

Abstract
Dysferlin-null A/J myofibers generate abnormal Ca2+ transients that are slightly reduced in amplitude compared to controls. These are further reduced in amplitude by hypoosmotic shock and often appear as Ca2+ waves (Lukyanenko et al., J. Physiol., 2017). Ca2+ waves are typically associated with Ca2+-induced Ca2+ release, or CICR, which can be myopathic. We tested the ability of a permeable Ca2+ chelator, BAPTA-AM, to inhibit CICR in injured dysferlin-null fibers and found that 10–50 nM BAPTA-AM suppressed all Ca2+ waves. The same concentrations of BAPTA-AM increased the amplitude of the Ca2+ transient in A/J fibers to wild type levels and protected transients against the loss of amplitude after hypoosmotic shock, as also seen in wild type fibers. Incubation with 10 nM BAPTA-AM led to intracellular BAPTA concentrations of ∼60 nM, as estimated with its fluorescent analog, Fluo-4AM. This should be sufficient to restore intracellular Ca2+ to levels seen in wild type muscle. Fluo-4AM was ∼10-fold less effective than BAPTA-AM, however, consistent with its lower affinity for Ca2+. EGTA, which has an affinity for Ca2+ similar to BAPTA, but with much slower kinetics of binding, was even less potent when introduced as the -AM derivative. By contrast, a dysferlin variant with GCaMP6fu in place of its C2A domain accumulated at triad junctions, like wild type dysferlin, and suppressed all abnormal Ca2+ signaling. GCaMP6fu introduced as a Venus chimera did not accumulate at junctions and failed to suppress abnormal Ca2+ signaling. Our results suggest that leak of Ca2+ into the triad junctional cleft underlies dysregulation of Ca2+ signaling in dysferlin-null myofibers, and that dysferlin’s C2A domain suppresses abnormal Ca2+ signaling and protects muscle against injury by binding Ca2+ in the cleft.
Keywords: CICR, Ca-induced Ca release, dysferlinopathy, GCaMP, BAPTA, injury, osmotic shock
Introduction
Dysferlin is an integral membrane protein of skeletal muscle that is missing in individuals with Limb Girdle Type 2B (LGMD2B), Miyoshi Myopathy (MMD1) and other, rarer muscular dystrophies (Aoki and Takahashi 1993; Urtizberea et al., 2008; Amato and Brown, 2011; Fanin and Angelini 2016). Studies of membrane repair in skeletal muscle have suggested that dysferlin plays a key role in the process (Bansal et al., 2003; Bansal and Campbell 2004; Ho et al., 2004; Glover and Brown 2007; Han and Campbell 2007), though its role may be secondary to that of other repair proteins (Lennon et al., 2003; Demonbreun et al., 2016; Demonbreun and McNally 2016; McDade et al., 2021). This is consistent with results from our laboratory showing that membrane repair after eccentric injury of skeletal muscle in vivo is not impaired by the absence of dysferlin (Roche et al., 2010), and that deficits in membrane integrity in dysferlin-null muscle are due in part to secondary effects associated with inflammation (Roche, et al., 2010; Roche et al., 2015). The subcellular localization of dysferlin, originally reported to be at the sarcolemma and in intracellular vesicles (Matsuda et al., 1999; Bansal et al., 2003; Cenacchi et al., 2005), has more recently been demonstrated to concentrate primarily in the transverse tubular membranes at triad junctions (Roche et al., 2011; Kerr et al., 2013), where the bulk of the protein is exposed to the sarcoplasm and the short C-terminal sequence is extracellular (Kerr et al., 2013).
This localization suggested to us that dysferlin might play a role in regulating Ca2+ signaling in healthy muscle, and that the dystrophic phenotypes that arise in its absence may be due in part to changes in the regulation of Ca2+, which are believed to be pathogenic in many diseases of skeletal muscle (e.g., Allen et al., 2010; Burr and Molkentin 2015; Kushnir et al., 2018; Mareedu et al., 2021). We tested this idea by studying the Ca2+ transients of dysferlin-null A/J myofibers in culture, before and after mild injury caused by a brief hypoosmotic shock. Our previous studies had shown that the transients generated in A/J fibers were ∼15% smaller than those in control myofibers or in fibers transfected to express dysferlin (Lukyanenko et al., 2017; Muriel et al., 2022). After osmotic shock, however, A/J fibers showed a precipitous drop in the amplitude of Ca2+ transients compared to controls. In addition, many shocked fibers showed spontaneous Ca2+ transients accompanied by Ca2+ sparks and waves, typically associated with Ca2+-induced Ca2+ release (CICR). These phenotypes were completely reversed by transfection of fibers to express dysferlin or by treatment of the fibers before and during hypoosmotic shock with drugs that block the L-type Ca2+ channels (LTCC; also known as dihydropyridine receptors, DHPR) and ryanodine receptors (RyR1) that mediate the release of Ca2+ (Lukyanenko et al., 2017). We interpreted these results to mean that dysferlin in healthy muscle suppresses CICR and that upon injury in the absence of dysferlin, CICR becomes an important contributor to Ca2+ signaling, potentially contributing to pathogenesis (Kerr et al., 2014).
Here we test this idea by examining the effects of a permeable form of BAPTA, a chelator with an affinity for Ca2+ of ∼160 nM. We incubated control and dysferlin-null A/J myofibers with very low concentrations of BAPTA-AM, insufficient to affect the levels of Ca2+ generated during a Ca2+ transient, and then studied the effects on the transients, before and after hypoosmotic shock injury. Our experiments show that, upon uptake into muscle fibers to a concentration we estimate at ∼60 nM, BAPTA-AM is sufficient to revert A/J fibers to the control Ca2+ signaling phenotype. We then tested two other Ca2+ chelators, Fluo-4 and EGTA, each introduced as their -AM derivatives. These reagents bind Ca2+ with lower affinity (Fluo-4) or slower kinetics (EGTA) and thus higher concentrations were needed to generate results similar to BAPTA’s. Finally, we examined myofibers expressing dysferlin containing GCaMP6fu (Helassa et al., 2016) in place of its most N-terminal C2 domain (GCaMP6fu-DYSF-ΔC2A). This molecule targets the triad junction like wild type (WT) dysferlin and like WT dysferlin it supports Ca2+ signaling at WT levels. In comparison, GCaMP6fu alone is less potent. Our results suggest that leak of Ca2+ into the triad junction underlies dysregulation of Ca2+ signaling in dysferlin-null myofibers. They further suggest that dysferlin’s ability to bind Ca2+ in the triad junction is sufficient to protect muscle from changes in Ca2+ signaling induced by injury or exercise.
Materials and methods
Ethical approval
All procedures involving mice complied with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1996). Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine.
Mice
Dysferlin-null (A/J) and control (A/JCr, C57Bl/6) mice were obtained from the Jackson Laboratory (A/J, C57Bl/6) or the National Cancer Institute, Frederick, MD (A/JCr) or bred at the University of Maryland, Baltimore (C57Bl/6). Mice were anesthetized with 2.5–4.5% isoflurane vaporized in oxygen and euthanized by cervical dislocation. Mice were 12–16 weeks of age at the time their tissues were studied.
Plasmid constructs and transfection
mVenus-dysferlin (N-terminal Venus) (Addgene plasmid 29,768) (Covian-Nares et al., 2010) was provided by The Jain Foundation (www.jain-foundation.org). The constructs carrying Venus-dysferlin and Venus-dysferlin missing the C2A domain (residues 1–107) have been reported (Kerr et al., 2013; Muriel et al., 2022). For insertion of the GCaMP6fu sequence (AddGene), we deleted the Venus moiety from Venus-dysferlin missing the C2A domain with NheI and KpnI and replaced it with GCaMP6fu using the same restriction sites. This placed the GCaMP6fu sequence in frame with the rest of the dysferlin ORF.
In vivo gene transfer via electroporation into FDB fibers was adapted from published methods (DiFranco et al., 2009), as described (Kerr et al., 2013; Lukyanenko et al., 2017; Muriel et al., 2022). Venus-dysferlin (V-Dysf), GCaMP6fu and their variants were visualized in cultured myofibers (see below) with a Zeiss Duo Laser Scanning Confocal System (Carl Zeiss, Thornwood, NY), equipped with a C-Apochromat 40×/1.20 W Korr objective. Fluorescence excitation and emission were at 488 and >505 nm, respectively, with the laser intensity attenuated to 1%.
Isolation of myofibers from FDB muscle
Mice were anesthetized and FDB muscles from both feet were harvested. A 2 week period was allowed after electroporation. Single myofibers were prepared in DMEM with 2% (wt/vol) BSA, 1 μl/ml gentamicin, and 2 mg/ml type II collagenase (Gibco, ThermoFisher, Waltham, MA) for 2 h at 37°C. Myofibers were kept for 12–14 h at 37°C and plated on 96-well plates coated with laminin (Sigma-Aldrich, St. Louis, MO) 1 h before experimentation. Fibers were then washed in normal Tyrode’s solution, pH 7.4, containing 140 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, 0.3 mM NaH2PO4, 5 mM HEPES, 5.5 mM glucose, 1.8 mM CaCl2.
Confocal imaging
Isolated FDB fibers were loaded for 45 min at 37°C with 4.4 µM Rhod-2AM in the presence of 0.25% Pluronic F-127 (both from ThermoFisher), diluted in culture medium, then washed with Tyrode’s solution. When BAPTA-AM, Fluo-4AM or EGTA-AM was used, it was included in the same solution at concentrations from 0 to 250 nM.
Rhod-2 was excited with the 560 nm laser line with the intensity attenuated to 0.5% and emission was monitored at >575 nm with a LP 575 filter. Fluo-4 or GCaMP6fu were excited by light at 488 nm (25 mW argon laser, intensity attenuated to 1%) and fluorescence was measured at wavelengths of >515 nm. Trains of voltage pulses transients were induced by field stimulation (1 Hz for 10 s) every 1 min for 5 min, as reported (Lukyanenko et al., 2017; Muriel et al., 2022). Perfusions and imaging were done in the dark.
Line-scan images were taken in the middle of myofibers at 1.9 ms per line at maximal aperture. ImageJ 1.31v (NIH, Bethesda, United States) averaged the profiles for every pixel over time and took the maximal value for each of 10 voltage pulses to calculate the mean maximal value of the Ca2+ transients, which were typically 225 pixels in width. The difference between maximal fluorescence intensity (F max) and background fluorescence (F o ), normalized to F o . is reported.
As the amplitudes of the Ca2+ transients in electroporated myofibers are higher than those in controls (Lukyanenko et al., 2017), we analyzed results from electroporated and non-electroporated samples separately, with differences analyzed with the paired Student t-test for the former, to compare regions that expressed the transgene with regions that did not, and a simple t test for the latter.
Osmotic shock injury
Osmotic shock injury (OSI) was induced as described (Lukyanenko et al., 2017). In brief, cultured FDB fibers were superfused with normal Tyrode’s solution and then for 60 s with a hypotonic Tyrode’s solution containing 70 mM NaCl. Cells were then perfused with isotonic Tyrode’s solution for 5 min. Experiments were performed at room temperature (21–23°C). Data were collected from myofibers cultured from 2 or more mice.
Calibration of Fluo-4
Calibration was done in the glass-bottom chambers used for culturing myofibers. Solutions containing known concentrations of Fluo-4 were added in the solution (mM): 10 HEPES, 264.2 KCl, 5 EGTA, 2.7 CaCl2 (to yield [Ca2+]free = 100 nM), 1.1 MgCl2 (to yield [Mg2+]free = 1 mM), pH 7.4. The concentrations were calculated with WEBMAXC STANDARD. Line-scan images through the solution were taken at 1.9 ms per line at maximal aperture. Averaged data from 5 experiments were used to build the calibration curve. We used identical conditions to scan fibers preloaded with 10 nM Fluo-4AM.
Immunoblotting
Immunoblotting for RyR used antibodies specific for RyR1 (ThermoFisher/Invitrogen), RyR2 (ProteinTech) and RyR3 (Millipore, ThermoFisher/Invitrogen), diluted 1:500. Gels were 3–8% Tris-Acetate with Tris-acetate-SDS running buffer (NuPAGE/Invitrogen). Secondary antibodies were HRP conjugates (anti-rabbit from Invitrogen; anti-mouse from Jackson ImmunoResearch), used at 1:10,000. Blots were visualized with SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher) and imaged with a BioRad ChemiDoc MP instrument.
Statistical analysis
Quantitative data are shown as mean ± SE. Statistical significance was determined with Student’s t test and Χ 2 analysis. A value of p < 0.05 was considered statistically significant.
Materials
BAPTA-AM and EGTA-AM were from MilliporeSigma. Fluo-4AM was from Invitrogen. Unless specified otherwise, all other chemicals were from Sigma-Aldrich.
Results
BAPTA
We first examined the effects of incubating dysferlin-null A/J myofibers with increasing concentrations of BAPTA-AM in the presence of 4.4 µM Rhod-2AM. We elicited Ca2+ transients with field stimulation and recorded the increase in Rhod-2 fluorescence, which tracks [Ca2+]i (Lukyanenko et al., 2017; Muriel et al., 2022). Figures 1, 2 show that incubation with ≥100 nM BAPTA-AM reduces the apparent amplitude of the Ca2+ transient, perhaps because the BAPTA generated by cleavage of the -AM moieties in the sarcoplasm accumulates to levels high enough to compete with the Rhod-2 and reduce its fluorescence during the transient. By contrast, concentrations of BAPTA-AM of ≤50 nM increase the amplitude of the Ca2+ transient by ∼15% (Figures 1C, 2A), to levels seen in dysferlin-positive, control A/JCr myofibers. This difference is significant (p < 0.05). DMSO, the vehicle, had no effect in the absence of the chelator (2.40 ± 0.10, n = 138 vs. 2.57 ± 0.07, n = 280, A/J fibers with DMSO present vs. DMSO absent, respectively, p = 0.09). These results show that low concentrations of BAPTA-AM enhance the amplitude of Ca2+ transients in A/J muscle fibers, restoring them to control levels.
FIGURE 1.
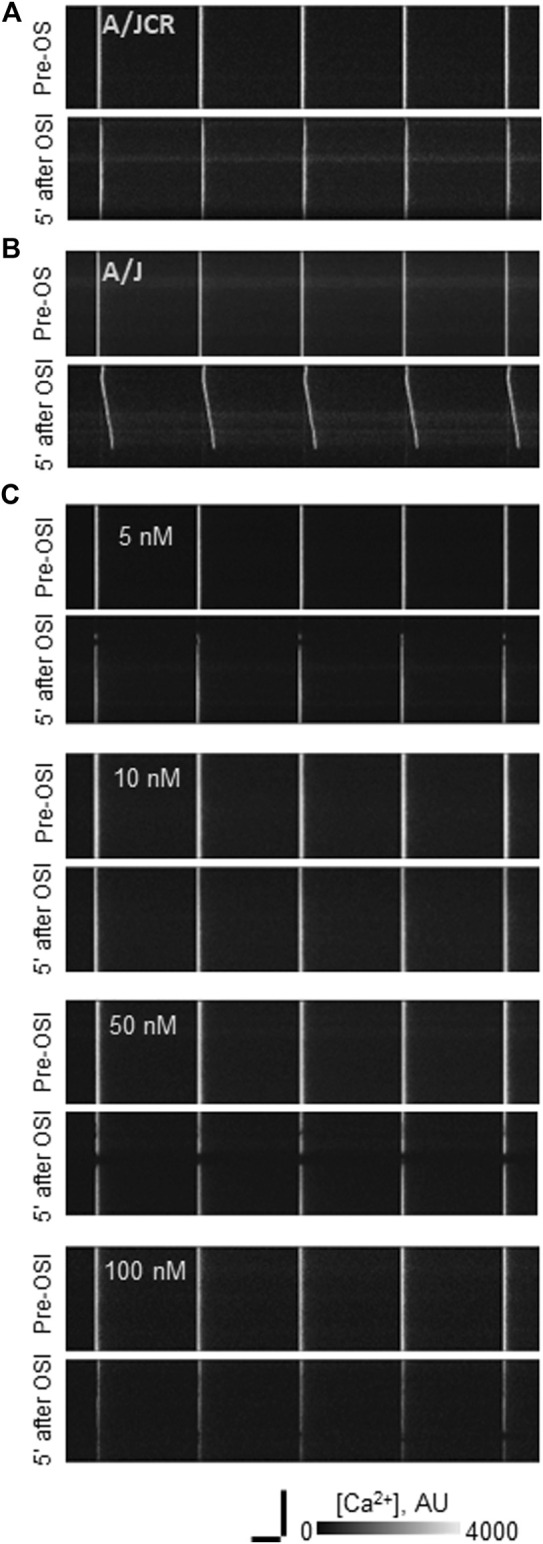
Effect of osmotic shock injury on Ca2+ transients in FDB fibers preloaded with different concentrations of BAPTA-AM. A/J or A/JCr myofibers were loaded with Rhod-2AM, with or without additional BAPTA-AM, subjected to 1 Hz stimulation, and imaged in line-scan mode under confocal optics (see Methods). Representative line-scan confocal images of voltage-induced Ca2+ transients in A/JCr (A) and sham (vehicle only) A/J [(B): A/J] fibers and in A/J fibers preloaded with BAPTA-AM at 5, 10, 50 and 100 nM (C). All examples in (B,C) were exposed to equal amounts of DMSO (1.5% by volume). All examples are shown before osmotic shock injury (OSI) and 5 min after OSI. Bars, 100 µm (vertical) and 250 ms (horizontal).
FIGURE 2.

Effect of osmotic shock injury on Ca2+ transients in FDB fibers preloaded with different concentrations of BAPTA-AM. Data from experiments like those shown in Figure 1 were analyzed. (A), averaged amplitudes of Ca2+ transients before OSI presented as (Fmax-F0)/F0. (B), averaged data for recovery of Ca2+ transients at 5 min after OSI. (C), averaged data for frequency of Ca2+ waves at 5 min after OSI. (D), % fibers that produced Ca2+ waves at 5 min after OSI. N is indicated in each bar. *. p < 0.05 compared to A/J (sham). **, p < 0.05 compared to A/JCr control. Student’s t test was used for A-C; Χ 2 was used for (D). VICT = Voltage-induced Ca2+ transient.
We next examined the effects of BAPTA-AM on the Ca2+ transients of dysferlin-null A/J myofibers after a brief osmotic shock injury (OSI). As above, we recorded the transients via Rhod-2 fluorescence in response to electrical stimulation. As we reported earlier (Kerr et al., 2013; Lukyanenko et al., 2017; Muriel et al., 2022), OSI significantly decreases the amplitude of Ca2+ transients of A/J fibers 5 min after injury, and the transients that appear are frequently accompanied by Ca2+ waves and spontaneous transients (Figures 1, 2B–D). In fibers incubated in 10 nM BAPTA-AM, however, the amplitudes of the transients were not reduced after osmotic shock injury (Figure 2B), and waves and spontaneous transients were reduced to control levels (Figures 1, 2C,D). These changes did not occur with DMSO alone (Figures 3B–D, A/J alone), suggesting that BAPTA mediates these effects.
FIGURE 3.

Effect of osmotic shock injury on Ca2+ transients in A/J FDB fibers preloaded with different concentrations of Fluo-4AM. As in Figure 2, but with myofibers loaded with Fluo-4AM. (A), averaged amplitudes of Ca2+ release before OSI presented as (Fmax-F0)/F0. (B), averaged data for recovery of Ca2+ transients at 5 min after OSI. (C), averaged data for frequency of Ca2+ waves at 5 min after OSI. (D), % fibers that produced Ca2+ waves at 5 min after OSI. Dashed lines represent values obtained with A/JCr fibers. *, p < 0.05 compared to A/J. N is indicated in each bar. Student’s t test was used for (A–C); Χ 2 was used for (D). VICT = Voltage-induced Ca2+ transient.
We studied the concentration dependence of the inhibition of abnormal Ca2+ signaling as a function of BAPTA-AM concentration. We found that 5 nM BAPTA-AM was not sufficient to protect the transient against loss of amplitude after OSI, but that concentrations of 10 and 50 nM were effective (Figures 2A,B). These results suggest that the presence of the chelator was sufficient to maintain normal Ca2+ signaling following OSI, mimicking the activity of dysferlin in this assay.
We next measured the effects of the -AM derivative of Fluo-4, a variant of BAPTA that fluoresces upon binding of Ca2+. Although Fluo-4AM partitions into the sarcoplasm and is cleaved by intracellular esterases much like BAPTA-AM (Paredes et al., 2008; Smith et al., 2018), ten-fold higher concentrations were required to mimic the effect of BAPTA-AM on the recovery of the Ca2+ transient after OSI and to reduce the appearance of Ca2+ waves (Figures 3B–D). Even at 250 nM, Fluo-4AM failed to reduce Ca2+ waves to the levels seen with 10 nM BAPTA-AM (compare Figures 3D–2D). Thus, Fluo-4AM is less effective than BAPTA-AM, perhaps because its affinity for Ca2+ is ∼2-fold poorer (Paredes et al., 2008). Fluo-4 is also larger, carries a negative charge and has a dielectric constant that is 20-fold higher than BAPTA’s, and so may accumulate in the sarcoplasm less efficiently than BAPTA.
Despite the differences between Fluo-4AM and BAPTA-AM, we used the former to approximate the intracellular concentration of BAPTA needed to reduce the abnormalities in Ca2+ signaling that we routinely assay. We measured the fluorescence intensity of intracellular Fluo-4 in myofibers incubated with 10 nM Fluo-4AM under conditions identical to those we used with BAPTA-AM and compared it to a standard curve, with the assumption that the concentration of free intracellular Ca2+ is ∼100 nM (e.g., López et al., 1983; Harkins et al., 1993; Head 1993; Pressmar et al., 1994; Konishi and Watanabe 1995; Baylor and Hollingworth 2007). For the standard curve, we measured the fluorescence intensities at different concentrations of the K+ salt of Fluo-4 in the presence of 100 nM Ca2+ and under identical confocal imaging conditions (see Methods). The results indicate that Fluo-4 reached concentrations in the sarcoplasm of ∼60 nM, or about 6 times higher than its concentration in the bath (Supplemental Figure S1). Given the significant differences between the effects of BAPTA-AM and Fluo-4AM, BAPTA may accumulate to levels significantly higher than 60 nM, thereby buffering sarcoplasmic free [Ca2+] after OSI to ≤100 nM.
EGTA-AM
Although the affinities of BAPTA and Fluo-4 for Ca2+ (in Mg2+-free conditions) are ∼160 and ∼370 nM, respectively, they both have relatively high “on” and “off” rates for Ca2+ and thus are able to chelate Ca2+ rapidly. The affinity of EGTA for Ca2+ is close to that of BAPTA, but its “on” and “off” rates for binding Ca2+ are ∼100-fold slower than BAPTA’s. We used EGTA-AM to test the idea that rapid, transient changes in the concentration of Ca2+, rather than changes in the equilibrium concentration alone, play a role in the abnormal Ca2+ signaling that we observe in A/J myofibers before and after OSI. Our results (Figure 4) show that incubating myofibers with concentrations of EGTA-AM up to 25-fold higher than the effective concentration of BAPTA-AM fails to protect the transient against disruption by OSI, although low concentrations are able to enhance the amplitude of the transient before injury. This suggests that the dysregulation of Ca2+ that alters the stability, but not the initial amplitude, of the Ca2+ transient in A/J fibers is due to rapid, transient changes in Ca2+ rather than the overall levels of Ca2+ in the cytoplasm.
FIGURE 4.

Effect of osmotic shock injury on Ca2+ transients in A/J FDB fibers preloaded with different concentrations of EGTA-AM. As in Figure 2, but with myofibers loaded with EGTA-AM. (A), averaged amplitudes of Ca2+ release before OSI presented as (Fmax-F0)/F0. (B), averaged data for recovery of Ca2+ transients at 5 min after OSI. (C), averaged data for frequency of Ca2+ waves at 5 min after OSI. (D), % fibers that produced Ca2+ waves at 5 min after OSI. Dashed lines represent values obtained with A/JCr fibers. *, p < 0.05 compared to A/J. N is indicated in each bar. Student’s t test was used for (A–C); Χ 2 was used for (D)
GCaMP6fu-DYSF-ΔC2A
Loading myofibers with BAPTA, Fluo-4 or EGTA, as we have done, can reduce the resting and peak levels of free Ca2+ in the sarcoplasm, which may only indirectly alter levels at the triad junction. We have postulated that abnormal Ca2+ signaling that follows injury of A/J myofibers is due to changes in Ca2+ at the triad junction that destabilize the LTCC-RyR1 couplons there, reducing normal Ca2+ release and promoting CICR. Here we test if suppression of local increases in Ca2+ at the triad junction is indeed sufficient to protect the Ca2+ transient from injury.
For these studies, we placed GCaMP6fu at the N-terminus of Venus-dysferlin lacking the C2A domain (GCaMP6fu-DYSF-ΔC2A; Figure 5A). We chose this GCaMP variant because, like the C2A domain (Abdullah et al., 2014; Wang et al., 2021), GCaMP6fu binds Ca2+ rapidly and with high affinity (Helassa et al., 2016). We found that, similar to the Venus construct of DYSF-ΔC2A, GCaMP6fu-DYSF-ΔC2A traffics normally to membranes at the level of the A-I junction (Figure 5B), concentrating in transverse tubules of the triad junction like both Dysf-ΔC2A and WT dysferlin (Muriel et al., 2022). By contrast, GCaMP6fu expressed as a Venus fusion protein (Figure 5A) distributes much more uniformly in the sarcoplasm and, like GFP itself, only accumulates at the level of Z-disks (Figure 5B). When A/J fibers expressing GCaMP6fu in the sarcoplasm or at the triad junction are loaded with Rhod-2 and electrically stimulated, both the GCaMP6fu moiety and Rhod-2 register the changes in Ca2+ concentration. The signals generated by Rhod-2 were brighter than those generated by GCaMP6fu, but we were generally able to use either for measurements of transient amplitudes and waves.
FIGURE 5.

Distributions of GCaMP6fu and GCaMP6fu-DYSF-ΔC2A in sarcoplasm. (A). Cartoon diagrams of chimeric structures, which include the CMV promoter, the GCaMP6fu reporter, and, for GCaMP6fu-DYSF-ΔC2A, the dysferlin ORF excluding most of the C2A domain (i.e., residues 108–2080) but including the remaining C2 domains B thought G (green hexagons), the Fer and DysF domains in the middle of the molecule (blue and pink outlined rectangles) and the transmembrane domain (blue rectangle near the C terminus; see Methods). (B). Subcellular distribution of Venus-GCaMP6fu and GCaMP6fu-DYSF-ΔC2A in transfected A/J myofibers. Plasmids were electroporated into A/J myofibers and imaged under confocal optics 2 weeks later. Double arrows: transverse tubules at level of triad junctions, as reported (Kerr et al., 2013; Muriel et al., 2022); single arrows: Z-disks, as reported (Muriel et al., 2022). Bars, 10 µm.
Remarkably, fibers expressing GCaMP6fu-DYSF-ΔC2A show an increase in the amplitude of the Ca2+ transient similar to that observed with BAPTA and that seen when WT dysferlin is restored to dysferlin-null fibers (Figures 6A,B). (Please note that the amplitudes of the transients in uninjured myofibers subjected to electroporation are higher than those studied without electroporation, as reported Lukyanenko et al., 2017). Moreover, the transient after OSI remains at control levels, similar to fibers expressing WT dysferlin (Figures 6A,C). In addition, fibers expressing GCaMP6fu-DYSF-ΔC2A rarely show Ca2+ waves (Figure 6D). These results are consistent across a wide range in the level of expression of the fusion protein (Figure 6E), suggesting that it is acting in a limited volume, i.e., the triad junction.
FIGURE 6.

Effect of osmotic shock injury on Ca2+ transients in A/J FDB fibers transfected with GCaMP6fu-Dysf-ΔC2A. Myofibers were transfected by electroporation. Two weeks later, they were loaded with Rhod-2AM and assayed as in Figure 2. (A), representative line-scan images of Ca2+ transients before and 5 min after OSI, as in Figure 1. (B), averaged amplitudes of Ca2+ release before OSI for A/J fibers transfected with Venus or with GCaMP6fu-Dysf-ΔC2A. (C), averaged data for recovery of Ca2+ transients from OSI for A/J fibers transfected with Venus or with GCaMP6fu-Dysf-ΔC2A at 5 min after OSI. Dashed line represents recovery in A/J fibers transfected with WT dysferlin. (D), averaged data for frequency of Ca2+ waves at 5 min after OSI. N is indicated in each bar. Student’s t test was used for (A–C); Χ 2 was used for (D) *, p < 0.05 compared to A/J fibers transfected with Venus. (E). Recovery from OSI and frequency of Ca2+ waves as a function of GCaMP6fu-DYSF-ΔC2A expression. GCaMP6fu-DYSF-ΔC2A levels were determined in AU by measuring the intensity of the GCaMP6fu fluorescence, after setting the background autofluorescence to 180 AU. For earlier data for A/J fibers expressing Venus- Dysf-ΔC2A, see Muriel et al., 2022.
By contrast, GCaMP6fu expressed as a Venus fusion protein fails to increase the amplitude of the transient in uninjured fibers (Figure 7B), and is somewhat less protective of the Ca2+ transients after OSI (e.g., Figures 7A,C). More strikingly, however, it fails to suppress the appearance of Ca2+ waves (Figure 7D). The differences with GCaMP6fu-DYSF-ΔC2A in amplitude and wave frequency after recovery from OSI were both statistically significant (p < 0.05). These results were far less consistent as a function of concentration than those of GCaMP6fu-DYSF-ΔC2A (Figure 7E; data obtained with Rhod-2, only). These results suggest that GCaMP6fu in the sarcoplasm acts as a weak Ca2+ chelator and thus is considerably less effective than GCaMP6fu-DYSF-ΔC2A in the triad junction.
FIGURE 7.

Effect of OSI on Ca2+ transients in A/J FDB fibers transfected with Venus-GCaMP6fu. Myofibers were transfected by electroporation. Two weeks later, they were loaded with Rhod-2AM and assayed as in Figure 2. (A), representative line-scan images of Ca2+ transients before and 5 min after OSI. (B), averaged amplitudes of Ca2+ release before OSI for A/J fibers transfected with Venus or with Venus-GCaMP6fu. (C), averaged data for recovery of Ca2+ transients from OSI for A/J fibers transfected with Venus or with Venus-GCaMP6fu at 5 min after OSI. The dashed line represents recovery in A/J fibers transfected with WT dysferlin. (D), averaged data for frequency of Ca2+ waves at 5 min after OSI. N is indicated in each bar. Student’s t test was used for (A–C); Χ 2 was used for (D) *, p < 0.05 compared to A/J fibers transfected with Venus. (E). Recovery from OSI and frequency of Ca2+ waves as a function of Venus-GCaMP6fu expression. Panels B–D show results obtained by imaging either Rhod-2 or GCaMP6fu fluorescence. Panel E shows results obtained only with Rhod-2. Venus-GCaMP6fu levels were determined in AU by measuring the intensity of the Venus fluorescence, after setting the background autofluorescence to 200 AU. For earlier data with A/J fibers expressing Venus alone, see Lukyanenko et al., 2017).
Our findings suggest that GCaMP6fu localized by DYSF-ΔC2A to the triad junction, but not as a cytoplasmic protein, can serve the function of the C2A domain of dysferlin in Ca2+ signaling. Thus, one of dysferlin’s likely roles in maintaining the health of skeletal muscle is to bind Ca2+ at triad junctions and thereby protect the muscle from injury.
Other ryanodine receptors
As the presence of non-junctional RyR isoforms could account for CICR in A/J muscle fibers, we used immunoblotting of muscle extracts to determine if RyR2 and RyR3 were expressed together with RyR1. The blots of extracts of Tibialis anterior muscles from A/J mice, which like FDB fibers are primarily fast twitch, showed RyR1 to be present at high levels but RyR2 and RyR3 to be undetectable (Figure 8). They were readily detectable in heart and brain, however (not shown). This is consistent with several reports in GEO Profiles which show much lower levels of RyR2 and RyR3 mRNAs than RyR1 mRNA in Tibialis anterior muscles (see also Futatsugi et al., 1995; Marziali et al., 1996). Thus, RyR2 and RyR3 are unlikely to contribute significantly to abnormal Ca2+ signaling in dysferlinopathic muscle.
FIGURE 8.
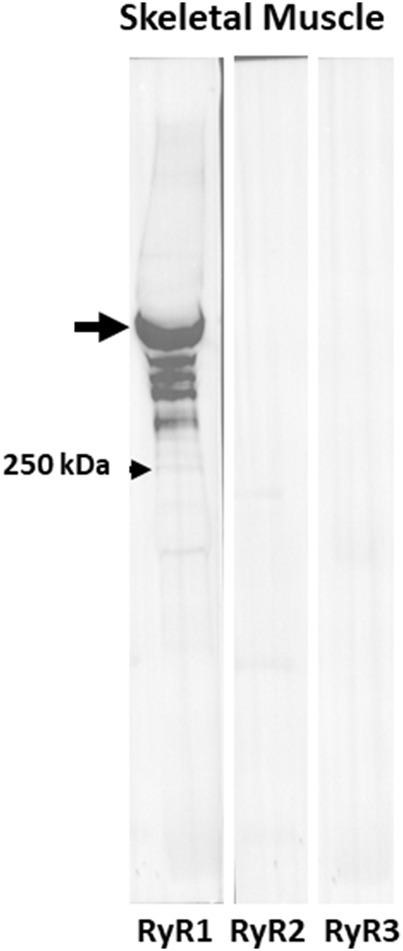
RyR1 but not RyR2 or RyR3 are expressed in fast twitch skeletal muscle. Immunoblots of extracts of Tibialis anterior muscles with antibodies to RyR1, RyR2, and RyR3. Only anti-RyR1 shows a strong band at ∼550 kDa.
Discussion
Previous reports from our laboratory showed that Ca2+ signaling is defective in dysferlin-null A/J muscle fibers, and that this phenotype can be reversed by the reintroduction of a Venus chimaera of WT dysferlin or by blocking the L-type Ca2+ channel (LTCC) and RyR1s (Lukyanenko et al., 2017). The defects include a small decrease in the amplitude of the Ca2+ transient in uninjured myofibers, a large decrease in transient amplitude in fibers injured by hypoosmotic shock, and the appearance of spontaneous Ca2+ transients, waves and sparks following shock. These features, which are typical of Ca2+-induced Ca2+ release (CICR), would be expected if the triad junctional regions of dysferlin-null muscle fibers experienced a small Ca2+ leak in the resting state that increased with injury. Although the leak in the resting state does not result in a detectable increase in [Ca2+]i (Kerr et al., 2013), it has been measured and modeled in healthy in muscle (Cully et al., 2018; Barclay and Launikonis, 2022). An increase in resting leak could explain the lower amplitude of the Ca2+ transients we observe in A/J myofibers, due to a decrease in the amount of Ca2+ available for release upon electrical stimulation. Furthermore, additional increases in sarcoplasmic Ca2+ levels caused by hypoosmotic shock could evince spontaneous Ca2+ release events via CICR. We hypothesized that BAPTA, introduced into the sarcoplasm at low levels as the -AM derivative, would chelate the Ca2+ responsible for CICR and thereby reduce the effect of osmotic shock on the Ca2+ transient and suppress Ca2+ waves. Here we show that incubation of A/J myofibers with 10 nM BAPTA-AM is indeed sufficient to protect A/J myofibers against the loss of transient amplitude and to suppress waves. Consistent with this, the -AM derivatives of other Ca2+ chelators with reduced abilities to bind Ca2+ were less effective than BAPTA-AM. Our studies with the Dysf-ΔC2A-GCaMP6fu chimera further suggest that the increase in Ca2+ that results in CICR-related dysregulation of Ca2+ signaling occurs primarily at or near the triad junction.
Our initial experiments utilized concentrations of BAPTA-AM that were considerably higher than 10 nM. These reduced the amplitude of the Ca2+ transients in uninjured fibers, measured with Rhod-2. It is likely that exposure of fibers to BAPTA-AM at concentrations ≥100 nM leads to sarcoplasmic concentrations of BAPTA that compete with Rhod-2 for free Ca2+ released following electrical stimulation. Remarkably, however, we found that concentrations of BAPTA-AM as low as 10 nM, much lower than concentrations used by other investigators (e.g., Jacquemond et al., 1991; Anderson and Meissner 1995; Gómez et al., 2006; Shkryl and Shirokova 2006; Ainbinder et al., 2015; Lamboley et al., 2015), were effective in suppressing abnormal Ca2+ signaling both before and after OSI.
As we could not directly measure BAPTA in the sarcoplasm, we used a close analog, Fluo-4AM, instead, and determined its concentration from its fluorescence after uptake, cleavage and equilibrium with sarcoplasmic Ca2+. We were obliged to perform these measurements with the aperture of our confocal microscope completely open, i.e., the identical conditions we used to observe myofibers, as we were unable to observe quantifiable fluorescence at low concentrations of Fluo-4 with the apertures consistent with confocal resolution. These conditions undoubtedly led to our inclusion of some out-of-focus fluorescence, too. Although we cannot compare the relative contributions to our measurements of out-of-focus fluorescence in myofibers and in solution, it is likely that our calibration, which indicates that Fluo-4 in the sarcoplasm reaches ∼60 nM in fibers incubated with 10 nM Fluo-4AM, underestimates the actual concentration of Fluo-4. This value should therefore be considered an estimate of the minimal concentration reached in myofibers exposed to 10 nM Fluo-4AM.
In contrast to Fluo-4AM, which carries a negative charge, BAPTA-AM is uncharged, suggesting that it should permeate the muscle cell membrane more efficiently. Nevertheless, if we assume that BAPTA accumulates like Fluo-4, and not considering the contribution of out-of-focus fluorescence to our calibration, intracellular concentrations of BAPTA should be ∼60 nM after myofibers are loaded with 10 nM BAPTA-AM. If the affinity of BAPTA for Ca2+ in the sarcoplasm is ∼160 nM (https://www.interchim.fr/ft/4/486103.pdf) and if we estimate [Ca2+]I after hypoosmotic shock in the absence of BAPTA is ∼120 nM, then BAPTA would be expected to reduce the resting [Ca2+]I after shock to ∼100 nM. A concentration of 120 nM is in the same range reported for the ability of Ca2+ to activate isolated RyR1, as well as RyR2 (Györke et al., 1994). CICR in the heart is also suppressed by BAPTA (Sham 1997) at concentrations sufficient to reduce [Ca2+]I from 120 nM to <100 nM (Lukyanenko and Györke 1999). Our results are consistent with the idea that even low concentrations of BAPTA-AM can suppress CICR and Ca2+ waves in injured dysferlin-null myofibers.
Our results further indicate that BAPTA has a significant effect on the Ca2+ transient in uninjured A/J fibers. The reduced amplitude of the Ca2+ transient that we observe in A/J myofibers before injury or treatment with BAPTA is likely due to the activation of RyR1 by sarcoplasmic Ca2+ and the consequent small depletion of the luminal Ca2+ stores in the terminal cisternae of the sarcoplasmic reticulum, both of which would be inhibited by chelation of sarcoplasmic Ca2+ by BAPTA. Notably, murine muscle expresses undetectable levels of the two other major forms of the RyR, RyR2 and RyR3 (Figure 8), consistent with the idea that any effect on Ca2+ release is mediated by RyR1. As noted above, RyR1 has been linked to CICR-associated pathogenesis in other diseases of muscle (Endo 2009; see also Kushnir et al., 2018). Our results suggest that it is likely to contribute to pathogenesis in dysferlinopathies as well.
As CICR mediated by the RyR1 in skeletal muscle is thought to be suppressed when the channels are well ordered in LTCC-RyR1 couplons at triad junctions, the absence of dysferlin may well lead to a “couplonopathy” (Ríos et al., 2015; Ríos 2018), in which couplon organization is weakened, making it susceptible to further disruption upon hypoosmotic shock and thus enabling RyR1-mediated CICR in injured muscle. Others have reported that triad junctional architecture is altered in dysferlinopathy (Barefield et al., 2021).
Our evidence suggests that the couplon can be stabilized not only by uniformly chelating sarcoplasmic Ca2+ with BAPTA, but also by placing a mutant form of dysferlin with an enhanced ability to bind Ca2+ in the cleft of the triad junction itself. These experiments took advantage of our earlier observation that dysferlin lacking its N-terminal C2 domain, C2A, trafficked normally to the transverse tubules but did not support normal Ca2+ signaling (Muriel et al., 2022). Notably, the C2A domain has several binding sites for Ca2+ with affinities in the micromolar range {Abdullah et al., 2014, Wang et al., 2021). Although DYSF-ΔC2A prevents the generation of Ca2+ waves, it does not restore the amplitude of the transient to control levels and it fails to preserve the amplitude after OSI. We therefore used DYSF-ΔC2A as a backbone to target a Ca2+ binding protein moiety specifically to the junctional cleft by replacing C2A with GCaMP6fu, a calmodulin-based fluorescent Ca2+ indicator with rapid binding kinetics and high affinity for Ca2+ (0.89 μM at room temperature (Helassa et al., 2016). We find that, like BAPTA, GCaMP6fu concentrated at triad junctions via Dysf-ΔC2A both enhances the amplitude of the Ca2+ transient to control levels in uninjured fibers and protects the transient from decreasing following OSI. GCaMP6fu expressed as a soluble protein in the sarcoplasm is inactive in the former and trends to lower activity in the latter. It also fails to suppress Ca2+ waves. This strongly suggests that chelating Ca2+ within the triad junction of dysferlin-null skeletal muscle is sufficient to suppress abnormal Ca2+ signaling associated with CICR. Considered together with our other results, it further suggests that CICR is triggered primarily by the local elevation of Ca2+ in the junctional cleft and not by more widespread changes in [Ca2+]i.
The similarities in the effects of GCaMP6fu-DYSF-ΔC2A and WT dysferlin indicate that a key role of the C2A domain in native dysferlin in the triad junction may be to bind Ca2+ rapidly and with high affinity, consistent with earlier reports of the activity of the isolated C2A domain (Abdullah et al., 2014; Wang et al., 2021), and that this alone is sufficient to suppress abnormal Ca2+ signaling following injury in wild type muscle. This activity may help to explain the fact that healthy muscle but not dysferlinopathic muscle recovers quickly from exercise-induced injuries, avoiding necrotic fiber death (Roche et al., 2008; Roche et al., 2010; see also Roman et al., 2021).
Because DYSF-ΔC2A largely suppresses Ca2+ waves (Muriel et al., 2022), we do not know if placing GCaMP6fu in the junctional cleft is sufficient to suppress waves. Further studies with additional constructs that also target the triad junction may be informative in this regard. Such studies may also permit us to use GCaMP-based methods to measure the concentrations of Ca2+ in the junctional cleft in healthy and dysferlin-null muscle, before and after injury.
Acknowledgments
We thank Andrea O’Neill and Ujwala Pimparkar for technical support and Dr. I. Dick for a useful discussion.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee, University of Maryland, Baltimore.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
Annual grants from the Jain Foundation have supported our work on dysferlin. Our research was supported by grants to RB from the NIH (RB: 2 RO1 AR064268) and from the Jain Foundation (Bellevue, WA), by funds generously provided to RB from the Department of Physiology, University of Maryland School of Medicine, and by resources from the Campus Microscopy and Imaging Facility (CMIF). LB is employed by Regeneron Pharmaceuticals.
Conflict of interest
LB is employed by Regeneron Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.1032447/full#supplementary-material
Calibration of [Fluo-4(K+)] in 100 nM free Ca2+. (A), Difference in the background fluorescence in A/J fibers pretreated with 1.5% DMSO or 1.5% DMSO containing 10 nM Fluo-4AM. N is indicated in each bar. (B), Calibration line for the 4 lowest concentrations of Fluo-4 (K+) of the 10 concentrations used for calibration. Open circle shows point for 23 AU. The same gain was used for each measurement. The solution contained (in mM): 5 EGTA, 2.7 CaCl2 (free, 100 nM), 1.1 MgCl2 (free, 1 mM), 10 HEPES, 264.2 KCl, pH = 7.4, super clean water. Calculations used WEBMAXC STANDARD.
References
- Abdullah N., Padmanarayana M., Marty N. J., ohnson C J. (2014). Quantitation of the calcium and membrane binding properties of the C2 domains of dysferlin. Biophys. J. 106, 382–389. 10.1016/j.bpj.2013.11.4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainbinder A., Boncompagni S., Protasi F., Dirksen R. T. (2015). Role of mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 57, 14–24. 10.1016/j.ceca.2014.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D. G., Gervasio O. L., Yeung E. W., Whitehead N. P. (2010). Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 88, 83–91. 10.1139/Y09-058 [DOI] [PubMed] [Google Scholar]
- Amato A. A., Brown R. H., (2011). Handb. Clin. Neurol. 101, 111–118. 10.1016/B978-0-08-045031-5.00007-4 [DOI] [PubMed] [Google Scholar]
- Anderson K., Meissner G. (1995). T-tubule depolarization-induced SR Ca2+ release is controlled by dihydropyridine receptor- and Ca2+-dependent mechanisms in cell homogenates from rabbit skeletal muscle. J. Gen. Physiol. 105, 363–383. 10.1085/jgp.105.3.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M., Takahashi T. (1993). in Dysferlinopathy. GeneReviews(®). Adam M. P., Ardinger H. H.. Editors (Seattle, WA: University of Washington; ). [PubMed] [Google Scholar]
- Bansal D., Campbell K. P. (2004). Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 14, 206–213. 10.1016/j.tcb.2004.03.001 [DOI] [PubMed] [Google Scholar]
- Bansal D., Miyake K., Vogel S. S., Groh S., Chen C. C., Williamson R., et al. (2003). Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172. 10.1038/nature01573 [DOI] [PubMed] [Google Scholar]
- Barclay C. J., Launikonis B. S. (2022). A mathematical model to quantify RYR Ca2+ leak and associated heat production in resting human skeletal muscle fibers. J. Gen. Physiol. 154, e202112994. 10.1085/jgp.202112994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield D. Y., Sell J. J., Tahtah I., Kearns S. D., McNally E. M., Demonbreun A. R. (2021). Loss of dysferlin or myoferlin results in differential defects in excitation-contraction coupling in mouse skeletal muscle. Sci. Rep. 11, 15865. 10.1038/s41598-021-95378-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor S. M., Hollingworth S. (2007). Simulation of Ca2+ movements within the sarcomere of fast-twitch mouse fibers stimulated by action potentials. J. Gen. Physiol. 130, 283–302. 10.1085/jgp.200709827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr A. R., Molkentin J. D. (2015). Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 22, 1402–1412. 10.1038/cdd.2015.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenacchi G., Fanin M., De Giorgi L. B., Angelini C. (2005). Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 58, 190–195. 10.1136/jcp.2004.018978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully T. R., Choi R. H., Bjorksten A. R., Stephenson D. G., Murphy R. M., Launikonis B. S. (2018). Junctional membrane Ca(2+) dynamics in human muscle fibers are altered by malignant hyperthermia causative RyR mutation. Proc. Natl. Acad. Sci. U. S. A. 115, 8215–8220. 10.1073/pnas.1800490115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demonbreun A. R., McNally E. M. (2016). Plasma membrane repair in health and disease. Curr. Top. Membr. 77, 67–96. 10.1016/bs.ctm.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demonbreun A. R., Quattrocelli M., Barefield D. Y., Allen M. V., Swanson K. E., McNally E. M. (2016). An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 213, 705–718. 10.1083/jcb.201512022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFranco M., Quinonez M., Capote J., Vergara J. (2009). DNA Transfection of Mammalian Skeletal Muscles using <em>in vivo</em> Electroporation. J. Vis. Exp., 1520. 10.3791/1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M. (2009). Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 89, 1153–1176. 10.1152/physrev.00040.2008 [DOI] [PubMed] [Google Scholar]
- Fanin M., Angelini C. (2016). Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerve 54, 821–835. 10.1002/mus.25367 [DOI] [PubMed] [Google Scholar]
- Futatsugi A., Kuwajima G., Mikoshiba K. (1995). Tissue-specific and developmentally regulated alternative splicing in mouse skeletal muscle ryanodine receptor mRNA. Biochem. J. 305 (2), 373–378. 10.1042/bj3050373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover L., Brown R. H., Jr. (2007). Dysferlin in membrane trafficking and patch repair. Traffic 8, 785–794. 10.1111/j.1600-0854.2007.00573.x [DOI] [PubMed] [Google Scholar]
- Gómez J., Neco P., DiFranco M., Vergara J., L. (2006). Calcium release domains in mammalian skeletal muscle studied with two-photon imaging and spot detection techniques. J. Gen. Physiol. 127, 623–637. 10.1085/jgp.200509475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke S., Vélez P., Suárez-Isla B., Fill M. (1994). Activation of single cardiac and skeletal ryanodine receptor channels by flash photolysis of caged Ca2+ . Biophys. J. 66, 1879–1886. 10.1016/S0006-3495(94)80981-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R., Campbell K. P. (2007). Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 19, 409–416. 10.1016/j.ceb.2007.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkins A. B., Kurebayashi N., Baylor S. M. (1993). Resting myoplasmic free calcium in frog skeletal muscle fibers estimated with fluo-3. Biophys. J. 65, 865–881. 10.1016/S0006-3495(93)81112-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head S. I. (1993). Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J. Physiol. 469, 11–19. 10.1113/jphysiol.1993.sp019801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helassa N., Podor B., Fine A., Török K. (2016). Design and mechanistic insight into ultrafast calcium indicators for monitoring intracellular calcium dynamics. Sci. Rep. 6, 38276. 10.1038/srep38276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M., Post C. M., Donahue L. R., Lidov H. G., Bronson R. T., Goolsby H., et al. (2004). Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum. Mol. Genet. 13, 1999–2010. 10.1093/hmg/ddh212 [DOI] [PubMed] [Google Scholar]
- Jacquemond V., Csernoch L., Klein M. G., Schneider M. F. (1991). Effects of low myoplasmic Mg2+ on calcium binding by parvalbumin and calcium uptake by the sarcoplasmic reticulum in frog skeletal muscle. J. Gen. Physiol. 60, 115–135. 10.1085/jgp.100.1.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. P., Ward C. W., Bloch R. J. (2014). Dysferlin at transverse tubules regulates Ca(2+) homeostasis in skeletal muscle. Front. Physiol. 5, 89. 10.3389/fphys.2014.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. P., Ziman A. P., Mueller A. L., Muriel J. M., Kleinhans-Welte E., Gumerson J. D., et al. (2013). Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc. Natl. Acad. Sci. U. S. A. 110, 20831–20836. 10.1073/pnas.1307960110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi M., Watanabe M. (1995). Resting cytoplasmic free Ca2+ concentration in frog skeletal muscle measured with fura-2 conJ.ugated to high molecular weight dextran. J. Gen. Physiol. 106, 1123–1150. 10.1085/jgp.106.6.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir A., Wajsberg B., Marks A. R. (2018). Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta. Mol. Cell Res. 1865, 1687–1697. 10.1016/j.bbamcr.2018.07.011 [DOI] [PubMed] [Google Scholar]
- Lamboley C. R., Kake Guena S. A., Touré F., Hébert C., Yaddaden L., Nadeau S., et al. (2015). New method for determining total calcium content in tissue applied to skeletal muscle with and without calsequestrin. J. Gen. Physiol. 145, 127–153. 10.1085/jgp.201411250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon N. J., Kho A., Bacskai B. J., Perlmutter S. L., Hyman T., Brown R. H., Jr. (2003). Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 278 (50), 50466–50473. 10.1074/jbc.M307247200 [DOI] [PubMed] [Google Scholar]
- López J. R., Alamo L., Caputo C., DiPolo R., Vergara S. (1983). Determination of ionic calcium in frog skeletal muscle fibers. Biophys. J. 43, 1–4. 10.1016/S0006-3495(83)84316-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V., Györke S. (1999). Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J. Physiol. 521 (3), 575–585. 10.1111/j.1469-7793.1999.00575.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V., Muriel J. M., Bloch R. J. (2017). Coupling of excitation to Ca(2+) release is modulated by dysferlin. J. Physiol. 595, 5191–5207. 10.1113/JP274515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareedu S., Million E. D., Duan D., Babu G. J. (2021). Abnormal calcium handling in Duchenne muscular dystrophy: Mechanisms and potential therapies. Front. Physiol. 12, 647010. 10.3389/fphys.2021.647010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marziali G., Rossi D., Giannini G., Charlesworth A., Sorrentino V. (1996). cDNA cloning reveals a tissue specific expression of alternatively spliced transcripts of the ryanodine receptor type 3 (RyR3) calcium release channel. FEBS Lett. 394, 76–82. 10.1016/0014-5793(96)00944-1 [DOI] [PubMed] [Google Scholar]
- Matsuda C., Aoki M., Hayashi Y. K., Ho M. F., Arahata K., Brown R. H., Jr. (1999). Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology 53, 1119–1122. 10.1212/wnl.53.5.1119 [DOI] [PubMed] [Google Scholar]
- McDade J. R., Naylor M. T., Michele D. E. (2021). Sarcolemma wounding activates dynamin-dependent endocytosis in striated muscle. FEBS J. 288, 160–174. 10.1111/febs.15556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriel J., Lukyanenko V., Kwiatkowski T., Bhattacharya S., Garman D., Weisleder N., et al. (2022). The C2 domains of dysferlin, roles in membrane localization, Ca(2+) signalling and sarcolemmal repair. J. Physiol. 600, 1953–1968. 10.1113/JP282648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes R. M., Etzler J. C., Watts L. T., Zheng W., Lechleiter J. D. (2008). Chemical calcium indicators. Methods 46, 143–151. 10.1016/j.ymeth.2008.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressmar J., Brinkmeier H., Seewald M. J., Naumann T., Rüdel R. (1994). Intracellular Ca2+ concentrations are not elevated in resting cultured muscle from Duchenne (DMD) patients and in MDX mouse muscle fibres. Pflugers Arch. 426, 499–505. 10.1007/BF00378527 [DOI] [PubMed] [Google Scholar]
- Ríos E. (2018). Calcium-induced release of calcium in muscle, 50 years of work and the emerging consensus. J. Gen. Physiol. 150, 521–537. 10.1085/jgp.201711959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos E., Figueroa L., Manno C., Kraeva N., Riazi S. (2015). The couplonopathies, A comparative approach to a class of diseases of skeletal and cardiac muscle. J. Gen. Physiol. 145, 459–474. 10.1085/jgp.201411321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J. A., Lovering R. M., Bloch R. J. (2008). Impaired recovery of dysferlin-null skeletal muscle after contraction-induced inJ.ury in vivo . Neuroreport 19, 1579–1584. 10.1097/WNR.0b013e328311ca35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J. A., Lovering R. M., Roche R., Ru L. W., Reed P. W., Bloch R. J. (2010). Extensive mononuclear infiltration and myogenesis characterize recovery of dysferlin-null skeletal muscle from contraction-induced inJ.uries. Am. J. Physiol. Cell Physiol. 298, C298–C312. 10.1152/ajpcell.00122.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J. A., Ru L. W., O'Neill A. M., Resneck W. G., Lovering R. M., Bloch R. J. (2011). Unmasking potential intracellular roles for dysferlin through improved immunolabeling methods. J. Histochem. Cytochem. 59, 964–975. 10.1369/0022155411423274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J. A., Tulapurkar M. E., Mueller A. L., van Rooijen N., Hasday J. D., Lovering R. M., et al. (2015). Myofiber damage precedes macrophage infiltration after in vivo inJ.ury in dysferlin-deficient A/J. mouse skeletal muscle. Am. J. Pathol. 185, 1686–1698. 10.1016/j.ajpath.2015.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman W., Pinheiro H., Pimentel M. R., Segalés J., Oliveira L. M., García-Dominguez E., et al. (2021). Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science 374, 3355–3359. 10.1126/science.abe5620 [DOI] [PubMed] [Google Scholar]
- Sham J. S. (1997). Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes, evidence for local Ca2+ signalling. J. Physiol. 500 (2), 285–295. 10.1113/jphysiol.1997.sp022020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkryl V. M., Shirokova N. (2006). Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J. Biol. Chem. 281, 1547–1554. 10.1074/jbc.M505024200 [DOI] [PubMed] [Google Scholar]
- Smith N. A., Kress B. T., Lu Y., Chandler-Militello D., Benraiss A., Nedergaard M. (2018). Fluorescent Ca(2+) indicators directly inhibit the Na, K-ATPase and disrupt cellular functions. Sci. Signal. 11, eaal2039. 10.1126/scisignal.aal2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urtizberea J. A., Bassez G., Leturcq F., Nguyen K., Krahn M., Levy N. (2008). Neurol. India 56, 289–297. 10.4103/0028-3886.43447 [DOI] [PubMed] [Google Scholar]
- Wang Y., Tadayon R., Santamaria L., Mercier P., Forristal C. J., Shaw G. S. (2021). Calcium binds and rigidifies the dysferlin C2A domain in a tightly coupled manner. Biochem. J. 478, 197–215. 10.1042/BCJ20200773 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Calibration of [Fluo-4(K+)] in 100 nM free Ca2+. (A), Difference in the background fluorescence in A/J fibers pretreated with 1.5% DMSO or 1.5% DMSO containing 10 nM Fluo-4AM. N is indicated in each bar. (B), Calibration line for the 4 lowest concentrations of Fluo-4 (K+) of the 10 concentrations used for calibration. Open circle shows point for 23 AU. The same gain was used for each measurement. The solution contained (in mM): 5 EGTA, 2.7 CaCl2 (free, 100 nM), 1.1 MgCl2 (free, 1 mM), 10 HEPES, 264.2 KCl, pH = 7.4, super clean water. Calculations used WEBMAXC STANDARD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.