

Abstract
To protect themselves from the constant threat of bacteriophage (phage) infection, bacteria have evolved diverse immune systems including restriction‐modification, CRISPR‐Cas, and many others. Here, we describe the discovery of a two‐protein transcriptional regulator module associated with hundreds of CBASS immune systems and demonstrate that this module drives the expression of its associated CBASS system in response to DNA damage. We show that the helix‐turn‐helix transcriptional repressor CapH binds the promoter region of its associated CBASS system to repress transcription until it is cleaved by the metallopeptidase CapP. CapP is activated in vitro by single‐stranded DNA, and in cells by DNA‐damaging drugs. Together, CapH and CapP drive increased expression of their associated CBASS system in response to DNA damage. We identify CapH‐ and CapP‐related proteins associated with diverse known and putative bacterial immune systems including DISARM and Pycsar antiphage operons. Overall, our data highlight a mechanism by which bacterial immune systems can sense and respond to a universal signal of cell stress, potentially enabling multiple immune systems to mount a coordinated defensive response against an invading pathogen.
Keywords: bacterial antiphage defense, DNA damage signaling, helix‐turn‐helix DNA binding protein, metallopeptidase
Subject Categories: DNA Replication, Recombination & Repair; Microbiology, Virology & Host Pathogen Interaction; Structural Biology
The CapH/CapP transcriptional regulator module exemplifies how bacterial anti‐phage immunity may be harnessed for other stress responses.
Introduction
In all organisms, survival depends on the ability of cells to sense and respond to both internal and external threats. In addition to environmental stress, bacteria are continually challenged by bacteriophages (phages) and have evolved a wide array of immune systems to protect themselves from phage infection and propagation. Many antiphage immune systems, including restriction‐modification and CRISPR‐Cas systems, specifically recognize and destroy foreign DNA to prevent phage replication (Makarova et al, 2013; Mohanraju et al, 2016). Other immune systems, termed abortive infection systems, sense phage infection and respond by killing the host cell, thereby preventing phage propagation and further infection in the bacterial community (Makarova et al, 2011a; Dy et al, 2014; Doron et al, 2018; Hampton et al, 2020). In many bacteria, multiple immune systems coexist in the so‐called “defense islands” (Makarova et al, 2011b; Doron et al, 2018) and may cooperate, with nonlethal systems acting as a first line of defense and abortive infection systems becoming activated only as a last resort (Bernheim & Sorek, 2020; Picton et al, 2021).
The widespread and functionally diverse CBASS antiphage immune systems use an abortive infection mechanism in which a cGAS/DncV‐like nucleotidyltransferase (CD‐NTase) is activated upon phage infection and synthesizes a cyclic oligonucleotide second messenger (Cohen et al, 2019; Whiteley et al, 2019; Lau et al, 2020; Ye et al, 2020). This molecule in turn activates one of a variety of effector proteins, including phospholipases, nucleases, and pore‐forming proteins, to kill the host cell (Severin et al, 2018; Cohen et al, 2019; Lau et al, 2020; Lowey et al, 2020). While the so‐called Type I CBASS systems encode only a CD‐NTase and a cell‐killing effector protein, the majority of CBASS systems encode ancillary proteins putatively involved in infection sensing and/or CD‐NTase activation (Burroughs et al, 2015; Millman et al, 2020). Type II CBASS systems encode two proteins, Cap2 and Cap3, that are related to eukaryotic ubiquitination machinery and are required for protection against phage (Cohen et al, 2019; preprint: Ledvina et al, 2022). Type III CBASS systems, meanwhile, encode peptide‐binding HORMA domain proteins (Cap7 and Cap8) that are proposed to bind specific peptides to sense infection and then activate their associated CD‐NTase (Ye et al, 2020).
All CBASS systems are thought to directly sense phage infection and respond by triggering cell death. Here, we identify a pair of transcriptional regulators, termed CapH and CapP, that are associated with hundreds of CBASS systems and upregulate CBASS expression in response to DNA damage. DNA damage is a universal stress signal in bacterial cells (Benler & Koonin, 2020) and has been shown to activate various bacterial stress responses, including the SOS response (Sassanfar & Roberts, 1990; Little, 1991). We show that CapH and CapP are structurally and functionally similar to regulators that mobilize prophages and integrative and conjugative elements (ICE elements) in response to DNA damage, and to activators of the DNA damage response in radiation‐resistant Deinococcus species. We also identify CapH‐ and CapP‐like regulators associated with a variety of known or putative bacterial immune systems, revealing that these proteins represent a conserved signaling module that regulates immune system expression in response to DNA damage across bacteria.
Results
Identification of capH and capP genes associated with CBASS systems
We previously showed that a Type III CBASS system from Escherichia coli strain MS115‐1 provides robust protection against bacteriophage λ through an abortive infection mechanism (Lau et al, 2020; Ye et al, 2020). Examination of this system's genomic neighborhood revealed a pair of genes directly upstream of the core CBASS genes and encoded on the opposite strand (i.e., sharing a promoter region with the core CBASS genes), that encode a predicted helix‐turn‐helix (HTH) DNA binding protein and a predicted Zn2+ metallopeptidase (Fig 1A). We term these two genes capH (CBASS‐associated protein, Helix‐turn‐helix) and capP (CBASS‐associated protein, Peptidase). In their position and orientation relative to the core CBASS genes, capH and capP are similar to capW, a transcriptional regulator associated with a distinct subset of CBASS systems (Blankenchip et al, 2022). BLAST searches revealed that CapP shares strong similarity to IrrE, a metallopeptidase that regulates the DNA damage response in Deinococcus by cleaving an HTH‐family transcription factor, DdrO (Vujičić‐Žagar et al, 2009; Ludanyi et al, 2014). DdrO normally binds the promoters of DNA damage response genes and suppresses their expression, but upon DNA damage, IrrE becomes activated and cleaves DdrO, releasing it from DNA and activating expression of the DNA damage response genes (Ludanyi et al, 2014; Blanchard et al, 2017; de Groot et al, 2019). The similarity of CapP to IrrE, and its association with the HTH protein CapH, suggested that CapH and CapP may functionally cooperate to control expression of their associated CBASS system.
Figure 1. Identification of CBASS‐associated genes capH and capP and role of CapH and CapP in the CBASS antiviral response.
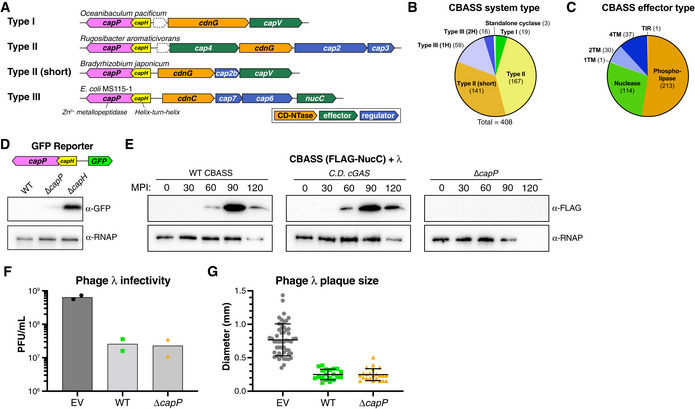
- Operon schematics of four representative CBASS systems with associated capH (yellow) and capP (pink) genes. See Table EV1 for all identified systems. For each system, core CBASS genes are colored as in the key: CD‐NTases orange, putative regulator(s) blue, and effector(s) green. Dotted outlines indicate unknown theoretical genes.
- Distribution of capH+capP‐associated CBASS systems, sorted by system type as defined by Millman et al (2020). Type III (1H) and Type III (2H) refer to Type III systems with one or two HORMA domain proteins, respectively.
- Distribution of capH+capP‐associated CBASS systems, sorted by effector type as defined by Millman et al (2020). 1TM, 2TM, and 4TM refer to effectors with one, two, or four predicted transmembrane segments, respectively.
- Top: Schematic of GFP expression reporter system, with the CBASS promoter, capH, and capP genes from E. coli MS115‐1 and the CBASS core genes replaced with GFP. Bottom: Western blot showing GFP expression in cells with the wild‐type GFP reporter or constructs lacking either capP or capH genes. α‐RNAP, anti‐RNA polymerase loading control. See full blot in Fig EV1A.
- Western blots of the CBASS expression reporter system with FLAG‐NucC (see Fig EV1B), showing FLAG‐NucC expression after infection with phage λ cI− (multiplicity of infection: 10). α‐RNAP, anti‐RNA polymerase loading control; MPI, minutes postinfection. Low RNAP expression at later time points is due to cell death.
- Quantitative plaque assay showing infectivity of λ cI− against cells containing no CBASS system (EV, empty vector), the wild‐type E. coli MS115‐1 CBASS system (WT), or a mutant system lacking capP (ΔcapP). Bars represent average of plaque forming units per ml of purified phage (PFU/ml), from duplicate experiments.
- Size of phage plaques for λ CI− infecting cells containing no CBASS system (EV, empty vector; n = 52), the wild‐type E. coli MS115‐1 CBASS system (WT; n = 20), or a mutant system lacking capP (ΔcapP; n = 23). Data are shown as average and standard deviation of all plaques counted in panel (F).
Source data are available online for this figure.
We systematically searched the genomic neighborhoods of 6,233 bacterial CBASS CD‐NTases (Cohen et al, 2019) for genes related to capP and identified 408 CBASS systems with a predicted Zn2+ metallopeptidase within 10 kb of the system's CD‐NTase gene. In these systems, CapP is most often annotated as a “domain of unknown function” (DUF) 955 or PFAM06114 protein. We manually inspected each system and identified a gene encoding a CapH homolog alongside capP in 393 of the 408 systems (in 70 cases, this gene is not annotated; Fig 1A; Table EV1). The remaining 15 systems encode an apparent fusion of CapH and CapP (Table EV1). In all cases, the capH and capP genes are encoded upstream of the core CBASS genes and on the opposite strand (Fig 1A). We identified capH and capP genes associated with Type I, Type II, and Type III CBASS systems that encode a variety of predicted effectors including phospholipases, transmembrane proteins, and endonucleases (Fig 1B and C; Table EV1). In 24 systems, capH and capP are encoded alongside a predicted σ70‐family σ factor (Table EV1).
To determine whether capH and capP control expression of their associated CBASS operon, we generated a reporter construct with capH, capP, and the promoter region of the CBASS system from E. coli MS115‐1, plus a gene encoding GFP (green fluorescent protein) in place of the core CBASS genes. When both capH and capP were present, the expression of GFP in uninfected log‐phase cells was too low for detection by anti‐GFP immunoblotting (Fig 1D). Expression was also nearly undetectable in a strain lacking capP, but we observed high GFP expression in a strain lacking capH (Figs 1D and EV1A). These data suggest that CapH is a transcriptional repressor for its associated CBASS system.
Figure EV1. CBASS expression reporter systems.
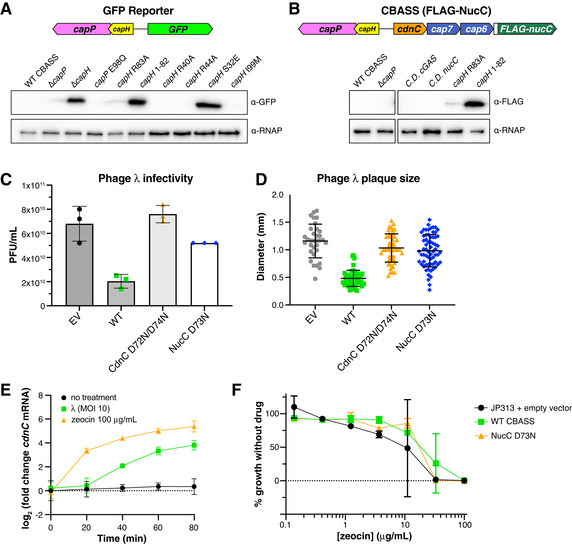
- Full Western blot for GFP reporter assay. α‐RNAP, anti‐RNA Polymerase loading control.
- Full Western blot for FLAG‐NucC expression reporter assay. α‐RNAP, anti-RNA Polymerase loading control.
- Quantitative plaque assay showing infectivity of λ cI− against cells containing no CBASS system (EV, empty vector), the wild‐type E. coli MS115‐1 CBASS system (WT), or mutant systems with catalytic‐dead CdnC (D72N/D74N) or NucC (D73N). Data are shown as average and standard deviation of plaque forming units per ml of purified phage (PFU/ml), from triplicate experiments. For NucC D73N, all three plates showed equal plaques (52 plaques at the tested dilution).
- Size of phage plaques for λ cI− infecting cells containing no CBASS system (EV, empty vector; n = 33), the wild‐type E. coli MS115‐1 CBASS system (WT; n = 46), or mutant systems with catalytic‐dead CdnC (D72N/D74N; n = 51) or NucC (D73N; n = 65). Data are shown as average and standard deviation of all plaques counted in panel (C).
- qRT–PCR for cdnC using JP313 cells containing the E. coli MS115‐1 CBASS operon on a plasmid under exponential growth conditions (black circles), after phage λ infection at MOI 10 (green squares), or after addition of zeocin at 100 μg/ml (orange triangles). Error bars indicate standard deviation from three technical replicates each of three biological replicates (nine total measurements per condition).
- Minimum inhibitory concentration (MIC) analysis of JP313 cells with either empty vector (black circles), E. coli MS115‐1 CBASS (green squares), or a mutant CBASS system with catalytic‐dead NucC (D73N; orange triangles). Data shown are the average and standard deviation of three biological replicates (see Materials and Methods).
Source data are available online for this figure.
Next, we generated a separate reporter construct encoding the full six‐gene CBASS system of E. coli MS115‐1, with a N‐terminal FLAG tag fused to the effector nuclease NucC (Figs 1E and EV1B). In agreement with our GFP reporter, the expression of FLAG‐NucC was undetectable by Western blot in uninfected cells (Fig 1E). FLAG‐NucC expression was also undetectable after deleting capP in this construct (Fig 1E). We were unable to delete capH in this construct, with all isolated clones lacking capH also lacking large regions of the core CBASS genes. Given that deletion of capH in our GFP reporter construct results in high expression of GFP, our inability to isolate a capH‐deleted version of the full CBASS system suggests that high expression of the system is toxic to host cells.
CapP controls CBASS expression but is not required for phage protection
Our reporter assays indicated that CapH likely acts as a transcriptional repressor for its associated CBASS operon. To determine the role of CapP in CBASS expression, we first used our FLAG‐NucC reporter system to test for CBASS expression changes upon infection with an obligately lytic variant of bacteriophage λ lacking the cI gene (λ cI−) (Rajagopala et al, 2011). With the wild‐type CBASS system encoding capH and capP, we observed a strong increase in FLAG‐NucC expression starting ~ 60 min after infection and peaking around 90 min after infection (Fig 1E). In the absence of capP, we observed no such increase in FLAG‐NucC expression (Fig 1E). We also observed increased FLAG‐NucC expression in a system with catalytically dead CD‐NTase (CdnC D72N/D74N), indicating that the observed expression changes do not depend on CBASS signaling (Fig 1E). These data suggest that CapP responds to phage infection by antagonizing CapH, resulting in a loss of repression and an increase in CBASS expression.
To test the role of CapH and CapP in phage protection, we compared the ability of wild‐type E. coli MS115‐1 CBASS and a mutant lacking capP to protect against λ cI− infection. We previously reported that when cloned into an IPTG‐inducible expression vector, the four core genes from E. coli MS115‐1 CBASS (cdnC [CD‐NTase], cap7 [HORMA], cap6 [TRIP13], and nucC) provide strong protection against λ cI− (Lau et al, 2020; Ye et al, 2020). We found that the native six‐gene operon encoding capH and capP also provides protection against λ cI−, as measured by both a reduction in viral plaque numbers (Fig 1F) and a reduction in plaque size compared with bacteria lacking CBASS (Fig 1G). While our prior study showed that the core CBASS genes under IPTG expression control reduced infection by λ cI− by over six logs (more than 106‐fold reduction in viral plaques) (Ye et al, 2020), the full system with capH and capP provides much more modest protection, with less than a two‐log reduction in viral plaques compared with a control strain (Fig 1F). Control infections with systems encoding catalytically dead CdnC (D72N/D74N) or NucC (D73N) (Fig EV1C and D), light microscopy analysis of infected cells (Fig EV2A and B), and bacterial growth curves (Fig EV2C) all confirmed that this protection was attributable to CBASS function.
Figure EV2. Phage infection of cells with E. coli MS115‐1 CBASS.
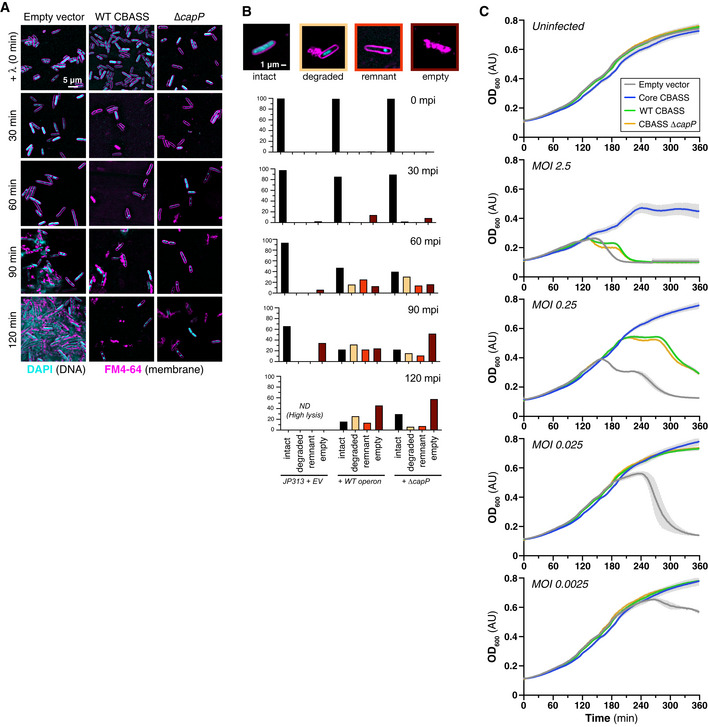
- Live‐cell fluorescence microscopy of λ cI− infecting cells containing no CBASS system (EV, empty vector), the wild‐type E. coli MS115‐1 CBASS system (WT), or a mutant system lacking capP (ΔcapP). DNA (DAPI) is colored cyan, and cell membranes (FM4‐64) are colored magenta.
- Quantification of fluorescence microscopy of λ cI− infecting cells containing no CBASS system (EV, empty vector), the wild‐type E. coli MS115‐1 CBASS system (WT), or a mutant system lacking capP (ΔcapP). Cells were manually quantified and assigned categories based on examples shown at top (n ≥ 30 cells for each strain and time point).
- Growth curves of JP313 cells containing plasmids with no insert (Empty vector; gray), the core four CBASS genes under a lac‐inducible promoter (Core CBASS; blue; Lau et al, 2020), the six‐gene CBASS system (WT CBASS; green), or the six‐gene CBASS system with capP deleted (CBASS ΔcapP; orange). All strains grew comparably when not infected with phage (Uninfected, top) and showed differing levels of protection when infected with phage λ at different multiplicity of infection (MOI). Data shown are the average and standard deviation of three independent measurements.
Source data are available online for this figure.
Unexpectedly, we found that a mutant CBASS system lacking capP and therefore unable to boost CBASS expression after infection (Fig 1E) provided protection against phage λ equivalent to the wild‐type system (Fig 1F and G). This finding suggests that the boost in CBASS expression mediated by capP comes too late to affect the course of an infection, and that the modest protection we observe is mediated by the low basal levels of CBASS proteins already present in these cells. To confirm that CBASS genes are expressed in uninfected cells, we used qRT–PCR to show that cdnC mRNA is present at ~ 2% of that of the abundant RNA polymerase gene rpoA (ΔC t = −4.8) at basal expression levels (Fig EV1E). As we observe for protein levels by Western blot, cdnC mRNA levels increase dramatically upon infection with λ cI− (Fig EV1E). Overall, these data show that while the native E. coli MS115‐1 CBASS system does provide modest antiphage protection, transcriptional regulation by CapH and CapP does not directly contribute to this protection. Thus, capH and capP may enable their associated CBASS system to play a defensive role other than front‐line protection against viral infection.
CapH binds the promoter region of its associated CBASS system
Our reporter assays suggested that CapH acts as a transcriptional repressor for its associated CBASS system, potentially by directly binding the CBASS promoter region. In E. coli MS115‐1 CBASS, the 182 bp region between the capH and cdnC genes contains strongly predicted promoter sequences for both forward (toward cdnC) and reverse (toward capH) transcription (Figs 2A and EV3A). We used fluorescence polarization to test CapP binding to a panel of 40‐bp DNAs covering this region, and identified two binding sites (Site 1 and Site 2) overlapping the predicted forward and reverse promoter sequences (Figs 2A and B, and EV3B and F). Both sites are conserved in the promoter regions of related CBASS operons (Fig EV3A). Site 2 possesses a perfect 8‐bp palindrome separated by 6 bp (Fig EV3A), but Site 1 does not possess any recognizable palindromes or tandem repeats. Nonetheless, CapH binding to both sites is best fit by a cooperative binding model with a Hill coefficient of ~ 2, suggesting cooperative binding of multiple CapH monomers to each site (Figs 2B and EV3B).
Figure 2. CapH binds the CBASS promoter region.
- Top: Schematic of the region between capH and cdnC genes in E. coli MS115‐1 CBASS with predicted −35, −10, and TSS (translation start site) sites for the top (forward) and bottom (reverse) strands, predicted using BPROM (Solovyev & Salamov, 2011). Bottom: Overlapping 40–41 bp DNAs tested for CapH binding by fluorescence polarization, denoted as either no binding (dotted line), weak/non‐cooperative binding (thin solid line), or strong/cooperative binding (thick solid line). The inferred boundaries of the two CapH binding sites (Site 1 and Site 2) are noted. See Fig EV3A–F for binding curves.
- Fluorescence polarization assay showing binding of E. coli MS115‐1 CapH (His6‐MBP tagged) to three 40‐bp DNAs: 1–40 (black circles), 31–70 (Site 1; green squares), and 81–120 (Site 2; orange triangles). Fit K d and Hill coefficient for each DNA is shown. Error bars indicate standard deviation from three technical replicates.
- Top: Domain schematic of E. coli MS115‐1 CapH, and truncation used for crystallization of the N‐terminal HTH domain (NTD; residues 2–67). Bottom: Crystal structure of the CapH NTD (blue), with bound DNA modeled from a structural overlay with a known HTH‐DNA complex structure (PDB ID 3CLC; McGeehan et al, 2008). Shown in sticks are three conserved residues (S32, R40, and R44) putatively involved in DNA binding. See Fig EV3G for sequence conservation of the CapH NTD.
- Fluorescence polarization assay showing binding of E. coli MS115‐1 CapH (His6‐MBP tagged; wild‐type or indicated point mutants) to the Site 2 DNA (bases 81–120 in panel (A)). Fit K d and Hill coefficient for each DNA is shown. Error bars indicate standard deviation from three technical replicates.
- GFP expression reporter assay showing loss of suppression upon mutation of CapH. α‐RNAP, anti‐RNA polymerase loading control. See full blot in Fig EV1A.
Source data are available online for this figure.
Figure EV3. CapH binds MS115‐1 CBASS intergenic region.
-
ASequence alignment of CBASS promoter regions in E. coli MS115‐1 (NCBI RefSeq GG771785.1), Cronobacter sakazakii strain cro3915C2 (NZ_NRJY01000012.1), Pseudomonas stutzeri strain KC NODE_1_length_951488_cov_16.453 (NZ_POUN01000001.1), Pseudomonas sp. RIT 412 RIT412_S3_7 (NZ_QBJA02000007.1), Pseudomonas sp. MF4836 (NZ_MVOL01000002.1), Burkholderia pseudomallei strain MSHR4301 (NZ_LXCN01000015.1), Ralstonia insidiosa strain WCHRI065162 (NZ_PKPC01000011.1), Thauera sp. K11 plasmid pTX1 (NZ_CP023440.1). Promoter sequences (−35, −10, and TSS) were predicted by the BPROM server (Solovyev & Salamov, 2011). Identified CapH‐binding Sites 1 and 2 are denoted by gray underlines, and the palindrome within Site 2 is denoted by gray arrows.
-
BDNA binding affinity (K d) and cooperativity (Hill coefficient) from fluorescence polarization measurements of CapH binding overlapping 40–41 bp DNAs spanning the E. coli MS115‐1 CBASS promoter region (Fig 2A).
-
C–FFluorescence polarization assays showing binding of E. coli MS115‐1 CapH (His6‐MBP tagged) to 40–41 bp DNAs spanning the E. coli MS115‐1 CBASS promoter region. Fit K d and Hill coefficient for each DNA is shown in panel (B). Error bars indicate standard deviation from three technical replicates.
-
GSchematic of the CapH NTD with sequence logo indicating the conservation of each residue in an alignment of 56 unique CBASS‐associated CapH proteins.
Source data are available online for this figure.
Sequence analysis and 3D structure predictions indicate that CapH possesses an N‐terminal helix‐turn‐helix (HTH) DNA binding domain followed by a short flexible linker and two conserved α‐helices that may constitute an oligomerization domain (Fig 2C). While we were unable to crystallize full‐length CapH, likely due to flexibility of the interdomain linker, we crystallized and determined a 1.02 Å‐resolution crystal structure of the protein's isolated HTH domain (residues 2–67; Fig 2C; Appendix Table S1). The CapH HTH domain forms a canonical HTH fold, and modeling a DNA‐bound complex based on known HTH‐DNA complexes revealed several conserved residues on the DNA‐binding face that that may be involved in DNA binding, including Ser32, Arg40, and Arg44 (Figs 2C and EV3G). We found that mutation of Ser32 completely eliminated CapH binding to the Site 2 DNA, and that mutation of either Arg40 or Arg44 reduced, but did not eliminate, binding (Fig 2D). We tested the same mutations in our GFP reporter system, and found that mutation of Ser32 resulted in high expression of GFP (Fig 2E). These data are consistent with CapH acting as a transcriptional repressor for its associated CBASS operon.
CapH oligomerization is required for DNA binding
In the Deinococcus DdrO‐IrrE system, DdrO forms a homodimer through its C‐terminal domain and this dimerization is required for DNA binding and transcriptional repression by the protein (Ludanyi et al, 2014; de Groot et al, 2019). To test whether CapH forms an oligomer, we used size exclusion chromatography coupled to multi‐angle light scattering (SEC‐MALS). We found that full‐length CapH forms an oligomer with an overall size consistent with either a homotrimer or a mixture of dimers and tetramers (Fig 3A and B; Appendix Fig S1A). The isolated C‐terminal region of CapH (residues 67–107; CapHCTD) forms a similar oligomer, while the isolated N‐terminal HTH domain is monomeric (Fig 3B). These data show that CapH oligomerizes through its C‐terminal domain. To determine the mechanism of oligomerization, we crystallized and determined a 1.75 Å‐resolution crystal structure of CapHCTD (Fig 3C; Appendix Table S1). In the structure, four CapHCTD protomers form a homotetramer with a dimer‐of‐dimers architecture. Each CapHCTD protomer forms two α‐helices that fold into a V shape, with the homodimer assembled by two protomers arranged antiparallel to one another with the V shapes interlocked. The CapHCTD homodimer is stabilized by a hydrophobic core comprising Phe81, Tyr85, Leu96, and Leu100 of each protomer (Fig 3C). The CapHCTD homodimer resembles the C‐terminal dimerization domains of other dimeric bacterial transcription factors, including Mycobacterium tuberculosis EspR, Bacillus subtilis SinR, and Citrobacter C.Csp231I (Lewis et al, 1998; Gangwar et al, 2014; Shevtsov et al, 2015). The CapHCTD homotetramer is assembled through a separate hydrophobic interface between the C‐terminal α‐helices of four protomers, involving residues Ile99 and Phe103 (Fig 3C).
Figure 3. CapH oligomerization is required for DNA binding.
- Domain schematic of E. coli MS115‐1 CapH, showing the truncations used for oligomeric state determination.
- Size exclusion chromatography coupled to multi‐angle light scattering (SEC‐MALS) determination of CapH oligomeric state. For each construct, measured molecular weight and inferred oligomer are indicated. MBP‐fused full‐length CapH (monomer MW = 57.0 kDa) is shown in brown; SUMO‐fused CapHCTD (monomer MW = 19.1 kDa) is yellow, and SUMO‐fused CapHNTD (monomer MW = 22.0 kDa) is cyan. See Appendix Fig S1A for SEC‐MALS analysis of untagged CapH.
- Crystal structure of the CapHCTD homotetramer. Residues comprising the hydrophobic core of each dimer (Phe81, Tyr85, Leu96, and Leu100) are shown as sticks, and residues comprising the hydrophobic tetramerization interface (Ile99 and Phe103) are shown as sticks and labeled. See Appendix Fig S1B for structure of the CapHCTD(I99M) homodimer.
- Fluorescence polarization assay showing binding of E. coli MS115‐1 CapH (His6‐MBP tagged; full‐length [black circles] or NTD [green squares]) to the Site 2 DNA. WT K d = 0.30 ± 0.08 μM, Hill coefficient = 2.0 ± 0.7; no binding detected for CapHNTD. Error bars indicate standard deviation from three technical replicates.
- Fluorescence polarization assay showing binding of E. coli MS115‐1 CapH (His6‐MBP tagged) to the Site 1 (bases 31–70) and Site 2 (bases 81–120) DNAs. Wild‐type CapH binding Site 1 is shown in orange triangles, and binding Site 2 is shown in blue diamonds. CapH(I99M) binding Site 1 is shown in black circles, and binding Site 2 is shown in green squares. Fit K d and Hill coefficient for each combination is shown. See Appendix Fig S1C for EMSA analysis of CapH WT and I99M binding to the Site 2 DNA. Error bars indicate standard deviation from three technical replicates.
Source data are available online for this figure.
During the course of structure determination for CapHCTD, we generated a construct with a mutation of Ile99 to methionine (CapHCTD(I99M)). We determined a 1.26 Å‐resolution structure of this mutant, revealing a CapH homodimer equivalent to our structure of wild‐type CapHCTD, but lacking the tetrameric assembly (Appendix Fig S1B). Consistent with this finding, SEC‐MALS showed that CapH(I99M) forms a stable homodimer in solution, rather than the larger oligomer observed with wild‐type CapH (Appendix Fig S1A). Thus, the I99M mutant disrupts CapH tetramerization, but not dimerization.
We next tested the role of CapH oligomerization in DNA binding. We used fluorescence polarization to compare the DNA binding affinity of full‐length wild‐type CapH to that of the CapH HTH domain (residues 2–67; CapHNTD), which forms a monomer; and to the CapH(I99M) mutant, which forms a homodimer. We observed no binding of CapHNTD to DNA, demonstrating that CapH oligomerization is required for DNA binding (Fig 3D). With both Site 1 and Site 2 DNAs, CapH(I99M) showed only a slight reduction in DNA binding affinity and cooperativity compared with wild‐type CapH (Fig 3E). An electrophoretic mobility shift assay (EMSA) with Site 2 DNA showed that while wild‐type CapH shows two shifted bands, CapH(I99M) shows only one (Appendix Fig S1C). Despite the reduced DNA binding affinity and inability to form tetramers, CapH(I99M) effectively suppressed expression of our GFP reporter system (Fig EV1A). Together, these data suggest that CapH can form tetramers and that tetramer formation does play a minor role in DNA binding, but tetramer formation is not necessary for high‐affinity DNA binding and suppression of CBASS gene expression.
The structure of CapP reveals an internal cysteine switch
In the Deinococcus DNA damage response pathway, cleavage of DdrO by the metallopeptidase IrrE results in loss of DNA binding by DdrO, enabling increased expression of DNA damage response genes (Ludanyi et al, 2014; de Groot et al, 2019). Our data on DNA binding and CBASS repression by CapH, and in particular the importance of CapH oligomerization for DNA binding, suggest a functional parallel between DdrO‐IrrE and CapH‐CapP. To better understand this relationship, we purified and determined a 1.35 Å‐resolution crystal structure of CapP from a CBASS system in Thauera sp. K11 (56% identical to E. coli MS115‐1 CapP; Fig EV4A). The overall structure of CapP resembles that of IrrE, with the protein folding into three domains: an N‐terminal Zn2+ metallopeptidase domain, a central linker domain with topology resembling a helix‐turn‐helix domain, and a C‐terminal GAF domain (Fig 4A and B). The N‐terminal domain closely resembles other HEexxH Zn2+ metallopeptidases including IrrE, with five α‐helices and a three‐stranded β‐sheet. A Zn2+ ion is coordinated in the conserved active site by residues His96, His100, and Glu129 (Fig 4C–E). The predicted active‐site glutamate residue, Glu97, is positioned close by but not directly coordinating the bound Zn2+ ion. Instead, a conserved cysteine residue, Cys113, completes the coordination of the bound Zn2+ ion. Cys113 is located on an insertion in the metallopeptidase domain, on a fourth β‐strand that drapes over the active site in the same position that substrate peptides bind in related Zn2+ metallopeptidases (Cerda‐Costa & Gomis‐Ruth, 2014). We term this region the cysteine switch loop, after the cysteine switch motif found in matrix metalloproteases. These enzymes are synthesized as inactive precursors with an N‐terminal domain bearing a conserved cysteine residue (the cysteine switch) that coordinates the active‐site Zn2+ ion and inhibits activity (Fig EV4B and C). The protease is only activated upon proteolytic cleavage and dissociation of the cysteine switch domain (Springman et al, 1990; Van Wart & Birkedal‐Hansen, 1990; Cerda‐Costa & Gomis‐Ruth, 2014). Of the 408 CapP proteins associated with CBASS systems, 134 (33%) possess the cysteine switch loop, including E. coli MS115‐1 CapP (Figs 4F and EV4D).
Figure EV4. CBASS‐associated CapP contains an internal cysteine switch.
- Operon schematic of the Thauera sp. K11 CBASS system, compared to the E. coli MS115‐1 system, with sequence identity between the two systems' CapH and CapP proteins indicated.
- Structure of Thauera sp. K11 CapP, with close‐up of its Zn2+ metallopeptidase domain (pink) with internal cysteine switch loop (blue) and cysteine switch residue (C113).
- Structure of human matrix metalloprotease MMP9 (PDB IF 1L6J; Elkins et al, 2002), with close‐up of its Zn2+ metallopeptidase domain (pink) and N‐terminal cysteine switch domain (blue) and cysteine switch residue (C99). The orientation of the Zn2+ metallopeptidase domains in panels (B) and (C) is identical.
- Evolutionary tree of 408 CBASS‐associated CapP proteins, colored by the presence (black) or absence (blue) of the internal cysteine switch.
Figure 4. Structure of CapP reveals an internal cysteine switch.
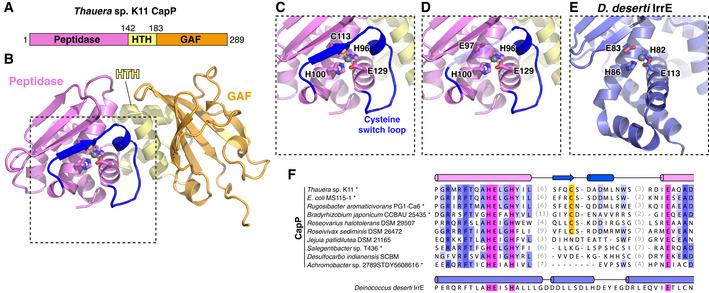
- Domain schematic of Thauera sp. K11 CapP, with N‐terminal Zn2+ peptidase domain pink, central HTH domain yellow, and C‐terminal GAF domain orange. See Fig EV4A for comparison of the Thauera sp. K11 and E. coli MS115‐1 CBASS operons.
- Structure of Thauera sp. K11 CapP with domains colored as in panel (A). Shown in sticks are active‐site residues H96, E97, H100, and E129, and a bound Zn2+ ion is shown as a gray sphere. Shown in blue is the internal cysteine switch loop, with C113 (shown as sticks) coordinating the bound Zn2+ ion.
- Close‐up view of the Thauera sp. K11 CapP active site. See Fig EV4B and C for comparison with a cysteine switch‐containing matrix metalloprotease.
- Close‐up view of the Thauera sp. K11 CapP active site as in panel (C), with transparent cysteine switch loop to show the position of the catalytic glutamate residue E97.
The presence of the cysteine switch loop in CapP suggests that the protein's peptidase activity is tightly controlled, perhaps by a conformational change that induces dissociation of the cysteine switch from the CapP active site and allows substrate binding. In Deinococcus IrrE, the protein's peptidase activity is thought to be activated upon DNA damage by the binding of an unknown ligand to the protein's C‐terminal GAF domain, although IrrE does not contain a cysteine switch loop (Vujičić‐Žagar et al, 2009). In other proteins, GAF domains are known to bind nucleotide‐based second messengers including cyclic GMP, which binds the GAF domain of phosphodiesterase 6C and allosterically regulates its enzymatic activity (Ho et al, 2000; Martinez et al, 2002a, 2002b, 2008; Gross‐Langenhoff et al, 2006; Levdikov et al, 2009). When we compared the structure of CapP to that of cyclic GMP‐bound phosphodiesterase 6C, we observed that CapP possesses a large number of surface‐exposed aromatic residues near the putative ligand‐binding site (Appendix Fig S2). If CapP is allosterically regulated through the GAF domain, these residues may be involved in the binding of nucleotide‐based ligand(s).
CapP cleaves CapH when stimulated by single‐stranded DNA
We next sought to directly test whether CapP cleaves CapH. Our initial tests using purified proteins in vitro showed no CapP‐mediated cleavage of CapH, so we instead developed an assay to detect CapP activity in E. coli cells. We coexpressed CapP with a fusion protein comprising CapH with an N‐terminal maltose binding protein (MBP) tag and a C‐terminal GFP tag, and used an anti‐GFP Western blot to detect CapP activity. We detected a minor band representing a CapP cleavage product in the presence of wild‐type CapP, but not when the CapP active site was mutated (Glu98 to glutamine; E98Q; Fig 5A). At ~ 32 kDa, this band likely represents a product of CapP cleavage near the C‐terminus of CapH.
Figure 5. CapP cleaves CapH and is stimulated by single‐stranded DNA.
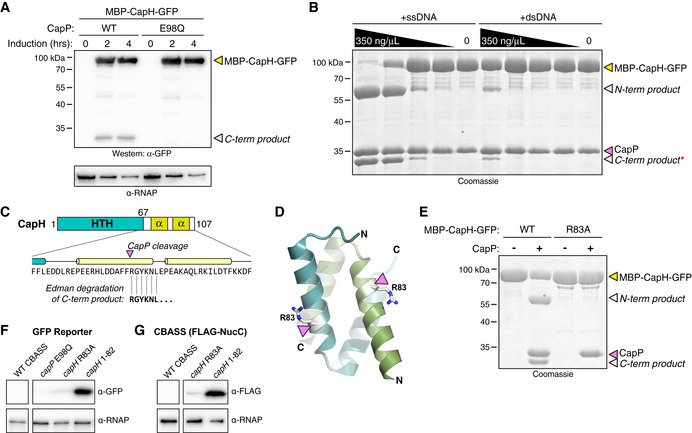
- Anti‐GFP Western blot showing coexpression in E. coli of an MBP‐CapH‐GFP fusion construct with wild‐type or catalytic‐dead (E98Q) CapP. Full‐length MBP‐CapH‐GFP is indicated with a yellow arrowhead, and the C‐terminal product of CapP cleavage is indicated with a white arrowhead. α‐RNAP, anti‐RNA polymerase loading control.
- In vitro cleavage of purified MBP‐CapH‐GFP (yellow arrowhead) into N‐terminal and C‐terminal products (white arrowheads) by CapP is stimulated by DNA. For both ssDNA and dsDNA, the highest concentration is 350 ng/μl, followed by three fivefold dilutions. Red asterisk indicates the band that was analyzed by Edman degradation. See Fig EV5 for analysis of DNA binding and sequence‐specificity for cleavage activation.
- Results of Edman degradation of CapH C‐terminal cleavage product (red asterisk from panel (B)), showing cleavage at residue R83. See Appendix Fig S3 for full data.
- Cartoon view of the CapHCTD 67‐107 dimer, with R83 colored white and shown as sticks.
- In vitro cleavage of MBP‐CapH‐GFP (wild‐type or R83A mutant) by CapP, in the presence of 10 μM ssDNA.
- GFP reporter assay showing effect of a CapP E98Q catalytic‐dead mutant, CapH R83A mutant, or removal of CapH residues 83–107 (capH 1–82) on GFP expression. α‐RNAP, anti‐RNA polymerase loading control. See full blot in Fig EV1A.
- CBASS expression reporter system with FLAG‐NucC, showing effect of a CapH R83A mutant, or removal of CapH residues 83–107 (capH 1–82) on FLAG‐NucC expression. α‐RNAP, anti‐RNA polymerase loading control. See full blot in Fig EV1B.
Source data are available online for this figure.
Since we observed CapP‐mediated CapH cleavage in cells, but not with purified proteins, we theorized that a soluble ligand present in cells is responsible for activating CapP. To test this idea, we performed in vitro cleavage assays with purified CapP and MBP‐CapH‐GFP in the presence of E. coli cell lysate. We observed robust activation of CapP in the presence of E. coli lysate, and stronger activation after the lysate was boiled and centrifuged to remove all proteins (Fig EV5A). By fractionating boiled cell lysate using anion‐exchange and size‐exclusion chromatography, we found that CapP was most likely activated by a large, negatively charged macromolecule (Fig EV5B). Based on this finding, we tested whether DNA and/or RNA could directly stimulate CapP. We found that single‐stranded DNA strongly activates CapP (Fig 5B), while double‐stranded DNA weakly activates CapP (Fig 5B), and single‐stranded RNA does not activate CapP (Fig EV5C). We found that single‐stranded DNA as short as 5 bases long stimulate CapP, and that pyrimidines—particularly thymine—have the strongest stimulatory effect (Fig EV5C). Finally, we found that CapP directly binds a single‐stranded DNA oligonucleotide in vitro, but not an equivalent‐length double‐stranded DNA (Fig EV5D). Furthermore, CapP strongly binds poly‐T DNA, weakly binds poly‐C, and does not bind poly‐A (Fig EV5E). These data support a model in which CapP's peptidase activity is stimulated by the binding of single‐stranded DNA, particularly T‐rich DNA.
Figure EV5. DNA‐mediated activation of CapP.
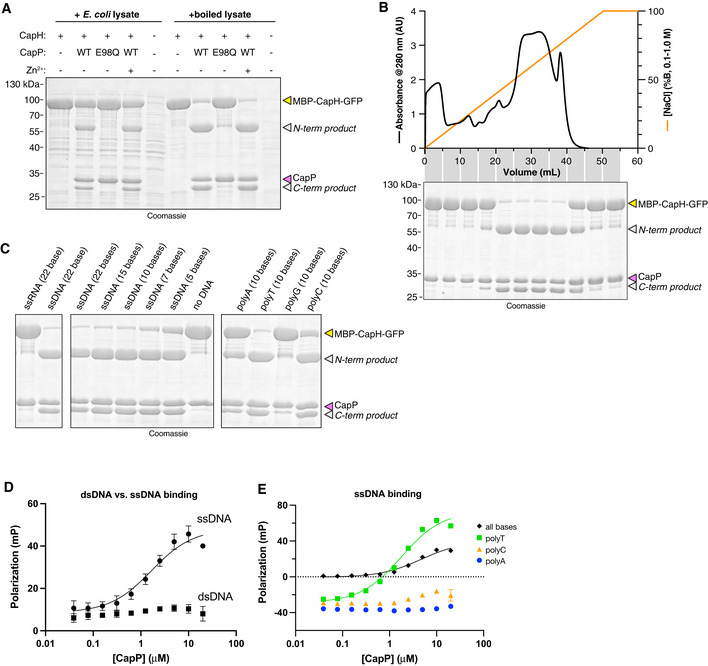
- In vitro cleavage assay with purified E. coli MS115‐1 CapP (wild‐type or catalytic‐dead E98Q mutant), MBP‐CapH‐GFP, and E. coli cell lysate.
- Top: UV absorbance (280 nm) of boiled E. coli cell lysate separated by HiTrap Q column. Gradient (orange line) goes from 100 mM NaCl (0% B) to 1 M NaCl (100% B). Bottom: Coomassie‐stained SDS–PAGE gel of cleavage assay with purified E. coli MS115‐1 CapP, MBP‐CapH‐GFP, and combined fractions from HiTrap Q column. A similar assay with a Superdex 75 gel filtration column showed high activity across the entire separation range.
- In vitro cleavage assay with purified E. coli MS115‐1 CapP, MBP‐CapH‐GFP, and the indicated nucleic acids.
- Fluorescence polarization DNA binding assay for E. coli MS115‐1 CapP and either single‐stranded DNA (circles and solid line; K d = 1.7 ± 0.6 μM) or double‐stranded DNA (squares; no binding detected). Error bars indicate standard deviation from three technical replicates.
- Fluorescence polarization DNA binding assay for E. coli MS115‐1 CapP and single‐stranded DNAs including a random sequence with all four bases (black diamonds; K d = 4.7 ± 1.2 μM), poly‐T (green squares; K d = 1.8 ± 0.3 μM), poly‐C (orange triangles; no binding detected), or poly‐A (blue circles; no binding detected). Error bars indicate standard deviation from three technical replicates.
Source data are available online for this figure.
We isolated the C‐terminal product from CapP‐mediated cleavage of MBP‐CapH‐GFP, and subjected it to Edman degradation to map the CapP cleavage site (Fig 5C; Appendix Fig S3). We identified the cleavage site as between Phe82 and Arg83, within the first α‐helix of the CapH C‐terminal dimerization domain (Fig 5C and D). Confirming this finding, we found that a CapH Arg83 to alanine (R83A) mutant is not cleaved by CapP in vitro (Fig 5E). Finally, we found that in cells, truncation of CapH at residue 82—mimicking CapP‐cleaved CapH—resulted in strong expression as measured by both our GFP reporter system (Fig 5F) and our FLAG‐NucC reporter (Fig 5G).
CapP is activated by DNA damage
Our data showing that CapP is directly stimulated by single‐stranded DNA in vitro suggests that in cells, it is activated by DNA damage. To test this idea, we coexpressed CapP and MBP‐CapH‐GFP in E. coli in the presence or absence of DNA damaging drugs. We found that in the presence of zeocin, a drug that induces DNA double‐strand breaks (Chankova et al, 2007), CapH cleavage was stimulated (Fig 6A). Using our FLAG‐NucC reporter system, we found that both zeocin and mitomycin C, which causes DNA double‐strand breaks through a separate mechanism (Kidane et al, 2004; Ayora et al, 2011), strongly stimulate CBASS expression within 60–120 min of adding the drugs (Fig 6B and C). qRT–PCR for cdnC mRNA confirms this finding, and shows that addition of zeocin boosts CBASS expression more strongly than phage λ infection (Fig EV1E). This boost in CBASS expression was not observed in cells lacking capP (Fig 6B and C). Thus, CapH and CapP mediate increased CBASS expression upon DNA damage, even in the absence of phage infection. Curiously, this increased CBASS expression does not result in increased cell death, as measured by minimum inhibitory concentration (MIC) analysis of zeocin for cells with and without the E. coli MS115‐1 CBASS system (Fig EV1F). This finding suggests that despite being highly expressed upon DNA damage, the CBASS system may still require a phage trigger to activate second messenger production and in turn activate NucC.
Figure 6. CapH and CapP induce CBASS expression in response to DNA damage.
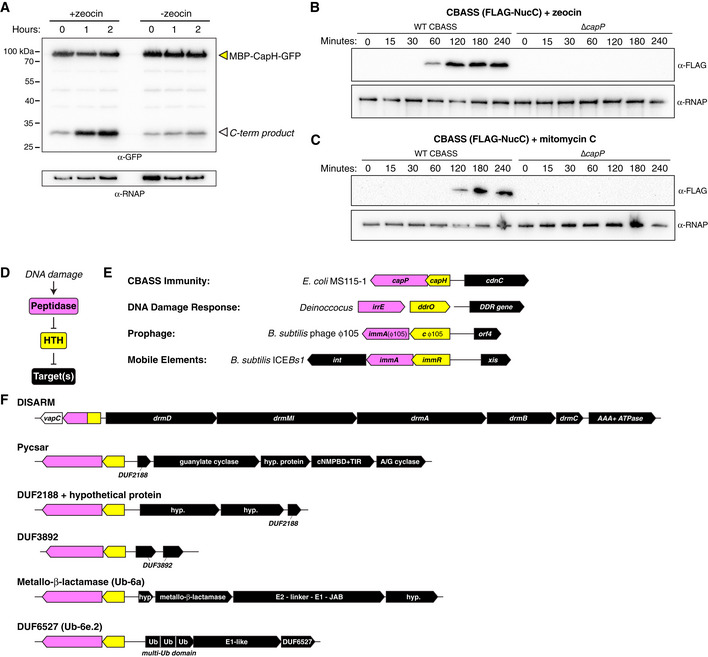
- Anti‐GFP Western blot showing coexpression in E. coli of an MBP‐CapH‐GFP fusion construct with wild‐type CapP after exposure to zeocin (100 μg/ml). Full‐length MBP‐CapH‐GFP is indicated with a yellow arrowhead, and the C‐terminal product of CapP cleavage is indicated with a white arrowhead. α‐RNAP, anti‐RNA polymerase loading control.
- Western blot of the CBASS expression reporter system with FLAG‐NucC, showing FLAG‐NucC expression after exposure to zeocin (100 μg/ml). α‐RNAP, anti‐RNA polymerase loading control.
- Western blot of the CBASS expression reporter system with FLAG‐NucC, showing FLAG‐NucC expression after exposure to mitomycin C (1 μg/ml). α‐RNAP, anti‐RNA polymerase loading control.
- Proposed signaling pathway for DNA damage‐responsive transcriptional control systems in bacteria.
- Diverse systems in bacteria that include an HTH transcriptional repressor (yellow) and a DNA damage‐activated Zn2+ metallopeptidase (pink) that targets the transcriptional repressor for cleavage.
- Known or likely bacterial defense systems associated with CapH (yellow) and CapP (pink)‐like genes. See Materials and Methods for accession numbers for each gene.
Source data are available online for this figure.
CapH and CapP are members of a broadly conserved family of DNA damage response proteins
CapH and CapP show strong structural and functional similarity to the Deinococcus proteins DdrO and IrrE, with both systems inducing expression of target genes upon DNA damage through metallopeptidase cleavage of a transcriptional repressor (Fig 6D and E). DNA damage is a universal signal of cell stress, and as such is a major signal to induce lysogenic phage (prophage) to switch to the lytic life cycle, and to induce mobility of integrative and conjugative elements (ICE elements) (Baek et al, 2003; Auchtung et al, 2005). Strikingly, the use of an HTH family transcriptional repressor coupled with a DNA damage‐stimulated metallopeptidase is shared in some prophages and ICE elements. For example, Bacillus subtilis mobile element ICEBs1 and prophage ɸ105 each encode an HTH‐family transcriptional repressor (ImmR and cɸ105, respectively) that strongly represses the expression of genes responsible for excision of these elements from the genome, and a Zn2+ metallopeptidase (ImmA) that cleaves the HTH protein upon DNA damage to relieve repression and induce excision (Fig 6E; Bose et al, 2008; Bose & Grossman, 2011).
The structural and functional parallels between CapH/CapP, DdrO/IrrE, and ImmR/ImmA suggest that these proteins represent a broadly conserved family of DNA damage‐responsive transcriptional regulators. We used the FlaGs (Flanking Genes) server to search for other instances of CapH/CapP‐like proteins and identify their associated operons (Saha et al, 2021). We identified a broad range of operons associated with capH‐ and capP‐like genes, with all of them sharing the location of capH and capP upstream of, and oriented on the opposite strand as, the associated operon (Fig 6F). Most of these systems represent known or putative defense systems, notably including a group of DISARM antiphage systems associated with a capH‐capP fusion gene, and a group of Pycsar systems associated with separate capH and capP genes. We identified three sets of operons encoding proteins of the DUF2188 or DUF3892 families, which are uncharacterized but have been previously linked to bacterial defense (Burroughs & Aravind, 2020). Both DUF2188 and DUF3892 proteins have also been identified in operons containing HTH and metallopeptidase genes, paralleling our findings (Burroughs & Aravind, 2020). We also identified operons encoding proteins related to eukaryotic ubiquitin signaling machinery, including the so‐called Ub‐6a systems that encode a predicted metallo‐β‐lactamase and a large protein with E2‐like, E1‐like, and JAB protease‐like domains (Iyer et al, 2006; Burroughs et al, 2009). Notably, this protein shares strong homology to the Cap2 protein in Type II CBASS systems (also classified as Ub‐6b systems), which conjugates the C‐terminus of its cognate CD‐NTase to an unknown target to regulate antiphage signaling (preprint: Ledvina et al, 2022). We also identified CapH‐ and CapP‐like proteins associated with Ub‐6e.2 systems, which encode a protein predicted to possess multiple ubiquitin‐like β‐grasp domains and an E1‐like protein, plus an uncharacterized protein (DUF6527; Fig 6F). Thus, CapH‐ and CapP‐like regulators are associated with a broad range of bacterial signaling pathways with known or predicted roles in antiphage or stress responses.
Discussion
Here, we identify a pair of proteins—CapH and CapP—that are associated with hundreds of CBASS antiphage systems, and show that they function together to regulate CBASS expression. In the basal state, the helix‐turn‐helix protein CapH forms oligomers and binds the promoter region of its associated CBASS system to repress transcription. With its distinctive cysteine switch motif, CapP is maintained in an inactive state in this mode. In E. coli MS115‐1 CBASS, the resulting low‐level basal expression of the CBASS core genes is apparently sufficient to provide modest protection against bacteriophage λ infection. Despite the extremely low levels of the core CBASS proteins in this repressed state, phage infection is sensed by the system's HORMA domain proteins, activating CdnC to produce a cyclic tri‐AMP second messenger. Cyclic tri‐AMP in turn activates NucC, which destroys the host genome to kill the cell and abort the infection (Fig 7A; Lau et al, 2020; Ye et al, 2020).
Figure 7. Model for CapH/CapP function in CBASS‐mediated immunity.
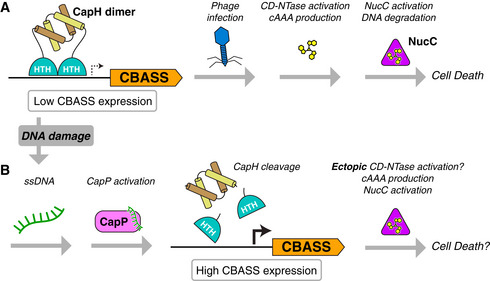
- In the basal state, CapH dimers (depicted) or tetramers (not shown) bind their cognate CBASS promoter and repress transcription. Low‐level CBASS expression is sufficient for detection of a lytic phage infection, CD‐NTase activation, and second messenger production (in E. coli MS115‐1 CBASS, cyclic tri‐AMP/cAAA), followed by effector activation (in E. coli MS115‐1, NucC) and cell death.
- Upon DNA damage, activation of other antiphage immune systems, or environmental stimuli, CapP is activated and cleaves CapH. CapH cleavage and release from the CBASS promoter mediates a dramatic increase in CBASS activation, resulting in ectopic CD‐NTase activation, second messenger production, effector activation, and possibly cell death.
We find that DNA damage strongly activates CapP‐mediated CapH cleavage through the production of T‐rich single‐stranded DNA, and that this activation results in a dramatic increase in CBASS expression. Since deletion of capP did not affect the system's ability to directly protect against bacteriophage λ infection, this expression boost likely plays a role other than primary response to phage infection. We propose that this pathway represents a second path for CBASS activation that directly responds to DNA damage instead of, or in addition to, phage infection. DNA damage is a universal signal of bacterial cell stress and is sensed by diverse stress response pathways including the SOS response, which is broadly similar but mechanistically distinct from the CapH/CapP system we describe. A mechanism to boost expression of immune systems upon DNA damage may enable these systems to sense and respond to a broader set of stimuli than they could without this regulation. For example, DNA damage could arise from the activation of a DNA‐targeting restriction‐modification or CRISPR‐Cas system after phage infection. If this damage is sufficiently severe or sustained, it could activate a CapH/CapP‐linked secondary immune system to aid the defensive response (Fig 7B). Alternatively, a CapH/CapP‐linked immune system could piggyback on the same stress signal that activates a latent prophage to become lytic, and suppress the production of virions by preemptively killing the host cell or otherwise restricting the phage.
Supporting the idea that DNA damage signaling could represent a broad mechanism for activation of bacterial immune systems, we identified a diverse set of known or putative defense systems associated with CapH‐ and CapP‐like regulators. These include groups of DISARM and Pycsar systems, which function similarly to restriction‐modification systems (Ofir et al, 2018) and CBASS systems (Tal et al, 2021), respectively. CapP and CapH also appear in operons encoding DUF2188 and DUF3892 proteins, which have both been predicted to participate in antiphage defense (Burroughs & Aravind, 2020). Finally, the association of CapH and CapP with uncharacterized operons encoding proteins related to ubiquitin signaling machinery suggests that these operons, too, may play a role in defense against phage infection or other stresses.
Our identification of a mechanism enabling a single defense system to respond to multiple stimuli parallels the recent discovery and characterization of BrxR/CapW transcriptional regulators, which are associated with a variety of immune systems including BREX and CBASS antiphage systems (Blankenchip et al, 2022;Luyten et al, 2022; Picton et al, 2022). CapW drives increased expression of its associated CBASS systems upon phage infection, but as with CapH/CapP‐associated systems, this increased expression is not specifically required for protection against lytic phage (Blankenchip et al, 2022). Similarly, BrxR controls expression of its associated BREX systems, but is not required for antiphage immunity (Luyten et al, 2022). While the activating signal of BrxR/CapW is not known, these data suggest that the protein controls activation of CBASS and BREX systems in response to signals other than phage infection. As with CapH and CapP, BrxR/CapW may enable its associated defense system to act as a second line of defense in coordination with restriction‐modification or CRISPR‐Cas systems (Luyten et al, 2022; Picton et al, 2022). More broadly, there may exist a range of signaling mechanisms that enable cross talk between distinct defense systems in bacteria, mediating these systems' cooperation and integration into a comprehensive, multifaceted immune system.
Materials and Methods
Bioinformatics
To comprehensively search CBASS systems, we exported the genomic DNA sequences within 10 kb of 6,233 previously reported CD‐NTases (Cohen et al, 2019) using the Integrated Microbial Genomes (IMG) database at the DOE Joint Genome Institute (https://img.jgi.doe.gov). We used NCBI Genome Workbench (https://www.ncbi.nlm.nih.gov/tools/gbench/) to perform custom TBLASTN searches for proteins related to E. coli MS115‐1 CapP (NCBI sequence ID WP_001290439.1; Table EV1). CBASS system type and effector assignments for each hit were taken from Cohen et al (2019) and manually updated. Each hit was manually inspected for the presence of CapH and CapP.
For identification of other bacterial defense systems with associated CapH‐ and CapP‐like regulators, we used the FlaGs (Flanking Genes) server (Saha et al, 2021) to search for groups of genes associated with CapP (using E. coli MS115‐1 CapP as a search sequence). We manually inspected each result to search for known or putative defense‐related genes. For Fig 6F, the noted DISARM system from Nocardia wallacei FMUON74 encodes a vapC‐like gene (NCBI accession # WP_187684394.1), a CapH‐CapP fusion‐like protein (WP_197986914.1), drmD (WP_187684395.1), drmMI (WP_187684396.1), drmA (WP_187684397.1), drmB (WP_187684398.1), drmC (WP_187684399.1), and a AAA+ ATPase (WP_232110603.1). The noted Pycsar system from Tsuneonella flava MS1‐4 encodes a CapP‐like protein (WP_102155989.1), a CapH‐like protein (WP_102155988.1), a DUF2188 protein (WP_007013875.1), a predicted guanylate cyclase (WP_102155986.1), a hypothetical protein (WP_123639961.1), a predicted cyclic nucleotide monophosphate binding domain (cNMPBD) plus TIR domain protein (WP_102155984.1), and a predicted adenylate/guanylate cyclase (WP_102155983.1). The noted DUF2188 + hypothetical protein system from Extensimonas perlucida HX2‐24 encodes a CapP‐like protein (WP_144728453.1), a CapH‐like protein (WP_144728455.1), two hypothetical proteins (WP_144728457.1 and WP_144728459.1), and a DUF2188 protein (WP_144729358.1). The noted DUF3892 system from Sulfitobacter sp. CW3 encodes a CapP‐like protein (WP_037275352.1), a CapH‐like protein (WP_037275354.1), and two DUF3892 proteins (WP_037275364.1 and WP_037275356.1). The noted metallo‐β‐lactamase (Ub‐6a) system from Methylobacterium oxalidis NBRC 107715 encodes a CapP‐like protein (WP_147028642.1), a CapH‐like protein (WP_147028643.1), a hypothetical protein (WP_147028644.1), a predicted metallo‐β‐lactamase (WP_147028653.1), a protein with predicted E2, E1, and JAB domains (WP_147028645.1), and a second hypothetical protein (WP_147028646.1). The noted DUF6527 (Ub‐6e.2) system from Mixta intestinalis SRCM103226 encodes a CapP‐like protein (WP_160622475.1), a CapH‐like protein (WP_048227226.1), a predicted multi‐ubiquitin domain protein (WP_053069300.1), a predicted E1‐like protein (WP_160622476.1), and a DUF6527 protein (WP_18149987.1).
Cloning, expression, and protein purification
Proteins were cloned into UC Berkeley Macrolab vector 2‐BT (Addgene #29666; encoding an N‐terminal TEV protease‐cleavable His6‐tag), 2‐ST (Addgene #29711, encoding an N‐terminal TEV protease‐cleavable His6‐SUMO tag), or 2‐CT (Addgene #29706, encoding an N‐terminal TEV protease‐cleavable His6‐MBP tag). Proteins used were as follows: E. coli MS115‐1 CapH (NCBI sequence ID WP_001515173.1), E. coli MS115‐1 CapP (NCBI sequence ID WP_001290439.1), and Thauera sp. K11 CapP (NCBI sequence ID WP_096453114.1). Proteins were expressed in E. coli strain Rosetta 2 (DE3) pLysS (EMD Millipore, Billerica, MA). Cultures were grown at 37°C to A600 = 0.5, then induced with 0.25 mM IPTG and shifted to 20°C for 15 h. Cells were harvested by centrifugation and resuspended in buffer A (20 mM Tris pH 7.5, 10% glycerol) plus 300 mM NaCl, 10 mM imidazole, and 5 mM β‐mercaptoethanol (10 μM ZnCl2 was added to buffers for CapP). Proteins were purified by Ni2+‐affinity (Ni‐NTA agarose, Qiagen) then passed over an anion‐exchange column (Hitrap Q HP, Cytiva) in Buffer A plus 5 mM β‐mercaptoethanol and 0.1–1 M NaCl, collecting flow‐through or peak fractions. Tags were cleaved with TEV protease (Tropea et al, 2009), and cleaved protein was passed over another Ni2+ column (collecting flow‐through fractions) to remove uncleaved protein, cleaved tags, and tagged TEV protease. The protein was passed over a size exclusion column (Superdex 200, Cytiva) in buffer GF (buffer A plus 300 mM NaCl and 1 mM dithiothreitol [DTT]), then concentrated by ultrafiltration (Amicon Ultra, EMD Millipore) to 10–20 mg/ml and stored at 4°C. For selenomethionine derivatization, protein expression was carried out in M9 minimal media supplemented with amino acids plus selenomethionine prior to IPTG induction (Van Duyne et al, 1993), and proteins were exchanged into buffer containing 1 mM tris(2‐carboxyethyl)phosphine (TCEP) after purification to maintain the selenomethionine residues in the reduced state.
Crystallization and structure determination
For crystallization of E. coli MS115‐1 CapHNTD (residues 2–67), protein was concentrated to 18 mg/ml in crystallization buffer (20 mM Tris–HCl pH 8.5, 150 mM NaCl, 1 mM DTT) then mixed 1:1 with well solution containing 0.1 M Ammonium acetate pH 4.5 and 2 M Ammonium sulfate in hanging‐drop format. Crystals were cryoprotected with an additional 25% sucrose and flash‐frozen in liquid nitrogen. Diffraction data to 1.02 Å resolution were collected at Advanced Light Source beamline 5.0.2 (see support statement below) and processed with the DIALS data‐processing pipeline (https://dials.github.io) (Beilsten‐Edmands et al, 2020). We determined the structure by molecular replacement in PHASER (McCoy et al, 2007), using an ideal α‐helix as a search model. We manually rebuilt the initial model in COOT (Emsley et al, 2010) and refined in phenix.refine (Afonine et al, 2012) using individual positional and anisotropic B‐factor refinement for nonhydrogen atoms, and riding hydrogens (Appendix Table S1).
For crystallization of E. coli MS115‐1 CapHCTD(I99M) (residues 67–107 with Ile99 to Met mutation), protein was concentrated to 8 mg/ml in crystallization buffer (20 mM Tris–HCl pH 8.5, 150 mM NaCl, 1 mM DTT) then mixed 1:1 with well solution containing 0.1 M HEPES pH 7.5, 25 mM MgCl2, and 30% PEG 550 monomethyl ether in hanging‐drop format. Crystals were cryoprotected with an additional 13% PEG 550 monomethyl ether and 10% glycerol, and flash‐frozen in liquid nitrogen. Diffraction data to 1.26 Å resolution were collected at Advanced Photon Source beamline 24ID‐C (see support statement below) and processed with the RAPD data‐processing pipeline, which uses XDS (Kabsch, 2010) for data indexing and reduction, AIMLESS (Evans & Murshudov, 2013) for scaling, and TRUNCATE (French & Wilson, 1978) for conversion to structure factors. We determined the structure by molecular replacement in PHASER, using an ideal α‐helix as a search model. We manually rebuilt the initial model in COOT, and refined in phenix.refine using individual positional and anisotropic B‐factor refinement for nonhydrogen atoms, and riding hydrogens (Appendix Table S1).
For crystallization of E. coli MS115‐1 CapHCTD (residues 67–107), protein was concentrated to 21 mg/ml in crystallization buffer (20 mM Tris–HCl pH 8.5, 150 mM NaCl, 1 mM DTT) then mixed 1:1 with well solution containing 0.1 M sodium citrate pH 3.0 and 1.6 M lithium sulfate in hanging‐drop format. Crystals were cryoprotected with an additional 20% ethylene glycol and flash‐frozen in liquid nitrogen. Diffraction data to 1.75 Å resolution were collected at Advanced Light Source beamline 5.0.2 (see support statement below) and processed with the DIALS data‐processing pipeline. We determined the structure by molecular replacement in PHASER, using the structure of CapHCTD(I99M) as a search model. We manually rebuilt the initial model in COOT, and refined in phenix.refine using individual positional and B‐factor refinement, and riding hydrogens (Appendix Table S1).
For crystallization of Thauera sp. K11 CapP, protein was concentrated to 9 mg/ml in crystallization buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, and 1 mM DTT) then mixed 1:1 with well solution containing 0.1 M CHES pH 9.5, 0.3 M NaCl, and 1.8 M lithium sulfate in hanging‐drop format. Crystals were cryoprotected with an additional 30% glycerol and flash‐frozen in liquid nitrogen. Diffraction data for both native and selenomethionine‐derivatized crystals were collected at the Stanford Synchrotron Radiation Lightsource beamline 9–2 (see support statement below) and processed with the autoxds data‐processing pipeline, which uses XDS for data indexing and reduction, AIMLESS for scaling, and TRUNCATE for conversion to structure factors. We determined the structure by single‐wavelength anomalous diffraction (SAD) methods in PHASER using a 1.6 Å resolution dataset from selenomethionine‐derivatized protein. We manually rebuilt the initial model in COOT, and refined against a 1.35 Å resolution native dataset in phenix.refine using individual positional and anisotropic B‐factor refinement, and riding hydrogens (Appendix Table S1).
Beamline support statements
ALS beamline 5.0.2
The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE‐AC02‐05CH11231. The Pilatus detector on 5.0.1. was funded under NIH grant S10OD021832. The ALS‐ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169.
APS beamline 24ID‐C
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
SSRL beamline 9‐2
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE‐AC02‐76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
SEC‐MALS
For characterization of oligomeric state by size exclusion chromatography coupled to multi‐angle light scattering (SEC‐MALS), 100 μl of purified protein/complex at 2–5 mg/ml was injected onto a Superdex 200 Increase 10/300 GL column (Cytiva) in a buffer containing 20 mM HEPES pH 7.5, 300 mM NaCl, 5% glycerol, and 1 mM DTT. Light scattering and refractive index profiles were collected by miniDAWN TREOS and Optilab T‐rEX detectors (Wyatt Technology), respectively, and molecular weight was calculated using ASTRA v. 8 software (Wyatt Technology).
DNA‐binding assays
For characterization of DNA binding by fluorescence polarization assays, 40–41 bp double‐stranded DNAs were produced by annealing complementary oligos, one of which was 5′‐6‐FAM labeled (the same oligonucleotide was used without annealing for ssDNA binding studies). Binding reactions (10 μl) contained 25 mM Tris pH 7.5, 50 mM NaCl, 5% glycerol, 5 mM MgCl2, 1 mM DTT, 0.01% Nonidet p40 substitute, 50 nM DNA, and the indicated amounts of His6‐MBP‐tagged protein. After a 10‐min incubation at room temperature, fluorescence polarization was read using a Tecan Infinite M1000 PRO fluorescence plate reader, and binding data were analyzed with Graphpad Prism v.9.2.0 using a single‐site binding model.
CapP cleavage assays
For detection of CapP activity in cells, E. coli MS115‐1 CapP was coexpressed with a construct of E. coli MS115‐1 CapH fused to an N‐terminal His6‐maltose binding protein (MBP) tag and a C‐terminal green fluorescent protein (GFP) tag (MBP‐CapH‐GFP) in E. coli Rosetta 2 (DE3) pLysS cells at 37°C. Protein expression was induced with 0.25 mM IPTG for 2–4 h, and then samples were removed for analysis by Western blot (see below). For CapP activity in cells in response to zeocin, E. coli Rosetta 2 (DE3) pLysS cells were grown at 37°C until reaching an OD600 of 0.3. Protein expression was induced with 0.25 mM IPTG for 1 h, and then zeocin was added at a concentration of 100 μg/ml. 500 μl of sample was taken at 1 and 2 h postantibiotic and centrifuged 10,000 g for 1 min. Pelleted cells were resuspended in 50 μl of 2xSDS sample buffer and analyzed by Western blot (see below).
For detection of CapP activity using purified proteins, 20 μl reactions containing 10 μM purified CapP and 10 μM purified MBP‐CapH‐GFP in a buffer containing 50 mM Tris pH 7.5, 300 mM NaCl and 5 μM ZnCl2 were incubated at 37°C for 2 h, then added to 20 μl 2xSDS sample buffer. 10 μl of each sample was loaded and separated by SDS–PAGE and Coomassie‐stained for visualization. For reactions containing E. coli cell lysate, log‐phage E. coli Rosetta 2 (DE3) pLysS cells were lysed by sonication in a minimum volume of buffer A, then centrifuged to remove cell debris. Optionally, the lysate was incubated in a boiling water bath for 10 min, then centrifuged again to remove denatured proteins. For the experiment shown in Fig EV5B, lysate was fractionated on a 5 ml HiTrap Q HP column (Cytiva). 10 μl of cell lysate was added to reactions with purified CapP and MBP‐CapH‐GFP. For reactions containing DNA, nucleic acid was added at a concentration of 350 μg/ml and subsequent serial fivefold dilutions, then incubated in the buffer as above for 2 h. For reactions containing DNA or RNA oligos, nucleic acid was added at a concentration of 10 μM, then incubated in the buffer as above for 2 h.
Edman degradation
For Edman degradation, MBP‐CapH‐GFP was treated with CapP plus single‐stranded DNA, separated by SDS–PAGE, then transferred to a PVDF membrane and visualized with Coomassie staining. The band representing the C‐terminal cleavage product of MBP‐CapH‐GFP was cut out and analyzed by Edman degradation at the UC Davis Molecular Structure Facility (http://msf.ucdavis.edu).
Western blots
For CapH repressor assays, cells containing plasmids with the MS115‐1 system were grown in 5 ml of LB plus the selection marker at 37°C until they reached an OD600 of 0.3–0.5. Cultures were adjusted to an OD600 of 0.3, and then an aliquot of 500 μl was removed and centrifuged for 1 min at 10,000 g to pellet the cells. The supernatant was removed, and the cells were resuspended in 50 μl of 2xSDS sample buffer (125 mM Tris pH 6.8, 20% Glycerol, 4% SDS, 200 mM DTT, 180 μM bromophenol blue). 10 μl of each sample was loaded onto a 12% SDS–PAGE gel for separation and transferred to a PVDF membrane using the Bio‐Rad Trans‐Blot Turbo Transfer System. Membranes were blocked for 1 h in 5% milk in TBST (100 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween‐20) at room temperature with shaking, then incubated with mouse anti‐FLAG M2 antibody (Sigma‐Aldrich #F3165) or mouse anti‐GFP antibody (Roche #11814460001) at 1:3,000 diluted in 5% milk in TBST for 1 h at room temperature with shaking. Membranes were washed three times with 10 ml of TBST then incubated with goat anti‐mouse IgG antibody conjugated to horseradish peroxidase (Millipore Sigma #12‐349) at 1:30,000 diluted in 5% milk in TBST for 1 h at room temperature with shaking. Membranes were again washed three times with 10 ml TBST then incubated with Pierce ECL Plus substrate for 2 min and then imaged on a Bio‐Rad ChemiDoc imager. Membranes were stripped with a solution of 200 mM glycine pH 2.2, 0.1% SDS, and 1% Tween 20 for 20 min at room temperature, then washed with TBST and reblocked with 5% milk overnight at 4°C. Membranes were reblotted using the same procedure as initial blot, but replacing primary antibody with anti‐RNA polymerase B mouse antibody (clone NT63; BioLegend #10019–878).
CBASS plasmid construction
The full CBASS operon was PCR‐amplified from E. coli MS115‐1 genomic DNA (BEI Resources #HM‐344), using NCBI nucleotide accession number CP073624.1 (E. coli AW1.7) as a reference because of ambiguous sequence in the nucleotide record for E. coli MS115‐1 (NCBI accession number GG771785.1). An insert spanning bases 2,861,237–2,866,476 of NCBI accession number CP073624.1 was cloned between the EcoRI and BamHI sites of plasmid pBR322 using isothermal assembly. PCR‐based mutagenesis was used for all point mutants, gene deletions, and to insert an N‐terminal FLAG tag and short linker (DYKDDDDK‐ASAS) at the N‐terminus of the nucC gene. For the GFP reporter strain, the region encoding the core CBASS genes was replaced by a gene encoding msfGFP.
DNA damage assays
For antibiotic treatment time course Western blots, E. coli JP313 (MC4100 araΔ714) (Economou et al, 1995) containing reporter plasmids were grown at 37°C until reaching an OD600 of 0.1. Cultures were moved to 30°C for 10 min, then antibiotics were added at a concentration of 100 μg/ml for zeocin and 1 μg/ml for mitomycin C. At each timepoint, 1 ml of culture was removed and resuspended in 2xSDS sample buffer with the volume adjusted to an equal concentration of cells per sample.
Bacteriophage infectivity
Phage λ cI‐ was amplified in E. coli JP313 by inoculating cultures grown in LB with 1 mM MgCl2 and 1 mM CaCl2 to OD600 = 0.3 with 100 μl of high‐titer phage. Cells were incubated with shaking at 37°C for 5 h or until clearing was observed. The lysed cultures were spun at 4,000 g for 20 min, the supernatant was collected and filtered with a 0.2 μm filter, and filtered supernatant was stored at 4°C. Phage was titered in JP313 cells. Cells were grown to OD600 = 0.3–0.5 at 37°C, then 500 μl was aliquoted into 5 ml Eppendorf tubes. 10 μl of phage was added to each 500 μl tube, at 10‐fold dilutions. Lysate was diluted in phage buffer (150 mM NaCl, 40 mM Tris pH 7.5, 10 mM MgSO4 plus 1 mM CaCl2). Cultures were incubated with phage for 20 min after which 4.5 ml of 0.35% top agar with LB was added to each culture and mixed, then poured onto LB plates. Plates were incubated at 37°C for 16 h, then plaques were counted. For plaque quantification assays, strains were grown from single colonies in 5 ml of LB with ampicillin (100 μg/ml) until reaching log phase (OD600 = 0.3–0.6). 500 μl of culture was transferred to a 5‐ml conical tube to which 10 μl of of λ cI− at a concentration of 1.6 × 1010 PFU/ml (and 10‐fold dilutions thereof) in phage buffer (150 mM NaCl, 40 mM Tris pH 7.5, 10 mM MgSO4 plus 1 mM CaCl2) was added. Tubes were incubated at room temperature for 20 min after which 4.5 ml of 0.35% top agar was added mixed, then poured onto LB plates containing carbenicillin. Plates were incubated at 37°C for 16 h, then plaques were counted.
For infection time course Western blots, E. coli JP313 cells containing reporter plasmids were grown at 37°C until reaching an OD600 of 0.3. Cultures were moved to 30°C for 10 min, then λ cI− was added at an MOI of 10. At each time point, 500 μl of sample was removed and centrifuged 10,000 g for 1 min. Cell pellets were resuspended in 50 μl 2xSDS sample buffer and analyzed by western blot.
Microscopy
For fluorescence microscopy imaging, each sample was grown as liquid culture at 30°C and induced with 0.2 mM IPTG 30 min before of imaging. Cells were infected with λ cI− at MOI 2.5 as 0 MPI. Cells were harvested at required time points and were briefly centrifuged (3,300 g for 30 s). After resuspending the cells with 20 μl volume, 5 μl of the samples was transferred onto an agarose pad containing 1% agarose and 20% LB medium for microscopy and stained with 1 μg/ml FM4‐64 and 2 mg/ml DAPI. Microscopy was performed using by a Deltavision Elite System (GE Healthcare). Cells were quantified through manual examination of at least three replicates per condition and 30–100 cells per condition.
qRT–PCR
Escherichia coli JP313 cells containing the WT plasmid in pBR322 were grown in 25 ml LB + Ampicillin in 125 ml flasks until log phase (OD600 0.3–0.5). Cells were diluted to an OD600 of 0.2 in a final volume of 25 ml, then zeocin (100 μg/ml), λ CI− (MOI 10), or LB at equivalent volumes were added to each flask. Initial time points were taken at this time. For each time point, 1 ml of cells was transferred to a 1.5‐ml Eppendorf tube and spun down 1 min at 10,000 g. Supernatant was removed and pellets were immediately flash‐frozen in liquid N2. Cells were grown at 30°C and time points were taken every 20 min in triplicate.
RNA extractions were performed using the Invitrogen PureLink RNA Mini Kit with on‐column DNase treatment (Qiagen). 1 μg of RNA was reverse transcribed to make cDNA using the Applied Biosystems High‐Capacity cDNA Reverse Transcription Kit. qPCR was performed in a 384‐well format with technical triplicates for each sample. Each reaction was composed of IDT PrimeTime Gene Expression Master Mix, 250 nM of each primer, 100 μM of each probe, and cDNA diluted 1:250 to a final volume of 10 μl. The following primers were synthesized by Integrated DNA Technologies:
RpoA_F: CGTGGCTTTGGCCATACTCT.
RpoA_R: ACGCCTTCTTTGGTGCTGTA.
RpoA probe: /56‐FAM/AGCGAATGA/ZEN/TTCCATCAGGTAGTCTGGC/3IABkFQ/.
CdnC_F: GGAACAGGCCAAGCGATTAC.
CdnC_R: AACGAGCGAAGAGCAGTTCC.
CdnC probe: /5Cy5/AGCAGAATA/TAO/CGGCGCAGTGCGT/3IAbRQSp/.
56‐FAM: 5′ 6‐FAM (Fluorescein); ZEN: ZEN internal quencher (Integrated DNA Technologies); 3IABkFQ: 3′ Iowa Black FQ quencher; 5Cy5: 5′ Cy5; TAO: TAO internal quencher (Integrated DNA Technologies); 3IAbRQSp: 3′ Iowa Black RQ quencher.
qPCR was performed using a Bio‐Rad CFX384 Touch Real‐Time PCR Detection System.
Electrophoretic mobility assay (EMSA)
Purified MBP‐tagged CapH was mixed with 100 nM 5′‐FAM‐labeled DNA at concentration stated in figures and twofold dilutions thereof, then incubated at room temperature for 20 min. Samples were loaded on to prerun 8% polyacrylamide gels (prerun 250 V for 60 min) made with 1× TBE pH 8.5 and run in 0.5 TBE pH 8.5 at 120 V for 80 min at 4°C. Gels were imaged using a Bio‐Rad ChemiDoc imager.
MIC assays
MIC assays were performed in a 96‐well format. 100 μl of LB + Ampicillin containing different concentrations of zeocin was added to each well in the top row of the plate. Concentrations of zeocin started from 100 μg/ml and were serially diluted at 1:3 across the plate in fresh LB + Amp. Cells containing either an empty vector, a plasmid with the 6‐gene E. coli MS115‐1 system, or a mutant system with catalytic‐dead NucC (D73N) were grown to OD600 = 0.1 in LB + Amp at 37°C with shaking. Cells were diluted and 100 μl of culture was added to each well such that each well contained ~ 2 × 105 CFU/ml. Each condition was run in triplicate. The plate was incubated at 37°C and OD600 readings were taken after 18 h of growth.
Author contributions
Rebecca K Lau: Conceptualization; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Eray Enustun: Formal analysis; investigation. Yajie Gu: Formal analysis; investigation. Justin V Nguyen: Formal analysis; investigation. Kevin D Corbett: Conceptualization; formal analysis; supervision; funding acquisition; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data EV and Appendix
Source Data Figure 1
Source Data Figure 2
Source Data Figure 3
Source Data Figure 5
Source Data Figure 6
PDF+
Acknowledgements
The authors thank John Schulze at the UC Davis Molecular Structure Facility for assistance with Edman Degradation, Michael Baughn for advice on qPCR experiment and primer design, and members of the Corbett laboratory and Aaron Whiteley for helpful feedback. K.D.C. acknowledges support from the National Institutes of Health (R35 GM144121). R.K.L. was supported by the UCSD Quantitative and Integrative Physiology Training Grant (NIH T32 GM127235) and an individual National Institutes of Health Predoctoral Fellowship (F31 GM137600).
The EMBO Journal (2022) 41: e111540
Data availability
The structural data produced in this study are available in the following databases: Primary X‐ray diffraction datasets: SBGrid Data Bank (https://data.sbgrid.org): E. coli MS115‐1 CapH NTD: #866; E. coli MS115‐1 CapH CTD: #867; E. coli MS115‐1 CapH CTD (I99M): #868; Thauera sp. K11 CapP: #864, 865. Reduced X‐ray diffraction datasets and refined structures: Protein Data Bank (http://www.wwpdb.org): E. coli MS115‐1 CapH NTD: 7T5U; E. coli MS115‐1 CapH CTD: 7T5W; E. coli MS115‐1 CapH CTD (I99M): 7T5V; Thauera sp. K11 CapP: 7T5T.
References
- Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD et al (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68: 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchtung JM, Lee CA, Monson RE, Lehman AP, Grossman AD (2005) Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc Natl Acad Sci USA 102: 12554–12559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayora S, Carrasco B, Cardenas PP, Cesar CE, Canas C, Yadav T, Marchisone C, Alonso JC (2011) Double‐strand break repair in bacteria: a view from Bacillus subtilis . FEMS Microbiol Rev 35: 1055–1081 [DOI] [PubMed] [Google Scholar]
- Baek K, Svenningsen S, Eisen H, Sneppen K, Brown S (2003) Single‐cell analysis of lambda immunity regulation. J Mol Biol 334: 363–372 [DOI] [PubMed] [Google Scholar]
- Beilsten‐Edmands J, Winter G, Gildea R, Parkhurst J, Waterman D, Evans G (2020) Scaling diffraction data in the DIALS software package: algorithms and new approaches for multi‐crystal scaling. Acta Crystallogr D Struct Biol 76: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benler S, Koonin EV (2020) Phage lysis‐lysogeny switches and programmed cell death: Danse macabre. Bioessays 42: e2000114 [DOI] [PubMed] [Google Scholar]
- Bernheim A, Sorek R (2020) The pan‐immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol 18: 113–119 [DOI] [PubMed] [Google Scholar]
- Blanchard L, Guérin P, Roche D, Cruveiller S, Pignol D, Vallenet D, Armengaud J, de Groot A (2017) Conservation and diversity of the IrrE/DdrO‐controlled radiation response in radiation‐resistant Deinococcus bacteria. Microbiologyopen 6: e00477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenchip CL, Nguyen JV, Lau RK, Ye Q, Gu Y, Corbett KD (2022) Control of bacterial anti‐phage signaling by a WYL domain transcription factor. Nucleic Acids Res 50: 5239–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose B, Grossman AD (2011) Regulation of horizontal gene transfer in Bacillus subtilis by activation of a conserved site‐specific protease. J Bacteriol 193: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose B, Auchtung JM, Lee CA, Grossman AD (2008) A conserved anti‐repressor controls horizontal gene transfer by proteolysis. Mol Microbiol 70: 570–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Aravind L (2020) Identification of uncharacterized components of prokaryotic immune systems and their diverse eukaryotic reformulations. J Bacteriol 202: e00365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Iyer LM, Aravind L (2009) Natural history of the E1‐like superfamily: Implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins 75: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Zhang D, Schäffer DE, Iyer LM, Aravind L (2015) Comparative genomic analyses reveal a vast, novel network of nucleotide‐centric systems in biological conflicts, immunity and signaling. Nucleic Acids Res 43: 10633–10654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda‐Costa N, Gomis‐Ruth FX (2014) Architecture and function of metallopeptidase catalytic domains. Protein Sci 23: 123–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chankova SG, Dimova E, Dimitrova M, Bryant PE (2007) Induction of DNA double‐strand breaks by zeocin in Chlamydomonas reinhardtii and the role of increased DNA double‐strand breaks rejoining in the formation of an adaptive response. Radiat Environ Biophys 46: 409–416 [DOI] [PubMed] [Google Scholar]
- Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer‐Shaanan Y, Kacen A, Doron S, Amitai G, Sorek R (2019) Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574: 691–695 [DOI] [PubMed] [Google Scholar]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, Sorek R (2018) Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359: eaar4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy RL, Richter C, Salmond GP, Fineran PC (2014) Remarkable mechanisms in microbes to resist phage infections. Annu Rev Virol 1: 307–331 [DOI] [PubMed] [Google Scholar]
- Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W (1995) SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83: 1171–1181 [DOI] [PubMed] [Google Scholar]
- Elkins PA, Ho YS, Smith WW, Janson CA, D'Alessio KJ, McQueney MS, Cummings MD, Romanic AM (2002) Structure of the C‐terminally truncated human ProMMP9, a gelatin‐binding matrix metalloproteinase. Acta Crystallogr D Biol Crystallogr 58: 1182–1192 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69: 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S, Wilson K (1978) On the treatment of negative intensity observations. Acta Crystallogr A Cryst Phys Diffr Theor Gen Crystallogr 34: 517–525 [Google Scholar]
- Gangwar SP, Meena SR, Saxena AK (2014) Comparison of four different crystal forms of the Mycobacterium tuberculosis ESX‐1 secreted protein regulator EspR. Acta Crystallogr F Struct Biol Commun 70: 433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot A, Siponen MI, Magerand R, Eugenie N, Martin‐Arevalillo R, Doloy J, Lemaire D, Brandelet G, Parcy F, Dumas R et al (2019) Crystal structure of the transcriptional repressor DdrO: insight into the metalloprotease/repressor‐controlled radiation response in Deinococcus . Nucleic Acids Res 47: 11403–11417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross‐Langenhoff M, Hofbauer K, Weber J, Schultz A, Schultz JE (2006) cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J Biol Chem 281: 2841–2846 [DOI] [PubMed] [Google Scholar]
- Hampton HG, Watson BNJ, Fineran PC (2020) The arms race between bacteria and their phage foes. Nature 577: 327–336 [DOI] [PubMed] [Google Scholar]
- Ho YS, Burden LM, Hurley JH (2000) Structure of the GAF domain, a ubiquitous signaling motif and a new class of cyclic GMP receptor. EMBO J 19: 5288–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Burroughs AM, Aravind L (2006) The prokaryotic antecedents of the ubiquitin‐signaling system and the early evolution of ubiquitin‐like β‐grasp domains. Genome Biol 7: R60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidane D, Sanchez H, Alonso JC, Graumann PL (2004) Visualization of DNA double‐strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol Microbiol 52: 1627–1639 [DOI] [PubMed] [Google Scholar]
- Lau RK, Ye Q, Birkholz EA, Berg KR, Patel L, Mathews IT, Watrous JD, Ego KM, Whiteley AT, Lowey B et al (2020) Structure and mechanism of a cyclic trinucleotide‐activated bacterial endonuclease mediating bacteriophage immunity. Mol Cell 77: 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledvina HE, Ye Q, Gu Y, Quan Y, Lau RK, Zhou H, Corbett KD, Whiteley AT (2022) cGASylation by a bacterial E1‐E2 fusion protein primes antiviral immune signaling. bioRxiv 10.1101/2022.03.31.486616 [PREPRINT] [DOI] [Google Scholar]
- Levdikov VM, Blagova E, Colledge VL, Lebedev AA, Williamson DC, Sonenshein AL, Wilkinson AJ (2009) Structural rearrangement accompanying ligand binding in the GAF domain of CodY from Bacillus subtilis . J Mol Biol 390: 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RJ, Brannigan JA, Offen WA, Smith I, Wilkinson AJ (1998) An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti‐repressor complex. J Mol Biol 283: 907–912 [DOI] [PubMed] [Google Scholar]
- Little JW (1991) Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73: 411–421 [DOI] [PubMed] [Google Scholar]
- Lowey B, Whiteley AT, Keszei AFA, Morehouse BR, Mathews IT, Antine SP, Cabrera VJ, Kashin D, Niemann P, Jain M et al (2020) CBASS immunity uses CARF‐related effectors to sense 3′‐5′‐ and 2′‐5'‐linked cyclic oligonucleotide signals and protect bacteria from phage infection. Cell 182: 38–49.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludanyi M, Blanchard L, Dulermo R, Brandelet G, Bellanger L, Pignol D, Lemaire D, de Groot A (2014) Radiation response in Deinococcus deserti: IrrE is a metalloprotease that cleaves repressor protein DdrO. Mol Microbiol 94: 434–449 [DOI] [PubMed] [Google Scholar]
- Luyten YA, Hausman DE, Young JC, Doyle LA, Higashi KM, Ubilla‐Rodriguez NC, Lambert AR, Arroyo CS, Forsberg KJ, Morgan RM et al (2022) Identification and characterization of the WYL BrxR protein and its gene as separable regulatory elements of a BREX phage restriction system. Nucleic Acids Res 50: 5171–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF et al (2011a) Evolution and classification of the CRISPR‐Cas systems. Nat Rev Microbiol 9: 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Snir S, Koonin EV (2011b) Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J Bacteriol 193: 6039–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Koonin EV (2013) Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res 41: 4360–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SE, Beavo JA, Hol WG (2002a) GAF domains: two‐billion‐year‐old molecular switches that bind cyclic nucleotides. Mol Interv 2: 317–323 [DOI] [PubMed] [Google Scholar]
- Martinez SE, Wu AY, Glavas NA, Tang XB, Turley S, Hol WG, Beavo JA (2002b) The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc Natl Acad Sci USA 99: 13260–13265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SE, Heikaus CC, Klevit RE, Beavo JA (2008) The structure of the GAF A domain from phosphodiesterase 6C reveals determinants of cGMP binding, a conserved binding surface, and a large cGMP‐dependent conformational change. J Biol Chem 283: 25913–25919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeehan JE, Streeter SD, Thresh SJ, Ball N, Ravelli RB, Kneale GG (2008) Structural analysis of the genetic switch that regulates the expression of restriction‐modification genes. Nucleic Acids Res 36: 4778–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millman A, Melamed S, Amitai G, Sorek R (2020) Diversity and classification of cyclic‐oligonucleotide‐based anti‐phage signalling systems. Nat Microbiol 5: 1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR‐Cas systems. Science 353: aad5147 [DOI] [PubMed] [Google Scholar]
- Ofir G, Melamed S, Sberro H, Mukamel Z, Silverman S, Yaakov G, Doron S, Sorek R (2018) DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol 3: 90-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton DM, Luyten YA, Morgan RD, Nelson A, Smith DL, Dryden DTF, Hinton JCD, Blower TR (2021) The phage defence Island of a multidrug resistant plasmid uses both BREX and type IV restriction for complementary protection from viruses. Nucleic Acids Res 49: 11257–11273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton DM, Harling‐Lee JD, Duffner SJ, Went SC, Morgan RM, Hinton JCD, Blower TR (2022) A widespread family of WYL‐domain transcriptional regulators co‐localizes with diverse phage defence systems and islands. Nucleic Acids Res 50: 5191–5207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Casjens S, Uetz P (2011) The protein interaction map of bacteriophage lambda. BMC Microbiol 11: 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha CK, Sanches Pires R, Brolin H, Delannoy M, Atkinson GC (2021) FlaGs and webFlaGs: discovering novel biology through the analysis of gene neighbourhood conservation. Bioinformatics 37: 1312–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassanfar M, Roberts JW (1990) Nature of the SOS‐inducing signal in Escherichia coli. The involvement of DNA replication. J Mol Biol 212: 79–96 [DOI] [PubMed] [Google Scholar]
- Severin GB, Ramliden MS, Hawver LA, Wang K, Pell ME, Kieninger AK, Khataokar A, O'Hara BJ, Behrmann LV, Neiditch MB et al (2018) Direct activation of a phospholipase by cyclic GMP‐AMP in El Tor Vibrio cholerae . Proc Natl Acad Sci USA 115: E6048–E6055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsov MB, Streeter SD, Thresh SJ, Swiderska A, McGeehan JE, Kneale GG (2015) Structural analysis of DNA binding by C.Csp231I, a member of a novel class of R‐M controller proteins regulating gene expression. Acta Crystallogr D Biol Crystallogr 71: 398–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V, Salamov A (2011) Automatic annotation of microbial genomes and metagenomic sequences. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies, Li RW (ed), pp 61–78. Hauppauge, NY: Nova Biomedical; [Google Scholar]
- Springman EB, Angleton EL, Birkedal‐Hansen H, Van Wart HE (1990) Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active‐site zinc complex in latency and a "cysteine switch" mechanism for activation. Proc Natl Acad Sci USA 87: 364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal N, Morehouse BR, Millman A, Stokar‐Avihail A, Avraham C, Fedorenko T, Yirmiya E, Herbst E, Brandis A, Mehlman T et al (2021) Cyclic CMP and cyclic UMP mediate bacterial immunity against phages. Cell 184: 5728–5739.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea JE, Cherry S, Waugh DS (2009) Expression and purification of soluble His(6)‐tagged TEV protease. In Methods in Molecular Biology, Doyle SA (ed), pp 297–307. Totowa, NJ: Humana Press; [DOI] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J (1993) Atomic structures of the human immunophilin FKBP‐12 complexes with FK506 and rapamycin. J Mol Biol 229: 105–124 [DOI] [PubMed] [Google Scholar]
- Van Wart HE, Birkedal‐Hansen H (1990) The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA 87: 5578–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujičić‐Žagar A, Dulermo R, Le Gorrec M, Vannier F, Servant P, Sommer S, de Groot A, Serre L (2009) Crystal structure of the IrrE protein, a central regulator of DNA damage repair in Deinococcaceae. J Mol Biol 386: 704–716 [DOI] [PubMed] [Google Scholar]
- Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, Danilchanka O, King DS, Lee ASY, Mekalanos JJ et al (2019) Bacterial cGAS‐like enzymes synthesize diverse nucleotide signals. Nature 567: 194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Lau RK, Mathews IT, Birkholz EA, Watrous JD, Azimi CS, Pogliano J, Jain M, Corbett KD (2020) HORMA domain proteins and a Trip13‐like ATPase regulate bacterial cGAS‐like enzymes to mediate bacteriophage immunity. Mol Cell 77: 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]