

Abstract
Decisions regarding the assignment of hormonal therapy for breast cancer are based solely upon the presence of nuclear estrogen receptors (ERs) in biopsied tumor tissue. This is despite the fact that the G-protein-coupled estrogen receptor (GPER) is linked to advanced breast cancer and is required for breast cancer stem cell survival; an observation which suggests that effective endocrine therapy should also target this receptor. Here, two ER/GPER-targeting proteolytic chimeras (UI-EP001 and UI-EP002) are described that effectively degrade ERα, ERβ and GPER. These chimeras form high affinity interactions with GPER and ER with binding dissociation constants of ~30 nM and 10–20 nM, respectively. Plasma membrane and intracellular GPERs and nuclear ERs were degraded by UI-EP001 and UI-EP002, but not by a partial PROTAC lacking its estrogen targeting domain. Pretreatment of cells with the proteasomal inhibitor, MG132, blocked UI-EP001 and UI-EP002 proteolysis, while the lysosomotrophic inhibitor, chloroquine, had no effect. Off-target activity was not observed against recombinant β1-adrenergic receptor or CXCR4. Target specificity was further demonstrated in human MCF-7 cells where both drugs effectively degraded ERα, ERβ, and GPER yet spared the progesterone receptor (PR). UI-EP001 and UI-EP002 induced cytotoxicity and G2/M cell cycle arrest in MCF-7 breast cancer and human SKBR3 (ERα-ERβ-GPER+) breast cancer cells but not human MDA-MB-231 breast cancer cells which do not express functional GPERs/ERs. These results suggest that it is possible to develop a receptor-based strategy of anti-estrogen treatment for breast cancer that targets both plasma membrane and intracellular estrogen receptors.
Keywords: PROTAC, estrogens receptor, GPER, endocrine therapy, breast cancer
Graphical Abstract
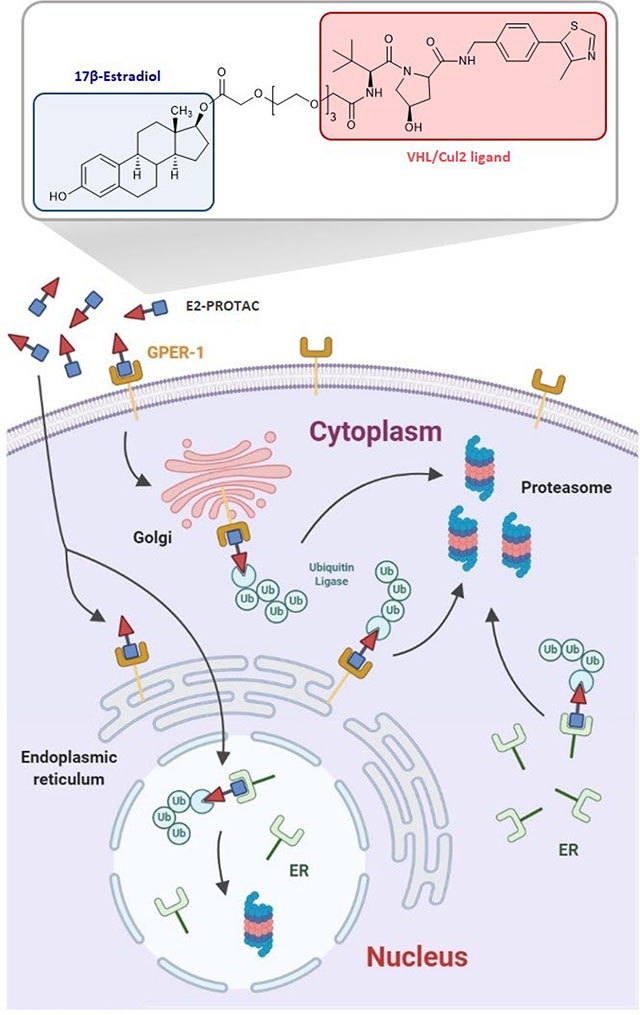
Introduction.
Epidemiological, clinical and preclinical evidence suggests that breast cancer is an estrogen-driven malignancy [1–5]. This explains the broad success of anti-estrogens as an effective adjuvant treatment for early stage ER (+) breast cancer [6–7]. Two classes of pharmacological agents are used to antagonize estrogen action: 1) compounds that inhibit aromatase (aromatase inhibitors, AIs), the enzyme that introconverts androgens to estrogens and 2) estrogen mimetics (ER antagonists) that competitively block the interaction of estrogen with the ER. In order to be effective, AIs or ER antagonists must be administered over a long-term drug schedule that lasts 3–5 years or longer. Ultimately, prolonged use of anti-estrogen therapy is associated with undesirable and sometimes intolerable side effects, including menopausal symptoms, osteoporosis, ostealgia and arthralgia [8]. In addition, long-term use of ER antagonists is linked with an increased risk of endometrial cancer [9–10] and thrombosis [11].
De-novo or acquired drug resistance further limits the use of anti-estrogen therapy, with resistance occurring in more than 20% of patients that are treated [12–15]. While de-novo resistance is attributed to intratumoral heterogeneity in ER expression at the time of diagnosis [16], acquired resistance reflects tumor heterogeneity that evolves due to the selective pressure applied by anti-estrogens during treatment. Examples include: a) the selection of mutations which result in a loss of drug-receptor interaction [17] or drive estrogen independent ER-mediated gene transactivation [18–19], b) epigenetic silencing of the ER promoter [20] and c) transcriptional upregulation of compensatory genes that drive estrogen-independent growth by regulating cell cycle activity [21] or signaling activity downstream of epidermal growth factor receptors (EGFRs) [22]. Yet another mechanism of anti-estrogen resistance is provided by the G-protein coupled estrogen receptor (GPER). This more recently appreciated estrogen receptor has clear importance for breast cancer biology and treatment [23]. Unlike nuclear estrogen receptors that primarily reside intracellularly and function as ligand-induced transcription factors, GPER is a Gs-coupled heptahelical transmembrane receptor that is located in the plasma membrane and intracellular membranes and promotes rapid pre-genomic actions, including activation of adenylyl cyclase [24] and transactivation of EGFRs [25]. Stimulation of GPER contributes to the activation of signaling effectors downstream of EGFRs such as Ras, PI3K, AKT, and Erk1/2 and are involved in cell proliferation, survival, invasion, and resistance to endocrine therapy [26]. Analysis of GPER expression in primary breast tumors demonstrates that its presence is linked with disease progression [27], survival of breast cancer stem cells [28], and tamoxifen resistance [29–31]. Furthermore, its expression is commonly retained in triple-negative breast cancers which lack ER, PR, and her-2/neu [32]. Thus, GPER broadens our ER-centric view of estrogen responsiveness [33] and disrupts the binary rubric that guides the rational assignment of adjuvant therapy for breast cancer. This is particularly important for patients receiving endocrine therapy because “partial” (tamoxifen) and “pure” (fulvestrant and raloxifene) ER antagonists function as GPER agonists [25,34].
Current clinical guidelines suggest the use of fulvestrant as a second-line therapy for endocrine-resistant breast cancer [35–36]. Fulvestrant is an estrogen mimetic that functions as a competitive inhibitor for ER but also acts as a selective ER degrader (SERD) [37]. Fulvestrant is the only FDA-approved drug in this class and upon binding ER, destabilizes its interaction with associated heat shock chaperone proteins resulting in the degradation of ER at the 26S proteasome [38]. In this manner, fulvestrant acts to reduce the bioavailability of its drug target, thus reducing the risk of secondary drug resistance. However, a major limitation of fulvestrant is its poor bioavailability following oral delivery; necessitating painful monthly intramuscular injections [39]. A revolutionary new method for selective degradation of the ER has been achieved by the use of PROteolytic TArgeting Chimeras (PROTACs) that utilize the ubiquitin-proteasome pathway [40–41]. In general, PROTACs are hetero-bifunctional compounds consisting of two functional binding domains that act to link a protein of interest directly to the ubiquitination machinery. They are comprised of a targeting domain that is coupled via a chemical spacer to an E3 ubiquitin ligase recognition motif. Upon binding, the PROTAC polyubiquitinylates its target and directs it towards the 26S proteasome for degradation. PROTACs offer the advantage that they have been used to target a broad spectrum of cytoplasmic, nuclear, and plasma membrane proteins [42], albeit efficient PROTAC degradation, requires a systematic approach to optimize the spatial positioning of the targeting and E3 ligase recruitment domains [43]. PROTACs provide the added benefit that they offer high specificity and processivity for their intended target protein; thus, effectively reducing the concentration of target molecules and decreasing the likelihood that drug-resistant mutants may evolve. This strategy has been used successfully to down-modulate ER and ER-PROTACs have been developed with targeting domains consisting of various estrogen derivatives including 17β-estradiol (17β-E2) [44], endoxifen [45], raloxifene [46], and tamoxifen [47]; each connected with different chemical spacers to protein or small molecule E3 ligase recruitment motifs. While ERα, ERβ and GPER differ in their binding affinities for natural and synthetic estrogens [48], they each show a high binding affinity for 17β-estradiol (E2) with dissociation constants measured in the low nanomolar range. However, PROTACs that target the ER have yet-to-been evaluated for their capacity to target GPER. A PROTAC approach for targeting GPER is particularly appealing since downmodulation of GPER from the plasma membrane occurs via a ubiquitin-proteasome degradation pathway [49].
As a first step towards developing GPER-specific PROTACs, we synthesized and characterized two E2 based PROTACs (UI-EP001 and UI-EP002) with regards to their capacity to interact with and degrade ERα, ERβ and GPER. These dual-specificity PROTACs were evaluated for their capacity to interact with the plasma membrane and intracellular estrogen receptors and promote ubiquitin-proteasome dependent degradation.
Materials and Methods
Molecular docking of E2-PROTAC to ERα or GPER.
Our previously established GPER homology models were used in the GLIDE (Schrödinger, Cambridge, MA) docking studies [50–51]. In these studies, a grid was first built around the previously established binding site of E2 using Receptor Grid Generation within GLIDE. UI-EP001 and UI-EP002 were drawn into Maestro 11.3 (Schrödinger, Cambridge, MA) and were prepared for docking using the LigPrep function within Schrödinger. The OPLS3 force field was used and possible ionization states at pH 7 ± 2 were generated using Epik; all other settings were kept at their default settings. GLIDE docking of UI-EP001 and UI-EP002 was then performed. Van der Waals radii were set to a scaling factor of 0.8 with a partial charge cutoff of 0.15. XP (extra precision) docking was performed with flexible ligand sampling with both sample nitrogen inversions and sample ring conformations turned on. Bias sampling for amides was set to penalize nonplanar conformations. Epik state penalties were added to the docking scores. For docking, 10,000 poses per ligand were kept for the initial phase of docking and the best 1,000 poses per ligand were kept for energy minimization using the OPLS3 force field. Post-docking minimization was performed and 100 poses per ligand were kept. All other settings were kept at their default settings. Both UI-EP001 and UI-EP002 were also docked within ERα in an analogous method. An ERα homodimer crystal structure was used (PDB:1A52 [52]) and prepared for docking by removing the bound E2 molecules and then using Receptor Grid Generation within GLIDE before docking studies with UI-EP001 and UI-EP002.
Synthesis of E2-PROTACs and E2-FITC.
Chemical synthesis of UI-EP001, UI-EP002, and E2-FITC was conducted according to the schematics in Scheme S1 and Scheme S4, respectively, the details of which are described in Supplementary Materials. 1H and 13C NMR spectra were recorded on a Bruker AVANCE AV-300 and Bruker AVANCE AV-500 instrument at 300 K. 1H-NMR spectra are reported in parts per million (ppm) downfield from tetramethylsilane (TMS). All 13CNMR spectra are reported in ppm and obtained with 1H decoupling. In the spectral data reported, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. MS analyses were carried out with Waters Q-Tof Premier mass spectrometer. UI-EP001 and UI-EP002 were purified by C18 reverse-phase preparative HPLC column with a Shimadzu Nexera X2 UHPLC System, with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in MeCN) as eluents.
Cells culture.
Human MCF-7, MDA-MB-231, HCC-1806, and SKBR3 breast carcinoma cells and human embryonic kidney 293 cells (HEK-293) were purchased from the American Tissue Culture Collection (Manassas, VA). HEK-293 cells that stably express hemagglutinin-tagged GPER (HA-GPER), β1-adrenergic receptor (HA-β1AR), and CXCR4 (HA-CXCR4) have been previously described [53]. All cell lines were cultured at 37°C in a humidified chamber containing 5% CO2 and in phenol red-free (1:1) DMEM/Ham’s F-12 medium (PRF-DF12) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and penicillin-streptomycin.
Steroids and inhibitors.
17β-estradiol (17β-E2) and aldosterone were purchased from Sigma-Aldrich (St.157 Louis, MO). The 26S-proteasome inhibitor, MG132 was purchased from Selleckchem (Pittsburgh, PA) and the lysosomotrophic agent, chloroquine diphosphate was purchased from Bio-Techne (Minneapolis, MN).
Antibodies.
GPER-specific antibodies were generated in New Zealand White Rabbits against a synthetic peptide derived from amino acids 1–62 from the N-terminus of the human GPER polypeptide (Pacific Immunology, Ramona, CA). Commercial antibodies included: rabbit anti-HA epitope antibody (Cell Signaling Technologies, Beverly, MA), mouse monoclonal ER-α (2Q418), PR (F-4) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and rabbit monoclonal ER-β antibody, clone 68–4 was purchased from EMD Millipore (Billerica, MA). Goat anti-rabbit Alexa-Fluor 594, goat anti-rabbit Alexa-Fluor 488 and goat anti-mouse Alexa-Fluor 488 secondary antibodies were purchased from Abcam (Cambridge, MA), Goat anti-rabbit IgG and goat-anti-mouse horseradish peroxidase (HRP)-conjugated antibodies were purchased from Southern Biotechnology (Birmingham, AL).
Plasmids.
Molecular clones encoding full-length human GPER [53] and ERα [54] under the transcriptional control of the CMV promoter have been described. The HiBiT tag (VSGWRLFKKIS) was inserted in-frame at the amino terminus of GPER by inversed PCR using forward 5′-GTTCAAGAAGATTAGCGATGTGACTTCCCAAGCC-3′ and reverse 5′-AGCCGCCAGCCGCTCACCATGTCTCTGCACCGTGC-3′ oligonucleotide primers and the Q5 mutagenesis kit (New England Biolabs, Salem, MA). A similar inverse PCR strategy was employed to insert the HiBiT tag at the carboxyl terminus of ER-α using forward 5′-TGGCGGCTGTTCAAGAAGATTAGCTGAGAGCTCCCTGGCGGA-3′ and reverse 5′-GCCGCTCACAG-AGCCTCCTCCACCGACTGTGGCAGGGAAACCC-3′ oligonucleotide primers.
Transient transfections and binary Nano-Bit ™ luminescence assays.
Total HiBiT-tagged GPER or ER estrogen receptors were detected in detergent-permeabilized cells by binary luminescence complementation with recombinant LgBit and substrate using the Nano-Glo HiBiT Extracellular Detection system kit (Promega Corporation, Madison, WI). In brief, HEK293 cells (0.75 × 106) were seeded in 35 mm tissue culture dishes and 24 hours later were transiently transfected with 50 ng of HiBiT-GPER or HiBiT-ERα and 950 ng of pcDNA3.1(+) zeo carrier plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The next day transfected cells were harvested by trypsinization and seeded at a density of 104 cells/well in 96-well poly-L183 lysine-coated Greiner white-bottomed microplates. On the following day, cell culture media was aspirated and replaced with 100 μL of serum-free PRF-DF12 containing E2-PROTAC, partial-PROTAC, or vehicle at various concentrations and for indicated time intervals at 37C. In some experiments, chloroquine (100 μM) or the proteasomal inhibitor, MG132 (10 μM) were included. Following treatment, total HiBiT-tagged receptor was measured in cells which were permeabilized in 0.05% Triton X-100 using HiBiT complementation reagent consisting of LgBiT protein and Nano-Glo® HiBiT substrate. Luminescence was measured using the Infinite 200 PRO multimode microplate reader from Tecan (Raleigh, NC) and reported as Relative Luminescent Units (RLU). All samples were measured in triplicate and expressed as the mean value plus or minus standard deviation.
Immunocytofluorescence.
Cells were seeded onto poly-L-lysine-coated, 18 mm glass coverslips at a density of 12,500 to 25,000 cells/cm2 in PRF-DF12 containing 5% FBS in 12-well cluster plates (CoStar, Corning, NY). The next day, cells were left untreated or treated at 37°C for 1 hour with 100 μM of UI-EP001, UI-EP002, or partial PROTAC in the presence or absence of chloroquine (100 μM) or the proteasomal inhibitor, MG132 (10 μM). Following treatment, plates were chilled on ice for 10 minutes and then labeled with GPER N-terminal peptide antibodies for 30 minutes at 4°C. Excess antibody was removed by washing with cold PBS and cells were then fixed in 4% paraformaldehyde in PBS for 5 minutes. Cells were then washed twice in PBS 199 and nonspecific antibody binding sites were blocked in PBS containing 5% bovine serum albumin (BSA) and 5% normal goat serum for 1 hour. Total receptor was measured in cells that were permeabilized in 0.1% Triton X-100 for 10 minutes prior to immunostaining. Fixed, permeabilized cells were washed twice in PBS and incubated in primary antibodies for 1 hour. Excess primary antibody was removed by washing in PBS and cells were then exposed to goat anti-rabbit or anti-mouse secondary antibodies for an additional hour. Following this second incubation, cells were washed once with PBS, once with tris-buffered saline prior, and then coverslips were mounted in Vecta-Shield anti-quench media containing DAPI (Vector Laboratories, Burlingame, CA). Immunofluorescence images were visualized with an Eclipse 80i microscope (Nikon, Inc., Melville, NY) equipped with a Nikon Plan Fluor 100 × 0.5–1.3 oil iris with differential interference contrast and epifluorescence capabilities using a Qimaging Retiga 2000R digital camera and Nikon imaging software (NIS-Elements-BR 3.0), Post-capture, images were processed with brightness/contrast adjustment using Photoshop CS2 (Adobe).
Competitive binding assays.
GPER binding was measured in an intact cell-based competitive binding assay using fluorescein-labeled estradiol (E2-FITC) as tracer as described with minor modifications [60]. SKBR3 cells were grown to near confluence in 175 cm2 flasks (Corning, NY, USA) and then placed into serum-free media overnight. Cells were detached in HBSS containing 5 mM EDTA and recovered by centrifugation at 150 g for 5 minutes. The cell pellet was washed twice in ice-cold HBSS containing 2 mM CaCl2 and 2 mM MgCl2 and cells were adjusted to a final concentration of 106 cells/ml in the same buffer. Cells (100 μL) were mixed with 100 nM E2-FITC and seeded into 96 well conical V-shaped bottom microtiter plates on ice containing an equal volume of either HBSS or various concentrations of 17β-E2, partial PROTAC, or E2-PROTAC. Samples were then incubated on ice for 30 min. Cells were pelleted by centrifugation at 4°C and washed once in HBSS with cations and analyzed in an Aurora FCM instrument (Cytek Biosciences, USA). At least 10,000 events per sample were analyzed using forward scatter versus side scatter dot-plot gating to resolve the primary cell population. The fluorescence intensity of cells in the fluorescein isothiocyanate (FITC) channel for each sample was recorded in log mode. Each condition was performed in triplicate and reported as median intensity fluorescence plus or minus standard deviation.
ER binding was measured using cytosolic fractions prepared from MCF-7 cells by fluorescence polarization. MCF-7 (106 cells) were detached in EDTA, cell homogenates were prepared using a Dounce homogenizer and subcellular fractions were isolated by differential centrifugation as previously described [24]. The protein concentration of each fraction was determined by bicinchoninic acid (BCA) assay (Pierce™ BCA protein assay kit, Thermo Fisher). For saturation binding assay of the probe, different230 concentrations of cytosolic fraction were diluted in assay buffer (10 mM Tris-HCl, pH 7.4; 50 mM KCl, 10% glycerol, 0.1 mM dithiothreitol (DTT), 1 ug/mL BGG, 10 nM protease inhibitor cocktail (Roche) to a final volume of 200 uL containing 10 nM E2-FITC. The mixture was incubated at ambient temperature for 1hour and interaction of the probe with cytosolic protein was determined by fluorescence polarization (FP). For competitive binding assay, an aliquot of cytosolic fraction and E2-FITC (10 nM final concentration) was mixed and incubated at 4°C for 1 hour. Then different concentrations of drugs and controls were added to the mixture. The mixture was incubated for an additional hour and then subjected to FP. All samples were measured in triplicate. Negative controls (only cytosolic fraction or only E2-FITC) and positive control (E2-FITC and cytosolic fraction at 100% binding without treatment) were included. Relative binding affinities were expressed as the percentage of FP recorded from treated wells compared to the positive control. FP values in millipolarization units (mP) were measured at ex/em wavelength of 485 nm/530 nm respectively using the Spectra Max plus 384 Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA) in 96-well, black, flat-bottom microplates (Greiner Bio-One North America, Inc., Monroe, NC). Kd values of the probe were determined by nonlinear regression fitting of the saturation curves, while IC50 values of treatment were determined by nonlinear regression fitting of the competition curves.
Cell cytotoxicity assays.
Cell viability was measured following E2-PROTAC treatment using the Presto-Blue Viability kit according to the manufacturer’s suggested protocol (Thermo Fisher Scientific). In brief, breast cancer cells were seeded in 96 well-plates at a density of 104 cells/well in growth media. On the following day, the contents of the 96 well plates were aspirated and replaced by treatment at different concentrations (ranging from 10–9 to 10–3 M), followed by the addition of media to bring the total volume to 200 μL/well. The untreated control group was incubated with 200 μL/well of fresh media. One day later, the media were aspirated and replaced by 90 μL media + 10 μL Prestoblue reagent, followed by incubation at 37°C for 1h. The fluorescence was excited at 560 nm and emission was recorded at 590 nm using a Spectra Max plus 384 Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA). Relative cell viability values were expressed as the percentage of the fluorescence recorded from wells containing treated cells compared to the control wells containing untreated cells.
Cell cycle analysis.
Cells were seeded in 6 well-plates at a density of 2.5×105 cells/well and then left untreated or exposed to partial PROTAC, UI-EP001 or UI-EP002 for 24 hours. Following treatment, cells were harvested by trypsinization and washed twice in ice-cold PBS. Cells were then fixed in 70% ethanol at 4°C for 30 minutes. Fixed cells were pelleted by centrifugation at 230 g for 5 minutes and the cell pellet was incubated in Krishan’s solution (3.8 mM sodium citrate (Fisher Scientific), 0.014 mM propidium iodide (AnaSpec, Fermont, Ca), 1% NP-40 (Sigma) and 2.0 mg/mL RNase A (Fisher Scientific)) for 30 minutes at 37°C. Cells were then centrifuged and washed in PBS prior to analysis using a FacScallibur flow cytometer. The data from flow cytometry were subjected to further analysis by CellQuest software version 3.3. DNA histograms generated indicate the fractions of the cell population in the sub-G1, G0-G1, S, or G2/M phase of the cell cycle.
Statistical analysis.
All data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA). The normality and homogeneity of variance of all data were analyzed by one-way analysis of variance (ANOVA). Each experiment was performed in triplicate and differences were considered significant if P < 0.05.
Results
Molecular docking analysis of ERα or GPER1 interaction with E2-PROTAC
Our previous studies of the GPER1 binding pocket and its interactions with E2 indicate that E2 can be modified using a PROTAC strategy and still maintain its binding efficacy [50–51]. Based on those homology modeling studies, the 17C-hydroxyl group is postulated to point outward of the GPER binding pocket between TM1 and TM7 and interact with N1182.62 and H3077.37. Our previous studies have shown that the attachment of a fluorophore at the 17C does not negatively impact GPER activity [53]. Therefore, two different PROTAC molecules were designed in silico to have a von Hippel Lindau, VHL, E3 ligand (VHL1/VHL032) attached to the 17-hydroxyl group via a polyethylene glycol (PEG) linker. An ester linkage to the 17-hydroxyl group was chosen for synthetic ease and two different lengths of linkers were chosen to explore the possible spatial constraints for GPER PROTACs. The first molecule, UI-EP001, was designed with a 14-atom linker between E2 and VHL1, and the second molecule, UI-EP002, was designed with a 32-atom linker between E2 and VHL1. The linker length for UI-EP001 was based on reported literature, where 13–14-atom linkers resulted in high degradation rates and selectivity of the target receptor, while the more elongated linker in UI-EP002 helped to increase the overall hydrophilicity and flexibility of the compound. Docking studies for UI-EP001 and UI-EP002 with GPER1 show that the original E2 binding pocket is maintained and that the linker exits the binding pocket between TM1 and TM7 (Figure 1). Based upon crystal structures of known PROTACs with their protein targets, both linker lengths would allow for UI-EP001 and UI-EP002 to interact with VHL E3 ligases while bound to GPER1 [55–56].
Figure 1. Modeling of PROTACs in GPER and ERα.
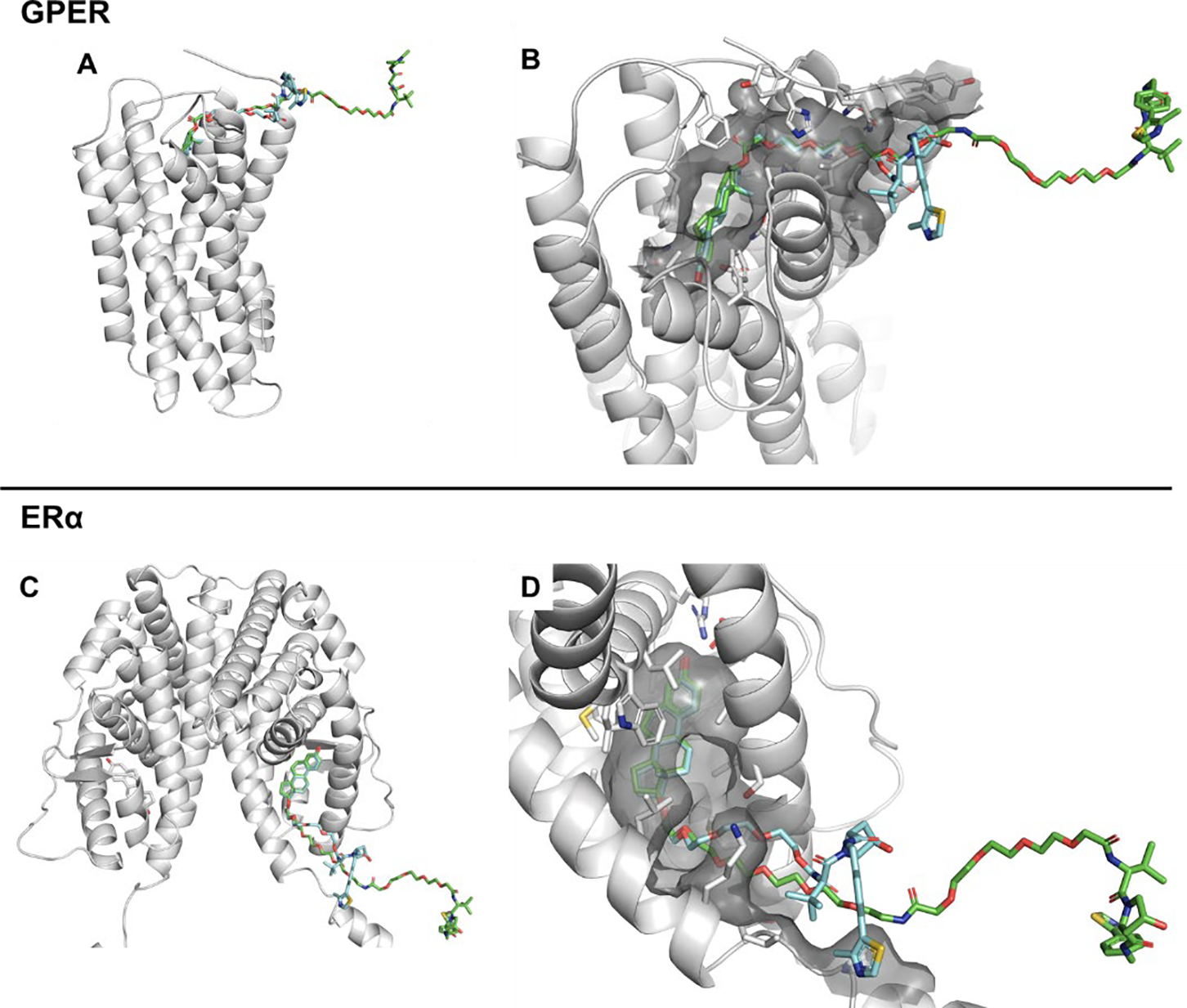
A) UI-EP001 (blue) and UI-EP002 (green) bound within a GPER homology model, which were designed to have the linker exiting the binding pocket with two varying lengths. B) GPER binding pocket (grey surface) showing the exit of UI-EP001 (blue) and UI-EP002 (green) from between TM1 and TM7. C) UI-EP001 (blue) and UI-EP002 (green) bound within the ERα ligand domain of an ERα homodimer (PDB:1A52) [51]. D) ERα binding pocket (grey surface) showing the exit of UI-EP001 (blue) and UI-EP002 (green).
While it is known that modification of the 17C-position of E2 is tolerated for GPER binding [57], it is unclear if the designed PROTAC molecules would favorably bind with ERα. Therefore, additional docking studies for UI-EP001 and UI-EP002 with ERα were performed using a crystal structure of E2 bound to an ERα ligand-binding domain (PDB:1A52) [52]. All the known interactions between E2 and ERα are maintained for both UI-EP001 and UI-EP002. In addition, the linker length of both molecules is sufficiently long enough for the VHL1 portion to interact with VHL E3 ligases.
The synthetic detail of partial PROTAC (compound 7) is schematized in scheme SI1. Linker 2 was prepared from triethylene glycol following a two-step process (scheme S1A). VHL E3 ligase ligand was synthesized following previously published literature (scheme S1B) [58–59]. Palladium-catalyzed cross-coupling between 4-bromobenzonitrile and 4-methylthiazole was carried out to achieve cyanide 3, which was subsequently treated with cobalt (II) chloride and sodium boron hydride to give primary amine 4. Compound 7 was achieved from 4 after amide coupling with Boc-Hyp-OH, Boc-Tle-OH and linker 2, subsequently. Conjugation between E2 and compound 7 was carried out under Steglich esterification to give UI-EP001 (compound 8) (scheme SI2). UI-EP002 (compound 10) was prepared by introducing a PEG8 linker in between E2 and compound 7. First, E2 and Fmoc-NH-PEG8-CH2CH2COOH underwent Steglich esterification, followed by treatment with trimethylamine to remove the Fmoc group and give compound 9. Finally, compound 10 was achieved from the conjugation between compound 9 and compound 7 with careful monitoring by HPLC for 72h (scheme SI3).
E2-PROTACs exhibited high-affinity specific binding to plasma membrane and intracellular estrogen receptors
GPERs and ERs each bind E2 with high affinity [48], yet each estrogen receptor resides in distinct subcellular compartments, which has important implications regarding their relative ability to interact with E2-PROTACs. GPER exhibits all the hallmarks of a plasma membrane receptor despite the fact that the bulk of the receptor is expressed in intracellular membranes [53]. In support of its role as a plasma membrane receptor, our prior studies have measured specific binding of 3H-E2 in sucrose density gradient enriched membrane homogenates suggesting a plasma membrane binding site [57]. To directly evaluate the binding of UI-EP0001 and UI-EP002 to GPER on the plasma membrane, competitive binding assays were performed using intact SKBR3 cells that express GPER, but not ERα or ERβ, using cell impermeant E2-FITC as a fluoro-tracer (Figure 2). GPER specific binding activity of E2-FITC was measured by differential saturation analysis, subtracting the binding activity of cells that express or do not express GPER on the cell surface (Figure 2A). Maximum GPER specific binding was reached near 100 nM E2-FITC (Figure 2B) and this concentration was selected for competitive binding experiments (Figure 2C). The concentration of E2 necessary to displace 50% of E2-FITC (IC50) was calculated as the dissociation constant (Kd) for E2 at 100 ± 1.5 nM of 17β-E2. This is similar to the IC50 value of 300 nM for E2 reported previously [60]. By comparison, the IC50 values of UI-EP001 and UI-EP002 were nearly 3-fold greater (RBA= 330%) than that measured for E2 (30.2 ± 0.9 nM and 30.2 ± 2.3 nM, respectively) (Table 1). This may, in part, be explained by the structural homology shared between E2-FITC and E2-PROTAC, which each contain a 17C-substituted hydroxyl. In contrast, a partial PROTAC consisting of the chemical spacer for UI-EP001 linked to the VHL E3 ubiquitin ligase recognition motif but lacking its E2-targeting domain was unable to compete for E2-FITC binding even at concentrations as high as 10 μM (Figure 2C).
Figure 2. E2-PROTACs act via high-affinity binding to GPER and ERs.
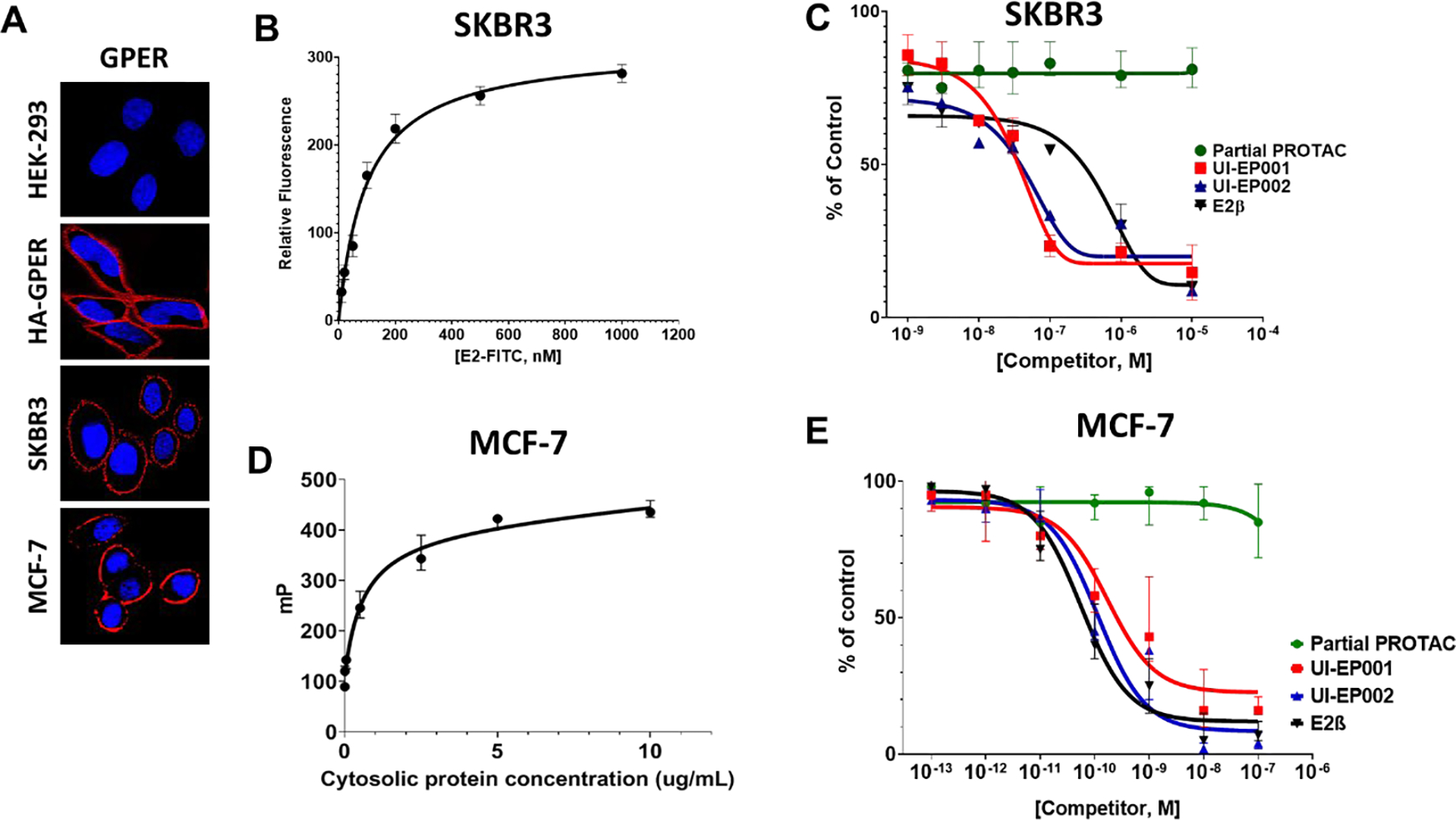
A) Intact HEK-293 (ER−, GPER−), HA-GPER (ER−, GPER+), SKBR3 (ER−, GPER+) and MCF-7 (ER+, GPER+) cells were labeled with rabbit GPER antibody at 4°C. Surface GPER was then visualized with Alexa 594-conjugated goat anti-rabbit IgG (red). B) Calculated specific binding as a function of E2-FITC concentration in intact SKBR3 cells. C) Specific binding of 17β-E2, UI-EP001, UI-EP002, and partial PROTAC in intact SKBR3 cells measured by a competitive binding assay using E2-FITC as a fluoro-tracer. D) Determination of E2-FITC total binding using 10 nM E2-FITC in the presence of increasing concentrations of cytosolic protein prepared from MCF-7 cells. E) Fluorescence polarization assay using E2-FITC to assess specific binding of E2, UI-EP001, UI-EP002, and partial PROTAC in cytosolic fractions prepared from MCF-7 cells. Mean values and SDs were derived from three independent experiments.
Table 1.
Binding efficacy of UI-EP001 and UI-EP002 for GPER and ERs.
| GPER | ERs | |||
|---|---|---|---|---|
| Compounds | IC50 (nM) | RBA% | IC50 (nM) | RBA% |
| Estradiol | 100 ± 1.5 | 100 | 5.6 ± 1.4 | 100.0 |
| UI-EP001 | 30.2 ± 0.9 | 331.1 | 17.2 ± 8.2 | 32.0 |
| UI-EP002 | 30.2 ± 2.3 | 331.1 | 11.4 ± 3.3 | 49.0 |
| Partial PROTAC | NA | NA | NA | NA |
Abbreviations: IC50, concentration of 50% inhibition; RBA, relative binding affinity calculated as the ratio of the IC50 value of 17β-E2 divided by the IC50 of the test compound. All IC50 values were measured from triplicate samples and are reported as the mean ± SD; NA, not achieved.
In contrast to GPER, ERα and ERβ are intracellular receptors that are concentrated in the cytoplasm and in the nucleus. Thus, the binding activities of UI-EP001 and UI-EP002 ERs were evaluated in cytosolic fractions isolated from MCF-7 breast cancer cells using fluorescence polarization (FP). A saturation analysis was performed using 10 nM E2-FITC and increasing concentrations of cytosolic protein (Figure 2D). Based upon near saturation (80%) FP values, competitive binding assays were performed using 600 μg/mL of cytosolic protein and E2 showed half-maximal displacement, IC50= 5.6 + 1.4 nM) (Figure 2E). By comparison, IC50 values for UI-EP001 and UI-EP002 were calculated at 17.2 ± 8.2 nM and 11.4 ± 3.3 nM, respectively. These results demonstrate that UI-EP001 and UI-EP002 display a slightly weaker binding affinity compared with E2, with RBAs of 32% and 49%, respectively (Table 1). Collectively, these competitive binding assays performed on intact cells and cytosolic fractions show that UI-EP001 and UI-EP002 bind plasma membrane and intracellular estrogen receptors GPER with high affinity in the low nanomolar range.
E2-PROTAC reduces the expression level of native and/or recombinant estrogen receptors
To evaluate the efficacy of UI-EP001 and UI-EP002 to degrade plasma membrane and intracellular estrogen receptors, a highly sensitive Nano-BiT binary luminescence complementation assay was employed that measures the interaction between HiBiT-tagged proteins and soluble Lg-BiT protein. Cells treated with UI-EP001 or UI-EP002 demonstrated a dose-dependent decrease of HiBiT-GPER or HiBiT-ERα (Figure 3A and B), while 100 μM of partial PROTAC did not show any significant decrease for either recombinant receptor within the tested concentration (Figure 3C). From these assays, a DC50 value (the concentration required for 50% protein degradation) was calculated for GPER (100 μM) and ERα (10–100 μM), respectively. Using these DC50 concentrations, the kinetics of UI-EP001 and UI-EP002 degradation of HiBiT-GPER and HiBiT-ER were measured. Both UI-EP001 and UI-EP002 induced time-dependent reduction in expression levels of either recombinant GPER or ER, with 50% reduction measured as early as 1 h (Figure 3D and E). In contrast, 83% of the receptor was present following treatment of cells with partial PROTAC for 8 hrs.
Figure 3. E2-PROTACs promote rapid loss of recombinant membrane and intracellular estrogen receptor.
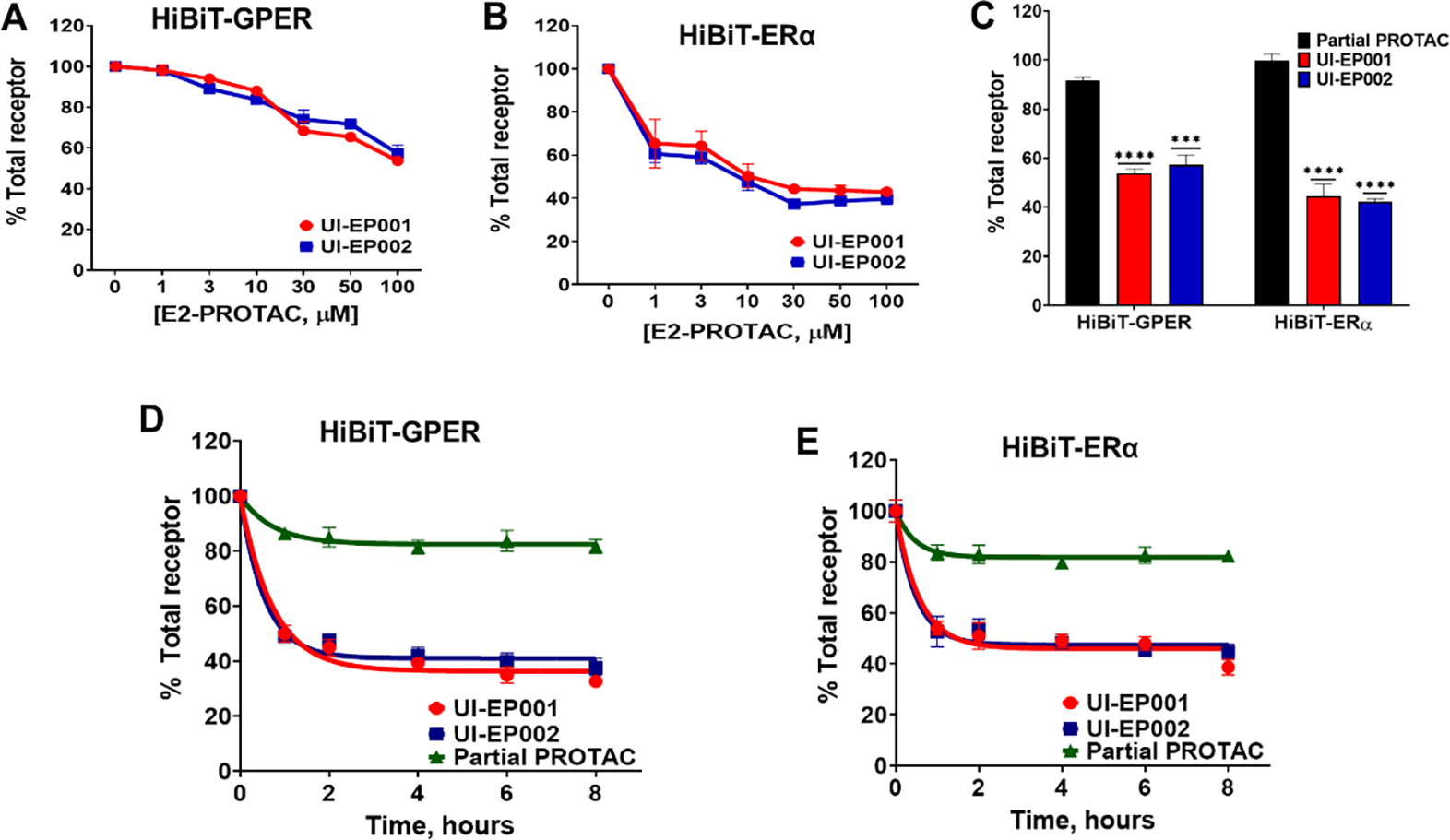
HEK-293 cells (ERα−, GPER−) were transiently transfected with 50 ng HiBiT-GPER or HiBiT-ER plus pcDNA3.1 ZEO carrier plasmid. Concentration-response curves of (A) HiBiT-GPER and (B) HiBiT-ERα were measured using extracellular LgBiT and luminescence substrate in detergent-permeabilized cells after treatment with increasing doses of either UI-EP001 or UI-EP002 for 1 hour. (C) Histogram depicting total HiBiT-GPER and HiBiT-ER following a 1-hour treatment at 100 μM drug or control. Kinetics of (D) HiBiT-GPER and (E) HiBiT-ERα after incubation of cells with 100 μM of UI-EP001, UI-EP002, and partial PROTAC for various incubation periods from 1 to 8 hours. Results shown represent the mean ± S.E. of three independent experiments. (***, P <0.0004; ****, P <0.0001; one-way ANOVA).
The specificity of UI-EP001 and UI-EP002 for GPER was determined by examining their capacity to degrade other GPCRs, including the β1-adrenergic receptor, β1AR and the chemokine C-X-C chemokine type 4 receptor, CXCR4 (Figure 4A). For this purpose, the relative efficacy of UI-EP001 or UI-EP002 or partial PROTAC was evaluated for their efficacy to degrade HA-GPER, -β1AR, or -CXCR4 in stably transfected HEK293 cell lines. By immunofluorescent analysis using HA-specific antibodies, specific degradation of HA-GPER was measured without detectable off-target degradation of either HA-β1AR or HA-CXCR4 (Figure 4A and B). Next, the specificity of UI-EP001 and UI-EP002 to degrade endogenous plasma membrane and intracellular estrogen receptors was assessed using human MCF-7 breast cancer cells which express ERα, ERβ, and GPER (Figure 4C and D). MCF-7 cells were exposed to either UI-EP001, UI-EP002, partial PROTAC for 1 hour or left untreated and then fixed, permeabilized and immunostained with specific antibodies for GPER, ERα, ERβ or progesterone receptor (PR). Clear site-specific reduction was observed for nuclear ERα and ERβ and intracellular GPER (Figure 4C and D). In contrast, treatment of MCF-7 cells with partial PROTAC did not impact ERα, ERβ, or GPER expression. Neither did UI-EP001 or UI-EP002 exhibit any detectable degradation of PR in MCF-7 cells further indicating the specificity of E2-PROTACs for estrogen receptors (Figure 4D). Moreover, UI-EP001 and UI-EP002, but not partial PROTAC, reduced plasma membrane and total GPER in human SKBR3 breast cancer cells that are negative for ERα and ERβ (Figure 4E). Similarly, E2-specific PROTAC-mediated degradation was observed at the surface of MCF-7 cells (data not shown).
Figure 4. E2-PROTACs selectively reduce the expression of native and recombinant GPER.
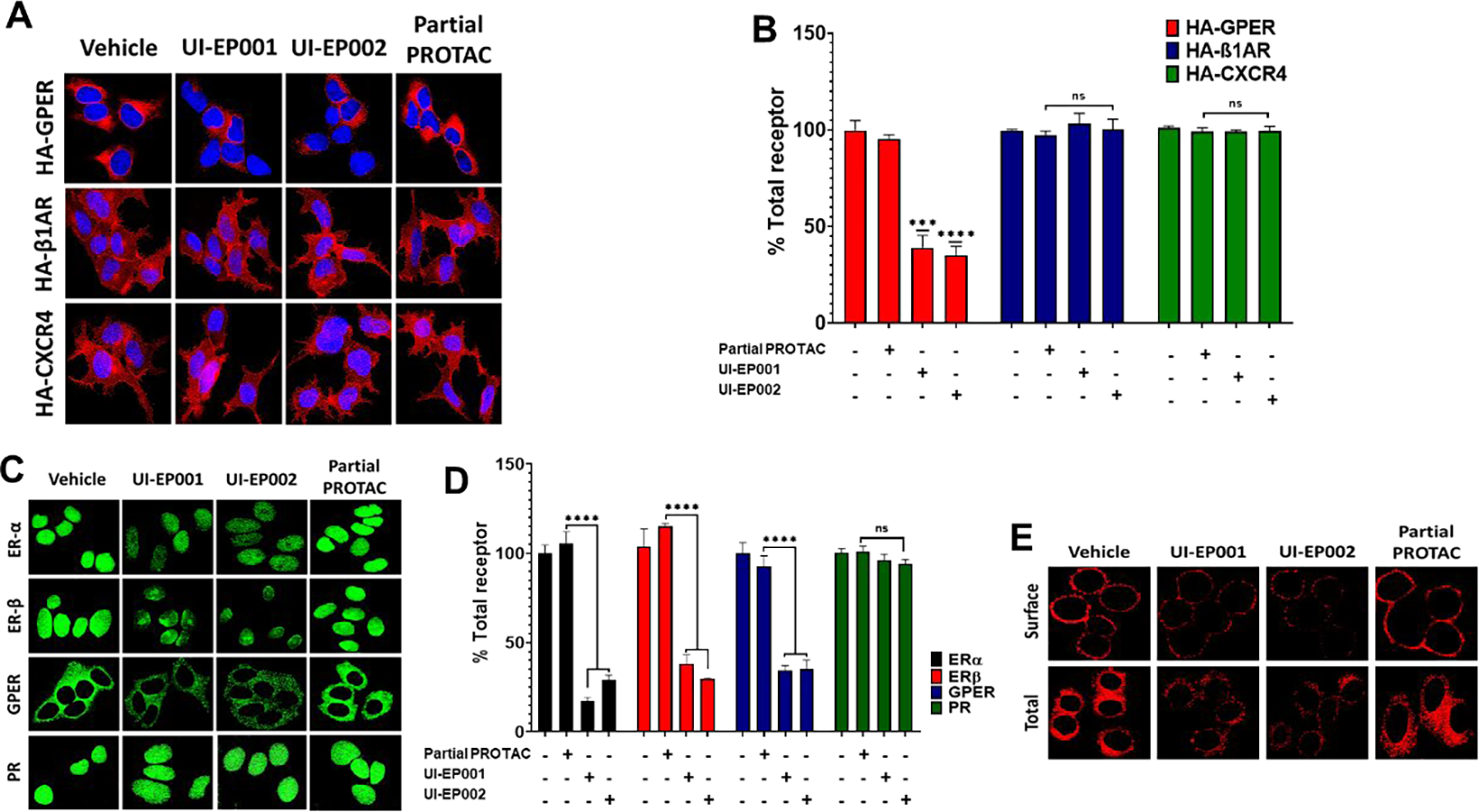
(A) HEK-293 cells stably expressing HA-GPER, HA-β1AR, or HA-CXCR4 were treated with 100 μM of UI-EP001, UI-EP002, and partial PROTAC for 1 hour. Fixed cells were permeabilized and then labeled with rabbit HA antibody and total receptors were then visualized using Alexa Fluor 594 anti-rabbit secondary antibody (red). (B) Corrected Total Red Fluorescence (CTRF) from images of HA-GPER, HA-β1AR, and HA-CXCR4 cells treated with either vehicle, UI-EP001, UI-EP002, and partial PROTAC was measured using Image J software from three different microscopic fields (***, P <0.0004; ****, P <0.0001; one-way ANOVA). (C) MCF-7 (ERα+, ERβ+, PR+, GPER+) cells were incubated at 37°C with vehicle, UI-EP001, UI-EP002, or partial PROTAC for 1 hour and then immunostained with mouse ERα, rabbit ERβ, mouse PR, and rabbit GPER antibodies and detected with either Alexa 488-conjugated anti-mouse IgG or Alexa 488-conjugated anti-rabbit IgG (green). (D) Quantification of results from images in (C) measured as CTRF. (E) SKBR3 (ERα+, ERβ−, GPER+) cells were incubated with vehicle (1% DMSO), 100 uM of UI-EP001, UI-EP002, or partial PROTAC for 1 hour. Surface and intracellular GPER were visualized in intact or detergent permeabilized cells, respectively using rabbit GPER antibodies and Alexa 594-conjugated goat anti-rabbit IgG (red).
The specificity of UI-EP001 and UI-EP002 for its intended targets was further assessed by examining the capacity of exogenous E2 to inhibit E2-PROTAC-mediated ER/GPER degradation (Figure 5A and B). Cells transfected with ER-HiBiT or GPER-HiBiT were treated with 100 μM of UI-EP001 and UI-EP002 alone or in combination with increasing doses of E2β or aldosterone (100 nM-100 μM) for 1 hour (Figure 5A and B). While E2 effectively inhibited UI-EP001 and UI-EP002 degradation, degradation of either estrogen receptor target was not measured in the presence of the control steroid, aldosterone (Figure 5C).
Figure 5. E2-PROTACs induce proteasome-dependent degradation of GPER and ERα.

HEK-293 cells transiently transfected with (A) HiBiT-GPER and (B) HiBiT-ERα were treated with 100 μM UI-EP001 in the presence of increasing concentrations of E2β or aldosterone. Total receptor was then measured by binary luminescence complementation in detergent-permeabilized cells. (C) Effects of 100 μM UI-EP001 alone or in combination with either 100 μM E2 or aldosterone in transiently transfected cells. **** Denotes statistical significance, P < 0.0001 or less. (D) Cells were treated with either (D) 10 μM MG132 or (E) 100 μM chloroquine in the presence or absence of 100 μM of UI-EP001 or UI-EP002 for 1 hour. Total receptor was then quantified by binary luminescence complementation. Representative luminescence data and mean values and SDs derived from three independent experiments are shown. (***, P <0.0004; **, P <0.001; ns, not significant; one-way ANOVA).
Taken together, these findings suggest that UI-EP001 and UI-EP002 function as estrogen receptor degraders (SERDs) that are capable of selectively degrading plasma membrane and intracellular estrogen receptors.
UI-EP001 and UI-EP002 degradation of ER and GPER occurs via the 26S- proteasome
Finally, we assessed whether UI-EP001 and UI-EP002 could promote the degradation of their target receptors via the ubiquitin-proteasome pathway. To conduct these experiments, HEK293 cells transiently expressing HiBiT-tagged estrogen receptors were pretreated with the proteasome inhibitor, MG132, the lysosomotrophic agent, chloroquine, or vehicle prior to exposure to UI-EP001 or UI-EP002, and then total receptor expression was assessed as above. While MG132 reversed UI-EP001 or UI-EP002 degradation of either GPER or ERα, chloroquine had no such effect (Figures 5D and E). This result is consistent with the fact that PROTAC-induced protein degradation occurs as the result of receptor polyubiquitination and proteolysis at the 26S-proteasome. Significantly, these results suggest that our first generation E2-PROTACs; UI-EP001 and UI-EP002, interact with estrogen receptors that reside at the plasma membrane and in intracellular compartments to rapidly promote the degradation of membrane (GPER) and nuclear (ERs) estrogen receptors at the 26S-proteasome.
Collectively, these studies imply that simultaneous engagement of estrogen receptors (ERα and GPER) and VHL E3 ubiquitin ligase by UI-EP001 and UI-EP002 is crucial for effective degradation.
E2-PROTAC inhibits breast cancer cell growth by inducing cell cycle arrest
To evaluate the cytotoxic potential of the E2-PROTACs, we measured the viability of four different human breast cancer cell lines with different estrogen receptor profiles following treatment with UI-EP001, UI-EP002, or partial PROTAC (Figure 7). In this study, we included: i) MCF-7 cells that express all three estrogen receptors (ERα+ ERβ+, GPER+), ii) SKBR3 cells that overexpress her2/neu (ERα−, ERβ−, GPER+), iii) HCC1806 cells, which are triple negative and GPER-positive (ERα−, ERβ−, GPER+) and iv) MDA-MB-231 cells, which are ERα-negative and express low levels of ERβ and GPER and do not elicit either ER [61] or GPER [25] -dependent signaling activity (ERα−, ERβ low, GPER low). Each breast cancer cell line was treated for 24 hours with various concentrations of E2-PROTAC or partial PROTAC and then cell viability was assessed using an Prestoblue assay that measures mitochondrial oxido-reductase enzymes capable of converting a tetrazolium salt to a formazan dye (summarized in Table 2). A dose-dependent decrease in cellular viability was measured in all breast cancer cell lines that were treated with either UI-EP001 or UI-EP002. For UI-EP001, IC50 values of 9.0, 10.9 and 34.9 μM were measured respectively in MCF-7, SKBR3 and HCC1806 breast cancer cell lines, which express GPER. Similar IC50 values were measured for UI-EP002 in these three cell lines (17.0, 11.1 and 7.4 μM), respectively. In contrast, IC50 values for UI-EP001 and UI-EP002 were approximately 100-fold higher (>100 μM) in MDA-MB-231 cells that express low concentrations of functional ERβ or GPER, with >80% cell survivability. These IC50 values were similar to those measured for cells treated with partial PROTAC. These data suggest that the elimination of membrane and intracellular estrogen receptors are critical factors in determining breast cancer cell viability.
Figure 7. E2-PROTACs promote estrogen receptor-dependent G2/M arrest.
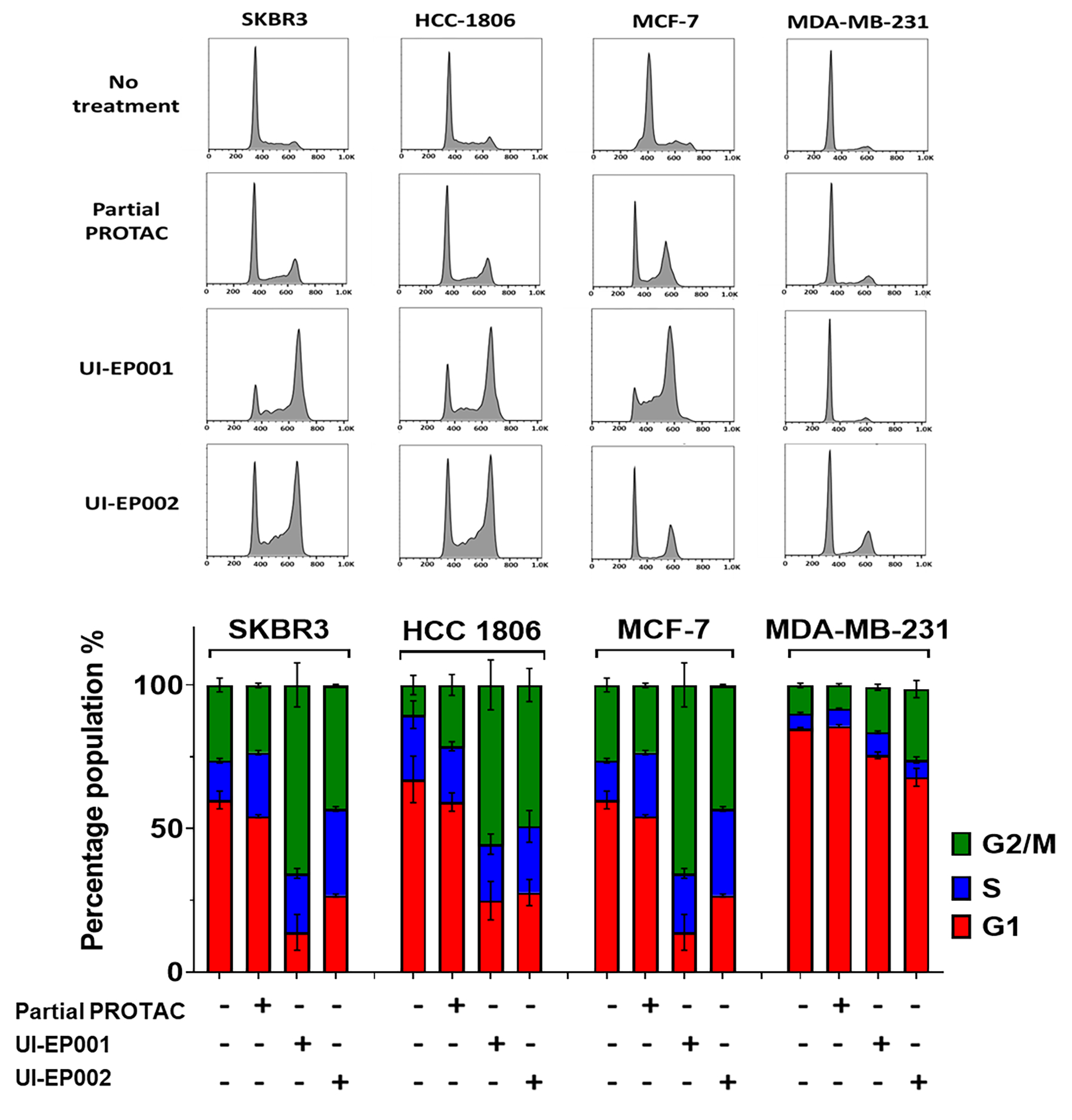
E2-PROTAC induced G2/M arrest in human breast cancer cell lines expressing membrane and/or intracellular estrogen receptors. SKBR3, HCC-1806, MCF-7 and MDA-MB-231 cells growing in 10 % FBS were treated with 10 μM of UI-EP001 or UI-EP002 or partial PROTAC for 24h. Fixed cells were permeabilized and dyed with propidium iodide (PI). Cell cycle data were acquired on a flow cytometer and analyzed using Flow-Jo software. Data are expressed as mean ± SEM (n=3). The percentage of cells in each phase were plotted using Prism 8.0.
Table 2.
IC50 values for UI-EP001 and UI-EP002 in cell viability assays
| UI-EP001 | UI-EP002 | |
|---|---|---|
| Cell line | IC50 (μM) | IC50 (μM) |
| MCF-7 | 9.0 ± 1.2 | 17.0 ± 1.6 |
| SKBR3 | 10.9 ± 1.2 | 11.1 ± 1.6 |
| HCC1806 | 34.9 ± 1.2 | 7.4 ± 1.3 |
| MDA-MB-231 | >100.0 | >100.0 |
Abbreviations: IC50, concentration of 50% inhibition; All IC50 values were measured from triplicate samples and are reported as the mean ± SD.
To investigate the biological effect associated with decreased cell viability, cell cycle analysis was performed on the same four cell lines using propidium iodide staining and measured by flow cytometry 24 hours after treatment of cells with 10 μM E2-PROTAC or partial PROTAC (Figure 7). Untreated cells from all four cell lines showed a typical cell cycle distribution pattern with the majority of the cells in G1 and a small percentage of cells in either S or G2/M. Following treatment with either UI-EP001 or UI-EP002, a dramatic increase in the G2/M peak with a significant reduction in G1, was observed in MCF-7, SKBR3 and HCC-1806 breast cancer cells. This shift was not observed in MDA-MB-231 cells that do not express ERα and express low amounts of functional ERβ or GPER, suggesting that E2-PROTACs induce cell cycle arrest at the G2/M phase by reducing either or both estrogen receptors. As expected, cells treated with partial PROTAC displayed a much less significant shift in the percentage of the total cell population between G1 and G2/M. These in vitro results strongly suggest that the degradation of ERα and/or GPER has the potential to be effective in vivo for treating not only ER+ but also ER− breast cancer.
Discussion
The carcinogenic effects of estrogen are manifested through cellular receptors that belong to the nuclear steroid hormone and G-protein coupled receptor superfamilies [33]. A number of ER-targeted PROTACs have been developed but they have not yet been tested for their capacity to degrade GPER. Here, proof-of-principle evidence is provided that a single estradiol-based PROTAC can target both receptor classes. Guided by in silico modeling and structure-activity relationship data, two first generation PROTACs (UI-EP001 and UI-EP002) were designed containing an estradiol-based targeting domain substituted at the 17C with ester-linked 14- or 32-atom length PEG spacers joined to the small molecule VHL E3 ubiquitin ligase recruitment motif; (S, R, S) AHPC. Each PROTAC exhibited a high binding affinity for nuclear ER and GPER and selective proteasome dependent degradative activity for GPER and ERs with no measurable detectable off-target activity. Most importantly, UI-EP001 and UI-EP002, exhibited target-specific cytotoxicity and G2/M arrest, desirable in-vitro characteristics for a first-generation pan-estrogen receptor PROTAC.
The PROTAC platform has been employed to selectively degrade a wide range of proteins that have therapeutic potential for human disease, including but not limited to; steroid hormone receptors (AR, ERα, ERRα, RAR) [62–64], Bromo- and Extra- Terminal (BET) family proteins [65–66], kinases [43,67], and microtubule associated proteins (Tau, TACC3) [68]. This group has recently been extended to include GPCRs [69]. The mere fact that PROTACs can efficiently target proteins with unique structural characteristics that are localized in distinct subcellular compartments demonstrates their exciting promise. However, the development of a PROTAC for a single protein target is not a trivial matter. Ultimately, target optimization is achieved by evaluating different targeting domains and/or warhead domains and making numerous fine adjustments that alter the length and composition of the chemical spacer as well as the overall stereochemistry of the entire hetero-bifunctional molecule. Theoretically, designing a single PROTAC capable of targeting two structurally and functionally distinct estrogen receptors with unique structures that reside in distinct physicochemical environments would pose a more challenging task but one that is necessary to treat ER+ breast cancers, since the majority of these tumors also express GPER. A broad assortment of ERα-PROTACs have been developed over the past decade but no single published study has demonstrated whether these compounds are also capable of targeting the highly related estrogen receptor, ERβ, or the more recently described plasma membrane estrogen receptor, GPER.
Pioneering ER-PROTACs contained peptidic warhead domains and consequently showed low cell permeability and dependence upon cellular enzymatic activity to effectively recruit the E3 ubiquitin ligase SCFβ-TCRP [70]. More recent second-generation ER-PROTACs employ ER antagonists in their targeting domain and demonstrate high cell permeability and degradative capacity at low nanomolar concentrations [46, 71–72]. These latter PROTACs may also target GPER as well as ER but perhaps to a lesser extent due to the anticipated reduced RBA of E2 compared to ER antagonists for either receptor. Assessment of drug-target interaction by experimental methodology is a critical step for evaluating target specificity and selectivity. However, in general, the binding properties of ER-PROTACs have not been measured, and in many instances target selectivity has not been evaluated. Both UI-EP001 and UI-EP002 show high affinity for both target estrogen receptors with Kd values calculated in the low nanomolar range suggesting that the 17C substituted E2 targeting domain interacts well with the ligand contact sites on both GPER and ER. Using 17C substituted E2-FITC and intact SKBR3 breast cancer cells that express GPER with no detectable ERα or ERβ by RT-PCR [73], we measured Kd values of 30.2 nM for UI-EP001 and UI-EP002. This is consistent with past evidence that 1, 3, 5(10)-estratriene-3, 17β- diol 17-hemisuccinate: BSA (E2-BSA) is capable of stimulating GPER-dependent activation of intracellular cAMP [53]. However, these findings are at odds with published work that showed that free E2, but not E2-BSA, was able to either displace 125I-16α-iodo-E2 bound to recombinant ERα or ERβ or induce a gel shift in electrophoretic mobility assays [74]. In our hands, free 17β-E2, UI-EP001 or UI-EP002 displaced E2-FITC bound to cytosolic fractions prepared from MCF-7 cell homogenates containing endogenous ERα and ERβ with Kd values measured at 5.6 nM, 17.2 nM and 11.4 nM, respectively. The reasons for the discrepancy between our findings and those of Stevis and colleagues are not clear but they may be related to both differences in the binding probe as well as the multi-valency of E2 for BSA relative to the 1:1 stoichiometry of E2 and FITC. One possibility is that the binding kinetics of E2-FITC for GPER are different compared to 125I-16α-iodo-E2, which is influencing the displacements of the probes. Nonetheless, these data suggest that our first generation E2-PROTACs possess the most important characteristic of any new drug, the capacity to interact specifically and with high affinity to its intended target.
By their design, PROTACs offer the promise as cancer therapeutics of selective protein-targeted degradation. With the intent of demonstrating target specificity for a given PROTAC, the gold standard assay is the use of mass spectroscopic analysis to provide a comparative global readout of the proteome of treated and untreated cells [75]. From a practical standpoint, target specificity can be best assessed by evaluating a difference in the survival of cancer cells that express or lack the cancer therapeutic target. This is particularly important for assessing a dual specificity PROTAC designed to selectively degrade ERs and GPER. In our study, target selectivity was evaluated using immunofluorescent, biochemical and biological methods. By immunofluorescent analysis, we were able to show selective site-specific decreases in ERα and ERβ within the nucleus and for GPER at both the cell surface and intracellularly. Degradation of intracellular GPER by UI-EP001 or UI-EP002 is consistent with our prior findings that showed GPER-dependent specific displacement of 3H-17β-E2 from crude membrane fractions as well as plasma membrane fractions enriched by sucrose density centrifugation [57]. Selectivity was demonstrated by the fact that no decrease was measured for PR, CXCR4 or β1AR in E2-PROTAC-treated cells. Likewise, by binary luminescence complementation, target selectivity was also demonstrated by showing that exogenous E2 but not aldosterone was capable of blocking E2-PROTAC mediated degradation of HiBiT-GPER or HiBiT-ER. Finally, target selectivity for UI-EP001 and UI-EP002, was shown in biological assays where we compared their relative effects on viability and cell cycle distribution in various breast cancer cell lines expressing different complements of estrogen receptors. Decreased viability and G2/M arrest were observed in MCF-7 cells that express all 3 estrogen receptors but not in MDA-MB-231 cells that lack ERα and express low levels of functional ERβ and GPER. Whether or not this was due to reduced ER or GPER, or both, is not clear in these cells. It is noteworthy that other investigators have evaluated the cytotoxicity of ER-PROTACs in ER-positive MCF-7, BT-474 and CAMA-1 breast cancer cell lines [76–77], and that these cells also express GPER [78–79]. Either E2-PROTAC in our study induced cytotoxicity and G2/M arrest following treatment of ER-negative, GPER-positive SKBR3 and HCC-1806 breast cancer cell lines that represent her-2/neu overexpressing and TNBC tumor immunophenotypes. While different chemotherapeutic agents demonstrate cell cycle arrest at different stages of the cell cycle, the observation that E2-PROTACs induce G2/M arrest is consistent with a prior report that the androgen receptor (AR)-PROTAC, ARD-61 also induces G2/M arrest [80]. In some regards it may appear somewhat surprising that UI-EP001 or UI-EP002 induce cytotoxic effects and cell cycle arrest due to the fact that a complete loss of either ER or GPER was not observed. One possible explanation for these results may be related to the fact that GPCRs generally show a hyperbolic relationship between ligand occupancy and receptor responses. This relationship is termed “fractional occupancy” and suggest why it may not be necessary to eliminate the total amount of GPER at the plasma membrane to observe a significant reduction in receptor activity. More importantly, our preliminary in vitro findings indicate that E2-PROTACs may have therapeutic benefit not only for breast cancers that express GPER but also for some ER-negative breast cancers that are considered to be untreatable by estrogen targeted therapy.
Arvinas, LLC has developed PROTACs targeting either the ER (ARV-471) or AR (ARV-110) that have shown promising success in preclinical mouse models of cancer. Both PROTACs are in phase І clinical trials (NCT03888612 and NCT04072952 in clinicaltrials.gov) for prostate and breast cancer, respectively [81–82]. PROTACs have several advantages that make them more appealing than current SERDs for the treatment of breast cancer [82]. Because PROTACs are catalytic in nature, they are ideally suited for overcoming one of the main limitations of endocrine therapy, acquired drug resistance by either overexpression or mutation (provided that the ubiquitination acceptor site is not altered). In fact, ARV-471 has been demonstrated to efficiently degrade clinically relevant ERα mutants (Y537S and D538G) in cell lines and in PDX models [81]. The targeting domain of ARV-471 is undisclosed and proprietary so it is not clear whether it is capable of also targeting GPER. Likewise, a number of other ER-PROTACs have been developed and it is also not clear whether or not these PROTACs target GPER. Because GPER is expressed in 60% of ER-positive breast cancers [83], linked with clinical variables that predict advanced cancer, commonly expressed in ER-negative breast cancer and TNBC, and associated with resistance to anti-estrogen therapy, it is critical to develop cancer therapeutics that target GPER. A pan-estrogen receptor PROTAC, such as the first generation E2-PROTACs, UI-EP001 and UI-EP002, may be ideally suited for this purpose and would complement existing anti-estrogen therapies.
Supplementary Material
Figure 6. E2-PROTACs induce estrogen receptor-specific cell cytotoxicity in human breast cancer cells.
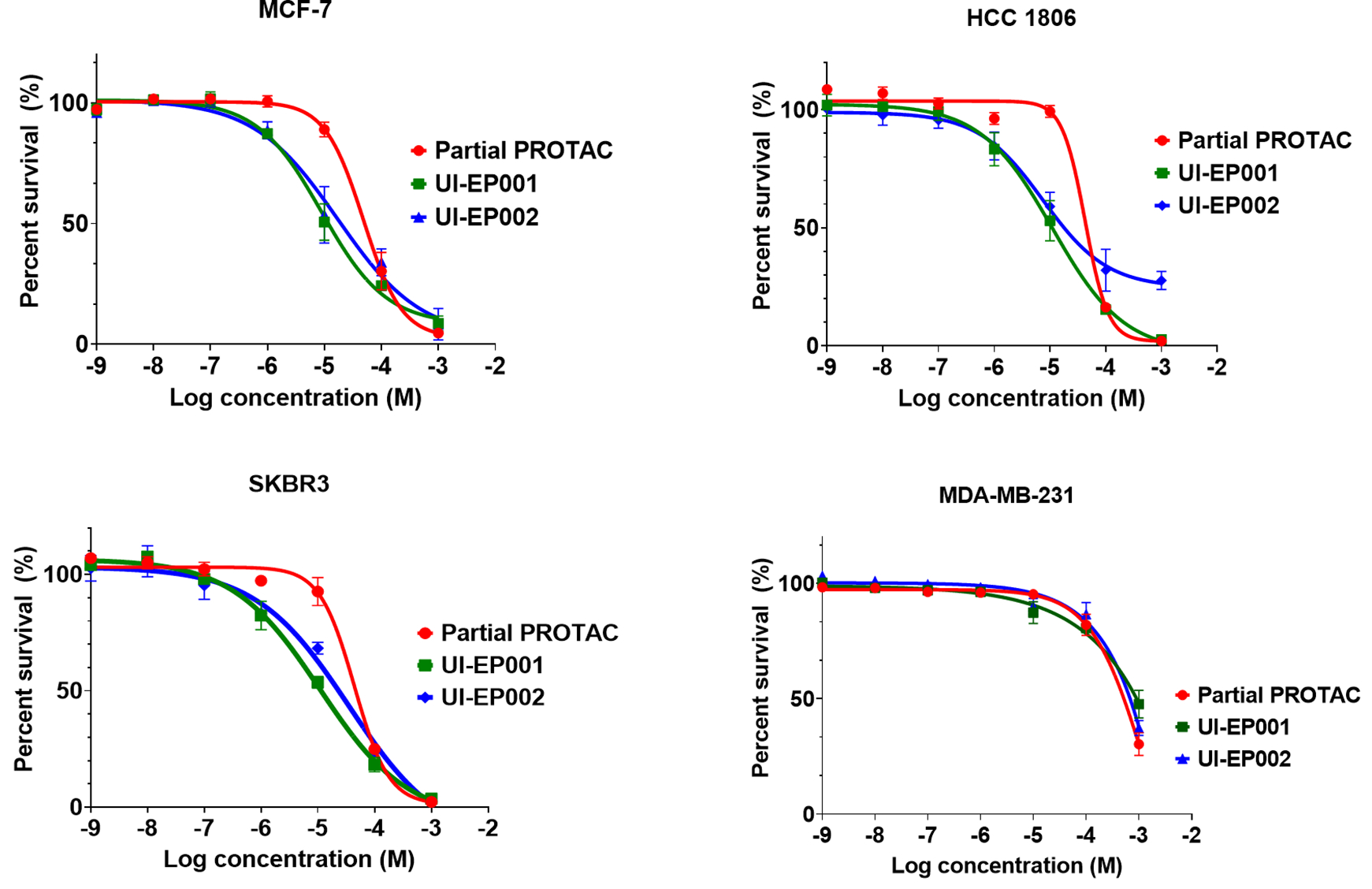
Cell viability of SKBR3, HCC-1806, MCF-7 and MDA-MB-231 breast cancer cells were measured after 24 hour treatment with increasing concentrations of either UI-EP001 or UI-EP002 or partial PROTAC. Results shown represent the mean ± S.E. of three independent experiments.
Acknowledgements
The data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. Research reported in this publication was supported by the Lyle and Sharon Bighley Chair of Pharmaceutical Sciences, a pilot award from the University of Iowa Breast Cancer Research Group and the National Cancer Institute of the National Institutes of Health under Award Number P30CA086862. The authors would like to thank Profs Jon Houtman, Michael Henry, George Weiner and Ronald Weigel for their helpful comments and advice.
Abbreviations.
- IC50
concentration of 50% inhibition; All IC50 values were measured from triplicate samples and are reported as the mean ± SD
- RBA
relative binding affinity calculated as the ratio of the IC50 value of 17β-E2 divided by the IC50 of the test compound
Footnotes
Supporting Information. Synthetic scheme and analyses of partial PROTAC, UI-EP001 and UI-EP-002 can be found in supporting information.
References:
- 1.Germain D Estrogen carcinogenesis in breast cancer. Endocrinol Metab Clin North Am 2011, 40, (3), 473–84, vii. [DOI] [PubMed] [Google Scholar]
- 2.Parl FF; Crooke PS; Plummer WD Jr.; Dupont WD Genomic-Epidemiologic Evidence That Estrogens Promote Breast Cancer Development. Cancer Epidemiol Biomarkers Prev 2018, 27, (8), 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothenberger NJ; Somasundaram A; Stabile LP The Role of the Estrogen Pathway in the Tumor Microenvironment. Int J Mol Sci 2018, 19, (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rugo HS; Rumble RB; Macrae E; Barton DL; Connolly HK; Dickler MN; Fallowfield L; Fowble B; Ingle JN; Jahanzeb M Endocrine therapy for hormone receptor-positive metastatic breast cancer: American Society of Clinical Oncology Guideline. Journal of Clinical Oncology 2016, 34, (25), 3069–3103. [DOI] [PubMed] [Google Scholar]
- 5.Yue W; Wang JP; Li Y; Fan P; Liu G; Zhang N; Conaway M; Wang H; Korach KS; Bocchinfuso W; Santen R Effects of estrogen on breast cancer development: Role of estrogen receptor independent mechanisms. Int J Cancer 2010, 127, (8), 1748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Placido S; Gallo C; De Laurentiis M; Bisagni G; Arpino G; Sarobba MG; Riccardi F; Russo A; Del Mastro L; Cogoni AA; Cognetti F; Gori S; Foglietta J; Frassoldati A; Amoroso D; Laudadio L; Moscetti L; Montemurro F; Verusio C; Bernardo A; Lorusso V; Gravina A; Moretti G; Lauria R; Lai A; Mocerino C; Rizzo S; Nuzzo F; Carlini P; Perrone F; Investigators GIM Adjuvant anastrozole versus exemestane versus letrozole, upfront or after 2 years of tamoxifen, in endocrine-sensitive breast cancer (FATA-GIM3): a randomised, phase 3 trial. Lancet Oncol 2018, 19, (4), 474–485. [DOI] [PubMed] [Google Scholar]
- 7.Tremont A; Lu J; Cole JT Endocrine Therapy for Early Breast Cancer: Updated Review. Ochsner J 2017, 17, (4), 405–411. [PMC free article] [PubMed] [Google Scholar]
- 8.Yang G; Nowsheen S; Aziz K; Georgakilas AG Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacol Ther 2013, 139, (3), 392–404. [DOI] [PubMed] [Google Scholar]
- 9.Hu R; Hilakivi-Clarke L; Clarke R Molecular mechanisms of tamoxifen-associated endometrial cancer (Review). Oncol Lett 2015, 9, (4), 1495–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones ME; van Leeuwen FE; Hoogendoorn WE; Mourits MJ; Hollema H; van Boven H; Press MF; Bernstein L; Swerdlow AJ Endometrial cancer survival after breast cancer in relation to tamoxifen treatment: pooled results from three countries. Breast Cancer Res 2012, 14, (3), R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosman F; Baz-Hecht M; Cushman M; Vardy MD; Cruz JD; Nieves JW; Zion M; Lindsay R Short-term effects of estrogen, tamoxifen and raloxifene on hemostasis: a randomized-controlled study and review of the literature. Thromb Res 2005, 116, (1), 1–13. [DOI] [PubMed] [Google Scholar]
- 12.Augusto TV; Correia-da-Silva G; Rodrigues CMP; Teixeira N; Amaral C Acquired resistance to aromatase inhibitors: where we stand! Endocr Relat Cancer 2018, 25, (5), R283–R301. [DOI] [PubMed] [Google Scholar]
- 13.Clarke R; Leonessa F; Welch JN; Skaar TC Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev 2001, 53, (1), 25–71. [PubMed] [Google Scholar]
- 14.Haque MM; Desai KV Pathways to Endocrine Therapy Resistance in Breast Cancer. Front Endocrinol (Lausanne) 2019, 10, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lei JT; Gou X; Seker S; Ellis MJ ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat 2019, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinhardt F; Franken A; Fehm T; Neubauer H Navigation through inter- and intratumoral heterogeneity of endocrine resistance mechanisms in breast cancer: A potential role for Liquid Biopsies? Tumour Biol 2017, 39, (11), 1010428317731511. [DOI] [PubMed] [Google Scholar]
- 17.Fan W; Chang J; Fu P Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med Chem 2015, 7, (12), 1511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barone I; Brusco L; Fuqua SA Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res 2010, 16, (10), 2702–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Becerra R; Santos N; Diaz L; Camacho J Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int J Mol Sci 2012, 14, (1), 108–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Achinger-Kawecka J; Valdes-Mora F; Luu PL; Giles KA; Caldon CE; Qu W; Nair S; Soto S; Locke WJ; Yeo-Teh NS; Gould CM; Du Q; Smith GC; Ramos IR; Fernandez KF; Hoon DS; Gee JMW; Stirzaker C; Clark SJ Epigenetic reprogramming at estrogen-receptor binding sites alters 3D chromatin landscape in endocrine-resistant breast cancer. Nat Commun 2020, 11, (1), 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thangavel C; Dean JL; Ertel A; Knudsen KE; Aldaz CM; Witkiewicz AK; Clarke R; Knudsen ES Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer 2011, 18, (3), 333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giuliano M; Schifp R; Osborne CK; Trivedi MV Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast 2011, 20 Suppl 3, S42–9. [DOI] [PubMed] [Google Scholar]
- 23.Rouhimoghadam M; Lu AS; Salem AK; Filardo EJ Therapeutic perspectives on the modulation of G-protein coupled estrogen receptor, GPER, function. Frontiers in Endocrinology 2020, 11, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filardo EJ Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol 2002, 80, (2), 231–8. [DOI] [PubMed] [Google Scholar]
- 25.Filardo EJ; Quinn JA; Bland KI; Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via transactivation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 2000, 14, (10), 1649–60. [DOI] [PubMed] [Google Scholar]
- 26.Pepermans RA; Prossnitz ER ERalpha-targeted endocrine therapy, resistance and the role of GPER. Steroids 2019, 152, 108493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filardo EJ; Graeber CT; Quinn JA; Resnick MB; Giri D; DeLellis RA; Steinhoff MM; Sabo E Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res 2006, 12, (21), 6359–66. [DOI] [PubMed] [Google Scholar]
- 28.Chan YT; Lai AC; Lin RJ; Wang YH; Wang YT; Chang WW; Wu HY; Lin YJ; Chang WY; Wu JC; Yu JC; Chen YJ; Yu AL GPER-induced signaling is essential for the survival of breast cancer stem cells. Int J Cancer 2020, 146, (6), 1674–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ignatov A; Ignatov T; Weissenborn C; Eggemann H; Bischoff J; Semczuk A; Roessner A; Costa SD; Kalinski T G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat 2011, 128, (2), 457–66. [DOI] [PubMed] [Google Scholar]
- 30.Rouhimoghadam M; Safarian S; Carroll JS; Sheibani N; Bidkhori G Tamoxifen-Induced Apoptosis of MCF-7 Cells via GPR30/PI3K/MAPKs Interactions: Verification by ODE Modeling and RNA Sequencing. Front Physiol 2018, 9, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin H; Zhu Q; Liu M; Tu G; Li Q; Yuan J; Wen S; Yang G GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int J Oncol 2017, 51, (4), 1191–1198. [DOI] [PubMed] [Google Scholar]
- 32.Steiman J; Peralta EA; Louis S; Kamel O Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am J Surg 2013, 206, (5), 698–703. [DOI] [PubMed] [Google Scholar]
- 33.Filardo EJ A role for G-protein coupled estrogen receptor (GPER) in estrogen-induced carcinogenesis: Dysregulated glandular homeostasis, survival and metastasis. J Steroid Biochem Mol Biol 2018, 176, 38–48. [DOI] [PubMed] [Google Scholar]
- 34.Petrie WK; Dennis MK; Hu C; Dai D; Arterburn JB; Smith HO; Hathaway HJ; Prossnitz ER G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int 2013, 2013, 472720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nathan MR; Schmid P A Review of Fulvestrant in Breast Cancer. Oncol Ther 2017, 5, (1), 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sammons S; Kornblum NS; Blackwell KL Fulvestrant-Based Combination Therapy for Second-Line Treatment of Hormone Receptor-Positive Advanced Breast Cancer. Target Oncol 2019, 14, (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pike AC; Brzozowski AM; Walton J; Hubbard RE; Thorsell AG; Li YL; Gustafsson JA; Carlquist M Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, (2), 145–53. [DOI] [PubMed] [Google Scholar]
- 38.Callige M; Richard-Foy H Ligand-induced estrogen receptor alpha degradation by the proteasome: new actors? Nucl Recept Signal 2006, 4, e004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson JF Fulvestrant (Faslodex) -- how to make a good drug better. Oncologist 2007, 12, (7), 774–84. [DOI] [PubMed] [Google Scholar]
- 40.Cyrus K; Wehenkel M; Choi EY; Han HJ; Lee H; Swanson H; Kim KB Impact of linker length on the activity of PROTACs. Molecular bioSystems 2011, 7, (2), 359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang X; Dixit VM Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res 2016, 26, (4), 484–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 2019, 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol 2018, 25, (1), 78–87 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Gonzalez A; Cyrus K; Salcius M; Kim K; Crews CM; Deshaies RJ; Sakamoto KM Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, (57), 7201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dragovich PS; Adhikari P; Blake RA; Blaquiere N; Chen J; Cheng YX; den Besten W; Han J; Hartman SJ; He J; He M; Rei Ingalla E; Kamath AV; Kleinheinz T; Lai T; Leipold DD; Li CS; Liu Q; Lu J; Lu Y; Meng F; Meng L; Ng C; Peng K; Lewis Phillips G; Pillow TH; Rowntree RK; Sadowsky JD; Sampath D; Staben L; Staben ST; Wai J; Wan K; Wang X; Wei B; Wertz IE; Xin J; Xu K; Yao H; Zang R; Zhang D; Zhou H; Zhao Y Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERalpha). Bioorg Med Chem Lett 2020, 30, (4), 126907. [DOI] [PubMed] [Google Scholar]
- 46.Hu J; Hu B; Wang M; Xu F; Miao B; Yang CY; Wang M; Liu Z; Hayes DF; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J Med Chem 2019, 62, (3), 1420–1442. [DOI] [PubMed] [Google Scholar]
- 47.Fan J; Liu K, Novel compounds having estrogen receptor alpha degradation activity and uses thereof. 2020. U.S. Patent Application No. 16/560,178.
- 48.Prossnitz ER; Arterburn JB International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev 2015, 67, (3), 505–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng SB; Quinn JA; Graeber CT; Filardo EJ Down-modulation of the G-protein-coupled estrogen 768 receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem 2011, 286, 22441–769 22455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnatt CK; Zhang Y A nuclear G protein-coupled estrogen receptor, GPER. Homology modeling studies toward its ligand-binding mode characterization. Computational approaches to nuclear receptors 2012, (30), 117. [Google Scholar]
- 51.Arnatt CK; Zhang Y G Protein-Coupled Estrogen Receptor (GPER) Agonist Dual Binding Mode Analyses toward Understanding of its Activation Mechanism: A Comparative Homology Modeling Approach. Mol Inform 2013, 32, (7), 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanenbaum DM; Wang Y; Williams SP; Sigler PB Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci U S A 1998, 95, (11), 5998–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filardo E; Quinn J; Pang Y; Graeber C; Shaw S; Dong J; Thomas P Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007, 148, (7), 3236–45. [DOI] [PubMed] [Google Scholar]
- 54.deConinck EC; McPherson LA; Weigel RJ Transcriptional regulation of estrogen receptor in breast carcinomas. Mol Cell Biol 1995, 15, (4), 2191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farnaby W; Koegl M; Roy MJ; Whitworth C; Diers E; Trainor N; Zollman D; Steurer S; Karolyi-Oezguer J; Riedmueller C; Gmaschitz T; Wachter J; Dank C; Galant M; Sharps B; Rumpel K; Traxler E; Gerstberger T; Schnitzer R; Petermann O; Greb P; Weinstabl H; Bader G; Zoephel A; Weiss-Puxbaum A; Ehrenhofer-Wolfer K; Wohrle S; Boehmelt G; Rinnenthal J; Arnhof H; Wiechens N; Wu MY; Owen-Hughes T; Ettmayer P; Pearson M; McConnell DB; Ciulli A BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol 2019, 15, (7), 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gadd MS; Testa A; Lucas X; Chan KH; Chen W; Lamont DJ; Zengerle M; Ciulli A Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 2017, 13, (5), 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas P; Pang Y; Filardo EJ; Dong J Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, (2), 624–32. [DOI] [PubMed] [Google Scholar]
- 58.Steinebach C; Kehm H; Lindner S; Vu LP; Kopff S; Lopez Marmol A; Weiler C; Wagner KG; Reichenzeller M; Kronke J; Gutschow M PROTAC-mediated crosstalk between E3 ligases. Chem Commun (Camb) 2019, 55, (12), 1821–1824. [DOI] [PubMed] [Google Scholar]
- 59.Madak JT; Cuthbertson CR; Chen W; Showalter HD; Neamati N Design, Synthesis, and Characterization of Brequinar Conjugates as Probes to Study DHODH Inhibition. Chemistry 2017, 23, (56), 13875–13878. [DOI] [PubMed] [Google Scholar]
- 60.Cao LY; Ren XM; Li CH; Zhang J; Qin WP; Yang Y; Wan B; Guo LH Bisphenol AF and Bisphenol B Exert Higher Estrogenic Effects than Bisphenol A via G Protein-Coupled Estrogen Receptor Pathway. Environ Sci Technol 2017, 51, (19), 11423–11430. [DOI] [PubMed] [Google Scholar]
- 61.Al-Bader M; Ford C; Al-Ayadhy B; Francis I Analysis of estrogen receptor isoforms and variants in breast cancer cell lines. Exp Ther Med 2011, 2, (3), 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itoh Y; Ishikawa M; Kitaguchi R; Sato S; Naito M; Hashimoto Y Development of target protein-selective degradation inducer for protein knockdown. Bioorg Med Chem 2011, 19, (10), 3229–41. [DOI] [PubMed] [Google Scholar]
- 63.Peng L; Zhang Z; Lei C; Li S; Ren X; Chang Y; Zhang Y; Xu Y; Ding K Identification of New Small-Molecule Inducers of Estrogen-related Receptor alpha (ERRalpha) Degradation. ACS Med Chem Lett 2019, 10, (5), 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneekloth AR; Pucheault M; Tae HS; Crews CM Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett 2008, 18, (22), 5904–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 2015, 22, (6), 755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, (6241), 1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl 2016, 55, (2), 807–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Silva MC; Ferguson FM; Cai Q; Donovan KA; Nandi G; Patnaik D; Zhang T; Huang HT; Lucente DE; Dickerson BC; Mitchison TJ; Fischer ES; Gray NS; Haggarty SJ Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Z; Lin Y; Song H; Qin X; Yu Z; Zhang Z; Dong G; Li X; Shi X; Du L; Zhao W; Li M First small-molecule PROTACs for G protein-coupled receptors: inducing alpha 1A-adrenergic receptor degradation. Acta Pharm Sin B 2020, 10, (9), 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakamoto KM; Kim KB; Verma R; Ransick A; Stein B; Crews CM; Deshaies RJ Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics 2003, 2, (12), 1350–8. [DOI] [PubMed] [Google Scholar]
- 71.Demizu Y; Okuhira K; Motoi H; Ohno A; Shoda T; Fukuhara K; Okuda H; Naito M; Kurihara M Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg Med Chem Lett 2012, 22, (4), 1793–6. [DOI] [PubMed] [Google Scholar]
- 72.Okuhira K; Demizu Y; Hattori T; Ohoka N; Shibata N; Nishimaki-Mogami T; Okuda H; Kurihara M; Naito M Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci 2013, 104, (11), 1492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vladusic EA; Hornby AE; Guerra-Vladusic FK; Lupu R Expression of estrogen receptor beta messenger RNA variant in breast cancer. Cancer Res 1998, 58, (2), 210–4. [PubMed] [Google Scholar]
- 74.Stevis PE; Deecher DC; Suhadolnik L; Mallis LM; Frail DE Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology 1999, 140, (11), 5455–8. [DOI] [PubMed] [Google Scholar]
- 75.Beveridge R; Stadlmann J; Penninger JM; Mechtler K A synthetic peptide library for benchmarking crosslinking-mass spectrometry search engines for proteins and protein complexes. Nat Commun 2020, 11, (1), 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kargbo RB PROTAC-Mediated Degradation of Estrogen Receptor in the Treatment of Cancer. ACS Med Chem Lett 2019, 10, (10), 1367–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang D; Baek SH; Ho A; Kim K Degradation of target protein in living cells by small-molecule proteolysis inducer. Bioorg Med Chem Lett 2004, 14, (3), 645–8. [DOI] [PubMed] [Google Scholar]
- 78.Molina L; Bustamante F; Ortloff A; Ramos I; Ehrenfeld P; Figueroa CD Continuous Exposure of Breast Cancer Cells to Tamoxifen Upregulates GPER-1 and Increases Cell Proliferation. Front Endocrinol (Lausanne) 2020, 11, 563165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou X; Wang S; Wang Z; Feng X; Liu P; Lv XB; Li F; Yu FX; Sun Y; Yuan H; Zhu H; Xiong Y; Lei QY; Guan KL Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest 2015, 125, (5), 2123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao L; Han X; Lu J; McEachern D; Wang S A highly potent PROTAC androgen receptor (AR) degrader ARD-61 effectively inhibits AR-positive breast cancer cell growth in vitro and tumor growth in vivo. Neoplasia 2020, 22, (10), 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flanagan J; Qian Y; Gough S; Andreoli M; Bookbinder M; Cadelina G; Bradley J; Rousseau E; Chandler J; Willard R ARV-471, an oral estrogen receptor PROTAC™ protein degrader for breast cancer. ARV 2018, 1000, 3. [Google Scholar]
- 82.Neklesa T; Snyder LB; Willard RR; Vitale N; Pizzano J; Gordon DA; Bookbinder M; Macaluso J; Dong H; Ferraro C ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol 2019, 37, (Suppl 7)), 259–259. [Google Scholar]
- 83.Filardo EJ; Quinn JA; Sabo E Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids 2008, 73, (9–10), 870–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.