

Abstract
The first total synthesis of isoindolinone (±)-entonalactam A (6), originally obtained from the fungus Entonaema sp., was achieved in 14 steps from commercially available 5-bromovanillin via benzophenone intermediates. Isoindolinone, phthalide, and benzophenone analogues of natural products were also synthesized. The monoamine oxidase (MAO) A and B inhibitory activities were tested. The isoindolinone derivative 30 exhibited inhibition of both MAO-A and -B (IC50 = 17.8 and 15.8 μM, respectively).
Introduction
The isoindolinone derivatives entonalactams A–C, daldinans A–G, and childinin C were previously isolated from fruiting bodies of the ascomycetes Entonaema sp., Daldinia concentrica, and D. childiae, commonly classified as Xylariaceae.1−4 These compounds comprise 3,5-dioxyisoindolinone with a 2′,3′-dioxy-5-methylphenyl group connected to C-8. Isoindolinone derivatives with this structural feature have only been isolated from fruiting bodies of the Xylariaceae family.
One of the proposed biosynthesis methods of entonalactams likely uses the formyl benzophenone derivatives daldinals A and B (1 and 2) as starting compounds (Scheme 1).1 The addition of ammonia generates the hemiaminals 3a and 3b. Dihydroxylated intermediates 4a and 4b are generated by intramolecular nucleophilic attack of the amine, and entonalactam C (5) is yielded by oxidation of 4b. Entonalactams A and B (6 and 7) are biosynthesized by reduction at C-8. The phthalide daldinolides A and B (8 and 9) were also previously isolated from fruiting bodies of D. concentrica, and their biosynthesis is similar to that of entonalactams. Oxidation of the formyl group of daldinals A and B (1 and 2) yields carboxylic acid intermediates (10a and 10b); then, intramolecular attack on the ketone to the carbonyl group generates daldinolides A and B (8 and 9).3 This biomimetic synthetic route via benzophenone intermediates is thus effective for generating a comprehensive range of natural isoindolinones and phthalides isolated from Xylariaceae.
Scheme 1. Proposed Biosynthesis of Isoindolinones and Phthalides.

Monoamine oxidase (MAO) is a highly important enzyme in neurodegeneration. There are two isoforms of MAO (MAO-A and MAO-B).5,6 The MAO-A inhibitor moclobemide is a moderately effective antidepressant drug,7 and the MAO-B inhibitors selegiline and resagiline are globally approved treatments for Parkinson’s disease.8 Kumar et al. reported the structure–activity relationships for MAO-A and -B inhibitors.9 In this report, isoindoline 1,3-dione, which shares a common structure with isoindolinones, and phthalide derivatives were evaluated. Substitution at C-5 of isoindoline 1,3-dione or phthalides increased the inhibitory activity of MAO-A and MAO-B.9 Thus, isoindolinones and phthalides isolated from Xylariaceae may show MAO inhibitory activity. In this study, we developed a synthetic strategy for the total synthesis of (±)-entonalactam A (6) and its analogues. The inhibitory activities of the synthesized compounds toward MAO-A and -B were evaluated.
Results and Discussion
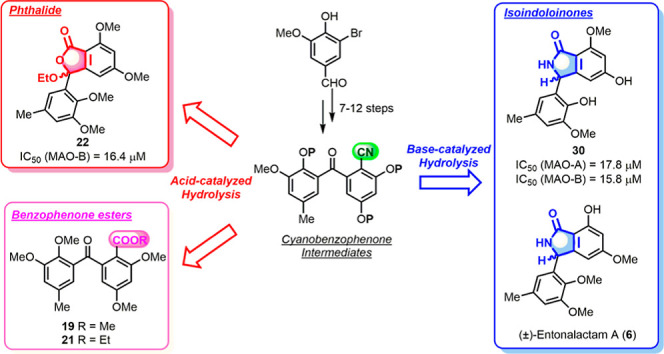
A retrosynthetic analysis of the isoindolinones and phthalides is presented in Figure 1. Entonalactams and daldinolides (3,5,10,11-tetraoxyisoindolinone or phthalides) can be generated from cyanobenzophenones with substituents at the corresponding locations using acid or base hydrolysis and cyclization. The rationale is as follows: hydrolysis of the cyano group with an acid or base yields a carboxylic acid via an amide. The carboxyl group then attacks the ketone of benzophenone and cyclizes to form a phthalide derivative.3,10 Alternatively, isoindolinone is formed when the amide attacks the ketone.11 Cyanobenzophenones are obtained from benzophenones through bromination and cyanation at C-2.12,13 The benzophenone scaffold can be assembled using two building blocks (3,5-substituted benzaldehyde and 2,3,5-substituted bromobenzene) linked by nucleophilic addition of a lithiated arene and subsequent oxidation.14
Figure 1.

Retrosynthetic study for entonalactams and daldinolides.
To probe the general feasibility of this approach, we first elaborated the synthesis of 5-O-methylentonalactam C (11) and 5-O-methyldaldinolide A (12) (Scheme 2). Starting from commercially available 5-bromovanillin, 2,3-dimethoxy-5-methylbromobenzene (13) was prepared according to the literature.14 Bromine–lithium exchange of 13 employing n-BuLi, followed by reaction with 3,5-dimethoxybenzaldehyde at −78 °C, gave benzhydrol (14) in 60% yield. Oxidation of the benzylic position of 14 with MnO2 provided the benzophenone derivative 15. The regioselective bromination at C-2 of 15 was accomplished by N-bromosuccinimide, providing 16 with 85% yield. The structure of bromobenzophenone (16) was confirmed by 1H NMR spectroscopic analysis, which showed four inequivalent aromatic protons (δH 7.04, d, J = 2.2 Hz, H-12, δH 6.90, d, J = 2.2 Hz, H-14, δH 6.51, d, J = 2.7 Hz, H-4, and δH 6.55, d, J = 2.7 Hz, H-6). These signal patterns suggested that this bromination occurred at C-2, whereas an equivalent proton signal (H-2 and H-6) would appear in 4-, 12-, or 14-brominated benzophenone derivatives. Compound 16 was then treated with copper (I) cyanide to yield cyanobenzophenone (17).
Scheme 2. Synthesis of 5-O-methylentonalactam C (11) and 5-O-methyldaldinolide A (12).

Cyclization to isoindolinones and phthalides was conducted by acid or base hydrolysis of the nitrile group. The carboxyl group of the intermediates spontaneously attacked the C-8 carbonyl group to provide the lactone, whereas the amide group attacked the C-8 carbonyl group to provide the lactam. Therefore, our objective to access diverse isoindolinones and phthalides in one operation was achieved. The effects of several conditions were screened (Table 1). The use of hydrogen chloride as the acid and acetonitrile as the solvent mainly yielded isoindolinone (11) along with 8-aminophthalide (18) as a minor product. Using methanol as the solvent provided benzophenone methyl ester 19, phthalide methyl ether 20, and 8-aminophthalide 18. Using ethanol instead of methanol as a solvent yielded benzophenone ethyl ester 21, phthalide ethyl ether 22, and 8-aminophthalide 18. Cyclization under acidic conditions did not yield the desired phthalide (12), but basic hydrolysis of 21 generated 5-O-methyldaldinolide A (12) (Figure 2). The use of sodium hydroxide as the base under reflux provided mainly 18 together with the desired 5-O-methylentonalactam C (11). Using potassium hydroxide as the base and conducting the reaction at room temperature yielded 11 selectively. Palladium-activated carbon (Pd/C)-catalyzed reduction substituted the 8-hydroxy group to hydrogen,15 providing 8-hydroisoindolinone (23) (Figure 3).
Table 1. Reaction Conditions for the Acid and Alkaline Hydrolysis of Cyanobenzophenone (17).

| products (yield, %) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | solvent | catalyst | temperature (°C) | time (h) | 11 | 18 | 19 | 20 | 21 | 22 |
| 1 | MeCN | HCl | r.t. | 12 | 53 | 13 | ||||
| 2 | MeOH | HCl | reflux | 4 | 10 | 11 | 5 | |||
| 3 | EtOH | HCl | reflux | 4 | 6 | 21 | 8 | |||
| 4 | EtOH | NaOH | reflux | 2 | 16 | 59 | ||||
| 5 | MeCN | KOH | r.t. | 12 | 82 | |||||
Figure 2.

Synthesis of the phthalide derivative by alkaline hydrolysis.
Figure 3.

Pd-catalyzed hydride reduction of 11.
Several considerations were taken into account regarding this cyclization reaction. Alkaline hydrolysis of cyano groups generated amide intermediates which attacked the carbonyl group to generate 8-hydroxyisoindolinone (11). Upon heating, the γ-hydroxylactam undergoes recyclization to a lactone, yielding 8-aminophthalide (18). Complications were observed under acidic conditions. Esterification by the corresponding alcoholic solvents generated benzophenone esters 19 and 21. Phthalide ethers 20 and 22 were generated by attack of methanol or ethanol to C-8, respectively. These results indicate that alkaline hydrolysis of cyanobenzophenone at room temperature is suitable for the selective synthesis of isoindolinones.
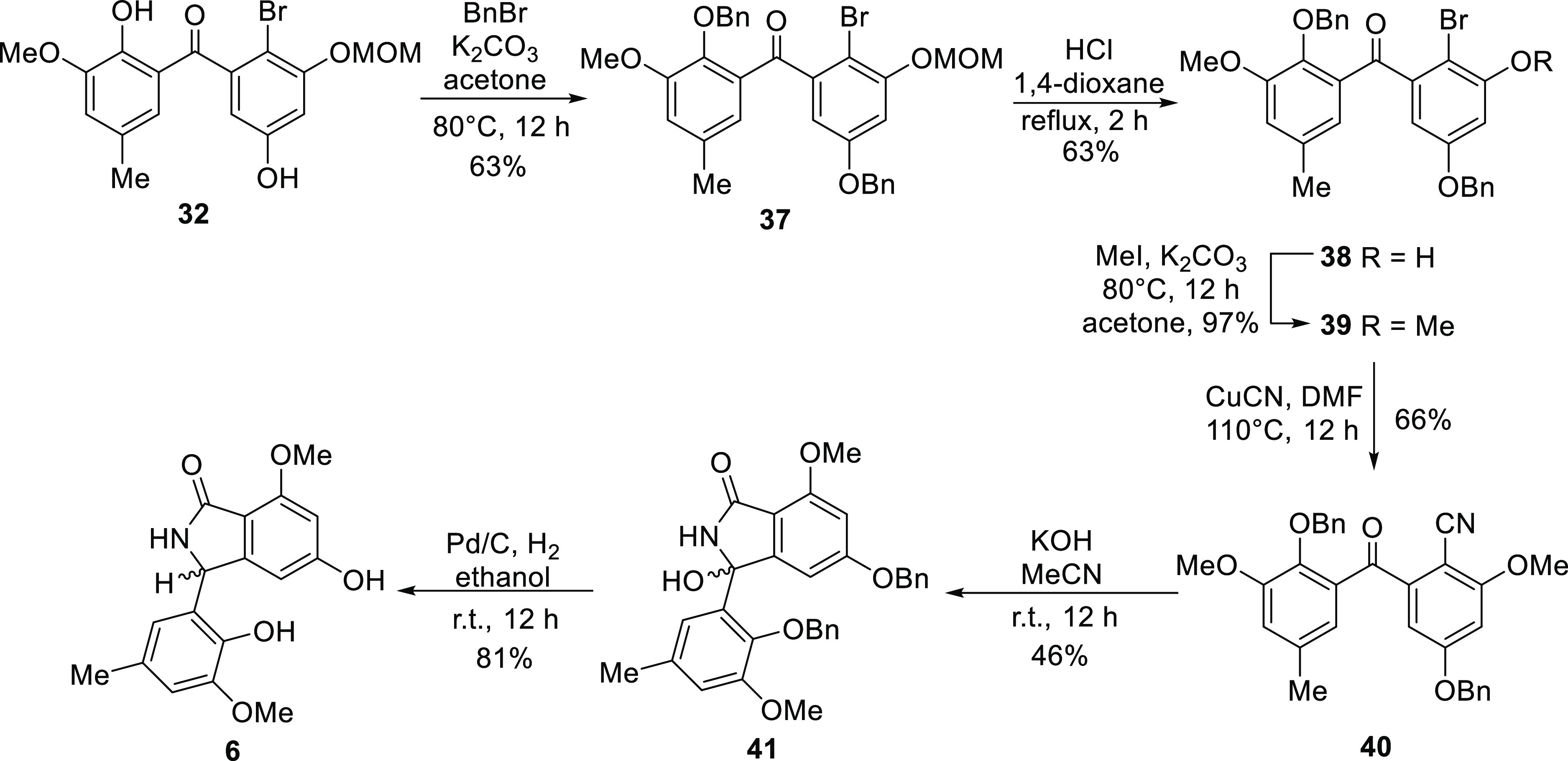
The abovementioned analysis led to the synthesis of entonalactams, as shown in Scheme 3. First, we prepared 3,5-di(benzyloxy)benzhydrol (24) from 13 and 3,5-di(benzyloxy)benzaldehyde. Oxidation, bromination, and cyanation were performed, as shown in Scheme 3, to yield benzophenone derivatives 25–27. 3,5-Di-O-benzylisoindolinone (28) was obtained in good yield from 27 using potassium hydroxide. Next, we attempted the total synthesis of (±)-entonalactam B (7). Compound 29 was generated by Pd/C-catalyzed reduction of 28, with the removal of two benzyl groups and substitution of the 8-hydroxy group to hydrogen. Protection of the 5-hydroxyl group of 29 by chloromethyl methyl ether was not successful due to the poor solubility of 29 in dichloromethane and other nonpolar solvents. Methylation of the 5-hydroxyl group by iodomethane in acetone provided 30 in low yield. Poorly soluble compounds generated at the end of the reaction sequence made it difficult to optimize conditions. Therefore, the synthesis was carried out by reconstituting the protecting group when the benzophenone derivative was soluble in nonpolar solvents (Scheme 4). Deprotection of bromobenzophenone (26) by boron trichloride yielded 3,5,10-trihydroxybromobenzophenone (31). Regioselective mono-methoxymethyl (MOM) protection by chloromethyl methyl ether provided undesired 3-O-MOM-benzophenone (32) instead of the desired 5-O-MOM-benzophenone (32′). Detailed 2D-NMR analysis revealed that the C-3 hydroxyl group of 32 was protected by a MOM group (Figure 4). Methylation and cyanation provided the corresponding benzophenone (33 and 34). The isoindolinone (35 and 36) was then obtained by cyclization and deprotection of the MOM group.
Scheme 3. Synthesis of the Isomer of Entonalactam B (30).
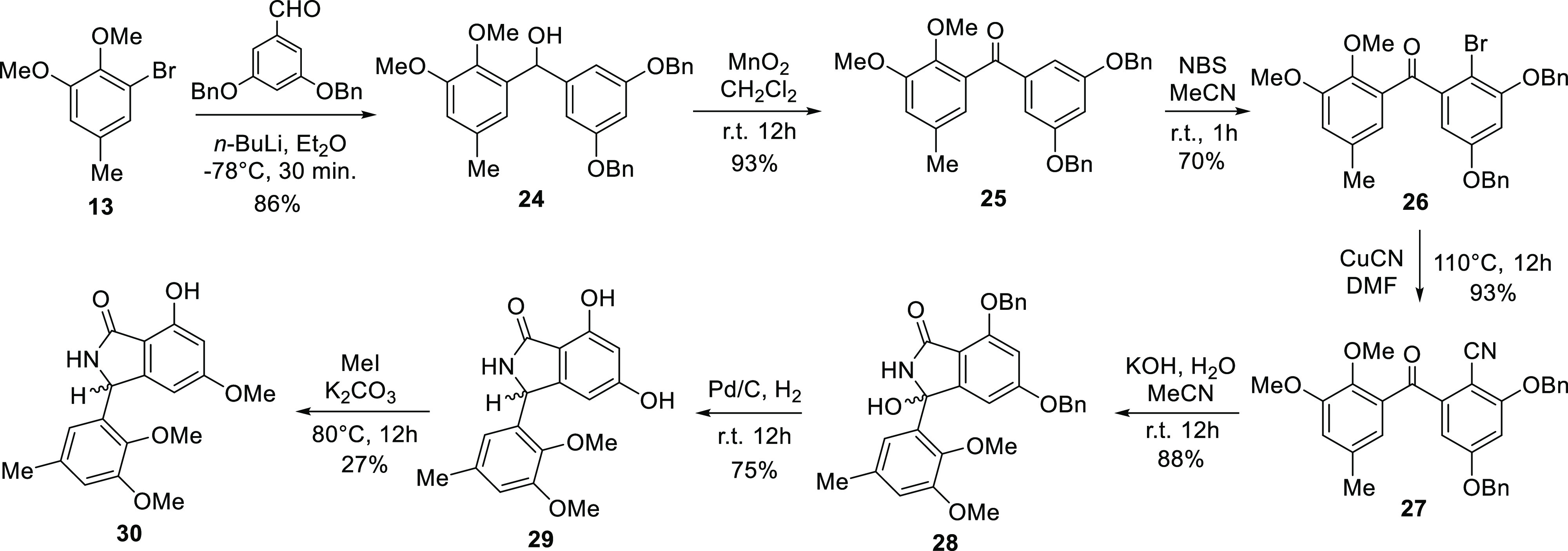
Scheme 4. Synthesis of the Isomer of Entonalactam C (36).
Figure 4.
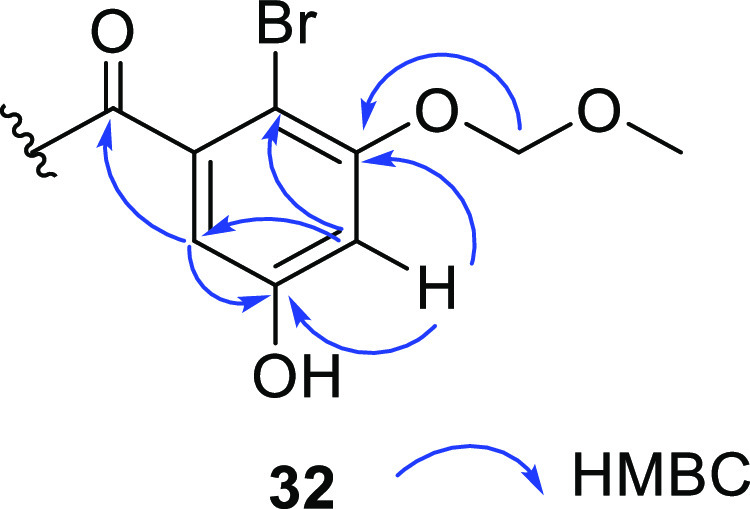
HMBC correlations of 32.
Although MOM protection did not generate the desired product (32′), this reaction selectively protected the 3-hydroxyl group. Therefore, the desired 3-methoxy-benzophenone was synthesized from 32 (Scheme 5). Benzylation of 32 protected the hydroxyl groups at C-5 and C-10 (37). Deprotection of the MOM groups with hydrogen chloride yielded 3-hydroxybenzophenone (38). The desired 3-methoxy benzophenone (39) was generated by methylation using iodomethane, followed by cyanation and base cyclization to provide the corresponding benzophenone and isoindolinone (40 and 41). Finally, Pd/C-catalyzed reduction yielded (±)-entonalactam A (6). The 1H, 13C, and MS spectroscopic data of synthesized 6 matched those reported for the natural product.1
Scheme 5. Synthesis of Entonalactam A (6).
The MAO-A and -B inhibitory activities of the synthesized isoindolinones, phthalides, and benzophenones were tested. As shown in Table 2, (±)-entonalactam A (6) showed slight inhibitory activity for both MAO-A and -B (IC50 = 100.0 and 66.8 μM, respectively). Compounds 15, 30, and 31 had MAO-A inhibitory activity IC50 values of 11.0, 17.8, and 14.8 μM, respectively. MAO-B inhibitory tests showed that 22, 30, and 37 were active (IC50 < 20.0 μM). Of the tested compounds, 30 exhibited both MAO-A and MAO-B inhibition.
Table 2. MAO-A and -B Inhibitory Activities of the Synthesized Compounds.
| MAO-A | MAO-B | MAO-A | MAO-B | ||
|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) | ||
| benzophenones | isoindolinones | ||||
| 15 | 11.0 | 71.9 | 6 | 100.0 | 66.8 |
| 16 | 62.1 | n.d. | 11 | n.d. | n.d. |
| 17 | n.d. | n.d. | 23 | n.d. | n.d. |
| 19 | n.d. | n.d. | 28 | n.d. | n.d. |
| 21 | 47.0 | 61.6 | 29 | 75.4 | n.d. |
| 25 | n.d. | n.d. | 30 | 17.8 | 15.8 |
| 26 | n.d. | n.d. | 35 | n.d. | n.d. |
| 27 | n.d. | n.d. | 36 | 31.9 | 38.4 |
| 31 | 14.8 | 43.7 | 41 | 22.2 | n.d. |
| 32 | 34.1 | 53.6 | |||
| 33 | 27.5 | n.d. | phthalides | ||
| 34 | 24.8 | n.d. | 12 | n.d. | 52.8 |
| 37 | 52.2 | 18.6 | 18 | n.d. | n.d. |
| 38 | 20.6 | 94.3 | 20 | 38.5 | 38.9 |
| 39 | n.d. | n.d. | 22 | 86.3 | 16.4 |
| 40 | n.d. | n.d. | |||
| pargylinea | 4.0 | 1.5 |
Pargyline was used as a positive control. n.d. means > 100 μM.
Conclusions
In conclusion, we developed a synthetic strategy for 3,5,10,11-tetraoxyisoindolinone and phthalide derivatives through their benzophenone intermediates. The total synthesis of (±)-entonalactam A (6) was achieved in 14 steps. The MAO-A and -B inhibitory activities of the synthesized compounds were tested, and 30 showed inhibition of both MAO-A and MAO-B. These results suggest that isoindolinone derivatives isolated from fungi belonging to the Xylariaceae family can be synthesized using this synthetic route to develop novel lead compounds for treating neurological disorders.
Experimental Section
General Experimental Procedures
All reagents and solvents were purchased from commercial suppliers and used without further purification. IR spectra were recorded with an IR Affinity-1S spectrophotometer (ATR, Shimazu Corp. Kyoto, Japan). 1D and 2D NMR spectra were measured at 298 K with a Varian 400 MR (400 MHz) spectrometer (Agilent Technologies Japan, Ltd., Tokyo, Japan) and a Bruker Avance NEO 400 MHz spectrometer (Bruker Japan K.K., Kanagawa, Japan) using tetramethylsilane as the internal standard. Low- and high-resolution EI and FABMS spectra were measured with a JMS-700 spectrometer (JEOL, Tokyo, Japan). Column chromatography was performed using Wakogel C-200 (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). Analytical and preparative HPLC was performed on a Jasco PU-4580 equipped with a Jasco UV-4570 detector (Jasco Corp., Tokyo, Japan) at 254 nm. Preparative HPLC columns were Inertsil diol columns (ϕ10 × 250 mm, 5 μm, GL Sciences Inc., Tokyo, Japan).
(2-Bromo-3,5-dimethoxyphenyl)(2,3-dimethoxy-5-methylphenyl)methanone (16)
Benzophenone 15 was prepared according to the literature.14 To a solution of 15 (328.3 mg, 1.04 mmol) in dry acetonitrile (50 mL) was added N-bromosuccinimide (203.4 mg, 1.14 mmol), and the solution was stirred for 6 h at room temperature. The reaction mixture was purified by silica gel column chromatography (n-Hex–EtOAc 4:1) to afford 16 as colorless oil (345.5 mg, 0.88 mmol 85%). IR (ATR) νmax: 1660 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.04 (d, J = 2.2 Hz, H-12), 6.90 (d, J = 2.2 Hz, H-14), 6.55 (d, J = 2.7 Hz, H-6), 6.51 (d, J = 2.7 Hz, H-4), 3.89 (s, 3-OMe), 3.85 (s, 5-OMe), 3.79 (s, 11-OMe), 3.58 (s, 10-OMe), 2.34 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 195.7 (C-8), 159.7 (C-5), 156.6 (C-3), 152.8 (C-10), 147.0 (C-11), 144.6 (C-7), 133.7 (C-9), 131.9 (C-13), 122.3 (C-14), 117.6 (C-12), 100.9 (C-4), 104.7 (C-6), 99.9 (C-2), 61.2 (10-OMe), 56.5 (3-OMe), 56.0 (5-OMe), 55.7 (11-OMe), 21.2 (13-Me); HRFABMS m/z: 547.1108 [M]+ (calcd for C30H2879BrO5, 547.1042).
2-(2,3-Dimethoxy-5-methylbenzoyl)-4,6-dimethoxybenzonitrile (17)
To a solution of 16 (200.0 mg, 0.51 mmol) in dry N,N-dimethylformamide (20 mL) was added copper (I) cyanide (136.1 mg, 1.52 mmol), and the solution was stirred for 12 h at 110 °C. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated to afford 17 as colorless oil (153.3 mg, 0.45 mmol 88%). IR (ATR) νmax: 2214, 1676 cm–1; 1H NMR (400 MHz, CDCl3): δ 6.91 (overlapped, H-12 and H-14), 6.64 (d, J = 2.2 Hz, H-4), 6.58 (d, J = 2.2 Hz, H-6), 3.94 (s, 3-OMe), 3.86 (s, 11-OMe), 3.82 (s, 5-OMe), 3.62 (s, 10-OMe), 2.34 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 193.9 (C-8), 163.6* (C-3), 163.5* (C-5), 152.4 (C-11), 146.3 (C-10), 145.9 (C-7), 134.1 (C-13), 132.0 (C-9), 121.6 (C-14), 117.4 (C-12), 114.9 (C-1), 107.1 (C-6), 100.3 (C-4), 92.4 (C-2), 61.6 (10-OMe), 56.4 (3-OMe), 55.9 (5-OMe), 55.9 (11-OMe), 21.2 (13-Me), * may be interchanged. HREIMS m/z: 341.1263 [M]+ (calcd for C19H19NO5, 341.1263).
Acid and Base Hydrolysis
Procedure A: To a solution of 17 (25.0 mg, 0.073 mmol) in dry acetonitrile (4.0 mL) was added 35% hydrochloric acid (2.0 mL), and the solution was stirred for 12 h at room temperature. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and purified by silica gel column chromatography (n-Hex–EtOAc 2:1 and 1:2) to afford 11 (14.1 mg, 0.039 mmol 53%) and 18 (3.4 mg, 0.0094 mmol, 13%).
Procedure B: To a solution of 17 (50.0 mg, 0.15 mmol) in dry methanol (10 mL) was added 35% hydrochloric acid (5.0 mL), and the solution was stirred for 4 h at 80 °C. Extraction and purification were performed as in procedure A to afford 18 (5.4 mg, 0.015 mmol 10%), 19 (5.9 mg, 0.015 mmol 11%), and 20 (2.6 mg, 0.0070 mmol 5%).
Procedure C: To a solution of 17 (30.0 mg, 0.09 mmol) in dry ethanol (6.0 mL) was added 35% hydrochloric acid (3.0 mL), and the solution was stirred for 4 h at 80 °C. Extraction and purification were performed as in procedure A to afford 18 (2.6 mg, 0.0050 mmol 6%), 21 (7.2 mg, 0.019 mmol 21%), and 22 (2.8 mg, 0.0070 mmol 8%).
Procedure D: To a solution of 17 (30.0 mg, 0.088 mmol) in dry ethanol (3.0 mL) was added 10 mol/L sodium hydroxide solution (3.0 mL), and the solution was stirred for 2 h under reflux. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and purified by silica gel column chromatography (n-Hex–EtOAc 1:1) to afford 18 (18.7 mg, 0.052 mmol 59%) and 11 (5.2 mg, 0.0014 mmol 16%).
Procedure E: To a solution of 17 (30.0 mg, 0.088 mmol) in dry acetonitrile (6.0 mL) was added 0.3 mol/L potassium hydroxide solution (0.6 mL), and the solution was stirred for 12 h at room temperature. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and purified by silica gel column chromatography (n-Hex–EtOAc 1:1) to afford 11 (26.2 mg, 0.073 mmol 82%).
(±)-3-(2,3-Dimethoxy-5-methylphenyl)-3-hydroxy-5,7-dimethoxyisoindolin-1-one (11)
Colorless oil; IR (ATR) νmax: 3387, 1676 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.37 (s, NH), 7.25 (d, J = 2.1 Hz, H-14), 6.82 (d, J = 2.1 Hz, H-12), 6.51 (br s, 8-OH), 6.48 (d, J = 1.9 Hz, H-4), 6.18 (d, J = 1.9 Hz, H-6), 3.82 (s, 3-OMe), 3.70 (s, 5-OMe and 11-OMe), 3.14 (s, 10-OMe), 2.28 (s, H-13-Me); 13C NMR (100 MHz, DMSO-d6): δ 167.8 (C-1), 164.4 (C-5), 157.8 (C-3), 156.3 (C-7), 152.7 (C-11), 144.3 (C-10), 134.8 (C-9), 132.1 (C-13), 120.2 (C-12), 114.1 (C-14), 111.9 (C-2), 99.4 (C-6), 99.0 (C-4), 84.5 (C-8), 59.7 (10-OMe), 56.1 (3-OMe), 56.1 (5-OMe), 56.1 (11-OMe), 21.7 (13-Me); HRFABMS m/z: 360.1458 [M + H]+ (calcd for C19H22NO6, 360.1447).
(±)-3-Amino-3-(2,3-dimethoxy-5-methylphenyl)-5,7-dimethoxyisobenzofuran-1(3H)-one (18)
White powder; IR (ATR) νmax: 3406, 1743 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 7.02 (d, J = 2.0 Hz, H-12), 6.88 (d, J = 2.0 Hz, H-14), 6.59 (d, J = 1.8 Hz, H-4), 6.30 (d, J = 1.8 Hz, H-6), 3.87 (s, 3-OMe), 3.77 (s, 5-OMe), 3.72 (s, 11-OMe), 3.67 (br s, NH2), 3.29 (s, 10-OMe), 2.26 (s, 13-Me); 13C NMR (100 MHz, DMSO-d6): δ 166.9 (C-1), 166.0 (C-5),158.8 (C-3), 156.2 (C-7), 153.0 (C-11), 145.0 (C-10), 132.8 (C-9), 132.7 (C-13), 120.1 (C-12), 114.9 (C-14), 108.5 (C-2), 99.9 (C-6), 99.6 (C-4), 97.2 (C-8), 60.0 (10-OMe), 56.4 (3-OMe), 56.3 (5-OMe), 56.2 (11-OMe), 21.6 (13-Me); HRFABMS m/z: 360.1425 [M + H]+ (calcd for C19H22NO6, 360.1447).
Methyl 2-(2,3-dimethoxy-5-methylbenzoyl)-4,6-dimethoxybenzoate (19)
Colorless oil; IR (ATR) νmax: 1732 cm–1; 1H NMR (400 MHz, CDCl3): δ 6.86 (d, J = 2.1 Hz, H-12), 6.84 (d, J = 2.1 Hz, H-14), 6.60 (d, J = 2.3 Hz, H-4), 6.57 (d, J = 2.3 Hz, H-6), 3.86 (s, 3-OMe and 11-OMe), 3.78 (s, 5-OMe), 3.69 (s, 2-COOMe), 3.66 (s, 10-OMe), 2.31 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 195.6 (C-8), 167.1 (C-1), 161.5 (C-5), 158.4 (C-3), 152.6 (C-11), 146.0 (C-10), 141.6 (C-7), 133.6 (C-13), 132.5 (C-9), 121.7 (C-14), 116.6 (C-12), 115.0 (C-2), 106.3 (C-6), 101.4 (C-4), 61.7 (10-OMe), 55.9* (11-OMe), 55.6 (5-OMe), 55.3* (3-OMe), 52.2 (1-COOMe), 21.2 (13-Me), *may be interchanged; HRFABMS m/z: 375.1428 [M + H]+ (calcd for C20H23O7, 375.1444).
(±)-3-(2,3-Dimethoxy-5-methylphenyl)-3,5,7-trimethoxyisobenzofuran-1(3H)-one (20)
Colorless oil; IR (ATR) νmax: 1761 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 2.2 Hz, H-14), 6.76 (d, J = 2.2 Hz, H-12), 6.45 (d, J = 1.9 Hz, H-4), 6.42 (d, J = 1.9 Hz, H-6), 3.96 (s, 3-OMe), 3.80* (s, 5-OMe), 3.79* (s, 11-OMe), 3.66 (s, 10-OMe), 3.25 (s, 8-OMe), 2.35 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 166.9 (C-5), 158.9 (C-3), 153.0** (C-7), 152.8** (C-11), 144.7 (C-10), 133.3 (C-9), 130.6 (C-13), 119.2 (C-14), 114.4 (C-12), 105.6 (C-8), 100.0 (C-4), 98.9 (C-6), 60.8 (10-OMe), 55.7* (11-OMe), 56.1 (3-OMe), 55.9* (5-OMe), 50.7 (8-OMe), 21.6 (13-Me) *, ** may be interchanged, two carbons (C-1 and C-2) were not observed. HRFABMS m/z: 375.1424 [M + H]+ (calcd for C20H23O7, 375.1444).
Ethyl 2-(2,3-dimethoxy-5-methylbenzoyl)-4,6-dimethoxybenzoate (21)
Colorless oil; IR (ATR) νmax: 1728 cm–1; 1H NMR (400 MHz, CDCl3): δ 6.87 (s, H-12), 6.87 (s, H-14), 6.60 (d, J = 2.2 Hz, H-4), 6.56 (d, J = 2.2 Hz, H-6), 4.17 (q, J = 7.2 Hz, COOCH2CH3), 3.86 (s, 3-OMe and 11-OMe), 3.77 (s, 5-OMe), 3.65 (s, 10-OMe), 2.32 (s, 13-Me), 1.32 (t, J = 7.2 Hz, COOCH2CH3); 13C NMR (100 MHz, CDCl3): δ 195.5 (C-8), 166.7 (C-1), 161.3 (C-5), 158.4 (C-3), 152.5 (C-11), 146.0 (C-10), 141.4 (C-7), 133.6 (C-13), 132.6 (C-9), 121.8 (C-14), 116.6 (C-12), 115.5 (C-2), 106.3 (C-6), 101.4 (C-4), 61.7 (10-OMe), 61.3 (COOCH2CH3), 56.3 (11-OMe), 55.9 (3-OMe), 55.6 (5-OMe), 21.2 (13-Me), 13.8 (COOCH2CH3); HRFABMS m/z: 388.1517 [M]+ (calcd for C21H24O7, 388.1522).
(±)-3-(2,3-Dimethoxy-5-methylphenyl)-3-ethoxy-5,7-dimethoxyisobenzofuran-1(3H)-one (22)
Colorless oil; IR (ATR) νmax: 1762 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.25 (d, J = 1.5 Hz, H-14), 6.74 (d, J = 1.5 Hz, H-12), 6.51 (d, J = 1.8 Hz, H-4), 6.41 (d, J = 1.8 Hz, H-6), 3.95 (s, 3-OMe), 3.85 (s, 5-OMe), 3.79 (s, 11-OMe), 3.68 (s, 10-OMe), 3.60 (dd, J = 9.0, 7.0 Hz, OCH2CH3), 3.29 (dd, J = 9.0, 7.0 Hz, OCH2CH3), 2.35 (s, 13-Me), 1.26 (t, J = 7.0 Hz, OCH2CH3); 13C NMR (100 MHz, CDCl3): δ 166.9 (C-5), 158.9 (C-3), 153.3 (C-7), 153.0 (C-11), 144.7 (C-10), 133.3 (C-9), 131.0 (C-13), 119.2 (C-14), 114.3 (C-12), 107.9 (C-2), 107.9 (C-8), 99.8 (C-4), 99.0 (C-6), 60.8 (10-OMe), 59.1 (OCH2CH3), 56.1 (3-OMe), 55.9 (5-OMe), 55.7 (11-OMe), 21.6 (13-Me), 15.4 (OCH2CH3), one carbon (C-1) was not observed; HREIMS m/z: 388.1525 [M]+ (calcd for C21H24O7, 388.1522).
3-(2,3-Dimethoxy-5-methylphenyl)-3-hydroxy-5,7-dimethoxyisobenzofuran-1(3H)-one (12)
To a solution of 21 (8.7 mg, 0.023 mmol) in dry methanol (5.4 mL) was added 1.0 mol/L sodium hydroxide solution (1.1 mL), and the solution was stirred for 1 h at room temperature. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and purified by silica gel column chromatography (n-Hex–EtOAc 1:1) to afford 12 (1.4 mg, 0.0038 mmol 17%) as colorless oil. IR (ATR) νmax: 3361, 1749 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.07 (br s, OH), 7.19 (s, H-14), 6.94 (s, H-12), 6.63 (s, H-6), 6.30 (s, H-4), 3.88 (s, 7-OMe), 3.77* (s, 3-OMe), 3.75* (s, 5-OMe), 3.22 (s, 11-OMe), 2.31 (s, 13-Me); 13C NMR signals were not observed; HREIMS m/z: 360.1203 [M]+ (calcd for C19H20O7, 360.1209).
(±)-3-(2,3-Dimethoxy-5-methylphenyl)-5,7-dimethoxyisoindolin-1-one (23)
The solution of 11 (7.0 mg 0.020 mmol) and palladium 10% on carbon (5.0 mg) in ethanol (3.0 mL) was stirred under hydrogen for 12 h at room temperature. The reaction mixture was then filtered through cerite, and the filtrate was concentrated. 23 was obtained as white powder (6.4 mg, 0.019 mmol 96%). Colorless oil; IR (ATR) νmax: 3017, 1678 cm–1; 1H NMR (400 MHz, CDCl3): δ 6.66 (s, H-14), 6.46 (s, H-12), 6.41 (s, H-4), 6.38 (s, H-6), 6.23 (s, NH), 5.90 (s, H-8), 3.93* (s, 3-OMe), 3.91* (s, 5-OMe), 3.76 (s, 10-OMe), 3.87 (s, 11-OMe); 13C NMR (100 MHz, CDCl3): δ 170.1 (C-1), 165.0 (C-5), 158.5 (C-3), 152.9 (C-11), 152.4 (C-7), 144.7 (C-10), 134.4 (C-13), 131.8 (C-9), 118.6 (C-14), 113.0 (C-12), 111.5 (C-2), 98.5 (C-4), 98.5 (C-6), 61.2 (10-OMe), 55.9 (11-OMe), 55.8* (3-OMe), 55.7* (5-OMe), 54.1 (C-8), 21.3 (13-Me), *may be interchanged; HRFABMS m/z: 344.1484 [M + H]+ (calcd for C19H22NO5, 344.1498).
(3,5-Bis(benzyloxy)phenyl) (2,3-dimethoxy-5-methylphenyl)methanol (24)
Under an atmosphere of argon, a solution of 13 (4.4 g, 18.9 mmol) in Et2O (50 mL) was cooled to −78 °C. n-BuLi (17.7 mL, 1.6 M in hexane, 28.3 mmol) was added dropwise via a syringe. After 15 min at −78 °C, a solution of 3,5-dibenzyloxybenzaldehyde (2.0 g, 6.3 mmol) in Et2O/THF 1:1 (10 mL) was slowly added and additionally stirred for 30 min. The stirred mixture was allowed to warm to room temperature and quenched by the addition of H2O (10 mL). The mixture was extracted with EtOAc, dried over anhydrous Na2SO4, and concentrated under reduced pressure. Purification of the residue by silica gel column chromatography was carried out. (n-Hex–EtOAc 2:1) yielded 24 (2.5 g, 5.4 mmol, 86%) as colorless oil. IR (ATR) νmax: 3412, 1593, 1456 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.29–7.49 (overlapped, 3-O–CH2–Ph and 5-O–CH2–Ph), 6.70 (d, J = 2.0 Hz, H-14), 6.67 (overlapped, H-2, H-6 and H-12), 6.51 (t, J = 2.3 Hz, H-4), 5.86 (d, J = 6.6 Hz, H-8), 5.01 (s, 3-O–CH2–Ph and 5-O–CH2–Ph), 3.84 (s, 11-OMe), 3.52 (s, 10-OMe), 3.03 (d, J = 6.6 Hz, 8-OH), 2.30 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 159.8 (C-3 and C-5), 152.3 (C-11), 146.8 (C-10), 144.1 (C-7), 136.9, 128.5, 127.9, 127.6 (3-O–CH2–Ph and 5-O–CH2–Ph), 136.6 (C-9), 133.9 (C-13), 120.2 (C-14), 112.8 (C-12), 105.5 (C-2 and C-6), 100.8 (C-4), 72.5 (C-8), 70.0 (3-O–CH2–Ph and 5-O–CH2–Ph), 60.5 (10-OMe), 55.7 (11-OMe), 21.4 (3-Me); HRFABMS m/z: 470.2097 [M]+ (calcd for C30H30O5, 470.2093).
(3,5-Bis(benzyloxy)phenyl)(2,3-dimethoxy-5-methylphenyl)methanone (25)
To a solution of 24 (4.4 g, 9.4 mmol) in CH2Cl2 (250 mL) was added MnO2 (32.6 g), and the solution was stirred for 12 h at room temperature. The catalyst was removed by filtration through cerite to afford 25 (4.1 g, 8.76 mmol, 93%) as colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.31–7.42 (overlapped, 3-O–CH2–Ph and 5-O–CH2–Ph), 7.09 (d, J = 2.3 Hz, H-2 and H-6), 6.85 (br s, H-14), 6.82 (br s, H-12), 6.67 (br s, H-4), 5.04 (s, 3-O–CH2–Ph and 5-O–CH2–Ph),3.89 (s, 11-OMe), 3.69 (s, 10-OMe), 2.33 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 196.1 (C-8), 159.8 (C-3 and C-5), 152.3 (C-10), 144.6 (C-11), 139.6 (C-7), 136.4, 128.6, 128.1, 127.7 (3-O–CH2–Ph and 5-O–CH2–Ph), 133.8 (C-9), 133.8 (C-13), 120.2 (C-14), 115.2 (C-12), 108.9 (C-2 and C-6), 107.1 (C-4), 70.3 (3-O–CH2–Ph and 5-O–CH2–Ph), 61.7 (10-OMe), 55.9 (11-OMe), 21.3 (13-Me); HRFABMS m/z: 469.2011 [M + H]+ (calcd for C30H29O5, 469.2015).
(3,5-Bis(benzyloxy)-2-bromophenyl)(2,3-dimethoxy-5-methylphenyl)methanone (26)
To a solution of 25 (2.5 g, 5.3 mmol) in dry acetonitrile (70 mL) was added N-bromosuccinimide (945 mg, 5.3 mmol), and the solution was stirred for 1 h at room temperature. The reaction mixture was purified by silica gel column chromatography (n-Hex–EtOAc 5:1) to afford 26 as colorless oil (2.0 g, 3.7 mmol 70%). IR (ATR) νmax: 1662 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.28–7.49 (overlapped, 3-O–CH2–Ph and 5-O–CH2–Ph), 7.05 (br s, H-12), 6.90 (br s, H-14), 6.66 (d, J = 2.7 Hz, H-6), 6.60 (br s, H-4), 5.13* (s, 5-O–CH2–Ph), 4.98* (s, 3-O–CH2–Ph), 3.89 (s, 11-OMe), 3.69 (s, 10-OMe), 2.33 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 195.6 (C-8), 158.6** (C-5), 155.7** (C-3), 152.7 (C-10), 147.0 (C-11), 144.7 (C-7), 136.1, 136.1, 128.6, 128.2, 128.0, 127.6, 127.1, 127.0 (3-O–CH2–Ph and 5-O–CH2–Ph), 133.7 (C-9), 131.9 (C-13), 122.2 (C-14), 117.6 (C-12), 106.4 (C-2), 106.4 (C-6), 101.0 (C-4), 71.0*** (3-O–CH2–Ph), 70.4*** (5-O–CH2–Ph), 61.1 (10-OMe), 56.0 (11-OMe), 21.2 (13-Me), *,**,*** may be interchanged; HRFABMS m/z: 547.1108 [M]+ (calcd for C30H2879BrO5, 547.1042).
2,4-Bis(benzyloxy)-6-(2,3-dimethoxy-5-methylbenzoyl)benzonitrile (27)
To a solution of 26 (600.0 mg, 1.1 mmol) in dry N,N-dimethylformamide (10 mL) was added copper (I) cyanide (292.9 mg, 3.3 mmol), and the solution was stirred for 12 h at 110 °C. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated. The filtrate was purified by silica gel column chromatography (n-Hex–EtOAc 3:1) to afford 27 as colorless oil (506.2 mg, 1.0 mmol 93%). IR (ATR) νmax: 2222, 1674 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.31–7.45 (overlapped, 3-O–CH2–Ph and 5-O–CH2–Ph), 6.90 (d, J = 2.0 Hz, H-12), 6.88 (d, J = 2.0 Hz, H-14), 6.71 (d, J = 2.2 Hz, H-4), 6.69 (d, J = 2.2 Hz, H-6), 5.20* (s, 3-O–CH2–Ph), 5.00* (s, 5-O–CH2–Ph), 3.86 (s, 11-OMe), 3.59 (s, 10-OMe), 2.34 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 193.9 (C-8), 162.5 (C-5), 162.4 (C-3), 152.4 (C-11), 146.2 (C-10), 145.7 (C-7), 135.3, 132.2, 128.7, 128.2, 127.7, 126.9 (3-O–CH2–Ph and 5-O–CH2–Ph), 134.1 (C-13), 132.1 (C-9), 121.5 (C-14), 117.2 (C-12), 114.6 (C-1), 108.6 (C-6), 102.7 (C-4), 93.2 (C-2), 71.0** (3-O–CH2–Ph), 70.7** (5-O-CH2-Ph), 61.6 (10-OMe), 55.9 (11-OMe), 21.2 (13-Me), *, ** may be interchanged; HRFABMS m/z: 494.1962 [M + H]+ (calcd for C31H28NO5, 494.1967).
(±)-5,7-Bis(benzyloxy)-3-(2,3-dimethoxy-5-methylphenyl)-3-hydroxyisoindolin-1-one (28)
To a solution of 27 (340.0 mg, 0.69 mmol) in dry acetonitrile (10 mL) was added 0.3 mol/L potassium hydroxide solution (6.8 mL), and the solution was stirred for 12 h at room temperature. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 to afford 28 (311.5 mg, 0.61 mmol 88%) as white powder. IR (ATR) νmax: 3356, 3203, 1687 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.42 (s, NH), 7.28–7.54 (overlapped, 3-O–CH2–Ph and 5-O–CH2–Ph), 7.25 (d, J = 2.1 Hz, H-14), 6.82 (d, J = 2.1 Hz, H-12), 6.66 (d, J = 1.9 Hz, H-4), 6.49 (br s, 8-OH), 6.32 (d, J = 1.9 Hz, H-6), 5.27 (d, J = 12.8 Hz, 5-O–CH2–Ph), 5.22 (d, J = 12.8 Hz, 5-O–CH2–Ph), 5.01 (d, J = 11.8 Hz, 3-O–CH2–Ph), 4.98 (d, J = 11.8 Hz, 3-O–CH2–Ph), 3.70 (s, 11-OMe), 3.09 (s, 10-OMe), 2.28 (s, 13-Me); 13C NMR (100 MHz, DMSO-d6): δ 167.7 (C-1), 163.2 (C-5), 156.7 (C-3), 156.4 (C-7), 152.7 (C-11), 144.3 (C-10), 137.5, 136.9, 128.8, 128.3, 128.1, 127.6 (5-O–CH2–Ph and 5-O–CH2–Ph), 134.7 (C-9), 132.1 (C-13), 120.2 (C-14), 114.1 (C-12), 112.8 (C-2), 101.5 (C-6), 100.8 (C-4), 84.5 (C-8), 70.1 (3-O–CH2–Ph), 70.1 (5-O–CH2–Ph), 59.6 (11-OMe), 56.1 (10-OMe), 21.7 (13-Me); HRFABMS m/z: 512.2071 [M + H]+ (calcd for C31H30NO6, 512.2073).
(±)-3-(2,3-Dimethoxy-5-methylphenyl)-5,7-dihydroxyisoindolin-1-one (29)
The solution of 28 (150.0 mg 0.29 mmol) and palladium 10% on carbon (75.0 mg) in ethanol (10 mL) was stirred under hydrogen for 2 h at room temperature. The reaction mixture was then filtered through cerite, and the filtrate was concentrated. The crude mixture was purified by silica gel column chromatography (CHCl3–MeOH 30:1) to afford 29 as white powder (73.0 mg, 0.23 mmol 79%). IR (ATR) νmax: 3277, 1651 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 9.85 (br s, 5-OH), 9.40 (br s, 3-OH), 8.33 (br s, NH), 6.80 (d, J = 2.1 Hz, H-12), 6.33 (d, J = 1.9 Hz, H-4), 6.16 (d, J = 2.1 Hz, H-14), 6.11 (d, J = 1.9 Hz, H-6), 5.72 (s, H-8), 3.81 (s, 11-OMe), 3.76 (s, 10-OMe), 2.18 (s, 13-Me); 13C NMR (100 MHz, DMSO-d6): δ 170.8 (C-1), 162.9 (C-5), 156.6 (C-3), 152.3* (C-11), 152.6* (C-7), 144.7 (C-10), 133.8** (C-13), 133.2** (C-9), 118.6 (C-14), 113.3 (C-12), 108.9 (C-2), 102.1*** (C-4), 102.0*** (C-6), 61.1 (10-OMe), 56.1 (11-OMe), 54.1 (C-8), 21.3 (13-Me); HRFABMS m/z: 316.1191 [M + H]+ (calcd for C17H17NO5, 316.1185).
(±)-3-(2,3-Dimethoxy-5-methylphenyl)-7-hydroxy-5-methoxyisoindolin-1-one (30)
To a solution of 29 (30.0 mg, 0.095 mmol) and potassium carbonate (32.1 mg, 0.29 mmol) in dry acetone (10 mL) was added iodomethane (27.0 mg, 0.19 mmol), and the solution was stirred for 12 h at 80 °C. The resulting mixture was concentrated. The crude mixture was purified by silica gel column chromatography (CHCl3–MeOH 100:1) and preparative HPLC (CHCl3–MeOH 200:1) to afford 30 as white powder (8.6 mg, 0.020 mmol 27%, tR 8.0 min); IR (ATR) νmax: 3182, 1683 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 9.61 (br s, 3-OH), 8.41 (br s, NH), 6.78 (br s, H-12), 6.31 (overlapped, H-4 and H-14), 6.16 (br s, H-6), 5.73 (s, H-8), 3.78 (s, 11-OMe), 3.73 (s, 10-OMe), 3.65 (s, 5-OMe), 2.15 (s, 13-Me); 13C NMR (100 MHz, DMSO-d6): δ 170.5 (C-1), 164.3 (C-5), 156.7 (C-3), 152.6 (C-11), 152.2 (C-7), 144.8 (C-10), 133.8 (C-13), 132.8 (C-9), 118.9 (C-14), 113.4 (C-12), 110.5 (C-2), 101.0 (C-4), 100.3 (C-6), 56.1 (11-OMe), 55.9 (5-OMe), 54.4 (C-8), 31.1 (10-OMe), 21.3 (13-Me); HRFABMS m/z: 330.1356 [M + H]+ (calcd for C18H20NO5, 330.1341).
(2-Bromo-3,5-dihydroxyphenyl)(2-hydroxy-3-methoxy-5-methylphenyl)methanone (31)
To a solution of 26 (2.0 g, 3.7 mmol) in dry dichloromethane (40 mL) was added boron trichloride in dichloromethane solution (1.0 mol/L, 12 mL, 12.0 mmol) dropwise at 0 °C. The solution was stirred for 1 h. The resulting mixture was diluted with water and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated. The crude mixture was purified by silica gel column chromatography (n-Hex–EtOAc 2:1) to afford 31 as yellow powder (1.2 g, 3.2 mmol 86%); IR (ATR) νmax: 3404, 1610 cm–1; 1H NMR (400 MHz, CDCl3): δ 11.41 (s, 10-OH), 10.47* (br s, 3-OH), 9.87* (br s, 5-OH), 7.11 (d, J = 2.0 Hz, H-12), 6.58 (d, J = 2.0 Hz, H-14), 6.54 (d, J = 2.7 Hz, H-4), 6.23 (d, J = 2.7 Hz, H-6), 3.80 (s, 11-OMe), 2.16 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 200.6 (C-8), 155.0 (C-3), 157.4 (C-5), 149.8 (C-10), 148.0 (C-11), 141.1 (C-7), 127.6 (C-13), 122.8 (C-14), 119.4 (C-12), 118.9 (C-9), 105.9 (C-6), 104.1 (C-4), 94.9 (C-2), 55.8 (11-OMe), 20.4 (13-Me); HRFABMS m/z: 351.9953 [M]+ (calcd for C15H1379BrO5, 351.9946).
(2-Bromo-5-hydroxy-3-(methoxymethoxy)phenyl) (2-hydroxy-3-methoxy-5-methylphenyl)methanone (32)
To a solution of 31 (300.0 mg, 0.82 mmol) and N,N-diisopropylethylamine (211.0 mg, 1.64 mmol) in dry dichloromethane (20 mL) was added chloromethyl methyl ether (77.3 mg, 0.90 mmol) dropwise at 0 °C. The solution was stirred for 1 h. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated. The crude mixture was purified by silica gel column chromatography (n-Hex–EtOAc 2:1) to afford 32 as yellow oil (171.0 mg, 0.43 mmol 53%); IR (ATR) νmax: 3419, 1631 cm–1; 1H NMR (400 MHz, CDCl3): δ 11.98 (s, 10-OH), 6.92 (br s, H-12), 6.84 (d, J = 2.7 Hz, H-4), 6.65 (br s, H-14), 6.46 (d, J = 2.7 Hz, H-6), 5.86 (br s, 5-OH), 5.28 (s, 3-O-CH2-OMe), 3.92 (s, 11-OMe), 3.55 (s, 3-O–CH2–OMe), 2.22 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 201.0 (C-8), 156.0 (C-5), 155.0 (C-3), 151.5 (C-10), 148.5 (C-11), 141.4 (C-7), 128.0 (C-13), 123.9 (C-14), 122.5 (C-9), 119.3 (C-12), 108.3 (C-6), 104.8 (C-4), 100.0 (C-2), 95.3 (3-O–CH2–OMe), 56.7* (3-O–CH2–OMe), 56.6* (11-OMe), 21.1 (13-Me), *may be interchanged; HRFABMS m/z: 397.0295 [M + H]+ (calcd for C17H1879BrO6, 397.0287).
(2-Bromo-5-methoxy-3-(methoxymethoxy)phenyl)(2,3-dimethoxy-5-methylphenyl)methanone (33)
To a solution of 32 (215.0 mg, 0.54 mmol) and potassium carbonate (374.0 mg, 2.7 mmol) in dry acetone (20 mL) was added iodomethane (27.0 mg, 0.19 mmol), and the solution was stirred for 12 h at 80 °C. The resulting mixture was concentrated and purified by silica gel column chromatography (n-Hex–EtOAc 3:1) afforded 33 as colorless oil (156.0 mg, 0.37 mmol 68%); IR (ATR) νmax: 1664 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.05 (d, J = 2.1 Hz, H-14), 6.91 (d, J = 2.1 Hz, H-12), 6.82 (d, J = 2.8 Hz, H-4), 6.57 (d, J = 2.8 Hz, H-6), 5.25 (s, 3-O–CH2–OMe), 3.86 (s, 11-OMe), 3.78 (s, 5-OMe), 3.58 (s, 10-OMe), 3.51 (s, 3-O–CH2–OMe), 2.34 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 195.6 (C-8), 159.5 (C-5), 154.5 (C-3), 152.8 (C-11), 147.0 (C-10), 144.6 (C-7), 133.7 (C-13), 131.9 (C-9), 122.3 (C-14), 117.6 (C-12), 106.9 (C-6), 104.3 (C-4), 100.9 (C-2), 95.3 (3-O–CH2–OMe), 61.1 (10-OMe), 56.4 (3-O–CH2–OMe), 56.0 (11-OMe), 55.7 (5-OMe), 21.2 (13-Me); HRFABMS m/z: 425.0580 [M + H]+ (calcd for C19H2279BrO6, 425.0600).
2-(2,3-Dimethoxy-5-methylbenzoyl)-4-methoxy-6-(methoxymethoxy)benzonitrile (34)
To a solution of 33 (156.0 mg, 0.37 mmol) in dry N,N-dimethylformamide (5.0 mL) was added copper (I) cyanide (163.3 mg, 1.8 mmol), and the solution was stirred for 48 h at 110 °C. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated. The filtrate was purified by silica gel column chromatography (n-Hex–EtOAc 2:1) to afford 34 as colorless oil (68.6 mg, 0.18 mmol 50%). IR (ATR) νmax: 2222, 1668 cm–1; 1H NMR (400 MHz, CDCl3): δ 6.92 (d, J = 2.1 Hz, H-12), 6.90 (d, J = 2.1 Hz, H-14), 6.89 (d, J = 2.3 Hz, H-4), 6.71 (d, J = 2.3 Hz, H-6), 5.31 (s, 3-O–CH2–OMe), 3.87 (s, 11-OMe), 3.82 (s, 5-OMe), 3.65 (s, 10-OMe), 3.54 (s, 3-O–CH2–OMe), 2.35 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 193.8 (C-8), 161.6 (C-3), 163.3 (C-5), 152.4 (C-11), 145.4 (C-7), 146.2 (C-10), 134.1 (C-13), 132.0 (C-9), 121.5 (C-14), 117.3 (C-12), 114.7 (C-1), 109.4 (C-6), 103.3 (C-4), 95.1 (3-O-CH2-OMe), 93.2 (C-2), 61.7 (10-OMe), 56.7 (3-O–CH2–OMe), 55.9 (5-OMe), 55.9 (11-OMe), 21.2 (13-Me); HRFABMS m/z: 371.1366 [M]+ (calcd for C20H21NO6, 371.1369).
3-(2,3-Dimethoxy-5-methylphenyl)-3-hydroxy-5-methoxy-7-(methoxymethoxy)isoindolin-1-one (35)
To a solution of 34 (68.4 mg, 0.19 mmol) in dry acetonitrile (10 mL) was added 0.3 mol/L potassium hydroxide solution (1.0 mL), and the solution was stirred for 12 h at room temperature. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 to afford 35 (67.9 mg, 0.18 mmol 94%) as white powder. IR (ATR) νmax: 3336, 3224, 1693 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 6.77 (br s, H-14), 6.72 (overlapped, H-6), 6.72 (overlapped, H-12), 6.62 (d, J = 2.0 Hz, H-4), 6.51 (br s, NH), 5.32 (s, 3-O-CH2-OMe), 4.73 (br s, 8-OH), 3.84 (s, 5-OMe), 3.81 (s, 10-OMe), 3.81 (s, 11-OMe), 3.52 (s, 3-O–CH2–OMe), 2.25 (s, 13-Me);; 13C NMR (100 MHz, DMSO-d6): δ 167.8 (C-1), 165.0 (C-11), 156.0 (C-3), 153.4 (C-7), 152.6 (C-5), 144.5 (C-10), 133.6 (C-13), 132.4 (C-9), 119.3 (C-14), 114.1 (C-12), 111.4 (C-2), 103.1 (C-6), 102.0 (C-4), 86.7 (C-8), 55.8 (5-OMe), 95.1 (3-O–CH2–OMe), 56.5 (3-O–CH2–OMe), 61.1 (10-OMe), 55.9 (11-OMe), 21.4 (13-Me); HRFABMS m/z: 390.1570 [M + H]+ (calcd for C20H24NO7, 390.1553).
3-(2,3-Dimethoxy-5-methylphenyl)-3,7-dihydroxy-5-methoxyisoindolin-1-one (36)
To a solution of 35 (10.0 mg, 0.026 mmol) in dry 1,4-dioxiane (3.0 mL) was added 10% hydrochloric acid (0.6 mL), and the solution was stirred for 12 h at room temperature. The resulting mixture was quenched with sodium bicarbonate solution and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated. The filtrate was purified by silica gel column chromatography (CHCl3–MeOH 50:1) to afford 36 as colorless oil (7.2 mg, 0.021 mmol 81%). IR (ATR) νmax: 3377, 3244, 1674 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 9.45 (br s, 3-OH), 8.49 (br s, NH), 7.25 (d, J = 2.1 Hz, H-14), 6.82 (d, J = 2.1 Hz, H-12), 6.54 (br s, 8-OH), 6.30 (d, J = 2.0 Hz, H-4), 6.11 (d, J = 2.0 Hz, H-6), 3.70 (s, 11-OMe), 3.65 (s, 5-OMe), 3.15 (s, 10-OMe), 2.28 (s, 13-Me); 13C NMR (100 MHz, DMSO-d6): δ 169.3 (C-1), 164.3 (C-5), 156.1 (C-3), 155.3 (C-7),152.8 (C-11), 144.3 (C-10), 134.7 (C-9), 132.2 (C-13), 120.0 (C-12), 114.1 (C-14), 110.2 (C-2), 101.5 (C-6), 100.0 (C-4), 85.2 (C-8), 59.6 (10-OMe), 56.0 (5-OMe), 56.0 (11-OMe), 21.7 (13-Me); HRFABMS m/z: 345.1217 [M]+ (calcd for C18H19NO6, 345.1212).
(5-(Benzyloxy)-2-bromo-3-(methoxymethoxy)phenyl) (2-(benzyloxy)-3-methoxy-5-methylphenyl)methanone (37)
To a solution of 32 (99.6 mg, 0.25 mmol) and potassium carbonate (173.0 mg, 1.3 mmol) in dry acetone (10 mL) was added benzyl bromide (213.8 mg, 1.3 mmol), and the solution was stirred for 12 h at 80 °C. The resulting mixture was concentrated and purified by silica gel column chromatography (n-Hex–EtOAc 5:1) to afford 37 as colorless oil (91.6 mg, 0.16 mmol 63%); IR (ATR) νmax: 1662 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.17–7.36 (overlapped, 5-O–CH2–Ph and 10-O–CH2–Ph), 7.07 (d, J = 2.3 Hz, H-14), 6.94 (d, J = 2.3 Hz, H-12), 6.79 (d, J = 2.8 Hz, H-4), 6.61 (d, J = 2.8 Hz, H-6), 4.89 (s, 5-O–CH2–Ph), 4.79 (s, 3-O–CH2–OMe), 3.86 (s, 11-OMe), 3.47 (s, 3-O–CH2–OMe), 2.36 (s, 13-Me), 5.15 (s, 10-O–CH2–Ph); 13C NMR (100 MHz, CDCl3): δ 195.4 (C-8), 158.5 (C-5), 154.7 (C-3), 152.8 (C-11), 145.6 (C-10), 144.2 (C-7), 137.4, 136.2, 128.6, 128.2, 128.0, 127.7, 127.6, 127.5 (5-O–CH2–Ph and 10-O-CH2-Ph), 134.0 (C-9), 132.5 (C-13), 122.6 (C-14), 117.7 (C-12), 108.3 (C-6), 105.1 (C-4), 101.4 (C-2), 95.3 (3-O–CH2–OMe), 74.9 (10-O–CH2–Ph), 70.4 (5-O–CH2–Ph), 56.4 (3-O–CH2–OMe), 56.0 (11-OMe), 21.3 (13-Me); HREIMS m/z: 576.1146 [M]+ (calcd for C31H2979BrO6, 576.1148).
(5-(Benzyloxy)-2-bromo-3-hydroxyphenyl) (2-(benzyloxy)-3-methoxy-5-methylphenyl)methanone (38)
To a solution of 37 (121.1 mg, 0.21 mmol) in dry 1,4-dioxiane (10.0 mL) was added 10% hydrochloric acid (1.7 mL), and the solution was stirred for 2 h under reflux. The resulting mixture was quenched with sodium bicarbonate solution and extracted with dichloromethane. The organic layer was dried over Na2SO4 and concentrated. The filtrate was purified by silica gel column chromatography (n-Hex–EtOAc 5:1) to afford 38 as colorless oil (70.0 mg, 0.13 mmol 63%). IR (ATR) νmax: 3392, 1660 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.12–7.35 (overlapped, 5-O–CH2–Ph and 10-O–CH2–Ph), 6.98 (d, J = 2.3 Hz, H-14), 6.93 (d, J = 2.3 Hz, H-12), 6.66 (d, J = 2.9 Hz, H-4), 6.57 (d, J = 2.9 Hz, H-6), 5.77 (s, 3-OH), 4.91 (s, 5-O–CH2–Ph), 4.78 (s, 10-O–CH2–Ph), 3.86 (s, 11-OMe), 2.35 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 195.1 (C-8), 158.8 (C-5), 153.5 (C-3), 152.7 (C-11), 145.3 (C-10), 142.3 (C-7), 137.1, 136.1, 128.6, 128.2, 128.1, 127.7, 127.6 (5-O–CH2–Ph and 10-O–CH2–Ph), 134.1 (C-9), 132.7 (C-13), 122.2 (C-14), 117.4 (C-12), 109.7 (C-6), 104.1 (C-4), 99.3 (C-2), 75.1 (10-O–CH2–Ph), 70.4 (5-O–CH2–Ph), 56.0 (11-OMe), 21.3 (13-Me); HREIMS m/z: 532.0877 [M]+ (calcd for C29H2579BrO5, 532.0885).
(5-(Benzyloxy)-2-bromo-3-methoxyphenyl) (2-(benzyloxy)-3-methoxy-5-methylphenyl)methanone (39)
To a solution of 38 (55.5 mg, 0.10 mmol) and potassium carbonate (71.8 mg, 0.52 mmol) in dry acetone (10 mL) was added iodomethane (146.6 mg, 1.0 mmol), and the solution was stirred for 3 h at 80 °C. The resulting mixture was concentrated and purified by silica gel column chromatography (n-Hex–EtOAc 4:1) to afford 39 as colorless oil (55.0 mg, 0.10 mmol 97%); IR (ATR) νmax: 1660 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.17–7.37 (overlapped, 5-O–CH2–Ph and 10-O–CH2–Ph), 7.06 (d, J = 2.3 Hz, H-14), 6.94 (d, J = 2.3 Hz, H-12), 6.55 (d, J = 2.8 Hz, H-4), 6.47 (d, J = 2.8 Hz, H-6), 4.89 (s, 5-O-CH2-Ph), 4.81 (s, 10-O-CH2-Ph), 3.86 (s, 11-OMe), 3.80 (s, 3-OMe), 2.35 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 195.6 (C-8), 156.6 (C-3), 158.8 (C-5), 152.8 (C-11), 145.6 (C-10), 144.2 (C-7), 137.4, 136.2, 128.6, 128.2, 128.0, 127.8, 127.7, 127.3 (5-O–CH2–Ph and 10-O–CH2–Ph), 133.9 (C-9), 132.4 (C-13), 122.6 (C-14), 106.0 (C-6), 117.7 (C-12), 101.7 (C-4), 100.5 (C-2), 74.8 (10-O–CH2–Ph), 70.4 (5-O–CH2–Ph), 56.4 (3-OMe), 56.0 (11-OMe), 21.2 (13-Me); HREIMS m/z: 546.1038 [M]+ (calcd for C30H2779BrO5, 546.1042).
(5-(Benzyloxy)-2-cyano-3-methoxyphenyl) (2-(benzyloxy)-3-methoxy-5-methylphenyl)methanone (40)
To a solution of 39 (32.5 mg, 0.078 mmol) in dry N,N-dimethylformamide (3.0 mL) was added copper (I) cyanide (34.5 mg, 0.39 mmol), and the solution was stirred for 12 h at 110 °C. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated. The filtrate was purified by silica gel column chromatography (n-Hex–EtOAc 3:1) to afford 40 as colorless oil (25.2 mg, 0.051 mmol 66%). IR (ATR) νmax: 2220, 1672 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.10–7.38 (overlapped, 5-O–CH2–Ph and 10-O–CH2–Ph), 6.94 (overlapped, H-12 and H-14), 6.64 (d, J = 2.3 Hz, H-4), 6.48 (d, J = 2.3 Hz, H-6), 4.95 (s, 5-O–CH2–Ph), 4.86 (s, 10-O–CH2–Ph), 3.88 (s, 11-OMe), 3.83 (s, 3-OMe), 2.36 (s, 13-Me); 13C NMR (100 MHz, CDCl3): δ 193.9 (C-8), 163.4 (C-5), 162.5 (C-3), 152.5 (C-11), 145.6 (C-10), 145.0 (C-7), 70.6 (5-O-CH2-Ph), 137.1, 135.4, 128.8, 128.6, 128.1, 127.7, 127.6, 127.6 (5-O–CH2–Ph and 10-O–CH2–Ph), 134.3 (C-9), 132.4 (C-13), 121.9 (C-14), 117.5 (C-12), 114.7 (C-1), 108.0 (C-6), 101.1 (C-4), 92.8 (C-2), 75.4 (10-O-CH2-Ph), 56.3 (3-OMe), 56.0 (11-OMe), 21.3 (13-Me); HREIMS m/z: 493.1891 [M]+ (calcd for C31H27NO5, 493.1889).
(±)-5-(Benzyloxy)-3-(2-(benzyloxy)-3-methoxy-5-methylphenyl)-3-hydroxy-7-methoxyisoindolin-1-one (41)
To a solution of 40 (24.0 mg, 0.49 mmol) in dry acetonitrile (6.0 mL) was added 0.3 mol/L potassium hydroxide solution (0.6 mL), and the solution was stirred for 12 h at room temperature. The resulting mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4 to afford 41 (11.4 mg, 0.022 mmol 46%) as white powder. IR (ATR) νmax: 3336, 3207, 1676 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.51 (s, NH), 7.32 (overlapped, H-14), 7.24–7.46 (overlapped, 5-O–CH2–Ph and 10-O–CH2–Ph), 6.97 (d, J = 2.3 Hz, H-12), 6.60 (br s, 8-OH), 6.58 (d, J = 1.9 Hz, H-4), 6.48 (d, J = 1.9 Hz, H-6), 5.09 (s, 5-O–CH2–Ph), 4.64 (d, J = 10.2 Hz, 10-O–CH2–Ph), 3.96 (d, J = 10.2 Hz, 10-O–CH2–Ph), 3.83* (s, 11-OMe), 3.80* (s, 3-OMe), 2.39 (s, 13-Me); 13C NMR (100 MHz, DMSO-d6): δ 167.7 (C-1), 163.5 (C-5), 158.0 (C-3), 156.4 (C-7), 152.8 (C-11), 143.4 (C-10), 138.2, 137.0, 128.8, 128.5, 128.4, 128.3, 128.3, 127.8 (5-O–CH2–Ph and 10-O–CH2–Ph), 135.2 (C-9), 132.5 (C-13), 120.3 (C-14), 114.2 (C-12), 112.2 (C-2), 100.5 (C-6), 99.8 (C-4), 84.7 (C-8), 73.7 (10-O–CH2–Ph), 70.1 (5-O–CH2–Ph), 56.2** (11-OMe), 56.0** (3-OMe), 21.7 (13-Me), *, ** may be interchanged; HRFABMS m/z: 511.2081 [M + H]+ (calcd for C31H30NO6, 512.2073).
(±)-Entonalactam A (6)
The solution of 41 (9.0 mg 0.018 mmol) and palladium 10% on carbon (5.0 mg) in ethanol (3.0 mL) was stirred under hydrogen for 12 h at room temperature. The reaction mixture was then filtered through cerite, and the filtrate was concentrated. The crude mixture was purified by silica gel column chromatography (CHCl3–MeOH 30:1) to afford 6 as white powder (4.5 mg, 0.014 mmol 81%). IR (ATR) νmax: 3315, 1656 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 6.28 (br s, H-4), 6.32 (br s, H-6), 5.77 (s, H-8), 6.69 (d, J = 2.1 Hz, H-12), 6.30 (d, J = 2.1 Hz, H-14), 9.95 (br s, 5-OH), 8.76 (br s, 10-OH), 3.76 (s, 3-OMe), 3.80 (s, 11-OMe), 2.13 (s, 13-Me), 8.10 (br s, NH); 13C NMR (100 MHz, DMSO-d6): δ 169.0 (C-1), 162.4 (C-5), 157.9 (C-3), 153.4 (C-7), 147.5 (C-11), 141.4 (C-10), 127.7 (C-13), 126.6 (C-9), 117.5 (C-14), 111.4 (C-12), 109.8 (C-2), 101.9 (C-6), 98.4 (C-4), 55.8 (11-OMe), 55.2 (3-OMe), 52.7 (C-8), 20.7 (13-Me); HREIMS m/z: 315.1106 [M]+ (calcd for C17H17NO5, 315.1107).
MAO-A and -B Inhibitory Assay
MAO-A and MAO-B inhibitory activities were assayed using the method in our previous report with slight modification.16 Human recombinant MAO-A solution (3 μL, M7316, Sigma-Aldrich, St. Louis, MO) or 7 μL of MAO-B solution (M7441, Sigma-Aldrich) was diluted with 1100 μL of potassium phosphate buffer (0.1 M, pH 7.4). Potassium phosphate buffer (140 μL), 8 μL of kynuramine (final concentration is 30 μM, Sigma-Aldrich) in potassium phosphate buffer, and 2 μL of a dimethyl sulfoxide (DMSO) inhibitor solution [final DMSO concentration of 1% (v/v)] were mixed and preincubated at 37 °C for 10 min. Diluted MAO-A or MAO-B solution (50 μL) was then added to each well. The reaction mixture was further incubated at 37 °C, and the reaction was stopped after 20 min by the addition of 75 μL of 2 M NaOH. The product generated by MAO-A or MAO-B, 4-quinolinol, is fluorescent and was measured at Ex 310 nm/Em 400 nm using a microplate reader (SPECTRA MAX M2, Molecular Devices, Tokyo, Japan). DMSO without the test compound was used as the negative control, and pargyline (Sigma-Aldrich) was used as a positive control.17 The IC50 values were estimated using Prism software (version 5.02; GraphPad, San Diego, CA).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06260.
Copies of NMR spectra of all of the compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Choomuenwai V.; Beattie K. D.; Healy P. C.; Andrews K. T.; Fechner N.; Davis R. A. Entonalactams A-C: Isoindolinone derivatives from an Australian rainforest fungus belonging to the genus Entonaema. Phytochemistry 2015, 117, 10–16. 10.1016/j.phytochem.2015.05.018. [DOI] [PubMed] [Google Scholar]
- Lee I. K.; Kim S. E.; Yeom J. H.; Ki D. W.; Lee M. S.; Song J. G.; Kim Y. S.; Seok S. J.; Yun B. S. Daldinan A, a novel isoindolinone antioxidant from the ascomycete Daldinia concentrica. J. Antibiot. 2012, 65, 95–97. 10.1038/ja.2011.109. [DOI] [PubMed] [Google Scholar]
- Kamauchi H.; Shiraishi Y.; Kojima A.; Kawazoe N.; Kinoshita K.; Koyama K. Isoindolinones, Phthalides, and a Naphthoquinone from the Fruiting Body of Daldinia concentrica. J. Nat. Prod. 2018, 81, 1290–1294. 10.1021/acs.jnatprod.7b00976. [DOI] [PubMed] [Google Scholar]
- Ki D. W.; Kim S. E.; Kim J. Y.; Song J. G.; Hwang B. S.; Lee I. K.; Yun B. S. Daldinans D–G, new isoindolinone antioxidants isolated from the ascomycete Daldinia concentrica. J. Nat. Med. 2022, 76, 476–481. 10.1007/s11418-021-01592-0. [DOI] [PubMed] [Google Scholar]
- Herraiz T.; Flores A.; Fernández L. Analysis of monoamine oxidase (MAO) enzymatic activity by high-performance liquid chromatography-diode array detection combined with an assay of oxidation with a peroxidase and its application to MAO inhibitors from foods and plants. J. Chromatogr., B 2018, 1073, 136–144. 10.1016/j.jchromb.2017.12.004. [DOI] [PubMed] [Google Scholar]
- Ugun-Klusek A.; Theodosi T. S.; Fitzgerald J. C.; Burté F.; Ufer C.; Boocock D. J.; Yu-Wai-Man P.; Bedford L.; Billett E. E. Monoamine oxidase-A promotes protective autophagy in human SH-SY5Y neuroblastoma cells through Bcl-2 phosphorylation. Redox Biol. 2019, 20, 167–181. 10.1016/j.redox.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiuccariello L.; Cooke R. G.; Miler L.; Levitan R. D.; Baker G. B.; Kish S. J.; Kolla N. J.; Rusjan P. M.; Houle S.; Wilson A. A.; Meyer J. H. Monoamine Oxidase-A Occupancy by Moclobemide and Phenelzine: Implications for the Development of Monoamine Oxidase Inhibitors. Int. J. Neuropsychopharmacol. 2016, 19, pyv078. 10.1093/ijnp/pyv078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereda E.; Cilia R.; Canesi M.; Tesei S.; Mariani C. B.; Zecchinelli A. L.; Pezzoli G. Efficacy of rasagiline and selegiline in Parkinson’s disease: a head-to-head 3-year retrospective case-control study. J. Neurol. 2017, 264, 1254–1263. 10.1007/s00415-017-8523-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Nair A. S.; Abdelgawad M. A.; Mathew B. Exploration of the Detailed Structure-Activity Relationships of Isatin and Their Isomers As Monoamine Oxidase Inhibitors. ACS Omega 2022, 7, 16244–16259. 10.1021/acsomega.2c01470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu X.; Chen J.; Deady L. W.; Smith C. L.; Baguley B. C.; Greenhalgh D.; Yang S.; Denny W. A. Synthesis and cytotoxic activity of N-[(alkylamino)alkyl]carboxamide derivatives of 7-oxo-7H-benz[de]anthracene, 7-oxo-7H-naphtho[1,2,3-de]quinoline, and 7-oxo-7H-benzo[e]perimidine. Bioorg. Med. Chem. 2005, 13, 3657–3665. 10.1016/j.bmc.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Di Mola A.; Macchia A.; Tedesco C.; Pierri G.; Palombi L.; Filosa R.; Massa A. Synthetic Strategies and Cascade Reactions of 2-Cyanobenzophenones for the Access to Diverse 3,3-Disubstituted Isoindolinones and 3-Aryl-3-Hydroxyisoindolinones. Chemistry Select 2019, 4, 4820–4826. 10.1002/slct.201901045. [DOI] [Google Scholar]
- Zhao W.; Feng X.; Ban S.; Lin W.; Li Q. Synthesis and biological activity of halophenols as potent antioxidant and cytoprotective agents. Bioorg. Med. Chem. Lett. 2010, 20, 4132–4134. 10.1016/j.bmcl.2010.05.068. [DOI] [PubMed] [Google Scholar]
- Reddy M. P.; Rao B.; Usharani V.; Dubey P. K. Novel and Improved Process for the Preparation of Citalopram. Asian J. Chem. 2011, 23, 1829–1832. [Google Scholar]
- Kamauchi H.; Hirata M.; Takao K.; Sugita Y. Synthesis and pharmacological evaluation of childinin E and several derivatives as anti-hyphal formation inhibitors against Candida albicans. Tetrahedron Lett. 2020, 61, 152588. 10.1016/j.tetlet.2020.152588. [DOI] [Google Scholar]
- Sagirova Z. R.; Starodubtseva E. V.; Turova O. V.; Vinogradov M. G. Hydrogenolysis of the C-O bond of hydroxylactams as a convenient method for the synthesis of substituted isoindolin-1-ones. Russ. Chem. Bull. 2013, 62, 1032–1037. 10.1007/s11172-013-0137-7. [DOI] [Google Scholar]
- Novaroli L.; Reist M.; Favre E.; Carotti A.; Catto M.; Carrupt P. A. Human recombinant monoamine oxidase B as reliable and efficient enzyme source for inhibitor screening. Bioorg. Med. Chem. 2005, 13, 6212–6217. 10.1016/j.bmc.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Thull U.; Carrupt P. A.; Testa B. Pargyline Analogues as Potent, Non-selective Monoamine Oxidase Inhibitors. Pharm. Pharmacol. Commun. 1998, 4, 579–581. 10.1111/j.2042-7158.1998.tb00678.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.