Table 2).
PEO1/OlaR cells show increased HR activity in a BRCA2-independent manner
Similar to PEO1/OlaJR, persistent RAD51 foci formation was also observed in the BRCA2-deficient PEO1/OlaR cells (Fig. 3a), consistent with previous research on this cell line [16]. In both OlapR cell lines, treatment with olaparib further increased the percentage of RAD51-positive cells (Fig. 3a). HR reporter assays also demonstrated similar increases in HR activity in OlapR lines with PARPi treatment, while their parental line did not show comparable HR activity (Fig. 3b). This was largely reversed by silencing RAD51, suggesting RAD51 dependency in HR activity for OlapR cells (Fig. 3b). RAD51 depletion also reversed PARPi resistance in both OlapR cells (Fig. 3c), highlighting an essential role of HR activity in PARPi resistance irrespective of BRCA2 functionality.
Restoration of HR involves a stepwise resolution of DSBs, which is initiated by resection forming single-strand DNAs (ssDNAs), RPA binding and recruitment of active γH2AX followed by loading of 53BP1 and BRCA1 to the site of DSBs [30]. As such, we observed an increase in 53BP1 foci formation in both PEO1 and its OlapR variants upon PARPi treatment (Supplementary Fig. 5a). Notably, PEO1/OlaR persistently showed abundant 53BP1-positive cells at baseline and with PARPi treatment, indicating high levels of DSBs, which is in line with its significant genomic instability (Supplementary Fig. 2 and Supplementary Table 2–3). We also performed neutral comet assays to examine DSBs. Increased tail moments were shown in both PEO1 and PEO1/OlaJR cells treated with PARPi, while no significant difference was observed in PEO1/OlaR (Supplementary Fig. 5b), suggesting efficient repair of DSBs in PEO1/OlaR. The elevated basal levels of 53BP1 foci in PEO1/OlaR would also indicate a higher dependency on HR in this cell line than in BRCA2-dependent PEO1/OlaJR, which again agrees with the RAD51 silencing data observed earlier (Fig. 3c).
While resistance in PEO1/OlaJR was clearly attributed to a re-expression of functional BRCA2, mechanisms of BRCA2-independent HR in PEO1/OlaR remained unclear. Towards that, we performed WES and immunoblotting analysis to examine HR regulators that are known to restore HR in BRCA1-deficient cancers, such as 53BP1 [31, 32], REV7 [33], and its negative regulator TRIP13 [34]. However, no loss of 53BP1, REV7, or amplification of TRIP13 was found in both OlapR lines (Supplementary Table 2), nor were their levels any different from parental PEO1 (Supplementary Fig. 4a). Other reversion mutations of non-BRCA HR genes, e.g., PALB2, RAD51C, and RAD51D [13, 35], were also not observed in OlapR cells (Supplementary Table 2).
A slower replication fork progression characterizes PARPi-resistant cells
PARPi resistance can derive from reduced replication fork speed in BRCA-deficient cells [36]. We thus performed DNA fiber assays to study the dynamics of replication forks. Replication fork progression was significantly slower in both OlapR lines than in parental PEO1 (Fig. 4a), indicating possible stalled replication forks in OlapR cells. Notably, the speeds of replication fork were unchanged between no treatment and olaparib in PEO1/OlaR, although both OlapR lines showed reduced fork speeds compared to their parental cells (Fig. 4a, left). In contrast, PARPi treatment increased fork speeds in parental PEO1 and PEO1/OlaJR cells, consistent with a previous report for PEO1 [36].
Fig. 4. PARPi-resistant PEO1 cell lines slow fork progression through increasing unscheduled origin firing.
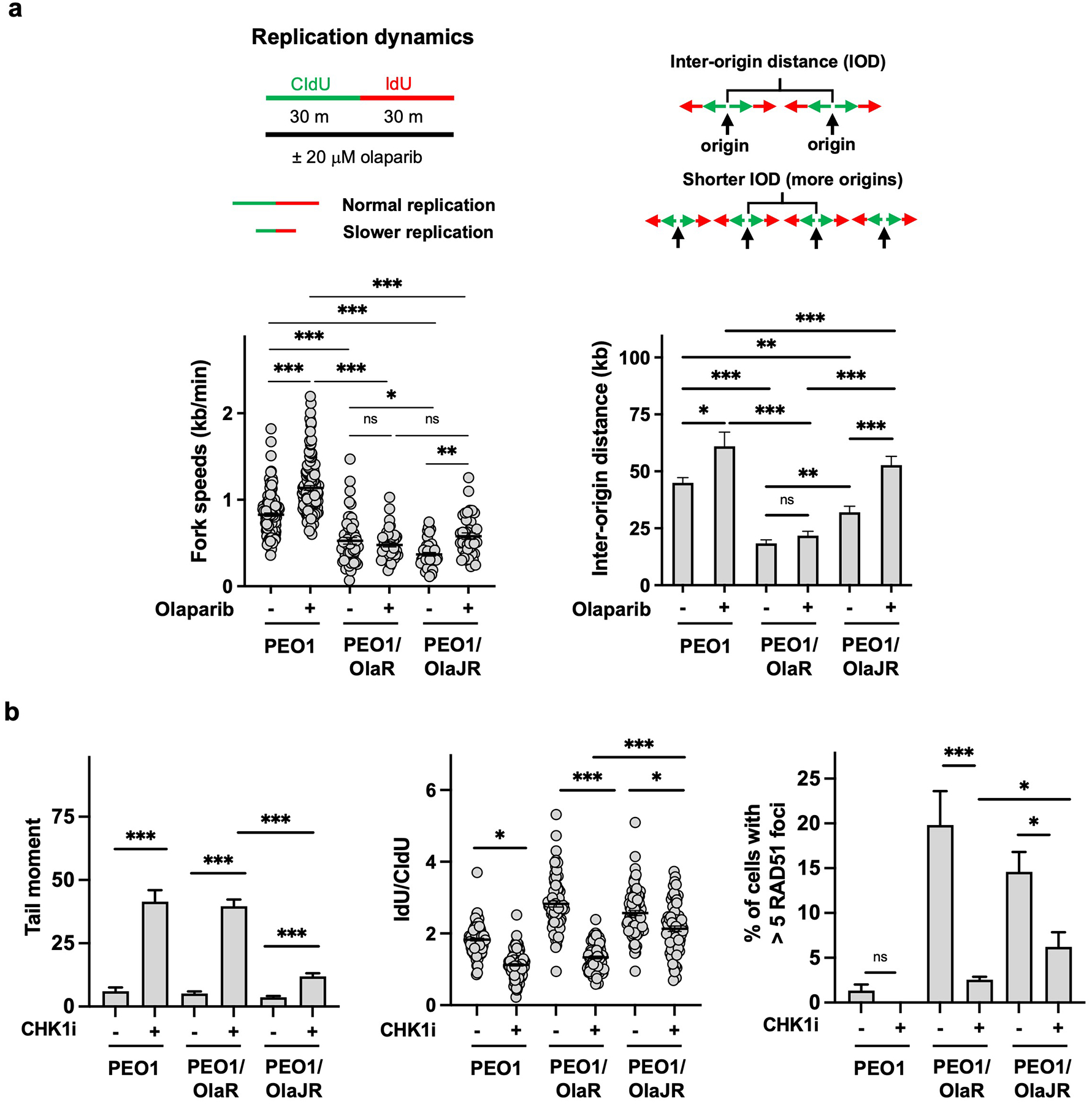
(a) DNA fiber assays were performed to study alterations in replication fork dynamics, as described in Materials and Methods. The schematics are shown above. Average replication fork speeds (bottom left) and inter-origin distances (bottom right) in each group are shown. (b) Cells were treated with or without CHK1 inhibitor (CHK1i) prexasertib and its effect on DNA damage quantitated by comet assay (left), fork stabilization assayed by DNA fiber assay (middle), and HR repair assessed by RAD51 foci (right). *, p < 0.05; ***, p < 0.001; ns, not significant.
Unscheduled origin firing is known to reduce fork speeds globally [37]. However, the inter-origin distances (IOD) were not substantially changed between untreated and PARPi-treated PEO1/OlaR cells despite shorter IOD seen in both OlapR lines at baseline (Fig. 4a, right). We, therefore, speculated that PEO1/OlaR may activate cell cycle checkpoint pathways to prevent unscheduled origin firings. Specifically, ATR/CHK1 pathway suppresses unscheduled origins through blocking the recruitment of CDC45 to the minichromosome maintenance 2–7 complex on replication forks [38] and its activation increases both HR repair and fork stability in HGSOC cells with acquired PARPi resistance [39]. As such, we found that CHK1 inhibition induced greater DNA damage (Fig. 4b, left) and fork degradation (Fig. 4b, middle) while decreasing RAD51 foci formation in PEO1/OlaR (Fig. 4b, right) compared to PEO1/OlaJR, suggesting PEO1/OlaR is highly dependent upon ATR/CHK1 signaling.
Loss of EZH2 or MUS81 stabilizes replication forks in PEO1/OlaR cells
Increased fork stability also contributes to PARPi resistance [39]. We treated OlapR cells with olaparib for 2 hours following CldU/IdU treatment to examine replication fork stabilization. No significant difference in fork degradation was observed between untreated and olaparib-treated groups in both OlapR lines, indicating intact fork stabilization, unlike parental PEO1 (Fig. 5a). Additionally, we used a replication inhibitor HU (2 mM) in the place of olaparib. As expected, HU treatment induced significant DNA resection activity in parental PEO1, while showing no effect on PEO1/OlaJR and displaying only a modest fork degradation in PEO1/OlaR compared to PEO1 (Supplementary Fig. 6), indicating substantial fork protection. Our results suggest that fork stabilization partly contributes to PARPi resistance, and this is more evident in BRCA2-proficient PEO1/OlaJR than in the BRCA2-deficient PEO1/OlaR.
Fig. 5. PARPi-resistant BRCA2-deficient cells increase fork protection via suppressing EZH2/MUS81 pathway.
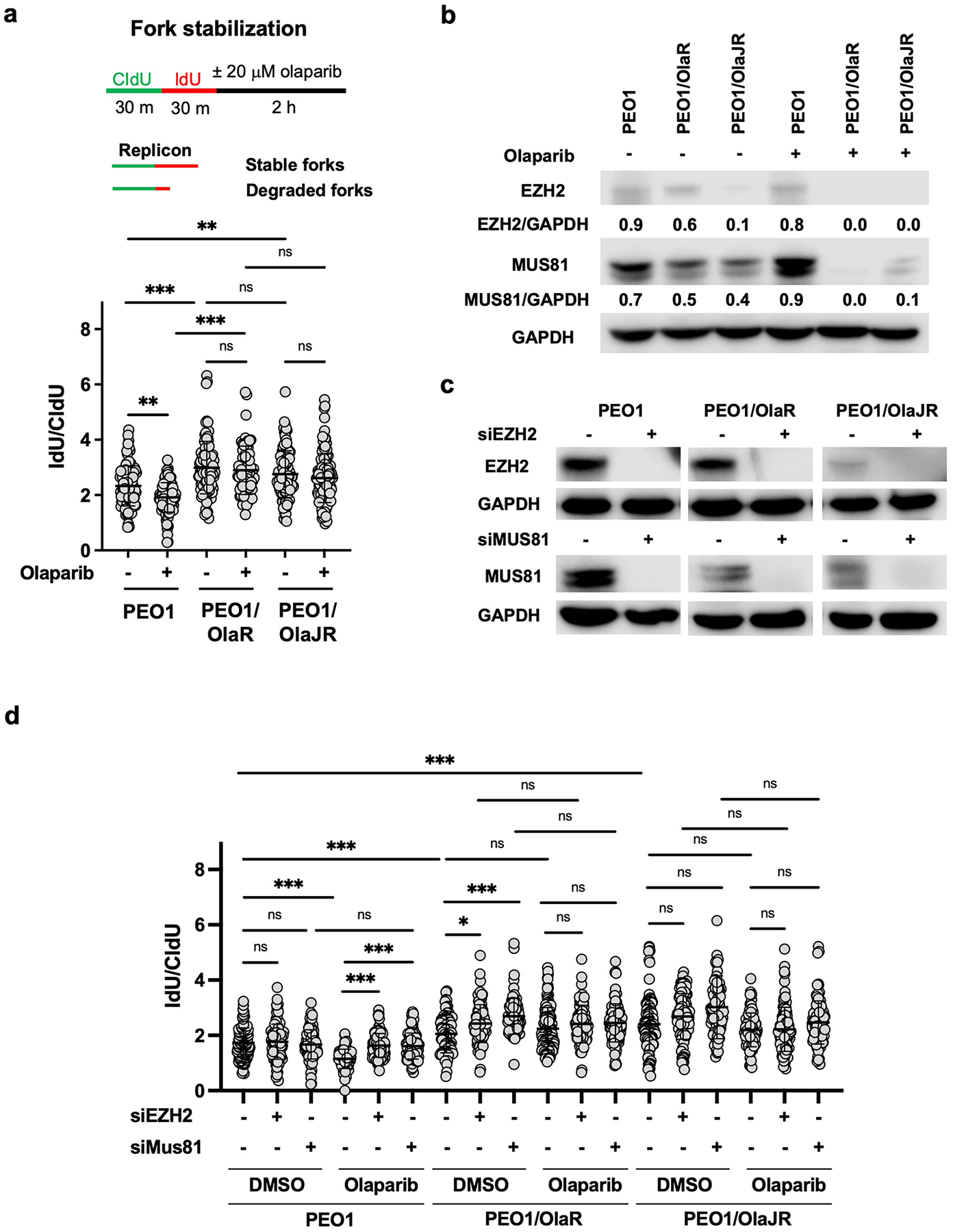
(a) Fork stabilization was determined by DNA fiber assay as described in Materials and Methods. The schematic is shown above. Plots of IdU/CldU tract length ratios for individual replication forks in treated cells are shown below the schematic. (b) Western blotting of EZH2, MUS81, and GAPDH in cells with or without olaparib treatment. Densitometric values of EZH2 and MUS81 relative to GAPDH are shown. (c) Immunoblotting of EZH2 and MUS81 in cells transfected with or without siRNAs against EZH2 or MUS81. (d) DNA fiber assays were performed to study replication fork protection as mentioned in (a). CldU/IdU pulse-labeling followed by a 2-hour olaparib (20 μM) with or without silencing of EZH2 or MUS81. Dot plots of IdU to CldU tract length ratios for individual replication forks in treated cells are shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Next, we investigated the effect of replication fork remodelers on OlapR cells. Data suggest that loss of EZH2 promotes PARPi resistance through reducing the recruitment of MUS81 at stalled forks, thus stabilizing the replication forks in BRCA2-deficient ovarian cancer cell lines [20]. Both OlapR lines showed lower baseline levels of EZH2 and MUS81 compared to their parental cells (Fig. 5b). Of note, levels of both proteins were further decreased upon olaparib treatment in OlapR cells, especially in PEO1/OlaR, while no obvious difference was observed in their parental cells (Fig. 5b). Therefore, we hypothesized that OlapR cells might have less need for EZH2/MUS81 cascade upon PARPi treatment, thus preventing fork collapse and DSBs. To test this hypothesis, we knocked down EZH2 or MUS81 using siRNAs (Fig. 5c) to study the importance of this pathway in OlapR cells. At baseline, depletion of EZH2 or MUS81 did not affect fork stability in PEO1 cells while reversing PARPi-induced fork degradation (Fig. 5d), which is in line with a previous report [20]. Consistent with the lower levels of these molecules observed in OlapR cells when treated with olaparib (Fig. 5b), no further advantage was found in fork stability in olaparib-treated cells when either EZH2 or MUS81 was silenced with siRNAs. Together, our findings suggest that loss of fork remodelers confers PARPi resistance in OlapR cells.
Enhanced ABCB1-mediated drug efflux activity confers PARPi resistance in PEO1/OlaJR
Besides replication fork protection, another resistance mechanism against PARPi involves drug efflux. Increased expression of drug efflux proteins like ABCB1 has been observed in PARPi-resistant cells in a BRCA2-dependent manner [40]. However, we noted elevated ABCB1 transcripts (Fig. 6a) and protein levels in both OlapR lines irrespective of their BRCA2 status (Fig. 6b). Notably, in de novo PARPi-resistant PEO4, only a weak expression of ABCB1 mRNA (Fig. 6a) and protein (Fig. 6b) was detected, suggesting that de novo and acquired resistance to PARPi might have differing ABCB1 dependencies.
Fig. 6. Enhanced ABCB1-mediated drug efflux activity contributes to acquired PARPi resistance.
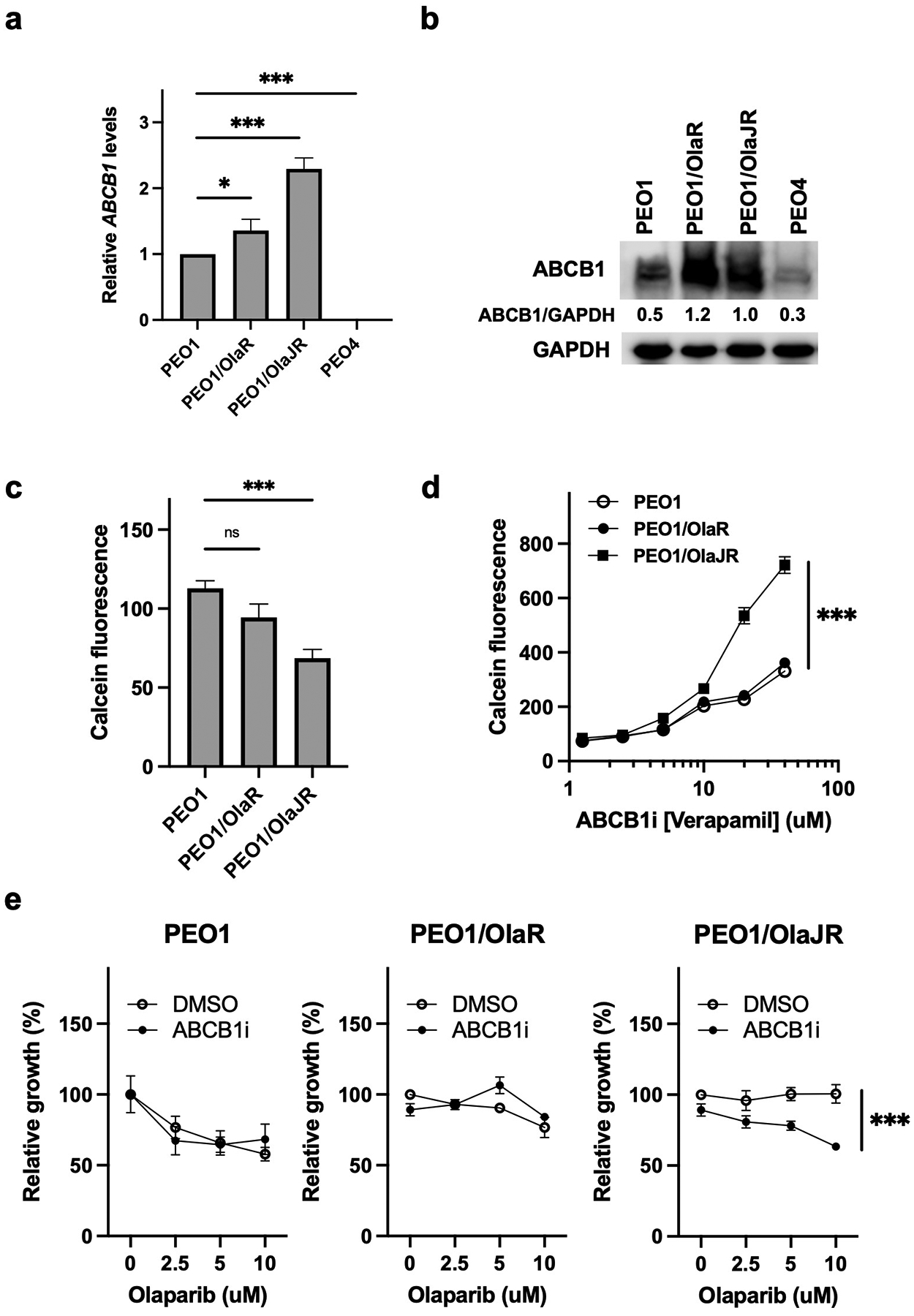
(a-b) Relative mRNA by qPCR (a) and protein levels of ABCB1 by immunoblotting analysis (b) in PEO1 and PARPi-resistant cell lines. (c-d) MDR activity assay was used to compare ABCB1-dependent efflux of the substrate CalceinAM in (c) untreated PEO1 and PARPi-resistant cell lines and (d) in the presence of 0–40 μM verapamil of above cell lines. (e) Cell growth was determined by XTT assays in cells treated with olaparib and/or ABCB1 inhibitor (ABCB1i, verapamil) at indicated doses for 72 hours. ***, p < 0.001.
Next, the multidrug drug resistance (MDR) activity assay was used to examine the activity of ABCB1 in cellular drug retention in OlapR cells. Decreased CalceinAM fluorescence was observed in PEO1/OlaJR but not in parental or PEO1/OlaR cells (Fig. 6c), indicating higher MDR activity in PEO1/OlaJR. Moreover, we used an ABCB1 inhibitor (ABCB1i, verapamil) to block the efflux pump activity. We found that ABCB1i not only induced a dose-dependent increase in CalceinAM fluorescence (Fig. 6d) but also re-sensitized PEO1/OlaJR to PARPi (Fig. 6e), which was again not observed in either PEO1 or PEO1/OlaR. These results indicate the activity of ABCB1 but not its expression level is a key factor of MDR-related PARPi resistance for PEO1/OlaJR.
Discussion
The effects of PARPi treatment schedules and dosages on the development of PARPi resistance remain elusive, while several preclinical [25, 31, 41, 42] and clinical studies [12, 35, 43] have reported multiple PARPi resistance mechanisms. In this study, we report that HR-competent PARPi-resistant variants develop distinct preferences in the resistance pathways via different treatment and selective pressures to be either BRCA2-deficient or -revertant.
PARPi resistant variants originated from the same parental BRCA2-deficient PEO1 cells. The resistant line, PEO1/OlaJR, which was established by initial higher doses and short-term PARPi exposure, develops acquired resistance by promoting drug efflux activity and re-expressing functional BRCA2 to restore HR. In contrast, PEO1/OlaR, developed by initial lower doses and long-term PARPi treatment, exhibited more complex mechanisms of PARPi resistance with a mesenchymal-like phenotype and restored HR repair as well as fork stabilization with constitutively activated ATR/CHK1 and suppressed EZH2/MUS81 pathways.
In line with this, previous data also suggest that PEO1/OlaR utilizes other survival mechanisms such as Wnt activation [16] and increased activity of euchromatic histone-lysine-N-methyltransferases 1 and 2 (EHMT1/2) that catalyzes dimethylation of histone H3 lysine 9 (H3K9me2) [44], which may partly explain BRCA2-independent HR restoration in PEO1/OlaR. EMT is a well-known phenomenon, related to metastasis and drug resistance in ovarian cancer [45–47] as well as in other cancers [25]. Accordingly, we noted increased mesenchymal markers N-cadherin and vimentin in mesenchymal-like PEO1/OlaR, consistent with a previous report in PARPi-resistant BRCA2-mutant breast cancer [41]. Thus, it is possible that N-cadherin may have contributed to PARPi resistance via activating pro-survival protein Bcl-2 [24, 25], requiring further investigation.
In HR repair, a functional BRCA2 is essential for RAD51 assembly [15], while other studies have identified putative pathways that enable robust RAD51 foci and HR in BRCA2-deficient cells [35, 48], though this involved targeted silencing of specific pathways. BRCA2 is also necessary for the replacement of RPA which binds and protects ssDNAs at stalled replication forks, and eventually replaced by RAD51 in a BRCA2-dependent manner [49]. While RAD51-dependent HR activity was observed in both OlapR cell lines, this was mediated in a BRCA2-redundant manner at least in the PEO1/OlaR variant. Silencing RAD51 partially re-sensitized this cell line to PARPi, while in the BRCA2-revertant PEO1/OlaJR, silencing either RAD51 or BRCA2 proved to be lethal.
As Bitler’s group previously demonstrated the loading of RPA and RAD51 to DSBs via activated Wnt and EHMT1/2, as a means of HR restoration in PEO1/OlaR [16, 50], we questioned if other pathways, e.g., the cell cycle checkpoint ATR/CHK1 signaling, which also regulates RAD51 recruitment to the sites of DNA damage [51] were involved in this process as well. We found that CHK1 inhibition induced greater DNA damage and reduction in RAD51 foci formation in PEO1/OlaR compared to PEO1/OlaJR, suggesting the involvement of ATR/CHK1 signaling in PARPi resistance in PEO1/OlaR. Also, others reported upregulation of ATR/CHK1 pathway in BRCA-mutant ovarian cancer cells as a means of acquired PARPi resistance [21, 52], supporting our notion. Together, these findings suggest that PEO1/OlaR exhibits a complex interplay of survival signaling activation and transcriptional programming to support its HR activity while PEO1/OlaJR has relatively fewer mechanisms of PARPi resistance.
Also, it is worth noting that both OlapR cell lines maintain substantial replication fork stability upon PARPi treatment, which may be explained by reliance on the EZH2/MUS81 axis. Rondinelli et al. reported that loss of EZH2 decreases the recruitment of MUS81 on stalled replication forks, thus preventing fork degradation and conferring PARPi resistance in a panel of BRCA2-deficient but not in BRCA1-deficient ovarian and breast cancer cell lines [20], suggesting this resistant mechanism is peculiar to BRCA2 deficiency. These data explain in part why the loss of either EZH2 or MUS81 exhibited increased fork stability at baseline in BRCA2-deficient PEO1/OlaR but not in BRCA2-proficient PEO1/OlaJR, though the latter also showed lower EZH2 and MUS81 levels relative to parental PEO1. Exploring potential molecules that negatively affect EZH2 and MUS81 proteins in these OlapR variants requires further studies.
Lastly, ABCB1-mediated drug efflux also plays a role in PARPi resistance as shown in BRCA2-deficient breast cancer models [40]. However, we found that only BRCA2-proficient PEO1/OlaJR displays increased drug efflux activity mediated by ABCB1, which was absent in its BRCA2-deficient counterpart PEO1/OlaR. Of note, though increased ABCB1 expression was observed in PEO1/OlaR cells, its inhibition failed to show any change in PARPi sensitivity or intracellular drug retention. Furthermore, the de novo PARPi-resistant PEO4 with BRCA2 restoration did not exhibit any apparent ABCB1 involvement, suggesting the drug efflux ability of PEO1/OlaJR may not entirely rely on BRCA2. These data further confirm the importance of assessing the activity before concluding the role of different pathways in PARPi resistance.
In summary, we demonstrate for the first time that PARPi resistance is governed by the nature of treatment strategies that preceded the development of resistance and this is again influenced by the BRCA2 status of the OlapR cells. This is particularly important in the clinic where it is not uncommon to have patients that have received multiple PARPis from different treatment strategies and combination therapies, reinforcing the complex biology of drug resistance in the clinical setting. Hence, it is possible that different treatment durations and strategies can influence the evolution of different resistance mechanisms that co-exist in the same patient or in different patients from the same PARPi treatment. Future studies for mining clinical specimens with different treatment schemes for PARPis need to be investigated for better therapeutic strategies in overcoming PARPi resistance in ovarian cancer.
Materials and Methods
Cell growth, invasion, immunoblotting, immunofluorescence staining, and neutral comet assays were detailed in Supplementary Materials and Methods.
Drug preparation
Olaparib (#S1060) and rucaparib (#S1098) were purchased from Selleck Chemicals, Houston, TX. Hydroxyurea (HU; #H8627) was from Sigma-Aldrich. ADH-1 (N-cadherin inhibitor; #HY-13541) and prexasertib (CHK1 inhibitor; #HY-18174) were from MedChemExpress (Monmouth Junction, NJ). 100 mM of olaparib, 10 mM of rucaparib, HU, ADH-1, and prexasertib were prepared as stocks in dimethyl sulfoxide (DMSO; #S-002-M, Sigma-Aldrich, Saint Louis, MO). All drugs were stored in aliquots at −80 °C until use.
Cell lines
PEO1 (BRCA2 mutation 5193C>G, Y1655X; #10032308–1VL) and PEO4 (BRCA2 reversion mutation 5193C>T, Y1655Y; #10032309–1VL) HGSOC cell lines were purchased from Sigma-Aldrich. To establish OlapR cells, we cultured PEO1 cells in gradually increasing olaparib concentrations from 0.5 μM to 40 μM over 15 weeks and confirmed their resistance to olaparib by a 3-day XTT assay (PEO1/OlaJR). The second OlapR line was also developed from PEO1 by Dr. Bitler’s group [16], where the cells were cultured by increasing concentrations of olaparib from 5 nM to 20 μM over 24 weeks, and four BRCA2-mutant clones were selectively isolated and mixed to produce PEO1/OlaR [16]. All cells were cultured in RPMI1640 medium with (+) l-glutamine supplemented with 10% fetal bovine serum (FBS), 0.01 mg/mL insulin, and 1% penicillin/streptomycin. PEO1/OlaR was routinely maintained at 2 μM of olaparib while PEO1/OlaJR at 20 μM of olaparib. Cells were cultured without olaparib for at least 3 days prior to experiments.
PCR amplification and Sanger sequencing
The exon11 of BRCA2 was amplified using primers that flank the target region 5’TACACAAA3’ (4963–4970 positions of reference mRNA sequence NM_000059.4 from GenBank). Two sets of primers BRC2For2 5’-AGATCACAGCTGCCCCAAAG-3’ and BRC2Rev2 5’-CAGCCTTAGCTTTTTACACAAGT-3’; BRC2For4 5’-CCATTGAGATCACAGCTGCC-3’ and BRC2Rev4 5’-ACTTCTGTGAGTCAGACTTCATT-3’ were used to generate 200–300 bp amplicons. The amplicons were further sequenced by Sanger sequencing on an applied biosystems sequencer, and the raw ABI trace files were analyzed using BioEdit™ software [53].
siRNA transfections
Cells were transfected with siRNA against BRCA2, RAD51, EZH2, MUS81, or scrambled control for 48 hours using DharmaFECT 1 siRNA Transfection Reagent (#T-2001–01, Horizon Discovery, Lafayette, CO) per manufacturer’s protocol and subjected to cell proliferation assays. The following ON-TARGETplus SMARTpool-Human siRNA oligonucleotides from Horizon Discovery were used: BRCA2 (#L-003462-00-0010), EZH2 (#L-004218-00-0005), RAD51 (#L-003530-00-0005), MUS81 (#L-016143-01-0005), and non-targeting control pool (#D-001810-01-05) as negative control. The target sequences of BRCA2 and RAD51 siRNAs are as follows: BRCA2, 5′-GAAACGGACUUGCUAUUUA-3′, 5′-GGUAUCAGAUGCUUCAUUA-3’, 5′-GAAGAAUGCAGGUUUAAUA-3’, 5′-UAAGGAACGUCAAGAGAUA-3’; RAD51, 5′-UAUCAUCGCCCAUGCAUCA-3′, 5′-CUAAUCAGGUGGUAGCUCA-3′, 5′-GCAGUGAUGUCCUGGAUAA-3’, 5′-CCAACGAUGUGAAGAAAUU-3’; EZH2, 5’-GAGGACGGCUUCCCAAUAA-3’, 5’-GCUGAAGCCUCAAUGUUUA-3’. 5’-UAACGGUGAUCACAGGAUA-3’, 5’-GCAAUUCUCGGUGUCAAA-3’; MUS81, 5’-CAGCCCUGGUGGAUCGAUA-3’, 5’-CAUUAAGUGUGGGCGUCUA-3’, 5’-UGACCCACACGGUGCGCAA-3’, 5’-CUCAGGAGCCCGAGUGAUA-3’.
Immunoprecipitation (IP) of BRCA2
The mouse anti-BRCA2 antibody purchased from Sigma (#OP95) was used for the IP of BRCA2 in cells. The immunoprecipitation was conducted using Recombinant Protein G Agarose following manufacturer’s recommendations (#15920010, Invitrogen).
Quantitative real-time PCR (qPCR) analysis
The mRNA expressions of ABCB1 gene were validated by qPCR. Total RNA of cells was isolated using RNeasy™ Micro kit (Qiagen, Germantown, MD). The cDNA was generated using the Superscript™ First-strand synthesis system (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s instructions. Primers specific for ABCB1 (Assay ID: Hs.PT.58.26805128) and for endogenous control GAPDH were obtained from Integrated DNA Technologies, Coralville, IA. qPCR was performed on an ABI ViiA7 Real-Time PCR system (Applied Biosystems, Carlsbad, CA) and analyzed with QuantStudio™ Real-Time PCR software.
HR reporter assay
HR repair activity was measured by a HR reporter assay [54, 55]. Briefly, cells were transfected with plasmid-carrying pDRGFP (#26475, Addgene, Cambridge, MA) and pCBASceI (#26477, Addgene) plasmid with Lipofectamine 2000 transfection reagent (#11668019, Thermo Fisher Scientific). After 72 hours, cells were collected and examined by Sony SA3800 Spectral Analyzer (Sony Biotechnology USA, San Jose, CA) to quantify the GFP-positive cells.
DNA fiber assay
DNA fiber assays were conducted to assess replication fork stabilization and progression as described before [56]. To measure replication fork dynamics, cells were labeled with 100 μM 5-chloro-2’-deoxyuridine (CldU; #C6891, Sigma-Aldrich) and 200 μM 5-Iodo-2′-deoxyuridine (IdU; #I7125, Sigma-Aldrich) for 30 minutes each with or without 20 μM olaparib. Replicating 1 μm roughly corresponds to 2.59 kb [37]. Also, cells were labeled with CldU and IdU for 30 minutes each, followed by treatment with or without 20 μM olaparib or 2 mM of HU for another 2 hours to examine replication fork stabilization. Cells were lysed with lysis buffer (1% sodium dodecyl sulfate, 100 mM Tris-HCl [pH 7.4], 50 mM EDTA). Labeled DNAs with CldU and IdU were stained with mouse anti-IdU primary antibody (1:250; #NBP2–44056, Novus Biological, Centennial, CO) and rat anti-CldU primary (1:200; #NB500–169, Novus Biological), respectively. Anti-rat Alexa Fluor 488 (1:250; #A-11006, Thermo Fisher Scientific) and anti-mouse Alexa 594 (1:250; #A-11005, Thermo Fisher Scientific) were used for secondary antibodies. Images were captured with a Zeiss LSM 780 confocal microscope. Fiber length was measured using ImageJ software.
MDR activity assay
Cells were incubated with 0.25 μM calceinAM ± verapamil (0–40 μM) for 15 minutes at 37 °C. Assessment of basal drug efflux activity and the influence of the ABCB1 inhibitor verapamil in parental PEO1 and its OlapR lines were performed using the Vybrant Multidrug Resistance Assay Kit (#V13180, Life Technologies, Carlsbad, CA) according to manufacturer’s instructions. Intracellular calceinAM fluorescence was measured by Synergy™ HTX Multi-Mode Microplate Reader with Gen5™ software (BioTek Instruments).
Statistical analysis
All experiments were performed in triplicate and randomly assigned to treatment groups. Data were analyzed using one-way ANOVA test and shown as mean ± SEM. All differences were considered statistically significant if p < 0.05. All statistical analyses were done using GraphPad Prism v.7.1 (GraphPad Software). Investigators were blinded during data collection and analysis.
Supplementary Material
Funding
This research was funded by the intramural research program of the CCR, NCI, NIH (ZIA BC011525 awarded to J.M.L.). This study was partly supported by an NIH/NCI grant (R37CA261987-01 awarded to B.G.B.).
Footnotes
Conflicts of interest
All the authors declare no conflicts of interest.
References
- 1.Howlader NNA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J et al. SEER Cancer Statistics Review, 1975–2018, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2018/, based on November 2020 SEER data submission, posted to the SEER web site, April 2021.
- 2.Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol 2014; 25: 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King MC, Marks JH, Mandell JB, New York Breast Cancer Study G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003; 302: 643–646. [DOI] [PubMed] [Google Scholar]
- 4.Tew WP, Lacchetti C, Ellis A, Maxian K, Banerjee S, Bookman M et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J Clin Oncol 2020; 38: 3468–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noordermeer SM, van Attikum H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol 2019; 29: 820–834. [DOI] [PubMed] [Google Scholar]
- 6.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008; 451: 1111–1115. [DOI] [PubMed] [Google Scholar]
- 7.Haynes B, Murai J, Lee JM. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat Rev 2018; 71: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JM, Gulley JL. Checkpoint and PARP inhibitors, for whom and when. Oncotarget 2017; 8: 95036–95037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drean A, Williamson CT, Brough R, Brandsma I, Menon M, Konde A et al. Modeling Therapy Resistance in BRCA1/2-Mutant Cancers. Mol Cancer Ther 2017; 16: 2022–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 2018; 71: 172–176. [DOI] [PubMed] [Google Scholar]
- 11.Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol 2021; 18: 773–791. [DOI] [PubMed] [Google Scholar]
- 12.Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov 2019; 9: 210–219. [DOI] [PubMed] [Google Scholar]
- 13.Pettitt SJ, Frankum JR, Punta M, Lise S, Alexander J, Chen Y et al. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov 2020; 10: 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol 2011; 29: 3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res 1999; 59: 3547–3551. [PubMed] [Google Scholar]
- 16.Yamamoto TM, McMellen A, Watson ZL, Aguilera J, Ferguson R, Nurmemmedov E et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol Carcinog 2019; 58: 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 2017; 68: 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 2017; 67: 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun 2017; 8: 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 2017; 19: 1371–1378. [DOI] [PubMed] [Google Scholar]
- 21.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 2020; 11: 3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez MK, Illuzzi G, Colomer C, Churchman M, Hollis RL, O’Connor MJ et al. Identifying and Overcoming Mechanisms of PARP Inhibitor Resistance in Homologous Recombination Repair-Deficient and Repair-Proficient High Grade Serous Ovarian Cancer Cells. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMullen M, Karakasis K, Madariaga A, Oza AM. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran NL, Adams DG, Vaillancourt RR, Heimark RL. Signal transduction from N-cadherin increases Bcl-2. Regulation of the phosphatidylinositol 3-kinase/Akt pathway by homophilic adhesion and actin cytoskeletal organization. J Biol Chem 2002; 277: 32905–32914. [DOI] [PubMed] [Google Scholar]
- 25.Allison Stewart C, Tong P, Cardnell RJ, Sen T, Li L, Gay CM et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017; 8: 28575–28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res 2009; 69: 6381–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng J, Xu J. RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins 2011; 79 Suppl 10: 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A 1995; 92: 2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JM, Gordon N, Trepel JB, Lee MJ, Yu M, Kohn EC. Development of a multiparameter flow cytometric assay as a potential biomarker for homologous recombination deficiency in women with high-grade serous ovarian cancer. J Transl Med 2015; 13: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer 2020; 19: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 2013; 3: 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015; 521: 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L et al. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol 2020; 22: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov 2017; 7: 984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell 2021; 81: 3227. [DOI] [PubMed] [Google Scholar]
- 37.Toledo L, Neelsen KJ, Lukas J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol Cell 2017; 66: 735–749. [DOI] [PubMed] [Google Scholar]
- 38.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 2017; 18: 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta N, Huang T, Horibata S, Lee J. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor–resistant cancer. Pharmacol Res 2022; 178: 106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaspers JE, Sol W, Kersbergen A, Schlicker A, Guyader C, Xu G et al. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res 2015; 75: 732–741. [DOI] [PubMed] [Google Scholar]
- 41.Ordonez LD, Hay T, McEwen R, Polanska UM, Hughes A, Delpuech O et al. Rapid activation of epithelial-mesenchymal transition drives PARP inhibitor resistance in Brca2-mutant mammary tumours. Oncotarget 2019; 10: 2586–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 2017; 31: 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Do KT, Kochupurakkal B, Kelland S, de Jonge A, Hedglin J, Powers A et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin Cancer Res 2021; 27: 4710–4716. [DOI] [PubMed] [Google Scholar]
- 44.Watson ZL, Yamamoto TM, McMellen A, Kim H, Hughes CJ, Wheeler LJ et al. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin Epigenetics 2019; 11: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang RY, Chung VY, Thiery JP. Targeting pathways contributing to epithelial-mesenchymal transition (EMT) in epithelial ovarian cancer. Curr Drug Targets 2012; 13: 1649–1653. [DOI] [PubMed] [Google Scholar]
- 46.Baribeau S, Chaudhry P, Parent S, Asselin E. Resveratrol inhibits cisplatin-induced epithelial-to-mesenchymal transition in ovarian cancer cell lines. PLoS One 2014; 9: e86987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012; 12: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clements KE, Schleicher EM, Thakar T, Hale A, Dhoonmoon A, Tolman NJ et al. Identification of regulators of poly-ADP-ribose polymerase inhibitor response through complementary CRISPR knockout and activation screens. Nat Commun 2020; 11: 6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhat KP, Cortez D. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 2018; 25: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Q, Zhu Q, Lu X, Du Y, Cao L, Shen C et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc Natl Acad Sci U S A 2017; 114: E6054–E6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 2005; 7: 195–201. [DOI] [PubMed] [Google Scholar]
- 52.Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C et al. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res 2017; 23: 3097–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hall T,A BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symposium Series 1999; 41: 95–98. [Google Scholar]
- 54.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13: 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richardson C, Moynahan ME, Jasin M. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 1998; 12: 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nair J, Huang TT, Murai J, Haynes B, Steeg PS, Pommier Y et al. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020; 39: 5520–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.