

Abstract
Decaprenylphosphoryl-β-d-ribose 2′-epimerase (DprE1) is a critical flavoenzyme in Mycobacterium tuberculosis, catalyzing a vital step in the production of lipoarabinomannan and arabinogalactan, both of which are essential for cell wall biosynthesis. Due to its periplasmic localization, DprE1 is a susceptible target, and several compounds with diverse scaffolds have been discovered that inhibit this enzyme, covalently or noncovalently. We evaluated a total of ∼1519 DprE1 inhibitors disclosed in the literature from 2009 to April 2022 by performing an in-depth analysis of physicochemical descriptors and absorption, distribution, metabolism, excretion, and toxicity (ADMET), to gain new insights into these properties in DprE1 inhibitors. Several molecular properties that should facilitate the design and optimization of future DprE1 inhibitors are described, allowing for the development of improved analogues targeting M. tuberculosis.
Introduction
Tuberculosis (TB) is an airborne illness caused by a single infectious agent, Mycobacterium tuberculosis (Mtb), continuing to be one of the world’s top ten infectious killers.1Mtb is predicted to infect around 2 billion people (mostly in the latent form), with a risk of individuals contracting the disease’s most aggressive form (generally 5–10% of the cases, predominantly among those with comorbidities such as diabetes or AIDS).2 Even though TB is usually treatable, it remains a worldwide concern. In 2020, over 1.3 million HIV-negative, together with 214000 HIV-positive individuals, died of TB. Worldwide, 9.9 million new cases of TB were reported in the same year, with men accounting for 56% of this total, 33% in adult women, and 11% in children.2 Additionally, the spread of multidrug-resistant (MDR) and extensively drug-resistant (XDR) tuberculosis and the simultaneous pandemic of HIV-TB coinfection, together with a deficient health care infrastructure and the lack of an effective vaccine, all contribute to the disease’s endurance.3,4 The current anti-Mtb pharmaceutical combination treatment, developed more than 40 years ago, consists of a four-drug regimen comprising isoniazid (1, INH), pyrazinamide (2, PYR), rifampicin (3, RFP) and ethambutol (4, EMB) (Figure 1).4,5 These therapies present disadvantages, including lengthy treatments, undesirable side effects, drug interactions, and poor patient compliance,6,7 in addition to the emergence of mycobacteria mutations conferring resistance to the drugs of this combination therapy. Consequently, the search for more effective drugs and therapy regimens has been critical in maintaining disease control.8
Figure 1.

Structural representations of the front-line TB drugs.
One strategy for treating tuberculosis has been to target the mycobacterial cell wall.9 The front-line TB drugs EMB and INH inhibit key enzymes involved in producing arabinogalactan and mycolic acids, which are noncovalently connected to a protein- and polysaccharide-based outer capsule.10−13 Numerous new drug families have been found by whole-cell screening that target essential proteins implicated in the construction of the cell wall components.9
DprE1, also known as decaprenylphosphoryl-β-d-ribose 2′-epimerase, is an indispensable flavoenzyme involved in forming the Mtb cell wall.14 It catalyzes the two-step epimerization of decaprenyl-phospho-ribose (DPR) to decaprenyl-phospho-arabinose (DPA), the precursor for arabinogalactan and lipoarabinomannan synthesis, in conjunction with decaprenylphosphoryl-d-2-keto erythro pentose reductase (DprE2, Figure 2-A).14−16 DprE1 initiates the first step of the epimerization process, where DPR is oxidized to the intermediate decaprenyl-phospho-2′-keto-d-arabinose (DPX), cofactored by flavin adenine dinucleotide (FAD), yielding FADH2. DprE2, which is NADH-dependent, subsequently converts DPX to DPA.17−19 The epimerization happens in the periplasmic region, which explains DprE1’s vulnerability as a target,19 making this flavoenzyme a promising target for developing novel therapeutic candidates to tackle TB. The druggable yet promiscuous nature of DprE1 has led to a significant number of DprE1 inhibitors with diverse molecular scaffolds and pharmacological profiles,20−25 as evidenced by an increasing number of publications on the subject. There have been 23 new classes of DprE1 inhibitors identified with antimycobacterial activity, and their different scaffolds are displayed in Tables 1 and 2. These inhibitors are divided into two types, according to their mechanism of action (MoA): (1) covalent binders, where five classes have been shown to irreversibly inhibit DprE1 by generating a covalent adduct with the C387 residue, and (2) noncovalent inhibitors, in which 17 reported classes were experimentally confirmed to act as competitive inhibitors (Figure 2B). Several DprE1 inhibitor reviews have been written during the past decade, covering both scaffold and docking studies.20−24,26,27
Figure 2.

(A) The DPA biosynthetic pathway: in the presence of the cofactor FAD, the DprE1 and DprE2 enzymes catalyze the epimerization of the 2′-OH group in DPR to form DPA. (B) Timeline of the discovery of the different DprE1 classes.
Table 1. Reported Compounds Targeting DprE1 Covalently.

MIC90.
Table 2. Reported Compounds Targeting DprE1 Noncovalently.

MIC99;
MIC50Ms
DprE1 Covalent Inhibitors
DprE1 was first discovered as a target of benzothiazinones, which inhibit the flavoenzyme DprE1 irreversibly through the generation of a covalent adduct with the amino acid Cys387. A fundamental similarity of the covalent DprE1 inhibitors is the presence of a nitro group on the molecule, which is required for its inhibition mechanism.28 Makarov and colleagues were the first to demonstrate this type of inhibition, in which they proved that benzothiazinones (BTZ) were capable of strongly suppressing DprE1 activity in vitro and in vivo. Compound BTZ043 (Figure 3) was shown to act as a prodrug in the presence of FADH2, where the nitro group on the benzothiazinone core is reduced to its nitroso derivative. The reactive nitroso form reacts with the thiol group on the Cys387 residue in DprE1, producing a semimercaptal bond with the amino acid residue and a covalent adduct that acts as a suicide substrate, irreversibly inhibiting the enzyme (Figure 3).29−35
Figure 3.
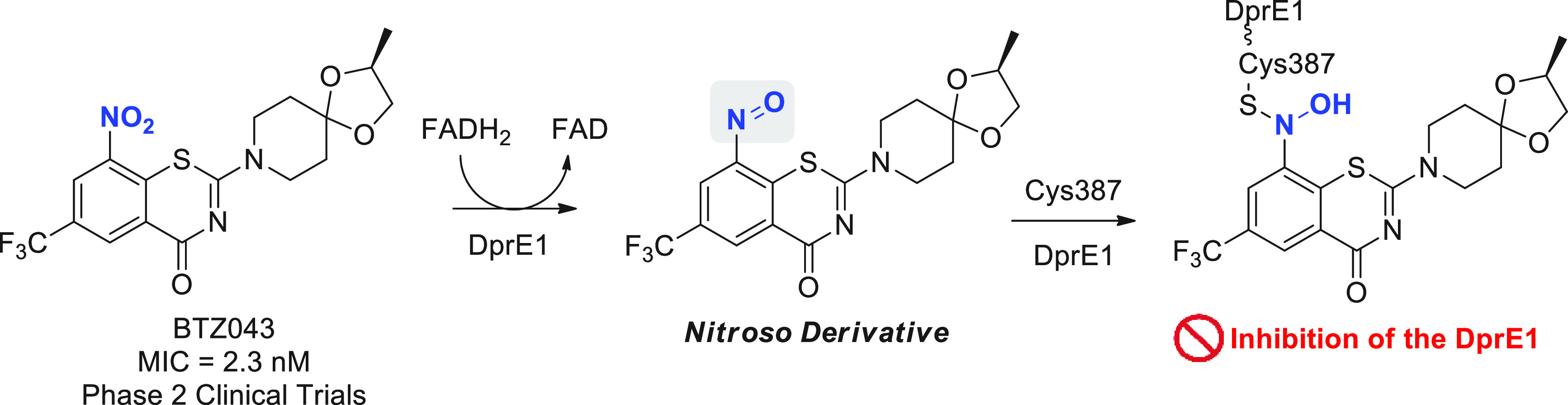
Bioactivation of BTZ043 by DprE1 and cofactor FADH2, and subsequent nucleophilic addition by Cys387.
Since then, more than ∼600 new nitrobenzothiazinones (BTZ; Table 1) have been described, and nearly 90% of these molecules proved active against Mtb (MIC < 10 μM).30−63 DprE1 has also been identified as the target of nitrobenzothiazoles (NBTO),64,65 dinitrobenzamides (DNBs),66 nitroquinoxalines (NQs),67 and 3-nitro-1,2,4-triazoles (NTZs),68 all of which interact as covalent inhibitors (Table 1).
DprE1 Noncovalent Inhibitors
Numerous scaffolds acting noncovalently also have been investigated for their activity against Mtb and are depicted in Table 2. These inhibitors include non-nitro BTZ analogues (NC BTZ),69−71 benzothiazoles (BTO),72,73 1,2,3-triazole-2-mercaptobenzothiazoles (2-S-BTO),74 1,4-azaindoles (AZA),75−80 benzimidazoles (BI),81 pyrazolopyridones (PP),82 4-aminoquinolone piperidine amides (4-AQ),83 2-carboxyquinoxaline derivatives (2-CQ),84 pyrrolothiadiazoles (PTD),85,86 morpholine-pyrimidines (MP),87N-alkyl-5-hydroxypyrimidinone carboxamides (NAHPC),88,89 hydantoins (HYD),90,91 benzodioxanes (BD),92 3,4-dihydrocarbostyril derivatives (CD),93−97 thiophene-arylamide compounds (TPA),98N-(4-hydroxy-3-mercaptonaphthalenyl) sulfonamides (NHMS),99 and avermectins (AVMT).100
Physicochemical and ADMET Properties of DprE1 Inhibitors
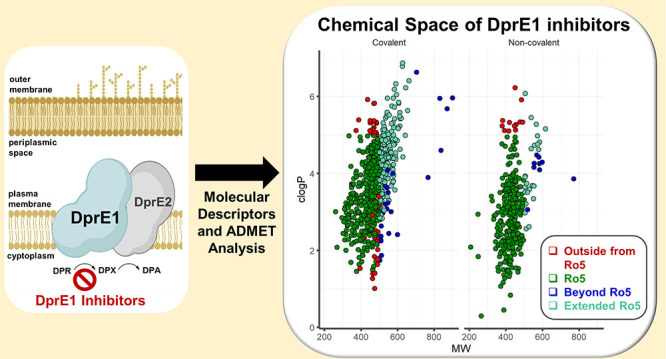
Numerous research groups have examined the connections between small molecules’ physicochemical (PC) descriptors, potency, and ADMET profile.102−104 PC descriptors can affect efficacy, safety, or metabolism. Numerous molecular descriptors have been shown to be useful in predicting ADMET characteristics and have been used to characterize a variety of molecular properties, including lipophilicity, molecular flexibility, hydrogen-bonding ability, and molecular weight.103,105 Additionally, small-molecule-based pharmacological candidates must be sufficiently permeable and soluble to allow experimental testing and have the capability to reach their site of action as well as to activate their main targets, for which the PC descriptors are critical.106 Research on the chemical space exploration of DprE1 inhibitors found a significant lipophilic character, establishing a different cluster from currently available tuberculosis medicines, as shown by principal component analysis from their physicochemical descriptor analysis.107 Thus, ongoing research is essential to gain new insights into the design and development of highly active covalent and noncovalent DprE1 inhibitors and guiding hit and lead optimization to produce nonhazardous small-molecule-based treatments against Mtb.
Data Collection and Preprocessing
To investigate the molecular diversity and ADMET properties of the DprE1 inhibitors disclosed in this review, we collected a data set of a total of 1519 structurally diverse molecules by reviewing the literature from the year 2009 to April 2022.28−101,108−119
The data set was split by two subsets, covalent (Cov) and noncovalent (NCov) binders, and then each compound was classified as active (MIC < 10 μM, Act) or not active (MIC ≥ 10 μM, NAct), following the MIC cutoff criteria adapted by the report of Makarov et al.26 The PC descriptors molecular weight (MW), lipophilicity through calculated partition coefficient (C log P), distribution coefficient at pH = 7.4 (C log D), intrinsic aqueous solubility (log S), hydrogen bond acceptors and donors (HBAs and HBDs), topological polar surface area (TPSA), number of rotatable bonds (ROTBS), and flexibility index (FInd) were computed by StarDrop v7.2.0.32905.120 The median (Md), mean (Mn), standard deviation (SD), Student t-test analyses were implemented and analyzed. Drug design oriented rules such as Lipinski’s Rule of 5 (Ro5),121 GSK’s 4/400 rule,122 and Pfizer’s 3/75 rule123 were also explored in this work. The ADMET predictions, CYP inhibition and metabolism, blood–brain barrier (BBB) penetration, plasma protein binding (PPB), P-glycoprotein (P-gp) substrate classification, and pan-assay interference compound (PAINS) count were obtained with StarDrop v7.2.0.32905,120,124−126 and structure alerts were processed through ChemBioServer 2.0.127 The generated raw data were then analyzed using manual R scripts in RStudio (Version 1.4.1106). Prior to processing, any observation with missing values was removed using the na.omit function, and the graphic figures were produced using the ggplot2 package.
QSAR Metrics to Evaluate Model Performance
When appropriate, an analysis of the predictive model was conducted in which numerous model performance metrics for a classification model were calculated. Internally, we used four measures: (1) accuracy (ACC), (2) precision, (3) sensitivity, and (4) specificity. The following equations show their corresponding definitions128
| 1 |
| 2 |
| 3 |
| 4 |
where TP are true positives, TN are true negatives, FP are false positives, and FN are false negatives.128
Correlations of MIC with IC50 DprE1 and with CC50
To establish a correlation between the DprE1 enzyme inhibition and the subsequent MIC between the different classes of inhibitors, we performed a Pearson’s correlation (between the experimental pIC50 DprE1 and pMIC values), and the results are described in Figure 4A. In both types of binders, a moderate to strong positive correlation is observed between DprE1 pIC50 and Mtb pMIC, with statistical analyses of our results showing a significant correlation (p < 0.0001), whereas noncovalent inhibitors display a higher positive correlation coefficient (n = 420, r = 0.647) than the covalent binders (n = 47, r = 0.539). These results reveal conclusively that antimycobacterial efficacy significantly depends on the inhibition of the flavoenzyme DprE1. Several of these inhibitors were investigated for cytotoxicity in various human cell lines, including A549,64,67,77,82,83 HeLa,37,39 HepG2,28,56,66,69,72,74,87,88,90−92,109,111 J-774,111 THP-1,60 and Vero cell lines.9,37−42,44,47−49,51,52,54,98,108,113,119 A small negative correlation (r = −0.291, p = 0.011, Figure 4B) was found between the experimental cytotoxicity concentrations (CC50) and MIC, for covalent binders. This observation suggests that even the most effective covalent binders appear to display a safe profile, encouraging the ongoing search for novel inhibitors. In contrast, for the noncovalent binders, a Pearson’s correlation analysis did not reveal the existence of a correlation between pCC50 and pMIC (not statistically significant, n = 42, r = 0.221, p = 0.161, Figure 4B).
Figure 4.

(A) Scatter plot of DprE1 pIC50 (−log10[IC50 (molar)]) versus pMIC (−log10[MIC (molar)]) against Mtb for covalent (left) and noncovalent (right) inhibitors and Pearson correlation coefficient between DprE1 pIC50 and pMIC. (B) Scatter plot of cytotoxicity pCC50 (−log10[CC50 (molar)]) versus pMIC for covalent (left) and noncovalent (right) inhibitors and Pearson correlation coefficient between pCC50 and pMIC.
The Impact of Nine Molecular Properties
The nine molecular properties for the active (≤10 μM, Act) and nonactive classes (>10 μM, Nact), considering separately the covalent and noncovalent molecules, are represented in Figure 5. We evaluated the significance of the difference between the means by a two-sided Student’s t-test.
Figure 5.

Physicochemical property distribution and statistics of the inhibitor (Act, in green) and noninhibitor (NAct, in red) classes. Each corresponding binding type (covalent (Cov) upper and noncovalent (NCov) above) is shown for MW, C log P, C log D, log S, HBA, HBD, TPSA, flexibility, and ROTBS. N indicates the total number of compounds considered in each analysis. A two-sided Student’s t-test was used to determine the statistical significance of active and inactive compounds, among those classified as covalent or noncovalent inhibitors, and the p values were evaluated (ns p-value >0.05, *p value <0.05, **p value <0.01, ***p value <0.001, ****p value <0.0001; Md, median; Mn, mean).
Molecular Weight
The molecular weight (MW) is a critical property in the development of small-molecule drugs.129,130 It has the potential to influence a variety of molecular processes, including absorption, blood–brain barrier (BBB) penetration, bile elimination rate, and interactions with biological targets, being frequently investigated as part of the process for compounds optimization.131,132
Covalent Inhibitors
The MW values for most of the drugs varied from 361.3 Da (10th percentile) to 558.5 Da (90th percentile), with a median MW value of 480.4 Da. Nonactive compounds generally had a lower molecular weight, with most molecules ranging from 332.9 Da (10th percentile) to 525.8 Da (90th percentile) and a median MW value of 408.4 Da. Comparison of the MW for active and nonactive candidates also showed that the set of actives had a higher mean MW (474.5 Da) than the set of nonactives (424.4 Da) by 50.1 Da, which proved statistically significant (p < 0.0001).
Noncovalent Inhibitors
The MW values for most of the compounds varied from 373.4 Da (10th percentile) to 500.2 Da (90th percentile), with a median MW value of 435.6 Da. Nonactive molecules had a lower molecular weight, with most of the molecules ranging from 333.1 Da (10th percentile) to 452.7 Da (90th percentile) and a median MW value of 383.3 Da. A comparison of the MW for active and nonactive candidates also showed that the set of active molecules had a higher mean MW (436.0 Da) than the set of nonactive molecules (391.7 Da) by 44.3 Da, which was statistically significant (p < 0.0001).
Lipophilicity
Lipophilicity, as indicated by the C log P and C log D values obtained here, is critical in defining key ADMET characteristics and potency. For instance, when lipophilicity levels are high, metabolism and solubility are more susceptible to being impaired, while low lipophilicity may increase permeability.133
Covalent Inhibitors
Covalent molecules in the active set displayed a similar C log P range, though right-shifted to higher lipophilicity, with 10th to 90th percentile values of 2.40 to 5.03 and a higher mean value of 3.77 versus 3.28 (p < 0.0001), compared to the nonactive counterparts (1.87 for the 10th percentile, 4.71 for the 90th percentile). Regarding the C log D values, the covalent binders in the active set showed a similar C log D range to the nonactive counterparts, with 10th to 90th percentile values of 2.18 to 3.77 and a slightly higher mean value of 2.99 versus 2.73 (p = 0.002), compared to the nonactive counterparts (1.83 for the 10th percentile, 3.60 for the 90th percentile).
Noncovalent Inhibitors
An opposite situation is observed in the noncovalent binders, although in this case there was no statistically significant difference in both C log P (p = 0.298) and C log D (p = 0.633) properties.
Intrinsic Aqueous Solubility (log S)
The intrinsic aqueous solubility (log S) of an ionizable molecule is defined as its concentration in saturated aqueous solution at a particular temperature.134
Covalent Inhibitors
A comparison of log S for active and nonactive inhibitors showed that the active set had a lower mean of log S (0.58) than the nonactive set (1.24, p < 0.0001).
Noncovalent Inhibitors
In contrast, an assessment of log S values for the noncovalent inhibitors was not significantly different between active and nonactive sets (p = 0.057).
Hydrogen Bond Acceptors and Donors
HBAs and HBDs are additional significant descriptors for drug discovery that relate to the polarity and permeability of compounds.131,135 For example, it was revealed that while the properties HBAs and MW have increased considerably over time, HBDs and lipophilicity have remained rather consistent.136 These data suggest that counting HBDs may be more significant for drug development than counting HBAs, in which a higher number of HBDs can lead to very poor solubility, permeability, and bioavailability.137
Covalent Inhibitors
The active covalent inhibitor set displayed more HBAs (from 7 (10th percentile) to 10 (90th percentile) with a median HBA value of 8), compared to the nonactive counterparts (from 5 (10th percentile) to 10 (90th percentile) with a median of 7). The higher mean value of 8.26 to the active compounds was shown to be statistically significant against the nonactive (x̅ = 7.43, p = 0.0007). The covalent active and nonactive inhibitors had a minimal number of HBDs, with the median value being 0 for the active and 1 for the nonactive sets. Comparison of the HBDs for active and nonactive candidates also showed that the active set had a lower mean HBD (0.27) than the nonactive set (0.66, p < 0.0001). A more significant number of HBAs in the active set is likely attributable to the enthalpic aspect of the binding process, in which H-bonding plays a crucial role in aligning the molecule/warhead to facilitate interaction with the active nucleophile site. Regarding the effect of HBDs leading to molecules with poor solubility, permeability, and bioavailability, the observation of a reduced number of HBDs in active compounds can be explained as the avoidance of a self-reaction of the molecules with their covalent warhead and the corresponding hydrogen-bond donor (e.g., −OH/NH/SH groups).
Noncovalent Inhibitors
The noncovalent active set displayed higher values of HBAs, from 5 (10th percentile) to 9 (90th percentile) with a median HBA value of 7, compared to the nonactive set (5, 10th percentile; 8, 90th percentile; 6, median). The higher mean value of 7.25 for the active compounds was shown to be statistically significant against the nonactive (x̅ = 6.23, p < 0.0001). Unlike the case for the covalent binders, the noncovalent inhibitors showed a higher number of HBDs, with a median value of 2 for the active set and 1 for the nonactive. A comparison of the HBD for active and nonactive sets also showed that the active set had a higher mean HBD (x̅ = 1.69) than the nonactive set (x̅ = 1.24, p < 0.0001). This result is expected, given that the H-bonding potential via HBD or HBA would be greater in noncovalent analogues for active versus inactive. The analysis shows that increasing HBA/HBD for noncovalent inhibitors can be a strategy to increase potency by increasing a stronger binding via H-bonding on the binding site rather than increasing lipophilicity (C log P values were found to be not statistically significant in the noncovalent set).
Topological Surface Area
The topological surface area (TPSA) is another descriptor of importance in permeability and oral bioavailability estimates connected to hydrogen bonding (N and O atom count).138
Covalent Inhibitors
TPSA values ranged from about 82.3 Å2 (10th percentile) to 137.7 Å2 (90th percentile), with a median value of 100.6 Å2 for the actives set. Nonactives are left-shifted to a lower value of TPSA, with values varying from 60.2 Å2 (10th percentile) to 138.0 Å2 (90th percentile) and a median TPSA value of 94.3 Å2. A comparison of the TPSA for active and nonactive candidates also showed a higher mean TPSA value of 104.4 Å2 for the active set, compared to the nonactive set (97.1 Å2, p = 0.027).
Noncovalent Inhibitors
TPSA values varied from about 61.8 Å2 (10th percentile) to 130.7 Å2 (90th percentile) with a median value of 85.4 Å2 for the active set. Nonactives are left-shifted to a lower value of TPSA, with values varying from 50.5 Å2 (10th percentile) to 112.6 Å2 (90th percentile), with a median TPSA value of 79.4 Å2. Comparison of the TPSA for active and nonactive candidates also showed that the active set had a higher mean TPSA value (93.0 Å2) than the nonactive set (79.4 Å2, p < 0.0001). Noncovalent binders have lower TPSA values than covalent inhibitors, both active and inactive. This result is likely due to the existing electrophilic warhead in the covalent binders (acrylamide or nitro), which increases this PC descriptor. Similarly demonstrated with HBAs and HBDs, we can observe the H-bonding role in affecting the potency of the different types of inhibitors.
Flexibility Index
The flexibility index (FInd) is described as the ratio of rotatable bonds to total bonds. No statistically significant difference was observed between the active and nonactive sets, for the covalent inhibitors (p = 0.34). The noncovalent active set displayed values of FInd from 0.12 (10th percentile) to 0.30 (90th percentile) with a median FInd value of 0.19, compared to the nonactive set (0.14, 10th percentile; 0.23, 90th percentile; 0.19, median). The higher mean value of 0.20 for the active compounds was shown to be statistically significant against the nonactives (x̅ = 0.19, p = 0.0002).
Number of Rotatable Bonds
Similarly, ROTBS for the covalent inhibitors were not significantly different between active and nonactive sets (p = 0.13), even though the means were quite similar between inhibitors (x̅ = 6.02) and noninhibitors (x̅ = 5.65). In contrast, for the noncovalent inhibitors the means for the ROTBS were statistically significant (p < 0.0001), with values of 6.62 for the active vs 5.56 for the nonactive sets. ROTBS values for most of the inhibitor set varied from 4 (10th percentile) to 10 (90th percentile) with a median ROTBS value of 6.
The analysis described above indicates that, for inactive covalent DprE1 inhibitors, it may be necessary to optimize the compounds by increasing MW, C log P, C log D, HBA, and TPSA while reducing log S and HBD, to match more closely the active set’s corresponding properties. Concerning reducing HBD, the presence of a hydrogen bond donor in a core with a chemically reactive warhead could lead to drug instability through self-reactivity (though less likely for in situ bioreductively activated warheads such as nitro heterocycles); therefore, this needs to be considered in line with the analysis. The inactive noncovalent DprE1 inhibitors indicate that compound optimization may benefit from increasing MW, HBA, HBD, TPSA, FInd, and ROTBS. This step change in properties will drive the enthalpic component of binding by enhancement of hydrogen bonding and enhancing the ligand conformation for optimal fit.
Impact of Physicochemical Properties of DprE1 inhibitors on Oral Absorption
Lipinski’s Rule of 5 (Ro5) indicates that if a molecule meets the criteria log P ≤ 5, MW ≤ 500 Da, HBAs (O + N atom count) ≤ 10 and HBDs (OH + NH count) ≤ 5, the compound is more likely to have membrane permeability and hence be more readily absorbed in the human digestive system via passive diffusion.121,139 These set limits were chosen to cover around 90% of the range for the four estimated PC descriptors, and the Ro5 is compromised when two or more criteria are exceeded.121,139 Our analysis reveals that 65.1% of active DprE1 inhibitors (n = 674/1035, MIC < 10 μM) do not show violations of the Ro5. Among the covalent binders, only 54.8% (381/695) fall inside the chemical space of Ro5, while a larger proportion is seen among the noncovalent binders [86.2% (293/340)] (Figure 6B). Within the covalent data set, NBTO, NQ, and NTZ score 100% for no Ro5 violations, while DNB and BTZ score 80.4% and 48.2%, respectively. The noncovalent data set proved more diverse, with 4-AQ, AZA, BI, BD, HYD, NAHPC, and PTD showing no violations (100%), and BTO (96.3%) > MP (96.2%) > CD (88.6%) > TPA (88.1%) > NMDS (80.0%) > PP (77.8%). 2-CQ and NC BTZ were the classes with a lower score of no violation (37.5, 27.3%) (Figure 6A). If we analyze both binding subsets which scored with one violation (25.6%, 265/1035), among covalent (33.4%, 232/695) and noncovalent (9.7%, 33/340) inhibitors, the classes of the covalent category were BTZ (38.6%) > DNB (12.8%) and those of the noncovalent were 2-CQ (62.5%) > NC BTZ (54.5%) > 2-S-BTO (33.3%) > PP (22.2%) > NMDS (20%) > CD (11.4%) > TPA (10.4%) > MP (3.8%) and BTO (3.7%) (Figure 6A). The main descriptor involved in Ro5 violations is MW, with a prevalence of 55.4% for the covalent and 42.6% for the noncovalent binders (Figure 6C). While analyzing the two subsets that scored at least two violations, we obtained for the covalent 11.8% (82/695) and for noncovalent 4.1% (14/340) (Figure 6B). The classes for the covalent inhibitors were BTZ (13.2%) > DNB (6.9%) and for the noncovalent counterparts AVMT (100%) > 2-S-BTO (66.7%) > NC BTZ (18.2%) and TPA (1.5%), respectively (Figure 6A). The most frequently used pair of PC descriptors in two Ro5 violations is MW–C log P for the covalent binders, with a frequency of 15.9%, and MW–HBA for noncovalent binders, 21.3%. The set MW-ClogP-HBA was found to be the most frequently violated for the compounds with three violations, with a score of 1.0% for the covalent binders (Figure 6-C). This finding was consistent with our PC descriptor analysis; nevertheless, covalent inhibitors exhibit higher molecular weight values and are more lipophilic than noncovalent binders. This property may impair oral bioavailability and should be considered during drug optimization.
Figure 6.

Lipinski’s Rule of 5 (Ro5): distribution of the number of Lipinski Ro5 violations for (A) each class of DprE1 inhibitors and (B) for the covalent and noncovalent category binders. (C) Ro5 violation types. (D) Physicochemical property space of covalent and noncovalent DprE1 inhibitors, with C log P as a function of MW.
Mapping bRo5 and eRo5 Space
Lipinski’s rules delineate chemical space with compounds that “are more likely to be orally absorbed” as one way to describe chemical space, whereas “possible to be orally absorbed” is regarding the extended Ro5 (eRo5) and beyond Ro5 (bRo5). Understanding the precise boundaries of this chemical space will increase the likelihood of creating cell-permeable and orally accessible ligands for more difficult targets.131,140,141 Of the 1035 active compounds in this study, a large proportion (23.9%, n = 247/1035) cluster into what can be considered as an extension of Ro5 space (0 ≤ log P ≤ 7.5; 500 Da < MW ≤ 700 Da; HBDs (OH + NH count) ≤ 5; TPSA ≤ 200 Å2; ROTBS ≤ 20) and a natural tail of the distribution of compounds is based on Ro5 properties. Among these, 32.1% (223/695) are from the covalent class while only 7.1% (24/340) are from the noncovalent class. For the covalent and noncovalent inhibitors, only 4.7% (33/695) and 2.9% (10/340), respectively, were observed in oral bRo5 space (0 < log P or >7.5; 700 Da < MW ≤ 3000 Da; HBDs (OH + NH count) > 5; TPSA > 200 Å2; ROTBS > 20). A small proportion of DprE1 inhibitors, 6.9% (71/1035), did not fall into any Ro5 chemical space, with the highest proportion, 8.3% (58/695), for covalent and 3.8% (13/340) for the noncovalent binders (Figure 6-D).
Distribution
The term “drug distribution” refers to how a substance is distributed across the body’s compartments. Certain factors, such as penetration through the central nervous system (CNS) or BBB, P-gp efflux, and PPB, can be adequately studied in silico. Additionally, since only the unbound (free) drug can interact with the target protein, the interaction of the drug with plasma proteins must be evaluated throughout the drug development process.142
Central Nervous System Penetration
For therapeutic CNS targets, good penetration is an essential requirement, but for non-CNS targets, the BBB penetration rate should be minimized to reduce potential neurotoxicity or adverse pharmacological events.143 StarDrop software uses the random forest classification model to classify if a molecule is a crossing or noncrossing of the BBB, while being one that employs descriptors compatible with the common fact that neutral molecules tend to penetrate the CNS more effectively than charged compounds and that cations normally permeate the CNS more effectively than anions.120,124,125 The predictive accuracy of BBB+ ranges from 80% to 100%, while that of BBB– ranges from 65% to 87%.120,124,125 Close to ∼99% of the total DprE1 inhibitors were predicted not to penetrate the CNS, with 100% within the covalent set and 96.8% for the noncovalent binders. Only 3.2% of the noncovalent inhibitors were found to have some BBB penetration, respectively NC BTZ 27.3% (3/11) > NHMS 20% (1/5) > MP 7.7% (2/26) > CD 6.8% (3/44) > AZA 5.3% (2/38) (Figure 7A).
Figure 7.

Analysis of some properties related to distribution: (A) Blood–brain barrier (BBB) penetration classification; (B) P-gp inhibitor classification; (C) plasma protein binding (PPB) classification.
P-gp Efflux System
P-gp is one of the most widely studied drug transporters to date, given the evidence of its presence in the majority of cells, including those of the intestinal mucosa and the BBB.122 We used the statistical model built-in to StarDrop v7.2.0.32905 to predict which DprE1 inhibitors could behave as P-gp substrates. It employs a random forest classification approach to classify compounds as probable or unlikely to be P-gp substrates. The model’s performance was evaluated on an independent test set of 51 chemicals, with 82% of nonsubstrates and 79% of substrates accurately categorized.124 A higher frequency of P-gp binders is predicted among the covalent inhibitors (80.4%), compared to the noncovalent class (65%). For the covalent inhibitor set, predictions point to P-gp substrates among the BTZ (88%) > NBTO (50%) > DNB (48%) and no P-gp substrates for the NTZ, NQ data sets (0%). For the noncovalent inhibitors set, predictions identify P-gp substrates among the AVMT, NC BTZ, PP, PTD (100%) > MP (92.3%) > 4-AQ (91.7%) > CD (90.9%) > 2-S-BTO (86.7%) > TPA (85.1%) > BTO (63%) > 2-CQ (50%) > BI (44.4%) > NHMS (40%) > AZA (23.7%) > NAHPC (11.1%) > HYD (7.9%) and no P-gp substrates for the BD (0%) (Figure 7B).
Plasma Protein Binding
The extent to which a drug binds to plasma proteins substantially affects its pharmacokinetic and pharmacodynamic effects. The drug’s efficacy will be proportional to the quantity of unbound drug in plasma. Additionally, the bound drug in plasma can operate as a reservoir for free drug clearance via various elimination pathways, lengthening the duration of action.144,145 From a QSAR model integrated into StarDrop v7.2.0.32905, Figure 7C categorizes and forecasts human PPB% (Hu PPB%) values for both covalent and noncovalent data sets. The model is a random forest that classifies the extent of plasma protein binding of test set substances as either “high” or “low” about the threshold above. Low-binding molecules are those that are less than 90% bound, and high-binding molecules are those that are more than 90% bound.120,124,125 It can be observed that both types of inhibitors display high binding capacity, with the noncovalent inhibitors scoring around 84.1% while the covalent inhibitors scored around 77.1%. For the covalent set, all inhibitors belonging to the NQ class displayed a high binding capacity (100%), this high tendency holding for DNB and BTZ classes (80.4% and 78.8%), while NBTO scores only around 18.2% and NTZ is predicted to have an irrelevant protein binding capacity (0%). For the noncovalent inhibitors, nine classes (2-CQ, 2-S-BTO, BD, BTO, BI, NAHPC, CD, NHMS, PP) were predicted to have a high binding capacity (100%), followed by 4-AQ (87.5%) > AZA (86.8%) > NC BTZ (81.8%) > PTD (75%) > HYD (71.1%) > TPA (67.2%) > MP (65.4%). AVMT is predicted to have an irrelevant protein binding capacity.
A comparison of the experimental PPB% values of the covalent and noncovalent binders to the StarDrop model results was performed, and the corresponding data sets are depicted in Tables 3 and 4. Regarding the covalent inhibitors, the literature examination returned a total of 21 compounds, including 16 BTZ44,146 and 5 NBTO64 molecules. The compounds with experimental data were employed to evaluate the performance metrics of the classification model from StarDrop, and the results and confusion matrix are displayed in Table 3. The obtained accuracy (ACC) rating of 81% indicates that the classification of ∼8 of every 10 molecules is correct. According to the StarDrop manual, the accuracy of the Plasma Protein Binding Classification (90%) model is 81%, which is consistent with the obtained predictions using covalent inhibitors. The precision value of 100% indicates that all the molecules predicted with high PPB% were correctly identified, and a sensitivity value of 80% reveals that 20% of the molecules with high PPB% were lost during the model application. The specificity value of 100% indicates that all molecules with low PPB% were accurately labeled.
Table 3. Experimental and Corresponding Predicted Plasma Protein Binding (PPB) Data, Confusion Matrix, and Performance Metrics Evaluating the PPB Data for Covalent Inhibitors.
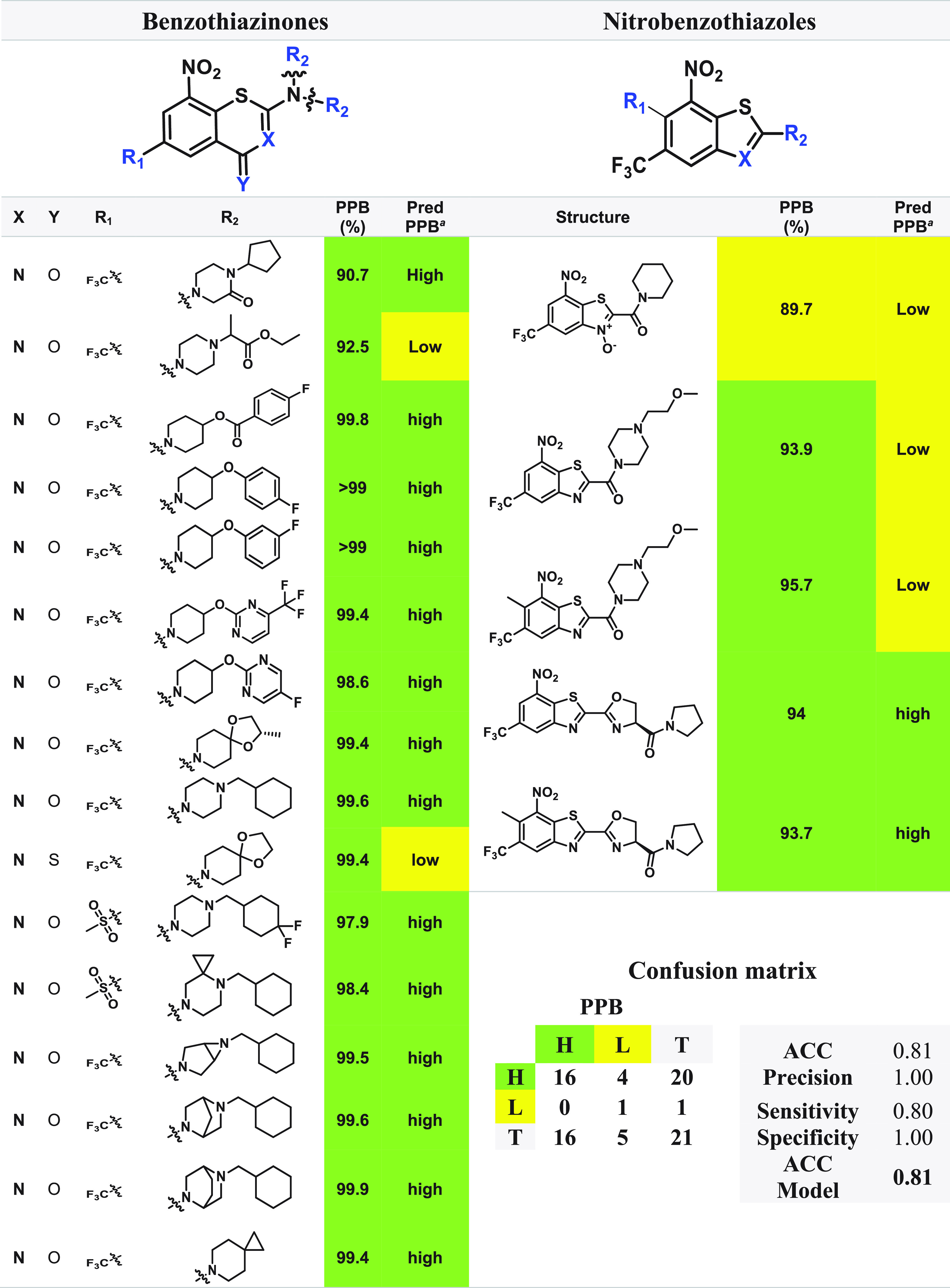
Pred PPB: predicted plasma protein binding computed by StarDrop v7.2.0.32905. Legend: H, high (in green); L, low (in yellow).
Table 4. Experimental and Corresponding Predicted PPB data, Confusion Matrix, and Performance Metrics Evaluating the PPB Data for Noncovalent Inhibitors.
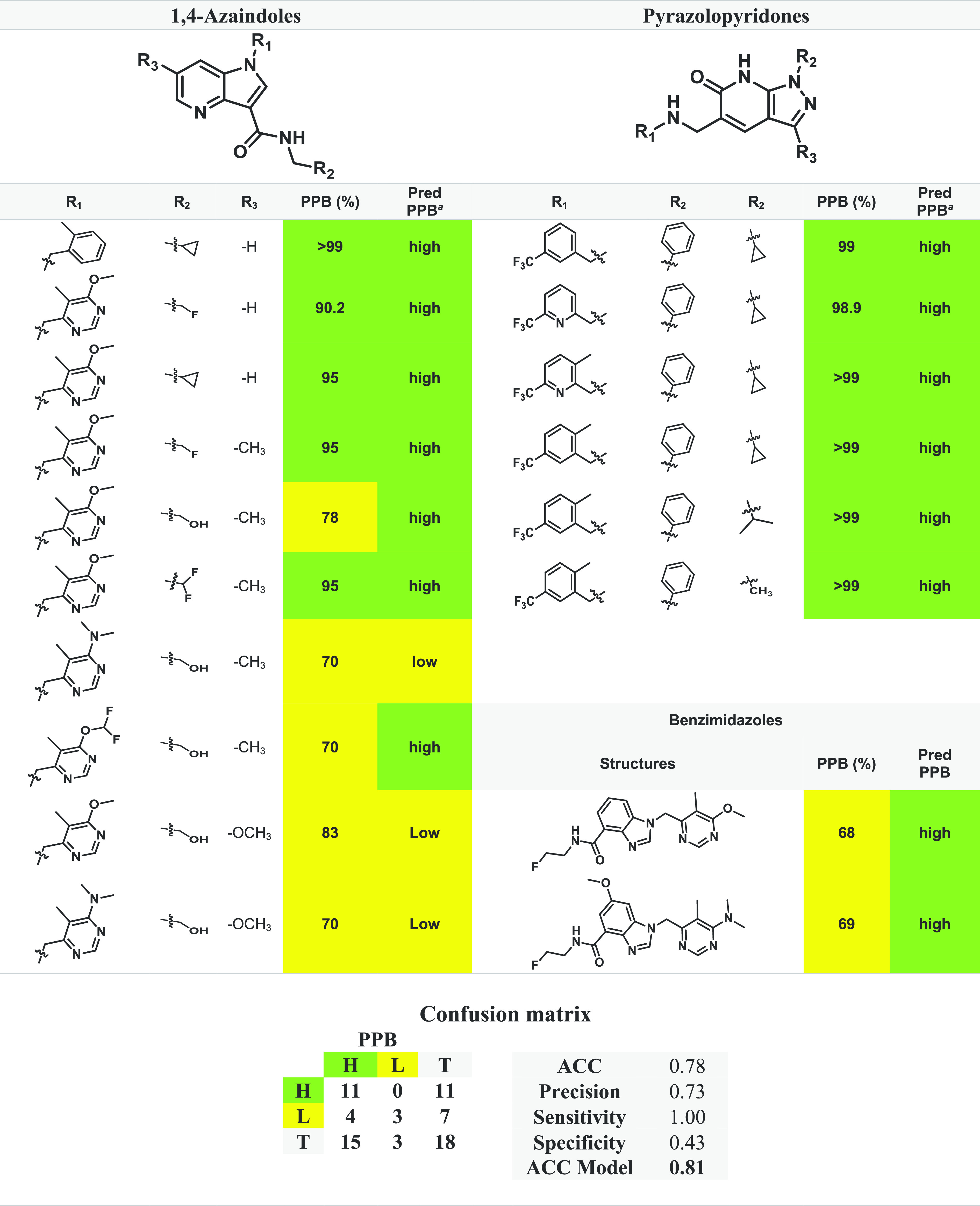
Pred PPB: predicted plasma protein binding computed by StarDrop v7.2.0.32905. Legend: H, high (in green); L, low (in yellow).
For the noncovalent inhibitors, the literature analysis provided a total of 18 compounds, including 10 AZA,78,79 6 PP,82 and 2 BI81 molecules. These compounds with experimental data were also employed to evaluate the performance metrics of the classification model from StarDrop, the results and confusion matrix being displayed in Table 4. The obtained accuracy (ACC) rating of 78% indicates that classification of ∼8 of every 10 molecules is correct, with a similar accuracy value being reported from StarDrop’s manual (81%). The precision value (73%) indicates that 73% of the molecules predicted with high PPB% were correctly identified, and a sensitivity value of 100% reveals that no false negative value was predicted. The specificity value of 43% indicates that 57% of the molecules with low PPB% were mislabeled as high PPB% (false positive).
High plasma protein binding restricts the distribution of xenobiotics from the blood to tissues, affecting their metabolism, also holding a significant role in drug–drug interactions. Therefore, a reasonable predictive model with high sensitivity is required to avoid losing high PPB molecules during prediction and a high precision to prevent excess false positive results. Both model analyses for the experimental data of each class were shown to have a high sensitivity (Cov = 0.80 and Ncov = 1) together with high precision (Cov = 1 and Ncov = 0.73). Even though the sample size is relatively small in both testing sets (N(Cov) = 21 and N(Ncov) = 18), this study using experimental data reveals that the StarDrop model can reasonably predict plasma protein binding for both covalent and noncovalent binders and can be fairly reliable to assist with the development of DprE1 inhibitors.
To summarize this section, our computed analyses, along with literature values, indicate that both covalent and noncovalent DprE1 inhibitors are projected to be nonpermeable to the blood–brain barrier and to have a moderate to high plasma protein binding affinity. Covalent DprE1 inhibitors are more likely to be possible substrates (80.4%) for P-gp substrate transporters than their noncovalent counterparts (65%).
Cytochromes P450 Metabolism
Six cytochromes P450 (s) alleles are particularly important in drug metabolism: CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4. They catalyze the oxidative metabolism of about 90% of human drugs and are the main determinants of the systemic clearance and bioavailability of these molecules.147,148 To evaluate which compounds in our data set might be CYP binders, we used the StarDrop WhichP450 module. Predictions of CYP isoform metabolism for each class of inhibitors are displayed in Figure 8. Calculations were conducted to determine the drug’s mean probability of being metabolized by related isoforms, indicating that it could be a candidate substrate. The computed predictions suggest that the DprE1 inhibitors would be metabolized mainly by the 3A4 isoform, with mean values of 54.4% and 47.6% of metabolism prediction for covalent and noncovalent binders, followed by the isoforms 2D6, 2C19, 2C9 (13.21%, 9.53%, and 8.86%) for the covalent inhibitors and 2C9, 2D6, 2C19 (15.09%, 12.16%, and 11.61%) for the noncovalent inhibitors. This set of molecules reveals a low proportion of metabolism from the 1A2, 2C8, and 2E1 isoforms (6.84%, 6.28%, and 0.83% for covalent inhibitors and 7.16%, 5.72%, and 0.66% for noncovalent inhibitors; Figure 8A). Regarding the corresponding moiety class for each isoform, AVMT was found to be the class with highest prediction to be metabolized by 3A4 isoform (83.0%) and BD the lowest (34.3%); NQ scored the highest prediction to be metabolized by the 1A2 isoform (19.67%) and AVMT the lowest (2.2%). CD showed the highest metabolism prediction (8.03%) for the 2C8 isoform and NQ the lowest (0.93%). HYD was the class with the highest probability to be metabolized by the 2C9 isoform (28.9%) and AVMT the lowest (3.0%). Regarding the 2C19 isoform, the HYD class was predicted to have the highest probability (15.7%) and AVMT the lowest (3.6%). For the 2D6 isoform, PP displayed the highest value (25.3%) and AVMT the lowest (1.4%). For the 2E1 isoform, NQ had the highest metabolism prediction (2.53%) and BI the lowest (0.13%, Figure 8B). These findings should be interpreted cautiously, as building appropriate prediction models is challenging due to the complicated chemical mechanisms underlying CYP metabolism,142 but they allow for a broad comparison of the various classes of compounds. These predictions contribute to our understanding of the role of the CYP superfamily in the metabolic stability of DprE1 inhibitors.
Figure 8.

CYP isoform metabolism for (A) all covalent and noncovalent inhibitors and (B) each corresponding class of the covalent and noncovalent inhibitors.
Safety Profile
In addition to bioavailability, the safety profile is important, since it details the harmful consequences associated with the chemical substances under study. The analysis of log P versus MW, GSK’s 4/400 rule (log P ≤ 4 and MW ≤ 400 Da)122 for the evaluation of ADMET liabilities, shows that 17.1% (n = 177/1035) of the active inhibitors fall in the more desirable category, with 15.7% for the covalent (n = 109/695) and 20.0% (n = 68/340) for noncovalent inhibitors (Figure 9A1). The prevalence of adverse toxicological outcomes can be assessed using Pfizer’s 3/75 rule,123 where log P > 3 and TPSA < 75 Å2 are related to the adverse effect of chemical compounds. Application to our data set reveals that 2.4% (n = 17/695) of the covalent and 22.9% (n = 78/340) of the noncovalent binders do not comply with the Pfizer 3/75 rule, meaning that they may exhibit increased toxicity (Figure 9A2). It is worth highlighting that the Pfizer 3/75 rule does not take into consideration the possible presence of mutagenic functional groups. For instance, nitro groups are often present in DprE1 covalent inhibitors, although drugs containing nitro groups have been linked to mutagenicity and genotoxicity.149 It is worth noting that various reports indicate that nitro-containing DprE1 inhibitors exhibit favorable metabolic, microsomal, and plasma stability and reduced toxicity.37
Figure 9.

(A1) MW as a function of C log P. The yellow area indicates conformity by GSK’s 4/400 Rule. (A2) TPSA as a function of C log P. The red area indicates the molecules that failed on Pfizer’s 3/75 Rule. (B) hERG inhibition. (C) AMES mutagenicity category.
hERG Inhibition
Throughout the drug development process, one of the most common undesirable side effects that contributes to a medicine’s failure is cardiac arrhythmias.150 Numerous forms of cardiovascular toxicity must be taken into account, as the promiscuous blocking of hERG cardiac potassium channels by small molecules poses a significant therapeutic challenge, with severe consequences for human health.151−153
The model implemented in StarDrop v7.2.0.32905 predicts that covalent DprE1 inhibitors exhibit the highest potential for hERG inhibition, with a mean pIC50 value of 6.16, with values ranging from 5.01 (10th percentile) to 7.21 (90th percentile) for most drugs, while the noncovalent binders varied from 4.28 (10th percentile) to 6.30 (90th percentile), with a lower mean of 5.26. In general, an experimental binding assay is indicated if the score is larger than 5, since the molecules are likely to display some toxicity linked to these cardiac potassium channels.151−153 We present a categorization histogram (Figure 9B) where it is shown that 90.4% of the covalent and 55.0% for the noncovalent inhibitors have pIC50 > 5. Both of these subsets also scored 58.7% (Cov) and 25.0% (NCov) of compounds with pIC50 > 6. Within the covalent inhibitors, 99.3% of the benzothiazinones have a pIC50 > 5, with 70.7% for pIC50 > 6 and 28.6% between 5 and 6. hERG inhibitions in BTZ have been observed previously, and further optimizations through SAR studies of three moieties (benzene ring, linker, and N-heterocycle) on the C-2 side chain of the BTZ scalffold have been performed, allowing identification of new lead compounds with reduced hERG liability (inhibition rate (IR) < 50% at 10 μM) without sacrificing antimycobacterial potency.49,54 The DNB class showed a smaller proportion for a predicted pIC50 > 6 (6.9%). A pIC50 of 5–6 is predicted for 100% of the inhibitors in both NQ and NTZ classes, followed by 68.2% for NBTO and 38.2% for DNB. Values of pIC50 below 5 are predicted for DNB (54.9%) > NBTO (31.8%) > BTZ (0.8%) inhibitors. Among the covalent binders, the NBTO class emerges as the one with lower predicted hERG inhibition. Predictions within the noncovalent inhibitors are pIC50 > 6, NC BTZ (81.8%) > 2-S-BTO (80%) > PTD (75%) > MP (65.4%) > CD (59.1%) > PP (55.6%) > 4-AQ (50%) > NHMS (20%). For pIC50 values in the range 5–6: AVMT, BI (100%) > BD (80%) > NHMS (60%) > AZA (57.9%) > 4-AQ (50%) > PP (44.4%) > CD (40.9%) > 2-CQ (37.5%) > MP (34.6%) > PTD (25%) > NAHPC (22.2%) > 2-S-BTO (20%) > NC BTZ (18.2%) > TPA (9.0%) > HYD (5.3%) > BTO (3.7%). pIC50 values below 5 are predicted for BTO (96.3%) > HYD (94.7%) > TPA (91.0%) > NAHPC (77.8%) > 2-CQ (62.5%) > AZA (42.1%) > BD, NHMS (20%).
1,4-Azaindoles scored 57.9% for pIC50 in the range 5–6 and 42.1% for pIC50 < 5. Reported hERG assays for this scaffold have shown no inhibition of the hERG channel at up to 33 μM (pIC50 < 4.48) concentrations,77,79 displaying a calculated absolute error (x̅ ± σ) of 0.40 ± 0.11 log IC50 comparing to the predicted values (Table 5). Although the hydantoin heterocycle is linked to potential cardiotoxicity,90,91 predictions for this scaffold point to 5.3% with pIC50 between 5 and 6 and 94.7% with pIC50 < 5. The calculated absolute error (x̅ ± σ) for the HYD class between the experimental and predicted data was in the range of 0.26 ± 0.14 log IC50, for the HYD compounds (Table 5). Thiophene-arylamide compounds showed a high proportion of predicted pIC50 < 5 (91.0%), which is in keeping with literature reports. In contrast, selected TPA compounds exhibited low inhibition profiles of the hERG channel (IC50 > 20 μM (pIC50 < 4.70)) across the series, indicating a low risk of blocking the cardiac potassium channel and causing QT prolongation.98 The calculated absolute error (x̅ ± σ) between the experimental and predicted data was in the range of 0.26 ± 0.14 log IC50, for the TPA compounds (Table 5). Predictions for the BI series placed 100% of the compounds within a pIC50 range of 5–6, though hERG channel assays indicated no major safety liabilities, with values of IC50 > 33 μM (pIC50 < 4.48).81 The calculated absolute error (x̅ ± σ) between the experimental and predicted data was in the range of 0.80 ± 0.11 log IC50, for the BI class, displaying the highest relative error (15.1 ± 1.8%) while using the predictive model (Table 5). For the benzothiazole group, an hERG assay showed that TCA1 has no activity at IC50 > 30 μM (pIC50 < 4.52),72 in keeping with the prediction of 96.3% for BTO, for pIC50 values below 5. For an evaluation of the prediction model between the noncovalent binders, it shows that it seems to vary between the different scaffolds, with the HYD class having the best predicted values and BI the poorest.
Table 5. Experimental and Corresponding Predicted hERG pIC50 Data of the Noncovalent Inhibitors.

Pred pIC50: predicted hERG pIC50 computed by StarDrop v7.2.0.32905.
AMES Mutagenicity
The AMES test is a biological assay used to determine the mutagenic potential of a chemical compound,154,155 which entails the activation of promutagens via mammalian metabolism.156,157 AMES mutagenicity predictions were computed using StarDrop modules’ toxicity models. The results yielded a high score of 96.0% (667/695) for covalent inhibitors, in contrast to the noncovalent binders, with only 32.5% (109/340) with an AMES positive prediction. This high value computed for covalent inhibitors was somewhat expected, since nitro-aromatics are generally associated with mutagenicity149,158—the nitro-aromatic moiety is a common motif in covalent DprE1 inhibitors. The results revealed predictions of AMES mutagenicity for NBTO and NQ (100%), followed by BTZ (98.8%) > DNB (80.4%) and, last, of the nonmutagenic nature of NTZ (0%). All tested experimental NBTOs were found to be AMES positive,65 agreeing with the predictions. This was rectified by the addition of a methyl group, adjacent to the nitro group of the NBTOs, affording the crowded benzothiazoles (cBTs), which tested AMES negative. Although experimental work has shown no indication of mutagenic or nitrosactive gene expression profiles following treatment with BTZ043, chemical proteomics showed evidence for induction of 60 genes, which was expected, as BTZs specifically target cell wall biogenesis. Therefore, concerns on the mutagenicity of the nitro group proved unfounded.69 The AMES test demonstrated that the DNPT did not generate mutations in S. typhimurium TA98 and TA100 strains, even with metabolic activation.101 For the noncovalent data set, it scored for mutagenicity of 34.9%, with 2-S-BTO (100%) > HYD (76.3%) > MP (69.2%) > BD (60%) > BI (55.6%) > PTD (50%) > NC BTZ (36.4%) > AZA (31.6%) > PP (22.2%) > BTO (18.5%) > CD (15.9%) > TPA (10.4%) > 4-AQ, AVMT, NAHPC (0%) (Figure 9C).
PAINS and Structural Alerts
Substructural warnings have become a common feature of the triage process in biological screening campaigns to identify pan-assay interference compounds (PAINS). PAINS generate false-positive assay responses as a result of their reactivity under assay circumstances,159 which may include covalent modification, metal chelation, autofluorescence, aggregation, and redox reactivity, among others.160−163 Certain structural motifs (“structural alerts”) may result in covalent alteration of proteins or DNA, inducing negative effects (hepatotoxicity, CYP inhibition, in vitro genotoxicity, carcinogenicity).160−163 We screened our data set for PAINS count with StarDrop that embeds the original PAINS definitions, and we show that only 7.5% (52/695) of the covalent inhibitors scored for detected PAINS, with DNB having the highest percentage (29.4%), followed by the BTZ class (3.9%). The identified structural alerts (SA) for the covalent subset were Anil_Di_alk_E (3.45%) > catechol (2.01%) > hydroquinone (1.01%) > Anil_Di_alk_C (0.86%) > aminothiazole (0.43%) > benzodioxane, Azo_A (0.14%). The noncovalent set scored a higher proportion than the covalent set for PAINS, with 13.4% (45/340). The classes that contained SA were 2-S-BTO, BD (100%) > BTO (74.1%) > PTD (25%) > NC BTZ (9.1%) > CD (4.5%) > HYD (2.6%). Aminothiazole (11.48%) was the most frequent SA detected, followed by catechol (3.29%) and some residual Anil_Di_alk_E and hydroquinone (0.60%), together with anil_Di_alk_C, azide and benzodioxane (0.30%) (Figure 10). It is important to emphasize that, as for PC descriptors, PAINS substructure searches must be used cautiously when picking candidates, as there have been numerous observed deviations to these principles.
Figure 10.

Matrix plot of structural alerts computed by ChemBioServer 2.0.
Conclusions
DprE1 has been established as a potential therapeutic target for inhibiting mycobacterial cell wall biosynthesis, in which this enzyme is a highly druggable target against M. tuberculosis, and various chemical scaffolds have been developed since its discovery. Twenty-three distinct scaffolds have been found to exhibit a high affinity for this enzyme, with varying antimycobacterial activity and DMPK profiles, and these inhibitors are divided into covalent and noncovalent binders.
The design of DprE1 inhibitors can be challenging; therefore, prediction of PC descriptors and ADMET properties for these molecules may aid in the design of new lead compounds. An extensive PC descriptor analysis indicates that for inactive covalent DprE1 it may be necessary to optimize the compounds by increasing MW, C log P, C log D, HBA and TPSA, while reducing log S and HBD to match the active set’s corresponding properties more closely. In contrast, for inactive noncovalent DprE1 inhibitors it may be required to optimize the compounds by increasing MW, HBA, HBD, TPSA, FInd, and ROTBS. All these changes are likely to enhance the enthalpic component of drug binding through enhanced hydrogen-bonding contacts with the enzyme. Covalent DprE1 inhibitors tend to violate the Ro5 more frequently than the noncovalent counterparts. However, only a small proportion fails the criteria of two or more violations, indicating that the DprE1 inhibitors are more likely to have membrane permeability and hence be more readily absorbed in the human digestive system via passive diffusion. Almost all DprE1 inhibitors were predicted to have no CNS penetration, with the entire covalent subgroup scoring no CNS penetration and a residual value for noncovalent binders, reducing the possibility of side effects on the CNS. On the other hand, DprE1 inhibitors, particularly covalent binders, may act as P-gp substrates, which must be closely evaluated during drug optimization.
CYP3A4 was the major predicted isoform to metabolize DprE1 inhibitors, followed by the isoforms 2D6 > 2C19 > 2C9 for the covalent inhibitors and 2C9 > 2D6 > 2C19 for the noncovalent inhibitors. These predictions contribute to our understanding of the role of the CYP superfamily in the metabolic stability of DprE1 inhibitors.
Toxicity end points were also examined, and the cardiovascular toxicity of the DprE1 inhibitors via hERG inhibition was observed to be higher in the covalent than in the noncovalent subset, this observation holding for a cardiotoxicity investigation. Experimental data show that optimizations can be made to improve this feature, as seen in the case of the hydantoin class. It is worth noting that other data with BTZ and TPA have shown no inhibition of the hERG potassium channel. Covalent inhibitors have scored in a higher proportion for mutagenic warnings than the noncovalent binders. This computed high value was expected, since nitro-aromatic molecules are known to be mutagenic. In terms of undesirable structural motifs (structural alerts and PAINS), DprE1 inhibitors have a small number of these substructures, with the noncovalent set scoring higher for PAINS than the covalent set.
In conclusion, several molecular properties that should facilitate the design and optimization of future DprE1 inhibitors were described, allowing for the development of novel compounds targeting M. tuberculosis. As a mere aside, we wish to emphasize that our study comparing predicted and experimental values reveal that software tools employed to predict specific DMPK parameters must be used with caution while optimizing a drug class.
Acknowledgments
P.S.M.A. and M.L.S.C. acknowledge Fundação para a Ciência e a Tecnologia (FCT)–Portugal for Grants SFRH/BD/130407/2017 and COVID/BD/152392/2022 and projects UIDB/04326/2020, UIDP/04326/2020 and LA/P/0101/2020 (CCMAR).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c05307.
Molecular formula strings (SMILES) and extensive data for 1519 DprE1 compounds in 57 variables (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Pai M.; Behr M. A.; Dowdy D.; Dheda K.; Divangahi M.; Boehme C. C.; Ginsberg A.; Swaminathan S.; Spigelman M.; Getahun H.; Menzies D.; Raviglione M. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 1–23. 10.1038/nrdp.2016.76. [DOI] [PubMed] [Google Scholar]
- WHO . Global Tuberculosis Report, 2021.
- Lechartier B.; Rybniker J.; Zumla A.; Cole S. T. Tuberculosis Drug Discovery in the Post-Post-Genomic Era. EMBO Mol. Med. 2014, 6, 158–168. 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A.; Nahid P.; Cole S. T. Advances in the Development of New Tuberculosis Drugs and Treatment Regimens. Nat. Rev. Drug Discovery 2013, 12, 388–404. 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Av-Gay Y. New Era of TB Drug Discovery and Its Impact on Disease Management. Curr. Treat. Options Infect. Dis. 2016, 8, 299–310. 10.1007/s40506-016-0098-0. [DOI] [Google Scholar]
- Horsburgh C. R.; Barry C. E. III; Lange C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- Conradie F.; Diacon A. H.; Ngubane N.; Howell P.; Everitt D.; Crook A. M.; Mendel C. M.; Egizi E.; Moreira J.; Timm J.; McHugh T. D.; Wills G. H.; Bateson A.; Hunt R.; Van Niekerk C.; Li M.; Olugbosi M.; Spigelman M. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. 10.1056/NEJMoa1901814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser K. J.; Rubin E. J. How Sisters Grow Apart: Mycobacterial Growth and Division. Nat. Rev. Microbiol. 2014, 12, 550–562. 10.1038/nrmicro3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi G.; Pasca M. R.; Chiarelli L. R.; Manina G.; Mattevi A.; Binda C. The DprE1 Enzyme, One of the Most Vulnerable Targets of Mycobacterium Tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. 10.1007/s00253-013-5218-x. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Kilburn J. Inhibition of Synthesis of Arabinogalactan by Ethambutol in Mycobacterium Smegmatis. Antimicrob. Agents Chemother. 1989, 33, 1493–1499. 10.1128/AAC.33.9.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quemard A.; Lacave C.; Laneelle G. Isoniazid Inhibition of Mycolic Acid Synthesis by Cell Extracts of Sensitive and Resistant Strains of Mycobacterium Aurum. Antimicrob. Agents Chemother. 1991, 35, 1035–1039. 10.1128/AAC.35.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchèze C.; Wang F.; Arai M.; Hazbón M. H.; Colangeli R.; Kremer L.; Weisbrod T. R.; Alland D.; Sacchettini J. C.; Jacobs W. R. Jr. Transfer of a Point Mutation in Mycobacterium Tuberculosis InhA Resolves the Target of Isoniazid. Nat. Med. 2006, 12, 1027–1029. 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- Yuan T.; Sampson N. S. Hit Generation in TB Drug Discovery: From Genome to Granuloma. Chem. Rev. 2018, 118, 1887–1916. 10.1021/acs.chemrev.7b00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikušová K.; Huang H.; Yagi T.; Holsters M.; Vereecke D.; D’Haeze W.; Scherman M. S.; Brennan P. J.; McNeil M. R.; Crick D. C. Decaprenylphosphoryl Arabinofuranose, the Donor of the D-Arabinofuranosyl Residues of Mycobacterial Arabinan, Is Formed via a Two-Step Epimerization of Decaprenylphosphoryl Ribose. J. Bacteriol. 2005, 187, 8020–8025. 10.1128/JB.187.23.8020-8025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolucka B. A. Biosynthesis of D-Arabinose in Mycobacteria - a Novel Bacterial Pathway with Implications for Antimycobacterial Therapy. FEBS J. 2008, 275, 2691–2711. 10.1111/j.1742-4658.2008.06395.x. [DOI] [PubMed] [Google Scholar]
- Lee B. S.; Pethe K. Therapeutic Potential of Promiscuous Targets in Mycobacterium Tuberculosis. Curr. Opin. Pharmacol. 2018, 42, 22–26. 10.1016/j.coph.2018.06.006. [DOI] [PubMed] [Google Scholar]
- Crellin P. K.; Brammananth R.; Coppel R. L. Decaprenylphosphoryl-β-D-Ribose 2’-Epimerase, the Target of Benzothiazinones and Dinitrobenzamides, Is an Essential Enzyme in Mycobacterium Smegmatis. PLoS One 2011, 6, e16869 10.1371/journal.pone.0016869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolly G. S.; Boldrin F.; Sala C.; Dhar N.; Hartkoorn R. C.; Ventura M.; Serafini A.; Mckinney J. D.; Manganelli R.; Cole S. T. Assessing the Essentiality of the Decaprenyl-Phospho-D-Arabinofuranose Pathway in Mycobacterium Tuberculosis Using Conditional Mutants. Mol. Microbiol. 2014, 92, 194–211. 10.1111/mmi.12546. [DOI] [PubMed] [Google Scholar]
- Brecik M.; Centárová I.; Mukherjee R.; Kolly G. S.; Huszár S.; Bobovska A.; Kilacskova E.; Mokośova V.; Svetlíkova Z.; Michal S.; Neres J.; Korduláková J.; Cole S. T.; Mikušová K. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- Piton J.; Foo C. S.-Y.; Cole S. T. Structural Studies of Mycobacterium Tuberculosis DprE1 Interacting with Its Inhibitors. Drug Discovery Today 2017, 22, 526–533. 10.1016/j.drudis.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Campaniço A.; Moreira R.; Lopes F. Drug Discovery in Tuberculosis. New Drug Targets and Antimycobacterial Agents. Eur. J. Med. Chem. 2018, 150, 525–545. 10.1016/j.ejmech.2018.03.020. [DOI] [PubMed] [Google Scholar]
- Chikhale R. V.; Barmade M. A.; Murumkar P. R.; Yadav M. R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. 10.1021/acs.jmedchem.8b00281. [DOI] [PubMed] [Google Scholar]
- Degiacomi G.; Belardinelli J. M.; Pasca M. R.; De Rossi E.; Riccardi G.; Chiarelli L. R. Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Appl. Sci. 2020, 10, 623. 10.3390/app10020623. [DOI] [Google Scholar]
- Huszár S.; Chibale K.; Singh V. The Quest for the Holy Grail: New Antitubercular Chemical Entities, Targets and Strategies. Drug Discovery Today 2020, 25, 772–780. 10.1016/j.drudis.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A.; Rudolph I.; Möllmann U.; Voigt K.; Chung C.-W.; Singh O. M. P.; Rees M.; Mendoza-Losana A.; Bates R.; Ballell L.; Batt S.; Veerapen N.; Fütterer K.; Besra G.; Imming P.; Argyrou A. Novel Insight into the Reaction of Nitro, Nitroso and Hydroxylamino Benzothiazinones and of Benzoxacinones with Mycobacterium Tuberculosis DprE1. Sci. Rep. 2018, 8, 13473. 10.1038/s41598-018-31316-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Salina E.; Reynolds R. C.; Phyo P.; Zin K.; Ekins S. Molecule Property Analyses of Active Compounds for Mycobacterium Tuberculosis. J. Med. Chem. 2020, 63, 8917–8955. 10.1021/acs.jmedchem.9b02075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S.; Trifonov L.; Yadav V. D.; Barry C. E. III; Boshoff H. I. Tuberculosis Drug Discovery: A Decade of Hit Assessment for Defined Targets. Front. Cell. Infect. Microbiol. 2021, 11, 1–23. 10.3389/fcimb.2021.611304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton J.; Vocat A.; Lupien A.; Foo C. S.; Ryabova O.; Makarov V.; Cole S. T. Structure-Based Drug Design and Characterization of Sulfonyl- Piperazine Benzothiazinone Inhibitors of DprE1 from Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00681-18 10.1128/AAC.00681-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer C.; Rengifo-Gonzalez M.; Hinner M. J.; Schneider P.; Makarov V.; Cole S. T.; Johnsson K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-β-D-Ribose 2′-Epimerase DprE1 of Mycobacterium Tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- Trefzer C.; Skovierová H.; Buroni S.; Bobovská A.; Nenci S.; Molteni E.; Pojer F.; Pasca M. R.; Makarov V.; Cole S. T.; Riccardi G.; Mikušová K.; Johnsson K. Benzothiazinones Are Suicide Inhibitors of Mycobacterial Decaprenylphosphoryl-β-D-Ribofuranose 2′-Oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- Tiwari R.; Moraski G. C.; Krchn V.; Miller P. A.; Colon-martinez M.; Herrero E.; Oliver A. G.; Miller M. J. Thiolates Chemically Induce Redox Activation of BTZ043 and Related Potent Nitroaromatic Anti-Tuberculosis Agents. J. Am. Chem. Soc. 2013, 135, 3539–3549. 10.1021/ja311058q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Krchnak V.; Brown S. N.; Miller M. J. Deuteration of BTZ043 Extends the Lifetime of Meisenheimer Intermediates to the Antituberculosis Nitroso Oxidation State. ACS Med. Chem. Lett. 2019, 10, 1462–1466. 10.1021/acsmedchemlett.9b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Möllmann U.; Ryabova O.; Saint-joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; Rossi E. De.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; Mckinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Benzothiazinones Kill Mycobacterium Tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–805. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a New Combination Therapy for Tuberculosis with next Generation Benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt S. M.; Jabeen T.; Bhowruth V.; Quill L.; Lund P. A.; Eggeling L.; Alderwick L. J.; Fütterer K.; Besra G. S. Structural Basis of Inhibition of Mycobacterium Tuberculosis DprE1 by Benzothiazinone Inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11354–11359. 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer R.; Neres J.; Piton J.; Dhar N.; van der Sar A.; Mukherjee R.; Laroche T.; Dyson P. J.; McKinney J. D.; Bitter W.; Makarov V.; Cole S. T. Fluorescent Benzothiazinone Analogues Efficiently and Selectively Label Dpre1 in Mycobacteria and Actinobacteria. ACS Chem. Biol. 2018, 13, 3184–3192. 10.1021/acschembio.8b00790. [DOI] [PubMed] [Google Scholar]
- Karoli T.; Becker B.; Zuegg J.; Möllmann U.; Ramu S.; Huang J. X.; Cooper M. A. Identification of Antitubercular Benzothiazinone Compounds by Ligand-Based Design. J. Med. Chem. 2012, 55, 7940–7944. 10.1021/jm3008882. [DOI] [PubMed] [Google Scholar]
- Gao C.; Ye T.-H.; Wang N.-Y.; Zeng X.-X.; Zhang L.-D.; Xiong Y.; You X.-Y.; Xia Y.; Xu Y.; Peng C.-T.; Zuo W.-Q.; Wei Y.; Yu L.-T. Synthesis and Structure-Activity Relationships Evaluation of Benzothiazinone Derivatives as Potential Anti-Tubercular Agents. Bioorg. Med. Chem. Lett. 2013, 23, 4919–4922. 10.1016/j.bmcl.2013.06.069. [DOI] [PubMed] [Google Scholar]
- Tiwari R.; Miller P. A.; Cho S.; Franzblau S. G.; Miller M. J. Syntheses and Antituberculosis Activity of 1,3-Benzothiazinone Sulfoxide and Sulfone Derived from BTZ043. ACS Med. Chem. Lett. 2015, 6, 128–133. 10.1021/ml5003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.; Peng C.; Shi Y.; You X.; Ran K.; Xiong L.; Ye T.-H.; Zhang L.; Wang N.; Zhu Y.-X.; Liu K.; Zuo W.; Yu L.; Wei Y. Benzothiazinethione Is a Potent Preclinical Candidate for the Treatment of Drug-Resistant Tuberculosis. Sci. Rep. 2016, 6, 29717. 10.1038/srep29717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Lv K.; Wang B.; Li L.; Wang B.; Liu M.; Guo H.; Wanga A.; Lu Y. Design, Synthesis and Antitubercular Evaluation of Benzothiazinones Containing an Oximido or Amino Nitrogen Heterocycle Moiety. RSC Adv. 2017, 7, 1480–1483. 10.1039/C6RA25712G. [DOI] [Google Scholar]
- Wang A.; Lv K.; Tao Z.; Gu J.; Fu L.; Liu M.; Wan B.; Franzblau S. G.; Ma C.; Ma X.; Han B.; Wang A.; Xu S.; Lu Y. Identification of Benzothiazinones Containing an Oxime Functional Moiety as New Anti-Tuberculosis Agents. Eur. J. Med. Chem. 2019, 181, 111595. 10.1016/j.ejmech.2019.111595. [DOI] [PubMed] [Google Scholar]
- Peng C.-T.; Gao C.; Wang N.-Y.; You X.-Y.; Zhang L.-D.; Zhu Y.-X.; Xv Y.; Zuo W.-Q.; Ran K.; Deng H.-X.; Lei Q.; Xiao K.-J.; Yu L.-T. Synthesis and Antitubercular Evaluation of 4-Carbonyl Piperazine Substituted 1,3-Benzothiazin-4-One Derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 1373–1376. 10.1016/j.bmcl.2015.02.061. [DOI] [PubMed] [Google Scholar]
- Xiong L.; Gao C.; Shi Y.-J.; Tao X.; Rong J.; Liu K.-L.; Peng C.-T.; Wang N.-Y.; Lei Q.; Zhang Y.-W.; Yu L.-T.; Wei Y.-Q. Identification of a New Series of Benzothiazinone Derivatives with Excellent Antitubercular Activity and Improved Pharmacokinetic Profile. RSC Adv. 2018, 8, 11163–11176. 10.1039/C8RA00720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Wang B.; Zhang X.; Batt S. M.; Besra G. S.; Zhang T.; Ma C.; Zhang D.; Lin Z.; Li G.; Huang H.; Lu Y. Identification of Novel Benzothiopyranone Compounds against Mycobacterium Tuberculosis through Scaffold Morphing from Benzothiazinones. Eur. J. Med. Chem. 2018, 160, 157–170. 10.1016/j.ejmech.2018.09.042. [DOI] [PubMed] [Google Scholar]
- Lv K.; Tao Z.; Liu Q.; Yang L.; Wang B.; Wu S.; Wang A.; Huang M.; Liu M.; Lu Y. Design, Synthesis and Antitubercular Evaluation of Benzothiazinones Containing a Piperidine Moiety. Eur. J. Med. Chem. 2018, 151, 1–8. 10.1016/j.ejmech.2018.03.060. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Howe M.; Aldrich C. C. Spirocyclic and Bicyclic 8-Nitrobenzothiazinones for Tuberculosis with Improved Physicochemical and Pharmacokinetic Properties. ACS Med. Chem. Lett. 2019, 10, 348–351. 10.1021/acsmedchemlett.8b00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv K.; You X.; Wang B.; Wei Z.; Chai Y.; Wang B.; Wang A.; Huang G.; Liu M.; Lu Y. Identification of Better Pharmacokinetic Benzothiazinone Derivatives as New Antitubercular Agents. ACS Med. Chem. Lett. 2017, 8, 636–641. 10.1021/acsmedchemlett.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv K.; Wang A.; Tao Z.; Fu L.; Liu H.; Wang B.; Ma C.; Wang H.; Ma X.; Han B.; Wang A.; Zhang K.; Liu M.; Lu Y. HERG Optimizations of IMB1603, Discovery of Alternative Benzothiazinones as New Antitubercular Agents. Eur. J. Med. Chem. 2019, 179, 208–217. 10.1016/j.ejmech.2019.06.053. [DOI] [PubMed] [Google Scholar]
- Ma X.; Han B.; Wang A.; Yang L.; Huang M.; Chowdhury K.; Gu J.; Zhang K.; Lv K. Identification of Benzothiazones Containing a Hexahydropyrrolo[3,4-c]Pyrrol Moiety as Antitubercular Agents against MDR-MTB. RSC Adv. 2020, 10, 14410–14414. 10.1039/D0RA00750A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.; Ma C.; Chai Y.; Liu X.; Lv K.; Fu L.; Wang B.; Jia X.; Liu M.; Lu Y. Identification of Benzothiazinones Containing 2-Benzyl-2,7-Diazaspiro [3.5]Nonane Moieties as New Antitubercular Agents. Eur. J. Med. Chem. 2020, 200, 112409. 10.1016/j.ejmech.2020.112409. [DOI] [PubMed] [Google Scholar]
- Wang A.; Lu Y.; Lv K.; Ma C.; Xu S.; Wang B.; Wang A.; Xia G.; Liu M. Design, Synthesis and Antimycobacterial Activity of New Benzothiazinones Inspired by Rifampicin/Rifapentine. Bioorg. Chem. 2020, 102, 104135. 10.1016/j.bioorg.2020.104135. [DOI] [PubMed] [Google Scholar]
- Liu L.; Kong C.; Fumagalli M.; Savková K.; Xu Y.; Huszár S.; Sammartino J. C.; Fan D.; Chiarelli L. R.; Mikušová K.; Sun Z.; Qiao C. Design, Synthesis and Evaluation of Covalent Inhibitors of DprE1 as Antitubercular Agents. Eur. J. Med. Chem. 2020, 208, 112773. 10.1016/j.ejmech.2020.112773. [DOI] [PubMed] [Google Scholar]
- Wang A.; Xu S.; Chai Y.; Xia G.; Wang B.; Lv K.; Ma C.; Wang D.; Wang A.; Qin X.; Liu M.; Lu Y. Design, Synthesis and Biological Activity of N-(Amino)Piperazine- Containing Benzothiazinones against Mycobacterium Tuberculosis. Eur. J. Med. Chem. 2021, 218, 113398. 10.1016/j.ejmech.2021.113398. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Sheng L.; Hegde P.; Li Y.; Aldrich C. C. 8-Cyanobenzothiazinone Analogs with Potent Antitubercular Activity. Med. Chem. Res. 2021, 30, 449–458. 10.1007/s00044-020-02676-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan D.; Wang B.; Stelitano G.; Savkov K.; Shi R.; Husz S.; Chiarelli L. R.; Lu Y.; Qiao C. Structural and Activity Relationships of 6-Sulfonyl-8-Nitrobenzothiazinones as Antitubercular Agents. J. Med. Chem. 2021, 64, 14526–14539. 10.1021/acs.jmedchem.1c01049. [DOI] [PubMed] [Google Scholar]
- Li P.; Wang B.; Fu L.; Guo K.; Ma C.; Wang B.; Lin Z.; Li G.; Huang H.; Lu Y. Identification of Novel Benzothiopyranones with Ester and Amide Motifs Derived from Active Metabolite as Promising Leads against Mycobacterium Tuberculosis. Eur. J. Med. Chem. 2021, 222, 113603. 10.1016/j.ejmech.2021.113603. [DOI] [PubMed] [Google Scholar]
- Shi R.; Wang B.; Stelitano G.; Wu X.; Shan Y.; Wu Y.; Wang X.; Chiarelli L. R.; Lu Y.; Qiao C. Development of 6-Methanesulfonyl-8-Nitrobenzothiazinone Based Antitubercular Agents. ACS Med. Chem. Lett. 2022, 13 (4), 593–598. 10.1021/acsmedchemlett.1c00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieferdecker S.; Bernal F. A.; Wojtas K. P.; Keiff F.; Li Y.; Dahse H.-M.; Kloss F. Development of Predictive Classification Models for Whole Cell Antimycobacterial Activity of Benzothiazinones. J. Med. Chem. 2022, 65, 6748–6763. 10.1021/acs.jmedchem.2c00098. [DOI] [PubMed] [Google Scholar]
- Richter A.; Narula G.; Rudolph I.; Seidel R. W.; Wagner C.; Av-gay Y.; Imming P. Efficient Synthesis of Benzothiazinone Analogues with Activity against Intracellular Mycobacterium Tuberculosis. ChemMedChem 2022, 17, e202100733 10.1002/cmdc.202100733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V. A.; Cole S. T.; Möllmann U. Patent EP2029583 B1. 2010.
- Pasca M. R.; Degiacomi G.; Riberio A. L. J. L.; Zara F.; Mori P. De.; Heym B.; Mirrione M.; Brerra R.; Pagani L.; Pucillo L.; Troupioti P.; Makarov V.; Cole S. T.; Riccardi G. Clinical Isolates of Mycobacterium Tuberculosis in Four European Hospitals Are Uniformly Susceptible to Benzothiazinones. Antimicrob. Agents Chemother. 2010, 54, 1616–1618. 10.1128/AAC.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neres J.; Pojer F.; Molteni E.; Chiarelli L. R.; Dhar N.; Boy-Röttger S.; Buroni S.; Fullam E.; Degiacomi G.; Lucarelli A. P.; Read R. J.; Zanoni G.; Edmondson D. E.; Rossi E. De.; Pasca M. R.; Mckinney J. D.; Dyson P. J.; Riccardi G.; Mattevi A.; Cole S. T.; Binda C. Structural Basis for Benzothiazinone-Mediated Killing of Mycobacterium Tuberculosis. Sci. Transl. Med. 2012, 4, 150ra121. 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landge S.; Mullick A. B.; Nagalapur K.; Neres J.; Subbulakshmi V.; Murugan K.; Ghosh A.; Sadler C.; Fellows M. D.; Humnabadkar V.; Mahadevaswamy J.; Vachaspati P.; Sharma S.; Kaur P.; Mallya M.; Rudrapatna S.; Awasthy D.; Sambandamurthy V. K.; Pojer F.; Cole S. T.; Balganesh T. S.; Ugarkar B. G.; Balasubramanian V.; Bandodkar B. S.; Panda M.; Ramachandran V. Discovery of Benzothiazoles as Antimycobacterial Agents: Synthesis, Structure-Activity Relationships and Binding Studies with Mycobacterium Tuberculosis Decaprenylphosphoryl-β-D-Ribose 2’-Oxidase. Bioorg. Med. Chem. 2015, 23, 7694–7710. 10.1016/j.bmc.2015.11.017. [DOI] [PubMed] [Google Scholar]
- Landge S.; Ramachandran V.; Kumar A.; Neres J.; Murugan K.; Sadler C.; Fellows M. D.; Humnabadkar V.; Vachaspati P.; Raichurkar A.; Sharma S.; Ravishankar S.; Guptha S.; Sambandamurthy V. K.; Balganesh T. S.; Ugarkar B. G.; Balasubramanian V.; Bandodkar B. S.; Panda M. Nitroarenes as Antitubercular Agents:Stereoelectronic Modulation to Mitigate Mutagenicity. ChemMedChem. 2016, 11, 331–339. 10.1002/cmdc.201500462. [DOI] [PubMed] [Google Scholar]
- Christophe T.; Jackson M.; Jeon H. K.; Fenistein D.; Contreras-Dominguez M.; Kim J.; Genovesio A.; Carralot J.-P.; Ewann F.; Hye E.; Pham H.; Lee S. Y.; Kang S.; Seo M. J.; Park E. J.; Shovierová H.; Pham H.; Riccardi G.; Nam J. Y.; Marsollier L.; Joly-Guillou M.-L.; Oh T.; Shin W. K.; No Z.; Nehrbass U.; Brosch R.; Cole S. T.; Brodin P. High Content Screening Identifies Decaprenyl- Phosphoribose 2’ Epimerase as a Target for Intracellular Antimycobacterial Inhibitors. PLOS Pathog. 2009, 5, e1000645 10.1371/journal.ppat.1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnet S.; Hartkoorn R. C.; Székely R.; Pató J.; Triccas J. A.; Schneider P.; Szántai-Kis C.; Or L.; Chambon M.; Ban D.; Bueno M.; Turcatti G.; Kéri G.; Cole S. T. Leads for Antitubercular Compounds from Kinase Inhibitor Library Screens. Tuberculosis 2010, 90, 354–360. 10.1016/j.tube.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Stanley S. A.; Grant S. S.; Kawate T.; Iwase N.; Shimizu M.; Wivagg C.; Silvis M.; Kazyanskaya E.; Aquadro J.; Golas A.; Fitzgerald M.; Dai H.; Zhang L.; Hung D. T. Identification of Novel Inhibitors of M. Tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol. 2012, 7, 1377–1384. 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Neres J.; Hartkoorn R. C.; Ryabova O. B.; Kazakova E.; Šarkan M.; Huszár S.; Piton J.; Kolly G. S.; Vocat A.; Conroy T. M.; Mikušová K.; Cole S. T. The 8-Pyrrole-Benzothiazinones Are Noncovalent Inhibitors of DprE1 from Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4446–4452. 10.1128/AAC.00778-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari R.; Miller P. A.; Chiarelli L. R.; Mori G.; Michal S.; Centárová I.; Cho S.; Mikuśova K.; Franzblau S. G.; Oliver A. G.; Miller M. J. Design, Syntheses, and Anti-TB Activity of 1,3-Benzothiazinone Azide and Click Chemistry Products Inspired by BTZ043. ACS Med. Chem. Lett. 2016, 7, 266–270. 10.1021/acsmedchemlett.5b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madikizela B.; Eckhardt T.; Goddard R.; Richter A.; Lins A.; Lehmann C.; Imming P.; Seidel R. W. Synthesis, Structural Characterization and Antimycobacterial Evaluation of Several Halogenated Non-Nitro Benzothiazinones. Med. Chem. Res. 2021, 30, 1523–1533. 10.1007/s00044-021-02735-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Sambandan D.; Halder R.; Wang J.; Batt S. M.; Weinrick B.; Ahmad I.; Yang P.; Zhang Y.; Kim J.; Hassani M.; Huszar S.; Trefzer C.; Ma Z.; Kaneko T.; Mdluli K. E.; Franzblau S.; Chatterjee A. K.; Johnsson K.; Mikusova K.; Besra G. S.; Fütterer K.; Robbins S. H.; Barnes S. W.; Walker J. R.; Jr W. R. J.; Schultz P. G. Identification of a Small Molecule with Activity against Drug-Resistant and Persistent Tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, e2510–e2517. 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhale R.; Menghani S.; Babu R.; Bansode R.; Bhargavi G.; Karodia N.; Rajasekharan M. V.; Paradkar A.; Khedekar P. Development of Selective DprE1 Inhibitors: Design, Synthesis, Crystal Structure and Antitubercular Activity of Benzothiazolylpyrimidine-5- Carboxamides. Eur. J. Med. Chem. 2015, 96, 30–46. 10.1016/j.ejmech.2015.04.011. [DOI] [PubMed] [Google Scholar]
- Mir F.; Shifi S.; Zaman M. S.; Kalia N. P.; Rajput V. S.; Mulakayala C.; Mulakayala N.; Khan I. A.; Alam M. S. Sulfur Rich 2-Mercaptobenzothiazole and 1,2,3-Triazole Conjugates as Novel Antitubercular Agents. Eur. J. Med. Chem. 2014, 76, 274–283. 10.1016/j.ejmech.2014.02.017. [DOI] [PubMed] [Google Scholar]
- No Z.; Kim J.; Brodin P. B.; Seo M. J.; Kim Y. M.; Cechetto J.; Jeon H.; Genovesio A.; Lee S.; Kang S.; Ewann F. A.; Nam J. Y.; Christophe T.; Fenistein D. P. C.; Jamung H.; Jiyeon J. Patent WO 2011/113606 A1, 2011.
- Barsanti P. A.; Hu C.; Jin J.; Keyes R.; Kucejko R.; Lin X.; Pan Y.; Pfister K. B.; Sendzik M.; Sutton J.; Wan L. Patent WO 2011/085990 A1, 2011.
- Shirude P. S.; Shandil R.; Sadler C.; Naik M.; Hosagrahara V.; Hameed S.; Shinde V.; Bathula C.; Humnabadkar V.; Kumar N.; Reddy J.; Panduga V.; Sharma S.; Ambady A.; Hegde N.; Whiteaker J.; McLaughlin R. E.; Gardner H.; Madhavapeddi P.; Ramachandran V.; Kaur P.; Narayan A.; Guptha S.; Awasthy D.; Narayan C.; Mahadevaswamy J.; Vishwas K.; Ahuja V.; Srivastava A.; Prabhakar K.; Bharath S.; Kale R.; Ramaiah M.; Choudhury N. R.; Sambandamurthy V. K.; Solapure S.; Iyer P. S.; Narayanan S.; Chatterji M. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium Tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56, 9701–9708. 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R. K.; Manjunatha M. R.; Sadler C.; Panda M.; Panduga V.; Reddy J.; Saralaya R.; Nanduri R.; Ambady A.; Ravishankar S.; Sambandamurthy V. K.; Humnabadkar V.; Jena L. K.; Suresh R. S.; Srivastava A.; Prabhakar K. R.; Whiteaker J.; Mclaughlin R. E.; Sharma S.; Cooper C. B.; Mdluli K.; Butler S.; Iyer P. S.; Narayanan S.; Chatterji M. Lead Optimization of 1,4-Azaindoles as Antimycobacterial Agents. J. Med. Chem. 2014, 57, 5728–5737. 10.1021/jm500571f. [DOI] [PubMed] [Google Scholar]
- Chatterji M.; Shandil R.; Manjunatha M. R.; Solapure S.; Ramachandran V.; Kumar N.; Saralaya R.; Panduga V.; Reddy J.; KR P.; Sharma S.; Sadler C.; Cooper C. B.; Mdluli K.; Iyer P. S.; Narayanan S.; Shirudec P. S. 1,4-Azaindole, a Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. 10.1128/AAC.03233-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/show/NCT03199339 (accessed on 19th July, 2022).
- Manjunatha M. R.; Shandil R.; Panda M.; Sadler C.; Ambady A.; Panduga V.; Kumar N.; Mahadevaswamy J.; Sreenivasaiah M.; Narayan A.; Guptha S.; Sharma S.; Sambandamurthy V. K.; Ramachandran V.; Mallya M.; Cooper C.; Mdluli K.; Butler S.; Tommasi R.; Iyer P. S.; Narayanan S.; Chatterji M.; Shirude P. S. Scaffold Morphing To Identify Novel DprE1 Inhibitors with Antimycobacterial Activity. ACS Med. Chem. Lett. 2019, 10, 1480–1485. 10.1021/acsmedchemlett.9b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda M.; Ramachandran S.; Ramachandran V.; Shirude P. S.; Humnabadkar V.; Nagalapur K.; Sharma S.; Kaur P.; Guptha S.; Narayan A.; Mahadevaswamy J.; Ambady A.; Hegde N.; Rudrapatna S. S.; Hosagrahara V. P.; Sambandamurthy V. K.; Raichurkar A. Discovery of Pyrazolopyridones as a Novel Class of Noncovalent DprE1 Inhibitor with Potent Anti-Mycobacterial Activity. J. Med. Chem. 2014, 57, 4761–4771. 10.1021/jm5002937. [DOI] [PubMed] [Google Scholar]
- Naik M.; Humnabadkar V.; Tantry S. J.; Panda M.; Narayan A.; Guptha S.; Panduga V.; Manjrekar P.; Jena L. K.; Koushik K.; Shanbhag G.; Jatheendranath S.; Manjunatha M. R.; Gorai G.; Bathula C.; Rudrapatna S.; Achar V.; Sharma S.; Ambady A.; Hegde N.; Mahadevaswamy J.; Kaur P.; Sambandamurthy V. K.; Awasthy D.; Narayan C.; Ravishankar S.; Madhavapeddi P.; Reddy J.; Prabhakar K.; Saralaya R.; Chatterji M.; Whiteaker J.; McLaughlin B.; Chiarelli L. R.; Riccardi G.; Pasca M. R.; Binda C.; Neres J.; Dhar N.; Signorino-Gelo F.; McKinney J. D.; Ramachandran V.; Shandil R.; Tommasi R.; Iyer P. S.; Narayanan S.; Hosagrahara V.; Kavanagh S.; Dinesh N.; Ghorpade S. R. 4-Aminoquinolone Piperidine Amides: Noncovalent Inhibitors of DprE1 with Long Residence Time and Potent Antimycobacterial Activity. J. Med. Chem. 2014, 57, 5419–5434. 10.1021/jm5005978. [DOI] [PubMed] [Google Scholar]
- Neres J.; Hartkoorn R. C.; Chiarelli L. R.; Gadupudi R.; Pasca M. R.; Mori G.; Farina D.; Savina S.; Makarov V.; Kolly G. S.; Molteni E.; Dhar N.; Ferrari S.; Brodin P.; Delorme V.; Landry V.; De A. L.; Ribeiro J. L.; Venturelli A.; Saxena P.; Pojer F.; Carta A.; Luciani R.; Porta A.; Zanoni G.; Rossi E. De.; Costi M. P.; Riccardi G.; Cole S. T. 2-Carboxyquinoxalines Kill Mycobacterium Tuberculosis through Non-Covalent Inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. 10.1021/cb5007163. [DOI] [PubMed] [Google Scholar]
- Ballell L.; Bates R. H.; Young R. J.; Alvarez-Gomez D.; Alvarez-Ruiz E.; Barroso V.; Blanco D.; Crespo B.; Escribano J.; González R.; Lozanom S.; Huss S.; Santos-Villarejo A.; Martín-Plaza J. J.; Mendoza A.; Rebollo-Lopez M. J.; Remuiñan-Blanco M.; Lavandera J. L.; Pérez-Herran E.; Gamo-Benito F. J.; García-Bustos J. F.; Barros D.; Castro J. P.; Cammack N. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem. 2013, 8, 313–321. 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt S. M.; Cacho Izquierdo M.; Castro Pichel J.; Stubbs C. J.; Vela-Glez Del Peral L.; Perez-Herran E.; Dhar N.; Mouzon B.; Rees M.; Hutchinson J. P.; Young R. J.; McKinney J. D.; Barros Aguirre D.; Ballell L.; Besra G. S.; Argyrou A. Whole Cell Target Engagement Identifies Novel Inhibitors of Mycobacterium Tuberculosis Decaprenylphosphoryl-β-D-ribose Oxidase. ACS Infect. Dis. 2015, 1, 615–626. 10.1021/acsinfecdis.5b00065. [DOI] [PubMed] [Google Scholar]
- Borthwick J. A.; Alemparte C.; Wall I.; Whitehurst B. C.; Argyrou A.; Burley G.; Dios-Anton P. De.; Guijarro L.; Monteiro M. C.; Ortega F.; Suckling C. J.; Pichel J. C.; Cacho M.; Young R. J. Mycobacterium Tuberculosis Decaprenylphosphoryl-β-D-ribose Oxidase Inhibitors: Expeditious Reconstruction of Suboptimal Hits into a Series with Potent in Vivo Activity. J. Med. Chem. 2020, 63, 2557–2576. 10.1021/acs.jmedchem.9b01561. [DOI] [PubMed] [Google Scholar]
- Oh S.; Park Y.; Engelhart C. A.; Wallach J. B.; Schnappinger D.; Arora K.; Manikkam M.; Gac B.; Wang H.; Murgolo N.; Olsen D. B.; Goodwin M.; Sutphin M.; Weiner D. M.; Via L. E.; Bosho H. I. M.; Barry C. E. III Discovery and Structure-Activity-Relationship Study of N-Alkyl-5- Hydroxypyrimidinone Carboxamides as Novel Antitubercular Agents Targeting Decaprenylphosphoryl-β-D-ribose 2’-Oxidase. J. Med. Chem. 2018, 61, 9952–9965. 10.1021/acs.jmedchem.8b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd V. A.; Mason J.; Hanumesh P.; Price J.; Russell C. J.; Webb T. R. 2-Substituted-4,5-Dihydroxypyrimidine-6-Carboxamide Antiviral Targeted Libraries. J. Comb. Chem. 2009, 11, 1100–1104. 10.1021/cc900111u. [DOI] [PubMed] [Google Scholar]
- Rogacki M. K.; Pitta E.; Balabon O.; Huss S.; Lopez-Roman E. M.; Argyrou A.; Blanco-Ruano D.; Cacho M.; Vande Velde C. M. L.; Augustyns K.; Ballell L.; Barros D.; Bates R. H.; Cunningham F.; Van der Veken P. Identification and Profiling of Hydantoins-A Novel Class of Potent Antimycobacterial DprE1 Inhibitors. J. Med. Chem. 2018, 61, 11221–11249. 10.1021/acs.jmedchem.8b01356. [DOI] [PubMed] [Google Scholar]
- Balabon O.; Pitta E.; Rogacki M. K.; Meiler E.; Casanueva R.; Guijarro L.; Huss S.; Lopez-Roman E. M.; Santos-Villarejo A.; Augustyns K.; Ballell L.; Aguirre D. B.; Bates R. H.; Cunningham F.; Cacho M.; Van der Veken P. Optimization of Hydantoins as Potent Antimycobacterial Decaprenylphosphoryl-β-D-Ribose Oxidase (DprE1) Inhibitors. J. Med. Chem. 2020, 63, 5367–5386. 10.1021/acs.jmedchem.0c00107. [DOI] [PubMed] [Google Scholar]
- Whitehurst B. C.; Young R. J.; Burley G. A.; Cacho M.; Torres P.; Whitehurst B. C.; Young R. J.; Burley G. A.; Cacho M.; Torres P.; Peral L. V.-G. Identification of 2-((2,3-Dihydrobenzo[b][1,4]Dioxin-6-Yl)Amino)-N- Phenylpropanamides as a Novel Class of Potent DprE1 Inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127192. 10.1016/j.bmcl.2020.127192. [DOI] [PubMed] [Google Scholar]
- Hariguchi N.; Chen X.; Hayashi Y.; Kawano Y.; Fujiwara M.; Matsuba M.; Shimizu H.; Ohba Y.; Nakamura I.; Kitamoto R.; Shinohara T.; Uematsu Y.; Ishikawa S.; Itotani M.; Haraguchi Y.; Takemura I.; Matsumoto M. OPC-167832, a Novel Carbostyril Derivative with Potent Antituberculosis Activity as a DprE1 Inhibitor. Antimicrob. Agents Chemother. 2020, 64, e02020-19 10.1128/AAC.02020-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H.; Kawano Y.; Ishikawa S.; Uematsu Y.; Shinohara T.; Itotani M.; Haraguchi Y.; Takemura I.; Kaneshige A.; Nakai Y.; Hariguchi N.; Hayashi Y.; Matsumoto M. Patent WO/2016/031255, 2016.
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting Physical in Drug Discovery II: The Impact of Chromatographic Hydrophobicity Measurements and Aromaticity. Drug Discovery Today 2011, 16, 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Https://Clinicaltrials.Gov/Ct2/Show/NCT03678688 (accessed on 25th July, 2022).
- Robertson G. T.; Ramey M. E.; Massoudi L. M.; Carter C. L.; Zimmerman M.; Kaya F.; Graham B. G.; Gruppo V.; Hastings C.; Woolhiser L. K.; Scott D. W. L.; Asay B. C.; Eshun-Wilson F.; Maidj E.; Podell B. K.; Vásquez J. J.; Lyons M. A.; Dartois V.; Lenaerts A. J. Comparative Analysis of Pharmacodynamics in the C3HeB/FeJ Mouse Tuberculosis Model for DprE1 Inhibitors TBA-7371, PBTZ169, and OPC-167832. Antimicrob. Agents Chemother. 2021, 65, e00583-21 10.1128/AAC.00583-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Batt S. M.; Wang B.; Fu L.; Qin R.; Lu Y.; Li G.; Besra G. S.; Huang H. Discovery of Novel Thiophene-Arylamide Derivatives as DprE1 Inhibitors with Potent Antimycobacterial Activities. J. Med. Chem. 2021, 64, 6241–6261. 10.1021/acs.jmedchem.1c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Yang L.; Chai X.; Lei Y.; Shah A.; Lu L.; Shen C.; Jiang D.; Wang Z.; Liu Z.; Xu L.; Wan K.; Zhang T.; Yin Y.; Li D.; Cao D.; Hou T. Discovery of Novel DprE1 Inhibitors via Computational Bioactivity Fingerprints and Structure-Based Virtual Screening. Acta Pharmacol. Sin. 2021, 1605. 10.1038/s41401-021-00779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezquerra-Aznárez M.; Degiacomi G.; Gašparovic H.; Stelitano G.; Sammartino C.; Korduláková J.; Governa P.; Manetti F.; Pasca M. R.; Ramón-García S. The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium Tuberculosis DprE1 Enzyme. Int. J. Mol. Sci. 2022, 23, 771. 10.3390/ijms23020771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabanovich G.; Dušek J.; Savková K.; Pavliš O.; Pávková I.; Korábečný J.; Kučera T.; Vlčková H. K.; Huszár S.; Konyariková Z.; Konečná K.; Jand’ourek O.; Stolaříková J.; Korduláková J.; Vávrová K.; Pávek P.; Klimešová V.; Hrabálek A.; MikušováJ K.; Roh J. Development of 3,5-Dinitrophenyl-Containing 1,2,4-Triazoles and Their Trifluoromethyl Analogues as Highly Efficient Antitubercular Agents Inhibiting Decaprenylphosphoryl-β-D-ribofuranose 2′- Oxidase. J. Med. Chem. 2019, 62, 8115–8139. 10.1021/acs.jmedchem.9b00912. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- Gola J.; Obrezanova O.; Champness E.; Segall M. ADMET Property Prediction : The State of the Art and Current Challenges. QSAR Comb. Sci. 2006, 25, 1172–1180. 10.1002/qsar.200610093. [DOI] [Google Scholar]
- Lagorce D.; Douguet D.; Miteva M. A.; Villoutreix B. O. Computational Analysis of Calculated Physicochemical and ADMET Properties of Protein-Protein Interaction Inhibitors. Sci. Rep. 2017, 7, 46277. 10.1038/srep46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beresford A. P.; Selick H. E.; Tarbit M. H. The Emerging Importance of Predictive ADME Simulation in Drug Discovery. Drug Discovery Today 2002, 7, 109–116. 10.1016/S1359-6446(01)02100-6. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Defining Optimum Lipophilicity and Molecular Weight Ranges for Drug Candidates—Molecular Weight Dependent Lower LogD Limits Based on Permeability. Bioorg. Med. Chem. Lett. 2009, 19, 2844–2851. 10.1016/j.bmcl.2009.03.109. [DOI] [PubMed] [Google Scholar]
- Chhabra S.; Kumar S.; Parkesh R. Chemical Space Exploration of DprE1 Inhibitors Using Chemoinformatics and Artificial Intelligence. ACS Omega 2021, 6, 14430–14441. 10.1021/acsomega.1c01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Lyu X.; Batt S. M.; Hsu M.; Harbut M. B.; Vilcheze C.; Cheng B.; Ajayi K.; Yang B.; Yang Y.; Guo H.; Lin C.; Gan F.; Wang C.; Franzblau S. G.; Jacobs W. R. Jr.; Besra G. S.; Johnson E. F.; Petrassi M.; Chatterjee A. K.; Fütterer K.; Wang F. Determinants of the Inhibition of DprE1 and CYP2C9 by Antitubercular Thiophenes. Angew. Chemie - Int. Ed. 2017, 56, 13011–13015. 10.1002/anie.201707324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawad J.; Bonde C. Design, Synthesis and Biological Evaluation of Some 2-(6-Nitrobenzo[d]Thiazol-2-Ylthio)-N-Benzyl-N-(6-Nitrobenzo[d]Thiazol-2-Yl)Acetamide Derivatives as Selective DprE1 Inhibitors. Synth. Commun. 2019, 49, 2696–2708. 10.1080/00397911.2019.1639756. [DOI] [Google Scholar]
- Liu J.; Dai H.; Wang B.; Liu H.; Tian Z.; Zhang Y. Exploring Disordered Loops in DprE1 Provides a Functional Site to Combat Drug-Resistance in Mycobacterium Strains. Eur. J. Med. Chem. 2022, 227, 113932. 10.1016/j.ejmech.2021.113932. [DOI] [PubMed] [Google Scholar]
- Munagala G.; Yempalla K. R.; Aithagani S. K.; Kalia N. P.; Ali F.; Ali I.; Rajput V. S.; Rani C.; Chib R.; Mehra R.; Nargotra A.; Khan I. A.; Vishwakarma R. A.; Singh P. P. Synthesis and Biological Evaluation of Substituted N-Alkylphenyl-3,5-Dinitrobenzamide Analogs as Anti-TB Agents. Med. Chem. Commun 2014, 5, 521–527. 10.1039/c3md00366c. [DOI] [Google Scholar]
- Li L.; Lv K.; Yang Y.; Sun J.; Tao Z.; Wang A.; Wang B.; Wang H.; Geng Y.; Liu M.; Guo H.; Lu Y. Identification of N-Benzyl 3,5-Dinitrobenzamides Derived from PBTZ169 as Antitubercular Agents. ACS Med. Chem. Lett. 2018, 9, 741–745. 10.1021/acsmedchemlett.8b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Lv K.; Li X.; Wang B.; Wang A.; Tao Z.; Geng Y.; Wang B.; Huang M.; Liu M.; Guo H.; Lu Y. Design, Synthesis and Antimycobacterial Activity of Novel Nitrobenzamide Derivatives. Chin. Chem. Lett. 2019, 30, 413–416. 10.1016/j.cclet.2018.08.005. [DOI] [Google Scholar]
- Goldman R. C.; Laughon B. E. Discovery and Validation of New Antitubercular Compounds as Potential Drug Leads and Probes. Tuberculosis 2009, 89, 331–333. 10.1016/j.tube.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthan S.; Faaleolea E. R.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Laughon B. E.; Maddry J. A.; Mehta A.; Rasmussen L.; Reynolds R. C.; Secrist J. A. III; Shindo N.; Showe D. N.; Sosa M. I.; Suling W. J.; White E. L. High-Throughput Screening for Inhibitors of Mycobacterium Tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddry J. A.; Ananthan S.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Maddox C.; Rasmussen L.; Reynolds R. C.; Secrist J. A. III; Sosa M. I.; White E. L.; Zhang W. Antituberculosis Activity of the Molecular Libraries Screening Center Network Library. Tuberculosis 2009, 89, 354–363. 10.1016/j.tube.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodige S.; Ravula P.; Gulipalli K. C.; Endoori S.; Cherukumalli P. K. R.; Sharath N. S. C. J.; Seelam N. Design, Synthesis, Antitubercular and Antibacterial Activities of Pyrrolo[3,2-b]Pyridine-3-Carboxamide Linked 2-Methoxypyridine Derivatives and in Silico Docking Studies. Synth. Commun. 2019, 49, 2219–2234. 10.1080/00397911.2019.1618874. [DOI] [Google Scholar]
- Hill A. P.; Young R. J. Getting Physical in Drug Discovery: A Contemporary Perspective on Solubility and Hydrophobicity. Drug Discovery Today 2010, 15, 648–655. 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Qin R.; Wang P.; Wang B.; Fu L.; Batt S. M.; Besra G. S.; Wu C.; Wang Y.; Huang H.; Lu Y.; Li G. Identification of Thiophene-Benzenesulfonamide Derivatives for the Treatment of Multidrug-Resistant Tuberculosis. Eur. J. Med. Chem. 2022, 231, 114145. 10.1016/j.ejmech.2022.114145. [DOI] [PubMed] [Google Scholar]
- StarDrop v7.2.0.32905, 2022.
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834. 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; Decrescenzo G. A.; Devraj R. V.; Ellsworth E.; Fobian Y. M.; Gibbs M. E.; Gilles R. W.; Greene N.; Huang E.; Krieger-Burke T.; Loesel J.; Wager T.; Whiteley L.; Zhang Y. Physiochemical Drug Properties Associated with in Vivo Toxicological Outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- ADME Models Reference; Optibrium Ltd.: 2011. [Google Scholar]
- StarDrop V6.3 Reference Guide; Optibrium Ltd.: 2016. [Google Scholar]
- Toxicity Prediction in StarDrop; Optibrium Ltd.: 2011. [Google Scholar]
- Athanasiadis E.; Cournia Z.; Spyrou G. ChemBioServer: A Web-Based Pipeline for Filtering, Clustering and Visualization of Chemical Compounds Used in Drug Discovery. Bioinformatics 2012, 28, 3002–3003. 10.1093/bioinformatics/bts551. [DOI] [PubMed] [Google Scholar]
- Kuhn M.; Johnson K.. Applied Predictive Modeling; Springer: 2013. [Google Scholar]
- Meanwell N. A. Improving Drug Candidates by Design: A Focus on Physicochemical Properties As a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Molecular Inflation, Attrition and the Rule of Five. Adv. Drug Delivery Rev. 2016, 101, 22–33. 10.1016/j.addr.2016.01.018. [DOI] [PubMed] [Google Scholar]
- Matsson P.; Doak B. C.; Over B.; Kihlberg J. Cell Permeability beyond the Rule of 5 ☆. Adv. Drug Delivery Rev. 2016, 101, 42–61. 10.1016/j.addr.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Tinworth C. P.; Young R. J. Facts, Patterns, and Principles in Drug Discovery: Appraising the Rule of 5 with Measured Physicochemical Data. J. Med. Chem. 2020, 63, 10091–10108. 10.1021/acs.jmedchem.9b01596. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in Drug Discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- Palmer D. S.; Llinàs A.; Morao I.; Day G. M.; Goodman J. M.; Glen R. C.; Mitchell J. B. O. Predicting Intrinsic Aqueous Solubility by a Thermodynamic Cycle. Mol. Pharmaceutics 2008, 5, 266–279. 10.1021/mp7000878. [DOI] [PubMed] [Google Scholar]
- Alex A.; Millan D. S.; Perez M.; Wakenhut F.; Whitlock G. A. Intramolecular Hydrogen Bonding to Improve Membrane Permeability and Absorption in beyond Rule of Five Chemical Space. Med. Chem. Commun 2011, 2, 669–674. 10.1039/c1md00093d. [DOI] [Google Scholar]
- Leeson P. D.; Davis A. M. Time-Related Differences in the Physical Property Profiles of Oral Drugs. J. Med. Chem. 2004, 47, 6338–6348. 10.1021/jm049717d. [DOI] [PubMed] [Google Scholar]
- Wenlock M. C.; Austin R. P.; Barton P.; Davis A. M.; Leeson P. D. A Comparison of Physiochemical Property Profiles of Development and Marketed Oral Drugs. J. Med. Chem. 2003, 46, 1250–1256. 10.1021/jm021053p. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Rule Offive in 2015 and beyond: Target and Ligand Structural Limitations, Ligand Chemistry Structure and Drug Discovery Project Decisions ☆. Adv. Drug Delivery Rev. 2016, 101, 34–41. 10.1016/j.addr.2016.04.029. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- Doak B. C.; Zheng J.; Dobritzsch D.; Kihlberg J. How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem. 2016, 59, 2312–2327. 10.1021/acs.jmedchem.5b01286. [DOI] [PubMed] [Google Scholar]
- Doak B. C.; Over B.; Giordanetto F.; Kihlberg J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Moroy G.; Martiny V. Y.; Vayer P.; Villoutreix B. O.; Miteva M. A. Toward in Silico Structure-Based ADMET Prediction in Drug Discovery. Drug Discovery Today 2012, 17, 44–55. 10.1016/j.drudis.2011.10.023. [DOI] [PubMed] [Google Scholar]
- Jeffrey P.; Summerfield S. Neurobiology of Disease Assessment of the Blood - Brain Barrier in CNS Drug Discovery. Neurobiol. Dis. 2010, 37, 33–37. 10.1016/j.nbd.2009.07.033. [DOI] [PubMed] [Google Scholar]
- Vallner J. J. Binding of Drugs by Albumin and Plasma Protein. J. Pharm. Sci. 1977, 66, 447–465. 10.1002/jps.2600660402. [DOI] [PubMed] [Google Scholar]
- Lambrinidis G.; Vallianatou T.; Tsantili-kakoulidou A. In Vitro, in Silico and Integrated Strategies for the Estimation of Plasma Protein Binding. A Review. Adv. Drug Delivery Rev. 2015, 86, 27–45. 10.1016/j.addr.2015.03.011. [DOI] [PubMed] [Google Scholar]
- Xiong L.; Gao C.; Shi Y.-J.; Tao X.; Peng C.-T.; Rong J.; Liu K.-L.; Lei Q.; Zhang Y.-W.; Wang N.-Y.; Yu L.-T. Metabolism of SKLB-TB1001, a Potent Antituberculosis Agent, in Animals. Antimicrob. Agents Chemother. 2018, 62, e02375-17 10.1128/AAC.02375-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger U. M.; Schwab M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Isvoran A.; Louet M.; Vladoiu D. L.; Craciun D.; Loriot M.-A.; Villoutreix B. O.; Miteva M. A. Pharmacogenomics of the Cytochrome P450 2C Family: Impacts of Amino Acid Variations on Drug Metabolism. Drug Discovery Today 2017, 22, 366–376. 10.1016/j.drudis.2016.09.015. [DOI] [PubMed] [Google Scholar]
- Nepali K.; Lee H.; Liou J.-P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. 10.1021/acs.jmedchem.8b00147. [DOI] [PubMed] [Google Scholar]
- Garrido A.; Lepailleur A.; Mignani S. M.; Dallemagne P.; Rochais C. HERG Toxicity Assessment: Useful Guidelines for Drug Design Amanda. Eur. J. Med. Chem. 2020, 195, 112290. 10.1016/j.ejmech.2020.112290. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Poluzzi E.; De Ponti F.; Recanatini M. Toward a Pharmacophore for Drugs Inducing the Long QT Syndrome: Insights from a CoMFA Study of HERG K+ Channel Blockers. J. Med. Chem. 2002, 45, 3844–3853. 10.1021/jm0208875. [DOI] [PubMed] [Google Scholar]
- Aronov A. M.; Goldman B. B. A Model for Identifying HERG K+ Channel Blockers. Bioorg. Med. Chem. 2004, 12, 2307–2315. 10.1016/j.bmc.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Raschi E.; Ceccarini L.; De Ponti F.; Recanatini M. HERG-Related Drug Toxicity and Models for Predicting HERG Liability and QT Prolongation. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1005–1021. 10.1517/17425250903055070. [DOI] [PubMed] [Google Scholar]
- Modi S.; Li J.; Malcomber S.; Moore C.; Scott A.; White A.; Carmichael P. Integrated in Silico Approaches for the Prediction of Ames Test Mutagenicity. J. Comput. Aided. Mol. Des. 2012, 26, 1017–1033. 10.1007/s10822-012-9595-5. [DOI] [PubMed] [Google Scholar]
- Benfenati E.; Golbamaki A.; Raitano G.; Roncaglioni A.; Manganelli S.; Lemke F.; Norinder U.; Piparo E. L.; Honma M.; Manganaro A.; Gini G.; Golbamaki A.; Raitano G.; Roncaglioni A.; Manganelli S. A Large Comparison of Integrated SAR/QSAR Models of the Ames Test for Mutagenicity Ames Test for Mutagenicity. SAR QSAR Environ. Res. 2018, 29, 591–611. 10.1080/1062936X.2018.1497702. [DOI] [PubMed] [Google Scholar]
- Hengstler J. G.; Oesch F. Ames Test 2001, 51–54. 10.1006/rwgn.2001.1543. [DOI] [Google Scholar]
- Föllmann W.; Degen G.; Oesch F.; Hengstler J. G.; Environment W.; Factors H.. Ames Test. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier: 2013; Vol. 1, pp 104–107. [Google Scholar]
- Purohit V.; Basu A. K. Mutagenicity of Nitroaromatic Compounds. Chem. Res. Toxicol. 2000, 13, 673–692. 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]
- Jasial S.; Hu Y.; Bajorah J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886. 10.1021/acs.jmedchem.7b00154. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Nissink J. W. M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017 - Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B. Broad Coverage of Commercially Available Lead-like Screening Space with Fewer than 350,000 Compounds. J. Chem. Inf. Model. 2013, 53, 39–55. 10.1021/ci300461a. [DOI] [PubMed] [Google Scholar]
- Capuzzi S. J.; Muratov E. N.; Tropsha A. Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference CompoundS. J. Chem. Inf. Model. 2017, 57, 417–427. 10.1021/acs.jcim.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.