

Abstract

As a large family of two-dimensional materials, MXenes have attracted intensive attention in recent years. For more functional applications, it is of great significance to determine new MXene members. Here, we theoretically expand the M elements of MXenes to the lanthanide series. Based on density functional theory calculations, the bare lanthanide-based carbides M2C (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) and the corresponding fluorine- and hydroxyl-terminated configurations are investigated. Most of the fluorine- and hydroxyl-terminated MXenes investigated are half-metals. Specifically, in the half-metallic Eu2CF2, the spin-down states show a band gap larger than 2 eV, implying this configuration’s potential applications in spin generation and injection. Both Gd2CT2 (T = F and OH) are magnetic semiconductors. The former shows an indirect band gap of 1.38 eV, while the latter presents a direct one of 0.882 eV. These two configurations also show large magnetic moments higher than 13.7 μB per unit cell. All the hydroxyl-terminated MXene members show relatively low work functions, with the lowest value of 1.46 eV determined in Tm2C(OH)2. These predicted electronic properties imply that the lanthanide-based MXenes could have potential applications in spintronics, information storage, near-infrared detectors, field effect transistors, and field emitter cathodes.
Introduction
With the enhanced degree of integration, conventional silicon-based electronics encounter increasing challenges. For instance, the behavior of the circuit with the chip size at the nanoscale becomes unstable due to the quantum tunneling effect. Moreover, the performance of silicon transistors degrades at high temperatures because of the small band gap and unsatisfactory thermal conductivity. Regarding the development of next-generation electronics, to find promising semiconducting materials to replace or combine with silicon has been demonstrated to be an effective approach.1 On the other hand, spintronics have been demonstrated to show many advantages over conventional semiconductor devices, such as nonvolatility, increased data processing speed, and decreased electric power consumption.2 In order to utilize the spin degree of freedom, the corresponding spintronic materials should possess high spin polarization.3 As a consequence, many efforts have been devoted to developing more semiconducting or spintronic materials.2,4,5 Combined with the increasing demands on paper displays and wearable electronics,6,7 two-dimensional (2D) semiconducting or spintronic films have attracted increasing attention. Regarding the 2D semiconductors, a number of configurations such as hexagonal boron nitride (h-BN),8 phosphorene,4 and molybdenum disulfide (MoS2)5,9 have been synthesized, but improvements are also required for these configurations. For example, the band gap of h-BN is extremely large (5.5 eV), and phosphorene is prone to chemical degradation upon exposure to ambient conditions.10 For 2D spintronic materials, an intrinsic 2D chromium triiodide CrI3 was synthesized through mechanical exfoliation,11,12 and a 2D ferromagnetic van der Waals crystal Cr2Ge2Te6 was also developed.13 However, these two synthesized 2D materials are magnetic metals with low Curie temperatures. Consequently, it is crucial to predict and synthesize more 2D semiconducting and spintronic materials.14−20
In 2011, a new family of 2D materials named MXenes was proposed,21 which is generally synthesized from the selective etching of layered parent Mn+1AXn phases (or denoted as MAX phases, where M denotes an early transition metal; A is an A-group element, such as Al and Si; X is C and/or N; and n is generally 1–3). More than 150 MAX phases have been synthesized,22,23 and the corresponding MXene family generally possesses versatile chemical elements and various configurations. Since hydrofluoric acid is usually adopted as etchant, the surface of MXene is normally functionalized by fluorine and hydroxyl groups.24 To date, more than 40 MXene members haven been synthesized,21,25−30 which show potential applications ranging from energy storage,31,32 electromagnetic interference shielding,33 reinforcement materials,34 catalysts,35−37 to sensors.37 Among these 2D carbides, most configurations are metallic.36 Only Mo2CTx38 and Mo2TiC2Tx39 present semiconducting-like behaviors in experiments, with Tx denoting surface functional group. Theoretically, several MXene members were determined to be semiconducting, such as M2CO2 (M = Sc, Ti, Zr, Hf, Mn and W), M2CF2 (Sc, Cr and Mo) and Sc2C(OH)2.40−48 Mn2CF2 was found to be a half-metal with a high Curie temperature of 520 K.44 M2C and M2N (M = Ti and V) were also predicted to show half-metallic or spin gapless semiconducting feature under strains.49 Obviously, it is feasible to tune the electronic properties of MXenes by varying their transition metals and surface groups.
Specially, MXenes with its transition metal in group IIIB generally show semiconducting characteristics regardless of functional groups, such as M2CT2 (M = Sc, Y, La and Lu; T = O, F and OH).50,51 M2C(OH)2 (M = Sc, Y and Lu) are the only three direct-band-gap semiconducting MXene members up to date.42,50 Sc2CO2 presents large out-of-plane polarization, which could have potential applications in piezoelectric and ferroelectric materials.52 Both Sc2CT2 (T = F and OH) and Lu2CT2 (T = F and OH) could be used in high-frequency semiconductor devices on the basis of their promising electron mobilities, where the electron mobility of Lu2C(OH)2 is nearly 10 times that of silicon.43,51 Moreover, the intermediate states Sc2C(OH)xO2–x among the transition from Sc2C(OH)2 to Sc2CO2 show magnetic semiconducting properties.54
Based on the excellent semiconducting and spintronic properties reported for the MXenes with group IIIB transition metals, here we expand the M element of MXene to the lanthanide series. Based on the first-principles calculations, 12 bare M2C MXenes (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm and Yb) are first studied. The left lanthanide-based M2C (M = La and Lu) have been reported previously.50,51 All of the structures investigated are found to be stabilized in the T-type configuration. Further, the structures, stabilities and electronic properties of corresponding fluorine- and hydroxyl-functionalized members are discussed. Most functionalized configurations are also predicted to be stable. The fluorine- and hydroxyl-functionalized structures are generally half-metallic except for M2CT2 (M = Ce, Tm, Gd and Yb). M2CT2 (M = Ce, Tm and Yb; T = F and OH) are magnetic metals. Gd2CT2 (T = F and OH) are magntic semiconductors with their band gaps are 1.38 and 0.882 eV, respectively. Moreover, the hydroxyl-terminated MXenes generally show relatively low work functions.
Computational Details
All the first-principles calculations are carried out using the Vienna Ab initio Simulation Package (VASP) code.55 The Perdew–Burke–Ernzerhof (PBE) scheme of generalized gradient approximation (GGA)56 is adopted for the exchange–correlation functional. A plane-wave cutoff energy of 500 eV based on the projector augmented-wave pseudopotential57 is utilized for describing the ion–electron interactions. All the structures are relaxed until the forces on each atom are less than 1.0 × 10–4 eV/Å, and the energy tolerance is set as 1.0 × 10–6 eV/unit cell. A Γ-centered 12 × 12 × 1 k-point mesh sampled in the hexagonal Brillouin zone (BZ) is adopted for structural optimization, and a 60 k-point grid along the high-symmetry routes of BZ is employed for plotting the electronic energy band. To eliminate the interactions of neighboring layers, a vacuum layer with its thickness larger than 15 Å is adopted. The layer thickness of MXene is chosen as the monomer thickness in its corresponding multilayer configuration, as reported in our previous work.58 To test the dynamic stability of MXenes, 4 × 4 × 1 supercells are considered for the phonon calculations from the density functional perturbation theory (DFPT)59 combined with the Phonopy60 and VASP55 software. The work function is calculated as the difference between the vacuum level and Fermi level of each configuration, and the vacuum level is obtained by plotting the average potential along the z-axis perpendicular to the MXene surface. All the calculations are based on the spin-polarization scheme. All the structures are visualized in the VESTA code.61
In order to test the thermodynamic stability of the functionalized configurations, the formation energy is calculated according to the following equation40
| 1 |
where Etot(M2CT2) denotes the total energy of the functionalized MXene, Etot(M2C) is the total energy of bare MXene, and Etot(T2) is the total energy of F2 or O2 + H2, as the fluorine and hydroxyl groups are studied in our work.
Results and Discussion
In order to investigate the lanthanide-based MXenes, their structures and stabilities are first studied. The 2D bare transition-metal carbides could be categorized as T- and H-types,62 and both types of structures are shown in Figure 1. For the T-type configuration shown in Figure 1a, both the top and bottom transition metals are located in the triangle centers of neighboring carbon atoms. Many reported MXenes appeared in this type;26 for the H-type structure, the transition-metal atoms in the top layer are located on the top sites of the bottom transition metals. The well-known MoS2 is stabilized in the H-type configuration.5
Figure 1.

(a,b) Side views of the T- and H-type M2C configurations. Blue and brown balls show the lanthanide and carbon atoms, respectively.
Based on the unit cells, the relative total energies between the T- and H-type M2C (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) are provided in Table S1 in Supporting Information. From the table, it is observed that the T-type configurations generally show lower total energies, which implies that all the bare M2C MXenes are stabilized in the T-type. The lattice parameters of all the T- and H-type M2C are listed in Table S2. The lattice parameters in M2C generally decrease with the increasing atomic number of M, which could be ascribed to M with a larger atomic number that has a smaller atomic radius.63 Further, the decomposition energies of the T-type M2C into its potential competing phases are studied and provided in Table S3. Most of the structures are metastable, which show negative decomposition energies, but their absolute values are smaller than 200 meV/atom.64 M2C (M = Sm, Dy, Er, and Tm) show positive decomposition energies, which imply that these configurations are thermodynamically stable. The dynamic stabilities of these T-type configurations are also evaluated. All the phonon dispersions of these configurations are calculated and provided in Figure 2. From this figure, the phonon frequencies in all the dispersions are observed to be positive, which implies that all the T-type M2C configurations are dynamically stable.43,59 In addition, there are evident phonon gaps in these phonon dispersions, which could be ascribed to the remarkable difference in the mass of carbon and lanthanide atoms. The evident phonon gaps might enable the applications of these M2C configurations in phononic crystals.65
Figure 2.

(a–l) Phonon dispersions of the T-type M2C (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes.
Based on the stable T-type configurations, spin-polarized band structures for all the M2C configurations are calculated,66,67 and the corresponding results are provided in Figure S1 in Supporting Information. From the figure, all the bare M2C MXenes are observed to be magnetic metals. The corresponding magnetic moments based on the unit cell of M2C and the magnetic contribution from the f orbital in each M are provided in Table S4. Apparently, the magnetism of M2C is mainly contributed by the f orbital. Further, the functionalized M2C MXenes are studied. The functional group on MXene is dependent on the type of etchant with the selective etching approach.21,26 Using hydrofluoric acid as the etchant, the surface of MXene is generally functionalized by the F and OH groups.21 Therefore, we focus on the fluorine and hydroxyl functional groups in this work. According to previous reports,40,42 the locations of functional groups could vary in different MXenes. To determine the stable functionalized structure, six possible configurations, as shown in Figure 3 are investigated. In this figure, the first and second rows present six different structural models of the fluorine- and hydroxyl-functionalized MXenes, respectively. To facilitate elaboration, the configurations from the left to the right side are denoted as model 1, 2, 3, 4, 5, and 6, respectively. In model 1, the functional groups are located on the top sites of the neighboring lanthanide atoms on both sides, while the groups are on the top sites of the bottom metals on both sides in model 2. In model 3, the functional groups are on the top sites of the central carbon atoms on both sides. In model 4, the functional groups are on the top sites of central carbon atoms on one side, which are on the top sites of the bottom metals on the opposite side. Regarding model 5, the groups are on the top sites of neighboring metals on one side, which are on the top sites of the hollow centers of neighboring metals on the other side. In the last model, the groups are on the top sites of neighboring metals on one side, which are on the top sites of the central carbon atoms on the opposite side. After structural relaxation, the relative total energies of these models for fluorine-functionalized MXenes are provided in Table 1, and the corresponding values for the hydroxyl-terminated structures are given in Table 2. From these tables, it is observed that model 2 shows the lowest energy for all the fluorine- and hydroxyl-functionalized configurations investigated. This stable configuration is similar to the structures of many other MXenes reported.42 Model 1 generally presents the highest energy, and model 4 shows a little higher value than that of model 2. Taking Ce2C(OH)2 as an example, the total energy of model 4 is only 0.180 eV higher than that of model 2.
Figure 3.

(a–f) Side views of models 1–6 of the fluorine-functionalized MXenes. (g–l) Side views of models 1–6 of the hydroxyl-terminated MXenes. Blue, brown, silver, red, and white balls show the lanthanide, carbon, fluorine, oxygen, and hydrogen atoms, respectively.
Table 1. Relative Total Energies (in eV) of Fluorine-Functionalized MXenes M2CF2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb).
| MXenes | model 1 | model 2 | model 3 | model 4 | model 5 | model 6 |
|---|---|---|---|---|---|---|
| Ce2CF2 | 3.16 | 0.00 | 0.714 | 0.353 | 1.42 | 1.89 |
| Pr2CF2 | 3.14 | 0.00 | 0.746 | 0.368 | 1.44 | 1.93 |
| Nd2CF2 | 3.13 | 0.00 | 0.779 | 0.382 | 1.46 | 1.96 |
| Sm2CF2 | 3.12 | 0.00 | 0.845 | 0.406 | 1.48 | 2.01 |
| Eu2CF2 | 3.15 | 0.00 | 0.890 | 0.425 | 1.49 | 2.05 |
| Gd2CF2 | 3.08 | 0.00 | 0.906 | 0.422 | 1.48 | 2.04 |
| Tb2CF2 | 3.13 | 0.00 | 0.928 | 0.426 | 1.46 | 2.04 |
| Dy2CF2 | 3.02 | 0.00 | 0.946 | 0.427 | 1.45 | 2.09 |
| Ho2CF2 | 2.99 | 0.00 | 0.972 | 0.426 | 1.43 | 2.04 |
| Er2CF2 | 3.05 | 0.00 | 1.08 | 0.517 | 1.51 | 2.13 |
| Tm2CF2 | 2.93 | 0.00 | 0.997 | 0.422 | 1.40 | 2.04 |
| Yb2CF2 | 4.50 | 0.00 | 0.547 | 0.256 | 2.15 | 2.61 |
Table 2. Relative Total Energies (in eV) of Hydroxyl-Terminated MXenes M2C(OH)2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb).
| MXenes | model 1 | model 2 | model 3 | model 4 | model 5 | model 6 |
|---|---|---|---|---|---|---|
| Ce2C(OH)2 | 2.60 | 0.00 | 0.368 | 0.180 | 1.26 | 1.48 |
| Pr2C(OH)2 | 2.63 | 0.00 | 0.399 | 0.192 | 1.28 | 1.52 |
| Nd2C(OH)2 | 2.65 | 0.00 | 0.433 | 0.204 | 1.54 | 1.29 |
| Sm2C(OH)2 | 2.66 | 0.00 | 0.469 | 0.232 | 1.32 | 1.60 |
| Eu2C(OH)2 | 2.69 | 0.00 | 0.553 | 0.245 | 1.33 | 1.62 |
| Gd2C(OH)2 | 2.68 | 0.00 | 0.610 | 0.274 | 1.35 | 1.66 |
| Tb2C(OH)2 | 2.63 | 0.00 | 0.639 | 0.255 | 1.32 | 1.66 |
| Dy2C(OH)2 | 2.64 | 0.00 | 0.564 | 0.261 | 1.34 | 1.67 |
| Ho2C(OH)2 | 2.67 | 0.00 | 0.759 | 0.300 | 1.34 | 1.67 |
| Er2C(OH)2 | 2.61 | 0.00 | 0.632 | 0.305 | 1.33 | 1.70 |
| Tm2C(OH)2 | 2.63 | 0.00 | 0.810 | 0.262 | 1.60 | 1.28 |
| Yb2C(OH)2 | 3.64 | 0.00 | 0.302 | 0.136 | 1.83 | 2.05 |
Based on model 2, the thermodynamic stabilities of these lanthanide-based MXenes are further examined. According to eq 1, the formation energies for all the functionalized configurations are calculated and provided in Table 3. From the table, the formation energies of all the fluorine- and hydroxyl-terminated MXenes are observed to be negative. The negative values with the largest and smallest magnitudes are determined in Gd2C(OH)2 and Yb2CF2, which are −35.4 and −12.3 eV, respectively. These values with large magnitudes imply that the lanthanide-based MXenes are prone to be terminated by the fluorine and hydroxyl groups in hydrofluoric acid.21,26 The formation energy of the hydroxyl-terminated MXene generally shows a larger magnitude than that of the fluorine-functionalized structure, which implicates that the fluorine group could transform into the hydroxyl one upon washing and/or storing in water.24 Moreover, the magnitude of the formation energy of M2CT2 (T = F and OH) generally increases, with its M from Ce to Gd, and then decreases from Gd to Yb. This behavior could be related to the occupation fraction of the f orbital of the lanthanide atom. Gd with half-full f electrons shows a stronger chemical bond with its surface group.
Table 3. Formation Energies Ef (in eV) of the Lanthanide-based MXenes Terminated by the Fluorine and Hydroxyl Groups Based on Their Unit Cells.
| M2CT2 | T = F | T = OH |
|---|---|---|
| M = Ce | –14.8 | –18.4 |
| M = Pr | –16.1 | –19.7 |
| M = Nd | –18.1 | –21.7 |
| M = Sm | –24.6 | –28.1 |
| M = Eu | –22.2 | –25.8 |
| M = Gd | –31.7 | –35.4 |
| M = Tb | –27.6 | –31.4 |
| M = Dy | –24.1 | –27.8 |
| M = Ho | –20.5 | –24.9 |
| M = Er | –19.1 | –22.8 |
| M = Tm | –13.4 | –20.4 |
| M = Yb | –12.3 | –16.1 |
Further, the dynamic stabilities of these functionalized configurations are studied. The phonon dispersions of M2CF2 and M2C(OH)2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) are provided in Figures 4 and 5, respectively. All of the phonon dispersions except for that of Ce2C(OH)2 are positive, implying that these configurations are dynamically stable. The hydroxyl-terminated structures generally show much higher frequencies than the corresponding fluorine-terminated members, which could be ascribed to the relatively light mass of the hydrogen element. Upon the same functional group, the highest phonon frequency also varies in different lanthanide-based MXenes. This is a reflection of different bond strengths in lanthanide-based MXenes as a stronger bond strength generally introduces a higher phonon frequency.58 Based on the discussions above, most of the lanthanide-based MXenes investigated are thermodynamically and dynamically stable, which could be realized in further experiments.
Figure 4.

(a–l) Phonon dispersions of M2CF2 MXenes (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb).
Figure 5.

(a–k) Phonon dispersions of M2C(OH)2 MXenes (M = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb).
To evaluate the magnetic stability of all the fluorine- and hydroxyl-terminated MXenes, nonmagnetic (NM), ferromagnetic (FM), and antiferromagnetic (AFM) states for the fluorine- and hydroxyl-terminated MXenes are examined.68−72 The structural diagrams of FM and AFM configurations are shown in Figure 6. The corresponding relative total energies for these NM, FM, and AFM configurations are presented in Table S5. Apparently, most FM configurations show the lowest total energies for these M2CF2 and M2C(OH)2 structures. Only Ho2CF2 and Dy2C(OH)2 show the lowest total energies in their AFM states. The energy difference between NM and FM is generally larger than that between FM and AFM. Eu2CF2 shows the largest energy difference between its FM and AFM states, which implies that this configuration might possess a high Curie temperature.
Figure 6.
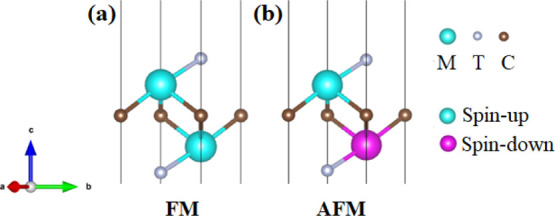
(a,b) Side views of the FM and AFM configurations for M2CT2 (T = F and OH) MXenes, respectively. Pink and green balls represent the M atoms with spin-up and spin-down electron configurations, respectively. Brown and small gray balls represent the carbon atom and the surface group T, respectively.
After checking the structural stabilities, the structural parameters are further investigated. The lattice parameters, layer thicknesses, and bond lengths for the fluorine-terminated MXenes are provided in Table 4, and the corresponding values for the hydroxyl-functionalized members are presented in Table 5. Apparently, the lattice parameters of both fluorine- and hydroxyl-terminated MXenes investigated here are generally larger than those of MXenes reported previously,42 which is due to the larger atomic radii of lanthanides than those of early transition metals. Moreover, the lattice parameters and bond lengths generally decrease with the increasing atomic number of lanthanides, which could be ascribed to the decreasing atomic radius of lanthanides.73 In particular, the Eu-containing MXenes are against this trend. Taking Eu2CF2 as an example, its lattice parameter of 3.74 Å is larger than that of 3.71 Å of its former Sm2CF2. The Eu–C bond is much larger than the other M–C bonds. Similar behaviors are also determined in the Eu2C(OH)2 MXene. These special structural parameters for Eu-containing MXenes could be caused by the special valence electron configuration of 4f75d06s2 in Eu. For the other lanthanide elements, except for Yb, the valence electrons are composed by 5d16s2 and varying numbers of 4f electrons. Generally, these predicted structural parameters will give some guidance for future synthesis and characterization of these lanthanide-based MXenes.
Table 4. Lattice Parameter a, Layer Thickness h, and Bond Lengths M–C and M–F (in Å) for M2CF2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes.
| MXenes | a | h | M–C | M–F |
|---|---|---|---|---|
| Ce2CF2 | 3.69 | 6.82 | 2.57 | 2.46 |
| Pr2CF2 | 3.68 | 6.77 | 2.56 | 2.43 |
| Nd2CF2 | 3.69 | 6.76 | 2.58 | 2.43 |
| Sm2CF2 | 3.71 | 6.71 | 2.60 | 2.41 |
| Eu2CF2 | 3.74 | 6.73 | 2.63 | 2.41 |
| Gd2CF2 | 3.65 | 6.65 | 2.53 | 2.41 |
| Tb2CF2 | 3.59 | 6.60 | 2.49 | 2.39 |
| Dy2CF2 | 3.56 | 6.58 | 2.48 | 2.37 |
| Ho2CF2 | 3.56 | 6.56 | 2.48 | 2.36 |
| Er2CF2 | 3.54 | 6.53 | 2.48 | 2.35 |
| Tm2CF2 | 3.52 | 6.51 | 2.47 | 2.33 |
| Yb2CF2 | 3.54 | 6.58 | 2.51 | 2.33 |
Table 5. Lattice Parameter a, Layer Thickness h, and Bond Lengths M–C and M–O (in Å) for M2C(OH)2 (M = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes.
| MXenes | a | h | M–C | M–O |
|---|---|---|---|---|
| Pr2C(OH)2 | 3.69 | 8.60 | 2.57 | 2.51 |
| Nd2C(OH)2 | 3.71 | 8.59 | 2.58 | 2.50 |
| Sm2C(OH)2 | 3.72 | 8.58 | 2.60 | 2.49 |
| Eu2C(OH)2 | 3.76 | 8.60 | 2.63 | 2.50 |
| Gd2C(OH)2 | 3.65 | 8.50 | 2.53 | 2.48 |
| Tb2C(OH)2 | 3.59 | 8.42 | 2.49 | 2.45 |
| Dy2C(OH)2 | 3.59 | 8.39 | 2.49 | 2.44 |
| Ho2C(OH)2 | 3.56 | 8.37 | 2.48 | 2.43 |
| Er2C(OH)2 | 3.55 | 8.35 | 2.47 | 2.41 |
| Tm2C(OH)2 | 3.53 | 8.33 | 2.47 | 2.39 |
| Yb2C(OH)2 | 3.54 | 8.42 | 2.51 | 2.39 |
As mentioned previously, MXenes with their M elements in group IIIB generally show promising semiconducting properties;42,43,50−54 thus, the electronic properties of these lanthanide-based MXenes are further investigated. Based on the GGA-PBE functional, all the electronic structures for the fluorine- and hydroxyl-functionalized MXenes are provided in Figures 7 and 8, respectively. For the fluorine-terminated configurations, Ho2CF2 is stabilized in its AFM states, as discussed previously, and its spin-up and spin-down states are degenerate in electronic structures. M2CF2 (M = Ce, Tm, and Yb) are FM metals, and both the spin-up and spin-down states intersect with their Fermi levels. For the hydroxyl-terminated MXenes, Dy2C(OH)2 is an AFM metal, while M2C(OH)2 (M = Tb, Er, Tm, and Yb) are FM metals. Gd2CT2 (T = F and OH) are the only two FM semiconducting members determined here. All the rest of the configurations are half-metals. For these half-metals, it is worth pointing out that the metal state of the half-metal is spin-up when the atomic number of M in M2CT2 is smaller than that of Gd. On the contrary, the metal state of half-metal is spin-down when the atomic number of M is larger than that of Gd. This behavior might be related to the occupation fractions of f orbital in these lanthanide atoms. For example, the band gap of the spin-down states in Eu2CF2 is as large as 2.39 eV. This high spin-polarization in these half-metals enables these configurations in spintronic devices, especially for pure spin generation and injection.74
Figure 7.

(a–l) Electronic energy bands of M2CF2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes based on the GGA-PBE functional. The Fermi level is set as 0 eV.
Figure 8.

(a–k) Electronic energy bands of M2C(OH)2 (M = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes based on the GGA-PBE functional. The Fermi level is set as 0 eV.
Based on the GGA-PBE functional, the band gaps in Gd2CF2 and Gd2C(OH)2 are 0.732 and 0.400 eV, respectively. Apparently, the band gap of Gd2C(OH)2 is smaller than that of the fluorine-terminated configuration. The band gap in Gd2CF2 is indirect, with the valence band maximum (VBM) in its electronic energy band located at the BZ center, and the corresponding conduction band minimums (CBMs) lie on the high-symmetry M point. Gd2C(OH)2 is a direct band gap semiconductor, with both its VBM and CBM located at the BZ center. As the GGA-PBE functional generally underestimates the band gaps for semiconductors, the more precise HSE06 functional is also adopted. The electronic energy bands of Gd2CT2 (T = F and OH) from HSE06 are provided in Figure 9. Both the band gaps of Gd2CF2 and Gd2C(OH)2 increase significantly after the HSE06 correction. For Gd2CF2, its band gap increases from the GGA of 0.732 to 1.381 eV. This value is a little higher than the band gap of bulk silicon, implying that Gd2CF2 is suitable for semiconductor devices. For Gd2C(OH)2, its band gap increases to 0.882 eV after correction. On the basis of the direct and appropriate band gap, Gd2C(OH)2 could have promising applications in infrared adsorption devices.75 Moreover, these two structures are intrinsic 2D magnetic semiconductors, which are rarely reported in previous works. The properties of magnetic semiconductors indicate that Gd2CT2 (T = F and OH) have potential applications in the channel materials of spin devices74 and quantum computing.76
Figure 9.

(a,b) Electronic energy bands of Gd2CF2 and Gd2C(OH)2 after the HSE06 correction. The Fermi energy is set as 0 eV.
In order to understand the origin of magnetism in these systems, the magnetic moments of the fluorine- and hydroxyl-terminated MXenes are calculated and listed in Tables 6 and 7, respectively. The projected density of states (PDOS) of M in these lanthanide-based M2CF2 and M2C(OH)2 MXenes are calculated and show in Figures S2 and S3, respectively. Apparently, the magnetism is correlated with the occupation fraction of f orbitals in these M atoms, and their spin-up and spin-down states are separated in those FM configurations. Based on the unit cells, the magnetic moments vary significantly among these lanthanide-based MXenes. Yb2C(OH)2 shows the lowest magnetic moment of 0.390 μB. The largest magnetic moment is determined in Gd2CF2, the value of which is as large as 13.8 μB. The total magnetisms in M2CF2 (M = Dy and Ho) and M2C(OH)2 is zero because their stable magnetic configurations are all in the AFM states. Apparently, the magnetism is correlated with the occupation fraction of f orbitals in these lanthanide atoms. Gd2CT2 (T = F and OH) generally show large magnetic moments because the f orbital in Gd is half-full. This behavior could be directly seen from the magnetic contributions of f orbitals listed in Tables 6 and 7. The f orbital of each Gd atom contributes 6.84 μB in both Gd2CT2 (T = F and OH). These high magnetic moments could enable these lanthanide-based MXenes’ applications in information storage, especially in 2D electronic devices.
Table 6. Magnetic Moments (in μB) of M2CF2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes.
| MXenes | total magnetization | M(f-orbital) |
|---|---|---|
| Ce2CF2 | 1.33 | 0.628 |
| Pr2CF2 | 3.75 | 1.88 |
| Nd2CF2 | 5.87 | 3.05 |
| Sm2CF2 | 10.1 | 5.31 |
| Eu2CF2 | 12.3 | 6.43 |
| Gd2CF2 | 13.8 | 6.84 |
| Tb2CF2 | 11.8 | 5.83 |
| Dy2CF2 | 9.77 | 4.77 |
| Ho2CF2 | 0.00 | ±3.69 |
| Er2CF2 | 5.72 | 2.67 |
| Tm2CF2 | 3.53 | 1.60 |
| Yb2CF2 | 0.458 | 0.190 |
Table 7. Magnetic Moments (in μB) of M2C(OH)2 (M = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb) MXenes.
| MXenes | total magnetization | M(f-orbital) |
|---|---|---|
| Pr2C(OH)2 | 3.68 | 1.85 |
| Nd2C(OH)2 | 5.82 | 3.02 |
| Sm2C(OH)2 | 10.1 | 5.29 |
| Eu2C(OH)2 | 12.3 | 6.42 |
| Gd2C(OH)2 | 13.7 | 6.84 |
| Tb2C(OH)2 | 11.8 | 5.86 |
| Dy2C(OH)2 | 0.00 | ±4.79 |
| Ho2C(OH)2 | 7.74 | 3.75 |
| Er2C(OH)2 | 5.69 | 2.69 |
| Tm2C(OH)2 | 3.48 | 1.60 |
| Yb2C(OH)2 | 0.390 | 0.161 |
As the hydroxyl-terminated MXenes have been demonstrated to show low work functions previously,53 the work functions of these lanthanide-based MXenes M2CT2 (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb; T = F and OH) are also investigated here. As shown in Figure 10, the hydroxyl-terminated MXenes generally show much lower work functions than the fluorine-terminated ones. The work functions of the fluorine-functionalized structures are in the range from 3.62 to 4.24 eV, with the minimum and maximum values determined in Pr2CF2 and Yb2CF2, respectively. With respect to the hydroxyl-terminated configurations, the corresponding values are in the range from 1.46 to 2.17 eV. These values are generally lower than 2.1 eV of the typical low work function material cesium.77 The lowest work function is determined in Tm2C(OH)2. Noteworthily, the work functions in several hydroxyl-terminated configurations are even lower than that in Sc2C(OH)2, which was found to be the lowest work function material reported previously.53 The low work function in these hydroxyl-terminated MXenes could be ascribed to the high potential of surface hydrogen atoms.78 Based on these low work functions, these lanthanide-based MXenes could have potential applications in field emitter cathodes.
Figure 10.

Work functions of M2CT2 MXenes (M = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb; T = F and OH).
Conclusions
In summary, the stabilities, structural parameters, and electronic properties of lanthanide-based MXenes have been investigated in this work. According to the formation energies and phonon dispersions calculated, most fluorine- and hydroxyl-terminated MXenes are stable. The fluorine- and hydroxyl-terminated MXenes are generally magnetic, and most of them are half-metals. High spin polarization has been determined in these half-metals. The spin-up states in the half-metal Eu2CF2 show a large band gap of 2.39 eV. Gd2CT2 (T = F and OH) are the only two magnetic semiconductors. From HSE06, it is observed that Gd2CF2 is an indirect semiconductor with a band gap of 1.38 eV, while Gd2C(OH)2 shows a direct band gap of 0.882 eV. The magnetisms in these lanthanide-based MXenes are mainly related to the occupation fractions of 4f orbitals in the lanthanide atoms. Based on the half-full 4f orbital of Gd, both the unit cells of Gd2CT2 (T = F and OH) show magnetic moments higher than 13.7 μB. Moreover, the hydroxyl-terminated structures generally show relatively low work functions. The work function in Tm2C(OH)2 is as low as 1.46 eV, which could be the lowest work function to the best of our knowledge. Based on these predicted magnetic half-metallic, and semiconducting properties, with low work functions, these lanthanide-based MXenes could have widespread potential applications such as in spintronics, quantum computation, near-infrared detectors, field effect transistors, information storage, and field emitter cathodes. We look forward to the synthesis of these lanthanide-based MXenes in future experiments.
Acknowledgments
The authors acknowledge the support by the Key R&D Projects of Zhejiang Province (nos 2022C01236, 2019C01060), International Partnership Program of Chinese Academy of Sciences (grant no. 174433KYSB20190019), Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (grant no. 2019R01003), National Natural Science Foundation of China (grant nos. 12004009, 52250005, 21875271, and U20B2021), the Anyang Institute of Technology, Anyang, Henan, Specialized Research Fund for the Doctoral (no. BSJ2021008), the Key Research Project of Henan Provincial Higher Education (grant no. 21A430001, 23A150048), and the Entrepreneurship Program of Foshan National Hi-tech Industrial Development Zone, Ningbo Natural Science Foundation (grant nos 2014A610006, 2016A610273, and 2019A610106).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03964.
Relative total energies between the T- and H-type M2C MXenes; lattice parameters for the T- and H-type M2C MXenes; potential competing phases and the corresponding decomposition energies for the T-type M2C MXenes; electronic energy bands for the T-type M2C MXenes based on the GGA-PBE functional and the corresponding magnetic moments and relative total energies between the NM, FM, and AFM states of the M2CT2 MXenes; and PDOS of M in M2CF2 and M2C(OH)2 MXenes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kim K.; Choi J.-Y.; Kim T.; Cho S.-H.; Chung H.-J. A role for graphene in silicon-based semiconductor devices. Nature 2011, 479, 338–344. 10.1038/nature10680. [DOI] [PubMed] [Google Scholar]
- Wolf S. A.; Awschalom D. D.; Buhrman R. A.; Daughton J. M.; von Molnár S.; Roukes M. L.; Chtchelkanova A. Y.; Treger D. M. Spintronics: A spin-based electronics vision for the future. Science 2001, 294, 1488. 10.1126/science.1065389. [DOI] [PubMed] [Google Scholar]
- Li X.; Yang J. First-principles design of spintronics materials. Natl. Sci. Rev. 2016, 3, 365–381. 10.1093/nsr/nww026. [DOI] [Google Scholar]
- Li L.; Yu Y.; Ye G. J.; Ge Q.; Ou X.; Wu H.; Feng D.; Chen X. H.; Zhang Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372. 10.1038/nnano.2014.35. [DOI] [PubMed] [Google Scholar]
- Mak K. F.; Lee C.; Hone J.; Shan J.; Heinz T. F. Atomically thin MoS2: A new direct-gap semiconductor. Phys. Rev. Lett. 2010, 105, 136805. 10.1103/physrevlett.105.136805. [DOI] [PubMed] [Google Scholar]
- Nomura K.; Ohta H.; Takagi A.; Kamiya T.; Hirano M.; Hosono H. Room-temperature fabrication of transparent flexible thin-film transistors using amorphous oxide semiconductors. Nature 2004, 432, 488. 10.1038/nature03090. [DOI] [PubMed] [Google Scholar]
- Huitema H. E. A.; Gelinck G. H.; van der Putten J. B. P. H.; Kuijk K. E.; Hart C. M.; Cantatore E.; Herwig P. T.; van Breemen A. J. J. M.; de Leeuw D. M. Plastic transistors in active-matrix displays. Nature 2001, 414, 599. 10.1038/414599a. [DOI] [PubMed] [Google Scholar]
- Novoselov K. S.; Jiang D.; Schedin F.; Booth T. J.; Khotkevich V. V.; Morozov S. V.; Geim A. K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. 2005, 102, 10451. 10.1073/pnas.0502848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zande A. M.; Huang P. Y.; Chenet D. A.; Berkelbach T. C.; You Y.; Lee G.-H.; Heinz T. F.; Reichman D. R.; Muller D. A.; Hone J. C. Grains and grain boundaries in highly crystalline monolayer molybdenum disulphide. Nat. Mater. 2013, 12, 554. 10.1038/nmat3633. [DOI] [PubMed] [Google Scholar]
- Wood J. D.; Wells S. A.; Jariwala D.; Chen K. S.; Cho E.; Sangwan V. K.; Liu X. L.; Lauhon L. J.; Marks T. J.; Hersam M. C. Effective passivation of exfoliated black phosphorus transistors against ambient degradation. Nano Lett. 2014, 14, 6964. 10.1021/nl5032293. [DOI] [PubMed] [Google Scholar]
- Huang B.; Clark G.; Navarro-Moratalla E.; Klein D. R.; Cheng R.; Seyler K. L.; Zhong D.; Schmidgall E.; McGuire M. A.; Cobden D. H.; et al. Layer-dependent ferromagnetism in a van der waals crystal down to the monolayer limit. Nature 2017, 546, 270. 10.1038/nature22391. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Yi Y.; Song T.; Clark G.; Huang B.; Shan Y.; Wu S.; Huang D.; Gao C.; Chen Z.; et al. Giant nonreciprocal second-harmonic generation from antiferromagnetic bilayer CrI3. Nature 2019, 572, 497. 10.1038/s41586-019-1445-3. [DOI] [PubMed] [Google Scholar]
- Gong C.; Li L.; Li Z.; Ji H.; Stern A.; Xia Y.; Cao T.; Bao W.; Wang C.; Wang Y.; et al. Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 2017, 546, 265. 10.1038/nature22060. [DOI] [PubMed] [Google Scholar]
- Mounet N.; Gibertini M.; Schwaller P.; Campi D.; Merkys A.; Marrazzo A.; Sohier T.; Castelli I. E.; Cepellotti A.; Pizzi G.; et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246. 10.1038/s41565-017-0035-5. [DOI] [PubMed] [Google Scholar]
- Li B.; Wan Z.; Wang C.; Chen P.; Huang B.; Cheng X.; Qian Q.; Li J.; Zhang Z.; Sun G.; et al. Van der Waals epitaxial growth of air-stable CrSe2 nanosheets with thickness-tunable magnetic order. Nat. Mater. 2021, 20, 818. 10.1038/s41563-021-00927-2. [DOI] [PubMed] [Google Scholar]
- Guan Z.; Ni S. Predicted 2D ferromagnetic Janus VSeTe monolayer with high Curie temperature, large valley polarization and magnetic crystal anisotropy. Nanoscale 2020, 12, 22735. 10.1039/d0nr04837b. [DOI] [PubMed] [Google Scholar]
- Guan Z.; Ni S. Strain-controllable high curie temperature, large valley polarization, and magnetic crystal anisotropy in a 2D ferromagnetic janus VSeTe monolayer. ACS Appl. Mater. Interfaces 2020, 12, 53067. 10.1021/acsami.0c13988. [DOI] [PubMed] [Google Scholar]
- Guan Z.; Ni S. Strain-controllable high curie temperature and magnetic crystal anisotropy in a 2D ferromagnetic semiconductive FeI3 monolayer. ACS Appl. Electron. Mater. 2021, 3, 3147. 10.1021/acsaelm.1c00363. [DOI] [PubMed] [Google Scholar]
- Guan Z.; Ni S. Prediction of high curie temperature, large magnetic crystal anisotropy, and carrier doping-induced half-metallicity in two-dimensional ferromagnetic FeX3 (X = F, Cl, Br, and I) monolayers. J. Phys. Chem. C 2021, 125, 16700. 10.1021/acs.jpcc.1c03915. [DOI] [Google Scholar]
- Zhang C.; Nie Y.; Sanvito S.; Du A. First-principles prediction of a room-temperature ferromagnetic janus VSSe monolayer with piezoelectricity, ferroelasticity, and large valley polarization. Nano Lett. 2019, 19, 1366. 10.1021/acs.nanolett.8b05050. [DOI] [PubMed] [Google Scholar]
- Naguib M.; Kurtoglu M.; Presser V.; Lu J.; Niu J.; Heon M.; Hultman L.; Gogotsi Y.; Barsoum M. W. Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248. 10.1002/adma.201102306. [DOI] [PubMed] [Google Scholar]
- Barsoum M. W.; Radovic M. Elastic and mechanical properties of the MAX phases. Annu. Rev. Mater. Res. 2011, 41, 195. 10.1146/annurev-matsci-062910-100448. [DOI] [Google Scholar]
- Gonzalez-Julian J. Processing of MAX phases: from synthesis to applications. J. Am. Ceram. Soc. 2021, 104, 659. 10.1111/jace.17544. [DOI] [Google Scholar]
- Xie Y.; Naguib M.; Mochalin V. N.; Barsoum M. W.; Gogotsi Y.; Yu X.; Nam K.-W.; Yang X.-Q.; Kolesnikov A. I.; Kent P. R. C. Role of surface structure on Li-ion energy storage capacity of two-dimensional transition-metal carbides. J. Am. Chem. Soc. 2014, 136, 6385. 10.1021/ja501520b. [DOI] [PubMed] [Google Scholar]
- Anasori B.; Lukatskaya M. R.; Gogotsi Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098. 10.1038/natrevmats.2016.98. [DOI] [Google Scholar]
- Naguib M.; Barsoum M. W.; Gogotsi Y. Ten years of progress in the synthesis and development of MXenes. Adv. Mater. 2021, 33, 21033931. 10.1002/adma.202103393. [DOI] [PubMed] [Google Scholar]
- Naguib M.; Halim J.; Lu J.; Cook K. M.; Hultman L.; Gogotsi Y.; Barsoum M. W. New two-dimensional niobium and vanadium carbides as promising materials for Li-ion batteries. J. Am. Chem. Soc. 2013, 135, 15966. 10.1021/ja405735d. [DOI] [PubMed] [Google Scholar]
- Naguib M.; Mochalin V. N.; Barsoum M. W.; Gogotsi Y. 25th anniversary article: MXenes: a new family of two-dimensional materials. Adv. Mater. 2014, 26, 992. 10.1002/adma.201304138. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Zha X.-H.; Chen F. Y.; Ye Q.; Eklund P.; Du S.; Huang Q. A two-dimensional zirconium carbide by selective etching of Al3C3 from nanolaminated Zr3Al3C5. Angew. Chem., Int. Ed. 2016, 55, 5008. 10.1002/anie.201510432. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Zha X.; Zhou X.; Chen F.; Gao G.; Wang S.; Shen C.; Chen T.; Zhi C.; Eklund P.; et al. Synthesis and electrochemical properties of two-dimensional hafnium carbide. ACS Nano 2017, 11, 3841. 10.1021/acsnano.7b00030. [DOI] [PubMed] [Google Scholar]
- Lukatskaya M. R.; Mashtalir O.; Ren C. E.; Dall’Agnese Y.; Rozier P.; Taberna P. L.; Naguib M.; Simon P.; Barsoum M. W.; Gogotsi Y. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science 2013, 341, 1502. 10.1126/science.1241488. [DOI] [PubMed] [Google Scholar]
- Ghidiu M.; Lukatskaya M. R.; Zhao M.-Q.; Gogotsi Y.; Barsoum M. W. Conductive two-dimensional titanium carbide ‘clay’ with high volumetric capacitance. Nature 2014, 516, 78. 10.1038/nature13970. [DOI] [PubMed] [Google Scholar]
- Shahzad F.; Alhabeb M.; Hatter C. B.; Anasori B.; Man Hong S.; Koo C. M.; Gogotsi Y. Electromagnetic interference shielding with 2D transition metal carbides (MXenes). Science 2016, 353, 1137. 10.1126/science.aag2421. [DOI] [PubMed] [Google Scholar]
- Ling Z.; Ren C. E.; Zhao M.-Q.; Yang J.; Giammarco J. M.; Qiu J.; Barsoum M. W.; Gogotsi Y. Flexible and conductive MXene films and nanocomposites with high capacitance. Proc. Natl. Acad. Sci. 2014, 111, 16676. 10.1073/pnas.1414215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhang Z.; Li J.; Zhao X.; Wu D.; Zhou Z. Ti2CO2 MXene: a highly active and selective photocatalyst for CO2 reduction. J. Mater. Chem. A 2017, 5, 12899. 10.1039/c7ta03557h. [DOI] [Google Scholar]
- Zhang X.; Lei J.; Wu D.; Zhao X.; Jing Y.; Zhou Z. A Ti-anchored Ti2CO2 monolayer (MXene) as a single-atom catalyst for CO oxidation. J. Mater. Chem. A 2016, 4, 4871. 10.1039/c6ta00554c. [DOI] [Google Scholar]
- Xu B.; Zhu M.; Zhang W.; Zhen X.; Pei Z.; Xue Q.; Zhi C.; Shi P. Ultrathin MXene-micropattern-based field-effect transistor for probing neural activity. Adv. Mater. 2016, 28, 3333. 10.1002/adma.201504657. [DOI] [PubMed] [Google Scholar]
- Halim J.; Kota S.; Lukatskaya M. R.; Naguib M.; Zhao M.-Q.; Moon E. J.; Pitock J.; Nanda J.; May S. J.; Gogotsi Y.; et al. Synthesis and characterization of 2D molybdenum carbide (MXene). Adv. Funct. Mater. 2016, 26, 3118. 10.1002/adfm.201505328. [DOI] [Google Scholar]
- Anasori B.; Shi C.; Moon E. J.; Xie Y.; Voigt C. A.; Kent P. R. C.; May S. J.; Billinge S. J. L.; Barsoum M. W.; Gogotsi Y. Control of electronic properties of 2D carbides (MXenes) by manipulating their transition metal layers. Nanoscale Horiz. 2016, 1, 227. 10.1039/c5nh00125k. [DOI] [PubMed] [Google Scholar]
- Khazaei M.; Arai M.; Sasaki T.; Chung C.-Y.; Venkataramanan N. S.; Estili M.; Sakka Y.; Kawazoe Y. Novel electronic and magnetic properties of two-dimensional transition metal carbides and nitrides. Adv. Funct. Mater. 2013, 23, 2185. 10.1002/adfm.201202502. [DOI] [Google Scholar]
- Zha X.-H.; Huang Q.; He J.; He H.; Zhai J.; Francisco J. S.; Du S. The thermal and electrical properties of the promising semiconductor MXene Hf2CO2. Sci. Rep. 2016, 6, 27971. 10.1038/srep27971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha X.-H.; Luo K.; Li Q.; Huang Q.; He J.; Wen X.; Du S. Role of the surface effect on the structural,electronic and mechanical properties of the carbide MXenes. Europhys. Lett. 2015, 111, 26007. 10.1209/0295-5075/111/26007. [DOI] [Google Scholar]
- Zha X.-H.; Zhou J.; Zhou Y.; Huang Q.; He J.; Francisco J. S.; Luo K.; Du S. Promising electron mobility and high thermal conductivity in Sc2CT2(T = F, OH) MXenes. Nanoscale 2016, 8, 6110. 10.1039/c5nr08639f. [DOI] [PubMed] [Google Scholar]
- He J.; Lyu P.; Nachtigall P. New two-dimensional Mn-based MXenes with room-temperature ferromagnetism and half-metallicity. J. Mater. Chem. C 2016, 4, 11143. 10.1039/c6tc03917k. [DOI] [Google Scholar]
- Zhou J.; Zha X.-H.; Yildizhan M.; Eklund P.; Xue J.; Liao M.; Persson P.; Du S.; Huang Q. Two-dimensional hydroxyl-functionalized and carbon-deficient scandium carbide, ScCxOH, a direct band gap semiconductor. ACS Nano 2019, 13, 1195. 10.1021/acsnano.8b06279. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Luo K.; Zha X.; Liu Z.; Bai X.; Huang Q.; Guo Z.; Lin C.-T.; Du S. Electronic and transport properties of Ti2CO2 MXene nanoribbons. J. Phys. Chem. C 2016, 120, 17143. 10.1021/acs.jpcc.6b06426. [DOI] [Google Scholar]
- Zhou Y.; Zhai G.; Yan T.; Huang Q.; Guo Z.; Lin C.-T.; Du S. Current rectification induced by V-doped and Sc-doped in Ti2CO2 devices. Comput. Mater. Sci. 2017, 138, 175. 10.1016/j.commatsci.2017.06.017. [DOI] [Google Scholar]
- Zhou Y.; Zhai G.; Yan T.; Francisco J. S.; Tian H.; Huang Q.; Du S. Effects of different surface functionalization and doping on the electronic transport properties of M2CTx-M2CO2 heterojunction devices. J. Phys. Chem. C 2018, 122, 14908. 10.1021/acs.jpcc.8b02026. [DOI] [Google Scholar]
- Gao G.; Ding G.; Li J.; Yao K.; Wu M.; Qian M. Monolayer MXenes: promising half-metals and spin gapless semiconductors. Nanoscale 2016, 8, 8986. 10.1039/c6nr01333c. [DOI] [PubMed] [Google Scholar]
- Hong L.; Klie R. F.; Öğüt S. First-principles study of size- and edge-dependent properties of MXene nanoribbons. Phys. Rev. B 2016, 93, 115412. 10.1103/physrevb.93.115412. [DOI] [Google Scholar]
- Bai X.; Zha X.-H.; Qiao Y.; Qiu N.; Zhang Y.; Luo K.; He J.; Li Q.; Huang Q.; Francisco J. S.; Lin S.; Du S. Two-dimensional semiconducting Lu2CT2 (T = F, OH) MXene with low work function and high carrier mobility. Nanoscale 2020, 12, 3795. 10.1039/c9nr10806h. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran A.; Mishra A.; Singh A. K. Ferroelectricity, antiferroelectricity, and ultrathin 2D electron/hole gas in multifunctional monolayer MXene. Nano Lett. 2017, 17, 3290. 10.1021/acs.nanolett.7b01035. [DOI] [PubMed] [Google Scholar]
- Khazaei M.; Arai M.; Sasaki T.; Ranjbar A.; Liang Y.; Yunoki S. OH-terminated two-dimensional transition metal carbides and nitrides as ultralow work function materials. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 92, 075411. 10.1103/physrevb.92.075411. [DOI] [Google Scholar]
- Zha X.-H.; Ren J.-C.; Feng L.; Bai X.; Luo K.; Zhang Y.; He J.; Huang Q.; Francisco J. S.; Du S. Bipolar magnetic semiconductors among intermediate states during the conversion from Sc2C(OH)2 to Sc2CO2 MXene. Nanoscale 2018, 10, 8763. 10.1039/c8nr01292j. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169. 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/physrevlett.77.3865. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953. 10.1103/physrevb.50.17953. [DOI] [PubMed] [Google Scholar]
- Zha X.-H.; Yin J.; Zhou Y.; Huang Q.; Luo K.; Lang J.; Francisco J. S.; He J.; Du S. Intrinsic structural, electrical, thermal, and mechanical properties of the promising conductor Mo2C MXene. J. Phys. Chem. C 2016, 120, 15082. 10.1021/acs.jpcc.6b04192. [DOI] [Google Scholar]
- Gonze X.; Lee C. Dynamical matrices, born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B: Condens. Matter Mater. Phys. 1997, 55, 10355. 10.1103/physrevb.55.10355. [DOI] [Google Scholar]
- Togo A.; Oba F.; Tanaka I. First-principles calculations of the ferroelastic transition between rutile-type andCaCl2-type SiO2 at high pressures. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 78, 134106. 10.1103/physrevb.78.134106. [DOI] [Google Scholar]
- Momma K.; Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272. 10.1107/s0021889811038970. [DOI] [Google Scholar]
- Chen C.; Ji X.; Xu K.; Zhang B.; Miao L.; Jiang J. Prediction of T- and H-phase two-dimensional transition-metal carbides/nitrides and their semiconducting-metallic phase transition. Chem. Phys. Chem. 2017, 18, 1897. 10.1002/cphc.201700111. [DOI] [PubMed] [Google Scholar]
- Slater J. C. Atomic radii in crystals. J. Chem. Phys. 1964, 41, 3199. 10.1063/1.1725697. [DOI] [Google Scholar]
- Ong S. P.; Wang L.; Kang B.; Ceder G. Li–Fe–P–O2 phase diagram from first principles calculations. Chem. Mater. 2008, 20, 1798. 10.1021/cm702327g. [DOI] [Google Scholar]
- Ilami M.; Ahmed R. J.; Petras A.; Beigzadeh B.; Marvi H. Magnetic needle steering in soft phantom tissue. Sci. Rep. 2020, 10, 2500. 10.1038/s41598-020-59275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu A. A.; Karazhanov S.; Kumar K.; Jose P. Platinum doped iron carbide for the hydrogen evolution reaction: the effects of charge transfer and magnetic moment by first-principles approach. Int. J. Hydrogen Energy 2020, 45, 31825. 10.1016/j.ijhydene.2020.08.163. [DOI] [Google Scholar]
- Banu A. A.; Sinthika S.; Premkumar S.; Vigneshwaran J.; Karazhanov S.; Jose S. P. DFT study of NH3 adsorption on 2D monolayer MXenes (M2C, M=Cr, Fe) via oxygen functionalization: Suitable materials for gas sensors. FlatChem 2022, 31, 100329. 10.1016/j.flatc.2021.100329. [DOI] [Google Scholar]
- Li S.; Ao Z.; Zhu J.; Ren J.; Yi J.; Wang G.; Liu W. Strain controlled ferromagnetic-antiferromagnetic transformation in Mn-doped silicene for information transformation devices. J. Phys. Chem. Lett. 2017, 8, 1484. 10.1021/acs.jpclett.7b00115. [DOI] [PubMed] [Google Scholar]
- Seixas L.; Carvalho A.; Castro Neto A. H. Atomically thin dilute magnetism in Co-doped phosphorene. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 91, 155138. 10.1103/physrevb.91.155138. [DOI] [Google Scholar]
- Pozzo M.; Alfè D.; Lacovig P.; Hofmann P.; Lizzit S.; Baraldi A. Thermal expansion of supported and freestanding graphene: lattice constant versus interatomic distance. Phys. Rev. Lett. 2011, 106, 135501. 10.1103/physrevlett.106.135501. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Fu Z.-M.; Yang Z. Tunable magnetic and electronic properties of the Cr-based MXene (Cr2C) with functional groups and doping. J. Magn. Magn. Mater. 2020, 514, 167141. 10.1016/j.jmmm.2020.167141. [DOI] [Google Scholar]
- Chen W.; Qu Y.; Yao L.; Hou X.; Shi X.; Pan H. Electronic, magnetic, catalytic, and electrochemical properties of two-dimensional Janus transition metal chalcogenides. J. Mater. Chem. A 2018, 6, 8021. 10.1039/c8ta01202d. [DOI] [Google Scholar]
- Slater J. C. Atomic Radii in Crystals. J. Chem. Phys. 1964, 41, 3199. 10.1063/1.1725697. [DOI] [Google Scholar]
- Li X.; Jinlong Y. First-principles design of spintronics materials. Natl. Sci. Rev. 2016, 3, 365. 10.1093/nsr/nww026. [DOI] [Google Scholar]
- Zha J.; Luo M.; Ye M.; Ahmed T.; Yu X.; Lien D.; He Q.; Lei D.; Ho J. C.; Bullock J.; et al. Infrared photodetectors based on 2D materials and nanophotonics. Adv. Funct. Mater. 2022, 32, 2111970. 10.1002/adfm.202111970. [DOI] [Google Scholar]
- Zhou Z. Chinese science education in schools and beyond. Natl. Sci. Rev. 2019, 6, 183. 10.1093/nsr/nwz017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang N.-D.; Kohn W. Theory of metal surfaces: work function. Phys. Rev. B: Solid State 1971, 3, 1215. 10.1103/physrevb.3.1215. [DOI] [Google Scholar]
- Zha X.-H.; Ma X.; Luo J.-T.; Fu C. Surface potential-determined performance of Ti3C2T2 (T=O, F, OH) and Zr3C2T2 (T=O, F, OH, S) MXenes as anode materials of sodium ion batteries. Nanoscale 2022, 14, 10549. 10.1039/d2nr02271k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.