

Abstract
Gulf War Illness (GWI), a disorder suffered by approximately 200,000 veterans of the first Gulf War, was caused by exposure to low-level organophosphate pesticides and nerve agents in combination with battlefield stress. To elucidate the mechanistic basis of the brain-related symptoms of GWI, human-induced pluripotent stem cells (hiPSCs) derived from veterans with or without GWI were differentiated into forebrain glutamatergic neurons and then exposed to a Gulf War (GW) relevant toxicant regimen consisting of a sarin analog and cortisol, a human stress hormone. Elevated levels of total and phosphorylated tau, reduced microtubule acetylation, altered mitochondrial dynamics/transport, and decreased neuronal activity were observed in neurons exposed to the toxicant regimen. Some of the data are consistent with the possibility that some veterans may have been predisposed to acquire GWI. Wistar rats exposed to a similar toxicant regimen showed a mild learning and memory deficit, as well as cell loss and tau pathology selectively in the CA3 region of the hippocampus. These cellular responses offer a mechanistic explanation for the memory loss suffered by veterans with GWI and provide a cell-based model for screening drugs and developing personalized therapies for these veterans.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-03942-3.
Keywords: Gulf War Illness, Human-induced pluripotent stem cells, Tau, Microtubule, Mitochondria, Neuronal activity, Memory
Introduction
Gulf War Illness (GWI) is a debilitating multi-symptom disease suffered by 30% of the nearly 700,000 United States veterans deployed during the 1991 Gulf War [1–3]. Symptoms include fatigue, musculoskeletal pain, gastrointestinal problems, and alterations in central nervous system (CNS) functioning [4]. CNS dysfunction includes short-term memory problems, poor attention/concentration, reduced information processing speeds, persistent headaches, and mood and sleep disturbances [5–7]. The etiology of the disease remains unclear, with no effective treatments yet available [8].
Although high-dose organophosphate exposure irreversibly inhibits acetylcholinesterase [9], growing evidence indicates that low-dose organophosphate exposure is responsible for the long-term cognitive deficits in GWI [10, 11], thus implicating novel biological targets. Studies have shown that GWI results from organophosphate pesticides and nerve agents combined with the stress of the battlefield, the latter of which can be mimicked in the laboratory by restraint of an animal’s mobility or introduction of a stress hormone [12, 13]. The most prevalent mechanistic hypothesis for GWI is that its symptoms result from a toxicant-induced self-perpetuating state of neuroinflammation [14, 15], while other studies indicate that microtubule-based abnormalities (including axonal transport deficits) might underlie the disease [16–18]. Rodents exposed to GW-relevant toxicants display neuronal microtubules with fewer associated proteins than normal, leading to reduced microtubule width [19]. This may be due to abnormal phosphorylation (and hence detachment) of fibrous microtubule-associated proteins as well as molecular motor proteins [20]. Our own studies have shown that a GW toxicant regimen consisting of a sarin analog and a stress hormone results in reduced microtubule acetylation in cultured rat neurons, as well as deficits in mitochondrial transport and dopamine release, both of which are corrected by pharmacologic restoration of normal microtubule acetylation [21].
Abnormalities in tau, a prominent fibrous microtubule-associated protein in the axon, contribute to many neurological disorders [22]. Aberrantly phosphorylated tau detaches from the microtubule and results in altered microtubule properties [23], while the abnormal tau, both in soluble form and aggregated into intracellular inclusions, produces a variety of toxic effects [24, 25]. Interestingly, auto-antibodies against tau are present at higher levels in peripheral blood serum from veterans with GWI relative to non-veteran controls [26, 27], and low-level organophosphate-based neuroinflammation can also induce tau hyperphosphorylation [28, 29]. Thus, potential tau pathologies in GWI might explain why a limited exposure to neurotoxicants led to a persistent disease condition that was not reversed upon termination of the exposure, and may reconcile mechanistic models based on either neuroinflammation or microtubule abnormalities. A predisposition to tauopathy might also explain a key mystery of GWI, which is why some veterans acquired the disease, while their similarly exposed colleagues did not.
Here, together with a rat model for GWI, we used glutamatergic neurons [30] differentiated from human-induced pluripotent stem cell (hiPSC) lines developed from veterans with or without GWI and banked in a biorepository [31] to delve deeper into the mechanistic basis of the disease. Our results reveal new information on the response of neurons and the nervous system to a GW toxicant regimen, specifically on microtubules, tau, mitochondria, neuronal activity, and memory. Together, our results shine new light on the mechanistic basis of the disease, provide insight into a potential predisposition of some veterans to acquire the disease, and set the stage for the development and testing of new therapies.
Materials and methods
1. hiPSC generation and culture. hiPSC lines were generated from peripheral blood mononuclear cells (PBMCs) isolated from GW veterans with or without GWI, according to the Kansas GWI case definition [4], and banked in the Boston Biorepository, Recruitment & Integrative Network (BBRAIN) for GWI. The hiPSCs were validated for their pluripotency by multiple methods, including morphology, immunostaining for pluripotency markers (TRA-1–60 and TRA-1–81 positive, and SSEA-1 negative), evaluation of karyotype, and ability to differentiate into the three germ layers. Three-germ layer differentiation was carried out as per the manufacturer’s instructions using the Human Pluripotent Stem Cell Functional Identification Kit (R&D Systems, SC027B), and cells were stained for markers of each germ layer: SOX17 (endoderm), Brachyury (mesoderm), and Otx2 (ectoderm).
All cells were cultured in 5% CO2 at 37 °C (see supplemental material for more details). Briefly, the hiPSCs were initially cultured on mouse embryonic fibroblast (MEF) feeder cells (Gibco, A34180) for 2–3 passages before transitioning to a feeder-free culture in mTeSR1 (Stem Cell Technologies, 85857) on ES-qualified Matrigel (Corning, 354277) diluted 1:75 in cold DMEM/F12. Before passaging or directed differentiation, hiPSC colonies were selected to remove areas of spontaneous differentiation. hiPSCs at 80% confluency were passaged weekly with Collagenase Type IV (Stem Cell Technologies, 07909) with 10 µM ROCK inhibitor Y-27632 (Tocris, 1254).
2. Neuronal differentiation and GW toxicant treatment. hiPSC lines from three healthy GW veterans (controls) and four veterans with GWI (cases) were differentiated into forebrain glutamatergic neurons by methods adapted with modifications from previously established protocols [32] (see supplemental material for more details). Briefly, hiPSC colonies were dissociated in Accutase (Stem Cell Technologies, 07920) and 3 million hiPSCs were cultured in one well (10,000 cells per microwell) of an AggreWell 800 plate (Stem Cell Technologies, 34811) using the STEMdiff SMADi Neural Induction Kit (Stem Cell Technologies, 08581) supplemented with 10 µM Y-27632 and low-bFGF (4 ng/ml). On day 5, embryoid bodies (EBs) were replated using a cut P-200 pipette tip and cultured in three wells of a 6-well plate coated with ES-Matrigel. The STEMdiff SMADi Neural Induction medium was gradually replaced with neural progenitor cell (NPC) medium (see supplemental material). On day 12, neural rosettes were isolated using the STEMdiff Neural Rosette Selection Reagent (Stem Cell Technologies, 05832), resuspended in NPC medium, and NPCs were plated on Matrigel (Corning, 354234) coated plates. On day 16, NPCs were replated onto the final culture platform pre-coated with 0.01% poly-l-ornithine (Sigma-Aldrich, P4957) and 10 µg/ml laminin (Invitrogen, 23017015). NPCs were detached via Accutase, plated in neural differentiation medium supplemented with 200 µM ascorbic acid (Sigma-Aldrich, A5960), 500 µM dibutyryl cAMP (Sigma-Aldrich, D0627), and 10 µM DAPT (Tocris, 2634), and cultured for 6 days before long-term culture in neuronal maturation medium supplemented with ascorbic acid, dibutyryl cAMP, 20 ng/ml BDNF (Peprotech, 450–02), and 20 ng/ml GDNF (Peprotech, 450–10) (see supplemental material for more details). After 3 weeks in neuronal maturation medium, mature neurons were treated with our GW toxicant regimen consisting of 3 days of 2 μM hydrocortisone (Cortisol) (Sigma-Aldrich, H0888), followed by 2 days of 2 μM Cortisol plus 200 nM diisopropyl fluorophosphate (DFP) (Sigma-Aldrich, D0879). After 2 days in Cortisol + DFP, one half of the medium was changed, and then, cells were studied 3 days later (8 days after first cortisol exposure).
3. Evaluation of cell viability and plasma membrane integrity in cells exposed to GW toxicants. On day 16 of neuronal differentiation, NPCs were replated at a density of 25,000 cells/well on clear 96 well flat-bottom plates and cultured for 4 weeks followed by treatment with GW toxicants according to the procedure described above, except that DFP was tested at various concentrations, namely 200 nM, 1 µM, and 2.5 µM, with all concentrations also including 2 μM cortisol exposure. Cells treated with our GW toxicant regimen were evaluated for their viability via the MTT assay (Abcam, ab211091) and the lactate dehydrogenase (LDH) assay (Invitrogen, C20300) according to the manufacturers’ instructions. The absorbance was read on a Tecan Infinite M200Pro spectral plate reader for optical density with subsequent analyses to calculate percent cytotoxicity. See supplemental material for more detailed methods.
4. Quantitative immunocytochemistry. On day 16 of neuronal differentiation, NPCs were plated at a density of 70,000 cells per well of a 24 well plate on round German glass coverslips (Neuvitro, GG12PRE) and cultured for 4 weeks followed by treatment with the GW toxicant regimen (2 μM Cortisol plus 200 nM DFP) according to the procedure described above. Cells were fixed in pre-warmed 4% paraformaldehyde (Electron Microscopy Sciences, 19202) in 0.1 M phosphate-buffered saline (PBS) for 10 min at room temperature. Fixed cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, X100) diluted in PBS for 10 min followed by blocking with 10% goat serum (Jackson ImmunoResearch, 005–000-121) diluted in PBS for 1 h at room temperature. Primary antibodies were diluted in PBS and incubated with cells overnight at 4 °C. The next day, secondary antibodies (diluted in PBS) were incubated with cells for one hour at room temperature protected from light. Cells were stained with 4,6-diamino-2-phenylindole (DAPI) (1:15,000; Invitrogen, D1306) in PBS for 5 min. Glass coverslips were mounted on microscope slides using Fluoro-Gel with Tris Buffer (Electron Microscopy Sciences, 17985–10). Primary antibodies used were rabbit anti-TauR1 (1:1000; donation from Nicholas Kanaan, Michigan State University), mouse anti-AT8 (1:500; Thermo Scientific, MN1020), chicken anti-Tbr1 (1:200; Millipore-Sigma, AB2261), mouse anti-NeuN (1:500; Abcam, 104224), rabbit anti-MAP2 (1:500; Millipore-Sigma, AB5622), rabbit anti-ßIII-tubulin (1:2000; BioLegend, 802001), mouse anti-acetylated tubulin (1:8000; Sigma-Aldrich, T6793), and rat anti-tyrosinated tubulin (1:2000; Millipore-Sigma, MAB1864). Appropriate goat secondary antibodies conjugated to Alexa Fluor 488, 555, and 647 were used at 1:1000 dilutions. Images were acquired on a Zeiss AxioObserver Z1 inverted microscope equipped with a Zeiss 20X/0.8 Plan-Apochromat air objective and AxioCam 506 mono-camera using the Zeiss Zen Blue software. All images from each replicate were acquired with identical exposure settings, did not contain pixel saturation, and showed minimal bleaching during image acquisition. Fluorescence intensity was quantified using the Zeiss Zen Blue software by tracing axon lengths and measuring the average fluorescence intensity per unit length of axon.
5. Extracellular tau ELISA analysis. Cell cultures were treated with 2 μM Cortisol plus 200 nM DFP, and their supernatant was collected with protease inhibitors (Thermo Scientific, 78430) and phosphatase inhibitors (Thermo Scientific, 78428) and analyzed fresh for extracellular human total tau (Invitrogen, KHB0041) and [pT231] human phosphorylated tau (Invitrogen, KHB8051) according to the manufacturer’s instructions. The absorbance was read on a Tecan Infinite M200Pro spectral plate reader for optical density with subsequent analyses to calculate the concentration of extracellular human total tau or [pT231] human phosphorylated tau in each sample. See supplemental material for more detailed methods.
6. Mitochondrial health, dynamics, and transport. On day 16 of neuronal differentiation, NPCs were replated at a density of 35,000 cells on 35 mm round glass bottomed dishes (Cellvis, D35-14–1.5-N) and cultured as described above. After application of the GW toxicant treatment (2 μM Cortisol plus 200 nM DFP) or vehicle, the cell permeable dye tetramethylrhodamine, ethyl ester (TMRE) (Abcam, ab113852) was added directly into the cell culture medium to a final concentration of 200 nM and incubated at 37 °C for 30 min. After incubation, dishes were washed with FluoroBrite DMEM (Gibco, A1896701) imaging medium pre-warmed to 37 °C, and then, fresh FluoroBrite DMEM was used for live-cell imaging in a heated chamber at 37 °C and 5% CO2. Dishes were imaged using a Zeiss AxioObserver Z1 inverted microscope equipped with a Zeiss 63X/1.4 Plan-Apochromat oil objective and a Photometrics Evolve 512 Delta EMCCD camera using the Zeiss Zen Blue software. Images were acquired every 1 s for 2 min at a fixed exposure time across conditions. Mitochondrial transport was analyzed using the MultiKymograph plugin for (Fiji) ImageJ. Mitochondrial fluorescence intensity was analyzed by tracing the circumference of individual mitochondria and dividing the mean arbitrary fluorescence by the area, while length was measured using the longest axis. Fusion and fission events were quantified by counting the number of events per 100 µm of axon per minute.
7. Intracellular HDAC6 activity. HDAC6 activity was measured using a fluorometric kit (Biovision, K466-100) on fresh lysates according to the manufacturer’s instructions. The assay utilizes deacetylase activity of HDAC6 toward a synthetic acetylated-peptide substrate resulting in the release of an AFC fluorophore, which can be quantified using a conventional microplate reader. In brief, cells treated with or without the GW toxicant regimen (2 μM Cortisol plus 200 nM DFP) were lysed in HDAC6 lysis buffer for 5 min on ice and centrifuged at 16,000 × g for 10 min at 4 °C, and then, the supernatant was used to quantify protein content via the Pierce BCA protein assay kit (Thermo Scientific, 23225). 3 μl of each sample was mixed with 50 µl of HDAC6 substrate mix and incubated at 37 °C for 30 min, followed by addition of 10 µl of Developer to stop the reaction and incubated at 37 °C for 10 min. Fluorescence was measured on a Tecan spectral plate reader at excitation/emission 380/490 nm in endpoint mode at 37 °C. Manufacturer’s instructions were followed to determine the amount of HDAC6 activity in each sample using a standard curve to plot the fluorescence obtained from the sample (ΔRFU) and calculate the HDAC6 activity according to the amount of protein loaded. The results are expressed as U per mg of protein, where U stands for the amount of HDAC6 required to deacetylate 1 pmol of HDAC6 substrate per minute under the assay conditions.
8. Neuronal activity analysis via Multi-Electrode Array (MEA) electrophysiology. On day 16 of neuronal differentiation, NPCs were replated onto Multiwell-MEA plates (Multi Channel Systems MCS GmbH, 24W300/30G-288) pre-coated overnight at room temperature with ~ 0.07% polyethyleneimine (PEI) (Sigma-Aldrich, 181978) diluted in borate buffer [for 50 ml: 155 mg boric acid (Sigma-Aldrich, B-0252), 237.5 mg borax (Sigma-Aldrich, B-9876), ddH2O to 50 ml]. Plates were washed with ddH2O and then thoroughly dried for 1 h before plating cells. Cells were plated at a density of 50,000 cells per well and differentiated according to the protocol described above for 1 week in neuronal differentiation medium and 1 week in neuronal maturation medium, both with 5 µg/ml laminin added weekly into the medium. Cells were then cultured in BrainPhys neuronal medium N2-A and SM1 Kit (Stem Cell Technologies, 05793) supplemented with ascorbic acid, dibutyryl cAMP, BDNF, GDNF, and weekly laminin, for 3 weeks before GW toxicant regimen treatment (2 μM Cortisol plus 200 nM DFP) and extracellular electrophysiological recordings. Spontaneous neuronal activity was recorded for 5 min using the Multiwell-Screen software (Multi Channel Systems MCS GmbH). Signals were sampled at 20 kHz and filtered with a 3500 Hz fourth-order low-pass Butterworth filter and a 1 Hz second-order high-pass Butterworth filter. Data analysis was performed using the Multiwell-Analyzer software (Multi Channel Systems MCS GmbH). Spikes were detected when the signal exceeded 6 standard deviations from the baseline noise. Bursts were detected when there were ≥ 4 spikes in 50 ms with 100 ms between bursts, with a maximum interval of 50 ms to start and end the burst. Network bursts were detected when there were six electrodes participating with three of them simultaneously participating. Spike and burst data were exported to Microsoft Excel for quantification. Neuronal activity was quantified for numerous spike, burst, and network burst parameters. An electrode was considered active with a minimum threshold of three spikes per minute. Actively bursting electrodes or active wells with network bursting needed at least one burst or network burst during the 5 min recording to be included for analysis. Neuronal activity was evaluated and confirmed pharmacologically by adding synaptic antagonists into the cell culture media. Activity was recorded before and 5–60 min after addition of the AMPA receptor antagonist CNQX (50 µM; Sigma-Aldrich, C239), NMDA receptor antagonist AP5 (50 µM; Sigma-Aldrich, A8054), GABA receptor antagonist Picrotoxin (50 µM; Sigma-Aldrich, 124–87-8), and sodium channel antagonist tetrodotoxin (TTX) (1 µM; Hello Bio, HB1034).
9. Extracellular glutamate analysis. Cell cultures were treated with the GW toxicant regimen (2 μM Cortisol plus 200 nM DFP) and their supernatant was collected with protease inhibitors and analyzed fresh for extracellular glutamate using the glutamate assay kit (Sigma-Aldrich, MAK004) according to the manufacturer’s instructions. See supplemental material for more detailed methods.
10. Rat Behavioral Studies. Male albino Wistar rats (Envigo RMS, Inc., Indianapolis, IN) approximately 2 months old were housed in pairs in a temperature-controlled room (25 °C), maintained on a 12 h light/dark cycle with free access to food (Teklad Rodent Diet 8604 pellets, Harlan, Madison, WI) and water. All animal procedures employed during this study were reviewed and approved by the Institutional Animal Care and Use Committee of Augusta University and are consistent with AAALAC guidelines. Measures were taken to minimize pain or discomfort in accordance with the Guide for the Care and Use of Laboratory Animals [33]. Efforts were made to minimize the total number of animals used while maintaining statistically valid group numbers. The drug dosing procedures for the in vivo experiments in this study were based on Koo et al. [13] where corticosterone (CORT) was administered to rats for several days to simulate physical stress and a single injection of DFP was administered to simulate nerve agent exposure. CORT is the stress hormone in rodents that corresponds to cortisol in humans. In our studies, 0.4 mg/kg CORT was administered in the drinking water (400 mg/L in 1.2% ethanol) for seven consecutive days based on each animal’s 24 h water consumption, and DFP 1.5 mg/kg was administered on day seven by intraperitoneal injection. Separate groups of control rats were administered drinking water (1.2% ethanol) for 7 days followed by a saline injection on day 7. Behavioral testing (i.e., day 1 of the water maze hidden platform acquisition trials, and day 1 habituation in the novel object recognition test, see task descriptions below) was initiated 48 h after the DFP or saline control injection.
10a. Morris water maze procedures. Test apparatus—water maze testing was conducted using a modification of procedures that we have published previously [34, 35] in a circular pool (diameter: 180 cm, height 76 cm) made of plastic (Bonar Plastics, Noonan, GA) with the inner surface painted black. The pool was filled to a depth of 35 cm of water (maintained at 25.0 ± 1.0 ºC) that covered an invisible (black) 10 cm round platform. The platform was submerged approximately 1 cm below the surface of the water and placed in the center of the northwest quadrant. The pool was located in a large room with diffuse lighting and a black curtain surrounded the pool that hid the experimenter from the test subjects. The curtain fully surrounded the pool with only narrow openings at the north and south quadrants and contained extra-maze geometric images (squares, triangles and circles) hung on the curtain to provide visual spatial cues to the subject. Swimming activity of each rat was monitored via a camera mounted overhead which relayed information including latency to find the platform, total distance traveled, time, and distance spent in each quadrant, to a video tracking system (Noldus EthoVision XT13®).
Hidden platform trials—for hidden platform trials, rats were given three trials per day for five consecutive days to locate and climb onto the hidden platform. A trial was initiated by placing the rat in the water directly facing the pool wall (i.e., nose approximately 2 cm from the wall) in one of the four quadrants. The daily order of entry into individual quadrants was pseudo-randomized, such that all four quadrants were used across the study. For each trial, the rat was allowed to swim a maximum of 90 s to find the platform. When successful, the rat was allowed a 20 s rest period on the platform. If unsuccessful within the allotted time period, the rat was given a score of 90 s and then physically placed on the platform and also allowed the 20 s rest period. In either case, the rat was given the next trial after an additional 40 min rest period (i.e., intertrial interval = 40.0 min).
Probe trial—After the last hidden platform test on day 5, a 60 s probe trial was performed where the platform was removed from the pool and the “spatial bias” for the previous platform location was assessed [36]. This was accomplished by measuring the percentage of time spent in the previous target quadrant and the total dwell time in the annulus-40, defined as a 40 cm-diameter target zone centered on the previous hidden platform location.
Reversal learning trials—24 days after the DFP injection and 17 days after the completion of the first round of hidden platform and probe trials, the platform was switched to the opposite quadrant (i.e., southwest) in the pool and a second series of “reversal learning” trials was conducted. Other than the location of the hidden platform, these trials were conducted in the same manner as the first round of testing (three trials per day for five consecutive days, plus a probe trial on day five).
10b. Spontaneous novel object recognition (NOR) task. The NOR task used in this study was adapted from [37] and modified from versions published previously in our laboratory [38–40] using 4 h delays between training and test sessions. Briefly, test subjects were acclimated to laboratory conditions (i.e., tail marking, daily handling, and weighing) for at least 3 days prior to experimentation. During experimentation, the animals were transported to the laboratory and acclimated for 30 min prior to initiating the experimental phase; the animals remained in the laboratory for 15 min following completion of the day’s testing.
Habituation—The animals were individually placed in a dimly lit (10-lx) training/testing environment (an opaque plastic chamber, 78.7 cm × 39.4 cm × 31.7 cm with bedding on the floor) for 10 min of chamber exploration. The NOR chamber was placed on a table positioned along the wall of the laboratory, and there were no room orienting cues or wall-mounted visual cues (except for the small B/W camera positioned above the NOR chamber). At the beginning of each series of NOR experiments, fresh bedding material was placed in the chamber prior to habituation and allowed to become saturated with animal odors. Animal droppings were removed between experimental sessions; however, the same bedding remained in the chamber for the remainder of each study (i.e., during training and testing), thus preventing any specific olfactory cues over the course of experimentation.
Training trial—twenty-four hours after the habituation session, the animals were again acclimated to the testing room, then placed in the testing chamber with their nose facing the center of a long wall, and allowed to explore two identical objects for 10 min. The animal’s behavior was observed and digitally recorded via a B/W camera located 69 cm above the chamber; the investigator sat quietly 10–15 ft. away from the NOR chamber.
Test trial—four hours after the training trial, two objects, one object identical to training (familiar) object, and a novel object were placed in the chamber, and the animal was allowed to explore both objects for 5 min. Experimental objects to be discriminated were a plastic multicolored Duplo-Lego blocks tower (12 cm in height, 6 cm in width) paired with ceramic conical-shaped green Christmas tree salt/pepper shakers (12 cm in height, 5 cm in diameter); all objects existed in duplicate. The objects were placed 19.3 cm from the walls of the chamber with a distance of 40 cm between the two objects. The role of familiar and novel object as well as chamber position of the object was randomly assigned across subjects and treatments, and objects were cleaned between sessions with a 50% ethanol solution to eliminate olfactory cues. The criteria for the observer to classify an object interaction as exploratory (investigative) behavior were direct interaction with nostrils or head positioning toward the object from a maximum distance of 2 cm. Physically climbing, rearing, and digging around an object was not scored as object exploration. The primary behavioral measure was time (s) spent investigating each object. A discrimination index (d2) was calculated on each test trial and was defined as the difference in time spent exploring the novel and familiar objects divided by the total exploration time for both objects: d2 index = (novel – familiar)/(novel + familiar). This measure is considered as an index of recognition memory and takes into account individual differences in the total amount of exploration time. For data inclusion, the rat had to explore each individual object for at least 4 s and spend a minimum of 12 s of total object exploration. Animals were tested only once, and object exploration time was scored live under blind testing methods (i.e., the investigator was unaware of treatment assignment). The test sessions were also digitally recorded for record-keeping purposes.
11. Immunohistochemistry analyses. Rats were anesthetized with isoflurane and euthanized by transcardial perfusion with PBS (pH 7.4) followed by 4% (wt/vol) paraformaldehyde in PBS. Whole brains were post-fixed overnight in 4% paraformaldehyde. Brains were then transferred to a cryoprotectant of 30% (wt/vol) sucrose and allowed to sink before cryosectioning. Whole 20 µm coronal cryosections were mounted in sequence onto charged microslides by the Histology Core Facility at Augusta University. For tau staining, slides were washed in PBS before incubation in sodium citrate buffer (10 mM tri-sodium citrate dihydrate, 0.05% Tween 20, pH 6.0) at 95 °C for 20 min. After washing in PBS, slides were blocked for 1 h at room temperature in 10% goat serum, 1% bovine serum albumin (Sigma-Aldrich, A7906), 0.5% Triton X-100, and glycine (7.5 mg in 1 ml, Fisher, BP381-5) diluted in PBS. Primary antibodies to tau (see immunocytochemistry above) were diluted in PBS with 10% of the blocking solution without glycine and incubated overnight at 4 °C. Appropriate goat secondary antibodies conjugated to Alexa Fluor 488, 555, and 647 were used at 1:1000 dilutions. Slides were mounted with glass coverslips (Corning, 2975–245) using Fluoro-Gel with Tris Buffer. Images were acquired on a Zeiss AxioObserver Z1 inverted microscope equipped with a Zeiss 20X/0.8 Plan-Apochromat air objective and AxioCam 506 mono-camera using the Zeiss Zen Blue software. All images from each replicate were acquired with identical exposure settings, did not contain pixel saturation, and showed minimal bleaching during image acquisition. Fluorescence intensity was quantified using Fiji (ImageJ) by drawing a box encompassing the CA1 or CA3 regions of the hippocampus, or in the prefrontal cortex, to measure the average fluorescence intensity, followed by dividing by the total number of DAPI-positive cells in the box.
12. Statistics. hiPSC-derived neurons and rat histology: all data analyses were conducted blind and presented as mean ± SEM. At least three independent repeats were conducted for each experiment. Data were arranged in Microsoft Excel before statistical analyses were performed using Prism 9 (GraphPad). All data sets were tested for normality using Shapiro–Wilk’s test, and outliers were excluded using the GraphPad ROUT method (a three-step process that includes a robust nonlinear regression, the residuals were analyzed for outliers, and an ordinary least-squares regression was performed on the remaining data) with a coefficient of Q = 0.1% set at a strict threshold to remove definitive outliers. The statistical tests used were one-way analysis of variance (ANOVA) followed by the appropriate Tukey, Dunnett, or Šídák post hoc test to account for multiple comparisons (Kruskal–Wallis test followed by Dunn’s multiple comparison test for data not normally distributed). An alpha of p < 0.05 was considered statistically significant.
Rat behavior statistical analysis was performed using SigmaPlot 11.2 and statistical significance was assessed using an alpha level of 0.05. Student’s t tests and analysis of variance (with repeated measures when indicated) were used as appropriate followed by the Student Newman–Keuls test for post hoc analysis after ANOVA comparisons. All results are expressed as the mean (± SEM).
Study approval
All human studies involving recruitment and participation of GW veterans were approved by the Institutional Review Board of Boston University. All participants provided written informed consent for use of their blood samples for reprogramming into hiPSCs prior to participation in the study. All patient demographic information was deidentified to protect patient privacy. All animal procedures employed during this study were reviewed and approved by the Institutional Animal Care and Use Committee of Augusta University and are consistent with AAALAC guidelines.
Results
Validated pluripotency of veteran-derived hiPSCs. We recently established multiple lines of hiPSCs generated from peripheral blood mononuclear cells (PBMCs) donated by veterans with or without GWI [31] and banked them in the BBRAIN biorepository. Demographic information for the GW veterans whose blood cells were reprogrammed into hiPSCs and used in this study is listed in Table 1. Phase-contrast imaging revealed that the hiPSCs displayed normal morphologies and formed colonies of densely packed cells (Fig. 1 a–c), which also positively stained for the nuclear marker DAPI (Fig. 1 d–f). Pluripotency was validated by three independent methods. First, the hiPSCs positively immunostained for TRA-1–60 and TRA-1–81, which are two surface markers of pluripotent stem cells, but stained negative for SSEA-1, which is not expressed on undifferentiated human stem cells (Fig. 1 g–i). Second, the hiPSCs have a normal karyotype, shown here for a male (Fig. 1 j). Third, the hiPSCs were able to differentiate into the three germ layers, and cells were positively immunostained for markers of each germ layer: SOX17 (endoderm), Brachyury (mesoderm), and Otx2 (ectoderm) (Fig. 1 k–m).
Table 1.
Demographic information for GW veterans who donated blood samples for reprogramming
| GW veterans with (case) or without GWI | Age | Memory problems |
|---|---|---|
| Control 1 | 58 | No |
| Control 2 | 69 | Yes |
| Control 3 | 63 | No |
| Case 1 | 57 | Yes |
| Case 2 | 54 | No |
| Case 3 | 59 | Yes |
| Case 4 | 54 | No |
Fig. 1.

Pluripotency validation of GW veteran-derived hiPSCs. a–c Phase-contrast images of the hiPSC colonies. Scale bar, 100 µm. d–f DAPI staining. g–i Immunostaining for the pluripotent stem cell surface markers TRA-1–60 and TRA-1–81, and negative staining for the differentiation marker SSEA-1. j The hiPSCs have a normal (male) karyotype. k–m Three-germ-layer differentiation immunostained for the germ layer specific markers Sox17 (endoderm), Brachyury (mesoderm), and Otx2 (ectoderm). Scale bar, 20 µm
Forebrain neuronal differentiation of veteran-derived hiPSCs. As shown schematically in Fig. 2a, hiPSCs were differentiated into forebrain glutamatergic neurons which positively immunostained for the neuronal markers MAP2, Tau, NeuN, and the forebrain glutamatergic marker Tbr1 (Fig. 2 b–d). Neuronal differentiation was compared across all the hiPSC lines to determine whether they had similar differentiation capabilities and could thus be compared to each other for experimental analyses. The ratio of the forebrain glutamatergic neuron marker Tbr1 to DAPI-stained nuclei was not significantly different across the lines, p = 0.0637 (Fig. 2 e). The neurons all exhibited spontaneous neuronal activity that was recorded on a Multiwell Multielectrode Array (MEA) that detected neuronal spiking and bursting and displayed the spike waveforms (Fig. 2 f–h). No differences in neuronal differentiation were observed at baseline among the lines, indicating that experimental results could be compared across the lines. Because epigenetic memories of the original cells are generally lost during the extensive cloning process as well as the prolonged de-differentiation and re-differentiation procedures during development of hiPSC lines [41], it was necessary to re-expose the hiPSC-derived neurons to the GW-relevant toxicants for our investigations.
Fig. 2.

Validation of forebrain glutamatergic neuronal differentiation from veteran-derived hiPSCs and GW toxicant regimen. a Schematic of the neuronal differentiation protocol with phase contrast and immunocytochemical images relevant to each stage. The large gray arrows represent the culture conditions for all experiments (top arrow) except for electrophysiology (bottom arrow). Scale bar for phase contrast images (100 µm) and for immunofluorescent image (20 µm). b–d Immunocytochemical staining for the neuronal markers MAP2, Tau, NeuN, and the forebrain glutamatergic marker Tbr1. Scale bar, 50 µm. e Bar graph shows quantification of the percent of Tbr1-positive cells per total DAPI-stained cells across the seven lines of hiPSC-derived glutamatergic neurons, analyzed using one-way ANOVA with Tukey’s post hoc test. f–h Representative images of spontaneous neuronal firing recorded from a Multiwell MultiElectrode Array. f Raster plot shows the total activity recorded for all 12 electrodes in a sample well. Black lines indicate detected spikes and red bars indicate detected bursts. g Representative spike and burst activity recorded on electrode 43 from F. h Spike waveforms recorded on electrode 43 from F. i, j All 7 hiPSC lines were differentiated into forebrain glutamatergic neurons and exposed to the GW toxicant regimen of 2 µM cortisol plus DFP at three different concentrations (200 nM, 1 µM, and 2.5 µM). The neurons were evaluated for percent cell membrane damage via the LDH assay (i) and the percent metabolic impairment (reduced cell viability) via the MTT assay (j). Each line of hiPSC-derived neurons was exposed to vehicle (veh) or cortisol plus DFP (C + D). Empty bars for vehicle represent no cell damage. All data are represented as mean ± SEM normalized to vehicle per line, and analyzed using one-way ANOVA with Dunnett’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Lowest DFP concentration does not impair cell viability and plasma membrane integrity in hiPSC-derived neurons. Previous studies in rodents and primary rat neurons have shown that the stress hormone corticosterone (CORT) exacerbates the effects of organophosphate exposure [12, 13, 21]. Here, we examined in the hiPSC-derived neurons the cytotoxicity of our GW toxicant regimen of the human stress hormone cortisol plus DFP, a sarin surrogate, across multiple concentrations. We exposed neurons to 2 µM cortisol, a concentration established in our recent studies [21], combined with three concentrations of DFP, 200 nM, 1 µM, and 2.5 µM (Fig. 2i, j); previously, the 200 nM DFP concentration was the highest dose tested in our primary rodent neuron cultures [21].
The LDH and MTT assays were used to measure cytotoxicity. LDH is a cytosolic enzyme that is released into the extracellular media upon cell membrane damage. None of the seven lines showed impaired membrane integrity at the 200 nM DFP concentration and only very low levels of cell membrane damage were observed at the higher 1 µM and 2.5 µM DFP concentrations (Fig. 2i). Only two out of seven lines showed significant cell membrane damage at any concentration compared to vehicle control, with significant percent cytotoxicity measured at 4.11 ± 1.34% (p = 0.037) for 1 µM DFP, and 5.71 ± 1.18% (p = 0.008) and 5.34 ± 1.25% (p = 0.013) for 2.5 µM DFP (Fig. 2i). All other lines at all other concentrations were not significantly different from control.
The MTT assay, which is based on the conversion of water soluble MTT to an insoluble formazan product, can be used as a measure of metabolic activity and cell viability; cells with impaired metabolism will have reduced completion of this reaction, which can be calculated as percent cytotoxicity. Only one out of seven lines showed significant cytotoxicity at the lowest 200 nM DFP concentration (25.55 ± 5.82%, p = 0.012), whereas 6/7 lines showed significant cytotoxicity at the 1 µM DFP and 2.5 µM DFP concentrations, ranging from 11.90 ± 2.94% (p = 0.015) cytotoxicity at 1 µM DFP to 64.47 ± 1.22% (p < 0.0001) cytotoxicity at 2.5 µM DFP (Fig. 2j).
hiPSC-derived neurons from veterans with GWI are more susceptible to develop tau pathology. For examination of tau phenotypes, hiPSC-derived neurons were cultured for more than 1 month to allow for maturation of neural networks before toxicant exposure. Quantitative immunocytochemistry was performed by tracing axons and cell bodies to measure arbitrary fluorescence units per area. The GW toxicant regimen of cortisol plus DFP increased total tau levels (recognized by the TauR1 antibody) in neurons derived from five of seven GW hiPSC lines, ranging from an increase of 1.30 ± 0.07 (p = 0.0014) to 1.74 ± 0.24 (p = 0.0091) times relative to vehicle control (Fig. 3a, c). In addition, the toxicants increased the levels of pSer202/pThr205 phosphorylated tau (recognized by the AT8 antibody) in five of seven hiPSC lines, ranging from an increase of 1.26 ± 0.09 (p = 0.044) to 1.75 ± 0.21 (p = 0.014) times relative to vehicle control (Fig. 3b, d). The increases in total tau and phosphorylated tau levels occurred in all four case lines but in only one out of three control lines. Since active release and uptake of tau have been implicated in the development of tau pathologies [42, 43], and because no overt cell death or plasma membrane damage was identified with 200 nM DFP treatment, we next examined the extracellular tau proteins detected in the media of the hiPSC-neuronal cultures. The neuronal cultures derived from some GW case lines consistently exhibited increased extracellular total tau [in one case line, 1.72 ± 0.03 times relative to vehicle (p < 0.0013)] and increased extracellular Thr231 phosphorylated tau [in two case lines, 1.26 ± 0.03 (p = 0.0003) and 2.05 ± 0.03 (p = 0.041) times relative to vehicle control], whereas none of the control lines showed elevations in extracellular tau (Fig. 3e, f). Taken together, these various results on tau suggest that hiPSC-derived neurons from veterans with GWI may be more susceptible to develop tau pathology than counterpart neurons from their healthy colleagues.
Fig. 3.

hiPSC-derived glutamatergic neurons from veterans with GWI are more susceptible to develop tau pathology after toxicant exposure. a, b Representative images of hiPSC-derived glutamatergic neurons from one control line and one case line exposed to 2 µM cortisol plus 200 nM DFP, and immunostained for total tau (TauR1) (a) and phosphorylated tau (AT8) (b). Scale bar, 50 µm. c, d Graphs show mean arbitrary fluorescence units (AFU) ± SEM normalized to vehicle per hiPSC line for all seven lines of hiPSC-derived neurons exposed to vehicle (veh) or cortisol plus DFP (C + D). Analyses were conducted using the Kruskal–Wallis test followed by Dunn’s post hoc test. e, f Extracellular cell culture supernatant was collected from all seven lines of hiPSC-derived neurons and measured for total tau or p[T231] phosphorylated tau via ELISA. Graphs show mean ± SEM normalized to vehicle per line. Analyses were conducted using one-way ANOVA followed by Šídák’s post hoc test. *p < 0.05, **p < 0.01
Reduced microtubule acetylation in hiPSC-derived neurons upon GW toxicant exposure. GW toxicant exposure was previously shown to lead to reduced microtubule acetylation in primary rat and hiPSC-derived neurons not derived from veterans [21]. Here, two GW hiPSC-neuronal groups, one control line and one case line, were randomly selected and examined for microtubule acetylation. A consistent reduction in the ratio of microtubule acetylation to total neuronal ßIII-tubulin was found in both groups, with levels decreasing to 0.82 ± 0.05 (p = 0.0274) and 0.82 ± 0.04 (p = 0.0122) times relative to vehicle per line (Fig. 4a, b). There was not a corresponding change in the ratio of tyrosinated tubulin to ßIII-tubulin, consistent with the toxicant effects being specific to microtubule acetylation.
Fig. 4.

Reduced microtubule acetylation in hiPSC-derived glutamatergic neurons upon GW toxicant exposure. a Representative images of hiPSC-derived glutamatergic neurons from one control line and one case line exposed to 2 µM cortisol plus 200 nM DFP, and immunostained for ßIII-tubulin (green), acetylated tubulin (red), and tyrosinated tubulin (white). Scale bar, 20 µm. b Graph shows the ratio of acetylated tubulin and tyrosinated tubulin to ßIII tubulin expressed as mean arbitrary fluorescence units (AFU) ± SEM normalized to vehicle per line of hiPSC-derived glutamatergic neurons exposed to vehicle or cortisol + DFP. Analyses were conducted using the Kruskal–Wallis test followed by Dunn’s post hoc test. c Quantification of HDAC6 activity from cell lysates of all seven lines of hiPSC-derived glutamatergic neurons exposed to vehicle (veh) or 2 µM cortisol plus 200 nM DFP (C + D) using an HDAC6 activity assay kit. Bars show mean HDAC6 activity ± SEM normalized to vehicle per line. Analyses were conducted using one-way ANOVA followed by Šídák’s post hoc test. *p < 0.05, **p < 0.01, ****p < 0.0001
To further investigate the potential mechanisms that may underlie reductions in microtubule acetylation, we quantified the activity of the histone deacetylase 6 (HDAC6) enzyme, which is the predominant tubulin deacetylase in neurons. We found a corresponding increase in HDAC6 activity in the same two hiPSC-neuronal lines when treated with GW toxicants, with increased levels of 5.36 ± 0.30 (p < 0.0001) and 6.44 ± 1.36 (p < 0.0001) times relative to vehicle control per line (Fig. 4c).
Altered mitochondrial health, dynamics, and transport in hiPSC-derived neurons exposed to GW toxicants. Mitochondrial transport defects were previously demonstrated in primary rodent neurons exposed to GW toxicants, and so studies here were limited to two hiPSC lines, one control and one case. TMRE added to cell culture media was taken up by mitochondria with an active membrane potential, which were then imaged live (Fig. 5a). Various parameters relevant to mitochondrial health, dynamics, and transport were then quantified. No significant differences were observed in the percent of mitochondria that were moving vs. stationary, percent anterograde vs retrograde movements, number of reversals, number of stops, time of movement, or instantaneous or average velocity. However, the GW toxicant regimen of cortisol plus DFP reduced mitochondrial fluorescence intensity, which indicates mitochondrial health, but intensity decreased significantly only in the case line to 0.68 ± 0.12 times relative to vehicle control, p = 0.045 (Fig. 5a, c). Additionally, a corresponding increase in mitochondrial length and number was observed. Mitochondrial length increased only in the case line to 1.54 ± 0.11 times relative to vehicle (p < 0.0001), while the number of mitochondria per 100 µm of axon increased in the control line to 1.18 ± 0.06 (p = 0.034) and in the case line to 1.42 ± 0.09 (p = 0.0004) times relative to vehicle per line (Fig. 5d, e). The toxicant regimen also significantly decreased run length, which is a measure of the distance traveled during each transport event, for both hiPSC lines, with length decreasing to 0.74 ± 0.08 (p = 0.0267) and 0.73 ± 0.06 (p = 0.0297) times relative to vehicle for the control line and case line, respectively (Fig. 5b, f). Finally, GW toxicant regimen exposure increased the number of mitochondrial fusion and fission events per 100 µm of axon per minute, with an increase in fusion in the case line to 2.98 ± 0.46 times relative to vehicle (p = 0.0216), and an increase in fission in the control line to 1.41 ± 0.14 (p = 0.008) and in the case line to 1.66 ± 0.14 (p = 0.0416) times relative to vehicle per line (Fig. 5 g, h). Together, these results suggest that exposure of the hiPSC-derived neurons to the GW toxicant regimen impairs mitochondrial health, dynamics, and transport. The fact that many of the alterations occur only in the case line suggests that these neurons are more susceptible to toxicant exposure than neurons from the control line.
Fig. 5.

Altered mitochondrial health, dynamics, and transport in hiPSC-derived glutamatergic neurons exposed to GW toxicants. hiPSC-derived glutamatergic neurons from one control line and one case line exposed to vehicle (veh) or 2 µM cortisol plus 200 nM DFP (C + D) were incubated with the cell permeable dye TMRE at 200 nM for 30 min to label mitochondria with an active membrane potential. a Representative live-cell images of individual mitochondria marked with the mitochondrial dye TMRE. Scale bar, 5 µm. b Representative kymographs depicting mitochondrial distance traveled over time. Scale bar, 10 µm. c Graph shows mean arbitrary TMRE fluorescence units (AFU) for individual axonal mitochondria in one control line and one case line exposed to vehicle or cortisol + DFP. d Bars show mean length for individual axonal mitochondria. e Quantification of the average number of mitochondria per 100 µm of axon. f Analysis of mean mitochondrial run length. g, h Graphs show mean mitochondrial fusion (g) and fission (h) events per 100 µm of axon per minute. All data are represented as mean ± SEM normalized to vehicle per line, and analyzed using the Kruskal–Wallis test followed by Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001
hiPSC-derived neurons exhibit reduced neuronal activity after toxicant exposure. hiPSC-derived neurons from all seven lines were cultured on multielectrode arrays (MEA, Multi Channel Systems) to record how the toxicant regimen might alter neuronal activity. The activity of the hiPSC-derived forebrain glutamatergic neurons was first validated by pharmacological administration into the culture media of antagonists to sodium channels and to NMDA/AMPA/GABA receptors. Spontaneous neuronal activity was recorded before and 5–60 min after addition of the AMPA receptor antagonist CNQX (50 µM), NMDA receptor antagonist AP5 (50 µM), GABA receptor antagonist Picrotoxin (50 µM), and sodium channel antagonist tetrodotoxin (TTX) (1 µM). Addition of CNQX and AP5 into the culture media reduced network burst frequency, which can be used to measure synchronous network synaptic activity across multiple electrodes, while addition of picrotoxin increased network burst frequency (data not shown). Addition of TTX abolished all neuronal activity (data not shown).
Next, the hiPSC-derived glutamatergic neurons were exposed to the GW toxicant regimen to examine toxicant-induced impairments in neuronal activity (Fig. 6a, b). Cortisol plus DFP reduced spontaneous neuronal activity for 6 out of 7 hiPSC lines; however, only one control line and one case line showed significant decreases in neuronal firing. The number of active electrodes per well decreased in the control line to 0.40 ± 0.11 (p = 0.0281) times relative to vehicle (Fig. 6c). Spike frequency, measured with at least three spikes per electrode per minute, was significantly reduced to 0.46 ± 0.13 (p < 0.0086) and 0.46 ± 0.05 (p < 0.0001) times relative to vehicle per line for the control and case lines, respectively (Fig. 6d). The GW toxicant regimen also reduced spike bursting, which is another measure of neuronal activity. In the same control and case lines, cortisol plus DFP significantly reduced the number of actively bursting electrodes per well to 0.61 ± 0.10 (p = 0.044) and 0.60 ± 0.23 (p = 0.046) times relative to vehicle per line, respectively (Fig. 6e). Burst frequency, measured as the number of bursts per electrode per minute, was reduced in two control and two case lines of hiPSC-derived neurons to 0.38 ± 0.17 (p = 0.0067), 0.27 ± 0.20 (p = 0.0322), 0.43 ± 0.21 (p = 0.0358), and 0.25 ± 0.09 (p = 0.0128) times relative to vehicle per line, respectively (Fig. 6f). The toxicant regimen reduced burst duration in control line 1 and case line 1 to 0.80 ± 0.08 (p = 0.047) and 0.66 ± 0.07 (p = 0.0015) times relative to vehicle per line (Fig. 6 g), and the number of spikes per burst in the same lines to 0.33 ± 0.08 (p = 0.0425) and 0.47 ± 0.04 (p = 0.0037) times relative to vehicle per line, respectively (Fig. 6h). We also examined network burst frequency, which measures the synchronous activity of a neuronal network across multiple electrodes at the circuit level. Although network bursts were not detected for every line, the GW toxicant regimen significantly reduced the network burst duration to 0.23 ± 0.03 (p = 0.0008) and the number of spikes per network burst to 0.33 ± 0.09 (p = 0.0465) times relative to vehicle only in a case line (Fig. 6k, l), suggesting potential disease susceptibility to toxicant exposure in GW veterans with GWI.
Fig. 6.

hiPSC-derived glutamatergic neurons exhibit reduced neuronal activity after GW toxicant exposure. Neurons were cultured on 24 well Multiwell-MEA plates with 12 electrodes/well from Multi Channel Systems. Spontaneous extracellular neuronal activity was recorded from all seven hiPSC-neuronal lines exposed to vehicle (veh) or 2 µM cortisol plus 200 nM DFP (C + D). a Representative raster plots show the neuronal activity recorded for all 12 electrodes in a sample well for 3 min. Black lines indicate detected spikes, red lines indicate detected bursts, and vertical blue shadows indicate detected network bursts. b Representative spike, burst, and network burst activity for 5 s recorded from hiPSC-derived glutamatergic neurons exposed to vehicle or C + D. c Graph shows the average number of active electrodes per well, defined as at least three spikes per electrode per minute. d Bars show the mean spike frequency quantified from active electrodes. e Graph shows the average number of actively bursting electrodes per well with at least one burst recorded. f Bars show the mean burst frequency quantified as the number of bursts per electrode per minute. g, h Graphs show the mean burst duration (g) and the number of spikes per burst (h). i-l Network bursts were quantified for one control line and one case line for the average number of wells with network bursts detected (i), mean network burst frequency measured as the number of network bursts per well per minute (j), network burst duration (k), and the number of spikes per network burst (l). m Cell culture supernatant was collected from two representative lines to measure the levels of extracellular glutamate via a glutamate detection kit. All data are represented as mean ± SEM normalized to vehicle per line. Analyses were conducted using the Kruskal–Wallis test followed by Dunn’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
In addition to reduced neuronal activity, we collected the cell culture supernatant to measure the amount of extracellular glutamate after toxicant exposure. The GW toxicant regimen reduced the levels of extracellular glutamate in representative control and case lines to 0.65 ± 0.16 (p = 0.036) and 0.31 ± 0.02 (p = 0.005) times relative to vehicle control for each line (Fig. 6m).
Mild learning and memory deficits identified in adult rats exposed to GW toxicants. To ascertain whether the GW toxicant regimen results in learning and memory deficits in an animal model, Morris water maze tests and spontaneous novel object recognition (NOR) analysis were performed. Figure 7 illustrates the acquisition curves for the vehicle and CORT + DFP-treated groups to locate a hidden platform in two phases of water maze testing. For the initial hidden platform trials (acquisition phase), both vehicle control and CORT + DFP-treated rats progressively learned to locate the hidden platform with increasing levels of efficiency over the course of the 5 days of testing as indicated by the decreasing slope of the acquisition curves [main effect of treatment, F(1, 28) = 0.6, p = 0.4; day, F(4,112) = 29.1, p < 0.001; treatment × day, F(4, 112) = 0.8, p = 0.5] (Fig. 7 a). Moreover, there were no significant differences in performance of the task between the treatment groups. Likewise, in the probe trials after the acquisition phase, there were no significant treatment-related differences in the percentage of time spent in the previous target quadrant or the time spent in the annulus−40 area (Fig. 7b).
Fig. 7.
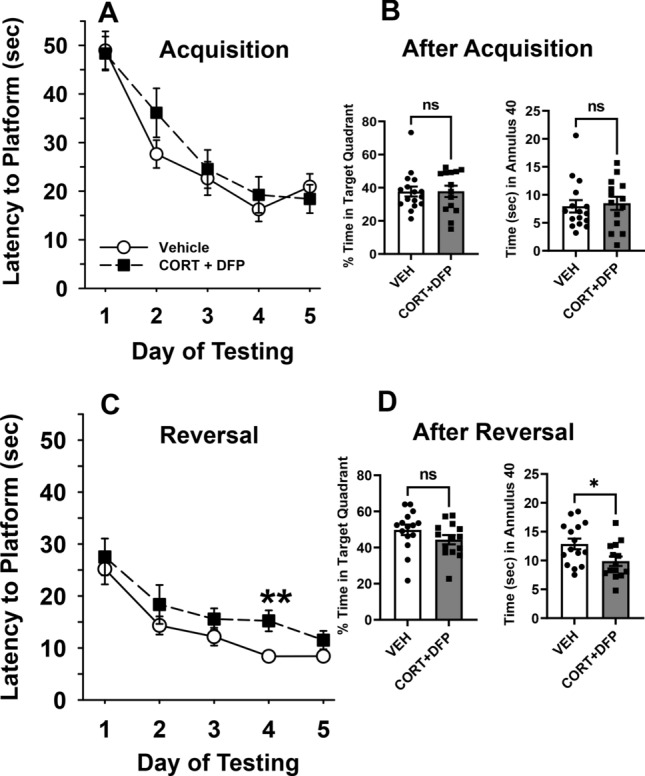
GW toxicants impair reversal learning and memory in adult rats. Effects of 7 days of exposure to CORT (corticosterone) plus a single injection of DFP 1.5 mg/kg on day seven on a water maze repeated acquisition procedure beginning 48 h after the injection. a The graph illustrates the results of the hidden platform tests that were conducted for the first five consecutive days of testing. Each point of the plotted curves represents the mean latency in seconds ± SEM for each testing day. b Effects of CORT + DFP on water maze probe trials. The graph illustrates the results of the probe trial tests that were conducted on day five after the last hidden platform trial in the first phase of testing. Bars represent mean percentage of times spent in the previous target quadrant and the mean times in seconds ± SEM spent in the annulus 40 with filled circles and squares representing individual rats. c The graph illustrates the results of reversal learning trials 24 days after the DFP injection and 17 days after the first phase of water maze testing. In these trials, the hidden platform was moved to a new quadrant location in the pool and 5 additional days of testing occurred. Each point of the plotted curves represents the mean latency in seconds ± SEM for each testing day. d Effects of CORT + DFP on water maze probe trials. The graph illustrates the results of the probe trial tests that were conducted on day five after the last hidden platform trial in the second (reversal learning) phase of testing. Bars represent mean percentage of times spent in the previous target quadrant and the mean times in seconds ± SEM spent in the annulus 40 with filled circles and squares representing individual rats. *p = 0.02, Vehicle vs CORT + DFP. N = 14–16. Analyses were conducted using ANOVA (with repeated measures when indicated) followed by the Student–Newman–Keuls post hoc test and Student’s t test. ns, not significant
Next, the hidden platform was moved to the opposite quadrant of the pool and a second phase of hidden platform trials (reversal trials) were conducted 24 days after the DFP injection and 17 days after the completion of the first round of hidden platform and probe trials. Again, both vehicle control and CORT + DFP-treated rats progressively learned to locate the hidden platform with increasing levels of efficiency over the course of the 5 days of testing as indicated by the decreasing slope of the reversal learning curves [main effect of treatment, F(1,28) = 4.9, p < 0.04; day, F(4,112) = 18.0, p < 0.001; treatment × day, F(4,112) = 1.5, p = 0.2] (Fig. 7c). In this case, there was a modest but statistically significant impairment in performance in the CORT + DFP-treated animals compared to vehicle controls, most notably on day 4 (post hoc comparison, p = 0.02). In the probe trial tests after the reversal learning trials, CORT + DFP-treated rats also spent significantly less time in the annulus−40 area (t = 2.4, p = 0.02) compared to controls (Fig. 7d), but not in the percentage of time spent in the larger target quadrant area.
For the NOR task, all rats met the inclusion criteria of exploring each individual object for at least 4 s and a minimum of 12 s of total object exploration in both the training (A/A) and testing (A/B) sessions. The effects of CORT + DFP treatment on the mean (± SEM) exploration times of the novel and familiar objects in the test (A/B) sessions are illustrated in Fig. 8a, and the calculated discrimination (d2) ratios (to account for individual variability in exploration times) for each subject are illustrated in Fig. 8b. Statistical analysis revealed the following: main effect of treatment [F(1,14) = 0.03, p = 0.9], object type [F(1,14) = 55.7, p < 0.001], and treatment-by-object type interaction [F(1,14) = 2.8, p = 0.1]. Post hoc analysis indicated that there was a significant preference for the novel object (p < 0.01) versus the familiar object in both treatment groups (Fig. 8a). For the analysis of the d2 ratios, there was no significant difference in the d2 ratios in the CORT + DFP treatment group versus the vehicle control animals, p = 0.0956 (Fig. 8b).
Fig. 8.
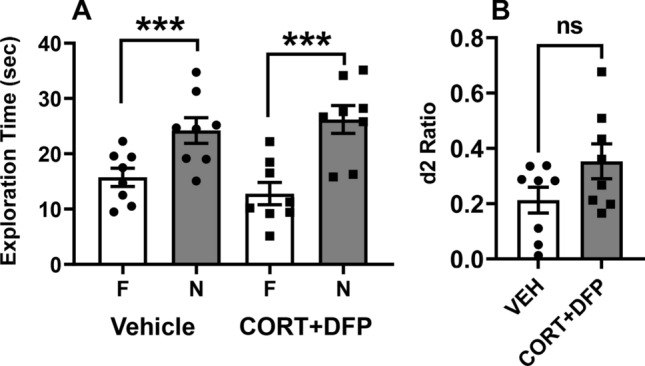
GW toxicants do not alter object recognition memory in adult rats. Effects of 7 days of exposure to CORT (corticosterone) plus a single injection of DFP 1.5 mg/kg on day 7 on performance of a spontaneous novel object recognition (NOR) procedure 48 h after the injection. a Bars on the graph represent the mean (± S.E.M.) exploration times of the familiar (F) and novel (N) objects after a 4 h delay. b The graph shows scatterplots representing discrimination (d2) ratios with filled circles and squares representing individual rats. d2 ratio = (novel—familiar)/(novel + familiar). ***p < 0.001 novel vs familiar object. N = 8 for each group. Analyses were conducted using ANOVA followed by the Student–Newman–Keuls post hoc test and Student’s t test. ns, not significant
GW toxicants induce tau pathology and cell loss in the CA3 region of the hippocampus. After exposure to the CORT + DFP toxicant regimen, rats were sacrificed, and the brains were processed for histological staining. Three regions of the brain involved in learning and memory and executive functioning relevant to the behavioral tasks were examined: the CA1 and CA3 regions of the hippocampus, and the prefrontal cortex (Fig. 9a). In the CA3 region of the hippocampus, rats exposed to CORT + DFP exhibited elevated levels of total tau (recognized by the TauR1 antibody) at 1.40 ± 0.05 times relative to control, p < 0.0001 (Fig. 9b), and elevated levels of pSer202/pThr205 phosphorylated tau (recognized by AT8 antibody) at 1.43 ± 0.06 times relative to control, p < 0.0001 (Fig. 9c), with fluorescence intensity normalized to the number of nuclei per region quantified. No significant differences in total tau or phosphorylated tau were obtained in the CA1 region of the hippocampus or in the prefrontal cortex. Additionally, the pathological tau phenotypes in the CA3 region of the hippocampus were accompanied by a significant reduction in the total number of cells per 100 µm2, decreasing to 0.80 ± 0.03 times relative to control, p = 0.0027 (Fig. 9d). We did not identify any cellular morphological alterations in the CA1 and CA3 regions of the hippocampus or the prefrontal cortex (data not shown). Together, these results suggest that the CA3 region of the hippocampus may be particularly vulnerable to the GW toxicants, which is consistent with imaging studies carried out in GW veterans [44–46].
Fig. 9.

GW toxicants induce tau pathology and cell loss selectively in the CA3 region of the hippocampus. Rats exposed to 7 days of CORT (corticosterone) plus a single injection of DFP 1.5 mg/kg on day seven were sacrificed 48 h after the injection for histological examination. a Representative immunohistochemical images show staining for total tau (TauR1), phosphorylated tau (AT8), and DAPI in the CA1 and CA3 regions of the hippocampus, and in the prefrontal cortex (PFC). Scale bar in CA1 and CA3, 100 µm; in PFC, 50 µm. b, c Graphs show mean arbitrary fluorescence units (AFU) ± SEM, relative to the number of nuclei and normalized to vehicle for total tau staining (b) and phosphorylated tau staining (c) for each brain region. d Quantification shows the mean number of DAPI-stained nuclei per 100µm2 ± SEM normalized to vehicle per each brain region. Histology was quantified from three mice from each group. Analyses were conducted using one-way ANOVA followed by Šídák’s post hoc test. **p < 0.01, ****p < 0.0001
Discussion
The community of researchers and physicians studying GWI in recent years has been funded by the United States Department of Defense to define exactly what the disease is, to elucidate the underlying causes of the disease, to ascertain whether certain veterans are predisposed to develop the disease and why, and most importantly to develop effective therapies for suffering veterans who have been without treatment for decades. While vertebrate animal models remain critical for system-level analyses including behavior, we have advocated for a cellular approach, so that mechanism-based therapies can be developed for the veterans. In addition, there is value in using human cells, because rodents lack significant genomic variation, rendering them unsuitable for tackling the question of why some soldiers developed the disease, while others did not, despite similar exposures. Although the total number of hiPSC lines derived from the GW veterans included in our current studies limits the statistical power of our conclusions, the present work provides a starting point for experimental studies using these cell lines. Larger genomic studies such as single-nucleotide polymorphism (SNP) investigations carried out on GW veteran-derived non-neural cells and tissues may provide insights into susceptible genetic variants, but the CNS-specific susceptibilities might not be fully revealed.
Our earlier work taking a cellular approach to GWI mainly used primary cultures of rat neurons and our present findings are entirely consistent with those earlier findings on deficits in both microtubule acetylation and mitochondrial transport in response to the GW toxicant regimen [21]. We also observed a reduction in mitochondrial membrane potential, increased mitochondrial length and density in axons, and more frequent mitochondrial fusion and fission. Reduced microtubule acetylation renders microtubules less suitable as substrates for key motor proteins involved in axonal transport, and hence might explain the diminished mitochondrial transport as well as decreased neurotransmitter release. Mitochondria are the primary site of energy production in the cell, and disruptions in mitochondrial health, dynamics, and transport have been implicated in many neurological disorders [47–49]. Studies have shown that organophosphate exposure directly affects many aspects of mitochondrial behavior [50–52], and these alterations might contribute to some of the cognitive symptoms of GWI, such as reduced processing speeds and chronic fatigue [6, 7, 11].
Here, for the first time, we showed that exposure to the GW toxicant regimen resulted in robust and consistent tau pathology in hiPSC-derived neurons, with elevated levels of total tau and phosphorylated tau inside and outside of the neuron. The elevated tau might account for the diminished microtubule acetylation, given that greater tau levels on microtubules correspond to reduced stability as does reduced acetylation. In the rat model, the same tau effects were documented in response to the toxicant regimen only in the CA3 region of the hippocampus, but not in the CA1 region or the prefrontal cortex. The toxicant regimen also resulted in loss of cells in the CA3 region of the hippocampus, but not in the CA1 region or the prefrontal cortex, further highlighting the selective vulnerability of the CA3 region to GW-relevant toxicants. Consistent with our findings, smaller hippocampal CA3 volumes have been identified in GW veterans with GWI via magnetic resonance imaging analyses [44–46]. Taken together with a mild memory deficit in these rats in response to the toxicant regimen, these results implicate tau abnormalities as a potential cause of the memory defects in GWI.
Our behavioral experiments using the Morris Water Maze and the NOR task, both of which have notable translational value for human neuropsychological disorders, were designed to examine hippocampal-dependent spatial reference memory as well as reversal learning with cognitive flexibility, and recognition memory, respectively. Moreover, while the hippocampus is considered important for performance of spatial learning tasks, the CA3 region has been specifically described as crucial for both memory acquisition and consolidation in a Morris water maze task in rodents [53]. The exposure produced modest but statistically significant impairments in the performance of reversal learning trials and probe trials (after the reversal learning phase). The deficits in the second phase of water maze testing were suggestive of alterations in cognitive flexibility, which is generally considered a subcomponent of executive function involving the shifting of strategies when the stimulus–response contingency changes [54]. Our results indicate that the behavioral deficit in exposed rats is specific to hippocampal-dependent spatial reference memory, but not recognition memory (tested in the NOR task). Platform re-locating in water maze tasks requires cognitive flexibility, which is suggested to be especially appropriate for extrapolation of learning and memory measures from rodent to human [55]. Correspondingly, deficits in visuospatial processing, learning and memory, and cognitive flexibility/executive function have been consistently identified in veterans with GWI [6, 11].
Various studies have shown that GW-relevant toxicants can impair neurotransmission [21, 56–59] and this disruption may contribute to some of the most prominent cognitive symptoms in GWI, such as reduced processing speeds and poor attention/concentration [7, 11, 60]. We observed robust electrophysiological impairments in neuronal activity in almost all of the hiPSC lines exposed to the toxicant regimen, including reduced activity at both the individual cell and circuit levels, as well as reduced extracellular release of glutamate. These results are consistent with studies on animals and on GW veterans that have identified altered connectivity and neurotransmission as important components of GWI [13, 56, 57, 61], as well as with our previous observations on impaired neurotransmitter release in rat neurons exposed the GW toxicant regimen [21]. We are eager to test whether these effects are due to the alterations in tau and/or microtubules that we have thus far identified.
Throughout our present studies, there were many instances in which the case lines displayed stronger phenotypes in response to the toxicant regimen than the control lines. While the small number of hiPSC lines limits the statistical power with which we can draw conclusions from these observations, we nevertheless find them compelling in light of the often discussed possibility of a predisposition of certain soldiers over others to acquire GWI. Especially compelling is the possibility that a predisposition to tauopathy might be key to which soldiers are most vulnerable. Such considerations may also be relevant to the future of veterans living with GWI, as the potential for tau-based degeneration increases with age. To test for genetic or epigenetic predisposition, we would ideally perform genome-wide association studies (GWAS) on much larger sample sizes (> 100) to identify genetic variants and SNPs associated with GWI.
In conclusion, the studies presented here represent the first experimental results with GW veteran-based hiPSC lines, which are available to the GWI community through our biorepository. The utility of these lines is multifold and accentuate the importance of a cellular approach to resolving the mechanistic basis of the disease. In addition, hiPSC lines will be instrumental for screening of drugs as well as providing an avenue toward personalized therapy for individual veterans.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants from the Department of Defense to Liang Qiang (GW160151) and Peter W. Baas (GW170023), and grants from the CURE program via Drexel University College of Medicine to Liang Qiang (CURE Grant SAP Number: 4100083087) and Peter W. Baas (CURE Grant SAP Number: 4100072545). Additional funding was provided by the National Institutes of Health to Liang Qiang (1R01NS115977) and Peter W. Baas (R01NS28785, R01NS118117 and R21AG068597). Philip L. Yates was partially supported by an Interdisciplinary and Translational Research Training Grant (T32-MH079785) to Drexel University from the National Institutes of Health. Ankita Patil was partially supported by a Dean’s Fellowship for Excellence in Collaborative or Themed Research from Drexel University. The work was further supported by a Department of Defense grant to the Boston Biorepository, Recruitment & Integrative Network (GW170055), directed by Kimberly A. Sullivan. The multielectrode device used for some of the experiments was purchased with funds provided by Drexel University’s Scholarly Material and Research Equipment Awards. We thank Nicholas Kanaan of Michigan State University for providing tau antibodies. We thank Gustavo Mostoslavsky and Marianne F. James of the Boston University Center for Regenerative Medicine and Barbara Corneo and Achchhe Patel from the Columbia University Stem Core Facility for assistance with the hiPSC lines. We also thank Hemalatha Muralidharan, Shrobona Guha, and Neha Mohan of Drexel University for contributing to the project.
Author contributions
PLY conducted all cell culture experiments, acquired and analyzed the data, created the figures, and wrote the manuscript. AP performed the live-cell imaging experiments and analysis. XS conducted the histological experiments and analysis. AN assisted with cell culture and immunocytochemical analysis. RG analyzed the live-cell imaging data. PC and WB performed the animal behavioral studies and analysis. EP conducted the HDAC6 experiments. AVT designed the animal behavioral studies and wrote the behavioral sections of the manuscript. KAS directed the biorepository that stored the hiPSC cell lines, assisted with designing the research studies, and edited the manuscript. PWB and LQ designed the research studies, assisted with data interpretation, and wrote the manuscript.
Funding
This work was supported by grants from the Department of Defense to Liang Qiang (GW160151) and Peter W. Baas (GW170023), and grants from the CURE program via Drexel University College of Medicine to Liang Qiang (CURE Grant SAP Number: 4100083087) and Peter W. Baas (CURE Grant SAP Number: 4100072545). Additional funding was provided by the National Institutes of Health to Liang Qiang (1R01NS115977) and Peter W. Baas (R01NS28785, R01NS118117 and R21AG068597). Philip L. Yates was partially supported by an Interdisciplinary and Translational Research Training Grant (T32-MH079785) to Drexel University from the National Institutes of Health. Ankita Patil was partially supported by a Dean’s Fellowship for Excellence in Collaborative or Themed Research from Drexel University. The work was further supported by a Department of Defense grant to the Boston Biorepository, Recruitment & Integrative Network (GW170055), directed by Kimberly A. Sullivan.
Availability of data and materials
The data that support the findings discussed here are available from the corresponding author upon reasonable request.
Declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethical approval
All human studies involving recruitment and participation of Gulf War veterans were approved by the Institutional Review Board of Boston University. All animal procedures employed during this study were reviewed and approved by the Institutional Animal Care and Use Committee of Augusta University and are consistent with AAALAC guidelines.
Consent to participate
All human participants provided written informed consent for use of their blood samples for reprogramming into hiPSCs prior to participation in the study.
Consent for publication
All human participants provided written informed consent for use of their data for publication after all patient demographic information was deidentified to protect patient privacy.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peter W. Baas and Liang Qiang are co-senior authors.
References
- 1.RAC, Gulf War Illness and the Health of Gulf War Veterans: Scientific Findings and Recommendations, in Research Advisory Committee on Gulf War Veterans’ Illnesses. 2008, U.S. Government Printing Office: Washington, D.C.
- 2.RAC, Gulf War Illness and the Health of Gulf War Veterans: Research Update and Recommendations, 2009–2013, in Research Advisory Committee on Gulf War Veterans’ Illnesses. 2014, U.S. Government Printing Office: Washington, D.C.
- 3.GWIRP, The Gulf War Illness Landscape, in Department of Defense Gulf War Illness Research Program. 2020.
- 4.Steele L. Prevalence and patterns of Gulf War illness in Kansas veterans: association of symptoms with characteristics of person, place, and time of military service. Am J Epidemiol. 2000;152(10):992–1002. doi: 10.1093/aje/152.10.992. [DOI] [PubMed] [Google Scholar]
- 5.White RF, et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: effects of toxicant exposures during deployment. Cortex. 2016;74:449–475. doi: 10.1016/j.cortex.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janulewicz PA, et al. Neuropsychological characteristics of Gulf War illness: A meta-analysis. PLoS One. 2017;12(5):e0177121. doi: 10.1371/journal.pone.0177121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeffrey MG, et al. Neuropsychological Findings in Gulf War Illness: a review. Front Psychol. 2019;10:2088. doi: 10.3389/fpsyg.2019.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickey B, Madhu LN, Shetty AK. Gulf War Illness: mechanisms underlying brain dysfunction and promising therapeutic strategies. Pharmacol Ther. 2020;220:107716. doi: 10.1016/j.pharmthera.2020.107716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colovic MB, et al. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol. 2013;11(3):315–335. doi: 10.2174/1570159X11311030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross SM, et al. Neurobehavioral problems following low-level exposure to organophosphate pesticides: a systematic and meta-analytic review. Crit Rev Toxicol. 2013;43(1):21–44. doi: 10.3109/10408444.2012.738645. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan K, et al. Neuropsychological functioning in military pesticide applicators from the Gulf War: effects on information processing speed, attention and visual memory. Neurotoxicol Teratol. 2018;65:1–13. doi: 10.1016/j.ntt.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 12.O'Callaghan JP, et al. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J Neurochem. 2015;133(5):708–721. doi: 10.1111/jnc.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koo BB, et al. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War Illness. Brain Behav Immun. 2018;67:42–46. doi: 10.1016/j.bbi.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locker AR, et al. Corticosterone primes the neuroinflammatory response to Gulf War Illness-relevant organophosphates independently of acetylcholinesterase inhibition. J Neurochem. 2017;142(3):444–455. doi: 10.1111/jnc.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alshelh Z, et al. In-vivo imaging of neuroinflammation in veterans with Gulf War illness. Brain Behav Immun. 2020;87:498–507. doi: 10.1016/j.bbi.2020.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prendergast MA, et al. Microtubule-associated targets in chlorpyrifos oxon hippocampal neurotoxicity. Neuroscience. 2007;146(1):330–339. doi: 10.1016/j.neuroscience.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gearhart DA, et al. Chlorpyrifos, chlorpyrifos-oxon, and diisopropylfluorophosphate inhibit kinesin-dependent microtubule motility. Toxicol Appl Pharmacol. 2007;218(1):20–29. doi: 10.1016/j.taap.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Grigoryan H, et al. Mass spectrometry identifies covalent binding of soman, sarin, chlorpyrifos oxon, diisopropyl fluorophosphate, and FP-biotin to tyrosines on tubulin: a potential mechanism of long term toxicity by organophosphorus agents. Chem Biol Interact. 2008;175(1–3):180–186. doi: 10.1016/j.cbi.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang W, et al. Mice treated with chlorpyrifos or chlorpyrifos oxon have organophosphorylated tubulin in the brain and disrupted microtubule structures, suggesting a role for tubulin in neurotoxicity associated with exposure to organophosphorus agents. Toxicol Sci. 2010;115(1):183–193. doi: 10.1093/toxsci/kfq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abou-Donia MB. Involvement of cytoskeletal proteins in the mechanisms of organophosphorus ester-induced delayed neurotoxicity. Clin Exp Pharmacol Physiol. 1995;22(5):358–359. doi: 10.1111/j.1440-1681.1995.tb02015.x. [DOI] [PubMed] [Google Scholar]
- 21.Rao AN, et al. Pharmacologically increasing microtubule acetylation corrects stress-exacerbated effects of organophosphates on neurons. Traffic. 2017;18(7):433–441. doi: 10.1111/tra.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 23.Baas PW, et al. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken) 2016;73(9):442–460. doi: 10.1002/cm.21286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12(6):609–622. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- 25.Kneynsberg A, et al. Axonal degeneration in tauopathies: disease relevance and underlying mechanisms. Front Neurosci. 2017;11:572. doi: 10.3389/fnins.2017.00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abou-Donia MB, et al. Screening for novel central nervous system biomarkers in veterans with Gulf War Illness. Neurotoxicol Teratol. 2017;61:36–46. doi: 10.1016/j.ntt.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Abou-Donia MB, et al. Using plasma autoantibodies of central nervous system proteins to distinguish veterans with gulf war illness from healthy and symptomatic controls. Brain Sci. 2020;10(9):610. doi: 10.3390/brainsci10090610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Luo X, et al. Tau hyperphosphorylation: a downstream effector of isoflurane-induced neuroinflammation in aged rodents. Med Hypotheses. 2014;82(1):94–96. doi: 10.1016/j.mehy.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Yoshiyama Y, et al. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J Alzheimers Dis. 2010;22(1):295–306. doi: 10.3233/JAD-2010-100681. [DOI] [PubMed] [Google Scholar]
- 30.Joyce MR, Holton KF. Neurotoxicity in Gulf War Illness and the potential role of glutamate. Neurotoxicology. 2020;80:60–70. doi: 10.1016/j.neuro.2020.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Qiang L, et al. Reprogramming cells from Gulf War veterans into neurons to study Gulf War illness. Neurology. 2017;88(20):1968–1975. doi: 10.1212/WNL.0000000000003938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals Guide for the Care and Use of Laboratory Animals in Guide for the Care and Use of Laboratory Animals. 2011, National Academies Press: Washington (DC).
- 34.Terry AV, Jr, et al. Chronic, intermittent exposure to chlorpyrifos in rats: protracted effects on axonal transport, neurotrophin receptors, cholinergic markers, and information processing. J Pharmacol Exp Ther. 2007;322(3):1117–1128. doi: 10.1124/jpet.107.125625. [DOI] [PubMed] [Google Scholar]
- 35.Callahan PM, et al. Positive allosteric modulator of alpha7 nicotinic-acetylcholine receptors, PNU-120596 augments the effects of donepezil on learning and memory in aged rodents and non-human primates. Neuropharmacology. 2013;67:201–212. doi: 10.1016/j.neuropharm.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11(1):47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 37.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. Behav Data Behav Brain Res. 1988;31(1):47–59. doi: 10.1016/0166-4328(88)90157-X. [DOI] [PubMed] [Google Scholar]
- 38.Callahan PM, et al. Tropisetron sensitizes alpha7 containing nicotinic receptors to low levels of acetylcholine in vitro and improves memory-related task performance in young and aged animals. Neuropharmacology. 2017;117:422–433. doi: 10.1016/j.neuropharm.2017.02.025. [DOI] [PubMed] [Google Scholar]
- 39.Poddar I, et al. Tropisetron enhances recognition memory in rats chronically treated with risperidone or quetiapine. Biochem Pharmacol. 2018;151:180–187. doi: 10.1016/j.bcp.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Callahan PM, Terry AV., Jr . Nicotinic Receptor Ligands and Novel Object Recognition. In: Ennaceur A, de Souza Silva MA, editors. Handbook of Object Novelty Recognition. London: Academic Press; 2018. [Google Scholar]
- 41.Nashun B, Hill PW, Hajkova P. Reprogramming of cell fate: epigenetic memory and the erasure of memories past. EMBO J. 2015;34(10):1296–1308. doi: 10.15252/embj.201490649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener. 2017;12(1):5. doi: 10.1186/s13024-016-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287(6):3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chao LL, et al. Effects of low-level sarin and cyclosarin exposure on hippocampal subfields in Gulf War Veterans. Neurotoxicology. 2014;44:263–269. doi: 10.1016/j.neuro.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chao LL, et al. Evidence of hippocampal structural alterations in Gulf War Veterans With predicted exposure to the Khamisiyah Plume. J Occup Environ Med. 2017;59(10):923–929. doi: 10.1097/JOM.0000000000001082. [DOI] [PubMed] [Google Scholar]
- 46.Chao LL, Zhang Y. Effects of low-level sarin and cyclosarin exposure on hippocampal microstructure in Gulf War Veterans. Neurotoxicol Teratol. 2018;68:36–46. doi: 10.1016/j.ntt.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 48.Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet. 2009;18(R2):R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu YJ, et al. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech Ageing Dev. 2020;186:111212. doi: 10.1016/j.mad.2020.111212. [DOI] [PubMed] [Google Scholar]
- 50.Middlemore-Risher ML, et al. Effects of chlorpyrifos and chlorpyrifos-oxon on the dynamics and movement of mitochondria in rat cortical neurons. J Pharmacol Exp Ther. 2011;339(2):341–349. doi: 10.1124/jpet.111.184762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koslik HJ, Hamilton G, Golomb BA. Mitochondrial dysfunction in Gulf War illness revealed by 31phosphorus magnetic resonance spectroscopy: a case—control study. PLoS One. 2014;9(3):e92887. doi: 10.1371/journal.pone.0092887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masoud A, Kiran R, Sandhir R. Impaired mitochondrial functions in organophosphate induced delayed neuropathy in rats. Cell Mol Neurobiol. 2009;29(8):1245–1255. doi: 10.1007/s10571-009-9420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Florian C, Roullet P. Hippocampal CA3-region is crucial for acquisition and memory consolidation in Morris water maze task in mice. Behav Brain Res. 2004;154(2):365–374. doi: 10.1016/j.bbr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Tanimura Y, Yang MC, Lewis MH. Procedural learning and cognitive flexibility in a mouse model of restricted, repetitive behaviour. Behav Brain Res. 2008;189(2):250–256. doi: 10.1016/j.bbr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 55.Saab BJ, Saab AM, Roder JC. Statistical and theoretical considerations for the platform re-location water maze. J Neurosci Methods. 2011;198(1):44–52. doi: 10.1016/j.jneumeth.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 56.Torres-Altoro MI, et al. Organophosphates dysregulate dopamine signaling, glutamatergic neurotransmission, and induce neuronal injury markers in striatum. J Neurochem. 2011;119(2):303–313. doi: 10.1111/j.1471-4159.2011.07428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gopinath KS, et al. Exploring brain mechanisms underlying Gulf War Illness with group ICA based analysis of fMRI resting state networks. Neurosci Lett. 2019;701:136–141. doi: 10.1016/j.neulet.2019.02.041. [DOI] [PubMed] [Google Scholar]
- 58.Macht VA, et al. Interactions between pyridostigmine bromide and stress on glutamatergic neurochemistry: insights from a rat model of Gulf War Illness. Neurobiol Stress. 2020;12:100210. doi: 10.1016/j.ynstr.2019.100210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown KA, Filipov NM, Wagner JJ. Dorsoventral-specific effects of nerve agent surrogate diisopropylfluorophosphate on synaptic transmission in the mouse hippocampus. J Pharmacol Exp Ther. 2020;373(1):10–23. doi: 10.1124/jpet.119.263053. [DOI] [PubMed] [Google Scholar]
- 60.Toomey R, et al. Neuropsychological functioning of US Gulf War veterans 10 years after the war. J Int Neuropsychol Soc. 2009;15(5):717–29. doi: 10.1017/S1355617709990294. [DOI] [PubMed] [Google Scholar]
- 61.Tillman GD, et al. Electrophysiological correlates of semantic memory retrieval in Gulf War Syndrome 2 patients. J Neurol Sci. 2017;373:66–72. doi: 10.1016/j.jns.2016.12.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings discussed here are available from the corresponding author upon reasonable request.