

Abstract
Background
The diagnostic challenges associated with the COVID‐19 pandemic resulted in rapid development of diagnostic test methods for detecting SARS‐CoV‐2 infection. Serology tests to detect the presence of antibodies to SARS‐CoV‐2 enable detection of past infection and may detect cases of SARS‐CoV‐2 infection that were missed by earlier diagnostic tests. Understanding the diagnostic accuracy of serology tests for SARS‐CoV‐2 infection may enable development of effective diagnostic and management pathways, inform public health management decisions and understanding of SARS‐CoV‐2 epidemiology.
Objectives
To assess the accuracy of antibody tests, firstly, to determine if a person presenting in the community, or in primary or secondary care has current SARS‐CoV‐2 infection according to time after onset of infection and, secondly, to determine if a person has previously been infected with SARS‐CoV‐2. Sources of heterogeneity investigated included: timing of test, test method, SARS‐CoV‐2 antigen used, test brand, and reference standard for non‐SARS‐CoV‐2 cases.
Search methods
The COVID‐19 Open Access Project living evidence database from the University of Bern (which includes daily updates from PubMed and Embase and preprints from medRxiv and bioRxiv) was searched on 30 September 2020. We included additional publications from the Evidence for Policy and Practice Information and Co‐ordinating Centre (EPPI‐Centre) ‘COVID‐19: Living map of the evidence’ and the Norwegian Institute of Public Health ’NIPH systematic and living map on COVID‐19 evidence’. We did not apply language restrictions.
Selection criteria
We included test accuracy studies of any design that evaluated commercially produced serology tests, targeting IgG, IgM, IgA alone, or in combination. Studies must have provided data for sensitivity, that could be allocated to a predefined time period after onset of symptoms, or after a positive RT‐PCR test. Small studies with fewer than 25 SARS‐CoV‐2 infection cases were excluded. We included any reference standard to define the presence or absence of SARS‐CoV‐2 (including reverse transcription polymerase chain reaction tests (RT‐PCR), clinical diagnostic criteria, and pre‐pandemic samples).
Data collection and analysis
We use standard screening procedures with three reviewers. Quality assessment (using the QUADAS‐2 tool) and numeric study results were extracted independently by two people. Other study characteristics were extracted by one reviewer and checked by a second. We present sensitivity and specificity with 95% confidence intervals (CIs) for each test and, for meta‐analysis, we fitted univariate random‐effects logistic regression models for sensitivity by eligible time period and for specificity by reference standard group. Heterogeneity was investigated by including indicator variables in the random‐effects logistic regression models. We tabulated results by test manufacturer and summarised results for tests that were evaluated in 200 or more samples and that met a modification of UK Medicines and Healthcare products Regulatory Agency (MHRA) target performance criteria.
Main results
We included 178 separate studies (described in 177 study reports, with 45 as pre‐prints) providing 527 test evaluations. The studies included 64,688 samples including 25,724 from people with confirmed SARS‐CoV‐2; most compared the accuracy of two or more assays (102/178, 57%). Participants with confirmed SARS‐CoV‐2 infection were most commonly hospital inpatients (78/178, 44%), and pre‐pandemic samples were used by 45% (81/178) to estimate specificity. Over two‐thirds of studies recruited participants based on known SARS‐CoV‐2 infection status (123/178, 69%). All studies were conducted prior to the introduction of SARS‐CoV‐2 vaccines and present data for naturally acquired antibody responses. Seventy‐nine percent (141/178) of studies reported sensitivity by week after symptom onset and 66% (117/178) for convalescent phase infection. Studies evaluated enzyme‐linked immunosorbent assays (ELISA) (165/527; 31%), chemiluminescent assays (CLIA) (167/527; 32%) or lateral flow assays (LFA) (188/527; 36%).
Risk of bias was high because of participant selection (172, 97%); application and interpretation of the index test (35, 20%); weaknesses in the reference standard (38, 21%); and issues related to participant flow and timing (148, 82%). We judged that there were high concerns about the applicability of the evidence related to participants in 170 (96%) studies, and about the applicability of the reference standard in 162 (91%) studies.
Average sensitivities for current SARS‐CoV‐2 infection increased by week after onset for all target antibodies. Average sensitivity for the combination of either IgG or IgM was 41.1% in week one (95% CI 38.1 to 44.2; 103 evaluations; 3881 samples, 1593 cases), 74.9% in week two (95% CI 72.4 to 77.3; 96 evaluations, 3948 samples, 2904 cases) and 88.0% by week three after onset of symptoms (95% CI 86.3 to 89.5; 103 evaluations, 2929 samples, 2571 cases). Average sensitivity during the convalescent phase of infection (up to a maximum of 100 days since onset of symptoms, where reported) was 89.8% for IgG (95% CI 88.5 to 90.9; 253 evaluations, 16,846 samples, 14,183 cases), 92.9% for IgG or IgM combined (95% CI 91.0 to 94.4; 108 evaluations, 3571 samples, 3206 cases) and 94.3% for total antibodies (95% CI 92.8 to 95.5; 58 evaluations, 7063 samples, 6652 cases). Average sensitivities for IgM alone followed a similar pattern but were of a lower test accuracy in every time slot.
Average specificities were consistently high and precise, particularly for pre‐pandemic samples which provide the least biased estimates of specificity (ranging from 98.6% for IgM to 99.8% for total antibodies).
Subgroup analyses suggested small differences in sensitivity and specificity by test technology however heterogeneity in study results, timing of sample collection, and smaller sample numbers in some groups made comparisons difficult. For IgG, CLIAs were the most sensitive (convalescent‐phase infection) and specific (pre‐pandemic samples) compared to both ELISAs and LFAs (P < 0.001 for differences across test methods). The antigen(s) used (whether from the Spike‐protein or nucleocapsid) appeared to have some effect on average sensitivity in the first weeks after onset but there was no clear evidence of an effect during convalescent‐phase infection.
Investigations of test performance by brand showed considerable variation in sensitivity between tests, and in results between studies evaluating the same test. For tests that were evaluated in 200 or more samples, the lower bound of the 95% CI for sensitivity was 90% or more for only a small number of tests (IgG, n = 5; IgG or IgM, n = 1; total antibodies, n = 4). More test brands met the MHRA minimum criteria for specificity of 98% or above (IgG, n = 16; IgG or IgM, n = 5; total antibodies, n = 7). Seven assays met the specified criteria for both sensitivity and specificity.
In a low‐prevalence (2%) setting, where antibody testing is used to diagnose COVID‐19 in people with symptoms but who have had a negative PCR test, we would anticipate that 1 (1 to 2) case would be missed and 8 (5 to 15) would be falsely positive in 1000 people undergoing IgG or IgM testing in week three after onset of SARS‐CoV‐2 infection.
In a seroprevalence survey, where prevalence of prior infection is 50%, we would anticipate that 51 (46 to 58) cases would be missed and 6 (5 to 7) would be falsely positive in 1000 people having IgG tests during the convalescent phase (21 to 100 days post‐symptom onset or post‐positive PCR) of SARS‐CoV‐2 infection.
Authors' conclusions
Some antibody tests could be a useful diagnostic tool for those in whom molecular‐ or antigen‐based tests have failed to detect the SARS‐CoV‐2 virus, including in those with ongoing symptoms of acute infection (from week three onwards) or those presenting with post‐acute sequelae of COVID‐19. However, antibody tests have an increasing likelihood of detecting an immune response to infection as time since onset of infection progresses and have demonstrated adequate performance for detection of prior infection for sero‐epidemiological purposes. The applicability of results for detection of vaccination‐induced antibodies is uncertain.
Keywords: Humans; Antibodies, Viral; COVID-19; COVID-19/diagnosis; COVID-19/epidemiology; COVID-19 Vaccines; Immunoglobulin G; Immunoglobulin M; Pandemics; SARS-CoV-2; Seroepidemiologic Studies
Plain language summary
What is the diagnostic accuracy of antibody tests for the detection of infection with the COVID‐19 virus?
Background
COVID‐19 is an infectious disease caused by the SARS‐CoV‐2 virus that spreads easily between people in a similar way to the common cold or ‘flu’. Most people with COVID‐19 have a mild‐to‐moderate respiratory illness, and some may have no symptoms (asymptomatic infection). Others experience severe symptoms and need specialist treatment and intensive care.
In response to COVID‐19 infection, the immune system develops proteins called antibodies that can attack the virus as it circulates in their blood. People who have been vaccinated against COVID‐19 also produce these antibodies against the virus. Tests are available to detect antibodies in peoples' blood, which may indicate that they currently have COVID‐19 or have had it previously, or it may indicate that they have been vaccinated (although this group was not the focus of this review).
Why are accurate tests important?
Accurate testing allows identification of people who need to isolate themselves to prevent the spread of infection, or who might need treatment for their infection. Failure of diagnostic tests to detect infection with COVID‐19 when it is present (a false negative result) may delay treatment and risk further spread of infection to others. Incorrect diagnosis of COVID‐19 when it is not present (a false positive result) may lead to unnecessary further testing, treatment, and isolation of the person and close contacts. Accurate identification of people who have previously had COVID‐19 is important in measuring disease spread and assessing the success of public health interventions.
To determine the accuracy of an antibody test in identifying COVID‐19, test results are compared in people known to have (or have had) COVID‐19 and in people known not to have (or have had) COVID‐19. The criteria used to determine whether people are known or not known to have COVID‐19 is called the ‘reference standard’. Many studies use a test called reverse transcriptase polymerase chain reaction (RT‐PCR) as the reference standard, with samples taken from the nose and throat. Additional tests that can be used include measuring symptoms, like coughing or high temperature, or ‘imaging’ tests like chest X‐rays. People known not to have COVID‐19 are sometimes identified from stored blood samples taken before COVID‐19 existed, or from patients with symptoms confirmed to be caused by other diseases.
What did the review study?
We wanted to find out whether antibody tests:
‐ are able to diagnose infection in people with or without symptoms of COVID‐19, and
‐ can be used to find out if someone has already had COVID‐19.
The studies we included in our review looked at three types of antibodies. Most commonly, antibody tests measure two types known as IgG and IgM, but some tests only measure a single type of antibody or different combinations of the three types of antibodies (IgA, IgG, IgM).
What did we do?
We looked for studies that measured the diagnostic accuracy of antibody tests to detect current or past COVID‐19 infection and compared them with reference standard criteria. Since there are many antibody tests available, we included studies assessing any antibody test compared with any reference standard. People could be tested in hospital or in the community. The people tested may have been confirmed to have, or not to have, COVID‐19 infection, or they may be suspected of having COVID‐19.
Study characteristics
We found 178 relevant studies. Studies took place in Europe (94), Asia (45), North America (35), Australia (2), and South America (2).
Seventy‐eight studies included people who were in hospital with suspected or confirmed COVID‐19 infection and 14 studies included people in community settings. Several studies included people from multiple settings (35) or did not report where the participants were recruited from (39).
One hundred and forty‐one studies included recent infection cases (mainly week 1 to week 3 after onset of symptoms), and many also included people tested later (from day 21 onwards after infection) (117).
Main results
In participants that had COVID‐19 and were tested one week after symptoms developed, antibody tests detected only 27% to 41% of infections. In week 2 after first symptoms, 64% to 79% of infections were detected, rising to 78% to 88% in week 3. Tests that specifically detected IgG or IgM antibodies were the most accurate and, when testing people from 21 days after first symptoms, they detected 93% of people with COVID‐19. Tests gave false positive results for 1% of those without COVID‐19.
Below we illustrate results for two different scenarios.
If 1000 people were tested for IgG or IgM antibodies during the third week after onset of symptoms and only 20 (2%) of them actually had COVID‐19:
‐ 26 people would test positive. Of these, 8 people (31%) would not have COVID‐19 (false positive result).
‐ 974 people would test negative. Of these, 2 people (0.2%) would actually have COVID‐19 (false negative result).
If 1000 people with no symptoms for COVID‐19 were tested for IgG antibodies and 500 (50%) of them had previously had COVID‐19 infection more than 21 days previously:
‐ 455 people would test positive. Of these, 6 people (1%) would not have been infected (false positive result).
‐ 545 people would test negative. Of these, 51 (9%) would actually have had a prior COVID‐19 infection (false negative result).
How reliable were the results of the studies of this review?
We have limited confidence in the evidence for several reasons. The number of samples contributed by studies for each week post‐symptom onset was often small, and there were sometimes problems with how studies were conducted. Participants included in the studies were often hospital patients who were more likely to have experienced severe symptoms of COVID‐19. The accuracy of antibody tests for detecting COVID‐19 in these patients may be different from the accuracy of the tests in people with mild or moderate symptoms. It is not possible to identify by how much the test results would differ in other populations.
Who do the results of this review apply to?
A high percentage of participants were in hospital with COVID‐19, so were likely to have more severe disease than people with COVID‐19 who were not hospitalised. Only a small number of studies assessed these tests in people with no symptoms. The results of the review may therefore be more applicable to those with severe disease than people with mild symptoms.
Studies frequently did not report whether participants had symptoms at the time samples were taken for testing making it difficult to fully separate test results for early‐phase infection as opposed to later‐phase infections.
The studies in our review assessed several test methods across a global population, therefore it is likely that test results would be similar in most areas of the world.
What are the implications of this review?
The review shows that antibody tests could have a useful role in detecting if someone has had COVID‐19, but the timing of test use is important. Some antibody tests may help to confirm COVID‐19 infection in people who have had symptoms for more than two weeks but who have been unable to confirm their infection using other methods. This is particularly useful if they are experiencing potentially serious symptoms that may be due to COVID‐19 as they may require specific treatment. Antibody tests may also be useful to determine how many people have had a previous COVID‐19 infection. We could not be certain about how well the tests work for people who have milder disease or no symptoms, or for detecting antibodies resulting from vaccination.
How up‐to‐date is this review?
This review updates our previous review. The evidence is up‐to‐date to September 2020.
Summary of findings
Summary of findings 1. What is the diagnostic accuracy of antibody tests, for the diagnosis of current or prior SARS‐CoV‐2 infection?
| Question | What is the diagnostic accuracy of antibody tests, for the diagnosis of current or prior SARS‐CoV‐2 infection? | |||||
| Population | Adults or children suspected of current SARS‐CoV‐2 infection or who may have had prior SARS‐CoV‐2 infection, including populations undergoing screening for SARS‐CoV‐2 such as asymptomatic contacts of confirmed COVID‐19 cases or community‐based testing | |||||
| Index test | Any commercially produced test for detecting antibodies to SARS‐CoV‐2, including:
|
|||||
| Target condition | Detection of:
|
|||||
| Reference standard |
Presence of current infection: RT‐PCR alone or combined with clinical diagnosis of COVID‐19 based on established guidelines or combinations of clinical features for RT‐PCR‐negative Presence of prior infection: RT‐PCR alone Absence of infection: pre‐pandemic sources of samples for testing, RT‐PCR‐negative samples from COVID‐suspects, from healthy participants or those with pre‐existing disease |
|||||
| Action |
|
|||||
| Limitations in the evidence | ||||||
| Risk of bias |
Participant selection: high risk of bias in 172 studies (99%), primarily because of selection for inclusion based on known disease status (i.e. separate recruitment of confirmed SARS‐CoV‐2 cases and non‐cases) Index test: high risk of bias in 35 studies (22%) because blinded index test interpretation was not implemented or the threshold to define test positivity was determined by analysing the data rather than prespecified Reference standard: high risk of bias in 39 studies (22%) because of inadequate reference standards for confirming absence of infection, e.g. reliance on a single negative RT‐PCR result in people with suspected COVID‐19, or no RT‐PCR testing reported in contemporaneous healthy or other disease non‐COVID‐19 groups, or because serology results in part determined the presence of infection Flow and timing: high risk of bias in 146 studies (84%) because of different reference standards used to verify presence or absence of infection, some participants with no reference standard, exclusions from analyses, and inclusion of multiple samples per participant |
|||||
| Concerns about applicability of the evidence |
Participants: high concerns in 171 studies (98%) because participants were unlikely to be similar to those in whom the test would be used in clinical practice, e.g. hospitalised confirmed cases of COVID‐19 or healthy or other disease non‐SARS‐CoV‐2 groups Index test: no studies rated as high concerns for applicability Reference standard: high concerns in 162 studies (93%), primarily because cases were defined based only on RT‐PCR‐positive results and did not consider clinically defined cases |
|||||
| Findings | ||||||
| ||||||
| Quantity of evidence | Number of studies | Total participants or samplesa | Total cases | |||
| 178 | 64,688 | 25,724 | ||||
|
Sensitivity (95% CI) N evaluations (TP/SARS‐CoV‐2 cases) |
Specificity (95%CI) N evaluations (TN/non‐SARS‐CoV‐2 cases) |
|||||
| Week 1 | Week 2 | Week 3 | Convalescent | Pre‐pandemic | ||
| Assays targeting IgG alone |
27.2 (24.9, 29.7) |
64.8 (62.1, 67.4) |
88.1 (86.6, 89.5) |
89.8** (88.5, 90.9) |
98.9** (98.6, 99.1) |
|
| 189 (2177/6679) | 202 (5883/9078) | 190 (4328/5027) | 253 (14,183/16,846) | 179 (37,385/38,090) | ||
| Assays targeting IgM alone |
29.5 (25.8, 33.6) |
64.6 (60.3, 68.7) |
78.3 (74.8, 81.4) |
71.2 (65.5, 76.2) |
98.6 (98.0, 99.1) |
|
| 126 (1770/4492) | 122 (3715/5577) | 118 (2416/3231) | 125 (4683/7124) | 83 (14,691/15,126) | ||
| Assays targeting either IgG or IgMb |
41.1 (38.1, 44.2) |
74.9 (72.4, 77.3) |
88.0* (86.3, 89.5) |
92.9 (91.0, 94.4) |
99.2* (98.5, 99.5) |
|
| 103 (1593/3881) | 96 (2904/3948) | 103 (2571/2929) | 108 (3206/3571) | 68 (8989/9262) | ||
| Assays targeting total antibodies |
37.7 (31.0, 44.9) |
79.4 (74.0, 83.9) |
90.9 (87.8, 93.2) |
94.3 (92.8, 95.5) |
99.8 (99.6, 99.9) |
|
| 27 (428/1010) | 29 (804/1030) | 33 (908/1016) | 58 (6652/7063) | 45 (12,166/12,207) | ||
| Antibody tests for diagnosis of current infection: Numbers applied to a hypothetical cohort of 1000 people, using summary data for the combination of IgG or IgM in week 3 after onset of infection for sensitivity and pre‐pandemic samples (denoted using * above) | ||||||
| Prevalence of current infection | TP (95% CI) | FP (95% CI) | FN (95% CI) | TN (95% CI) | PPV (%) | 1‐NPV (%) |
| 1% | 9 (9, 9) | 8 (5, 15) | 1 (1, 1) | 982 (975, 985) | 53 | 0.1 |
| 2% | 18 (17, 18) | 8 (5, 15) | 2 (2, 3) | 972 (965, 975) | 69 | 0.2 |
| 5% | 46 (46, 47) | 8 (5, 14) | 4 (3, 5) | 942 (936, 945) | 85 | 0.6 |
| Antibody tests for diagnosis of prior infection: Numbers applied to a hypothetical cohort of 1000 people, using summary data for IgG alone during the convalescent phase of infection for sensitivity and pre‐pandemic samples (denoted using ** above) | ||||||
| Prevalence of prior infection | TP (95% CI) | FP (95% CI) | FN (95% CI) | TN (95% CI) | PPV (%) | 1‐NPV (%) |
| 20% | 180 (177, 182) | 9 (7, 11) | 20 (18, 23) | 791 (789, 793) | 95 | 2.5 |
| 50% | 449 (443, 455) | 6 (5, 7) | 51 (46, 58) | 494 (493, 496) | 99 | 9.4 |
| *Data applied to hypothetical cohort with current infection. ** Data applied to hypothetical cohort with prior infection. | ||||||
CGIA: colloidal gold immunoassays CI: confidence interval CLIA: chemiluminescence immunoassays ELISA: enzyme‐linked immunosorbent assays FIA: fluorescence‐labelled immunochromatographic assays FN: false negative FP: false positive RT‐PCR: reverse transcription polymerase chain reaction TN: true negative TP: true positive
aSamples counted once per study; results per antibody and time period were counted per test evaluated (i.e. could be counted more than once per study)
bPositive if either IgG‐ or IgM‐positive
Background
We are creating and maintaining a suite of living systematic reviews to cover the roles of tests and characteristics in the diagnosis of coronavirus disease (COVID‐19). This review summarises evidence of the accuracy of COVID‐19 antibody tests; both laboratory‐based tests and rapid tests using a lateral flow format.
Target condition being diagnosed
COVID‐19 is the disease caused by infection with the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). The key target conditions for this suite of reviews are current SARS‐CoV‐2 infection, current COVID‐19 disease, and past SARS‐CoV‐2 infection. The COVID‐19 antibody tests included in this review primarily concern the identification of previous SARS‐CoV‐2 infection, however, we also consider their use for identification of current infection in the immediate days and weeks after onset.
For current infection, the severity of the disease is of importance. SARS‐CoV‐2 infection can be asymptomatic (no symptoms); mild or moderate (symptoms such as fever, cough, aches, lethargy but without difficulty breathing at rest); severe (symptoms with breathlessness and increased respiratory rate indicative of pneumonia); or critical (requiring respiratory support due to severe acute respiratory syndrome (SARS) or acute respiratory distress syndrome (ARDS)). People with COVID‐19 pneumonia (severe or critical disease) require different patient management, and it is important to be able to identify them. There is no consideration that antibody tests are able to distinguish severity of disease, thus, in this review, we consider their role for detecting SARS‐CoV‐2 infection of any severity (asymptomatic or symptomatic).
In the context of test evaluation, and throughout this review, we use the term 'reference standard' to denote the best available method (test or tests) for diagnosing the target condition, as opposed to other uses of the term in diagnostic virology (such as reference methods or reference materials). Clinicians typically diagnose current SARS‐CoV‐2 infection through direct detection of viral nucleic acid in respiratory tract specimens (e.g., nasopharyngeal swabs). The most frequently used tool to do this are nucleic acid amplification‐based tests such as reverse‐transcriptase polymerase chain reaction (RT‐PCR). The RT‐PCR carries a very small risk of false‐positive results for infection and a higher risk of false‐negative results. False‐positive results may result from failures in sampling or laboratory protocols (e.g. mislabelling), contamination during sampling or processing, or low‐level reactions during PCR (Healy 2021; Mayers 2020). As for other reviews in this series, we consider the upper bound on the possible false‐positive rate of RT‐PCR of less than 0.077%. This estimate is based on population prevalence surveys showing RT‐PCR positivity rates (comprising both true‐positive and false‐positive results) of 0.44% (95% credible interval: 0.22% to 0.76%) (August 2020; ONS 2020), and 0.077% (0.065%, 0.092%) (June to July 2020; Riley 2020 React‐1 study).
False‐negative rates for RT‐PCR have been estimated by looking at individuals with symptoms who initially test negative, but positive on a subsequent test. These rates have been estimated to be as high as 20% to 30% in the first week of symptom onset (Arevalo‐Rodriguez 2020a; Kucirka 2020; Yang 2020a; Zhao 2020). Including probable SARS‐CoV‐2 infection cases within the target condition, as defined by internationally recognised clinical guidelines for diagnosis of SARS‐CoV‐2, will partially mitigate missed cases due to false‐negative RT‐PCR results but risk over‐classification of COVID‐19 when it is not in fact present. Both the World Health Organization (WHO) and the Chinese Center for Disease Control and Prevention (China CDC) have produced case definitions for ‘probable cases of SARS‐COV‐2 infection’ that include RT‐PCR‐negative cases that display other convincing clinical evidence (Appendix 1). The most recent case definition from the China CDC includes positive antibody tests. Confirming an acute clinical diagnosis using an antibody test requires detectable virus‐specific IgM and IgG in serum, or detectable virus‐specific IgG, or a 4‐fold or greater increase in titration to be observed during convalescence compared with the acute phase. The U.S. Centers for Disease Control and Prevention (US CDC) guidelines consider the presence of SARS‐CoV‐2 specific antibodies in serum, plasma, or whole blood to provide supportive rather than confirmatory laboratory evidence of infection (CDC 2021b).
For the presence of both current or prior SARS‐CoV‐2 infection, we require a confirmed positive RT‐PCR result or a clearly documented application of clinical guideline‐based diagnosis of symptomatic COVID‐19 in those who were RT‐PCR negative.
For the absence of current SARS‐CoV‐2, a number of reference standards may be used:
stored samples obtained prior to the initial spread of SARS‐CoV‐2 (or ‘pre‐pandemic’ samples); these samples may be from healthy volunteers, or from individuals with other respiratory infections,
contemporaneous samples from healthy individuals, such as blood donors, or from those with other respiratory infections (preferably with confirmation of the absence of SARS‐CoV‐2 infection by RT‐PCR),
contemporaneous samples obtained from RT‐PCR‐negative individuals suspected of having COVID‐19, usually based on signs and symptoms of infection.
Positive serology results in pre‐pandemic samples can be considered as truly false positive, however, for contemporaneous samples from individuals considered not to have SARS‐CoV‐2 infection, there is a risk that some positive results do indicate a current or previous SARS‐CoV‐2 infection. Test manufacturers also carry out test evaluations in confounder or cross‐reactivity panels of samples from individuals with other types of laboratory‐confirmed respiratory infection or with conditions that produce antibodies that might cause false‐positive results on a SARS‐CoV‐2 assay. Although we did not set out to systematically evaluate results in cross‐reactivity panels, we have included these results separately where available.
Index test(s)
This review evaluates serology tests to measure antibodies to the SARS‐CoV‐2 virus. Antibodies are formed by the body's immune system in response to infections, and can be detected in whole blood, plasma, serum, urine or saliva, although the latter two are not applicable for detection of a response to SARS‐CoV‐2 infection. The antibodies produced are largely specific to a particular virus, and therefore can be used to differentiate between infections. There are three types of binding antibodies created in response to infection ‐ IgA, IgG, and IgM ‐ these effectively alert the body’s immune system to the presence of a foreign pathogen. Neutralising antibodies (or Nabs) are antibodies that act to prevent the virus from further replication; they are less easily measured compared to IgA, IgG or IgM and are not used to diagnose the presence or absence of current or prior SARS‐CoV‐2 infection.
Antibody tests available for laboratory use include enzyme‐linked immunosorbent assay (ELISA or EIA) methods, or more advanced chemiluminescence immunoassays (CLIA). These laboratory‐based tests require relatively specialised equipment and biosafety procedures and not only detect the presence or absence of antibodies but quantitatively measure antibody levels or titres. Laboratory‐independent, point‐of‐care lateral flow assays for antibody detection use disposable devices, akin to a pregnancy test, that use a minimal amount of blood on a testing strip. Antibody detection is indicated by visible lines appearing on the test strip, or through fluorescence, which can be detected using a reader device. Many of these tests are known as colloidal gold‐based immunoassays, as they use SARS‐CoV‐2 antigen conjugated to gold nanoparticles.
All serological assays use purified SARS‐CoV‐2 proteins (typically the nucleocapsid ‘N’‐ or spike ‘S’‐protein, or more specific subunits such as S1, S2 or the receptor binding domain [RBD] on the S1 subunit) to target virus‐specific IgA, IgG or IgM (Figure 1). Many tests assess the presence of both IgG and IgM. IgM typically rises quickly with infection and declines soon after an infection is cleared; IgG persists for longer but is reported to wane during the late convalescent period (3 to 6 months post‐infection) (ECDC 2021). Alternatively, tests may combine IgA with IgG, or measure all antibodies (IgA, IgG, and IgM). The implications from the choice of antigen or protein used in an assay have become increasingly important with the advent of vaccination and waning natural immunity from prior SARS‐CoV‐2 infections. Infection‐induced antibodies may arise in response to the N‐ and S‐ proteins and are therefore potentially detectable by any assay using either of these proteins. Because vaccines are designed to induce antibodies to the spike‐protein or RBD, a positive result on an S‐based assay (that uses the specific viral protein target that was used in the vaccine) could indicate either prior infection or vaccine‐induced antibodies. The US CDC have provided guidelines for interpretation of results, particularly tests for IgG, in vaccinated and non‐vaccinated individuals according to the antigenic target (CDC 2021b). This review includes studies conducted prior to the introduction of vaccines against COVID‐19 and therefore can only consider how well antibody tests are able to detect prior natural infection with SARS‐CoV‐2.
1.

SARS‐CoV‐2 diagram
The production and nature of neutralising antibodies is known to be affected by SARS‐CoV‐2 variants (Greaney 2022), however, the extent to which the development of binding antibodies is affected by variants is as yet unclear (Junker 2022; Yadav 2022). Viral mutations could also lead to changes in viral proteins that may in turn affect the accuracy of serological tests that were developed using viral proteins without those mutations (FDA 2021a). The FDA have not as yet listed any serological assay as being impacted by genetic variation (FDA 2021a).
Following the emergence of COVID‐19, there has been prolific industry activity to develop accurate antibody tests. FIND (which is a global non‐profit alliance for diagnostics) and the Johns Hopkins Centre for Health Security have maintained online lists of these and other molecular‐based tests for SARS‐CoV‐2. At the time of writing (24 March 2022), FIND listed 298 commercially available antibody tests. Regulatory approval in the European Union (EU; CE‐IVD) had been awarded to 223 on the list, whereas in China only two had been approved, and 37 by the US Food and Drug Administration (FDA). For a period of time (16 March to 11 May 2020), the FDA allowed marketing of antibody tests in the USA without formal regulatory approval, the intention being to allow tests to quickly be made available while the manufacturers prepared their applications for Emergency Use Authorisation (EUA). As a consequence, hundreds of tests were placed on the market, many from manufacturers with no track record in developing in vitro devices and often with insufficient validation. FDA policy changes were soon implemented and, by early July 2020, 56 serological assays had already been added to the FDAs’ ‘do not distribute list’. A comprehensive case study review of the experience in the US and lessons to be learned for future pandemics is provided by West and colleagues (West 2021).
Clinical pathway
For the first iteration of this review, we considered four possible use cases for antibody tests:
Diagnosis of acute suspected COVID‐19;
Serial testing to assess immune response in patients with severe disease;
Identification of prior infection as a possible indicator of immunity to further infection;
In seroprevalence surveys for public health management purposes.
Based primarily on data from hospital inpatients, we showed that for diagnosis of symptomatic COVID‐19, antibody tests had very low sensitivity in the first week following onset of symptoms, rising in the second week, but only exceeding 90% in the third week after onset (Deeks 2020a). For the detection of prior infection, few studies with longer term follow‐up were available, however, there was some indication that antibody tests could have a useful role for detecting previous SARS‐CoV‐2 infection if used 15 days or more after the onset of symptoms (Deeks 2020a).
Two years into the COVID‐19 pandemic, the potential for re‐infection and rising vaccination rates necessarily affects the way in which antibody tests might be used. Below we reconsider possible use case scenarios, also taking into consideration an update of the US CDC guidelines for antibody testing which sets out potential indications for antibody testing and interpretation of results in the current landscape (CDC 2021b), however, as previously stated, we are not able to consider how well serology tests can detect vaccination‐induced antibodies:
Diagnosis of acute COVID‐19 (current infection) in those with negative RT‐PCR results. The CDC suggest that a positive antibody test at least seven days after the onset of infection could suggest the presence of current SARS‐CoV‐2 infection where earlier antibody test results were negative (CDC 2021b), implying that antibody tests are only useful for diagnosis in individuals with no evidence of prior infection or in those who have not been vaccinated. In vaccinated individuals, an N‐based antibody assay could be used to identify the emergence of infection‐related antibodies, however, an earlier negative result would be required to confirm the presence of a newly acquired SARS‐CoV‐2 infection.
To assist diagnosis where patients present with a multisystem inflammatory syndrome (MIS) or other post‐acute sequelae of COVID‐19 (current or previous infection) (CDC 2021b). MIS in children or in adults typically arises within four weeks of a SARS‐CoV‐2 infection and can occur in individuals who had no obvious signs or symptoms of COVID‐19 during the acute phase of infection (CDC 2020a; CDC 2021a). Serologic testing could be used to support the diagnosis of a prior SARS‐CoV‐2 infection having led to MIS in those with no previous RT‐PCR test, with a negative previous RT‐PCR test, or in those who are negative on RT‐PCR at the time of presentation.
For seroprevalence surveys for epidemiological or public health management purposes (previous infection). Understanding the prevalence of detectable antibodies resulting from infection and or vaccination can serve a number of purposes (Bonanni 2021):
To retrospectively determine the size of an outbreak,
To identify how much infection has spread in a population under study, either overall or in specific subgroups, for example by age,
To estimate the prevalence of mild and asymptomatic infection,
To inform estimation of infection fatality rates and vaccine effectiveness, and
To estimate the proportion of the population who may be protected against infection, or at least protected against developing severe COVID‐19, in the future.
This information can be used in a number of ways not least to inform public health containment (or alternatively de‐escalation) strategies or to identify groups for targeted vaccination policies. Differentiating the prevalence of infection‐acquired antibodies from those resulting from vaccination would require the use of both N‐ and S‐based serologic assays. Although rapid tests are used for seroprevalence purposes (e.g. REACT‐2 surveys in the UK (Ward 2022)), quantitative serological assays that measure antibody titres are needed to allow antibody kinetics to be examined over time and facilitate understanding of the role of antibodies in immunity from further infection.
Two additional use cases during current infection include:
4. Serial testing for monitoring immune response in patients with severe disease.
5. To select currently infected, seronegative COVID‐19 patients who are at high risk of progression to severe COVID‐19 for monoclonal antibody treatments such as casirivimab or imdevimab (Agarwal 2020) or bebtelovimab for the Omicron variant (FDA 2022).
Use case 4 is a monitoring rather than diagnostic use case and use case 5 is a stratified medicine scenario. Both use cases would require comparison with a reference standard test of antibody response, rather than evidence of infection; as such these use cases will not be further considered in this review.
Prior test(s)
Prior testing depends on the purpose of the test. Where antibody testing is proposed to assist with acute diagnosis of infection or for diagnosis of longer‐term sequelae from COVID‐19 (use cases 1 and 2), we anticipate that patients would be symptomatic and most likely have undergone RT‐PCR testing and possibly computed tomography (CT) imaging with other laboratory markers used as needed. For the identification of prior SARS‐CoV‐2 infection (use case 3), individuals may have undergone rapid antigen testing or RT‐PCR if symptomatic or if exposure to a confirmed case was suspected. However prior testing will not necessarily influence the likelihood of any subsequent antibody testing.
Alternative test(s)
This review is one of a suite of reviews that cover the range of tests and characteristics being considered in the management of COVID‐19 (Deeks 2020b; Leeflang 2021; McInnes 2020), five of which have already been published (Dinnes 2021; Islam 2021; Stegeman 2020; Struyf 2021), including one previous iteration of this review (Deeks 2020a). Full details of the alternative tests and evidence of their accuracy is summarised in these reviews. As we have previously established that antibody tests may only have a role for diagnosis of acute current infection when other tests are negative or inconclusive, they are not further described here.
Rationale
It is essential to understand the clinical accuracy of tests and diagnostic features to identify the best way they can be used in different settings to develop effective diagnostic and management pathways. The suite of Cochrane’s 'living systematic reviews' summarises evidence on the clinical accuracy of different tests and diagnostic features, grouped according to the research questions and settings that we are aware of. Estimates of accuracy from these reviews will help inform diagnosis, screening, isolation, and patient management decisions.
Summary of the previous version of the review
The first iteration of this review (Deeks 2020a) included 57 publications reporting 54 separate study cohorts with 15,976 samples, including 8526 from cases with SARS‐CoV‐2 infection. Data for 25 commercial tests and 25 inhouse assays were evaluated. Studies were primarily conducted in Asia (n = 38, 70%) and over half (n = 28, 52%) were only available as preprints. We identified several methodological limitations including use of multi‐group designs (n = 29, 54%) or inclusion of only SARS‐CoV‐2 cases (n = 19, 35%), lack of blinding of the index test (n = 49, 91%) and reference standard (n = 29, 54%), differential verification (n = 22, 41%), and the lack of clarity about participant numbers, characteristics and study exclusions (n = 47, 87%). Most studies (n = 44, 81%) only included people hospitalised due to suspected or confirmed COVID‐19.
We observed substantial heterogeneity in sensitivities of IgA, IgM and IgG antibodies, or combinations thereof, for results aggregated across different time periods post‐symptom onset (range 0% to 100% for all target antibodies). Main results were therefore based on studies that stratified results by time since symptom onset (n = 38, 70%); the numbers of individuals contributing data within each study for each time period were small and usually not based on tracking the same groups of patients over time. Pooled results for IgG, IgM, IgA, total antibodies and IgG/IgM all showed low sensitivity during the first week since onset of symptoms (all less than 30.1%), rising in the second week and reaching their highest values in the third week. The combination of IgG/IgM had a sensitivity of 30.1% (95% CI 21.4 to 40.7; 9 evaluations, 259 samples) for 1 to 7 days, 72.2% (95% CI 63.5 to 79.5; 9 evaluations, 608 samples) for 8 to 14 days, and 91.4% (95% CI 87.0 to 94.4; 9 evaluations, 692 samples) for 15 to 21 days. Estimates of accuracy beyond three weeks were based on smaller sample sizes and fewer studies. For 21 to 35 days, pooled sensitivities for IgG/IgM were 96.0% (95% CI 90.6 to 98.3). There were insufficient studies to estimate the sensitivity of tests beyond 35 days post‐symptom onset. Summary specificities (provided in 35 studies) exceeded 98% for all target antibodies with confidence intervals no more than two percentage points wide. False‐positive results were more common where COVID‐19 had been suspected and ruled out, but numbers were small and the difference was within the range expected by chance. Analyses showed small differences in sensitivity between assay type, but methodological concerns and sparse data prevent comparisons between test brands.
The review concluded that antibody tests have no role for the diagnosis of acute COVID‐19 in the early weeks after symptom onset but may complement other testing in individuals presenting later (after 14 days), when RT‐PCR tests are negative, or are not done. Antibody tests seemed likely to be useful for detecting previous SARS‐CoV‐2 infection, however, at that time the duration of antibody rises was unknown, and very little data beyond 35 days post‐symptom onset, or from individuals in the community with milder or no symptoms of COVID‐19, was identified.
Changes in the evidence base since the previous version
There has been a considerable increase in the number of available evaluations of antibody assays, primarily from symptomatic populations but with some studies including asymptomatic individuals. This iteration of the review restricts study inclusion to evaluations of commercially produced tests and to those reporting sensitivities according to time after onset of infection, primarily defined as time from symptom onset. Results for specificity are presented separately for pre‐pandemic and for different groups of contemporaneously collected samples (from either people tested because of suspicion of COVID‐19, people with other confirmed respiratory infections or other conditions, or from healthy individuals). The number of test brands with available data has increased as has the amount of data by week after symptom onset (up to day 35). We have also been able to analyse data for those in the convalescent phase of infection (defined as 21 days or more after symptom onset, or 14 days or more after a positive PCR test) and for those reported as asymptomatic at the time of testing. Studies mostly continue to rely on a single RT‐PCR result to confirm the presence or absence of infection, however, we have been able to conduct subgroup analyses to investigate the effect of different index test methods (ELISA, CLIA or lateral flow assay) and antigens used for both sensitivity and specificity. Results by test brand in convalescent individuals are considered according to the UK Medicines and Healthcare products Regulatory Agency (MHRA) target product profiles for COVID‐19 diagnostics (i.e. acceptable performance criterion of sensitivity ≥ 98% and specificity ≥ 98% (MHRA 2021b) as a benchmark against which to consider test performance.
The volume of literature on the accuracy of antibody tests has increased substantially since the last iteration of this review. This has allowed us to generate more precise estimates of accuracy for specific diagnostic test applications and stratified by important clinical subgroups. However, antibody tests have not had the widespread use that was predicted at the beginning of the pandemic. Although antibody tests are potentially useful for certain use cases as defined in the Clinical Pathway, we do not currently have any plans to further update this review. Vaccination for SARS‐CoV‐2 infection was introduced shortly following the search cut‐off of this review. This review therefore provides a summary of diagnostic test accuracy for antibody tests for naturally‐acquired SARS‐CoV‐2 infection.
This review follows a generic protocol that covers six Cochrane COVID‐19 diagnostic test accuracy reviews (Deeks 2020b). The ‘Background’, ‘Objectives’ and ‘Methods’ sections of this review therefore use some text that was originally published in the protocol (Deeks 2020b), in the previous iteration of this review (Deeks 2020a) and text that overlaps some of our other reviews (Dinnes 2021; Struyf 2021).
Objectives
To assess the diagnostic accuracy of antibody tests to determine if a person presenting in the community or in primary or secondary care has current SARS‐CoV‐2 infection according to time after onset of infection.
To assess the diagnostic accuracy of antibody tests to determine if a person has previously been infected with SARS‐CoV‐2.
Secondary objectives
Where data were available, we investigated the accuracy (either by stratified analysis or meta‐regression) according to:
time after onset of symptoms in periods of one week for the first five weeks, and for prior infection or convalescent phase from 21 days after onset of symptoms;
test method (ELISA, CLIA, LFA);
SARS‐CoV‐2 antigen used (N‐based, S‐based, total antibodies);
test brand;
reference standard for non‐SARS‐CoV‐2 cases (pre‐pandemic versus contemporaneous controls with or without the use of RT‐PCR to confirm absence of infection).
We had planned to investigate the effect of both study design and setting for recruitment of cases, however, the majority of studies used two‐or multi‐group designs and primarily included hospital inpatients or did not report the source of RT‐PCR‐positive samples, precluding the conduct of this planned analysis.
Methods
Criteria for considering studies for this review
Types of studies
We applied broad eligibility criteria in order to include all patient groups and all variations of a test (that is, if the patient population was unclear, we included the study).
We included studies of all designs that produced estimates of test accuracy or provided data from which estimates could be computed, including the following.
Studies restricted to participants confirmed to have (or to have had) the target condition (to estimate sensitivity)
Single‐group studies, which recruited participants before disease status had been ascertained
Multi‐group studies, where people with and without the target condition were recruited separately (often referred to as two‐gate or diagnostic case‐control studies)
Studies based on either patients or samples
We excluded studies from which we could not extract data to compute sensitivity (i.e. studies reporting data to allow calculation only of specificity were excluded). All studies had to provide data for sensitivity that could be allocated to a predefined time slot after onset of symptoms, or after a positive RT‐PCR test (see Data extraction and management).
We excluded small studies with fewer than 25 samples from those with confirmed SARS‐CoV‐2 (irrespective of the number of samples from non‐SARS‐CoV‐2 cases, for studies with both diseased and non‐diseased participants). For studies with more than 25 samples from those with SARS‐CoV‐2 but fewer than 25 samples from non‐ SARS‐CoV‐2 cases, only the sensitivity estimates were eligible. Although the size threshold of 25 is arbitrary, our requirement for studies to present results according to time after onset of infection means that smaller studies could frequently contribute only very small numbers of samples to any eligible time period, leading to unreliable estimates of sensitivity. Our sample size threshold aims to reduce this, however, some studies with smaller total numbers of samples do contribute < 25 samples to any one time period after symptom onset.
We included studies reported in published articles and as preprints.
Participants
We included studies recruiting people presenting with suspicion of current or prior SARS‐CoV‐2 infection or those recruiting populations where tests were used to screen for disease (for example, contact tracing or community screening).
We also included studies that recruited people either known to have SARS‐CoV‐2 infection or known not to have SARS‐CoV‐2 infection (multi‐group studies).
Index tests
For this version of the review, we included studies evaluating any commercially produced test for detecting antibodies to SARS‐CoV‐2, including laboratory‐based methods and tests designed to be used at point‐of‐care. Test methods include the following:
Laboratory‐based:
Enzyme‐linked immunosorbent assays (ELISA);
Chemiluminescence immunoassays (CLIA).
Rapid tests:
Lateral flow assays, including both colloidal gold or fluorescence‐labelled immunochromatographic assays (CGIA or FIA)
Studies evaluating inhouse assays or ‘laboratory‐developed tests’ were excluded.
Target conditions
The target conditions were the identification of:
current SARS‐CoV‐2 infection (symptomatic for COVID‐19);
previous SARS‐CoV‐2 infection (in convalescent [post‐symptomatic] or asymptomatic cases).
Reference standards
We anticipated that studies would use a range of reference standards to define both the presence and absence of SARS‐CoV‐2 infection, as set out under Target conditions.
For the presence of SARS‐CoV‐2, we accepted positive nucleic acid amplification test results (e.g. RT‐PCR) or a clinical guideline‐based diagnosis of COVID‐19 for those who were RT‐PCR‐negative but had high clinical suspicion.
For the absence of SARS‐CoV‐2 we included:
‘Pre‐pandemic’ stored samples obtained prior to the initial spread of SARS‐CoV‐2;
Contemporaneous samples from healthy individuals, such as blood donors, or from those with other confirmed respiratory infections, with or without confirmation of absence of SARS‐CoV‐2 infection by RT‐PCR;
Contemporaneous samples obtained from RT‐PCR‐negative individuals suspected of having COVID‐19.
Studies using serology‐based reference standards such as ELISA (for example, to evaluate the performance of rapid antibody tests) were not eligible for inclusion because these studies can only consider how well included tests estimate antibody response and are likely to overestimate accuracy for diagnosis of SARS‐COV‐2.
For the Quality Assessment tool for Diagnostic Accuracy Studies (QUADAS‐2; Whiting 2011), we categorised each method of defining the presence of SARS‐CoV‐2 according to the risk of bias (the chances that it would misclassify participants as not having SARS‐CoV‐2) and whether it defined SARS‐CoV‐2 in an appropriate way that reflected cases encountered in practice. Likewise, we considered the risk of bias in definitions of the absence of infection, and whether the definition reflected those who, in practice, would be tested.
Search methods for identification of studies
Electronic searches
The previous iteration of this review included records from electronic searches up to 27 April 2020 (Deeks 2020a). This section documents additional searches undertaken for the current iteration of this living review (first update version) up to 30 September 2020. All included studies were identified on or before 30 September 2020. Where studies originally identified as preprints were subsequently published, both publications were included in the reference list and, in some cases, may have a study ID of 2021.
COVID‐19 Open Access Project living evidence database from the University of Bern
We used the COVID‐19 Open Access Project living evidence database from the University of Bern (www.ispm.unibe.ch) (last feed obtained for this review on 30 September 2020) (COVID‐19 Open Access Project 2021). The database was constructed from daily (Monday to Friday) systematic searches of Embase via OVID, MEDLINE via PubMed, bioRxiv and medRxiv. The strategies as described on the ISPM website are described here (https://ispmbern.github.io/covid-19/living-review/collectingdata.html). See Appendix 2.
Due to the increased volume of literature since 25 May 2020, we have used artificial intelligence text analysis to retrieve more relevant records from the COVID‐19 Open Access Project living evidence database; prior to that date all records retrieved were screened manually. We used three iterations of manual screening for any one of the first set of COVID‐19 DTA (diagnostic test accuracy) reviews from the period up to 25 May 2020 (title and abstract screening, followed by full‐text review) to build and test a generic classifier that would identify records more likely to report test accuracy data based on their title and abstract information (see Appendix 3 for further details). All references from the COVID‐19 Open Access Project living evidence database from 25 May 2020 onwards were run against the classifier and references labelled as potentially relevant by the classifier were then screened manually and tagged according to the COVID‐19 DTA review(s) to which they related.
Other electronic sources
We checked our search results against two additional repositories of COVID‐19 publications up to 30 September 2020:
the Evidence for Policy and Practice Information and Co‐ordinating Centre (EPPI‐Centre) ‘COVID‐19: Living map of the evidence’ (eppi.ioe.ac.uk/COVID19_MAP/covid_map_v4.html);
the Norwegian Institute of Public Health’ NIPH systematic and living map on COVID‐19 evidence’ (www.nornesk.no/forskningskart/NIPH_diagnosisMap.html).
Both repositories allow their contents to be filtered according to studies potentially relating to diagnosis, and both agreed to provide us with updates of new diagnosis studies.
Searching other resources
We did not perform additional searches in other resources.
We did not apply any language restrictions.
Data collection and analysis
Selection of studies
A team of experienced systematic review authors from the University of Birmingham screened the titles and abstracts of all records retrieved from the literature searches following the application of artificial intelligence text analysis (described in Electronic searches). Two review authors independently screened titles and abstracts in Covidence. A third senior review author resolved any disagreements. Potentially relevant publications were obtained and independently assessed in Covidence by two review authors. Disagreements were resolved through consensus, with the inclusion of a third, senior reviewer if required. Records that were excluded at full‐text stage were documented, including the reasons for their exclusion.
Up to 30 September 2020, screening was conducted across all Cochrane COVID‐19 DTA biomarker reviews (molecular, antigen or antibody tests), using tagging of records according to the review(s) for which they might be eligible.
Data extraction and management
One review author carried out data extraction, which was checked by a second review author. Items that we extracted are listed in Appendix 4.
Both review authors independently performed data extraction of 2 x 2 contingency tables of the number of true positives, false positives, false negatives, and true negatives. They resolved disagreements by discussion.
Where possible, we extracted 2 x 2 tables according to time since onset of symptoms. In order to examine test sensitivity in the immediate period after onset of infection, we predefined groups of interest by week for the first five weeks after onset of symptoms (‘week 1’ being day 1‐7, ‘week 2’ day 8‐14, ‘week 3’ day 15‐21, ‘week 4’ day 22‐28, and ‘week 5’ day 29‐35 post‐symptom onset). Where the data presented did not exactly match these categorisations, we entered data in the time group that had the greatest overlap with our groupings, for example, day 1‐10 would be included as ‘week 1’ and day 11‐20 as ‘week 3’. We also extracted data for a broader category of convalescent phase of infection. We defined ‘convalescent data’ as samples collected 21 days or more from onset of symptoms or 14 days or more after a positive PCR result that did not fit into our criteria for ‘week 4’ or ‘week 5’, i.e. the time interval was longer than 10 days (e.g. studies providing data for samples collected between 21 and 40 days after onset of symptoms would be categorised as ‘convalescent’). Studies presenting data from up to three days prior to these thresholds (e.g. day 18 onwards) were also grouped as ‘convalescent’, as were studies reporting any data with a starting point any time after these thresholds (e.g. data from 28, 35 or 50 days after a positive PCR result were all considered as convalescent). There was no overlap in data categorised as ‘week 4’ or ‘week 5’ after onset of symptoms and data categorised as ‘convalescent data’.
Where possible, we separately extracted data related to each type of antibody (IgA, IgG and IgM, respectively), or combinations thereof (IgG or IgM, IgA or IgM, IgA or IgG, where a positive test result is defined as either or both antibodies detected). We also extracted data on total antibodies, where this was reported.
We encourage study authors to contact us regarding missing details on the included studies (coviddta@contacts.bham.ac.uk).
Assessment of methodological quality
Two review authors independently assessed risk of bias and applicability concerns using the QUADAS‐2 checklist tailored to this review (Appendix 5; Whiting 2011). The two review authors resolved any disagreements by discussion or sought advice from a third, senior review author.
Ideally, studies should prospectively recruit a representative sample of participants presenting with signs and symptoms of COVID‐19, either in community or primary care settings or in a hospital setting, and they should clearly record the time of testing after the onset of symptoms. Studies should perform antibody tests in their intended use setting, using appropriate sample types as described in the 'Instructions for use' sheet (e.g. finger prick blood for tests being evaluated for use as point‐of‐care tests), and tests should be performed by relevant personnel (e.g. healthcare workers), and should be interpreted blinded to the final diagnosis (COVID‐19 or not). Serology samples should be taken at time points that reflect the intended use (either whilst symptomatic for diagnosis of infection, or during a convalescent period (after resolution of symptoms) for diagnosis of previous infection). The reference standard diagnosis should be blinded to the result of the antibody test and should not incorporate the result of the index test or any other serology test. If the reference standard includes clinical diagnosis of COVID‐19, then established criteria should be used. Studies including samples from participants known not to have COVID‐19 should use pre‐pandemic sources, contemporaneous samples from asymptomatic contacts or people with no clinical suspicion of COVID‐19 with at least one RT‐PCR‐negative test result, or contemporaneous samples from those suspected of COVID‐19 based on signs or symptoms with at least two RT‐PCR‐negative tests. Data should be reported for all study participants (flow and timing domain), including those where the result of the antibody test was inconclusive, or participants in whom the final diagnosis of COVID‐19 was uncertain, and any delay between application of the index test and reference standard that could introduce bias (for example, because of changing disease status between time points) should be considered. If studies obtained multiple samples for testing over time from the same study participants, then they should disaggregate results by time post‐symptom onset.
Statistical analysis and data synthesis
The first iteration of this review clearly demonstrated the strong relationship of sensitivity with time, particularly in the first weeks after onset of infection. For this updated review, we do not present ‘overall’ estimates of sensitivity across all time periods but instead present results by target antibody or combination of antibodies by week after onset of symptoms (up to five weeks, where reported), and, for the first time were able to compute average estimates of sensitivity by target antibody for participants who are more likely to have reached a convalescent phase of infection (i.e. > 21 days after onset of symptoms), and for those who were reported as asymptomatic at the time of infection. The cut‐off of 21 days should be taken as only indicative of test accuracy for detection of prior infection, as some participants in the included studies would have been hospitalised for prolonged periods and are likely to reflect those with more severe and long‐lasting symptoms.
We grouped data by study and antibody test so that studies that evaluated multiple index tests in the same participants were included multiple times. We present estimates of sensitivity and specificity for each antibody (or combination of antibodies) using paired forest plots, and also summarised them in tables as appropriate.
For analysis purposes, unlike in most diagnostic test accuracy (DTA) reviews, we considered estimates of sensitivity and specificity separately because many of the included studies presented only estimates of sensitivity. Estimates of specificity were typically exceptionally high, thus the correlation between sensitivity and specificity across studies was unlikely to be high (Macaskill 2010; Takwoingi 2017).
Where we were able to perform meta‐analysis, we fitted random‐effects logistic regression models separately for sensitivity and specificity using the melogit command in Stata v17.0 (Stata). In a small number of instances, the random‐effects logistic regression analyses failed to converge (mostly this was where individual studies had specificities of 100%), and we have instead computed estimates and confidence intervals by summing the counts of true positive, false positive, false negative and true negative across 2 x 2 tables. These analyses are clearly marked in the tables. We present all estimates with 95% confidence intervals. Where sensitivity or specificity was calculated directly or by summing across the 2 x 2 tables, exact (Clopper‐Pearson) 95% binomial confidence intervals (CI) were presented.
Investigations of heterogeneity
We investigated sources of heterogeneity in two ways. First, for analysis of sensitivity for time since onset of symptoms, we extracted data by week and extended the random‐effects logistic regression model to include indicator variables for each week. Because of a strong relationship between time since onset of symptoms and sensitivity found in the previous version of this review (Deeks 2020a) and also in this version, we elected to fit all subsequent models for investigation of heterogeneity in sensitivity separately for each week. Note that the convalescent‐phase data were not included in this model and were considered separately. We excluded studies for which stratified data were not available at this stage.
The random‐effects logistic regression for specificity was also extended to include indicator variables for the type of reference standard and source of participants who did not have COVID‐19. Because we anticipated a strong relationship between reference standard type and specificity, it was decided to fit subsequent models for investigation of heterogeneity in specificity separately for each reference standard type. Note that the cross‐reactivity/confounder panel data was not included in this model and was considered separately.
We investigated heterogeneity related to test technology and antigen by including indicator variables in the random‐effects logistic regression model for each of these covariates separately. Categories such as ‘other’ or ‘unclear’ were not included as indicator variables since it is not logical to make comparisons to an unknown category. Sensitivities and specificities in this case were pooled by relevant subgroups. Models with and without a covariate were compared using likelihood ratio tests to obtain P values. We present estimates from these models by test technology or antigen for sensitivity during the convalescent phase of infection and for each week up to the third week since onset of symptoms. We did not fit models to compare sensitivities/specificities by test brand due to the small number of studies available.
Sensitivity analyses
We planned to undertake sensitivity analyses by excluding:
Unpublished studies;
Studies identified only from industry 'Instructions for use' documentation;
Studies using sample banks or spiked or contrived samples;
Studies with inadequate reference standards, for example, lack of a clear definition of clinical criteria used to diagnose the presence of COVID‐19.
For previous infection, we also planned to assess increasing lengths of time since symptoms cleared.
In this version of the review, we did not undertake any of these analyses because we did not include any unpublished studies, company documents, and no study used spiked samples. We investigated differences in reference standards used and time after onset of symptoms as part of the primary analyses.
Assessment of reporting bias
We made no formal assessment of reporting bias.
Summary of findings
We summarised key findings in a 'Table 1' table indicating the strength of evidence for each test and findings.
Updating
We are aware that additional potentially eligible studies have been published since the search date of 30 September 2020, however, because tests for diagnosis of the presence of current SARS‐CoV‐2 infection have a much higher priority for pandemic management, this review has not been prioritised for a further update in the immediate future. Although it is likely that more recently published studies will be relevant to the use cases we have explored, we would not expect significant changes to our conclusions.
Results
Results of the search
We screened 37,742 unique references (published or preprints) for inclusion in the complete suite of reviews to assist in the diagnosis of COVID‐19 (Deeks 2020b; McInnes 2020). Of 1749 records selected for further assessment for inclusion in any of the six reviews, we assessed 362 full‐text reports for inclusion in this review. We also identified a further 33 published versions of preprint reports, taking the total number of full‐text publications reviewed to 395. See Figure 2 for the PRISMA flow diagram of search and eligibility results (McInnes 2018; Moher 2009).
2.
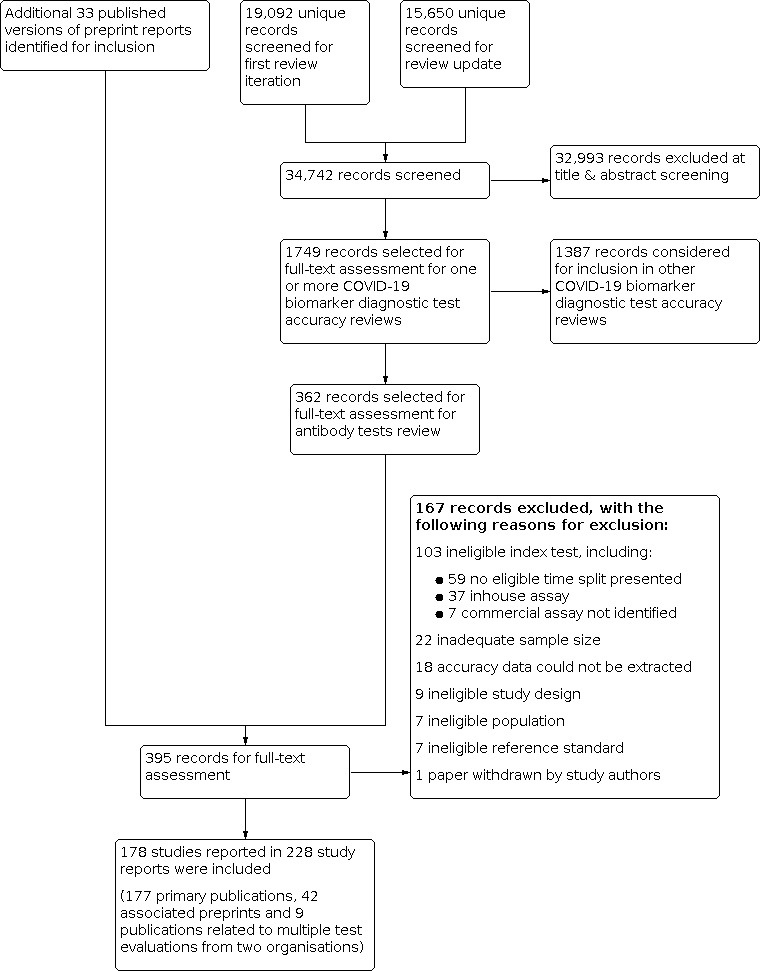
Study flow diagram
We included 177 primary study reports and 51 secondary publications, to make a total of 228 study reports included in this review (42 studies had both preprint and published versions and two organisations conducted multiple test evaluations with separate reports that were included as two primary reports and 9 additional related publications). We excluded 167 publications. Exclusions were mainly related to the index test (n = 108, including 59 that did not present serology results in an eligible time split, 37 that evaluated an inhouse instead of a commercial assay, and 7 that did not identify the assay being evaluated), because the sample size was inadequate (n < 25) (n = 22), or because we were unable to extract accuracy data from the study report (e.g. antibody levels over time were presented with no underlying numbers at given time points) (n = 18). The reasons for exclusion of all 167 publications are provided in the Characteristics of excluded studies.
We contacted the authors of 25 study reports for further information (Chaudhuri 2020 [A]; Conklin 2020 [A]; Decru 2020 [A]; Dortet 2021 [A]; GeurtsvanKessel 2020 [A]; Harritshoej 2021 [A]; Huang 2020a; Huang 2020b; Jung 2020a; Korte 2021 [A]; Krishnamurthy 2020; Liu 2021; MacMullan 2020 [A]; Manalac 2020 [A]; Merrill 2020 [A]; Naaber 2020 [A]; Paiva 2021 [A]; Patel 2020; Prazuck 2020 [A]; Rudolf 2020 [A]; Ruetalo 2020 [A]; Schnurra 2020 [A]; Sun 2020; Valdivia 2020 [A]; Weidner 2020 [A]) and received replies and the requested information with five exceptions (Huang 2020a; Krishnamurthy 2020; Merrill 2020 [A]; Sun 2020; Valdivia 2020 [A]).
The 177 primary study reports relate to 178 separate studies providing 527 test evaluations. Of the 177 study reports, 23 studies were available only as preprints. Please note when naming studies, we use the letters (a) and (b) in lower case letters and brackets at the end of the publication year (2020(a), 2020(b)) to indicate multiple studies from the same publication, and the letters [A], [B], [C] etc. in square brackets to indicate data on different tests evaluated in the same study.
Description of included studies
The 178 studies include a total of 64,688 samples with 25,724 samples from cases of SARS‐CoV‐2. These calculations are based on the total number of either samples or participants as reported in the original study reports and not on accuracy data extracted for any particular eligible time slot. Because studies did not consistently report the number of participants who provided samples for analysis, in this review, we frequently refer to the number of samples as opposed to participants.
Summary study characteristics are presented in Table 2 with further details of study design and index test details in Appendix 6 and Appendix 7. The median sample size across the 178 included studies is 185 (interquartile range [IQR] 92 to 386) and the median number of samples from people with SARS‐CoV‐2 is 94 (IQR 47 to 168). The majority (n = 94) of studies were conducted in Europe, 45 in Asia (including 26 from China), 35 in North America, two in Australia, and two in South America.
1. Description of studies.
| Participants |
Studies (percentage) (n = 178 studies) |
|
| Sample size1 | Total (no. cases) | 64,688 (25,724) |
| Median sample size (IQR) | 185 (92, 386); range 16 to 5565 |
|
| Median number of SARS‐CoV‐2 cases (IQR) | 94 (47, 168); Range 12 to 1853 |
|
| Continent | Asia | 45 (25) |
| Europe | 94 (53) | |
| North America | 35 (20) | |
| South America | 2 (1) | |
| Australia | 2 (1) | |
| Setting (SARS‐CoV‐2 cases only) | Hospital inpatient | 78 (44) |
| Hospital outpatient | 5 (3) | |
| Emergency departments | 6 (3) | |
| Community | 14 (8) | |
| Quarantine (COVID‐19 suspects) | 1 (1) | |
| Mixed | 35 (20) | |
| Unclear | 39 (22) | |
| Patient group (SARS‐CoV‐2 cases only) | Acute | 45 (25) |
| Acute and asymptomatic | 7 (4) | |
| Acute and convalescent | 77 (43) | |
| Convalescent | 40 (40) | |
| Convalescent and asymptomatic | 3 (2) | |
| Mixed | 6 (3) | |
| Study design | ||
| Recruitment structure | Single group, SARS‐CoV‐2 cases only | 48 (27) |
| Single group, both SARS‐CoV‐2 cases and non‐cases | 5 (3) | |
| Two or more groups, both SARS‐CoV‐2 cases and non‐cases | 124 (70) | |
| Unclear | 1 (1) | |
| Reference standards | ||
| For COVID‐19 cases | All RT‐PCR‐positive | 162 (91) |
| China criteria including RT‐PCR‐negative patients | 7 (4) | |
| Other criteria including RT‐PCR‐negative patients | 4 (2) | |
| Other criteria | 1 (1) | |
| Mixed | 2 (1) | |
| Unclear | 2 (1) | |
| For non‐COVID‐19 cases (n = 180 control groups from 130 studies) | Denominator = 180 | |
| Pre‐pandemic | 81 (45) | |
| Contemporaneous COVID‐19 suspects (RT‐PCR‐negative) | 21 (12) | |
| Contemporaneous healthy or other disease (RT‐PCR‐negative) | 16 (9) | |
| Contemporaneous healthy or other disease (no RT‐PCR reported) | 14 (8) | |
| Cross‐reactivity or confounder panel (any time period) | 31 (17) | |
| Mixed | 17 (9) | |
| Reference standard controls detail for non‐SARS‐CoV‐2 cases | ||
| Pre‐pandemic (n = 81) | Healthy | 27 (34) |
| Healthy and other disease | 50 (63) | |
| Other disease | 1 (1) | |
| Not specified | 2 (3) | |
| Suspected of COVID‐19 (n = 21) | Double PCR‐negative | 6 (29) |
| Single PCR‐negative | 15 (71) | |
| Current RT‐PCR‐negative (n = 16) | Healthy | 3 (19) |
| Healthy and other disease | 2 (13) | |
| Other disease | 9 (56) | |
| Current other disease (RT‐PCR‐negative) | 1 (6) | |
| Not specified | 1 (6) | |
| Current untested (n = 14) | Healthy | 10 (71) |
| Healthy and other disease | 4 (29) | |
| Cross‐reactivity (n = 31) | Pre‐pandemic | 11 (35) |
| Concurrent | 12 (39) | |
| Mixed timing | 4 (13) | |
| Timing not specified | 4 (13) | |
| Mixed (n = 17) | Mixed | 17 (100) |
| Tests | ||
| Number of assays per study (n = 178) | 1 | 76 (43) |
| 2 | 34 (19) | |
| 3 | 32 (18) | |
| 4 | 23 (13) | |
| 5 | 9 (5) | |
| 6 | 6 (3) | |
| 7 | 7 (4) | |
| 8 | 4 (2) | |
| More than 8* | 13 (7) | |
| Test technology (n = 527) | ELISA | 165 (31) |
| CLIA | 167 (32) | |
| LFA | 188 (36) | |
| Other/unclear | 7 (1) | |
| Antigen used (n = 522) | N‐based | 161 (31) |
| S‐based, including | 213 (40) | |
| S1‐based | 89 (17) | |
| RBD | 42 (8) | |
| S‐based (not further specified) | 82 (16) | |
| N‐ and S‐based | 96 (18) | |
| 2019‐nCoV | 3 (1) | |
| Unclear | 54 (10) | |
|
1Based on total number reported per study and does not relate to any particular time slot; the numbers reported in primary studies could be either samples or participants *Number of assays was 10 in 2 studies, 11 in 2 studies, 12 in 2 studies, 13 in 1 study, 14 in 1 study and 16 in 1 study. | ||
Ab: antibody CDC: Center for Disease Control and Prevention CGIA: colloidal gold immunoassay CLIA: chemiluminescence immunoassay ELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay IQR: interquartile range IIFT: indirect immunofluorescence assay IQR: interquartile ratio LFA: lateral flow assay LIPS: luciferase immunoprecipitation system max: maximum min: minimum N‐based: nucleocapsid protein RBD: receptor binding domain RT‐PCR: reverse transcription polymerase chain reaction S‐based: spike‐protein S‐flow: flow‐cytometry assay WHO: World Health Organization
Participant characteristics
In almost half of studies (n = 78; 44%), cases with SARS‐CoV‐2 were hospital inpatients, 14 studies included cases from community settings (8%), and small numbers of studies included hospital outpatients (n = 5; 3%), patients from emergency departments (n = 6; 3%) or quarantine settings (n = 1; 1%) (Table 2). The remaining studies recruited participants from multiple settings (n = 35; 20%) or did not clearly report the participant source (n = 39; 22%). All studies were identified before the introduction of vaccination for SARS‐CoV‐2 infection, therefore, none of the participants had developed antibodies as a result of vaccination.
One hundred and forty‐one studies reported data for cases by week and 117 included cases of convalescent‐phase infection. Fourteen studies included cases of asymptomatic infection. The age of included cases ranged between 1 and 102 years (reported in 121 studies). The mean or median age ranged from 32 to 82 years (reported in 85 studies), and 20% to 100% of participants were male (reported in 93 studies). Full details are in the Characteristics of included studies table.
Study designs
Only six studies used a single group or ‘single gate’ design, whereby participants were included regardless of SARS‐CoV‐2 status. The majority of studies (n = 123; 69%) used a two‐ or multi‐group design with separate selection of confirmed SARS‐CoV‐2 cases and healthy participants or participants with another disease or infection other than SARS‐CoV‐2. A single group of confirmed SARS‐CoV‐2 cases was included in 48 studies (27%), thus only allowing estimation of sensitivity and, in one, we were unable to determine the study design used.
Index tests
In total, the 178 studies reported on a total of 527 index test evaluations (counting each test brand once per study). Seventy‐six studies (43%) evaluated a single test brand and 102 compared the accuracy of two or more assays.
Studies evaluated ELISA (n = 165 evaluations; 31%), CLIA (n = 167; 32%) and lateral flow assays (LFAs, n = 188; 36%), including nine evaluations of fluorescent immunoassays (FIAs), 136 evaluations of colloidal gold‐based immunoassays (CGIAs), and 43 where the assay format could not be determined either from the primary study report or the manufacturer’s product insert. The combined 332 laboratory‐based evaluations (ELISA and CLIAs) included 59 different tests produced by 41 different commercial companies. The 188 LFA‐based evaluations included 102 different tests produced by 89 different commercial companies (five were FIAs, 60 were CGIAs and, for 37 assays, the format could not be determined). One study evaluated an LFA produced by Hangzhou with unlabelled packaging (Doherty Institute 2020 [B]), and one study evaluated an LFA with no known manufacturer (Conklin 2020 [H]).
Reference standards
One hundred and sixty‐two of 178 studies (91%) defined the presence of infection based on a positive RT‐PCR test, seven (4%) used the China CDC criteria including RT‐PCR negatives, and five used other clinical criteria. In the five remaining studies, the reference standard used other criteria (1%), was mixed (1%) or was not clearly defined (1%).
Data from the 130 studies reporting specificity data was extracted as 180 separate control groups (i.e. 50 studies reported more than one source of controls). Pre‐pandemic sources were used for 81 control groups (45%), and contemporaneous participants in 51 (28%). The contemporaneous control groups included participants suspected of having COVID‐19 but found to be RT‐PCR‐negative (n = 21; 12%), or healthy participants or those with other respiratory infections either RT‐PCR‐negative for SARS‐CoV‐2 (n = 16; 9%), or with no RT‐PCR reported (n = 14; 8%). Thirty‐one studies (17%) reported results for deliberately assembled confounder or cross‐reactivity panels (including from pre‐pandemic and contemporaneous sources), and 17 (9%) reported results in groups defined using multiple reference standards.
Methodological quality of included studies
We report the overall methodological quality assessed using the QUADAS‐2 tool for all included studies (n = 178) in Figure 3 (Whiting 2011). See Appendix 8 for study‐level ratings by quality.
3.
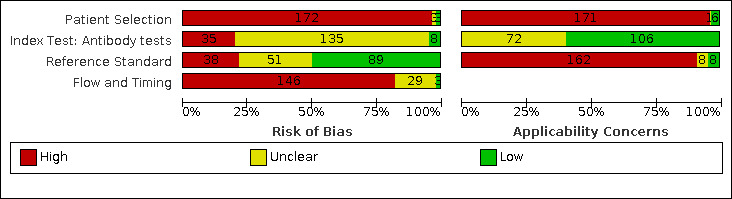
Risk of bias and applicability concerns graph: review authors' judgements about each domain presented as percentages across included studies
Overall, we judged risk of bias to be high in 172 (97%) studies concerning how participants were selected, 35 (20%) related to application of the index test, 38 (21%) through concerns about the reference standard and 146 (82%) for issues related to participant flow and timing. No study had low risk in all four domains. We judged that there were high concerns about the applicability of the evidence related to participants in 170 (96%) studies, and about the applicability of the reference standard in 162 (91%) studies. Explanations of how we have reached these judgements are given below and in the Characteristics of included studies table.
Participant selection
For participant selection, we judged three studies to be at low risk of bias and three to be of unclear risk. For the remaining 172 (97%) studies, we found high risk of bias because of non‐random or non‐consecutive sampling (n = 6), separate recruitment of confirmed SARS‐CoV‐2 cases and non‐cases (n = 123), inclusion of only confirmed SARS‐CoV‐2 cases (n = 48), inappropriate exclusions (n = 4), or inappropriate inclusions (n = 61). Numbers per group are not mutually exclusive.
We had high concerns about the applicability of the selection of participants in 171 studies (96%), meaning that the participants who were recruited were unlikely to be similar to those in whom the test would be used in clinical practice. This was frequently because studies only recruited hospitalised, confirmed cases of COVID‐19, often with severe symptoms or recruited healthy or other disease non‐COVID‐19 groups.
Index tests
Thirty‐five studies had high risk of bias for the index test domain because they explicitly reported that they had undertaken the index test with knowledge of whether individuals did or did not have COVID‐19, i.e. blinding was not implemented (n = 27), the threshold to define test positivity was determined by analysing the data, rather than it being predetermined (n = 6), or for both reasons (n = 2). In 135 studies, the risk of bias could not be judged; this was because blinding of the index test interpretation was not clearly reported (n = 132) and/or the threshold prespecification was not reported (n = 15). We judged only eight studies to have implemented the index test in a way that protected against the risk of bias.
In 106 studies (60%), we judged the test to be implemented as it would be in practice and, in 72, the applicability of the test application and interpretation could not be judged, either because of the use of mixed‐sample types or insufficient information was provided about the test operator and interpretation.
Reference standards
We judged 89 studies (50%) to have used an appropriate reference standard and implemented it in ways that prevented bias. In 38 studies, there was a risk of misclassification, as they had used a single, negative RT‐PCR result to define the absence of disease in people with suspected COVID‐19, did not report any RT‐PCR testing to confirm COVID‐19 status for contemporaneous healthy or other disease non‐COVID‐19 groups, or used serology results in part to determine the reference standard diagnosis, thus risking incorporation bias. We judged 51 studies as having unclear risk of bias because of unclear descriptions of the reference standards used (e.g. unclear description of the time period during which samples for non‐COVID cases were obtained; n = 32), insufficient information about blinding of the reference standard interpretation to the index test (n = 24), or possible incorporation of the index test result in the reference standard diagnosis (n = 3); these numbers are not mutually exclusive.
We had high concerns about the applicability of the reference standard used to define the presence of SARS‐CoV‐2 in 162 studies (91%), primarily because cases were defined based only on RT‐PCR‐positive results and did not consider clinically defined cases. Eight studies (4%) reported inadequate detail to assess the applicability of the reference standard and, in only the remaining eight studies, the reference standard was considered to be equivalent to WHO or China CDC definitions of COVID‐19, and therefore of low concern.
Flow and timing
One hundred and forty‐six (82%) studies were at high risk of bias due to using different reference standards to verify the presence and absence of SARS‐CoV‐2 infection (n = 105), not all participants receiving a reference standard test (n = 48), participants being excluded from the analysis (n = 47), or the inclusion of multiple samples per participant (n = 64). In 136 (76%) of these studies, we could not make judgements on one or more of these issues, primarily due to lack of clarity around participant inclusion and exclusion from analyses. Three studies were judged as being at low risk of bias for this domain and, in 29 (16%), there was inadequate detail to rule out these risks of bias.
Findings
The 178 included studies reported 527 test evaluations, with up to a maximum of 16 different tests evaluated using the same samples within a single study (Table 2). To incorporate all results from all tests, we have treated results from different tests within a study as separate data points. This leads to individual samples being included multiple times in some analyses. The numbers of true positives, false positives, COVID‐19 samples and non‐COVID samples are based on test result counts.
Below we present detailed results for the most commonly reported target antibodies (IgM, IgG, the combination of IgM or IgG, or total antibodies) for sensitivity by week after symptom onset (Table 3), for convalescent‐phase infection (Table 4) and for asymptomatic infection (Table 5), and for specificity by reference standard for non‐SARS‐CoV‐2 cases (Table 6). Forest plots of summary results for sensitivity and specificity are given in Figure 4 and Figure 5. Forest plots of individual study results by target antibody and by time after symptom onset are given in Appendix 9 (sensitivity by week after symptom onset), Appendix 10 (sensitivity for convalescent‐phase infection), Appendix 11 (sensitivity for asymptomatic infection), and for specificity by reference standard in Appendix 12. Results of analyses of specificity in cross‐reactivity/confounder panels are reported in Appendix 13.
2. Sensitivity by week after onset of symptoms (IgG, IgM, total Ab).
|
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
|||||
| Target |
Days 1‐7 (week 1) |
Days 8‐14 (week 2) |
Days 15‐21 (week 3) |
Days 22‐28 (week 4) |
Days 29‐35 (week 5) |
| IgGa | 189 (2177/6679) | 202 (5883/9078) | 190 (4328/5027) | 42 (828/940) | 21 (482/531) |
|
27.2 (24.9, 29.7) |
64.8 (62.1, 67.4) |
88.1 (86.6, 89.5) |
92.6 (90.5, 94.3) |
93.5 (90.8, 95.4) |
|
| IgMa | 126 (1770/4492) | 122 (3715/5577) | 118 (2416/3231) | 23 (220/411) | 9 (128/220) |
|
29.5 (25.8, 33.6) |
64.6 (60.3, 68.7) |
78.3 (74.8, 81.4) |
63.8 (56.5, 70.6) |
59.8 (50.5, 68.5) |
|
| IgG/IgMa | 103 (1593/3881) | 96 (2904/3948) | 103 (2571/2929) | 28 (649/734) | 14 (208/225) |
|
41.1 (38.1, 44.2) |
74.9 (72.4, 77.3) |
88.0 (86.3, 89.5) |
91.3 (88.8, 93.3) |
94.4 (90.7, 96.7) |
|
| Total antibodies (Ab)a | 27 (428/1010) | 29 (804/1030) | 33 (908/1016) | 7 (208/233) | 6 (139/147) |
|
37.7 (31.0, 44.9) |
79.4 (74.0, 83.9) |
90.9 (87.8, 93.2) |
94.1 (89.9 96.6) |
97.3 (93.8, 98.8) |
|
| aP values for comparisons across weeks < 0.0001 | |||||
CI: confidence interval
3. Sensitivity and heterogeneity investigations for convalescent phase infection (IgG, IgM, total Ab).
| Overall result |
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
||||
| IgG | IgM | IgG or IgM | Total Ab | ||
| 253 (14,183/16,846) | 125 (4683/7124) | 108 (3206/3571) | 58 (6652/7063) | ||
|
89.8 (88.5, 90.9) |
71.2 (65.5, 76.2) |
92.9 (91.0, 94.4) |
94.3 (92.8, 95.5) |
||
| Subgroup analyses | |||||
| By test method | ELISA | 77 (4642/5888) | 18 (721/1138) | 6 (146/161) | 10 (1631/1729) |
|
89.4 (87.0, 91.3) |
72.4 (56.8, 83.9) |
93.4 (83.6, 97.5) |
95.2 (91.5, 97.3) |
||
| CLIA | 76 (4666/5135) | 17 (431/678) | 4 (69/71) | 47 (5002/5315) | |
|
92.4 (90.6, 93.9) |
76.2 (61.2, 86.7) |
98.2 (89.9, 99.7) |
94.0 (92.3, 95.4) |
||
| Lateral flow/CGIA/FIA | 96 (4791/5734) | 88 (3496/5250) | 96 (2940/3288) | ‐ | |
|
86.9 (84.4, 89.1) |
69.9 (62.9, 76.0) |
92.3 (90.3, 93.9) |
‐ | ||
| Comparison between groupsa | P < 0.001 | P = 0.704 | P = 0.194 | P = 0.49 | |
| By antigen used | N‐based | 74 (4272/5308) | 25 (782/1297) | 15 (393/436) | 36 (3752/4009) |
|
89.7 (87.3, 91.7) |
65.4 (50.7, 77.7) |
92.5 (86.5, 96.0) |
93.3 (91.2, 94.9) |
||
| S‐based | 95 (5650/6403) | 24 (1041/1465) | 26 (1016/1126) | 22 (2900/3054) | |
|
90.4 (88.4, 92.0) |
77.9 (65.7, 86.6) |
92.7 (89.0, 95.3) |
95.6 (93.6, 97.0) |
||
| N‐ and S‐based | 54 (3122/3657) | 50 (1902/3137) | 27 (710/797) | ‐ | |
|
90.1 (87.2, 92.3) |
64.6 (54.5, 73.6) |
92.6 (88.3, 95.4) |
‐ | ||
| Unclear/not reported | 30 (1139/1406) | 26 (958/1225) | 33 (796/873) | ‐ | |
|
86.8 (81.2, 90.9) |
79.0 (71.1, 85.2) |
93.7 (90.1, 96.1) |
‐ | ||
| Comparison between groupsa,b | P = 0.897 | P = 0.201 | P = 1 | P = 0.075 | |
| S‐based assays by subgroup | S‐based (no further detail) | 42 (2978/3440) | 16 (426/678) | 24 (862/978) | 1 (24/28) |
|
88.3 (85.2, 90.8) |
76.3 (66.0, 84.3) |
92.9 (88.3, 95.8) |
86.4 (54.6, 97.1) |
||
| S1‐based | 42 (2069/2295) | ‐ | 2 (233/252) | 3 (225/235) | |
|
91.4 (88.9, 93.4) |
‐ |
96.1 (80.9, 99.3) |
95.9 (89.5, 98.4) |
||
| RBD | 11 (603/668) | 8 (615/787) | 5 (170/189) | 18 (2651/2791) | |
|
91.1 (85.6, 94.6) |
79.2 (65.2, 88.5) |
90.7 (76.2, 96.7) |
95.8 (93.7, 97.2) |
||
| Comparison between groupsa | 0.1980 | 0.7130 | 0.6717 | 0.3710 | |
CGIA: colloidal gold immunoassay CI: confidence interval CLIA: chemiluminescent immunoassayELISA: enzyme linked immunosorbent assayFIA: fluorescence immunoassay
aP values generated using the likelihood ratio test comparing the model for each target antibody including a covariate for each variable (test type, antigen or S‐antigen) to the model without the covariate
bexcluding 'unclear/not reported' group
4. Sensitivity in asymptomatic populations (IgG, IgM, total Ab).
|
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
|||
| Target | Days 0‐14 post‐RT‐PCR‐positive | Days > 14 post‐RT‐PCR‐positive | Timing unknown |
| IgG | 9 (96/208) | 10 (85/111) | 9 (82/155) |
|
49.8 (25.7, 73.9) |
78.2 (61.5, 88.9) |
28.4 (10.7, 56.9) |
|
| IgM | 6 (55/144) | 1 (7/27) | 2 (22/28) |
|
42.9 (19.5, 70.0) |
25.9a (11.1, 46.3)b |
78.6 (59.8, 90.0) |
|
| IgA | 3 (41/64) | 1 (27/27) | ‐ |
|
64.1 (51.7, 74.8) |
100a (87.2, 100)c |
‐ | |
| IgG/IgM | 2 (28/68) | ‐ | 2 (51/81) |
|
41.2 (30.2, 53.1) |
‐ |
63.0 (52.0, 72.7) |
|
| Total antibodies (Ab) | 4 (35/52) | 2 (33/38) | 2 (6/20) |
|
67.1 (45.8, 83.1) |
95.5 (7.2, 100) |
30.0 (14.2, 52.7) |
|
Ab: antibody
CI: confidence interval
RT‐PCR: reverse transcription polymerase chain reaction
aEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables
b95% exact binomial confidence interval
c97.5% one‐sided exact binomial confidence interval
5. Specificity for non‐COVID cases by reference standard (IgG, IgM, total Ab).
|
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
||||||
| Target | Pre‐pandemic | Suspected of COVID‐19 (PCR‐negative) |
Current healthy/ other disease (RT‐PCR‐negative) |
Current untested | Other/ mixed unclear | Comparison between groupsa |
| IgG | 179 (37,385/38,090) | 19 (1496/1569) | 29 (7239/7336) | 16 (2514/2561) | 28 (7319/7384) | |
|
98.9 (98.6, 99.1) |
97.8 (96.5, 98.6) |
98.5 (97.9, 98.9) |
98.6 (97.8, 99.1) |
98.4 (97.4, 99.1) |
P = 0.006 | |
| IgM | 83 (14,691/15,126) | 9 (532/597) | 20 (2652/2735) | 10 (2145/2195) | 19 (7088/7153) | |
|
98.6 (98.0, 99.1) |
96.0 (92.9, 97.8) |
97.9 (96.7, 98.6) |
98.1 (96.6, 98.9) |
98.3 (96.9, 99.1) |
P < 0.001 | |
| IgG/IgM | 68 (8989/9262) | 18 (1796/1887) | 7 (348/359) | 6 (705/713) | 20 (1809/1887) | |
|
99.2 (98.5, 99.5) |
98.2 (96.3, 99.1) |
97.9 (94.4, 99.2) |
98.4 (94.8, 99.5) |
97.2 (95.2, 98.4) |
P = 0.012 | |
|
Total antibodies (Ab) |
45 (12,166/12,207) | 4 (534/540) | 4 (364/364) | 3 (1329/1329) | 3 (5373/5388) | |
|
99.8 (99.6, 99.9) |
99.5 (97.9, 99.9) |
100a (99.0, 100)** |
100a (99.7, 100)** |
99.4 (97.2, 99.9) |
P = 0.056 | |
Ab: antibody CI: confidence interval PCR: polymerase chain reaction RT‐PCR: reverse transcription polymerase chain reaction
aP values were generated using the likelihood ratio test by comparing the model including a covariate for each reference standard group to the model without the covariate; the "Other/mixed/unclear" group was not included in the comparison.
bEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables
c97.5% one‐sided exact binomial confidence interval
4.

Summary forest plot of average sensitivities by time after symptom onset
5.

Summary forest plot of average specificities by reference standard for defining absence of COVID‐19 infection
Forest plots of individual study data for IgA alone or combined with IgG or IgM are provided in Appendix 14, with results of analyses briefly described below and tabulated in Appendix 15 (sensitivity) and Appendix 16 (specificity).
Results of heterogeneity investigations for IgG, IgM or total antibodies are tabulated in Table 4 for sensitivity during convalescent‐phase infection, Table 7 for specificity, and in Table 8 and Table 9 for sensitivity by week after onset.
6. Specificity by test method (IgG, IgM, total Ab).
| Target | Subgroup |
Test groups (true positives/COVID cases) Specificity (95% CI) |
||||
| Pre‐pandemic | Suspected of COVID‐19 |
Current RT‐PCR‐ negative |
Current untested | Other/mixed unclear | ||
| IgG | ELISA | 55 (9999/10,336) | 6 (585/609) | 4 (298/308) | 9 (739/745) | 9 (544/562) |
|
98.4 (97.7, 98.9) |
96.9 (92.5, 98.7) |
97.0 (91.9, 98.9) |
99.2 (98.2, 99.7) |
97.2 (94.1, 98.7) |
||
| CLIA | 55 (16,413/16,545) | 9 (640/661) | 11 (5854/5899) | 2 (820/829) | 3 (132/135) | |
|
99.5 (99.2, 99.7) |
97.2 (93.8, 98.8) |
99.3 (98.6, 99.7) |
98.9 (97.5, 99.5) |
98.0 (91.5, 99.6) |
||
| Lateral flow/CGIA/FIA | 68 (10,657/10,889) | 4 (271/299) | 14 (1087/1129) | 5 (955/987) | 15 (1393/1425) | |
|
98.7 (98.2, 99.1) |
90.7 (77.2, 96.5) |
97.3 (95.1, 98.5) |
97.5 (95.2, 98.7) |
98.5 (97.1, 99.2) |
||
| Comparison between groupsa | P < 0.001 | P = 0.172 | P = 0.01 | P = 0.099 | P = 0.452 | |
| IgM | ELISA | 14 (2743/2840) | 2 (206/213) | 2 (97/98) | 4 (617/620) | 2 (354/360) |
|
98.1 (95.7, 99.2) |
97.1 (81.6, 99.6) |
99.1 (92.3, 99.9) |
99.6 (98.3, 99.9) |
97.2 (87.6, 99.4) |
||
| CLIA | 10 (4232/4298) | 3 (79/84) | 4 (1471/1510) | 1 (583/586) | 1 (72/72) | |
|
99.2 (97.7, 99.7) |
96.1 (78.0, 99.4) |
98.1 (95.3, 99.2) |
99.5 (97.6, 99.9) |
99.7 (97.6, 100) |
||
| Lateral flow/CGIA/FIA | 58 (7398/7668) | 4 (247/300) | 14 (1084/1127) | 5 (945/989) | 18 (2424/2494) | |
|
98.3 (97.3, 98.9) |
80.9 (51.6, 94.4) |
97.1 (94.9, 98.4) |
97.2 (93.4, 98.9) |
97.1 (95.5, 98.1) |
||
| Comparison between groupsa | P = 0.412 | P = 0.205 | P = 0.436 | P = 0.075 | P = 0.048 | |
| IgG/IgM | ELISA | 6 (1269/1294) | ‐ | ‐ | 3 (316/320) | ‐ |
|
99.2 (95.9, 99.9) |
‐ | ‐ |
99.2 (94.9, 99.9) |
‐ | ||
| CLIA | 1 (40/40) | 2 (180/188) | ‐ | ‐ | 2 (304/307) | |
|
100b (91.2, 100)c |
97.3 (28.3, 100) |
‐ | ‐ |
99.3 (96.0, 99.9) |
||
| Lateral flow/CGIA/FIA | 60 (7180/7428) | 13 (1404/1477) | 7 (348/359) | 3 (389/393) | 18 (1505/1580) | |
|
98.5 (97.4, 99.2) |
99.3 (94.7, 99.9) |
96.9 (94.6, 98.3) |
99.2 (95.8, 99.8) |
96.7 (94.5, 98.0) |
||
| Comparison between groups | P = 0.397 | P = 0.578 | ‐ | P = 0.948 | P = 0.085 | |
| Total antibodies (Ab) | ELISA | 8 (2009/2020) | 1 (50/50) | ‐ | 1 (300/300) | 1 (97/100) |
|
99.6 (98.7, 99.9) |
100b (92.9, 100)c |
‐ |
100b (98.8, 100)c |
97.0b (91.5, 99.4)d |
||
| CLIA | 36 (9903/9931) | 3 (484/490) | 4 (364/364) | 2 (1029/1029) | 1 (26/26) | |
|
99.9 (99.7, 99.9) |
98.8 (97.3, 99.4) |
100b (99.0, 100)c |
100b (99.6, 100)c |
100b (86.7, 100)c |
||
| Lateral flow/CGIA/ FIA | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ | ‐ | ‐ | ‐ | ‐ | ||
| Comparison between groups | P = 0.183 | ‐ | ‐ | P = 0.948 | ‐ | |
CI: confidence interval CGIA: colloidal gold immunoassay CLIA: chemiluminescent immunoassay ELISA: enzyme linked immunosorbent assay FIA: fluorescence immunoassay RT‐PCR: reverse transcription polymerase chain reaction
aP values were generated using the likelihood ratio test comparing the model for each reference standard group including a covariate for test method to the model without the covariate
bEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables
c97.5% one‐sided exact binomial confidence interval
d95% exact binomial confidence interval
7. Sensitivity by test method by week after onset (IgG, IgM, total Ab).
| Target | Test method | Week 1 | Week 2 | Week 3 |
|
Test groups (true positives/COVID cases) Sensitivity (95% CI) | ||||
| IgG | By test method | |||
| ELISA | 56 (487/1780) | 62 (1783/2991) | 54 (1227/1416) | |
|
21.8 (17.1, 27.4) |
63.7 (58.7, 68.4) |
89.6 (86.5, 92.1) |
||
| CLIA | 46 (480/1616) | 51 (1541/2382) | 43 (1125/1282) | |
|
28.0 (22.0, 34.9) |
66.1 (60.8, 71.0) |
87.4 (83.5, 90.5) |
||
| Lateral flow/CGIA/ FIA | 83 (881/2816) | 85 (2122/3171) | 90 (1901/2236) | |
|
28.1 (23.5, 33.3) |
67.6 (63.6, 71.5) |
87.1 (84.3, 89.4) |
||
| Comparison between groupsa | P = 0.178 | P = 0.461 | P = 0.38 | |
| IgM | By test method | |||
| ELISA | 21 (670/208) | 21 (802/1217) | 16 (367/461) | |
|
29.3 (21.6, 38.4) |
68.2 (57.1, 77.5) |
84.5 (73.5, 91.4) |
||
| CLIA | 19 (208/536) | 17 (590/889) | 14 (488/613) | |
|
35.0 (25.5, 45.7) |
64.2 (51.3, 75.4) |
78.7 (65.0, 88.1) |
||
| Lateral flow/CGIA/FIA | 82 (1020/2819) | 82 (1970/3099) | 87 (1548/2142) | |
|
32.6 (28.1, 37.4) |
63.4 (57.6, 68.9) |
76.9 (71.4, 81.7) |
||
| Comparison between groupsa | P = 0.69 | P = 0.74 | P = 0.422 | |
| IgG/IgM | By test method | |||
| ELISA | 8 (67/197) | 8 (376/514) | 8 (225/237) | |
|
33.8 (21.6, 48.6) |
72.9 (64.5, 79.9) |
95.9 (91.0, 98.2) |
||
| CLIA | 4 (78/173) | 4 (224/286) | 2 (173/178) | |
|
43.9 (25.0, 64.8) |
75.9 (64.1, 84.7) |
97.3 (89.6, 99.3) |
||
| Lateral flow/CGIA/FIA | 90 (1439/3470) | 83 (2275/3115) | 91 (2127/2454) | |
|
40.5 (36.2, 44.9) |
74.6 (71.9, 77.2) |
88.7 (86.3, 90.7) |
||
| Comparison between groupsa | P = 0.634 | P = 0.886 | P = 0.006 | |
| Total antibodies (Ab) | By test method | |||
| ELISA | 6 (158/292) | 8 (304/342) | 7 (209/219) | |
|
56.5 (37.4, 73.8) |
88.5 (80.1, 93.6) |
96.4 (91.4, 98.6) |
||
| CLIA | 19 (252/660) | 20 (474/648) | 25 (685/782) | |
|
34.6 (24.3, 46.6) |
76.0 (68.2, 82.4) |
88.4 (83.6, 92.0) |
||
| Lateral flow/CGIA/FIA | 7 (103/272) | ‐ | ‐ | |
|
39.2 (7.9, 82.9) |
‐ | ‐ | ||
| Comparison between groupsa | P = 0.181 | P = 0.029 | P = 0.013 | |
CI: confidence interval CGIA: colloidal gold immunoassay CLIA: chemiluminescent immunoassay ELISA: enzyme linked immunosorbent assay FIA: fluorescence immunoassay
aP values were generated using the likelihood ratio test comparing the model for each target antibody by week after onset including a covariate for test method to the model without the covariate; comparison does not include the ‘unclear/not reported’ test method category
8. Sensitivity by antigen type by week after onset (IgG, IgM, total Ab).
| Target | Antigen | Week 1 | Week 2 | Week 3 |
|
Test groups (true positives/COVID cases) Sensitivity (95% CI) | ||||
| IgG | N‐based | 52 (553/1928) | 61 (1715/2688) | 53 (1173/1307) |
|
26.2 (20.9, 32.2) |
66.7 (61.9, 71.1) |
91.2 (88.5, 93.2) |
||
| S‐based | 64 (577/2236) | 65 (1864/3222) | 65 (1433/1717) | |
|
21.1 (17.0, 26.0) |
59.8 (54.9, 64.5) |
85.4 (82.2, 88.2) |
||
| N‐ and S‐based | 43 (765/1509) | 47 (1752/2272) | 40 (1168/1323) | |
|
37.7 (30.4, 45.5) |
76.7 (72.1, 80.8) |
89.2 (86.0, 91.8) |
||
| Unclear/not reported | 30 (282/1006) | 29 (552/896) | 32 (554/680) | |
|
22.6 (14.2, 34.1) |
61.2 (52.0, 69.6) |
82.5 (76.6, 87.2) |
||
| Comparison between groupsa | P = 0.001 | P < 0.0001 | P = 0.01 | |
| IgM | N‐based | 31 (311/1062) | 33 (952/1607) | 26 (442/630) |
|
25.4 (18.9, 33.1) |
57.8 (47.5, 67.5) |
72.2 (61.1, 81.1) |
||
| S‐based | 24 (365/905) | 21 (846/1116) | 22 (573/675) | |
|
37.8 (28.6, 48.1) |
78.2 (67.7, 86.1) |
89.0 (81.8, 93.5) |
||
| N‐ and S‐based | 43 (754/1529) | 41 (1456/2060) | 39 (966/1292) | |
|
35.1 (28.2, 42.8) |
66.3 (57.3, 74.2) |
76.5 (68.4, 83.0) |
||
| Unclear/not reported | 28 (340/996) | 27 (461/794) | 31 (435/634) | |
|
29.6 (20.9, 40.1) |
61.2 (52.5, 69.3) |
76.0 (66.6, 83.4) |
||
| Comparison between groupsa | P = 0.084 | P = 0.025 | P = 0.011 | |
| IgG/IgM | N‐based | 22 (294/836) | 21 (742/1042) | 21 (507/549) |
|
34.0 (27.7, 40.9) |
71.2 (65.8, 76.1) |
94.9 (91.3, 97.0) |
||
| S‐based | 24 (431/1016) | 22 (705/921) | 26 (810/920) | |
|
41.8 (35.4, 48.5) |
77.3 (72.4, 81.6) |
90.6 (86.0, 93.7) |
||
| N‐ and S‐based | 25 (377/829) | 23 (717/947) | 20 (539/628) | |
|
44.6 (37.9, 51.5) |
76.7 (71.8, 81.0) |
87.1 (80.6, 91.7) |
||
| Unclear/not reported | 31 (469/1162) | 30 (740/1038) | 35 (681/788) | |
|
37.9 (28.5, 48.2) |
73.4 (68.7, 77.7) |
87.3 (83.2, 90.5) |
||
| Comparison between groupsa | P = 0.058 | P = 0.166 | P = 0.032 | |
| Total antibodies (Ab) | N‐based | 15 (169/535) | 17 (432/608) | 20 (502/565) |
|
28.9 (19.1, 41.2) |
74.6 (66.2, 81.5) |
90.8 (85.5, 94.3) |
||
| S‐based | 12 (259/475) | 12 (372/422) | 13 (406/451) | |
|
54.6 (40.1, 68.4) |
86.5 (78.9, 91.7) |
91.0 (84.5, 95.0) |
||
| N‐ and S‐based | ‐ | ‐ | ‐ | |
| ‐ | ‐ | ‐ | ||
| Comparison between groupsa | P = 0.011 | P = 0.033 | P = 0.942 | |
Ab: antibody CI: confidence interval CGIA: colloidal gold immunoassay CLIA: chemiluminescent immunoassay ELISA: enzyme linked immunosorbent assay FIA: fluorescence immunoassay N: nucleocapsid protein S: spike‐protein
aP values were generated using the likelihood ratio test comparing the model for each target antibody by week after onset including a covariate for antigen type to the model without the covariate; for each comparison, the 'unclear/not reported' category was not included
Results of primary analyses
Sensitivity by week after onset of symptoms
Table 3 and Figure 4 present results of meta‐analyses of sensitivity by week after onset of symptoms. The number of evaluations (and of samples) for the first three weeks after onset of symptoms ranged from 189 to 202 evaluations (5027 to 9078 samples) for IgG, 118 to 126 evaluations (3231 to 5577 samples) for IgM, 96 to 103 evaluations (2929 to 3948 samples) for the combination of IgG or IgM, and 28 to 33 evaluations (1016 to 1071 samples) for total antibodies detection. The number of evaluations contributing to average sensitivity calculations for week 4 and week 5 after onset of symptoms were considerably lower but are nevertheless based on a relatively large number of samples (from 297 to 940 samples in week 4 and 179 to 531 samples in week 5).
The forest plots of individual study data by week (Appendix 9) show considerable heterogeneity between studies in the first two weeks after onset, particularly for IgG but also for IgM, which substantially reduces by week three. Results for the combination of IgG or IgM and for total antibodies for detection of SARS‐CoV‐2 show similarly high levels of heterogeneity in the first week after onset, reducing slightly by week two and with much more consistent results between studies by week three. The strength of the relationship of sensitivity with time shows exceptionally high levels of statistical significance (P < 0.0001).
The results for IgG, IgG or IgM, and total antibodies showed the same general pattern over the first five weeks, with low sensitivity for detection of SARS‐CoV‐2 infection when tests were used in the first week since onset of symptoms, rising in the second and third week, and reaching their highest values in the fifth week (the latter values based on relatively less data).
For IgG, average sensitivities across the five weeks were 27.2% (95% CI 24.9 to 29.7; week 1), 64.8% (95% CI 62.1 to 67.4; week 2), 88.1% (95% CI 86.6 to 89.5; week 3), 92.6% (95% CI 90.5 to 94.3; week 4), and 93.5% (95% CI 90.8 to 95.4; week 5).
For IgG or IgM combined, average sensitivities were 41.1% (95% CI 38.1 to 44.2; week 1), 74.9% (95% CI 72.4 to 77.3; week 2), 88.0% (95% CI 86.3 to 89.5; week 3), 91.3% (95% CI 88.8 to 93.3; week 4), and 94.4% (95% CI 90.7 to 96.7; week 5).
For total antibodies, average sensitivities were 37.7% (95% CI 31.0 to 44.9; week 1), 79.4% (95% CI 74.0 to 83.9; week 2), 90.9% (95% CI 87.8 to 93.2; week 3), 94.1% (8998 to 96.6; week 4), and 97.3% (93.8 to 98.8; week 5). Average sensitivities for total antibodies for detecting infection were similar to those for the IgG or IgM combination (Table 3; Figure 4).
The results for IgM alone confirm the expected pattern over the five‐week period, with lower sensitivity when tests were used in the first week since symptom onset, reaching their highest value in the third week, and then declining in the fourth and fifth week after onset (Figure 4). For IgM, average sensitivities across the five weeks were 29.5% (95% CI 25.8 to 33.6; week 1), 64.6% (95% CI 60.3 to 68.7; week 2), 78.3% (95% CI 74.8 to 81.4; week 3), 63.8% (95% CI 56.5 to 70.6; week 4), and 59.8% (95% CI 50.5 to 68.5; week 5) (Table 3).
Sensitivity for convalescent phase of infection
Table 4 and Figure 4 present results of meta‐analyses of sensitivity for each target antibody for samples obtained during the convalescent period after infection (from day 21 after symptom onset, see Data extraction and management for detailed definition). The data contributing to these analyses do not overlap with those for the analyses by week after symptom onset. The forest plots of individual study data by target antibody show considerable heterogeneity between studies, particularly for IgM (Appendix 10).
Average sensitivities per target antibody were very much line with those reported for week three after onset of infection: 89.8% for IgG (95% CI 88.5 to 90.9; based on 253 evaluations and 16,846 samples), 71.2% for IgM (95% CI 65.5 to 76.2; 125 evaluations comprising 7124 samples), 92.9% for IgG or IgM combined (95% CI 91.0 to 94.4; 108 evaluations comprising 3571 samples) and 94.3% for total antibodies (95% CI 92.8 to 95.5; 58 evaluations comprising 7063 samples).
Sensitivity for IgA alone or combined with other antibodies
Fewer evaluations of IgA‐based assays were identified, particularly for IgA combined with IgM or IgG (Appendix 14; Appendix 15). For IgA alone, average sensitivities were higher than those calculated for either IgG or IgM alone in weeks one and two after onset of symptoms, but similar to those for IgG or IgM combined: 44.3% in week one (95% CI 36.22 to 52.8; 24 evaluations, comprising 1079 samples), 72.9% in week 2 (95% CI 65.6 to 79.2; 22 evaluations, 1181 samples). By week three after onset of symptoms, IgA sensitivity was in the range of that observed for IgG alone or for IgG or IgM combined: 86.6% in week 3 (95% CI 81.2 to 90.6; 19 evaluations, 501 samples) and 82.3% for convalescent‐phase infection (95% CI 70.0 to 90.3; 22 evaluations, 1257 samples). Results for IgA were primarily driven by data for a single test as discussed below (Appendix 14).
Sensitivity for asymptomatic infection
Very small numbers of samples (fewer than 208 samples from cases of SARS‐CoV‐2 infection for all evaluated time points and antibody targets) were obtained from participants reported as asymptomatic at the time of SARS‐CoV‐2 infection, and it is not possible to make clear statements about assay sensitivities in this group (Table 5; Appendix 11).
Specificity by reference standard for non‐SARS‐CoV‐2 cases
We estimated antibody test specificity from 180 non‐COVID‐19 control groups reported in 130 studies. Average specificity was calculated separately for each prespecified control group according to the reference standard used to define the absence of SARS‐CoV‐2. The forest plots of individual study data (Appendix 12) show consistently low heterogeneity in study estimates of specificity across studies (with a very small number of outliers), target antibodies and all control groups apart from the cross‐reactivity/confounder panel data.
Pre‐pandemic
The majority of studies used samples obtained during the pre‐pandemic period to estimate assay specificity; these data arguably reflect the true specificity of the tests because the possibility of false‐positive results from undiagnosed SARS‐CoV‐2 infection is removed. Results of the meta‐analyses for pre‐pandemic samples show specificity exceeding 98% for all antibody types, with precise estimates (confidence intervals up to 1.1 percentage points wide) (Table 6; Figure 5).
Average specificities per target antibody were: 98.9% for IgG (95% CI 98.6% to 99.1%; based on 179 evaluations and 38,090 samples), 98.6% for IgM (95% CI 98.0 to 99.1; 83 evaluations, 15,126 samples), 99.2% for the combination of IgG or IgM (95% CI 98.5 to 99.5; 68 evaluations, 9262 samples) and 99.8% for total antibodies (95% CI 99.6 to 99.9; 45 evaluations, 12,207 samples).
Specificity for IgA assays was consistently lower than for other target antibodies, e.g. summary specificity in pre‐pandemic samples for IgA alone was 93.8% (95% CI 90.5 to 96.0; 17 evaluations,1711 samples) (Appendix 16).
We anticipated that specificities based on samples obtained during the time period of the pandemic might show higher false‐positive rates, however, this was not consistently reflected in the data despite the high numbers of samples (Table 6). Average specificities calculated for all reference standard groups apart from the ‘suspected of COVID‐19’ RT‐PCR‐negative group were broadly consistent with those for pre‐pandemic samples (differences in specificity less than 1%) (Table 6). The forest plots of individual data (Appendix 12) suggested greater heterogeneity in specificities in each group of contemporaneously collected samples compared to pre‐pandemic sources.
RT‐PCR‐negative
Results of meta‐analyses for antibody‐test specificity for the RT‐PCR‐negative COVID‐19 suspect group suggested marginally lower average specificities (differences of between 0.7% and 2.6%) for IgG alone (97.8%, 95% CI 96.5 to 98.6; 19 evaluations,1569 samples) and for IgM alone (96.0%, 95% CI 92.9 to 97.8; 9 evaluations, 597 samples). Although the number of samples contributing to these analyses was much lower than for the pre‐pandemic group, the confidence intervals for both estimates did not overlap those for the pre‐pandemic estimates (Table 6; Figure 5).
Cross‐reactivity/confounder
Results for antibody test specificity for the cross‐reactivity/confounder group showed broadly similar results for IgG and for total antibodies (Appendix 13), however, average estimates for IgM alone (97.0%, 95% CI 95.1 to 98.2; 44 evaluations, 2625 samples) and for IgG or IgM combined (96.8%, 95% CI 94.4 to 98.2; 36 evaluations, 2175 samples) were lower than those calculated for the pre‐pandemic group.
Heterogeneity investigations
Heterogeneity investigations focused primarily on identifying any effects from test technology, antigen used and test brand. For ease of presentation, we focused on results for sensitivity based on convalescent‐phase data (Table 4) and for specificity we primarily focused on results using pre‐pandemic samples (Table 7 and Table 10). Forest plots of individual study data are organised by test method to facilitate visual comparisons (Appendix 10; Appendix 12). Results for sensitivity using data by week after symptom onset are presented in Table 8 and Table 9.
9. Specificity by antigen type (IgG, IgM, total Ab).
|
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
||||||
| Target |
Antigen subgroup |
Pre‐pandemic | Suspected of COVID‐19 |
Current RT‐PCR negative |
Current untested | Other/mixed unclear |
| IgG | N‐based | 55 (13,929/14,159) | 7 (542/559) | 6 (834/840) | 4 (697/705) | 6 (508/526) |
|
99.1 (98.7, 99.4) |
97.3 (94.5, 98.7) |
99.4 (97.9, 99.8) |
99.0 (97.6, 99.6) |
97.6 (93.4, 99.2) |
||
| S‐based | 66 (14,331/14,615) | 7 (696/722) | 11 (4569/4604) | 7 (521/525) | 12 (836/854) | |
|
98.9 (98.4, 99.2) |
96.9 (94.0, 98.4) |
98.4 (96.6, 99.2) |
99.3 (98.0, 99.8) |
98.4 (96.4, 99.3) |
||
| N‐ and S‐based | 37 (7325/7449) | 3 (197/204) | 9 (1661/1706) | 5 (1296/1331) | 5 (5550/5573) | |
|
99.0 (98.4, 99.4) |
95.8 (87.7, 98.7) |
98.0 (95.7, 99.1) |
97.9 (95.9, 98.9) |
98.7 (96.0, 99.6) |
||
| Unclear/not reported | 21 (1800/1867) | 2 (61/84) | 3 (175/186) | ‐ | 5 (425/431) | |
|
98.8 (97.0, 99.5) |
73.4 (53.3, 87.0) |
96.5 (83.9, 99.3) |
‐ |
98.8 (96.2, 99.6) |
||
| Comparison between groupsa | P = 0.594 | P = 0.804 | P = 0.276 | P = 0.167 | P = 0.748 | |
| IgM | N‐based | 22 (5564/5674) | 3 (258/268) | 2 (99/99) | 3 (455/464) | 5 (476/501) |
|
98.4 (96.9, 99.2) |
96.3 (93.2, 98.0) |
100b (96.3, 100)** |
98.6 (95.0, 99.6) |
95.9 (89.9, 98.4) |
||
| S‐based | 16 (2800/2870) | 1 (38/40) | 6 (720/746) | 2 (400/400) | 4 (379/387) | |
|
98.3 (96.3, 99.2) |
95.0 (82.1, 98.7) |
96.9 (93.2, 98.6) |
100b (99.1, 100)c |
98.4 (94.6, 99.5) |
||
| N‐ and S‐based | 28 (4957/5114) | 3 (194/204) | 9 (1655/1704) | 5 (1290/1331) | 5 (5809/5834) | |
|
98.9 (97.8, 99.5) |
95.1 (91.1, 97.3) |
97.8 (95.5, 99.0) |
98.4 (95.5, 99.4) |
99.0 (97.2, 99.6) |
||
| Unclear/not reported | 17 (1370/1468) | 2 (42/85) | 3 (178/186) | ‐ | 5 (424/431) | |
|
97.2 (93.9, 98.8) |
47.3 (9.9, 88.0) |
95.7 (91.6, 97.8) |
‐ |
98.6 (95.7, 99.6) |
||
| Comparison between groupsa | P = 0.653 | P = 0.803 | P = 0.089 | P = 0.04 | P = 0.173 | |
| IgG or IgM | N‐based | 11 (2595/2637) | 3 (236/236) | 1 (4/4) | 4 (361/368) | 5 (416/451) |
|
99.2 (96.7, 99.8) |
100b (98.4, 100)c |
100b (39.8, 100)c |
98.1 (96.1, 99.1) |
94.1 (87.9, 97.2) |
||
| S‐based | 16 (3748/3800) | 7 (994/1018) | 3 (231/238) | 2 (344/345) | 5 (468/479) | |
|
99.4 (97.9, 99.8) |
99.0 (95.3, 99.8) |
97.1 (94.0, 98.6) |
99.7 (97.9, 100) |
98.0 (95.2, 99.2) |
||
| N‐ and S‐based | 19 (1106/1170) | 4 (385/406) | 1 (38/39) | ‐ | 4 (491/513) | |
|
98.8 (96.1, 99.6) |
95.6 (82.8, 99.0) |
97.4 (83.9, 99.6) |
‐ |
96.3 (91.5, 98.5) |
||
| Unclear/not reported | 21 (1347/1455) | 4 (181/227) | 2 (75/78) | ‐ | 6 (434/444) | |
|
97.6 (94.4, 99.0) |
79.7a (73.9, 84.8)d |
96.2 (88.7, 98.8) |
‐ |
98.3 (94.6, 99.5) |
||
| Comparison between groupsa | P = 1 | P = 0.021 | P = 0.617 | ‐ | P = 0.236 | |
| Total Ab | N‐based | 26 (5808/5824) | 3 (303/306) | 2 (277/277) | 2 (1029/1029) | 1 (26/26) |
|
99.9 (99.6, 99.9) |
99.0 (97.0, 99.7) |
100b (98.7, 100)c |
100b (99.6, 100)c |
100b (86.8, 100)c |
||
| S‐based | 19 (6358/6383) | 1 (231/234) | 2 (87/87) | 1 (300/300) | 1 (97/100) | |
|
99.8 (99.4, 99.9) |
98.7 (96.1, 99.6) |
100b (95.8, 100)c |
100a (98.8, 100)c |
97.0 (91.1, 99.0) |
||
| N‐ and S‐based | ‐ | ‐ | ‐ | ‐ | 1 (5250/5262) | |
| ‐ | ‐ | ‐ | ‐ |
99.8 (99.6, 99.9) |
||
| Unclear/not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ | ‐ | ‐ | ‐ | ‐ | ||
| Comparison between groupsa | P = 0.525 | P = 0.741 | ‐ | ‐ | ‐ | |
CI: confidence interval N: nucleocapsid protein RT‐PCR: reverse transcription polymerase chain reaction S: spike‐protein
aP values were generated using the likelihood ratio test comparing the model including a covariate for test method to the model without the covariate for each reference standard group per target antibody; for each comparison, the 'unclear/not reported' group was not included
bEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables
c97.5% one‐sided exact binomial confidence interval
d95% exact binomial confidence interval
Sensitivity by technology (test method)
We investigated the heterogeneity in sensitivity estimates according to three main types of test technology: two laboratory‐based methods (ELISA and CLIA) and lateral flow devices (grouping CGIAs and FIAs together). Table 4 shows that most of available data for laboratory‐based assays is for IgG alone (77 evaluations comprising 5888 samples for ELISA and 76 evaluations comprising 5135 samples for CLIAs) or for total antibody assays (10 evaluations with 1729 samples for ELISA and 47 evaluations with 5315 samples for CLIA). IgM alone or the combination of IgG or IgM for detection of SARS‐CoV‐2 infection was evaluated in between four and 18 evaluations with 71 to 1138 samples for laboratory‐based assays. In contrast, lateral flow devices primarily targeted IgM or IgG either alone or in combination (between 88 and 96 evaluations with 3288 to 5734 samples), and none targeted total antibodies (i.e. the combination of IgG, IgM or IgA) during the convalescent phase of infection. Table 4 suggests a trend towards higher sensitivities for laboratory‐based assays (particularly CLIAs) compared to rapid lateral flow‐based tests, however, small sample numbers in some groups are likely to have affected the ability of the model to identify true differences between test methods.
For IgG alone, the average sensitivity for CLIAs for detecting SARS‐CoV‐2 infection was 92.4% (95% CI 90.6 to 93.9; 76 evaluations, 5135 samples), 89.4% for ELISA‐based assays (95% CI 87.0 to 91.3; 77 evaluations, 5888 samples), and 86.9% for lateral flow assays (95% CI 84.4 to 89.1; 96 evaluations, 5734 samples) (P = 0.0008 for the difference in sensitivity) (Table 4).
A similar magnitude of differences in average sensitivities for IgM was observed but with greater heterogeneity in individual study results and smaller numbers of samples for the laboratory‐based assays (Table 4): average sensitivities were 76.2% for CLIAs (95% CI 61.2 to 86.7; 17 evaluations, 678 samples), 72.4% for ELISAs (95% CI 56.8 to 83.9; 18 evaluations, 1138 samples) and 69.9% for lateral flow assays (95% CI 62.9 to 76.0; 88 evaluations, 5250 samples) (P = 0.70 for the difference in sensitivity).
For total antibodies, ELISA‐ and CLIA‐based assays appear to perform similarly with average sensitivities 95.2% (95% CI 91.5 to 97.3; 10 evaluations, 1729 samples) and 94.0% (95% CI 92.3 to 95.4; 47 evaluations, 5315 samples).
A similar pattern of results was observed for investigations of the effect of test method by week after symptom onset (Table 8).
Specificity by technology (test method)
For both IgG alone and for IgM alone, results for the pre‐pandemic group suggested that test technology had only a marginal effect on average specificities (differences within 1.1 percentage points) (Table 7). Larger differences were observed for average specificities based on contemporaneously collected samples; between 1.0 to 7.0 percentage points differences for IgG and 2.0 to 17.0 percentage points differences for IgM, and the biggest differences observed in the ‘suspected of COVID‐19’ group and the lowest average specificities for lateral flow‐based tests compared to the two laboratory‐based methods. The observed differences are likely to be influenced by the number of available evaluations and samples between groups.
For IgG alone, CLIAs had the highest average specificities across all groups apart from the ‘current untested’ group (Table 7). Average specificity for CLIAs using pre‐pandemic samples was 99.5% (95% CI 99.2 to 99.7; 55 evaluations, 16,545 samples), compared to 98.4% for ELISAs (95% CI 97.7, 98.9; 55 evaluations, 10,336 samples) and 98.7% for LFAs (95% CI 98.2, 99.1; 68 evaluations, 10,889 samples); P < 0.001 for the difference between test methods.
For IgM alone, ELISAs had the highest average specificities across the most of the reference standard groups (Table 7), however, very small numbers of evaluations and samples were available for some groups. Using pre‐pandemic samples, average specificity for CLIAs was 99.2% (95% CI 97.7, 99.7; 10 evaluations, 4298 samples), 98.1% for ELISAs (95% CI 95.7 to 99.2; 14 evaluations, 2840 samples) and 98.3% for LFAs (95% CI 97.3 to 98.9; 58 evaluations, 7668 samples); P = 0.41 for the difference between test methods.
For the combination of IgM or IgG, the number of evaluations and samples per test technology was smaller and it was not possible to identify clear patterns in results between reference groups (Table 7). Average specificities for pre‐pandemic samples were: 99.2% for ELISAs (95% CI 95.9 to 99.9; 6 evaluations, 1294 samples), 100% for CLIA in a single evaluation (95% CI 91.2 to 100; 40 samples), and 98.5% for lateral flow‐based assays (95% CI 97.4 to 99.2; 60 evaluations, 7428 samples); P = 0.40 for the difference between test methods.
For total antibody assays, no clear differences in specificity by test technology were identified (Table 7). For pre‐pandemic samples, average specificities were: 99.6% for ELISA (95% CI 98.7 to 99.9; 8 evaluations, 2020 samples) and 99.9% for CLIA (95% CI 99.7 to 99.9; 36 evaluations, 9931 samples).
Sensitivity by antigen
Analyses by type of antigen (Spike‐protein [S‐based], nucleoprotein [N‐based] or both [N‐ and S‐based]) suggested no clear differences in average sensitivities between assays targeting IgG alone, IgG or IgM combined, or total antibodies for samples collected during the convalescent phase of infection (Table 4). For example, for IgG alone, the sensitivity of N‐based assays was 89.7% (95% CI 87.3 to 91.7; 74 evaluations, 5308 samples), for S‐based assays 90.4% (95% CI 88.4 to 92.0; 95 evaluations, 6403 samples), and for assays using both N‐ and S‐based antigens 90.1% (95% CI 87.2% to 92.3%; 54 evaluations, 3657 samples) (P = 0.88 for difference between groups; Table 4). The same pattern was observed for IgG or IgM, combined and for total antibody assays.
For IgM alone, there was a suggestion of higher average sensitivity for S‐based assays using convalescent data compared to the other groups although the difference was within that which could be expected by chance (P = 0.20). Average sensitivity for S‐based assays was 77.9% (95% CI 65.7 to 86.6; 24 evaluations, 1465 samples) compared to 65.4% for N‐based (95% CI 50.7 to 77.7; 25 evaluations, 1297 samples), and 64.6% (95% CI 54.5 to 73.6; 50 evaluations, 3137 samples) (Table 4).
A further specified sensitivity analysis of S‐based assays according to the use of the S1 subunit or RBD did not identify any clear patterns in sensitivity (Table 4). This analysis was not repeated for results by week after onset of symptoms.
Results by week after onset of symptoms suggested a possible effect on IgG assay sensitivity according to the antigen used (Table 9). For example, in week two after onset, average sensitivity for assays using N‐ and S‐based antigens was 76.7% (95% CI 72.1 to 80.8; 47 evaluations, 2272 samples) compared to 66.7% for N‐based (95% CI 61.9 to 71.1; 61 evaluations, 2688 samples), and 59.8% for S‐based assays (95% CI 54.9 to 64.5; 65 evaluations 3222 samples) (P < 0.001). The differences in average sensitivities was less in week three after onset: sensitivity 85.4% for S‐based assays (95% CI 82.2 to 88.2%; 65 evaluations, 1717 samples), 91.2% for N‐based assays (95% CI 88.5 to 93.2; 53 evaluations, 1307 samples), and 89.2% for N‐ and S‐based assays (95% CI 86.0% to 91.8%; 40 evaluations, 1323 samples) (P = 0.01; Table 9).
For IgM alone, assays that used the Spike‐protein were on average more sensitive than those incorporating the nucleoprotein in each of the first three weeks after onset of symptoms; for example, in week 2 after onset, average sensitivity was 78.2% for S‐based assays (95% CI 67.7 to 86.1; based on 21 evaluations comprising 1116 samples), compared to 57.8% for N‐based assays (95% CI 47.5 to 67.5; 33 evaluations, 1607 samples), and 66.3% (95% CI 57.3, 74.2; 41 evaluations, 2060 samples) for N‐ and S‐based assays (P = 0.02; Table 9).
For the combination of IgG or IgM, and for total antibodies, fewer and smaller differences in average sensitivity by antigen type were observed (Table 9).
Specificity by antigen
Type of antigen used in the antibody assays did not have a strong effect on average specificities for either IgG alone or for IgM alone (Table 10).
For pre‐pandemic samples, average specificities for IgG alone were 99.1% for N‐based tests (95% CI 98.7 to 99.4; 55 evaluations, 14,159 samples) and 98.9% for S‐based tests (95% CI 98.4 to 99.2; 66 evaluations, 14,615 samples). Results for assays using both N and S antigens were also slightly lower than those for N‐based tests: average specificity 99.0% (95% CI 98.4 to 99.4; 37 evaluations, 7449 samples). A generally similar pattern was seen across the other reference standard groups (Table 10).
For IgM alone, average specificities using pre‐pandemic samples were: 98.4% for N‐based tests (95% CI 96.9 to 99.2; 22 evaluations, 5674 samples), 98.3% for S‐based tests (95% CI 96.3 to 99.2; 16 evaluations, 2870 samples), and 98.9% (95% CI 97.8 to 99.5; 28 evaluations, 5114 samples) for assays using both N and S antigens. Some differences in average specificities by antigen type were observed for the other reference standard groups, however, fewer evaluations and samples in some groups are likely to have contributed to observed differences (Table 10).
No differences in average specificities by antigen were identified for assays detecting IgG or IgM, or total antibodies using pre‐pandemic samples (Table 10). Some differences were noted for IgG or IgM assays for other reference standard groups; however, again, fewer evaluations and samples in some groups are likely to have contributed to observed differences.
Sensitivity and specificity by test brand
We identified 112 test brands with data for the sensitivity of IgG, IgM, or IgG or IgM combined (79 lateral flow assays, 17 ELISAs, 12 CLIAs, and 4 other laboratory‐based tests), and 12 test brands with data for the sensitivity of total antibody detection (3 ELISAs, eight CLIAs, and 1 other laboratory‐based assay) for detection of prior SARS‐CoV‐2 during the convalescent phase of infection. Because of the large number of test brands, we have tabulated results for sensitivity and specificity for every test brand according to test technology and timing of testing for sensitivity in Appendices, as described below. Results of meta‐analyses are reported where possible, and results of individual studies used for brands with only one available evaluation. Caution is required in the interpretation of these data as many are based only on single studies with small sample sizes. We present confidence intervals to quantify the uncertainty in the estimates. Note that although in this section we present summary estimates for sensitivity and/or specificity, we frequently observed considerable heterogeneity between studies.
Forest plots of individual study results by target antibody and time after onset (for sensitivity) and by reference group (for specificity) are presented in Appendices organised by test method (laboratory‐based followed by lateral flow‐based tests), and then in alphabetical order by manufacturer. Refer to Appendix 9 for forest plots of sensitivity by week after onset, Appendix 10 for sensitivity for convalescent‐phase infection, and Appendix 12 for specificity by reference group.
Results by test brand (either based on meta‐analysis or for individual studies if only one per study per brand) are also reported in Appendices. Sensitivities during the convalescent phase are reported in Appendix 17 (for IgG, IgM and IgG or IgM) and Appendix 18 (total antibodies), and specificities in pre‐pandemic samples are reported in Appendix 19 (for IgG, IgM and IgG or IgM) and Appendix 20 (total antibodies; also reports data for other reference groups). Sensitivities by test brand by week after onset (weeks 1 to week 3) are reported in Appendix 21 (IgG alone), Appendix 22 (IgG or IgM), Appendix 23 (total antibodies) and Appendix 24 (IgM). Specificities by test brand for additional reference groups (not pre‐pandemic) are reported in Appendix 25 (IgG alone), Appendix 26 (IgG or IgM), and Appendix 27 (IgM). Sensitivities and specificities by test brand for IgA alone or combined with other target antibodies are given in Appendix 28 and Appendix 29.
We have used UK MHRA minimum performance targets for IgG and total antibody assays as set out in target product profiles (TPPs) that cover point of care (MHRA 2021a) and laboratory‐based enzyme‐immunoassays (MHRA 2021b) to aid interpretation of data. The TPPs recommend that both clinical sensitivity and specificity should be at least 98% (with 95% CIs 96% to 100%), each based on testing at least 200 samples (collected 20 days or more after the appearance of first symptoms for sensitivity and based on pre‐pandemic samples collected at least 6 months before the known appearance of the virus for specificity). Because the 98% sensitivity target is very high and unlikely to be achievable for many tests unless evaluated against a serological reference standard, we instead highlight test brands for which the lower bound of the 95% CI for sensitivity is 90% or more. We have also extended these criteria to include results for the combination of IgG or IgM as well as for IgG alone and for total antibody detection. Results for test brands that meet these pre‐set criteria for either acceptable sensitivity or specificity (or both) are reported in Table 11 along with their respective sensitivities or specificities, even if these did not meet the pre‐set criteria.
10. Sensitivity and specificity by brand (IgG, IgG or IgM, total Ab).
|
Target antibody Method |
Test brand | Test name | Antigen |
95% CI > 90%a (Yes/No) |
Convalescent N evaluations (N samples) |
Sensitivity (%, 95% CI) | 95%CI > 96%b |
Pre‐pandemic N evaluations (N samples) |
Specificity (%, 95% CI)c |
| IgG alone | |||||||||
| CLIA | Autobio Diagnostics | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | S | Y | 1 (271/273) | 99.3(97.4, 99.9) | N | no data | |
| LFA | Qingdao HIGHTOP | SARS‐CoV‐2 IgG/IgM Ab Rapid Test | N, S | Y | 1 (216/229) | 94.3 (90.5, 96.9) | N | 1 (149/150) | 99.3 (96.3, 100) |
| LFA | Sure Biotech | SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | N, S | Y | 2 (226/235) | 96.2 (92.8, 98.0) | N | 2 (370/378) | 98.7 (87.2, 99.9) |
| CLIA | Abbott Diagnostics | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG/IgM | N | Y | 33 (1824/1977) | 92.5 (90.3, 94.3) | Y | 24 (7460/7483) | 99.7 (99.5, 99.8) |
| CLIA | Shenzhen YHLO Biotech | YHLO iFlash IgG/IgM assay | N, S | Y | 5 (260/268) | 97.0 (94.1, 98.5) | Y | 2 (657/661) | 99.4 (98.4, 99.8) |
| LFA | Augurix SA | SimtomaX Corona Check IgG | N, S | N | 1 (124/220) | 56.4 (49.5, 63.0) | Y | 1 (267/268) | 99.6 (97.9, 100) |
| LFA | bioMerieux | Vidas SARS‐CoV‐2 IgG | S (RBD) | N | 2 (101/107) | 94.4 (88.1, 97.5) | Y | 2 (1084/1085) | 99.9 (99.3, 100) |
| LFA | Biopanda | COVID‐19 Rapid Ab test | N, S | N | 1 (102/163) | 62.6 (54.7, 70.0) | Y | 1 (499/500) | 99.8 (98.9, 1.00) |
| LFA | Dynamiker Biotechnology | 2019‐nCOV IgG Rapid Test | N | < 100 | 1 (72/75) | 96.0(88.8, 99.2) | Y | 1 (395/403) | 98.0(96.1, 99.1) |
| LFA | Fortress Diagnostics | COVID‐19 Total Ab | S | N | 1 (258/307) | 84.0 (79.5, 88.0) | Y | 1 (493/500) | 98.6 (97.1, 99.4) |
| LFA | SD Biosensor | COVID‐19 IgG Duo | N | < 100 | 4 (50/69) | 74.2 (52.1, 88.3) | Y | 4 (1125/1127) | 99.8 (96.2, 100) |
| ELISA | Beijing Wantai | ELISA IgG assay | S | < 100 | 2 (57/58) | 98.3 (90.8, 99.9) | Y | 1 (195/197) | 99.0(96.4, 99.9) |
| ELISA | Eagle Biosciences | COVID‐19 IgG Quantitative ELISA | N | N | 1 (539/1134) | 47.5 (44.6, 50.5) | Y | 1 (429/437) | 98.2 (96.4, 99.2) |
| ELISA | Epitope Diagnostics | EDI nCov COVID‐19 IgM ELISA kit | N | N | 13 (780/934) | 87.7 (78.1, 93.4) | Y | 13 (2892/3014) | 98.0 (96.2, 99.0) |
| ELISA | EUROIMMUN | anti‐SARS‐COV‐2 IgG ELISA | S (S1) | N | 41 (2200/2442) | 90.8 (88.6, 92.7) | Y | 29 (4998/5144) | 98.4 (97.5, 98.9) |
| CLIA | Abbott Diagnostics | Abbott Alinity anti‐SARS‐CoV‐2 nucleocapsid IgG | N | N | 2 (149/163) | 91.3 (83.8, 95.5) | Y | 2 (636/640) | 99.4 (98.3, 99.8) |
| CLIA | Beckman Coulter | Beckman Coulter ‐ Access SARS‐CoV‐2 IgG | S (RBD) | < 100 | 2 (78/94) | 92.4 (38.8, 99.6) | Y | 1 (396/399) | 99.2(97.8, 99.8) |
| CLIA | DiaSorin | LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA | S | N | 21 (1523/1735) | 88.1 (84.1, 91.2) | Y | 16 (4290/4367) | 98.6 (97.8, 99.1) |
| CLIA | Ortho Clinical Diagnostics | VITROS Anti‐SARS‐Cov‐2 Total assay IgG | S (S1) | N | 3 (201/221) | 92.3 (80.8, 97.1) | Y | 3 (1139/1141) | 99.8(99.3, 100) |
| CLIA | SNIBE | MAGLUMI 2019‐nCoV IgG kits | N, S | N | 7 (325/369) | 89.9 (83.3, 94.1) | Y | 7 (1801/1818) | 99.1 (98.5, 99.4) |
| Other | ET Healthcare | Pylon 3D automated immunoassay system IgG | N, S | < 100 | 2 (37/37) | 100(90.5, 100) | Y | 1 (316/320) | 98.8(96.8, 99.7) |
| IgG or IgM | |||||||||
| LFA | SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgG/IgM | S | Y | 3 (248/257) | 96.5 (93.4, 98.2) | Y | 2 (497/500) | 99.4 (98.2, 99.8) |
| LFA | Guangzhou Wondfo | SARS‐CoV‐2 Antibody Test | S | N | 6 (211/265) | 85.1 (69.0, 93.6) | Y | 4 (1644/1648) | 99.8 (98.8, 100) |
| LFA | SD Biosensor | COVID‐19 IgG Duo | N | < 100 | 1 (6/7) | 85.7(42.1, 99.6) | Y | 1 (933/942) | 99.0(98.2, 99.6) |
| LFA | NG Biotech | NG‐Test IgG COVID‐19 | N | N | no data | Y | 2 (274/276) | 99.3 (97.2, 99.8) | |
| ELISA | Epitope Diagnostics Inc. | EDI nCov COVID‐19 IgM ELISA kit | N | < 100 | 2 (42/50) | 84.0 (71.1, 91.8) | Y | 4 (1159/1184) | 97.9 (96.9, 98.6) |
| Total Ab | |||||||||
| ELISA | Beijing Wantai | ELISA Total‐Ab assay | S (RBD) | Y | 8 (1562/1649) | 95.7 (92.6, 97.5) | Y | 8 (2009/2020) | 99.5 (99.0, 99.7) |
| CLIA | Ortho Clinical Diagnostics | VITROS anti‐SARS‐Cov‐2 Total assay | S (S1) | Y | 2 (192/200) | 96.0 (92.2, 98.0) | Y | 2 (995/997) | 99.8 (98.5, 100) |
| CLIA | Roche Diagnostics | Elecsys anti‐SARS‐CoV‐2 antibody assay | N | Y | 34 (3669/3916) | 93.4 (91.1, 95.1) | Y | 25 (5569/5579) | 99.8 (99.7, 99.9) |
| CLIA | Siemens Healthcare | Siemens Atellica Total‐Ab assay | S (RBD) | Y | 7 (979/1009) | 96.7 (94.2 98.1) | Y | 6 (2435/2439) | 99.9 (99.3, 100) |
| CLIA | Xiamen InnoDx | 2019‐nCoV antibody test kit Total‐Ab | S (RBD) | < 100 | 1 (37/38) | 97.4(86.2, 99.9) | Y | 1 (267/270) | 98.9(96.8, 99.8) |
| CLIA | Siemens Healthcare | Siemens Vista Total‐Ab assay | S (RBD) | N | 1 (94/116) | 81.0 (72.7, 87.7) | Y | 1 (596/596) | 100 (99.4, 100) |
| Other | Luminex | SARS‐CoV‐2 MIA Total Ab | N | < 100 | 1 (19/19) | 100(82.4, 100) | Y | 1 (254/256) | 99.2(97.2, 99.9) |
| a pre‐set criteria for sensitivity were assay evaluation in ≥ 200 samples and lower bound of 95% CI for sensitivity was 90% or higher b pre‐set criteria for specificity were assay evaluation in ≥ 200 samples, point estimate for specificity ≥ 98% and lower bound of 95% CI was 96% or higher c sensitivity and specificity estimates per brand are not necessarily paired estimates from the same set of studies (i.e. were not calculated from the same meta‐analytic model) but were calculated separately using univariate analyses such that there may be differences in the set of studies contributing to sensitivity and to specificity for any given test brand (although there will be overlap between them). | |||||||||
CI: confidence interval CGIA: colloidal gold immunoassay CLIA: chemiluminescent immunoassay ELISA: enzyme linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assay N: nucleocapsid antigen RBD: receptor binding domain RT‐PCR: reverse transcription polymerase chain reaction S: soluble antigen
IgG alone by test brand: sensitivity during convalescent phase of infection and specificity in pre‐pandemic samples
Data for the sensitivity of IgG alone was reported for 96 test brands, with sensitivities or summary sensitivities ranging from 25% to 100% (Appendix 17) (see Appendix 9 for plots of individual study results). Two‐thirds of assays (63/96) were evaluated in a single study, and sensitivity estimates were based on more than 100 samples for only around a third (35/96), and on more than 200 samples for only 21% (20/96) of assays (Appendix 17). Specificities for IgG alone were reported for 72 test brands using pre‐pandemic samples and ranged from 75% to 100% (Appendix 19). Almost three‐quarters (52/72) were evaluated in a single study, and specificity estimates were based on more than 200 samples for only 31 assays (43%). Results by individual study are reported in Appendix 12).
Using MHRA minimum performance standards, the point estimate for sensitivity met or exceeded the 98% target for sensitivity based on more than 200 samples for only one assay (Table 11); 99.3% (95% CI 97.4% to 99.9%) for the Autobio Diagnostics CLIA Microparticles (1 evaluation, 273 COVID‐19 samples). No specificity estimate based on pre‐pandemic samples was available for this assay. For a further four assays, the lower bound of the 95% CI around the sensitivity estimate was 90% or more (the Qingdao HIGHTOP and Sure Biotech LFAs, and the Abbott Architect CLIA and Shenzhen YHLO Biotech iFlash CLIA assay); sensitivities ranged from 92.5% to 97.0% (Table 11).
A total of 18 assays met MHRA minimum standards for specificity in pre‐pandemic samples, including two of the four assays (both CLIAs) meeting our pre‐set criteria for sensitivity. Summary specificities were 99.7% for Abbott Architect (95% CI 99.5 to 99.8; 24 evaluations, 7483 samples) and 99.4% for Shenzhen YHLO Biotech iFLash (95% CI 98.4 to 99.8; 2 evaluations, 661 samples) (Table 11). Of the remaining 16 assays meeting minimum standards for specificity, sensitivities for convalescent‐phase infection were based on at least 100 samples (and therefore are more reliable) for only 11 assays; these included four LFAs, three ELISAs and four CLIAs. Sensitivities ranged from 47.5% (95% CI 44.6 to 50.5) for the Eagle Biosciences COVID‐19 IgG Quantitative ELISA (1 evaluation, 1134 samples) to 94.4% (95% CI 88.1 to 97.5) for the bioMerieux Vidas LFA (2 evaluations, 107 samples); point estimates for sensitivity exceeded 90% for a total of four assays (Table 11).
IgG or IgM by test brand: sensitivity during convalescent phase of infection and specificity in pre‐pandemic samples
Data for the sensitivity of IgG or IgM combined was reported for 73 test brands, with sensitivities ranging from 57.5% to 100% (Appendix 19). Three‐quarters of assays (54/73) were evaluated in a single study, and sensitivity estimates were based on more than 100 samples for only 9 assays (12%), and on more than 200 samples for only two assays (3%). The vast majority of assays (85%) were rapid point‐of‐care rather than laboratory‐based tests. Specificities for IgG or IgM were reported for 44 test brands using pre‐pandemic samples, with specificities ranging from 65% to 100% (Appendix 18). Almost three‐quarters (31/44) of test brands were evaluated in a single study, and specificity estimates were based on more than 200 samples for only 12 assays (28%).
Only one assay met our prespecified criteria for sensitivity (Table 11); the average sensitivity for the SureScreen Diagnostics Rapid test was 96.5% (95% CI 93.4%, 98.2%, based on three evaluations with 257 samples).
A total of five assays met MHRA minimum standards for specificity in pre‐pandemic samples, including the SureScreen LFA (average specificity 99.4%, 95% CI 98.2 to 99.8; 2 evaluations, 500 samples). Of the remaining four assays, only one had the sensitivity estimate based on more than 100 convalescent samples (Table 11). Average sensitivity for the Guangzhou Wondfo LFA was 85.1%; the 95% CI reflecting the small number of samples for this assay (95% CI 69.0 to 93.6; 6 evaluations, 265 samples) (Appendix 10), with average specificity of 99.8% (95% CI 98.8 to 100; 4 evaluations, 1648 samples).
Total antibodies by test brand: sensitivity during convalescent phase of infection and specificity in pre‐pandemic samples
Data for the sensitivity of total antibody detection was reported for 12 test brands, all of which were laboratory‐based assays; sensitivities ranged from 81.0% to 100% (Appendix 20), but only five assays were evaluated in more than 100 samples and four in more than 200 samples (Appendix 10). Specificities for total antibodies were reported for eight test brands using pre‐pandemic samples, with specificities ranging from 82% to 100% (Appendix 20). All but one assay was evaluated in more than 200 samples (Appendix 12).
Four assays met our prespecified criteria for sensitivity (Table 11). Average sensitivities ranged from 93.4% (95% CI 91.1% to 95.1%) for Roche Diagnostics Elecsys anti‐SARS‐CoV‐2 Total Ab CLIA (34 evaluations, 3916 samples) to 96.7% (95% CI 94.2 to 98.1) for Siemens Atellica Total‐Ab assay (7 evaluations, 1009 cases).
All seven assays evaluated in 200 or more samples met MHRA minimum standards for specificity. Average specificities for the test brands meeting our pre‐set criteria for sensitivity ranged between 99.5% (95% CI 99.0 to 99.7) for the Beijing Wantai Total‐Ab ELISA (8 evaluations, 2020 samples) and 99.9% (95% CI 99.3 to 100) for the Siemens Atellica Total‐Ab CLIA (6 evaluations, 2439 samples).
Of the remaining three, only one had sensitivity estimates for convalescent‐phase infection based on at least 100 samples (Table 11). Specificity for the Siemens Vista Total‐Ab assay was 100% (95% CI 99.4 to 100; 1 evaluation, 596 samples) and sensitivity was 81.0% (95% CI 72.7 to 87.7; 1 evaluation, 116 samples).
IgM by test brand: sensitivity during convalescent phase of infection and specificity in pre‐pandemic samples
The sensitivity of IgM detection was reported for 80 assays during the convalescent phase of infection including 23 assays with 100 or more samples and 12 with 200 or more samples (9 LFAs, 1 ELISA and 1 CLIA assay (Appendix 10)). The specificity of IgM in pre‐pandemic samples was available for 59 assays, 38 evaluated in 100 or more samples and 23 in 200 or more samples (Appendix 12).
Of those 12 assays with at least 200 samples, sensitivities ranged from 27.3% to 90.6%. We did not apply MHRA TPP criteria to IgM assays, however, the point estimate for sensitivity for three LFAs exceeded 80% (assays from MEDSan, NTBIO Diagnostics and Xiamen Biotime) and for two LFA assays exceeded 90% (Appendix 24):
average sensitivity 90.2% (95% CI 85.7 to 93.4; 2 evaluations, 235 samples) for the Sure Biotech rapid test;
sensitivity 90.6% (95% CI 86.0 to 94.1; 1 evaluation, 224 samples) for the bioMerieux Vidas LFA.
Specificities in pre‐pandemic samples for these five assays exceeded 99% for two assays (Sure Biotech and BioMerieux LFAs) and ranged between 94.4% and 96.8% for the other three (Appendix 27).
Direct comparisons of test brands in convalescent‐phase infection: IgG, IgG or IgM, or total antibodies
A total of 67 studies reported the comparison of two or more test brands targeting IgG alone, IgG or IgM combined or total antibodies during convalescent infection. Of these, only 13 studies included at least 100 samples per test brand (Table 12). Two studies compared LFAs only (Flower 2020 [A]; Rudolf 2020 [A]), 10 studies compared laboratory‐based tests (Chaudhuri 2020 [A]; DomBourian 2020 [A]; Gudbjartsson 2020 [A]; Harritshoej 2021 [A]; Horber 2020 [A]; Kaltenbach 2020 [A]; Korte 2021 [A]; MacMullan 2020 [A]; NSAE 2020 [A]; Patel 2020), and one included both LFAs and laboratory‐based tests (Weidner 2020 [A]). Although essentially reporting direct comparisons of tests, some studies reported different sample numbers per assays, either because of insufficient sample numbers to conduct all tests, variation in test failures between brands, or because some assays were taken forward for further evaluation on more samples based on preliminary results.
11. Direct comparisons of test brands: convalescent phase IgG, IgG or IgM or total Ab (with around 100 samples per brand).
| Study | Setting (cases) | Test method | New combined name | Antigen | Target | TP/D+ | Sensitivity | Range in percentage points |
| Comparison of lateral flow assays | ||||||||
| Flower 2020 [A] | Community | CGIA | Guangzhou Wondfo ‐ SARS‐CoV‐2 Ab | S‐based | IgG or IgM | 75/99a | 75.8% | 26.4 |
| Flower 2020 [B] | Community | CGIA | Zhejiang Orient‐Gene IgG/IgM | N‐ and S‐based | IgG | 113/127 | 89.0% | |
| Flower 2020 [C] | Community | Not detailed | Fortress Diagnostics ‐ COVID‐19 Total Ab | S‐based | IgG | 258/307 | 84.0% | |
| Flower 2020 [D] | Community | Not detailed | Biopanda ‐ COVID‐19 Rapid Ab test | N‐ and S‐based | IgG | 102/163 | 62.6% | |
| Flower 2020 [E] | Community | Not detailed | Mologic ‐ IgG COVID‐19 | N‐ and S‐based | IgG | 99/148 | 66.9% | |
| Rudolf 2020 [A] | Unclear | CGIA | CTK OnSite COVID‐19 IgG/IgM | S‐based | IgG | 158/212 | 74.5% | 40.6 |
| Rudolf 2020 [B] | Unclear | CGIA | Sure Biotech ‐ SARS‐CoV‐2 IgM/IgG Ab | N‐ and S‐based | IgG | 216/224 | 96.4% | |
| Rudolf 2020 [C] | Unclear | CGIA | Augurix SimtomaX Corona Check | N‐ and S‐based | IgG | 124/220 | 56.4% | |
| Rudolf 2020 [D] | Unclear | Not detailed | TAmiRNA SARS‐CoV‐2 Ab | S1‐based | IgG or IgM | 203/222 | 91.4% | |
| Rudolf 2020 [E] | Unclear | Not detailed | NTBIO One Step IgG/IgM | Unclear | IgG | 188/219 | 85.8% | |
| Rudolf 2020 [F] | Unclear | Not detailed | MEXACARE QuickTestCorona IgG/IgM | N‐ and S‐based | IgG | 190/224 | 84.8% | |
| Rudolf 2020 [G] | Unclear | CGIA | Xiamen Biotime SARS‐Cov‐2 IgG/IgM | Unclear | IgG | 183/200 | 91.5% | |
| Rudolf 2020 [H] | Unclear | Not detailed | Inzek ‐ BIOZEK COVID‐19 IgG/IgM | Unclear | IgG | 106/115 | 92.2% | |
| Rudolf 2020 [I] | Unclear | CGIA | MEDsan COVID19 IgG/IgM | N‐ and S‐based | IgG | 201/227 | 88.5% | |
| Rudolf 2020 [J] | Unclear | Not detailed | Qingdao HIGHTOP IgM/IgG | N‐ and S‐based | IgG | 216/229 | 94.3% | |
| Rudolf 2020 [K] | Unclear | Not detailed | Hangzhou Biotest ‐ RightSign IgG/IgM | S‐based | IgG | 130/134 | 97.0% | |
| Weidner 2020 [E] | Community | CGIA | MEDsan COVID19 IgG/IgM | N‐ and S‐based | IgG or IgM | 92/99a | 92.9% | 4.1 |
| Weidner 2020 [F] | Community | CGIA | Wantai SARS‐CoV‐2 Ab rapid assay | RBD | IgG or IgM | 87/98a | 88.8% | |
| Comparison of laboratory‐based tests | ||||||||
| Chaudhuri 2020 [A] | Mixed | CLIA | Diasorin ‐ LIAISON SARS‐CoV‐2 | S‐based | IgG | 313/379 | 82.6% | 6.9 |
| Chaudhuri 2020 [B] | Mixed | ELISA | Zydus Covid Kavach IgG | Unclear | IgG | 287/379 | 75.7% | |
| DomBourian 2020 [A] | Unclear | ELISA | Epitope Diagnostics ‐ EDI nCov COVID‐19 | N‐based | IgG | 84/99a | 84.8% | 7.0 |
| DomBourian 2020 [B] | Unclear | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 90/98a | 91.8% | |
| Gudbjartsson 2020 [A] | Community | CLIA | Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab | N‐based | Total ab | 1120/1215 | 92.2% | 46.6 |
| Gudbjartsson 2020 [B] | Community | ELISA | Wantai ELISA Total‐Ab assay | RBD | Total ab | 1143/1215 | 94.1% | |
| Gudbjartsson 2020 [C] | Community | ELISA | EDI/Eagle COVID‐19 IgG/IgM | N‐based | IgG | 539/1134 | 47.5% | |
| Harritshoej 2021 [A] | Community | ELISA | Wantai ELISA Total‐Ab assay | RBD | Total ab | 120/123 | 97.6% | 16.6 |
| Harritshoej 2021 [B] | Community | CLIA | Ortho Clinical VITROS Anti‐SARS‐Cov‐2 | S‐based | IgG | 118/123 | 95.9% | |
| Harritshoej 2021 [C] | Community | CLIA | Siemens Atellica Total‐Ab assay | RBD | Total ab | 117/121 | 96.7% | |
| Harritshoej 2021 [D] | Community | CLIA | Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab | N‐based | Total ab | 118/123 | 95.9% | |
| Harritshoej 2021 [E] | Community | CLIA | YHLO SARS‐CoV‐2 iFlash IgG/IgM assay | N‐ and S‐based | IgG | 118/123 | 95.9% | |
| Harritshoej 2021 [F] | Community | CLIA | Abbott Architect anti‐SARS‐CoV‐2 IgG | N‐based | IgG | 115/123 | 93.5% | |
| Harritshoej 2021 [G] | Community | CLIA | Abbott Alinity anti‐SARS‐CoV‐2 IgG | N‐based | IgG | 115/123 | 93.5% | |
| Harritshoej 2021 [H] | Community | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 102/123 | 82.9% | |
| Harritshoej 2021 [I] | Community | CLIA | Snibe Diagnostic ‐ MAGLUMI 2019‐nCoV | N‐ and S‐based | IgG | 101/122 | 82.8% | |
| Harritshoej 2021 [J] | Community | CLIA | Diasorin ‐ LIAISON SARS‐CoV‐2 | S‐based | IgG | 110/123 | 89.4% | |
| Harritshoej 2021 [L] | Community | CLIA | Ortho Clinical VITROS Anti‐SARS‐Cov‐2 | S1‐based | Total ab | 120/123 | 97.6% | |
| Harritshoej 2021 [M] | Community | CLIA | Siemens Vista Total‐Ab assay | RBD | Total ab | 94/116 | 81.0% | |
| Horber 2020 [A] | Hospital inpatient | CLIA | Siemens Atellica Total‐Ab assay | RBD | Total ab | 128/132 | 97.0% | 7.6 |
| Horber 2020 [B] | Hospital inpatient | CLIA | Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab | N‐based | Total ab | 118/132 | 89.4% | |
| Horber 2020 [C] | Hospital inpatient | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 126/132 | 95.5% | |
| Kaltenbach 2020 [B] | Community | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 226/239 | 94.6% | 5.1 |
| Kaltenbach 2020 [C] | Community | ELISA | Epitope Diagnostics ‐ EDI nCov COVID‐19 | N‐based | IgG | 214/239 | 89.5% | |
| Korte 2021 [A] | Unclear | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 132/141 | 93.6% | 7.8 |
| Korte 2021 [C] | Unclear | ELISA | Epitope Diagnostics ‐ EDI nCov COVID‐19 | N‐based | IgG | 121/141 | 85.8% | |
| MacMullan 2020 [B] | Unclear | ELISA | Gold Standard SARS‐CoV‐2 IgG ELISA | N‐based | IgG | 85/123 | 69.1% | 21.1 |
| MacMullan 2020 [D] | Unclear | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 111/123 | 90.2% | |
| NSAE 2020 [A] | Hospital inpatient | CLIA | Abbott Architect anti‐SARS‐CoV‐2 IgG | N‐based | IgG | 458/490 | 93.5% | 4.9 |
| NSAE 2020 [B] | Hospital inpatient | CLIA | Diasorin ‐ LIAISON SARS‐CoV‐2 | S‐based | IgG | 468/490 | 95.5% | |
| NSAE 2020 [C] | Hospital inpatient | CLIA | Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab | N‐based | Total Ab | 481/490 | 98.2% | |
| NSAE 2020 [D] | Hospital inpatient | CLIA | Siemens Atellica Total‐Ab assay | RBD | Total Ab | 482/490 | 98.4% | |
| Patel 2021 [A] | Community | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | S1‐based | IgG | 127/146 | 87.0% | 17.5 |
| Patel 2021 [B] | Community | ELISA | Epitope Diagnostics ‐ EDI nCov COVID‐19 | N‐based | IgG | 115/146 | 78.8% | |
| Patel 2021 [C] | Community | ELISA | ImmunoDiagnostics SARS‐CoV‐2 IgG | N‐based | IgG | 107/140 | 76.4% | |
| Patel 2021 [D] | Community | CLIA | Abbott Architect anti‐SARS‐CoV‐2 IgG | N‐based | IgG | 135/146 | 92.5% | |
| Patel 2021 [E] | Community | CLIA | Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab | N‐based | Total ab | 201/214 | 93.9% | |
| Weidner 2020 [A] | Community | ELISA | EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | N‐based | IgG | 179/197 | 90.9% | 10.0 |
| Weidner 2020 [B] | Community | ELISA | Wantai ELISA Total‐Ab assay | RBD | Total ab | 98/100 | 98.0% | |
| Weidner 2020 [C] | Community | CLIA | Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab | N‐based | Total ab | 94/99 | 94.9% | |
| Weidner 2020 [D] | Community | CLIA | Diasorin ‐ LIAISON SARS‐CoV‐2 | S‐based | IgG | 88/100 | 88.0% | |
Ab: antibody CGIA: colloidal gold immunoassay CLIA: chemiluminescent immunoassay D+: number of positive cases included in the analysis ELISA: enzyme‐linked immunosorbent assay TP: true positive aIn principle the cut‐off for inclusion was evaluation in at least 100 samples, however, to allow inclusion of as many studies with direct test comparisons as possible, we reported studies reporting evaluation in 98 samples or more.
Table 12 shows variations in sensitivity of LFAs between 4.1 and 40.6 percentage points and in sensitivity of laboratory‐based tests between 4.9 to 46.6 percentage points. It is likely that a combination of test method and antigen contributed to observed variations, but it is not possible to disentangle any effect because of small numbers of studies.
Sensitivity by test brand – week 1 to week 3 after onset of symptoms
In this section, we consider the evidence for individual test brands by week after onset of symptoms, with a particular focus on those with 200 or more samples in a particular time period (Appendix 21 (IgG alone), Appendix 22 (IgG or IgM), Appendix 23 (total antibodies) and Appendix 24 (IgM).
During week one after onset of symptoms, heterogeneity in results and average sensitivities below 50% were observed for almost all test brands and target antibodies. One assay (a protein microarray from Vibrant America) outperformed the rest (sensitivity for IgG alone 93% and for IgM alone 97.6%), however, this assay was evaluated in only a single study with limited information about study participants and tests performed by the company, so it is not clear whether results will be reproducible.
In week two after onset, excluding the Vibrant America assay (which demonstrated 100% sensitivity for both IgG alone and IgM alone), 21 assays were evaluated in 200 or more samples (11 for IgG alone, 4 IgM alone, 4 for IgG or IgM combined and 2 for total antibody detection). Considerable variability in results remained, particularly for the detection of IgG alone (average sensitivities ranged from 54.4% [EUROIMMUN ELISA; 32 evaluations, 1407 samples] to 78.8% [Epitope EDI ELISA, 11 evaluations, 455 samples]) and for IgM alone (average sensitivity from 58.3% [Epitope EDI ELISA; 7 evaluations, 381 samples) to 80.4% (Wantai ELISA; 4 evaluations, 315 samples)]. The best performing rapid tests for IgM alone were from NG Biotech (78.6% sensitivity219 samples) and the Hangzhou RightSign assay (77.5% sensitivity, 218 samples). More consistent results were observed for tests detecting IgG or IgM combined; average sensitivities ranged between 73.9% (Guangzhou Wondfo LFA; 6 evaluations, 245 samples) and 85.0% (Zhejiang Orient‐Gene Biotech LFA; 5 evaluations, 195 samples). Two total antibody assays with results in more than 200 samples reported average sensitivities of 72.0% (Roche Elecsys CLIA; 16 evaluations, 544 samples) and 88.5% (Beijing Wantai ELISA Total‐Ab assay; 8 evaluations, 342 samples).
For week three after onset of symptoms, five laboratory‐based assays for IgG detection had data based on 200 or more samples. More consistent results were observed with average sensitivities ranging from 81.8% (95% CI 70.3% to 89.5%; 10 evaluations, 303 samples) for the Diasorin LIAISON CLIA to 95.9% (95% CI 92.2% to 97.8%; 2 evaluations, 217 samples) for the Bioscience Co (Chongqing) CLIA. The latter brand was also the only one with results for IgM in more than 200 samples; average sensitivity was 76.2% (95% CI 60.4% to 87.0%; 2 evaluations, 217 samples). None of the test brands had data based on more than 200 samples for IgG or IgM combined, but two had average sensitivities for total antibodies of 89.8 (95% CI 85.3 to 93.1) (Roche Elecsys CLIA; 18 evaluations, 529 samples), and 96.4 (95% CI 89.9 to 98.8) (Wantai ELISA Total‐Ab; 6 evaluations, 198 samples).
Specificity by test brand – other reference groups
For IgG alone, the specificity of 38 assays was evaluated only in pre‐pandemic samples; 80 assays had data from other reference groups, of which only 12 were evaluated in more than 200 non‐COVID‐19 samples (Appendix 25). Nine of the 12 assays also had specificity estimates based on more than 200 pre‐pandemic samples, including: LFAs from BioMerieux, VivaChek Biotech, and SD Biosensor; the EUROMIMMUN IgG ELISA; CLIAs from Abbott Diagnostics (using either Alinity or Architect platforms), DiaSorin, SNIBE, and Shenzhen YHLO.
The three remaining assays with specificity data based on at least 200 samples from non‐pre‐pandemic reference groups included the:
Beijing Diagreat LFA; specificity 95.5% (95% CI 93.5% to 97.0%, based on one evaluation and 600 samples), and
Zhuhai Livzon IgG ELISA; specificity 99.1% (95% CI 96.4% to 99.8%, two evaluations and 220 samples).
Both were evaluated in contemporaneously collected samples from healthy or other disease controls (no RT‐PCR reported).
The third assay was evaluated in non‐COVID‐19 samples collected from multiple sources:
Vibrant America ‐ Vibrant COVID‐19 Ab, specificity 99.8% (95% CI 99.6% to 99.9%, 1 evaluation with 5262 samples).
For total antibodies, the specificity of three assays was evaluated only in pre‐pandemic samples; eight assays had data from other reference groups, of which only two had specificity estimates on at least 200 samples (Appendix 20). The Xiamen Wantai total ab CLIA was evaluated in 234 samples in a single evaluation; specificity was 98.7% (95% CI 96.3% to 99.7%). Specificity reported for the total antibodies for the Vibrant America assay was the same as for IgG alone; 99.8% (95% CI 99.6% to 99.9%) in 5262 samples.
Data for specificity based on other reference groups by test brand for IgG or IgM combined and for IgM alone are reported in Appendix 26 and Appendix 27. Only a small number of assays had any data based on 200 or more samples (three for IgG or IgM, combined and 10 for IgM alone).
IgA alone or combined with IgM or IgG by test brand: sensitivity and specificity
Data for IgA alone or combined with IgM or IgG was primarily driven by the EUROIMMUN IgA ELISA (Appendix 28 and Appendix 29). Insufficient data were available to make any meaningful comparison between tests.
Discussion
This is the updated version of a Cochrane living review summarising the accuracy of antibody tests for detecting current or previous SARS‐CoV‐2 infection. This version of the review is based on published studies or studies available as preprints up until 30 September 2020. The speed of development and publication of studies for COVID‐19 antibody tests is unprecedented, making it difficult for any living review to keep on top of emerging literature. The landscape in which antibody tests are used has also changed considerably since we published the first version of the review. Rapid antigen tests are considerably better at identifying SARS‐CoV‐2 infection early compared to serology‐based tests and are now in widespread use, and laboratory capacity for conducting RT‐PCR tests has expanded exponentially. The trajectory of primary humoral response to infection is now more understood, with IgG known to begin to wane around eight weeks after infection (Post 2021) with mixed evidence for detectable IgG at six to eight months after infection (HIQA 2021). Although the presence of IgG is thought to most likely reflect immunity to SARS‐CoV‐2, immunity is more appropriately measured using the presence of neutralising antibodies or T cell or B cell responses. The successful development and widespread adoption of vaccines against SARS‐CoV‐2 have led to COVID‐19 ‘vaccine passports’ based on evidencing of vaccination status rather than the presence of antibodies as originally proposed. These factors limit the usefulness of serology tests for identification of prior infection as a possible indicator of immunity to further infection. Current use cases for antibody tests are likely to be limited to seroprevalence surveys or, in a limited proportion of cases, detection of current infection to assist with diagnosis of COVID‐19 or post‐acute sequelae of COVID‐19. Although we are aware of additional eligible studies published since our search, this review provides a comprehensive and significant overview of the effect of timing of testing on assay sensitivity for detecting current or prior natural infection with SARS‐CoV‐2, of reference group on specificity, and of the effect of factors such as test method and antigen used.
The studies included in this version are largely from Europe (n = 94), evaluating tests from European universities and manufacturers, although many were also conducted in Asia (n = 45) and North America (n = 35). Whilst some of the included studies were early phase reports, the commercial nature of the tests evaluated means there was more consistent application of the tests, such as following instructions for use (IFU) and less reliance on data‐driven thresholds. There are still only a small number of field‐based studies that evaluated rapid tests as point‐of‐care tests, with the majority of assays having been carried out by technical experts in laboratories, utilising samples that are easily available to the research team, with multiple samples obtained from the same participants. A large proportion of participants with confirmed SARS‐CoV‐2 are likely to have been severely ill hospitalised patients, and very few of our included studies included community‐based cases (n = 14).
For non‐COVID‐19 groups, most studies recruited healthy or other disease participants. The majority of studies (n = 132) did not clearly report blinding of the index test used, whilst 35 were at high risk of bias because of lack of blinding of the index test interpretation to participants’ COVID‐19 status. Very few studies (n = 8) implemented the test in a way that prevented a risk of bias. These limitations explain much of the rating for high risk of bias and concerns about applicability in this review. Many of these issues make it likely that the accuracy of tests, when used in clinical care, will be lower than that observed here. Only five evaluations recruited patients in clinical pathways before it was established whether they had COVID‐19. This is more likely to produce results that reflect clinical practice, and we encourage future evaluations to consider this study design.
A concern with the previous version of this review was the high likelihood of selective reporting of results, particularly by manufacturers. Although for this review iteration we excluded studies that did not disclose test brands, it does appear to have become less of a problem with only seven studies excluded on this basis. Our decision to exclude evaluations of ‘inhouse’ assays also reduced the likelihood of selective reporting of results at ‘optimal’ thresholds. Unlike randomised controlled trials of interventions, there are no requirements for test accuracy studies to be prospectively registered on study registers, nor to publish their findings. Many industry studies are only briefly described on 'Information for use' documents included with the tests, and study reports submitted to regulators are regarded as confidential. We are also aware that there are independent studies undertaken by National Public Health bodies, some of which have been submitted to FIND's data tracking tool for speedy data sharing (https://www.finddx.org/covid-19/pipeline/). We plead for greater transparency and full publication in this field and continue to encourage laboratories to submit data and reports via FIND's portal.
Summary of main results
We summarise 10 key findings from this review.
Evaluations of many antibody tests on the market are not available as publications or even as preprints. This review has evaluated data on 124 commercial assays, potentially representing a significant proportion of the 270 antibody tests listed by FIND in November 2021. We did not, however, systematically assess whether all assays evaluated in our set of included studies remain on the market, nor whether any amendments to assay kits have been made since the evaluations were published.
The design and execution of the current studies limits the strength of conclusions that we are able to draw. Nearly all studies sampled cases with and without SARS‐CoV‐2 infection separately, and methods for selecting participants were not described. Eighty‐nine studies reported blinded reference standard interpretation and only eight clearly blinded index test interpretation. There is also a risk of reference standard misclassification especially for absence of SARS‐CoV‐2 infection in contemporaneously collected samples relying on a single negative RT‐PCR test result (the possibility of missing true cases of infection leading to apparent ‘false positive’ results) or relying on samples from ‘healthy’ blood donors with no apparent confirmation of absence of infection.
Many studies only applied tests in laboratory settings on plasma or serum, whilst they are also approved for use as point‐of‐care tests using (capillary) whole blood. From these data, it is not possible to ascertain the clinical accuracy of these tests in lower resource and more accessible settings.
Data for sensitivity strengthens the results from the previous version of this review, showing a strong trend towards increased sensitivity of antibody tests over time from onset of symptoms. The ability of antibody tests to detect SARS‐CoV‐2 infection is very low in the first week after onset of symptoms (for example for IgG or IgM combined, the average sensitivity was 41.1%, 95% CI 38.1 to 44.2), and is only moderate in the second week (74.9%, 95% CI 72.4 to 77.3). By week three after onset, however, average sensitivity for IgG or IgM combined was 88.0% (95% CI 86.3 to 89.5) (compared to just 78.3% for IgM alone, 95% CI 74.8 to 81.4). Average sensitivity during the convalescent phase of infection (up to a maximum of 100 days since onset of symptoms) was 89.8% for IgG (95% CI 88.5 to 90.9), 92.9% for IgG or IgM combined (95% CI 91.0 to 94.4), and 94.3% for total antibodies (95% CI 92.8 to 95.5). These estimates are now based on thousands of samples, however, it is likely that, in the early weeks after onset of symptoms, many participants may have remained hospitalised with COVID‐19 at the time of sampling. To some degree, the observed results might represent those at the more severe end of the disease spectrum, however, this population likely includes both immune‐competent individuals (who might be expected to show higher antibody responses) and immunosuppressed individuals and it is not possible to determine the extent to which observed results are representative of those with milder forms of COVID‐19.
Data for IgA as target antibody are based on smaller numbers of samples but suggest a similar pattern as for other antibodies, with average sensitivity for IgA alone exceeding 80% from week 3 onwards. Data for the combination of IgA with IgG were limited suggesting increase in sensitivity over time.
Average specificities were consistently high and precise, especially for pre‐pandemic samples which provide the least biased estimates of specificity. Average specificities were between 98.6% (for IgM) and 99.8% (for total antibodies) with 95% CIs spanning between 0.3 and 1.1 percentage points. All reference groups, except those including samples from those suspected of COVID‐19 and those based exclusively on samples deliberately selected for cross‐reactivity or confounder panels (which reflect analytical rather than clinical specificity), were broadly consistent with the specificity estimates for pre‐pandemic samples (differences in specificity were less than 1%). There is some evidence of greater heterogeneity in specificities from samples other than pre‐pandemic sources.
Some differences were noted by test technology, however, heterogeneity in study results, timing of sample collection, and smaller sample numbers in some groups complicate interpretation of results. For IgG assays, both types of laboratory‐based test appeared to be more sensitive on average than rapid tests: CLIA methods were marginally more sensitive (92.4%, 95% CI 90.6 to 93.9) than ELISA (89.4%, 95% CI 87.0 to 91.3) or lateral flow assays (86.9%, 95% CI 84.4 to 89.1) (P = 0.0008). Similar patterns were observed for IgM and combination antibodies. Based on results for pre‐pandemic samples, CLIAs may also be the most specific test method, however differences were marginal.
For assays that included IgG as a target antibody (alone or combined with other antibodies), there was no clear evidence of differences in sensitivity according to the antigen used (N‐ or S‐) during the convalescent phase of infection, although a trend towards higher average sensitivity for S‐based assays targeting IgM (average sensitivity 78%) compared to assays using N‐ alone or both N‐ and S‐ antigens (average sensitivities around 65%) was suggested. It is possible that the antigen used has a stronger effect on sensitivity in the first weeks after onset, (the highest sensitivity for assays targeting IgM was observed for S‐based assays, and highest sensitivity for assays targeting IgG was observed for those using both N‐ and S‐protein as opposed to those using either N‐ or S‐protein alone), however, this requires confirmation by direct comparison. Because the studies included in this review were conducted before vaccines against COVID‐19 were available, we could not directly address test performance for vaccination‐induced antibodies. The apparent lack of consistent effect from antigen type on the sensitivity of IgG assays might suggest similar sensitivities for S‐based assays for detection of vaccination‐induced antibodies as for those resulting from natural infection, but this would need to be confirmed by a review of relevant studies. It is also possible that mutations to the viral genome after 2020 could affect the accuracy of antibody tests that were developed using the original SARS‐CoV‐2 variant unless the proteins used in these assays are updated by the manufacturers.
Investigations of test performance by brand showed considerable variation in sensitivity between tests, and variability in results between studies evaluating the same test. None of the test brands in our review fully meet UK MHRA target performance criteria for sensitivity or specificity (both should be 98% or more, established in at least 200 samples). Using a modified version of the performance criteria, we identified a small number of tests that were evaluated in 200 or more samples and for which the lower bound of the 95% CI for sensitivity was 90% or more: five tests targeting IgG alone, one targeting IgG or IgM combined, and four total antibody assays. Larger numbers of test brands met the MHRA minimum criteria for specificity, however: 16 for IgG, five for IgG or IgM, and seven for total antibodies. Only seven antibody tests met these modified criteria for both sensitivity and specificity: two CLIAs for IgG alone (Abbott Architect and Shenzhen YHLO iFlash), one LFA targeting IgG or IgM combined (SureScreen Diagnostics rapid test), and three CLIAs and one ELISA targeting total antibodies (Ortho Clinical Diagnostics VITROS Total assay, Roche Elecsys, Siemens Atellica Total‐Ab assay, and the Beijing Wantai ELISA Total‐Ab assay).
A limited number of evaluations investigated antibody assays using samples from asymptomatic participants; however, small sample sizes limit interpretation of results. A similar effect from time after diagnosis was observed, with lower sensitivity for IgG assays within two weeks of a positive RT‐PCR result (49.8%; 95% CI 25.7 to 73.9; 208 samples), increasing to 78.2% (95% CI 61.5 to 88.9; 111 samples) 14 or more days after positive PCR.
Strengths and weaknesses of the review
Our review used a broad search screening all articles concerning COVID‐19. We undertook all screening and eligibility assessments, QUADAS‐2 assessments (Whiting 2011), and data extraction of study findings independently and in duplicate. Whilst we thus have reasonable confidence in the completeness and accuracy of the findings up until the search date, should errors be noted please inform us at coviddta@contacts.bham.ac.uk.
We have identified two main weaknesses in our review. Firstly, while we have tried to address the question of identification of current as opposed to prior SARS‐CoV‐2 infection by using time since onset of symptoms, studies that reported results beyond the first two weeks of infection often did not report whether participants' symptoms had resolved (and thus they were in a convalescent state) when serology samples were taken. Where data were reported by week after onset, we were therefore frequently unable to clearly distinguish between studies that evaluated the accuracy of antibody tests to identify current infection from past infection. Similarly, our definition of convalescent phase (or prior) infection was based on either 21 or more days after symptom onset or 14 or more days after a positive RT‐PCR result, and we were not able to consider later definitions of prior infection (e.g. two, three or four months after onset of symptoms or infection). It is also important to note that many studies did not report the maximum time after onset of infection; the longest time from onset to sampling that was reported was approximately 100 days (e.g. Butterfield 2021 [A]; Flower 2020 [A]; Gudbjartsson 2020 [A]).
The second main weakness of this review is the length of time elapsed between the last search (September 2020) and the publication of the review. It is not possible for us to quantify the number of eligible studies that have been published during the interim period. Nevertheless, we have conducted a scoping search to map available systematic reviews on the same topic. We identified a total of 17 reviews (two available only as preprint), five of which either have the same search cut‐off date as this review (De Carvalho 2021; Macedo 2022) or a more recent search cut‐off (Chua 2021; Gracienta 2022; Makoah 2022), the most recent being April 2021 (Makoah 2022). The number of studies included in the five reviews ranged from 10 (De Carvalho 2021) to 58 (Makoah 2022), and, where reported, the total number of samples included ranged from 2824 (De Carvalho 2021) to 13,650 (Macedo 2022). All five studies had narrower review questions, e.g. evaluating only LFAs (Gracienta 2022), only assays authorised in the review authors’ country (Chua 2021) or restricting inclusion to cohort studies only (De Carvalho 2021). Only two reviews considered the effect of time from onset of symptoms on sensitivity: Makoah 2022 considering only week one after onset, and Macedo 2022 considering weeks one, two and three onwards, but only for the combination of IgG or IgM. In summary, although there are other reviews with more recent search cut‐offs than our review and that include more recently published studies, we have not been able to identify any other review that provides such a comprehensive oversight of the effect of time on the sensitivity of serological assays for detection of natural infection with SARS‐CoV‐2.
Additional flaws reflect weaknesses in the primary studies and their reporting. Many studies omitted descriptions of sample recruitment, and key aspects of study design and execution. Studies frequently did not differentiate between ‘participants’ and ‘samples’, and we have therefore had to treat studies that describe their data as being based on 'samples' as if the samples were individual patients. Our separation of sensitivity data into distinct time periods after onset of symptoms should have minimised any effect from individual patients contributing multiple samples to each time slot. Quality assessment and data extraction were frequently hindered by poor quality reporting, particularly in regard to participant recruitment and application of the index test. Greater adherence to the STARD reporting guideline for diagnostic accuracy studies (Bossuyt 2015) and use of STARD participant flow diagrams is needed.
For this iteration of the review, we identified more studies with direct comparisons of tests (102 studies with two or more tests evaluated); however, there were still not enough test comparisons in common across studies to allow us to make meaningful direct comparisons of test brands. We have instead relied on indirect and informal comparison between test brands, identifying assays with the best performance from those with at least 200 samples and highlighting studies with direct comparisons of tests in at least 100 samples. Although historically less common for DTA studies than for intervention studies, network meta‐analyses to compare the accuracy of tests are increasingly being conducted (Veroniki 2022), and are likely to provide the best approach to compare the sensitivity of different brands of antibody tests in relevant time periods after onset of symptoms. Such a review should consider using the recently published extension of QUADAS‐2 to evaluate the accuracy of comparative test accuracy studies (QUADAS‐C; Yang 2021).
Applicability of findings to the review question
In the background, we outlined five possible roles for antibody testing, two of which we did not further consider in this review (serial testing to assess immune response and selection of seronegative COVID‐19 patients for monoclonal antibody treatments). We here consider the evidence for the remaining three use cases:
Diagnosis of infection in patients presenting with symptoms of suspected COVID‐19, particularly where molecular testing had failed to detect the virus and if earlier antibody test results (soon after onset of symptoms) were negative. It is unclear how generalisable our results for weeks one to three after symptom onset are to people who present either in the community or in hospital settings with a negative RT‐PCR result but ongoing and potentially concerning symptoms. A large proportion of studies included in this review iteration collected data from patients in the acute phase of disease in hospital inpatient settings, with less than 15% clearly recruiting individuals from community or emergency care settings. As noted in the Index test(s) section, where COVID‐19 vaccination rates are high, antibody tests that can distinguish between antibodies to the N‐ and S‐ proteins would be needed to distinguish SARS‐CoV‐2 infection from vaccination induced antibodies. Because the studies included in this review were conducted before vaccines against COVID‐19 were available, we have not been able to assess test performance for vaccination‐induced antibodies compared to antibodies from infection.
To assist diagnosis when patients present with a multi‐system inflammatory syndrome or other post‐acute sequelae of COVID‐19, and no clear diagnosis of SARS‐COV‐2 infection in the immediately preceding weeks, including individuals with mild or no symptoms of COVID‐19 during the acute phase (current or previous infection). Our results are likely to be applicable to this use case scenario, with the caveat that individuals who were asymptomatic or had only mild symptoms at the onset of infection may not have mounted the same level of antibody response by week three or week four as the participants included in the studies in our review, many of whom were hospital inpatients.
In seroprevalence surveys to estimate the prevalence of detectable antibodies resulting from infection in a community at any given point in time. Our results for convalescent‐phase infection may be applicable to this use case, bearing in mind the caveats around how we have been able to define convalescent or ‘prior’ infection and the lack of very long‐term follow‐up in the studies included. Because IgG persists for the longest time after infection, results for IgG assays are likely to be the most relevant for this use case. We found some evidence to suggest that quantitative assays, especially CLIAs are more sensitive than rapid LFAs. Our heterogeneity investigations according to the antigen or protein used in the test kit suggested no obvious effect on IgG assay sensitivity during the convalescent period, i.e. assays using N‐ alone, S‐ alone or N‐ and S‐proteins had on average similarly sensitivities (around 90%). These results imply that as long as at least three weeks have passed since symptom onset, antibody tests have the potential to detect around 90% of those infected. With a maximum reported participant follow‐up of only around 100 days, we are not able to comment on the duration of time that this level of sensitivity is maintained after infection, nor could we directly address the accuracy of tests for detecting vaccination‐induced antibodies. The choice of test (or tests) for seroprevalence surveys and the specific antigens used in those tests will dictate whether or not previous infection can be differentiated from vaccination response, as per CDC guidelines (CDC 2021b). Sensitivity varies between test brands, however. Although we have not captured all available evaluations of all available test brands, the included direct comparisons of tests have shown variations in sensitivity of as much as 40.6 percentage points between 11 different LFAs (Rudolf 2020 [A]) or 46.6 percentage points between three laboratory‐based assays (Gudbjartsson 2020 [A]). Even restricting to comparison of assays using the same protein does not necessarily reduce the difference in sensitivity. High specificity of tests is essential in seroprevalence testing, which appears likely for many of the tests included in this review. The suitability of pre‐pandemic samples to establish specificity requires further discussion. We found specificity for IgG assays was on average one percentage point lower for tests evaluated in those where COVID‐19 was ruled out after initially being suspected (‘suspected COVID‐19’ group) compared to pre‐pandemic samples. This either reflects misclassification as not having SARS‐CoV‐2 infection, or a true lower specificity in those presenting with symptoms.
Illustration of predicted effect of antibody testing by current or prior SARS‐CoV‐2 infection
We illustrate our results for two different scenarios.
Firstly, for antibody testing used in a diagnostic context, we use IgG or IgM data in week three after onset (sensitivity 92.9%, 95% CI 91.0 to 94.4) and average specificity in pre‐pandemic samples (99.2%, 95% CI 98.5 to 99.5). We have computed predictive values, and the numbers of true positives, false positives, false negatives and true negatives in a hypothetical cohort of 1000 people at a COVID‐19 prevalence of 2% (a value that might reflect antibody testing used to diagnose COVID‐19 in people with symptoms but who have had a negative PCR test). In this scenario, the positive predictive value is estimated as 69.2% (95% CI 48.2, 85.7), the negative predictive value as 99.8% (95% CI 99.3 to 100). Of 1000 people undergoing testing, we would anticipate eight false positives (95% CI 5 to 15) and two false negatives (95% CI 2 to 3).
Secondly, in a higher prevalence setting, where we wanted to understand how many people in a community had previously been infected with SARS‐CoV‐2, we use results for IgG during the later phase of infection (average sensitivity 89.8%, 95% CI 88.5 to 90.9) and average specificity using pre‐pandemic samples (98.9%, 95% CI 98.6 to 99.1). In this scenario, the positive predictive value is estimated as 98.7% (95% CI 97.2 to 99.5), the negative predictive value as 90.6% (95% CI 87.9 to 93.0). Of 1000 people undergoing testing, we would anticipate six false positives (95% CI 5 to 7) and 51 false negatives (95% CI 46 to 58).
Predictions at alternative prevalences of infection are provided in Table 1.
Authors' conclusions
Implications for practice.
Diagnosis of acute suspected COVID‐19 in patients with negative RT‐PCR results (use cases 1 and 2)
Based on this analysis, in patients presenting with symptoms of acute suspected COVID‐19, antibody tests have no role on their own as the primary test to use in the diagnosis of COVID‐19 when patients present during the first week since onset of symptoms, as their sensitivity is too low.
However, antibody tests have an increasing likelihood of detecting immune response to the infection as time since onset of symptoms progresses. Some antibody tests, therefore, could be a useful diagnostic tool for those with ongoing symptoms of acute infection but in whom molecular‐ or antigen‐based tests have failed to detect the SARS‐COV‐2 virus, particularly if they also had negative serological results early in the course of infection. Antibody tests are also likely to be a useful diagnostic aid in those presenting with post‐acute sequelae of COVID‐19, who may have been asymptomatic or had only mild symptoms at the onset of infection. Much of the data that we have reflects detection of antibody response in hospitalised patients but may be generalisable to those who do not require hospital admission.
Assessment of previous SARS‐CoV‐2 infection (use cases 2 and 3)
The data analysed in the review suggest that antibody tests are likely to have a useful role for detecting previous SARS‐CoV‐2 infection, for example, for sero‐epidemiological purposes, if used during what we defined as the convalescent phase of infection (day 21 onwards). Again, this conclusion needs to be cautioned by the relatively poor study quality, the applicability of the study settings and lack of availability of COVID‐19 vaccination at the time the studies were conducted, the typically small sample sizes of individual studies and restricted number of tests that have undergone evaluation.
Implications for research.
Although further research into the accuracy of antibody tests for diagnosis of acute SARS‐CoV‐2 infection is unlikely to be necessary, there is preliminary evidence for superior accuracy of some assays that could warrant further investigation. Any such studies should include participants who experience mild symptoms, or who were asymptomatic at the time of testing, and should clearly disaggregate test sensitivity by time since onset of symptoms. There is a lack of data about antibody test accuracy in those suspected of MIS, however, there is evidence that the majority of patients with MIS have detectable IgG antibodies on presentation (Kumar 2021; Patel 2021).
In regard to detection of prior infection, much of our data is based on cross‐sectional studies with samples collected from day 21 after onset of symptoms or day 14 after a positive PCR onwards, rather than all samples being collected at a longer time after the acute infection period. Ideally, longitudinal studies that sample from the same patients at several time points over a lengthy period of time are needed to fully understand how time since onset of infection affects test performance, and the extent to which type of test (laboratory‐based versus rapid test) affects accuracy. There remains a need for tests intended for use for seroprevalence purposes to be properly validated in the population in which the test is intended to be used.
Any future study should adhere to the methodological standard for test accuracy studies, for example, as set out by Doust and colleagues (Doust 2021) or by the UK Royal Statistical Society Working Group on Diagnostic tests (RSS 2021), and should adhere to standard requirements for the reporting of a test accuracy study (Bossuyt 2015). Test performance should be evaluated in consecutive individuals who are recruited from a representative clinical population and with due consideration to optimal sample size to estimate both sensitivity and specificity, in order to reflect the likely performance of the tests in practice. Studies should ensure that the test is used as intended (i.e. in the right setting, on the right specimens, by the intended test operator (whether at‐home self‐collection or by healthcare workers). We encourage investigators to utilise blinding in their study designs, such that index tests are undertaken without knowledge of the reference standard diagnosis and, likewise, reference standards are determined without knowledge of the index test findings.
It is also important to have good data upon which to compare tests, the strongest comparisons being made by testing the same participants multiple times with different tests. Whilst it is possible for this to be undertaken in prospective studies, it is easier to undertake in laboratory‐based studies utilising serum banks, which will compromise the applicability of the absolute estimates of test accuracy but provide some information about comparability. Tests utilising novel technologies, such as protein microarrays, should be directly compared with the best performing alternative tests in order for relative performance to be put in context.
What's new
| Date | Event | Description |
|---|---|---|
| 1 September 2022 | New search has been performed | Review updated to include studies available up to 30 September 2020. This is the first update of the review. |
| 1 September 2022 | New citation required and conclusions have changed | This iteration of the review restricts study inclusion to evaluations of commercially produced tests and to those reporting sensitivities according to time after onset of infection, primarily defined as time from symptom onset. The number of test brands with available data has increased as has the amount of data by week after symptom onset (up to day 35). We have also been able to analyse data for those in the convalescent phase of infection (defined as 21 days or more after symptom onset, or 14 days or more after a positive PCR test) and for those reported as asymptomatic at the time of testing. |
History
Review first published: Issue 6, 2020
Acknowledgements
Members of the Cochrane COVID‐19 Diagnostic Test Accuracy Review Group include:
the project team (Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MMG, Spijker R, Hooft L, Van den Bruel A, McInnes MDF, Emperador D, Dittrich S, Cunningham J);
the systematic review teams for each review: Molecular, antigen, and antibody tests (Arevalo‐Rodriguez I, Buitrago DC, Ciapponi A, Domen J, Dretzke J, Mateos M, Nyaaba N, Sharma P, Taylor M, Taylor‐Phillips S, Van Wyk S, Fox T, Geppert J, Bigio J, Sulis G, Hettiarachchi D, Mathangasinghe Y, Weeratunga P, Wickramasinghe D, Buckley B, Bergman H, Probyn K, Sguassero Y, Henschke N, Villanueva G, Cogo E, Hamel C, Petkovic J)
signs and symptoms (Stuyf T, Domen J, Horn S)
routine laboratory markers (Yang B, Langendam M, Ochodo E, Guleid F, Holtman G, Verbakel J, Wang J, Stegeman I)
imaging tests (Salameh JP, McGrath TA, Van der Pol CB, Frank RA, Prager R, Hare SS, Dennie C, Jenniskens K, Korevaar DA, Cohen JF, Van de Wijgert J, Damen JAAG, Wang J);
Thanks to the wider team of systematic reviewers from the University of Birmingham, UK who assisted with title and abstract screening across the entire suite of reviews for the diagnosis of COVID‐19 prior to the publication of the first iteration of this review.
Cochrane Infectious Diseases supported the authors in the development of this review update. The following people conducted the editorial process for this review update:
Sign‐off Editor (final editorial decision): Michael Brown, Michigan State University College of Human Medicine, USA
Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Joey Kwong, Cochrane Central Editorial Service
Editorial Assistant (conducted editorial policy checks and supported editorial team): Lisa Wydrzynski, Cochrane Central Editorial Service
Copy Editor (copy‐editing and production): Anne Lethaby, Cochrane Production Service
Peer‐reviewers (provided comments and recommended an editorial decision): Jeannette Guarner, Emory University (clinical/content review); Jessica Watson, University of Bristol (clinical/content review); Patricia R Slev, Immunology Division, ARUP Laboratories; Department of Pathology, University of Utah School of Medicine, Salt Lake City, UT, USA (clinical/content review); Robert Walton, Cochrane UK (summary versions review); Cochrane Diagnostic Test Accuracy Reviews Editorial Team (methods review), Robin Featherstone, Cochrane Central Editorial Service (search review). One additional peer reviewer provided consumer review but chose not to be publicly acknowledged.
The editorial base of Cochrane Infectious Diseases is funded by UK aid from the UK Government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK Government’s official policies.
We would also like to thank all corresponding authors who provided additional information regarding their studies.
Tilly Fox is supported by the Research, Evidence and Development Initiative (READ‐It). READ‐It (project number 300342‐104) is funded by UK aid from the UK government; however, the views expressed do not necessarily reflect the UK government’s official policies. Jonathan Deeks is a UK National Institute for Health and Care Research (NIHR) Senior Investigator Emeritus. Yemisi Takwoingi is supported by a NIHR Postdoctoral Fellowship. Jonathan Deeks, Jacqueline Dinnes, Yemisi Takwoingi, and Clare Davenport are supported by the NIHR Birmingham Biomedical Research Centre. Sian Taylor‐Phillips is supported by an NIHR Career Development Fellowship. This paper presents independent research supported by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Appendices
Appendix 1. Summary of World Health Organization and Chinese National Health Commission Guidelines for the diagnosis of SARS‐CoV‐2
Table A: World Health Organization guidelines for the diagnosis of SARS‐CoV‐2a
Includes laboratory testing guidelines and global surveillance guidelines
| Date range (2020) | Definition of confirmed case | Definition of confirmed non‐case | Definition of suspect case | Definition of probable case | Role of serology in testing |
| 10‐30 January | 10‐30 January: no documentation to define at this time (before first date of global guidelines)
31 January onwards: a confirmed case is a person with laboratory confirmation of COVID‐19 infection, irrespective of clinical signs and symptoms. No prescribed test in laboratory guidelines, suggested tests from 10 January include broad coronavirus RT‐PCR (with sequencing of precise virus in test positives), whole genome sequencing, broad coronavirus serology on paired samples, microscopy, culture. (Lab 10 January) Four suggested tests from 17 January: broad coronavirus RT‐PCR (with sequencing of precise virus in test positives), NAAT for SARS‐CoV‐2 when it becomes available, whole genome sequencing, and broad coronavirus serology on paired samples States that once specific NAAT assays are developed and validated, confirmation will be based on specific detection of unique sequences of viral nucleic acid by RT‐PCR |
None stated | No definition of 'suspect case' at this time, but case definitions for surveillance are defined as a combination of symptoms and exposure, with more severe symptoms requiring less evidence for exposure | No definition at this time | Serological testing may be useful to confirm immunologic response to a pathogen from a specific viral group, e.g. coronavirus. Best results from serologic testing requires the collection of paired serum samples (in the acute and convalescent phase) from cases under investigation. |
| 31 January‐26 February | None stated | Suspect case defined as combination of symptoms and exposure, with more severe symptoms requiring less evidence for exposure | A suspect case with inconclusive laboratory results or is test‐positive using a pan‐coronavirus assay without laboratory evidence of other respiratory pathogens (global 31 January) | ||
| 27 February‐1 March | None stated | Suspect case defined as combination of symptoms and exposure, with more severe symptoms requiring less evidence for exposure, OR defined by symptoms requiring hospitalisation and an absence of alternative explanation | A suspected case with inconclusive laboratory results (global 27 February) | ||
| 2 March‐19 March | A person with laboratory confirmation of COVID‐19 infection, irrespective of clinical signs and symptoms (global 31 January, 27 February, 20 March)
Laboratory confirmation of cases by NAAT specific to SAR‐CoV‐2 such as real‐time reverse‐transcription polymerase chain reaction (rRT‐PCR) with confirmation by nucleic acid sequencing when necessary. The viral genes targeted so far include the N, E, S and RdRP genes. In areas with no known COVID‐19 virus circulation confirmation requires:
Discordant results should be resampled. In areas where COVID‐19 virus is widely spread, a simpler algorithm might be adopted (e.g. RT‐PCR of a single discriminatory target) |
One or more negative results do not rule out the possibility of COVID‐19 virus infection. | In cases where NAAT assays are negative and there is a strong epidemiological link to COVID‐19 infection, paired serum samples (in the acute and convalescent phase) could support diagnosis once validated serology tests are available. Serological assays will play an important role in research and surveillance but are not currently recommended for case detection. |
||
| 19 March‐present | Probable case A suspect case for whom testing for the COVID‐19 virus is inconclusive OR A suspect case for whom testing could not be performed for any reason | ||||
| NAAT: nucleic acids amplification test; RT‐PCR: reverse transcription polymerase chain reaction; Source: WHO 2020 . | |||||
aSource data from Laboratory testing of 2019 novel coronavirus (2019‐nCoV) in suspected human cases: interim guidance, World Health Organization (2020), 10 January, 17January, 2nd March, 19 March, 21st March, and Global surveillance for COVID‐19 caused by human infection with COVID‐19 virus, interim guidance, 31st January, 27 February, and 20 March
Table B: Summary of Chinese National Health Commission guidelines for diagnosis and treatment for novel coronavirus pneumonia (trial versions 1‐7)
| Dates in effect | Definition of confirmed case | Definition of confirmed non‐case | Definition of suspect case | Role of serology in testing |
| 16‐17 January 2020 (version 1) | Cases (not confirmed cases) defined as virus genome highly homologous to coronaviruses | Not defined | Observation cases: defined as combination of exposure in Wuhan and symptoms focused on pneumonia, leukopenia and lack of improvement | No role |
| 18 January‐2 March (versions 2, 3, 4, 5, 5 revised, and 6) | Suspect cases with either
|
Suspect cases can be ruled out after 2 consecutive negative respiratory tract nucleic acid tests taken at least 24‐hours apart. | Suspect cases: combination of exposure (such as residence in/travel to Wuhan or exposure to a confirmed case within 14 days of onset) AND clinical features (such as symptoms: fever, respiratory symptoms, and tests: chest imaging, white blood cell and lymphocyte count). Exact definition varies slightly with version. | No role |
| 3 March‐present (version 7) | Suspect cases with either
|
Suspect cases can be ruled out after 2 negative NAATs, taken at least 24‐hours apart, and the NCP virus‐specific IgM and IgG are negative after 7 days from onset | Suspect cases: combination of exposure (such as residence in/travel to Wuhan or exposure to a confirmed case within 14 days of onset) AND clinical features (such as symptoms: fever, respiratory symptoms, and tests: chest imaging, white blood cell and lymphocyte count) | Part of definition of cases and confirmed non‐cases |
| NAAT: nucleic acids amplification test; NCP: novel coronavirus pneumonia; RT‐PCR: reverse transcription polymerase chain reaction; Source: CDC China 2020 . | ||||
Appendix 2. COVID‐19 Open Access Project living evidence database from the University of Bern
The following information is taken from the University of Bern website (see: ispmbern.github.io/covid-19/living-review/collectingdata.html).
The register is updated daily and CSV file downloads are made available.
MEDLINE
29 April to 29 October 2020
("coronavirus"[MH] OR "coronavirus infections"[MH] OR "coronavirus"[TW] OR "corona virus"[TW] OR "HCoV"[TW] OR "nCov"[TW] OR "covid"[TW] OR "covid19"[TW] OR "Severe Acute Respiratory Syndrome Coronavirus 2"[TW] OR "SARS‐CoV2"[TW] OR "SARS‐CoV 2"[TW] OR "SARS Coronavirus 2"[TW] OR "MERS‐CoV"[TW]) AND (2019/1/1:3000[PDAT])
26 March to 28 April 2020
(\"Wuhan coronavirus\" [Supplementary Concept] OR \"COVID‐19\" OR \"2019 ncov\"[tiab] OR ((\"novel coronavirus\"[tiab] OR \"new coronavirus\"[tiab]) AND (wuhan[tiab] OR 2019[tiab])) OR 2019‐nCoV[All Fields] OR (wuhan[tiab] AND coronavirus[tiab])))))
1 January to 25 March 2020
("Wuhan coronavirus" [Supplementary Concept] OR "COVID‐19" OR "2019 ncov"[tiab] OR (("novel coronavirus"[tiab] OR "new coronavirus"[tiab]) AND (wuhan[tiab] OR 2019[tiab])) OR 2019‐nCoV[All Fields] OR (wuhan[tiab] AND coronavirus[tiab])))))
Embase
01 May to 29 October 2020
(SARS coronavirus/ or middle east respiratory syndrome/ or severe acute respiratory syndrome/ or (coronavirus* or corona virus* or HCoV* or ncov* or covid or covid19 or sars‐cov* or sarscov* or Sars‐coronavirus* or Severe Acute Respiratory Syndrome Coronavirus*).mp.) and 20191201:20301231.(dc)
26 March to 30 April 2020
(nCoV or 2019‐nCoV or ((new or novel or wuhan) adj3 coronavirus) or covid19 or covid‐19 or SARS‐CoV‐2).mp
1 January to 25 March 2020
ncov OR (wuhan AND corona) OR COVID
bioRxiv and medRxiv pre‐print servers
1 April 2020 to 08 March 2021
From 1 April 2020 onwards the curated BioRxiv/MedRxiv dataset was retrieved (connect.medrxiv.org/relate/content/181)
26 to 29 March 2020
ncov or corona or wuhan or COVID or SARS‐CoV‐2
1 January 2020 to 25 March 2020
ncov or corona or wuhan or COVID
Appendix 3. Search classification model
From 25 May to 30 September 2020: generic classifier covering all types of test
We needed a more efficient approach to keep up with the rapidly increasing volume of COVID‐19 literature. A classification model for COVID‐19 diagnostic studies was built with the model building function within Eppi Reviewer, which uses the standard SGCClassifier in Scikit‐learn on word trigrams. As outputs, new documents receive a percentage (from the predict_proba function) where scores close to 100 indicate a high probability of belonging to the class ‘relevant document’ and scores close to 0 indicate a low probability of belonging to the class ‘relevant document’. We used three iterations of manual screening (title and abstract screening, followed by full text review) to build and test classifiers. The final included studies were used as relevant documents, while the remainder of the COVID‐19 studies were used as irrelevant documents. The classifier was trained on the first round of selected articles, and tested and retrained on the second round of selected articles. Testing on the second round of selected articles revealed poor positive predictive value but 100% sensitivity at a cut‐off of 10. The poor positive predictive value is mainly due to the broad scope of our topic (all diagnostic studies in COVID‐19), poor reporting in abstracts, and a small set of included documents. The model was retrained using the articles selected of the second and third rounds of screening, which added a considerable number of additional documents. This led to a large increase in positive predictive value, at the cost of a lower sensitivity, which led us to reduce the cut‐off to 5. The largest proportion of documents had a score between 0‐5. This set did not contain any of the relevant documents. This version of the classifier with a cut‐off 5 was used in subsequent rounds and accounted for approximately 80% of the screening burden.
Appendix 4. Data extraction items
| Patient sampling items | Patient characteristics and setting items | Index test items | Reference standard items | Flow and timing items | Notes items |
| A0 Test type (antibody/antigen etc) | COVID patients (or all patients if single group study) | ||||
| A1 Purpose | B1 Setting | D1.1 Test name | E1 Reference standard for cases including threshold | F1 What was the time interval between index and reference tests? | G1: Funding |
| A2 Design (and description of groups labelled [1] [2] …) | B2 Location (include name of institution if available) | D1.2 Manufacturer | E2 Samples used | F2 Did all patients receive the same reference standard? | G2: Publication status |
| A3 Recruitment | B3 Country | D1.3 Antibody targets | E3 Timing of reference standard (preferably since symptom onset only, if not from a different time points) | F3 Missing data | G3: Source (preprint or journal name) |
| A4 Were cases recruited prospectively or retrospectively? | B4 Dates | D1.4 Antigens used | E4 Was it blind to index test? | F4 Uninterpretable results | G4: Study author COI (including any manufacturer affiliations) |
| A5 Sample size (virus/COVID cases) | B5 Symptoms and severity | D1.5 Point‐of‐care or laboratory (is the test designed to be used at point‐of‐care or in laboratory, and was it used as point‐of‐care or in laboratory)? | E5 Did it incorporate index test? | F5 Indeterminate results | G5 Comment |
| A6 Inclusion and exclusion criteria | B6 Demographics | D1.6 Test method | F6 Samples or patients | ||
| A7 Comment | B7 Exposure history | D1.7 When were samples taken (preferably since symptom onset only, if not from a different time points)? | E6 Reference standard for non‐cases | F7 Comment | |
| B8 Comment | D1.8 Samples used | E7 Samples used | |||
| Non‐COVID patients (if additional groups) | D1.9 Who applied the test | E8 Timing of reference standard (preferably since symptom onset only, if not from a different time points) | |||
| C1.1 Group name | D1.10 How was positive defined? | E9 Was it blind to index test? | |||
| C1.2 Source and time | D1.11 Blinded to reference standard | E10 Did it incorporate index test? | |||
| C1.3 Characteristics | D1.12 Threshold predefined | E11 Comment | |||
| C2.1 Group name | D1.13 Comment | ||||
| C2.2 Source and time | |||||
| C2.3 Characteristics | |||||
| C4 Comment |
Appendix 5. Criteria for assessment of study quality (QUADAS‐2)
| DOMAIN: PARTICIPANT SELECTION | |
| Was a consecutive or random sample of patients enrolled? | This will be similar for all index tests, target conditions, and populations. YES: if a study explicitly stated that all participants within a certain time frame were included; that this was done consecutively; or that a random selection was done. NO: if it was clear that a different selection procedure was employed; for example, selection based on clinician's preference, or based on institutions. UNCLEAR: if the selection procedure was not clear or not reported. |
| Was a case‐control design avoided? | This will be similar for all index tests, target conditions, and populations. YES: if a study explicitly stated that all participants came from the same group of (suspected) patients. NO: if it was clear that a different selection procedure was employed for the participants depending on their COVID‐19 status or SARS‐CoV‐2 infection status; or if only participants with SARS‐CoV‐2 infection were included UNCLEAR: if the selection procedure was not clear or not reported. |
| Did the study avoid inappropriate exclusions? | Studies may have excluded patients, or selected patients in such a way that they avoided including those who were difficult to diagnose or likely to be borderline. Although the inclusion and exclusion criteria will be different for the different index tests, inappropriate exclusions and inclusions will be similar for all index tests: for example, only elderly patients excluded, or children (as sampling may be more difficult). This needs to be addressed on a case‐to‐case basis. YES: if a high proportion of eligible patients was included without clear selection. NO: if a high proportion of eligible patients was excluded without providing a reason; if, in a retrospective study, participants without index test or reference standard results were excluded. UNCLEAR: if the exclusion criteria were not reported. |
| Did the study avoid inappropriate inclusions? | Some laboratory studies may have intentionally included groups of patients in whom the accuracy was likely to differ, such as those with particularly low or high viral loads, or who had other diseases, such that the sample over‐represented these groups. This needs to be addressed on a case‐to‐case basis. Artificial spiked samples are a clear example. YES: if samples included were likely to be representative of the spectrum of disease. NO: if the study oversampled patients with particular characteristics likely to affect estimates of accuracy. UNCLEAR: if the exclusion criteria were not reported. |
| Could the selection of patients have introduced bias? | HIGH: if one or more signalling questions were answered with NO, as any deviation from the selection process may lead to bias. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Is there concern that the included participant s do not match the review question? | HIGH: for two‐group studies that included healthy or other disease controls, whether pre‐pandemic or contemporaneous; studies that only included people with COVID‐19 (whether reverse transcription polymerase chain reaction (RT‐PCR)‐confirmed only, participants meeting official guideline criteria). LOW: for single‐group studies recruiting participants with signs and symptoms of COVID‐19; or for two‐group studies where control groups suspected of COVID‐19 were separately recruited. UNCLEAR: if a description about the participants was lacking. |
| DOMAIN: INDEX TESTS | |
| Were the index test results interpreted without knowledge of the results of the reference standard? | YES: if blinding was explicitly stated or index test was recorded before the results from the reference standard were available. NO: if it was explicitly stated that the index test results were interpreted with knowledge of the results of the reference standard. UNCLEAR: if blinding was unclearly reported. |
| If a threshold was used, was it prespecified? | YES: if the test was dichotomous by nature, or if the threshold was stated in the methods section, or if study authors stated that the threshold as recommended by the manufacturer was used. NO: if a receiver‐operating‐characteristic curve was drawn or multiple threshold reported in the results section; and the final result was based on one of these thresholds. UNCLEAR: if threshold selection was not clearly reported. |
| Could the conduct or interpretation of the index test have introduced bias? | HIGH: if one or more signalling questions were answered with NO, as even in a laboratory situation knowledge of the reference standard may lead to bias. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Is there concern that the index test, its conduct, or interpretation differ from the review question? | For evaluations of laboratory‐based tests, HIGH: if tests were built inhouse, or if commercially available tests using SARS‐Cov antigens instead of SARS‐CoV‐2‐specific antigens. LOW: most other laboratory evaluations. UNCLEAR: name of the test was withheld. For evaluations of lateral flow assays, HIGH: if tests were built inhouse; if only serum or plasma instead of fingerprick or whole blood samples were used; if test evaluated in laboratory settings rather than at the point of care. LOW: commercially available tests, using whole blood or fingerprick samples, and that were conducted in the intended setting for the test (i.e. point‐of‐care). UNCLEAR: name of the test was withheld; mixed sample types; or did not report the evaluation setting. |
| DOMAIN: REFERENCE STANDARD | |
| Is the reference standard likely to correctly classify the target condition? | We will define acceptable reference standards using a consensus process once the list of reference standards that have been used has been obtained from the eligible studies. For COVID‐19 cases: YES: RT‐PCR; confirmed or suspected case using official criteria (WHO, CDC) or a clearly set out combination of signs/symptoms/exposure. NO: RT‐PCR not used, or if inadequate combination of clinical characteristics used in PCR negatives, e.g. computed tomography alone. UNCLEAR: if definition of COVID‐19 was not reported. For absence of COVID‐19: YES: if at least 2 negative RT‐PCR results reported if suspected COVID‐19 based on signs/symptoms; single‐negative RT‐PCR test for asymptomatic contacts or contemporaneous controls with no clinical suspicion of COVID‐19; only pre‐pandemic sources of control samples used. NO: single RT‐PCR or number of negative RT‐PCRs not reported for COVID‐19 suspects; no RT‐PCR reported (untested) for asymptomatic contacts or contemporaneous controls. UNCLEAR: if timing of control samples (pre‐pandemic or contemporaneous) was not reported. |
| Were the reference standard results interpreted without knowledge of the results of the index test? | YES: if it was explicitly stated that the reference standard results were interpreted without knowledge of the results of the index test, or if the result of the index test was obtained after the reference standard. NO: if it was explicitly stated that the reference standard results were interpreted with knowledge of the results of the index test or if the index test was used to make the final diagnosis. UNCLEAR: if blinding was unclearly reported. |
| Did the definition of the reference standard incorporate results from the index test(s)? | YES: if results from the index test were a component of the reference standard definition. NO: if the reference standard did not incorporate the index standard test. UNCLEAR: if it was unclear whether the results of the index test formed part of the reference standard. |
| Could the conduct or interpretation of the reference standard have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
| Is there concern that the target condition as defined by the reference standard does not match the review question? | Applicability was judged primarily on the definition of disease‐positive. HIGH: if RT‐PCR alone used to define cases. LOW: if clinical criteria, including RT‐PCR, were used to define cases, regardless of whether official criteria were used, as long as the criteria were explicitly described. UNCLEAR: if definition of COVID‐19 cases was not provided, including if some clinically diagnosed cases were included but the clinical criteria used were not described. |
| DOMAIN: FLOW AND TIMING | |
| Did all participants receive the same reference standard? | YES: if all participants received the same reference standard (clearly no differential verification). NO: if (part of) the index test‐positives or index test‐negatives received a different reference standard. UNCLEAR: if it was not reported. |
| Were all participants included in the analysis? | YES: if it is clear that all eligible participants were included in the analyses. NO: if after the inclusion/exclusion process, participants were removed from the analyses for different reasons: no reference standard done, no index test done, intermediate results of both index test or reference standard, indeterminate results of both index test or reference standard, samples unusable. UNCLEAR: if it is not possible to determine whether all participants were included (e.g. from a STARD style participant flow diagram) |
| Did all participants receive a reference standard? | YES: if all participants received a reference standard (clearly no partial verification). NO: if only (part of) the index test‐positives or index test‐negatives received the complete reference standard. UNCLEAR: if it was not reported. |
| Were results presented per participant? | YES: if either only one sample per participant (regardless of disaggregation of results over time), or if multiple samples per participant but results are disaggregated by time period (at least week by week). NO: if multiple samples per participant and results are not disaggregated by time period. UNCLEAR: if it is not possible to tell whether results presented are per participant or per sample. |
| Could the participant flow have introduced bias? | HIGH: if one or more signalling questions were answered with NO. LOW: if all signalling questions were answered with YES. UNCLEAR: all other instances. |
CDC: Centers for Disease ControlICU: intensive care unit; RT‐PCR: real‐time polymerase chain reaction; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; STARD: standards for reporting of diagnostic accuracy studies ;WHO: World Health Organization
CDC: Centers for Disease Control ICU: intensive care unit RT‐PCR: real‐time polymerase chain reaction SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2 STARD: standards for reporting of diagnostic accuracy studies WHO: World Health Organization
Appendix 6. Summary details of study design and participants
|
Study (source) N samples (cases) Setting (for cases) Country (recruitment dates) |
Design Inclusion criteria |
Reference standard (disease‐positive) | Reference standard (disease‐negative) |
|
Adams 2020 (Preprint); 834 (270) Unclear; UK (not stated; conducted subsequent to 26 March 2020) |
Two‐group design with sensitivity and specificity [1] Confirmed (RT‐PCR‐positive) COVID‐19 (n = 270 samples from 124 patients) [2] Pre‐pandemic bio‐banked serum samples from three sources (from 2018 to pre‐December 2019) (n = 564 samples). | RT‐PCR; commercial assay Samples: Respiratory samples |
Pre‐pandemic controls |
|
Alvim 2020 (Pre‐print); 437 (437) Unclear; Brazil (Not stated) |
Single‐group analysis for sensitivity. [1] Confirmed COVID patients (437 samples); all symptomatic with >= 2 of: loss of taste or smell, fever, shortness of breath, diarrhoea, headache, extreme tiredness, dry cough, sore throat, runny or stuffy nose, or muscle aches | RT‐PCR, threshold not stated Samples: Unclear |
NA |
|
Andrey 2020a [A] (Published paper); 57 (12) Hospital inpatient; Switzerland (April 2020) |
Two‐group study to determine sensitivity and specificity ‐ [1] 46 RT‐PCR–confirmed COVID‐19 cases hospitalised at the University Hospitals of Geneva [2] 45 unmatched control blood samples from asymptomatic donors without known exposure to SARS‐CoV‐2 | RT‐PCR ‐ multiple assays used Samples: Nasopharyngeal secretions in 45/46; On one occasion, the RT‐PCR was carried out on a bronchial aspirate. |
No RT‐PCR testing; stated that this did not meet the institutional standard for testing Asymptomatic donors without known exposure to SARS‐CoV‐2 |
|
Andrey 2020b [A] (Published paper); 91 (41) Hospital inpatient; Switzerland (April 2020) |
Two‐group study to estimate sensitivity and specificity [1] RT‐PCR‐confirmed samples from hospitalised patients (n = 41) [2] unmatched asymptomatic donors (n = 50) | RT‐PCR ‐ multiple assays used. Samples: Not clearly stated |
Asymptomatic, untested |
|
Bartolini 2020 [A] (Pre‐print); 151 (151) community; Italy (3rd to 27th of April 2020) |
RT‐PCR‐positive asymptomatic/mildly symptomatic healthcare workers (n = 151) | SARS‐CoV‐2 RT‐PCR Samples: nasopharyngeal or oropharyngeal samples |
NA |
|
Beavis 2020 (Published paper); 168 (82) Hospital inpatient; USA (March to May 2020) |
Two‐group design to assess sensitivity and specificity: [1] COVID‐19 PCR+ve patients (n = 82) [2] COVID‐19 PCR‐ve patients (n = 86); Ambulatory and pre‐pandemic. | RT‐PCR Samples: Nasopharyngeal and nasal mid‐turbinate |
RT‐PCR or pre‐pandemic |
|
Bernasconi 2020 (Published paper); 243 (67) Hospital A&E; Switzerland (March‐April 2020) |
Three groups for two comparisons (COVID cases versus same time period controls or pre‐pandemic controls): [1] & [2] clinical suspicion of acute airway infection presenting to ED; COVID‐19 positive (n = 67); COVID‐19 negative (n = 76) [3] COVID‐19‐negative historical pre‐pandemic controls (n = 100) | RT‐PCR ‐ commercial assay
Diagnosis of COVID‐19 was based on clinical, microbiological and radiological criteria according to inhouse, national and international recommendations and guidelines. Samples: nasopharyngeal swab samples (transportation medium ESwab, Copan Italia, Brescia, Italy) or nasopharyngeal fluid |
[2] COVID‐19‐negative patients, RT‐PCR (Seegene Inc., Seoul, Republic of Korea) [3] pre‐pandemic controls; samples collected between May and October 2018 |
|
Bettencourt 2020 (Published letter); 66 (66) Hospital inpatient; Portugal (Not stated) |
Single‐group study to estimate sensitivity [1] Confirmed COVID cases (66 patients); diagnosis was based on clinical evaluation and positive RT‐PCR SARS Cov‐2 identification. Patients in the recovery phase of infection, after the resolution of symptoms and a negative result for the first RT‐PCR test | RT‐PCR, threshold not stated Samples: Not stated |
NA |
|
Bond 2020 (Published paper); 1400 (91) Hospital in and outpatients; Australia (first semester of 2020.) |
Multi‐group study estimating both sensitivity and specificity. [1A] Symptomatic RT‐PCR‐confirmed COVID‐19 cases (n = 91 patients) [1B] Symptomatic COVID‐19‐negative on single RT‐PCR (n = 1217 patients) [2] Pre‐pandemic controls obtained in 2018 (n = 56 patients) Group 1B only used to assess specificity for one test | Group [1A]: commercial assay followed by an unspecified confirmatory test at the state reference lab Samples: Upper and/or lower respiratory tract specimens |
Group [1B]: Single negative RT‐PCR Group [3]: no testing, pre‐pandemic sera |
|
Boukli 2020 [A] (Published letter); 217 (117) Mixed; included hospital inpatients; France (Not stated) |
Multi‐group study to estimate sensitivity and specificity, including: [1] PCR‐positive Covid‐19 patients in intensive care unit (76 samples from 49 patients) [2] PCR‐positive Covid‐19 patients, described as 'unselected' (68 samples from 68 patients) [3] 'unselected' pre‐pandemic samples (n = 40) [4] pre‐pandemic samples from cases with other infection (n = 60) Results were presented for group [1] and [2] combined, and separately for group [3] and [4]. Reported results suggest that not all samples were tested with both assays. | RT‐PCR; no further details Samples: Not stated |
RT‐PCR‐negative Pre‐pandemic samples used |
|
Brochot 2020 [A] (Published paper); 168 (78) Mixed; Hospital in and outpatients; France (Not stated) |
Multi‐group study estimating sensitivity and specificity including: [1] patients hospitalised for COVID‐19 (n = 20); [2] non‐hospitalised patients but PCR‐confirmed with SARS‐CoV‐2 (n = 58); [3] patients participating in screening campaigns, also described as outpatients with no history of SARS‐CoV‐2 infection (n = 62); [4] and samples from patients with a history of other seasonal coronavirus infections (n = 28). Study focused mainly on agreement between evaluated assays; data could be extracted for samples with PCR+ result (from group [1] and group [2]) at two time points and for non‐COVID‐19 cases (group [3]) | PCR; no further details Samples: Nasopharyngeal swab |
PCR for group 3. Pre‐pandemec for group 4 |
|
Bryan 2020a (Pre‐print); 41 (41) Mixed; Hospital in and outpatients; USA (Not stated) |
1‐group study to estimate sensitivity for diagnosis of acute Covid‐19 [1] PCR‐positive Covid cases (245) | qRT‐PCR Samples: Nasopharyngeal swabs |
NA |
|
Bundschuh 2020 (Published paper); 560 (104) Hospital inpatient; Austria (15th of March 2020 to 10th of April 2020) |
Three‐group study: [1] RT‐PCR‐positive COVID‐19 patients admitted for treatment at two tertiary hospitals (n = 64) [2] Healthy blood donors (pre‐pandemic, n = 200) [3] Medical intensive care patients (pre‐pandemic, n = 256) | RT‐PCR Samples: respiratory specimens |
[2] and [3] Pre‐pandemic |
|
Butterfield 2021 [A] (Published paper); 164 (42) Unclear; Jamaica (Not stated) |
Multi‐group study estimating sensitivity and specificity: [1] SARS‐CoV‐2 real‐time PCR‐positive patients (42 samples from 37 patients) [2] Pre‐pandemic samples from patients with viral infections (n = 102) or [3] attending routine antenatal testing (n = 20) [Further group of 17 PCR+ patients provided whole blood and serum samples to allow comparison of Trillium IgG/IgM rapid test results by sample type; not further considered due to small sample numbers] | Real time PCR using Charite Berlin protocol (Corman 2020) Samples: Not stated |
Pre‐pandemic |
|
Candel 2020 (Published paper); 35 (35) Hospital inpatients; Spain (April 27th to April 29th, 2020) |
Two‐group study: [1] randomly selected SARS‐CoV‐2 RT‐PCR‐confirmed patients (n = 35) [2] healthy volunteers with no history of COVID‐19 symptoms and negative SARS‐CoV‐2 RT‐PCR (n = 5) Group [2] excluded from review as < 25 controls | SARS‐CoV‐2‐positive RT‐PCR for pharyngeal swabs; threshold not stated Samples: pharyngeal swabs |
NA |
|
Carozzi 2020 [A] (Pre‐print); 430 (135) Hospital inpatients; Italy ([1a] Not stated) |
Multi‐group study to assess sensitivity and specificity [1] Covid‐positive [1a] Clinical hospitalised COVID‐19 cases (n = 135) [1b] PCR +ve healthcare workers (n = 33) [2] Non‐Covid [2a] Pre‐pandemic (n = 295) [2b] Suspected healthy healthcare workers (n = 17,065) Group [1b] and [2b] were not eligible for our review as [1b] was pre‐selected and [2b] had no reference standard. | RT‐real time PCR Samples: Not stated for [1a] |
[2a] Pre‐pandemic |
|
Carta 2020 (Published paper); 65 (54) Community; Italy (PCR March 29 to April 22, 2020; follow‐up for 2 months after) |
Single‐group study to assess sensitivity and specificity (n = 65) All residents (symptomatic and asymptomatic) of a long‐term care facility | RT‐PCR; commercial assay Samples: Oropharyngeal and nasopharyngeal swabs |
As for cases (single‐group study) |
|
Case 2020 [A] (Published paper); 40 (40) Unclear; USA (Not stated) |
1‐group study to estimate sensitivity for diagnosis of acute Covid‐19 [1] PCR‐confirmed Covid‐19 patients (20 patients, 42 samples) | PCR Samples: Not stated |
NA |
|
Caturegli 2020 (Published paper); 628 (60) Hospital inpatients; United States (11 Mar to 12 Apr, 2020) |
Multi‐group study estimating both sensitivity and specificity. Group [1] and [2] were hospitalised adults investigated for COVID‐19 selected from a cohort of patients with at least one NAT result (n = 11,066) and with available residual serum samples (n = NR): [1] COVID‐19 cases, including PCR‐confirmed (n = 50, including 38 with single positive result) and clinically defined PCR‐negative based on medical record review (n = 10) [2]: Symptomatic patients with negative PCR (n = 55, including 43 with single negative result) [3] Laboratory controls including healthy lab employees and patients with polyclonal activation of antibody response (n = 513; 325 pre‐pandemic and 188 contemporaneous). [A fourth cohort of 524 individuals with NAT results but no serology result was reported but did not provide accuracy data] | RT‐PCR test AND clinical evaluation based on clinical record review (risk factors, signs and symptoms on presentation, radiologic findings, comorbidities, smoking and alcohol history, BMI, reason for repeated NAAT testing (as applicable), and complications during hospital stay. No formalised combination of findings to indicate COVID‐19 was reported. Samples: Nasopharyngeal swabs |
Group [2]: RT‐PCR (as above) Group [3]: pre‐pandemic and contemporaneous (no testing) |
|
Cervia 2020 (Pre‐print); 56 (56) Mixed; Switzerland (Not stated) |
Study reported two cohorts, only one of which was eligible for this review. [1] Single‐group study to estimate sensitivity in patients with RT‐qPCR‐confirmed SARS‐CoV‐2 infection (n = 56) [(2) Second cohort of SARS‐CoV‐2‐exposed healthcare workers (HCW) with or without symptoms, RT‐qPCR positive or negative (n = 109) excluded as inhouse (ineligible) assay used] (Also mentioned evaluation in serum samples collected prior to the COVID‐19 pandemic but results were not reported) | RT‐qPCR; multiple assays Samples: NP |
0 |
|
Chan 2020a (Published paper); 208 (78) Hospital inpatients; USA (Not stated) |
Multi‐group study to assess sensitivity and specificity [1] Covid subjects (n = 144) [1a] Admitted PCR‐positive samples (n = 78) for clinical performance study [1b] Archived PCR‐positive samples (n = 66) for method comparison study [2] Non Covid subjects (n = 130) [2a] non‐SARS‐CoV‐2 respiratory viral samples (n = 25) [2b] Other viral positive samples (n = 52) [2c] Pre‐pandemic samples (n = 53) [1b] excluded from review as no time pso or post‐PCR+ reported | RT‐PCR, threshold not stated Samples: Not stated |
[2a] [2b] Not stated, possibly pre‐pandemic [2c] Pre‐pandemic |
|
Charlton 2020 [A] (Published paper); 158 (46) Mixed; Hospital inpatients and outpatients; Alberta, Canada (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (28 patients, 46 samples) [2] Pre‐pandemic non‐COVID (50 samples) [3] Cross‐reactivity non‐COVID samples [62 samples: pre‐pandemic (n = 15) and concurrent (n = 47)] | rRT‐PCR, threshold not reported Samples: Nasopharyngeal swab (27/28) or endotracheal aspirate (1/28) |
[2] Pre‐pandemic [3] Pre‐pandemic or inhouse rRT‐PCR on nasopharyngeal swab testing |
|
Charpentier 2020 [A] (Published paper); 208 (88) Mixed (in and outpatients); France (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (88 samples from 54 patients) [2] Pre‐pandemic non‐COVID‐samples (120 samples) [2a] Samples for testing as part of routine clinical care (n = 56) [2b] Serum samples corresponding to a cross‐reactivity panel (n = 64) [3] Healthcare workers who presented with clinical symptoms during the epidemic period for whom SARS‐CoV‐2 RT‐PCR was negative or not carried out (n = 54) Group [3] was excluded from the review as no test accuracy data are reported. | [1] RT‐PCR, threshold not stated Samples: Nasopharyngal samples |
[2] Pre‐pandemic samples (before November 2019) [3] RT‐PCR or no reference standard |
|
Chaudhuri 2020 [A] (Published paper); 563 (379) Mixed; India (from March 2020) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (368 patients with 379 samples); i) Suspected COVID‐19 patients enrolled at the time of RT‐PCR testing at the screening center and ii) RT‐PCR‐confirmed COVID‐19 positive patients admitted at one of the clinical sites [2] Pre‐pandemic non‐COVID samples from pregnant women enrolled in a pregnancy cohort. (n = 184) | RT‐PCR at an approved laboratory as per the National Testing Strategy of India; threshold not reported Samples: Not stated |
Pre‐pandemic |
|
Chen 2020 [A] (Published paper); 540 (346) Hospital inpatients; Taiwan (23 January 2020 to 31 May 2020) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (74 patients, n = 346 samples) [2] Non‐COVID samples (n = 194) including: [2a] Current patients with acute respiratory infection (n = 120) [2b] Current patients with presence of auto‐antibodies (n = 36) [2c] Pre‐pandemic samples with presence of antigens/antibodies (n = 38) | SARS‐CoV‐2 qRT‐PCR assay; If the result of the first sample was negative for SARS‐CoV‐2, an additional SARS‐CoV‐ 2 qRT‐PCR assay for another respiratory tract sample from the patient was performed to minimise the risk of false‐negative results using the qRT‐PCR assay. Samples: Respiratory tract specimens |
Current patients with acute respiratory infections: tested negative ≥ 2 times using SARS‐CoV‐2 qRT‐PCR Current patients with auto‐antibodies: not tested Pre‐pandemic samples |
|
Chew 2020 (Published paper); 340 (177) Hospital inpatients; Singapore (30th March 2020 to 15th May 2020) |
Two‐group study: [1] Symptomatic COVID‐19 patients selected on the basis of a positive SARS‐CoV‐2 rRT‐PCR from a respiratory sample (n = 177) [2] Negative controls were samples taken from patients prior to December 2019. These included patients with and without other positive serological tests (n = 163) | Two PCR assays were used during this time period (Fortitude, MirXES, Singapore, and cobas® SARS‐COV‐2, Roche Diagnostics, USA). No threshold reported Samples: respiratory samples |
None ‐ "negative samples collected prior to December 2019 were assumed to be negative as SARS‐CoV‐2 was first identified late in 2019". |
|
Chong 2021 (Published paper); 63 (63) Hospital inpatients; Japan (March and April 2020) |
Single‐group study to estimate sensitivity only [1] Confirmed COVID patients admitted to Kyushu University Hospital (63 samples from 18 patients) | Real‐time PCR assay performed by the Japanese Institute of Health according to the manual for the detection of pathogen 2019‐nCoV; threshold not stated Samples: nasal and pharyngeal swab specimens |
NA |
|
Clarke 2020 (Published letter); 121 (79) Dialysis patients; UK (April 27 and May 7, 2020) |
Single‐group study to estimate sensitivity and specificity
Routine screening of haemodialysis patients for the development of symptoms or a fever prior to each session; symptomatic were tested using RT‐PCR Excluded: [3] Concurrent, untested, asymptomatic patients (n = 235); high‐risk group without reference standard |
Rt‐PCR as per PHE guidelines using certification marked assays with primers directed to the nucleocapsid or RNA‐dependent RNA polymerase genes. Threshold not stated Samples: nasopharyngeal swab specimens |
RT‐PCR as for cases (single‐group study) |
|
Conklin 2020 [A] (Published paper); 372 (312) Mixed; hospital inpatients, plasma donors; [1] and [2] Maryland, USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients, convalescent and asymptomatic for at least 28 days. Human immunodeficiency virus (HIV) and hepatitis C virus (HCV) negative (n = 40) [2] Confirmed COVID samples, longitudinal testing (47 patients with 272 samples) [3] Pre‐pandemic non‐COVID challenge samples (60 patients); presenting to the Johns Hopkins Hospital Emergency Department with symptoms of an acute respiratory tract infection between January 2016 and June of 2019; known to represent infections with other respiratory viruses (rhinoviruses A, B, and C and/or coronavirus 229E, HKU1, and NL63 OC43) | [1] and [2] SARS‐CoV‐2 RT‐PCR, threshold not stated Samples: [1] and [2] Not stated |
[3] Pre‐pandemic |
|
Costa 2020 (Published letter); 134 (38) Mixed; hospital inpatients and outpatients; Brazil (Not stated) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 122) [1a] RT‐PCR‐positive (n = 106) [1b] negative RT‐PCR but a clinical COVID‐19 diagnosis based on highly suggestive symptoms and chest computed tomography (CT) findings (n = 16) [2] Non‐COVID samples (96 historical blood donation samples, Table 3 reported 100 though) | [1] RT‐PCR; adapted protocol, targeting E gene as first‐line screening tool, followed by confirmatory testing with N‐gene assay
[1b] 14/16 RT‐PCR‐ve patients had a second negative RT‐PCR. Clinical COVID‐19 diagnosis based on highly suggestive symptoms and chest computed tomography (CT) findings. Samples: [1] Respiratory samples were obtained from both the nasopharynx and oropharynx using rayon swabs. |
Pre‐pandemic |
|
Coste 2021 [A] (Pre‐print); 182 (113) Unclear; Switzerland (Not stated) |
Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 178) Group [2]: Pre‐pandemic controls (n = 404) [Some assays only had preliminary evaluation results for subgroup of 113 COVID‐19 samples and 69 non‐COVID samples] | RT‐PCR test (no more details available) Samples: Not stated |
No testing (pre‐pandemic samples) |
|
Criscuolo 2020 [A] (Pre‐print); 131 (46) Hospital inpatients; Italy (Not stated) |
Two‐group study estimating both sensitivity and specificity Group [1]: Lab‐confirmed cases of SARS‐CoV‐2 infection (n = 46) Group [2]: Pre‐pandemic controls (n = 85) For Group [1], lab confirmation likely refers to PCR test, but this was not explicitly stated. | Apparently RT‐PCR (the authors only reported "lab‐confirmation"). No more details available Samples: Not stated |
Pre‐pandemic samples ‐ no testing |
|
Dave 2020 (Published paper); 100 (100) Hospital inpatients; India (April 2020‐May 2020 (2 months)) |
Single‐group study to assess sensitivity: [1] RT‐PCR‐positive patients admitted to a tertiary care hospital | RT‐PCR‐positive; according to the protocols by National Institute of Virology, Pune Samples: Nasopharyngeal/oropharyngeal swab |
NA |
|
Decru 2020 [A] (Published letter); 72 (33) Unclear; Belgium (Not stated) |
Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 26 patients, 33 samples) Group [2]: PCR‐negative patients without clinical suspicion of COVID‐19 (n = 39 patients/samples) | RT‐PCR test (no further details available) Samples: Not stated |
RT‐PCR test (no further details available) |
|
Delliere 2020 [A] (Published paper); 146 (106) Unclear; included hospital inpatients; France (Not stated) |
Two‐group study: [1] COVID‐19 positive patients (n = 102, 106 samples) [2] Pre‐pandemic patients (n = 42; 14 occupational health patients with no known disease; 28 hospitalised patients with previous pulmonary infection, rhinovirus, metapneumovirus, influenza A, syncytial respiratory virus, recent malaria, antibodies against cytomegalovirus or Epstein‐Barr, HIV, hepatitis B, toxoplasmosis, rheumatic fever) | SARS‐COV‐2 RT‐PCR; commercial assay Samples: Not stated |
None ‐ pre‐pandemic |
|
Doherty Institute 2020 [A] (Published report); 229 (137) Unclear; Australia (Not stated) |
[1] Sensitivity group: patients with SARS‐CoV‐2 detected by RT‐PCR from upper and/or lower respiratory tract specimens (n = 91 patients, 137 samples) [2] Specificity group: (n = 92 people, 92 samples) [2a] patients with infections with the potential for cross‐reactivity in serological assays, namely (i) patients with respiratory viral infections, including seasonal coronavirus infections and (ii) patients with other acute infections (e.g. dengue; CMV; EBV) (n = 36 patients, 36 samples) ; [2b] representative sample of the Victorian population collected in 2018 and 2019 (‘pre‐pandemic controls’) (n = 56 people, 56 samples) | SARS‐CoV‐2 RT‐PCR; commercial assay
In addition, all positive samples had SARS‐CoV‐2 detected at VIDRL where testing was first conducted using an in‐house assay for the SARS‐CoV‐2 RdRp gene. If positive,
subsequent testing for the SARS‐CoV‐2 E gene was performed, using previously published primers. Samples: Upper and/or lower respiratory tract specimens |
[2a] Unclear for other diseases [2b] NA for pre‐pandemic controls |
|
DomBourian 2020 [A] (Published paper); 228 (102) Unclear; USA (March 2020.) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 convalescent plasma samples (n = 102); confirmed PCR‐positive for SARS‐CoV‐2 and were symptom‐free for at least 14 days prior to plasma donation and met all standard blood donation criteria per FDA requirements [2] non‐COVID samples (n = 126) [2a] Current non‐COVID, respiratory pathogen panel (RPP)‐positive samples (n = 20); were confirmed PCR‐negative for SARS‐CoV‐2; [2b] Pre‐pandemic samples (n = 106) | PCR, threshold not stated Samples: Not stated |
[2a] PCR (unclear how many negative tests) [2b] Pre‐pandemic (before November 2019) |
|
Dora 2020 (Published paper (Brief report)); 150 (26) Community; California, USA (5 to 12 June 2020) |
Two‐group study to estimate sensitivity and specificity
[1] Residents in SACC skilled nursing facility or WLA skilled nursing facility or in designated COVID‐19 recovery unit (CRU) with PCR test result. [2] Patients from the community who were diagnosed with COVID‐19 by RT‐PCR and treated in the acute care hospital and transferred to the CRU |
Nasopharyngeal RT‐PCR; commercial assay. Repeated approx. weekly on each ward and discontinued when all ward residents tested negative; threshold not stated Samples: Nasopharyngeal |
Same as for cases |
|
Dortet 2020 (Published paper); 306 (256) Mixed; Hospital inpatient and outpatient; France (March 11–23 2020) |
[1] RT‐PCR‐confirmed COVID‐19 patients (n = 101, 256 sera samples) [2] Non‐COVID‐19 controls (n = 50: 22 healthy volunteers, 24 pre‐pandemic; 4 RT‐PCR negative with common coronaviruses) | Real‐time RT‐PCR Samples: Nasopharyngeal samples (eSwabs™‐Virocult, Copan, Italy) | Pre‐pandemic (September/October 2017) (n = 24); RT‐PCR negative for SARS‐COV‐2 (n = 4); no respiratory symptoms for healthy volunteers (n = 22) |
|
Dortet 2021 [A] (Published paper); 504 (250) Hospital inpatients; France (11 March to 3 April 2020) |
Two‐group design with separate estimates of sensitivity and specificity: [1] COVID‐19‐positive patients (sample size = 250 from 159 patients) [2] Pre‐pandemic patients with other infections (sample size = 254) | RT‐PCR using Charite Berlin protocol (Corman 2020) Samples: Nasopharyngeal swabs |
Pre‐pandemic |
|
Du 2021 (Published paper); 252 (26) Unclear; USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 (n = 107) [2] Pre‐pandemic non‐COVID (n = 226) [2a] Healthy donor samples (n = 138) [2b] Cross‐reaction challenge samples (n = 88) | RT‐PCR Samples: Not stated |
[2] Pre‐pandemic (time not stated for all sources) |
|
Egger 2020 [A] (Published paper); 560 (104) Hospital inpatients; Austria (15th of March ‐10th of April 2020) |
[1] confirmed COVID‐19 patients (64 patients, 104 samples) [2] Healthy blood donors (n = 200) and [3] ICU patients (n = 256) | SARS‐CoV‐2 RT‐PCR Samples: respiratory specimens |
[2] and [3] Pre‐pandemic samples |
|
Fafi‐Kremer 2020 (Published paper); 160 (160) Community; France (6‐8 April 2020) |
Single‐group study estimating sensitivity only: [1] hospital staff with mild PCR‐confirmed COVID‐19 (n = 160); included doctors, nurses, physiotherapists, dentists, medical students, orderlies, hospital assistants, and hospital administrative staff | PCR test (not further specified) Samples: Not stated |
NA |
|
Favresse 2020a (Published letter); 176 (97) Unclear; Belgium (Not stated) |
Two groups of samples: [1] patients with a confirmed RT‐PCR SARS‐CoV‐2 diagnosis (n = 97 patients, 140 samples) [2] Non‐SARS‐CoV‐2 sera collected prior to the COVID‐19 pandemic with potential cross‐reactions (n = 79) | RT‐PCR; commercial assay Samples: respiratory samples (nasopharyngeal swab samples) |
Pre‐pandemic |
|
Favresse 2020b (Published letter); 150 (150) Unclear; Belgium (21 March to 25 May 2020) |
Single‐group study estimating sensitivity: [1] PCR‐confirmed COVID‐19 patients (n = 94, providing 150 serum samples) | RT‐PCR; commercial assay Samples: Nasopharyngeal swabs |
NA |
|
Fenwick 2021 [A] (Published paper); 158 (93) Hospital inpatients; Switzerland (Not stated) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (93 sera) [2] Non‐COVID samples (n = 65); pre‐pandemic including 18 samples from patients documented positive for a human coronavirus (E229, OC43, HKU1, or NL63) RT‐PCR | SARS‐CoV‐2 PCR, threshold not stated Samples: Not stated |
Pre‐pandemic |
|
Flinck 2021 [A] (Published paper); 244 (40) Mixed; Hospital in and outpatients; Finland (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients [1a] Inpatients (120 samples from 13 patients) for seroconversion [1b] Convalescent outpatients (n = 35) [2] Non‐COVID control samples [2a] Pre‐pandemic healthy (n = 161) [2b] Cross‐reaction samples (pre‐pandemic and current) (n = 43) | [1] RT‐PCR; multiple assays using Charite Berlin protocol (Corman 2020) Samples: Not stated |
[2a] Pre‐pandemic [2b] Pre‐pandemic or not stated |
|
Flower 2020 [A] (Published paper); 814 (314) Community; UK (1 to 29 May 2020.) |
Two‐group design with separate estimates of sensitivity and specificity: [1] Adult NHS workers (clinical or non‐clinical) who had previously tested positive for SARS‐CoV‐2 by PCR, but not hospitalised (276 patients with 314 samples); 21 d from symptom onset or positive swab test (whichever was earlier) [2] Pre‐pandemic healthy controls (n = 500); serum samples from Airwaves study (UK police officers) | For sensitivity, tests were compared against two standards:
(A) PCR‐confirmed clinical disease (via swab testing) and
(B) positivity in patients with either a positive S‐ELISA and/or hybrid DABA in the laboratory Samples: (A) Swab, no further details; (B) not stated |
Pre‐pandemic |
|
Fragkou 2020 (Published paper); 16 (16) Hospital inpatients; Greece (30th March 2020 and 6th April 2020) |
Single‐group sensitivity only [1] Hospitalised symptomatic confirmed COVID‐19 cases (n = 26) Excluded: [2] Asymptomatic healthcare volunteers with negative rRT‐PCR (n = 18) |
rRT‐PCR for SARS‐CoV‐2; commercial assay Samples: Nasopharyngeal and/or oropharyngeal swabs; lower respiratory tract samples (e.g. bronchoalveolar lavage or aspirates, sputum, etc.) were also accepted. |
|
|
Fujigaki 2020 [A] (Pre‐print); 29 (29) Hospital inpatients; Japan (28 February to 15 April 2020) |
Single‐group study estimating sensitivity: residual samples from PCR‐confirmed COVID‐19 patients (n = 29, providing 99 samples) | RT‐PCR test (no more details available) Samples: Nasopharyngeal swabs |
|
|
Gao 2020a (Published letter); 38 (38) Inpatient; Fuyang, China (22 Jan to 28 Feb 2020) |
Single group (cases); Inpatient cohort of COVID‐19 patients confirmed by New Coronavirus Pneumonia Prevention and Control Program (5th edition) published by the National Health Commission of China. Non‐Covid cases: NA |
New Coronavirus Pneumonia Prevention and Control Program (5th edition) published by the National Health Commission of China; Not reported (not reported) | NA |
|
Gao 2020b [A] (Published paper); 37 (37) Hospital inpatient; Shijiazhuang, Hebei, China (Jan 21 to Feb 24 2020) |
Single group (cases); Confirmed COVID‐19 cases (n = 22). Non‐Covid cases: NA | RT‐PCR assay; commercial assay; Nasal and pharyngeal swab specimens (Not stated (during hospital stay)) | NA |
|
Garnett 2020 (Published paper); 136 (79) Unclear; USA (Not stated) |
[1] patients previously diagnosed with COVID‐19 by RT‐PCR or by molecular methods at other local laboratories (n = 79) [2] healthy volunteers with no known exposure, travel history, or symptoms of COVID‐19 (n = 57) Excluded [3] patients previously tested to be negative for SARS‐CoV‐2 by RT‐PCR, but positive for another respiratory viral infection by molecular analysis (n = 14) | RT‐PCR. Threshold not stated (samples were tested on different days and by different operators) Samples: Not stated |
[2] RT‐PCR negative and no known exposure, travel history, or symptoms of COVID‐19 (samples were tested on different days and by different operators) |
|
GeurtsvanKessel 2020 [A] (Published paper); 386 (229) Mixed; hospital inpatients and outpatients; Netherlands (Not stated) |
Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 229 samples); published report included 107 patients (187 samples) with virus neutralisation antibodies detected by PRNT50 (all PRNT >= 20); Supplementary Data file included data for a further 42 samples with PRNT < 20; Group [2]: Patients with other infections (n = 147 reportedly included but results for 157 samples reported in Supplementary Data file and in Table 2 of published report for EUROIMMUN assays only) | RT‐PCR test (no more details available) Samples: Not stated |
Group [2]: Other infection or condition controls (timing not reported) no testing |
|
Graham 2021 (Published letter); 94 (94) Community; UK (June 2020) |
Single‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (PCR+) (n = 94); all residents of 4 UK Nursing Homes with rt‐PCR results available and informed consent. [2] PCR‐ residents (n = 147) PCR‐ residents (n = 147) were not included in our review as they did not have an adequate reference standard (PCR tests performed too late or not correctly swabbed). | RT‐PCR testing for all residents, with re‐testing one week later in those testing negative Samples: Oropharyngeal and nasal swabs |
NA |
|
Gudbjartsson 2020 [A] (Published paper); 2325 (1853) Community; Iceland (3 April to 8 July 2020) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID cases (1237 patients; 2102 samples; possible overlap of patients between [1a] and [1b]) [1a] Hospitalised (48 patients; 249 samples) [1b] Recovered (1215 patients; 1853 samples) [2] PCR‐ or not tested [2a] Pre‐pandemic: 2017 (n = 472) [2b] Early 2020 (n = 470) [2c] Health Care (n = 18,609) [2d] Reykjavik (n = 4843) [2e] Vestmannaeyjar (n = 663) [2f] Quarantine (n = 4222) Only groups [1b] and [2a] provided eligible data | RT‐PCR performed either at Landspitali – The National University Hospital of Iceland (LUH) or deCODE using similar qPCR methods Samples: Not stated |
[2a] Pre‐pandemic |
|
Guedez‐Lopez 2020 [A] (Published paper); 165 (101) Mixed; hospital inpatients and outpatients; Spain (8th March to 2nd April 2020) |
Two single‐group studies and a single group of controls to estimate sensitivity and specificity: [1] healthcare workers at Hospital Universitario La Paz, who attended the occupational health consultation for the first time between the 24th March and the 2nd of April referring symptoms compatible with COVID‐19 (n = 95; 55 cases) [2] patients randomly selected who were admitted to the Emergency Department of the Hospital with positive RT‐qPCR or high clinical suspicion of COVID‐19 (n = 50; 46 cases) [3] Pre‐pandemic patients (n = 20) | [1] and [2] RT‐qPCR (which detects N, S, E, Orf1ab and RdRp genes); selected RT‐qPCR commercial kits routinely used for diagnosis of COVID‐19 Samples: Nasopharyngeal swabs |
[1] and [2] as for cases [3] Pre‐pandemic |
|
Haljasmagi 2020 (Published letter); 52 (26) Hospital inpatient; Estonia (Not stated) |
Two‐group study estimating sensitivity and specificity: [1] PCR‐confirmed hospitalised Covid‐19 patients (n = 26) [2] Healthy controls without recent infection or Covid‐19 symptoms (fever or cough) for last month) (n = 26) | PCR; no further details Samples: Unclear |
Unclear, no SARS‐CoV‐2 testing reported |
|
Hamilton 2020 (Published letter); 263 (263) Hospital inpatient; UK (not reported) |
A multi‐group study with three groups to estimate sensitivity and specificity: [1] patients with laboratory‐confirmed or clinically suspected COVID‐19 enrolled into DISCOVER study (n = 149): [1a] 114 PCR+ hospitalised COVID patients; [1b] 35 PCR‐, clinically diagnosed hospitalised COVID patients); [2] healthcare workers at North Bristol NHS Trust with laboratory‐confirmed COVID‐19 (n = 114); [3] pre‐pandemic respiratory infection controls (n = 20). Group [3] not eligible for our review as < 25 samples leaving a "single‐group study to estimate sensitivity". | [1a] and [2] RT‐PCR
[1b] "Clinical diagnosis" Samples: Not reported |
NA |
|
Harritshoej 2021 [A] (Published paper); 1640 (150) Community; Denmark (from 3 May 2020) |
Multiple‐group study to assess sensitivity and specificity: [1] Convalescent patients with previous SARS‐CoV‐2 infection (n = 150) identified in the Danish Microbiology Database from February 2020 to April 2020 that were contacted and responded; [2] Pre‐pandemic healthy controls (for determination of clinical sensitivity); samples drawn during the influenza seasons of 2017–2018 and 2018–2019; [3] Pre‐pandemic patients with autoimmune diseases and acute viral infections (for determination of cross‐reactivity). | SARS‐CoV‐2 PCR, no further details Samples: Not reported |
[2] and [3] Pre‐pandemic |
|
Haselmann 2020 [A] (Published paper); 51 (26) Mixed; hospital inpatients and outpatients, community; Germany (Not stated) |
Multi‐group study to estimate sensitivity and specificity: [1] PCR‐confirmed Covid‐19 cases, after end of quarantine (outpatient or home‐based, including 6 asymptomatic) or hospitalisation (including 5 ICU cases) (n = 26) [2] Atypical respiratory infection within last 3 months and PCR‐negative for SARS‐CoV‐2 or not tested (n = 11) [3] Other respiratory viral infection diagnosed (n = 1) [4] Chronic disease (e.g. autoimmune disease) (n = 7) [5] Contact of a Covid‐19 patient but negative PCR and no symptoms (n = 2) [6] Healthy controls (n = 4) | qRT‐PCR; no further details Samples: Not stated |
[2] qRT‐PCR (for some unreported number) [3] Unclear (likely qRT‐PCR as confirmed with other infection) [4] Unclear [5] qRT‐PCR [5] Unclear |
|
Herroelen 2020 [A] (Published paper); 228 (171) Mixed; hospital inpatients and outpatients, community; Belgium (Inpatients = March 1 to April 27, 2020; healthcare workers unclear) |
[1] subjects with PCR‐confirmed SARS‐CoV‐2 infection, composed of 71 patients hospitalised for COVID‐19 pneumonia and 64 healthcare workers with paucisymptomatic infections (n = 135 patients, 171 samples) [2] pre‐pandemic serum samples obtained from patients with PCR‐confirmed infection by other HCoV respiratory viruses (n = 7), other pathogens and viruses (n = 42) or presence of auto‐immune antibodies (n = 8) (n = 57 samples) [3] healthcare workers who presented with WHO‐listed COVID‐19 symptoms but were not tested by PCR (n = 84, 84 samples) (this group not used in sensitivity/specificity analyses and not extracted. This group was also not mentioned in the published version) | PCR; commercial assay. Threshold not reported. Samples: nasopharyngeal swab |
Pre‐pandemic |
|
Hoffman 2020 (Published paper); 133 (29) Unclear; Sweden (not reported) |
(1) PCR‐confirmed COVID‐19 patients or convalescents (n = 29) (2) healthy volunteers with no known history of SARS‐CoV‐2 infection/COVID‐19 (n = 24) (3) pre‐pandemic anonymous blood donor sera from healthy adults (n = 80) [also reported 20 serum samples from babies (6–12 months) collected before or during 2018; not included in review] | PCR‐confirmed ‐ no further details Samples: not reported |
[2] No reference standard [3] Pre‐pandemic sera |
|
Hogan 2020a [A] (Published paper); 79 (17) Hospital inpatient; Kansas, USA (April 15 to June 1, 2020) |
Two‐group study to estimate sensitivity and specificity Samples collected as part of routine medical management of inpatients at a university hospital: [1] All available serum samples from hospitalised PCR‐positive patients [2] Hospital inpatients who had tested negative for SARS‐CoV‐2 by an RT‐PCR assay within 48 hours prior to collection (randomly sampled) |
FDA EUA RT‐PCR; commercial assays Samples: Nasopharyngeal swabs collected in either UTM or PBS |
As for cases |
|
Horber 2020 [A] (Published paper); 255 (132) Hospital inpatient; Germany (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Hospitalised confirmed COVID patients (186 samples from 58 patients) [2] Non‐COVID samples (n = 123) [2a] Pre‐pandemic samples collected before December 2019 (n = 88) [2b] Samples with potential cross‐reactive antibodies (n = 35) | rt‐PCR, threshold not stated Samples: Oro‐ and/or nasopharyngeal swab |
[2a] Pre‐pandemic [2b] Not stated. Other laboratory‐confirmed acute infections |
|
Hu 2020a (Pre‐print); 993 (993) Inpatient; Chongqing, China (Jan 23 to Mar 3) |
Single group (cases); Confirmed COVID‐19 patients (221); Chinese CDC guidelines (Trial Version 6); included RT‐PCR |
Chinese CDC guidelines (Trial Version 6); included RT‐PCR; Not stated (repeat PCR undertaken during hospitalisation) | NA |
|
Hu 2020b [A] (Pre‐print); 178 (68) Unclear; China (January and February 2020) |
Single or multi‐group study estimating sensitivity and specificity (design unclear), including participants who visited Huangshi Central Hospital during specified time period, described as [1] PCR‐positive COVID‐19 group (n = 68) [2] Suspected Covid group (PCR‐negative but with fever and other respiratory symptoms) (n = 9) [3] Group with other diseases and negative PCR (n = 101) Study authors considered group [2] and [3] as disease‐negative | RT‐PCR detecting open reading frame lab (ORFlab) and nucleocapsid protein (N) genes Samples: Not stated |
As for cases; unclear if single or > 1 negative PCR result |
|
Hubbard 2021 [A] (Published paper); 601 (216) Community; New Hampshire, USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Remnant serum/plasma specimens collected for clinical purposes from hospitalised patients with and without confirmed SARS‐CoV‐2 infection. (PCR+ 193 samples from 20 patients; PCR‐ 215 samples from 155 patients) [2] Convalescent plasma donors with paired serum & plasma specimens collected >= 14 d after resolution of symptoms (n = 23). [3] Remnant pre‐pandemic serum/plasma specimens collected for clinical purposes (170 samples) |
[1] RT‐PCR; multiple assays
[2] Not clear; possibly as above Samples: Not stated |
[1] as for cases [3] Pre‐pandemic |
|
Imai 2020 (Published paper); 160 (112) Hospital inpatient; Japan (February 11 to March 31, 2020) |
Two‐group study estimating sensitivity and specificity: [1] Patients with laboratory‐confirmed COVID‐19, including 74 symptomatic and 38 asymptomatic (total n = 112) [2] Pre‐pandemic samples from patients at Saitama Medical University Hospital (n = 48) | RT‐qPCR for SARS‐CoV2 Samples: pharyngeal and nasopharyngeal swabs |
Pre‐pandemic |
|
Jaaskelainen 2020 (Published letter); 84 (37) Hospital inpatient; Finland (Not stated) |
Multi‐group study to assess sensitivity and specificity: [1] COVID‐19‐positive patients (n = 47) [2] Non‐COVID‐19 (n = 37 patients); diagnosed with seasonal human coronaviruses or other respiratory viruses or viral infections, either pre‐pandemic or RT‐PCR negative for SARS‐CoV‐2 [3] Probable COVID‐19 patients (according to WHO definition) who had tested negative for SARS‐CoV‐2 by NAT (n = 13) | RT‐PCR Samples: nasopharyngeal swabs |
Pre‐pandemic or RT‐PCR |
|
Jin 2020 (Published paper); 98 (43) Hospital inpatients; Hangzhou, China (Jan to Mar 4, 2020) |
Two or more groups; [1] Laboratory‐confirmed COVID‐19 patients (n = 43); results reported separately for 27 patients while still PCR‐positive and for 34 patients after becoming PCR‐negative (excluded by review team) [2] Patients admitted with suspected SARS‐CoV‐2 infection, discharged with 2 x RT‐PCR‐negative results with an interval of 24 h and who quarantined at home (n = 33). Considered as disease‐negative for review purposes as clearly described as diseased ruled out. |
RT‐PCR ; Oral swab or sputum specimens (during hospital stay) | [2] 2 x RT‐PCR‐negative results with an interval of 24 h and who quarantined at home (n = 33) |
|
Jung 2020a (Published paper); 76 (19) Unclear; includes hospital inpatients; Texas, USA (Not specified (before Aug 2020)) |
Multi‐group study to establish sensitivity and specificity
[1] Confirmed COVID‐19 patients repeatedly assessed using PCR and with a known date of symptom onset (n = 19 with eligible time splits) [2] Non‐COVID cases with negative rt‐PCR test for SARS‐CoV‐2 (38 samples) for "clinical specificity" [3] Non‐COVID cases with other diseases (cross‐reaction panel, 19 samples) for "analytical specificity" |
RT‐PCR or TMA (Transcription‐mediated amplification) Samples: Not stated |
Negative PCR; or for cross‐reactivity no known exposure, travel history, or symptoms of COVID‐19 |
|
Kaltenbach 2020 [A] (Pre‐print); 606 (341) Community; Switzerland (11th April 2020 to 22nd April 2020) |
Three‐group study to estimate sensitivity and specificity: [1] Symptomatic and post‐symptomatic PCR‐confirmed Covid‐19 patients (n = 341) [2] PCR‐negative symptomatic patients (n = 115) [3] Pre‐pandemic blood donor controls (n = 150) | RT‐PCR Samples: Unclear |
[2] PCR [3] Pre‐pandemic |
|
Kaneko 2021 (Published paper); 187 (87) Hospital inpatient; Japan (March to May 2020) |
Two‐group study to estimate sensitivity and specificity [1] patients with acute COVID‐19 infection confirmed by RT‐PCR who were admitted between March and May 2020 (87 samples from 51 patients) [2] Pre‐pandemic controls (patients with other diseases admitted in Aug‐Sept 2019) (n = 100) | RT‐PCR for SARS‐CoV‐2 in accordance with the nationally recommended method in Japan. Samples: Pharyngeal and nasopharyngeal swabs |
Pre‐pandemic, other disease |
|
Knauer 2020 [A] (Published letter); 265 (125) Unclear; Canada (Not stated) |
Two‐group to estimate sensitivity and specificity [1] Confirmed COVID cases (NAAT‐positive) [2] Suspected COVID, NAAT‐negative patients | RT‐PCR; commercial assay threshold not stated but included inconclusive results Samples: Not stated |
As for cases |
|
Ko 2021 (Published letter); 52 (52) Mixed; hospital inpatients and outpatients; Korea (Not stated) |
Single‐group study to estimate sensitivity only [1] Confirmed COVID cases (51 samples from 29 patients) | Confirmed by RT‐PCR Samples: Not stated |
NA |
|
Kohmer 2020a [A] (Published paper); 50 (33) Mixed (in‐ and outpatient); Germany (Not stated) |
Multi‐group study estimating both sensitivity and specificity Group [1] PCR‐confirmed COVID‐19 cases (n = 33) Group [2]: Other known infections (SARS, other coronaviruses, EBV, CMV) (n = 17) | Group [1]: PCR (not further specified) Samples: Not stated |
Group [2]: No testing |
|
Kohmer 2020b [A] (Published paper); 82 (45) Mixed (in‐ and outpatient); Germany (Not stated) |
Multi‐group study estimating both sensitivity and specificity. Group [1]: Symptomatic PCR‐confirmed COVID‐19 cases (n = 45) Group [2]: Other known infections (other coronaviruses, EBV, CMV) including some pre‐pandemic (n = 37); review team excluded 6 samples with serologically confirmed SARS‐COV‐2 infection based on PRNT | Groups [1A] and [1B]: PCR (not further specified) Samples: Not stated |
Group [2] and [3]: No testing |
|
Korte 2021 [A] (Published letter); 159 (159) Unclear; Switzerland (Not stated) |
Single‐group study to estimate sensitivity [1] Confirmed COVID patients with a history of a positive SARS‐CoV‐2 PCR test (159 patients) | SARS‐CoV‐2 PCR test Samples: Not stated |
NA |
|
Kowitdamrong 2020 [A] (Published paper); 384 (213) Unclear; Thailand (March 10 to May 31, 2020) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID cases (118 patients with 213 samples); defined as those that tested positive for SARS‐CoV‐2 RNA on RT‐PCR of combined nasopharyngeal and throat swab [2a] patients under investigation (PUI) for COVID‐19 with RT‐PCR results that were negative for SARS‐CoV‐2 (n = 49); [2b] Concurrent patients with other respiratory infections (n = 20); (dengue, HBV, HCV, HIV, mumps, measles, rubella, EBV, CMV, VZV, HSV, and treponema); [2c] Healthy volunteers in the laboratory (pre‐pandemic?) (n = 20); [2d] Pre‐pandemic healthy blood donors (n = 82) | RT‐PCR targeting ORF1a/b and E genes specific to SARS‐CoV‐2 and pan‐sarbecovirus, respectively Samples: combined nasopharyngeal and throat swab (NT) samples |
[2a] RT‐PCR results that were negative for SARS‐CoV‐2 [2b] Not stated/ None? [2c] Pre‐pandemic (prior February 2020) [2d] Pre‐pandemic (prior February 2020) |
|
Krishnamurthy 2020 (Published paper); 5565 (303) Unclear; USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 cases (303 samples); SARS‐CoV2 microbiological confirmation from respiratory samples by PCR across multiple healthcare centres [2] Non‐COVID samples (5262 samples) [2a] Pre‐pandemic healthy controls (n = 4502) [2b] Other disease controls including auto‐immune and infectious diseases (n = 464) [2c] SARS‐COV‐2 negative PCR (n = 296); patients with SARS‐COV‐2 negative PCR results from respiratory samples by PCR across multiple healthcare centres | Microbial RT‐PCR, threshold not stated Samples: NP swab results |
[2a] Pre‐pandemic (Jan‐April 2019) [2b] Unclear [2c] Negative PCR for SARS‐COV‐2 (unclear if at least 2 negative PCR tests) |
|
Lassauniere 2020 [A] (Pre‐print); 112 (30) Intensive care; Hillerod, Denmark (Not stated) |
Two or more groups COVID‐19 PCR‐positive patients (n = 30) admitted to intensive care Non‐Covid cases: blood donors (n = 10); acute viral respiratory tract infections with other coronaviruses (n = 5) or non‐coronaviruses (n = 45); dengue virus (n = 9), cytomegalovirus (CMV; n = 2) and Epstein Barr virus (EBV; n = 10). One patient was positive for both CMV and EBV. |
Viral nucleic acid detection (no further detail) in hospital patients Samples: Respiratory (during hospital stay) |
Pre‐pandemic (n = 82) |
|
Lau 2020a (Pre‐print); 696 (338) Unclear; included hospital inpatients; Singapore (April to May 2020) |
Two‐group study to estimate sensitivity and specificity for diagnosis of acute and convalescent‐phase Covid [1] PCR‐positive Covid‐19 cases (n = 338) [1] cases of suspected or confirmed SARS‐CoV‐2 infection (n = 338) [2] Healthy healthcare workers (laboratory staff and frontline healthcare workers) with no suspicion for Covid‐19 (n = 294) [3] Samples positive for other antibodies including: dengue (n = 46), anti‐HCV (n = 3), HBsAg (n = 8), anti‐HBc IgM (n = 2), RF (n = 5) | RT‐PCR Samples: Not stated |
[2] None; unclear if pre‐pandemic [3] Unclear |
|
Lau 2020b (Pre‐print); 994 (280) Unclear; included hospital inpatients; Singapore (April to June 2020) |
[1] PCR‐confirmed Covid‐19 cases (n = 280 patients providing 415 samples) [2] Healthy healthcare worker controls (n = 597); 315 with annual Southern Hemisphere influenza vaccination 4 weeks prior [3] Antibody‐positive for different diseases: dengue (n = 74), hepatitis C (n = 3), hepatitis B (n = 12), syphilis (n = 1), antinuclear antibody (n = 16) double‐stranded DNA antibody (n = 4), RF (n = 7) | PCR; no further details Samples: Not stated |
[2] None; unclear if pre‐pandemic [3] Unclear |
|
Lau 2020c (Published paper); 1168 (353) Unclear; hospital, no further detail; Singapore (April‐June 2020) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients, residual leftover sera (n = 353) [2] Non‐COVID controls [2a] Current healthy healthcare workers (HCWs) without symptoms of upper respiratory tract infection/fever and two serial antibody testing 14 days apart (n = 262) [2b] pre‐pandemic samples from our staff health screening (HS) program in 2018 (n = 718) [2c] Cross‐reactivity panel (229/262 HCW volunteers from [2a] with recent influenza vaccination and 97 samples positive for dengue fever or other antibodies) Group [2a] and parts of [2c] were excluded from our review as they did not have an eligible reference standard. | RT‐PCR; commercial assay Samples: Not stated |
[2a] NA as excluded from review [2b] Pre‐pandemic (2018) [2c] Not stated for the additional 97 cross‐reactivity samples. |
|
Lau 2020d (Published paper); 785 (70) Unclear; hospital, no further detail; Singapore (April to June 2020) |
Multi‐group study to estimate sensitivity and specificity [1] Test group ‐ Confirmed COVID patients, residual leftover sera (n = 415) [2] Control group (n = 715) [2a] Non‐COVID control Current healthy healthcare workers (HCWs) (n = 597); (no self‐reported respiratory symptoms) [2b] Cross‐reactivity group ‐ 315 HCWs from group [2a] who received their annual influenza vaccination 4 weeks prior to testing and 118 non‐Covid patients who had antibody‐positive samples [dengue, hepatitis C (HCV), hepatitis B (HBV), syphilis, antinuclear antibody (ANA), double‐stranded DNA antibody (anti ds‐DNA), RF] from ambulatory patients (n = 433) | RT‐PCR Samples: Not stated |
[2a] Untested, no reported respiratory symptoms [2b] Pre‐pandemic, unclear for 74 dengue patients |
|
Li 2020 [A] (Pre‐print); 49 (49) Hospital inpatient; China (December 2019 to February 2020) |
Single‐group study estimating sensitivity alone: 49 NAT confirmed 2019‐nCoV infected patients (hospitalised patients) | NAT; no further details Samples: not reported |
NA |
|
Lippi 2020 [A] (Published letter); 48 (48) Inpatients; Verona, Italy (Not stated) |
Single group (cases); Participants with suspected COVID‐19; subgroup of cases (48/131 pts) with available data on days post‐symptom onset data could be included. | RT‐PCR; commercial assay; Samples: OP, NP swabs (during hospitalisation) |
NA |
|
Liu 2020a (Pre‐print); 358 (153) Inpatients; Hubei, China (Feb 6 to 14) |
Two or more groups: [1] Confirmed (153) or suspected (85) COVID‐19 [2] Non‐Covid cases: Ordinary' patients (70) and randomly sampled healthy blood donors (50); timing not reported, presumed to be contemporaneous |
RT‐PCR; commercial assay Clinical diagnosis according to General Office of National Health Committee guideline (5th ed); Pharyngeal (clinical diagnosis presumed on admission) |
Ordinary' patients (70) and randomly sampled healthy blood donors (50); timing not reported, presumed to be contemporaneous |
|
Liu 2020b [A] (Published paper); 314 (214) Inpatient, Hubei, China (Jan 18 to Feb 26) |
Two or more groups: RT‐PCR‐confirmed Covid‐19 cases (n = 214) Non‐Covid cases: Healthy blood donors, presumed to be contemporaneous (n = 100) |
RT‐PCR; pharyngeal (median 15 days post‐symptom onset (range, 0–55 days) | Healthy blood donors, presumed to be contemporaneous (n = 100) |
|
Liu 2020c (Published paper); 476 (206) Hospital inpatient; China (January 18 to April 4, 2020.) |
Two‐group study to estimate sensitivity and specificity [1] Test group ‐ Confirmed COVID patients, serum from hospitalised patients (n = 206) [2] Control group (n = 270) – Non‐Covid pre‐pandemic healthy donors | RT‐ commercial assay Samples: [1] pharyngeal swab specimens |
[2] Untested, pre‐pandemic healthy blood donors who donated blood in May 2019 |
|
Liu 2021 (Published paper); 151 (111) Hospital inpatients; China (Feb 3 to Mar 13 2020) |
[1] Confirmed COVID inpatients (n = 111) [1a] Symptomatic cases (n = 81) [1b] Asymptomatic cases (n = 30) [2] Non‐COVID patients (suspected COVID with multiple negative PCR tests) (n = 40) | Real‐time RT‐PCR; Ct <= 38 defined as a positive test Samples: nasopharyngeal swab |
Classed as "Non‐COVID‐2 control" based on clinical judgement as well as multiple negative RT‐PCR tests |
|
Loconsole 2020 (Published paper); 819 (148) Emergency Department; Italy (2020‐03‐23 to 2020‐04‐21) |
Prospective cohort study (n = 819)
Single‐group study to estimate sensitivity and specificity Consecutive patients presenting to the Emergency Department between 23 March and 21 April 2020: [1] PCR‐positive patients ‐ 148 [1a] < 7 days of symptoms ‐ 99 [1b] > 7 days of symptoms ‐ 44 [1c] Asymptomatic patients ‐ 5 [2] PCR‐negative patients ‐ 671 |
Real‐time PCR kit; commercial assay. Results were considered positive when two or three genes were identified. WHO real‐time RT‐PCR protocol used to confirm results when samples resulted in positive for one gene Samples: Nasal and pharyngeal swabs |
Same as for cases |
|
Long 2020 (Pre‐print); 363 (285) Inpatients; Chongqing, China (Feb 2020) |
Two or more groups; RT‐PCR+ve confirmed cases (n = 285). No further detail of inclusion or exclusion criteria. Additional cohorts reported but not extracted included: [A] follow‐up cohort in RT‐PCR+ve confirmed cases sampling every 3 days (n = 63 subset of cross‐sectional study); did not provide accuracy data [B] Study reported data for a cohort of RT‐PCR‐ve suspects (n = 52) which allowed specificity to be extracted |
RT‐PCR ; nasal and pharyngeal swabs (during hospital stay) | NA [Study reported data for a cohort of RT‐PCR‐ve suspects (n = 52) which allowed specificity to be extracted] |
|
Lou 2020 [A] (Pre‐print); 380 (80) inpatient; Hangzhou, China (Jan 19 to Feb 9 2020) |
Two or more groups; Confirmed Covid‐19 cases according to New Coronavirus Pneumonia Prevention and Control Program (6th edition) published by the National Health Commission of China (n = 80) Non‐COVID cases: Healthy people enrolled from the community, presumed contemporaneous selection (n = 300) |
CDC guideline (6th ed). Confirmed case should meet three criteria: 1) fever and/or respiratory symptoms; 2) abnormal lung imaging findings; and 3) positive result of the nucleic acid of SARS‐CoV‐2. No information on threshold; deep sputum samples' (on admission) | Healthy people enrolled from the community, presumed contemporaneous selection (n = 300) |
|
Lynch 2021 (Published paper); 613 (533) Mixed; hospital inpatients and outpatients; USA (Not specified) |
Multi‐group study
Remnant serum or plasma samples from routine clinical laboratory testing [1] COVID +ve patients [1a] ICU patients [1b] non‐ICU patients [1c] convalescent plasma donors (non‐ICU) [2] pre‐pandemic controls |
RT‐PCR Samples: nasopharyngeal swabs |
Pre‐pandemic samples, prior to June 2018 |
|
MacMullan 2020 [A] (Published paper); 199 (123) Unclear; USA (April to July 2020) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID samples from symptomatic participants collected more than 21 days post‐symptom onset, PCR‐positive (n = 123) [2] Current, PCR‐negative patients (n = 83) [3] Pre‐pandemic controls: serum samples collected prior to November (September?) 2019 (n = 76) Group [2] excluded from our review as test accuracy outcomes could not be read from Figure 1. | [1] Curative’s oral fluid PCR test, positive for viral RNA was determined as below 35 cycle thresholds (CT). Samples: Oral fluid |
[2] Curative’s oral fluid PCR test, positive for viral RNA was determined as below 35 cycle threshold (CT). [3] Pre‐pandemic |
|
Mairesse 2020 [A] (Published paper); 253 (178) Unclear; Belgium (May 15 to 30, 2020) |
Two groups [1] COVID‐19 confirmed patients ‐ n = 154 [2] Non‐SARS‐CoV‐2 sera ‐ n = 75 | Confirmed RT‐PCR and with COVID‐19 symptoms Samples: Serum |
Pre‐pandemic |
|
Manalac 2020 [A] (Published paper); 956 (31) Unclear; patients or HCWs; USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Covid‐19 patients or healthcare workers with RT‐PCR‐confirmed and/or clinical assessment indicated SARS‐CoV‐2 infections (n = 97) [2] Non‐COVID samples (n = 1062) [2a] Concurrent, negative controls with no RT‐PCR results nor clinical assessment indicating SARS‐CoV‐2 infections (n = 137) [2b] Concurrent cross‐reactivity panel with positive serology test results of other infectious diseases or autoimmunity (n = 78) [2c] Pre‐pandemic samples with other diseases (n = 847), e.g. rheumatoid diseases, thyroid cancer, and therapeutic drug monitoring No relevant test accuracy results reported for group [2a]. | [1] RT‐PCR‐confirmed and/or clinical assessment indicated SARS‐CoV‐2 infections, threshold not stated; clinical criteria not stated Samples: [1] nasopharyngeal swab |
[2b] Concurrent, not tested [2c] Pre‐pandemic |
|
Marlet 2020 [A] (Published paper); 102 (13) Hospital inpatient; France (April 8th and May 11th 2020) |
Two‐group study to estimate sensitivity and specificity Retrospective study [1] Confirmed COVID‐19 hospital inpatients (63 samples) [2] Pre‐pandemic controls with other diseases (89 patients) The prospective study was not eligible for our review as < 25 COVID cases. 203 patients: Covid‐negative (n = 181), Covid‐positive (n = 22) | SARS‐CoV‐2 RT‐PCR; multiple assays. Among the positive RT‐PCR results, inconclusive RT‐PCR results defined as positive only for one gene (E, ORF1ab or N). Samples: Respiratory samples |
Pre‐pandemic |
|
Martinaud 2020 (Published paper); 682 (123) Unclear; includes hospital inpatients; [1] France (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 161) [1a] Confirmed COVID patients with a medical record of the date of symptomatic onset admitted to Military Medical Center for "clinical sensitivity" experiment (n = 101) [1b] Confirmed COVID patients positive by RT‐PCR and more than 3 weeks after the symptoms onset for "analytical accuracy" experiment (n = 60) [2] Cross‐reactivity panel (n = 59); positive for IgG and IgM against dengue virus and Chikungunya virus, for HBsAg or anti−HCV, RF, monoclonal proteins, Abs against malaria, Abs against syphilis, IgG and IgM against EBV and IgG against CMV [3] Pre‐pandemic, healthy donors (n = 500) | SARS‐CoV‐2 infection was confirmed by PCR in samples from the respiratory tract according to French guidelines, threshold not stated Samples: samples from the respiratory tract (nasopharyngeal) |
[2] Unclear [3] Pre‐pandemic |
|
McAulay 2020 [A] (Published paper); 457 (352) Hospital inpatient; USA (Not stated) |
[1] RT‐PCR‐positive COVID‐19 patients (predominantly hospitalised (n = 62 patients, 352 samples, Seattle cohort) [2] Specificity group: 74 pre‐pandemic clinical serum specimens and 31 “cross‐reactivity challenge” specimens (27 from individuals with a history of seasonal coronavirus infection within 3 years prior to collection and 4 specimens reactive for RF, HIV‐1 antibody, HAV total antibody, HBV core total antibody and surface antibody, HCV antibody and/or HSV2 antibody) (n = 105 people) | RT‐PCR ("RT‐PCR‐confirmed COVID‐19") Samples: Not stated |
Pre‐pandemic and “cross‐reactivity challenge” specimens determined by a syndromic respiratory PCR test |
|
Merrill 2020 [A] (Published paper); 210 (36) Unclear; hospital, no further detail; Iowa, USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐patients (54 specimens from 32 unique patients) [2] Suspected COVID cases and/or potential cross‐reactives with negative PCR (n = 35) [3] Pre‐pandemic samples (n = 139) | rt‐PCR, threshold not stated Samples: Not stated |
[2] rt‐PCR, threshold not stated [3] Pre‐pandemic |
|
Montesinos 2020 [A] (Published paper); 200 (128) Unclear; Belgium (Not stated) |
Three‐group study to estimate sensitivity and specificity: [1] COVID‐19 patients confirmed by RT‐qPCR and CT‐scans (n = 128) [2] Negative controls. Stored sera from Jan 2018 to Aug 2019 (n = 62) included samples with a potential cross‐reaction to the SARS‐CoV‐2 immunoassays, namely, EBV infection (n = 5), CMV infection (n = 11), M. pneumoniae infection (n = 8), parvovirus infection (n = 1), HBV infection (n = 1), bartonella henselae infection (n = 1), brucella spp. infection (n = 1), autoimmune pathologies (anti‐DNA, n = 1; anti‐PL12, n = 1; anti Scl‐70, n = 1) [3] Sera from healthy volunteers (n = 10) obtained during the epidemic period (April 2020). | RT‐PCR and CT scan; commercial assays No further detail regarding how CT contributed to diagnosis Samples: Not stated |
[2] Pre‐pandemic stored samples with known (non‐COVID) diagnoses [3] contemporaneous healthy; no reference standard reported to confirm absence of disease |
|
Muecksch 2020 [A] (Pre‐print); 97 (97) Hospital outpatients; United Kingdom (Scotland) (Not stated) |
Single‐group study, sensitivity only [1] Prior RT‐PCR‐diagnosed SARS‐CoV‐2‐positive (non‐hospitalised, relatively mildly symptomatic) | RT‐PCR Samples: Not stated |
NA |
|
Naaber 2020 [A] (Published paper); () Mixed; Hospital inpatient and outpatient, contacts; Estonia (April 28 and May 07 2020.) |
Multi‐group study to assess sensitivity and specificity of 7 commercial antibody tests [1] Confirmed COVID patients (n = 97); at least one week pso or post RT‐PCR +ve [1a] asymptomatic (n = 20) [1b] symptoms score 1‐6 (n = 43) [1c] symptoms score 7‐14 (n = 34) [2] Pre‐pandemic healthy controls (n = 100) | RT‐PCR Samples: Not stated |
Pre‐pandemic healthy (date not stated) |
|
Nagasawa 2020 [A] (Published paper); 45 (45) Hospital inpatient; Japan (April 12 to May 8 2020) |
Multi‐group study to estimate sensitivity and specificity of 3 ELISA kits [1] Confirmed COVID patients (45 samples from 26 patients) [1a] Moderate COVID patients (n = 19) [1b] Severe COVID patients (n = 7) [2] Controls, not eligible for our review | RT‐PCR performed at SRL laboratory (Tokyo, Japan), threshold not stated Samples: naso‐pharynx swab specimens |
NA |
|
Nayak 2021 (Published paper); 42 (42) Unclear; India (Not stated) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 patients, n = 42; PCR‐positive at the time of initial diagnosis, and PCR‐negative when recruited for this study [2] Pre‐pandemic controls n = 22 Group [2] not eligible for our review as < 25 samples. | RT‐PCR; under the Government of India guidelines for COVID19 diagnosis. (ICMR‐NIV, 2020)
Threshold not stated Samples: Nasopharyngeal and throat swabs |
NA |
|
Ng 2020 [A] (Pre‐print); 1574 (43) Mixed; Hospital inpatient and outpatient; United States (March‐April 2020) |
Multi‐group study estimating sensitivity and specificity. (Possible that [1] and [2] could be considered as a single group, but recruitment was not sufficiently clearly described) [1] RT‐PCR‐confirmed COVID‐19 cases (n = 43 patients) [2]: SARS‐CoV‐2 PCR‐negative UCSF patients (indication for PCR testing was not reported but implied COVID‐19 suspects) (n = 163 patients for test [A] and 39 patients for test [B]) [3]: Pre‐pandemic controls collected by Abbott Laboratories (US blood donors) (n = 1013 for test [A], n = 1492 for test [B]) Two additional cohorts evaluated for seroprevalence survey not extracted for this review, including [4] patients hospitalised for indications other than COVID‐19 respiratory disease (March‐April 2020) (n = 387, and [5] contemporaneous blood donors (n = 1000) | RT‐PCR test (no more details available) Samples: NP and/or OP |
Group [2]: RT‐PCR (no more details available) Group [3]: Pre‐pandemic |
|
Nguyen 2020 (Published letter); 99 (99) Hospital inpatient; France (Not stated) |
Single‐group study to estimate sensitivity only [1] Confirmed COVID cases with severe SARS‐Cov‐2 infection (hospitalised, ICU) (n = 99) | Positive for SARS‐Cov‐2 using routine RT‐PCR methodology, threshold not stated Samples: Not stated |
NA |
|
Nicol 2020 [A] (Published paper); 293 (141) Hospital inpatient; [1] France (Not stated) |
[1] patients with RT‐PCR‐confirmed SARS‐CoV‐2 infection (n = 82 patients, 141 samples) [2] patients with symptoms consistent with COVID‐19 but RT‐PCR‐negative (clinical diagnosis of pneumonia of unknown aetiology) (n = 52 patients, 57 samples) [3] Pre‐pandemic control group specimens (n = 50 samples) [4] Samples with Pathogen potentially cross‐reactive with SARS‐CoV‐2 (n = 25 samples) [5] Samples from pregnant women (n = 10) [6] Samples from patients with positive RF (n = 10) | [1] RT‐PCR Samples: [1] Not reported |
[2] RT‐PCR for pneumonia PCR‐negative controls [3] pre‐pandemic [4]‐[6] None (no RT‐PCR detection performed) |
|
Nilles 2020 [A] (Pre‐print); 300 (68) Hospital inpatient; USA (March 30 to May 4, 2020) |
[1] patients that had been hospitalised at the Brigham and Women’s Hospital testing positive by SARS‐CoV‐2 RT‐PCR (n = 28 patients, 68 samples) [2] Pre‐pandemic controls with and without recent respiratory infections (n = 232 patients/samples) | RT‐PCR Samples: Not stated |
Pre‐pandemic |
|
NSAE 2020 [A] (Published report); 1530 (536) Mixed; HCWs or plasma donors; UK (Not stated) |
Multi‐group study to assess sensitivity and specificity [1] Covid patients with a previous positive SARS‐CoV‐2 RT‐PCR nose/throat swab, with blood samples taken ≥ 20 days post‐symptom onset (n = 536) [1a] Healthcare workers and patients from Oxford University Hospital (n = 158) [1b] Volunteer plasma donors (n = 378) [2] Healthy individuals 30‐50 years old (n = 994) | RT‐PCR, threshold not stated. Samples: Nose/throat swab |
Pre‐pandemic |
|
Ong 2020 [A] (Published paper); 278 (99) Hospital inpatient; Netherlands (17 March 2020 to 10 April 2020) |
[1] COVID‐19 positive patients presenting to a teaching hospital with respiratory symptoms that were suspected of respiratory tract infection. (N = 99) [2] COVID‐19 negative patients presenting to a teaching hospital with respiratory symptoms that were suspected of respiratory tract infection. (N = 129) [3] randomly selected historical patient control sera (N = 50) | PCR (referred to as PCR in Supplementary materials and as NAT in paper); multiple assays Samples: from the oral cavity and subsequently from the nasal cavity using the same nasopharyngeal swab |
[2] Same as for cases [3] Historic controls = pre‐pandemic |
|
Padoan 2020a (Published paper); 87 (87) inpatients; Padova, Italy (Mar 18 to Mar 26, 2020) |
Single group (cases); Hospitalised patients with confirmed COVID‐19 (n = 37). | RT‐PCR; NP (Not stated) | NA |
|
Padoan 2020b [A] (Published paper); 70 (70) Unclear; Italy (Not stated) |
Single‐group study to estimate sensitivity: [1] adult patients with PCR‐confirmed COVID‐19 (total N was not reported, 51 assessed for IgM and 19 assessed for IgA; any overlap between patient groups was not reported). The report contains two groups of COVID‐19 patients, one was assessed for IgM using CLIA and the other for IgA using ELISA. There was no non‐COVID‐19 or healthy control group. [1] Severely sick adult COVID‐19 (rRT‐PCR‐confirmed) patients longitudinally assessed for IgM using CLIA (n = 51) [2] Severely sick adult COVID‐19 (rRT‐PCR‐confirmed) patients longitudinally assessed for IgA using ELISA (n = 19) | rRT‐PCR, no further details Samples: Not stated |
Not applicable |
|
Paiva 2021 [A] (Published paper); 1300 (113) Community; USA (After 12 March 2020) |
[1] RT‐PCR‐positive COVID‐19 cases (n = 71, 113 samples) [2] Healthy individuals; pre‐employment screening (n = 126) [3] Samples positive for other viruses and pathogens (to test cross‐reaction of the assays) (n = 119) [4] Serum or plasma samples collected before the pandemic started in the United States (n = 942) | RT‐PCR; multiple assays Samples: nasopharyngeal swabs |
[2] healthy controls sampled early March 2020; before the first COVID‐19 case was diagnosed in the Lifespan Health System
[3] viral respiratory pathogen nucleic acid test [4] pre‐pandemic (before January 2020) [2] ‐ [4] The patients whose samples were reactive [positive result on index test] were followed by medical record review to ensure that they did not have COVID‐19. |
|
Pan 2020a (Published paper); 134 (134) Inpatients; Wuhan, China (Symptom onset Jan 7 to Feb 18)) |
Single group (cases); COVID‐19 patients according to CDC guideline (5th ed); confirmed by PCR (67) or clinical diagnosis (37) |
RT‐PCR; commercial assay Clinical diagnosis according to CDC guideline (5th ed), i.e. RT‐PCR‐negative but with viral pneumonia by radiography; throat swabs (Not stated) |
NA |
|
Pape 2021 [A] (Published paper); 275 (57) Mixed; Hospital in‐ and outpatient; Germany (Not stated) |
[1] SARS‐ CoV‐2‐positive sera were collected from PCR‐confirmed symptomatic COVID‐19 patients (n = 29, 57 samples, cohort C) [2] Pre‐pandemic negative control serum samples collected for various serological testing before the start of the SARS‐CoV‐2 outbreak (n = 218) ‐ healthy donors (n = 105, cohort B), patients that tested positive for common cold corona viruses several months before the blood sample was taken (n = 34, all four types of ccCoV represented; cohort A), patients with diagnosed mycoplasma pneumoniae (n = 22; cohort Z), EBV or CMV infection (n = 57, cohort E) | RT‐PCR Samples: Not stated |
Pre‐pandemic |
|
Patel 2021 [A] (Published paper); 1313 (214) Community plasma donors; USA (April 2020‐ July 2020) |
Two‐group study to assess sensitivity and specificity for commercially available serology assays [1] Covid‐19 convalescent plasma donors (n = 214 potential); convenience sample with a documented history of a positive molecular assay test result for SARS‐CoV‐2 infection and met standard self‐reported eligibility criteria for blood donation [2] Pre‐pandemic samples (n = 1099); identity‐unlinked HIV serosurvey conducted in 2016 among adult patients attending the Johns Hopkins Hospital Emergency Department | Positive molecular assay test Samples: Not stated. |
Pre‐pandemic samples. |
|
Pere 2020 (Published paper); 217 (100) Hospital inpatient; France (Not specified) |
Multi‐group study to establish sensitivity and specificity [1] Confirmed COVID‐19 patients [1a] COVID‐19 convalescent health care workers with a history of positive SARS‐CoV‐2 RT‐PCR at least 1 month before serology testing (n = 100) [1b] Hospitalised patients from COVID+ area (n = 63) [2] Non‐COVID patients [2a] Pre‐pandemic, other diseases (n = 117); left over sera from pre‐epidemic period (collected from October 2019 to January 2020), available at the virology laboratory [2b] Hospitalised patients from COVID‐free area (n = 96) Groups [1b] and [2b] were not eligible for our review. | [1a] COVID‐19 by RT‐PCR, threshold not stated Samples: Not stated |
[2a] Pre‐pandemic |
|
Perez‐Garcia 2020(a) (Published paper); 251 (151) Mixed; mainly inpatient; [2] and [3] Spain (March 1 to April 6, 2020; February 9 to April 2, 2020) |
[1] randomly selected group of pre‐pandemic patients who had a serum sample taken for other serologic studies (n = 100) [2] patients admitted to the Emergency department with suspicion of COVID‐19 and PCR‐positive for SARS‐CoV‐2 (n = 90) [3] patients admitted for at least 5 days with a clinical and radiological diagnosis of pneumonia of unknown aetiology, PCR‐negative for SARS‐CoV‐2 (n = 61) | [2] and [3] RT‐PCR; commercial assays
[3] Clinical diagnosis of COVID‐19 with negative PCR for SARS‐CoV‐2. Criteria for diagnosis not stated Samples: [2] and [3] Unclear ‐ "clinical samples" |
[1] Pre‐pandemic |
|
Perez‐Garcia 2020(b) (Pre‐print); 63 (63) Inpatient hospital (9 Feb and 2 Apr) |
Single group (cases); Patients admitted with a clinical and radiological diagnosis of pneumonia of unknown aetiology but RT‐PCR‐ve (n = 63) | Clinical diagnosis of COVID‐19 (no further detail); all PCR‐negative; not reported (not reported) | NA |
|
Perez‐Garcia 2021 [A] (Published paper); 140 (80) Hospital inpatient; Spain (2020‐03‐01 to 2020‐04‐28) |
Two‐group study, to assess sensitivity and specificity [1] Symptomatic patients admitted to the Emergency department between March 1 and April 28, 2020, with suspicion of COVID‐19 and confirmation by PCR (n = 80) [2] Pre‐pandemic control group (other diseases) (n = 60); patients with a sample taken for other serologic studies | RT‐PCR, threshold not stated Samples: Not stated |
Pre‐pandemic |
|
Pfluger 2020 [A] (Published paper); 395 (75) Hospital inpatient; Germany (March and April 2020.) |
Two‐group study to estimate sensitivity and specificity [1] Covid patients (positive RT‐PCR) in March and April of 2020) (n = 75) [2] Retained samples of a pre‐pandemic blood donor cohort collected (n = 320) | Positive RT‐PCR; multiple assays Samples: Naso‐pharyngeal swab |
Pre‐pandemic |
|
PHE 2020 [A] (Published report); 599 (100) Community; UK (before late April 2020.) |
Multi‐group study to assess sensitivity and specificity [1] Convalescent PCR‐confirmed covid cases with sufficient volume of serum to cover multiple assays: ([A] n = 100 (with 7 samples later excluded as PCR‐negative, so 93), [B] n = 100, [C] n = 100, [D] n = 100, [E] n = 100) [2] Non‐Covid patients ([A] n = 499, [B] n = 499, [C] n = 499, [D] n = 499, [E] n = 499) [2a] Historical serum samples collected before December 2019 ([A] n = 399, [B] n = 399, [C] n = 399, [D] n = 399, [E] n = 399) [2b] Confounder serum samples collected before December 2019 (confounder and RIPL samples) ([A] n = 100, [B] n = 100, [C] n = 100, [D] n = 100, [E] n = 100) | RT‐PCR, threshold not stated Samples: swab sample |
Pre‐pandemic |
|
Phipps 2020 (Pre‐print); 173 (76) Hospital inpatient; USA (not stated) |
Multi‐group study, including: [1] Single group of suspected COVID‐19 cases with available prior or same day PCR swab test result (n = 173) Excluded from current review: additional groups included to assess analytical specificity: [2] Healthy blood donors (n = 656, 240 pre‐pandemic and 416 from 2020) [3] Patients with SLE (n = 29) [4] Patients with rheumatoid arthritis (n = 20) [5] Patients with previous positive respiratory viral PCR panel (n = 90) | [1] RT‐PCR; commercial assay or
[2] isothermal PCR; Abbott ID NOW COVID‐19 assay Samples: nasopharyngeal swab |
Single negative PCR for absence of disease |
|
Pickering 2020 [A] (Published paper); 160 (110) Unclear; UK (4 March ‐ 21 April 2020) |
Two‐group study [1] RT‐PCR‐positive COVID‐19 patients ‐ venous serum samples collected at St Thomas’ Hospital, London (N = 87 patients, 110 samples) [2] pre‐Covid‐19 pandemic control samples (n = 50 samples, 50 patients) Further 200(?) samples were used for extended validations of 4 LFIAs. |
Real‐time RT‐PCR Samples: Not stated |
Pre‐pandemic |
|
Pollan 2020 (Published paper); 66 (24) Unclear; Spain (Not stated) |
2‐group study to estimate sensitivity and specificity for diagnosis of acute Covid 1] PCR‐confirmed Covid‐19 cases with serum samples (n = 82) 2] Pre‐pandemic serum samples for diagnosis of other pathogens (n = 42) [Study was reported as part of a wider seroprevalence survey; a second validation study of another immunoassay was also reported but not eligible for inclusion] | RT‐PCR Samples: Not stated |
Pre‐pandemic |
|
Prazuck 2020 [A] (Published paper); 427 (284) Hospital inpatient; France (March, 18, 2020 to April 10, 2020) |
[1] Suspected COVID‐19 patients who went to the hospital infectious diseases unit for a diagnostic consultation (n = 381) (patients whose symptoms, such as headache, fatigue, fever or respiratory signs suggest a COVID infection) [1a] RT‐PCR‐positive for COVID‐19 (n = 238) [1b] RT‐PCR‐negative for COVID‐19 (n = 143) | RT‐PCR assays for the detection of SARS‐CoV‐2 Samples: Nasopharyngeal (NP) swab specimens |
Real‐time RT‐PCR assays for the detection of SARS‐CoV‐2 |
|
Qian 2020 (Published paper); 2071 (513) Hospital inpatient; China (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Covid patients, n = 565 [1a] PCR+ COVID patients, n = 513 [1b] Suspected COVID patients with typical epidemiological history, clinical symptoms and featured chest CT images, n = 52 (54?) [2] Controls, n = 1558 [2a] Hospitalised patients (concurrent, other diseases, PCR‐ for SARS‐COV‐2), n = 972 [2b] Normal population (untested), n = 586 Group [1b] had no time pso reported so was not eligible for our review. | [1a] RT‐PCR, threshold not stated Samples: Not stated |
[2a] 972 hospitalised controls tested negative by RT‐PCR [2b] 586 normal population controls received no reference standard |
|
Qiu 2020 (Published paper); 864 (475) Community; China (January 20 2020 to March 12 2020) |
Two‐group study to estimate sensitivity and specificity: [1] Confirmed Covid cases n = 475 [2] Non‐COVID controls, concurrent non‐COVID patients n = 389 | RT‐qPCR Samples: Throat swabs |
As for case |
|
Ragnesola 2020 (Published paper); 63 (63) Unclear; plasma donors; New York, USA (Not stated) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients, convalescent plasma donor samples (n = 63); self‐reported, documented COVID‐19 disease by positive SARS‐CoV‐2 RT‐PCR test and complete resolution of symptoms at least 14 days prior to donation, and otherwise met all criteria for donating blood consistent with FDA’s policy on the Collection of COVID19 Convalescent Plasma [2] Pre‐pandemic samples (n = 10) Group [2] had < 25 samples and was excluded from our review. | Self‐reported documented COVID19 disease by positive SARS‐CoV‐2 RT‐PCR test (manufacturer and documentation not provided from referring institution of CP donors), threshold not stated Samples: Not stated |
NA |
|
Renard 2021 [A] (Published paper); 1670 (405) Mixed; hospital inpatients and outpatients; France (March 31 to June 2, 2020) |
Multi‐group study estimating sensitivity and specificity: [1] Symptomatic patients from three hospitals (inpatient and outpatients; RT‐PCR‐positive, N = 405 samples, n = 142 patients [2] Covid‐negative controls, N = 989 patients, pre‐pandemic healthy donors [3] Serum cross‐reactivity pre‐pandemic samples, n = 276 | RT‐PCR, threshold not stated Samples: Not stated |
[2] Pre‐pandemic (before September 2019) [3] Pre‐pandemic (timing unclear) |
|
Rijkers 2020 (Published paper); 62 (62) Hospital inpatient; The Netherlands (March 2020–May 2020) |
Single‐group study to estimate sensitivity only [1] Confirmed COVID patients (n = 62); [1a] Severe Covid‐19 group (n = 38); positive RT‐PCR and were hospitalised, both ICU and non ICU [1b] Mild Covid‐19 group (n = 24); hospital personnel (both from clinical departments as well as laboratory departments) who developed fever, coughing, and/or dyspnoea and had positive RT‐PCR and were non‐hospitalised with mild disease | RT‐PCR, threshold not stated Samples: [1a] Nasopharyngeal swabs [1b] Not stated |
NA |
|
Rode 2021 [A] (Published paper); 60 (60) Hospital inpatient; Croatia (Not stated) |
Single‐group study to estimate sensitivity [1] Subjects who had positive RT‐PCR and were hospitalised (n = 21, 60 samples) | RT‐qPCR; Charité protocol. Threshold not stated Samples: Combined nasopharyngeal and oropharyngeal swabs |
NA |
|
Rudolf 2020 [A] (Pre‐print); 976 (366) Community; Switzerland (first wave in Switzerland) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID samples; Serum of our previously described positive (SERO‐BL‐positive) cohort of study participants testing PCR‐positive for SARS‐CoV‐2 during the initial wave of COVID‐19 infections (n = 366) [2] Non‐COVID samples [2a] Blood donor samples from influenza seasons 2016/17 and 2017/18 (n = 500) [2b] Samples which tested PCR‐negative for SARS‐CoV‐2 from (SERO‐BL‐negative) cohort (n = 110) | [1] PCR‐positive for SARS‐CoV‐2 Samples: Not stated |
[2a] Pre‐pandemic [2b] PCR‐negative for SARS‐CoV‐2 |
|
Ruetalo 2020 [A] (Pre‐print); 46 (46) Unclear; Germany (April 04 to May 12, 2020) |
Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients [1a] Non‐hospitalised COVID‐patients (n = 49), 46 PCR+, 3 symptomatic close contacts who were potential blood donors for reconvalescent plasma therapy [1b] one hospitalised, convalescent COVID patient (2 samples) [2] Healthy donors (n = 4) Group [2] excluded from our review as < 25 samples. Group [1b] not included as no information on time pso or time post‐PCR+. [1a] 3 symptomatic close contacts excluded as not PCR‐confirmed | RT‐PCR Samples: Not stated |
NA |
|
Schnurra 2020 [A] (Published paper); 57 (57) Unclear; Germany (Not stated) |
Single‐group study to estimate sensitivity only [1] Confirmed COVID patients (73 sera from 57 patients) | Positive SARS CoV‐2 RNA test, threshold not stated Samples: Not stated |
NA |
|
Serre‐Miranda 2021 [A] (Published paper); 162 (126) Hospital inpatient; Portugal (Not stated) |
Two‐group study to estimate sensitivity and specificity [1] SARS‐CoV‐2‐infected inpatients (126 samples from 89 patients) [2] banked human plasma samples from 2 pre‐COVID‐19 pandemic studies (36 samples) [2a] Healthy (n = 25) and [2b] HIV and other viral diseases (n = 11) | [1] RT‐qPCR at a reference laboratory; threshold not stated Samples: Not stated |
[2] Pre‐pandemic |
|
Shamsollahi 2020 (Published paper); 312 (83) Quarantine (COVID suspects); Iran (Not stated) |
[1] 114 RT PCR‐confirmed COVID‐19 patients in hospitals affiliated to Tehran University of Medical Sciences in 2020 [2] 198 frozen serum specimens taken from healthy people in summer and autumn 2019 (pre‐COVID‐19) From group [1], time split 0‐19 days pso was excluded from our review (n = 31). | RT‐PCR ‐ no threshold reported Samples: not stated |
Pre‐pandemic blood samples. No report of these being tested by RT‐PCR |
|
Shen 2020a (Published paper); 150 (97) Quarantine; China (January 20, 2020 to February 2, 2020) |
Single‐group study to estimate sensitivity and specificity: Patients with fever or respiratory symptoms suspected as having COVID‐19 based on China CDC guideline (v5) (including 97 RT‐PCR‐confirmed) [A separate cohort of 26 healthy blood donors were tested ‐ not included in main analysis] | RT‐PCR at referral laboratory; not further described Samples: Nasopharyngeal and oropharyngeal swab samples | As above |
|
Shen 2020b (Published paper); 130 (70) Hospital inpatient; [1] and [2] China (Not stated) |
Study 1: [1] RT‐qPCR‐confirmed COVID‐19 cases (n = 45) [2] clinically‐confirmed but RT‐qPCR‐negative COVID‐19 patients (n = 25) [3] patients with non‐coronaviral respiratory illness (n = 10) (2 confirmed for influenza A virus, 3 confirmed for influenza B virus, 3 confirmed for respiratory syncytial virus and 2 confirmed for adenovirus) [4] negative control, consisted of 50 sera samples collected from 50 healthy people assessed by physical examination (n = 50) | [1] RT‐PCR
[2] Clinically‐confirmed but RT‐qPCR‐negative Samples: [1] and [2] Not stated |
[3] Not stated [4] Pre‐pandemic |
|
Soleimani 2021 (Published paper); 276 (176) Hospital inpatient; Belgium (25 February to 10 March 2020) |
Multi‐group study to estimate sensitivity and specificity [1] Symptomatic and hospitalised patients positive RT‐qPCR tests and characteristic radiological lung patterns such as ground‐glass opacity and/or bilateral involvement (176 samples obtained from 125 patients) [2] Non‐COVID (100 samples) [2a] Pre‐pandemic healthy (n = 40) with no known confounding factors [2b] Pre‐pandemic, other diseases (n = 40); supposedly confounding factors known to interfere with serological assays such as autoimmune Ab and infectious diseases Ab [2c] Asymptomatic subjects in overlapping period of Flu epidemic and COVID‐19 outbreak March 2020 (n = 20) | RT‐qPCR and radiographic criteria (bilateral chest
involvement and/or ground‐glass opacity [GGO] identified by X‐ray or computed tomography [CT] scan) Samples: nasopharyngeal swab samples |
[2a] and [2b] Pre‐pandemic [2c] Current asymptomatic, untested |
|
Sterlin 2021 [A] (Pre‐print); 214 (214) Hospital inpatient; France (March 22 to April 24, 2020) |
Group [1]: PCR‐confirmed adult COVID‐19 cases (n = 135) Group [2]: Age‐ and sex‐matched healthy donors (n = 20) Group [3]: 10 cases with CT scan displaying features suggesting a COVID‐19 infection and tested positive for the presence of serum anti‐SARS‐CoV‐2 antibodies Group [2] was excluded from the review as < 25 controls. Group [3] was excluded as < 10 cases and no test accuracy outcomes | RT‐PCR assay (no more details available) Samples: Nasopharyngeal swabs |
NA |
|
Suhandynata 2020a (Published paper); 289 (54) Hospital inpatient; USA (Not stated) |
Multiple‐group study to estimate sensitivity and specificity: [1] laboratory‐confirmed COVID‐19 patients (n = 54); [2] patients PCR‐positive on a respiratory panel nucleic acid (RPNA) test for other infections (n = 21), [3] patients with positive for antinuclear antibodies (ANA) or anti‐double stranded DNA (dsDNA) (n = 24) [4] HIV‐positive patients (n = 10), [5] apparently healthy subjects (no respiratory symptoms per self‐report) (n = 78), [6] pre‐pandemic samples (n = 102) | Not stated, EUA NAT that had been clinically validated in the laboratory Samples: not stated |
Not stated for group [2] to [5]; group [6] was pre‐pandemic |
|
Suhandynata 2020b [A] (Published paper); 204 (25) Unclear; California, USA (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 60) [2] Non‐COVID subjects (n = 179) [2a] Current, other diseases (n = 22); PCR‐positive on a respiratory panel nucleic acid (RPNA) test infections other than SARS‐CoV‐2 [2b] Current, positive for other antibodies, DNA or IgM/IgG (n = 27) [2c] Current, apparently healthy subjects (n = 20), (no respiratory symptoms per self‐report) [2d] Pre‐pandemic samples (n = 110) | NAT; clinically validated and EUA listing Threshold not stated. Samples: Not stated |
[2a], [2b] ePlex PCR (GenMark ePlex); detects adenovirus (A‐F), coronavirus (229E, HKU1, NL63, OC42), human metapneumovirus, human rhinovirus/enterovirus, influenza A, B and C, influenza 2009 H1N1, parainfluenza (1‐4), respiratory syncytial virus (A and B), chlamydia pneumoniae and mycoplasma pneumoniae. [2c] Untested (no respiratory symptoms per self‐report) [2d] Pre‐pandemic |
|
Sun 2020 (Published paper); 209 (209) Hospital inpatient; China (23 January to 27 February 2020) |
Two‐group study to estimate sensitivity and specificity [1] Hospitalised COVID‐19 cases in two designated hospitals (209 samples from 35 patients) [2] Healthy close contacts (n = 21) Group [2] excluded from review as < 25 samples | Laboratory confirmed by >= 1 of: isolation of virus, RT‐PCR or a genome sequence that matches SARS‐CoV‐2 Samples: Respiratory specimens |
NA |
|
Sweeney 2020 (Pre‐print); 601 (301) Unclear; UK (Not stated) |
[1] PCR‐confirmed SARS‐CoV‐2‐positive individuals (n = 301) [2] Pre‐pandemic stored serum samples [unclear if diseased or not] (n = 200) [3] Pre‐pandemic stored acute and convalescent confounder samples from individuals with a range of viral, bacterial and fungal pathogens (n = 100) | RT‐PCR (AusDiagnostics); threshold not stated (reference PHE 2020 rapid assessment) Samples: Unclear |
Pre‐pandemic |
|
Tan 2020 [A] (Published paper); 333 (170) Hospital inpatient; Singapore (30 March 2020 and 15 May 2020) |
[1] Inpatients with >= 1 RT‐PCR‐positive result (n = 170) [2] Pre‐pandemic (n = 163) [2a] Healthy controls (n = 60) [2b] Cross‐reactivity group (n = 103) | RT‐PCR; commercial assay Samples: Respiratory samples |
Pre‐pandemic. |
|
Tang 2020 [A] (Published paper); 256 (103) Hospital inpatient; USA (No information) |
Multiple‐group study estimating sensitivity and specificity: [1] residual serum samples from patients with laboratory‐confirmed COVID‐19 infection and physician ordered completed blood count (n = 48, providing 103 samples) [2] PCR‐negative COVID‐19 suspects (n = 80); [3] pre‐pandemic serum (n = 50) [4] PCR‐negative, with other confirmed coronavirus (HKU1, NL63, and 229E) (n = 5) or influenza A or B (n = 4) [5] serum from patients with potentially interfering antibodies (n = 14; CMV IgG (n = 5), EBV VCA IgG (n = 3) or IgM (n = 3) or both (n = 2), RF+ (n = 1)) | RT‐PCR using one of three platforms Samples: nasopharyngeal (NP) swabs, oropharyngeal (OP) swabs, or lower respiratory tract specimens (only latter used with Diasorin Simplexa) |
COVID‐19 suspects – same as for cases Unclear reference for other interfering antibody samples (n = 14); pre‐pandemic for remaining 50 |
|
Theel 2020 [A] (Published paper); 476 (213) Mixed; Hospital inpatient and outpatient; USA (March and April 2020) |
[1] serum samples from patients with confirmed COVID‐19 (n = 56, 224 samples) [2] healthy donor sera from 2018 (n = 149 samples) [3] cross‐reactivity serum panel collected in early 2020 (n = 105 samples, see comments) In group [1], 11 samples from outpatients would be excluded from our review as taken 0‐7 days post‐positive PCR. | SARS‐CoV‐2 RT‐PCR assay (laboratory‐developed or commercially available FDA EUA) Samples: nasopharyngeal swab |
[2] Pre‐pandemic [3] Not stated |
|
Thijsen 2020 (Published letter); 43 (27) Hospital inpatient; Netherlands (Not stated) |
Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 27) Group [2]: Healthy controls (n = 16) | RT‐PCR (no more details available) Samples: Not stated |
Not stated, but likely no testing |
|
Trabaud 2020 [A] (Published paper); 66 (66) Hospital in‐ and outpatient; France (Not stated) |
Single‐group study to estimate sensitivity [1] RT‐PCR‐confirmed infection (N = 68, 82 samples) [1a] hospitalised patients (N = 40) [1b] non‐hospitalised healthcare workers (N = 28) | RT‐PCR, threshold not stated Samples: Not stated |
NA |
|
Traugott 2020 [A] (Published paper); 177 (77) Hospital inpatient; Austria (27th February to 30th March 2020) |
Multi‐group study estimating sensitivity and specificity [1] Symptomatic patients with acute PCR‐confirmed COVID‐19 infection (n = 77) [2] Symptomatic patients with negative PCR results (n = 30) [3] Healthy volunteers with negative PCR (n = 30) [4] Stored samples from individuals with previous PCR‐confirmed coronavirus OC43 infection (n = 10); interval from infection to sampling of 4 to 1452 days [5] Pre‐pandemic samples from patients with pneumonia (n = 30) | RT‐PCR; WHO‐recommended primers Samples: Nasopharyngeal swab/respiratory secretion samples |
[2] RT‐PCR [3] RT‐PCR [4] Unclear ('stored') [5] Pre‐pandemic |
|
Tre‐Hardy 2021 [A] (Published paper); 125 (44) Hospital inpatient; Belgium (April 16 to 20, 2020) |
Retrospective two‐group analysis to estimate sensitivity and specificity (n = 125) [1] patients with mild, severe or critical infection. Patients were considered positive according to the results of the RT‐qPCR (n = 44) [2] Pre‐pandemic patients (n = 81) [2a] Cross‐reactivity panel with other viral, bacterial, parasitic or autoimmune pathologies (n = 75) [2b] Healthy subjects (n = 6); no history of known auto‐immune pathologies and without any acute infection of viral or bacterial origin | RT‐qPCR, threshold not stated Samples: Respiratory samples |
Pre‐pandemic |
|
Tuaillon 2020 [A] (Pre‐print); 58 (38) Hospital inpatient; France (from 18 March 2020 (ongoing)) |
Two‐group study to estimate sensitivity and specificity, including: [1] Hospitalised patients with PCR‐proven or suspected COVID‐19 Infection (PCR‐negative were excluded), n = 38 samples Additional group of pre‐pandemic controls not included in review [2] Pre‐pandemic controls (samples collected in 2017‐2018 from patients care in the Department of Infectious Diseases), n = 20 | RT‐PCR; no details Samples: Not stated |
Pre‐pandemic |
|
Valdivia 2020 [A] (Published paper); 110 (90) Hospital inpatient; Spain (March 5 and April 30, 2020) |
Two‐group study to assess sensitivity and specificity: [1] Laboratory‐confirmed SARS‐CoV‐2 infection (n = 90) [2] Pre‐pandemic controls (n = 20); healthy individuals in 2019, 10 of which had prior endemic coronavirus infection | SARS‐COV2‐RT PCR Samples: Not stated |
Pre‐pandemic |
|
Van Elslande 2020a [A] (Published paper); 197 (94) Hospital inpatient; Belgium (March and April 2020) |
Multiple‐group design with separate estimates of sensitivity and specificity [1] Symptomatic PCR‐confirmed COVID‐19 cases with available residual samples (n = 94) [2] Pre‐pandemic patients with a respiratory infection who had a PCR test for respiratory pathogens (n = 49) [3] Pre‐pandemic other infections (patients with confirmed non‐SARS‐CoV‐2 coronavirus infection) (n = 14) [4] Pre‐pandemic other infections (patients with antigens against other pathogens (e.g. CMV, EBV, HIV) from routine serology testing) (n = 40) | RT‐PCR; described as 'inhouse method complying with the WHO guidelines' Samples: Nasopharygeal swabs in UTM |
[2] Pre‐pandemic [3] PCR [4] Antibody test |
|
Van Elslande 2020b [A] (Published paper); 346 (233) Hospital inpatient; Belgium (Not stated) |
Multi‐group study to assess sensitivity and specificity [1] Covid‐positive (n = 233 samples, 114 patients) [2] Covid‐negative, pre‐pandemic (n = 113) [2a] Pre‐pandemic respiratory infection (n = 49) [2b] Pre‐pandemic coronavirus (n = 24) [2c] Pre‐pandemic other infections (n = 40) | RT‐PCR; described as 'in‐house method complying with the WHO guidelines', threshold not stated Samples: Nasopharygeal swabs (UTM, Copan, Italy) |
Pre‐pandemic |
|
Velay 2020 [A] (Published paper); 325 (198) Hospital inpatient; France (2020 April) |
Multi‐group analysis to estimate sensitivity and specificity (n = 325) [1a] PCR‐confirmed hospital patients (n = 55) [1b] PCR‐confirmed healthcare workers (n = 143) [2a] Pre‐pandemic healthy blood donors (n = 100) [2b] Cross‐reactivity negative controls (n = 27); (n = 20) anti‐hCoV‐positive, (n = 2) anti‐influenza A virus‐positive, (n = 1) anti‐rhinovirus‐positive, (n = 2) RF‐positive, (n = 2) ANA‐positive | RT‐PCR according to current guidelines (Institut Pasteur, Paris, France; WHO technical guidance) Samples: Nasopharyngeal |
Pre‐pandemic |
|
Veyrenche 2021 [A] (Published paper); 45 (45) Hospital inpatient; France (14 March to 11 April 2020) |
Two‐group study to assess sensitivity and specificity [1] Hospital inpatients with RT‐PCR‐confirmed SARS‐CoV‐2 infection. Any disease severity (n = 45) [2] Non‐COVID controls (n = 20) Group [2] not eligible for our review as < 25 samples leaving a single‐group study to estimate sensitivity only | RT‐PCR; commercial assay Samples: Nasopharyngeal |
NA |
| Wang 2020a (Published paper); 375 (141) Inpatient; Nanchong, China (25th January to 16th March) | Single‐group study to estimate sensitivity and specificity in patients who visited the hospital with respiratory complaints during January to March 2020 (n = 375) | New Coronavirus Pneumonia Prevention and Control Program (7th edition) definition; specifically: [1] RT‐PCR; commercial assay [2] RT‐PCR‐negative with characteristic CT changes of the lungs | As for diseased |
|
Weidner 2020 [A] (Published paper); 100 (100) Community; Austria (Not stated) |
Single‐group study estimating sensitivity Used serum samples from convalescent plasma donors with Nucleic Acid Test (NAT)‐confirmed COVID‐19 (n = 100) | Nucleic Acid Test NAAT (no more details available) Samples: Nasopharyngeal swabs or pharyngeal swabs |
NA |
|
Wellinghausen 2020a [A] (Published paper); 67 (67) Mixed; Hospital outpatient or community; Germany (March 24th to May 6th 2020) |
Single‐group analysis to assess sensitivity [1] Covid patients (n = 67 samples from 58 patients); PCR+ at least 7 days before serum collection [1a] Symptomatic Covid outpatients (n = 60 samples from 51 patients) [1b] Asymptomatic Covid patients (n = 7 samples from 7 patients) | RT‐PCR; multiple assays used; threshold not stated Samples: nasopharyngeal swabs |
NA |
|
Wellinghausen 2020b (Published paper); 126 (126) Hospital outpatients or community; Germany (March 24th to May 6th 2020) |
Single‐group study to assess sensitivity [1] Covid patients PCR+ at least 7 days before serum collection (n = 137) [1a] Symptomatic outpatients (n = 111) [1b] Asymptomatic, PCR‐confirmed contacts (n = 26) | RT‐PCR; multiple assays used; threshold not stated Samples: nasopharyngeal swabs |
NA |
|
Whitman 2020a [A] (Published paper); 287 (128) Mixed; hospital inpatients and outpatients; USA (Not stated) |
Multi‐group study to assess sensitivity and specificity [1] In or outpatients with symptomatic infection, positive SARS‐CoV‐2 RT–PCR and remnant plasma and serum samples (n = 128 samples from 79 patients) [2] Non‐Covid patients (n = 159) [2a] Pre‐pandemic blood donors (n = 108) [2b] Concurrent patients from 2020 with detection of other respiratory viruses (n = 41) [2c] Concurrent, SARS‐COV‐2 PCR‐negative, no other viruses detected (n = 10) | RT‐PCR, threshold not stated Samples: Nasopharyngeal or oropharyngeal swabs |
[2a] Pre‐pandemic [2b] RT‐PCR‐negative or none [2c] RT‐PCR‐negative |
|
Whitman 2020b [A] (Pre‐print); 104 (44) Inpatient; USA (Not stated) |
Two‐group study estimating sensitivity and specificity [1] SARS‐CoV‐2 RT‐PCR‐positive (n = 44) [2] pre‐pandemic asymptomatic adults (n = 30) [3] pre‐pandemic other infection controls with febrile and/or respiratory illness (n = 30) | RT‐PCR; threshold NR Samples: Not stated |
[2] + [3] |
|
Wolff 2020 [A] (Published paper); 207 (111) Unclear; Belgium (Not stated) |
Multi‐group study to estimate sensitivity and specificity [1] PCR‐confirmed Covid patients (n = 111) [1a] symptomatic (mild to moderate or severe) Covid (n = 87) [1b] asymptomatic Covid screened due to close contacts with COVID‐19 patients (n = 24) [2] Pre‐pandemic, non‐Covid (n = 96) | qRT‐PCR; commercial assay Samples: nasopharyngeal swabs, threshold not stated Samples: Not stated |
Pre‐pandemic |
|
Wu 2020 [A] (Published paper); 74 (16) Hospital inpatient; Taiwan (January 23rd to April 25, 2020) |
Two‐group study to estimate sensitivity and specificity, however, it appeared that all participants met Taiwanese reporting criteria for COVID‐19: [1] PCR‐confirmed symptomatic and hospitalised Covid‐19 patients (n = 16) [2] Inpatients with respiratory tract infection or fever but 2 negative PCR results for SARS‐CoV‐2 (n = 58) (All patients meeting criteria for testing were simultaneously evaluated for SARS‐CoV‐2 and influenza A/B; if both PCR results were negative, an additional SARS‐CoV‐2 test was performed using a second sample from the suspected COVID patient) | rRT‐PCR Samples: Throat or lower respiratory specimens (OP, NP, sputum, gargling) |
As for disease‐positive |
|
Xiang 2020a (Published paper); 150 (85) Hospital patients (likely inpatients but not explicit); Wuhan, China (Jan 19 to Mar 2, 2020) |
Two or more groups: [1] RT‐PCR‐confirmed cases (n = 85) [2] Suspected cases with Covid‐19 pneumonia manifestations (and in some cases exposure) but had all had two or more negative RT‐PCR and none were positive (and protocol is to retest RT‐PCR negatives every 1‐2 days) (n = 24). Classed as disease‐positive for review purposes. Non‐Covid cases: [3] Contemporaneous control group of healthy blood donors (hospital staff) or patients with other diseases in the same hospital (all PCR‐negative) (n = 60) |
[1] RT‐PCR [2] Symptoms, Covid‐19 pneumonia manifestations and PCR‐negative at least twice | [3] Contemporaneous control group of healthy blood donors (hospital staff) or patients with other diseases in the same hospital (all PCR‐negative) (n = 60) |
|
Xiao 2020a (Published paper); 34 (34) Inpatients; Wuhan, China (Feb 1 to 29) |
Single‐group (cases); confirmed cases of COVID‐19 according to Chinese CDC (5th ed) (34) | Covid‐19 according to CDC diagnosis and treatment guideline (5th ed); not stated (not stated) | NA |
|
Xiao 2020b [A] (Pre‐print); 75 (75) Hospital inpatient; China (January 23, 2020‐April 1, 2020) |
Three‐group study to estimate sensitivity according to symptomatic status:
Patients who tested positive for the SARS‐CoV‐2 virus or had serum antibodies to the virus (1) 23 asymptomatic cases (no self‐perceived or clinically recognisable symptoms from admission to 14 days post‐discharge) (2) 33 pre‐symptomatic cases (asymptomatic on admission, but showed symptoms during hospitalisation) (3) 19 age‐matched symptomatic cases; Discharge criteria for recovered patients: 1) normal temperature for more than 3 days; 2) respiratory symptoms significantly improved, significant absorption of pulmonary lesions on CT; 3) two consecutive negative RNA tests in 24 h |
RT‐PCR or antibody tests for SARS‐CoV‐2 (not described), as per Chinese NHC guidelines (version 6) Samples: respiratory specimens for COVID‐19 confirmation; anal swabs also obtained (appeared from Figure that fewer anal swabs obtained compared to respiratory) |
NA |
|
Yang 2020 [A] (Published paper); 696 (120) Mixed; inpatient and ED; United States (6th March to 4th April 2020) |
Multi‐group study to estimate sensitivity and specificity: [1] patients presenting to ED and displaying signs and symptoms suspicious for COVID 19 (n = 87, only 42 PCR‐confirmed cases providing 120 samples could be included in the review) [2] Pre‐pandemic ED patients (sample number = 320; unclear patient number) [3] Pre‐pandemic healthy blood donors (n = 256) Groups [2] and [3] used for different assays [Additional cohorts were reported but not extracted] | RT‐PCR; multiple assays used; threshold not stated Samples: nasopharyngeal swabs |
[2] and [3] Pre‐pandemic controls [4] [5] unclear [6] PCR+ for other infection |
|
Yongchen 2020 (Published paper); 21 (21) Mixed; Jiangsu, China (Jan 25 to Mar 18, 2020) |
Single group (cases); Participants with COVID‐19 (n = 16) and asymptomatic carriers (n = 5). | RT‐PCR ‐ confirmed after two sequential positive respiratory tract sample results; Throat swabs (throat swab samples collected every 1‐2 days) | NA |
|
Zhang 2020a [A] (Published paper); 1730 (574) Mixed; hospital inpatients and outpatients, community, contacts; China (December 2019 to March 2020) |
Multi‐group study to assess sensitivity and specificity [1] Clinically confirmed Covid patients (572 samples) [1a] Confirmed hospitalised cases (338 samples from 164 patients) [1b] Follow‐up cases (234 samples from 234 patients) [2] Non‐Covid cases (n = 996) [2a] Healthy controls (n = 600) [2b] With other diseases, respiratory disease (n = 57), orthopaedic disease (n = 8), hepatobiliary disease (n = 48), gynaecological disease (n = 50), auto‐immune disease (n = 10), endocrine disease (n = 41), dermal disease (n = 18), nervous system disease (n = 13), kidney disease (n = 32), digestive disease (n = 64), cardiovascular disease (n = 24), blood disease (n = 21), other disease (n = 10) (n = 396) [3] Suspected COVID patients, close contact with COVID patients (162 samples from 154 patients) | [1] Clinically defined, criteria not described
Possibly RNA test and CT image?
[3] RNA test and characteristic CT image Samples: Not stated |
[2] Not stated, none [3] No fever, negative result of RNA detection, and no abnormal in CT image. |
|
Zhang 2020b [A] (Published paper); 112 (112) Hospital inpatient; China (February 1 to February 29, 2020) |
Single‐group study to estimate sensitivity only: [1] COVID‐19 patients (all RT‐PCR‐positive) (who were diagnosed based on the New Coronavirus Pneumonia Prevention and Control Program (4th edition) published by the National Health Commission of China, with positive results for severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) using quantitative RT‐PCR (qRT‐PCR) with samples from the respiratory tract) | RT ‐ RT‐PCR; commercial assay Samples: Nasopharynx and oropharynx swabs |
NA |
|
Zhao 2020a [A] (Published letter); 386 (173) Inpatients; Shenzhen, China (Jan 11 to Feb 9) |
Two or more groups Confirmed RT‐PCR‐positive COVID‐19 cases (173) . Non‐Covid cases: Pre‐pandemic healthy individuals (213) |
RT‐PCR; Respiratory (not stated) | Pre‐pandemic healthy individuals (213) |
|
Footnotes A&E: Accident and Emergency department ANA: antinuclear antibody BMI: body mass index CMV: cytamegalovirus CRU: cardiorespiratory unit CT: computerised tomography DABA: double antigen binding assay DNA: deoxyribonucleic acid Ds‐DNA: double‐stranded deoxyribonucleic acid EBV: Epstein‐Barr virus ED: emergency department ELISA: enzyme linked immunosorbent assay HBC: hemoglobin count HBsAg: hepatitis B surface antigen HBV: hepatitis B virus HCV: hepatitis C virus HCW: health care worker HIV: human immunodeficiency virus HS: health screening HSV: herpes simplex virus ICU: intensive care unit NA: nucleic acid NAAT: nucleic acid amplification test NAT: nucleid acid test NHC: national health commission NHS: National Health Service NP: nasopharyngeal NR: not reported OP: oropharyngeal PBS: phosphate‐buffered saline PHE: Public Health England PRNT: plaque reduction neutralization test PUI: person under investigation RF: rheumatoid factor RNA: ribonucleic acid RPP: respiratory pathogen panel RT: reverse transcription RT‐PCR: reverse transcription polymerase chain reaction SARS: severe acute respiratory syndrome TMA: transcription‐mediated amplification UTM: universal transport medium VIDRL: victorian infectious diseases reference laboratory VZV: varicella zoster virus |
|||
Appendix 7. Summary details of index tests per study
| Study Test methods | Index test (manufacturer) | Antigens used | Antibodies measured | Index sample timing (days pso) | Sample type |
|
Adams 2020 Laboratory only; ELISA |
Mologic ‐ IgG COVID‐19 ELISA | NP and S2 antigens | IgG | [1] post‐symptom onset (range 1 to 54 d based on Figure 3) < 7: n = 16 (6%) (not reported but back‐calculated from Table 3B) ≥ 7‐14, n = 32 ≥ 14‐21, n = 45 ≥ 21‐28, n = 58 ≥ 28‐35, n = 30 ≥ 35, n = 29, 11% asymptomatic, n = 60 Also reported day post‐PCR (see time interval between index and reference standard) | Serum |
|
Alvim 2020 Laboratory only; ELISA |
Euroimmun ‐ anti‐SARS‐COV‐2 IgG ELISA (#EI 2606‐9601 G) | S1 subunit | IgG | Samples collected from D0 after symptom onset, up to 98 d after symptom onset 0‐5 d pso: n = 33 6‐10 d pso: n = 42 11‐15 d pso: n = 83 16‐20 d pso: n = 62 21‐25 d pso: n = 56 26‐30 d pso: n = 54 31‐98 d pso: n = 107 | Unclear |
|
Andrey 2020a [A] Andrey 2020a [B] Laboratory and LFA; [A] LFA; [B] ELISA |
[A] Augurix ‐ SARS‐CoV‐2 IgM/IgG RDT [B] Euroimmun AG‐ SARS‐CoV‐2 IgG ELISA (# EI 2606‐9601 G) | [A] Not stated [B] S1 domain | [A] IgM and/or IgG [B] IgG | Median 10 d (IQR 5‐15 d) post‐+ve PCR results: 0‐6 d post‐+ve PCR: n = 20 7‐14 d post‐+ve PCR: n = 14 > 14 d post‐+ve PCR: n = 12 | [A] 20 µL whole blood & 10 µL plasma applied in parallel [B] Plasma |
|
Andrey 2020b [A] Andrey 2020b [B] Andrey 2020b [C] Laboratory and LFA; [A]‐[B] LFA; [C] ELISA |
[A] NTBIO Diagnostics In (test name not stated) [B] Zhejiang Orient‐Gene Biotech Co. Ltd (test name not stated) [C] MEDsan GmbH RDT (test name not stated) [D] Euroimmun IgG ELISA (# EI 2606‐9601 G) | [A] Full Spike‐protein [B] Not stated [C] N‐ and S‐based [D] S1 domain | [A] ‐[C] IgM and/or IgG [D] IgG | Median 22 d (IQR 13–31 d) post‐+ve PCR: 0‐14 d post‐PCR+: n = 14 > 14 d post‐PCR+: n = 27 | [A]‐[C] Whole blood and plasma [D] Plasma |
|
Bartolini 2020 [A] Bartolini 2020 [B] LFA only; [A] CGIA; [B] LFA |
[A] KHB® Diagnostic Kit for SARS‐ CoV‐2 IgM/IgG Antibody (Colloidal Gold) [B] Cellex qSARS‐CoV‐ 2 IgG/IgM Cassette Rapid Test | Not stated ‐ SARS‐CoV‐2 conjugate | [A] IgG; IgM [B] IgG; IgM | All 0‐45 d post‐+ve RT‐PCR ([1]/[2]) 0‐5 d: 3/151 (2/1) 6‐10 d: 3/151 (3/0) 11‐15 d: 16/151 (3/13) 16‐20 d: 28/151 (6/22) 21‐25 d: 43/151 (19/24) 26‐30 d: 19/151 (2/17) 31‐35 d: 25/151 (0/25) 36‐40 d: 11/151 (0/11) 41‐45 d: 3/151 (0/3) | Plasma |
|
Beavis 2020 Laboratory only; ELISA |
EUROIMMUN Anti‐SARS‐CoV‐2 ELISA IgG and IgA assays | S1 domain | IgG, IgA | 0 to 49 d after PCR testing | Serum or EDTA plasma |
|
Bernasconi 2020 LFA only; LFA |
Maccura Biotechnology LFIA SARS‐CoV‐2 IgM/IgG | S‐ and N‐ proteins | IgM, IgG | [1] COVID‐19‐+ve patients (n = 67): < 7 d onset (n = 21), ≥ 7 d onset (n = 46) Figure 1 in paper reported on 135 samples from 1‐31 d pso. [2] COVID‐19‐negative patients ‐ not stated | Not stated |
|
Bettencourt 2020 LFA only; LFA |
Biozec COVID‐19 IgM/IgG Rapid Test LFIA | Not stated | IgM and IgG | Mean 20.5 d (18–23) pso | Not stated |
|
Bond 2020 Laboratory only; ELISA |
Study evaluated multiple assays; timing pso provided only for one of them: EUROIMMUN Anti‐SARS‐CoV‐2 ELISA (IgA, IgG) | S1 domain | IgA, IgG | Any time point (229 samples); > 14 d (157 samples) | Serum |
|
Boukli 2020 [A] Boukli 2020 [B] Laboratory only; [A, B] CLIA |
[A] DiaSorin ‐ Liaison SARS‐CoV‐2 S1/S2 IgG assay [B] Abbott Diagnostics ‐ Alinity I SARS‐CoV‐2 IgG assay | [A] Recombinant S1 and S2‐proteins; [B] N antigen | [A] and [B] IgG | Not stated Day 1 to day 30 pso | Group [2], [3], [4] serum Group [1] plasma; samples stored at ‐20 or ‐80°C |
|
Brochot 2020 [A] Brochot 2020 [B] Brochot 2020 [C] Brochot 2020 [D] Brochot 2020 [E] Laboratory only; [A to C] ELISA; [D, E] CLIA |
Assays identified only by manufacturer: [A] Abbott; [B] Biorad; [C] Euroimmun; [D] Liaison; [E] Wantai. | [A] nucleocapsid; [B] nucleocapsid; [C] spike 1; [D] spike1/2; [E] RBD |
[A] IgG; [B] total antibodies; [C] IgG; [D] IgG; [E] total antibodies. |
Time pso not given; number of samples by time post‐PCR+ given only for day 31‐50 (n = 21) and > 50 d (n = 14) | Serum |
|
Bryan 2020a Laboratory only; CLIA |
Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG | Nucleocapsid | IgG | < 7 d pso: 24/41 7‐10 d pso: 10/41 11‐14 d pso: 2/41 > 14 d pso: 5/41 | Serum or plasma |
|
Bundschuh 2020 Laboratory only; ELISA |
Epitope Diagnostics Inc. ‐ EDI™ Novel Coronavirus COVID‐19 IgM and IgG ELISA kit | N‐protein of SARS‐CoV‐2 | IgM, IgG | < 5 d ‐ 22 d pso (COVID‐19 patients) Results were reported for 4 time bands: < 5 d 34/104 samples; 5‐10 d 35/104 samples; > 10‐15 d 17/104 samples; > 15‐22 d 18/104 samples. |
Plasma (EDTA and lithium‐heparin anticoagulated blood were collected and plasma aliquots were frozen at −80 °C until further analysis) |
|
Butterfield 2021 [A] Butterfield 2021 [B] Butterfield 2021 [C] Butterfield 2021 [D] Butterfield 2021 [E] Butterfield 2021 [F] Laboratory and LFA; [A‐C] CLIA, [D‐E] ELISA, [F] LFA |
[A] Roche Elecsys1 Anti‐SARS‐CoV‐2, [B] Abbott Architect SARS‐CoV‐2 IgM, [C] Abbott Architect SARS‐CoV‐2 IgG, [D] Euroimmun SARS‐CoV‐2 IgA, [E] Euroimmun SARS‐CoV‐2 IgG ELISA, [F] Trillium IgG/IgM rapid assays. | Not stated | [A] Total Ab; [B] IgM; [C] IgG; [D] IgA; [E] IgG; [F] IgG/IgM. |
Symptomatic: 6–103 d pso; Asymptomatic: 20–69 d post‐PCR+. |
Blood samples collected in tubes without anticoagulant |
|
Candel 2020 LFA only; CGIA |
Autobio Diagnostics Co Anti‐SARS‐CoV‐2 Rapid Test | Spike | IgM, IgG | Mean 28 d pso (SD: 8.7) Range 16‐48 d pso: 16‐21 d pso: 8/35 22‐28 d pso: 14/35 > 28 d pso: 13/35 | Whole blood |
|
Carozzi 2020 [A] Carozzi 2020 [B]LFA only; [A] [B] LFA |
[A] Screen Italia ‐ Screen Test Covid‐19 2019‐nCOV IgG/IgM [B] Zhejiang Orient Gene Biotech Co ‐ COVID‐19 IgG/IgM rapid test cassette | Not stated | [A] [B] IgG/IgM | 14+ d post‐PCR | [1a] [2a] Serum |
|
Carta 2020 Laboratory only; CLIA |
Shenzen New Industries Biomedical Engineering Co (SNIBE) MAGLUMI 2019‐nCoV IgG and IgM | [A] S and N‐proteins | [A] IgG/IgM | Day 32 (28–36) and 49 (47–50) post‐PCR +ve | Serum |
|
Case 2020 [A] Case 2020 [B] Laboratory only; [A] [B] ELISA |
[A] Euroimmun ‐ Anti‐SARS‐CoV‐2 IgG ELISA [B] Epitope IgG ELISA | [A] SARS‐CoV‐2 S‐protein [B] Not stated | [A] and [B] IgG | 5‐7 d pso: 5/40 (13%) 8‐14 d pso: 23/40 (50%) 15‐20 d pso: 12/40 (30%) 2 not stated | Serum |
|
Caturegli 2020 Laboratory only; ELISA |
EUROIMMUN AG ‐ Anti‐SARS‐CoV‐2 ELISA IgG and IgA assays | [A], [B]: S1 domain | [A]: IgG [B]: IgA | Multiple samples taken from each patient at various points in time, from 0 to 59 d pso | Residual serum samples |
|
Cervia 2020 Laboratory only; ELISA |
EUROIMMUN ‐ Anti‐SARS‐CoV‐2 ELISA IgG and IgA assays | SARS‐CoV‐2 Spike‐protein (S1) | IgA, IgG | [1] mean 16.4 d (median 13 d) for the mild group and approx day 2 to day 48; mean 20.9 d (median 16 d) for the severe group since symptom onset | Serum |
|
Chan 2020a Laboratory only; CLIA |
[A] Roche diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 antibody assay [B] EuroImmun ‐ anti‐SARS‐CoV‐2 IgG ELISA | [A] N‐protein [B] Not stated | [A] Total antibody [B] IgA/IgG | [1a] 0‐13 d post‐PCR + (n = 40) ≥ 14 d post‐PCR + (n = 38) | Serum and plasma |
|
Charlton 2020 [A] Charlton 2020 [B] Charlton 2020 [C] Charlton 2020 [D] Charlton 2020 [E] Charlton 2020 [F] Charlton 2020 [G] Charlton 2020 [H] Charlton 2020 [I] Charlton 2020 [J] Charlton 2020 [K] Charlton 2020 [L] Laboratory and LFA; [A, D, F] CLIA; [B, C, E] ELISA; [G ‐ L] LFA |
[A] Abbott Laboratories ‐ SARS‐CoV‐2 IgG assay [B] Epitope Diagnostics Inc ‐ EDI novel coronavirus COVID‐19 IgM and IgG ELISA [C] DRG International Inc., supplied by Bio‐Rad ‐ a novel coronavirus COVID‐19 IgM and IgG assay [D] DiaSorin ‐ SARS‐CoV‐2 S1/S2 IgG [E] Euroimmun ‐ anti‐SARS‐CoV‐2 ELISA IgA and IgG assay [F] Roche Diagnostics ‐ anti‐SARS‐CoV‐2 [G] BTNX ‐ Rapid Response [H] Biolidics Limited ‐ 2019 nCoV IgM/IgG detection kit [I] Anhui Deep Blue Medical Technology Co ‐ SARS‐CoV‐2 IgG/IgM Ab test kit [J] Genrui Biotech Inc ‐ Novel Coronavirus IgG/IgM test kit [K] Getein Biotech Inc ‐ One Step Test for Novel Coronavirus [L] Innovita Biological Technology Co ‐ 2019‐nCoV Ab test | [A] N‐protein
[B] RBD and Spike‐protein
[C] N‐proteins and peptides
[D] S1 and S2 domains
[E] S1 domain [F] N antigen [G], [I], [J], [L] Target unspecified [H] Recombinant protein, target unspecified [K] Recombinant nucleocapsid and Spike‐proteins |
[A] IgG [B] IgM and IgG [C] IgM and IgG [D] IgG [E] IgA and IgG [F] Total antibodies (including IgG) [G]‐[L] IgM and IgG | [A]‐[L] 0‐14 d pso 21/42 15‐21 d pso 11/42 > 21 d pso 10/42 | [A]‐[L] Serum (all kits assessed using same patient samples from single‐use aliquots) [G]‐[L] Cross‐reactivity panel [3] was not assessed on the POCTs. |
|
Charpentier 2020 [A] Charpentier 2020 [B] Charpentier 2020 [C] Laboratory and LFA; [A, B] LFA; [C] CLIA |
[A] AAZ ‐ Covid‐Presto® test rapid Covid‐19 IgG/IgM [B] NG Biotech ‐ NG‐Test® IgM‐IgG COVID‐19 [C] Abbott SARS‐CoV‐2 IgG kit | [A]‐[C] Not stated | [A] IgG and IgM [B] IgG and IgM [C] IgG | [A] 88 samples between day 4 and day 42 after onset of symptoms 4‐9 d pso: 18/88 10‐14 d pso: 33/88 15‐42 d pso: 37/88 [B] Subgroup of 59 samples among the 88 samples between d 7 and 28 after onset of symptoms 7‐9 d pso: 6/59 10‐14 d pso: 22/59 15‐28 d pso: 31/59 [C] 57 samples: 7‐9 d pso: 6/57 10‐14 d pso: 22/57 > 14 d pso: 29/57 | [A]‐[C] Serum |
|
Chaudhuri 2020 [A] Chaudhuri 2020 [B] Laboratory only; [A] CLIA; [B] ELISA |
[A] Diasorin LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA [B] Zydus ‐ Covid Kavach IgG ELISA | [A] S1/S2 domains of the Spike‐protein [B] Specific antigenic epitope(s) of the inactivated virus in the Kavach assay are not defined | [A] IgG [B] IgG | 20‐72 d of illness in symptomatic or RT‐PCR positivity in asymptomatic individuals; duration of illness bimodal due to study design: The means of the sampling window distributions were 23.5 and 49.3 d respectively. | Serum or plasma |
|
Chen 2020 [A] Chen 2020 [B] Chen 2020 [C] Chen 2020 [D] Chen 2020 [E] Laboratory and LFA; [A, B] CLIA; [C]‐[E] LFAs |
[A] Roche Elecsys® Anti‐SARS‐CoV‐2 Test [B] Abbott SARS‐CoV‐2 IgG [C] Guangzhou Wondfo Biotech ‐ Wondfo SARS‐CoV‐2 Antibody Test [D] TONYAR Biotech Inc ‐ ASK COVID‐19 IgG/IgM Rapid Test [E] Dynamiker Biotechnology ‐ Dynamiker 2019‐nCoV IgG/IgM Rapid Test | [A] N‐protein [B] N‐protein [C] Spike‐protein [D] Spike‐protein [E] N‐protein | [A] Total antibodies (including IgG) [B] IgG [C] Total antibodies [D] IgG and IgM [E] IgG and IgM | Median 7 d pso (range 1‐93 d pso) Mean 11.4 (SD 14.8) d pso 0‐7 d pso: 61/346 8‐14 d pso: 73/346 15‐21 d pso: 61/346 22‐28 d pso: 64/346 29‐35 d pso: 32/346 36‐93 d pso: 55/346 | [A]‐[E] Serum |
|
Chew 2020 Laboratory only; CLIA |
Abbott Architect SARS‐CoV‐2 IgG assay | IgG raised against the N‐protein of SARS‐CoV‐2 | IgG | COVID cases stratified according to time from onset of clinical illness to testing: (≤ 6 d, 81/177 7‐13 d, 39/177 14‐20 d 25/177, and ≥ 21 d 32/177) | Residual sera |
|
Chong 2021 LFA only; CGIA |
Hangzhou Alltest Biotech ‐ 2019‐nCoV IgG/IgM Rapid Test Cassette | N‐protein | IgG and IgM | 1‐33 d pso or post‐+ve PCR for asymptomatic cases: 1‐6 d: 8/63 samples 7‐13 d: 35/63 samples 14‐20 d: 11/63 samples 21‐33 d: 9/63 samples | Serum samples, remnant |
|
Clarke 2020 Laboratory only; CLIA |
Abbott SARS‐CoV‐2 IgG assay | Nucleocapsid‐protein antigen | IgG | [1] Mean 34 +/‐ 6.4 d, median 22 (range 14–34) d after PCR testing [2] Median time between tests was 23 (14–35) d [3] Asymptomatic | Serum |
|
Conklin 2020 [A] Conklin 2020 [B] Conklin 2020 [C] Conklin 2020 [D] Conklin 2020 [E] Conklin 2020 [F] Conklin 2020 [G] Conklin 2020 [H] Conklin 2020 [I] Conklin 2020 [J] Conklin 2020 [K] Conklin 2020 [L] Conklin 2020 [M] Conklin 2020 [N] Conklin 2020 [O] Conklin 2020 [P] Laboratory and LFA; [A to H, J to O] LFAs; [I, P] ELISA |
[A] Hangzhou AllTest Biotech ‐ AllTest 2019‐nCoV IgG/IgM [B] AYTU Biosciences ‐ AYTU COVID‐19 IgG/IgM [C] Alfa Scientific Designs Inc. ‐ Clarity COVID‐19 IgG/IgM [D] Hangzhou Biotest Biotech ‐ RightSign IgG/IgM [E] W.H.P.M., Inc. ‐ Covisure COVID‐19 IgM/IgG [F] DNA Link ‐ AccuFind COVID19 [G] Nirmidas Biotech IgM/IgG [H] Unknown manufacturer ‐ Ready Result [I] Epitope Diagnostics ‐ EDI nCov COVID‐19 IgM/IgG [J] Safecare Biotech (Hangszhou) ‐ Safecare IgG/IgM Rapid Test [K] Sensing Self ‐ Rapid IgG/IgM [L] Intelligent Endoscopy ‐ Smart Screen [M] TBG Biotechnology Corp. ‐ SARS‐CoV‐2 IgG/IgM Rapid Test [N] Guangzhou Wondfo ‐ SARS‐CoV‐2 Ab [O] Zeus Scientific ‐ SARS‐CoV‐2 IgM/IgG rapid test [P] Euroimmun ‐ anti‐SARS‐COV‐2 IgG | [A] N, S [B] N, S [C] N, S [D] RBD [E] Not stated [F] Not stated [G] S [H] N, S [I] Not stated [J] Not stated [K] N, S [L] Not stated [M] Not stated [N] Not stated [O] N, S [P] Not stated | [A] to [O] [P] IgG | [1] 45 d (standard deviation [SD], +/‐7.5 d (at least 28 d asymptomatic) Figure 2a reported "> 26 d" [2] Median 6 (IQR 4‐8) post‐symptom onset; Dataset S1 reported range from ‐2 to 36 d pso. | [1] and [2] Plasma [3] Serum |
|
Costa 2020 LFA only; CGIA |
Guangzhou Wondfo ‐ SARS‐CoV‐2 Ab | Not stated | IgG and IgM | [1a] PCR+ inpatients Mean 10.7 (range 4‐23) d pso PCR+ outpatients Mean 32.0 (range 16‐42) d pso All PCR+ patients: < 14 d: 38/106 14+ d pso: 59/106 Unknown: 9/106 [1b] PCR‐ inpatients Mean 8 (range 2‐15) d pso | Plasma |
|
Coste 2021 [A] Coste 2021 [B] Coste 2021 [C] Coste 2021 [D] Coste 2021 [E] Coste 2021 [F] Coste 2021 [G] Coste 2021 [H] Coste 2021 [I] Coste 2021 [J] Coste 2021 [K] Coste 2021 [L] Coste 2021 [M] Coste 2021 [N] Coste 2021 [O] Laboratory and LFA; [A to I] ELISA; [J to L] LFAs; [M to O] CLIA |
[A] Epitope Diagnostics ‐ EDI™ Novel Coronavirus COVID‐19 IgG ELISA Kit [B] EUROIMMUN ‐ Anti‐SARS‐CoV‐2 ELISA (IgG) [C] EUROIMMUN ‐ Anti‐SARS‐CoV‐2 ELISA (IgA) [D] ImmunoDiagnostics SARS‐CoV‐2 NP IgG ELISA Kit [E] ImmunoDiagnostics SARS‐CoV‐2 NP IgM ELISA Kit [F] Vircell COVID‐19 ELISA IgG [G] Vircell COVID‐19 ELISA IgM+IgA [H] Creative Diagnostic ‐ SARS‐CoV‐2 IgG ELISA Kit [I] Creative Diagnostic ‐ SARS‐CoV‐2 IgM ELISA Kit [J] Dynamiker ‐ 2019‐nCOV IgG/IgM Rapid Test [K] Nal Von Minden ‐ NADAL® COVID‐19 IgG/IgM Test [L] Augurix Diagnostics ‐ One Step Test for Novel Coronavirus (2019‐nCoV) IgM/IgG Antibody [M] Diasorin LIAISON® SARS‐CoV‐2 IgG kit [N] SNIBE Diagnostic ‐ MAGLUMI™ 2019‐nCoV IgG and IgM [O] Roche ‐ Elecsys anti‐SARS‐CoV‐2 | [A] N‐protein [B] S1 domain [C] S1 domain [D, E] N‐protein [F, G] N‐ and S‐ proteins [H] Whole virus lysate [I] N‐ and S‐proteins [J] N‐protein [K] S‐protein [L] N‐ and S‐proteins [M] S1 and S2 domains of the Spike‐protein [N] N‐ and S‐proteins [O] N‐protein | [A] IgG, IgM [B] IgG [C] IgA [D] IgG [E] IgM [F] IgG [G] IgM, IgA [H] IgG [I] IgM [J] IgG, IgM [K] IgG, IgM [L] IgG, IgM [M] IgG [N] IgG [O] Total Ig | Group [1]: during the first 2 months post‐symptom onset. No more details available | Serum |
|
Criscuolo 2020 [A] Criscuolo 2020 [B] Laboratory only; [A, B] CLIA |
[A] Roche Diagnostics ‐ Elecsys Anti‐SARS‐CoV‐2 [B] DiaSorin ‐ LIAISON® SARS‐CoV‐2 69 S1/S2 IgG assay | [A] N‐protein [B] S1 and S2 domains of the Spike‐protein | [A] Total antibodies [B] IgG | For each patient: one serum sample collected at hospital admission and another one 15 d later. Time since symptom onset not reported | Serum |
|
Dave 2020 LFA only; LFA |
SIDAK Life Care ‐ Antibody‐based rapid card test | Not reported | IgM and IgG | d of illness for all 100 patients (74/100 were asymptomatic so must be days post‐+ve PCR?): 0‐7 (n = 23); 8‐14 (n = 27); 15‐21 (n = 36); > 21 (n = 14) | Whole blood (2 drops) |
|
Decru 2020 [A] Decru 2020 [B] Decru 2020 [C] Decru 2020 [D] LFA only; [A to D] CGIA |
[A] Multi‐G single lane (MultiG1, lot NCP‐20030181) [B] MultiG dual lane (MultiG2, lot COV1452003C) [C] Orient Gene Biotech COVID‐19 IgM/IgG Rapid Test Cassette (lot 2003318) [D] SureScreen Diagnostics COVID‐19 Coronavirus Rapid Test Cassette (lot COV20030120) | Not stated | IgG and IgM | 23‐65 d pso; (data by week provided by authors) day 23‐28: 3, 9% day 29‐35: 5, 14% day > 35: 25, 71% | Whole blood, plasma |
|
Delliere 2020 [A] Delliere 2020 [B] Laboratory and LFA; [A] CGIA; [B] CLIA |
[A] Orient Gene Biotech COVID‐19 IgG/IgM Rapid Test Cassette [B] Abbott SARS‐CoV‐2 IgG | [A] and [B] Not stated | [A] IgG, IgM [B] IgG | [A] and [B] ≥ 4 d (4‐40, median = 18) since onset of symptoms or +ve PCR for asymptomatic patients | [A] and [B] Serum (stored at –20°C upon use) |
|
Doherty Institute 2020 [A] Doherty Institute 2020 [B] Doherty Institute 2020 [C] Doherty Institute 2020 [D] Doherty Institute 2020 [E] Doherty Institute 2020 [F] Doherty Institute 2020 [G] Laboratory and LFA; [A to F] CGIA; [G] ELISA |
[A] Hangzhou Alltest IgG/IgM Rapid Test [B] Hangzhou unlabelled packaging (see comments) [C] Wondfo SARS‐CoV‐2 Antibody Test [D] Hightop SARS‐COV‐2 IgM/IgG Antibody Rapid Test [E] OnSite COVID‐19 IgG/IgM Rapid Test [F] VivaDiag COVID‐19 IgM/IgG Rapid Test [G] EUROIMMUN Anti‐SARS‐CoV‐2 ELISA (IgA) (IgG) | [1‐6] The specific SARS‐CoV‐2 recombinant antigen(s) incorporated into the assay were not described in the manufacturers' information [G] Not stated | [A] IgG, IgM
[B] IgG, IgM
[C] Total Ab [D] IgG, IgM [E] IgG, IgM [F] IgG, IgM [G] IgA, IgG |
0‐> 30 d pso 0‐3 d pso: 23/137 (16.8%) samples 4‐8 d pso: 28/137 (20.4%) samples 9‐14 d pso: 21/137 (15.3%) samples 15‐20 d pso: 8/137 (5.8%) samples 21‐30 d pso: 27/137 (19.7%) samples > 30 d pso: 30/137 (21.9%) samples | Serum |
|
DomBourian 2020 [A] DomBourian 2020 [B] Laboratory only; [A] [B] ELISA |
[A] Epitope Diagnostics Inc EDI™ Novel Coronavirus COVID‐19 IgG ELISA kit [B] Euroimmun Anti‐SARS‐CoV‐2 ELISA (IgG) | [A] nucleocapsid antigen [B] S1 domain, including RBD | [A] and [B] IgG | At least 14 d symptom‐free | Plasma or serum |
|
Dora 2020 Laboratory only; CLIA |
DiaSorin ‐ LIAISON SARS‐CoV‐2 S1/S2 IgG | S1/S2 Spike‐protein | IgG | 46–76 d after their initial diagnosis | Not stated |
|
Dortet 2020 LFA only; CGIA |
NG Biotech ‐ NG‐Test IgM‐IgG COVID All‐in‐one | The assay contains anti‐human IgM and anti‐human IgG as the capture reagent, and SARS‐CoV‐2 (N‐protein) antigen gold particles as the detection reagent. | IgM, IgG | For 97 patients, sera were available from the first day of hospitalisation, when nasopharyngeal sampling was performed for RT–PCR testing, until the eleventh day of hospitalisation. Most sera were sampled between day 0–15 after the onset of symptoms (85.5%, 219/256) but later sera, up to day 31, were also available. | Serum; capillary blood for healthy volunteers |
|
Dortet 2021 [A] Dortet 2021 [B] LFA only; [A, B] CGIA |
[A] NG Biotech SA, Guipry, France; [B] Autobio Diagnostic Co; Ltd, Zhengshou, China [C] Avioq Bio‐Tech Co; Ltd, Shandong, China [D] Nal Von Minden GmbH; Ltd, Moers, Germany [E] Biosynex SWISS SA, Fribourg, Switzerland; [F] Innovita Biological Technology Co.; Ltd, Hebei, China [G] Biolidics Co, Ltd, Mapex, Singapore; [H] Vedal Lab SA, Alençon, France; [I] Wondfo Biotech Co, Ltd, Guangzhou, China; [J] Wondfo Biotech Co, Ltd, Guangzhou, China ‐ [A] NG‐Test IgG‐IgM COVID‐19; [B] anti‐SARS‐CoV‐2 rapid test; [C] a novel coronavirus 2019 (2019‐nCoV) antibody IgG/IgM test; [D] Nadal COVID‐19 IgG/IgM test; [E] Biosynex COVID‐19 BSS; [F] 2019‐nCoV Ab test; [G] 2019‐nCoV IgG/IgM test; [H] COVID‐19‐Check‐1; [I] Finecare SARS‐CoV‐2 antibody test; [J] Wondfo SARS‐CoV‐2 antibody test | [A] NP and SP; [B] Not reported; [C] Not reported; [D] Not reported; [E] Not reported; [F] NP and SP; [G] Not reported; [H] Not reported; [I] Not reported; [J] Not reported. | [A] IgG and IgM; [B] IgG and IgM; [C] IgG and IgM; [D] IgG and IgM; [E] IgG and IgM; [F] IgG and IgM; [G] IgG and IgM; [H] IgG and IgM; [I] Total antibody; [J] Total antibody (IgM/IgG as 1 line). | 0‐9 d pso; 101/250 10‐14 d pso; 86/250 15‐32 d pso. 63/250 Most serum samples were obtained on d 0 to 15 (85.5%; 219/256) after symptoms appeared, although serum samples from later dates (up to day 31) were also available. | Serum |
|
Du 2021 Laboratory only; Method not clear |
DiaCarta ‐ QuantiVirus™ anti‐SARS‐CoV‐2 IgG test | Spike‐protein 1 (S1) RBD | IgG | 0‐7 d pso: 13/107 8‐14 d pso: 13/107 > 14 d pso: 81/107 | [1] Serum [2a] Serum and plasma [2b] Serum |
|
Egger 2020 [A] Egger 2020 [B] Laboratory only; [A] CLIA; [B] ELISA |
[A] Roche Diagnostics ‐ Elecsys Anti‐SARS‐CoV‐2 assay [B] Epitope Diagnostics Inc EDI™ Novel Coronavirus COVID‐19 IgM (reagent lot number P630C) and IgG (reagent lot number P621C) | [A] and [B] recombinant nucleocapsid protein (N) | [A] IgA, IgM, IgG (total SARS‐CoV‐2 antibody assay [IgA, IgM, and IgG] detecting predominantly, but not exclusively, IgG) [B] IgM or IgG | < 5 d to > 15‐22 d since symptom onset < 5 d pso: 34/104 5‐10 d pso: 35/104 11‐15 d pso: 17/104 16‐22 d pso: 18/104 | [A] and [B] Plasma |
|
Fafi‐Kremer 2020 LFA only; CGIA |
Biosynex ‐ COVID‐19 BSS IgG/IgM | S‐protein | IgG and IgM | Time between symptom onset and sample collection: median 24 d (IQR 21 to 28) 13‐20 d: 29/160 (18%) 21‐27 d: 83/160 (52%) 28‐41 d: 48/160 (30%) | Serum |
|
Favresse 2020a Laboratory only; CLIA |
Roche Diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 | SARS‐CoV‐2 nucleocapsid | total antibodies (including IgG) | 0‐ ≥ 28 d after +ve RT‐PCR test 0‐6 d post‐PCR+: 45/140 7‐13 d post PCR+: 35/140 14‐20 d post PCR+: 24/140 21‐27 d post PCR+: 15/140 28+ d post PCR+: 21/140 0‐ > 28 d after onset of symptoms 0‐6 d pso: 22/129 7‐13 d pso: 28/129 14‐20 d pso: 26/129 21‐27 d pso: 23/129 28+ d pso: 30/129 11 missing data on time pso | Serum samples |
|
Favresse 2020b Laboratory only; CLIA |
Roche Diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 | N‐protein | Total antibodies | Range day 0 to 63: 0‐2 d: 15 (10%); 3‐5 d: 6 (4%); 6‐8 d: 14 (9.3%); 9‐11 d: 10 (6.7%); 12‐14 d: 13 (8.7%); 15‐17 d: 14 (9.3%); 18‐20 d: 7 (4.7%); 21‐23 d: 19 (12.7%); 24‐30 d: 16 (10.7%); 31‐40 d: 15 (10%); 41‐63 d: 16 (10.7%) | Serum or plasma (n for each not reported) |
|
Fenwick 2021 [A] Fenwick 2021 [B] Fenwick 2021 [C] Fenwick 2021 [D] Fenwick 2021 [E] Laboratory only; [A, B] ELISA; [C to E] CLIA |
[A] Euroimmun ‐ no name given [B] Epitope Diagnostics ‐ no name given [C] Diasorin ‐ LIAISON SARS‐CoV‐2 IgG kit [D] Snibe ‐ MAGLUMI 2019‐nCoV IgG and IgM kits [E] Roche ‐ Elecsys anti‐SARS‐CoV‐2 assay | [A] S1‐protein [B] N‐protein [C] S1‐protein [D] N‐protein and S antigen peptide [E] N‐protein | [A] IgG [B] IgG [C] IgG [D] IgG [E] pan‐Ig | 0 to 33 d post‐onset of the symptoms: 0‐5 d pso: 8/93 6‐10 d pso: 19/93 11‐15 d pso: 37/93 16‐33 d pso: 29/93 | Serum |
|
Flinck 2021 [A] Flinck 2021 [B] Laboratory only; [A, B] CLIA |
[A] Roche Diagnostics ‐ Elecsys® Anti–SARS‐CoV‐2 test [B] DiaSorin ‐ LIAISON® SARS‐CoV‐2 S1/S2 IgG | [A] N‐protein [B] Spike‐protein S1 and S2 antigens | [A] Total antibodies [B] IgG | [1a] Not stated [3‐40 d pso (figure 1) for 83/120 samples] [1b] At least 16 d after +ve NAAT | [1a] Residual EDTA plasma, stored −20 °C [1b] Residual plasma/serum samples [2a] Serum samples stored at −20 °C [2b] Plasma/serum samples |
|
Flower 2020 [A] Flower 2020 [B] Flower 2020 [C] Flower 2020 [D] Flower 2020 [E] LFA only; [A to E] LFAs |
[A] Guangzhou Wondfo Biotech ‐ Wondfo SARS‐CoV‐2 Antibody test [B] Menarini Zhejiang Orient Gene LFA [C] Fortress Diagnostics COVID‐19 TOTAL Ab Device [D] Biopanda COVID‐19 Rapid Antibody test [E] Biosure (Mologic) ‐ Biosure COVID‐1 Antibody Self‐Test | [A] S; [B] S1, S2 and N; [C] S; [D] S and N; [E] N. | [A] IgG/M combined; [B] IgG & M; [C] IgG & M; [D] IgG & M; [E] IgG only. | After 21 d of symptom onset; median (q1, q3) duration = 44 (35‐53) d range 21–100 d | Finger‐prick capillary blood; provided on the same day venous whole blood and serum samples for laboratory analysis |
|
Fragkou 2020 LFA only; FIA |
Lansion Biotechnology Co (COVID‐19) IgG/IgM Test Kit | Not stated | IgG and IgM | < 7 d: 5/26 7‐14 d: 11/26 > 14 d: 10/26 | Capillary whole blood |
|
Fujigaki 2020 [A] Fujigaki 2020 [B] Fujigaki 2020 [C] LFA only; [A to C] CGIA |
[A] Hangzhou AllTest Biotech Co ‐ 2019‐nCoV IgG/IgM Rapid Test Cassette [B] SD BIOSENSOR ‐ COVID‐19 IgM/IgG Duo [C] Vazyme Biotech Co ‐ 2019‐nCoV IgG/IgM Detection Kit | All tests: Unclear | All tests: IgM, IgG | Day 0 to 35; Day 0‐7: 18 patients; 27 samples Day 8‐14: 22 patients; 39 samples Day 15‐21 18 patients; 28 samples Day > 21 4 patients; 5 samples | Serum (residual and frozen prior to testing) |
|
Gao 2020a LFA only; LFA |
CGIA (Innovita Biological Technology Co) | Not reported | IgG, IgM | Day 0 to > 14 | Serum |
|
Gao 2020b [A] Gao 2020b [B] Gao 2020b [C] Laboratory and LFA; [A] Laboratory [B] CGIA [C] Laboratory |
[A] CLIA (Beier Bioengineering Company, Beijing) [B] CGIA (Beier Bioengineering Company, Beijing) [C] ELISA (Beier Bioengineering Company, Beijing) | [A], [B], and [C] all S‐ and N‐based | IgG and IgM ([A] ≥ 8 arbitrary unit (AU)/mL; [B] Visible line; [C] method to calculate threshold reported) | Day 1 to 24 | Serum |
|
Garnett 2020 Laboratory only; CLIA |
Ortho Clinical Diagnostics ‐ Vitros (VITROS®) Anti‐SARS‐Cov‐2 Total assay | Solid‐phase SARS‐CoV‐2 spike‐protein antigen | Total IgG and IgM | 0 to 35 d after onset of symptoms for 55 COVID patients (methods say 0‐35 d after +ve PCR, possibly for all 79 COVID patients) Categorised as < 3 d pso: 17/55, 4‐7 d pso: 7/55; 8‐13 d pso: 8/55; and > 13 d since first reported symptom: 23/55. | Serum and plasma |
|
GeurtsvanKessel 2020 [A] GeurtsvanKessel 2020 [B] GeurtsvanKessel 2020 [C] GeurtsvanKessel 2020 [D] GeurtsvanKessel 2020 [E] GeurtsvanKessel 2020 [F] GeurtsvanKessel 2020 [G] GeurtsvanKessel 2020 [H] Laboratory and LFA; [A to D] ELISA; [E] CLIA; [F to H] CGIA |
[A], [B]: Beijing Wantai SARS‐CoV‐2 total Ig ELISA; SARS‐CoV‐2 IgM ELISA [C], [D]: EUROIMMUN Anti‐SARS‐CoV‐2 IgG ELISA assay; IgA ELISA [E] DiaSorin ‐ LIAISON SARS‐CoV‐2 S1/S2 IgG [F] InTec Products ‐ Rapid SARS‐CoV‐2 Antibody (IgM/IgG) Test (Test lots S2020021505 and GJ20030288) [G] Cellex Inc ‐ qSARS‐CoV‐2 IgG/IgM Cassette Rapid Test (GICA) (Test lot 20200416WI5513C) [H] OrientGene Biotech / Healgen ‐ COVID‐19 IgG/IgM Rapid Test Cassette (Test lot 2003309) | [A], [B]: RBD [C], [D]: S1 domain [E]: S1 and S2 domains of the Spike‐protein [F]: S and N‐proteins [G]: N‐protein [H]: S and N‐proteins | [A]: Total IgG [B]: IgM [C]: IgG [D]: IgA [E]: IgG [F]‐[H]: IgM, IgG | Median 16 d pso (calculated from Suppl Data file), range 4 to 73 d The number of samples tested varied for each assay, and results were presented per sample but not per patient, so a clear breakdown by time is hard. | Serum (COVID‐19 cases); serum or plasma (non‐COVID‐19 samples) |
|
Gudbjartsson 2020 [A] Gudbjartsson 2020 [B] Gudbjartsson 2020 [C] Laboratory only; [A] ECLIA; [B]‐[C] ELISA |
[A] Roche Elecsys chemiluminescence assay [B] Wantai ELISA [C] EDI ELISA | [A] Nucleocapsid (anti‐N) [B] Spike 1 RBD (anti‐S1‐RBD) [C] Nucleocapsid (anti‐N) | [A] Total antibodies [B] Total antibodies [C] IgG | [1b] at least two weeks from qPCR diagnosis and one week after end of symptoms; (text and Figure 2 in paper stated "25 d after diagnosis" for the earliest time point) and again on July 1, on average 100 d after diagnosis with qPCR (487/1215 recovered patients with at least 2 samples at least 30 d apart); up to 4 months after PCR+ | Serum |
|
Guedez‐Lopez 2020 [A] Guedez‐Lopez 2020 [B] Guedez‐Lopez 2020 [C] LFA only; [A to C] LFAs |
[A] T & D Diagnostics ‐ Sienna 2019‐nCoV IgG/IgM Rapid Test [B] Wondfo ‐ SARS‐CoV‐2 Antibody Test [C] Prometheus Bio Inc ‐ Prometheus 2019‐nCoV IgG/IgM Test | Not reported | [A] IgG and IgM separately [B] Total antibody [C] IgG and IgM separately | [1] Median 5 (range 1–24) d pso [2] Median 11 (range 3–18) d pso [1a] and [2a] Early stage (first week): n = 41 intermediate stage (second week: n = 48 late stage (third week) n = 9 | Serum |
|
Haljasmagi 2020 Laboratory only; ELISA |
Euroimmun ‐ Anti‐SARS‐CoV‐2 IgG ELISA | S1 | IgG | Median 16 d (range 8 to 37d) Day 8‐14 after infection: 9/26 (35%) Day 15‐21 after infection: 11/26 (42%) Day 22+ after infection: 6/26 (23%) | Plasma |
|
Hamilton 2020 Laboratory only; CLIA |
Abbott Architect SARS‐CoV‐2 IgG assay | Not reported | IgG | [1] Time was calculated from reported symptom onset date. Median time unclear. < 5 d pso: 18/149 5‐9 d pso: 57/149 10‐14 d pso: 28/149 15‐20 d pso: 14/149 > 20 d pso: 32/149 > 42 d pso: 30/149 [2] Timing was calculated from the time of the +ve PCR test. Median time to test 45 d (range 32‐51 d) | EDTA plasma (fresh or stored at −80 C) |
|
Harritshoej 2021 [A] Harritshoej 2021 [B] Harritshoej 2021 [C] Harritshoej 2021 [D] Harritshoej 2021 [E] Harritshoej 2021 [F] Harritshoej 2021 [G] Harritshoej 2021 [H] Harritshoej 2021 [I] Harritshoej 2021 [J] Harritshoej 2021 [K] Harritshoej 2021 [L] Harritshoej 2021 [M] Laboratory only; [A, H, K] ELISA; [B to G, I, J, L, M] CLIAs |
[A] Wantai ELISA Total‐Ab assay; [B] Ortho Clinical Diagnostics ‐ Vitros Total‐Ab assay; [C] Siemens Atellica Total‐Ab assay; [D] Roche Elecsys Total‐Ab assay; [E] YHLO iFlash IgG assay; [F] Abbott Architect IgG assay; [G] Abbott Alinity IgG assay; [H] Euroimmun ELISA IgG assay; [I] Snibe DIagnostic ‐ Maglumi IgG assay; [J] DiaSorin Liaison XL IgG assay; [K] Wantai ELISA IgM assay; [L] Ortho Clinical VITROS Anti‐SARS‐Cov‐2 Total Ab [M] Siemens Vista Total‐Ab assay; | [A] S RBD; [B] S1 RBD; [C] S1 RBD; [D] N; [E] N and S [F] N; [G] N; [H] S1 RBD; [I] N and S [J] S; [K] S [L] S1 [M] RBD | [A, C, D, L, M] Total Ab; [B, E‐J] IgG; [K] IgM | pso: 0‐7 (n = 0); > 7‐14 (n = 7); > 14‐21 (n = 13); > 21‐42 (n = 49); > 42 (n = 71); Unknown (n = 10). Corrected data from corresponding author stated 123 samples > 21 d pso. | [1] Serum [2] Plasma [3] Not stated |
|
Haselmann 2020 [A] Haselmann 2020 [B] Haselmann 2020 [C] Laboratory only; [A to B] ELISA; [C] CLIAs |
[A] Euroimmun anti‐SARS‐CoV‐2 IgG ELISA (Lot:E200429AG) [B] Epitope Diagnostics EDI Novel Coronavirus COVID‐19 IgG ELISA (Lot:P745U) [C] Roche Elecsys Anti‐SARSCoV‐2 (Lot:496298) | [A] S1 domain of viral spike‐protein [B] Full length N‐protein [C] Recombinant protein representing the nucleocapsid antigen | [A] IgG [B] IgG [C] IgG | Unclear Median 29 d pso (range 10‐47) Day 10‐14: 5, 19% Day 15‐21: 5, 19% Day 22‐28: 2, 8% Day 29‐35: 7, 27% Day 36‐42: 2, 8% Day > 42: 5, 19% [from Suppl Table 3] | Serum (n = 26) and plasma (n = 13) |
|
Herroelen 2020 [A] Herroelen 2020 [B] Herroelen 2020 [C] Herroelen 2020 [D] Herroelen 2020 [E] Herroelen 2020 [F] Herroelen 2020 [G] Laboratory and LFA; [A, B] CGIA; [C to E] ELISA; [F, G] CLIA |
[A] Zhejiang Orient Gene Biotech Co ‐ COVID‐19 IgG/IgM Rapid Test [B] Innovita Biological Technology 2019‐nCoV Ab Test [C] Beijing Wantai Biological Pharmacy Enterprise Wantai SARS‐COV‐2 Ab ELISA [D] EUROIMMUN AG Anti‐SARS‐CoV‐2 IgG and IgA assays [E] EUROIMMUN AG Anti‐SARS‐ CoV‐2‐NCP (IgG) assay [F] Roche Diagnostics ‐ Elecsys Anti‐SARS‐CoV‐2 assay [G] DiaSorin ‐ LIAISON SARS‐CoV‐2 S1/S2 IgG | [A] recombinant N‐ and S‐proteins [B] undisclosed SARS‐CoV‐2 epitopes [C] RBD domain of the S1‐protein [D] S1‐protein [E] N‐protein [F] N‐protein [G] S1/S2‐proteins | [A, B] IgM, IgG
[C] Total Ab [D] IgA, IgG [E] IgG [F] TotaL Ab [G] IgG |
Inpatients ‐ 0 to 39 d pso
HCWs ‐ 11 to 54 d pso < 10 d pso: 53/171 10‐20 d pso: 42/171 > 20 d pso: 76/171 |
[A]‐[G] Serum |
|
Hoffman 2020 LFA only; LFA |
Zhejiang Orient Gene Biotech ‐ COVID‐19 IgG/IgM Rapid Test Cassette [GCCOV‐402a, Lot: 2003242] | Not stated | SARS‐CoV‐2‐specific antibodies IgG/IgM | 9‐17 d pso (n = 10); 18‐29 d (n = 19) | Capillary blood samples or serum |
|
Hogan 2020a [A] Hogan 2020a [B] Hogan 2020a [C] Laboratory only; [A to C] CLIA |
[A] DiaSorin ‐ Liaison SARS‐CoV‐2 S1/S2 IgG [B] Roche Diagnostics ‐ Elecsys anti‐SARS CoV‐2 total antibody [C] Beckman Coulter ‐ Access SARS‐CoV‐2 IgG | [A] S1 and S2 subunits of the spike‐protein [B] N‐protein [C] RBD of the S1‐protein | [A] IgG [B] Total antibodies [C] IgG | 1‐45 d overall (median: 9) post PCR+: 0‐6 d (median 5) post PCR+: 17/51 7‐13 d (median 9) post PCR+: 17/51 14+ d (median 18) post PCR+: 17/51 Combined samples were represented by the day farthest from the patient’s +ve PCR test. | Residual serum |
|
Horber 2020 [A] Horber 2020 [B] Horber 2020 [C] Laboratory only; [A, B] CLIA; [C] ELISA. |
[A] Siemens Healthineers SARS‐CoV‐2 Total (COV2T) [B] Roche Diagnostics Elecsys anti‐SARS‐CoV‐2 [C] Euroimmun ‐ SARS‐CoV‐2‐ELISA (IgG) | [A] S1‐protein RBD [B] N‐protein [C] S1 spike‐protein | [A] Total antibodies [B] Total antibodies [C] IgG | Median time between +ve PCR result and blood sample collection was 19 d (interquartile range: 12–29 d). 0‐6 d post PCR+: 23/186 7‐13 d post PCR+: 31/186 14+ d post PCR+: 132/186 | Plasma |
|
Hu 2020a Laboratory only; Laboratory |
Magnetic MCLIA kit (Bioscience Co., Ltd (Chongqing, China)) | N‐ and S‐based | IgM, IgG | Day 1 to > 37 | Serum |
|
Hu 2020b [A] Hu 2020b [B] Laboratory only; [A, B] CLIA |
[A, B] Shenzhen YHLO Biotech ‐ SARS‐COV‐2 IgM and IgG CLIA kit | N‐protein, Spike‐protein | [A] IgM [B] IgG |
< 7 d pso: 12/68 (18%) 7‐14 d pso: 25/66 (37%) > 14 d pso: 31/68 (46%) | Serum |
|
Hubbard 2021 [A] Hubbard 2021 [B] Laboratory only; [A, B] CLIA |
[A] Abbott SARS‐CoV‐2 IgG assay [B] Roche Elecsys Anti–SARS‐CoV‐2 assay | [A] N‐protein [B] N‐protein | [A] IgG [B] Total antibodies | [1a] Not stated (could work out from Figure 1 and Table 2: 14+ d post‐PCR+: 10/193) [1b] Convalescent (14+ d symptom‐free) | Remnant serum and plasma |
|
Imai 2020 LFA only; CGIA |
Artron ‐ One Step nCov (COVID‐19) IgM/IgG Antibody Test | Not stated | IgM, IgG | Day of admission and during hospitalisation (within 1 week (n = 90), 1–2 weeks (n = 25), and > 2 weeks after onset (n = 24) | Serum |
|
Jaaskelainen 2020 Laboratory only; ELISA |
Euroimmun SARS‐CoV‐2 IgG ELISA and IgA ELISA | S1‐protein | IgA, IgG | 1‐23 d pso | Serum |
|
Jin 2020 Laboratory only; Laboratory |
SARS CoV‐2 IgM and IgG CLIA kits (Shenzhen YHLO Biotech Co) | N‐ and S‐based | IgM, IgG | Day 1 to 55 | Serum |
|
Jung 2020a Laboratory only; ELISA |
Ansh Laboratories SARS‐CoV2 IgG ELISA and IgM ELISA | IgG ‐ N‐ and S‐based IgM ‐ anti‐human IgM capture antibody | IgG, IgM | [2a] < 6 d pso: n = 10, 6‐14 d pso: n = 9, > 14 d pso: n = 24 for [A] and n = 22 for [B]. | Peripheral venous blood |
|
Kaltenbach 2020 [A] Kaltenbach 2020 [B] Kaltenbach 2020 [C] Laboratory only; [A to C] ELISA |
[A] Euroimmun Anti‐SARS‐CoV‐2‐ELISA‐IgA (# EI 2606‐9601 A) [B] Euroimmun Anti‐SARS‐CoV‐2‐ELISA‐IgG (# EI 2606‐9601 G) [C] Epitope Diagnostics EDI Novel Coronavirus COVID‐19 IgM ELISA kit (# KT‐ 114 1033) and IgG ELISA (# KT‐1032) | Unclear | [A] IgA [B] IgG [C] IgM, IgG | <= 14 d: 54/345 (16%) 15‐20 d: 52/345 (15%) ≥ 12 d: 239/345 (69%) | [A] and [2] Serum [3] and [4] Serum and plasma |
|
Kaneko 2021 LFA only; CGIA |
Innovita ‐ 2019‐nCoV Ab Test Cassette (Colloidal Gold) | Not described | IgM/IgG | Different time points 0‐4 d pso: 2/87 4‐7 d pso: 6/87 8‐14 d pso: 38/87 15‐28 d pso: 23/87 > 28 d pso: 18/87 | Serum |
|
Knauer 2020 [A] Knauer 2020 [B] Knauer 2020 [C] Laboratory only; [A, E] CLIA; [B to D] ELISA |
[A] DiaSorin SARS‐CoV‐2 S1/S2 IgG [B] EUROIMMUN Anti‐SARS‐CoV‐2 IgG [C] EUROIMMUN Anti‐SARS‐CoV‐2 IgA [D] Epitope Diagnostics Novel Coronavirus COVID‐19 IgM [E] Roche Elecsys Anti‐SARS‐CoV‐2 Total Assay | [A] S1/S2 from test name [B]‐[E] Not stated | [A] IgG [B] IgG [C] IgA [D] IgM [E] Total antibodies | < 7 d after +ve NAAT, 8‐14 d after +ve NAAT, > 14 d after +ve NAAT, 28 d post‐+ve NAAT (n = 11 to 61) | Residual plasma samples |
|
Ko 2021 LFA only; CGIA |
Wells Bio ‐ careUS COVID‐19 IgM/IgG | SARS‐CoV‐2 spike‐protein | IgG, IgM | Range 4‐56 d pso 4‐6 d pso: 3/52 7‐13 d pso: 6/52 14‐20 d pso: 6/52 21‐27 d pso: 10/52 28+ d pso: 27/52 | Serum |
|
Kohmer 2020a [A] Kohmer 2020a [B] Kohmer 2020a [C] Laboratory and LFA; [A] CLIA; [B] ELISA; [C] CGIA |
[A] Vircell COVID‐19 ELISA IgG [B] Euroimmun SARS‐CoV‐2 IgG ELISA [C] Assure Tech (Hangzhou) Co FaStep (COVID‐19 IgG/IgM) rapid test cassettes | [A] S and N [B] S‐protein [C] Not stated | [A] IgG [B] IgG [C] IgM and IgG | Time since symptom onset not reported For Group [1], time since PCR done: 17/33 (52%) collected 5‐9 d after PCR; 16/33 (48%) collected 10‐18 d after PCR. | Serum |
|
Kohmer 2020b [A] Kohmer 2020b [B] Kohmer 2020b [C] Kohmer 2020b [D] Kohmer 2020b [E] Kohmer 2020b [F] Laboratory only; [A, C, E] CLIA; [B, D, F] ELISA |
[A] Abbott SARS‐CoV‐2 IgG [B] Roche Diagnostics Elecsys Anti‐SARS‐CoV‐2 [C] DiaSorin Liaison SARS‐CoV‐2 S1/S2 IgG [D] Vircell COVID‐19 VIRCLIA IgG MONOTEST [E] Euroimmun Anti‐SARS‐CoV‐2 ELISA (IgG) [F] Virotech Diagnostics Virotech SARS‐CoV‐2 ELISA IgG | [A] N‐protein [B] N‐protein [C] S1 and S2‐protein [D] S1 and N‐protein [E] S1‐protein [F] N‐protein | [A, C to F] IgG [B]: total antibody | No info regarding time since symptom onset Group [1A]: collected 2‐49 d after PCR‐+ve test. Not stated for the others | Serum or plasma |
|
Korte 2021 [A] Korte 2021 [B] Korte 2021 [C]Laboratory only; [A]‐[C] ELISA |
[A] and [B] Euroimmun anti‐SP IgG and IgA [C] Epitope Diagnostics anti‐NC IgG | [A] Spike‐protein [B] Spike‐protein [C] Nucleocapsid‐protein | [A] IgG [B] IgA [C] IgG | Tests performed weekly in first month and after another four weeks in the second month Range: 2‐10 weeks after a +ve SARS‐CoV‐2 PCR test | Not stated |
|
Kowitdamrong 2020 [A] Kowitdamrong 2020 [B] Laboratory only; [A] and [B] ELISA |
[A] EUROIMMUN anti‐SARS‐CoV‐2 ELISA IgA kit [B] EUROIMMUN anti‐SARS‐CoV‐2 ELISA IgG kit | [A] and [B] S1‐protein | [A] IgA [B] IgG | 0‐3 d pso: 37/213 4‐7 d pso: 49/213 8‐14 d pso: 45/213 15‐28 d pso: 21/213 > 28 d pso: 61/213 | Plasma and serum |
|
Krishnamurthy 2020 Laboratory only; Other; protein microarray |
Vibrant America ‐ Vibrant COVID‐19 Ab | S1‐glycoprotein RBD S2‐glycoprotein Nucleoprotein | IgM, IgA and IgG | < 7 d 7‐14 d > 14 d | Serum |
|
Lassauniere 2020 [A] Lassauniere 2020 [B] Lassauniere 2020 [C] Lassauniere 2020 [D] Lassauniere 2020 [E] Lassauniere 2020 [F] Lassauniere 2020 [G] Laboratory and LFA; [A] to [C] Laboratory [D] to [I] LFA |
[A] Wantai ELISA Total‐Ab assay [B] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [C]EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA [D] Dynamiker Biotech 2019‐nCOV IgG/IgM Rapid Test [E] CTK Biotech ‐ OnSiteTM COVID‐19 IgG/IgM Rapid Test [F] Autobio Diagnostics Anti‐SARS‐CoV‐2 Rapid Test [G] Artron Labs Coronavirus Diseases 2019 (COVID‐19) IgM/IgG Antibody Test | [A] S‐based [B] and [C] S1‐based [D] to [I] Not stated | [A] Total antibodies [B] IgG and [C] IgM [D] to [I] IgG/IgM | Day 7 to ≥ 21 | Serum |
|
Lau 2020a Laboratory only; CLIA |
Abbott Architect SARS‐CoV‐2 IgG assay | Undisclosed epitope on the viral nucleocapsid | IgG | Timing was post‐PCR+ve: 0‐7 d: 155/266 (58%) 7‐14 d: 57/266 (21%) 14‐21 d: 22/266 (8%) ≥ 21 d: 32/266 (12%) | Serum |
|
Lau 2020b Laboratory only; CLIA |
Roche Diagnostics ‐ ELECSYS anti‐SARS‐CoV‐2 assay. | Biotinylated SARS CoV‐2 specific recombinant antigens and SARS‐CoV‐2 specific recombinant antigens labelled with ruthenium | Unclear Not stated; appeared to be total Ab | Timing reported was post‐PCR+ve 0‐7 d: 189/349 (54%) 7‐13 d: 90/349 (26%) 14‐20 d: 34/349 (10%) ≥ 21 d: 36/349 (10%) | Serum |
|
Lau 2020c Laboratory only; CLIA |
Abbott SARS‐CoV‐2‐IgG | Undisclosed epitope on the viral nucleocapsid | IgG | 0‐6 d post‐PCR+: 172/279 7‐13 d post‐PCR+: 47/279 14+ d post‐PCR+: 60/279 | Serum |
|
Lau 2020d Laboratory only; CLIA |
Roche Elecsys anti‐SARS‐CoV‐2 assay | Undisclosed epitope | Total antibodies | 0‐6 d: 189 7‐13 d: 90 ≥ 14 d: 70 | Serum |
|
Li 2020 [A] Laboratory only; [A, B] ELISA |
[A] Hotgen Biotech ‐ S‐protein based IgG/IgM ELISA [B] Livzon Group ‐ N‐protein based IgG/IgM ELISA | [A] S (Spike) [B] N‐(Nucleocapsid) protein | [A, B]: IgM, IgG | Day 2 to 45 pso; 40 samples collected at < 10 d; up to 41 samples > 10 d | Serum |
|
Lippi 2020 [A] Lippi 2020 [B] Laboratory only; [A] and [B] Laboratory |
[A] MAGLUMI 2019‐nCoV CLIAs (Snibe DIagnostics ‐Shenzhen New Industries Biomedical Engineering Co., Ltd,) [B] ELISAs (Euroimmun AG, Luebeck, Germany) | [A] N‐ and S‐based [B] Not stated | [A] IgM or IgG [B] IgA or IgG | Day < 5 to 21 | [A] Serum or plasma [B] Not stated |
|
Liu 2020a Laboratory only; Laboratory |
Zhuhai Livzon ‐ 2019‐nCoV IgM/IgG ELISA | N‐based | IgM, IgG (threshold set in healthy control sample) | Day 0 to ≥ 16 | Serum |
|
Liu 2020b [A] Liu 2020b [B] Laboratory only; Laboratory |
[A] ELISA (Hotgen, Beijing, China) [B] ELISA (Lizhu, Zhuhai, China) | [A] S‐based [B] N‐based | IgM, IgG | Day 0 to 30 | Serum |
|
Liu 2020c Laboratory only; CLIA |
Xiamen InnoDx Biotech CMIA | RBD, S‐protein | IgM, total antibodies | Symptom onset 0‐7 d pso: 26/206 8‐14 d pso: 70/206 15‐21 d pso: 72/206 > 21 d pso: 38/206 | [1] Serum [2] Serum |
|
Liu 2021 Laboratory only; CLIA |
Hunan Yuanjing Biotechnology Co ‐ "COVID‐19 IgG Detection Kits" | SARS‐CoV‐2 spike receptor‐binding domain (S‐RBD) and N Spike‐protein as antigens | IgM, IgG | [1a] First sample (n = 81): Median 7 d (range 4, 14) pso [1b] First sample (n = 30): Median 8 d (range 7, 9) after the +ve RT‐PCR test detection. [2] Median 9.5 (range 5, 12) day pso (n = 40) | Serum |
|
Loconsole 2020 LFA only; CGIA |
Vivacheck Biotech ‐ SARS‐CoV‐2 VivaDiag™ | Not stated | IgM and/or IgG | [1] PCR +ve patients ‐ 148 [1a] < 7 d of symptoms ‐ 99 [1b] > 7 d of symptoms ‐ 44 [1c] Asymptomatic patients ‐ 5 | Plasma or whole blood |
|
Long 2020 Laboratory only; Laboratory |
Magnetic CLIA (Bioscience (Chongqing) Co., Ltd) | N‐ and S‐based | IgM, IgG (threshold not described) | Day 2 to ≥ 23 | Serum |
|
Liu 2020b [A] Liu 2020b [B] Laboratory only; Laboratory |
[A] ELISA (Hotgen, Beijing, China) [B] ELISA (Lizhu, Zhuhai, China) | [A] S‐based [B] N‐based | IgM, IgG | Day 0 to 29 | Serum |
|
Liu 2020c Laboratory only; CLIA |
Xiamen InnoDx Biotech CMIA | RBD, S‐protein | IgM, total antibodies | 1‐70 d pso [1a] week 2 [1b] week 4 or later [1c] two time periods ‐ 21‐40 d and 41‐70 d | Plasma or serum |
|
Liu 2021 Laboratory only; CLIA |
Hunan Yuanjing Biotechnology Co ‐ "COVID‐19 IgG Detection Kits" | SARS‐CoV‐2 spike receptor‐binding domain (S‐RBD) and N spike‐protein as antigens | IgM, IgG | > 21 d pso | Serum |
|
MacMullan 2020 [A] MacMullan 2020 [B] MacMullan 2020 [C] MacMullan 2020 [D] Laboratory only; All ELISA |
[A] Gold Standard Diagnostics SARS‐CoV‐2 IgA ELISA (GSD01‐1029 IgA) [B] Gold Standard Diagnostics SARS‐CoV‐2 IgG ELISA (GSD01‐1028 IgG) [C] EuroImmun SARS‐CoV‐2 IgA ELISA (EI 2606‐9620 IgA) [D] EuroImmun SARS‐CoV‐2 IgG ELISA (EI 2606‐9620 IgG) | [A, B] Nucleocapsid [C, D] Spike | [A, C] IgA [B, D] IgG | > 21 days pso | Serum |
|
Mairesse 2020 [A] Mairesse 2020[B] Laboratory only; [A, B] CLIA |
[A, B] YHLO biotechnology co ‐ iFlash® anti‐SARS‐CoV‐2 IgM, IgG assays | S and N | IgM and IgG | 0‐6 d pso: n = 45 7‐13 d pso: n = 35 14‐20 d pso: n = 37 21‐27 d pso: n = 29 > 28 d: n = 32 | Serum stored at ‐20 c |
|
Manalac 2020 [A] Manalac 2020 [B] Laboratory only; [A] CLIA; [B] ELISA |
[A] Abbott Architect anti‐SARS‐CoV‐2 CMIA IgG [B] Euroimmun anti‐SARS‐CoV‐2 ELISA IgA and IgG assays | [A] N‐protein [B] S1 domain of viral Spike‐protein | [A] IgG [B] IgA, IgG | 14‐21 d pso: n = 4 > 21 d pso: n = 27 Unknown: n = 66 <= 10 d post‐PCR+: n = 8 > 10 d post‐PCR+: n = 48 Unknown: n = 41 | Abstract stated "Plasma" [2c] Remnant serum |
|
Marlet 2020 [A] Marlet 2020 [B] Marlet 2020 [C] Marlet 2020 [D] Laboratory only; [A, C, D] ELISA; [B] CLIA |
[A] Euroimmun ELISA SARS‐CoV‐2 IgG, [B] Abbott SARS‐CoV‐2 IgG, [C] Wantai SARS‐CoV‐2 Ab ELISA [D] DiaPro COVID‐19 IgG Confirmation | [A] S1 [B] N [C] S (RBD) [D] S1, S2, N | [A] IgG [B] IgG [C] Total antibody [D] IgG | 2–36 d after the onset of symptoms: 7‐13 d pso: 13 samples 14+ d pso: 45 samples | Plasma |
|
Martinaud 2020 Laboratory only; Other; solid‐phase photometric immunoassay |
Quotient ‐ MosaiQ™ COVID‐19 antibody microarray | Spike S1‐protein | IgM and IgG | [1a} < 14 d pso: 38/101 14‐20 d pso: 33/101 > 20 d pso: 30/101 [1b] > 20 d pso: 60 samples | Serum |
|
McAulay 2020 [A] McAulay 2020 [B] McAulay 2020 [C] McAulay 2020 [D] Laboratory and LFA; [A to C] LFA; [D] CLIA |
[A] BTNX IncRapid ResponseTM COVID‐19 Test Cassette Kit1 [B] ACON Laboratories SARS‐COV‐2 IgG/IgM Rapid Test [C] SD BIOSENSOR Standard Q COVID‐19 IgM/IgG Duo [D] Abbott SARS‐CoV‐2 IgG immunoassay Second iteration of test [A] also evaluated | [A] Not stated [B] Not stated [C] Not stated [D] Not stated [E] Not stated | [A] IgM/IgG [B] IgM/IgG [C] IgM/IgG [D] IgG (this kit is supplied as individual IgM and IgG cartridges; only the IgG cartridges were evaluated in this study) [E] IgG | 1 to 31 d pso (Supplementary Table S1) < 7 d pso: 154/352 7‐13 d pso: 103/352 14‐31 d pso: 95/352 | [1] Mixed: 250 plasma, 77 serum, and 21 whole blood specimens [2] Pre‐pandemic serum Cross‐reactivity samples: not stated |
|
Merrill 2020 [A] Merrill 2020 [B] Laboratory only; [A, B] CLIA |
[A] DiaSorin Liaison SARS‐CoV‐2 S1/S2 IgG [B] Roche Diagnostics Elecsys Anti‐SARS‐COV‐2 assay | [A] S1 and S2 domains of the spike (S)‐protein [B] Nucleocapsid (N)‐protein | [A] IgG [B] total antibodies (IgG, IgM, IgA) | < 7 d pso: 5/54 7‐13 d pso: 12/54 > 13 d pso: 12/54 Unknown: 12/54 Asymptomatic: 13/54 < 7 d post‐PCR+: 35/54 7‐13 d post‐PCR+: 13/54 > 13 d post‐PCR+: 6/54 | Plasma samples (lithium heparin and EDTA) |
|
Montesinos 2020 [A] Montesinos 2020 [B] Montesinos 2020 [C] Montesinos 2020 [D] Montesinos 2020 [E] Montesinos 2020 [F] Montesinos 2020 [G] Laboratory and LFA; [A, E, G] LFA; [B to D] ELISA; [F] CLIA |
[A] Avioq Bio‐Tech ‐ 2019‐nCov Antibody IgG/IgM [B] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA [C] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [D] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA or IgG [E] LaboOn Time rapid test cassette [F] Snibe Diagnostic ‐ MAGLUMI 2019‐nCoV IgG/IgM [G] ZenTech ‐ QuickZen COVID‐19 IgM/IgG | [A] Unclear [B to C] S1‐based [D] 2019‐nCoV [E] N‐ and S‐based [F] RBD | [A, E, F, G] IgG or IgM [B] IgA [C] IgG [D] IgA or IgG | Day 0 to > 15; no further details | Serum |
|
Muecksch 2020 [A] Muecksch 2020 [B] Muecksch 2020 [C] Muecksch 2020 [D] Laboratory only; [A to D] CLIA |
[A] Abbott SARS‐CoV‐2 IgG assay [B] DiaSorin SARS‐CoV‐2 IgG assay [C] Roche Anti‐SARS‐CoV total antibody assay [D] Siemens SARS‐CoV‐2 total antibody assay | [A] N‐protein [B] S‐protein [C] N‐protein [D] RBD of S‐protein | [A] IgG [B] IgG [C] total antibody [D] total antibody | Visit [1] (baseline) avg. 40.8 d post‐PCR +ve (range 24‐61 d), Visit [2] (two weeks post‐baseline) avg. 55.1 d post‐PCR +ve (range 40‐79 d), Visit [3] (four weeks post‐baseline) avg. 69.8 d post‐PCR +ve (range 55‐95 d), Visit [4] (8 weeks post‐baseline) avg. 98.4 d (85‐110 d). | Convalescent serum |
|
Naaber 2020 [A] Naaber 2020 [B] Naaber 2020 [C] Naaber 2020 [D] Naaber 2020 [E] Naaber 2020 [F] Naaber 2020 [G] Laboratory and LFA; [A, C, D, F] CLIA; [B, E] ELISA; [G] CGIA |
[A] SNIBE (Shenzhen New Industries Biomedical Engineering Co) MAGLUMI 2019‐nCoV IgG [B] EUROIMMUN AG SARS‐CoV‐2 ELISA IgG [C] Abbott Laboratories SARS‐CoV‐2 IgG [D] Roche Diagnostics Elecsys Anti‐SARS‐CoV‐2 [E] Epitope Diagnostics Inc EDI Novel Coronavirus COVID‐19 IgG ELISA [F] DiaSorin LIAISON SARS‐CoV‐2 S1/S2 IgG [G] SD BioSensor Inc STANDARD™ Q COVID‐19 IgM/IgG Duo Test | [A] not specified [B] S1 [C] N‐protein [D] N‐protein [E] N and S‐protein [F] S1 and S2 [G] N‐protein | [A] IgG [B] IgG [C] IgG [D] Total antibody [E] IgG [F] IgG [G] IgG | At least one week pso or post‐PCR +ve Median 28 (range 7–57) d to test 7‐14 d, n = 20 15‐30 d, n = 35 31‐57 d, n = 42 | Serum |
|
Nagasawa 2020 [A] Nagasawa 2020 [B] Nagasawa 2020 [C] LFA only; [A to C] CGIA |
[A] INNOVITA Biological Technology ‐ 2019‐nCoV Ab Test [B] Zhejiang Orient Gene Biotech ‐ COVID‐19 IgG/IgM Rapid Cassette Test [C] Hangzhou AllTest Biotech ‐ 2019‐nCoV IgG/IgM Rapid Test Casette | [A], [B], [C] Unclear | [A] IgG/IgM [B] IgG/IgM [C] IgG/IgM | 1‐29 d pso 1‐5 d pso: 1/45 6‐10 d pso: 10/45 11‐15 d pso: 19/45 16‐20 d pso: 9/45 21‐29 d pso: 6/45 | Serum |
|
Nayak 2021 Laboratory only; ELISA |
Zydus diagnostics‐ COVID‐Kavach ELISA test kit | Not stated | IgG | 25‐84 d post‐PCR +ve | Plasma |
|
Ng 2020 [A] Ng 2020 [B] Laboratory only; [A, B] CLIA |
[A] Abbott Laboratories ‐ Architect SARS‐CoV‐2 IgG assay [B] Abbott Laboratories ‐ Architect SARS‐CoV‐2 IgM assay (reported as prototype; not currently commercially available) | [A] N‐protein [B] S‐protein | [A] IgG [B] IgM | Day 1 to at least day 49 pso (Figure 2 D and E) [A] n samples by d pso: 41 (10%) day 1‐7 (from 16 patients) 106 (25%) day 8‐14 (from 24 patients) 113 (27%) day 15‐21 (from 21 patients) 163 (38%) day 22+ (up to 49) (from 18 patients) [B]: 26/346 (8%) day 1‐7 pso; 91/346 (26%) day 8‐14 pso; 83/346 (24%) day 15‐21 pso; 146/346 (42%) day 22+ pso | Serum, plasma |
|
Nguyen 2020 LFA only; CGIA |
BIOSYNEX COVID‐19 BSS (IgG/IgM)® | Not stated | IgG/IgM | 17.9 ± 8.2 d since pso 0‐10 d: n = 18 11‐20 d: n = 45 21+ d: n = 35 1 sample unclear (only 98 samples in Fig. 1a) | Finger‐prick blood |
|
Nicol 2020 [A] Nicol 2020 [B] Nicol 2020 [C] Nicol 2020 [D] Laboratory and LFA; [A] CLIA; [B to D] ELISA; [E] CGIA |
[A] Abbott Diagnostics SARS‐CoV‐2 CLIA IgG assay [B] Euroimmun Anti‐SARS‐CoV‐2 ELISA IgG/IgA assays [C] NG Biotech Laboratoires LFIA NG‐Test® IgG‐IgM COVID‐19 | [A] SARS‐CoV‐2 nucleoprotein (NP) [B] Recombinant S1 structural protein ‐ assay detects antibodies against the viral Spike‐protein [C] SARS‐CoV‐2 nucleoprotein | [A] IgG [B] IgG, IgA [C] IgG, IgM | [A]‐[C] 0‐> 15 d after onset of symptoms 0‐7 d pso 32/141 8‐14 d pso 29/141 15+ d pso 80/141 [2] median time between symptom onset and sera: 9.5 d 0‐7 d pso 24/57 8‐14 d pso 15/57 15+ d pso 18/57 | [A]‐[C] Serum |
|
Nilles 2020 [A] Nilles 2020 [B] Laboratory only; [A] CLIA; [B] ELISA |
[A] Roche Diagnostics ‐ Elecsys Anti‐SARS‐CoV‐2 immunoassay [B] Epitope Diagnostics ‐ EDI New Coronavirus COVID‐19 IgG or IgM ELISAs | [A] nucleocapsid (NC) antigen [B] IgG against the NC antigen [C] IgM against an unspecified antigen | [A] IgG [B] IgG, IgM | Mean 10.5 d (SD 6.0 d) post‐RT‐PCR confirmation; 16.1 d (SD 5.4 d) pso 8‐14 d pso: 30/68 15‐21 d pso: 29/68 > 21 d pso: 9/68 | Serum or plasma |
|
NSAE 2020 [A] NSAE 2020 [B] NSAE 2020 [C] NSAE 2020 [D] Laboratory only; [A to D] CLIA |
[A] Abbott Diagnostics SARS‐CoV‐2 Immunoassay [B] Roche Diagnostics Elecsys Anti‐SARS‐CoV‐2 [C] DiaSorin LIAISON SARS‐CoV‐2 S1/S2 IgG [D] Siemens Healthineers SARS‐CoV‐2 Total (COV2T) | [A] N‐protein [B] N‐protein [C] S1/S2‐protein [D] S1‐protein, RBD | [A] IgG [B] Total antibody [C] IgG [D]Total antibody | [1] ≥ 20 d pso [1a] Median 37 (range 20‐73) d pso [1b] All ≥ 28 d pso Median 44 (range 32‐82) d post‐PCR+ [1] Secondary analyses ≥ 30 d pso (n = 490) | Serum and plasma |
|
Ong 2020 [A] Ong 2020 [B] Laboratory and LFA; [A] CGIA; [B] ELISA |
[A] Orient Gene Biotech COVID‐19 IgG/IgM Rapid Test Cassette [B] Wantai SARS‐CoV‐2 Ab ELISA kit | [A] Not stated [B] Not stated | [A] IgG/IgM [B] Total antibody | Median time pso 7 d (IQR 4‐14 d) for all 228 (+ve and negative) patients < 7 d: 39/99 cases 7+ d: 52/99 cases 14+ d: 14/99 cases 7‐13 d: 38/99 cases Unclear: 8/99 cases | [1] and [2] Plasma samples [3] Serum |
|
Padoan 2020a Laboratory only; Laboratory |
CLIA ‐ MAGLUMI™ 2000 Plus nCoV (Snibe Diagnostics; Shenzhen New Industries Biomedical Engineering Co., Ltd) | Not stated | IgM (1.0 Au/mL); IgG (1.1 Au/mL) | Day 0 to ≥ 13 | Serum |
|
Padoan 2020b [A] Padoan 2020b [B] Laboratory only; [A] CLIA; [B] ELISA |
[A] SNIBE diagnostics MAGLUMI 2000 Plus [B] Euroimmun ELISA | [A] S‐antigen and N‐protein [B] S1‐specific IgA | [A] IgM (IgG also measured, limited details in supplementary information) [B] IgA (IgG also measured, results not reported) | From the onset of symptoms (fever) to 6 weeks after | Not stated |
|
Paiva 2021 [A] Paiva 2021 [B] Paiva 2021 [C] Laboratory and LFA; [A, B] CGIA; [C] CLIA |
[A] Wondfo SARS‐CoV‐2 Total Antibody Test [B] SD Biosensor STANDARD Q COVID‐ 19 IgM/IgG Duo Test [C] Abbott Diagnostics SARS‐CoV‐2 IgG test | [A] Spike‐protein [B] Not stated [C] N‐protein | [A] Total antibody [B] IgM/IgG [C] IgG | [1] 1‐35 (38) d pso (mean 11.2) [2] NA [3] Unclear [4] NA | [A, B, C] serum (n = 16) or plasma (n = 97) for [1], serum for [2] and [3], plasma for [4] |
|
Pan 2020a LFA only; LFA |
CGIA (Zhuhai Livzon Diagnositic Inc) | Not described | IgM, IgG | Day 1 to ≥ 15 | Serum or plasma |
|
Pape 2021 [A] Pape 2021 [B] Laboratory only; [A, B] ELISA |
[A] Euroimmun Anti‐SARS‐CoV‐2‐ELISA (IgA) [B] Euroimmun Anti‐SARS‐ CoV‐2‐ELISA (IgG) | S1 domain of the viral spike‐protein | IgA, IgG | 5‐27 d pso 5‐11 d pso: 17/57 11‐14 d pso: 24/57 15‐27 d pso: 16/57 | Serum |
|
Patel 2021 [A] Patel 2021 [B] Patel 2021 [C] Patel 2021 [D] Patel 2021 [E] Laboratory only; [A to C] ELISA; [D, E] CLIA |
[A] Euroimmun ‐ Anti‐SARS‐CoV‐2 ELISA (IgG) [B] Epitope Diagnostics ‐ EDI nCov COVID‐19 IgG ELISA kit [C] ImmunoDiagnostics Limited ‐ SARS‐CoV‐2 NP IgG ELISA kit [D] Abbott Laboratories Inc ‐ Architect SARS‐CoV‐2 IgG assay [E] Roche Diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 | [A] Spike 1‐protein [B] N‐protein [C] N‐protein [D] N‐protein [E] N‐protein | [A] IgG [B] IgG [C] IgG [D] IgG [E] Total Antibodies | 38‐57 d post‐PCR + | Plasma/serum |
|
Pere 2020 Laboratory only; CLIA |
Abbott SARS‐CoV‐2 IgG assay | SARS‐CoV‐2 nucleoprotein | IgG | [1a] Cases ‐ 39.5 (median) d after PCR, at least 1 month after COVID diagnosis | Serum |
|
Perez‐Garcia 2020(a) LFA only; CGIA |
Hangzhou AllTest Biotech ‐ AllTest COV‐19 IgG/IgM kit | Unclear | IgG, IgM | [1] NA (pre‐pandemic) [2] median (IQR) d from symptom onset = 17 (9‐25) <= 7 d pso: 19/90 8‐14 d pso: 21/90 15‐21 d pso: 15/90 22‐28 d pso: 20/90 28 d pso: 15/90 [3] median (IQR) d from symptom onset = 17 (15‐20) <= 7 d pso: 0/61 8‐14 d pso: 15/61 15‐21 d pso: 31/61 22‐28 d pso: 14/61 28 d pso: 1/61 | Serum |
|
Perez‐Garcia 2020(b) LFA only; LFA |
CGIA ‐ AllTest COV‐19 IgG / IgM kit (AllTest Biotech, Hangzhou, China)) | Not reported | IgG and IgM (visible line for either) | Day 8 to ≥ 14 | |
|
Perez‐Garcia 2021 [A] Perez‐Garcia 2021 [B] Perez‐Garcia 2021 [C] Perez‐Garcia 2021 [D] Perez‐Garcia 2021 [E] Perez‐Garcia 2021 [F] Laboratory and LFA; [A to C] CGIA; [D] ELISA; [E, F] CLIA |
[A] Hangzhou Alltest ‐ 2019‐nCoV IgG/IgM [B] Innovita Biological ‐ 2019‐nCoV Ab test [C] Epigentek SeroFlash IgM/IgG [D] DiaPro COVID‐19 IgG Confirmation [E] Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab [F] Siemens Atellica Total‐Ab assay | [A] [E] N [B] [C] [D] N & S [F] S1 | [A]‐[D] IgM and/or IgG [E] [F] Total antibodies (IgM + IgG) | 0‐7 d from onset of symptoms n = 18 8‐14 d from onset of symptoms n = 21 > 14 d from onset of symptoms n = 41 | Serum |
|
Pfluger 2020 [A] Pfluger 2020 [B] Pfluger 2020 [C] Pfluger 2020 [D] Pfluger 2020 [E] Laboratory only; [A, D] ELISA; [B, C, E] CLIA |
[A] EUROIMMUN Anti‐SARS‐CoV‐2 ELISA (IgG) [B] DiaSorin LIAISON SARS‐CoV‐2 S1/S2 IgG [C] Roche Diagnostics Elecsys Anti‐SARS‐CoV‐2 [D] Beijing Wantai Biological Pharmacy EnterpriseWANTAI SARS‐CoV‐2 Ab ELISA [E] Siemens Healthcare Atellica IM SARS‐CoV‐2 Total (COV2T) | [A] S1‐domain, spike‐protein [B] S1 and S2‐protein [C] N‐protein [D] RBD [E] Spike‐protein | [A] IgG [B] IgG [C] Total Ab [D] Total Ab [E] Total Ab | Mean time pso was 11.4 d (± 6.6), range 1‐38 d 1‐10 d n = 37 11‐15 d n = 22 16‐38 d pso n = 16 | Plasma/serum |
|
PHE 2020 [A] PHE 2020 [B] PHE 2020 [C] PHE 2020 [D] PHE 2020 [E] PHE 2020 [F] PHE 2020 [G] PHE 2020 [H] Laboratory only; [A] ELISA; [B to H] CLIA |
[A] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [B] Abbott Architect anti‐SARS‐CoV‐2 IgG [C] Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab [D] Ortho Clinical VITROS Anti‐SARS‐Cov‐2 IgG [E] Siemens Atellica Total‐Ab assay [F] Ortho Clinical VITROS Anti‐SARS‐Cov‐2 Total Ab [G] Diasorin ‐ LIAISON SARS‐CoV‐2 [H] Beckman Coulter ‐ Access SARS‐CoV‐2 IgG | [A, F] S1‐based [B, C] N‐based [D, G] S‐based [E, H] RBD | [A, B, D, G, H] IgG [C, E, F] Total antibody | 0 to 50 d pso <= 10 d pso: 11‐14 samples 11‐20 d pso: 4‐12 samples 21‐30 d pso: 35‐37 samples 31‐40 d pso: 30‐32 samples 51‐50 d pso: 9‐10 samples For some samples, the interval was d from hospital admission to sample collection: [A] [D] n = 14 [B] [C] [E] n = 0 | Serum |
|
Phipps 2020 Laboratory only; CLIA |
Abbott ‐SARS‐CoV‐2 IgG (Abbott 06R86) testing | SARS‐CoV‐2 N‐protein | IgM or IgG | Figure 3 in paper showed samples collected between day 0 and day c45 | Plasma |
|
Pickering 2020 [A] Pickering 2020 [B] Pickering 2020 [C] Pickering 2020 [D] Pickering 2020 [E] Pickering 2020 [F] Pickering 2020 [G] Pickering 2020 [H] Pickering 2020 [I] Pickering 2020 [J] Laboratory and LFA; [A] to [G] CGIA; [H] CLIA; [I], [J] ELISA |
[A] AccuBiotech ‐ Accu‐Tell COVID‐19 IgG/IgM [B] Anhui Deep Blue IgG/IgM [C] Biohit Healthcare IgM/IgG [D] GenBody COVID‐19 IgM/IgG [E] Spring Healthcare IgM/IgG rapid test [F] SureScreen Diagnostics ‐ COVID‐19 Coronavirus Rapid Test [G] Jiangsu Medomics Combined Ab [H] Watmind Medical SARS‐CoV‐2 [I] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA [J] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | [A to G] Not reported [H] total antibody against SARS‐CoV‐2 [I] SARS‐CoV‐2 S1‐protein [J] SARS‐CoV‐2 S1‐protein |
[A to G] IgG, IgM [H] Total antibody (IgG, IgM, IgA) [I] IgA [J] IgG | 1 to 30 d after onset of self‐reported symptoms < 10 d pso: 38/110 samples 10+ d pso: 72/110 samples < 14 d pso: 56/110 samples 14+ d pso: 54/110 samples 20+ d pso: 28/110 samples 10‐14 d pso: 18/110 samples 14‐20 d pso: 26/110 samples 10‐20 d pso: 44/110 samples | Venous serum samples |
|
Pollan 2020 Laboratory only; CLIA |
Abbott Architect ‐ SARS‐CoV‐2 IgG | SARS‐CoV‐2 nucleoprotein | IgG | All PCR+ were ≥ 10 d pso (n = 82), 58 (71%) ≥ 14 d pso | Serum samples |
|
Prazuck 2020 [A] Prazuck 2020 [B] LFA only; [A] [B] CGIA |
[A] AAZ‐LMB COVID‐PRESTO [B] AAZ‐LMB COVID‐DUO | [A] and [B] recombinant COVID‐19 antigens labelled with colloidal gold | [A] and [B] IgM, IgG | For [1a] 0‐ > 15 d post‐onset
[test A] 0‐5 d pso: 20/150 6‐10 d pso: 43/150 11‐15 d pso: 39/150 15‐31 d pso: 48/150 [test B] 0‐5 d pso: 14/134 6‐10 d pso: 42/134 11‐15 d pso: 44/134 15‐31 d pso: 34/134 For [1b] 24 hours to 8 d from onset of symptoms (median 2 d; range 1–8 d) |
Capillary whole blood |
|
Qian 2020 Laboratory only; CLIA |
Shenzhen YHLO Biotech Co ‐ SARS‐CoV‐2 IgM and IgG | SARS‐CoV‐2 N‐protein and spike‐protein | IgM and IgG | [1a] < 7 d pso, n = 63 7‐14 d pso, n = 99 > 14 d pso, n = 351 | Serum |
|
Qiu 2020 Laboratory only; CLIA |
Autobio Diagnostics Co ‐ SARS‐CoV‐2 IgG and IgM CLIA | [A] and [B] S‐protein | [A] IgG [B] IgM | 1 to 87 d pso 1‐10 d pso: 66/409 11‐20 d pso: 70/409 21+ d pso: 273/409 | Serum |
|
Ragnesola 2020 LFA only; CGIA |
Hangzhou Clongene Biotech Co., Ltd., Hangzhou, China ‐ Clungene® SARS‐CoV‐2 IgG/IgM Rapid Test Cassettes | Receptor‐binding domain (RBD) of the S‐ and N‐protein | IgM/IgG | Symptom‐free for at least 14 d so at least 14 d post‐PCR+ | Plasma |
|
Renard 2021 [A] Renard 2021 [B] Laboratory only; [A] [B] Enzyme Linked Fluorescent Assay (ELFA) |
[A] bioMerieux Vidas SARS‐CoV‐2 IgM (423833) [B] bioMerieux Vidas SARS‐CoV‐2 IgG (423834) | [A] [B] RBD of Spike‐Protein | [A] IgM [B] IgG | 0 to 32+ 1‐65 d pso (n = 105):
0‐7 d: n = 22
8‐15 d: n = 29
16‐23 d: n = 26
24‐31 d: n = 18
≥ 32 d: n = 10 0‐65 d post‐PCR +ve (n = 232): 0‐7 d: n = 110 8‐15 d: n = 60 16‐23 d: n = 38 24‐31 d: n = 13 ≥ 32 d: n = 11 |
Serum or plasma |
|
Rijkers 2020 Laboratory only; ELISA |
Beijing SARS‐CoV‐2 total antibody ELISA (catalog number WS1096) | [A] RBD antigen of SARS‐CoV‐2 [B] S1 domain | [A] Total antibodies [B] SARS‐CoV‐2 IgG and/or IgA | Serial blood sampling (3 times per week) was started a median of 2 d (range, 1–7 d) after +ve RT‐PCR. | Serum |
|
Rode 2021 [A] Rode 2021 [B] Rode 2021 [C] Laboratory and LFA; [A, B] ELISA; [C] CGIA |
[A] Euroimmun Anti‐SARS‐CoV‐2 IgA ELISA [B] Euroimmun Anti‐SARS‐CoV‐2 IgG ELISA [C] Maccura Biotechnology Co SARS‐CoV‐2 IgM/IgG Antibody Assay Kit | [A.B] S1 antigen [C] N/S antigen | [A, B] IgA and IgG [C] IgM and IgG | Range 0‐22 d pso: 0‐3 d pso: n = 11, 4‐7 d pso: n = 17, 8‐11 d pso: n = 18, ≥ 12 d from onset of illness: n = 14 | Serum |
|
Rudolf 2020 [A] Rudolf 2020 [B] Rudolf 2020 [C] Rudolf 2020 [D] Rudolf 2020 [E] Rudolf 2020 [F] Rudolf 2020 [G] Rudolf 2020 [H] Rudolf 2020 [I] Rudolf 2020 [J] Rudolf 2020 [K] Laboratory and LFA; [A to C, G, I] CGIA; [D to F, H, J, K] LFA |
[A] CTK OnSite COVID‐19 IgG/IgM (LOT F0507R1C00) [B] Sure Biotech ‐ SARS‐CoV‐2 IgM/IgG Ab (LOT COV1252006A) [C] Augurix SimtomaX Corona Check (LOT GGM20089W) [D] TAmiRNA SARS‐CoV‐2 Ab (LOT 20200428) [E] NTBIO One Step IgG/IgM (LOT V02009201) [F] EXACARE QuickTestCorona IgG/IgM (LOT MC0000102) [G] Xiamen Biotime SARS‐Cov‐2 IgG/IgM (LOT X2003602) [H] Inzek ‐ BIOZEK COVID‐19 IgG/IgM (LOT BNCP40200080) [I] MEDsan COVID19 IgG/IgM (LOT 20200325) [J] Qingdao HIGHTOP IgM/IgG (LOT COV1252003C) [K] Hangzhou Biotest ‐ RightSign IgG/IgM (LOT COV20040013) | [A] Spike [B] S1, S2, RBD [C] RBD, N‐protein [D] S1 [E] Not stated [F] RBD, N‐protein [G] Not stated [H] Not stated [I] Not stated [J] Spike, N‐protein [K] Spike | All tests IgG, IgM [D] IgM/IgG (single band) |
Wide range of d pso; numbers differed between tests. | [J] Whole blood, serum and plasma All other tests: serum and plasma |
|
Ruetalo 2020 [A] Ruetalo 2020 [B] Ruetalo 2020 [C] Laboratory only; [A, B] ELISA; [C] CLIA |
[A] Euroimmun SARS‐CoV‐2‐ELISA (IgG) [B] Mediagnost S1 RBD SARS‐CoV‐2 IgG, IgA, IgM [C] Roche Elecsys anti‐SARS‐CoV‐2 | [A] S1‐based [B] Spike RBD [C] N‐protein | [A] IgG [B] IgG, IgA, IgM [C] IgG + IgM | The time from +ve SARS‐CoV‐2 test to blood sampling was 14‐64 d (median 45 d). | Serum samples were stored at ‐80°C. |
|
Schnurra 2020 [A] Schnurra 2020 [B] Schnurra 2020 [C] Schnurra 2020 [D] Schnurra 2020 [E] Schnurra 2020 [F] Schnurra 2020 [G] Laboratory only; [A, B, G] CLIA; [C to F] ELISA |
[A] Roche Elecsys Anti‐SARS‐CoV‐2 [B] Abbott Architect SARS‐CoV‐2 IgG [C] Novatec Novalisa SARS‐CoV‐2 IgG ELISA [D] Virotech SARS‐CoV‐2 IgG ELISA [E] Euroimmun Anti‐SARS‐CoV‐2‐ELISA (IgG) [F] Mediagnost AntiSARS CoV‐2 ELISA [G] Siemens Atellica IM COV2T | [A] N‐protein [B] N‐protein [C] N‐protein [D] N‐protein [E] S1‐glycoprotein [F] RBD of S1‐glycoprotein [G] RBD of S1‐glycoprotein | [A] IgM, IgG and other Ig antibody bridging [B] IgG [C] IgG [D] IgG [E] IgG [F] IgG [G] IgM, IgG and other Ig antibody bridging | Between 2 and 10 weeks pso or viral RNA testing (additional data provided by author showed range from 14 to 70 d post‐PCR test): 2‐3 weeks after PCR+: n = 25 4‐10 weeks after PCR+: n = 48 | Serum samples |
|
Serre‐Miranda 2021 [A] Serre‐Miranda 2021 [B] Serre‐Miranda 2021 [C] Serre‐Miranda 2021 [D] Serre‐Miranda 2021 [E] Serre‐Miranda 2021 [F] Serre‐Miranda 2021 [G] Serre‐Miranda 2021 [H] Serre‐Miranda 2021 [I] Serre‐Miranda 2021 [J] Serre‐Miranda 2021 [K] Serre‐Miranda 2021 [L] Laboratory and LFA; [A, D] CLIA; [B, C] ELISA; [E to G, I, J, L] CGIA; [H, K] LFA |
[A] Abbott Architect anti‐SARS‐CoV‐2 IgG (no. 06R86) [B] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG (no. EI 2606‐9601 G) [C] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA (no. EI 2606‐9601 A) [D] Snibe Diagnostic ‐ MAGLUMI 2019‐nCoV IgG/IgM (no. 130219016M) [E] Cellex qSARS‐CoV‐ 2 IgG/IgM (no. WI5513C) [F] Getein One Step Test (no. CG2057) [G] Innovita Biological ‐ 2019‐nCoV Ab test [H] Liming Bio StrongStep1 IgM/IgG [I] Leccurate ‐ SARS‐CoV‐2 [J] Jiangsu Medomics Combined Ab [K] Render COVID‐19 IgM/IgG (no. K‐20‐RC‐CoV‐2) [L] SD Biosensor IgM/IgG Duo (no. Q‐NCOV‐01D) | [A] N‐protein [B, C] S1‐protein [D to H] S antigen and N‐protein [I to L] Not specified | [A] IgG [B] IgG [C] IgA [D] IgM [E] IgG [F] IgM/IgG [G] Total antibodies [H to L] IgM/IgG | Days since symptom onset: Numbers varied per test: < 10 d: 12‐24 samples; 10–15 d: 12‐33 samples; 16–21 d: 20‐34 samples; > 21 d: 16‐35 samples | Plasma |
|
Shamsollahi 2020 LFA only; CGIA |
VivaChek Inc VivaDiag COVID‐19 IgM/IgG | Not stated | IgM, IgG | 5‐53 d (mean: 27.9) pso 0‐19 d pso: 31/114 (27.2%) 20‐39 d pso 65/114 (57.0%) 40+ d pso: 18/114 (15.8%) | Whole blood; frozen pre‐pandemic serum |
|
Shen 2020a LFA only; CGIA |
Shanghai Outdo Biotech ‐ SARS‐Cov‐2 IgM/IgG (LOT: 20200101) | S, M, and N‐proteins of COVID‐19 | SARS‐Cov‐2 IgM/IgG | At time of consultation: Time since symptom onset for COVID 19‐+ve cases: 0‐7 d = 40/97 (41.2%) 8‐14 d = 33/97 (34.0%) ≥ 15 d = 24/97 (24.7%) Since symptom onset for COVID 19‐negative: 0‐7 d = 50/53 (94.3%) 8‐14 d = 3/53 (5.7%) | Serum |
|
Shen 2020b LFA only; CGIA |
Shanghai Outdo Biotech ‐ SARS‐CoV‐2 IgM GICA kit | N and S | IgM | 4–14 d pso | Serum |
|
Soleimani 2021 Laboratory only; CLIA |
Snibe MAGLUMI 2019‐Novel Coronavirus (nCoV) Kit | N and S‐proteins | IgG and/or IgM | 0 to 4 d pso n = 21, 5 to 9 d pso n = 50, 10 to 14 d pso n = 61, 15 to 25 d pso n = 44 | Serum |
|
Sterlin 2021 [A] Sterlin 2021 [B] Laboratory only; Other; Multi‐Antigen Serology Panel |
Genalyte Inc ‐ Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel | [A] S1 RBD, S1, S2 [B] N‐based |
IgA, IgM, IgG | Multiple samples obtained from each patient (214 samples from 135 patients): 48 samples collected 1‐7 d pso: 8‐14 d pso: 81/214 15‐21 d pso: 39/214 22‐28 d pso: 20/214 > 28 d pso: 26/214 | Serum |
|
Suhandynata 2020a Laboratory only; CLIA |
Diazyme ‐ DZ‐LITE 2019‐nCoV IgG, IgM (CLIA) Assay Kits (Cat # 130219015M; Cat # 130219016M) | SARS‐CoV‐2 N and S‐proteins | IgM, IgG, IgM or IgG | Unclear, data by time were in relation to first +ve PCR result | Serum or plasma |
|
Suhandynata 2020b [A] Suhandynata 2020b [B] Suhandynata 2020b [C] Laboratory only; [A to C] CLIA |
[A] Diazyme DZ‐LITE 2019‐nCoV IgG, IgM (CLIA) Assay Kits (Cat # 130219015M; Cat # 130219016M) [B] Roche Elecsys Anti‐SARS‐CoV‐2 total Ig (Ref # 09203079190) [C] Abbott SARS‐CoV‐2 IgG (Ref # 06R8620) reagent kit | Not stated | [A] IgG, IgM [B] Total antibodies [C] IgG | ≤ 7 d post‐PCR+ (n = 43), 8–14 d post‐PCR+ (n = 31), ≥ 15 d post‐PCR+ (n = 25) | Plasma (Li‐Heparin or K‐EDTA) and serum samples |
|
Sun 2020 Laboratory only; ELISA |
RayBiotech ‐ Covid‐19 human ELISA (IgG, IgA, IgM; Cat. no. IEQ‐CoVS1RBD‐IgG; Cat. No. IE‐CoVS1RBD‐IgA; Cat. No. IE‐CoVS1RBD‐IgM | RBD (from cat. No.) | IgG, IgA, IgM | Serum samples were collected prospectively from cases every 3 d from hospitalisation until the date of discharge from hospital. | Serum |
|
Sweeney 2020 LFA only; Lateral flow immunoassay |
Surescreen Diagnostics ‐ SureScreen LFIA | "detecting antibodies to SARS‐CoV‐2 Spike‐proteins" | IgM/IgG | [1] 14+ d post‐onset of symptoms: 301/301 (100%), of which: 14‐19 d post‐onset of symptoms: 97/301 (32%) 20+ d post‐onset of symptoms: 204/301 (68%) | Serum |
|
Tan 2020 [A] Tan 2020 [B] Laboratory only; [A] [B] CLIA |
[A] Roche Elecsys Anti‐SARS‐CoV‐2 assay [B] Abbott Architect Anti‐SARS‐CoV‐2 assay | [A] N‐protein [B] N‐protein | [A] Total Antibodies (IgG and IgM) [B] IgG | < 7 d pso (n = 80) 7‐13 d pso (n = 37) 14‐20 d pso (n = 21) ≥ 21 d pso (n = 32) | Serum |
|
Tang 2020 [A] Tang 2020 [B] Tang 2020 [C] Laboratory only; [A, C] CLIA; [B] ELISA |
[A] Abbott SARS‐CoV‐2 IgG assay [B] EUROIMMUN SARS‐CoV‐2 IgG assay [C] Roche Elecsys Anti‐SARS‐CoV‐2 | [A] N‐protein [B] S1 domain [C] N‐protein from SARS‐CoV‐2 | [A] and [B] IgG [C] total Ab | Day 0 to ≥ 14 d pso | Plasma (stated in Discussion) |
|
Theel 2020 [A] Theel 2020 [B] Theel 2020 [C] Theel 2020 [D] Laboratory only; [A], [B] ELISA; [C], [D] CLIA |
[A] Abbott Architect anti‐SARS‐CoV‐2 IgG [B] Epitope Diagnostics ‐ EDI nCov COVID‐19 IgM/IgG [C] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [D] Ortho Clinical VITROS Anti‐SARS‐Cov‐2 IgG | [A] S1‐protein [B] N‐protein from SARS‐CoV‐2 [C] SARS‐CoV‐2 nucleocapsid antigen [D] SARS‐CoV‐2 spike antigen | [A‐D] IgG | 33 inpatients (190 samples) 0‐7 d pso: 38 8‐14 d pso: 91 15‐26 d pso: 61 23 outpatients (34 samples): 0‐7 d post‐PCR+: 11 (excluded from review) 20‐31 d post‐PCR+: 23 | [1] Serum [2] Serum [3] Serum [4] Serum |
|
Thijsen 2020 Laboratory only; ELISA |
EUROIMMUN ‐ Anti‐SARS‐CoV‐2 ELISA IgG, IgA | S1 domain | IgG, IgA | 6‐32 d pso | Not stated (likely serum or plasma) |
|
Trabaud 2020 [A] Trabaud 2020 [B] Trabaud 2020 [C] Trabaud 2020 [D] Trabaud 2020 [E] Trabaud 2020 [F] Trabaud 2020 [G] Trabaud 2020 [H] Laboratory only; [A, C, E, F] CLIA; [B] Enzyme Linked Fluorescent Assay (ELFA); [D, G, H] ELISA |
[A] Diasorin ‐ LIAISON SARS‐CoV‐2 [B] bioMerieux ‐ Vidas SARS‐CoV‐2 IgG [C] Siemens Atellica Total‐Ab assay [D] Wantai ELISA Total‐Ab assay [E] Abbott Diagnostics Architect anti‐SARS‐CoV‐2 IgG [F] Roche Diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 Ab [G] Bio‐Rad Laboratories Platelia SARS‐CoV‐2 Total Ab [H] Epitope Diagnostics ‐ EDI nCov COVID‐19 IgM/IgG | [A] S1 and S2 [B] S1 and peptide [C] RBD [D] RBD [E‐H] N‐protein | [A,B,E,H] IgG [C,D,F,G] Total antibody | Range 4 to 52 d pso: <= 15 d pso (n = 16) 16‐20 d pso (n = 21) > 20 d pso (n = 45) | All serum/plasma |
|
Traugott 2020 [A] Traugott 2020 [B] Traugott 2020 [C] Traugott 2020 [D] Traugott 2020 [E] Traugott 2020 [F] Laboratory and LFA; [A] to [D] ELISA; [E], [F] LFA |
[A] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA [B] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [C] Wantai Biological Pharmacy SARS‐CoV‐2 IgM ELISA [D] Wantai Biological Pharmacy ELISA Total‐Ab assay [E] Wantai Biological Pharmacy SARS‐CoV‐2 Ab rapid assay [F] Hangzhou Alltest ‐ 2019‐nCoV IgG/IgM | [A], [B] S1 domain [C], [D] Spike‐protein RBD [E], [F] Not stated | [A] IgA [B] IgG [C] IgM [D] Total antibody [E] Total antibody [F] IgG/IgM | PCR+ cases only: 1‐5 d pso: 30 (39%) 6‐10 d pso: 25 (32%) 11‐29 d pso: 22 (29%) | Serum or plasma |
|
Tre‐Hardy 2021 [A] Tre‐Hardy 2021 [B] Laboratory only; [A] CLIA; [B] ELISA |
[A] Diasorin LIAISON SARS‐CoV‐2 IgG [B] Euroimmun anti‐SARS‐CoV‐2 ELISA IgG | [A] S1 and S2 subunits [B] S1 subunit | [A] IgG [B] IgG | ≥ 14 d post‐PCR + | Serum stored in the laboratory serum biobank at ≤−20 °C |
|
Tuaillon 2020 [A] Tuaillon 2020 [B] Tuaillon 2020 [C] Tuaillon 2020 [D] Tuaillon 2020 [E] Tuaillon 2020 [F] Tuaillon 2020 [G] Tuaillon 2020 [H] Tuaillon 2020 [I] Tuaillon 2020 [J] Laboratory and LFA; [A] to [F] LFAs; [G to J] ELISAs |
[A] Zhuhai Livzon Pharmaceutical Group ‐ 2019‐nCoV IgM/IgG [B] UNscience Biotechnology ‐ COVID‐19 IgG/IgM [C] Chongqing iSIA BIO‐Technology ‐ 2019‐nCoV IgM/IgG kit [D] Guangdong Hecin Biotech ‐ 2019‐nCoV IgM kit [E] AccuBiotech ‐ Accu‐Tell COVID‐19 IgG/IgM [F] Acro Biotech ‐ 2019‐nCoV IgM/IgG [G] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA [H] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [I] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA or IgG [J] ID.Vet ‐ ID Screen SARS‐CoV‐2‐N IgG Indirect ELISA | [A] to [F] not stated [G to I] recombinant subunit protein 1 (S1) [J] N‐protein | [A] to [C], [E] to [F] IgM, IgG, IgM or IgG
[D] IgM [G to I] IgA, IgG [J] IgG |
[1] 1‐6 d (n = 9), 7‐14 d (n = 14), ≥ 15 d (n = 15) from the onset of symptoms | Plasma |
|
Valdivia 2020 [A] Valdivia 2020 [B] Valdivia 2020 [C] Valdivia 2020 [D] Laboratory only; [A, C] CLIA; [B, D] ELISA |
[A] Diasorin ‐ LIAISON SARS‐CoV‐2 [B] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [C] Snibe Diagnostic ‐ MAGLUMI 2019‐nCoV IgG/IgM [D] Vircell ‐ COVID‐19 ELISA IgG | [A] S‐protein [B] S1 domain [C] N‐protein [D] S1 and N‐protein | [A][B][C][D] IgG | Samples were stored for a maximum 1 month from point of collection | Serum |
|
Van Elslande 2020a [A] Van Elslande 2020a [B] Van Elslande 2020a [C] Van Elslande 2020a [D] Van Elslande 2020a [E] Van Elslande 2020a [F] Van Elslande 2020a [G] Van Elslande 2020a [H] Laboratory and LFA; [A] to [G] LFA; [H] ELISA |
[A] Hangzhou Clungene SARS‐CoV‐2 IgG/IgM [B] Zhejiang Orient‐Gene IgG/IgM [C] VivaChek Biotech ‐ VivaDiag COVID‐19 IgM/IgG [D] Liming Bio‐Products StrongStep1 IgM/IgG [E] Dynamiker Biotechnology 2019‐nCoV IgG/IgM Rapid Test [F] Multi‐G MGA 2019‐nCoV IgG/IgM Rapid test cassette [G] Prima Lab COVID‐19 IgG/IgM Rapid test [H] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | [A] Recombinant envelope antigens [B] Recombinant antigens [C] Recombinant antigen [D] Recombinant antigen [E] N‐protein [F] N‐protein [G] COVID‐19 antigen [H] S1‐protein | [A] IgG/IgM [B] IgG/IgM [C] IgG/IgM [D] IgG/IgM [E] IgG/IgM [F] IgG/IgM [G] IgG/IgM [H] IgG/IgA | Day 0‐6 pso: 37 (24%) Day 7‐13 pso: 78 (51%) Day 14‐25 pso: 38 (25%) | Serum or plasma |
|
Van Elslande 2020b [A] Van Elslande 2020b [B] Van Elslande 2020b [C] Van Elslande 2020b [D] Van Elslande 2020b [E] Van Elslande 2020b [F] Van Elslande 2020b [G] Laboratory only; [A, B, E, F] CLIA; [C, D, G] ELISA |
[A] Roche Diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 Ab [B] Abbott Diagnostics Architect anti‐SARS‐CoV‐2 IgG [C] EUROIMMUN ‐ (N‐based) anti‐SARS‐COV‐2 IgG [D] Mikrogen IgG anti‐N [E] Snibe Diagnostic ‐ MAGLUMI 2019‐nCoV IgG/IgM [F] Diasorin ‐ LIAISON SARS‐CoV‐2 [G] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG | [A] N‐protein [B] N‐protein [C] N‐protein [D] N‐protein [E] N and S‐protein [F] S‐protein (S1 and S2) [G] S1‐protein | [A] Total Ig antibodies [B]‐[G] IgG | 0‐6 d pso, n = 43 7‐13 d pso, n = 98 14‐17 d pso, n = 42 18‐21 d pso, n = 16 22‐27 d pso, n = 13 28‐37 d pso, n = 11 | Serum |
|
Velay 2020 [A] Velay 2020 [B] Velay 2020 [C] Velay 2020 [D] Laboratory and LFA; [A, B] LFA, [C, D] ELISA |
[A] Biosynex COVID‐19 BSS [B] Servibio/VEDALAB COVID‐19 Sign IgM/IgG [C] Euroimmun ELISA anti–SARS‐CoV‐2 IgA and IgG [D] Epitope Diagnostics EDI™ novel coronavirus COVID‐19 IgM and IgG | [A] N‐protein [B] S1‐protein | [A] IgM and IgG [B] IgM and IgG [C] IgA and IgG [D] IgM and IgG | [1a] Serum samples were collected at a median of 7 days pso (range, 0–31 days pso) [1b] 24 days pso (range, 15–39 days pso) | Serum and plasma |
|
Veyrenche 2021 [A] Veyrenche 2021 [B] Veyrenche 2021 [C] Veyrenche 2021 [D] Laboratory and LFA; [A] CLIA; [B] ELISA; [C] CGIA; [D] LFA |
[A] Abbott Diagnostics Architect anti‐SARS‐CoV‐2 IgG [B] Theradiag COVID‐19 THERA02 IgM assay [C] SureScreen Diagnostics ‐ COVID‐19 Coronavirus Rapid Test [D] Syzbio SARS‐CoV‐2 IgM/IgG Antibody Assay Kit | [A] N‐protein [B] S‐protein [C] S‐protein [D] Not stated | [A] IgG [B] IgM [C] IgG [D] IgM and/or IgG | Day 1‐20 pso 1‐7 d (n = 22) 7‐14 d (n = 14) 14‐20 d (n = 9) | Plasma |
|
Wang 2020a Laboratory |
Xiamen Wantai ‐ Total Ab CMIA | RBD | Total Ab | Day 0 to > 20 days pso; 0‐10 days after symptom onset: 61 (43%) 11‐20 days after symptom onset: 72 (51%) 21+ days after symptom onset: 8 (6%) |
Serum |
|
Weidner 2020 [A] Weidner 2020 [B] Weidner 2020 [C] Weidner 2020 [D] Weidner 2020 [E] Weidner 2020 [F] Laboratory and LFA; [A to C] ELISA; [D, E] CLIA; [F, G] LFA |
[A] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [B] Wantai ELISA Total‐Ab assay [C] Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab [D] Diasorin ‐ LIAISON SARS‐CoV‐2 [E] MEDsan COVID19 IgG/IgM [F] Wantai SARS‐CoV‐2 Ab rapid assay | [A] S1 domain [B] N‐protein [C] Not stated [D] N‐protein [E] S1 and S2 domains of the spike‐protein [F] Not stated | [A] IgG [B] IgG [C] Total antibodies? [D] Total antibodies [E] IgG [F] IgM, IgG | Samples collected between 26 and 61 d pso (median 47 d, standard deviation 6.6 d) | Serum, plasma |
|
Wellinghausen 2020a [A] Wellinghausen 2020a [B] Wellinghausen 2020a [C] Wellinghausen 2020a [D] Wellinghausen 2020a [E] Laboratory only; [A, B] ELISA; [C to E] CLIA |
[A] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [B] Epitope Diagnostics ‐ EDI nCov COVID‐19 IgM/IgG [C] Diasorin ‐ LIAISON SARS‐CoV‐2 [D] Abbott Diagnostics ‐ Architect anti‐SARS‐CoV‐2 IgG [E] Roche Diagnostics ‐ Elecsys anti‐SARS‐CoV‐2 Ab | [A] S1‐protein [B] N‐protein [C] S1 and S2‐protein [D] N‐protein [E] N‐protein | [A] IgG [B] IgG [C] IgG [D] IgG [E] Total Ab | [1a] 10 to 54 d pso (median 24 d) day 10‐20 (n = 11), day 21 to 54 (n = 40) plus 3 patients with three follow‐up serum samples each [1b] 9‐56 d post‐PCR+ | Serum |
|
Wellinghausen 2020b Laboratory only; ELISA |
Euroimmun ‐ Anti‐SARS‐CoV‐2‐ELISA IgG | S1‐protein | IgG | All had recovered at the time point of blood collection. [1a] Day 10‐20 pso, n = 11; Day 21‐68 pso, n = 100; [1b] Day 9‐20 post‐PCR+, n = 10; day 21‐56 post‐PCR+, n = 16; day 28‐56 post‐PCR+, n = 14. | Serum |
|
Whitman 2020a [A] Whitman 2020a [B] Whitman 2020a [C] Whitman 2020a [D] Whitman 2020a [E] Whitman 2020a [F] Whitman 2020a [G] Whitman 2020a [H] Whitman 2020a [I] Whitman 2020a [J] Whitman 2020a [K] Laboratory and LFA; [A]‐[J] LFAs; [K] ELISA |
[A] BioMedomics ‐ COVID‐19 IgM‐IgG Rapid Test (51‐002‐20) [B] Bioperfectus Technologies ‐ PerfectPOC nCoV (SARS CoV‐2) IgM/IgG (SC30201 W) [C] Decombio Biotechnology ‐ nCoV (SARS‐CoV‐2) IgM/IgG Combo [D] DeepBlue Medical Technology ‐ COVID‐19 (SARS CoV‐2) IgG/IgM Ab [E] Innovita Biological Technology ‐ 2019‐nCoV Ab test [F] Hangzhou Biotest/Premier Biotech ‐ RightSign IgG/IgM (INGMMC42S) [G] Sure Biotech ‐ SARS‐CoV‐2 IgM/IgG Ab (VC01210 3) [H] UCP Bioscience ‐ Covid‐19 IgG/IgM (U‐CoV102) [I] VivaChek Biotech ‐ VivaDiag COVID‐19 IgM/IgG (VID35‐08‐011) [J] Guangzhou Wondfo ‐ SARS‐CoV‐2 Ab (W195) [K] Epitope Diagnostics ‐ EDI nCov COVID‐19 IgM/IgG (KT‐1033; kt‐1032) | [A] RBD [B] N and S [C] Not stated [D] Not stated [E] N and S [F] S‐based [G] N and S [H] Not stated [I, J] S‐based [K] N | [A]‐[I][K] IgM and/or IgG [J] Total Ab | 0‐ > 20 d pso 1‐5 d pso: n = 28 6‐10 d pso: n = 36 11‐15 d pso: n = 34 16‐20 d pso: n = 19 > 20 d pso: n = 11 | Plasma or serum |
|
Whitman 2020b [A] Whitman 2020b [B] Whitman 2020b [C] LFA only; [A to C] LFA |
[A] SD Biosensor ‐ Standard Q COVID‐19 IgM/IgG Duo (KT1032; lot P630C) [B] Biolidics ‐ 2019‐nCoV IgG/IgM antibody detection kit (CBB‐F015016‐V; V2020 0330) [C] Biomedomics ‐ COVID‐19 IgM and IgG Rapid Test (51‐002‐20; lot 20200, 22702, 20200, 32103) ‐ See below | [A] N‐based [B] N‐ and S‐based [C] S‐based | IgG, IgM | Not stated | Serum/plasma |
|
Wolff 2020 [A] Wolff 2020 [B] Wolff 2020 [C] Wolff 2020 [D] Wolff 2020 [E] Wolff 2020 [F] Laboratory only; [A, B] CLIA; [C; D] ELISA; [E, F] enzyme linked fluorescence assay (ELFA) |
[A] Roche Diagnostics Elecsys Anti‐SARS CoV‐2 [B] Diasorin Liaison SARS‐CoV‐2 S1/S2 IgG [C] Euroimmun Anti‐SARS CoV‐2 IgG ELISA [D] Euroimmun Anti‐SARS CoV‐2 IgA ELISA [E] BioMerieux VIDAS Anti‐SARS CoV‐2 IgG [F] BioMerieux VIDAS Anti‐SARS CoV‐2 IgM | [A] N‐protein [B] S1/S2‐protein [C] [D] S1‐protein [E] [F] S‐protein | [A] IgM/IgG (total antibodies including IgG) [B] IgG [C] IgG [D] IgA [E] IgG [F] IgM | [1a] 0‐54 d pso [1b] 0‐15 d post‐PCR + 0–7 d post‐symptoms or post‐+PCR: n = 35 8–14 d post‐symptoms or post‐+PCR: n = 31 > 15 d post‐symptoms or post‐+PCR: n = 45 | Serum |
|
Wu 2020 [A] Wu 2020 [B] Wu 2020 [C] Wu 2020 [D] LFA only; [A to D] LFAs |
[A] Hangzhou ALLTEST 2019‐nCoV IgG/IgM Rapid Test [B] Dynamiker Biotechnology 2019‐nCoV IgG/IgM Rapid Test [C] TONYAR Biotech Inc. ASK COVID‐19 IgG/IgM Rapid Test [D] Wondfo Biotech Co SARS‐CoV‐2 Antibody Test | [A] Nucleocapsid [B] Nucleocapsid [C] Spike [D] Not described | [A] IgG/IgM [B] IgG/IgM [C] IgG/IgM [D] Total antibody | [1] Day 1‐14 pso: 46/99 (46%) Day 15‐21 pso: 23/99 (23%) > Day 21 pso: 30/99 (30%) [2] Day 1‐14 pso: 37/58 (64%) Day 15‐21 pso: 11/58 (19%) > Day 21 pso: 10/58 (17%) | Serum |
|
Xiang 2020a Laboratory only; Laboratory |
ELISA (ELISA kits, Zuhai Livzon Inc, number of IgM: 20200308, IgG: 20200308) | N‐based | IgG IgM | Day 0 to > 21 | Serum |
|
Xiao 2020a Laboratory only; Laboratory |
CLIA (Shenzhen YHLO Biotechnology Co. Ltd.) | Not described | IgM, IgG (<= 10 AU/mL) | Day 1 to 49 | Blood |
|
Xiao 2020b [A] Xiao 2020b [B] Xiao 2020b [C] Xiao 2020b [D] Laboratory only; [A] CLIA; [B to D] ELISA |
[A] Wantai Biological Pharmacy Enterprise CLIA Total‐Ab assay [B] Wantai Biological Pharmacy Enterprise SARS‐CoV‐2 IgG ELISA [C] Wantai Biological Pharmacy Enterprise SARS‐CoV‐2 IgM ELISA [D] Wantai Biological Pharmacy Enterprise ELISA IgA assay | [A] RBD [B to C] S‐based |
SARS‐CoV‐2 specific total antibody (Ab), IgG, IgA, and IgM | Day 0 to 65 pso (or post‐admission for asymptomatic group). Number of patients with samples obtained per week varied (total (asymptomatic/presymptomatic/symptomatic)): day 1‐7 48/75 (17/23/8); day 8‐14 38/75 (9/17/22); day 15‐30 48/75 (10/21/19); day 31‐65 64/75 (17/28/19) | Plasma |
|
Yang 2020 [A] Yang 2020 [B] Laboratory only; [A] cyclic enhanced fluorescence assay (CEFA); [B] microsphere immunoassay (MIA) |
[A] ET Healthcare Pylon COVID‐19 IgM and IgG assays [B] Luminex Corporation SARS‐CoV‐2 | [A] S‐RBD and recombinant N‐protein [B] recombinant N‐protein | [A] IgG, IgM, IgG or and IgM [B] Total antibody | 0 to > 32 d pso, of the 120 samples from 42 PCR+ cases: 8, 7% day 0‐3 33, 28% day 4‐7 42, 35% day 8‐14 15, 13% day 15‐20 21, 18%, day 21‐32 1, 0.8% day > 32 | Serum |
|
Yongchen 2020 LFA only; LFA |
CGIA (Innovita Co., Ltd, China) | N‐ and S‐based | IgG, IgM (coloured line) | Day 8 to 42 | Serum |
|
Zhang 2020a [A] Zhang 2020a [B] LFA only; [A, B] FIA |
[A] Beijing Diagreat Biotechnolog 2019‐nCoV IgM Antibody Determination Kit [B] Beijing Diagreat Biotechnolog 2019‐nCoV IgG Antibody Determination Kit | [A] [B] S1 and N‐protein | [A] IgM [B] IgG | [1] 0‐70 d of onset of fever [1a] < 15 d pso: n = 9 15‐21 d pso (n = 38) > 21 d pso (n = 291) [1b] > 21 d pso: n = 234 [3] Asymptomatic | Whole blood |
|
Zhang 2020b [A] Zhang 2020b [B] Zhang 2020b [C] Laboratory only; [A to C] CLIA |
[A] YHLO SARS‐CoV‐2 iFlash IgM assay [B] YHLO SARS‐CoV‐2 iFlash IgG assay [C] YHLO SARS‐CoV‐2 iFlash IgG/IgM assay | N and S based | IgM, IgG | Range < 10 to 49 d pso; data presented by time period Serological antibody tests were performed at different times post–disease onset: < 10 d, 10‐20 d, 20‐30 d, 30‐40 d, 40‐50 d | Not stated; presume serum from Introduction/Discussion |
|
Zhao 2020a [A] Zhao 2020a [B] Zhao 2020a [C] Laboratory only; Laboratory |
[A] Beijing Wantai ‐ SARS‐CoV‐2 Ab ELISA [B] Beijing Wantai ‐ SARS‐CoV‐2 IgM ELISA [C] Beijing Wantai ‐ SARS‐CoV‐2 IgG ELISA |
[A,B] RBD [C] N‐based |
[A] Ab, [B] IgM, [C] IgG | Day 1 to 39 | Plasma |
Footnotes
Ab: antibodyANA: antinucelar antibodyCEFA: cyclic enhanced fluorescence assayCGIA: colloidal gold immunoassayCLIA: chemiluminescent immunoassayCT: computerised tomographyDNA: deoxyribonucelic acidds‐DNA: doube‐stranded deoxyribonucelic aciddays pso: days since symptom onsetECLIA: electro‐chemiluminescence ImmunoassayEDTA: ethylennediamine tetraacetic acidELFA: enzyme‐linked fluorescent assayELISA: enzyme linked immunosorbent assayEUA: emergency use authorization FIA: fluorescence immunoassayHBc: hepatitis B core antibodyHBsAg: hepatitis B surface antigenHCW: health care workedHS: health screeningIQR: interquartile rangeLFA: lateral flow assayLFIA: lateral flow immunoassayMCLIA: magnetic chemiluminescent immunoassayNAAT: nucleic acid amplification testNAT: nucleic acid testNP: nasopharyngealNR: not reportedOP: oropharyngealPCR: polymerase chain reactionPOCT: point of care testpso: post symptom onsetQ1/3: quartile 1 and quartile 3RF: rheumatoid factorRT: reverse transcriptionSD: standard deviationSLE: systemic lupus erythematosus
a Please note that square brackets indicate different tests within one study.
Appendix 8. Study level assessments of study quality
6.

Risk of bias and applicability concerns summary: review authors' judgements about each domain for each included study
Appendix 9. Forest plots of sensitivity by week after symptom onset (IgM, IgG, IgM or IgG, total Ab)
7.

Sensitivity by brand by week post‐symptom onset (IgG)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
8.

Sensitivity by brand by week post‐symptom onset (IgM)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
9.

Sensitivity by brand by week post‐symptom onset (IgG or IgM)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
10.

Sensitivity by brand by week post‐symptom onset (total antibodies).
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
Appendix 10. Forest plots of sensitivity in convalescent phase infection (IgM, IgG, IgM or IgG, total Ab)
11.

Sensitivity by brand for convalescent‐phase infection (IgG)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
12.

Sensitivity by brand for convalescent‐phase infection (IgM)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
13.

Sensitivity by brand for convalescent‐phase infection (IgG or IgM)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
14.

Sensitivity by brand for convalescent‐phase infection (total antibodies)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
Appendix 11. Forest plots of sensitivity in asymptomatic infection (IgM, IgG, IgM or IgG, total Ab)
15.

Sensitivity by brand for asymptomatic infection by time post‐symptom onset (IgG, IgM, IgG or IgM, total antibodies and IgA)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
Appendix 12. Forest plots of specificity according to reference standard (IgM, IgG, IgM or IgG, total Ab)
16.

Specificity by brand (IgG: pre‐pandemic, suspected of COVID‐19, current RT‐PCR‐negative, current untested, other/mixed/unclear, cross‐reactivity/confounder)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
17.

Specificity by brand (IgM: pre‐pandemic, suspected of COVID‐19, current RT‐PCR‐negative, current untested, other/mixed/unclear, cross‐reactivity/confounder)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
18.

Specificity by brand (IgG or IgM: pre‐pandemic, suspected of COVID‐19, current RT‐PCR‐negative, current untested, other/mixed/unclear, cross‐reactivity/confounder)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
19.

Specificity by brand (Total antibodies: pre‐pandemic, suspected of COVID‐19, current RT‐PCR‐negative, current untested, other/mixed/unclear, cross‐reactivity/confounder)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
Appendix 13. Specificity data for cross‐reactivity/confounder panel
|
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
||||||
| IgG | IgM | IgG or IgM | Total Ab | IgA | IgA or IgG | |
| Overall result | 96 (6036/6176) | 44 (2492/2625) | 36 (2047/2175) | 22 (1407/1411) | 5 (278/311) | 2 (203/219) |
| 98.6 (98.0, 99.0) |
97.0 (95.1, 98.2) |
96.8 (94.4, 98.2) |
99.8 (98.6, 100) |
89.1 (80.6, 94.2) |
92.7 (88.4, 95.5) |
|
| By test method | ||||||
| ELISA | 28 (1573/1619) | 8 (495/503) | 3 (261/270) | 3 (158/158) | 5 (278/311) | 2 (203/219) |
| 98.0 (96.5, 98.8) |
98.8 (96.6, 99.6) |
96.9 (85.4, 99.4) |
100a (97.7, 100)** |
89.1 (80.6, 94.2) |
92.7 (88.4, 95.5) |
|
| CLIA | 34 (2465/2514) | 4 (202/204) | 1 (20/20) | 16 (1249/1253) | ||
| 98.9 (98.1, 99.4) |
99.4 (96.2, 99.9) |
100a (83.2, 100)** |
99.8 (98.5, 100) |
|||
| Lateral flow/CGIA/FIA | 34 (1998/2043) | 32 (1795/1918) | 31 (1707/1826) | |||
| 98.6 (97.6, 99.2) |
95.4 (92.8, 97.0) |
96.4 (93.7, 98.0) |
||||
| COMPARISON | 0.2587 | 0.0097 | 0.4655 | |||
| By antigen | ||||||
| N‐based | 29 (1889/1916) | 11 (540/559) | 6 (389/397) | 12 (786/787) | ||
| 99.1 (98.3, 99.5) |
97.5 (93.9, 99.0) |
99.0 (95.1, 99.8) |
99.7 (98.2, 99.9) |
|||
| S‐based | 38 (2668/2738) | 8 (719/746) | 10 (652/671) | 10 (656/657) | 5 (278/311) | 2 (203/219) |
| 98.3 (97.3, 99.0) |
97.5 (93.0, 99.1) |
99.0 (96.2, 99.7) |
99.8 (98.3, 100) |
89.1 (80.6, 94.2) |
92.7 (88.4, 95.5) |
|
| N‐ and S‐based | 17 (877/895) | 15 (718/755) | 10 (500/542) | |||
| 98.9 (97.4, 99.5) |
97.7 (94.5, 99.1) |
95.9 (88.5, 98.6) |
||||
| Unclear/not reported | 12 (602/627) | 10 (515/565) | 10 (506/565) | |||
| 96.9 (94.0, 98.4) |
94.0 (87.2, 97.3) |
90.9 (85.6, 94.4) |
||||
| 0.2515 | 0.9853 | 0.1613 | 0.6053 | |||
|
aP value obtained using ‘meqrlogit’; P < 0.0001 (comparison excluded ‘other/mixed/unclear’ group) *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval CI: confidence interval CGIA: colloidal gold immunoassayCLIA: chemiluminescent immunoassayELISA: enzyme‐linked immunoassayFIA: fluorescence immunoassay | ||||||
Appendix 14. Forest plots for detection of IgA alone or combined with IgM or IgG
20.

Sensitivity by brand by week post‐symptom onset (IgA, IgA or IgG, IgA or IgM)
CLIA: chemiluminescence immunoassay CGIA: colloidal gold immunoassay FIA: fluorescence immunoassay
Appendix 15. Sensitivity ‐ all time periods (IgA alone or combined with IgM or IgG)
|
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
||||||
| Target |
Days 1‐7 (week 1) |
Days 8‐14 (week 2) |
Days 15‐21 (week 3) |
Days 22‐28 (week 4) |
Days 29‐35 (week 5) |
Convalescent |
| IgAa | 24 (525/1079) | 22 (874/1181) | 19 (424/501) | 5 (52/63) | 1 (4/4) | 22 (959/1257) |
| 44.3 (36.2, 52.8) |
72.9 (65.6, 79.2) |
86.6 (81.2, 90.6) |
89.1 (79.2, 94.6) |
100a (39.8, 100)* |
82.3 (70.0, 90.3) |
|
| IgA/IgGa | 6 (87/167) | 5 (141/170) | 5 (118/125) | 2 (73/74) | 1 (4/4) | 4 (149/157) |
| 48.8 (36.0, 61.8) |
80.8 (70.1, 88.4) |
95.6 (89.9, 98.1) |
99.2 (94.1, 99.9) |
100a (39.8, 100)** |
94.9 (90.1, 97.4) |
|
| IgA/IgM | 1 (49/52) | 1 (36/36) | 1 (43/44) | |||
| 94.2 (83.6, 98.1) |
100a (90.3, 100)** |
97.7a (88.0, 99.9) | ||||
|
aP value for comparison across week to week 5 obtained using ‘melogit’; both P < 0.0001 bEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval CI: confidence interval | ||||||
Appendix 16. Specificity (IgA alone or combined with IgM or IgG)
|
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
|||
| Target | Pre‐pandemic | Mixed groups | Other groups |
| IgA a | 17 (1603/1711) | 6 (5573/5949) | |
| 93.8 (90.5, 96.0) |
91.9 (68.5, 98.3) |
Suspected of COVID‐19 4 (310/385) 91.5 (84.0, 95.7) |
|
|
Current untested 2 (197/204) 96.0 (89.4, 98.6) |
|||
| IgA/IgG a | 2 (101/106) | 3 (197/256) | |
| 97.6 (66.8, 99.9) |
77.8 (68.8, 84.7) |
||
| IgA/IgM | 1 (26/56) | ||
| 46.4a (33.0, 60.3)* |
|||
|
aP value for comparison across week to week 5 obtained using ‘melogit’; both P < 0.0001 bEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval CI: confidence interval | |||
Appendix 17. Sensitivity data by test brand (IgG, IgM alone or combined, convalescent phase)
| IgG | IgM | IgG or IgM | |||||
| Test brand | Test name | Evaluations (TP/TP + FN) | Sensitivity (95% CI) | Evaluations (TP/TP + FN) | Sensitivity (95% CI) | Evaluations (TP/TP + FN) | Sensitivity (95% CI) |
| Lateral flow assays | |||||||
| AAZ‐LM CGIA |
Covid‐Duo test rapid Covid‐19 IgG/IgM | 1 (22/22) | 100a (84.6, 100)** | ||||
| AccuBiotech CGIA |
Accu‐Tell COVID‐19 IgG/IgM Cassette | 1 (27/28) | 96.4a (81.7, 99.9)* | ||||
| ACON Laboratories | SARS‐COV‐2 IgG/IgM Rapid Test | 1 (34/37) | 91.9a (78.1, 98.3)* | 1 (143/146) | 97.9a (94.1, 99.6)* | 1 (34/37) | 91.9a (78.1, 98.3)* |
| Alfa Scientific | CLARITY COVID‐19 IgG/IgM Antibody Test | 1 (26/40) | 65.0a (48.3, 79.4)* | 1 (33/40) | 82.5a (67.2, 92.7)* | 1 (39/40) | 97.5a (86.8, 99.9)* |
| Anhui Deep Blue | SARS‐CoV‐2 IgG/IgM Ab test kit | 1 (9/10) | 90.0a (55.5, 99.7)* | 1 (10/10) | 100a (69.2, 100)** | 2 (38/38) | 100a (90.7, 100)** |
| Artron CGIA |
Artron COVID‐19 IgG/IgM | 1 (8/8) | 100a (63.1, 100)** | ||||
| Augurix Diagnostics | One Step Test for Novel Coronavirus (2019‐nCoV) IgG/IgM Antibody | 1 (41/44) | 93.2a(81.3, 98.6)* | 1 (6/33) | 18.2a (7.0, 35.5)* | ||
| Augurix Diagnostics | Augurix SARS‐CoV‐2 IgG/IgM RDT | 1 (12/12) | 100a (73.5, 100)** | 1 (1/12) | 8.3a (0.2, 38.5)* | ||
| Augurix Diagnostics | SimtomaX Corona Check IgG/IgM | 1 (124/220) | 56.4a (49.5, 63.0)* | 1 (160/224) | 71.4a (65.0, 77.2)* | ||
| Autobio Diagnostics CGIA |
Anti‐SARS‐CoV‐2 Rapid Test IgG/IgM | 1 (13/13) | 100a (75.3, 100)** | 1 (9/13) | 69.2a (38.6, 90.9)* | 2 (21/21) | 100a (83.9, 100)** |
| Avioq Bio‐Tech CGIA |
Novel Coronavirus (2019‐nCov) Antibody IgG/IgM Assay Kit | 1 (31/33) | 93.9a (79.8, 99.3)* | ||||
| AYTU Biosciences | COVID‐19 IgG/IgM Rapid Test Cassette | 1 (31/40) | 77.5a (61.5, 89.2)* | 1 (18/40) | 45.0a (29.3, 61.5)* | 1 (33/40) | 82.5a (67.2, 92.7)* |
| Beijing Diagreat Biotechnology FIA |
2019‐nCoV IgG/IgM Antibody Determination Kit | 1 (468/525) | 89.1a (86.2, 91.7)* | 1 (269/525) | 51.2a (46.9, 55.6)* | ||
| Beijing Wantai | Wantai Ab rapid assay | 1 (87/98) | 88.8a (80.8, 94.3)* | ||||
| Biohit Healthcare CGIA |
SARS‐CoV‐2 IgG/IgM ANTIBODY TEST KIT | 1 (27/28) | 96.4a (81.7, 99.9)* | ||||
| Biolidics | 2019 nCoV IgG/IgM detection kit | 2 (15/17) | 88.2 (63.2, 97.0) | 2 (8/17) | 47.0 (25.5, 69.7) | 2 (16/17) | 94.1 (68.0, 99.2) |
| BioMedomics | COVID‐19 IgG/IgM Rapid Test | 2 (15/18) | 83.3 (59.1, 94.5) | 3 (27/30) | 90.0 (73.2, 96.7) | 2 (15/18) | 83.3(59.1, 94.5) |
| bioMerieux FIA |
Vidas SARS‐CoV‐2 IgG/IgM | 2 (101/107) | 94.4 (88.1, 97.5) | 1 (203/224) | 90.6a (86.0, 94.1)* | ||
| Biopanda | COVID‐19 Rapid Ab test | 1 (102/163) | 62.6 (54.7, 70.0) | ||||
| Bioperfectus Technologies | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgG/IgM Rapid Test Kit | 1 (9/10) | 90.0a (55.5, 99.7)* | 1 (1/6) | 16.7a(0.4, 64.1)* | 1 (10/10) | 100a (69.2, 100)* |
| Biosynex | COVID‐19 BSS IgG/IgM | 2 (147/195) | 84.5 (58.2, 95.5) | 2 (24/39) | 61.5 (45.6, 75.3) | 3 (119/125) | 95.2 (89.7, 97.8) |
| BTNX | Rapid Response™ COVID‐19 Test Cassette (BTNX Inc.) Kit1 IgG/IgM | 2 (44/47) | 93.6 (82.0, 97.9) | 2 (38/42) | 90.5(77.2, 97.4) | 2 (45/47) | 95.7 (84.5, 98.9) |
| Cellex | qSARS‐CoV‐ 2 IgG/IgM Cassette Rapid Test | 2 (19/23) | 82.6 (61.8, 93.3) | 2 (26/42) | 56.9 (27.5, 82.1) | 2 (19/23) | 82.6 (61.8, 93.3) |
| Clungene Biotech | Clungene SARS‐CoV‐2 IgG/IgM Rapid Test Cassettes | 1 (55/63) | 87.3a (76.5, 94.4)* | 1 (160/212) | 75.5a (69.1, 81.1)* | 1 (56/63) | 88.9a (78.4, 95.4)* |
| CTK Biotech CGIA |
OnSite COVID‐19 IgG/IgM Rapid Test | 2 (181/242) | 74.8 (68.8, 79.9) | 1 (60/220) | 27.3a (21.5, 33.7)* | 1 (8/8) | 100a (63.1, 100)** |
| Decombio Biotechnology | Novel Coronavirus (SARS‐CoV‐2) IgG/IgM Combo Rapid Test‐Cassette | 1 (10/11) | 90.9a (58.7, 99.8)* | 1 (10/11) | 90.9a (58.7, 99.8)* | 1 (10/11) | 90.9a (58.7, 99.8)* |
| DeepBlue Medical CGIA |
COVID‐19 (SARS CoV‐2) IgG/IgM Antibody Test Kit (Colloidal Gold) | 1 (9/11) | 81.8a (48.2, 97.7)* | 1 (8/11) | 72.7a (39.0, 94.0)* | 1 (10/11) | 90.9a (58.7, 99.8)* |
| DNA Link | AccuFind COVID19 IgG/IgM | 1 (38/40) | 95.0a (83.1, 99.4)* | 1 (33/40) | 82.5a (67.2, 92.7)* | 1 (39/40) | 97.5a (86.8, 99.9)* |
| Dynamiker Biotechnology | 2019‐nCOV IgG/IgM Rapid Test | 1 (72/75) | 96.0a (88.8, 99.2)* | 1 (72/74) | 97.3a (90.6, 99.7)* | 3 (87/93) | 97.1 (58.3, 99.9) |
| Fortress Diagnostics | COVID‐19 Total Ab | 1 (258/307) | 84.0 (79.5, 88.0) | ||||
| GenBody CGIA |
GenBody COVID‐19 IgG/IgM | 1 (25/28) | 89.3a (71.8, 97.7)* | ||||
| Genrui Biotech | Novel Coronavirus IgG/IgM test kit | 1 (10/10) | 100a (69.2, 100)** | 1 (10/10) | 100a (69.2, 100)** | 1 (10/10) | 100a (69.2, 100)** |
| Getein Biotech CGIA |
One Step Test for Novel Coronavirus IgG/IgM | 1 (9/10) | 90.0a (55.5, 99.7)* | 1 (0/10) | 0a (0, 30.8)** | 2 (25/28) | 89.3 (71.5, 96.5) |
| Guangzhou Wondfo | Wondfo SARS‐CoV‐2 Antibody Test | 6 (211/265) | 85.1 (69.0, 93.6) | ||||
| Hangzhou Alltest CGIA |
Unlabelled packaging IgG/IgM | 1 (25/30) | 83.3a (65.3, 94.4)* | 1 (17/30) | 56.7a (37.4, 74.5)* | 1 (25/30) | 83.3a (65.3, 94.4)* |
| Hangzhou Alltest CGIA |
2019‐nCoV IgG/IgM Rapid Test Cassette | 4 (97/109) | 89.0 (81.6, 93.6) | 4 (36/137) | 11.6 (0.6, 72.8) | 4 (123/135) | 91.4 (83.7, 95.6) |
| Hangzhou Biotest (distributed by Premier Biotech) | The RightsignTM COVID‐19 IgG/IgM Rapid Test Casette / Lumiratek (also distributed as COVID‐19 IgG/IgM Rapid Test Casette and CoronaChek IgG/IgM) | 3 (175/185) | 93.6 (84.5, 97.5) | 3 (55/82) | 67.1 (56.2, 76.4) | 2 (48/51) | 94.1 (83.3, 98.1) |
| Hightop Biotech CGIA |
Hightop SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | 1 (28/30) | 93.3a (77.9, 99.2)* | 1 (141/160) | 88.1a (82.1, 92.7)* | 1 (28/30) | 93.3a (77.9, 99.2)* |
| Innovita Biological Technology CGIA | 2019‐nCoV Ab test IgG/IgM | 6 (117/152) | 77.2 (64.4, 86.4) | 6 (172/335) | 60.8 (46.0, 73.8) | 5 (106/134) | 79.1 (71.4, 85.2) |
| InTec CGIA |
Rapid SARS‐CoV‐2 Antibody (IgG/IgM) Test | 1 (5/7) | 71.4a (29.0, 96.3)* | 1 (6/7) | 85.7a (42.1, 99.6)* | 1 (6/7) | 85.7a (42.1, 99.6)* |
| Intelligent Endoscopy | Smart screen IgG/IgM | 1 (10/40) | 25.0a (12.7, 41.2)* | 1 (24/40) | 60.0a (43.3, 75.1)* | 1 (26/40) | 65.0a (48.3, 79.4)* |
| Inzec | BIOZEK COVID‐19 IgG/IgM | 1 (106/115) | 92.2a (85.7, 96.4)* | 1 (26/115) | 22.6a (15.3, 31.3)* | ||
| Jiangsu Medomics CGIA |
Rapid IgM‐IgG combined antibody test kit for SARS‐CoV‐2 | 1 (13/18) | 72.2a (46.5, 90.3)* | 1 (13/16) | 81.3a (54.4, 96.0)* | 2 (41/46) | 89.8 (68.3, 97.3) |
| LabOn Time | LaboOn Time rapid test cassette IgG/IgM | 1 (31/33) | 93.9a (79.8, 99.3)* | ||||
| Leccurate CGIA |
SARS‐CoV‐2 antibody test IgG/IgM | 1 (29/35) | 82.9a (66.4, 93.4)* | 1 (13/20) | 65.0a (40.0, 84.6)* | 1 (12/16) | 75.0a (47.6, 92.7)* |
| Liming Bio | StrongStep SARS‐CoV‐2 IgG/IgM | 1 (29/35) | 82.9a (66.4, 93.4)* | 1 (12/18) | 66.7a (41.0, 86.7)* | 1 (29/35) | 82.9a (66.4, 93.4)* |
| MEDsan | MEDsan COVID‐19 IgG/IgM Rapid Test | 2 (227/254) | 89.4 (84.9, 92.6) | 2 (207/254) | 81.5 (76.2, 85.8) | 1 (92/99) | 92.9a (86.0, 97.1)* |
| MEXACARE | QuickTestCorona COVID‐19 IgG/IgM | 1 (190/224) | 84.8a (79.4, 89.3)* | 1 (26/115) | 22.6a (15.3, 31.3)* | ||
| Mologic | IgG COVID‐19 | 1 (35/50) | 70.0a (55.4, 82.1)* | ||||
| Multi‐G | 2019‐nCoV IgG/IgM Rapid Test Cassette single lane | 1 (32/33) | 97.0a (84.2, 99.9)* | 1 (15/33) | 45.5a (28.1, 63.6)* | 1 (24/25) | 96.0a (79.6, 99.9)* |
| Multi‐G | 2019‐nCoV IgG/IgM Rapid Test Cassette dual lane | 1 (32/33) | 97.0a (84.2, 99.9)* | 1 (28/33) | 84.8a (68.1, 94.9)* | 1 (24/25) | 96.0a (79.6, 99.9)* |
| Nal Von Minden | NADAL COVID‐19 IgG/IgM Test | 1 (26/31) | 83.9a (66.3, 94.5)* | 1 (30/32) | 93.8a (79.2, 99.2)* | ||
| Nirmidas Biotech | COVID‐19 (SARS‐CoV‐2) IgG/IgM Antibody Detection Kit | 1 (33/40) | 82.5a (67.2, 92.7)* | 1 (33/40) | 82.5a (67.2, 92.7)* | 1 (37/40) | 92.5a (79.6, 98.4)* |
| NTBIO® Diagnostics | NTBIO One Step Rapid Test ‐ COVID‐19 IgG/IgM Antibody Test | 1 (188/219) | 85.8a (80.5, 90.2)* | 1 (167/200) | 83.5a (77.6, 88.4)* | ||
| Qingdao HIGHTOP | SARS‐CoV‐2 IgG/IgM Ab Rapid Test | 1 (216/229) | 94.3a (90.5, 96.9)* | 1 (30/46) | 65.2a (49.8, 78.6)* | ||
| Render | COVID‐19 IgG/IgM Test | 1 (15/20) | 75.0a (50.9, 91.3)* | 1 (34/65) | 52.3a (39.5, 64.9)* | 1 (16/20) | 80.0a (56.3, 94.3)* |
| Safecare Biotech | SafeCare Bio‐Tech COVID‐19 IgG/IgM Rapid Test Device | 1 (37/40) | 92.5a (79.6, 98.4)* | 1 (31/40) | 77.5a (61.5, 89.2)* | 1 (38/40) | 95.0a (83.1, 99.4)* |
| Screen Italia | Screen Test Covid‐19 2019‐nCOV IgG/IgM | 1 (79/109) | 72.5a (63.1, 80.6)* | 1 (89/109) | 81.7a (73.1, 88.4)* | 1 (108/109) | 99.1a (95.0, 100.0)* |
| SD Biosensor | COVID‐19 IgG/IgM Duo | 4 (50/69) | 74.2 (52.1, 88.3) | 3 (17/23) | 63.5 (5.4, 98.1) | 1 (6/7) | 85.7a (42.1, 99.6)* |
| Sensing Self | Covid‐19 Rapid IgG/IgM combined Antibody assay | 1 (35/40) | 87.5a (73.2, 95.8)* | 1 (6/40) | 15.0a (5.7, 29.8)* | 1 (35/40) | 87.5a (73.2, 95.8)* |
| Servibio/VEDALAB | COVID‐19 Sign IgG/IgM | 1 (33/42) | 78.6a (63.2, 89.7)* | ||||
| SIDAK Life Care | Sidak Covid 19 Antibody IgG/IgM Rapid Test Kit | 1 (9/14) | 64.3a (35.1, 87.2)* | 1 (17/33) | 51.5a (33.5, 69.2)* | 1 (9/14) | 64.3a (35.1, 87.2)* |
| Spring Healthcare Services CGIA |
COVID‐19 Spring IgG/IgM Rapid Test Cassette | 1 (28/28) | 100a (87.7, 100)** | ||||
| Sure Biotech | SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | 2 (226/235) | 96.2 (92.8, 98.0) | 2 (212/235) | 90.2 (85.7, 93.4) | 1 (10/11) | 90.9a (58.7, 99.8)* |
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgG/IgM | 1 (31/33) | 93.9a (79.8, 99.3)* | 1 (16/19) | 84.2a (60.4, 96.6)* | 3 (248/257) | 96.5 (93.4, 98.2) |
| TAmiRNA | SARS‐CoV‐2 Antibody Lateral Flow Test IgG/IgM | 1 (203/222) | 91.4a (87.0, 94.8)* | ||||
| TBG Biotechnology | TBG SARS‐CoV‐2 IgG/IgM Rapid Test Kit | 1 (38/40) | 95.0a (83.1, 99.4)* | 1 (35/40) | 87.5a (73.2, 95.8)** | 1 (38/40) | 95.0a (83.1, 99.4)* |
| TONYAR Biotech | ASK COVID‐19 IgG/IgM Rapid Test | 2 (83/85) | 97.6 (91.1, 99.4) | ||||
| Trillium | IgG/IgM rapid assay | 1 (20/28) | 71.4a (51.3, 86.8)* | 1 (19/28) | 67.9a (47.6, 84.1)* | ||
| UCP Biosciences | Coronavirus IgG/IgM Antibody (COVID‐19) Test Cassette | 1 (9/11) | 81.8a (48.2, 97.7)* | 1 (21/47) | 44.7a (30.2, 59.9)* | 1 (10/11) | 90.9a (58.7, 99.8)* |
| Unknown manufacturer | Ready Result IgG/IgM | 1 (35/38) | 92.1a (78.6, 98.3)* | 1 (33/38) | 86.8a (71.9, 95.6)* | 1 (38/39) | 97.4a (86.5, 99.9)* |
| Vazyme Biotech | 2019‐nCoV IgG/IgM Detection Kit | 1 (4/4) | 100a (39.8, 100)** | 1 (9/11) | 81.8a (48.2, 97.7)* | ||
| VivaChek Biotech CGIA |
VivaDiag COVID‐19 IgG/IgM Rapid Test | 3 (67/105) | 72.8 (49.0, 88.1) | 2 (35/39) | 89.7 (75.7, 96.1) | 2 (43/75) | 68.7 (32.9, 90.8) |
| W.H.P.M. | COVISURE COVID‐19 IgG/IgM Rapid Test | 1 (26/38) | 68.4a (51.3, 82.5)* | 1 (25/38) | 65.8a (48.6, 80.4)* | 1 (26/38) | 68.4a (51.3, 82.5)* |
| Wells Bio | CareUS COVID‐19 IgG/IgM | 1 (35/37) | 94.6a (81.8, 99.3)* | 1 (33/38) | 86.8a (71.9, 95.6)* | ||
| Xiamen Biotime | SARS‐Cov‐2 IgG/IgM Rapid Qualitative Test | 1 (183/200) | 91.5a (86.7, 95.0)* | 1 (167/200) | 83.5a (77.6, 88.4)* | ||
| ZenTech CGIA |
QuickZen COVID‐19 IgG/IgM | 1 (30/33) | 90.9a (75.7, 98.1)* | ||||
| Zeus Scientific | ZEUS SARS‐CoV‐2 IgG/IgM rapid test | 1 (22/40) | 55.0a (38.5, 70.7)* | 1 (14/40) | 35.0a (20.6, 51.7)* | 1 (23/40) | 57.5a (40.9, 73.0)* |
| Zhejiang Orient‐Gene CGIA |
COVID‐19 IgG rapid test cassette | 7 (322/355) | 90.7 (87.2, 93.3) | 6 (170/310) | 63.7 (25.7, 89.9) | 5 (184/194) | 94.8 (90.7, 97.2) |
| ELISA | |||||||
| Beijing Wantai | Wantai ELISA IgG/IgM assay | 3 (63/91) | 58.4 (32.1, 80.6) | ||||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgG/IgM EIA | 1 (10/10) | 100a (69.2, 100)** | 1 (5/10) | 50.0a (18.7, 81.3)* | 1 10/10) | 100a (69.2, 100)** |
| Creative Diagnostics | SARS‐CoV‐2 IgG/IgM ELISA Kit | 1 (38/41) | 92.7a (80.1, 98.5)* | 1 (38/41) | 92.7a (80.1, 98.5)* | ||
| EDI/Eagle Biosciences | COVID‐19 IgG/IgM Quantitative ELISA | 1 (539/1134) | 47.5a (44.6, 50.5)* | 1 (100/123) | 81.3a (73.3, 87.8)* | ||
| Epitope Diagnostics | EDITM Novel Coronavirus COVID‐19 IgG/IgM ELISA kit | 13 (780/934) | 87.7 (78.1, 93.4) | 7 (340/662) | 61.3 (33.2, 83.4) | 2 (42/50) | 84.0 (71.1, 91.8) |
| Euroimmun | anti‐SARS‐COV‐2 IgG | 41 (2200/2442) |
90.8 (88.6, 92.7) | ||||
| Euroimmun | anti‐SARS‐COV‐2 IgG (N‐based) | 2 (85/100) | 84.8 (37.3, 99.8) | ||||
| Gold Standard Diagnostics (GSD) | Gold Standard SARS‐CoV‐2 IgG ELISA | 1 (85/123) | 69.1a (60.1, 77.1)* | ||||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgG/IgM ELISA | 1 (39/45) | 86.7 (73.2, 94.9) | 1 (40/45) | 88.9a (75.9, 96.3)* | 1 (41/45) | 91.1a (78.8, 97.5)* |
| ImmunoDiagnostics | SARS‐CoV‐2 NP IgG/IgM ELISA Kit | 2 (151/186) | 87.9 (64.8, 96.6) | 1 (43/46) | 93.5a (82.1, 98.6)* | ||
| Mediagnost | Mediagnost Anti‐SARS‐CoV‐2 ELISA IgG/IgM | 2 (84/94) | 89.4(81.3, 94.2) | 1 (14/16) | 87.5a (61.7, 98.4)* | ||
| Mikrogen | Mikrogen IgG anti‐N | 1 (24/24) | 100a (85.8, 100)** | ||||
| Novatek | Novatec Novalisa SARS‐CoV‐2 IgG ELISA | 1 (38/48) | 79.2a (65.0, 89.5)* | ||||
| Vircell | COVID‐19 ELISA IgG | 2 (56/60) | 93.3 (83.5, 97.5) | ||||
| Virotech | Virotec(h SARS‐CoV‐2 ELISA IgG | 2 (45/64) | 70.3a (58.1, 80.2)* | ||||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG/IgM ELISA | 2 (91/104) | 85.6 (77.4, 91.1) | 2 (78/104) | 81.1 (66.8, 90.1) | 1 (43/45) | 95.6a (84.9, 99.5)* |
| Zydus diagnostics | Covid Kavach IgG ELISA | 2 (320/421) | 76.0 (71.7, 79.8) | ||||
| CLIA | |||||||
| Abbott Diagnostics | Abbott Alinity anti‐SARS‐CoV‐2 nucleocapsid IgG | 2 (149/163) | 91.3 (83.8, 95.5) | ||||
| Abbott Diagnostics | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG/IgM | 33 (1824/1977) | 92.5 (90.3, 94.3) | 2 (32/38) | 85.1a (64.5, 94.7)* | ||
| Autobio Diagnostics | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | 1 (271/273) | 99.3a (97.4, 99.9)* | 1 (53/62) | 85.5a (74.2, 93.1)* | ||
| Beckman Coulter | Access SARS‐CoV‐2 IgG | 2 (78/94) | 92.4 (38.8, 99.6) | ||||
| Beijing Wantai | Wantai CLIA IgG/IgM assay | 1 (47/47) | 100a (92.5, 100)* | 1 (4/8) | 50.0a (15.7, 84.3)* | ||
| Bioscience Co | Bioscience Co (Chongqing) SARS‐CoV‐2 IgG/IgM | 1 (13/13) | 100a (75.3, 100)** | 1 (12/13) | 92.3 (64.0, 99.8) | 1 (13/13) | 100a (75.3, 100)** |
| DiaSorin | LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA | 21 (1523/1735) | 88.1 (84.1, 91.2) | ||||
| Diazyme | Diazyme DZ‐LITE 2019‐nCoV IgG/IgM (CLIA) Assay Kit | 2 (41/43) | 95.3 (83.2, 98.8) | 2 (20/21) | 95.2 (72.9, 99.3) | 1 (18/18) | 100a (81.5, 100)** |
| Ortho Clinical Diagnostics | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay IgG | 3 (201/221) | 92.3 (80.8, 97.1) | ||||
| Shenzhen YHLO Biotech | YHLO iFlash IgG/IgM assay | 5 (260/268) | 97.0 (94.1, 98.5) | 5 (109/184) | 68.7 (44.6, 85.7) | 1 (8/8) | 100a (63.1, 100)** |
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgG/IgM kits | 7 (325/369) | 89.9 (83.3, 94.1) | 5 (174/299) | 63.6 (37.4, 83.7) | 1 (30/32) | 93.8a (79.2, 99.2)* |
| Xiamen InnoDx Biotech | Novel coronavirus (2019‐nCoV) antibody test kit IgM | 1 (31/61) | 50.8a (37.7, 63.9)* | ||||
| Other/unclear | |||||||
| ET Healthcare | Pylon 3D automated immunoassay system IgG/IgM | 2 (37/37) | 100a (90.5, 100)** | 2 (35/58) | 71.2 (28.3, 93.9) | 1 (21/21) | 100a (83.9, 100)** |
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgG (S‐based) | 1 (24/26) | 92.3 (74.9, 99.1) | ||||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgG (N‐based) | 1 (23/26) | 88.5 (69.8, 97.6) | ||||
| Quotient | MosaiQ COVID‐19 antibody microarray IgG/IgM | 1 (30/30) | 100a (88.4, 100)** | ||||
|
a Estimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervalCLIA: chemiluminescence immunoassayELISA: enzyme‐linked immunosorbent assayFN: false negativeTP: true positive | |||||||
Appendix 18. Sensitivity data by test brand (total Ab, convalescent phase)
| Test brand | Test name |
Convalescent Test groups (true positives/COVID cases) Sensitivity (95% CI) |
|
| ELISA | |||
| Beijing Wantai | Wantai ELISA Total‐Ab assay | 8 (1562/1649) | 95.7 (92.6, 97.5) |
| Bio‐Rad Laboratories | BioRad Platelia SARS‐CoV‐2 Total Ab | 1 (39/45) | 86.7a (73.2, 94.9)* |
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 Total Ab assay | 1 (20/21) | 95.2a (76.2, 99.9)* |
| CLIA | |||
| Beijing Wantai | Wantai CLIA Total‐Ab assay | 1 (46/47) | 97.9a (88.7, 99.9)* |
| Ortho Clinical Diagnostics | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay Total Ab | 2 (192/200) | 96.0 (92.2, 98.0) |
| Roche Diagnostics | Elecsys anti‐SARS‐CoV‐2 antibody assay Total Ab | 34 (3669/3916) | 93.4 (91.1, 95.1) |
| Siemens Healthcare | Siemens Atellica Total‐Ab assay | 7 (979/1009) | 96.7 (94.2 98.1) |
| Siemens Healthcare | Siemens Vista Total‐Ab assay | 1 (94/116) | 81.0a (72.7, 87.7)* |
| Watmind Medical | SARS‐CoV‐2 Ab Diagnostic Test Kit | 1 (24/28) | 85.7a (67.3, 96.0)* |
| Xiamen InnoDx Biotech | Novel coronavirus (2019‐nCoV) antibody test kit Total‐Ab | 1 (37/38) | 97.4a (86.2, 99.9)* |
| Xiamen Wantai | Total Ab CMIA | 1 (7/8) | 87.5a (47.3, 99.7)* |
| Other/unclear | |||
| Luminex Corporation | SARS‐CoV‐2 MIA Total Ab | 1 (19/19) | 100a (82.4, 100)** |
|
aEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervalCLIA: chemiluminescent immunoassayCMIA: chemiluminescent microparticle immunoassayELISA: enzyme‐linked immunosorbent assay | |||
Appendix 19. Specificity data by test brand (IgG, IgM alone or combined, pre‐pandemic)
| Pre‐pandemic IgG | Pre‐pandemic IgM | Pre‐pandemic IgM or IgG | ||
| Test brand | Test name | Specificity (95% CI) | Specificity (95% CI) | Specificity (95% CI) |
| Lateral flow | ||||
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgG/IgM | 1 (55/56) | 1 (56/56) | 1 (55/56) |
| LFA | 98.2a (90.4, 100)* | 100a (93.6, 100)** | 98.2a (90.4, 100)* | |
| AccuBiotech Co., Ltd. | Accu‐Tell COVID‐19 IgG/IgM Cassette | 1 (49/50) | ||
| CGIA | 98.0a (89.4, 100)* | |||
| ACON Laboratories | SARS‐COV‐2 IgG Rapid Test | 1 (74/74) | ||
| LFA | 100a (95.1, 100)** | |||
| Anhui Deep Blue Medical Technology Co | SARS‐CoV‐2 IgG Ab test kit | 1 (50/50) | 1 (50/50) | 2 (91/100) |
| CGIA | 100a (92.9, 100)** | 100a (92.9, 100)** | 97.6(32.0, 100) | |
| Artron, Burnaby, Canada | Artron COVID‐19 IgG | 1 (50/50) | 1 (49/50) | 2 (66/67) |
| LFA | 100a (92.9, 100)** | 98.0a (89.4, 100)* | 98.5 (90.2, 99.8) | |
| Augurix Diagnostics | One Step Test for Novel Coronavirus (2019‐nCoV) IgG Antibody | 1 (67/67) | 1 (68/68) | |
| LFA | 100a (94.6, 100)** | 100a (94.7, 100)** | ||
| Augurix SA (CH) | SimtomaX Corona Check IgG | 1 (267/268) | 1 (267/268) | |
| LFA | 99.6a (97.9, 100)* | 99.6a (97.9, 100)* | ||
| Autobio Diagnostics Co. Zhengzhou, China | Anti‐SARS‐CoV‐2 Rapid Test IgG | 1 (247/253) | 1 (243/253) | 2 (271/285) |
| CGIA | 97.6a (94.9, 99.1)* | 96.0a (92.9, 98.1)* | 95.1 (91.9, 97.1) | |
| Biohit Healthcare Co., Ltd. | SARS‐CoV‐2 IgG/IgM ANTIBODY TEST KIT | 1 (193/200) | ||
| CGIA | 96.5a (92.9, 98.6)* | |||
| Biolidics Limited | 2019 nCoV IgG detection kit | 2 (110/110) | 1 (49/50) | 1 (50/50) |
| LFA | 100a (96.7, 100)** | 98.0a (89.4, 99.9)* | 100a (92.9, 100)** | |
| BioMedomics Inc, Morrisville, NC, USA | COVID‐19 IgG Rapid Test | 2 (163/167) | 1 (94/107) | 1 (93/107) |
| LFA | 97.7 (90.1, 99.5) | 87.9a (80.1, 93.4) | 86.9a (79.0, 92.7)* | |
| BioMerieux, France | Vidas SARS‐CoV‐2 IgG | 2 (1084/1085) | 2 (402/404) | |
| FIA | 99.9 (99.3, 100) | 99.5 (98.0, 99.9) | ||
| Biopanda | COVID‐19 Rapid Ab test | 1 (499/500) | ||
| LFA | 99.8 (98.9, 1.00) | |||
| Bioperfectus Technologies Co | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgG Rapid Test Kit | 1 (102/104) | 1 (101/104) | 1 (99/104) |
| LFA | 98.1a (93.2, 99.8)* | 97.1a (91.8, 99.4)* | 95.2a (89.1, 98.4)* | |
| Biosynex, Illkirch‐Graffenstaden, France | COVID‐19 BSS IgG | 1 (99/100) | 1 (99/100) | |
| LFA | 99.0a (94.6, 100)* | 99.0a (94.6, 100)* | ||
| BTNX Inc. | Rapid ResponseTM COVID‐19 Test Cassette (BTNX Inc.) Kit1 IgG | 2 (121/123) | 1 (50/50) | 1 (50/50) |
| LFA | 98.4 (93.7, 99.6) | 100a (92.9, 100)** | 100a (92.9, 100)** | |
| Cellex | Cellex qSARS‐CoV‐ 2 IgG Cassette Rapid Test | 1 (22/25) | 1 (19/25) | 1 (19/25) |
| LFA | 88.0a (68.8, 97.5)* | 76.0a (54.8, 90.6)* | 76.0a (54.9, 90.6)* | |
| CTK Biotech Inc. | OnSite COVID‐19 IgG Rapid Test | 1 (246/251) | 1 (245/251) | 1 (32/32) |
| CGIA | 98.0a (95.4, 99.4)* | 97.6a (94.9, 99.1)* | 100a (89.1, 100)** | |
| DecombioBiotechnology Co Ltd, Beijing | Novel Coronavirus (SARS‐CoV‐2) IgG Combo Rapid Test‐Cassette | 1 (98/107) | 1 (97/107) | 1 (96/107) |
| LFA | 91.6a (84.6, 96.1)* | 90.7a (83.5, 95.4)* | 89.7a (82.3, 94.8)* | |
| DeepBlue Medical Technology Co Ltd, Anhui, China | COVID‐19 (SARS CoV‐2) IgG Antibody Test Kit (Colloidal Gold) | 1 (107/108) | 1 (91/108) | 1 (91/108) |
| CGIA | 99.1a (94.9, 100)* | 84.3a (76.0, 90.6)* | 84.3a (76.0, 90.6)* | |
| DiaCarta Inc | QuantiVirus anti‐SARS‐CoV‐2 IgG test | 1 (134/138) | ||
| FIA | 97.1a (92.7, 99.2)* | |||
| Dynamiker Biotechnology (Tianjin) Co Ltd., China | Dynamiker Biotechnology ‐ 2019‐nCOV IgG Rapid Test | 1 (395/403) | 1 (385/401) | 1 (32/32) |
| LFA | 98.0a (96.1, 99.1)* | 96.0a (93.6, 97.7)* | 100a (89.1, 100)** | |
| Epigentek Group, New York, USA | SeroFlash SARS‐CoV‐2 IgG/IgM | 1 (58/60) | ||
| LFA | 96.7a (88.5, 99.6)* | |||
| Fortress Diagnostics | COVID‐19 Total Ab | 1 (493/500) | ||
| LFA | 98.6a (97.1, 99.4)* | |||
| GenBody Inc. | GenBody COVID‐19 IgG/IgM | 1 (50/50) | ||
| CGIA | 100a (92.9, 100)** | |||
| Genrui Biotech Inc | Novel Coronavirus IgG test kit | 1 (50/50) | 1 (50/50) | 1 (50/50) |
| LFA | 100a (92.9, 100)** | 100a (92.9, 100)** | 100a (92.9, 100)** | |
| Getein Biotech Inc | One Step Test for Novel Coronavirus IgG | 1 (50/50) | 1 (50/50) | 2 (50/51) |
| CGIA | 100a (92.9, 100)** | 100a (92.9, 100)** | 98.0a (89.6, 100)* | |
| Guangzhou Wondfo Biotech Co., Ltd., China | Wondfo SARS‐CoV‐2 Antibody | 4 (1644/1648) | ||
| LFA | 99.8 (98.8, 100) | |||
| Hangzhou Alltest Biotech Co. Ltd. | 2019‐nCoV IgG Rapid Test Cassette | 1 (100/100) | 1 (100/100) | 2 (160/160) |
| LFA | 100a (96.4, 100)** | 100a (96.4, 100)** | 100a (97.7, 100)** | |
| Hangzhou Biotest Biotech Co Ltd (CN) | The RightSignTM COVID‐19 IgG Rapid Test Cassette / Lumiratek | 1 (160/163) | 1 (152/163) | |
| CGIA | 98.2a (94.7, 99.6)* | 93.3a (88.2, 96.6)* | ||
| Innovita Biological Technology Co., Ltd., Beijing, China | 2019‐nCoV Ab test IgG | 5 (348/349) | 5 (345/349) | 5 (304/309) |
| LFA | 99.8 (88.3, 100) | 99.8 (76.2, 100) | 98.9 (93.5, 99.8) | |
| Inzec | BIOZEK COVID‐19 IgG/IgM | 1 (125/135) | 1 (131/135) | |
| LFA | 92.6a (86.8, 96.4)* | 97.0a (92.6, 99.2)* | ||
| Jiangsu Medomics Medical Technology Co. Ltd. | Rapid IgM‐IgG Combined Antibody Test Kit for SARS‐ CoV‐2 IgG | 1 (23/24) | 1 (24/24) | 2 (159/166) |
| CGIA | 95.8a (78.9, 99.9)* | 100a (85.8, 100)** | 95.8(91.4, 98.0) | |
| Leccurate | SARS‐CoV‐2 antibody test IgG | 1 (25/25) | 1 (24/25) | 1 (24/25) |
| CGIA | 100a (86.3, 100)** | 96.0a (79.6, 99.9)* | 96.0a (79.6, 99.9)* | |
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgG | 1 (38/38) | 1 (38/38) | 1 (38/38) |
| LFA | 100a (90.7, 100)** | 100a (90.7, 100)** | 100a (90.7, 100)** | |
| Maccura Biotechnology, Chengdu, China | Maccura LFIA SARS‐CoV‐2 IgG/IgM | 1 (90/100) | ||
| LFA | 90.0a (82.4, 95.1)* | |||
| MEDsan GmbH, Biological Health Solutions, Hamburg, Germany | MEDsan COVID‐19 IgG Rapid Test | 1 (142/150) | 1 (144/150) | |
| LFA | 94.7a (89.8, 97.7)* | 96.0a (91.5, 98.5)* | ||
| Mologic | IgG COVID‐19 | 1 (486/500) | ||
| LFA | 97.2a (95.3, 98.5)* | |||
| Nal Von Minden | NADAL COVID‐19 IgG Test | 1 (68/68) | 1 (66/67) | |
| LFA | 100a (94.7, 100)** | 98.5a (92.0, 100)* | ||
| NG Biotech, Guipry, France | NG‐Test IgG COVID‐19 | 2 (272/276) | 3 (322/326) | 2 (274/276) |
| CGIA | 98.0 (89.5, 99.6) | 98.4 (92.9, 99.7) | 99.3 (97.2, 99.8) | |
| NTBIO® Diagnostics Inc. (CA) | NTBIO One Step Rapid Test ‐ COVID‐19 IgG Antibody Test | 1 (238/267) | 1 (253/268) | |
| LFA | 89.1a (84.8, 92.6)* | 94.4a (90.9, 96.8)* | ||
| Premier Biotech, Minneapolis, MN, USA | COVID‐19 IgG Rapid Test Cassette | 1 (107/108) | 1 (106/108) | 1 (105/108) |
| LFA | 99.1a (94.9, 100)* | 98.1a (93.5, 99.8)* | 97.2a (92.1, 99.4)* | |
| Prometheus Bio Inc., Zhejiang, China | Prometheus Bio ‐ 2019‐nCoV IgG | 1 (15/20) | 1 (16/20) | 1 (13/20) |
| LFA | 75.0a (50.9, 91.3)* | 80.0a (56.3, 94.3)* | 65.0a (40.8, 84.6)* | |
| Qingdao HIGHTOP Biotech Co., Ltd. (CN) | SARS‐CoV‐2 IgG Ab Rapid Test | 1 (149/150) | 1 (150/150) | |
| 99.3a (96.3, 100)* | 100a (97.6, 100)** | |||
| Render | COVID‐19 IgG Test | 1 (26/26) | 1 (26/26) | 1 (27/27) |
| LFA | 100a (86.8, 100)** | 100a (86.8, 100)** | 100a (87.2, 100)** | |
| Screen Italia S.r.l | Screen Test Covid‐19 2019‐nCOV IgG | 1 (281/295) | 1 (253/295) | 1 (244/295) |
| LFA | 95.3a (92.2, 97.4)* | 85.8a (81.2, 89.5)* | 82.7a (77.9, 86,8)* | |
| SD Biosensor, Gyeonggi‐do, Korea | COVID‐19 IgG Duo | 4 (1125/1127) | 2 (957/967) | 1 (933/942) |
| LFA | 99.8 (96.2, 100) | 99.0 (98.1, 99.4) | 99.0a (98.2, 99.6)* | |
| Servibio/VEDALAB, France, Alençon | COVID‐19 Sign IgG | 1 (83/100) | 1 (78/100) | |
| LFA | 83.0a (74.2, 89.8)* | 78.0a (68.6, 85.7)* | ||
| Spring Healthcare Services AG | COVID‐19 Spring IgG/IgM Rapid Test Cassette | 1 (142/145) | ||
| CGIA | 97.9a (94.1, 99.6)* | |||
| Sure Biotech, New York, NY, USA; Wan Chai, Hong Kong | SARS‐CoV‐2 IgG Antibody Rapid Test | 2 (370/378) | 2 (376/378) | 1 (108/108) |
| LFA | 98.7 (87.2, 99.9) | 99.5 (98.1, 99.9) | 100a (96.6, 100)** | |
| SureScreen Diagnostics Co., Ltd. | COVID‐19 Coronavirus Rapid Test Cassette IgG | 2 (497/500) | ||
| LFA | 99.4 (98.2, 99.8) | |||
| T&D Diagnostics, Sienna, Halifax, Nova Scotia, Canada | Sienna 2019‐nCoV IgG Rapid Test | 1 (20/20) | 1 (19/20) | 1 (19/20) |
| LFA | 100a (83.2, 100)** | 95.0a (75.1, 99.9)* | 95.0a (71.1, 99.9)* | |
| TAmiRNA GmbH (AT) | SARS‐CoV‐2 Antibody Lateral Flow Test IgG/IgM | 1 (260/265) | ||
| LFA | 98.1a (95.7, 99.4)* | |||
| Trillium | Trillium IgG rapid assay | 1 (89/90) | 1 (87/90) | |
| LFA | 98.9a (94.0, 100)* | 96.7a (90.6, 99.3)* | ||
| UCP Biosciences, San Jose, CA, USAKong | Coronavirus IgG Antibody (COVID‐19) Test Cassette | 1 (105/107) | 1 (105/107) | 1 (105/107) |
| LFA | 98.1a (93.4, 99.8)* | 98.1a (93.4, 99.8)* | 99.1a (94.9,100)* | |
| Unknown manufacturer | Ready Result IgG | |||
| LFA | ||||
| VivaChek Biotech Co, Hangzhou, China | VivaDiag COVID‐19 IgG Rapid Test | 2 (293/297) | 2 (290/297) | 2 (292/297) |
| CGIA | 99.4 (77.8, 100) | 97.7 (93.0, 99.3) | 99.4 (69.0, 100) | |
| W.H.P.M., Inc. | COVISURE COVID‐19 IgG Rapid Test | |||
| LFA | ||||
| Xiamen Biotime Biotechnology Co., Ltd. (CN) | SARS‐Cov‐2 IgG Rapid Qualitative Test | 1 (178/186) | 1 (180/186) | |
| LFA | 95.7a (91.7, 98.1)* | 96.8a (93.1, 98.8)* | ||
| Zeus Scientific, Inc. | ZEUS SARS‐CoV‐2 IgG/IgM rapid test | |||
| LFA | ||||
| Zhejiang Orient‐Gene Biotech Co. Ltd., Huzhou, China | COVID‐19 IgG rapid test cassette | 4 (893/931) | 3 (416/431) | 5 (482/523) |
| LFA | 96.3 (92.7, 98.2) | 97.9a (90.5, 99.6)* | 97.8 (88.2, 99.6) | |
| ELISA | ||||
| Beijing Wantai Biological Pharmacy Enterprise Co | Wantai ELISA IgG assay | 1 (195/197) | 2 (606/613) | |
| 99.0a (96.4, 99.9)* | 98.9 (97.6, 99.5) | |||
| Bio‐Rad Laboratories, Inc. | Novel coronavirus COVID‐19 IgG assay | 1 (50/50) | 1 (50/50) | 1 (50/50) |
| 100a (92.9, 100)** | 100a (92.9, 100)** | 100a (92.9, 100)** | ||
| Creative Diagnostics | SARS‐CoV‐2 IgG ELISA Kit | 1 (68/69) | 1 (25/69) | |
| 98.6a (92.2, 100)* | 36.2a (25.0, 48.7)* | |||
| Dia.Pro Diagnostic Bioprobes, Sesto San Giovanni, Italy | DiaPro COVID‐19 IgG Confirmation | 1 (88/89) | 1 (60/60) | |
| 98.9a (93.9, 100)* | 100a (94.0, 100)** | |||
| Eagle Biosciences, Amherst, NH, United States | COVID‐19 IgG Quantitative ELISA | 1 (429/437) | 1 (426/434) | |
| 98.2a (96.4, 99.2)* | 98.2a (96.4, 99.2)* | |||
| Epitope Diagnostics Inc. | EDI Novel Coronavirus COVID‐19 IgM ELISA kit | 13 (2892/3014) | 8 (1589/1607) | 4 (1159/1184) |
| 98.0 (96.2, 99.0) | 98.9 (98.2,99.3) | 97.9 (96.9, 98.6) | ||
| Euroimmun AG, Lübeck, Germany | anti‐SARS‐COV‐2 IgG | 29 (4998/5144) | ||
| 98.4 (97.5, 98.9) | ||||
| Euroimmun AG, Lübeck, Germany | anti‐SARS‐COV‐2 IgG (N‐based) | 1 (55/56) | ||
| 98.2a (90.4, 100)* | ||||
| Gold Standard Diagnostics (GSD) | Gold Standard SARS‐CoV‐2 IgG ELISA | 1 (76/76) | ||
| 100a (95.3, 100)** | ||||
| ImmunoDiagnostics Limited, Sha Tin, Hong Kong | SARS‐CoV‐2 NP IgG ELISA Kit | 2 (334/372) | 1 (47/67) | |
| 89.5 (27.4, 99.5) | 70.1a (57.7, 80.7)* | |||
| Mikrogen, Neuried, Germany | Mikrogen IgG anti‐N | |||
| Mologic | IgG COVID‐19 ELISA | 1 (549/564) | ||
| 97.3a (95.7, 98.5)* | ||||
| Vircell | COVID‐19 ELISA IgG | 2 (82/84) | ||
| 97.6(91.0, 99.4) | ||||
| Zydus diagnostics, Calida Healthcare Limited | Covid Kavach IgG ELISA | 1 (183/184) | ||
| 99.5a (97.0, 100)* | ||||
| CLIA | ||||
| Abbott Diagnostics, Rungis, France | Abbott Alinity anti‐SARS‐CoV‐2 nucleocapsid IgG | 2 (636/640) | ||
| 99.4 (98.3, 99.8) | ||||
| Abbott Diagnostics, Rungis, France | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG | 24 (7460/7483) | 2 (5691/1597) | |
| 99.7 (99.5, 99.8) | 99.6(99.2, 99.8) | |||
| Autobio Diagnostics Co. Zhengzhou, China | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | |||
| Beckman Coulter | Beckman Coulter ‐ Access SARS‐CoV‐2 IgG | 1 (396/399) | ||
| 99.2a (97.8, 99.8)* | ||||
| Bioscience Co (Chongqing) | Bioscience Co (Chongqing) SARS‐CoVCoV‐2 IgG/IgM | |||
| DiaSorin S.p.A., Saluggia, Italy | Diasorin LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA | 16 (4290/4367) | ||
| 98.6 (97.8, 99.1) | ||||
| Diazyme | Diazyme DZ‐LITE 2019‐nCoV IgG (CLIA) Assay Kit | 1 (108/110) | 1 (109/110) | |
| 98.2a (93.6, 99.8)* | 99.1a (95.0, 100)* | |||
| Hunan Yuanjing Biotechnology Co., Ltd. | COVID‐19 IgG Detection Kits | |||
| MEXACARE GmbH (DE) | QuickTestCorona COVID‐19 IgG | 1 (241/246) | 1 (225/246) | |
| 98.0a (95.3, 99.3)* | 91.5a (87.2, 94.6)* | |||
| Ortho Clinical Diagnostics, Pencoed, UK | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay IgG | 3 (1139/1141) | ||
| 99.8(99.3, 100) | ||||
| Shenzhen YHLO Biotech Co., Ltd | YHLO iFlash IgG assay | 2 (657/661) | 2 (657/660) | |
| 99.4 (98.4, 99.8) | 99.5 (98.6, 99.6) | |||
| SNIBE Shenzhen New Industries Biomedical | MAGLUMI 2019‐nCoV IgG kits | 7 (1801/1818) | 4 (1607/1661) | 1 (40/40) |
| 99.1 (98.5, 99.4) | 96.9 (95.0, 98.0) | 100a (91.2, 100)** | ||
| Xiamen InnoDx Biotech Co., Ltd., China (Xiamen, China) | Novel coronavirus (2019‐nCoV) antibody test kit IgM | 1 (268/270) | ||
| 99.3a (97.3, 99.9)* | ||||
| Unclear/unknown | ||||
| ET Healthcare, Palo Alto, CA | Pylon 3D automated immunoassay system IgG | 1 (316/320) | 1 (318/320) | |
| 98.8a (96.8, 99.7)* | 99.4a (97.8, 99.9)* | |||
| Quotient | MosaiQ COVID‐19 antibody microarray IgM | 1 (500/500) | ||
| 100a (99.3, 100)** | ||||
|
a Estimates and confidence intervals by summing the counts of true negative and false positive across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCGIA: colloidal gold immunoassayCI: confidence immunoassayCLIA: chemiluminescence immunoassayELISA: enzyme‐linked immunosorbent assayFIA: fluorescence immunoassayLFA: lateral flow assay | ||||
Appendix 20. Specificity data by test brand (total Ab, all reference groups)
| Test name | Test name | Pre‐pandemic | Suspected of COVID‐19 | Current RT‐PCR‐negative | Current untested | Other/mixed unclear | Cross‐reactivity/confounder panels |
| Test brand | Test brand | Specificity (95% CI) | Test groups (true negatives/non‐COVID cases) | ||||
| ELISA | |||||||
| Beijing Wantai | Wantai ELISA Total‐Ab assay | 8 (2009/2020) | 1 (50/50) | 1 (300/300) | 1 (97/100) | 2 (130/130) | |
| 99.5 (99.0, 99.7) |
100a (92.9, 100)** |
100a (98.8, 100)** |
97.0a (91.5, 99.4)* |
100a (97.2, 100)** |
|||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 Total‐Ab assay | 1 (28/28) | |||||
| 100a (87.7, 100)** |
|||||||
| CLIA | |||||||
| Ortho Clinical | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay Total‐Ab | 2 (995/997) | 1 (57/57) | 2 (168/169) | |||
| 99.8 (98.5, 100) |
100a (93.7, 100)** |
99.4 (95.9, 99.9) |
|||||
| Roche Diagnostics | Elecsys anti‐SARS‐CoV‐2 antibody assay | 25 (5569/5579) | 3 (331/336) | 4 (307/307) | 2 (1029/1029) | 1 (26/26) 2 (99/99) |
13 (836/839) |
| 99.8 (99.7, 99.9) |
98.5 (96.5, 99.4) |
100a (98.8, 100)** |
100a (99.6, 100)** |
100a (96.3, 100)** |
99.8 (97.6, 100) |
||
| Siemens Healthcare | Siemens Atellica Total‐Ab assay | 6 (2435/2439) | 3 (210/210) | ||||
| 99.9 (99.3, 100) |
100a (98.3, 100)** |
||||||
| Siemens Healthcare | Siemens Vista Total‐Ab assay | 1 (596/596) | 1 (35/35) | ||||
| 100a (99.4, 100)** |
100a (90.0, 100)** |
||||||
| Watmind Medical | SARS‐CoV‐2 Ab Diagnostic Test Kit | 1 (41/50) | |||||
| 82.0a (68.6, 91.4)* |
|||||||
| Xiamen Wantai | Total Ab CMIA | 1 (231/234) | |||||
| 98.7a (96.3, 99.7)* |
|||||||
| Xiamen InnoDx | Novel coronavirus (2019‐nCoV) antibody test kit Total‐Ab | 1 (267/270) | |||||
| 98.9a (96.8, 99.8)* |
|||||||
| Other/unclear | |||||||
| Luminex | SARS‐CoV‐2 MIA Total Ab |
1 (254/256) | |||||
| 99.2a (97.2, 99.9)* |
|||||||
| Vibrant America | Vibrant COVID‐19 Ab Total‐Ab | 1 (5250/5262) | |||||
| 99.8a (99.6, 99.9)* |
|||||||
|
aEstimates and confidence intervals by summing the counts of true negative and false positive across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervalsCLIA: chemiluminescence immunoassayELISA: enzyme‐linked immunosorbent assay MIA: magnetic immunoassay RT‐PCR: reverse transcription polymerase chain reaction | |||||||
Appendix 21. Sensitivity data by test brand (IgG alone, by week after onset)
| Test brand | Test name | Week 1 | Week 2 | Week 3 |
| Lateral flow assays |
Evaluations (TP/TP + FN) Sensitivity (95% CI) |
|||
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgG | 1 (13/18) | 1 (31/33) | |
| LFA | 72.2a (46.5, 90.3)* |
93.9a (79.8, 99.3)* |
||
| AccuBiotech | Accu‐Tell COVID‐19 IgG Cassette | 1 (0/9) | 1 (5/14) | 1 (12/15) |
| CGIA | 0a (0, 33.6)** |
35.7a (12.8, 64.9)* |
80.0a (51.9, 95.7)* |
|
| ACON Laboratories | SARS‐COV‐2 IgG Rapid Test | 1 (56/58) | ||
| LFA | 96.6a (88.1, 99.6)* |
|||
| Acro Biotech | 2019‐nCoV IgG Rapid Test | 1 (0/9) | 1 (10/14) | 1 (12/15) |
| 0a (0, 33.6)** |
71.4a (41.9, 91.6)* |
80.0a (51.9, 95.7)* |
||
| Anhui Deep Blue | SARS‐CoV‐2 IgG Ab test kit | 1 (7/10) | ||
| LFA | 70.0a (34.8, 93.3)* |
|||
| Artron | COVID‐19 IgG | 1 (2/53) | 1 (2/25) | 1 (15/24) |
| LFA | 3.8a (0.5, 13.0)* |
8.0a (1.0, 26.0)* |
62.5a (40.6, 81.2)* |
|
| Augurix Diagnostics | One Step Test for Novel Coronavirus (2019‐nCoV) IgG Antibody | 1 (4/18) | 1 (24/32) | |
| LFA | 22.2a (6.4, 47.6)* |
75.0a (56.6, 88.5)* |
||
| Augurix Diagnostics | SimtomaX Corona Check IgG | 1 (37/59) | ||
| LFA | 62.7a (49.1, 75.0)* |
|||
| Autobio Diagnostics | Anti‐SARS‐CoV‐2 Rapid Test IgG | 1(44/100) | 1 (71/85) | 1 (8/8) |
| CGIA | 44.0a (34.1, 54.3)* |
83.5a (73.9, 90.7)* |
100a (63.1, 100)** |
|
| Beijing Beier Bioengineering | 2019 nCov IgG | 1 (2/10) | 1 (6/13) | 1 (11/14) |
| CGIA | 20.0a (2.5, 55.6)* |
46.2a (19.2, 74.9)* |
78.6a (49.2, 95.3)* |
|
| Beijing Diagreat Biotechnology | 2019‐nCoV IgG Antibody Determination Kit | 1 (33/38) | ||
| FIA | 86.8a (71.9, 95.6)* |
|||
| Biolidics | 2019 nCoV IgG detection kit | 1 (0/7) | 1 (24/34) | 1 (8/10) |
| LFA | 0a (0, 41.0)** |
70.6a (52.5, 84.9)* |
80.0a (44.4, 97.5)* |
|
| BioMedomics | COVID‐19 IgG Rapid Test | 2 (25/70) | 2 (43/67) | 1 (14/19) |
| LFA | 18.1 (1.1, 81.8) |
64.2 (52.1, 74.7) |
73.7a (48.8, 90.9)* |
|
| BioMerieux | Vidas SARS‐CoV‐2 IgG | 2 (27/57) | 2 (47/60) | 2 (55/59) |
| FIA | 46.0 (29.1, 63.8) |
78.4 (65.7, 87.3) |
93.2 (83.3, 97.4) |
|
| Bioperfectus Technologies | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgG Rapid Test Kit | 1 (30/63) | 1 (27/34) | 1 (14/19) |
| LFA | 47.6a (34.9, 60.6)* |
79.4a (62.1, 91.3)* |
73.7a (48.8, 90.9)* |
|
| Biosynex | COVID‐19 BSS IgG | 1 (10/18) | 1 (40/45) | |
| LFA | 55.6a (30.8, 78.5)* |
88.9a (75.9, 96.3)* |
||
| BTNX | Rapid Response™ COVID‐19 Test Cassette (BTNX Inc.) Kit1 IgG | 1 (24/154) | 1 (49/103) | 2 (63/68) |
| LFA | 15.6a (10.2, 22.3)* |
47.6a (37.6, 57.6)* |
90.4 (61.3, 98.2) |
|
| Cellex | qSARS‐CoV‐ 2 IgG Cassette Rapid Test | 2 (10/28) | 2 (51/67) | 2 (51/58) |
| LFA | 33.3 (10.1, 68.9) |
76.1 (64.5, 84.8) |
87.9 (76.8, 94.1) |
|
| Chongqing iSIA BIO‐Technology | 2019‐nCoV IgG Diagnostic Test Kit | 1 (0/9) | 1 (4/14) | 1 (12/15) |
| LFA | 0a (0. 33.6)** |
28.6a (8.4, 58.1)* |
80.0a (51.9, 95.7)* |
|
| Clungene Biotech | Clungene SARS‐CoV‐2 IgG Rapid Test Cassettes | 1 (11/37) | 1 (47/78) | 1 (37/38) |
| LFA | 29.7a (15.9, 47.0)* |
60.3a (48.5, 71.2)* |
97.4a (86.2, 99.9)* |
|
| CTK Biotech | OnSite COVID‐19 IgG Rapid Test | 1 (6/51) | 1 (6/21) | 2 (46/65) |
| CGIA | 11.8a (4.4, 23.9)* |
28.6a (11.3, 52.2)* |
70.8 (58.7, 80.5) |
|
| DecombioBiotechnology | Novel Coronavirus (SARS‐CoV‐2) IgG Combo Rapid Test‐Cassette | 1 (31/62) | 1 (29/33) | 1 (14/18) |
| LFA | 50.0a (37.0, 63.0)* |
87.9a (71.8, 96.6)* |
77.8a (52.4, 93.6)* |
|
| DeepBlue Medical | COVID‐19 (SARS CoV‐2) IgG Antibody Test Kit (Colloidal Gold) | 1 (24/64) | 1 (21/34) | 1 (15/19) |
| CGIA | 37.5a (25.7, 50.5)* |
61.8a (43.6, 77.8)* |
78.9a (54.4, 93.9)* |
|
| DiaCarta | QuantiVirus anti‐SARS‐CoV‐2 IgG test | 1 (6/13) | 1 (8/13) | |
| FIA | 46.2a (19.2, 74.9)* |
61.5a (31.6, 86.1)* |
||
| DNA Link | AccuFind COVID19 IgG | 1 (24/48) | 1 (40/42) | 1 (9/9) |
| LFA | 50.0a (35.2, 64.8)* |
95.2a (83.8, 99.4)* |
100a (66.4, 100)** |
|
| Dynamiker Biotechnology | 2019‐nCOV IgG Rapid Test | 2 (22/65) | 2 (97/154) | 1 (36/38) |
| LFA | 33.8 (23.4, 46.1) |
63.0 (55.1, 70.2) |
94.7a (82.3, 99.4)* |
|
| Genrui Biotech | Novel Coronavirus IgG test kit | 1 (7/10) | ||
| LFA | 70.0a (34.8, 93.3)* |
|||
| Getein Biotech | One Step Test for Novel Coronavirus IgG | 1 (7/10) | ||
| CGIA | 70.0a (34.8, 93.3)* |
|||
| Hangzhou Alltest | unlabelled packaging IgG | 1 (12/51) | 1 (15/21) | 1 (6/8) |
| CGIA | 23.5a (12.8, 37.5)* |
71.4a (47.8, 88.7)* |
75.0a (34.9, 96.8)* |
|
| Hangzhou Alltest | 2019‐nCoV IgG Rapid Test Cassette | 5 (25/129) | 7 (94/153) | 7 (111/114) |
| CGIA | 19.4 (13.4, 27.1) |
64.8 (48.8, 78.0) |
97.9 (86.0, 99.7) |
|
| Hangzhou Biotest | RightSign IgM/IgG (also distributed as CoronaChek IgG and Premier Biotech Covid‐19 IgG) | 2 (35/126) | 2 (144/218) | 3 (68/78) |
| 26.9 (14.8, 43.7) |
66.1 (59.5, 72.0) |
89.4 (67.4, 97.2) |
||
| Innovita Biological Technology | 2019‐nCoV Ab test IgG | 7 (71/183) | 6 (107/145) | 6 (93/116) |
| CGIA | 38.8 (32.0, 46.0) |
75.2 (59.7, 86.1) |
80.2 (71.9, 86.5) |
|
| InTec | Rapid SARS‐CoV‐2 Antibody (IgG) Test | 1 (8/14) | 1 (42/47) | 1 (13/16) |
| CGIA | 57.1a (28.9, 82.3)* |
89.4a (76.9, 96.5)* |
81.3a (54.4, 96.0)* |
|
| Inzek | BIOZEK COVID‐19 IgG/IgM | 2 (80/95) | ||
| LFA | 84.2 (75.4, 90.3) |
|||
| Jiangsu Medomics | Rapid IgM‐IgG Combined Antibody Test Kit for SARS‐ CoV‐2 IgG | 1 (6/12) | 1 (6/12) | 1 (15/20) |
| CGIA | 50.0a (21.1, 78.9)* |
50.0a (21.1, 78.9)* |
75.0a (50.9, 91.3)* |
|
| Lansion Biotechnology | (COVID‐19) IgG Test Kit | 1 (5/5) | 1 (8/11) | |
| FIA | 100a (47.8, 100)** |
72.7a (39.0, 94.0)* |
||
| Leccurate | SARS‐CoV‐2 antibody test IgG | 1 (10/14) | 1 (13/20) | 1 (12/16) |
| CGIA | 71.4a (41.9, 91.6)* |
65.0a (40.8, 84.6)* |
75.0a (47.6, 92.7)* |
|
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgG | 2 (25/61) | 2 (71/111) | 2 (64/72) |
| LFA | 41.5 (27.6, 57.0) |
64.0 (54.6, 72.3) |
91.1 (67.5, 98.1) |
|
| Maccura Biotechnology | Maccura LFIA SARS‐CoV‐2 IgG | 2 (8/49) | 1 (11/18) | 1 (13/14) |
| CGIA | 16.3 (8.4, 29.4) |
61.1a (35.7, 82.7)* |
92.9a (66.1, 99.8)* |
|
| Hightop Biotech | Hightop SARS‐CoV‐2 IgG Antibody Rapid Test | 1 (7/51) | 1 (13/21) | 1 (7/8) |
| CGIA | 13.7a (5.7, 26.3)* |
61.9a (38.4, 81.9)* |
87.5a (47.3, 99.7)* |
|
| Multi‐G | MGA 2019‐nCoV IgG Rapid test cassette | 1 (11/37) | 1 (51/78) | 1 (37/38) |
| LFA | 29.7a (15.9, 47.0)* |
65.4a (53.8, 75.8)* |
97.4a (86.2, 99.9)* |
|
| Nal Von Minden | NADAL COVID‐19 IgG Test | 1 (6/18) | 1 (27/35) | |
| LFA | 33.3a (13.3, 59.0)* |
77.1a (59.9, 89.6)* |
||
| NG Biotech | NG‐Test IgG COVID‐19 | 3 (78/273) | 4 (163/219) | |
| CGIA | 28.6 (23.5, 34.2) |
74.4 (68.2 79.8) |
||
| Nirmidas Biotech | COVID‐19 (SARS‐CoV‐2) IgG Antibody Detection Kit | 1 (24/73) | 1 (44/52) | 1 (18/19) |
| LFA | 32.9a (22.3, 44.9)* |
84.6a (71.9, 93.1)* |
94.7a (74.0, 99.9)* |
|
| MEDsan | COVID‐19 IgG Rapid Test | 1 (53/64) | ||
| LFA | 82.8 (71.3, 91.1) | |||
| NTBIO® Diagnostics | NTBIO One Step Rapid Test ‐ COVID‐19 IgG Antibody Test | 1 (52/59) | ||
| 88.1a (77.1, 95.1)* |
||||
| Prima Lab | Prima COVID‐19 IgG Rapid test | 1 (15/37) | 1 (56/78) | 1 (38/38) |
| LFA | 40.5a (24.8, 57.9)* |
71.8a (60.5, 81.4)* |
100a (90.7, 100)** |
|
| Prometheus Bio | Prometheus Bio ‐ 2019‐nCoV IgG | 1 (14/35) | 1 (7/11) | 1 (1/2) |
| LFA | 40.0a (23.9, 57.9)* |
63.6a (30.8, 89.1)* |
50.0a (1.3, 98.7)* |
|
| Qingdao HIGHTOP | SARS‐CoV‐2 IgG Ab Rapid Test | 1 (57/64) | ||
| 89.1a (78.8, 95.5)* |
||||
| Render | COVID‐19 IgG Test | 1 (6/16) | 1 (18/28) | 1 (17/24) |
| LFA | 37.5a (15.2, 64.6)* |
64.3a (44.1, 81.4)* |
70.8a (48.9, 87.4)* |
|
| SD Biosensor | COVID‐19 IgG Duo | 4 (14/62) | 5 (86/143) | 2 (33/38) |
| LFA | 20.7 (8.1, 43.6) |
60.1 (51.9, 67.8) |
86.8 (72.0, 94.4) |
|
| Sensing Self | Covid‐19 Rapid IgG combined Antibody assay | 1 (3/28) | 1 (83/136) | 1 (20/21) |
| LFA | 10.7a (2.3, 28.2)* |
61.0a (52.3, 69.3)* |
95.2a (76.2, 99.9)* |
|
| SIDAK Life Care | Sidak Covid 19 Antibody IgG Rapid Test Kit | 1 (0/23) | 1 (9/27) | 1 (12/36) |
| LFA | 0a (0, 14.8)** |
33.3a (16.5, 54.0)* |
33.3a (18.6, 51.0)* |
|
| Sure Biotech | SARS‐CoV‐2 IgG Antibody Rapid Test | 1 (24/63) | 1 (25/34) | 2 (70/80) |
| LFA | 38.1a (26.1, 51.2)* |
73.5a (55.6, 87.1)* |
86.3 (70.4, 94.4) |
|
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgG | 1 (4/22) | 1 (4/14) | 1 (6/9) |
| LFA | 18.2a (5.2, 40.3)* |
28.6a (8.4, 58.1)* |
66.7a (29.9, 92.5)* |
|
| Syzbio Biotech | SARS‐CoV‐2 IgG Antibody Assay Kit | 1 (4/22) | 1 (3/14) | 1 (5/9) |
| LFA | 18.2a (5.2, 40.3)* |
21.4a (4.7, 50.8)* |
55.6a (21.2, 86.3)* |
|
| T&D Diagnostics | Sienna 2019‐nCoV IgG Rapid Test | 1 (11/41) | 1 (33/48) | 1 (7/9) |
| LFA | 26.8a (14.2, 42.9)* |
68.8a (53.7, 81.3)* |
77.8a (40.0, 97.2)* |
|
| TBG Biotechnology | TBG SARS‐CoV‐2 IgG Rapid Test Kit | 1 (1/6) | 1 (44/69) | 1 (9/9) |
| LFA | 16.7a (0.4, 64.1)* |
63.8a (51.3, 75.0)* |
100a (66.4, 100)** |
|
| UCP Biosciences | Coronavirus IgG Antibody (COVID‐19) Test Cassette | 1 (25/64) | 1 (25/34) | 1 (14/19) |
| LFA | 39.1a (27.1, 52.1)* |
73.5a (55.6, 87.1)* |
73.7a (48.8, 90.9)* |
|
| UNscience Biotechnology | COVID‐19 IgG Rapid Test Kit | 1 (0/9) | 1 (10/14) | 1 (13/15) |
| LFA | 0a (0, 33.6)** |
71.4a (41.9, 91.6)* |
86.7a (59.5, 98.3)* |
|
| VivaChek Biotech | VivaDiag COVID‐19 IgG Rapid Test | 3 (50/148) | 3 (84/129) | 3 (56/65) |
| CGIA | 31.8 (17.8, 50.2) |
66.9 (52.8, 78.6) |
85.3 (67.4, 94.3) |
|
| Wells Bio | careUS COVID‐19 IgG | 1 (0/3) | 1 (3/6) | 1 (6/6) |
| LFA | 0a (0, 70.8)** |
50.0a (11.8, 88.2)* |
100a (54.1, 100)** |
|
| Xiamen Biotime | SARS‐Cov‐2 IgG/IgM | 1 (43/52) | ||
| LFA | 82.7a (69.7, 91.8)* | |||
| Zhejiang Orient‐Gene | COVID‐19 IgG rapid test cassette | 4 (54/115) | 5 (140/185) | 5 (128/139) |
| CGIA | 47.0 (38.0, 56.1) |
75.7 (69.0, 81.3) |
92.1 (86.3, 95.6) |
|
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG | 2 (5/45) | 2 (27/48) | 1 (13/15) |
| LFA | 11.1 (4.7, 24.1) |
56.3 (42.1, 69.5) |
86.7a (59.5, 98.3)* |
|
| ELISA | ||||
| Abbott Diagnostics | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG | 1 (15/32) | 1 (20/29) | |
| 46.9a (29.1, 65.3)* |
69.0a (49.2, 84.7)* |
|||
| Ash Laboratories | Ash Laboratories SARS‐CoV2 IgG ELISA Immunoassay | 1 (3/10) | 1 (9/9) | |
| 30.0a (6.7, 65.2)* |
100a (66.4, 100)** |
|||
| Beijing Beier Bioengineerin | Beijing Beier Bioengineering ‐ 2019 nCov IgG | 1 (4/10) | 1 (8/13) | 1 (12/14) |
| 40.0a (12.2, 73.8)* |
61.5a (31.6, 86.1)* |
85.7a (57.2, 98.2)* |
||
| Beijing Wantai | Wantai ELISA IgG assay | 3 (43/157) | 3 (194/290) | 1 (33/37) |
| 31.2 (18.6, 47.2) |
70.6 (56.0, 81.9) |
89.2a (74.6, 97.0)* |
||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgG assay | 1 (10/11) | ||
| 90.9a (58.7, 99.8)* |
||||
| Creative Diagnostics | SARS‐CoV‐2 IgG ELISA Kit | 1 (6/19) | 1 (27/38) | |
| 31.6a (12.6, 56.6)* |
71.1a (54.1 84.6)* |
|||
| Dia.Pro Diagnostic Bioprobes | DiaPro COVID‐19 IgG Confirmation | 1 (7/13) | ||
| 53.8a (25.1, 80.8)* |
||||
| Epitope Diagnostics | EDITM Novel Coronavirus COVID‐19 IgG ELISA kit | 7 (106/256) | 11 (301/455) | 10 (179/196) |
| 20.2 (4.2, 59.5) |
77.0 (62.1, 87.2) |
94.0 (84.3, 97.9) |
||
| Euroimmun | anti‐SARS‐COV‐2 IgG | 28 (161/913) | 32 (726/1407) | 28 (528/631) |
| 13.0 (8.4, 19.4) |
54.4 (47.7, 61.0) |
88.5 (82.8, 92.5) |
||
| Euroimmun | anti‐SARS‐COV‐2 IgG (N‐based) | 2 (42/95) | 1 (70/98) | 2 (94/100) |
| 44.2 (34.0, 54.8) | 71.4 (61.4, 80.1) | 94.0 (87.3, 97.3) | ||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgG ELISA | 2 (30/62) | 1 (60/92) | 1 (51/55) |
| 48.4 (36.3, 60.7) |
65.2a (54.6, 74.9)* |
92.7a (82.4, 98.0)* |
||
| ID.Vet | ID Screen SARS‐CoV‐2‐N IgG Indirect ELISA | 1 (0/9) | 1 (7/14) | 1 (14/15) |
| 0a (0, 33.6)** |
50.0a (23.0, 76.9)* |
93.3a (68.1, 99.8)* |
||
| ImmunoDiagnostics | SARS‐CoV‐2 NP IgG ELISA Kit | 1 (11/19) | 1 (31/38) | |
| 57.9a (33.5, 79.7)* |
81.6a (65.7, 92.3)* |
|||
| Mediagnost | Mediagnost Anti‐SARS‐CoV‐2 ELISA IgG | 1 (18/25) | ||
| 72.0a (50.6, 87.9)* |
||||
| Mikrogen | Mikrogen IgG anti‐N | 1 (13/43) | 1 (66/98) | 1 (54/58) |
| 30.2a (17.2, 46.1)* |
67.3a (57.1, 76.5)* |
93.1a (83.3, 98.1)* |
||
| Mologic | IgG COVID‐19 ELISA | 1 (12/16) | 1 (25/32) | 1 (43/45) |
| 75.0a (47.6, 92.7)* |
78.1a (60.0, 90.7)* |
95.6a (84.9, 99.5)* |
||
| Novatek | Novatec Novalisa SARS‐CoV‐2 IgG ELISA | 1 (20/25) | ||
| 80.0a (59.3, 93.2)* |
||||
| RayBiotech | Covid‐19 human ELISA IgG | 1 (1/28) | 1 (20/29) | |
| 3.6a (0.1, 18.3)* |
69.0a (49.2, 84.7)* |
|||
| Vazyme Biotech | 2019‐nCoV IgG Detection Kit | 1 (5/18) | 1 (14/22) | 1 (17/18) |
| 27.8a (9.7, 53.5)* |
63.6a (40.7, 82.8)* |
94.4a (72.7, 99.9)* |
||
| Vircell | COVID‐19 ELISA IgG | 1 (9/19) | 2 (67/77) | |
| 47.4a (24.4, 71.1)* |
87.0 (77.5, 92.9) |
|||
| Virotech | Virotech SARS‐CoV‐2 ELISA IgG | 1 (18/25) | ||
| 72.0a (50.6, 87.9)* |
||||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG ELISA | 4 (29/93) | 3 (163/288) | 2 (95/112) |
| 31.2 (22.6, 41.3) |
56.6 (50.8, 62.2) |
84.8 (76.9, 90.4) |
||
| CLIA | ||||
| Abbott Diagnostics | Abbott Alinity anti‐SARS‐CoV‐2 nucleocapsid IgG | 1 (13/29) | 1 (54/75) | |
| 44.8a (26.4, 64.3)* |
72.0a (60.4, 81.8) |
|||
| Abbott Diagnostics | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG | 15 (134/676) | 19 (430/718) | 19 (409/460) |
| 21.0 (13.4, 31.2) |
61.3 (52.4, 69.5) |
88.2 (84.0, 91.4) |
||
| Autobio Diagnostics | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | 1 (30/66) | 1 (55/70) | |
| 45.5a (33.1, 58.2)* |
78.6a (67.1, 87.5)* |
|||
| Beckman Coulter | Access SARS‐CoV‐2 IgG | 1 (6/12) | ||
| 50.0a (21.1, 78.9)* |
||||
| Beijing Beier Bioengineering | 2019 nCov IgG | 1 (4/10) | 1 (6/13) | 1 (9/14) |
| 40.0a (12.2, 73.8)* |
46.2a (19.2, 74.9)* |
64.3a (35.1, 87.2)* |
||
| Beijing Wantai | Wantai CLIA IgG assay | 1 (17/31) | 1 (22/29) | 1 (38/38) |
| 54.8a (36.0, 72.7)* |
75.9a (56.5, 89.7)* |
100a (90.7, 100)** |
||
| Bioscience Co | Bioscience Co (Chongqing) SARS‐CoVCoV‐2 IgG | 2 (43/89) | 2 (212/269) | 2 (208/217) |
| 48.3 (38.1, 58.6) |
78.8 (73.5, 83.2) |
95.9 (92.2, 97.8) |
||
| DiaSorin | LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA | 10 (93/301) | 10 (280/496) | 10 (251/303) |
| 28.6 (20.8, 38.0) |
56.8 (51.5, 61.9) |
81.8 (70.3, 89.5) |
||
| Hangzhou Biotest | The RightSignTM COVID‐19 IgG Rapid Test Cassette / Lumiratek | 1 (31/36) | ||
| 86.1a (70.5, 95.3)* |
||||
| MEXACARE | QuickTestCorona COVID‐19 IgG | 1 (48/61) | ||
| 78.7a (66.3, 88.1)* |
||||
| Ortho Clinical Diagnostics | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay IgG | 1 (1/38) | 1 (35/91) | 1 (2/4) |
| 2.6a (0.1, 13.8)* |
38.5a (28.4, 49.2)* |
50.0a ( 6.8, 93.2)* |
||
| Shenzhen YHLO | YHLO iFlash IgG assay | 6 (78/135) | 5 (174/188) | 4 (67/74) |
| 52.5 (27.0, 76.7) |
96.3 (80.1, 99.4) |
90.5 (81.5, 95.4) |
||
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgG kits | 8 (69/240) | 10 (318/486) | 4 (132/141) |
| 27.0 (18.5, 37.5) |
65.6 (60.4, 70.5) |
93.6 (88.2, 96.6) |
||
| Vircell | COVID‐19 VIRCLIA IgG MONOTEST | 1 (12/17) | 1 (16/16) | |
| 70.6a (44.0, 89.7) |
100a (79.4, 100)** |
|||
| Other/unclear | ||||
| ET Healthcare | Pylon 3D automated immunoassay system IgG | 1 (8/41) | 1 (28/42) | 1 (13/15) |
| 19.5a (8.8, 34.9)* |
66.7a (50.5, 80.4)* |
86.7a (59.5, 98.3)* |
||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgG (N‐based) | 1 (7/48) | 1 (34/81) | 1 (29/39) |
| 14.6 (6.1, 27.8) | 42.0 (31.1, 53.5) | 74.4 (57.9, 87.0) | ||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgG (S‐based) | 1 (7/48) | 1 (45/81) | 1 (33/39) |
| 14.6 (6.1, 27.8) | 55.6 (44.1, 66.6) | 84.6 (69.5, 94.1) | ||
| Vibrant America | Vibrant COVID‐19 Ab IgG | 1 (307/330) | 1 (330/330) | |
| 93.0a (89.7, 95.5)* | 100a (98.9, 100)** | |||
|
a Estimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCGIA: colloidal gold immunoassayCI: confidence intervalsCLIA: chemiluminescence immunossayDNA: deoxyribonucleic acidELISA: enzyme‐linked immunoassayFIA: fluorescence immunoassayFN: false negativeLFA: lateral flow assayLFIA: lateral flow immunoassayTP: true positive | ||||
Appendix 22. Sensitivity data by test brand (IgG or IgM, by week after onset)
| Test brand | Test name | Week 1 | Week 2 | Week 3 |
| Lateral flow assays |
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
|||
| AAZ‐LMB | Covid‐Duo test rapid Covid‐19 IgG/IgM | 1 (5/14) | 1 (59/86) | 1 (26/26) |
| CGIA | 35.7a (12.8, 64.9)* |
68.6a (57.7, 78.2)* |
100a (86.8, 100)** |
|
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgG/IgM | 2 (17/38) | 2 (84/125) | 1 (24/24) |
| CGIA | 42.8 (4.2, 9.3) |
85.3 (34.7, 98.4) |
100a (85.8, 100)** |
|
| AccuBiotech | Accu‐Tell COVID‐19 IgG/IgM Cassette | 2(30/47) | 1 (11/14) | 2 (51/59) |
| CGIA | 42.4 (6.1, 89.2) |
78.6a (49.2, 95.3)* |
86.4 (75.2, 93.1) |
|
| ACON Laboratories | SARS‐COV‐2 IgG/IgM Rapid Test | 1 (56/58) | ||
| LFA | 96.6a (88.1, 99.6)* |
|||
| Acro Biotech | 2019‐nCoV IgG/IgM Rapid Test | 1 (0/9) | 1 (13/14) | 1 (12/15) |
| LFA | 0a (0, 33.6)** |
92.9a (66.1, 99.8)* |
80.0a (51.9, 95.7)* |
|
| Anhui Deep Blue | SARS‐CoV‐2 IgG/IgM Ab test kit | 1 (30/38) | 2 (46/54) | |
| LFA | 78.9a (62.7, 90.4)* |
85.2 (73.1, 92.4) |
||
| Artron | Artron COVID‐19 IgG/IgM | 1 (25/90) | 2 (17/32) | 2 (35/39) |
| CGIA | 27.8a (18.9, 38.2)* |
53.1 (36.1, 69.4) |
89.9 (73.3, 96.6) |
|
| Assure Tech | FaStep (COVID‐19 IgG/IgM) rapid test cassettes | 1 (10/16) | 1 (15/16) | |
| LFA | 62.5a (35.4, 84.8)* |
93.8a (69.8, 99.8)* |
||
| Autobio Diagnostics | Anti‐SARS‐CoV‐2 Rapid Test IgG/IgM | 1 (52/100) | 2 (80/92) | 2 (22/23) |
| CGIA | 52.0a (41.8, 62.1)* |
87.0 (78.4, 92.4) |
95.7a (74.8, 99.4)* |
|
| Avioq Bio‐Tech | Novel Coronavirus (2019‐nCov) Antibody IgG/IgM Assay Kit | 1 (8/29) | 1 (46/62) | |
| CGIA | 27.6a (12.7, 47.2)* |
74.2a (61.5, 84.5)* |
||
| Biohit Healthcare | SARS‐CoV‐2 IgG/IgM ANTIBODY TEST KIT | 1 (22/38) | 1 (34/44) | |
| CGIA | 57.9a (40.8, 73.7)* |
77.3a (62.2, 88.5)* |
||
| Biolidics | 2019 nCoV IgG/IgM detection kit | 1 (0/7) | 1 (24/26) | 1 (8/10) |
| LFA | 0a (0, 41.0)** |
92.3a (74.9, 99.1)* |
80.0a (44.4, 97.5)* |
|
| BioMedomics | COVID‐19 IgG/IgM Rapid Test | 2 (32/70) | 2 (48/57) | 1 (17/19) |
| LFA | 45.7 (34.5, 57.4) |
84.2 (72.4, 91.6) |
89.5a (66.9, 98.7)* |
|
| Bioperfectus Technologies | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgG/IgM Rapid Test Kit | 1 (38/63) | 1 (30/37) | 1 (17/21) |
| LFA | 60.3a (47.2, 72.4)* |
81.1a (64.8, 92.0)* |
81.0a (58.1, 94.6)* |
|
| Biosynex | COVID‐19 BSS IgG/IgM | 2 (22/48) | 2 (40/46) | 3 (164/171) |
| LFA | 50.0 (21.7, 78.3) |
86.9 (71.5, 94.6) |
95.9 (61.7, 98.0) |
|
| BTNX | Rapid ResponseTM COVID‐19 Test Cassette (BTNX Inc.) Kit1 IgG/IgM | 1 (33/154) | 1 (64/103) | 2 (64/68) |
| LFA | 21.4a (15.2, 28.8)* |
62.1a (52.0, 71.5)* |
93.4 (77.4, 98.3) |
|
| Cellex | Cellex qSARS‐CoV‐ 2 IgG/IgM Cassette Rapid Test | 2 (12/28) | 2 (54/67) | 3 (51/58) |
| LFA | 41.8 (15.8, 73.4) |
80.6 (69.4, 88.4) |
87.9 (76.8, 94.1) |
|
| Chongqing iSIA BIO‐Technology | 2019‐nCoV IgG/IgM Diagnostic Test Kit | 1 (0/9) | 1 (7/14) | 1 (14/15) |
| LFA | 0a (0, 33.6)** |
50.0a (23.0, 77.0)* |
93.3a (68.1, 99.8)* |
|
| Clungene Biotech | Clungene SARS‐CoV‐2 IgG/IgM Rapid Test Cassettes | 1 (13/37) | 1 (50/78) | 1 (37/38) |
| LFA | 35.1a (20.2, 52.5)* |
64.1a (52.4, 74.7)* |
97.4a (86.2, 99.9)* |
|
| CTK Biotech | OnSite COVID‐19 IgG/IgM Rapid Test | 1 (5/7) | 1 (14/15) | |
| CGIA | 71.4a (29.0, 96.3)* |
93.3a (68.1, 99.8)* |
||
| DecombioBiotechnology | Novel Coronavirus (SARS‐CoV‐2) IgG/IgM Combo Rapid Test‐Cassette | 1 (32/62) | 1 (29/34) | 1 (14/16) |
| LFA | 51.6a (38.6, 64.5)* |
85.3a (68.9, 95.0)* |
87.5a (61.7, 98.4)* |
|
| DeepBlue Medical | COVID‐19 (SARS CoV‐2) IgG/IgM Antibody Test Kit (Colloidal Gold) | 1 (40/64) | 1 (28/37) | 1 (17/21) |
| CGIA | 62.5a (49.5, 74.3)* |
75.7a (58.8, 88.2)* |
81.0a (58.1, 94.6)* |
|
| Dynamiker Biotechnology | Dynamiker Biotechnology ‐ 2019‐nCOV IgG/IgM Rapid Test | 3 (54/144) | 3 (100/158) | 4 (118/136) |
| LFA | 37.6 (29.6, 46.3) |
63.3 (55.5, 70.4) |
90.7 (77.2, 96.6) |
|
| Epigentek Group | SeroFlash SARS‐CoV‐2 IgG/IgM | 1 (4/18) | 1 (11/14) | |
| LFA | 22.2a (6.4, 47.6)* |
78.6a (49.2, 95.3) |
||
| GenBody | GenBody COVID‐19 IgG/IgM | 1 (13/38) | 1 (31/44) | |
| CGIA | 34.2a (19.6, 51.4)* |
70.5a (54.8, 83.2)* |
||
| Genrui Biotech | Novel Coronavirus IgG/IgM test kit | 1 (8/10) | ||
| LFA | 80.0a (44.4, 97.5)* |
|||
| Getein Biotech | One Step Test for Novel Coronavirus Total Ab | 1 (8/13) | 1 (8/13) | 2 (27/32) |
| CGIA | 61.5a (31.6, 86.1)* |
61.5a (31.6, 86.1)* |
84.4 (67.5, 93.3) |
|
| Guangzhou Wondfo | Wondfo SARS‐CoV‐2 Antibody Test | 6 (106/280) | 6 (181/245) | 5 (103/117) |
| LFA | 29.8 (14.4, 51.7) |
73.9 (68.0, 79.0) |
88.0 (80.8, 92.8) |
|
| Hangzhou | Hangzhou unlabelled packaging IgG/IgM | 1 (13/51) | 1 (15/21) | 1 (6/8) |
| CGIA | 25.5a (14.3, 39.6)* |
71.4a (47.8, 88.7)* |
75.0a (34.9, 96.8)* |
|
| Hangzhou Alltest | 2019‐nCoV IgG/IgM Rapid Test Cassette | 4 (43/134) | 4 (45/73) | 5 (85/88) |
| CGIA | 30.6 (19.1, 45.1) |
63.1 (45.8, 77.5) |
96.6 (90.0, 98.9) |
|
| Hangzhou Biotest (also distributed by Premier Biotech) | RightSign IgM/IgG (distributed as COVID‐19 IgG/IgM Rapid Test Cassette) | 1 (35/63) | 1 (29/33) | 1 (17/21) |
| LFA | 55.6a (42.5, 68.1)* |
87.9a (71.8, 96.6)* |
81.0a (58.1, 94.6)* |
|
| Hightop Biotech | Hightop SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | 1 (8/51) | 1 (15/21) | 1 (7/8) |
| CGIA | 15.7a (7.0, 28.6)* |
71.4a (47.8, 88.7)* |
87.5a (47.3, 99.7)* |
|
| Innovita Biological Technology | 2019‐nCoV Ab test IgG/IgM | 5 (75/169) | 4 (72/106) | 5 (89/113) |
| CGIA | 44.4 (37.1, 51.9) |
67.9 (58.5, 76.1) |
78.8 (70.3, 85.3) |
|
| InTec | Rapid SARS‐CoV‐2 Antibody (IgG/IgM) Test | 1 (9/14) | 1 (44/47) | 1 (13/16) |
| CGIA | 64.3a (35.1, 87.2)* |
93.6a (82.5, 98.7)* |
81.3a (54.4, 96.0)* |
|
| Jiangsu Medomics | Rapid IgM‐IgG Combined Antibody Test Kit for SARS‐ CoV‐2 IgG/IgM | 2 (28/50) | 1 (7/12) | 2 (50/64) |
| CGIA | 56.0 (42.1, 69.0) |
58.3a (27.7, 84.8)* |
78.1 (66.4, 86.6) |
|
| LabOn Time | LaboOn Time rapid test cassette IgG/IgM | 1 (11/29) | 1 (47/62) | |
| LFA | 37.9a (20.7, 57.7)* |
75.8a (63.3, 85.8)* |
||
| Leccurate | SARS‐CoV‐2 antibody test IgG/IgM | 1 (11/14) | 1 (13/20) | 1 (15/20) |
| CGIA | 78.6a (49.2, 95.3)* |
65.0a (40.8, 84.6)* |
75.0a (50.9, 91.3)* |
|
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgG/IgM | 2 (28/61) | 2 (74/111) | 2 (65/72) |
| LFA | 47.4 (29.1, 66.4) |
66.7 (57.4, 74.8) |
91.8 (72.7, 97.9) |
|
| Maccura Biotechnology | Maccura LFIA SARS‐CoV‐2 IgG/IgM | 1 (9/21) | ||
| CGIA | 42.9a (21.8, 66.0)* |
|||
| Multi‐G | Multi‐G MGA 2019‐nCoV IgG/IgM Rapid test cassette | 1 (16/37) | 1 (56/78) | 1 (37/38) |
| LFA | 43.2a (27.1, 60.5)* |
71.8a (60.5, 81.4)* |
97.4a (86.2, 99.9)* |
|
| NG Biotech | NG‐Test IgG/IgM COVID‐19 | 3 (97/273) | 4 (172/219) | 1 (29/34) |
| CGIA | 36.5 (28.6, 45.1) |
78.6 (67.3, 86.8) |
85.3a (68.9, 95.0)* |
|
| Not stated | KHB Diagnostic Kit for SARS‐ CoV‐2 IgG/IgM Antibody | 1 (2/6) |
||
| CGIA | 33.3a (4.3, 77.7)* |
|||
| Prima Lab | Prima COVID‐19 IgG/IgM Rapid test | 1 (21/37) | 1 (62/78) | 1 (38/38) |
| LFA | 56.8a (39.5, 72.9)* |
79.5a (68.8, 87.8)* |
100a (90.7, 100)** |
|
| Prometheus Bio | Prometheus Bio ‐ 2019‐nCoV IgG/IgM | 1 (24/35) | 1 (10/11) | 1 (2/2) |
| LFA | 68.6a (50.7, 83.1)* |
90.9a (58.7, 99.8)* |
100a (15.8, 100)** |
|
| Render | COVID‐19 IgG/IgM Test | 1 (8/24) | 1 (18/28) | 1 (17/24) |
| LFA | 33.3a (15.6, 55.3)* |
64.3a (44.1, 81.4)* |
70.8a (48.9, 87.4)* |
|
| SD Biosensor | COVID‐19 IgG/IgM Duo | 2 (3/18) | 1 (24/37) | |
| LFA | 16.7 (5.5, 40.9) |
64.9a (47.5, 79.8)* |
||
| Servibio/VEDALAB | COVID‐19 Sign IgG/IgM | 1 (12/30) | 1 (12/17) | 1 (24/27) |
| LFA | 40.0a (22.7, 59.4)* |
70.6a (44.0, 89.7)* |
88.9a (70.8, 97.6)* |
|
| Shanghai Outdo Biotech | SARS‐CoV‐2 IgG/IgM GICA kit | 1 (22/40) | 1 (24/57) | 1 (23/47) |
| CGIA | 55.0a (38.5, 70.7)* |
42.1a (29.1, 55.9)* |
48.9a (34.1, 63.9)* |
|
| SIDAK Life Care | Sidak Covid 19 Antibody IgG/IgM Rapid Test Kit | 1 (2/23) | 1 (20/27) | 1 (30/36) |
| LFA | 8.7a (1.1, 28.0)* |
74.1a (53.7, 88.9)* |
83.3a (67.2, 93.6)* |
|
| Spring Healthcare Services | COVID‐19 Spring IgG/IgM Rapid Test Cassette | 1 (29/38) | 1 (36/44) | |
| CGIA | 76.3a (59.8, 88.6)* |
81.8a (67.3, 91.8)* |
||
| Sure Biotech | SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | 1 (24/63) | 1 (25/35) | 1 (15/16) |
| LFA | 38.1a (26.1, 51.2)* |
71.4a (53.7, 85.4)* |
93.8a (69.8, 99.8)* |
|
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgG/IgM | 2 (41/60) | 1 (10/14) | 3 (129/150) |
| LFA | 68.3 (55.6, 78.8) |
71.4a (41.9, 91.6)* |
85.0 (74.3, 91.8) |
|
| Syzbio Biotech | Syzbio SARS‐CoV‐2 IgG/IgM Antibody Assay Kit | 1 (14/22) | 1 (9/13) | 1 (6/10) |
| LFA | 63.6a (40.7, 82.8)* |
69.2a (38.6, 90.9)* |
60.0a (26.2, 87.8)* |
|
| T&D Diagnostics | Sienna 2019‐nCoV IgG/IgM Rapid Test | 1 (15/41) | 1 (39/48) | 1 (9/9) |
| LFA | 36.6a (22.1, 53.1)* |
81.3a (67.4, 91.1)* |
100a (66.4, 100)** |
|
| TAmiRNA | SARS‐CoV‐2 Antibody Lateral Flow Test IgG/IgM | 1 (49/58) | ||
| LFA | 84.5a (72.6, 92.7)* |
|||
| TONYAR Biotech | ASK COVID‐19 IgG/IgM Rapid Test | 2 (46/107) | 1 (54/73) | 2 (70/100) |
| LFA | 43.0 (34.0, 52.5) |
74.0a (62.4, 83.5)* |
69.0a (44.2, 86.1)* |
|
| UCP Biosciences | Coronavirus IgG/IgM Antibody (COVID‐19) Test Cassette |
1 (28/64) | 1 (27/37) | 1 (15/18) |
| LFA | 43.8a (31.4, 56.7)* |
73.0a (55.9, 86.2)* |
83.3a (58.6, 96.4)* |
|
| UNscience Biotechnology | COVID‐19 IgG/IgM Rapid Test Kit | 1 (2/9) | 1 (10/14) | 1 (13/15) |
| LFA | 22.2a (2.8, 60.0)* |
71.4a (41.9, 91.6)* |
86.7a (59.5, 98.3)* |
|
| VivaChek Biotech | VivaDiag COVID‐19 IgG/IgM Rapid Test | 3 (66/196) | 2 (76/116) | 2 (52/56) |
| CGIA | 34.8 (23.8, 47.8) |
65.5 (56.4, 73.6) |
93.0 (76.8, 98.1) |
|
| Beijing Wantai | Wantai Ab rapid assay | 1 (6/30) | 1 (20/25) | 1 (22/22) |
| LFA | 20.0a (7.7, 38.6)* |
80.0a (59.3, 93.2)* |
100a (84.6, 100)** |
|
| ZenTech | QuickZen COVID‐19 IgG/IgM | 1 (10/29) | 1 (48/62) | |
| CGIA | 34.5a (17.9, 54.3)* |
77.4a (65.0, 87.1)* |
||
| Zhejiang Orient‐Gene Biotech | COVID‐19 IgG/IgM rapid test cassette | 5 (79/154) | 5 (158/195) | 4 (123/130) |
| CGIA | 53.1 (38.0, 67.6) |
85.0 (69.6, 93.3) |
94.6 (89.1, 97.4) |
|
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG/IgM | 2 (8/45) | 2 (41/48) | 1 (14/15) |
| LFA | 17.8 (9.1, 31.7) |
84.9 (67.7, 93.8) |
93.3a (68.1, 99.8)* |
|
| ELISA | ||||
| Beijing Wantai | Wantai ELISA IgG/IgM assay | 1 (12/24) | 1 (64/80) | 1 (33/37) |
| 50.0a (29.1, 70.9)* | 80.0a (69.6, 88.1)* |
89.2a (74.6, 97.0)* |
||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgG/IgM assay | 1 (10/11) | ||
| 90.9a (58.7, 99.8)* |
||||
| Dia.Pro Diagnostic Bioprobes | DiaPro COVID‐19 IgG/IgM Confirmation | 1 (9/16) | 1 (18/20) | |
| 56.3a (29.9, 80.2)* |
90.0a (68.3, 98.8)* |
|||
| Epitope Diagnostics | EDITM Novel Coronavirus COVID‐19 IgG/IgM ELISA kit | 3 (22/96) | 3 (72/120) | 4 (77/79) |
| 18.5 (5.2, 48.5) |
60.0 (51.0, 68.4) |
97.5 (90.4, 99.4) |
||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgG/IgM ELISA | 1 (10/22) | 1 (72/92) | 1 (53/55) |
| 45.5 (24.4, 67.8) | 78.3 (68.4, 86.2) | 96.4 (87.5, 99.6) | ||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG/IgM ELISA | 2 (14/39) | 2 (150/202) | 1 (52/55) |
| 35.9 (22.5, 51.9) |
74.3 (67.8, 79.8) |
94.5a (84.9, 98.9)* |
||
| CLIA | ||||
| Bioscience | Bioscience Co (Chongqing) SARS‐CoVCoV‐2 IgG/IgM | 1 (34/67) | 1 (124/149) | 1 (131/134) |
| 50.7a (38.2, 63.2)* |
83.2a (76.2, 88.8)* |
97.8a (93.6, 99.5)* |
||
| Shenzhen YHLO | YHLO iFlash IgG/IgM assay | 1 (5/7) | 1 (8/14) | |
| 71.4a (29.0, 96.3)* |
57.1a (28.9, 82.3)* |
|||
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgG/IgM kits | 2 (39/99) | 2 (92/123) | 1 (42/44) |
| 33.0 (15.1, 57.7) |
74.8 (66.4, 81.7) |
95.5a (84.5, 99.4)* |
||
| Other/unclear | ||||
| ET Healthcare | Pylon 3D automated immunoassay system IgG/IgM | 1 (9/41) | 1 (29/33) | 1 (14/27) |
| 22.0a (10.6, 37.6)* |
87.9a (71.8, 96.6)* |
51.9a (31.9, 71.3)* |
||
| Quotient | MosaiQ COVID‐19 antibody microarray IgG/IgM | 1 (32/33) | ||
| 97.0a (84.2, 99.9)* |
||||
|
aEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval CGIA: colloidal gold immunoassay CI: confidence interval CLIA: chemiluminescence immunoassay ELISA: enzyme linked immusorbent assay LFA: lateral flow assay | ||||
Appendix 23. Sensitivity data by test brand (total Ab, by week after onset)
| Test name | Test name | Week 1 | Week 2 | Week 3 |
| ELISA |
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
|||
| Beijing Wantai | Wantai ELISA Total‐Ab assay | 7 (175/323) | 8 (304/342) | 6 (190/198) |
| 56.2 (44.5, 67.3) |
88.5 (79.5, 93.9) |
96.4 (89.9, 98.8) |
||
| Bio‐Rad Laboratories | BioRad Platelia SARS‐CoV‐2 Total Ab | 1 (19/21) | ||
| 90.5a (69.6, 98.8)* |
||||
| CLIA | ||||
| Beijing Wantai | Wantai CLIA Total‐Ab assay | 1 (17/31) | 1 (19/29) | 1 (38/38) |
| 54.8a (36.0, 72.7)* |
65.5a (45.7, 82.1)* |
100a (90.7, 100)* |
||
| Ortho Clinical Diagnostics | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay Total Ab | 1 (12/24) | 1 (8/8) | 1 (8/12) |
| 50.0a (29.1, 70.9)* |
100a (63.1, 100)** |
66.7a (34.9, 90.1)* |
||
| Roche Diagnostics | Elecsys anti‐SARS‐CoV‐2 antibody assay Total Ab | 13 (131/457) | 16 (380/544) | 18 (469/529) |
| 26.0 (16.2, 38.9) |
72.0 (64.7, 78.2) |
89.8 (85.3, 93.1) |
||
| Siemens Healthcare | Siemens Atellica Total‐Ab assay | 2 (15/42) | 2 (19/26) | 3 (40/53) |
| 35.7 (22.8, 51.1) |
73.0 (53.3, 86.6) |
75.5 (62.2, 85.2) |
||
| Watmind Medical | SARS‐CoV‐2 Ab Diagnostic Test Kit | 1 (14/38) | 1 (29/44) | |
| 36.8a (21.8, 54.0)* |
65.9a (50.1, 79.5)* |
|||
| Xiamen InnoDx Biotech | Novel coronavirus (2019‐nCoV) antibody test kit Total‐Ab | 1 (14/26) | 1 (67/70) | 1 (69/72) |
| 53.8a (33.4, 73.4)* |
95.7a (88.0, 99.1)* |
95.8a (88.3, 99.1)* |
||
| Xiamen Wantai | Total Ab CMIA | 1 (58/61) | 1 (70/72) | |
| 95.1a (86.3, 99.0)* |
97.2a (90.3, 99.7)* |
|||
| Other/unclear | ||||
| Luminex Corporation | SARS‐CoV‐2 MIA Total Ab |
1 (9/39) | 1 (26/40) | 1 (14/15) |
| 23.1a (11.1, 39.3)* |
65.0a (48.3, 79.4)* |
93.3a (68.1, 99.8)* |
||
|
aEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervalsCLIA: chemiluminescence immunoassayCMIA: chemiluminsent microparticle immunoassayELISA: enzyme linked immunosorbent assayMIA: magnetic immunoassay | ||||
Appendix 24. Sensitivity data by test brand (IgM alone, by week after onset)
| Test name | Test name | Week 1 | Week 2 | Week 3 |
| Test brand | Test brand | Test groups (true positives/COVID cases) | Sensitivity (95% CI) | |
| Lateral flow assays | ||||
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgM | 1 (12/18) | 1 (29/33) | |
| LFA | 66.7a (41.0, 86.7)* |
87.9a (71.8, 96.6)* |
||
| AccuBiotech | Accu‐Tell COVID‐19 IgM Cassette IgM | 1 (1/9) | 1 (11/14) | 1 (14/15) |
| CGIA | 11.1a (0.3, 48.2)* |
78.6a (49.2, 95.3)* |
93.3a (68.1, 99.8)* |
|
| ACON Laboratories | SARS‐COV‐2 IgM Rapid Test | 1 (54/58) | ||
| LFA | 93.1a (83.3, 98.1)* |
|||
| Acro Biotech | 2019‐nCoV IgM Rapid Test | 1 (0/9) | 1 (6/14) | 1 (11/15) |
| LFA | 0a (0, 33.6)** |
42.9a (17.7, 71.1)* |
73.3a (44.9, 92.2)* |
|
| Anhui Deep Blue | SARS‐CoV‐2 IgM Ab test kit | 1 (7/10) | ||
| LFA | 70.0a (34.8, 93.3)* |
|||
| Artron | Artron COVID‐19 IgM | 1 (25/90) | 1 (12/25) | 1 (23/24) |
| LFA | 27.8a (18.9, 38.2)* |
48.0a (27.8, 68.7)* |
95.8a (78.9, 99.9)* |
|
| Augurix Diagnostics | One Step Test for Novel Coronavirus (2019‐nCoV) IgM Antibody | 1 (2/18) | 1 (1/33) | |
| LFA | 11.1a (1.4, 35.7)* |
3.0a (0.1, 15.8)* |
||
| Augurix | SimtomaX Corona Check IgM | 1 (13/59) | ||
| LFA | 22.0a (12.3, 34.7)* |
|||
| Autobio Diagnostics | Anti‐SARS‐CoV‐2 Rapid Test IgM | 1 (46/100) | 1 (69/85) | 2 (64/78) |
| CGIA | 46.0a (36.0, 56.3)* |
81.2a (71.3, 88.8)* |
82.1 (71.9, 89.1) |
|
| Beijing Beier Bioengineerin | Beijing Beier Bioengineering – 2019 nCov IgM | 1 (5/10) | 1 (5/13) | 1 (9/14) |
| CGIA | 50.0a (18.7, 81.3)* |
38.5a (13.9, 68.4)* |
64.3a (35.2, 87.2)* |
|
| Beijing Diagreat Biotechnology | 2019‐nCoV IgM Antibody Determination Kit | 1 (30/38) | ||
| FIA | 78.9a (62.7, 90.4)* |
|||
| Biolidics | 2019 nCoV IgM detection kit | 1 (0/7) | 1 (11/34) | 1 (2/10) |
| LFA | 0a (0, 41.0)* |
32.4a (17.4, 50.5)* |
20.0a (2.5, 55.6)* |
|
| BioMedomics | COVID‐19 IgM Rapid Test | 3 (60/142) | 3 (86/115) | 2 (37/42) |
| LFA | 42.2 (34.4, 50.5) |
74.8 (60.2, 85.4) |
88.1 (74.4, 95.0) |
|
| bibioMerieux | Vidas SARS‐CoV‐2 IgM | 2 (21/57) | 2 (49/60) | 1 (26/26) |
| LFA | 36.8 (25.4, 50.0) |
81.7 (69.8, 89.5) |
100a (86.8, 100)** |
|
| Bioperfectus Technologies | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgM Rapid Test Kit | 1 (37/63) | 1 (28/34) | 1 (16/19) |
| LFA | 58.7a (45.6, 71.0)* |
82.4a (65.5, 93.2)* |
84.2a (60.4, 96.6)* |
|
| Biosynex | COVID‐19 BSS IgM | 1 (11/18) | 1 (37/45) | |
| LFA | 61.1a (35.7, 82.7)* |
82.2a (67.9, 92.0)* |
||
| BTNX | Rapid ResponseTM COVID‐19 Test Cassette (BTNX Inc.) Kit1 IgM | 1 (23/24) | 1 (44/103) | 2 (35/68) |
| LFA | 95.8a (78.9, 99.9)* |
42.7a (33.0, 52.8)* |
51.5 (39.7, 63.1) |
|
| Cellex | Cellex qSARS‐CoV‐ 2 IgM Cassette Rapid Test | 2 (12/28) | 2 (37/67) | 3 (40/58) |
| LFA | 41.8 (15.8, 73.4) |
58.9 (37.8, 77.2) |
69.8 (37.3, 90.0) |
|
| Chongqing iSIA BIO‐Technology | 2019‐nCoV IgM Diagnostic Test Kit | 1 (0/9) | 1 (7/14) | 1 (14/15) |
| LFA | 0a (0, 33.6)** |
50.0a (23.0, 77.0)* |
93.3a (68.1, 99.8)* |
|
| Clungene Biotech | Clungene SARS‐CoV‐2 IgM Rapid Test Cassettes | 1 (6/37) | 1 (33/78) | 1 (21/38) |
| LFA | 16.2a (6.2, 32.0)* |
42.3a (31.2, 54.0)* |
55.3a (38.3, 71.4)* |
|
| CTK Biotech | OnSite COVID‐19 IgM Rapid Test | 1 (47/57) | ||
| CGIA | 82.5a (70.1, 91.3)* |
|||
| DecombioBiotechnology | Novel Coronavirus (SARS‐CoV‐2) IgM Combo Rapid Test‐Cassette | 1 (32/62) | 1 (29/33) | 1 (14/18) |
| LFA | 51.6a (38.6, 64.5)* |
87.9a (71.8, 96.6)* |
77.8a (52.4, 93.6)* |
|
| DeepBlue Medical Technology |
COVID‐19 (SARS CoV‐2) IgM Antibody Test Kit (Colloidal Gold) | 1 (40/64) | 1 (28/34) | 1 (16/19) |
| CGIA | 62.5a (49.5, 74.3)* |
82.4a (65.5, 93.2)* |
84.2a (60.4, 96.6)* |
|
| DNA Link | AccuFind COVID‐19 IgM | 1 (35/48) | 1 (9/9) | |
| LFA | 72.9a (58.2, 84.7)* |
100a (66.4, 100)** |
||
| Dynamiker Biotechnology | Dynamiker Biotechnology ‐ 2019‐nCOV IgM Rapid Test | 2 (31/65) | 2 (105/153) | 1 (37/38) |
| LFA | 47.7 (35.9, 59.7) |
68.6 (60.9, 75.5) |
97.4a (86.2, 99.9)* |
|
| Genrui Biotech | Novel Coronavirus IgM test kit | 1 (8/10) | ||
| LFA | 80.0a (44.4, 97.5)* |
|||
| Guangdong Hecin Biotech | 2019‐nCoV IgM Antibody Test Kit | 1 (1/9) | 1 (11/14) | 1 (13/15) |
| LFA | 11.1a (0.3, 48.2)* |
78.6a (49.2, 95.3)* |
86.7a (59.5, 98.3)* |
|
| Hangzhou | Hangzhou unlabelled packaging IgM | 1 (7/51) | 1 (5/21) | 1 (4/8) |
| CGIA | 13.7a (5.7, 26.2)* |
23.8a (8.2, 47.2)* |
50.0a (15.7, 84.3)* |
|
| Hangzhou Alltest | 2019‐nCoV IgM Rapid Test Cassette | 5 (19/129) | 7 (31/153) | 7 (44/114) |
| CGIA | 14.7 (9.6, 21.9) |
19.8 (11.8, 31.5) |
37.0 (25.4, 50.2) |
|
| Hangzhou Biotest | RightSign IgM/IgG (also distributed as CoronaChek IgG and Premier Biotech Covid‐19 IgG) | 2 (50/126) | 2 (169/218) | 3 (72/78) |
| 38.6 (19.2, 62.5) |
77.5 (71.5, 82.6) |
92.3 (82.3, 96.9) |
||
| Hightop Biotech | Hightop SARS‐CoV‐2 IgM Antibody Rapid Test | 1 (5/51) | 1 (10/21) | 1 (6/8) |
| CGIA | 9.8a (3.3, 21.4)* |
47.6a (25.7, 70.2)* |
75.0a (34.9, 96.8)* |
|
| Innovita Biological Technology | 2019‐nCoV Ab test IgM | 7 (55/184) | 6 (84/143) | 6 (70/118) |
| CGIA | 29.9 (23.5, 37.2) |
58.0 (38.5, 75.3) |
60.0 (41.0, 76.5) |
|
| InTec | Rapid SARS‐CoV‐2 Antibody (IgM) Test | 1 (5/14) | 1 (43/47) | 1 (11/16) |
| CGIA | 35.7a (12.8, 64.9)* |
91.5a (79.6, 97.6)* |
68.8a (41.3, 89.0)* |
|
| Inzec | BIOZEK COVID‐19 IgG/IgM | 1 (5/29) | ||
| LFA | 17.2a (5.8, 35.8)* |
|||
| Jiangsu Medomics | Rapid IgM‐IgG Combined Antibody Test Kit for SARS‐ CoV‐2 IgM | 1 (6/12) | 1 (5/12) | 1 (15/20) |
| CGIA | 50.0a (21.1, 78.9)* |
41.7a (15.2, 72.3)* |
75.0a (50.9, 91.3)* |
|
| Lansion Biotechnology | (COVID‐19) IgM Test Kit | 1 (3/5) | 1 (8/11) | |
| FIA | 60.0a (14.7, 94.7)* |
72.7a (39.0, 94.0)* |
||
| Leccurate | SARS‐CoV‐2 antibody test IgM | 1 (9/14) | 1 (13/20) | 1 (15/20) |
| CGIA | 64.3a (35.1, 87.2)* |
65.0a (40.8, 84.6)* |
75.0a (50.9, 91.3)* |
|
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgM | 2 (17/61) | 2 (47/111) | 2 (45/72) |
| LFA | 27.5 (6.9, 66.0) |
46.9 (26.9, 68.1) |
63.6 (43.4, 80.0) |
|
| Maccura Biotechnology | Maccura LFIA SARS‐CoV‐2 IgM | 2 (16/49) | 1 (13/18) | 1 (14/14) |
| CGIA | 32.7 (21.1, 46.8) |
72.2a (46.5, 90.3)* |
100a (76.8, 100)** |
|
| MEDsan | MEDsan COVID‐19 IgM Rapid Test | 1 (49/64) | ||
| LFA | 76.6 (64.3, 86.2) | |||
| Multi‐G | Multi‐G MGA 2019‐nCoV IgM Rapid test cassette | 1 (10/37) | 1 (35/78) | 1 (22/38) |
| LFA | 27.0a (13.8, 44.1)* |
44.9a (33.6, 56.6)* |
57.9a (40.8, 73.7)* |
|
| Nal Von Minden | NADAL COVID‐19 IgM Test | 1 (7/17) | 1 (30/35) | |
| LFA | 41.2a (18.4, 67.1)* |
85.7a (69.7, 95.2)* |
||
| NG Biotech | NG‐Test IgM COVID‐19 | 3 (97/273) | 4 (172/219) | |
| CGIA | 36.5 (28.6, 45.1) | 78.6 (67.3, 86.8) | ||
| Nirmidas Biotech | Nirmidas COVID‐19 (SARS‐CoV‐2) IgM Antibody Detection Kit | 1 (46/73) | 1 (52/52) | 1 (19/19) |
| LFA | 63.0a (50.9, 74.0)* |
100a (93.2, 100)** |
100a (82.4, 100)* |
|
| Not stated | KHB Diagnostic Kit for SARS‐ CoV‐2 IgM Antibody | 1 (1/6) | ||
| CGIA | 16.7a (0.4, 64.1)* |
|||
| NTBIO® Diagnostics | NTBIO One Step Rapid Test ‐ COVID‐19 IgM Antibody Test | 1 (29/59) | ||
| 49.2a (35.9, 62.5)* |
||||
| Prima Lab | Prima COVID‐19 IgM Rapid test | 1 (16/37) | 1 (44/78) | 1 (26/38) |
| LFA | 43.2a (27.1, 60.5)* |
56.4a (44.7, 67.6)* |
68.4a (51.3, 82.5)* |
|
| Prometheus Bio | Prometheus Bio ‐ 2019‐nCoV IgM | 1 (20/35) | 1 (10/11) | 1 (2/2) |
| LFA | 57.1a (39.4, 73.7)* |
90.9a (58.7, 99.8)* |
100a (15.8, 100)** |
|
| Qingdao HIGHTOP Biotech | SARS‐CoV‐2 IgM Ab Rapid Test | 1 (55/64) | ||
| 85.9a (75.0, 93.4)* |
||||
| Render | COVID‐19 IgM Test | 1 (6/16) | 1 (13/28) | 1 (15/24) |
| LFA | 37.5a (15.2, 64.6)* |
46.4a (27.5, 66.1)* |
62.5a (40.6, 81.2)* |
|
| SD Biosensor | COVID‐19 IgM Duo | 4 (16/62) | 4 (84/130) | 2 (32/38) |
| LFA | 21.1 (6.1, 52.2) |
64.6 (56.0, 72.3) |
84.2 (69.0, 92.7) |
|
| Sensing Self | Covid‐19 Rapid IgM combined Antibody assay | 1 (4/28) | 1 (89/136) | 1 (21/21) |
| LFA | 14.3a (4.0, 32.7)* |
65.4a (56.8, 73.4)* |
100a (83.9, 100)** |
|
| SIDAK Life Care | Sidak Covid 19 Antibody IgM Rapid Test Kit | 1 (2/23) | 1 (11/27) | 1 (8/36) |
| LFA | 8.7a (1.1, 28.0)* |
40.7a (22.4, 61.2)* |
22.2a (10.1, 39.2)* |
|
| Sure Biotech | SARS‐CoV‐2 IgM Antibody Rapid Test | 1 (18/63) | 1 (22/34) | 2 (68/80) |
| LFA | 28.6a (17.9, 41.3)* |
64.7a (46.5, 80.3)* |
85.0 (75.4, 91.3) |
|
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgM | 1 (14/22) | 1 (10/14) | 1 (7/9) |
| LFA | 63.6a (40.7, 82.8)* |
71.4a (41.9, 91.6)* |
77.8a (40.0, 97.2)* |
|
| Syzbio Biotech | Syzbio SARS‐CoV‐2 IgM Antibody Assay Kit | 1 (14/22) | 1 (9/14) | 1 (6/9) |
| LFA | 63.6a (40.7, 82.8)* |
64.3a (35.1, 87.2)* |
66.7a (29.9, 92.5)* |
|
| T&D Diagnostics | Sienna 2019‐nCoV IgM Rapid Test | 1 (13/41) | 1 (33/48) | 1 (8/9) |
| LFA | 31.7a (18.1, 48.1)* |
68.8a (53.7, 81.3)* |
88.9a (51.8, 99.7)* |
|
| TBG Biotechnology | TBG SARS‐CoV‐2 IgM Rapid Test Kit | 1 (2/6) | 1 (31/46) | 1 (19/19) |
| LFA | 33.3a (4.3, 77.7)* |
67.4a (52.0, 80.5)* |
100a (82.4, 100)** |
|
| UCP Biosciences | Coronavirus IgM Antibody (COVID‐19) Test Cassette |
1 (28/64) | 1 (26/34) | 1 (15/19) |
| LFA | 43.8a (31.4, 56.7)* |
76.5a (58.8, 89.3)* |
78.9a (54.4, 93.9)* |
|
| UNscience Biotechnology | COVID‐19 IgM Rapid Test Kit | 1 (2/9) | 1 (5/14) | 1 (12/15) |
| LFA | 22.2a (2.8, 60.0)* |
35.7a (12.8, 64.9)* |
80.0a (51.9, 95.7)* |
|
| VivaChek Biotech | VivaDiag COVID‐19 IgM Rapid Test | 2 (42/97) | 2 (76/108) | 2 (52/57) |
| LFA | 43.3 (33.8, 53.3) |
74.8 (54.6, 88.0) |
91.7 (68.9, 98.2) |
|
| Wells Bio | careUS COVID‐19 IgM | 1 (0/3) | 1 (4/6) | 1 (6/6) |
| LFA | 0a (0, 70.8)** |
66.7a (22.3, 95.7)* |
100a (54.1, 100)** |
|
| Xiamen Biotime Biotechnology | SARS‐Cov‐2 IgM Rapid Qualitative Test | 1 (46/52) | ||
| LFA | 88.5a (76.6, 95.6)* | |||
| Zhejiang Orient‐Gene Biotech | COVID‐19 IgM rapid test cassette | 4 (63/115) | 5 (150/186) | 5 (125/139) |
| CGIA | 54.7 (43.4, 65.5) |
81.6 (72.0, 88.5) |
90.4 (82.1, 95.1) |
|
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgM | 2 (6/45) | 2 (31/48) | 1 (12/15) |
| LFA | 13.3 (6.1, 26.7) |
64.6 (50.2, 76.7) |
80.0a (51.9, 95.7)* |
|
| ELISA | ||||
| Ash Laboratories | Ash Laboratories SARS‐CoV2 IgM ELISA Immunoassay | 1 (1/10) | 1 (7/9) | 1 (7/9) |
| 10.0a (0.3, 44.5)* |
77.8a (40.0, 97.2)* |
77.8a (40.0, 97.2)* |
||
| Beijing Beier Bioengineerin | Beijing Beier Bioengineering – 2019 nCov IgM | 1 (4/10) | 1 (1/13) | 1 (6/14) |
| 40.0a (12.2, 73.8)* |
7.7a (0.2, 36.0)* |
42.9a (17.7, 71.1)* |
||
| Beijing Wantai | Wantai ELISA IgM assay | 4 (58/187) | 4 (247/315) | 2 (52/59) |
| 31.0 (24.8, 38.0) |
80.4 (71.6, 87.0) |
94.5 (43.0, 99.7) |
||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgM assay | 1 (7/11) | ||
| 63.6a (30.8, 89.1)* |
||||
| Creative Diagnostics | SARS‐CoV‐2 IgM ELISA Kit | 1 (12/19) | 1 (37/38) | |
| 63.2a (38.4, 83.7)* |
97.4a (86.2, 99.9)* |
|||
| Epitope Diagnostics | EDITM Novel Coronavirus COVID‐19 IgM ELISA kit | 5 (43/217) | 7 (205/381) | 7 (127/163) |
| 10.7 (3.1, 30.9) |
58.3 (43.9, 71.4) |
88.8 (69.6, 96.5) |
||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgM ELISA | 2 (30/62) | 1 (64/92) | 1 (53/55) |
| 48.4 (36.3, 60.7) |
69.6a (59.1, 78.7)* |
96.4a (87.5, 99.6)* |
||
| ImmunoDiagnostics | SARS‐CoV‐2 NP IgM ELISA Kit | 1 (10/19) | 1 (34/38) | |
| 52.6a (28.9, 75.6)* |
89.5a (75.2, 97.1)* |
|||
| Theradiag | ELISA COVID‐19 THERA02 IgM assay | 1 (11/22) | 1 (7/14) | 1 (6/9) |
| 50.0a (28.2, 71.8)* |
50.0a (23.0, 77.0)* |
66.7a (29.9, 92.5)* |
||
| Vazyme Biotech | 2019‐nCoV IgM Detection Kit | 1 (2/18) | 1 (4/22) | 1 (4/18) |
| 11.1a (1.4, 34.7)* |
18.2a (5.2, 40.3)* |
22.2a (6.4, 47.6)* |
||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgM ELISA | 4 (29/93) | 3 (188/288) | 2 (95/112) |
| 31.2 (22.6, 41.3) |
65.3 (59.6, 70.6) |
84.8 (76.9, 90.4) |
||
| CLIA | ||||
| Autobio Diagnostics | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | 1 (34/66) | 1 (58/70) | |
| 51.5a (38.9, 64.0)* |
82.9a (72.0, 90.8)* |
|||
| Beijing Beier Bioengineerin | Beijing Beier Bioengineering – 2019 nCov IgM | 1 (4/10) | 1 (4/13) | 1 (6/14) |
| 40.0a (12.2, 73.8)* |
30.8a (9.1, 61.4)* |
42.9a (17.7, 71.1)* |
||
| Bioscience | Bioscience Co (Chongqing) SARS‐CoVCoV‐2 IgM | 2 (28/88) | 2 (175/269) | 2 (167/217) |
| 31.8 (23.0, 42.2) |
65.1 (59.2, 70.5) |
76.2 (60.4, 87.0) |
||
| Beijing Wantai | Wantai CLIA IgM assay | 1 (8/31) | 1 (12/29) | 1 (26/38) |
| 25.8a (11.9, 44.6)* |
41.4a (23.5, 61.1)* |
68.4a (51.3, 82.5)* |
||
| Hangzhou Biotest | The RightSignTM COVID‐19 IgM Rapid Test Cassette / Lumiratek | 1 (33/36) | ||
| 91.7a (77.5, 98.2)* |
||||
| MEXACARE | QuickTestCorona COVID‐19 IgM | 1 (49/61) | ||
| 80.3a (68.2, 89.4)* |
||||
| Shenzhen YHLO | YHLO iFlash IgM assay | 6 (76/135) | 5 (144/188) | 4 (55/73) |
| 48.1 (20.7, 76.7) |
77.0 (48.7, 92.2) |
75.3 (64.2, 83.9) |
||
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgM kits | 7 (52/197) | 7 (205/327) | 4 (123/147) |
| 23.8 (14.1, 37.3) |
60.9 (51.7, 69.4) |
83.4 (74.3, 89.7) |
||
| Xiamen InnoDx Biotech | Novel coronavirus (2019‐nCoV) antibody test kit IgM | 1 (9/26) | 1 (45/70) | 1 (62/72) |
| 34.6a (17.2, 55.7)* |
64.3a (51.9, 75.4)* |
86.1a (75.9, 93.1)* |
||
| Other/unclear | ||||
| ET Healthcare | Pylon 3D automated immunoassay system IgM | 1 (6/41) | 1 (23/42) | 1 (13/15) |
| 14.6a (5.6, 29.2)* |
54.8a (38.7, 70.2)* |
86.7a (59.5, 98.3)* |
||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgM (S‐based) | 1 (7/48) | ||
| 14.6 (6.1, 27.8) | ||||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgM (N‐based) | 1 (7/48) | ||
| 14.6 (6.1, 27.8) | ||||
| Vibrant America | Vibrant COVID‐19 Ab IgM | 1 (322/330) | 1 (330/330) | |
| 97.6a (95.3, 98.9)* |
100a (98.9, 100)** |
|||
|
aEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervals CGIA: colloidal gold immunoassayCLIA: chemiluminescence immunoassay DNA: deoxyribonucleic acidELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assayMIA: magnetic immunoassay RT‐PCR: reverse transcription polymerase chain reaction | ||||
Appendix 25. Specificity data by test brand (IgG alone, all other reference groups)
| Test name | Test name | Suspected of COVID‐19 | Current RT‐PCR‐negative | Current untested | Other/mixed unclear | Cross‐reactivity/confounder panels |
| Test brand | Test brand |
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
||||
| Lateral flow/colloidal gold | ||||||
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgG | 1 (63/64) | ||||
| LFA | 98.4a (91.6, 100)* |
|||||
| ACON Laboratories | SARS‐COV‐2 IgG Rapid Test | 1 (29/31) | ||||
| LFA | 93.5a (78.6, 99.2)* |
|||||
| Alfa Scientific | CLARITY COVID‐19 IgG Antibody Test | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Augurix | Augurix SARS‐CoV‐2 IgG RDT | 1 (44/45) | ||||
| LFA | 97.8a (88.2, 99.9)* |
|||||
| Augurix | SimtomaX Corona Check IgG | 1 (106/107) | ||||
| LFA | 99.1a (94.9, 100)* |
|||||
| Avioq Bio‐Tech | Novel Coronavirus (2019‐nCov) Antibody IgG Assay Kit | 1 (69/72) | ||||
| CGIA | 95.8a (88.3, 99.1)* |
|||||
| AYTU Biosciences | AYTU COVID‐19 IgG Rapid Test Cassette | 1 (59/60) | ||||
| LFA | 98.3a (91.1, 100)* |
|||||
| Beijing Diagreat | 2019‐nCoV IgG Antibody Determination Kit | 1 (157/160) | 1 (573/600) | |||
| FIA | 98.1a (94.6, 99.6)* |
95.5a (93.5, 97.0)* |
||||
| BioMedomics | COVID‐19 IgG Rapid Test | 1 (47/51) | ||||
| LFA | 92.1a (81.1, 97.8)* |
|||||
| BioMerieux | Vidas SARS‐CoV‐2 IgG | 1 (259/261) | ||||
| FIA | 99.2a (97.3, 99.9)* |
|||||
| Bioperfectus | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgG Rapid Test Kit | 1 (39/45) | ||||
| LFA | 86.7a (73.2, 94.9)* |
|||||
| Biosynex | COVID‐19 BSS IgG | 1 (27/27) | ||||
| LFA | 100a (87.2, 100)** |
|||||
| BTNX | Rapid ResponseTM COVID‐19 Test Cassette (BTNX Inc.) Kit1 IgG | 1 (31/31) | ||||
| LFA | 100a (88.8, 100)** |
|||||
| Cellex | Cellex qSARS‐CoV‐ 2 IgG Cassette Rapid Test | 1 (79/79) | ||||
| LFA | 100a (95.4, 100)** |
|||||
| Clungene Biotech | Clungene SARS‐CoV‐2 IgG Rapid Test Cassettes | 1 (101/103) | ||||
| LFA | 98.1a (93.2, 99.8)* |
|||||
| CTK Biotech | OnSite COVID‐19 IgG Rapid Test | 1 (100/102) | 1 (91/92) | |||
| CGIA | 98.0a (93.1, 99.8)* |
98.9a (94.1, 100)* |
||||
| DecombioBiotechnology | Novel Coronavirus (SARS‐CoV‐2) IgG Combo Rapid Test‐Cassette | 1 (49/51) | ||||
| LFA | 96.1a (86.5, 99.5)* |
|||||
| DeepBlue Medical | COVID‐19 (SARS CoV‐2) IgG Antibody Test Kit (Colloidal Gold) | 1 (44/51) | ||||
| CGIA | 86.3a (73.7, 94.3)* |
|||||
| DiaCarta | QuantiVirus anti‐SARS‐CoV‐2 IgG test | 1 (88/88) | ||||
| FIA | 100a (95.9, 100)** |
|||||
| DNA Link | AccuFind COVID19 IgG | 1 (59/59) | ||||
| LFA | 100a (93.9, 100)** |
|||||
| Dynamiker Biotechnology | Dynamiker Biotechnology ‐ 2019‐nCOV IgG Rapid Test | 1 (102/103) | ||||
| LFA | 99.0a (94.7, 100)* |
|||||
| Hangzhou | Hangzhou unlabelled packaging IgG | 1 (90/92) | ||||
| CGIA | 97.8a (92.4, 99.7)* |
|||||
| Hangzhou Alltest | 2019‐nCoV IgG Rapid Test Cassette | 2 (192/192) | 1 (60/60) | |||
| LFA | 100a (98.1, 100)** |
100a (94.0, 100)** |
||||
| Hangzhou Biotest | RightSign IgG (Also distributed as CoronaChek IgG and Premier Biotech Covid‐19 IgG) | 1 (60/65) | 2 (110/111) | |||
| LFA | 92.3a (83.0, 97.5)* |
99.1 (93.9, 99.9) |
||||
| Hightop Biotech | Hightop SARS‐CoV‐2 IgG Antibody Rapid Test | 1 (92/92) | ||||
| CGIA | 100a (96.1, 100)** |
|||||
| Innovita Biological | 2019‐nCoV Ab test IgG | 1 (26/28) | ||||
| LFA | 92.9a (76.5, 99.1)* |
|||||
| InTec | Rapid SARS‐CoV‐2 Antibody (IgG) Test | 1 (76/79) | ||||
| CGIA | 96.2a (89.3, 99.2)* |
|||||
| Intelligent Endoscopy | Smart screen IgG | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Inzec | BIOZEK COVID‐19 IgG/IgM | 1 (51/55) | ||||
| LFA | 92.7a (82.4, 98.0)* |
|||||
| LabOn Time | LaboOn Time rapid test cassette IgG | 1 (72/72) | ||||
| LFA | 100a (95.0, 100)** |
|||||
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgG | 1 (102/103) | ||||
| LFA | 99.0a (94.7, 100)* |
|||||
| MEDsan GmbH | MEDsan COVID‐19 IgG Rapid Test | 1 (101/110) | 1 (49/50) | |||
| LFA | 91.8a (85.0, 96.2) |
98.0a (89.4, 99.9)* |
||||
| Multi‐G | Multi‐G MGA 2019‐nCoV IgG Rapid test cassette | 1 (100/103) | ||||
| LFA | 97.1a (91.7, 99.4)* |
|||||
| Multi‐G | 2019‐nCoV IgG Rapid Test Cassette dual lane | 1 (38/39) | ||||
| LFA | 97.4a (86.5, 99.9)* |
|||||
| Multi‐G | 2019‐nCoV IgG Rapid Test Cassette single lane | 1 (39/39) | ||||
| LFA | 100a (91.0, 100)** |
|||||
| NG Biotech | NG‐Test IgG COVID‐19 | 1 (53/55) | 1 (28/28) | |||
| CGIA | 96.4a (87.5, 99.6)* |
100a (87.7, 100)* |
||||
| Nirmidas Biotech | Nirmidas COVID‐19 (SARS‐CoV‐2) IgG Antibody Detection Kit | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| NTBIO® Diagnostics | NTBIO One Step Rapid Test ‐ COVID‐19 IgG Antibody Test | 1 (98/108) | ||||
| 90.7a (83.6, 95.5)* |
||||||
| Prima Lab | Prima COVID‐19 IgG Rapid test | 1 (93/103) | ||||
| LFA | 90.3a (82.9, 95.2)* |
|||||
| Prometheus Bio | Prometheus Bio ‐ 2019‐nCoV IgG | 1 (24/40) | ||||
| LFA | 60.0a (43.3, 75.1)* |
|||||
| Qingdao HIGHTOP | SARS‐CoV‐2 IgG Ab Rapid Test | 1 (111/111) | ||||
| 100a (96.7, 100)** |
||||||
| Safecare Biotech | SafeCare Bio‐Tech COVID‐19 IgG Rapid Test Device | 1 (54/60) | ||||
| LFA | 90.0a (79.5, 96.2)* |
|||||
| SD Biosensor | COVID‐19 IgG Duo | 1 (240/242) | ||||
| LFA | 99.2a (97.0, 99.9)* |
|||||
| Sensing Self | Covid‐19 Rapid IgG combined Antibody assay | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Servibio/VEDALAB | COVID‐19 Sign IgG | 1 (27/27) | ||||
| LFA | 100a (87.2, 100)** |
|||||
| Sure Biotech | SARS‐CoV‐2 IgG Antibody Rapid Test | 1 (107/110) | 1 (51/51) | |||
| LFA | 97.3a (92.2, 99.4)* |
100a (93.0, 100)** |
||||
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgG | 1 (38/39) | ||||
| LFA | 97.4a (86.5, 99.9)* |
|||||
| T&D Diagnostics | Sienna 2019‐nCoV IgG Rapid Test | 1 (37/44) | ||||
| LFA | 84.1a (69.9, 93.4)* |
|||||
| TBG Biotechnology | TBG SARS‐CoV‐2 IgG Rapid Test Kit | 1 (57/59) | ||||
| LFA | 96.6a (88.3, 99.6* |
|||||
| UCP Biosciences | Coronavirus IgG Antibody (COVID‐19) Test Cassette |
1 (49/51) | ||||
| LFA | 96.1a (86.5, 99.5)* |
|||||
| Unknown manufacturer | Ready Result IgG | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| VivaChek Biotech | VivaDiag COVID‐19 IgG Rapid Test | 2 (193/195) | 1 (47/48) | |||
| CGIA | 99.0 (96.0, 99.7) |
97.9a (88.9, 99.9)* |
||||
| W.H.P.M. | COVISURE COVID‐19 IgG Rapid Test | 1 (57/59) | ||||
| LFA | 96.6a (88.3, 99.6)* |
|||||
| Xiamen Biotime | SARS‐Cov‐2 IgG Rapid Qualitative Test | 1 (91/96) | ||||
| LFA | 94.8a (88.3, 98.3)* |
|||||
| Zeus Scientific | ZEUS SARS‐CoV‐2 IgG/IgM rapid test | 1 (60/60) |
||||
| LFA | 100a (94.0, 100)** |
|||||
| Zhejiang Orient‐Gene | COVID‐19 IgG rapid test cassette | 1 (39/39) | 1 (49/50) | 1 (96/103) | 1 (78/79) | |
| LFA | 100a (91.0, 100)** |
98.0a (89.4, 99.9)* |
93.2a (86.5, 97.2)* |
98.7a (93.1, 100)* |
||
| ELISA | ||||||
| Ash Laboratories | Ash Laboratories SARS‐CoV2 IgG ELISA Immunoassay | 1 (36/38) | 1 (19/19) | |||
| 94.7a (82.3, 99.4)* |
100a (82.4, 100)** |
|||||
| Beijing Wantai | Wantai ELISA IgG assay | 1 (100/100) | 1 (145/146) | |||
| 100a (96.4, 100)** |
99.3a (96.2, 100)* |
|||||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgG assay | 1 (59/62) | ||||
| 95.2a (86.5, 99.0)* |
||||||
| Epitope Diagnostics | EDI Novel Coronavirus COVID‐19 IgM ELISA kit | 1 (107/110) | 1 (104/105) | 1 (21/25) | 5 (169/178) | |
| 97.3a (92.2, 99.4)* |
99.0a (94.8, 100)* |
84.0a (63.9, 95.5)* |
97.1 (87.8, 99.4) |
|||
| Euroimmun | anti‐SARS‐COV‐2 IgG | 5 (478/499) | 1 (101/105) | 5 (321/325) | 8 (523/537) | 16 (915/.938) |
| 95.8 (93.6, 97.2) |
96.2 (90.5, 99.0) |
98.8 (96.8, 99.5) |
97.4 (95.6, 98.5) |
97.7 (96.1, 98.7) |
||
| Euroimmun | anti‐SARS‐COV‐2 IgG (N‐based) | 1 (107/113) | ||||
| 94.7a (88.8, 98.0)* |
||||||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgG ELISA | 1 (100/100) | ||||
| 100a (96.4, 100)** |
||||||
| Mikrogen | Mikrogen IgG anti‐N | 1 (109/113) | ||||
| 96.5a (91.2, 99.0)* |
||||||
| Vircell | COVID‐19 ELISA IgG | 1 (19/19) | ||||
| 100a (82.4, 100)** |
||||||
| Virotech | Virotech SARS‐CoV‐2 ELISA IgG | 1 (31/31) | ||||
| 100a (88.8, 100)** |
||||||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG ELISA | 1 (57/60) | 2 (218/220) | |||
| 95.0a (86.1, 99.0)* |
99.1 (96.4, 99.8) |
|||||
| CLIA | ||||||
| Abbott Diagnostics | Abbott Alinity anti‐SARS‐CoV‐2 nucleocapsid IgG | 2(134/135) | ||||
| 99.3 (94.9, 99.9) |
||||||
| Abbott Diagnostics | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG | 5 (382/394) | 5 (673/675) | 1 (239/243) | 1 (73/73) | 13 (1060/1070) |
| 98.2 (92.0, 99.6) |
99.7 (98.8, 99.9) |
98.4a (95.8, 99.5)* |
100a (95.1, 100)** |
99.1 (97.9, 99.6) |
||
| Autobio Diagnostics | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | 1 (3777/3789) | ||||
| 99.7a (99.4, 99.8)* |
||||||
| Beckman Coulter | Beckman Coulter ‐ Access SARS‐CoV‐2 IgG | 1 (62/62) | 1 (97/100) | |||
| 100a (94.2, 100)** |
97.0a (91.5, 99.4)* |
|||||
| DiaSorin | Diasorin LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA | 1 (179/183) | 2 (184/186) | 1 (29/30) | 10 (642/671) | |
| 97.8a (94.5, 99.4)* |
98.9 (95.8, 99.7) |
96.7a (82.8, 99.9)* |
97.9 (94.2, 99.3) |
|||
| Diazyme | Diazyme DZ‐LITE 2019‐nCoV IgG (CLIA) Assay Kit | 1 (49/49) | ||||
| 100a (92.7, 100)** |
||||||
| Hunan Yuanjing | COVID‐19 IgG Detection Kits | 1 (39/40) | ||||
| 97.5a (86.8, 99.9)* |
||||||
| MEXACARE GmbH | QuickTestCorona COVID‐19 IgG | 1 (108/109) | ||||
| 99.1a (95.0, 100)* |
||||||
| Ortho Clinical Diagnostics | Vitros (VITROS) Anti‐SARS‐Cov‐2 Total assay IgG | 1 (105/105) | 2 (174/175) | |||
| 100a (96.5, 100)** |
99.4 (96.1, 99.9) |
|||||
| Shenzhen YHLO | YHLO iFlash IgG assay | 1 (30/33) | 2 (1053/1082) | 1 (581/586) | 1 (74/75) | |
| 90.9a (75.7, 98.1)* |
97.3a (96.2, 98.1)* |
99.1a (98.0, 99.7)* |
98.7a (92.8, 100)* |
|||
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgG kits | 1 (10/11) | 1 (72/72) | 4 (207/211) | ||
| 90.9a (58.7, 99.8)* |
100a (95.0, 100)** |
98.6 (85.2, 99.9) |
||||
| Vircell | COVID‐19 VIRCLIA IgG MONOTEST | 1 (31/33) | ||||
| 93.9a (79.8, 99.3)* |
||||||
| Unclear/unknown | ||||||
| Vibrant America | Vibrant COVID‐19 Ab IgG | 1 (5250/5262) | ||||
| 99.8a (99.6, 99.9)* |
||||||
|
aEstimates and confidence intervals by summing the counts of true‐negative and false‐positive across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervals CGIA: colloidal gold immunoassayCLIA: chemiluminescence immunoassay DNA: deoxyribonucleic acidELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assayMIA: magnetic immunoassay RT‐PCR: reverse transcription polymerase chain reaction | ||||||
Appendix 26. Specificity data by test brand (IgG or IgM, all other reference groups)
| Test name | Test name | Suspected of COVID‐19 | Current RT‐PCR‐negative | Current untested | Other/mixed unclear | Cross‐reactivity/confounder panels |
| Test brand | Test brand |
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
||||
| Lateral flow/colloidal gold | ||||||
| AAZ‐LMB | Covid‐Duo test rapid Covid‐19 IgG/IgM | 1 (71/71) | ||||
| CGIA | 100a (94.9, 100)** |
|||||
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgG/IgM | 1 (72/72) | 1 (63/64) | |||
| CGIA | 100a (95.0, 100)** |
98.4a (91.6, 100)* |
||||
| Alfa Scientific | CLARITY COVID‐19 IgG/IgM Antibody Test | 1 (54/60) | ||||
| LFA | 90.0a (79.5, 96.2)* |
|||||
| Assure Tech | FaStep (COVID‐19 IgG/IgM) rapid test cassettes | 1 (13/13) | ||||
| LFA | 100a (75.3, 100)** |
|||||
| Avioq Bio‐Tech | Novel Coronavirus (2019‐nCov) Antibody IgG/IgM Assay Kit | 1 (69/72) | ||||
| CGIA | 95.8a (88.3, 99.1)* |
|||||
| AYTU Biosciences | AYTU COVID‐19 IgG/IgM Rapid Test Cassette | 1 (59/60) | ||||
| LFA | 98.3a (91.1, 100)* |
|||||
| BioMedomics | COVID‐19 IgG/IgM Rapid Test | 1 (40/51) | ||||
| LFA | 78.4a (64.7, 88.7)* |
|||||
| Bioperfectus Technologies | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgG/IgM Rapid Test Kit | 1 (37/45) | ||||
| LFA | 82.2a (67.9, 92.0)* |
|||||
| Cellex | Cellex qSARS‐CoV‐ 2 IgG/IgM Cassette Rapid Test | 1 (79/79) | ||||
| LFA | 100a (95.4, 100)* |
|||||
| Clungene Biotech | Clungene SARS‐CoV‐2 IgG/IgM Rapid Test Cassettes | 1 (93/103) | ||||
| LFA | 90.3a (82.9, 95.2)* |
|||||
| CTK Biotech | OnSite COVID‐19 IgG/IgM Rapid Test | 1 (88/92) | ||||
| CGIA | 95.7a (89.2, 98.8)* |
|||||
| DecombioBiotechnology | Novel Coronavirus (SARS‐CoV‐2) IgG/IgM Combo Rapid Test‐Cassette | 1 (45/51) | ||||
| LFA | 88.2a (76.1, 95.6)* |
|||||
| DeepBlue Medical | COVID‐19 (SARS CoV‐2) IgG/IgM Antibody Test Kit (Colloidal Gold) | 1 (31/57) | ||||
| CGIA | 54.4a (40.7, 67.6)* |
|||||
| DNA Link | AccuFind COVID19 IgG/IgM | 1 (47/59) | ||||
| LFA | 79.7a (67.2, 89.0)* |
|||||
| Dynamiker Biotechnology | Dynamiker Biotechnology ‐ 2019‐nCOV IgG/IgM Rapid Test | 2 (178/178) | 1 (98/103) | 1 (74/74) | ||
| LFA | 100a (97.9, 100)** |
95.1a (89.0, 98.4)* |
100a (95.1, 100)** |
|||
| Guangzhou Wondfo | Wondfo SARS‐CoV‐2 Antibody | 3 (212/222) | 1 (244/245) | 1 (90/92) | 3 (172/173) | |
| CGIA | 95.5a (91.9, 97.8)* |
99.6a (97.7, 100)* |
97.8a (92.4, 99.7)* |
99.4 (96.0, 99.9) |
||
| Hangzhou | Hangzhou unlabelled packaging IgG/IgM | 1 (87/92) | ||||
| CGIA | 94.6a (87.8, 98.2)* |
|||||
| Hangzhou Alltest | 2019‐nCoV IgG/IgM Rapid Test Cassette | 1 (58/58) | 1 (89/92) | 1 (58/60) | ||
| LFA | 100a (93.8, 100)** |
96.7a (90.8, 99.3)* |
96.7a (88.5, 99.6)* |
|||
| Hangzhou Biotest | RightSign IgM/IgG (also distributed as CoronaChek IgG and Premier Biotech Covid‐19 IgG) | 2 (109/111) | ||||
| LFA | 98.3 (89.9, 99.8) | |||||
| Hightop Biotech | Hightop SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | 1 (92/92) | ||||
| CGIA | 100a (96.1, 100)** |
|||||
| Innovita Biological | 2019‐nCoV Ab test IgG/IgM | 1 (25/28) | ||||
| LFA | 89.3a (71.8, 97.7)* |
|||||
| InTec | Rapid SARS‐CoV‐2 Antibody (IgG/IgM) Test | 1 (67/79) | ||||
| CGIA | 84.8a (75.0, 91.9)* |
|||||
| Intelligent Endoscopy | Smart screen IgG/IgM | 1 (55/60) | ||||
| LFA | 91.7a (81.6, 97.2)* |
|||||
| LabOn Time | LaboOn Time rapid test cassette IgG/IgM | 1 (72/72) | ||||
| LFA | 100a (95.0, 100)** |
|||||
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgG/IgM | 1 (101/103) | ||||
| LFA | 98.1a (93.2, 99.8)* |
|||||
| Maccura Biotechnology | Maccura LFIA SARS‐CoV‐2 IgG/IgM | 1 (67/76) | ||||
| LFA | 88.2a (78.7, 94.4)* |
|||||
| Multi‐G | Multi‐G MGA 2019‐nCoV IgG/IgM Rapid test cassette | 1 (91/103) | ||||
| LFA | 88.3a (80.5, 93.8)* |
|||||
| Multi‐G | 2019‐nCoV IgG/IgM Rapid Test Cassette dual lane | 1 (38/39) | ||||
| LFA | 97.4a (86.5, 99.9)* |
|||||
| Multi‐G | 2019‐nCoV IgG/IgM Rapid Test Cassette single lane | 1 (37/39) | ||||
| LFA | 94.9a (82.7, 99.4)* |
|||||
| NG Biotech | NG‐Test IgG/IgM COVID‐19 | 1 (4/4) | 1 (22/22) | 1 (50/50) | ||
| CGIA | 100a (39.7, 100)** |
100a (84.6, 100)** |
100a (92.9, 100)** |
|||
| Nirmidas Biotech | Nirmidas COVID‐19 (SARS‐CoV‐2) IgG/IgM Antibody Detection Kit | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Prima Lab | Prima COVID‐19 IgG/IgM Rapid test | 1 (88/103) | ||||
| LFA | 85.4a (77.1, 91.6)* |
|||||
| Prometheus Bio | Prometheus Bio ‐ 2019‐nCoV IgG/IgM | 1 (5/40) | ||||
| LFA | 12.5a (4.2, 26.8)* |
|||||
| Safecare Biotech | SafeCare Bio‐Tech COVID‐19 IgG/IgM Rapid Test Device | 1 (54/60) | ||||
| LFA | 90.0a (79.5, 96.2)* |
|||||
| SD Biosensor | COVID‐19 IgG/IgM Duo | 1 (123/126) | ||||
| LFA | 97.6a (93.2, 99.5)* |
|||||
| Sensing Self | Covid‐19 Rapid IgG/IgM combined Antibody assay | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Shanghai Outdo | SARS‐CoV‐2 IgG/IgM GICA kit | 1 (51/53) | ||||
| CGIA | 96.2a (87.0, 99.5)* |
|||||
| Sure Biotech Kong |
SARS‐CoV‐2 IgG/IgM Antibody Rapid Test | 1 (51/51) | ||||
| LFA | 100a (93.0, 100)** |
|||||
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgG/IgM | 1 (39/39) | ||||
| LFA | 100a (91.0, 100)** |
|||||
| T&D Diagnostics | Sienna 2019‐nCoV IgG/IgM Rapid Test | 1 (33/44) | ||||
| LFA | 75.0a (59.7, 86.8)* |
|||||
| TAmiRNA | SARS‐CoV‐2 Antibody Lateral Flow Test IgG/IgM | 1 (103/106) | ||||
| LFA | 97.2a (92.0, 99.4)* |
|||||
| TBG Biotechnology | TBG SARS‐CoV‐2 IgG/IgM Rapid Test Kit | 1 (52/59) | ||||
| LFA | 88.1a (77.1, 95.1)* |
|||||
| TONYAR Biotech | ASK COVID‐19 IgG/IgM Rapid Test | 2 (178/178) | 1 (74/74) | |||
| LFA | 100a (97.9, 100)** |
100a (95.1, 100)** |
||||
| UCP Biosciences | Coronavirus IgG/IgM Antibody (COVID‐19) Test Cassette |
1 (48/51) | ||||
| LFA | 94.1a (83.8, 98.8)* |
|||||
| Unknown manufacturer | Ready Result IgG/IgM | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| VivaChek Biotech | VivaDiag COVID‐19 IgG/IgM Rapid Test | 1 (565/578) | 1 (89/93) | 3 (845/866) | 1 (44/48) | |
| CGIA | 97.8a (96.2, 98.8)* |
95.7a (89.4, 98.8)* |
97.6 (96.3, 98.4) |
91.7a (80.0, 97.7)* |
||
| W.H.P.M. | COVISURE COVID‐19 IgG/IgM Rapid Test | 1 (56/59) | ||||
| LFA | 94.9a (85.9, 98.9)* |
|||||
| Wantai Biological | Wantai Ab rapid assay | 1 (98/100) | ||||
| LFA | 98.0a (93.0, 99.8)* |
|||||
| Zeus Scientific | ZEUS SARS‐CoV‐2 IgG/IgM rapid test | 1 (58/60) | ||||
| LFA | 96.7a (88.5, 99.6)* |
|||||
| Zhejiang Orient‐Gene | COVID‐19 IgG/IgM rapid test cassette | 1 (126/129) | 1 (38/39) | 1 (94/103) | 1 (69/79) | |
| CGIA | 97.7a (93.4, 99.5)* |
97.4a (86.5, 99.9)* |
91.3a (84.1, 95.9)* |
87.3a (78.0, 93.8)* |
||
| ELISA | ||||||
| Beijing Wantai | Wantai ELISA IgG/IgM assay | 1 (143/146) | ||||
| 97.9a (94.1, 99.6)* |
||||||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgG/IgM assay | 1 (58/62) | ||||
| 93.5a (84.3, 98.2)* |
||||||
| Epitope Diagnostics | EDI Novel Coronavirus COVID‐19 IgG/IgM ELISA kit | 1 (60/62) | ||||
| 96.8a (88.8, 99.6)* |
||||||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgG/IgM ELISA | 1 (100/100) | ||||
| 100a (96.4, 100)* |
||||||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgG/IgM ELISA | 2 (216/220) | ||||
| 98.8 (88.5, 99.9) |
||||||
| CLIA | ||||||
| Bioscience | Bioscience Co (Chongqing) SARS‐CoVCoV‐2 IgG/IgM | 1 (141/148) | ||||
| 95.3a (90.5, 98.1)* |
||||||
| Hunan Yuanjing | COVID‐19 IgG/IgM Detection Kits | 1 (39/40) | ||||
| 97.5a (86.8, 99.9)* |
||||||
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgG/IgM kits | 1 (72/72) | 1 (20/20) | |||
| 100a (95.0, 100)** |
100a (83.2, 100)** |
|||||
|
aEstimates and confidence intervals by summing the counts of true negative and false positive across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervals CGIA: colloidal gold immunoassayCLIA: chemiluminescence immunoassay DNA: deoxyribonucleic acidELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assayMIA: magnetic immunoassay RT‐PCR: reverse transcription polymerase chain reaction | ||||||
Appendix 27. Specificity data by test brand (IgM alone, all other reference groups)
| Test name | Test name | Suspected of COVID‐19 | Current RT‐PCR negative | Current untested | Other/ mixed unclear | Cross‐reactivity/ confounder panels |
| Test brand | Test brand |
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
||||
| Lateral flow/colloidal gold | ||||||
| AAZ‐LMB | Covid‐Presto test rapid Covid‐19 IgM | 1 (64/64) | ||||
| LFA | 100a (94.4, 100)** |
|||||
| Alfa Scientific Designs | CLARITY COVID‐19 IgM Antibody Test | 1 (54/60) | ||||
| LFA | 90.0a (79.5, 96.2)* |
|||||
| Augurix | Augurix SARS‐CoV‐2 IgM RDT | 1 (45/45) | ||||
| LFA | 100a (92.1, 100)** |
|||||
| Augurix | SimtomaX Corona Check IgM | 1 (105/105) | ||||
| LFA | 100a (96.5, 100* |
|||||
| Avioq Bio‐Tech | Novel Coronavirus (2019‐nCov) Antibody IgM Assay Kit | 1 (69/72) | ||||
| CGIA | 95.8a (88.3, 99.1)* |
|||||
| AYTU Biosciences | AYTU COVID‐19 IgM Rapid Test Cassette | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Beijing Diagreat | 2019‐nCoV IgM Antibody Determination Kit | 1 (153/160) | 1 (564/600) | |||
| FIA | 95.6a (91.2, 98.2)* |
94.0a (91.8, 95.8)* |
||||
| BioMedomics | COVID‐19 IgM Rapid Test | 1 (43/51) | ||||
| LFA | 84.3a (71.4, 93.0)* |
|||||
| BioMerieux | Vidas SARS‐CoV‐2 IgM | 1 (266/276) | ||||
| FIA | 96.4a (93.4, 98.2)* |
|||||
| Bioperfectus | PerfectPOC Novel Corona Virus (SARS CoV‐2) IgM Rapid Test Kit | 1 (40/45) | ||||
| LFA | 88.9a (75.9, 96.3)* |
|||||
| Biosynex | COVID‐19 BSS IgM | 1 (24/27) | ||||
| LFA | 88.9a (70.8, 97.6)* |
|||||
| Cellex | Cellex qSARS‐CoV‐ 2 IgM Cassette Rapid Test | 1 (79/79) | ||||
| LFA | 100a (95.4, 100)** |
|||||
| Clungene Biotech | Clungene SARS‐CoV‐2 IgM Rapid Test Cassettes | 1 (153/162) | ||||
| LFA | 94.4a (89.7, 97.4)* |
|||||
| CTK Biotech | OnSite COVID‐19 IgM Rapid Test | 1 (99/102) | 1 (89/92) | |||
| CGIA | 97.1a (91.6, 99.4)* |
96.7a (90.8, 99.3)* |
||||
| DecombioBiotechnology | Novel Coronavirus (SARS‐CoV‐2) IgM Combo Rapid Test‐Cassette | 1 (46/51) | ||||
| LFA | 90.2a (78.6, 96.7)* |
|||||
| DeepBlue Medical | COVID‐19 (SARS CoV‐2) IgM Antibody Test Kit (Colloidal Gold) | 1 (37/51) | ||||
| CGIA | 72.5a (58.3, 84.1)* |
|||||
| DNA Link | AccuFind COVID19 IgM | 1 (47/59) | ||||
| LFA | 79.7a (67.2, 89.0)* |
|||||
| Dynamiker Biotechnology | Dynamiker Biotechnology ‐ 2019‐nCOV IgM Rapid Test | 1 (98/103) | ||||
| LFA | 95.1a (89.0, 98.4)* |
|||||
| Hangzhou | Hangzhou unlabelled packaging IgM | 1 (89/92) | ||||
| CGIA | 96.7a (90.8, 99.3)* |
|||||
| Hangzhou Alltest | 2019‐nCoV IgM Rapid Test Cassette | 2 (188/192) | 1 (58/60) | |||
| LFA | 97.9 (94.6, 99.2) |
96.7a (88.5, 99.6)* |
||||
| Hangzhou Biotest | RightSign IgM/IgG (Also distributed as CoronaChek and Premier Biotech Covid‐19 IgM/IgG) | 1 (58/65) | 2 (111/111) | |||
| LFA | 89.2a (79.1, 95.6)* |
100a (96.7, 100)** |
||||
| Hightop Biotech | Hightop SARS‐CoV‐2 IgM Antibody Rapid Test | 1 (92/92) | ||||
| CGIA | 100a (96.1, 100)** |
|||||
| Innovita Biological | 2019‐nCoV Ab test IgM | 1 (26/28) | ||||
| LFA | 92.9a (76.5, 99.1)* |
|||||
| InTec | Rapid SARS‐CoV‐2 Antibody (IgM) Test | 1 (68/79) | ||||
| CGIA | 86.1a (76.5, 92.8)* |
|||||
| Intelligent Endoscopy | Smart screen IgM | 1 (55/60) | ||||
| LFA | 91.7a (81.6, 97.2)* |
|||||
| Inzek | BIOZEK COVID‐19 IgG/IgM | 1 (53/55) | ||||
| LFA | 96.4a (87.5, 99.6)* |
|||||
| LabOn Time | LaboOn Time rapid test cassette IgM | 1 (72/72) | ||||
| LFA | 100a (95.0, 100)** |
|||||
| Liming Bio | StrongStep1 SARS‐CoV‐2 IgM | 1 (102/103) | ||||
| LFA | 99.0a (94.7, 100)* |
|||||
| MEDsan GmbH | MEDsan COVID‐19 IgM Rapid Test | 1 (101/110) | 1 (49/50) | |||
| LFA | 91.8a (85.0, 96.2)* |
98.0a (89.4, 99.9)* |
||||
| Multi‐G | Multi‐G MGA 2019‐nCoV IgM Rapid test cassette | 1 (94/103) | ||||
| LFA | 91.3a (84.1, 95.9)* |
|||||
| Multi‐G | 2019‐nCoV IgM Rapid Test Cassette dual lane | 1 (39/39) | ||||
| LFA | 100a (91.0, 100)** |
|||||
| Multi‐G | 2019‐nCoV IgM Rapid Test Cassette single lane | 1 (37/39) | ||||
| LFA | 94.9a (82.7, 99.4)* |
|||||
| NG Biotech | NG‐Test IgM COVID‐19 | 2 (65/73) | ||||
| CGIA | 89.0 (79.2, 94.5) |
|||||
| Nirmidas Biotech | Nirmidas COVID‐19 (SARS‐CoV‐2) IgM Antibody Detection Kit | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| NTBIO® Diagnostics | NTBIO One Step Rapid Test ‐ COVID‐19 IgM Antibody Test | 1 (102/108) | ||||
| 94.4a (88.3, 97.9)* |
||||||
| Prima Lab | Prima COVID‐19 IgM Rapid test | 1 (96/103) | ||||
| LFA | 93.2a (86.5, 97.2)* |
|||||
| Prometheus Bio | Prometheus Bio ‐ 2019‐nCoV IgM | 1 (7/41) | ||||
| LFA | 17.1a (7.2, 32.1)* |
|||||
| Qingdao HIGHTOP Biotech | SARS‐CoV‐2 IgM Ab Rapid Test | 1 (111/111) | ||||
| 100a (96.7, 100)** |
||||||
| Safecare Biotech | SafeCare Bio‐Tech COVID‐19 IgM Rapid Test Device | 1 (59/60) | ||||
| LFA | 98.3a (91.1, 100)* |
|||||
| SD Biosensor | COVID‐19 IgM Duo | 1 (238/244) | ||||
| LFA | 97.5a (94.7, 99.1)* |
|||||
| Sensing Self | Covid‐19 Rapid IgM combined Antibody assay | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)** |
|||||
| Servibio/VEDALAB | COVID‐19 Sign IgM | 1 (24/27) | ||||
| LFA | 88.9a (70.8, 97.6)* |
|||||
| Shanghai Outdo | COVID‐19 IgM‐IgG Rapid Test IgM | 1 (10/10) | ||||
| LFA | 100a (69.2, 100)** |
|||||
| Sure Biotech | SARS‐CoV‐2 IgM Antibody Rapid Test | 1 (107/110) | 1 (51/51) | |||
| LFA | 97.3a (92.2, 99.4)* |
100a (93.0, 100)** |
||||
| SureScreen Diagnostics | COVID‐19 Coronavirus Rapid Test Cassette IgM | 1 (39/39) | ||||
| LFA | 100a (91.0, 100)** |
|||||
| T&D Diagnostics | Sienna 2019‐nCoV IgM Rapid Test | 1 (35/44) | ||||
| LFA | 79.5a (64.7, 90.2)* |
|||||
| TBG Biotechnology | TBG SARS‐CoV‐2 IgM Rapid Test Kit | 1 (52/59) | ||||
| LFA | 88.1a (77.1, 95.1)* |
|||||
| UCP Biosciences | Coronavirus IgM Antibody (COVID‐19) Test Cassette |
1 (48/51) | ||||
| LFA | 94.1a (83.8, 98.8)* |
|||||
| Unknown manufacturer | Ready Result IgM | 1 (60/60) | ||||
| LFA | 100a (94.0, 100)* |
|||||
| VivaChek Biotech | VivaDiag COVID‐19 IgM Rapid Test | 2 (193/195) | 1 (44/48) | |||
| CGIA | 99.0 (94.3, 99.9) |
91.7a (80.0, 97.7)* |
||||
| W.H.P.M. | COVISURE COVID‐19 IgM Rapid Test | 1 (56/59) | ||||
| LFA | 94.9a (85.9, 98.9)* |
|||||
| Xiamen Biotime | SARS‐Cov‐2 IgM Rapid Qualitative Test | 1 (94/96) | ||||
| LFA | 97.9a (92.7, 99.7)* |
|||||
| Zeus Scientific | ZEUS SARS‐CoV‐2 IgG/IgM rapid test | 1 (58/60) | ||||
| LFA | 96.7a (88.5, 99.6)* |
|||||
| Zhejiang Orient‐Gene | COVID‐19 IgM rapid test cassette | 1 (39/39) | 1 (49/50) | 1 (98/103) | 1 (70/79) | |
| LFA | 100a (91.0, 100)** |
98.0a (89.4, 99.9)* |
95.1a (88.9, 98.0)* |
88.6a (79.5, 94.7)* |
||
| ELISA | ||||||
| Ash Laboratories | Ash Laboratories SARS‐CoV2 IgM ELISA Immunoassay | 1 (37/38) | 1 (19/19) | |||
| 97.4a (86.2, 99.9)* |
100a (82.4, 100)** |
|||||
| Beijing Wantai | Wantai ELISA IgM assay | 1 (300/300) | 1 (97/100) | 2 (222/224) | ||
| 100a (98.8, 100)** |
97.0a (91.5, 99.4)* |
99.1 (96.5, 99.8) |
||||
| Bio‐Rad Laboratories | Novel coronavirus COVID‐19 IgM assay | 1 (58/60) | ||||
| 96.7a (88.5, 99.6)* |
||||||
| Epitope Diagnostics | EDI Novel Coronavirus COVID‐19 IgM ELISA kit | 2 (206/213) | 4 (196/200) | |||
| 96.7 (93.3, 98.4) |
98.0 (94.8, 99.2) |
|||||
| Hotgen | Beijing Hotgen SARS‐CoV‐2 IgM ELISA | 1 (100/100) | ||||
| 100a (96.4, 100)** |
||||||
| Zhuhai Livzon | Zhuhai Livzon SARS‐CoV‐2 IgM ELISA | 1 (60/60) | 2 (217/220) | |||
| 100a (94.0, 100)** |
98.8 (93.1, 99.8) |
|||||
| CLIA | ||||||
| Abbott Diagnostics | Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgM | 1 (39/39) | 1 (73/73) | |||
| 100a (91.0, 100)** |
100 (95.1, 100) | |||||
| Autobio Diagnostics | SARS‐CoV‐2 CLIA Microparticles IgM/IgG | 1 (377/389) | ||||
| 96.9a (94.7, 98.4)* |
||||||
| Diazyme | Diazyme DZ‐LITE 2019‐nCoV IgM (CLIA) Assay Kit | 1 (234/235) | 1 (49/49) | |||
| 99.6a (97.7, 100)** |
100a (92.7, 100)** |
|||||
| Hunan Yuanjing | COVID‐19 IgM Detection Kits | 1 (38/40) | ||||
| 95.0a (83.1, 99.4)* |
||||||
| MEXACARE GmbH | QuickTestCorona COVID‐19 IgM | 1 (100/109) | ||||
| 91.7a (84.9, 96.2)* |
||||||
| Shenzhen YHLO | YHLO iFlash IgM assay | 1 (30/33) | 2 (1055/1082) | 1 (583/586) | 1 (73/75) | |
| 90.9a (76.7, 98.1)* |
97.5 (96.4, 98.3) |
99.4a (98.5, 99.9)* |
97.3a (90.7, 99.7)* |
|||
| SNIBE Shenzhen | MAGLUMI 2019‐nCoV IgM kits | 1 (11/11) | 1 (72/72) | 2 (80/80) | ||
| 100a (71.5, 100)** |
100a (95.0, 100)** |
100a (95.5, 100)** |
||||
| Other/unclear | ||||||
| Quotient | MosaiQ COVID‐19 antibody microarray IgM | 1 (59/59) | ||||
| 100a (93.9, 100)* |
||||||
| Vibrant America | Vibrant COVID‐19 Ab IgM | 1 (5252/5262) | ||||
| 99.8a (99.7, 99.9)* |
||||||
|
aEstimates and confidence intervals by summing the counts of true negative and false positive across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervals CGIA: colloidal gold immunoassayCLIA: chemiluminescence immunoassay DNA: deoxyribonucleic acidELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assayMIA: magnetic immunoassay RT‐PCR: reverse transcription polymerase chain reaction | ||||||
Appendix 28. Sensitivity data by test brand (IgA alone or combined with IgG or IgM, all data)
| Test name | Test name | Week 1 | Week 2 | Week 3 | Convalescent |
| Test brand | Test brand |
Test groups (true positives/COVID cases) Sensitivity (95% CI) |
|||
| Assays targeting IgA alone | |||||
| ELISA | |||||
| Euroimmun | Anti‐SARS‐COV‐2 IgA | 19 (260/587) | 18 (490/660) | 16 (334/385) | 17 (866/989) |
| 44.0 (34.0, 54.6) |
76.7 (69.8, 82.4) |
92.7 (85.0, 96.6) |
87.7 (84.5, 90.3) |
||
| Gold Standard Diagnostics | Gold Standard SARS‐CoV‐2 IgA ELISA | 1 (18/123) | |||
| 14.6a (8.9, 22.1)* |
|||||
| Mediagnost | Mediagnost Anti‐SARS‐CoV‐2 ELISA IgA | 1 (28/46) | |||
| 60.9a (45.4, 74.9)* |
|||||
| RayBiotech | Covid‐19 human ELISA IgA | 1 (1/35) | |||
| 2.9a (0.1, 14.9)* |
|||||
| CLIA | |||||
| Beijing Wantai | Wantai CLIA IgA assay | 1 (18/31) | 1 (25/29) | 1 (36/38) | 1 (39/47) |
| 58.1a (39.1, 75.5)* |
86.2a (68.3, 96.1)* |
94.7a (82.3, 99.4)* |
83.0a (69.2, 92.4)* |
||
| Other/unclear | |||||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgA (S‐based) | 1 (15/48) | 1 (41/81) | 1 (28/39) | 1 (6/26) |
| 31.2 (18.7, 46.3) | 50.6 (39.3, 61.9) | 71.8 (55.1, 85.0) | 23.1 (9.0, 43.6) | ||
| Genalyte | Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel IgA (N‐based) | 1 (11/48) | 1 (42/81) | 1 (26/39) | |
| 22.9 (12.0, 37.3) | 51.9 (40.5, 63.1) | 66.7 (49.8, 80.9) | 1 (2/26) | ||
| Vibrant America | Vibrant COVID‐19 Ab IgA | 1 (220/330) | 1 (276/330) | 7.7 (0.9, 25.1) | |
| 66.7a (61.3, 71.7)* |
83.6a (79.2, 87.5)* |
||||
| Assays targeting IgA or IgG | |||||
| Euroimmun | anti‐SARS‐COV‐2 IgA/IgG ELISA | 6 (87/167) | 5 (141/170) | 5 (118/125) | 4 (149/157) |
| 49.6 (35.1, 64.0) |
82.9 (76.5, 87.9) |
94.4 (88.7, 97.3) |
94.9 (90.1, 97.4) |
||
| Assays targeting IgA or IgM | |||||
| Vircell | COVID‐19 ELISA IgM+IgA | 1 (49/52) | 1 (36/36) | 1 (43/44) | |
| 94.2a (84.1, 98.8)* |
100a (90.3, 100)** |
97.7a (88.0, 99.9)* |
|||
|
aEstimates and confidence intervals by summing the counts of true positive and false negative across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervals CGIA: colloidal gold immunoassayCLIA: chemiluminescence immunoassay DNA: deoxyribonucleic acidELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assayMIA: magnetic immunoassay RT‐PCR: reverse transcription polymerase chain reaction | |||||
Appendix 29. Specificity data by test brand (IgA alone or combined with IgG or IgM, all reference groups)
| Test brand | Test name | Pre‐pandemic | Suspected of COVID‐19 | Current RT‐PCR‐negative | Current untested | Other/mixed unclear | Cross‐reactivity/confounder panels |
|
Test groups (true negatives/non‐COVID cases) Specificity (95% CI) |
|||||||
| Assays targeting IgA alone | |||||||
| ELISA | |||||||
| Euroimmun | Anti‐SARS‐COV‐2 IgA | 16 (1527/1635) | 4 (310/385) | 2 (197/204) | 5 (316/387) | 5 (278/311) | |
| 94.0 (91.6, 95.7) |
86.4 (74.4, 93.3) |
93.9 (64.4, 99.2) |
81.8 (75.9, 86.5) |
89.1 (80.6, 94.2) |
|||
| Gold Standard Diagnostics | Gold Standard SARS‐CoV‐2 IgA ELISA | 1 (76/76) | |||||
| 100a (95.3, 100)** |
|||||||
| Other/unclear | |||||||
| Vibrant America | Vibrant COVID‐19 Ab IgA | 1 (5257/5262) | |||||
| 99.9a (99.8, 100)* |
|||||||
| Assays targeting IgA or IgG | |||||||
| Euroimmun | anti‐SARS‐COV‐2 IgA/IgG | 2 (101/106) | 3 (197/256) | 2 (203/219) | |||
| 97.6 (66.8, 99.9) |
77.8 (68.8, 84.7) |
92.7 (88.4, 95.5) |
|||||
| Assays targeting IgA or IgM | |||||||
| Vircell | COVID‐19 ELISA IgM+IgA IgA/IgM | 1 (26/56) | |||||
| 46.4a (33.0, 60.3)* |
|||||||
|
aEstimates and confidence intervals by summing the counts of true negative and false positive across 2 x 2 tables *95% exact binomial confidence interval **97.5% one‐sided exact binomial confidence interval Ab: antibodyCI: confidence intervals CGIA: colloidal gold immunoassayCLIA: chemiluminescence immunoassayELISA: enzyme‐linked immunosorbent assay FIA: fluorescence immunoassay LFA: lateral flow assay | |||||||
Data
Presented below are all the data for all of the tests entered into the review.
Tests. Data tables by test.
1. Test.
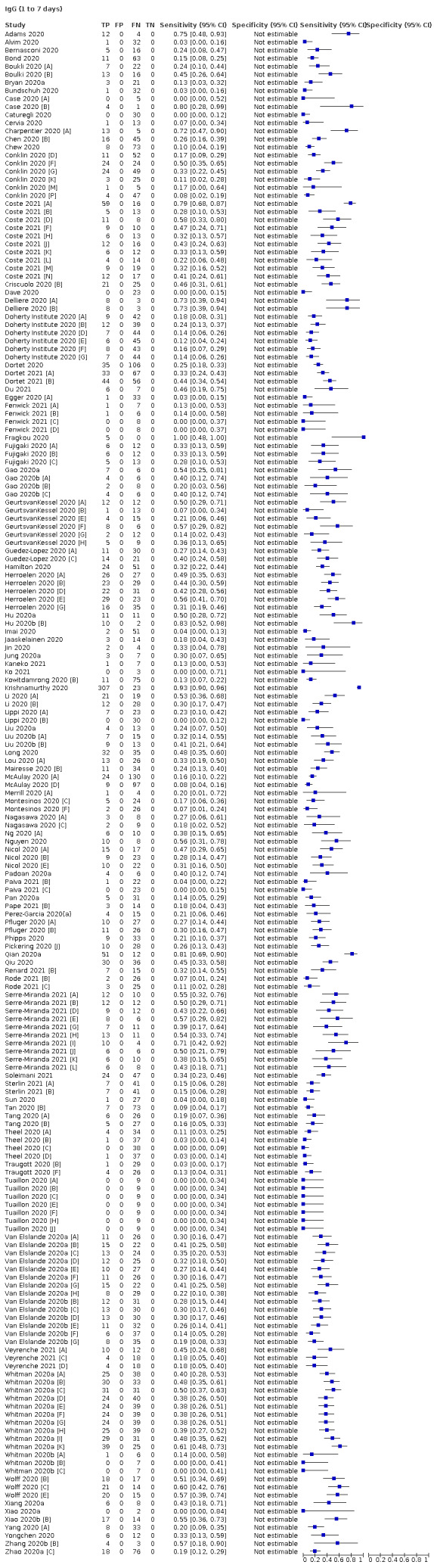
IgG (1 to 7 days)
2. Test.
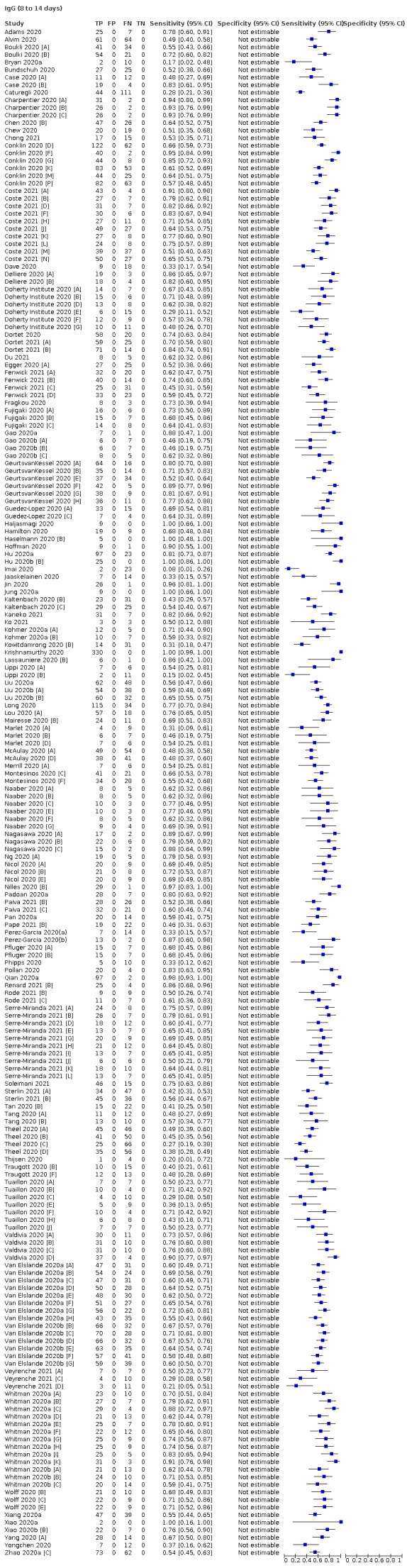
IgG (8 to 14 days)
3. Test.
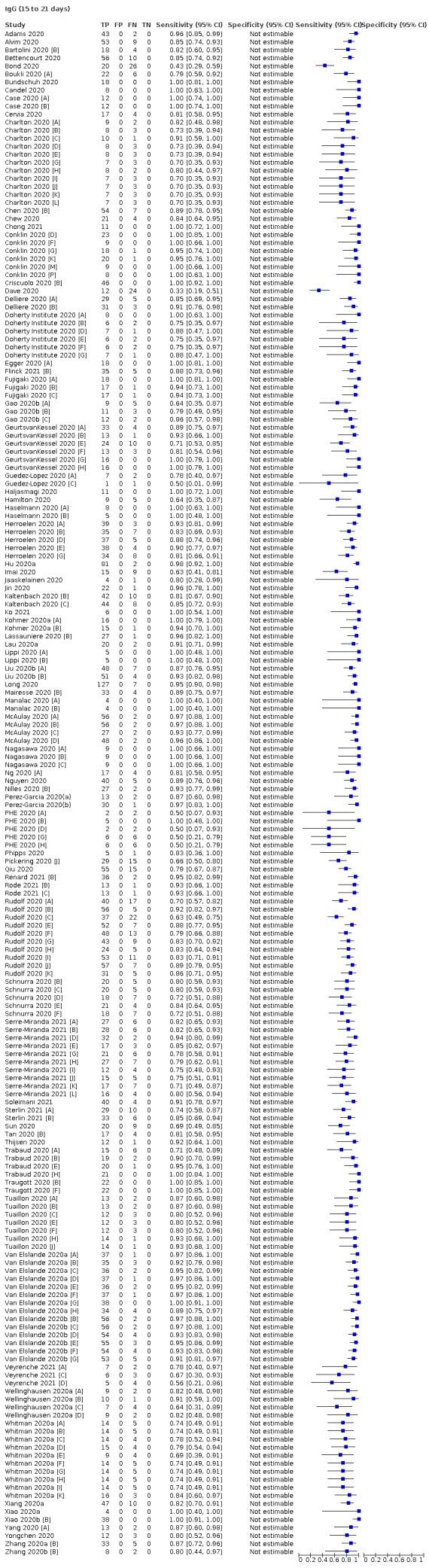
IgG (15 to 21 days)
4. Test.
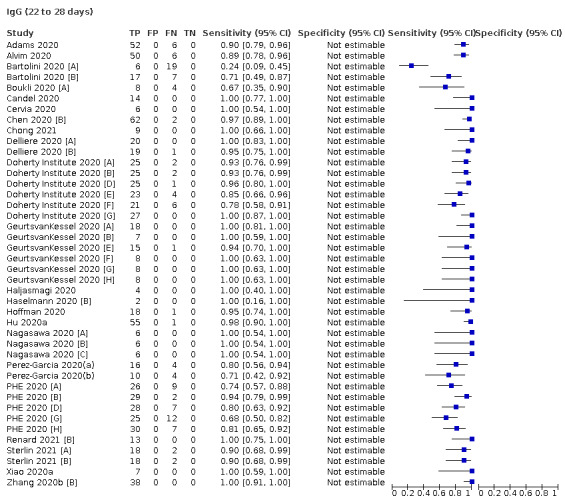
IgG (22 to 28 days)
5. Test.

IgG (29 to 35 days)
6. Test.
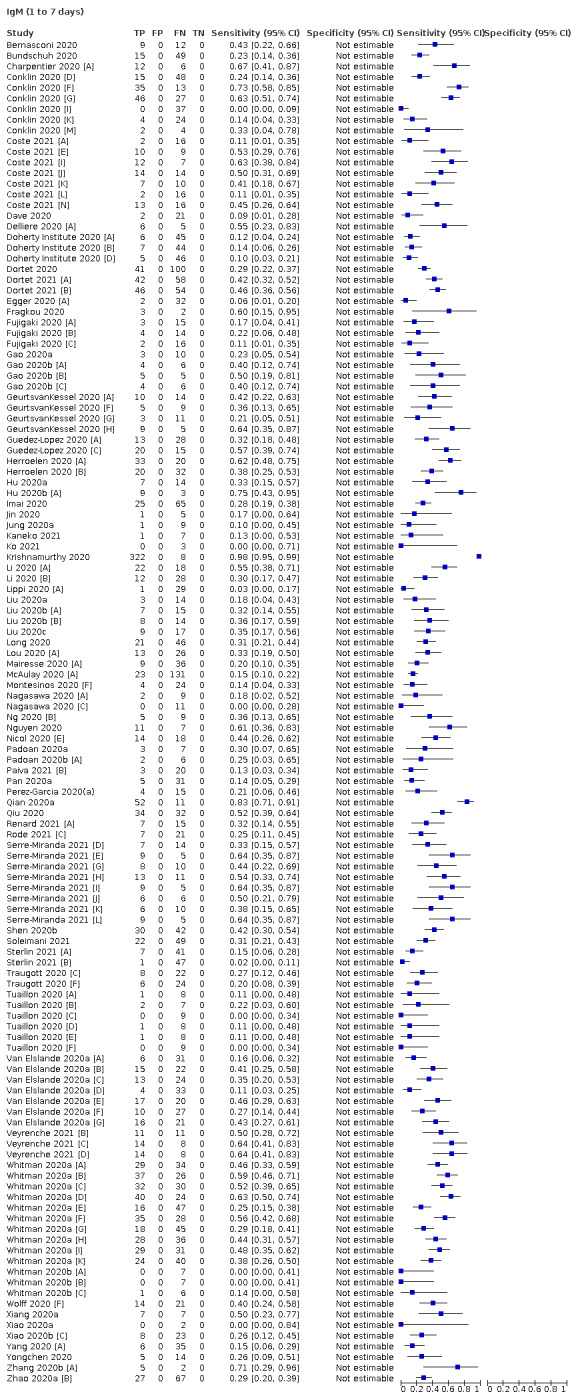
IgM (1 to 7 days)
7. Test.
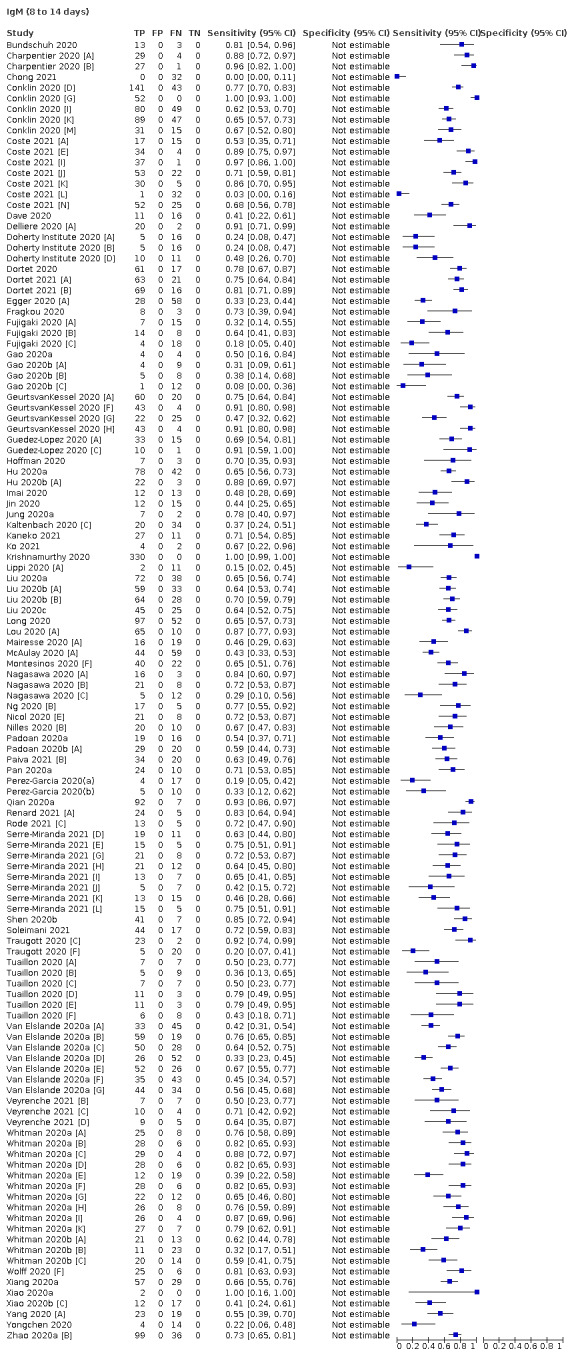
IgM (8 to 14 days)
8. Test.
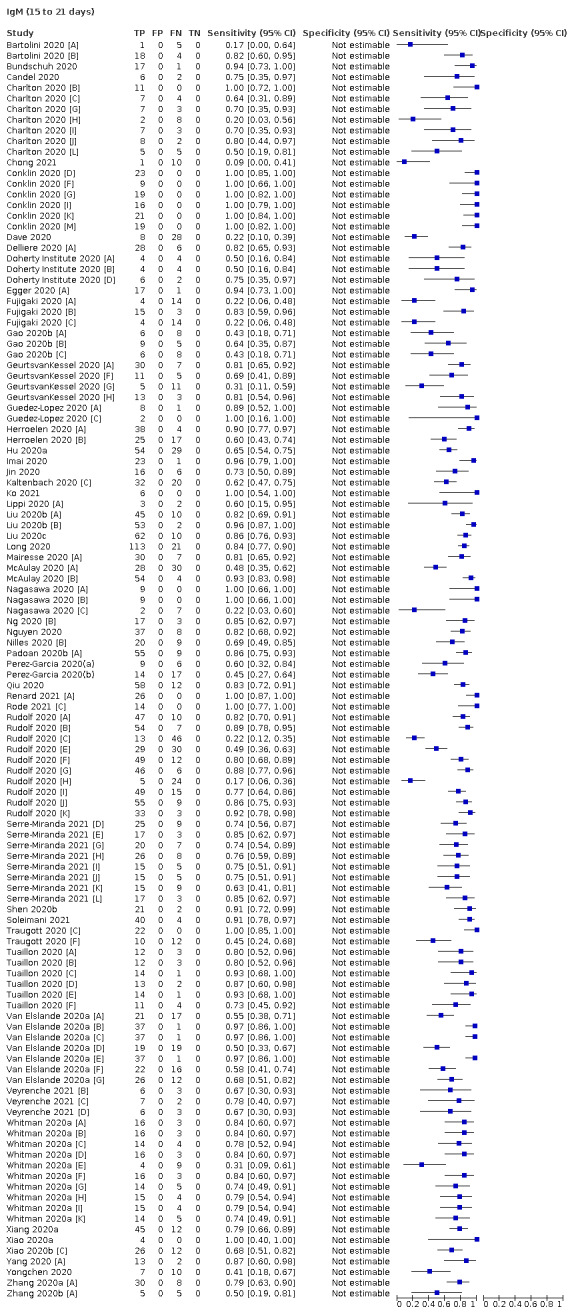
IgM (15 to 21 days)
9. Test.
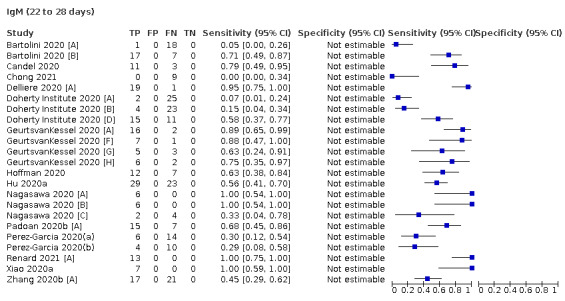
IgM (22 to 28 days)
10. Test.
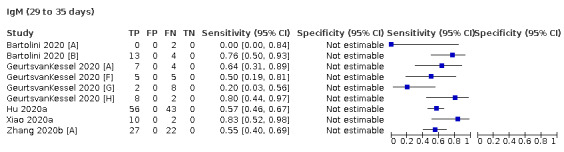
IgM (29 to 35 days)
11. Test.
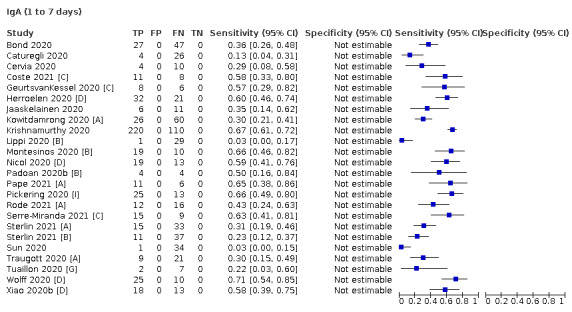
IgA (1 to 7 days)
12. Test.
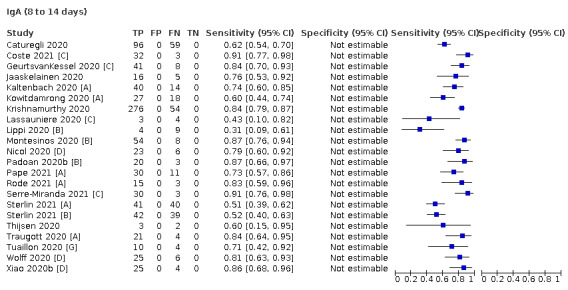
IgA (8 to 14 days)
13. Test.
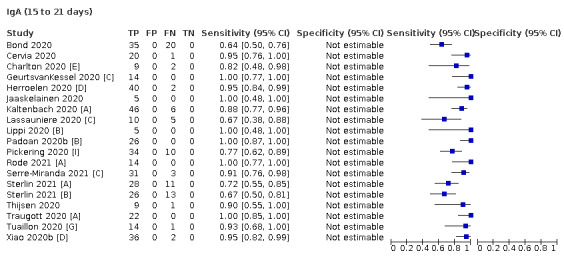
IgA (15 to 21 days)
14. Test.
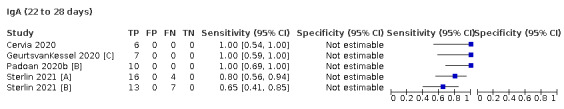
IgA (22 to 28 days)
15. Test.

IgA (29 to 35 days)
16. Test.
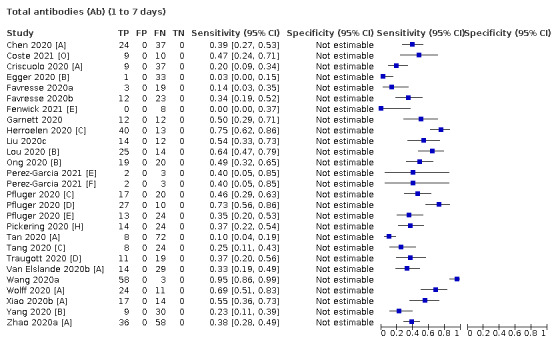
Total antibodies (Ab) (1 to 7 days)
17. Test.
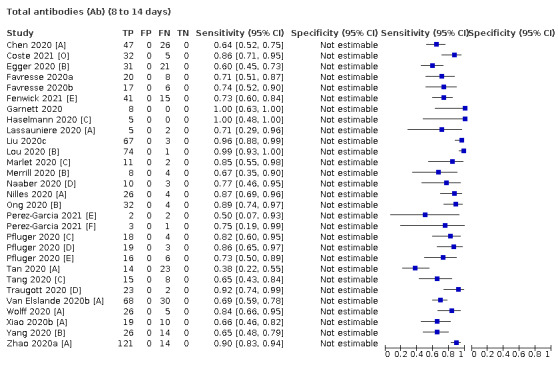
Total antibodies (Ab) (8 to 14 days)
18. Test.
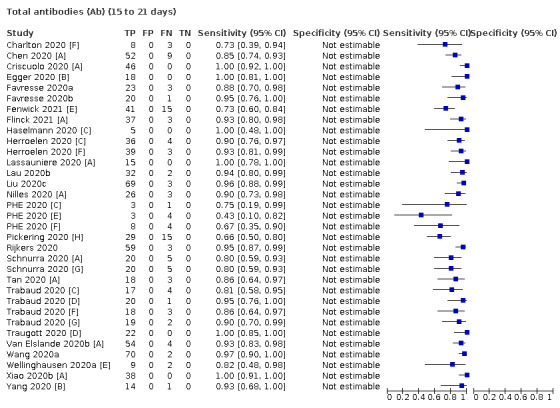
Total antibodies (Ab) (15 to 21 days)
19. Test.

Total antibodies (Ab) (22 to 28 days)
20. Test.
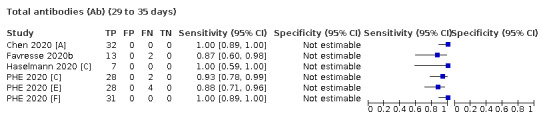
Total antibodies (Ab) (29 to 35 days)
21. Test.
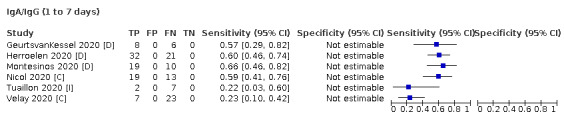
IgA/IgG (1 to 7 days)
22. Test.
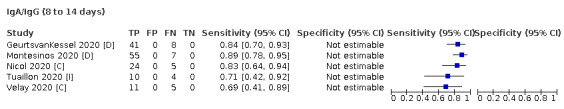
IgA/IgG (8 to 14 days)
23. Test.

IgA/IgG (15 to 21 days)
24. Test.
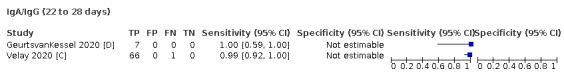
IgA/IgG (22 to 28 days)
25. Test.

IgA/IgG (29 to 35 days)
26. Test.

IgA/IgM (1 to 7 days)
27. Test.

IgA/IgM (8 to 15 days)
28. Test.
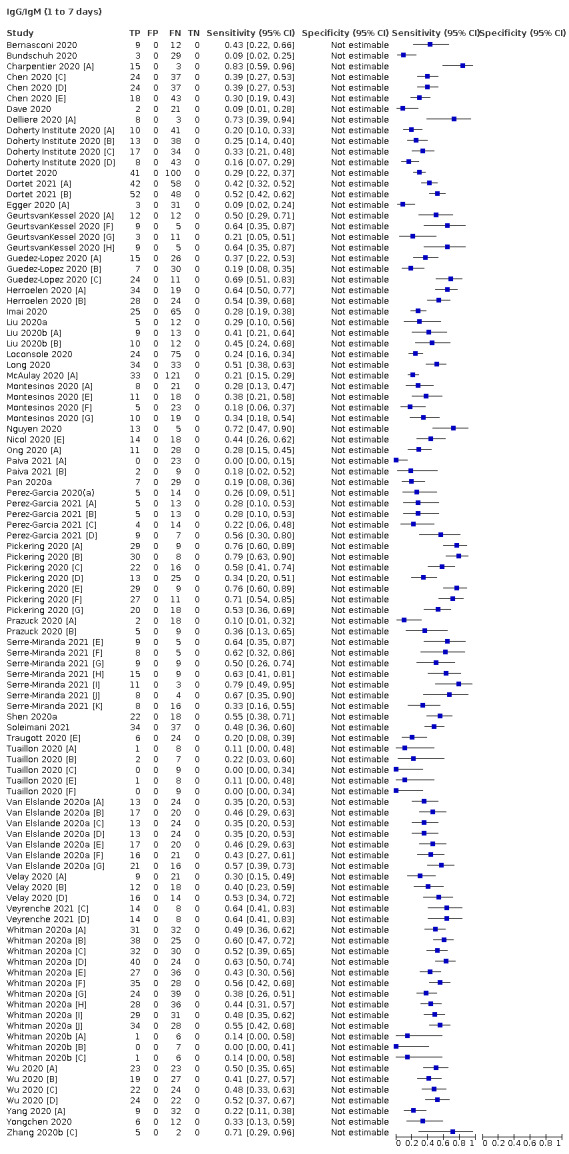
IgG/IgM (1 to 7 days)
29. Test.
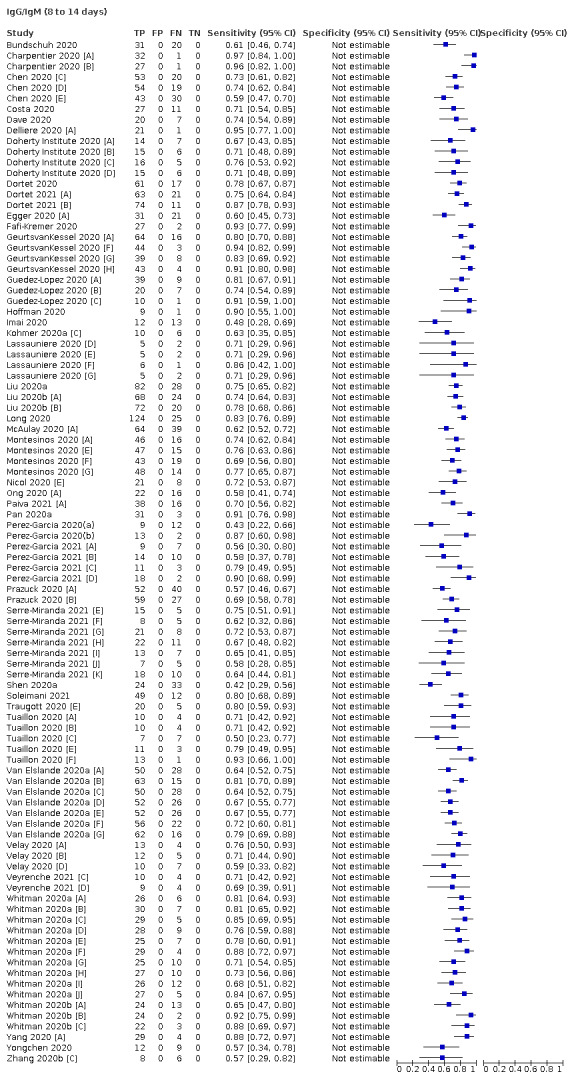
IgG/IgM (8 to 14 days)
30. Test.
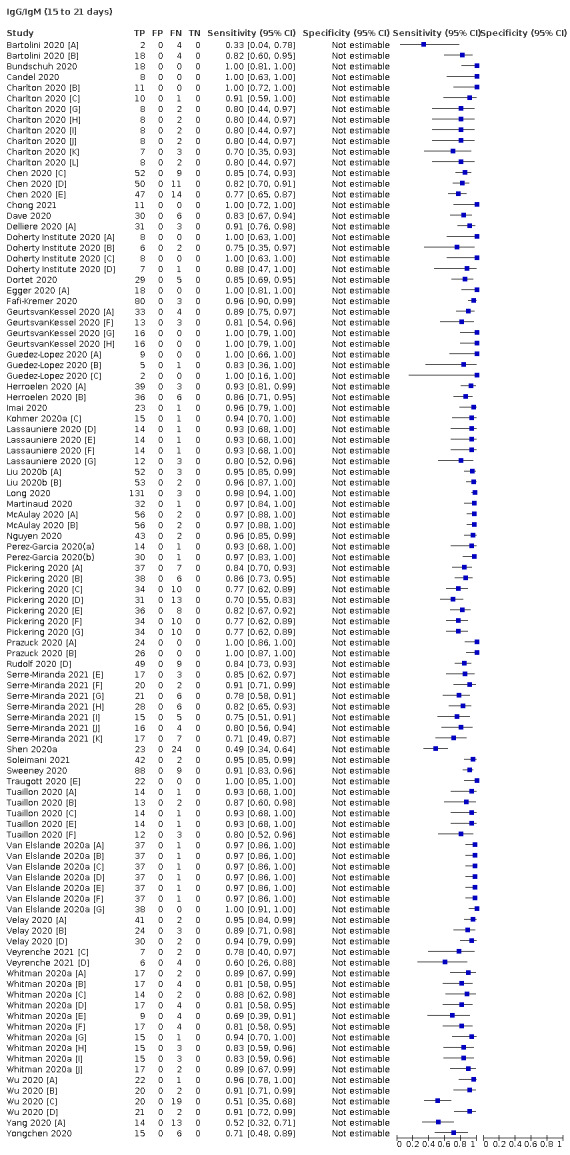
IgG/IgM (15 to 21 days)
31. Test.
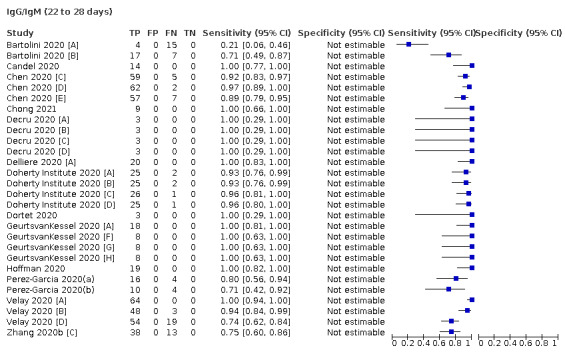
IgG/IgM (22 to 28 days)
32. Test.
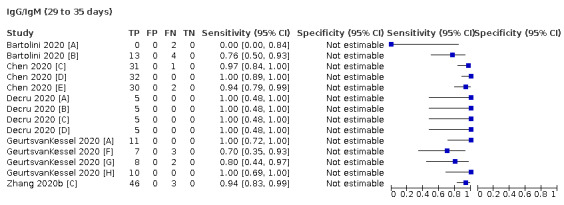
IgG/IgM (29 to 35 days)
33. Test.
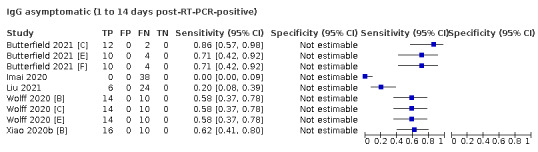
IgG asymptomatic (1 to 14 days post‐RT‐PCR‐positive)
34. Test.
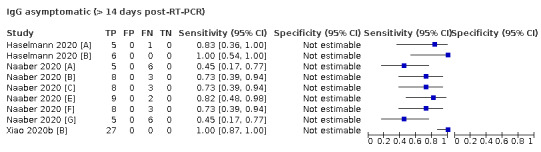
IgG asymptomatic (> 14 days post‐RT‐PCR)
35. Test.
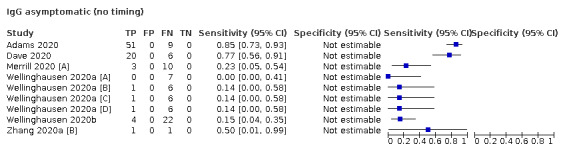
IgG asymptomatic (no timing)
36. Test.

IgM asymptomatic (1 to 14 days post‐RT‐PCR‐positive)
37. Test.

IgM asymptomatic (> 14 days post‐RT‐PCR)
38. Test.

IgM asymptomatic (no timing)
39. Test.

IgG/IgM asymptomatic (1 to 14 days post‐RT‐PCR‐positive)
40. Test.

IgG/IgM asymptomatic (no timing)
41. Test.
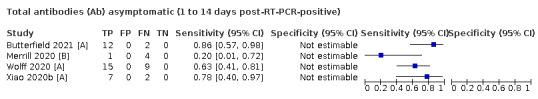
Total antibodies (Ab) asymptomatic (1 to 14 days post‐RT‐PCR‐positive)
42. Test.

Total antibodies (Ab) asymptomatic (> 14 days post‐RT‐PCR)
43. Test.

Total antibodies (Ab) asymptomatic (no timing)
44. Test.

IgA asymptomatic (1 to 14 days post‐RT‐PCR‐positive)
45. Test.

IgA asymptomatic (> 14 days post‐RT‐PCR)
46. Test.
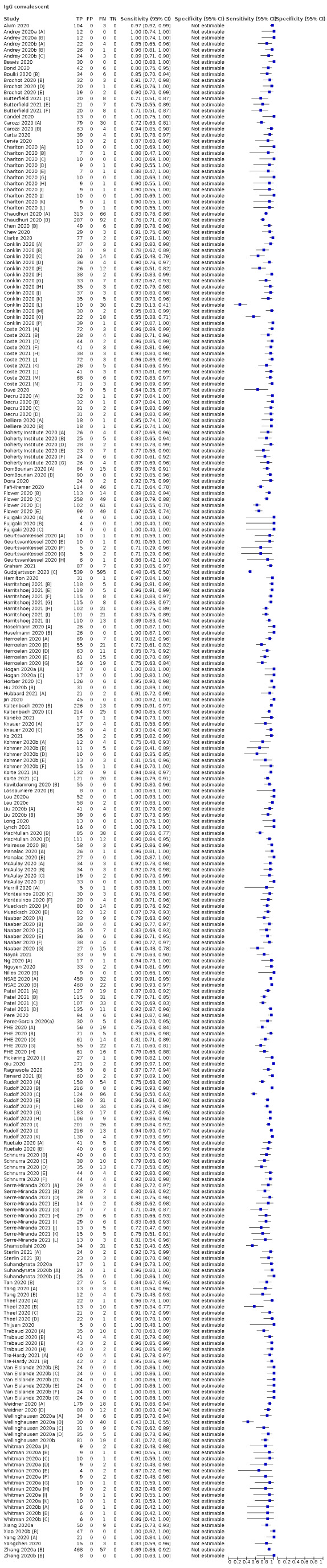
IgG convalescent
47. Test.
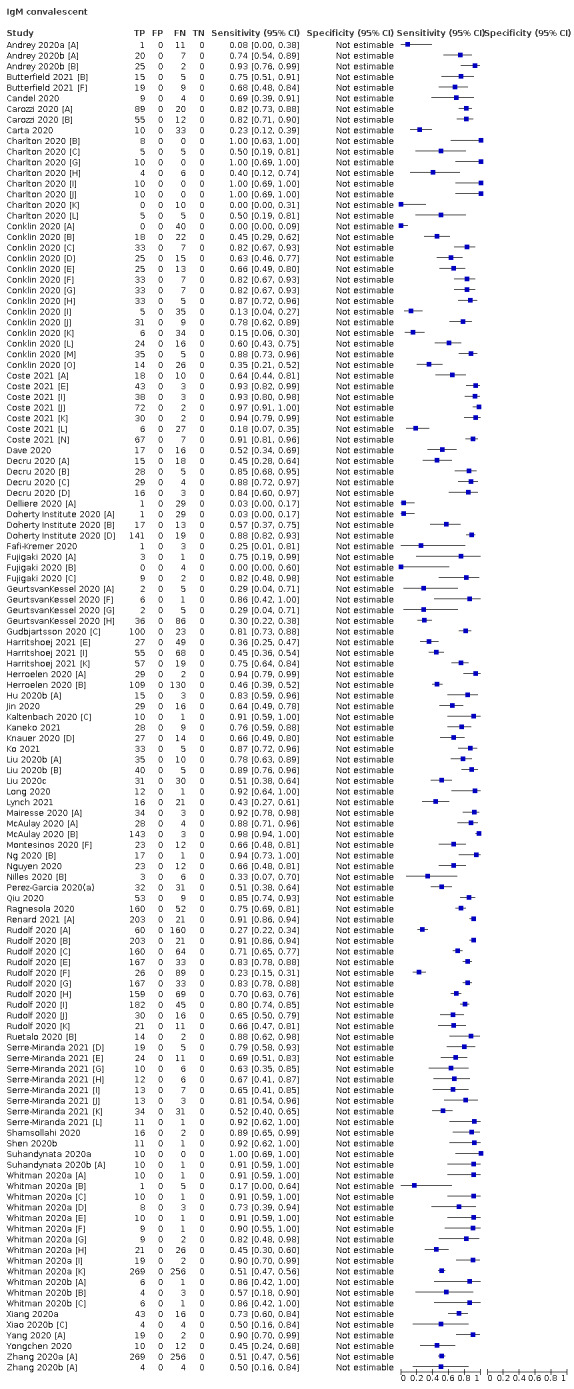
IgM convalescent
48. Test.
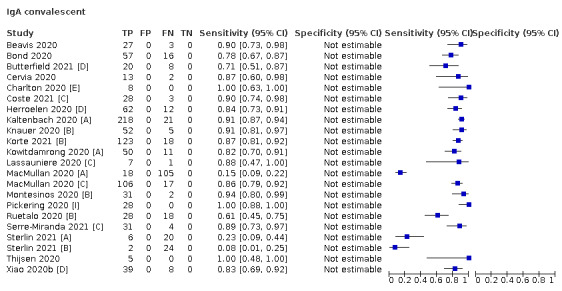
IgA convalescent
49. Test.

IgA/IgG convalescent
50. Test.
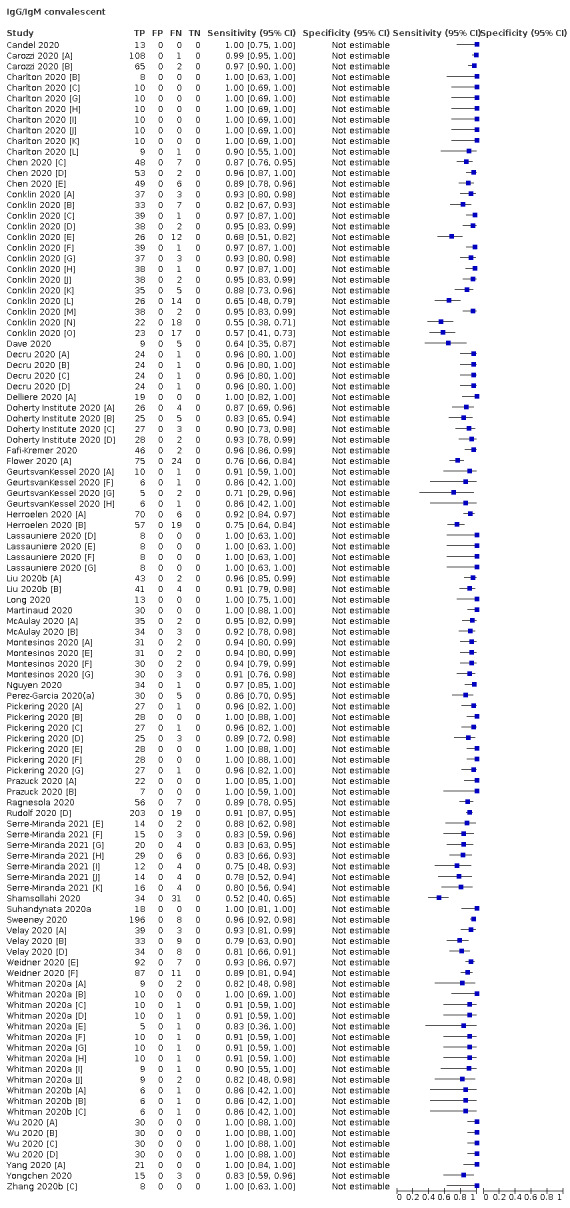
IgG/IgM convalescent
51. Test.

IgA/IgM convalescent
52. Test.
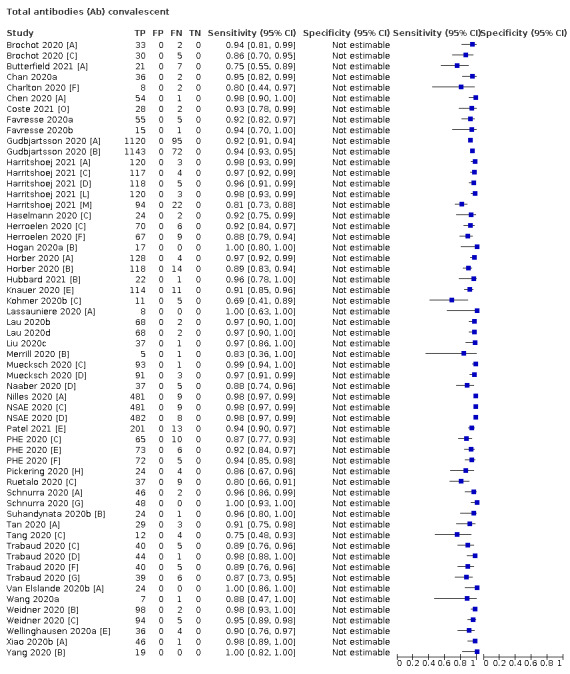
Total antibodies (Ab) convalescent
53. Test.
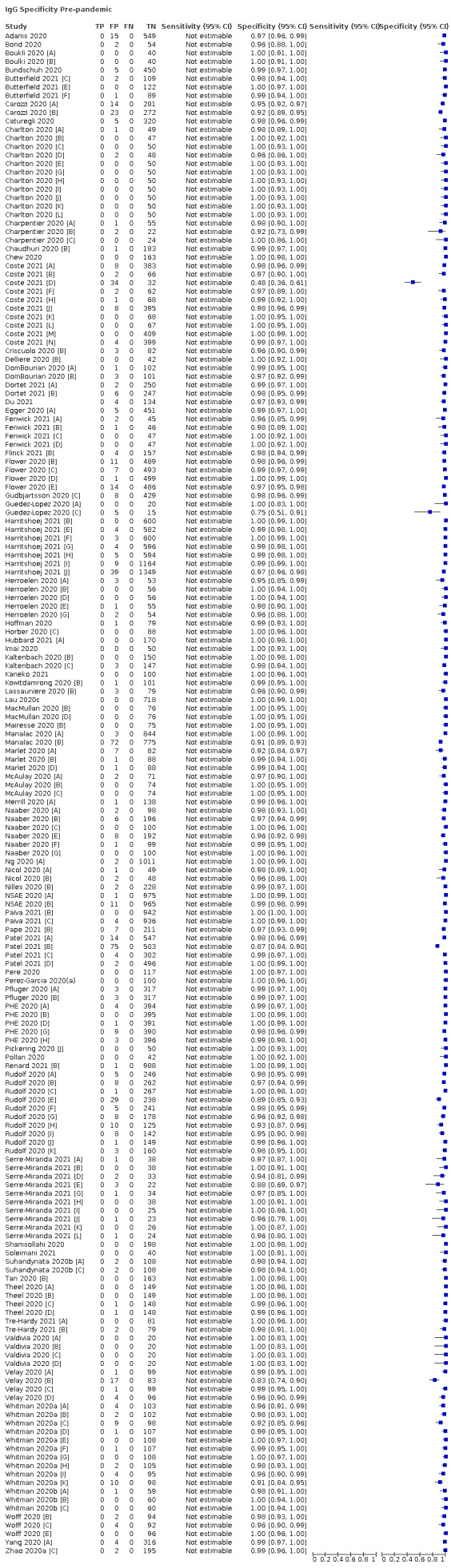
IgG Specificity Pre‐pandemic
54. Test.
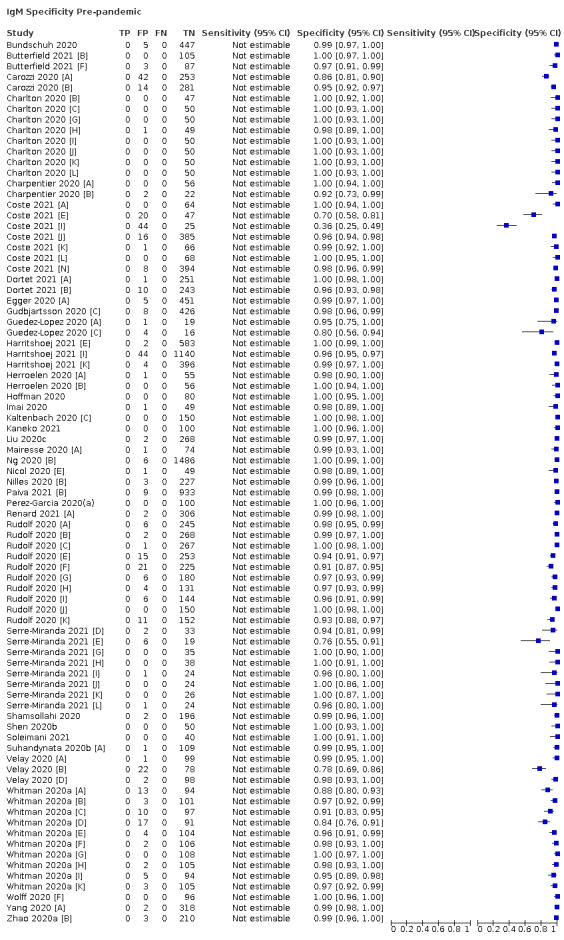
IgM Specificity Pre‐pandemic
55. Test.
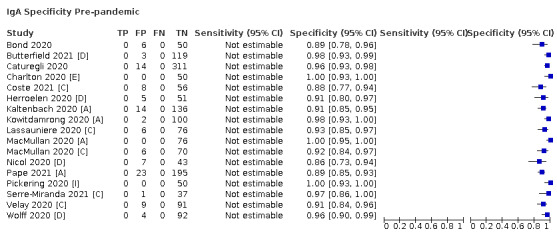
IgA Specificity Pre‐pandemic
56. Test.
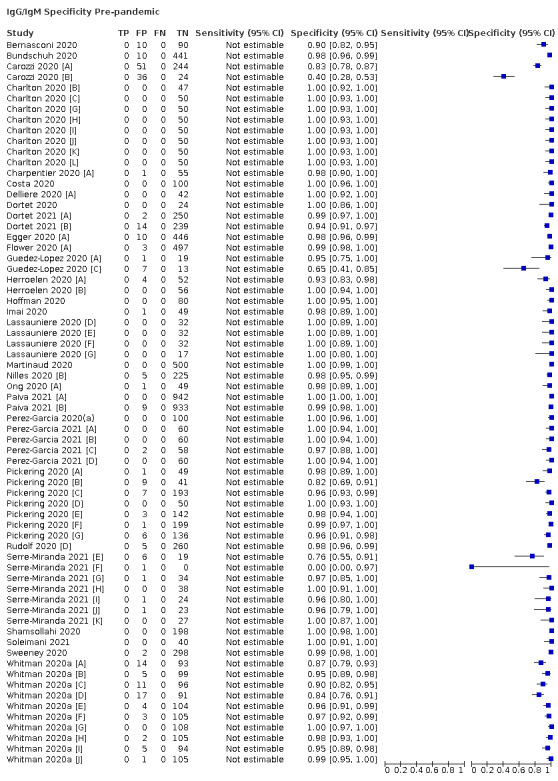
IgG/IgM Specificity Pre‐pandemic
57. Test.

IgA/IgM Specificity Pre‐pandemic
58. Test.

IgA/IgG Specificity Pre‐pandemic
59. Test.
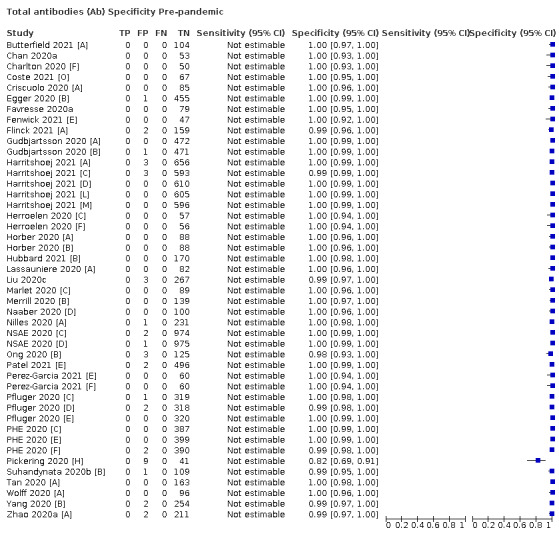
Total antibodies (Ab) Specificity Pre‐pandemic
60. Test.
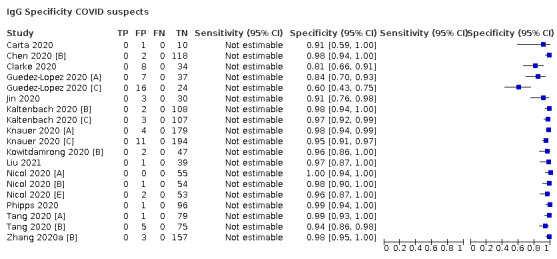
IgG Specificity COVID suspects
61. Test.
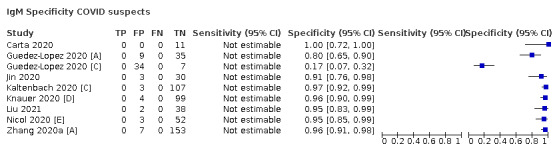
IgM Specificity COVID suspects
62. Test.
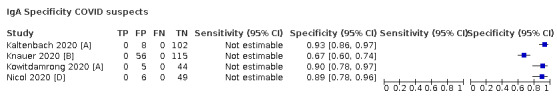
IgA Specificity COVID suspects
63. Test.
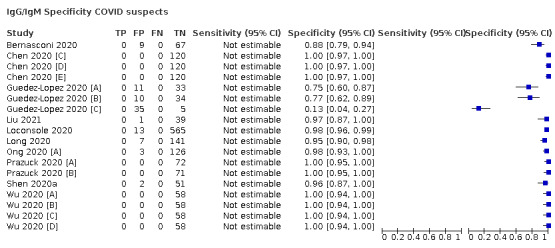
IgG/IgM Specificity COVID suspects
64. Test.

Total antibodies (Ab) Specificity COVID suspects
65. Test.
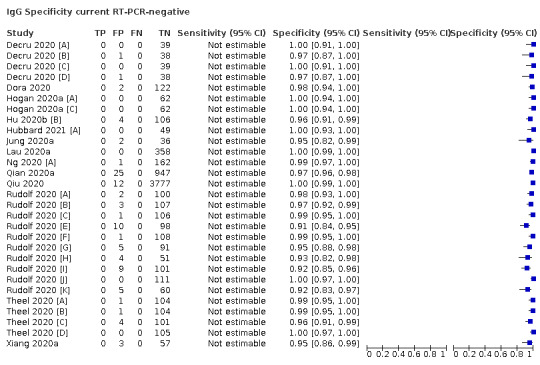
IgG Specificity current RT‐PCR‐negative
66. Test.
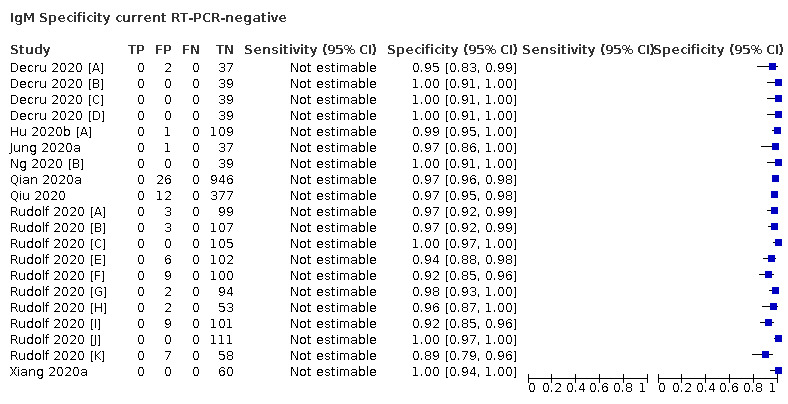
IgM Specificity current RT‐PCR‐negative
67. Test.

IgG/IgM Specificity current RT‐PCR‐negative
68. Test.

Total antibodies (Ab) Specificity current RT‐PCR‐negative
69. Test.
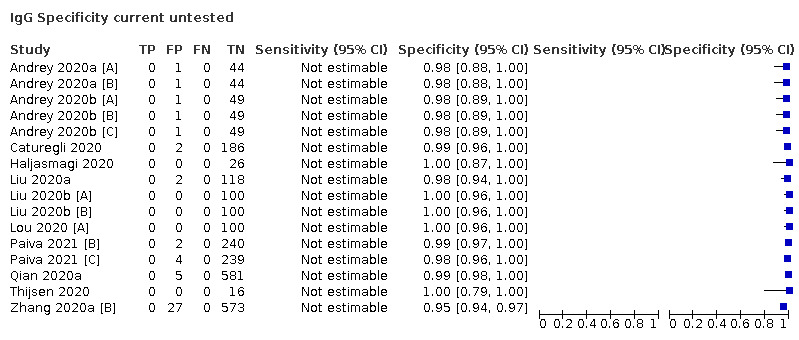
IgG Specificity current untested
70. Test.
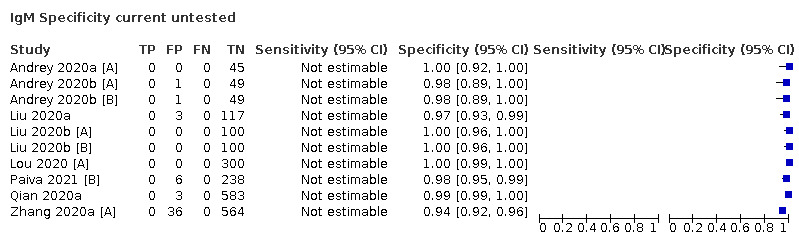
IgM Specificity current untested
71. Test.

IgA Specificity current untested
72. Test.
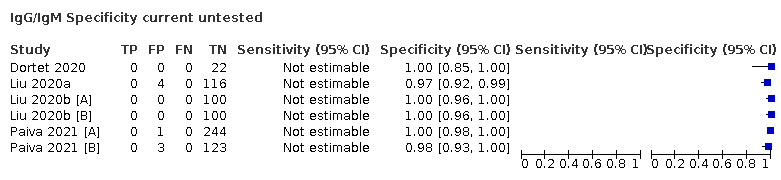
IgG/IgM Specificity current untested
73. Test.

Total antibodies (Ab) Specificity current untested
74. Test.
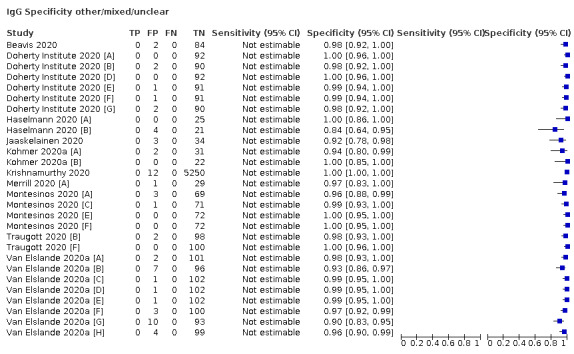
IgG Specificity other/mixed/unclear
75. Test.
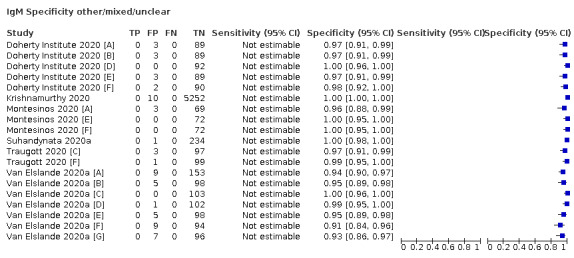
IgM Specificity other/mixed/unclear
76. Test.

IgA Specificity other/mixed/unclear
77. Test.

IgA/IgG Specificity other/mixed/unclear
78. Test.
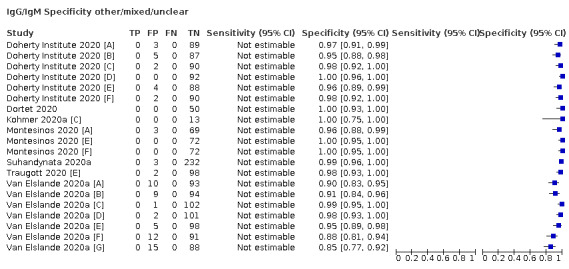
IgG/IgM Specificity other/mixed/unclear
79. Test.
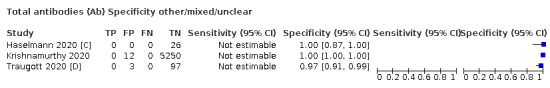
Total antibodies (Ab) Specificity other/mixed/unclear
80. Test.
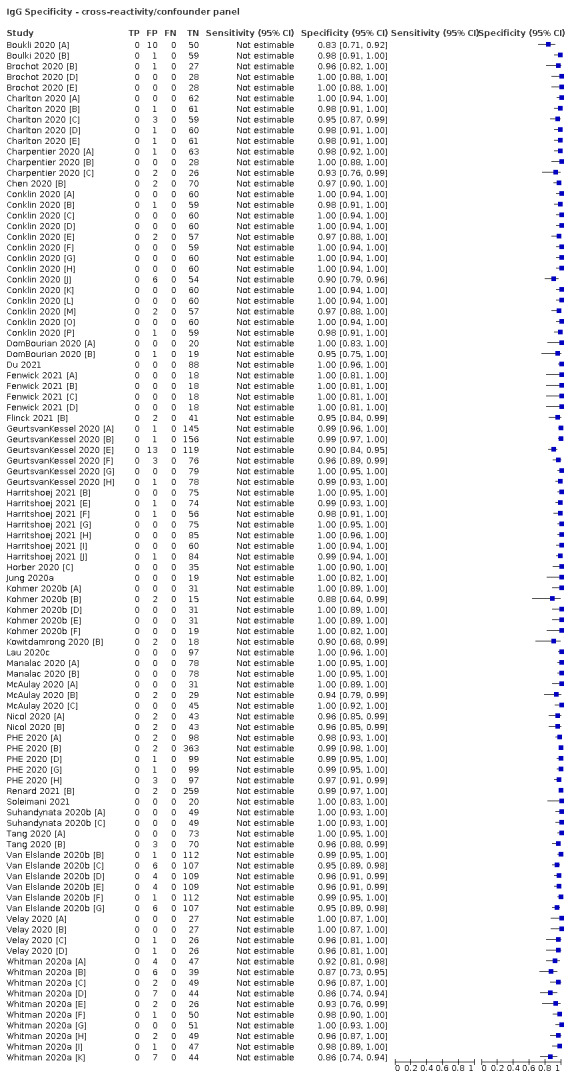
IgG Specificity ‐ cross‐reactivity/confounder panel
81. Test.
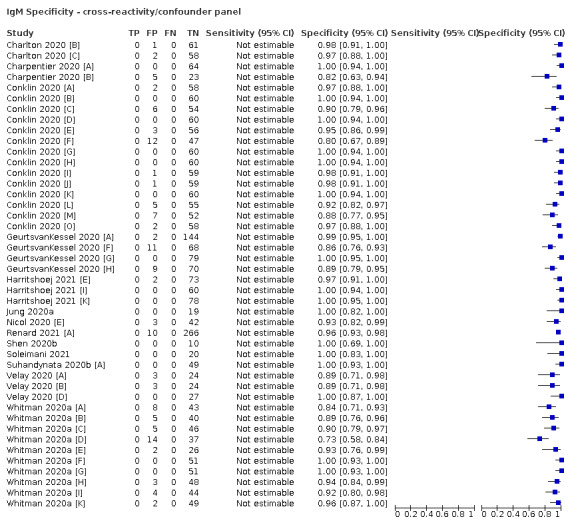
IgM Specificity ‐ cross‐reactivity/confounder panel
82. Test.
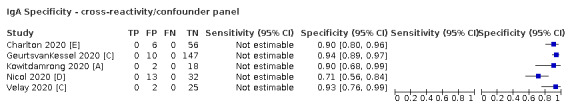
IgA Specificity ‐ cross‐reactivity/confounder panel
83. Test.
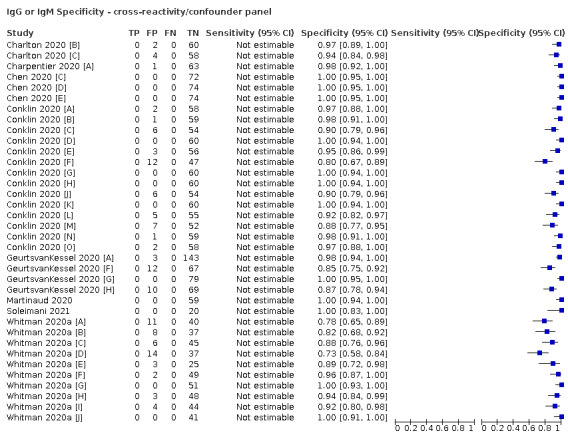
IgG or IgM Specificity ‐ cross‐reactivity/confounder panel
84. Test.

IgA or IgG Specificity ‐ cross‐reactivity/confounder panel
85. Test.
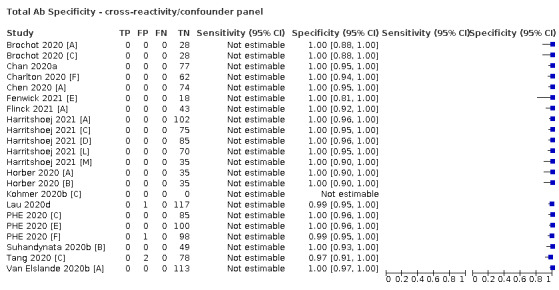
Total Ab Specificity ‐ cross‐reactivity/confounder panel
86. Test.
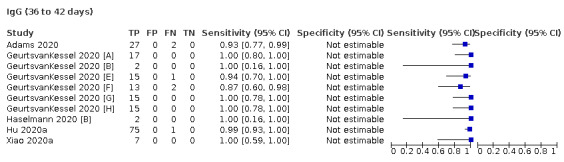
IgG (36 to 42 days)
87. Test.
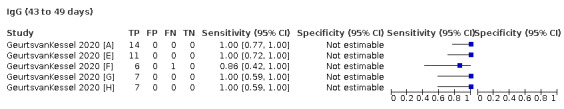
IgG (43 to 49 days)
88. Test.

IgG (50‐56 days)
89. Test.
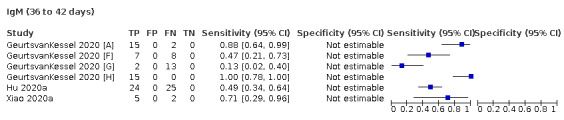
IgM (36 to 42 days)
90. Test.
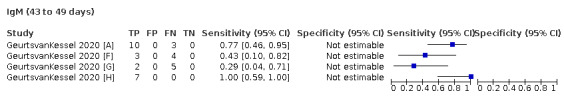
IgM (43 to 49 days)
91. Test.

IgM (50 to 56 days)
92. Test.

IgA (36 to 42 days)
93. Test.

IgA/IgG (36 to 42 days)
94. Test.
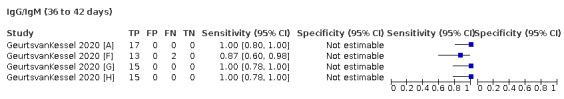
IgG/IgM (36 to 42 days)
95. Test.
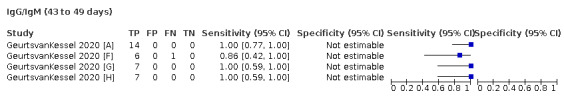
IgG/IgM (43 to 49 days)
96. Test.

IgG/IgM (50 to 56 days)
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adams 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase COVID‐19 infection Design: Two‐group design with sensitivity and specificity [1] Confirmed (RT‐PCR‐positive) COVID‐19 (n = 270 samples from 124 patients) [2] Pre‐pandemic bio‐banked serum samples from three sources (from 2018 to pre‐December 2019) (n = 564 samples) Recruitment: Unclear Prospective or retrospective: Cases prospectively enrolled Sample size: 834 (270) samples Further detail: Not further described |
||
| Patient characteristics and setting | Setting: Unclear (early validation conducted on inpatient samples but not clear for final validation study which included asymptomatic cases) Location: South West London Pathology Laboratory at St George’s University Hospital, London Country: UK Dates: Not stated; conducted subsequent to 26 March 2020 Symptoms and severity: 209/270 samples (77%) from 90/124 patients reporting symptoms of COVID‐19 61/270 samples (23%) from 34/124 individuals who were asymptomatic at first swab collection Demographics: age: range, 26‐88 years Sex: 74/124 (60%) male Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic Source: Hospital controls from 2018 to pre‐December 2019. Bio‐banked serum samples from Liverpool School of Tropical Medicine, Mologic, and St George’s University of London Characteristics: Not stated |
||
| Index tests | Test name: IgG COVID‐19 ELISA Manufacturer: Mologic Antibody: IgG Antigen target: NP and S2 antigens Evaluation setting: Laboratory‐based (South West London Pathology (SWLP) microbiology laboratory at St George’s, University Hospitals NHS Foundation Trust (SGHFT)) Test method: ELISA Timing of samples: [1] post‐symptom onset (range 1 to 54 days based on Fig 3) < 7: n = 16 (6%) (not reported but back‐calculated from Tabl 2B) >= 7‐14, n = 32, 12% >= 14‐21, n = 45, 17% >= 21‐28, n = 58, 21% >= 28‐35, n = 30, 11% >= 35, n = 29, 11% asymptomatic, n = 60, 22% [2] previously in hospital. Timing of samples: [1] post‐symptom onset (range 1 to 54 days based on Fig 3) < 7: n = 16 (6%) >= 7‐14, n = 32, 12% >=14 ‐ 21, n = 45, 17% >= 21‐28, n = 58, 21% >= 28‐35, n = 30, 11% >= 35, n = 29, 11% asymptomatic, n = 60, 22% Samples used: serum Test operator: Not stated Definition of test positivity: results were considered positive 'if they were 10% above the cut off value'; multiple thresholds reported in Suppl Appendix Blinding reported: Unclear Threshold predefined: Unclear |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Altona Diagnostics RealStar®SARS‐CoV‐2 RT‐PCR Kit detecting S and E genes from extracted RNA Samples used: Respiratory samples Timing of reference standard: Not reported Blinded to index test: Yes Incorporated index test: no Definition of non‐COVID cases: Pre‐pandemic controls Samples used: bio‐banked serum samples Timing of reference standard: Pre‐pandemic controls Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Serum samples obtained between 0 to 42 days post‐RT‐PCR
n = 53, 0‐7 days.
n = 215, ≥ 8 days.
n = 197, ≥ 10 days.
n = 159, 14‐42 days. All patients received same reference standard: No (different for cases and controls) Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples mainly (with a few results by patients) |
||
| Comparative | |||
| Notes | Funding: UK Department for International Development and Wellcome Trust Publication status: Pre‐print (not peer reviewed) Source: medRxiv Author COI: COI declared: SK is a member of the Scientific Advisory Committee for Foundation for Innovative New Diagnostics (FIND) a not‐for‐profit that produces global guidance on affordable diagnostics. The views expressed here are personal opinions and do not represent the recommendations of FIND. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Alvim 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of COVID‐19 current acute‐phase infection and current convalescent‐phase infection Design: This paper describes the validation of an in‐house test. Data were extracted only for the commercially available test. This is a single‐group analysis for sensitivity. [1] Confirmed COVID patients (437 samples) Recruitment: Not stated Prospective or retrospective: [1] Prospectively for samples collected at UFRJ COVID Screening and Diagnostic Center; unclear for samples collected at the State Hematology Institute Hemorio Sample size: 437 (437) Further detail: [1] Only symptomatic subjects who presented at least two of the following symptoms were included: loss of taste or smell, fever, shortness of breath, diarrhoea, headache, extreme tiredness, dry cough, sore throat, runny or stuffy nose, or muscle aches. PCR‐positive individuals who were followed along time |
||
| Patient characteristics and setting | Setting:
Not stated Location: State Hematology Institute Hemorio and UFRJ COVID Screening and Diagnostic Center, Federal University of Rio de Janeiro (UFRJ) Country: Brazil Dates: Not stated Symptoms and severity: Clinical characteristics of the patients not documented Demographics: Not available Exposure history: Not available Non‐Covid group 1: NA |
||
| Index tests | Test name: This paper primarily describes an in‐house assay. Data extraction was only performed for the commercially available test.
anti‐SARS‐COV‐2 IgG ELISA (#EI 2606‐9601 G) Manufacturer: Euroimmun Antibody: IgG Antigen target: S1 subunit of the spike‐protein of SARS‐COV‐2 Evaluation setting: Laboratory Test method: ELISA Timing of samples: Samples collected from D0 after symptom onset, up to 98 days after symptom onset 0‐5 days pso: n = 33 6‐10 days pso: n = 42 11‐15 days pso: n = 83 16‐20 days pso: n = 62 21‐25 days pso: n = 56 26‐30 days pso: n = 54 31‐98 days pso: n = 107 Samples used: Unclear Test operator: Not stated Definition of test positivity: Not stated (as per manufacturer's instructions) Blinding reported: Unclear (no as only COVID cases tested) Threshold predefined: As per manufacturer’s instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Unclear Timing of reference standard: Not stated Blinded to index test: yes, performed prior to index test Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: yes (more data reported in study that are not included in our review, e.g. non‐COVID samples for specificity results) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples (individuals who had tested positive by PCR and were followed over time) |
||
| Comparative | |||
| Notes | Funding: This work was supported by Senai CETIQT, Senai DN and CTG, and by the Brazilian research funding agencies Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento
Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Instituto Serrapilheira. DASR was supported by a fellowship from CNPq (DTI‐A; 401209/2020‐2). Publication status: Pre‐print (not peer reviewed) Source: medRxiv Pre‐print Author COI: We declare no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Andrey 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of COVID‐19 current acute‐phase infection and current convalescent‐phase infection Design: Two‐group study to determine sensitivity and specificity: [1] 46 real time RT‐PCR–confirmed COVID‐19 cases; [2] 45 unmatched control blood samples from asymptomatic donors without known exposure to SARS‐CoV‐2 Recruitment: [1] unclear [2] unmatched 1:1 case‐control study Prospective or retrospective: Unclear (prospectively as blood samples were analysed within 72 hours of sampling) Sample size: 91 (46) of which 57 (12) were eligible for our review Further detail: [1] COVID patients: RT‐PCR–confirmed COVID‐19 cases hospitalised at the University Hospitals of Geneva [2] Controls: Healthy blood donors, asymptomatic, no known exposure to SARS‐COV‐2, age 18‐65 years old, absence of known acute or chronic infection and without history of cancer, diabetes, or haematological disorders, as well as cardiovascular, autoimmune, inflammatory, chronic kidney or neurological disease |
||
| Patient characteristics and setting | Setting: hospital inpatients Location: University Hospitals of Geneva Country: Switzerland Dates: April 2020 Symptoms and severity: Stated moderate‐to‐severe critical COVID as mild COVID patients were not admitted to hospital. Demographics: Age ‐ median 66 years old, IQR 50.5 to 76) Males (n = 28, 60.9%) Exposure history: Unclear ‐ not stated Non‐Covid group 1: Healthy controls Source: University Hospitals of Geneva, April 2020 Characteristics: Median 47 years old, IQR 39.5‐55, 9 males (20%) Non‐Covid group 2: Asymptomatic donors without known exposure to SARS‐CoV‐2, who were not tested by RT‐PCR, since they did not meet the testing criteria of our institution These healthy donors met the blood donation criteria at our institution: age 18‐65 years old, absence of known acute or chronic infection and without history of cancer, diabetes, or haematological disorders, as well as cardiovascular, autoimmune, inflammatory, chronic kidney or neurological disease. Source: NA Characteristics: NA |
||
| Index tests | Test name: [A] Augurix SARS‐CoV‐2 IgM/IgG RDT [B] SARS‐CoV‐2 IgG ELISA (# EI 2606‐9601 G) Manufacturer: [A] Augurix, GaDia [B] Euroimmun AG, Lübeck, Germany Antibody: [A] IgM and/or IgG [B] IgG Antigen target: [A] Not stated [B] S1‐domain of the spike‐protein Evaluation setting: [A] POC performed in lab [B] Laboratory Test method: [A] Immunochromatographic cassette test (lateral flow test) [B] ELISA Timing of samples: Median 10 days (IQR 5‐15 days) post‐positive PCR results: 0‐6 days post‐positive PCR: n = 20 7‐14 days post‐positive PCR: n = 14 > 14 days post‐positive PCR: n = 12 Samples used: [A] 20 µL of whole blood (as a proxy for one capillary blood drop) and 10 µL of plasma were applied in parallel for each sample. [B] Plasma All analyses were performed within 72 hours of blood sampling. Test operator: [A] and [B] Lab personnel Definition of test positivity: [A] Test lines [B] The quantitative results (ratios) were then expressed in arbitrary units and interpreted following the cut‐offs derived from our validation study: OD ratio: < 0.5 = negative, ≥ 0.5 and < 1.5 = indeterminate, ≥ 1.5 = positive. Blinding reported: [A] Yes [B] Not stated, possibly yes Threshold predefined: [A] Yes [B] Cut‐offs derived from validation study |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR ‐ eMAG (bioMérieux, France)
and the Charité RT‐PCR protocol or the BD SARS‐CoV‐2 reagent kit for the BD Max system (Becton, Dickinson and Co, US) Cobas 6800 SARS‐CoV‐2 RT‐PCR (Roche). Threshold not stated Samples used: Nasopharyngeal secretions in 45/46; On one occasion, the RT‐PCR was carried out on a bronchial aspirate. Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test (as case‐control study) Incorporated index test: No Definition of non‐COVID cases: Non‐cases did not have RT‐PCR testing as it was stated that this did not meet the institutional standard for testing inclusion. Asymptomatic donors without known exposure to SARS‐CoV‐2 Samples used: None (untested) Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1] Among COVID‐19 patients, the median delay between a SARS‐CoV‐2 RT‐PCR diagnostic test and serology testing was 10 days (IQR 5‐15 days).
[2] Not stated All patients received same reference standard: No Missing data: Not stated for study analyses but 34/46 COVID samples excluded from review analyses Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients (Leftovers from blood specimens (whole blood and plasma) from single patients or controls, collected at a single time point) |
||
| Comparative | |||
| Notes | Funding:
Augurix RDTs were provided by Mr P. Ducret (GaDia, Switzerland). GaDia had no role in the study design and realisation nor in results interpretation.
This work was supported by the Division of Laboratory Medicine, HUG and the Geneva Centre for Emerging Viral Diseases. Publication status: Published paper Source: European Journal of Clinical Investigation Author COI: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Andrey 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Andrey 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of COVID‐19 current acute‐phase infection and current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] RT PCR confirmed samples from hospitalised patients (n = 41) [2] unmatched asymptomatic donors (n = 50) Recruitment: [1] Unclear [2] unmatched Prospective or retrospective: Unclear (prospectively as samples not frozen) Sample size: 91 (41) Further detail: No specific inclusion/exclusion criteria noted. [1] real‐time (RT)‐PCR confirmed COVID‐19 cases hospitalised at the University Hospitals of Geneva [2] Asymptomatic blood donors obtained during the same period (April 2020) Exclusion criteria not stated |
||
| Patient characteristics and setting | Setting: hospital inpatients Location: University Hospitals of Geneva, Switzerland Country: Switzerland Dates: April 2020 Symptoms and severity: Details on clinical characteristics not stated All hospitalised Demographics: Median 71 years old, (IQR 63–76) Males (n = 32, 78.1%) Exposure history: Not stated Non‐Covid group 1: Healthy controls Source: Asymptomatic donors, University Hospitals of Geneva, April 2020 Characteristics: Median 47 years old, IQR 40–55 Males (n = 11, 22.0%) Healthy donors, asymptomatic Non‐Covid group 2: NA |
||
| Index tests | Test name:
[A] NTBIO RDT (test name not stated)
[B] Orient‐Gene RDT (test name not stated)
[C] MEDsan RDT (test name not stated)
[D] Euroimmun IgG ELISA (# EI 2606‐9601 G) Manufacturer: [A] NTBIO Diagnostics Inc, Surrey, British Columbia, Canada [B] Zhejiang Orient‐Gene Biotech Co. Ltd., Huzhou, China [C] MEDsan GmbH, Biological Health Solutions, Hamburg, Germany [D] Euroimmun AG, Lübeck, Germany Antibody: [A]‐[C] IgM and/or IgG [D] IgG Antigen target: [A] Full spike‐protein [B] Not stated [C] N‐ and S‐based [D] S1 domain of spike‐protein Evaluation setting: [A]‐[C] POC performed in a laboratory environment [D] Laboratory Test method: [A]‐[C] Immunochromatographic method, lateral flow assay [D] ELISA Timing of samples: Median 22 days (IQR 13–31 days) post‐positive PCR: 0‐14 days post‐PCR+: n = 14 > 14 days post‐PCR+: n = 27 Samples used: [A]‐[C] Whole blood and plasma [D] Plasma Test operator: Lab personnel (technical assistance acknowledged) Definition of test positivity: [A]‐[C] Test lines [D] The quantitative results (ratios) obtained were then expressed in arbitrary units and interpreted following the recently published proposed cut‐offs derived from our local validation process: OD ratio: < 0.5 = negative, ≥ 0.5 and < 1.5 = indeterminate, and ≥ 1.5 = positive Blinding reported: Not explicitly stated Threshold predefined: Yes [A]‐[C] Visual‐based [D] Cut‐offs derived from our local validation process (recently published) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR ‐ eMAG (bioMérieux, France) and the Charité RT‐PCR protocol or BD SARS‐CoV‐2 reagent kit for the BD Max system (Becton, Dickinson and Co, US) Cobas 6800 SARS‐CoV‐2 RT‐PCR (Roche). Samples used: Not clearly stated Timing of reference standard: Unclear Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: Asymptomatic, untested Samples used: None as untested Timing of reference standard: Not stated (untested, asymptomatic) Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: The median delay between a positive SARS‐CoV‐2 RT‐PCR and serology testing was 22 days (IQR 13–31 days). All patients received same reference standard: No ([1] PCR‐tested, [2] Asymptomatic) Missing data: yes (1 invalid result for test [B] excluded; 14 samples taken 0‐14 days post‐PCR+ not eligible for review) Uninterpretable results: yes, 1 invalid result for test [B] excluded Indeterminate results: [A]‐[C] No indeterminate range [D] Indeterminate ELISA IgG results were considered to be negative for the test performances. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This work was supported by the Division of Laboratory Medicine, Geneva University Hospitals, Geneva,
Switzerland and the Centre for Emerging Viral Diseases, Geneva University Hospitals and Faculty of Medicine,
Geneva, Switzerland.
We thank Christine Kopp and Thomas Büeler (Swiss Red Cross) and Didier Trono (Swiss
National COVID‐19 Science Task Force and EPFL) for providing the rapid tests. Publication status: Published paper Source: Journal of Clinical Medicine Author COI: None stated The authors declared no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Andrey 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Andrey 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Bartolini 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Evaluation of two different immunochromatographic (IC) rapid tests for detection of IgG and IgM against SARS‐CoV‐2 Design: RT‐PCR‐positive asymptomatic/mildly symptomatic healthcare workers (n = 151) Recruitment: All 151 RT‐PCR‐positive cases from a mass screening of 10,945 asymptomatic/mildly symptomatic healthcare workers at the Local Health Unit of Bologna taking part in a seroprevalence study (surveillance programme established by the Emilia‐Romagna Region). Due the contacts caused by the type of work done, some of them were considered much more exposed to risk of infection; for this reason they had previously been submitted to RT‐PCR. Prospective or retrospective: Prospectively in a mass screening programme (surveillance programme). The samples were collected after informed consent was given. Sample size: 151 (151) of which 35 (35) were tested with test [1] and 116 (116) tested with test [2] Further detail: RT‐PCR‐positive |
||
| Patient characteristics and setting | Setting: Healthcare workers Location: Local Health Unit of Bologna Country: Italy Dates: Serological tests done between 3rd to 27th of April 2020 RT‐PCR tests done 0‐45 days before Symptoms and severity: asymptomatic/mildly symptomatic Demographics: Not stated Exposure history: Not stated (Due the contacts caused by the type of work done, some of them were considered much more exposed to risk of infection) Non‐Covid group 1: NA |
||
| Index tests | Test name: [1] KHB® Diagnostic Kit for SARS‐CoV‐2 IgM/IgG Antibody (Colloidal Gold) [2] Cellex qSARS‐CoV‐ 2 IgG/IgM Cassette Rapid Test Manufacturer: Not stated Antibody: [1] IgG. IgM [2] IgG. IgM Antigen target: Not stated ‐ SARS‐CoV‐2 conjugate Evaluation setting: [1] POC, used in a laboratory [2] POC, used in a laboratory Test method: [1] lateral flow chromatographic immunoassay (Colloidal Gold) [2] lateral flow chromatographic immunoassay Timing of samples: 0‐45 days after positive RT‐PCR ([1]/[2]) Samples used: plasma Test operator: Laboratory personnel Definition of test positivity: Both tests: The presence of the captured immunocomplex is visible due to its precipitation in a coloured red band. Blinding reported: No ‐ all samples RT‐PCR‐positive Threshold predefined: Yes, as per manufacturer, visual |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 RT‐PCR Samples used: nasopharyngeal or oropharyngeal samples Timing of reference standard: Not stated (mostly asymptomatic patients) Blinded to index test: Yes ‐ prior Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: 0‐45 days All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Pre‐print (not peer reviewed) Source: medRxiv preprint doi: https://doi.org/10.1101/2020.05.28.20116046 Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Bartolini 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Beavis 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: Two‐group design to assess sensitivity and specificity: [1] COVID‐19 PCR+ve patients (n = 82) [2] COVID‐19 PCR‐ve patients (n = 86) Recruitment: unclear Prospective or retrospective: retrospective Sample size: 168(82) Further detail: Inclusion [1] Samples RT‐PCR‐positive for SARS‐CoV‐2. [2] Samples RT‐PCR‐negative for SARS‐CoV‐2. Ambulatory and pre‐pandemic. Exclusion: [1] [2] not stated |
||
| Patient characteristics and setting | Setting: Clinical Laboratories Location: University of Chicago Medicine Country: USA Dates: March to May 2020 Symptoms and severity: not stated Demographics: not stated Exposure history: not stated Non‐Covid group 1: COVID‐19 PCR‐ve patients Source: 70 samples collected from ambulatory patients at the University of Chicago from March to May 2020. 16 collected in early 2019, pre‐pandemic Characteristics: 28 of these patients had tested positive for common coronavirus strains (HKU1 n = 6, NL63 n = 10, OC43 n = 9, 229E n = 1, and both 229E and OC43 n = 1). |
||
| Index tests | Test name: [A] EUROIMMUN Anti‐SARS‐CoV‐2 ELISA IgG Assay [B] EUROIMMUN Anti‐SARS‐CoV‐2 ELISA IgA Assay Manufacturer: [A] [B] EuroImmun Antibody: [A] IgG [B] IgA Antigen target: [A] [B] S1 domain Evaluation setting: laboratory test Test method: Enzyme‐Linked Immunosorbent Assay (ELISA) Timing of samples: 0 to 49 days after PCR testing Samples used: serum or EDTA plasma Test operator: not stated Definition of test positivity: Ratio ≥ 1.1 positive, Ratio ≥ 0.8 to < 1.0 borderline, < 0.8 negative Blinding reported: unclear Threshold predefined: unclear |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Nasopharyngeal and nasal mid‐turbinate Timing of reference standard: not stated Blinded to index test: yes, prior Incorporated index test: no Definition of non‐COVID cases: RT‐PCR or pre‐pandemic Samples used: Nasopharyngeal and nasal mid‐turbinate Timing of reference standard: not stated Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: 0‐49 days post‐PCR All patients received same reference standard: not stated Missing data: not stated Uninterpretable results: not stated Indeterminate results: borderline results were considered positive for analysis Unit of analysis: samples |
||
| Comparative | |||
| Notes | Funding: study was internally funded Publication status: Published paper Source: Journal of Clinical Virology Author COI: All authors declared no conflict of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Bernasconi 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection of SARS‐CoV‐2
3‐group study to estimate sensitivity and specificity for diagnosis of acute disease and convalescent infection Design: Three groups for two comparisons (COVID cases versus same time period controls or pre‐pandemic controls): [1] COVID‐19 positive ED patients (n = 67) [2] COVID‐19 negative ED patients (with clinical suspicion of acute airway infection) (n = 76, see comment) [3] COVID‐19 negative historical pre‐pandemic controls (n = 100) Recruitment: Unclear [1] and [2] Symptomatic patients presenting to the emergency department of 1 hospital; patients’ enrolment was based on clinical suspicion of acute airway infection. [3] SARS‐CoV‐2 seronegative samples collected between May and October 2018 Prospective or retrospective: Pandemic cases/controls, prospective (consent was obtained from participants). Pre‐pandemic controls, retrospective Sample size: Same time period comparison [1 and [2]: 143 (67) All 3 groups: 243 (67) patients with 315 (135) samples of which 243 (67) samples were included in our analysis Further detail: ED cases/controls: "symptomatic patients presenting to the ED of the Kantonsspital Aarau, Switzerland from March to April 2020... Patients’ enrolment was based on clinical suspicion of acute airway infection." Pre‐pandemic controls: Unclear.("SARS‐CoV‐2 seronegative samples collected between May and October 2018") |
||
| Patient characteristics and setting | Setting: Emergency department Location: Kantonsspital Aarau AG, Tellstrasse 25, 5001 Aarau, Switzerland Country: Switzerland Dates: March‐April 2020 Symptoms and severity: "symptomatic" Demographics: Not reported by COVID‐19 status. Whole ED case/control population (n = 143): Sex Male 84 (59%), female 59 (41%) Age, Median, years 69 (range 22‐95 years) Exposure history: not stated Non‐Covid group 1: [2] Contemporaneous ED controls Source: Emergency department, March‐April 2020 Characteristics: Not reported by COVID‐19 status Whole ED case/control population (n = 143): Sex Male 84 (59%), female 59 (41%) Age, Median, years 69 (range 22‐95 years) Non‐Covid group 2: [3] Pre‐pandemic controls Source: Source unclear, May‐October 2018 Characteristics: Not stated |
||
| Index tests | Test name: Maccura LFIA
SARS‐CoV‐2 IgM/IgG Manufacturer: Maccura Biotechnology, Chengdu, China Antibody: IgM, IgG Antigen target: recombinant spike and nucleocapsid proteins of the SARS‐CoV‐2 Evaluation setting: POC, tested at ED and during hospitalisation (figure 1) Test method: lateral flow immunochromatography assay (LFIA) Timing of samples: [1] COVID‐19‐positive patients (n = 67): < 7 days onset (n = 21), ≥ 7 days onset (n = 46) Fig 1 reported on 135 samples from 1‐31 days pso. [2] COVID‐19 negative patients ‐ not stated Timing of samples: [1] COVID‐19‐positive patients (n = 67): < 7 days onset (n = 21), ≥ 7 days onset (n = 46) Fig 1 reported on 135 samples from 1‐31 days pso. [2] COVID‐19 negative patients ‐ not stated Samples used: not stated Test operator: not stated Definition of test positivity: not stated Blinding reported: Yes, since antibody/RT‐PCR tests were done in parallel and antibody test results are faster than RT‐PCR Unclear for samples taken during hospitalisation Threshold predefined: not stated (visually based) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (Seegene Inc., Seoul, Republic of Korea)
Diagnosis of COVID‐19 was based on clinical, microbiological and radiological criteria according to in‐house, national and international recommendations and guidelines. Samples used: nasopharyngeal swab samples (transportation medium ESwab, Copan Italia, Brescia, Italy) or nasopharyngeal fluid Timing of reference standard: [1] COVID‐19‐positive patients (n = 67): < 7 days onset (n = 21), ≥ 7 days onset (n = 46) Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: [2] COVID‐19 negative patients, RT‐PCR (Seegene Inc., Seoul, Republic of Korea) [3] pre‐pandemic controls Samples used: [2] COVID‐19 negative patients, nasopharyngeal swab samples or nasopharyngeal fluid [3] pre‐pandemic controls, not stated Timing of reference standard: [2] COVID‐19‐positive patients ‐ not stated [3] pre‐pandemic controls Blinded to index test: [2] Not stated [3] yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1] and [2] LFIA and molecular testing for SARS‐CoV‐2 by RT‐PCR … was done in parallel for 67 samples.
Unclear for the remaining 72 samples (215 samples from 143 patients)
[3] Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: None Indeterminate results: Not stated Unit of analysis: Samples for [1] and [2] (215 samples of 143 ED patients) |
||
| Comparative | |||
| Notes | Funding: None declared Publication status: Published letter Source: Clinical Chemistry & Laboratory Medicine Author COI: Authors stated no conflict of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Bettencourt 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Single‐group study to estimate sensitivity [1] Confirmed COVID cases (66 patients) Recruitment: 66 consecutive patients in a real‐life study performed in a hospital partially devoted to COVID‐19 infection Prospective or retrospective: Prospectively Sample size: 66 (66) Further detail: Inclusion: Patients with COVID‐19 disease, which diagnosis was based on clinical evaluation and positive RT ‐PCR SARS‐CoV‐2 identification. Patients in the recovery phase of infection, after the resolution of symptoms and a negative result for the first RT‐PCR test Exclusion: Not stated |
||
| Patient characteristics and setting | Setting: Convalescent, hospital inpatients Location: Hospital partially devoted to COVID‐19 infection (Hospital CUF Porto, Faculdade de Medicina da UP, Unidade de Investigação Cardiovascular da FMUP, Portugal) Country: Portugal Dates: Not stated Symptoms and severity: 37 mild disease, 26 moderate disease, 3 severe disease. Overall median time of symptoms was 7 days. Demographics: median age was 59.5 years (44–70). 32/66 women Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: Biozec COVID‐19 IgM/IgG Rapid Test lateral flow immunoassay (LFIA) Manufacturer: Biozec (Inzec) Antibody: IgM and IgG Antigen target: Not stated Evaluation setting: POCT, unclear how performed Test method: Lateral flow immunoassay Timing of samples: Mean 20.5 days (18–23) pso Samples used: Not stated Test operator: Not stated Definition of test positivity: Visually based Blinding reported: Not stated, possibly no as only COVID cases included Threshold predefined: Performed according to the manufacturer’s instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: yes Missing data: Not stated (no results for IgM reported though) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published letter Source: Journal of Infection Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Bond 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnostic performance evaluation for multiple COVID‐19 tests Design: Multi‐group study estimating both sensitivity and specificity. [1A] Symptomatic RT‐PCR confirmed COVID‐19 cases (n = 91 patients) [1B] Symptomatic COVID‐19‐negative on single RT‐PCR (n = 1217 patients) [2] Pre‐pandemic controls obtained in 2018 (n = 56 patients) Group 1B only used to assess specificity for one test Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 1400 (91) Note: the total sample size reported above only applied to one test, for which sera from 1217 COVID‐19 negative subjects were used to further assess specificity. Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Both in‐ or outpatients. All serum samples were collected in a tertiary hospital or a state reference laboratory; mild cases were not hospitalised. Location: Royal Melbourne Hospital and Victorian Infectious Diseases Reference Laboratory Country: Australia Dates: Dates not reported but likely collected in the first semester of 2020 Symptoms and severity: 71 mild (not hospitalised), 17 moderate (hospitalised, non‐ICU) and 3 severe cases (ICU). Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Group [1B] symptomatic COVID‐19 negative Source: Subjects presenting to the hospital emergency department between Feb 6th and Apr 15th, 2020 Characteristics: Not stated Non‐Covid group 2: Group [2] ‐ 56 pre‐pandemic controls obtained in 2018 Source: Pre‐pandemic specimens collected in 2018 Characteristics: Not stated |
||
| Index tests | Study evaluated multiple assays; timing pso provided only for one of them; remainder were excluded Test name: EUROIMMUN Anti‐SARS‐CoV‐2 ELISA (IgA or IgG) [Assays from [A] CTK Biotech Inc. (China), [B] VivaChek Biotech (Hangzhou) Co. Ltd. (China), [C] Hangzhou Alltest Biotech Co. Ltd. (China), [D] Guangzhou Wondfo Biotech Co. Ltd. (China), [E] Hightop Biotech (China) all excluded] Manufacturer: [F] & [G] EUROIMMUN AG Manufacturer: EUROIMMUN AG Antibody: IgA or IgG Antigen target: S1 domain of the spike‐protein Evaluation setting: lab test, done in lab Test method: Enzyme‐Linked Immunosorbent Assay (ELISA) Timing of samples: Any time point (229 samples); > 14 days (157 samples) Samples used: Serum Test operator: Not stated Threshold: ratio < 0.8, negative result; (2) ratio ≥ 0.8 to < 1.1, borderline result; and (3) ratio ≥ 1.1, positive result Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Group [1A]: Coronavirus Typing Assay (AusDiagnostics) followed by an unspecified confirmatory test at the state reference laboratory Samples used: Upper and/or lower respiratory tract specimens Timing of reference standard: Not stated but likely done before index test Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: Group [1B]: Single negative RT‐PCR Group [3]: no testing, pre‐pandemic sera Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Unclear Uninterpretable results: Unclear Indeterminate results: Unclear |
||
| Comparative | |||
| Notes | Funding: Work supported by a grant from the NHMRC Medical Research Future Fund. Some authors are recipients of the following: Investigator Grant from the National Health and Medical Research Council (NHMRC) of Australia; NHMRC Practitioner Fellowship; NHMRC Postgraduate Scholarship. Publication status: Published article Source: Academic journal Author COI: None |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Boukli 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection, and current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity, including: [1] PCR‐positive Covid‐19 patients in intensive care unit (76 samples from 49 patients) [2] PCR‐positive Covid‐19 patients, described as 'unselected' (68 samples from 68 patients) [3] 'unselected' pre‐pandemic samples (n = 40) [4] pre‐pandemic samples from cases with other infection (n = 60) Results are presented for group [1] and [2] combined, and separately for group [3] and [4]. Reported results suggest that not all samples were tested with both assays. Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 217 (117) Further detail: Not further described |
||
| Patient characteristics and setting | Setting: Mixed; included hospital inpatient (Intensive care unit) and an unspecified setting Location: Saint‐Antoine Hospital, Paris Country: France Dates: Not stated Symptoms and severity: Not stated; 68/144 COVID‐19 samples from individuals in ICU Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic other infection Source: Pre‐pandemic Characteristics: Coronavirus 229E, NL63, OC43 (n = 10); primary CMV (n = 5); primary EBV (n = 10); acute HAV (n = 5); acute HBV (n = 4); acute HCV (n = 3); acute HEV (n = 5); acute HIV (n = 5); influenza A/B (n = 10); acute malaria (n = 3) Non‐Covid group 2: Pre‐pandemic 'unselected' samples Source: Pre‐pandemic Characteristics: No further details |
||
| Index tests | Test name: [A] Liaison SARS‐CoV‐2 S1/S2 IgG assay; [B] Alinity I SARS‐CoV‐2 IgG assay Manufacturer: [A] DiaSorin, Antony, France [B] Abbott Diagnostics, Rungis, France Antibody: [A] and [B] IgG Antigen target: [A] Recombinant S1 and S2 proteins; [B] capsid antigen Evaluation setting: Laboratory based Test method: CLIA Timing of samples: Not stated Day 1 to day 30 pso Samples used: Group [2], [3], [4] serum, Group [1] plasma; samples stored at ‐20 or ‐80°C Test operator: Not stated Definition of test positivity: [A] Negative was defined as < 12 absorbance units (AU)/mL, positive as > 15 AU/mL, and equivocal as 12 to 15 AU/mL; [B] positive was defined as > 1.4 index, negative was defined as < 1.4 index Equivocal results were re‐tested Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; no further details Samples used: Not stated Timing of reference standard: During hospital stay in 49 cases. The rest unclear Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: RT‐PCR‐negative Pre‐pandemic samples used Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No (pre‐pandemic did not have SARS‐CoV‐2 PCR) Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published letter Source: Journal of Clinical Microbiology Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Boulki 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Brochot 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase COVID‐19 infection Design: Multi‐group study estimating sensitivity and specificity including: [1] patients hospitalised for COVID‐19 (n = 20); [2] non‐hospitalised patients but PCR confirmed with SARS‐CoV‐2 (n = 58); [3] patients participating in screening campaigns, also described as outpatients with no history of SARS‐CoV‐2 infection (n = 62); [4] and samples from patients with a history of other seasonal coronavirus infections (n = 28). Study focused mainly on agreement between evaluated assays; data could be extracted for samples with PCR+ result (from group [1] and group [2]) at two time points and for non‐COVID‐19 cases (group [3]) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 168 (78) Further detail: Not further described |
||
| Patient characteristics and setting | Setting: Hospital in patients, outpatients and community screening Location: Amiens University medical Center, Amiens Country: France Dates: Not stated Symptoms and severity: Not stated Demographics: Available in the supplement. Could not open the file*** Exposure history: Not stated Non‐Covid group 1: Outpatients with no history of SARS‐CoV‐2 ***Review authors think that some of these have had Covid but have not had it PCR‐confirmed ‐ if you look at Fig 2, quite a number of samples have positive serology results, too many to all be false positives. What we did in this case, was to report the group in item A2 (as publication authors have done) but then we did not use the data because there was apparently no reference standard for them. Source: During pandemic Characteristics: Non‐Covid group 2: Other human coronavirus infections Source: Not clearly described; may be pre‐pandemic Characteristics: Not stated |
||
| Index tests | Test name: Assays identified only by manufacturer:
[A] Abbott; [B] Biorad; [C] Euroimmun; [D] Liaison; [E] Wantai Manufacturer: [A] Abbott; [B] Biorad; [C] Euroimmun; [D] Liaison; [E] Wantai Antibody: [A] IgG; [B] total antibodies; [C] IgG; [D] IgG; [E] total antibodies Antigen target: [A] nucleocapsid; [B] nucleocapsid; [C] spike 1; [D] spike1/2; [E] receptor binding domain Evaluation setting: Laboratory Test method: [A] CLIA; [B] ELISA; [C] ELISA; [D] CLIA; [E] ELISA Timing of samples: Time pso not given; number of samples by time post‐PCR+ given only for day 31‐50 (n = 21) and > 50 days (n = 14) Samples used: Serum Test operator: Not stated Definition of test positivity: Positivity thresholds were as follows: [A] Abbott >= 1.4; [B] Biorad >= 1; [C] Euroimmun >= 1.1; [D] Liaison >= 15; [E] Wantai >= 1 Samples with a 'doubtful' signal were tested a second time; if the same result was obtained, result was considered negative Blinding reported: Not stated Threshold predefined: Yes, manufacturer defined thresholds used |
||
| Target condition and reference standard(s) | Reference standard: PCR; no further details Samples used: Nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: Not stated Definition of non‐COVID cases: PCR for group 3. Pre‐pandemic for group 4 Samples used: Nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: Not stated Yes; conducted first (and was basis for selection of samples for testing) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Not stated; referred to selection of 'samples' but also stated that longitudinal data not available (Discussion) |
||
| Comparative | |||
| Notes | Funding: This work was supported by a grant from the Amiens University Medical Center Publication status: Published paper Source: Journal of Clinical Virology Author COI: Authors declared no COI present |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Brochot 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Brochot 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Brochot 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Brochot 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Bryan 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Single‐group study to estimate sensitivity for diagnosis of acute Covid‐19 Design: [1] PCR‐positive Covid cases (245) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 245 (245) of which 41 (41) had extractable outcome data from Fig 2 Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Hospital inpatients (n = 194), emergency department (n = 39), outpatients (n = 12) Location: University of Washington Medicine Hospitals, Seattle, Washington Country: USA Dates: Unclear Symptoms and severity: Unclear (8/245 asymptomatic at the time of initial PCR result) Demographics: Sex: 147/245 (60%) male Age: 10‐20: 1/245 (0.4%) 20‐29: 12/245 (4.9%) 30‐39: 17/245 (6.9%) 40‐49: 27/245 (11.0%) 50‐59: 42/245 (17.1%) 60‐69: 46/245 (18.8%) 70‐79: 52/245 (21.2%) 80‐89: 30/245 (12.2%) 90‐99: 18/245 (7.3%) Exposure history: Unclear Non‐Covid group 1: NA |
||
| Index tests | Test name: Abbott Architect anti‐SARS‐CoV‐2 nucleocapsid IgG Manufacturer: Abbott, USA Antibody: IgG Antigen target: Nucleocapsid Evaluation setting: Laboratory Test method: chemiluminescent microparticle immunoassay (CMIA) Timing of samples: < 7 days post‐symptom onset: 24/41 7‐10 days post‐symptom onset: 10/41 11‐14 days post‐symptom onset: 2/41 > 14 days post‐symptom onset: 5/41 Samples used: Serum or plasma Test operator: Unclear Definition of test positivity: Manufacturer's suggested cut‐off of 1.40 was used for seropositivity Blinding reported: Unclear (but study only included cases) Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: qRT‐PCR Samples used: Nasopharyngeal swabs Timing of reference standard: Unclear Blinded to index test: yes, performed before index test Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Unclear |
||
| Comparative | |||
| Notes | Funding: This work was supported by the Department of Laboratory Medicine at the University of Washington Medical Center. Publication status: Pre‐print (not peer reviewed) Source: medRxiv Author COI: ALG reported personal fees from Abbott Molecular, outside of the submitted work. AB, SLF, MAG, GP, AC, MHW, CM, KRJ, PCM reported no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Bundschuh 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: The aim of this study was to evaluate the effectiveness of the EDI ELISA test for the detection of SARS‐CoV‐2 IgM and IgG antibodies in human plasma.
3‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: Three‐group study: [1] RT‐PCR‐positive COVID‐19 patients admitted for treatment at two tertiary hospitals (n = 64) [2] Healthy blood donors (pre‐pandemic, n = 200) [3] Medical intensive care patients (pre‐pandemic, n = 256) Recruitment: [1] SARS‐CoV‐2 RT‐PCR (from respiratory specimens) confirmed COVID‐19 patients that were treated in one of the two tertiary care hospitals. Blood samples for clinical routine that were sent to central laboratory were included in the present study (frozen, leftover plasma). [2] First 200 consecutive EDTA plasma samples from our previously described cohort of healthy blood donors [3] 256 consecutive baseline EDTA plasma samples of patients admitted to the medical intensive care unit of the Konventhospital Barmherzige Brueder Linz, Austria Prospective or retrospective: [1] Unclear [2] and [3] retrospective Sample size: 520 (64) patients with 560 (104) samples Further detail: [1] All COVID‐19 patients admitted for treatment at two tertiary hospitals. Criteria unclear [2] Healthy blood donors. [3] Medical intensive care patients |
||
| Patient characteristics and setting | Setting: Hospital inpatients, two tertiary care hospitals Location: Konventhospital Barmherzige Brueder Linz and Ordensklinikum Linz Barmherzige Schwestern in Linz, Austria Country: Austria Dates: Between 15th of March 2020 and 10th of April 2020 Symptoms and severity: Not stated Demographics: 64 patients (53 males, 11 females), median age 65 years (range 14–95, IQR 56–87, years) Exposure history: Not stated Non‐Covid group 1: [2] Healthy blood donors Source: Recruited at the Red Cross organisation in Linz, Austria from January 31st to February 13th 2008 Characteristics: 3% immune‐compromised Non‐Covid group 2: [3] Intensive care patients Source: Intensive care unit of the Konventhospital Barmherzige Brueder Linz Characteristics: Intensive care patients |
||
| Index tests | Test name: EDI Novel Coronavirus COVID‐19 IgM and IgG ELISA kit Manufacturer: Epitope Diagnostics Inc. Antibody: IgM, IgG Antigen target: nucleocapsid protein of SARS‐CoV‐2 Evaluation setting: Laboratory (ELISA), used in laboratory Test method: Enzyme‐Linked Immunosorbent Assay (ELISA) Timing of samples: < 5 days‐22 days after symptom onset (COVID‐19 patients). Results were reported for 4 time bands Samples used: Plasma Test operator: Laboratory staff Definition of test positivity: Single run: If the patient sample OD (optical density) was below the negative cut‐off the result was reported negative (‐); If the patient sample OD was above the negative cut‐off but below the positive cut‐off the result was reported borderline (+‐); If the patient sample OD was above the positive cut‐off the patient was reported as positive (+). Blinding reported: Not stated Threshold predefined: following the manufacturers instruction |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: respiratory specimens Timing of reference standard: Not stated Blinded to index test: Yes, done prior index test Incorporated index test: No. Different specimens and tests Definition of non‐COVID cases: [2] and [3] pre‐pandemic Samples used: [2] and [3] pre‐pandemic Timing of reference standard: [2] and [3] pre‐pandemic Blinded to index test: Yes, done prior index test Incorporated index test: No, pre‐pandemic samples |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: If the patient sample was above the negative cut‐off but below the positive cut‐off the result was reported borderline ‐ these have not been extracted to the 2 x 2 sensitivity/specificity tables, and have accordingly been subtracted from group denominators. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Published paper Source: Clinica Chimica Acta Author COI: The authors declared that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Butterfield 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase COVID‐19 Design: Multi‐group study estimating sensitivity and specificity: [1] SARS‐CoV‐2 real‐time PCR‐positive patients (42 samples from 37 patients) [2] Pre‐pandemic samples from patients with viral infections (n = 102) or [3] attending routine antenatal testing (n = 20) Recruitment: Unclear Prospective or retrospective: Unclear Sample size: Patients: 159 (37); samples: 164 (42) Further detail: No further details reported |
||
| Patient characteristics and setting | Setting: Jamaica National Influenza Centre. No further details available Location: Jamaica National Influenza Centre Country: Jamaica Dates: Not stated Symptoms and severity: Disease severity for PCR+ was classified according to WHO criteria: 34/42 (81%) moderate, severe or critical; 8 (19%) asymptomatic or mild Demographics: Age and sex not reported Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic patients with viral infections Source: Pre‐pandemic (University of the West Indies Virology Laboratory) Characteristics: Influenza A/B, parainfluenza, EBV, CMV, HTLV I/II, DENV, CHIKV, ZIKV, HBV, HCV, Parvovirus B19 Non‐Covid group 2: [3] Healthy donors Source: Pre‐pandemic (University of the West Indies Virology Laboratory) Characteristics: Routine antenatal testing |
||
| Index tests | Test name: [A] Roche Elecsys1 Anti‐SARS‐CoV‐2, [B] Abbott Architect SARS‐CoV‐2 IgM, [C] Abbott Architect SARS‐CoV‐2 IgG, [D] Euroimmun SARS‐CoV‐2 IgA, [E] Euroimmun SARS‐CoV‐2 IgG ELISA, [F] Trillium IgG/IgM rapid assays Manufacturer: [A] Roche [B] Abbott [C] Abbott [D] Euroimmun [E] Euroimmun [F] Trillium Antibody: [A] Total Ab; [B] IgM; [C] IgG; [D] IgA; [E] IgG; [F] IgG/IgM Antigen target: Not stated Evaluation setting: [A‐E] Laboratory, [F] POC; all evaluations were laboratory‐based Test method: [A‐C] CLIA, [D‐E] ELISA, [F] Lateral flow assay Timing of samples: Symptomatic: 6–103 days pso; Asymptomatic: 20–69 days post‐PCR+ Samples used: Blood samples collected in tubes without anticoagulant Test operator: Not stated Definition of test positivity: As per manufacturer's instructions. For Euroimmun assays, borderline index values were considered negative. Blinding reported: Not stated Threshold predefined: Yes (as per manufacturer's instructions) |
||
| Target condition and reference standard(s) | Reference standard: Real time PCR using Charite Berlin protocol (Corman 2020) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Serum Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: Yes: number of samples for specificity estimates ranged from 90 to 122, either due to lack of sample volume or limited number of test kits; fewer test results also provided for Architect IgM (reason not given) Uninterpretable results: not reported Indeterminate results: For Euroimmun assays, borderline index values were considered negative. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. Publication status: agencies in the public, commercial, or not‐for‐profit sectors Source: International Journal of Infectious Diseases Author COI: All authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Butterfield 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Butterfield 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Butterfield 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Butterfield 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Butterfield 2021 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Candel 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: To analyse the accuracy of a point‐of‐care SARS‐CoV‐2 IgM and/or IgG rapid test for the diagnosis of COVID‐19, and to correlate this pattern of immune response with the severity of disease.
2‐group study to estimate sensitivity and specificity for diagnosis of active disease and identification of previous disease.
Only 1 group (sensitivity only) included in our review Design: Two‐group study: [1] randomly selected SARS‐CoV‐2 RT‐PCR confirmed patients (n = 35) [2] healthy volunteers with no history of COVID‐19 symptoms and negative SARS‐CoV‐2 RT‐PCR (n = 5) Group [2] excluded from review as <25 controls. Recruitment: [1] randomly selected SARS‐CoV‐2 RT‐PCR confirmed patients, admitted to IFEMA Field Hospital between April 27th and April 29th, 2020 [2] source of recruitment unclear Prospective or retrospective: Prospective Sample size: 40 (35) of which 35 (35) were eligible for our review Further detail: [1] positive RT‐PCR for pharyngeal swabs [2] healthy, nonsymptomatic, negative RT‐PCR |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: 1400‐bed field hospital set up at IFEMA (Institución Ferial de Madrid/Ferial Institution of Madrid) Country: Spain Dates: Recruitment April 27th to April 29th, 2020 Symptoms and severity: Mild = 3; Moderate = 9; Severe = 21; Critical = 2 12 (34.3%) mild‐moderate 23 (65.7%) severe‐critical 31/35 (88.6%) bilateral pneumonia Demographics: Female 21/35; mean age 58.2 years (COVID‐19 positive patients only) Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: Autobio rapid lateral‐flow point‐of‐care antibody test
Anti‐SARS‐CoV‐2 Rapid Test Manufacturer: Autobio Diagnostics Co. Zhengzhou, China Antibody: IgM, IgG Antigen target: SARS‐CoV‐2 recombinant spike‐protein antigen Evaluation setting: POC, used as POC Test method: Lateral flow immunoassay (colloidal gold) (CGIA) Timing of samples: The average time from the first day of reported symptoms to the lateral flow test was 28 days (SD: 8.7). The ranges were similar between the mild‐moderate cases (minimum: 17 days; maximum: 45 days) and the severe‐critical (minimum: 16 days; maximum: 48 days). Samples used: Whole blood Test operator: Not stated Definition of test positivity: According to the manufacturer instructions, IgG band reading rendered either negative or positive results. On the other hand, IgM band was classified as either negative, positive or weak positive depending on the intensity of the band staining. IgM‐positive, IgG‐positive and either IgM or IgG‐positive band staining were counted as positive results for the rapid test. A picture of every rapid test was taken at the manufacturer’s established time of reading. Test results were evaluated by two operators. In case of disagreement, a third operator was requested. Blinding reported: Not stated, but unlikely ‐ controls were healthy volunteers whereas cases were inpatients Threshold predefined: Visual, interpreted as per manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 positive RT‐PCR for pharyngeal swabs; threshold not stated Samples used: pharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes ‐ index test was done 16‐48 days after symptom onset Incorporated index test: No ‐ index test was done 16‐48 days after symptom onset Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated [2] Not stated All patients received same reference standard: Yes ‐ RT‐PCR Missing data: None Uninterpretable results: Not reported Indeterminate results: Not reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None to declare Publication status: Published Source: Journal: Revista Española de Quimioterapia (Official Journal of the Spanish Society of Chemotherapy) Author COI: The authors declared that they had no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Carozzi 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Multi‐group study to assess sensitivity and specificity [1] Covid‐positive [1a] Clinical hospitalised COVID‐19 cases (n = 135) [1b] PCR +ve healthcare workers (n = 33) [2] Non‐Covid [2a] Pre‐pandemic (n = 295) [2b] Suspected healthy healthcare workers (n = 17,065) Group [1b] and [2b] were not eligible for our review as [1b] was pre‐selected and [2b] had no reference standard. Recruitment: Not stated for [1a] and [2a] Prospective or retrospective: [1a] Unclear [2a] Retrospective [1b] [2b] Prospective for HCW seroprevalence survey Sample size: 17,528 (168) but 430 (135) included in our review Further detail: Inclusion: [1a] Clinical hospitalised cases with PCR +ve test at advanced stages of disease [2] Pre‐pandemic serum samples Exclusion: [1] [2] Not stated |
||
| Patient characteristics and setting | Setting: [1a Hospital inpatients Location: [1a] University Hospitals throughout Tuscany: [sA] AOUS, Siena ([n = 26) [sB] AOUC, Florence (n = 41) [sC] AOUP, Pisa ( n = 68) Country: Italy Dates: [1a] Not stated Symptoms and severity: [1a] Hospitalised, reported signs and symptoms since 10‐14 days Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic Source: Site F: Fondazione Toscana Gabriele Monasterio (FTGM) in Pisa and Massa. 2013‐2014 n = 200 November to February n = 95 July and August Characteristics: 145 women, 150 men aged 50‐70 years Non‐Covid group 2: NA |
||
| Index tests | Test name: [A] Screen Test Covid‐19 2019‐nCOV IgG/IgM [B] COVID‐19 IgG/IgM rapid test cassette Manufacturer: [A] Screen Italia S.r.l [B] Zhejiang Orient Gene Biotech Co., Ltd Antibody: [A] [B] IgG/IgM Antigen target: Not stated Evaluation setting: POCT used in lab Test method: [A] [B] Lateral Flow test Timing of samples: 14+ days post‐PCR Samples used: [1a] [2a] Serum Test operator: Staff in six laboratory departments of the participating institutions Definition of test positivity: IgG‐positive: presence of the expected control line and of a line at the IgG position only IgM‐positive: presence of the expected control line and of a line at the IgM position only IgG and IgM‐positive: Presence of the expected control line and of two lines at the IgG and IgM positions, respectively. Blinding reported: Not stated Threshold predefined: Yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: RT‐real time PCR Samples used: Not stated for [1a] Timing of reference standard: Not stated Blinded to index test: [1a] Yes, prior Incorporated index test: No Definition of non‐COVID cases: [2a] Pre‐pandemic Samples used: NA as pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1a] > 14 days All patients received same reference standard: No Missing data: yes, we excluded groups [1b] and [2b] from our review. Not all 135 samples from [1a] were tested with both rapid tests. Uninterpretable results: A test was considered invalid in the absence of a control line, none declared. Indeterminate results: Readings were considered doubtful if a shade, not classifiable as a clear line, appeared at the IgG or/and IgM positions. [A] 25/295 doubtful for IgM [B] 5/295 doubtful for IgM Number of doubtful results for IgG not reported Unit of analysis: [1a] Not stated [2a] Patients |
||
| Comparative | |||
| Notes | Funding: No external funding received, rapid tests provided by the Health Regional Department Publication status: Pre‐print (not peer reviewed) Source: medRxiv Pre‐print Author COI: Authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Carozzi 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Carta 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Single‐group study to assess sensitivity and specificity (n = 65) [1] Covid positive residents (n = 54) [2] Covid negative residents (n = 11) Recruitment: All residents in a long‐term care facility Prospective or retrospective: Prospective Sample size: 65 (54) Further detail: Inclusion: All the guests (symptomatic and asymptomatic) of a long‐term care facility. [1] Residents who tested positive for SARS‐CoV‐2 infection on RT‐PCR during any of three tests [2] Residents who tested negative for SARS‐CoV‐2 infection on all of three RT‐PCR tests Exclusion: [1] [2] No exclusion criteria; all residents included |
||
| Patient characteristics and setting | Setting: Long‐term care facility, all convalescent Location: Vicenza district Country: Italy Dates: PCR test performed between March 29 and April 22, 2020. Follow‐up for 2 months after outbreak Symptoms and severity: Symptomatic and asymptomatic, including 11 cases of fatal infection Demographics: 52/65 female, average age 82 years (range 56‐97 years) 26 not self‐sufficient Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] MAGLUMI 2019‐nCoV IgG and IgM Manufacturer: [A] [B] Shenzen New Industries Biomedical Engineering Co., SNIBE Diagnostic, Shenzen, PR China Antibody: [A] IgG/IgM Antigen target: [A] spike‐protein and nucleocapsid region Evaluation setting: Laboratory used in laboratory Test method: [A] CLIA Timing of samples: Day 32 (28–36) and 49 (47–50) post‐PCR +ve Samples used: Serum Test operator: Not stated, possibly Medicina di Laboratorio, AULSS 8 Berica, Viale Rodolfi, Vicenza, Italy Definition of test positivity: [A] IgG antibodies were considered negative < 0.90 AU/mL, grey‐zone 0.90‐1.10 AU/mL and positive >= 1.10 AU/mL [B] IgM antibodies were considered positive >= 1.00 AU/mL, negative < 1.00 AU/mL. Blinding reported: Not clear Threshold predefined: Yes, according to manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR on Cobas 6800 RT‐PCR System (Roche Diagnostics GmbH, Mannheim, Germany)
When two gene targets were found both positive, or even if only one target was found, but the patient had characteristic symptoms, the test was considered positive. Samples used: Oropharyngeal and nasopharyngeal swabs Timing of reference standard: Start of outbreak at long‐term care facility then on days 20, 32 and 49 Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: RT‐PCR on Cobas 6800 RT‐PCR System (Roche Diagnostics GmbH, Mannheim, Germany) When two gene targets were found both positive, or even if only one target was found, but the patient had characteristic symptoms, the test was considered positive. Samples used: Oropharyngeal and nasopharyngeal swabs Timing of reference standard: Start of outbreak at long‐term care facility then on days 20 and 32. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: 32 (28–36) and 49 (47–50) days All patients received same reference standard: Yes Missing data: Among 65 residents, 54 tested positive for COVID‐19 on the first swab but 11 of these patients subsequently died. Uninterpretable results: Not stated Indeterminate results: Grey zone for IgG antibody detection results, 0.90‐1.10 AU/mL, but no indeterminate results reported Unit of analysis: Samples (one sample on day 32 and one sample on day 49) |
||
| Comparative | |||
| Notes | Funding: None declared Publication status: Published paper Source: De Gruyter Diagnosis Author COI: Authors stated no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Case 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Single‐group study to estimate sensitivity for diagnosis of acute Covid‐19 Design: [1] PCR‐confirmed Covid‐19 patients (20 patients, 42 samples) Recruitment: Not stated Prospective or retrospective: Not stated Sample size: 42 (42) of which 40 (40) were eligible for our review |
||
| Patient characteristics and setting | Setting: Not stated Location: Not stated Country: USA Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] Anti‐SARS‐CoV‐2 IgG ELISA [B] Epitope IgG ELISA Manufacturer: [A] Euroimmun, Germany [B] Epitope Antibody: [A] and [B] IgG Antigen target: [A] SARS‐CoV‐2 S‐protein [B] Not stated Evaluation setting: [A] and [B] Laboratory Test method: [A] and [B] ELISA Timing of samples: 5‐7 days post‐symptom onset: 5/40 (13%) 8‐14 days post‐symptom onset: 23/40 (50%) 15‐20 days post‐symptom onset: 12/40 (30%) 2 not stated Samples used: Serum Test operator: Unclear Definition of test positivity: Index value positive if >= 1.1, as per manufacturer Blinding reported: Unclear |
||
| Target condition and reference standard(s) | Reference standard: PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, done before index test Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: Nothing mentioned Uninterpretable results: Nothing mentioned Indeterminate results: Nothing mentioned [A] 1 indeterminate results (0.8‐1.1) [B] 1 indeterminate results (0.8‐1.1) Unit of analysis: Samples (40 samples from 18 or 19 patients) |
||
| Comparative | |||
| Notes | Funding: This study was supported by NIH contracts and grants (75N93019C00062, HHSN272201700060C, R01 AI127828, R37 AI059371, and U01 AI151810), the Defense Advanced Research Project Agency (HR001117S0019), and gifts from Washington University in Saint Louis. J.B.C. is supported by a Helen Hay Whitney Foundation postdoctoral fellowship.
The Diamond laboratory has received unrelated funding under sponsored research agreements from Moderna and Emergent BioSolutions. Publication status: Pre‐print (not peer reviewed) Now published Source: bioRxiv Journal "Cell Host & Microbe" Author COI: M.S.D. is a consultant for Inbios, Vir Biotechnology, NGM Biopharmaceuticals, and on the Scientific Advisory Board of Moderna. D.C. and H.W.V. are employees of Vir Biotechnology Inc. and may hold shares in Vir Biotechnology Inc. S.P.J.W. and P.W.R. have filed a disclosure with Washington University for the recombinant VSV. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Case 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Caturegli 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of COVID‐19 diagnostic test Design: Multi‐group study estimating both sensitivity and specificity Group [1] and [2] were hospitalised adults investigated for COVID‐19 selected from a cohort of patients with at least one NAT result (n = 11,066) and with available residual serum samples (n = NR): [1] COVID‐19 cases, including PCR‐confirmed (n = 50, including 38 with single positive result) and clinically defined PCR‐negative based on medical record review (n = 10) [2]: Symptomatic patients with negative PCR (n = 55, including 43 with single negative result) [3] Laboratory controls including healthy lab employees and patients with polyclonal activation of antibody response (n = 513; 325 pre‐pandemic and 188 contemporaneous) Recruitment: Convenience Prospective or retrospective: Retrospective Sample size: Hospitalised COVID suspects: 115 (60) Full sample: 628 (60) |
||
| Patient characteristics and setting | Setting: Mixed
Groups [1] and [2]: Inpatient service of a tertiary hospital
Group [3] healthy and patients Location: Johns Hopkins Hospital, Baltimore, MD Country: United States Dates: 11 Mar to 12 Apr, 2020 Symptoms and severity: Group [1]: All symptomatic individuals. No clear details on severity but likely moderate to critical because they were all hospitalised and some developed ARDS. Demographics: Age, median (IQR): 59 (48‐70) Sex: 43/60 (72%) male Exposure history: 21/60 (35%) had travel history 20/60 (33%) had sick contacts 5/60 (8%) were healthcare workers Non‐Covid group 1: Group [2]: Symptomatic patients with negative PCR Source: Hospitalised patients who underwent one or more PCR tests for SARS‐CoV‐2 between 11 Mar and 12 Apr, 2020 Characteristics: Age, median (IQR): 61 (47‐69) Sex: 22/60 (40%) male All symptomatic, with fever (31%), cough (55%), shortness of breath (47%) the most common symptoms Non‐Covid group 2: Group [3]: non‐COVID controls (pre‐pandemic and contemporaneous) Source: Lab stocked samples mostly collected during the pre‐pandemic period (n = 327), except for 188 samples that were obtained in 2020. |
||
| Index tests | Test name: Anti‐SARS‐CoV‐2 ELISA IgG and IgA Manufacturer: EUROIMMUN AG Antibody: IgG, IgA Antigen target: S1 domain of the spike‐protein Evaluation setting: Lab tests, done in lab Test method: Enzyme‐linked immunosorbent assay (ELISA) Timing of samples: Multiple samples taken from each patient at various points in time, from 0 to 59 days after symptom onset Samples used: Residual serum samples Test operator: Not stated Definition of test positivity: positive if ratio > 1.1 Also reported threshold derived based on collected data (not extracted) Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR test (no further details available ‐ unclear whether more than one assay was used to test patients) AND clinical evaluation based on clinical record review (risk factors, signs and symptoms on presentation, radiologic findings, comorbidities, smoking and alcohol history, BMI, reason for repeated NAAT testing (as applicable), and complications during hospital stay. No formalised combination of findings to indicate COVID‐19 was reported. Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated; duration of symptoms on clinical presentation was 7 days (range 4 to 7) for cases and 3 days (range 1 to 7) for non‐COVID patients Blinded to index test: For PCR, yes but record review was post hoc Incorporated index test: No Definition of non‐COVID cases: Group [2]: RT‐PCR (as above) Group [3]: pre‐pandemic and contemporaneous (no testing) Samples used: Group [2]: Nasopharyngeal swab Group [3]: NA Timing of reference standard: Group [2]: Not stated Group [3]: NA Blinded to index test: Group [2]: Yes (done earlier) Group [3]: NA Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated. Only time from symptom onset for index test was available. All patients received same reference standard: No Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: The study was funded internally by the Clinical Immunology Laboratory of the Department of Pathology, Johns Hopkins Hospital. Publication status: Published article Source: Academic journal Author COI: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Cervia 2020.
| Study characteristics | |||
| Patient Sampling | Purpose:
Diagnosis of acute and convalescent‐phase infection Design: Study reported two cohorts, only one of which was eligible for this review. [1] Single‐group study to estimate sensitivity in patients with RT‐qPCR‐confirmed SARS‐CoV‐2 infection (n = 56) Recruitment: Unclear Prospective or retrospective: Prospective (Following written informed consent, patients and healthcare workers were recruited for sampling of blood and mucosal secretions) Sample size: [1] 56 (56) ([2] 109 (21)) Further detail: No further details |
||
| Patient characteristics and setting | Setting: Mixed Location: Not stated; authors' institution University Hospital Zurich (USZ) Country: Switzerland Dates: Not stated Symptoms and severity: WHO criteria: mild 19, 34% (mild illness and mild pneumonia); severe 37, 66% (severe pneumonia and acute respiratory distress syndrome) Outpatient: 10/19 mild cases and 0/37 severe cases Demographics: median 61 y (IQR 48, 77), 31 (55%) male Mild: median age 49 (IQR 34‐60) years, 8/19 (42%) male; Severe: median age 68 (IQR 57‐79) years, 23/37 (62%) male Exposure history: Not stated Non‐Covid group 1: Mentioned but results not documented |
||
| Index tests | Test name: Euroimmun SARS‐CoV‐2 IgA and IgG immunoassay (no product code reported) Manufacturer: Euroimmun Antibody: IgA, IgG Antigen target: SARS‐CoV‐2 spike‐protein (S1) Evaluation setting: Laboratory Test method: ELISA Timing of samples: [1] mean 16.4 days (median 13 days) for the mild group and approx day 2 to day 48; mean 20.9 days (median 16 days) for the severe group since symptom onset Samples used: serum (usable data were not reported for mucosal samples (tears, nasal fluid, saliva)) Test operator: Not stated Definition of test positivity: serum IgA: optical density (OD) ratios of 1.1–2.0 were considered borderline‐positive; values above 2.0 positive serum IgG: OD ratios of 0.8–1.1 were considered borderline‐positive and values above 1.1 positive. Blinding reported: Not stated Threshold predefined: As per manufacturer; IgA: OD > 2.0, IgG OD > 1.1 |
||
| Target condition and reference standard(s) | Reference standard: RT‐qPCR, TaqMan SARS‐CoV‐2 Assay Kit v2 (Thermo Fischer), the 2019‐nCoV CDC qPCR Probe Assay (2019‐nCov CDC EUA Kit; Integrated DNA Technologies, Inc.), or the Roche Cobas SARS‐CoV‐2 Test CE‐IVD (Roche) according to manufacturers' instructions Samples used: NP Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes (different RT‐PCR assays) Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: patients |
||
| Comparative | |||
| Notes | Funding: Academy of Medical Sciences fellowships, the Young Talents in Clinical Research Fellowship by the Swiss Academy of Medical Sciences and Bangerter Foundation, the Swiss National Science Foundation, the Clinical Research Priority Program of the University of Zurich for the CRPP CYTIMM‐Z, and a grant of the Innovation Fund of the University Hospital Zurich Publication status: Pre‐print Source: bioRxiv Author COI: The authors declared no competing financial interests related to this work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Chan 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Multi‐group study to assess sensitivity and specificity [1] Covid subjects (n = 144) [1a] Admitted PCR‐positive samples (n = 78) for clinical performance study [1b] Archived PCR‐positive samples (n = 66) for method comparison study [2] Non covid subjects (n = 130) [2a] non‐SARS‐CoV‐2 respiratory viral samples (n = 25) [2b] Other viral positive samples (n = 52) [2c] Pre‐pandemic samples (n = 53) [1b] excluded from review as no time pso or post‐PCR+ reported. Recruitment: [1a] Admitted PCR‐positive patients who had routine metabolic profiles and serologies ordered for clinical care [1b] Archived samples from the validation studies for the EuroImmun Ab assay [2a] [2b] Not stated, likely samples from storage [2c] Pre‐pandemic samples (41 from a reference range study prior to 2018 and 12 from a banked respiratory viral panel from early 2019) Prospective or retrospective: Retrospective Sample size: 274 (144) of which 208 (78) were eligible for review Further detail: Inclusion: [1a] PCR‐positive for SARS‐CoV‐2 admitted to hospital [1b] Archived PCR‐positive samples from the validation studies for the EuroImmun Ab assay [2a] Positive for non‐SARS‐CoV‐2 respiratory infection (coronaviruses: HKU1 n = 5, NL63 n = 7, OC43 n = 7, 229E n = 2, OC43 + 229E CV n = 1, Rhinovirus n = 2) [2b] Positive for other viruses (HIV n = 20, HepB n = 15, HCV n = 17) [2c] Pre‐pandemic (41 from a reference range study prior to 2018 and 12 from a banked respiratory viral panel from early 2019) Exclusion: [1a] [1b] [2a] [2b] [2c] Not stated |
||
| Patient characteristics and setting | Setting: [1a] Hospital inpatients Location: Chemistry and Immunology Laboratories, University of Chicago Hospitals, Chicago, IL Country: USA Dates: Not stated Symptoms and severity: [1a] All hospitalised Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] non‐SARS‐CoV‐2 respiratory viral samples Source: University of Chicago Hospitals, Chicago, IL; time not stated Characteristics: HKU1 CV n = 5, NL63 CV n = 7, OC43 CV n = 7, 229E CV n = 2, OC43 CV + 229E CV n = 1, Rhinovirus n = 2 Non‐Covid group 2: [2b] Other viral positive samples [2c] Pre‐pandemic samples Source: [2b] University of Chicago Hospitals, Chicago, IL; time not stated [2c] University of Chicago Hospitals, Chicago, IL, 41 prior to 2018, 12 from early 2019 Characteristics: [2b] HIV n = 20, HepB n = 15, HCV n = 17 [2c] Not stated (41 from a reference range study prior to 2018 and 12 from a banked respiratory viral panel from early 2019) |
||
| Index tests | Test name: [A] Elecsys anti‐SARS‐CoV‐2 antibody assay
[B] EuroImmun IgG antibody assay (anti‐SARS‐CoV‐2 ELISA) Manufacturer: [A] Roche diagnostics [B] Euroimmun Antibody: [A] Total antibody [B] IgA/IgG Antigen target: [A] Nucleocapsid protein [B] Not stated Evaluation setting: Laboratory test used in laboratory setting Test method: [A] ECLIA [B] ELISA Timing of samples: [1a] 0‐13 days post‐PCR + (n = 40) >= 14 days post‐PCR + (n = 38) Samples used: Serum and plasma Test operator: Clinical chemistry staff at the University of Chicago Definition of test positivity: [A] COI >= 1.0 positive, COI < 1.0 negative [B] Not stated Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: [2a] [2b] not stated, possibly pre‐pandemic [2c] pre‐pandemic Samples used: [2a] [2b] unclear [2c] pre‐pandemic Timing of reference standard: [2a] [2b] unclear [2c] pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1a] 0‐13 days post‐PCR + (n = 40)
>= 14 days post‐PCR + (n = 38) All patients received same reference standard: No Missing data: [1b] excluded from review as well as 40 samples from [1a] < 14 days post‐PCR+ Uninterpretable results: Not stated Indeterminate results: Not stated, no indeterminate threshold Unit of analysis: Unclear |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: American Journal of Clinical Pathology Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Charlton 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (28 patients, 46 samples) [2] Pre‐pandemic non‐COVID (50 samples) [3] Cross‐reactivity non‐COVID samples [62 samples: pre‐pandemic (n = 15) and concurrent (n = 47)] Recruitment: [1] Hospitalised (or ambulatory) patients confirmed to be positive for SARS‐CoV‐2 upon nasopharyngeal swab or endotracheal aspirate testing by rRT‐PCR [2] Negative samples were retrieved from bio‐banked sera stored at the public health laboratory (Alberta Precision Laboratories) in Alberta collected before 1 November 2019. [3] Convalescent phase sera (either retrieved from stored sera or prospectively collected) Prospective or retrospective: [1] Unclear [2] Retrospective [3] Prospective and retrospective Sample size: 158 (46) samples Further detail: [1] Patients who tested positive for SARS‐CoV‐2 by rRT‐PCR [2] Serum samples stored prior to 1 November 2019 [3] Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients (26/28; 93%) and ambulatory (2/28; 7%) Location: Not stated [history taken by Alberta Health Services Communicable Diseases Team (Public Health)] Country: Alberta, Canada Dates: Not stated Symptoms and severity: 2/28 (7%) ambulatory 26/28 (93%) hospitalised 9/28 ICU 7/28 Need for mechanical ventilation 1/28 Pulmonary embolism 26/28 COVID pneumonia 1/28 No COVID pneumonia 1/28 Unknown 13/28 acute respiratory distress syndrome 14/28 no acute respiratory distress syndrome 1/28 unknown Demographics: Mean age of patients was 70.1 (median 73; range, 34 to 102 years), 12/28 (43%) female Exposure history: Travel‐related exposures ‐ yes 4 (14%) (USA n = 2; United Arab Emirates n = 1; within Canada n = 1) ‐ no 23 (82%) ‐ unknown 1 (4%) Contact with traveller ‐ yes 6 (21%) ‐ no 21 (75%) ‐ unknown 1 (4%) Infection related to outbreak in long‐term‐care/continuing care facility 9 (32%) Non‐Covid group 1: [2] Pre‐pandemic controls Source: Bio‐banked sera stored at the public health laboratory (Alberta Precision Laboratories) in Alberta collected before 1 November 2019. Characteristics: Not stated Non‐Covid group 2: [3] Cross‐reactivity samples Source: 15 sera were collected prior to the first case of SARS‐CoV‐2 diagnosis in Alberta, and 47 were collected after the first case of SARS‐CoV‐2 was detected in Alberta. Characteristics: The sera were from patients who had tested negative for COVID‐19 by in‐house rRT‐PCR but positive for other viruses as follows (with the number of sera used): ‐ influenza A virus (n = 5), ‐ influenza B virus (n = 5), ‐ respiratory syncytial virus (RSVA, n = 6; RSVB, n = 1), ‐ rhinovirus/enterovirus (n = 6), ‐ human metapneumovirus (HMPV; n = 5), ‐ parainfluenza virus (PIV‐1 and PIV‐4; n = 4), ‐ CoV‐229E (n = 6), ‐ CoV‐NL63 (n = 11), ‐ CoV‐OC43 (n = 7), or ‐ CoV‐HKU1 (n = 7). One patient was positive for multiple viruses (RSVA and enterovirus/rhinovirus). |
||
| Index tests | Test name: [A] SARS‐CoV‐2 IgG assay [B] EDI novel coronavirus COVID‐19 IgM and IgG ELISA [C] a novel coronavirus COVID‐19 IgM and IgG assay [D] SARS‐CoV‐2 S1/S2 IgG [E] anti‐SARS‐CoV‐2 ELISA IgA and IgG assay [F] anti‐SARS‐CoV‐2 [G] Rapid Response [H] 2019 nCoV IgM/IgG detection kit [I] SARS‐CoV‐2 IgG/IgM Ab test kit [J] Novel coronavirus IgG/IgM test kit [K] One Step Test for novel coronavirus [L] 2019‐nCoV Ab test Manufacturer: [A] Abbott Laboratories, Abbott Park, IL, USA [B] Epitope Diagnostics Inc., supplied by Affinity Diagnostics Corp., Toronto, ON, Canada [C] DRG International Inc., supplied by Bio‐Rad, Hercules, CA, USA [D] DiaSorin, Stillwater, MN, USA [E] Euroimmun, Mississauga, ON, Canada [F] Roche Diagnostics, Indianapolis, IN, USA [G] BTNX, Markham, Ontario, Canada [H] Biolidics Limited, Singapore [I] Anhui Deep Blue Medical Technology Co., Ltd., Anhui, China [J] Genrui; Genrui Biotech Inc., Shenzhen, China [K] Getein Biotech Inc., Nanjing, China [L] Innovita Biological Technology Co. Ltd., Qian’an, Hebei, China Antibody: [A] IgG [B] IgM and IgG [C] IgM and IgG [D] IgG [E] IgA and IgG [F] Total antibodies (including IgG) [G]‐[L] IgM and IgG Antigen target: [A] Recombinant antigen nucleocapsid protein [B] Recombinant antigens of the RBD and spike‐protein [C] Antibodies recognising recombinant nucleocapsid proteins and peptides [D] IgG antibodies directed against the S1 and S2 domains of the spike‐protein [E] Recombinant S1 domain of the structural protein [F] Recombinant protein representing the nucleocapsid antigen [G], [I], [J], [L] Target unspecified [H] Recombinant protein, target unspecified [K] Recombinant nucleocapsid and spike proteins Evaluation setting: [A]‐[F] Lab test used in lab [G]‐[L] POCT used in lab Test method: [A] chemiluminescent microparticle immunoassay [CMIA] [B] ELISA [C] ELISA [D] chemiluminescence immunoassay [CLIA] [E] ELISA [F] electrochemiluminescence immunoassay (ECLIA) [G]‐[L] Lateral flow test Timing of samples: [A]‐[L] 0‐14 days pso 21/42 15‐21 days pso 11/42 > 21 days pso 10/42 Samples used: [A]‐[L] Serum (all kits assessed using same patient samples from single‐use aliquots). Samples collected, spun down (3000 rpm for 10 min), aliquoted into single‐use aliquots, and frozen at ‐80°C until the time of testing [G]‐[L] Cross‐reactivity panel [3] was not assessed on the POCTs. Test operator: [A]‐[L] Lab personnel Results read independently by two laboratorians; in case of discrepancy, a third laboratorian reading was used as an arbitrator (+/‐/‐ was considered equivocal, +/‐/+ was considered positive). Definition of test positivity: [A]‐[F] as per manufacturer specifications using cut‐offs as described in the package inserts. All values greater than the published cut‐off were considered positive. [G]‐[L] any banding detected for either IgM or IgG. Faint banding was considered positive. Assays where the control line was absent were considered invalid. Testing was performed as per manufacturer specifications. Blinding reported: not stated Threshold predefined: [A]‐[F] yes as per manufacturer specifications [G]‐[L] Yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: rRT‐PCR, threshold not reported Samples used: Nasopharyngeal swab (27/28) or endotracheal aspirate (1/28) Timing of reference standard: All dates of symptom onset were reported earlier than the date of diagnostic sample collection (mean, 16 days [range, 2 to 48 days]). Blinded to index test: yes, done prior Incorporated index test: no Definition of non‐COVID cases: [2] Pre‐pandemic [3] Pre‐pandemic or in‐house rRT‐PCR on nasopharyngeal swab testing Samples used: [2] Pre‐pandemic [3] Pre‐pandemic, otherwise nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: yes, done prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1] Not stated [time of PCR positivity was 5.3 days after date of symptom onset on average (range, 0 to 19 days)].
[2] Not stated
[3] The time from an RPP‐positive result to serum collection ranged from 11 to 135 days (mean 45 days) from the date of the original RPP result. All patients received same reference standard: No Missing data: yes (see Tables 3 and 4) Uninterpretable results: Two invalid samples observed for Affinity and for Euroimmun and one for Getein BioTech LFA (all excluded) Indeterminate results: Yes; number of equivocal results reported per test; these can be considered either as index‐positive or negative Unit of analysis: Patients; 11 COVID‐19‐positive patients had serum collected at multiple time periods; however, only one sample per patient was used per time interval to calculate assay sensitivity. When more than one serum sample from the same individual was within a given time interval, only the most recently collected serum sample was included. |
||
| Comparative | |||
| Notes | Funding: We also thank the following manufacturers for supplying kits for analysis: Abbott, Affinity, Bio‐Rad, DiaSorin, Euroimmun, Roche, BTNX, Biolidics, Deep Blue, Genrui, Getein BioTech, and Innovita. Publication status: Published paper Source: Journal of Clinical Microbiology Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Charlton 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charlton 2020 [L].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charpentier 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (88 samples from 54 patients) [2] Pre‐pandemic non‐COVID‐samples (120 samples) [2a] Samples for testing as part of routine clinical care (n = 56) [2b] Serum samples corresponding to a cross‐reactivity panel (n = 64) Recruitment: [1]‐[3] Samples collected in the Virology Laboratory of Bichat‐Claude Bernard and Saint‐Louis University‐Hospitals both in Paris, France Prospective or retrospective: [1] and [3] unclear [2] retrospective Sample size: 262 (88) samples of which 208 (88) were eligible for our review Further detail: [1] Patients with a confirmed COVID‐19 diagnosis by a positive nasopharyngeal sample RT‐PCR [2] Collected before November 2019 |
||
| Patient characteristics and setting | Setting: Hospital inpatients (40/54) and outpatients (14/54) (mixed) Location: Virology Laboratory of Bichat‐Claude Bernard and Saint‐Louis University‐Hospitals both in Paris, France. Country: France Dates: Not stated Symptoms and severity: 54 patients: 29 hospitalised in intensive care, 11 hospitalised in infectious diseases, so 74% with severe infections 14 outpatients Demographics: Median age was 52 years (range: 27–80), 36 were males. Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic Source: Virology Laboratory of Bichat‐Claude Bernard and Saint‐Louis University‐Hospitals both in Paris, France. All collected before November 2019 [2a] Samples for testing as part of routine clinical care (n = 56) [2b] Serum samples corresponding to a cross‐reactivity panel (n = 64): Coronaviruses (HKU1, NL63, 229E and OC43; n = 20), malarial (n = 26), respiratory viruses (influenza A [n = 2], influenza B [n = 1], respiratory syncytial virus [n = 2], metapneumovirus [n = 1], rhinovirus [n = 1]), sera with acute CMV infection (n = 2), acute EBV infection (n = 1), HIV‐HBV co‐infection (n = 1), acute parvovirus B19 infection (n = 1), toxoplasma (n = 1). Samples containing auto‐antibodies (4 rheumatoid factor and 1 systemic lupus erythematosus) Non‐Covid group 2: Suspected COVID‐19, negative or no RT‐PCR (not included in review) Source: Virology Laboratory of Bichat‐Claude Bernard and Saint‐Louis University‐Hospitals both in Paris, France. Time not stated Characteristics: 54 healthcare workers who presented with clinical symptoms during the epidemic period for whom SARS‐CoV‐2 RT‐PCR was negative or not carried out |
||
| Index tests | Test name: [A] Covid‐Presto® test rapid Covid‐19 IgG/IgM [B] NG‐Test® IgM‐IgG COVID‐19 [C] Abbott SARS‐CoV‐2 IgG kit Manufacturer: [A] AAZ, Boulogne‐Billancourt, France [B] NG Biotech, Guipry, France [C] Abbott, IL, USA Antibody: [A] IgG and IgM [B] IgG and IgM [C] IgG Antigen target: [A]‐[C] Not stated Evaluation setting: [A] and [B] POC test used in lab [C] Lab test used in lab Test method: [A] and [B] Lateral flow test [C] Chemiluminescent microparticle immunoassay Timing of samples: [A] 88 samples between day 4 and day 42 after onset of symptoms. 4‐9 days pso: 18/88 10‐14 days pso: 33/88 15‐42 days pso: 37/88 [B] Subgroup of 59 samples among the 88 samples between days 7 and 28 after onset of symptoms 7‐9 days pso: 6/59 10‐14 days pso: 22/59 15‐28 days pso: 31/59 [C] 57 samples: 7‐9 days pso: 6/57 10‐14 days pso: 22/57 >14 days pso: 29/57 Samples used: [A]‐[C] Serum Test operator: [A]‐[C] Lab personnel Definition of test positivity: [A] and [B] According to manufacturer’s instructions; results were read and interpreted 10 min after depositing serum. [C] The assay cut‐off is an index of 1.40 and the assigned grey zone is comprised between 1.12 and 1.68. Blinding reported: Not stated Threshold predefined: [A] and [B] yes, visual‐based [C] yes, according to manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: [1] RT‐PCR, threshold not stated Samples used: Nasopharyngeal samples Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: [2] Pre‐pandemic samples (before November 2019) [3] RT‐PCR or no reference standard Samples used: [2] Pre‐pandemic samples (before November 2019) [3] Not stated or no reference standard Timing of reference standard: [2] and [3] Not stated Blinded to index test: [2] and [3] yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: yes (sensitivity for [B] in 59/88 samples; sensitivity of for [C] in 57/88 samples; specificity for [B] and [C] in 52/120 samples) Uninterpretable results: Not stated Indeterminate results: yes [one sample was positive in the grey zone with Abbott SARS‐CoV‐2 IgG assay (index: 1.45)] Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Journal of Clinical Virology Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Charpentier 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Charpentier 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Chaudhuri 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase disease Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (368 patients with 379 samples) [2] Pre‐pandemic non‐COVID samples (n = 184) Recruitment: [1] The participants for this study were derived from a longitudinal cohort of COVID‐19‐positive participants known as the Department of Biotechnology (DBT) India COVID‐19 Consortium cohort with ongoing recruitment from March 2020 at eight clinical sites in the Delhi‐National Capital Region, India. [2] Sera samples collected in the pre‐pandemic period (184 from pregnant women enrolled in a pregnancy cohort). Prospective or retrospective: [1] Department of Biotechnology(DBT) India COVID‐19 Consortium cohort: prospective [2] Retrospective (stored samples) Sample size: 563 (379) samples Further detail: [1] For longitudinal cohort study: i) Suspected COVID‐19 patients enrolled at the time of RT‐PCR testing at the screening centre and ii) RT‐PCR confirmed COVID‐19 positive patients admitted at one of the clinical sites For the present study: sera/plasma samples collected ≥ 20 days of illness or following RT‐PCR positivity [2] Sera samples collected in the pre‐pandemic period (before September 2019.) from pregnant women enrolled in a pregnancy cohort |
||
| Patient characteristics and setting | Setting: Convalescent, setting not stated Location: 8 clinical sites in the Delhi‐National Capital Region, India [Department of Biotechnology (DBT) India COVID‐19 Consortium cohort] Country: India Dates: from March 2020 Symptoms and severity: 83.7% symptomatic 16.3% asymptomatic (text says 14%?) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic healthy Source: Collected before September 2019 from pregnant women enrolled in a pregnancy cohort. Characteristics: 184/184 pregnant women |
||
| Index tests | Test name: [A] Diasorin LIAISON SARS‐CoV‐2 S1/S1 IgG CLIA [B] Covid Kavach IgG ELISA Manufacturer: [A] Diasorin [B] Zydus Antibody: [A] IgG [B] IgG Antigen target: [A] S1/S2 domains of the spike‐protein [B] specific antigenic epitope(s) of the inactivated virus in the Kavach assay were not defined Evaluation setting: [A] and [B] Lab tests performed in lab Test method: [A] Chemiluminescence assay (CLIA) [B] ELISA Timing of samples: 20‐72 days of illness in symptomatic or RT‐PCR positivity in asymptomatic individuals; duration of illness bimodal due to study design: The means of the sampling window distributions were 23.5 and 49.3 days respectively. Samples used: Serum or plasma Test operator: [A] and [B] Lab personnel Definition of test positivity: [A] The tests were considered positive when the IgG concentration was ≥ 15 AU/mL, negative when the concentration was < 12 AU/mL and equivocal when the concentration was > 12 and < 15 AU/mL. Equivocal samples were considered negative for sensitivity analysis. [B] The kit suggests interpretation of the results by a two‐pronged method, based on OD value and P/N (Positive/Negative Ratio). When both read‐outs are in agreement, then the sample is considered positive or negative. The manufacturer’s instruction does not mention interpretation for samples with a read‐out not in agreement for the two criteria. We considered such results negative. Blinding reported: Not stated Threshold predefined: [A] IgG concentration (AU/mL) as per manufacturer's instructions [B] OD value and P/N (Positive/Negative Ratio) as per manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: The testing by RT‐PCR was done at an approved laboratory as per the National Testing Strategy of India; threshold not reported Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: The specificity of DiaSorin could not be evaluated due to limited availability of pre‐pandemic negative sera. Uninterpretable results: Not stated Indeterminate results: [A] Seven samples were reported as indeterminate by DiaSorin CLIA. Equivocal samples were considered negative for sensitivity analysis. [B] 6 samples were indeterminate in Zydus Kavach test and excluded from the study; and 23 (not 25, corrected by author) samples were positive only by one condition (cut‐off, P/N ratio) by Zydus Kavach. Unit of analysis: Samples (11 patients with 2 samples) |
||
| Comparative | |||
| Notes | Funding: We deeply thank the Department of Biotechnology, Government of India for supporting the consortium. We are grateful to the leadership and administration of all partner institutions in the consortium for their help and support. We thank all the clinical, laboratory and data management staff for their contributions to this work and the consortium at large. Publication status: Published paper Source: Journal of Clinical Virology Author COI: No conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | No | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Chaudhuri 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Chen 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (74 patients, n = 346 samples) [2] Non‐COVID samples (n = 194) [2a] Current patients with acute respiratory infection (n = 120) [2b] Current patients with presence of auto‐antibodies (n = 36) [2c] Pre‐pandemic samples with presence of antigens/antibodies (n = 38) Recruitment: [1] Consecutively qRT‐PCR‐confirmed COVID‐19 patients who were treated at six participating hospitals between 23 January 2020 and 31 May 2020 [2] Not stated [Hospitalised patients with an acute respiratory infection (ARI) who tested negative at least 2 times using qRT‐PCR with or without confirmed aetiology for ARI, treated between January 31 and May 31, 2020; patients with auto‐antibodies (1‐31 May 2020) or patients showing presence of specific microbiological antigens or antibodies, treated between 1 August and 31 December 2019] Prospective or retrospective: Retrospective Sample size: 540 (346) Further detail: [1] qRT‐PCR‐confirmed COVID‐19 patients who were treated at six participating hospitals between 23 January 2020 and 31 May 2020 [2] Not stated [Hospitalised patients with an acute respiratory infection (ARI) who tested negative at least 2 times using qRT‐PCR with or without confirmed aetiology for ARI, treated between January 31 and May 31, 2020; patients with auto‐antibodies (1‐31 May 2020) or patients showing presence of specific microbiological antigens or antibodies, treated between 1 August and 31 December 2019] |
||
| Patient characteristics and setting | Setting: Hospital inpatients (in Taiwan, all qRT‐PCR confirmed patients are mandatorily hospitalised) Location: 6 hospitals: National Taiwan University Hospital, National Cheng Kung University Hospital, Tao Yuan General Hospital, Ministry of Health and Welfare, Changhua Christian Hospital, Nantou Hospital, Ministry of Health and Welfare, and China Medical University Hospital. Country: Taiwan Dates: 23 January 2020 to 31 May 2020 Symptoms and severity: All 74 enrolled COVID‐19 patients reported at least one COVID‐19‐compatible symptom. Lower respiratory tract symptoms were the predominant symptom at the time of diagnosis (66.2%), followed by upper airway symptoms (62.2%), and fever (45.9%). 28 (37.8%) patients developed pneumonia during hospitalisation, among whom five (6.8%) required ventilator support and intensive care. 1/74 received ECMO support Demographics: Mean patient age was 38.5 years (SD, 16.2 years). 41 (55.4%) patients were men and 67 (90.5%) of them had no significant comorbid or surgical condition. Exposure history: Not stated Non‐Covid group 1: [2] Non‐COVID patients Source: [2a] Treated between 31 January and 31 May 2020, source not stated [2b] 1 May to 31 May 2020, source not stated [2c] Treated between 1 August and 31 December 2019, source not stated Characteristics: [2a] Acute respiratory infection and negative rt‐PCR without other confirmed aetiologies (n = 70); Acute respiratory infection and negative rt‐PCR with microbiological aetiologies (n = 50): Coronavirus n = 3 Cytomegalovirus (CMV) n = 18 CMV and herpes simplex virus (HPV) n = 2 CMV and HPV and Epstein‐Barr virus (EBV) n = 1 HSV n = 1 EBV n = 5 Mycoplasma pneumoniae n = 5 Chlamydophila trachomatis n = 5 Respiratory syncytial virus n = 2 Influenza A n = 4 Influenza B n = 4 [2b] Patients showing the presence of any specific auto‐antibodies (n = 36) [2c] Pre‐pandemic patients showing the presence of specific antigens/antibodies (n = 38): Mycoplasma pneumoniae n = 15 Chlamydophila pneumophila n = 5 EBV n = 10 Respiratory syncytial virus n = 1 Influenza A n = 3 Influenza B n = 4 |
||
| Index tests | Test name: [A] Roche Elecsys® Anti‐SARS‐CoV‐2 Test [B] Abbott SARS‐CoV‐2 IgG [C] Wondfo SARS‐CoV‐2 Antibody Test [D] ASK COVID‐19 IgG/IgM Rapid Test [E] Dynamiker 2019‐nCoV IgG/IgM Rapid Test Manufacturer: [A] Roche Diagnostics Basel, Switzerland [B] Abbott Laboratories, IL, USA [C] Guangzhou Wondfo Biotech Co., Ltd., China [D] TONYAR Biotech Inc. Taiwan [E] Dynamiker Biotechnology [Tianjin] Antibody: [A] Total antibodies (including IgG) [B] IgG [C] Total antibodies [D] IgG and IgM [E] IgG and IgM Antigen target: [A] N‐protein [B] N‐protein [C] spike‐protein [D] spike‐protein [E] N‐protein Evaluation setting: [A] Lab test used in lab [B] Lab test used in lab [C] POCT used in lab [D] POCT used in lab [E] POCT used in lab Test method: [A] Electrochemiluminescence immunoassay [B] Chemiluminescent microparticle immunoassay [C]‐[E] Lateral flow tests Timing of samples: Median 7 days pso (range 1‐93 days pso) Mean 11.4 (SD 14.8) days pso 0‐7 days pso: 61/346 8‐14 days pso: 73/346 15‐21 days pso: 61/346 22‐28 days pso: 64/346 29‐35 days pso: 32/346 36‐93 days pso: 55/346 Samples used: [A]‐[E] Serum (Residual blood samples; the serum of the collected blood samples was stored at −20°C before testing) Test operator: Not stated (possibly lab personnel) Definition of test positivity: [A] and [B] Test results were interpreted as positive if the electrochemiluminescent signal value of the Roche Test (cut‐off index, COI) ≧ 1.0, or the chemiluminescent signal value of the Abbott Test (index [sample/calibrator], S/C) ≧ 1.4, as manufacturers’ instructions [C]‐[E] Positive results were interpreted as the presence of control line and either IgG or IgM test line for ASK Test and Dynamiker Test, or control line and total antibody test line in Wondfo Test. A weakly positive result (any shade of colour in the test lines) of an antibody rapid testing was considered positive according to the manufacturers’ instructions. Blinding reported: Not stated Threshold predefined: [A]‐[E] yes |
||
| Target condition and reference standard(s) | Reference standard: In Taiwan, the respiratory tract specimens from patients who meet the reporting criteria for COVID‐19 have to be submitted to virology laboratories validated and associated with the Centers for Diseases Control of Taiwan (Taiwan CDC) for SARS‐CoV‐2 qRT‐PCR assay. Three sets of primers and probes targeting the SARS‐CoV‐2 envelope (E), nucleocapsid (N), and RNA‐dependent RNA polymerase (RdRp) genes were used. If the result of the first sample was negative for SARS‐CoV‐2, an additional SARS‐CoV‐ 2 qRT‐PCR assay for another respiratory tract sample
from the patient suggested of having COVID‐19 was performed to minimise the risk of false‐negative results using the qRT‐PCR assay. Samples used: Respiratory tract specimens Timing of reference standard: Not stated Blinded to index test: yes, prior Incorporated index test: no Definition of non‐COVID cases: Current patients with acute respiratory infections: tested negative ≥ 2 times using SARS‐CoV‐2 qRT‐PCR Current patients with auto‐antibodies: not tested Pre‐pandemic samples Samples used: Not stated (possibly as cases) or not tested Timing of reference standard: Not stated or not tested Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples (48 patients had sequential serum samples; 1 to 38 samples per patient, median 4 samples) |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Emerging Microbes & Infections Author COI: No potential conflict of interest was reported by the authors. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Chen 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Chen 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Chen 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Chen 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Chew 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: This study aimed to evaluate the diagnostic performance of the Abbott Architect SARS‐CoV‐2 IgG assay in COVID‐19 patients compared with pre‐pandemic controls.
2‐group study to estimate sensitivity and specificity for diagnosis of active disease and identification of previous disease Design: Two‐group study: [1] Symptomatic COVID‐19 patients selected on the basis of a positive SARS‐CoV‐2 rRT‐PCR from a respiratory sample (n = 177) [2] Negative controls were samples taken from patients prior to December 2019. These included patients with and without other positive serological tests (n = 163) Recruitment: Unclear whether all cases included ‐ "We prospectively identified confirmed COVID‐19 patients presenting at and admitted to our institution from 30th March 2020 to 15th May 2020". Prospective or retrospective: [1] prospective [2] retrospective Sample size: 340 (177) Further detail: [1] COVID‐19 patients selected on the basis of a positive SARS‐CoV‐2 rRT‐PCR from a respiratory sample. Patients who were asymptomatic at the time of PCR testing for contact screening purposes could not be stratified according to time from onset of illness and were excluded. [2] Unclear |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: National University Hospital, 5 Lower Kent Ridge Road, 11907, Singapore Country: Singapore Dates: 30th March 2020 to 15th May 2020 Symptoms and severity: Not stated, other than that patients who were asymptomatic at the time of PCR testing for contact screening purposes could not be stratified according to time from onset of illness and were excluded Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic controls Source: Negative controls were samples taken from patients prior to December 2019 Characteristics: Not stated. See comment Non‐Covid group 2: NA |
||
| Index tests | Test name: Abbott Architect SARS‐CoV‐2 IgG assay Manufacturer: Abbott Diagnostics, Chicago, USA Antibody: IgG Antigen target: IgG raised against the nucleocapsid protein of SARS‐CoV‐2 Evaluation setting: Laboratory, used in laboratory Test method: chemiluminescent immunoassay Timing of samples: COVID cases stratified according to time from onset of clinical illness to testing: (≤ 6 days, 81/177 7‐13 days, 39/177 14‐20 days 25/177, and ≥ 21 days 32/177) Samples used: Residual sera Test operator: Not stated Definition of test positivity: A signal/cut‐off (S/CO) ratio of >= 1.4 was interpreted as reactive and an S/CO ratio of < 1.4 was interpreted as non‐reactive. Also used alternate cut‐offs of 1.0 and 0.8 Blinding reported: Not stated Threshold predefined: A signal/cut‐off (S/CO) ratio of >= 1.4 was interpreted as reactive and an S/CO ratio of < 1.4 was interpreted as non‐reactive (Results also extracted for alternative lower cut‐off values. No for cutoffs 1.0 and 0.8) |
||
| Target condition and reference standard(s) | Reference standard: Two PCR assays were used during this time period (Fortitude, MirXES, Singapore, and cobas® SARS‐COV‐2, Roche Diagnostics, USA). No threshold reported Samples used: respiratory samples Timing of reference standard: Not stated Blinded to index test: Presumably, as cases selected on basis of reference test result Incorporated index test: No Definition of non‐COVID cases: None ‐ "negative samples collected prior to December 2019 were assumed to be negative as SARS‐CoV‐2 was first identified late in 2019". Samples used: pre‐pandemic Timing of reference standard: pre‐pandemic Blinded to index test: Yes, historical samples Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No. For COVID cases, there were two different PCR assays in use while historical controls included patients with and without other positive serological tests and were assumed to be COVID‐negative. Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No external funding was received for this study. Temasek Holdings Pte Ltd sponsored the laboratory testing kits used in this study. Publication status: Article in press; now published Source: Clinical Microbiology and Infection Author COI: One author (PT) received grants paid to the National University Hospital from Roche, Johnson & Johnson, Sanofi Pasteur, GlaxoSmithKline, and Shionogi. All other authors had no conflicts of interest to declare. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Chong 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Single‐group study to estimate sensitivity only [1] Confirmed COVID patients (63 samples from 18 patients) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 63 (63) samples from 18 (18) patients Further detail: Patients diagnosed with COVID‐19 on the basis of a positive rt‐PCR and admitted to Kyushu University Hospital (Fukuoka, Japan) Exclusion criteria not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: University Hospital, Fukuoka, Japan Country: Japan Dates: March and April 2020 Symptoms and severity: 5 asymptomatic 8 mild 3 severe 2 critical Demographics: Age: Mean 48.3 years (range 23‐69 years) Sex: 10 female, 8 male Exposure history: Not stated Non‐Covid group 1: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: [A] 2019‐nCoV IgG/IgM Rapid Test Cassette Manufacturer: [A] Hangzhou Alltest Biotech Co. Ltd. Antibody: IgG and IgM Antigen target: Nucleocapsid protein Evaluation setting: POCT performed retrospectively in a laboratory Test method: Immunochromatographic assay Timing of samples: 1‐33 days post‐symptom onset or post‐positive PCR for asymptomatic cases: 1‐6 days: 8/63 samples 7‐13 days: 35/63 samples 14‐20 days: 11/63 samples 21‐33 days: 9/63 samples Samples used: Serum samples, remaining from other biochemical tests (retrospective analysis) Test operator: Not stated Definition of test positivity: The presence of anti‐SARS‐CoV‐2 IgM and/or IgG antibodies was separately indicated by a red line in the corresponding area of the device. Blinding reported: Not stated but only included COVID patients Threshold predefined: yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: real‐time PCR assay performed by the Japanese Institute of Health according to the manual for the detection of pathogen 2019‐nCoV; threshold not stated Samples used: nasal and pharyngeal swab specimens Timing of reference standard: Not stated Blinded to index test: Yes, prior index test Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Influenza & Other Respiratory Viruses Author COI: "We have no financial conflicts of interest to declare." |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Clarke 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection or prior infection Design: Single‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 79) [2] Suspected COVID, PCR‐negative patients (n = 42) [3] Concurrent, untested, asymptomatic patients (n = 235) Group [3] not eligible for our review as a high‐risk group without reference standard Recruitment: Patients receiving dialysis within two units affiliated with Imperial College Renal and Transplant Centre between April 27 and May 7, 2020 who were routinely screened for the development of symptoms or a fever prior to each haemodialysis session Prospective or retrospective: Prospective Sample size: 356 (79) of which 121 (79) are eligible for our review Further detail: Inclusion: Patients receiving dialysis within two units affiliated with Imperial College Renal and Transplant Centre between April 27 and May 7, 2020 Exclusion from analysis: No informed consent |
||
| Patient characteristics and setting | Setting: Seroprevalence screening Location: Imperial College Renal and Transplant Centre, London, UK Country: UK Dates: April 27 and May 7, 2020 Symptoms and severity: All symptomatic Demographics: Patients with end‐stage kidney disease receiving haemodialysis (n = 79) Age: Median 65 (range 54–73) years Sex: 26 (32.9%) women Ethnicity: Black 9 (11.4%) White 19 (24.1%) Indoasian 38 (48.1%) Other 13 (16.5%) Immunosuppressed 8 (10.1%) Exposure history: Exposure within dialysis units Non‐Covid group 1: [2] Suspected COVID, PCR‐negative Source: Imperial College Renal and Transplant Centre, London, UK between April 27 and May 7, 2020 Characteristics: Patients with end‐stage kidney disease receiving haemodialysis with COVID symptoms (n = 42) Age: Median 62 (range 51–74) years Sex: 20 (47.6%) women Ethnicity: Black 8 (19.0%) White 9 (21.4%) Indoasian 19 (45.2%) Other 6 (14.2%) Immunosuppressed 4 (9.5%) Exposure within dialysis units Non‐Covid group 2: [3] Concurrent asymptomatic (untested) Source: Imperial College Renal and Transplant Centre, London, UK between April 27 and May 7, 2020 Characteristics: Patients with end‐stage kidney disease receiving haemodialysis without COVID symptoms (n = 235) Age: Median 68 (range 54–73) years Sex: 84 (35.7%) women Ethnicity: Black 29 (12.3%) White 62 (26.4%) Indoasian 97 (41.2%) Other 47 (20.0%) Immunosuppressed 43 (18.3%) Exposure within dialysis units |
||
| Index tests | Test name: [A] Abbott SARS‐CoV‐2 IgG assay Manufacturer: [A] Abbott Antibody: IgG Antigen target: Nucleocapsid‐protein antigen Evaluation setting: Lab test performed in lab Test method: Automated (Architect system) two‐step chemiluminescent microparticle immunoassay (CLIA) Timing of samples: [1] Mean 34+/‐6.4 days, median 22 (range 14–34) days after PCR testing [2] Median time between tests was 23 (14–35) days [3] Asymptomatic Samples used: Serum Test operator: Staff working in the Department of Infection and Immunity, North West London Pathology NHS Trust. Definition of test positivity: The index (sample/control) is calculated by comparing relative light units in the sample to the calibrator relative light units. Samples were interpreted as positive or negative according to the manufacturer’s instructions, with a cut‐off index value of 1.4. Blinding reported: Not stated Threshold predefined: yes, according to the manufacturer's instructions (S/C index) |
||
| Target condition and reference standard(s) | Reference standard: Routine screening of patients for the development of symptoms or a fever occurred prior to each haemodialysis session from March 9. Symptomatic patients received real‐time RT‐PCR assay of nasopharyngeal swab specimens following either routine screening or acute presentation; RT‐PCR was carried out as per PHE guidelines using certification marked assays with primers directed to the nucleocapsid or RNA‐dependent RNA polymerase genes. Threshold not stated Samples used: nasopharyngeal swab specimens Timing of reference standard: Not stated Blinded to index test: yes, prior Incorporated index test: no Definition of non‐COVID cases: [2] Routine screening of patients for the development of symptoms or a fever occurred prior to each haemodialysis session from March 9. Real‐time RT‐PCR assay of nasopharyngeal swab specimens following either routine screening or acute presentation; RT‐PCR was carried out as per PHE guidelines using certification marked assays with primers directed to the nucleocapsid or RNA‐dependent RNA polymerase genes. Threshold not stated [3] Routine screening of patients for the development of symptoms or a fever occurred prior to each haemodialysis session from March 9 (no PCR test) Samples used: [2] nasopharyngeal swab specimens [3] None Timing of reference standard: [2] Not stated [3] No reference standard as no symptoms Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1] Mean 34+/‐6.4 days, median 22 (range 14–34) days after PCR testing [2] Median time between tests was 23 (14–35) days [3] No reference standard All patients received same reference standard: yes for [1] and [2]; no reference standard for [3] Missing data: Exclusion of 235 PCR‐untested patients (group [3]) Uninterpretable results: None Indeterminate results: 3 of 356 (0.84%) patients had a borderline antibody result that was within +/‐20% of the cut‐off index for a positive result. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This research is supported by the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. Publication status: Published paper (rapid communication) Source: Journal of the American Society of Nephrology (JASN) Author COI: Dr. Liz Lightstone reported grants from Roche, outside the submitted work. M. Griffith reported an educational grant from Vifor Pharmaceuticals for £400 to attend the American Society of Nephrology 2019, outside the submitted work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Conklin 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients, convalescent (n = 40) [2] Confirmed COVID samples, longitudinal testing (47 patients with 272 samples) [3] Pre‐pandemic non‐COVID challenge samples (60 patients) Recruitment: [1] Not stated [2] Not stated [3] These samples came from a study of patients presenting to the Johns Hopkins Hospital Emergency Department with symptoms of an acute respiratory tract infection between January 2016 and June of 2019 as part of the Johns Hopkins Center for Influenza Research and Surveillance study. Prospective or retrospective: [1] Unclear [2] Unclear [3] Retrospective Sample size: 372 (312) Further detail: [1] RT‐PCR positive for SARS‐CoV‐2 and asymptomatic for at least 28 days. Human immunodeficiency virus (HIV) and hepatitis C virus (HCV) negative [2] Hospitalised SARS‐CoV‐2 RT‐PCR‐confirmed patients [3] Patients presenting to the Johns Hopkins Hospital Emergency Department with symptoms of an acute respiratory tract infection between January 2016 and June of 2019. Samples that were known to represent infections with other respiratory viruses (rhinoviruses A, B, and C and/or coronavirus 229E, HKU1, and NL63 OC43) |
||
| Patient characteristics and setting | Setting: [1] Convalescent plasma donors (community?) [2] Hospital inpatients Location: [1] and [2] Not stated (Johns Hopkins University School of Medicine, Baltimore?) Samples evaluated were from the Baltimore‐Washington region of the United States. Country: [1] and [2] Maryland, USA Dates: [1] and [2] Not stated Symptoms and severity: [1] Convalescent, asymptomatic since at least 28 days [2] Fever 34 (72%) Cough 29 (62%) Difficulty breathing 24 (51%) Muscle/body pain 14 (30%) Chills 9 (19%) Weakness/fatigue 7 (15%) Sore throat 6 (13%) Other 31 (66%) Demographics: [1] Not stated [2] Age: Median 62 (IQR 44‐80) years 29 (62%) male Black/African American 23 (49%) White/Caucasian 17 (36%) Hispanic/Latino 4 (9%) Asian 2 (4%) Other 1 (2%) Exposure history: [1] and [2] Not stated Non‐Covid group 1: [3] Pre‐pandemic challenge Source: Johns Hopkins Hospital Emergency Department between January 2016 and June of 2019 Characteristics: Samples that were known to represent infections with other respiratory viruses (rhinoviruses A, B, and C and/or coronavirus 229E, HKU1, and NL63 OC43). Non‐Covid group 2: NA |
||
| Index tests | Test name: [A] AllTest
[B] AYTU
[C] Clarity
[D] RightSign [E] Covisure [F] DNA Link [G] Nirmidas [H] Ready Result [I] EDI IgM ELISA [J] SafeCare [K] Sensing Self [L] Smart Screen [M] TBG SARS‐CoV‐2 IgG/IgM [N] Wondfo SARS‐CoV‐2 Ab [O] Zeus SARS‐CoV‐2 IgM/IgG [P] Euroimmun Anti‐SARS‐CoV‐2 IgG ELISA Manufacturer: [A] Hangzhou AllTest Biotech Co., Ltd. [B] AYTU Biosciences [C] Alfa Scientific Designs Inc. [D] Hangzhou Biotest Biotech Co., Ltd. [E] W.H.P.M., Inc. [F] Not stated [G] Nirmidas Biotech, Inc., and Lows Health [H] Hangzhou Biotest Biotech Co., Ltd. [I] Epitope Diagnostics, San Diego, CA [J] Safecare Biotech (Hangszhou) Co., Ltd. [K] Sensing Self, PTE. Ltd. [L] Intelligent Endoscopy [M] TBG Biotechnology Corp. [N] Wondfo Biotechnology [O] Zeus Scientific, Inc. [P] Euroimmun, Mountain Lakes, NJ Antibody: [A] IgM, IgG [B] IgM, IgG [C] IgM, IgG [D] IgM, IgG [E] IgM, IgG [F] IgM, IgG [G] IgM, IgG [H] IgM, IgG [I] IgM [J] IgM, IgG [K] IgM, IgG [L] IgM, IgG [M] IgM, IgG [N] IgM/IgG combined [O] IgM, IgG [P] IgG Antigen target: [A] N, S [B] N, S [C] N, S [D] RBD [E] Not stated [F] Not stated [G] S [H] N, S [I] Not stated [J] Not stated [K] N, S [L] Not stated [M] Not stated [N] Not stated [O] N, S [P] Not stated Evaluation setting: All POC tests apart from [I] and [P] Lab‐based Test method: [A] Lateral flow tests apart from [I] and [P] ELISAs Timing of samples: [1] 45 days (standard deviation [SD], +/‐7.5 days (at least 28 days asymptomatic). Figure 2a says "> 26 days" [2] Median 6 (IQR 4‐8) post‐symptom onset; Data Set S1 reported range from ‐2 to 36 days pso. Samples used: [1] and [2] Plasma [3] Serum Test operator: Not stated Definition of test positivity: [A]‐[O] All LFAs were performed according to the manufacturers’ protocols. Any detectable band (IgM and/or IgG) was considered a positive result. All LFAs, except Wondfo, had separate bands for IgM and IgG detection. Results were considered invalid when the control band was not visible. [P] and [Q] per the manufacturers’ protocols Blinding reported: Not stated Threshold predefined: Yes, visual‐based or as per manufacturer's protocols [I] and [P] |
||
| Target condition and reference standard(s) | Reference standard: [1] and [2] SARS‐CoV‐2 RT‐PCR, threshold not stated Samples used: [1] and [2] Not stated Timing of reference standard: [1] and [2] Not stated Blinded to index test: [1] and [2] yes, prior Incorporated index test: [1] and [2] no Definition of non‐COVID cases: [3] Pre‐pandemic Samples used: [3] Pre‐pandemic Timing of reference standard: [3] Pre‐pandemic Blinded to index test: [3] yes, prior Incorporated index test: [3] no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: yes (see Figure 2a, test [E] Covisure 3 invalid results, test [H] Premier Biotech 2 invalid results, test [F] DNA Link 1 no data; test [M] TBG 1 no data) Uninterpretable results: yes (3 invalid results for test [E], 2 invalid results for test [H], Figure 2a) Indeterminate results: Not stated Unit of analysis: [1] and [2] Patients [3] Samples |
||
| Comparative | |||
| Notes | Funding: The study was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). Research reported in this publication was supported by the following research awards: from the NIAID, UM1‐AI068613, R01AI120938, and R01AI128779; from the National Institute of
Biomedical Imaging and Bioengineering, U54EB007958; from the National Heart, Lung, and Blood Institute of the National Institutes of Health, 1K23HL151826‐01. The work described here was supported in part by NIAID contract HHSN272201400007C awarded to the Johns Hopkins Center for Influenza Research and Surveillance (JHCEIRS). Publication status: Published paper Source: Journal of Clinical Microbiology Author COI: E.M.B. is a member of the United States Food and Drug Administration (FDA) Blood Products Advisory Committee. Any views or opinions that are expressed in this article are ours, based on our own scientific expertise and professional judgment; they do not necessarily represent the views of either the Blood Products Advisory Committee or the formal position of FDA and also do not bind or otherwise obligate or commit either Advisory Committee or the Agency to the views expressed. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Conklin 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [L].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [M].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [N].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [O].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Conklin 2020 [P].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Costa 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection (only time split 4‐13 days pso was eligible for our review though) Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 122) [1a] rt‐PCR‐positive (n = 106) [1b] negative RT‐PCR but a clinical COVID‐19 diagnosis (n = 16) [2] Non‐COVID samples (96 historical blood donation samples, Table 3 specified 100 though) Recruitment: [1] Not stated (2 Brazilian hospitals) [2] Not stated Prospective or retrospective: [1] Prospective [2] Retrospective Sample size: 218 (122) of which 134 (38) are eligible for our review. Further detail: [1a] rt‐PCR‐positive [1b] rt‐PCR‐negative with clinical COVID diagnosis based on highly suggestive symptoms and chest computed tomography (CT) findings [2] historical (February 2019) blood donors Exclusion criteria not stated |
||
| Patient characteristics and setting | Setting: Mixed (inpatients and outpatients) Location: 2 Brazilian hospitals: Hospital das Clínicas da Faculdade de Medicina da Universidade de S˜ao Paulo (HC‐FMUSP; [1b]) and Hospital Sírio‐Libanes (HSL; [1a, inpatients]). Both hospitals are located in Sao Paulo. Country: Brazil Dates: Not stated Symptoms and severity: 75 inpatients and 47 outpatients Numbers (%) for 59 PCR+ inpatients, 47 PCT+ outpatients and 16 PCR‐ inpatients: Fever 34 (60); 27 (61); 13 (81) Cough 38 (67); 35 (79); 16 (100) Coryza 7 (12); 10 (23); 1 (6) Sore throat 6 (11;) 16 (36); 1 (6) Dyspnoea 30 (53); 12 (27); 15 (94) Myalgia 6 (11); 18 (41); 3 (19) Asthenia 6 (11); 8 (18); NA Headache 4 (7); 27 (61); 2 (13) GI symptoms* 5 (9); 17 (38); 3 (19) Haemoptysis 3 (5); NA; NA Dysgeusia 1 (1.8); 2 (4.5); 2 (13) Anosmia NA; 7 (15; ) 2(13) All 16 RT‐PCR‐negative patients had pneumonia, 6/16 (38%) were intubated. Demographics: [1a] 59 PCR+ inpatients Age median 61 (range 32‐90) years Male 41 (70%) [1a] 47 PCR+ outpatients (healthcare workers) Age median 44 (range 21‐62) years Male 20 (43%) [1b] 16 PCR‐ inpatients Age median 55 (range 36‐77) years Male 6 (38%) Exposure history: [1a] 47/106 were healthcare workers [1b] Not stated Non‐Covid group 1: [2] Pre‐pandemic controls Source: Blood donors; February 2019 Characteristics: Not stated (blood donors, so possibly healthy) |
||
| Index tests | Test name: [A] Not stated [B] Not stated Manufacturer: [A] Euroimmun‐ Lübeck, Germany [B] Wondfo‐China Antibody: [A] IgA and IgG [B] IgG and IgM Antigen target: [A] anti‐SARS‐CoV‐2 S1 IgG and IgA [B] Not stated Evaluation setting: [A] Lab test performed in lab [B] POCT, unclear where performed (plasma samples) Test method: [A] ELISA [B] Rapid chromatographic immunoassays; Anti‐SARS‐CoV‐2 antibodies present in the sample bind to recombinant antigens coated on colloidal gold particles and form an antigen‐antibody/colloidal gold complex. Timing of samples: [1a] PCR+ inpatients Mean 10.7 (range 4‐23) days pso PCR+ outpatients Mean 32.0 (range 16‐42) days pso All PCR+ patients: < 14 days: 38/106 14+ days pso: 59/106 Unknown: 9/106 [1b] PCR‐ inpatients Mean 8 (range 2‐15 ) days pso Samples used: Plasma Test operator: Not stated Definition of test positivity: [A] Results were interpreted according to the manufacturer’s recommendation: a ratio < 0.8 as negative, between 0.8 and 1.1 as borderline, and ≥ 1.1 as positive. [B] The result was read in 15 minutes by three people that had received appropriate training. The colour change was compared to the assay standard. Blinding reported: not stated Threshold predefined: [A] yes, according to the manufacturer's recommendation [B] yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: [1] RT‐PCR. RNA was extracted from clinical samples with an automated method using magnetic beads (sample Preparation System RNA, Abbott, Illinois, USA). SARS‐CoV‐2 RNA reverse transcription, amplification, and detection were performed using an adapted protocol, as described elsewhere. An assay detecting the E gene was used as the first‐line screening tool, followed by confirmatory testing with an assay detecting the N gene. Threshold not stated. [1b] 14/16 RT‐PCR‐negative patients had a second negative RT‐PCR. Clinical COVID‐19 diagnosis based on highly suggestive symptoms and chest computed tomography (CT) findings. Samples used: [1] Respiratory samples were obtained from both the nasopharynx and oropharynx using rayon swabs. Timing of reference standard: Not stated Blinded to index test: yes, prior Incorporated index test: no Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic (February 2019) Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: yes as only 38/122 COVID cases included in our review Uninterpretable results: Not stated Indeterminate results: Not stated (there seemed to be some borderline results for test [A] in Figure 1, see supplement) Unit of analysis: [1] Patients [2] Unclear |
||
| Comparative | |||
| Notes | Funding: Internal funding from the Hospital das Clínicas of University of S˜ao Paulo, Brazil. Publication status: Published paper (Short Communication) Source: Journal of Clinical Virology Author COI: The authors reported no declarations of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Coste 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of multiple tests for COVID‐19 diagnosis Design: Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 178); Group [2]: Pre‐pandemic controls (n = 404) [Some assays only had preliminary evaluation results for subgroup of 113 COVID‐19 samples and 69 non‐COVID samples] Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 582 (178); 182 (113) in preliminary evaluation Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear Location: Lausanne University Hospital Country: Switzerland Dates: Not stated Symptoms and severity: Not available Demographics: Not available Exposure history: Not available Non‐Covid group 1: Pre‐pandemic controls Source: Lab stocked samples obtained before Nov 1st, 2019 from patients with multiple infectious or autoimmune diseases. Characteristics: Other infections/conditions documented in supplementary table, including 129 nonspecific, 17 herpes simplex virus 1 and 2, 18 respiratory syncytial virus, 22 Epstein‐Barr virus, 33 cytomegalovirus, 27 mumps and/or measles virus, 14 parvovirus B19, 17 rubella virus, 45 influenza A, B or RSV, plus others (varicella‐zoster virus, human immunodeficiency virus, hepatitis virus A, B, C, D, and E, and some rheumatoid factors, or auto‐antibodies (anti‐PR3, ‐PR4, SCL70, SCL71)). Preliminary evaluation controls included 18 HCov, 12 lupus, and 39 nonspecific |
||
| Index tests | Test name: [A] EDI™ Novel Coronavirus COVID‐19 IgG ELISA Kit [B] EDI™ Novel Coronavirus COVID‐19 IgM ELISA Kit [C] Anti‐SARS‐CoV‐2 ELISA (IgG) [D] Anti‐SARS‐CoV‐2 ELISA (IgA) [E] SARS‐CoV‐2 NP IgG ELISA Kit [F] SARS‐CoV‐2 NP IgM ELISA Kit [G] COVID‐19 ELISA IgG [H] COVID‐19 ELISA IgM+IgA [I] SARS‐CoV‐2 IgG ELISA Kit [J] SARS‐CoV‐2 IgM ELISA Kit [K] 2019‐nCOV IgG/IgM Rapid Test [L] NADAL® COVID‐19 IgG/IgM Test [M] One Step Test for Novel Coronavirus (2019‐nCoV) IgM/IgG Antibody [N] ISON® SARS‐CoV‐2 IgG kit [O] MAGLUMITM 2019‐nCoV IgG + IgM [P] Elecsys anti‐SARS‐CoV‐2 Manufacturer: [A] Epitope Diagnostics, USA [B] Epitope Diagnostics, USA [C] EUROIMMUN AG, Germany [D] EUROIMMUN AG, Germany [E] Immunodiagnostics limited, Hong Kong [F] Immunodiagnostics limited, Hong Kong [G] Vircell, Spain [H] Vircell, Spain [I] Creative Diagnostics, USA [J] Creative Diagnostics, USA [K] Dynamiker, China [L] Nal Von Minden, Germany [M] Augurix Diagnostics, Switzerland/China [N] Diasorin, Italy [O] Snibe, China [P] Snibe, China [Q] Roche, Germany Antibody: [A] IgG [B] IgM [C] IgG [D] IgA [E] IgG [F] IgM [G] IgG [H] IgM, IgA [I] IgG [J] IgM [K] IgG, IgM [L] IgG, IgM [M] IgG, IgM [N] IgG [O] IgG, IgM [P] Total antibody Antigen target: [A] N‐protein [B] N‐protein [C] S1 domain of the spike‐protein [D] S1 domain of the spike‐protein [E] N‐protein [F] N‐protein [G] N and S‐proteins [H] N and S‐proteins [I] Whole virus lysate [J] N and S‐proteins [K] N‐protein [L] S‐protein [M] N and S‐proteins [N] S1 and S2 domains of the spike‐protein [O] N and S‐proteins [P] N‐protein Evaluation setting: [A] ‐ [J] and [N] ‐ [Q]: Lab tests; likely done in lab but not explicitly stated. [K] ‐ [M]: POC tests, unclear where they were performed. Test method: [A] ‐ [J]: Enzyme‐linked immunosorbent assay (ELISA) [K] ‐ [M]: Lateral flow assay [N] ‐ [O]: Chemiluminescence immunoassay (CLIA) [P]: Electrochemiluminescence immunoassay (ECLIA) Timing of samples: Obtained during the first 2 months post‐symptom onset. No more details available Samples used: Serum Test operator: Not stated Definition of test positivity: As per manufacturer (no more details available) Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR test (no more details available) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: No testing (pre‐pandemic samples) Samples used: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No (type of RT‐PCR unknown for cases; controls were pre‐pandemic samples) Missing data: Apparently no. However, only some tests were done on all samples. Selection for full evaluation was based on: i) sensitivity and specificity performance of the preliminary evaluation, ii) protein detected (anti‐N: ED IgG ELISA and Dynamiker IgG/IgM; anti‐S: Diasorin IgG CLIA, anti N+S: Snibe IgG/IgM CLIA) iii) availability of the kits on 15th April 2020 in Switzerland, iv) specific detection of IgG and/or IgM or IgA, and v) compatibility of the kits to most laboratory needs (e.g. median to low samples volumes per day and extended expiration days upon kits opening). "Despite its good performance, the ECLIA from Roche was selected as it detects pan‐180 Ig, which is not the most appropriate for infectious serology diagnostic". Uninterpretable results: None Indeterminate results: Yes, varied by test but the number of indeterminate results for each test was unclear due to contradictory numbers in supplementary tables [A] Epitope Diagnostics IgG: 0/178, 13/404 (full evaluation); 3/113, 4/69 (preliminary evaluation); more missing from preliminary evaluation compared to full [B] Epitope Diagnostics IgM: 12/178, 5/404 (full evaluation) [C] EUROIMMUN IgG: 8/113; 1/69 [D] EUROIMMUN IgA: 8/113; 5/69 [E] Immunodiagnostics limited, IgG: 1 extra sample reported for D+; 3/69 missing for [D]‐[F] Immunodiagnostics limited, IgM: 2 extra samples reported for D+; 2/69 missing for [D]‐ [G] Vircell, IgG: 3/113, 5/69 missing [H] Vircell, IgM+IgA: 7/113, 13/69 missing [I] and [J] Creative Diagnostics: 0 indeterminate [K] Dynamiker: preliminary evaluation 2/113 for IgG and 5/113 for IgM missing, but for full evaluation there was 1 extra sample reported for IgG (179 instead of 178), and only 2/178 missing for IgM (for disease‐negative there was 1/404 missing for IgG and 3/404 missing for IgM); more missing from preliminary evaluation compared to full [L] Nal Von Minden: IgG 6/113, 1/69 missing; IgM 7/113, 2/69 missing [M] Augurix Diagnostics IgM 8/113; 2/69; IgM 18/113; 1/69 [N] Diasorin, Italy: preliminary evaluation dataset showed 6/113 and 2/69 indeterminate; full evaluation showed 3/178 D+ missing but 5 extra results for D‐ (409 instead of 404); more missing from preliminary evaluation compared to full [O] Snibe, IgG: full evaluation 2/178, 1/404 missing; preliminary evaluation showed 6/113, 1/69 missing; more missing from preliminary evaluation compared to full Snibe, IgM: full evaluation 1/178, 2/404 missing; preliminary evaluation showed 5/113, 3/69 missing; more missing from preliminary evaluation compared to full [P] Roche pan‐IgG: 6/113, 2/69 missing Unit of analysis: Unclear ‐ referred to 'patients' but did not describe if 1 sample per patient |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Pre‐print article Source: Pre‐print server (medXriv) Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Coste 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [L].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [M].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [N].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Coste 2021 [O].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Criscuolo 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of two antibody tests for SARS‐CoV‐2 infection Design: Two‐group study estimating both sensitivity and specificity Group [1]: Lab‐confirmed cases of SARS‐CoV‐2 infection (n = 46). Group [2]: Pre‐pandemic controls (n = 85) For Group [1], lab confirmation likely referred to PCR test, but this was not explicitly stated. Recruitment: Random (approach not explained) Prospective or retrospective: Retrospective Sample size: 131 (46) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Inpatient service (all patients were hospitalised) Location: IRCCS San Raffaele Hospital, Milan Country: Italy Dates: Not stated Symptoms and severity: Not stated, all admitted Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Group [2]: Pre‐pandemic controls Source: Lab stocked samples collected between 2012 and 2018 Characteristics: Not stated |
||
| Index tests | Test name: [A] Elecsys Anti‐SARS‐CoV‐2 [B] LIAISON® SARS‐CoV‐2 69 S1/S2 IgG assay Manufacturer: [A] Roche Diagnostics [B] DiaSorin, Italy Antibody: [A] Total antibodies [B] IgG Antigen target: [A] N‐protein [B] S1 and S2 domains of the spike‐protein Evaluation setting: [A], [B]: Lab tests, done in lab Test method: [A] Electrochemiluminescence immunoassay (ECLIA) [B] Chemiluminescence immunoassay (CLIA) Timing of samples: For each patient: one serum sample collected at hospital admission and another one 15 days later. Time since symptom onset not reported. Samples used: Serum Test operator: Not stated Definition of test positivity: [A] Positive if COI >= 1 [B] Positive if > 15 AU/mL; undetermined if 12‐15 AU/mL; negative if < 12 AU/mL Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: Apparently RT‐PCR (the authors only reported "lab‐confirmation"). No more details available Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic samples ‐ no testing Samples used: NA Timing of reference standard: NA Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Unclear: all cases (Group [1]) were lab‐confirmed but various assays were likely used. Missing data: None Uninterpretable results: None Indeterminate results: Yes, 1 for test [B] on pre‐pandemic samples (Group [2]); none reported for cases. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Pre‐print article Source: Pre‐print server (medRxiv) Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Criscuolo 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Dave 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of COVID‐19 infection and prognostication Design: Single‐group study to assess sensitivity [1] RT‐PCR‐positive patients admitted to a tertiary care hospital Recruitment: All RT‐PCR‐positive COVID 19 patients (both symptomatic and asymptomatic) above the age of 18 y admitted in various wards of a dedicated Corona hospital from April 2020 to May 2020 Prospective or retrospective: Unclear Sample size: 100 (100) Further detail: Inclusion: All RT‐PCR‐positive COVID‐19 patients (both symptomatic and asymptomatic) above the age of 18 y Exclusion: (i) patients on steroids, immunosuppressants and chemotherapy (ii) PLHA and other immune‐deficiency diseases (iii) non‐consenting patients |
||
| Patient characteristics and setting | Setting: Patients admitted to a tertiary care hospital (dedicated COVID hospital) Location: RNT Medical College, Udaipur, Rajasthan Country: India Dates: April 2020‐May 2020 (2 months) Symptoms and severity: 76 asymptomatic; 17 mild to moderate; 7 severe Demographics: Male 45; female 55 Mean age 37 years Exposure history: Not reported Non‐Covid group 1: NA |
||
| Index tests | Test name: Antibody‐based rapid card test (no specific name provided) Manufacturer: SIDAK Life Care Antibody: IgM and IgG Antigen target: Not reported Evaluation setting: POC unclear where used Test method: Lateral flow method (immune chromatographic assay) Timing of samples: days of illness for all 100 patients (74/100 were asymptomatic so must be days post‐positive PCR?): 0‐7 (n = 23) 8‐14 (n = 27) 15‐21 (n = 36) > 21 (n = 14) Samples used: Whole blood (2 drops) Test operator: Not reported Definition of test positivity: (a) Along with C band, if band at zone 1, indicates the presence of IgM (b) Along with C band, if band at zone 2, indicates the presence of IgG (c) Along with C band, if band at zone 1 and 2, indicates the presence of both IgM and IgG Blinding reported: No Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR‐positive; according to the protocols by National Institute of Virology, Pune Samples used: Nasopharyngeal/oropharyngeal swab Timing of reference standard: Not stated Blinded to index test: Yes (done prior to the index test) Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not reported All patients received same reference standard: Yes Missing data: Not reported Uninterpretable results: Not reported Indeterminate results: Not reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published article Source: Journal of Indian Academy of Clinical Medicine Author COI: No COI declaration |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | No | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Decru 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of multiple rapid tests for diagnosis of convalescent‐phase COVID‐19 infection Design: Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 26 patients, 33 samples) Group [2]: PCR‐negative patients without clinical suspicion of COVID‐19 (n = 39 patients/samples) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 72 (33) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear Location: University Hospitals, Leuven Country: Belgium Dates: Not stated Symptoms and severity: All symptomatic individuals. No further details available (table footnote described one patient as having fever and compatible CT but no respiratory symptoms) Demographics: Age, median (IQR): 67 y (33‐92 y) Exposure history: Not stated Non‐Covid group 1: Group [2]: PCR‐negative patients without clinical suspicion of COVID‐19 Source: Not stated; negative PCR was within previous 7 days Characteristics: Not stated |
||
| Index tests | Test name: [A] MultiG single lane (MultiG1, lot NCP‐20030181) [B] MultiG dual lane (MultiG2, lot COV1452003C) [C] COVID‐19 IgM/IgG Rapid Test Cassette (lot 2003318) [D] COVID‐19 Coronavirus Rapid Test Cassette (lot COV20030120) Manufacturer: [A], [B]: Multi‐G, Belgium [C]: Orient Gene Biotech, China [D]: SureScreen Diagnostics Antibody: IgG and IgM Antigen target: Not stated Evaluation setting: All POC tests, but likely done in lab Test method: All lateral flow immunoassays (LFA) Timing of samples: 23‐65 days after symptom onset; (data by week provided by authors) day 23‐28: 3, 9% day 29‐35: 5, 14% day > 35: 25, 71% Samples used: Whole blood, plasma Test operator: Not stated Definition of test positivity: Not stated (but likely visual‐based) Blinding reported: Unclear Threshold predefined: Visual line |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR test (no further details available) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: RT‐PCR test (no further details available) Samples used: Not stated Timing of reference standard: Test done in the last 7 days before enrolment in the study Blinded to index test: Yes (done earlier) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: 21‐62 days from first RT‐PCR‐positive All patients received same reference standard: Yes (for the purpose of this item we considered any RT‐PCR to be adequate and 'the same') Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: The authors declared no specific funding was received. Publication status: Published letter Source: Clinical Chemistry & Laboratory Medicine Author COI: Authors stated no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Decru 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Decru 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Decru 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Delliere 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To evaluate the COVID‐19 IgG/IgM Rapid Test Cassette (Orient Gene Biotech, Zhejiang, China) and compare it to simultaneous CMIA IgG testing by the Abbott SARS‐CoV‐2 IgG (ASIA) on Architect Abbott Instrument i2000SR
2‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: Two‐group study: [1] COVID‐19‐positive patients (n = 102, 106 samples) [2] Pre‐pandemic patients (n = 42; 14 occupational health patients with no known disease; 28 hospitalised patients with previous pulmonary infection, rhinovirus, metapneumovirus, influenza A, syncytial respiratory virus, recent malaria,antibodies against cytomegalovirus or Epstein‐Barr, HIV, hepatitis B, toxoplasmosis, rheumatic fever) Recruitment: Unclear Prospective or retrospective: [1] Unclear; [2] retrospective Sample size: 142 (102) patients with 146 (106) samples Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Not stated
35/102 hospitalised in a medical unit
28/102 hospitalised in ICU
The remaining 39/102 possibly not inpatients (2 asymptomatic and 37 mild symptoms) Location: Hôpital Saint‐Louis, Département des Agents Infectieux, 1 avenue Claude Vellefaux, 75010 Paris, France Country: France Dates: Not stated Symptoms and severity: asymptomatic (n = 2), mild (n = 37), severe symptoms requiring hospitalisation in medical unit (n = 35), critical symptoms requiring hospitalisation in intensive care unit (n = 28) Demographics: Mean age of the patient population was 52 (± 16) years; male = 59/102 (57.8%) Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic controls Source: Not stated Characteristics: No known disease 14/42 Hospitalised patients 28/42: (previous pulmonary infection with endemic coronavirus 16/28; rhinovirus 1/28;metapneumovirus 1/28; influenza A 1/28; syncytial respiratory virus 1/28; recent infection with malaria 3/28; IgM antibodies (Ab) against cytomegalovirus 2/28; IgM Ab against Epstein‐Barr virus 2/28; IgG against HIV 1/28; hepatitis B virus 1/28; toxoplasmosis 1/28; high levels of rheumatic factor 2/28) |
||
| Index tests | Test name: [A] Orient Gene COVID‐19 IgG/IgM Rapid Test Cassette [B] Abbott SARS‐CoV‐2 IgG Manufacturer: [A] Orient Gene Biotech, Zhejiang, China [B] Abbott, Illinois, USA Antibody: [A] IgG, IgM [B] IgG Antigen target: [A] and [B] Not stated Evaluation setting: [A] POC, but performed on residual samples in laboratory [B] Performed in lab (on Architect Abbott Instrument i2000SR) Test method: [A] lateral flow assay (LFA) [B] CMIA Timing of samples: [A] and [B] ≥ 4 days (4‐40, median = 18) since onset of symptoms or positive PCR for asymptomatic patients Samples used: [A] and [B] Serum (stored at –20°C upon use) Test operator: [A] All Orient Gene test results were performed and read after 10 min by two clinical microbiologists unblinded regarding the sample group. Indeterminate readings were to be read by a third microbiologist. [B] Tests processed by microbiologists on Architect Abbott Instrument i2000SR Definition of test positivity: [A] The result is read at 10 minutes. The cassette displays a blue control band that turns red when the test has been performed correctly. IgG and IgM are represented by two separated bands and are read visually. All Orient Gene test results were performed and read after 10 min by two clinical microbiologists unblinded regarding the sample group. Indeterminate readings were to be read by a third microbiologist. [B] manufacturer’s recommended cut‐off of 1.4 Blinding reported: [A] microbiologists unblinded regarding the sample group [B] Unclear Threshold predefined: [A] yes, visual [B] yes, manufacturer’s recommended cut‐off of 1.4 |
||
| Target condition and reference standard(s) | Reference standard: SARS‐COV‐2 RT‐PCR (Cobas® SARS‐CoV‐2 Test, Roche, Meylan, France) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: None ‐ pre‐pandemic Samples used: pre‐pandemic Timing of reference standard: pre‐pandemic Blinded to index test: pre‐pandemic Incorporated index test: pre‐pandemic |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No. Cases had RT‐PCR, controls untested pre‐pandemic Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: COVID‐19 cases = 106 samples from 102 patients (4 patients with 2 consecutive sera) |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Accepted manuscript posted online 9 June 2020; now published Source: Journal of Clinical Microbiology Author COI: The authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Delliere 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Doherty Institute 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: An updated report evaluating the diagnostic performance of three serological point‐of‐care tests and comparing these with two POC tests and one EIA test included in a previous report
Multi‐group study to estimate sensitivity and specificity for diagnosis of active disease or identification of previous disease. Design: [1] Sensitivity group: patients with SARS‐CoV‐2 detected by RT‐PCR from upper and/or lower respiratory tract specimens. (n = 91 patients, 137 samples) [2] Specificity group: (n = 92 people, 92 samples) [2a] patients with infections with the potential for cross‐reactivity in serological assays, namely (i) patients with respiratory viral infections, including seasonal coronavirus infections and (ii) patients with other acute infections (e.g. dengue; CMV; EBV) (n = 36 patients, 36 samples) [2b] representative sample of the Victorian population collected in 2018 and 2019 (‘pre‐pandemic controls’) (n = 56 people, 56 samples) Recruitment: All serum samples were obtained from a tertiary hospital (Royal Melbourne Hospital, RMH) or the state reference laboratory for virology (Victorian Infectious Diseases Reference Laboratory, VIDRL). Prospective or retrospective: Retrospective Sample size: Patients: 183 (91) Samples: 229 (137) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Tertiary hospital and state reference laboratory Location: tertiary hospital (Royal Melbourne Hospital, RMH) or the state reference laboratory for virology (Victorian Infectious Diseases Reference Laboratory, VIDRL) Country: Australia Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] Other non‐COVID infections Source: Other diseases, dates not stated; pre‐pandemic controls 2018‐2019 Characteristics: Not stated Non‐Covid group 2: [2b] Pre‐pandemic controls Source: 2018‐2019 Characteristics: NR |
||
| Index tests | Test name: [A] Hangzhou Alltest IgG/IgM Rapid Test [B] Hangzhou unlabelled packaging (see comments) [C] Wondfo SARS‐CoV‐2 Antibody Test [D] Hightop SARS‐COV‐2 IgM/IgG Antibody Rapid Test [E] OnSite COVID‐19 IgG/IgM Rapid Test [F] VivaDiag COVID‐19 IgM/IgG Rapid Test [G] EUROIMMUN Anti‐SARS‐CoV‐2 ELISA (IgA) (IgG) Manufacturer: Not reported, but as per test names Antibody: [A] to [F] IgG, IgM, (NB assay [C] does not differentiate between antibody class, with only a single test line indicative of a positive test IgM/IgG) [G] IgA, IgG Antigen target: [A to F] The specific SARS‐CoV‐2 recombinant antigen(s) incorporated into the assay were not described in the manufacturers' information [G] Not stated Evaluation setting: [A to F] POC, used in laboratory; [G] Laboratory, used in laboratory Test method: [A to F] Lateral flow immunoassay (colloidal gold) (CGIA); [G] ELISA Timing of samples: 0 ‐> 30 days post‐symptom onset 0‐3 days pso: 23/137 (16.8%) samples 4‐8 days pso: 28/137 (20.4%) samples 9‐14 days pso: 21/137 (15.3%) samples 15‐20 days pso: 8/137 (5.8%) samples 21‐30 days pso: 27/137 (19.7%) samples > 30 days pso: 30/137 (21.9%) samples Samples used: Serum Test operator: [A to F] three laboratory research technicians, all of whom had undergone previous training in the use of lateral flow assays [G] Not reported Definition of test positivity: [A, B, D to F] Visible lines for IgG/IgM; [C] single test line indicative of a positive test (IgM/IgG); [G] Not reported [C] and [D] Testing was undertaken in duplicate, with a third test undertaken for discordant results.The majority result (i.e. 2/3) was taken as the final result, any faint line present at test termination was considered a positive result. Blinding reported: Yes. Threshold predefined: [A to F] Visible lines, interpreted as per manufacturer's instructions for use; [G] Not reported |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 detected using the Coronavirus Typing assay (AusDiagnostics, Mascot, NSW) ‐ a two‐step, hemi‐nested multiplex tandem PCR
In addition, all positive samples had SARS‐CoV‐2 detected at VIDRL where testing was first conducted using an in‐house assay for the SARS‐CoV‐2 RdRp gene. If positive, subsequent testing for the SARS‐CoV‐2 E gene was performed, using previously published primers. Samples used: Upper and/or lower respiratory tract specimens Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: [2a] Unclear for other diseases, [2b] NA for pre‐pandemic controls Samples used: [2a] Unclear for other diseases, [2b] NA for pre‐pandemic controls Timing of reference standard: [2a] Unclear for other diseases, [2b] NA for pre‐pandemic controls Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No ‐ pre‐pandemic controls included Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: One sample was excluded from testing in the Hightop SARS‐CoV‐2 IgM/IgG Antibody Rapid Test assay as results were discordant and insufficient test kits remained to test in triplicate. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Report of post‐market validation (Report prepared for Office of Health Protection, Commonwealth Government of Australia, and the Therapeutics Goods Administration (TGA) of Australia) Source: Doherty Institute Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Doherty Institute 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Doherty Institute 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Doherty Institute 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Doherty Institute 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Doherty Institute 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Doherty Institute 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
DomBourian 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 convalescent plasma samples (n = 102) [2] Non‐COVID samples (n = 126) [2a] Current non‐COVID, respiratory pathogen panel (RPP)‐positive samples (n = 20); [2b] Pre‐pandemic samples (n = 106) Recruitment: Not stated Prospective or retrospective: [1] and [2a] Unclear (possibly retrospective) [2b] Retrospective Sample size: 228 (102) samples Further detail: [1] SARS‐CoV‐2 PCR‐positive donors from the Children's Hospital Colorado CCP donor programme; eligible individuals for the CCP donor programme were confirmed PCR‐positive for SARS‐CoV‐2 and were symptom‐free for at least 14 days prior to plasma donation and met all standard blood donation criteria per FDA requirements. [2a] Residual samples from patients who had tested positive for one of the respiratory viral pathogens and who were confirmed to be PCR‐negative for SARS‐CoV‐2 [2b] Pre‐pandemic samples that were collected prior to November 2019 |
||
| Patient characteristics and setting | Setting: Convalescent plasma donors Location: Children's Hospital Colorado's CCP donor programme, Aurora, Colorado Country: USA Dates: Children's Hospital Colorado's CCP donor programme was registered with the FDA as eligible to collect CCP on March 31, 2020. Symptoms and severity: Symptom‐free for at least 14 days Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] Current cross reaction challenge Source: Not stated (current) Characteristics: Tested positive for one of the respiratory viral pathogens (adenovirus; human metapneumovirus [HMPV]; influenza virus A hemagglutinin [H] subtypes H1, H3, and 2009 H1N1; influenza virus B; respiratory syncytial virus; coronaviruses NL63, OC43, 229E, and HKU1; human rhinovirus/enterovirus; parainfluenza types 1–4; Bordetella pertussis; mycoplasma pneumonia; and chlamydophila pneumonia) Non‐Covid group 2: [2b] Pre‐pandemic Source: Source not stated; collected prior to November 2019 Characteristics: Not stated |
||
| Index tests | Test name: [A] EDI™ Novel Coronavirus COVID‐19 IgG ELISA kit [B] Euroimmun Anti‐SARS‐CoV‐2 ELISA (IgG) Manufacturer: [A] Epitope Diagnostics Inc. (EDI) (San Diego, CA) [B] Euroimmun (Lubeck, Germany) Antibody: [A] and [B] IgG Antigen target: [A] nucleocapsid antigen [B] S1 domain, including the receptor binding domain (RBD) of the SARS‐CoV‐2 spike‐protein Evaluation setting: [A] and [B] Lab test, unclear setting Test method: [A] and [B] ELISA Timing of samples: At least 14 days symptom‐free Samples used: Plasma or serum Test operator: Not stated Definition of test positivity: [A] For the EDI assay, positive, negative and borderline results were calculated based on the average optical density (OD450) value for the negative control assayed in triplicate for the specific assay. The positive and negative cut‐off values were calculated using the formula: positive cut‐off = 1.1 x (xNC + 0.18) and negative cut‐off = 0.9 x (xNC + 0.18), where xNC is the average OD450 of triplicate negative control OD values. Samples that had OD450 values that fell between positive and negative cut‐off values were reported as borderline. [B] The Euroimmun assay was interpreted based on the ratio of the sample OD450 to the calibrator OD450. Samples with a ratio of less than 0.8 were deemed negative, samples with a ratio of greater than 1.1 were positive, and OD450 values between 0.8 and 1.1 were reported as borderline. Blinding reported: Not stated Threshold predefined: Yes, for this study, the assays were used per the manufacturers' specifications. |
||
| Target condition and reference standard(s) | Reference standard: PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior index test Incorporated index test: No Definition of non‐COVID cases: [2a] PCR (unclear how many negative tests) [2b] Pre‐pandemic (before November 2019) Samples used: [2a] Not stated [2b] Pre‐pandemic Timing of reference standard: [2a] Not stated [2b] Pre‐pandemic Blinded to index test: Yes, prior index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: [A] 6 borderline results [B] 6 borderline results Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: The Departments of Pediatrics and Pathology at the University of Colorado School of Medicine, and Children's Hospital Colorado Publication status: Published paper Source: Journal of Immunological Methods Author COI: All authors declared that they had no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
DomBourian 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Dora 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection/prior infection (to identify potentially missed cases of COVID‐19 during serial surveillance testing) Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID cases (n = 26) [2] Current non‐COVID cases (n = 124) Recruitment: All eligible residents in the skilled nursing facilities (SACC and WLA) and additional patients from the community who were diagnosed with COVID‐19 by RT‐PCR and treated in the acute care hospital were transferred to the CRU. Prospective or retrospective: Prospective Sample size: 150 (26) Further detail: Inclusion: a) Residents in SACC skilled nursing facility or WLA skilled nursing facility or in designated COVID‐19 recovery unit (CRU) with PCR test result b) Patients from the community who were diagnosed with COVID‐19 by RT‐PCR and treated in the acute care hospital and transferred to the CRU Exclusion: 1 death; 1 received convalescent plasma; 25 refused blood draw, hospice care, discharged to community |
||
| Patient characteristics and setting | Setting: Skilled Nursing Facility or designated COVID‐19 recovery unit Location: Veterans Affairs Greater Los Angeles Healthcare System West Los Angeles (WLA) campus or from a satellite campus (Sepulveda Ambulatory Care Center, SACC) or designated COVID‐19 recovery unit, Los Angeles, California Country: USA Dates: Repeated PCR testing between 28 March 2020 and 18 May 2020 Serological testing: 5 to 12 June 2020 Symptoms and severity: 20 symptomatic; 6 asymptomatic Demographics: Age: median 75 (IQR 69‐78) years Sex: 26 (100%) males Black or African‐American 9 (35%) White 11 (42%) Native Hawaiian or Pacific Islander 1 (4%) Multiple races 1 (4%) Not reported 4 (15%) Hispanic 3 (12%) Exposure history: 17 nursing home 9 Not stated (community) Non‐Covid group 1: [2] Current PCR‐negative Source: Skilled nursing facility at the Veterans Affairs Greater Los Angeles Healthcare System West Los Angeles (WLA) campus or from a satellite campus (Sepulveda Ambulatory Care Center, SACC) Repeated PCR testing between 28 March 2020 and 18 May 2020 Serological testing: 5 to 12 June 2020 Characteristics: Age: median 74 (IQR 69‐83) years Sex: 122 (98%) males Asian 3 (2%) Black or African‐American 51 (41%) White 61 (49%) Native Hawaiian or Pacific Islander 1 (1%) Multiple races 1 (1%) Not reported 7 (6%) Hispanic 14 (11%) |
||
| Index tests | Test name: LIAISON SARS‐CoV‐2 S1/S2 IgG Manufacturer:DiaSorin Antibody: IgG Antigen target: S1/S2 spike‐protein Evaluation setting: Not stated Test method: Not stated Timing of samples: 46–76 days after their initial diagnosis Samples used: Not stated Test operator: Not stated Definition of test positivity: Not stated Blinding reported: Not stated Threshold predefined: Not stated |
||
| Target condition and reference standard(s) | Reference standard: nasopharyngeal RT‐PCR (Roche COBAS 6800) for SARS‐CoV‐2, repeated approximately weekly on each ward and discontinued when all ward residents tested negative; threshold not stated Samples used: Nasopharyngeal Timing of reference standard: Not stated (symptom‐based testing from 28‐30 March 2020; serial testing the weeks of 6 April, 13 April and 20 April 2020; surveillance testing on 11 May and 18 May) Blinded to index test: Yes, prior index test Incorporated index test: No Definition of non‐COVID cases: nasopharyngeal RT‐PCR (Roche COBAS 6800) for SARS‐CoV‐2, repeated approximately weekly on each ward and discontinued when all ward residents tested negative; threshold not stated Samples used: Nasopharyngeal Timing of reference standard: nasopharyngeal RT‐PCR (Roche COBAS 6800) for SARS‐CoV‐2, repeated approximately weekly on each ward and discontinued when all ward residents tested negative; threshold not stated Blinded to index test: Yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1] 46–76 days after their initial diagnosis [2] Serial PCR testing from 28 March to 18 May 2020; antibody testing 5‐12 June 2020 All patients received same reference standard: yes Missing data: no Uninterpretable results: no Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper (Brief report) Source: Clinical Infectious Diseases Author COI: No reported conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Dortet 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: to assess the rapid test's diagnostic accuracy and clinical utility for patient management
2‐group study to estimate sensitivity and specificity for diagnosis of active disease/identification of previous disease Design: [1] RT‐PCR‐confirmed COVID‐19 patients (n = 101, 256 sera samples) [2] Non‐COVID‐19 controls (n = 50: 22 healthy volunteers, 24 pre‐pandemic; 4 RT‐PCR‐negative with common coronaviruses) Recruitment: Unclear Prospective or retrospective: [1] Prospective at time of COVID‐specific consultation or ER attendance. RT‐PCR samples taken at same attendance as serum [2] Retrospective in 24 pre‐pandemic samples, prospective in 22 healthy volunteers, and unclear for 4 PCR‐negative samples Sample size: 151 (101) patients with 306 (256) samples Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Inpatients and ER consultations Location: Hôpital Bicêtre, AP‐HP; Le Kremlin‐Bicêtre, France; Hôpital Paul‐Brousse, AP‐HP; Villejuif, France Country: France Dates: March 11–23 2020 Symptoms and severity: 17.8% (18/101) were discharged 72.3% (72/101) were hospitalised in a dedicated COVID ward 10.9% (11/101) were critically ill and required immediate hospitalisation in the ICU Demographics: male/female ratio was 1.46; median age was 58 years (IQR, 35‐61) Exposure history: Not stated Non‐Covid group 1: Non‐COVID‐19 controls Source: 22 healthy volunteers and 4 RT‐PCR‐negative with common coronaviruses = contemporaneous; 24 pre‐pandemic = September‐October 2017 Characteristics: Not stated for 24 pre‐pandemic samples 4 from patients with respiratory symptoms that were RT–PCR‐negative for SARS‐CoV‐2 but positive for common coronaviruses (Coronavirus HKU1 (n = 2), NL63 (n = 1), 229E (n = 1)), recent common coronavirus infections in the past 3‐months; 22 healthy volunteers without any respiratory symptoms |
||
| Index tests | Test name: NG‐Test IgM‐IgG COVID All‐in‐one Manufacturer: NG Biotech, Guipry, France Antibody: IgM, IgG Antigen target: Nucleocapsid protein Evaluation setting: POC, applied POC for healthy volunteers but unclear for the other participants (retrospective analysis from stored sera) Test method: lateral flow immunoassay Timing of samples: For 97 patients, days 1‐11 after hospitalisation Most sera were sampled between day 0–15 after the onset of symptoms (85.5%, 219/256) Samples used: Serum for [1] and [2], pre‐pandemic and PCR‐negative samples or a drop of blood (after finger puncture) for [2, healthy volunteers] Test operator: Unclear Definition of test positivity: Results were read after 15 minutes according to the manufacturer’s recommendations, visual‐based Blinding reported: Not stated Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: Real‐time RT‐PCR targeting RNA‐dependent RNA polymerase and E genes Samples used: Nasopharyngeal samples Timing of reference standard: The average time between the onset of symptoms and receiving an RT‐PCR result was 5.4 (± 0.4) days Blinded to index test: Yes, done prior index test Incorporated index test: No Definition of non‐COVID cases: Mixed Pre‐pandemic (September/October 2017) (n = 24) RT‐PCR‐negative for SARS‐COV‐2 (n = 4) No respiratory symptoms for healthy volunteers (n = 22) Samples used: None Timing of reference standard: NA for pre‐pandemic samples and healthy volunteers Blinded to index test: Unclear for healthy volunteers (tested directly using a drop of whole blood) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: done during same consultation for 97 COVID‐19 samples, unclear for the remaining samples All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This research was supported by Assistance Publique–Hôpitaux de Paris (APHP), Médecins Sans Frontières (MSF), and by a Grant from the French Defence Innovation Agency (AID). We acknowledge NG Biotech for providing free testing devices. Publication status: Submitted to Lancet Infectious Diseases; now published article Source: Editorial Manager® and ProduXion Manager® from Aries Systems Corporation Journal: Emerging Microbes and Infections Author COI: The authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Dortet 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of COVID‐19 Design: Two‐group design with separate estimates of sensitivity and specificity: [1] COVID‐19‐positive patients (sample size = 250 from 159 patients) [2] Pre‐pandemic patients with other infections (sample size = 254) Recruitment: [1] Serum samples from COVID‐19 patients from 2 university hospitals located in the south of Paris were randomly selected from the BIOCOVID‐19 biobank. [2] Serum samples collected prior to December 2019 were selected, which had previously tested positive for a separate agent or pathology that could potentially interfere with SARS‐CoV‐2 testing results. Prospective or retrospective: Retrospective Sample size: Samples: 504 (250) Patients: 413 (159) Further detail: No further details ([1] Patients with documented RT‐PCR‐positive results for SARS‐CoV‐2 using nasopharyngeal swabs from 2 hospitals in the south of Paris [2] Serum samples collected prior to December 2019 were selected, which had previously tested positive for a separate agent or pathology that could potentially interfere with SARS‐CoV‐2 testing results) |
||
| Patient characteristics and setting | Setting: Patients from two university hospitals
4.4% (7/159) were discharged after their initial visit to the emergency room (ER), and 95.6% (152/159) were hospitalised.
Over the study period, 44.1% (67/152) of patients required intensive care unit (ICU) care while hospitalised. Location: Bicêtre and Paul Brousse Hospitals, Paris (BIOCOVID‐19 biobank) Country: France Dates: 11 March to 3 April 2020 Symptoms and severity: No standard classification of severity was provided. 4.4% (7/159) were discharged after their initial visit to the emergency room (ER), and 95.6% (152/159) were hospitalised. Over the study period, 44.1% (67/152) of patients required intensive care unit (ICU) care while hospitalised. The overall death rate among hospitalised patients was 19.1% (29/152): 10.5% (9/85) among non‐ICU patients and 29.9% (20/67) among ICU patients. Demographics: COVID patients Median age ‐ 62.9 years (range 12.8 to 97.6 years) Male:female ratio = 100:59 Exposure history: Not reported Non‐Covid group 1: Pre‐pandemic patients with other infections Source: Serum samples collected before December 2019 and tested positive for another pathogen Characteristics: another coronavirus (n = 11), other viral and parasitic infections (including Epstein‐Barr virus [EBV], cytomegalovirus [CMV], rubeola virus, and toxoplasma) (n = 129), a rheumatoid factor (n = 3), IgG (n = 6) and IgM (n = 3) hyperglobulinaemia, malaria (n = 5), or a positive Treponema pallidum hemagglutination assay (TPHA) (n = 97) |
||
| Index tests | Test name: [A] NG‐Test IgG‐IgM COVID‐19; [B] anti‐SARS‐CoV‐2 rapid test; [C](2019‐nCoV) antibody IgG/IgM test; [D] Nadal COVID‐19 IgG/IgM test; [E] Biosynex COVID‐19 BSS; [F] 2019‐nCoV Ab test; [G] 2019‐nCoV IgG/IgM test; [H] COVID‐19‐Check‐1; [I] Finecare SARS‐CoV‐2 antibody test; [J] Wondfo SARS‐CoV‐2 antibody test. Manufacturer: [A] NG Biotech SA, Guipry, France; [B] Autobio Diagnostic Co; Ltd, Zhengshou, China; [C] Avioq Bio‐Tech Co; Ltd, Shandong, China; [D] Nal Von Minden GmbH; Ltd, Moers, Germany; [E] Biosynex SWISS SA, Fribourg, Switzerland; [F] Innovita Biological Technology Co.; Ltd, Hebei, China; [G] Biolidics Co, Ltd, Mapex, Singapore; [H] Vedal Lab SA, Alençon, France; [I] Wondfo Biotech Co, Ltd, Guangzhou, China; [J] Wondfo Biotech Co, Ltd, Guangzhou. Antibody: [A] IgG and IgM; [B] IgG and IgM; [C] IgG and IgM; [D] IgG and IgM; [E] IgG and IgM; [F] IgG and IgM; [G] IgG and IgM; [H] IgG and IgM; [I] Total antibody; [J] Total antibody. Antigen target: [A] N and S; [B] Not reported; [C] Not reported; [D] Not reported; [E] Not reported; [F] N and S; [G] Not reported; [H] Not reported; [I] Not reported; [J] Not reported. Evaluation setting: POC performed in lab Test method: [A] lateral flow assay, colloidal gold; [B] lateral flow assay, colloidal gold; Timing of samples: 0‐9 days pso; 101/250 10‐14 days pso; 86/250 15‐32 days pso. 63/250 Most serum samples were obtained on days 0 to 15 (85.5%; 219/256) after symptoms appeared, although serum samples from later dates (up to day 31) were also available. Samples used: Serum; test performed at room temperature. Two boxes (the total number of boxes not provided) containing samples were stored at 4 degrees Celsius. Test operator: Trained laboratory technicians; two independent readers read the test. Definition of test positivity: By the intensity of lines: 0 ‐ non reactive; 1 ‐ very weak, but definitely reactive; 2 ‐ medium to high reactivity; U ‐ undetermined (values were not recorded when a control line did not appear, and tests were subsequently repeated). Blinding reported: Yes (Selected serum samples were randomly placed in working boxes so as not to bias the technicians’ interpretation of results. Two sets of these boxes were prepared and stored at 4°C prior to being used). Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR targeting the RNA‐dependent RNA polymerase and E genes (eSwabs‐Virocult; Copan, Italy); using Charite Berlin protocol (Corman 2020) Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: unclear All patients received same reference standard: No Missing data: Yes (number of samples tested is < 250 for COVID cases and < 254 non‐COVID cases for most tests; test [G] was evaluated on only half of the total serum samples collection, as only 250 tests were received: results for 167/250 COVID cases and 79/254 pre‐pandemic control samples) Uninterpretable results: Yes (1 for test [C], 3 for test [H]) Indeterminate results: If three readings were different, the result was reported as unknown (see also "Missing data") Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This research was supported by Assistance Publique‐Hôpitaux de Paris (APHP), Médecins Sans Frontières (MSF), and a grant from the French Defense Innovation Agency (AID). Publication status: Published article Source: Journal of Clinical Microbiology Author COI: Authors declared no conflicts of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Dortet 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Du 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 (n = 107) [2] Pre‐pandemic non‐COVID (n = 226) [2a] Healthy donor samples (n = 138) [2b] Cross‐reaction challenge samples (n = 88) Recruitment: [1] COVID‐19 patient serum samples were acquired from ProMedDx (Norton, MA) and University of California and VA Healthcare System. [2a] Healthy donor EDTA K2 plasma samples were purchased from Golden West Biosolutions (Temecula, CA) in 2019 prior to the outbreak of COVID‐19. COVID‐19 negative EDTA K2 plasma samples were also obtained from University of Florida Department of Radiation Oncology in 2017. Healthy donor serum samples were purchased from Innovative Research, LLC (Plymouth, MN). [2b] Patient serum samples positive for IgG to HBV/HCV/HIV/RSV were purchased from Antibody Systems, Inc (Hurst, TX). Patient serum samples positive for IgG to HAV/CMV/EBV/Rubella/Influenza B were purchased from ProMedDx (Norton, MA). Patient serum samples positive for IgG to Influenza A were purchased from Dx Biosamples, LLC (San Diego, CA). Prospective or retrospective: Retrospective Sample size: 333 (107) of which 252 (26) with eligible time splits Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Not stated Location: ProMedDx (Norton, MA) and University of California and VA Healthcare System Country: USA Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic healthy or cross‐reactivity Source: [2a] Healthy donor EDTA K2 plasma samples were purchased from Golden West Biosolutions (Temecula, CA) in 2019 prior to the outbreak of COVID‐19. COVID‐19 negative EDTA K2 plasma samples were also obtained from University of Florida Department of Radiation Oncology in 2017. Healthy donor serum samples were purchased from Innovative Research, LLC (Plymouth, MN). [2b] Patient serum samples positive for IgG to HBV/HCV/HIV/RSV were purchased from Antibody Systems, Inc (Hurst, TX). Patient serum samples positive for IgG to HAV/CMV/EBV/Rubella/Influenza B were purchased from ProMedDx (Norton, MA). Patient serum samples positive for IgG to Influenza A were purchased from Dx Biosamples, LLC (San Diego, CA). Characteristics: [2a] Healthy donors (n = 138) [2b] Cross‐reactivity (n = 88) HIV n = 4 Hepatitis A virus n = 7 Hepatitis B virus n = 4 Hepatitis C virus n = 4 Respiratory syncytial virus n = 5 Influenza A n = 5 Influenza B n = 13 Cytomegalovirus n = 16 Epstein‐Barr virus n = 13 Rubella n = 17 Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: QuantiVirus™ anti‐SARS‐CoV‐2 IgG test Manufacturer: DiaCarta Inc, 2600 Hilltop Dr. Richmond, CA 94806, United States Antibody: IgG Antigen target: spike‐protein 1 (S1) RBD Evaluation setting: Laboratory test performed in lab Test method: Fluorescence immunoassay Phycoerythrin fluorescence of each well in a 96‐well microplate was measured on Luminex 200 or MAGPIX® instrument for Median Fluorescence Intensity (MFI) Timing of samples: 0‐7 days pso: 13/107 8‐14 days pso: 13/107 > 14 days pso: 81/107 Samples used: [1] Serum [2a] Serum and plasma [2b] Serum Test operator: Lab personnel Definition of test positivity: Median Fluorescence Intensity (MFI). Interpretation of the testing results was performed by calculating the MFI ratio of each sample to the average MFI of two blank wells. Blinding reported: Not stated Threshold predefined: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: [2] Pre‐pandemic (time not stated for all sources) Samples used: [2] Pre‐pandemic Timing of reference standard: [2] Pre‐pandemic Blinded to index test: yes, prior index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Journal of Virological Methods Author COI: The authors reported no declarations of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Egger 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of SARS‐CoV‐2
3‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: [1] confirmed COVID‐19 patients (64 patients, 104 samples) [2] Healthy blood donors (n = 200) and [3] ICU patients (n = 256) Recruitment: [1] Between 15th of March 2020 and 10th of April 2020, of all SARS‐CoV‐2 RT‐PCR (from respiratory specimens) confirmed COVID‐19 patients, that were treated in one of the two tertiary care hospitals, Konventhospital Barmherzige Brueder Linz and Ordensklinikum Linz Barmherzige Schwestern in Linz Austria, blood samples for clinical routine that were sent to central laboratory were included in the present study. [2] Cohorts of 200 consecutive plasma samples from healthy blood donors and [3] 256 consecutive plasma samples of ICU patients from Linz Intensive Care Unit (LICU) study were recruited prior to COVID‐19 outbreak (Dec 3, 2019). Prospective or retrospective: [1] prospective for COVID patients, [2] and [3] retrospective for healthy blood donors/ICU patients Sample size: 520 (64) patients; 560 (104) samples Further detail: Unclear |
||
| Patient characteristics and setting | Setting: Inpatients (Konventhospital Barmherzige Brueder Linz and Ordensklinikum Linz Barmherzige Schwestern) Location: Konventhospital Barmherzige Brueder Linz and Ordensklinikum Linz Barmherzige Schwestern, Linz Country: Austria Dates: COVID patients: 15th of March‐10th of April 2020 Symptoms and severity: unclear Demographics: unclear Exposure history: unclear Non‐Covid group 1: [2] Pre‐pandemic healthy blood donors Source: recruited at Red Cross organisation in Linz Austria ‐ 31 Jan‐13 Feb 2008 Characteristics: Cohort of healthy blood donors, 200 consecutive plasma samples that were stored ‐80degrees C and had 1 freeze/thaw cycle Non‐Covid group 2: [3] Pre‐pandemic ICU patients Source: Medical intensive care unit of the Konventhospital Barmherzige Brueder Linz, Austria, recruited from August 9th 2009 to February 8th 2010 Characteristics: Cohort of the Linz Intensive Care Unit (LICU) study; baseline samples of patients admitted to medical ICU of Konventhospital Barmherzige Brueder Linz, Austria, between 9 Aug 2009 and 8 Feb 2010, plasma aliquots store ‐80degrees C and one freeze/thaw cycle |
||
| Index tests | Test name: [A] Elecsys Anti‐SARS‐CoV‐2 assay [B] EDI Novel Coronavirus COVID‐19 IgM (reagent lot number P630C) and IgG (reagent lot number P621C) enzyme linked immunosorbent assay Manufacturer: [A] Roche Diagnostics [B] Epitope Diagnostics Inc., San Diego, CA, USA Antibody: [A] IgA, IgM, IgG (total SARS‐CoV‐2 antibody assay [IgA, IgM, and IgG] detecting predominantly, but not exclusively, IgG) [B] IgM or IgG Antigen target: [A] and [B] recombinant nucleocapsid protein (N) Evaluation setting: laboratory Test method: [A] electrochemiluminescence immunoassay (fully automated) [B] ELISA Timing of samples: < 5 days to > 15‐22 days since symptom onset < 5 days pso: 34/104 5‐10 days pso: 35/104 11‐15 days pso: 17/104 16‐22 days pso: 18/104 Samples used: [A] and [B] plasma Test operator: [A] and [B] unclear (seemed to be lab personnel) Definition of test positivity: [A] COI > or = 1.0 positive; COI < 1.0 negative [results were reported as numeric values in form of a cut‐off index (COI; signal sample/cut‐off) as well as in form of a qualitative results non‐reactive (COI < 1.0; negative) and reactive (COI ≥ 1.0; positive]. [B] Single run: If the patient sample optical density (OD) was below the positive cut‐off, the result was reported negative; If the patients sample OD was equal or above the positive cut‐off the patient was reported as positive. Blinding reported: [A] and [B] Not stated Threshold predefined: [A] yes (performed following the manufacturer’s instructions) [B] unclear (did not use the thresholds as reported in the IFU) |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 RT‐PCR Samples used: respiratory specimens Timing of reference standard: not reported Blinded to index test: Yes, prior index test Incorporated index test: no Definition of non‐COVID cases: [2] and [3] pre‐pandemic samples Samples used: [2] and [3] NA as pre‐pandemic samples Timing of reference standard: [2] January 31st to February 13th 2008 [3] August 9th 2009 to February 8th 2010 Blinded to index test: yes, done prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: unclear All patients received same reference standard: no Missing data: Not reported Uninterpretable results: Not reported Indeterminate results: Not reported Unit of analysis: samples for COVID patients, patients for healthy blood donors/ICU patients |
||
| Comparative | |||
| Notes | Funding: Roche Diagnostics provided reagents for Elecsys® Anti‐SARS‐CoV‐2 measurements free of charge.
Benjamin Dieplinger and Thomas Mueller have received speaking fees from Roche Diagnostics. Publication status: published Source: Clinica Chimica Acta Author COI: The authors declared that they had no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Egger 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Fafi‐Kremer 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of antibody kinetics in individuals who had recovered from mild COVID‐19 Design: Single‐group study estimating sensitivity only: [1] hospital staff with mild PCR‐confirmed COVID‐19 (n = 160); included doctors, nurses, physiotherapists, dentists, medical students, orderlies, hospital assistants, and hospital administrative staff Recruitment: Likely consecutive; described including 'all' eligible staff within specific dates Prospective or retrospective: Not explicitly stated, but likely prospective Sample size: 160 (160) Further detail: Inclusion was conditional of informed consent. Excluded 2 patients who were hospitalised |
||
| Patient characteristics and setting | Setting: Tertiary hospital staff; cluster infected following outbreak
Suggested code as 'outpatient' or 'community' as they were not inpatient; or even as contacts or outbreak investigation (need new covariate) Location: Strasbourg University Hospitals Country: France Dates: 6‐8 April 2020 Symptoms and severity: Severity: all described as mild disease; symptoms classified as minor 5 (3%) or major 155/160 (97%) (cough, fever, dyspnoea, anosmia and ageusia) Demographics: Age, median (IQR): 32 (26‐44) Sex: 50/160 (31.2%) male Exposure history: Contact with COVID‐19 patients: yes 74/160 (46.3%), no 80/160 (50%), missing 6/160 (3.7%). Level of exposure to COVID‐19 patients: none 10/75 (13.5%), some 27/75 (36.5%), high 37/75 (50%). Non‐Covid group 1: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: COVID‐19 BSS IgG/IgM
[Data for a second in‐house test (flow cytometry based) were reported but not included in the review] Manufacturer: Biosynex Antibody: IgG and IgM Antigen target: S‐protein Evaluation setting: POC test, evaluated in lab setting Test method: Lateral flow assay Timing of samples: Time between symptom onset and sample collection: median 24 days (IQR 21 to 28) 13‐20 days: 29/160 (18%) 21‐27 days: 83/160 (52%) 28‐41 days: 48/160 (30%) Samples used: Serum Test operator: Not stated; presume lab scientist Definition of test positivity: Not stated Blinding reported: Not stated Threshold predefined: Yes (visual result) |
||
| Target condition and reference standard(s) | Reference standard: PCR test (not further specified) Samples used: Not stated Timing of reference standard: Time from symptom onset to PCR‐positive in days, median (IQR): 2 (1‐4) Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Approx 3 weeks (based on median times reported) All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No specific funds for this work, but the labs where the study was done receive funding from multiple sources (Institut Pasteur, ANRS, Sidaction, Vaccine Research Institute, Labex IBEID, TIMTAMDEN, CHIKViro‐Immuno, Gilead HIV cure programme, French Ministry of Higher Education‐Research‐Innovation, Strasbourg University Hospitals). Publication status: Published article Source: Academic journal Author COI: One author is founder and CSO of TheraVectys; four other authors hold a provisional patent on the S‐flow assay; one author reported grants and/or personal fees from Mylan, ViiV Healthcare, Gilead, Abbvie. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Favresse 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: external validation of a new electrochemiluminescent immunoassay (ECLIA) test that allows the detection of total antibodies
2‐group study to estimate sensitivity and specificity for diagnosis of active disease/identification of previous disease Design: Two groups of samples: [1] patients with a confirmed RT‐PCR SARS‐CoV‐2 diagnosis (n = 97 patients, 140 samples) [2] Non‐SARS‐CoV‐2 sera collected prior to the COVID‐19 pandemic with potential cross‐reactions (n = 79) Recruitment: Retrospective, no further information Prospective or retrospective: Retrospective Sample size: 219 (140) samples, 176 (97) patients Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Unclear ‐ probably hospital inpatients because of multiple samples for patients Location: Clinique Saint‐Luc Bouge (SLBO, Namur, Belgium) Country: Belgium Dates: Unclear. "This retrospective study was conducted from May 6 to 12, 2020", but not clear whether these were recruitment dates Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic controls Source: Between January 2019 and December 2019. Source not stated Characteristics: Potential cross‐reactions (cross‐reactivity test group) were also analysed. Samples in this group included: positive antinuclear antibodies (n = 5), antithyroglobulin antibody (n = 1), anti‐Treponema pallidum antibodies (n = 2), antistreptolysin O (n = 1), antithyroid peroxidase antibodies (n = 4), chikungunya antibody (n = 1), direct Coombs (n = 1), hepatitis B antigen (n = 4), hepatitis C antibodies (n = 7), hepatitis E antibodies (n = 4), HIV antibodies (n = 2), IgA chlamydia pneumoniae (n = 1), IgG chlamydia trachomatis (n = 1), IgG Coxiella burneti (n = 2), IgM Borrelia (n = 1), IgM Coxiella burnetii (n = 1), IgM cytomegalovirus (n = 5), IgM Epstein‐Barr virus viral capsid (n = 5), IgM mycoplasma pneumoniae (n = 6), IgM parvovirus B19 (n = 7), IgM toxoplasma gondii (n = 5), influenza antibodies (n = 6), irregular agglutinins (n = 2), and rheumatoid factor (n = 5). |
||
| Index tests | Test name: Elecsys anti‐SARS‐CoV‐2 Manufacturer: Roche Diagnostics Antibody: total antibodies (including IgG) Antigen target: SARS‐ CoV‐2 nucleocapsid Evaluation setting: Laboratory test conducted in the laboratory Test method: electrochemiluminescent immunoassay (ECLIA) Timing of samples: 0‐ ≥ 28 days after positive RT‐PCR test, 0‐6 days post‐PCR+: 45/140 7‐13 days post‐PCR+: 35/140 14‐20 days post‐PCR+: 24/140 21‐27 days post‐PCR+: 15/140 28+ days post‐PCR+: 21/140 0‐ > 28 days after onset of symptoms 0‐6 days pso: 22/129 7‐13 days pso: 28/129 14‐20 days pso: 26/129 21‐27 days pso: 23/129 28+ days pso: 30/129 11 missing data on time pso Samples used: Serum samples Test operator: Laboratory personnel Definition of test positivity: Two thresholds reported: [A] According to the manufacturer, a result < 1.0 is considered negative while a result ≥ 1.0 is considered positive [B] optimal cut‐off provided by ROC curve analyses (i.e. > 0.165) Blinding reported: Not stated Threshold predefined: [A] The test result is given as a cut‐off index (COI). According to the manufacturer, a result < 1.0 is considered negative while a result ≥ 1.0 is considered positive. [B] No for optimised cut‐off |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR performed on the LightCycler® 480 Instrument II using the LightMix® Modular SARS‐CoV‐2 E‐gene set (Roche Diagnostics®) Samples used: respiratory samples (nasopharyngeal swab samples) Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Not stated Timing of reference standard: Pre‐pandemic Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: 0‐ ≥ 28 days All patients received same reference standard: No, controls were pre‐pandemic Missing data: Among the 97 patients (140 samples), data about time of symptom onset were available for 92 patients (129 samples). Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Roche Diagnostics provided the kits for the validation. Publication status: Published letter Source: Clinical Chemistry, Volume 66, Issue 8, August 2020, Pages 1104–6 Author COI: J. Douxfils, personal fees from Diagnostica Stago, Roche, Roche Diagnostics, Daiichi‐Sankyo, and Portola, outside the submitted work. J. Douxfils, chief executive officer and founder of QUALIblood sa |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Favresse 2020b.
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of the longitudinal kinetics of anti‐SARS‐CoV‐2 antibodies since symptom onset (acute and convalescent‐phase infection) Design: Single‐group study estimating sensitivity: [1] PCR‐confirmed COVID‐19 patients (n = 94, providing 150 serum samples) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 94 (94; 150 samples) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear
Study performed in a hospital, but unclear if all patients were admitted to an inpatient ward Location: Clinique St‐Luc Bouge, Namur Country: Belgium Dates: 21 March to 25 May 2020 Symptoms and severity: All patients had at least one symptom. Reported symptoms: fever (68.1%), cough (60.4%), fatigue (58.2%), difficulty breathing (45.1%), muscle aches (31.9%), chest pain (6.6%), sore throat (6.6%), and anosmia (6.6%) No details on severity available Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Elecsys anti‐SARS‐CoV‐2 [product code not reported] Manufacturer: Roche Diagnostics Antibody: Total antibodies Antigen target: N‐protein Evaluation setting: Lab test, done in lab Test method: Electrochemiluminescence immunoassay (ECLIA) Timing of samples: Range day 0 to 63: 0‐2 days: 15 (10%); 3‐5 days: 6 (4%); 6‐8 days: 14 (9.3%); 9‐11 days: 10 (6.7%); 12‐14 days: 13 (8.7%); 15‐17 days: 14 (9.3%); 18‐20 days: 7 (4.7%); 21‐23 days: 19 (12.7%); 24‐30 days: 16 (10.7%); 31‐40 days: 15 (10%); 41‐63 days: 16 (10.7%). Samples used: Serum or plasma (n for each not reported); collected into serum‐gel tubes (BD Vacutainer® 8.5 mL tubes, Becton Dickinson, New Jersey, USA) or lithium‐heparin plasma tubes (BD Vacutainer® 4.0 mL tubes) according to standardised operating procedure Test operator: Not stated (presume lab staff); sera and plasma samples stored at −20 °C and thawed 1 h at room temperature on the day of the analysis Definition of test positivity: Positive if COI ≥ 1.0 Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; LightCycler® 480 Instrument II (Roche Diagnostics®) using the LightMix® Modular SARS‐CoV E‐gene set Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Unclear ‐ 5 serum samples were excluded because only one sample per patient per time category was used Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Roche provided the kits for the validation. Publication status: Published letter Source: Clinical Chemistry & Laboratory Medicine Author COI: One of the authors is chief executive officer and founder of QUALIblood sa and reported personal fees from Diagnostica Stago, Roche, Roche Diagnostics, Daiichi‐Sankyo, and Portola, outside the submitted work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Fenwick 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute/sub‐acute‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (93 sera) [2] Non‐COVID samples (n = 65); pre‐pandemic including 18 samples from patients documented positive for a human coronavirus (E229, OC43, HKU1, or NL63) RT‐PCR Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 158 (93) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: [1] Hospital inpatients Location: Not stated (Lausanne University Hospital, Lausanne?) Country: Switzerland Dates: Not stated Symptoms and severity: Hospitalised patients with severe‐to‐moderate symptoms Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic Source: Sampled before November 2019, source not stated Characteristics: 18/65 samples from patients documented positive for a human coronavirus (E229, OC43, HKU1, or NL63) RT‐PCR. Part of the diverse set of 108 patient sera used for study 2 (which included an additional 43 pre‐pandemic patient samples). This diverse set of 108 samples consisted of sera from pregnant women (n = 14), pre‐pandemic coronavirus‐infected donors (OC43, E229, NL63, and HKU1; n = 19), patients with infectious diseases (HIV, rubella, HSV1, HSV2, RSV, CMV, EBV, influenza, and varicella; (n = 57)), and patients with auto‐immune diseases, including lupus (n = 18). |
||
| Index tests | Test name: [A] Not stated [B] Not stated [C] LIAISON SARS‐CoV‐2 IgG kit [D] MAGLUMI 2019‐nCoV IgG and IgM kits [E] Elecsys anti‐SARS‐CoV‐2 assay Manufacturer: [A] Euroimmun [B] Epitope Diagnostis [C] Diasorin [D] Snibe [E] Roche Antibody: [A] IgG [B] IgG [C] IgG [D] IgG [E] pan‐Ig Antigen target: [A] S1‐protein [B] N‐protein [C] S1‐protein [D] N‐protein and S antigen peptide (the Snibe assay was grouped with the N‐protein assays in our analysis since it contained only a portion of the S1‐protein) [E] N‐protein Evaluation setting: [A] Lab test performed in lab [B] Lab test performed in lab [C] Lab test performed in lab [D] Lab test performed in lab [E] Lab test performed in lab Test method: [A] ELISA [B] ELISA [C] CLIA [D] CLIA [E] ECLIA Timing of samples: 0 to 33 days post‐onset of the symptoms: 0‐5 days pso: 8/93 6‐10 days pso: 19/93 11‐15 days pso: 37/93 16‐33 days pso: 29/93 Samples used: Serum Test operator: Service of Immunology and Allergy and Service of Microbiology at the Lausanne University Hospital Definition of test positivity: Not stated Blinding reported: Unclear Threshold predefined: ELISA and CLIA were performed according to the manufacturers’ instructions. Optical densities (OD) were measured with a microplate reader (800 TSI, BioTek, USA). Each sample was measured in duplicate. |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Before November 2019 Blinded to index test: yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Small differences in the number of sera tested across assays were due to the insufficient volume of some samples. [A] 89/93 COVID samples [B] 90/93 COVID samples Uninterpretable results: Not stated Indeterminate results: Not stated (according to Figure 4, there must have been intermediate results for tests [A], [B] and [C]) Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | Funding: Funding for this project was provided through the Lausanne University Hospital, through the Swiss Vaccine Research Institute and through the Coronavirus Accelerated R&D in Europe (CARE) IMI project. Publication status: Published paper Source: Journal of Virology Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Fenwick 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Fenwick 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Fenwick 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Fenwick 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Flinck 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients [1a] Inpatients (120 samples from 13 patients) for seroconversion [1b] Convalescent outpatients (n = 35) [2] Non‐COVID control samples [2a] Pre‐pandemic healthy (n = 161) [2b] Cross‐reaction samples (pre‐pandemic and current) (n = 43) Recruitment: [1a] Residual plasma samples from patients admitted to Tampere University Hospital or other communal hospitals in Fimlab Laboratories operation region [1b] serum samples from the COVID‐19 NAAT positive outpatients were traced and collected for evaluation. All patients had had respiratory tract symptoms [2a] Stored samples from the Chitosan study before the COVID‐19 era [2b] Follow‐up plasma/serum samples from patients with other diseases. EBV‐, HBcAb‐, and ANA‐positive samples collected in year 2019. RF‐positive samples collected in year 2017. The samples from other coronavirus and influenza A/B patients had been collected in April–May 2020. Prospective or retrospective: Retrospective Sample size: 359 (155) samples of which 244 (40) had extractable results for our review Further detail: Not stated |
||
| Patient characteristics and setting | Setting: [1a] Hospital inpatients [1b] Hospital outpatients Location: [1a] and [1b] Tampere University Hospital or other communal hospitals in Fimlab Laboratories operation region, Tampere Country: Finland Dates: Not stated Symptoms and severity: [1a] aggravated COVID‐19 respiratory tract symptoms, i.e. difficulty breathing [1b] All these patients had had respiratory tract symptoms including rhinitis, cough, sore throat, chest pain, and/or difficulty breathing, with or without fever Demographics: [1a] Age 55 years (median), range 20–79; 8/13 males [1b] Age 47 years (median), range 11–95; 12/35 males Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic healthy Source: [2a] Part of the Chitosan study before the COVID‐19 era (cited study published in 2005) Characteristics: [2a] Apparently healthy adults [age 45 years (mean), range 32–65; 72 males] with mildly to moderately increased total cholesterol Non‐Covid group 2: [2b] Cross‐reaction panel Source: EBV‐, HBcAb‐, and ANA‐positive samples had been collected in year 2019, and RF‐positive samples in year 2017 before the COVID‐19 pandemic The samples from other coronavirus and influenza A/B patients had been collected in April–May 2020 Characteristics: Human coronavirus OC43: n = 13 Human coronavirus NL63: n = 2 Human coronavirus: 229E: n = 1 Human coronavirus OC43 and human bocavirus: n = 1 Influenza A virus: n = 5 Influenza A and B virus: n = 1 Acute Epstein‐Barr virus: n = 5 Hepatitis B core antibody positive: n = 5 Antinuclear antibody positive: n = 5 Rheumatoid factor positive: n = 5 |
||
| Index tests | Test name: [A] Elecsys® Anti–SARS‐CoV‐2 test [B] LIAISON® SARS‐CoV‐2 S1/S2 IgG Manufacturer: [A] Roche Diagnostics GmbH, Mannheim, Germany [B] DiaSorin S.p.A., Saluggia, Italy Antibody: [A] Total antibodies [B] IgG Antigen target: [A] N‐protein [B] spike‐protein S1 and S2 antigens Evaluation setting: [A] and [B] Lab test performed in lab Test method: [A] Not stated (should be ECLIA) [B] Not stated (should be CLIA) Timing of samples: [1a] Not stated [3‐40 days pso (figure 1) for 83/120 samples] [1b] At least 16 days after positive NAAT Samples used: [1a] Residual EDTA plasma, stored −20 °C [1b] Residual plasma/serum samples [2a] Serum samples stored at −20 °C [2b] Plasma/serum samples Test operator: Lab personnel (Fimlab Laboratories, Tampere, Finland) Definition of test positivity: [A] COI = 1 (Fig 1) [B] Not stated (AU/mL) Blinding reported: Not stated Threshold predefined: Not stated |
||
| Target condition and reference standard(s) | Reference standard: [1] In‐house real‐time reverse‐transcription (RT)‐PCR test detecting E‐gene target sequence (using Charite Berlin protocol; Corman 2020); Allplex™ 2019‐nCoV Assay (Seegene Inc., Seoul, South Korea) detecting target sequences E, N, and RdRp; or Abbott RealTime SARS‐CoV‐2 Assay (Abbott Laboratories, Abbott Park, IL) detecting target sequences N and RdRp. The used RT‐PCR method had been chosen based on the availability. The primary COVID‐19 diagnosis was based on 1 RT‐PCR result. Samples used: Not stated Timing of reference standard: [1a] Not stated [1b] At least 16 days before index test Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: [2a] Pre‐pandemic [2b] Pre‐pandemic or not stated Samples used: [2a] Pre‐pandemic [2b] Pre‐pandemic or not stated Timing of reference standard: [2a] Pre‐pandemic [2b] Pre‐pandemic or not stated Blinded to index test: yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1a] Not stated [1b] At least 16 days [2] Not stated All patients received same reference standard: No Missing data: yes (only 83 of 120 samples from seroconversion panel analysed) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: [1a] Samples [1b] Patients [2a] Patients [2b] Patients |
||
| Comparative | |||
| Notes | Funding: The study was supported by Tampere Tuberculosis Foundation and Competitive State Research Financing of Expert Responsibility area of Tampere. Publication status: Published paper Source: Diagnostic Microbiology and Infectious Disease Author COI: No conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Flinck 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Flower 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of convalescent‐phase COVID‐19 in patients who did not require hospitalisation Design: Two‐group design with separate estimates of sensitivity and specificity: [1] Adult NHS workers (clinical or non‐clinical) who had previously tested positive for SARS‐CoV‐2 by PCR, but not hospitalised (276 patients with 314 samples); all 21d from symptom onset or positive swab test (whichever was earlier) and not hospitalised. [2] Pre‐pandemic healthy controls (n = 500) Recruitment: [1] Participants (adult NHS workers across 4 hospitals in 2 London NHS trusts) were enrolled once they were at least 21 days from the onset of symptoms, or positive swab test (whichever was earlier); Study advertisement through trust communications [2] Sera from pre‐pandemic healthy controls as part of the Airwaves study from UK police personnel Prospective or retrospective: [1] Prospective; [2] Retrospective Sample size: COVID patients: n = 276 (314 samples); Controls: n = 500 [NB Only data from Phase I used as Phase II used ELISA‐based reference standard] |
||
| Patient characteristics and setting | Setting: Workers from four hospitals in two London NHS trusts (patients who did not need admission; at least 21 days from the symptom onset or PCR‐positive, whichever was earlier); coded as community Location: Four hospitals in two London NHS trusts Country: UK Dates: Cases were recruited between 1 and 29 May 2020. Symptoms and severity: self‐assessed severity based on its effect on daily life: Asymptomatic (7), mild (56), moderate (163), severe but not hospitalised (87) Demographics: Age: median (q1, q3) = 37 (29‐47); female n = 221, Total n = 315 Exposure history: Not stated (all NHS staff) Non‐Covid group 1: pre‐pandemic controls Source: Serum samples from Airwaves study (UK police officers) ‐ pre‐pandemic (prior to August 2019; specified November 2019 though) Characteristics: Not stated (police officers) |
||
| Index tests | Test name: Phase I:
(A) Wondfo SARS‐CoV‐2 Antibody test (lateral flow method);
(B) Menarini Zheijang Orient Gene (lateral flow);
(C) Fortress Diagnostics COVID‐19 TOTAL Ab Device;
(D) Biopanda COVID‐19 Rapid Antibody test;
(E) Biosure COVID‐1 Antibody Self‐Test. Manufacturer: (A) Guangzhou Wondfo Biotech (Guangzhou, China); (B) Menarini Zhejiang Orient Gene Biotech Co Ltd; (C) Fortress Diagnostics; (D) Biopanda; (E) Biosure (Mologic). Antibody: (A) IgG/M combined; (B) IgG & M; (C) IgG & M; (D) IgG & M; (E) IgG only. Antigen target: (A) S; (B) S1, S2 and N; (C) S; D) S and N; (E) N Evaluation setting: POCT performed as POCT and in lab for comparison Test method: Lateral flow immunoassay Timing of samples: After 21 days of symptom onset; median (q1, q3) duration = 44 (35‐53) days; range 21–100 days Samples used: LFIA self‐tests with finger‐prick capillary blood; provided on the same day venous whole blood and serum samples for laboratory analysis. *Review team chose serum samples tested in laboratory for main analyses as largest number of samples per test Test operator: Self‐test (participant interpretation) and observed by a member of the team (trained interpreter observation), finger prick participant self‐read and finger prick trained observer‐read Lab test on serum and whole blood samples: Initially, scoring was performed independently by two technicians, but this practice ceased after inter‐rater scoring was found to be almost perfect by 7‐point categorical score (Kappa = 0.81) Definition of test positivity: By the presence of IgG band (if two separate lines are there for IgG and IgM separately, n = 3 kits OR if only one line to detect IgG only, n = 1 kit) or presence of combined IgG + IgM band (n = 1 kit). Manufacturer instructions were followed. Intensity of the result band(s) from 0 (negative) to 6 according to a standardised scoring system on a visual guide. Invalid tests were repeated. A photograph of the completed test was emailed to the study team. For consistency, in the three kits which had separate IgM and IgG bands ([B], [C], [D]), only IgG was counted as a positive result (i.e. ‘MG’ or ‘G’ but not ‘M’, distinct from manufacturer guidance). [E] Commercial Biosure kit comes in box with device holder and reading card. Clinic self‐tests in this study were performed with the device alone. Blinding reported: Yes for lab analyses, no for self‐test Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: For sensitivity, tests were compared against two standards:
(A) PCR‐confirmed clinical disease (via swab testing) and
(B) positivity in patients with either a positive S‐ELISA and/or hybrid DABA in the laboratory Samples used: (A) Swab, no further details (B) not stated Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear (at least 21 days from the symptom onset or PCR‐positive, whichever was earlier) All patients received same reference standard: No Missing data: Yes (not all patients were included in the analysis) Not all 276 participants received all index tests: 238/276 received one POCT, 38/276 received two different POCTs (314 finger prick tests and 314 sera for lab tests). Also missing data for whole blood analyses Uninterpretable results: Not reported (Possibly none as invalid tests were repeated) Indeterminate results: Invalid tests were repeated. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by funding from The Department of Health and Social Care (DHSC) and NIHR Biomedical Research Centre of Imperial College NHS Trust. GC is supported by an NIHR Professorship. WB is the Action Medical Research Professor. AD is an NIHR senior investigator. DA is an Emeritus NIHR Senior Investigator. HW is an NIHR Senior Investigator. RC holds IPR on the hybrid DABA and this work was supported by UKRI/MRC grant (reference is MC_PC_19078). The sponsor is Imperial College London.
The funders had no role in the production of this manuscript. Publication status: Published paper Source: Thorax Author COI: All authors have completed the ICMJE uniform disclosure form at www. icmje. org/ coi_ disclosure. pdf and declared: no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | |||
| If a threshold was used, was it pre‐specified? | |||
| Could the conduct or interpretation of the index test have introduced bias? | |||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Flower 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | Test name: Phase I:
(A) Wondfo SARS‐CoV‐2 Antibody test (lateral flow method);
(B) Menarini Zheijang Orient Gene (lateral flow);
(C) Fortress Diagnostics COVID‐19 TOTAL Ab Device;
(D) Biopanda COVID‐19 Rapid Antibody test;
(E) Biosure COVID‐1 Antibody Self‐Test.
Manufacturer: (A) Guangzhou Wondfo Biotech (Guangzhou, China); (B) Menarini Zhejiang Orient Gene Biotech Co Ltd; (C) Fortress Diagnostics; (D) Biopanda; (E) Biosure (Mologic). Antibody: (A) IgG/M combined; (B) IgG & M; (C) IgG & M; (D) IgG & M; (E) IgG only. Antigen target: (A) S; (B) S1, S2 and N; (C) S; D) S and N; (E) N. Evaluation setting: POCT performed as POCT and in lab for comparison Test method: Lateral flow immunoassay Timing of samples: After 21 days of symptom onset; median (q1, q3) duration = 44 (35‐53) days; range 21–100 days Samples used: LFIA self‐tests with finger‐prick capillary blood; provided on the same day venous whole blood and serum samples for laboratory analysis Test operator: Self‐test (participant interpretation) and observed by a member of the team (trained interpreter observation), finger prick participant self‐read and finger prick trained observer‐read Lab test on serum and whole blood samples: Initially, scoring was performed independently by two technicians, but this practice ceased after inter‐rater scoring was found to be almost perfect by 7‐point categorical score (Kappa = 0.81) Definition of test positivity: By the presence of IgG band (if two separate lines are there for IgG and IgM separately, n=3 kits OR if only one line to detect IgG only, n=1 kit) or presence of combined IgG + IgM band (n=1 kit). Manufacturer instructions were followed. Intensity of the result band(s) from 0 (negative) to 6 according to a standardised scoring system on a visual guide. Invalid tests were repeated. A photograph of the completed test was emailed to the study team. For consistency, in the three kits which had separate IgM and IgG bands ([B], [C], [D]), only IgG was counted as a positive result (i.e. ‘MG’ or ‘G’ but not ‘M’, distinct from manufacturer guidance). [E] Commercial Biosure kit comes in box with device holder and reading card. Clinic self‐tests in this study were performed with the device alone. Blinding reported: Yes for lab analyses, no for self‐test Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: For sensitivity, tests were compared against two standards:
(A) PCR‐confirmed clinical disease (via swab testing) and
(B) positivity in patients with either a positive S‐ELISA and/or hybrid DABA in the laboratory Samples used: (A) Swab, no further details (B) not stated Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | |||
| If a threshold was used, was it pre‐specified? | |||
| Could the conduct or interpretation of the index test have introduced bias? | |||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Flower 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | Test name: Phase I:
(A) Wondfo SARS‐CoV‐2 Antibody test (lateral flow method);
(B) Menarini Zheijang Orient Gene (lateral flow);
(C) Fortress Diagnostics COVID‐19 TOTAL Ab Device;
(D) Biopanda COVID‐19 Rapid Antibody test;
(E) Biosure COVID‐1 Antibody Self‐Test. Manufacturer: (A) Guangzhou Wondfo Biotech (Guangzhou, China); (B) Menarini Zhejiang Orient Gene Biotech Co Ltd; (C) Fortress Diagnostics; (D) Biopanda; (E) Biosure (Mologic). Antibody: (A) IgG/M combined; (B) IgG & M; (C) IgG & M; (D) IgG & M; (E) IgG only. Antigen target: (A) S; (B) S1, S2 and N; (C) S; D) S and N; (E) N. Evaluation setting: POCT performed as POCT and in lab for comparison Test method: Lateral flow immunoassay Timing of samples: After 21 days of symptom onset; median (q1, q3) duration = 44 (35‐53) days; range 21–100 days Samples used: LFIA self‐tests with finger‐prick capillary blood; provided on the same day venous whole blood and serum samples for laboratory analysis Test operator: Self‐test (participant interpretation) and observed by a member of the team (trained interpreter observation), finger prick participant self‐read and finger prick trained observer‐read Lab test on serum and whole blood samples: Initially, scoring was performed independently by two technicians, but this practice ceased after inter‐rater scoring was found to be almost perfect by 7‐point categorical score (Kappa = 0.81). Definition of test positivity: By the presence of IgG band (if two separate lines are there for IgG and IgM separately, n=3 kits OR if only one line to detect IgG only, n=1 kit) or presence of combined IgG + IgM band (n=1 kit). Manufacturer instructions were followed. Intensity of the result band(s) from 0 (negative) to 6 according to a standardised scoring system on a visual guide. Invalid tests were repeated. A photograph of the completed test was emailed to the study team. For consistency, in the three kits which had separate IgM and IgG bands ([B], [C], [D]), only IgG was counted as a positive result (i.e. ‘MG’ or ‘G’ but not ‘M’, distinct from manufacturer guidance). [E] Commercial Biosure kit comes in box with device holder and reading card. Clinic self‐tests in this study were performed with the device alone. Blinding reported: Yes for lab analyses, no for self‐test Threshold predefined: Yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | |||
| If a threshold was used, was it pre‐specified? | |||
| Could the conduct or interpretation of the index test have introduced bias? | |||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Flower 2020 [D].
| Study characteristics | |||
| Patient Sampling | |||
| Patient characteristics and setting | |||
| Index tests | Test name: Phase I:
(A) Wondfo SARS‐CoV‐2 Antibody test (lateral flow method);
(B) Menarini Zheijang Orient Gene (lateral flow);
(C) Fortress Diagnostics COVID‐19 TOTAL Ab Device;
(D) Biopanda COVID‐19 Rapid Antibody test;
(E) Biosure COVID‐1 Antibody Self‐Test. Manufacturer: (A) Guangzhou Wondfo Biotech (Guangzhou, China); (B) Menarini Zhejiang Orient Gene Biotech Co Ltd; (C) Fortress Diagnostics; (D) Biopanda; (E) Biosure (Mologic). Antibody: (A) IgG/M combined; (B) IgG & M; (C) IgG & M; (D) IgG & M; (E) IgG only. Antigen target: (A) S; (B) S1, S2 and N; (C) S; (D) S and N; (E) N. Evaluation setting: POCT performed as POCT and in lab for comparison Test method: Lateral flow immunoassay Timing of samples: After 21 days of symptom onset; median (q1, q3) duration = 44 (35‐53) days; range 21–100 days Samples used: LFIA self‐tests with finger‐prick capillary blood; provided on the same day venous whole blood and serum samples for laboratory analysis Test operator: Self‐test (participant interpretation) and observed by a member of the team (trained interpreter observation), finger prick participant self‐read and finger prick trained observer‐read Lab test on serum and whole blood samples: Initially, scoring was performed independently by two technicians, but this practice ceased after inter‐rater scoring was found to be almost perfect by 7‐point categorical score (Kappa = 0.81) Definition of test positivity: By the presence of IgG band (if two separate lines are there for IgG and IgM separately, n=3 kits OR if only one line to detect IgG only, n=1 kit) or presence of combined IgG + IgM band (n=1 kit). Manufacturer instructions were followed. Intensity of the result band(s) from 0 (negative) to 6 according to a standardised scoring system on a visual guide. Invalid tests were repeated. A photograph of the completed test was emailed to the study team. For consistency, in the three kits which had separate IgM and IgG bands ([B], [C], [D]), only IgG was counted as a positive result (i.e. ‘MG’ or ‘G’ but not ‘M’, distinct from manufacturer guidance). [E] Commercial Biosure kit comes in box with device holder and reading card. Clinic self‐tests in this study were performed with the device alone. Blinding reported: Yes for lab analyses, no for self‐test Threshold predefined: Yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | |||
| If a threshold was used, was it pre‐specified? | |||
| Could the conduct or interpretation of the index test have introduced bias? | |||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Flower 2020 [E].
| Study characteristics | |||
| Patient Sampling | |||
| Patient characteristics and setting | |||
| Index tests | Test name: Phase I:
(A) Wondfo SARS‐CoV‐2 Antibody test (lateral flow method);
(B) Menarini Zheijang Orient Gene (lateral flow);
(C) Fortress Diagnostics COVID‐19 TOTAL Ab Device;
(D) Biopanda COVID‐19 Rapid Antibody test;
(E) Biosure COVID‐1 Antibody Self‐Test. Manufacturer: (A) Guangzhou Wondfo Biotech (Guangzhou, China); (B) Menarini Zhejiang Orient Gene Biotech Co Ltd; (C) Fortress Diagnostics; (D) Biopanda; (E) Biosure (Mologic). Antibody: (A) IgG/M combined; (B) IgG & M; (C) IgG & M; (D) IgG & M; (E) IgG only. Antigen target: (A) S; (B) S1, S2 and N; (C) S; D) S and N; (E) N. Evaluation setting: POCT performed as POCT and in lab for comparison Test method: Lateral flow immunoassay Timing of samples: After 21 days of symptom onset; median (q1, q3) duration = 44 (35‐53) days; range 21–100 days Samples used: LFIA self‐tests with finger‐prick capillary blood; provided on the same day venous whole blood and serum samples for laboratory analysis Test operator: Self‐test (participant interpretation) and observed by a member of the team (trained interpreter observation), finger prick participant self‐read and finger prick trained observer‐read Lab test on serum and whole blood samples: Initially, scoring was performed independently by two technicians, but this practice ceased after inter‐rater scoring was found to be almost perfect by 7‐point categorical score (Kappa = 0.81) Definition of test positivity: By the presence of IgG band (if two separate lines are there for IgG and IgM separately, n=3 kits OR if only one line to detect IgG only, n=1 kit) or presence of combined IgG + IgM band (n=1 kit). Manufacturer instructions were followed. Intensity of the result band(s) from 0 (negative) to 6 according to a standardised scoring system on a visual guide. Invalid tests were repeated. A photograph of the completed test was emailed to the study team. For consistency, in the three kits which had separate IgM and IgG bands ([B], [C], [D]), only IgG was counted as a positive result (i.e. ‘MG’ or ‘G’ but not ‘M’, distinct from manufacturer guidance). [E] Commercial Biosure kit comes in box with device holder and reading card. Clinic self‐tests in this study were performed with the device alone. Blinding reported: Yes for lab analyses, no for self‐test Threshold predefined: Yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Fragkou 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Hospitalised confirmed COVID‐19 cases (n = 26) [2] Asymptomatic healthcare volunteers with negative rRT‐PCR (n = 18) Group [2] had < 25 samples and was excluded from our review. Recruitment: Not stated Prospective or retrospective: Prospective Sample size: 44 (26) of which 16 (16) were eligible for our review Further detail: [1] Hospitalised symptomatic patients with rRTPCR‐confirmed COVID‐19 infection [2] Hospital asymptomatic volunteers, with no clinical symptoms for the past month, with negative SARS‐CoV‐2 rRT‐PCR at the day of sampling and no reported “close contact” history (based on the ECDC definitions for confirmed cases and close contacts) [1] and [2] adults (≥ 18 years old) No additional exclusion criteria were applied. |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Attikon University Hospital, National and Kapodistrian University of Athens, Athens, Greece Country: Greece Dates: 30th March 2020 and 6th April 2020 Symptoms and severity: Mild: 8/26; moderate: 8/26; severe and/or critical: 10/26 Demographics: 65.9 ± 15.4 years old, male 57.7% 0‐7 days pso (n = 5): 81.6 ± 11.8 years 7‐14 days pso (n = 11): 68.2 ± 9.4 years > 14 days pso (n = 10): 55.5 ± 15.2 years Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic healthy Source: Asymptomatic healthcare volunteers from a tertiary teaching hospital between 30th March 2020 and 6th April 2020 Characteristics: Adults; hospital staff; no clinical symptoms for the past month, with negative SARS‐CoV‐2 rRT‐PCR at the day of sampling and no reported “close contact” history (based on the ECDC definitions for confirmed cases and close contacts) 45.6 ± 10.1 years old, male 33.3% |
||
| Index tests | Test name: (COVID‐19) IgG/IgM Test Kit Manufacturer: Lansion Biotechnology Co., Ltd. (Nanjing, PR China) Antibody: IgG and IgM Antigen target: Not stated Evaluation setting: POCT performed as POC (actual clinical setting) Test method: dry fluorescence immunoassay via a portable analyser Timing of samples: < 7 days: 5/26 7‐14 days: 11/26 > 14 days: 10/26 Samples used: Capillary whole blood: finger‐prick, 5 μL of whole blood was collected in a micropipette and delivered on a test strip. Test operator: Not stated Definition of test positivity: Manufacturer’s cut‐off ≥ 0.04 mIU/mL for both IgG and IgM antibodies; cut‐off of IgM ≥ 0.05 mIU/mL and IgG ≥ 0.10 mIU/mL; cut‐off of IgM ≥ 0.08 mIU/mL and IgG ≥ 0.19 mIU/mL Blinding reported: Not stated Threshold predefined: yes for manufacturer's cut‐off, no for the other cut‐offs |
||
| Target condition and reference standard(s) | Reference standard: rRT‐PCR for SARS‐CoV‐2: using the VIASURE SARS‐CoV‐2 Real Time PCR Detection Kit (CerTest Biotec SL, Zaragoza, Spain) Samples used: Nasopharyngeal and/or oropharyngeal swabs; lower respiratory tract samples (e.g. bronchoalveolar lavage or aspirates, sputum, etc.) were also accepted. Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: rRT‐PCR for SARS‐CoV‐2 and clinical symptoms, using the VIASURE SARS‐CoV‐2 Real Time PCR Detection Kit (CerTest Biotec SL, Zaragoza, Spain) Samples used: Nasopharyngeal and/or oropharyngeal swabs Timing of reference standard: negative SARS‐CoV‐2 rRT‐PCR at the day of sampling Blinded to index test: unclear Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1] Not stated [2] Same day All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: No Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: For PCF: Supported by Doctorate scholarship by the State Scholarships Foundation (IKY), Partnership Agreement (PA) 2014‐2020, co‐financed by Greece and the European Union (European Social Fund‐ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning 2014‐2020"
Consumables, test strips and the reader were provided for free by Lansion Biotech. Publication status: Published paper Source: In Vivo Author COI: The authors declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Fujigaki 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of three antibody tests for identification of acute and convalescent‐phase SARS‐CoV‐2 infection Design: Single‐group study estimating sensitivity: residual samples from PCR‐confirmed COVID‐19 patients (n = 29, providing 99 samples) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 29 (29) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Inpatient setting (all hospitalised) Location: Fujita Health University Hospital,Toyoake, Aichi Country: Japan Dates: 28 February to 15 April 2020 Symptoms and severity: Not stated (however, all patients were hospitalised, so likely had symptoms) Demographics: Mean age 52.9 y (SD 21.9); 14, 48% male Exposure history: Not stated |
||
| Index tests | Test name: [A] 2019‐nCoV IgG/IgM Rapid Test Cassette [B] COVID‐19 IgM/IgG Duo [C] 2019‐nCoV IgG/IgM Detection Kit Manufacturer: [A] Hangzhou AllTest Biotech Co., Ltd., China [B] SD BIOSENSOR, Korea [C] Vazyme Biotech Co., Ltd., China Antibody: All tests: IgM, IgG Antigen target: All tests: Unclear Evaluation setting: All tests: POC tests, likely done in lab (samples were residual and had been frozen) Test method: All tests: Lateral flow immunoassay ‐ colloidal gold (CGIA) Timing of samples: Day 0 to 35; day 0‐7: 18 patients; 27 samples day 8‐14: 22 patients; 39 samples day 15‐21 18 patients; 28 samples day > 21 4 patients; 5 samples Samples used: Serum (residual and frozen prior to testing) Test operator: Not stated Definition of test positivity: As per manufacturer: visual‐based Blinding reported: Not stated Threshold predefined: Yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR test (no more details available) Samples used: Nasopharyngeal swabs Timing of reference standard: At the time or prior to hospital admission (not further specified) Blinded to index test: Yes (done earlier) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes, although different RT‐PCR assays could have been used. Missing data: None reported; some participants provided only one sample while others provided up to 12. Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Presented per sample in the paper; extracted on a per patient basis by review team using Fig 1 and Fig 2 and including one sample per patient per week post‐symptom onset (any positive result over‐rode negative results in same week) |
||
| Comparative | |||
| Notes | Funding: No funding reported
Nichirei Biosciences Inc. and Shionogi & Co., Ltd respectively provided the 2019‐nCoV IgG/IgM Rapid Test Cassette and COVID‐19 IgM/IgG Duo kits and the 2019‐nCoV IgG/IgM Detection Kit. Publication status: Pre‐print article Source: Pre‐print server (medRxiv) Author COI: One of the authors received immunochromatographic anti‐SARS‐CoV‐2 antibody detection kits from Nichirei Biosciences Inc. and Shionogi & Co., Ltd, none of which was related to this work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Fujigaki 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Fujigaki 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Gao 2020a.
| Study characteristics | |||
| Patient Sampling | Single‐group study estimating sensitivity [1] Patients with confirmed COVID‐19 (n = 38) Recruitment: unclear Sample size (virus/COVID cases): 38 (38) Inclusion and exclusion criteria: COVID‐19 confirmed by New Coronavirus Pneumonia Prevention and Control Program (5th edition) published by the National Health Commission of China | ||
| Patient characteristics and setting | Setting: hospital inpatient Location: Second People's Hospital of Fuyang Country: China Dates: 22 January 2020‐28 February 2020 Symptoms and severity: 3/38 described as in severe or critical conditions; 35/38 described as mild cases Sex: 55.3% (21/38) male Age: median age 40.5 years (IQR 31.0‐49.5 years), range 15‐75 years Exposure history: NR | ||
| Index tests | Test name: Colloidal Gold Antibodies Test Manufacturer: Innovita Biological Technology Co., Ltd Ab targets: IgM, IgG Antigens used: NR Test method: CGIA Timing of samples: days 0‐15+ Samples used: serum Test operators: NR Definition of test positivity: visible line Blinded to reference standard: NR Threshold predefined: yes | ||
| Target condition and reference standard(s) | Reference standard for cases: participants met the criteria of the New Coronavirus Pneumonia Prevention and Control Program (5th edition) published by the National Health Commission of China. Samples used: NR Timing of reference standard: NR Blinded to index test: yes Incorporated index test: no | ||
| Flow and timing | Time interval between index and reference tests: NR Results presented by time period: yes: 0‐7 days (n = 13), 8‐14 days (n = 8) and ≥ 15 days (n = 23) after onset of symptoms All participants received the same reference standard: yes Missing data: NR Uninterpretable results: NR Indeterminate results: NR Unit of analysis: results reported for participants. 38 participants included and 76 serum samples collected in total from these 38 participants. Median number of samples collected from each participant was 8. | ||
| Comparative | |||
| Notes | Funding: The Science and Technology Bureau of Fuyang Publication status: accepted manuscript (peer reviewed) Source: Journal of Medical Virology Study author COI: none reported | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Gao 2020b [A].
| Study characteristics | |||
| Patient Sampling | Single‐group study recruiting patients estimating sensitivity [1] confirmed COVID‐19 cases Recruitment: consecutive (inferred). From all confirmed cases admitted to hospital Prospective or retrospective recruitment of cases: retrospective (appeared) Sample size (virus/COVID cases): 22 participants (corresponding to 37 samples) Inclusion and exclusion criteria: not clearly defined; described all participants having typical ground‐glass opacity of the lung on CT but not clear if this was part of eligibility | ||
| Patient characteristics and setting | Setting: hospital inpatient Location: Fifth Hospital of Shijiazhuang Country: China Dates: from 21 January‐24 February 2020 Symptoms and severity: typical ground‐glass opacity in lung was observed in CT scan results of all participants. At the time the paper was written all participants had recovered and been discharged from hospital. Sex: 14/22 male (64%) Age: 40 (4‐72) years Exposure history: 11 participants had recent history of travel to epidemic areas, and the remaining 10 had close contacts with their family members, who were confirmed to be infected by 2019‐nCoV. | ||
| Index tests |
Gao 2020b [A] is test [A] from the following entry: Test name: [A] CLIA; [B] GICA; [C] ELISA Manufacturer: Beier Bioengineering Company (Beijing, China) Ab targets: IgG and IgM Antigens used: spike (S) and nucleocapsid (N) proteins of 2019‐nCoV Test method: [A] CLIA; [B] GICA; [C] ELISA Timing of samples: [1] early stage (1‐7 days pso) 10/37 samples (27%), [2] middle stage (8‐14 days pso) 13/37 samples (35%); [3] late stage (14‐24 days pso) 14/37 samples (38%) Samples used: serum Test operators: laboratory staff Definition of test positivity: [A] samples with an concentration ≥ 8 arbitrary unit (AU)/mL were considered positive. [B] Visible line [C] The absorbance at 450 nm (A450 nm) of each well was determined and the cut‐off value was 0.10 + A negative control. A value > cut‐off value was considered a positive result. Blinded to reference standard: NR Threshold predefined: [A] samples with an concentration ≥ 8 arbitrary unit (AU)/mL were considered positive. [B] Positive results showed the appearance of both control line and testing line. [C] The absorbance at 450 nm (A450 nm) of each well was determined and the cut‐off value was 0.10 + A negative control. A value > cut‐off value was considered a positive result. |
||
| Target condition and reference standard(s) | Reference standard for cases: RT‐PCR assay (2019‐nCoV RNA Test Kit, Daan Gene Company, China) Samples used: nasal and pharyngeal swab specimens Timing of reference standard: on admission (most likely) Blinded to index test: yes, index tests performed on already‐confirmed cases (inferred) Incorporated index test: no Reference standard for non‐cases: NA | ||
| Flow and timing | Time interval between index and reference tests: NR Results presented by time period: yes All participants received the same reference standard: yes Missing data: timing of reference standard test Uninterpretable results: Indeterminate results: Unit of analysis: samples | ||
| Comparative | |||
| Notes | Funding: NR Publication status: published letter Source: Chinese Medical Journal Study author COI: none | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Gao 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Index tests |
Gao 2020b [B] is test [B] from the following entry: Test name: [A] CLIA; [B] GICA; [C] ELISA Manufacturer: Beier Bioengineering Company (Beijing, China) Ab targets: IgG and IgM Antigens used: spike (S) and nucleocapsid (N) proteins of 2019‐nCoV Test method: [A] CLIA; [B] GICA; [C] ELISA Timing of samples: [1] early stage (1‐7 days pso) 10/37 samples (27%), [2] middle stage (8‐14 days pso) 13/37 samples (35%); [3] late stage (14‐24 days pso) 14/37 samples (38%) Samples used: serum Test operators: laboratory staff Definition of test positivity: [A] samples with an concentration ≥ 8 arbitrary unit (AU)/mL were considered positive. [B] Visible line [C] The absorbance at 450 nm (A450 nm) of each well was determined and the cut‐off value was 0.10 + A negative control. A value > cut‐off value was considered a positive result. Blinded to reference standard: NR Threshold predefined: [A] samples with an concentration ≥ 8 arbitrary unit (AU)/mL were considered positive. [B] Positive results showed the appearance of both control line and testing line. [C] The absorbance at 450 nm (A450 nm) of each well was determined and the cut‐off value was 0.10 + A negative control. A value > cut‐off value was considered a positive result. |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Comparative | |||
| Notes | |||
Gao 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Index tests |
Gao 2020b [C] is test [C] from the following entry: Test name: [A] CLIA; [B] GICA; [C] ELISA Manufacturer: Beier Bioengineering Company (Beijing, China) Ab targets: IgG and IgM Antigens used: spike (S) and nucleocapsid (N) proteins of 2019‐nCoV Test method: [A] CLIA; [B] GICA; [C] ELISA Timing of samples: [1] early stage (1‐7 days pso) 10/37 samples (27%), [2] middle stage (8‐14 days pso) 13/37 samples (35%); [3] late stage (14‐24 days pso) 14/37 samples (38%) Samples used: serum Test operators: laboratory staff Definition of test positivity: [A] samples with an concentration ≥ 8 arbitrary unit (AU)/mL were considered positive. [B] Visible line [C] The absorbance at 450 nm (A450 nm) of each well was determined and the cut‐off value was 0.10 + A negative control. A value > cut‐off value was considered a positive result. Blinded to reference standard: NR Threshold predefined: [A] samples with an concentration ≥ 8 arbitrary unit (AU)/mL were considered positive. [B] Positive results showed the appearance of both control line and testing line. [C] The absorbance at 450 nm (A450 nm) of each well was determined and the cut‐off value was 0.10 + A negative control. A value > cut‐off value was considered a positive result. |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment (Gao 2020b [A]) | ||
| Comparative | |||
| Notes | |||
Garnett 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Validation of an automated platform, the Vitros Anti‐SARS‐CoV‐2 Total antibody assay, for screening of previous exposure to SARS‐CoV‐2 in our patient population. Comparison serum analysis of known COVID‐19 patients, healthy controls and COVID‐19‐negative but positive for another respiratory viral infection.
3‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: [1] patients previously diagnosed with COVID‐19 by RT‐PCR (n = 79) [2] healthy volunteers with no known exposure, travel history, or symptoms of COVID‐19 (n = 57) [3] patients previously tested to be negative for SARS‐CoV‐2 by RT‐PCR, but positive for another respiratory viral infection by molecular analysis (n = 14) Group [3] was excluded from our review as it contained < 25 samples. Recruitment: [1] and [2] Specimens for validation were obtained with informed consent from healthy volunteers and known patients with COVID‐19 under an approved protocol from our local institutional review board. [3] Unclear Prospective or retrospective: [1], [2] and [3] Unclear Sample size: 150 (79) of which 136 (79) were eligible for our review Further detail: [1] Previously diagnosed with COVID‐19 by reverse transcription–polymerase chain reaction (RT‐PCR) methods at our hospital or by molecular methods at other local laboratories within our large academic medical centre [2] Negative for SARS CoV‐2 by RT‐PCR and who had no known exposure, travel history, or symptoms of COVID‐19 [3] Known to be positive for other viruses by molecular testing (including influenza A virus, influenza B virus, respiratory syncytial virus, adenovirus, rhinovirus, or other coronaviruses) but negative for SARS‐CoV‐2 by RT‐PCR |
||
| Patient characteristics and setting | Setting: Unclear. Location: Known positive patients were previously diagnosed with COVID‐19 by RT‐PCR methods at Texas Children’s Hospital clinical laboratories, or at other institutions in the Texas Medical Center This sentence changed in the published paper to "Known positive patients were previously diagnosed with COVID‐19 by reverse transcription–polymerase chain reaction (RT‐PCR) methods at our hospital or by molecular methods at other local laboratories within our large academic medical center." Houston, Texas from affiliations Country: USA Dates: Not stated Symptoms and severity: Patients "were RT‐PCR‐positive for SARS CoV‐2 and/or admitted to the COVID ICU" Not stated in publication Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Healthy controls Source: Not stated Characteristics: healthy volunteers who were negative for SARS CoV‐2 by RT‐PCR and who had no known exposure, travel history, or symptoms of COVID‐19 |
||
| Index tests | Test name: Vitros (VITROS®) Anti‐SARS‐Cov‐2 Total assay Manufacturer: Ortho Clinical Diagnostics, Raritan, NJ Publication only stated "Ortho Clinical Diagnostics", no city or state Antibody: total IgG and IgM Antigen target: solid‐phase SARS‐CoV‐2 spike‐protein antigen Evaluation setting: Laboratory test ‐ Vitros (VITROS®) Anti‐SARS‐Cov‐2 Total assay used on the Vitros 5600 automated chemistry analyser (Ortho Clinical Diagnostics, Raritan, NJ) Test method: Unclear. "The Vitros (VITROS®) Anti‐SARS‐Cov‐2 Total assay (CoV2T, Ortho Clinical Diagnostics, Raritan, NJ) detects total IgG and IgM directed against SARS‐Cov‐2, and was evaluated for use on the Vitros 5600 automated chemistry analyzer (Ortho Clinical Diagnostics, Raritan, NJ). The CoV2T assay uses a solid‐phase SARS‐CoV‐2 spike‐protein antigen to capture antibodies in the patient specimen, and horseradish peroxidase‐labelled recombinant SARS‐CoV‐2 antigen as a detection reagent." Agree that the test method was unclear from the text. I have checked online and it seems to be a CLIA method. Timing of samples: 0 to 35 days after onset of symptoms for 55 COVID patients (methods say 0‐35 days after positive PCR, possibly for all 79 COVID patients) Categorised as: < 3 days pso: 17/55, 4‐7 days pso: 7/55, 8‐13 days pso: 8/55 and > 13 days since first reported symptom: 23/55 Samples used: Serum and plasma Test operator: Laboratory personnel Definition of test positivity: The assay is qualitative and reports results as reactive or nonreactive based on a manufacturer‐defined signal/cut‐off (s/c) ratio of 1.00 as the decision limit Blinding reported: Unclear Threshold predefined: manufacturer‐defined signal/cut‐off (s/c) ratio of 1.00 as the decision limit |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR. Threshold not stated (samples were tested on different days and by different operators) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes ‐ prior to index test Incorporated index test: No Definition of non‐COVID cases: [2] healthy volunteers who were negative for SARS CoV‐2 by RT‐PCR and who had no known exposure, travel history, or symptoms of COVID‐19 (samples were tested on different days and by different operators) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: None 55/79 included in seroconversion study ("Seroconversion in our patient population was assessed by correlation of chart review of 55 patients known to be positive for SARS‐CoV‐2 by RT‐PCR and known date of symptom onset…") so 24/79 no data on symptom onset? Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: EG and JJ were supported by the Ching Nan Ou Fellowship Endowment. Some of the validation kits used in this study were provided by Ortho Clinical Diagnostics, but they maintained no involvement in study design or validation, and were not privy to any of the data or interpretation. Publication status: Pre‐print (not peer reviewed); now published paper Source: medRxiv preprint doi: https://doi.org/10.1101/2020.06.09.20126474 Journal (American Journal of Clinical Pathology) Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
GeurtsvanKessel 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of multiple diagnostic tests for acute and convalescent‐phase COVID‐19 and evaluation of antibody kinetics Design: Two‐group study estimating both sensitivity and specificity Group [1]: PCR‐confirmed COVID‐19 cases (n = 229 samples); published report included 107 patients (187 samples) with virus neutralisation antibodies detected by PRNT50 (all PRNT >= 20); Supplementary Data file included data for a further 42 samples with PRNT < 20 Group [2]: Patients with other infections (n = 147 reportedly included but results for 157 samples reported in Supplementary Data file and in Tabl 1 of published report for EUROIMMUN assays only) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 254 (107) Samples: 386 (229); as reported in Supplementary Data file Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Mixed; outpatient and inpatient (all COVID‐19 patients admitted to Erasmus MC were asked for permission to use their clinical data and left‐over patient material for COVID‐19 research purposes) Location: Erasmus Medical Center, Rotterdam Country: Netherlands Dates: Not stated Symptoms and severity: Of 229 samples in Supplementary Data file: 71, 31% mild (non‐hospitalised); 55, 24% moderate (hospitalised); 103, 45% ICU Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Group [2]: Patients with exposure to human coronaviruses (HCoV‐229E, NL63 or OC43), SARS, MERS), or with a range of other respiratory viruses (adenovirus, human metapneumovirus, influenza A/B, RSV A/B, rhinovirus, bocavirus, parainfluenza virus 1 and 3, enterovirus, EBV, CMV) Source: Lab stocked samples. Collection period was not stated but likely pre‐pandemic. Characteristics: No more details available |
||
| Index tests | Test name: [A] Wantai SARS‐CoV‐2 total Ig ELISA [B] Wantai SARS‐CoV‐2 IgM ELISA [C] Euroimmun Anti‐SARS‐CoV‐2 IgG ELISA assay [D] Euroimmun Anti‐SARS‐CoV‐2 IgA ELISA assay [E] LIAISON SARS‐CoV‐2 S1/S2 IgG [F] Rapid SARS‐CoV‐2 Antibody (IgM/IgG) Test (Test lots S2020021505 and GJ20030288) [G] (GICA) (Test lot 20200416WI5513C) [H] COVID‐19 IgG/IgM Rapid Test Cassette (Test lot 2003309) Manufacturer: [A], [B]: Beijing Wantai Biological Pharmacy Enterprise Co., Ltd., China [C], [D]: EUROIMMUN Medizinische Labordiagnostika AG, Lübeck, Germany [E] DiaSorin, Saluggia, Italy [F] InTec Products Inc. [G] Cellex Inc. [H] OrientGene Biotech / Healgan, China Antibody: [A]: Total IgG [B]: IgM [C]: IgG [D]: IgA [E]: IgG [F] ‐ [H]: IgM, IgG Antigen target: [A], [B]: RBD [C], [D]: S1 domain of the spike‐protein [E]: S1 and S2 domains of the spike‐protein [F]: S and N proteins [G]: N‐protein [H]: S and N proteins Evaluation setting: [A]‐[E]: Lab tests, done in lab [F]‐[H]: POC tests, likely done in lab. Test method: [A]‐[D]: Enzyme‐linked immunosorbent assay (ELISA) [E]: Chemiluminescence immunoassay (CLIA) [F]‐[H]: Lateral flow immunoassay (CGIA) Timing of samples: Median 16 days pso (calculated from Suppl Data file), range 4 to 73 days The number of samples tested varied for each assay, and results were presented per sample but not per patient, so a clear breakdown by time was hard. Samples used: Serum (COVID‐19 cases); serum or plasma (non‐COVID‐19 samples) Test operator: Not stated; presumably lab staff as all specimen were stored at −20 °C until use Definition of test positivity: [A, B] Wantai ELISAs, OD ratio > 1; [C,D] Euroimmun ELISAs, OD ratio > 1.1; [E] DiaSorin Liaison IgG > 15 AU/mL; [F to H] presence of visible lines. For assay [E] samples with values between 12 and 15 on initial test, were retested as per manufacturer IFU, and considered positive if value >= 12 for a second time. Blinding reported: Unclear Threshold predefined: Yes as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR test (no more details available) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: Group [2]: Other infection or condition controls (timing not reported) no testing Samples used: Not stated Timing of reference standard: 2.3 weeks prior to serum collection "Sera (for index test) were collected from 2–3 weeks upon the respiratory infection, and during the acute phase of CMV or EBV." Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No ‐ multiple assays were likely used to test patients from Group [1]. Group [2] received no reference standard (unclear if pre‐pandemic or not) Missing data: The number of samples tested per assay reportedly varied due to limited sample volume and limited availability of the LFAs at the time of the evaluation; however some discrepancies between data reported in paper (Tabl 1) and data provided in Suppl Data file (Fig 1) could not be explained by limited sample volume (e.g. LFAs reported for 9 control samples in published report but 79 samples in data file). Of 229 available samples from COVID‐19 cases and 157 samples from non‐COVID‐19 cases, results were available for: [A] 229/146 [B] 227/146 [C + D] 90/157 [E] 202/137 [F to H] 131/79 (NB Tabl 1 of paper reported data for 98 control samples for all LFAs) Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work partially was funded through EU COVID‐19 grant RECOVER. Publication status: Published article Source: Nature Communications Author COI: Authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
GeurtsvanKessel 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
GeurtsvanKessel 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
GeurtsvanKessel 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
GeurtsvanKessel 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
GeurtsvanKessel 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
GeurtsvanKessel 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
GeurtsvanKessel 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Graham 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of prior infection (sero‐prevalence in nursing home residents) Design: Single‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (PCR+) (n = 94) [2] PCR‐ residents (n = 147) PCR‐ residents (n = 147) were not included in our review as they did not have an adequate reference standard (PCR tests performed too late or not correctly swabbed). Recruitment: Testing was performed as part of an outbreak investigation with Public Health England and verbal consent obtained from residents (or their relative/friend as appropriate) who had a RT‐PCR result available. All residents available and consenting to testing from 4 UK nursing homes Prospective or retrospective: Prospective Sample size: 241 (94) samples of which 94 (94) samples were eligible for our review Further detail: All residents of 4 UK Nursing Homes with rt‐PCR results available and informed consent [1] All rt‐PCR‐positive residents |
||
| Patient characteristics and setting | Setting: Convalescent (Nursing home residents) Location: 4 UK Nursing Homes (West London Nursing Homes) Country: UK Dates: June 2020 Symptoms and severity: Convalescent (around 2 months after outbreak) [Of 158 PCR+ residents, 43% had no identifiable symptoms in the preceding two‐week period. 35% of antibody‐positive residents (62 of 173) had been asymptomatic in the two‐week ascertainment window prior to PCR testing during the outbreak. Not stated for the 94 included COVID cases] Demographics: Not stated (high‐dependency nursing home residents) Exposure history: All nursing home residents |
||
| Index tests | Test name: Abbott Architect nucleocapsid IgG assay Manufacturer: Abbott Antibody: IgG Antigen target: N‐protein Evaluation setting: Lab test performed in lab Test method: Not stated Timing of samples: Not stated (convalescent, around 2 months after diagnosis) Samples used: Serum Test operator: Not stated (as part of an outbreak investigation with Public Health England) Definition of test positivity: Not stated (samples with binding ratios near to the cut‐off were confirmed on an in‐house receptor binding domain double antigen bridging assay to determine final status) Blinding reported: Not stated Threshold predefined: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR testing for all residents, with re‐testing one week later in those testing negative Samples used: Oropharyngeal and nasal swabs Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Around 2 months (PCR+ in April 2020, index test in June 2020) All patients received same reference standard: yes Missing data: yes (147 PCR‐ residents not included in our review) Uninterpretable results: Not stated Indeterminate results: Samples with binding ratios near to the cut‐off were confirmed on an in‐house receptor binding domain double antigen bridging assay to determine final status. Number not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: UK DRI Centre for Care Research and Technology for funding the work Publication status: Published letter Source: Journal of Infection Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Gudbjartsson 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection, current convalescent‐phase infection, and prior infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID cases (1237 patients; 2102 samples; possible overlap of patients between [1a] and [1b]) [1a] Hospitalised (48 patients; 249 samples) [1b] Recovered (1215 patients; 1853 samples) [2] PCR‐ or not tested [2a] Pre‐pandemic: 2017 (n = 472) [2b] Early 2020 (n = 470) [2c] Health Care (n = 18,609) [2d] Reykjavik (n = 4843) [2e] Vestmannaeyjar (n = 663) [2f] Quarantine (n = 4222) Only groups [1b] and [2a] were eligible for our review. Recruitment: [1] From February 28 to May 1, 1797 patients were found to be SARS‐CoV‐2 positive by qPCR. We collected samples from a group of hospitalised qPCR‐positive persons and invited all qPCR‐positive persons who had recovered from infection to donate samples, both shortly after recovery and again approximately 3 months after recovery (a total of 2102 samples from 1237 persons). [1a] 48 out of 101 (48%) hospitalised Icelandic COVID‐19 patients during their hospital admission The most common reason for missing samples was that the patient had been discharged before commencement of the study, followed by the patient not consenting to participate in the study. [1b] We invited all qPCR‐positive persons to give a blood sample after recovery (defined as at least two weeks from qPCR diagnosis and one week after end of symptoms) and again on July 1, on average 100 days after diagnosis with qPCR. Non‐participation was because of refusal or inability to participate because of health or geographic constraints. [2a] Persons participating in the deCODE health study in the year 2017 [2b] Persons participating in the deCODE health study from February 18 through March 9 2020 [2c], [2d], [2e] Persons who had neither tested qPCR‐positive nor been quarantined to evaluate seroprevalence outside quarantine and the spread of the virus in Iceland (the Health Care, Reykjavik, and Vestmannaeyjar sample groups) [2f] Samples from quarantined persons who had not tested qPCR‐positive Prospective or retrospective: [1a], [1b], [2c], [2d], [2e], [2f] Prospective [2a], [2b] Retrospective Sample size: 30,576 (1237) people of which 2325 (1853) samples from 1687 (1215) patients were eligible for our review. Further detail: Inclusion criteria: either that the person had not been tested positive with qPCR (Neg/NA) (group [2]) or that the person had been positive with qPCR assay (positive) (group 1]) |
||
| Patient characteristics and setting | Setting: [1b] Convalescent/community Location: Former inpatients or outpatients of Landspitali ‐ The National University Hospital of Iceland (LUH), Reykjavik. Country: Iceland Dates: [1b] 3 April to 8 July 2020 Symptoms and severity: [1b] Not stated (1215 of 1797 PCR+ COVID patients included in [1b]: Of the 1797 confirmed COVID patients, 1746 (97.2%) were treated as outpatients while the remaining 51 (2.8%) patients were admitted to hospital at the time of diagnosis. Now all recovered with at least 1 week without symptoms) Demographics: [1b] 48% male Age: Mean 43 (SD 16) years Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic Source: Persons participating in the deCODE health study in the year 2017 (2 January to 4 December 2017) Characteristics: 41% male Age: mean 57 (SD 16) years |
||
| Index tests | Test name: Name not stated
[A] Roche Elecsys chemiluminescence assay
[B] Wantai ELISA
[C] EDI ELISA
[D] EDI ELISA
[E] Euroimmun ELISA
[F] Euroimmun ELISA Manufacturer: [A] Roche International, Basel, Switzerland [B] Wantai/Nordic BioSite, Täby, Sweden [C] EDI/Eagle Biosciences, Amherst, NH, United States [D] EDI/Eagle Biosciences, Amherst, NH, United States [E] Euroimmun AG, Luebeck, Germany [F] Euroimmun AG, Luebeck, Germany Antibody: [A] Total antibodies [B] Total antibodies [C] IgG [D] IgM [E] IgG [F] IgA Antigen target: [A] Nucleocapsid (anti‐N) [B] Spike 1 RBD (anti‐S1‐RBD) [C] Nucleocapsid (anti‐N) [D] Nucleocapsid (anti‐N) [E] Spike subunit 1 (anti‐S1) [F] Spike subunit 1 (anti‐S1) Evaluation setting: [A]‐[F] Lab tests performed in lab Test method: [A] ECLIA [B]‐[F] ELISA Timing of samples: [1b] at least two weeks from qPCR diagnosis and one week after end of symptoms; (text and Fig 2 stated "25 days after diagnosis" for the earliest time point) and again on July 1, on average 100 days after diagnosis with qPCR (487/1215 recovered patients with at least 2 samples at least 30 days apart); up to 4 months after PCR+ Samples used: Serum samples were frozen in aliquots at ‐80°C. Test operator: Not stated Definition of test positivity: All measurements were done according to manufacturer ‘s instructions. The ELISA results are expressed as optical density (OD) and the ECLIA results as log light emission. [C] and [D] For the IgG and IgM anti‐N assays, we ran four negative controls per 96 well plate and subtracted the mean OD of the negative controls from the OD. After subtraction of the negative controls, the manufacturer recommended OD thresholds for positive results were 0.198 for the IgG anti‐N assay [C] and 0.11 for the IgM anti‐N assay [D]. [A] and [B] The manufacturer recommended OD thresholds for positive results were 1 (0 for log(OD)) for the pan‐Ig anti‐N assay [A] and 0.19 for the pan‐Ig anti‐S1‐RBD assay [B]. [E] and [F] For the IgG and IgA anti‐S1 assays, the manufacturer recommended using two negative controls and two calibrator samples per plate and declaring samples positive if they have greater OD than the difference of the mean OD for the calibrator samples minus the mean OD for the negative control samples. The mean threshold was 0.33 for the IgG anti‐S1 assay [E] and 0.36 for the IgA anti‐S1 assay [F]. Blinding reported: Not stated Threshold predefined: yes, thresholds for positivity were supplied by the assay manufacturers. |
||
| Target condition and reference standard(s) | Reference standard: Testing for SARS‐CoV‐2 was performed either at Landspitali – The National University Hospital of Iceland (LUH) or deCODE using similar qPCR methods.
LUH: WHO recommended screening method: single probe pan‐screening assay for betacoronaviruses, followed by confirmatory measurements for all positive samples using an nCoV‐2019 specific assay
The broad betacoronavirus assay is based on probes for a conserved region of the E‐gene, whereas confirmatory testing assays were done using either nCoV2019 specific probes for the RdRp gene or the TaqMan™ Fast Virus 1‐step Master Mix, 2019‐nCoV Assay kits v1 from Thermo Fisher.
Samples in the E‐gene screening assay with Ct < 35 were considered strong positive and went for confirmatory testing using RdRp, whereas samples with Ct values between 35‐37 were considered weak positive and were confirmed using the TaqMan™ Fast Virus method.
Samples with Ct values from 37‐40 were classified as inconclusive and were tested again to confirm their status.
deCODE: SARS‐CoV‐2 screening was performed using qPCR assays in either a singleplex (Method 1) or a multiplex (method 2) format, respectively.
Method 1 uses the three probe TaqMan™ Fast Virus 1‐step Master Mix, 2019‐nCoV Assay kits v1 and 2019‐nCov control kit from Thermo Fisher.
Method 2 uses the TaqPath™ COVID‐19 CE‐IVD RT‐PCR kit from Thermo Fisher.
Results criteria for methods 1 and 2:
Samples with FAM™ dye Ct 7 values < 37 in at least two of three assays were classified as positive.
Samples with FAM™ dye Ct values between 37 and 40 were classified as inconclusive and their testing repeated.
If repeated testing gave the same result with at least two probes the sample was classified as positive.
If repeated testing gave positive results for only one probe the test was considered inconclusive and a new sample from the subject was requested.
Samples with undetected FAM™ dye Ct values or values equal to 40 in all three assays were classified as negative if the human RNaseP assay was positive (VIC™ dye Ct < 40). Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: [2a] Pre‐pandemic Samples used: [2a] Pre‐pandemic Timing of reference standard: [2a] Pre‐pandemic Blinded to index test: [2a] Pre‐pandemic, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1b] at least two weeks from qPCR diagnosis
[2] Not stated All patients received same reference standard: No Missing data: yes (see Table S3: only 1134/1215 and 437/472 samples tested with test [C]; only 1145/1215 and 434/472 samples tested with test [D]), results for tests [E] and [F] not reported, groups [1a], [2b], [2c], [2d]. [2e] and [2f] excluded from review Uninterpretable results: Not stated Indeterminate results: No intermediate results as per manufacturer's instructions Unit of analysis: [1b] Samples, but for persons with multiple samples, only the results for the most recently obtained sample were used. [2a] Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: New England Journal of Medicine Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Gudbjartsson 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Gudbjartsson 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Guedez‐Lopez 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute COVID‐19 Design: Multiple groups design to estimate sensitivity and specificity: [1] healthcare workers at Hospital Universitario La Paz, who attended the occupational health consultation for the first time between the 24th March and the 2nd of April referring symptoms compatible with COVID‐19 (n = 95) [1a] PCR+ for SARS‐COV‐2 (n = 55); [1b] PCR‐ for SARS‐COV‐2 (n = 40); [2] patients randomly selected who were admitted to the Emergency Department of the Hospital with positive RT‐qPCR or high clinical suspicion of COVID‐19 (n = 50); [2a] PCR+ for SARS‐COV‐2 (n = 46); [2b] PCR‐ for SARS‐COV‐2 (n = 4); [3] Pre‐pandemic patients (n = 20). Recruitment: [1] Healthcare workers at Hospital Universitario La Paz, who attended the occupational health consultation for the first time between the 24th March and the 2nd of April referring symptoms compatible with COVID‐19 [2] Randomly selected patients who were admitted to the Emergency Department of the Hospital with positive RT‐qPCR or high clinical suspicion of COVID‐19 [3] Randomly selected patients from 2018 Prospective or retrospective: [1] and [2] Prospective [3] Retrospective Sample size: 165 (101) Further detail: [1] Healthcare workers at Hospital Universitario La Paz, who attended the occupational health consultation for the first time between the 24th March and the 2nd of April referring symptoms compatible with COVID‐19 [2] Patients who were admitted to the Emergency Department of the Hospital with positive RT‐qPCR or high clinical suspicion of COVID‐19 [3] No further details |
||
| Patient characteristics and setting | Setting: [2a] patients attending accident and emergency department, ([2] 47/50 later hospitalised); [1a] Healthcare workers who attended the occupational health consultation for the first time ([1] 93/95 outpatients and 2/95 hospitalised); No separate data for PCR+ cases ([1a] and [2a]) Location: [1a] and [2a] Hospital Universitario La Paz (Madrid, Spain) Country: Spain Dates: [1a] and [2a] Serum samples collected between 8th March and 2nd April 2020 [1a] Attended occupational health consultation between 24th March and the 2nd of April 2020 Symptoms and severity: Only reported for all 95 and 50 patients with suspected COVID‐19, not for 50 and 46 rtPCR+ patients separately: [1] Hospitalised 2/95; pneumonia 12/95 [2] Hospitalised 47/50; pneumonia 48/50 Demographics: Only reported for all 95 and 50 patients with suspected COVID‐19, not for 50 and 46 rtPCR+ patients separately: [1] Healthcare workers; 74/95 female; median age 43 (range 21–79) years [2] ER admissions; 23/50 female; median age 50 (range 28–98) years Exposure history: [1a] Healthcare workers [2a] Unclear Non‐Covid group 1: [3] Pre‐pandemic patients Source: Pre‐pandemic (from 2018), Hospital Universitario La Paz (Madrid, Spain) Characteristics: Not reported (randomly selected patients, so possibly other diseases) Non‐Covid group 2: [1b] and [2b] Suspected COVID patients with negative PCR result Source: Hospital Universitario La Paz (Madrid, Spain), occupational health consultation or ER admissions; 8th of March and the 2nd of April 2020 Characteristics: [1b] 40 healthcare workers [2b] 4 patients admitted to ER department. Futher demographics only reported for all 95 and 50 patients with suspected COVID‐19, not for 40 and 4 rtPCR‐ patients separately: [1] Healthcare workers; 74/95 female; median age 43 (range 21–79) years [2] ER admissions; 23/50 female; median age 50 (range 28–98) years |
||
| Index tests | Test name: [A] Sienna 2019‐nCoV IgG/IgM Rapid Test [B] Wondfo, SARS‐CoV‐2 Antibody Test [C] Prometheus, 2019‐nCoV IgG/IgM Test Manufacturer: [A] T&D Diagnostics, Sienna, Halifax, Nova Scotia, Canada; [B] Wondfo, Luogang District, Guangzhou, China; [C] Prometheus Bio Inc., Zhejiang, China Antibody: [A] IgG and IgM separately; [B] Total antibody; [C] IgG and IgM separately Antigen target: Not reported Evaluation setting: Designed as POC. Unclear where it was performed Test method: Lateral flow assay (immunochromatographic assay) Timing of samples: [1] Median 5 (range 1–24) days pso [2] Median 11 (range 3–18) days pso [1a] and [2a] Early stage (first week): n = 41; intermediate stage (second week: n = 48; late stage (third week) n = 9 Samples used: Serum Test operator: Unclear. Interpretation was done by two observers. Definition of test positivity: After a short time (no longer than 20 min), the interpretation of the results was done by two observers based on appearance of a coloured band according to manufacturer's protocol. Weakly positive results (appearance of a blurred band) was considered as a positive result according to the manufacturer's protocol. Blinding reported: Unclear Threshold predefined: Yes, according to manufacturer’s protocol |
||
| Target condition and reference standard(s) | Reference standard: [1a] and [1b] RT‐qPCR (which detects N, S, E, Orf1ab and RdRp genes); no further details
RNA was extracted using an automated system and analysed using selected RT‐qPCR commercial kits routinely used for diagnosis of COVID‐19. Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated (all COVID suspects in [1] Median 0 (range 0–17) days before index test; All COVID suspects in [2] Median 4 (range 0–13) days before index test) Blinded to index test: Unclear for Group [1a] and [2a] Incorporated index test: No Definition of non‐COVID cases: 1b] and [2b] RT‐qPCR (which detects N, S, E, Orf1ab and RdRp genes); no further details RNA was extracted using an automated system and analysed using selected RT‐qPCR commercial kits routinely used for diagnosis of COVID‐19. [3] Pre‐pandemic Samples used: [1b] and [2b] Nasopharyngeal swabs [3] Pre‐pandemic Timing of reference standard: All COVID suspects [1] Median 0 (range 0–17) days before index test All COVID suspects in [2] Median 4 (range 0–13) days before index test; [3] Pre‐pandemic Blinded to index test: [1b] and [2b] Unclear [3] yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Group [1]: serum samples and nasopharyngeal swabs were collected at the same time in 82 patients, while in the other 13 patients, the average time since the nasopharyngeal swab collection and the serum extraction was 7.5 days; median 0 (range 0‐17) days. Group [2] in 48 patients, serum samples were taken days after the swab collection in an average time of 4.3 days, while in two patients, both samples were collected at the same time; median 4 (range 0‐13) days. All patients received same reference standard: Yes for [1] and [2]; no for [3] Missing data: 20 extra serum samples of randomly selected patients from 2018 not tested with Wondfo® test [B] due to lack of reagents 89 samples, which belonged to the group of healthcare workers, were tested with the three ICT assays, 28 samples (6 from the first and 22 from the second group of patients) were tested with Sienna® [A] and Wondfo® [B} and the other 28 samples from the second group of patients were tested only with Sienna [A]. Uninterpretable results: Not reported Indeterminate results: Weakly positive results (appearance of a blurred band) was considered as a positive result according to the manufacturer's protocol Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not reported Publication status: Published article Source: European Journal of Clinical Microbiology & Infectious Diseases Author COI: The authors declared that they had no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Guedez‐Lopez 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Guedez‐Lopez 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Haljasmagi 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Detection of acute and convalescent‐phase SARS‐CoV‐2 antibodies Design: Two‐group study estimating sensitivity and specificity: [1] PCR‐confirmed hospitalised Covid‐19 patients (n = 26) [2] Healthy controls without recent infection or Covid‐19 symptoms (fever or cough) for last month) (n = 26) Recruitment: Unclear Prospective or retrospective: Unclear Sample size: 52 (26) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: Tartu University Hospital Country: Estonia Dates: Not stated Symptoms and severity: Not described Demographics: Median age 62 y (range 33‐91 y); 18, 51% male [calculated from Suppl Tabl 1] Exposure history: Not described Non‐Covid group 1: Contemporaneous apparently healthy controls Source: Unclear Characteristics: Without recent infection or Covid‐19 symptoms (fever or cough) for previous month; age range 23‐54 years No further details |
||
| Index tests | Test name: Anti‐SARS‐CoV‐2 IgG ELISA Manufacturer: Euroimmun, Germany Antibody: IgG Antigen target: S1 Evaluation setting: Laboratory, laboratory Test method: ELISA Timing of samples: median 16 days (range 8 to 37 d) Day 8‐14 after infection: 9/26 (35%) Day 15‐21 after infection: 11/26 (42%) Day 22+ after infection: 6/26 (23%) Samples used: Plasma Test operator: Unclear Definition of test positivity: "According to the manufacturer’s recommendations, a ratio < 0.8 is considered negative, ≥ 0.8 and < 1.1 borderline, and ≥ 1.1 positive." Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: PCR; no further details Samples used: Unclear Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No Definition of non‐COVID cases: Unclear, no SARS‐CoV‐2 testing reported Samples used: Unclear, possibly none Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No (controls had different reference) Missing data: Nothing mentioned Uninterpretable results: Nothing mentioned Indeterminate results: Nothing mentioned Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: "The study was supported by the Estonian Research Council grants [#] (PP) and [#] (K.K.)" Publication status: Published letter Source: European Journal of Immunology Author COI: The authors declared no commercial or financial conflict of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Hamilton 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: A multi‐group study with three groups to estimate sensitivity and specificity: [1] patients with laboratory confirmed or clinically suspected COVID‐19 enrolled into DISCOVER study (n = 149): [1a] 114 PCR+ hospitalised COVID patients; [1b] 35 PCR‐, clinically diagnosed hospitalised COVID patients); [2] healthcare workers at North Bristol NHS Trust with laboratory confirmed COVID‐19 (n = 114); [3] pre‐pandemic respiratory infection controls (n = 20). Group [3] not eligible for our review as < 25 samples leaving a "Single‐group study to estimate sensitivity". Recruitment: [1] For the DISCOVER cohort, patients with confirmed (PCR+) and suspected (PCR‐) COVID‐19 were prospectively recruited and samples were taken on admission; [2] all healthcare worker who had received a positive PCR for SARS‐CoV‐2 at the PHE South West regional virology laboratory and went on to have antibody testing as part of as part of NHS England’s strategy for healthcare worker antibody testing; [3] pre‐pandemic plasma samples of patients with respiratory infection from an established tissue bank (pleural investigation database). Prospective or retrospective: Group [1] prospective; Group [2] unclear; Group [3] retrospective. Sample size: 283 (263) samples of which 263 (263) were eligible for our review Further detail: [1] Patients with COVID‐19 enrolled into the DISCOVER study (PCR+ or clinically diagnosed); [2] Healthcare worker at North Bristol NHS Trust with laboratory confirmed COVID‐19 (positive PCR for SARS‐CoV‐2) and antibody testing as part of as part of NHS England’s strategy for healthcare worker antibody testing; [3] Pre‐pandemic plasma samples of patients with respiratory infection from the Pleural Investigation Database. No further details on exclusions but [1] 18 excluded from DISCOVER cohort; [2] 52 healthcare workers excluded. |
||
| Patient characteristics and setting | Setting: [1] Hospitalised patients with COVID‐19 [2] Convalescent (majority had not been hospitalised) Location: [1] An NHS hospital in the UK (Southmead Hospital, Bristol) [2] North Bristol NHS Trust ‐ PCR performed in the PHE southwest regional virology lab Country: UK Dates: Not reported Symptoms and severity: [1] mixed severity (all hospitalised);13 patients (8%) intensive care; 15 patients (9%) died; [2] Predominantly mild COVID‐19 (aware of fewer than 5 hospitalised patients). Demographics: [1] Median age 58 years, sex not reported; [2] Age or sex not reported Exposure history: [1] Not reported [2] Healthcare workers Non‐Covid group 1: NA |
||
| Index tests | Test name: Abbott Architect SARS‐CoV‐2 IgG assay Manufacturer: Abbott Antibody: IgG Antigen target: Not reported Evaluation setting: Laboratory Test method: Not reported (Architect platform) Timing of samples: [1] Time was calculated from reported symptom onset date. Median time unclear. < 5 days pso: 18/149 5‐9 days pso: 57/149 10‐14 days pso: 28/149 15‐20 days pso: 14/149 > 20 days pso: 32/149 > 42 days pso: 30/149 [2] Timing was calculated from the time of the positive PCR test. Median time to test 45 days (range 32‐51 days) Samples used: EDTA plasma (either fresh or stored at −80 C) Test operator: Not reported Definition of test positivity: According to manufacturer protocol Blinding reported: Not reported Threshold predefined: Not reported |
||
| Target condition and reference standard(s) | Reference standard: [1a] and [2] RT‐PCR [1b] "Clinical diagnosis" Samples used: Not reported Timing of reference standard: Not reported Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: [1] Not reported [2] Median time 45 days (range 32‐51 days) post‐positive PCR: > 20 days: 114/114 > 42 days: 66/114 All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not reported Indeterminate results: Not reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not reported Publication status: Published letter Source: Journal of Infection Author COI: Not reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Harritshoej 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To diagnose convalescent SARS‐CoV‐2 infection Design: Multiple group study to assess sensitivity and specificity: [1] Convalescent patients with previous SARS‐CoV‐2 infection (n = 150); [2] Pre‐pandemic healthy controls (for determination of clinical sensitivity); [3] Pre‐pandemic patients with auto‐immune diseases and acute viral infections (for determination of cross‐reactivity). NB: the same set of PCR+ samples were tested across all assays, however different control sample sets were tested across assays and laboratories with minimum overlap, i.e. specificities were not from head to head comparisons. Recruitment: [1] A total of 3692 individuals were contacted via public secure mail and 639 persons responded. Only the first 150 consecutively collected serum samples from 3 May 2020 were chosen without any further selection. [2] and [3] Not stated Prospective or retrospective: Retrospective Sample size: Total sample size unclear (150 COVID cases) of which 123 samples with time pso > 21 days were eligible for our review Further detail: No further details on exclusions Inclusion: [1] convalescent patients in the Capital Region of Denmark with a confirmed SARS‐CoV‐2 NAAT result that were identified in the Danish Microbiology Database from February 2020 to April 2020 that were contacted and responded. [2] Archived plasma samples from regional pre‐COVID‐19 blood donations drawn during the influenza seasons of 2017–2018 and 2018–2019 [3] patients with unspecified auto‐immune diseases or archived local samples from patients with acute infections of cytomegalovirus (CMV) or Epstein‐Barr virus (EBV) or other acute viral respiratory infections (respiratory syncytial virus, influenza A and B viruses, and adenovirus) based on positive IgM serology obtained prior to January 2020 |
||
| Patient characteristics and setting | Setting: Convalescent samples (hospitalised and non‐hospitalised) Location: Patients were recruited from Capital Region of Denmark based on the Danish Microbiology Database. Country: Denmark Dates: Diagnosis was made from February 2020 to April 2020. Subsequently the samples were obtained from 3 May 2020 Symptoms and severity: Available for 149 patients only: No symptoms (n = 6, 4%); Mild (at home, well) (n = 37, 24.8%); Moderate (home, bedridden) (n = 75, 50.3%); Severe (hospitalised) (n = 2, 1.3%); Critical (assisted ventilation) (n = 29, 19.5%). Demographics: Median age (q1‐q3) = 54 (43‐64), range = 18‐83 years; male (n = 52), female (n = 97) [*1 missing value] Exposure history: Not reported Non‐Covid group 1: [2] Pre‐pandemic healthy controls (for determination of clinical sensitivity) Source: Archived plasma samples from regional pre‐COVID‐19 blood donations drawn during the influenza seasons of 2017–2018 and 2018–2019 Characteristics: Unclear (healthy blood donors) Non‐Covid group 2: [3] Pre‐pandemic patients with auto‐immune diseases and acute viral infections (for determination of cross‐reactivity) Source: Samples obtained before January 2020 Characteristics: Patients with unspecified auto‐immune diseases (n = 10 to 131) [10 samples were pooled and tested across all assays. The non‐pooled samples were tested in selected assays] Patients with acute infections of cytomegalovirus (CMV) or Epstein‐Barr virus (EBV) or other acute viral respiratory infections (respiratory syncytial virus, influenza A and B viruses, and adenovirus) based on positive IgM serology (n = 10 to 37) |
||
| Index tests | Test name: A] Wantai ELISA Total‐Ab assay; B] Ortho CD Vitros IgG assay; C] Siemens Atellica Total‐Ab assay; D] Roche Elecsys Total‐Ab assay; E] YHLO iFlash IgG or IgM assay; F] Abbott Architect IgG assay; G] Abbott Alinity IgG assay; H] Euroimmun ELISA IgG assay; I] Snibe Maglumi IgG/IgM assay; J] DiaSorin Liaison XL IgG assay; K] Wantai ELISA IgM assay; L] Ortho CD Vitros Total‐Ab assay; M] Siemens Vista Total‐Ab assay; Manufacturer: [A] and [K] Wantai, Beijing, China; [B] and [L] Ortho Clinical Diagnostics, Pencoed, UK; [C] and [M] Siemens Healthcare, Tarrytown, NY, USA; [D] Roche Diagnostics, Mannheim, Germany; [E] YHLO Biotechnology, Shenzhen, China; [F] Abbott, Abbott Park, IL, USA; [H] Euroimmun, Lubeck, Germany; [I] Snibe, Shenzhen, China; [J] DiaSorin, Saluggia, Italy; Antibody: [A, C, D, L, M] Total‐Ab; [B, E, F, G, H, J] IgG [E, I, K] IgM; Antigen target: [A, C, K, M] RBD; [D, F, G] N‐based; [E, I] N‐, S‐based [B,H.J.L] S‐based Evaluation setting: Designed and performed as laboratory Test method: [A, H, K] ELISA; [B, C, , E, F, G, I, J] CLIA Timing of samples: PSO: 0‐7 (n = 0); > 7‐14 (n = 7); > 14‐21 (n = 13); > 21‐42 (n = 49); > 42 (n = 71); Unknown (n = 10) Corrected data from corresponding author say 123 samples > 21 days pso. Samples used: [1] Serum; [2] Plasma; [3] Not stated Test operator: Experienced technicians from 16 participating laboratories Definition of test positivity: According to manufacturers' guidelines in all tests except CUH‐NOVO test, where ROC analysis (prioritising sensitivity) was used to define positivity: [A, B, K, L] Negative, < 1.1 (S/CO); Positive >= 1.1 (S/CO); [C, D] Negative, < 1.0 COI; positive >= 1.0 COI; [E] Negative, < 10 AU/mL (IgG), < 8 AU/mL (IgM); Positive >= 10 AU/mL (IgG), >= 8 AU/mL (IgM); [F, G] Negative, < 1.4 (S/C); Positive >= 1.4 (S/C); [H] Negative, < 0.8; Borderline, >= 0.8 to < 1.1; Positive >= 1.1; [I] Negative, < 1.0; Positive >= 1.0; [J] Negative, < 12 AU/mL; Equivocal, 12‐15 AU/mL; Positive >= 15 AU/mL; [M] Negative, < 1000 QU; Positive >= 1000 QU; Blinding reported: Unclear Threshold predefined: According to manufacturers' guidelines in all tests except CUH‐NOVO test, where ROC analysis (prioritising sensitivity) were used to define positivity |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 PCR, no further details Samples used: Not reported Timing of reference standard: Not reported Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: [2] and [3] Pre‐pandemic Samples used: [2] and [3] Pre‐pandemic Timing of reference standard: [2] and [3] Pre‐pandemic Blinded to index test: Yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Time from positive PCR:
0‐7 (n = 1);
> 7‐14 (n = 15);
> 14‐21 (n = 22);
> 21‐42 (n = 90);
> 42 (n = 21);
Unknown (n = 1). All patients received same reference standard: No Missing data: Yes (see numbers in Tabl 3) Uninterpretable results: Not reported Indeterminate results: Borderline results of Euroimmun ELISA [K] and DiaSorin Liaison XL [M] assays were interpreted as negative. Unit of analysis: Patients (for group [3], 10 samples were pooled and tested across all assays) |
||
| Comparative | |||
| Notes | Funding: The development of the CUH‐NOVO SARS‐CoV‐2 total‐Ab ELISA was financially supported by grants from the Carlsberg Foundation (CF20‐0045) and the Novo Nordisk Foundation (205A0063505). Publication status: Published article Source: Journal of Clinical Microbiology Author COI: R. B. Dessau reported personal fees from a Roche Diagnostics advisory board meeting in 2018 outside this work. All other authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Harritshoej 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [L].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Harritshoej 2021 [M].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Haselmann 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Detection of antibodies in primarily convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity: [1] PCR‐confirmed Covid‐19 cases, after end of quarantine (outpatient or home‐based, including 6 asymptomatic) or hospitalisation (including 5 ICU cases) (n = 26) [2] Atypical respiratory infection within last 3 months and PCR‐negative for SARS‐CoV‐2 or not tested (n = 11) [3] Other respiratory viral infection diagnosed (n = 1) [4] Chronic disease (e.g. auto‐immune disease) (n = 7) [5] Contact of a Covid‐19 patient but negative PCR and no symptoms (n = 2) [6] Healthy controls (n = 4) Recruitment: Unclear Prospective or retrospective: Prospective Sample size: 51 (26) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Mixed; home‐based or outpatient (quarantining patients); hospital inpatient Location: University Medical Center Mannheim, Medical Faculty Mannheim, University of Heidelberg Country: Germany Dates: Unclear Symptoms and severity: Cases only: Asymptomatic 6, 23%; mild 9, 35%; severe 8, 31% Treated at home 5, 19%; outpatient 13, 50%%; inpatient 3, 12%; ICU 5, 19% Immunocompromised 0 [from Suppl Tabl 2] Demographics: Total sample: Age: median 48.0 years, range 20‐73 years Sex: 18/51 male (68%) Exposure history: Unclear Non‐Covid group 1: [2] Atypical respiratory infection, PCR‐negative or not tested [3] Other respiratory viral infection diagnosed [4] Chronic disease (e.g. auto‐immune disease) [5] Asymptomatic Covid‐19 contact; PCR‐negative [6] Healthy controls Source: [2] Last 3 months [3] Not reported [4] Not reported [5] Not reported [6] Not reported Characteristics: [2] Not reported [3] Not reported [4] Diabetes type I (n = 1), Hashimoto disease (n = 2); rituximab Rx (n = 1); not reported (n = 3) [5] Not reported [6] Not reported |
||
| Index tests | Test name: [A] anti‐SARS‐CoV‐2 IgG ELISA (Lot:E200429AG) [B] EDI Novel Coronavirus COVID‐19 IgG ELISA (Lot:P745U) [C] Elecsys Anti‐SARSCoV‐2 (Lot:496298) Manufacturer: [A] Euroimmun, Germany [B] Epitope Diagnostics, United States [C] Roche, Germany Additional assays were evaluated but sample numbers did not meet review minimum. Antibody: All reported as IgG assays, however, Roche Elecsys is a Total Ab assay; author stated result was recorded as IgG only Antigen target: [A] S1 domain of viral spike‐protein [B] Full‐length nucleocapsid protein [C] Recombinant protein representing the nucleocapsid antigen Evaluation setting: [A] Laboratory [B] Laboratory [C] Laboratory Test method: [A] ELISA; [B] ELISA; [C] Electrochemiluminescence immunoassay (ECLIA) Timing of samples: Unclear; median 29 days pso (range 10‐47) Day 10‐14: 5, 19%; day 15‐21: 5, 19%; day 22‐28: 2, 8%; day 29‐35: 7, 27%; day 36‐42: 2, 8%; day > 42: 5, 19% [from Suppl Table 3] Samples used: Serum (n = 26) and plasma (n = 13) Test operator: Unclear Definition of test positivity: [A] Ratio of sample absorbance divided by calibrator absorbance ≥ 1.1 [B] The cut‐offs used for interpretation of assay results (positive, negative and borderline) have to be calculated according to a provided formula and therefore might differ in every run. [C] Cut‐off index ≥ 1.0 Blinding reported: Unclear Threshold predefined: [A] As manufacturer [B] Calculated according to a manufacturer formula and therefore might differ every run [C] As manufacturer |
||
| Target condition and reference standard(s) | Reference standard: qRT‐PCR; no further details Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: [2] qRT‐PCR (for some unreported number) [3] Unclear (likely qRT‐PCR as confirmed with other infection) [4] Unclear [5] qRT‐PCR [5] Unclear Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unknown All patients received same reference standard: Unclear Missing data: Nothing mentioned Uninterpretable results: Nothing mentioned Indeterminate results: Nothing mentioned Unit of analysis: Patients ‐ described in terms of patients and no suggestion of multiple samples per patient |
||
| Comparative | |||
| Notes | Funding: Authors reported no specific funding received Publication status: Published paper Source: Clinica Chimica Acta Author COI: The authors declared that they had no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Haselmann 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Haselmann 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Herroelen 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: A comparative analysis of analytical sensitivity was performed of seven commercial SARS‐CoV‐2 serology assays on 171 sera from 135 subjects with PCR‐confirmed SARS‐CoV‐2 infection, composed of 71 patients hospitalised for COVID‐19 pneumonia and 64 healthcare workers with paucisymptomatic infections. Specificity was verified on 57 pre‐pandemic samples.
2‐group study to estimate sensitivity and specificity for diagnosis of active disease/identification of previous disease Design: [1] subjects with PCR‐confirmed SARS‐CoV‐2 infection, composed of 71 patients hospitalised for COVID‐19 pneumonia and 64 healthcare workers with paucisymptomatic infections (n = 135 patients, 171 samples) [2] pre‐pandemic serum samples obtained from patients with PCR‐confirmed infection by other HCoV respiratory viruses (n = 7), other pathogens and viruses (n = 42) or presence of auto‐immune antibodies (n = 8) (n = 57 samples) [3] healthcare workers who presented WHO‐listed COVID‐19 symptoms but were not tested by PCR (n = 84, 84 samples) (this group not used in sensitivity/specificity analyses and not extracted. This group was also not mentioned in the published version) Recruitment: Unclear Prospective or retrospective: Unclear, but probably mixed. No informed consent from the hospitalised COVID‐19 patients (so likely serum samples already available = retrospective), but with written informed consent from participants with paucisymptomatic and suspected SARS‐CoV‐2 infections (so prospective). Pre‐pandemic samples = retrospective Sample size: 276 (135) patients with 312 (171) samples of which 228 (171) samples were included in this review (the excluded group [3] was not mentioned in the published version). Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatient and home‐quarantined Location: Inpatients at AZ Delta General Hospital in Roeselare, Belgium Country: Belgium Dates: Inpatients = March 1 to April 27, 2020 ; healthcare workers unclear Symptoms and severity: 71/135 = inpatients admitted for severe COVID‐19 pneumonia ; PCR‐confirmed SARS‐CoV‐2 infections; very high level of suspicion of COVID‐19 pneumonia on chest CT (CO‐RADS score = 5) 64/135 = healthcare workers with PCR‐confirmed SARS‐CoV‐2 infection with mild (n = 61) or no (n = 3) WHO‐listed COVID‐19 symptoms: myalgia (present in 62.5%), fever (60.9%), dry cough (56.2%), dyspnoea (40.6%), severe fatigue (35.9%), headaches (30.0%), loss of smell or taste (26.6%) or diarrhoea (18.8%). These patients were home‐quarantined without the need for hospitalisation. Demographics: Inpatients = 48 males (median age 65 years, IQR 53‐80) and 23 females (median age 79 years, IQR 67‐86) Health care workers = Not reported Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic cross‐reactivity Source: Pre‐pandemic Characteristics: PCR‐confirmed infection by other HCoV respiratory viruses (n = 7; HCoV 229E, n = 1; HCoV HKU1, n = 3; HCoV OC43, n = 2; HCoV OC43 + adenovirus, n = 1); other pathogens and viruses (n = 42); presence of auto‐immune antibodies (n = 8) |
||
| Index tests | Test name: [A] COVID‐19 IgG/IgM Rapid Test [B] Innovita 2019‐nCoV Ab Test [C] Wantai SARS‐COV‐2 Ab ELISA [D] Anti‐SARS‐CoV‐2 IgG and IgA assays [E] Anti‐SARS‐ CoV‐2‐NCP (IgG) assay [F] Elecsys Anti‐SARS‐CoV‐2 assay for Cobas e601 module [G] LIAISON SARS‐CoV‐2 S1/S2 IgG Manufacturer: [A] Zhejiang Orient Gene Biotech Co., Ltd., Zhejiang, China [B] Innovita Biological Technology Co., Ltd., Beijing, China [C] Beijing Wantai Biological Pharmacy Enterprise, Beijing, China [D] EUROIMMUN AG (a PerkinElmer Company, Luebeck, Germany) [E] EUROIMMUN AG (a PerkinElmer Company, Luebeck, Germany) [F] Roche Diagnostics, Basel, Switzerland [G] DiaSorin, Saluggia, Italy Antibody: [A] IgM and IgG antibodies to recombinant N‐ and S‐proteins [B] IgM and IgG antibodies to undisclosed SARS‐CoV‐2 epitopes [C] all antibody isotypes (IgM, IgA, IgG) against the RBD domain of the S1‐protein [D] IgA and IgG antibodies against the S1‐protein [E] IgG to the N‐protein [F] all antibody isotypes (IgM, IgA, IgG) against the N‐protein [G] IgG antibodies against S1/S2 proteins Antigen target: [A] recombinant N‐ and S‐proteins [B] undisclosed SARS‐CoV‐2 epitopes [C] RBD domain of the S1‐protein [D] S1‐protein [E] N‐protein [F] N‐protein [G] S1/S2 proteins Evaluation setting: [A] POC, assessed in laboratory [B] POC, assessed in laboratory [C] Laboratory test, assessed in laboratory [D] Laboratory test, assessed in laboratory [E] Laboratory test, assessed in laboratory [F] Laboratory test, assessed in laboratory [G] Laboratory test, assessed in laboratory Test method: [A] solid phase immunochromatographic assay [B] colloidal gold lateral flow assay [C] ELISA double‐antigen sandwich immunoassay [D] indirect ELISA [E] indirect ELISA [F] Electrochemiluminescence immunoassay (CLIA) [G] Electrochemiluminescence immunoassay (CLIA) Timing of samples: Inpatients = Serum samples ranged from 0 to 39 days after patient‐reported symptom onset. Healthcare workers = Serum samples ranged from 11 to 54 days after patient‐reported symptom onset < 10 days pso: 53/171 10‐20 days pso: 42/171 > 20 days pso: 76/171 Samples used: [A]‐[G] Serum Test operator: [A]‐[G] Laboratory personnel Definition of test positivity: [A] considered positive if a line was observed for either IgM, IgG or both [B] considered positive if a line was observed for either IgM, IgG or both [C] Samples with a cut‐off ratio (absorbance of the sample at 459 nm divided by 0.19 higher than 0.9 were considered positive, classifying gray zone results 0.9‐1.1 as positive. [D] cut‐off = 0.8 units, classifying gray zone results 0.8‐1.1 units as positive [E] cut‐off = 0.8 units, classifying gray zone results 0.8‐1.1 units as positive [F] cut‐off = 1 Cut‐off Index [G] cut‐off = 12 AU/mL, classifying gray zone results between 12 and 15 AU/mL as positive Blinding reported: Unclear Threshold predefined: All serology assays were used according to the manufacturers’ protocol using the cut‐offs specified. |
||
| Target condition and reference standard(s) | Reference standard: PCR: Allplex 2019‐nCoV assay (Seegene, Seoul, Korea) for E/N/RdRP genes on nasopharyngeal swab Threshold not reported Samples used: nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: Done prior index test Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: yes (specificity results for most tests for only 56 of 57 samples, also missing samples for sensitivity) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by a private donation by board members of Fagron (Nazareth, Belgium), a healthcare company, to RADar, the teaching and education initiative of AZ Delta General Hospital, to be used as unconditional research grant for data collection, collaborative collaboration and open access publication. The sponsor had no influence on the study design, data interpretation and drafting of the manuscript. Publication status: Pre‐print (not peer reviewed); now published Source: medRxiv preprint doi: https://doi.org/10.1101/2020.06.09.20124719 Journal (American Journal of Clinical Pathology) Author COI: The authors declared no conflict of interest. Not stated in published version |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Herroelen 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Herroelen 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Herroelen 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Herroelen 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Herroelen 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Herroelen 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Hoffman 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: (1) PCR‐confirmed COVID‐19 patients or convalescents (n = 29) (2) healthy volunteers with no known history of SARS‐CoV‐2 infection/COVID‐19 (n = 24), (3) pre‐pandemic anonymous blood donor sera from healthy adults (n = 80) [also reported 20 serum samples from babies (6–12 months) collected before or during 2018; not included in review] Recruitment: Not reported Prospective or retrospective: Not reported Sample size: 133 (29) Further detail: no more details available |
||
| Patient characteristics and setting | Setting: Unclear Location: Not reported; author institution is Uppsala University Hospital, Uppsala Country: Sweden Dates: not reported Symptoms and severity: not reported Demographics: not reported Exposure history: not reported Non‐Covid group 1: (2) healthy volunteers Source: unclear; appeared to be contemporaneous Characteristics: Not reported Non‐Covid group 2: Pre‐pandemic serum samples Source: Before or during 2018; Uppsala Biobank Characteristics: Not reported; healthy |
||
| Index tests | Test name: COVID‐19 IgG/IgM Rapid Test Cassette [GCCOV‐402a, Lot: 2003242] Manufacturer: Zhejiang Orient Gene Biotech Co Ltd, Huzhou, Zhejiang, China Antibody: SARS‐CoV‐2‐specific antibodies IgG/IgM Antigen target: not stated Evaluation setting: POC test; evaluated in laboratory Test method: LFA Timing of samples: 9‐17 days pso (n = 10); 18‐29 days (n = 19) Samples used: Capillary blood samples or serum Test operator: not reported Definition of test positivity: Visible line Blinding reported: not reported Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: PCR‐confirmed ‐ no further details Samples used: not reported Timing of reference standard: not reported Blinded to index test: Not reported; likely conducted first Incorporated index test: No Definition of non‐COVID cases: [2] No reference standard [3] Pre‐pandemic sera Samples used: Serum Timing of reference standard: [2] Not reported [3] 2018 Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: not reported All patients received same reference standard: No Missing data: none reported Uninterpretable results: none reported Indeterminate results: none reported Unit of analysis: patients (1 sample per patient) |
||
| Comparative | |||
| Notes | Funding: This work was supported by the Swedish Research Council (VR, grant numbers 2016‐02596, 2017‐05807 and 2018‐
02569). The rapid tests that have enabled this study were donated to us by the Swedish company Noviral AB (organization number: 559175‐7942). Publication status: published paper (published online 14 April 2020) Source: Infection Ecology & Epidemiology Author COI: The authors declared no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Hogan 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID cases (51 samples) [2] Non‐COVID samples (62 samples), current PCR‐ Recruitment: Between April 15 and June 1, 2020, residual serum samples ordered for routine medical management of inpatients at the University of Kansas Hospital Samples were collected for two groups: [1] serum samples from patients who tested positive for SARS‐CoV‐2 by an RT‐PCR assay; [2] serum samples from randomly selected patients who had tested negative for SARS‐CoV‐2 by an RT‐PCR assay within 48 hours prior to collection. All available serum samples from PCR‐positive patients and randomly selected PCR‐negative patients that were older than 18 years with adequate residual volume for parallel testing were included. Prospective or retrospective: Retrospective Sample size: 113 (51) of which 79 (17) were eligible for our review. Further detail: [1] Hospital inpatients who tested positive for SARS‐CoV‐2 by an RT‐PCR assay, [2] Hospital inpatients who had tested negative for SARS‐CoV‐2 by an RT‐PCR assay within 48 hours prior to collection. [1] and [2] older than 18 years with adequate residual volume for parallel testing |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: University of Kansas Hospital, Kansas City Country: Kansas, USA Dates: Between April 15 and June 1, 2020 Symptoms and severity: Not stated (all hospitalised, likely "greater average patient acuity") Demographics: 0‐6 days post‐PCR+ (n = 17); 71% (12/17) female; median age 71 (IQR 52‐77) years 7‐13 days post‐PCR+ (n = 17); 53% (9/17) female; median age 64 (IQR 42‐74) years 14+ days post‐PCR+ (n = 17); 53% (9/17) female; median age 64 (IQR 55‐69) years Exposure history: Not stated Non‐Covid group 1: [2] Current non‐COVID patients with other diseases Source: University of Kansas Hospital, Kansas City, Kansas (USA) between April 15 and June 1, 2020 Characteristics: Hospital inpatients, adults: 63% (39/62) female; median 53 (IQR 35‐70) years; patient samples representative of the current local circulating viruses among individuals with healthcare contacts |
||
| Index tests | Test name: [A] Liaison SARS‐CoV‐2 S1/S2 IgG [B] Elecsys anti‐SARS CoV‐2 total antibody [C] Access SARS‐CoV‐2 IgG Manufacturer: [A] DiaSorin S.p.A., Saluggia, Italy [B] Roche Diagnostics, Rotkreuz, Switzerland [C] Beckman Coulter, Inc., Minnesota, USA Antibody: [A] IgG [B] Total antibodies [C] IgG Antigen target: [A] S1 and S2 subunits of the spike‐protein [B] N‐protein [C] receptor binding domain (RBD) of the S1‐protein Evaluation setting: Lab tests performed in lab Test method: [A] indirect CLIA [B] ECLIA [C] CLIA Timing of samples: 1‐45 days overall (median: 9) post‐PCR+: 0‐6 days (median 5) post‐PCR+: 17/51 7‐13 days (median 9) post‐PCR+: 17/51 14+ days (median 18) post‐PCR+: 17/51 Combined samples were represented by the day farthest from the patient’s positive PCR test. Samples used: Residual serum samples were centrifuged, aliquoted, and frozen at ‐30 °C for 1 to 46 days. Samples were sequentially thawed and maintained at 2‐8 °C for < 14 days prior to testing. Test operator: Clinical Laboratory Scientists Definition of test positivity: [A] Reported in arbitrary units per millilitre (AU/mL). A result of < 15 was considered negative while a result of ≥ 15.0 was considered positive. [B] Results were expressed as a cut‐off index (COI). A result of < 1.0 was considered non‐reactive while a result of ≥ 1.0 was considered reactive. [C] The light signal was compared to the cut‐off value and was expressed as a signal to cut‐off ratio (S/CO). A result of < 0.8 was interpreted as non‐reactive while a result of ≥ 1.0 was considered reactive. Results between 0.8 and 1.0 (inclusive) were considered equivocal. For the purposes of analysis, equivocal results were treated as negative. Blinding reported: No, clinical Laboratory Scientists were not specifically blinded to the clinical status or PCR results of the patients. Threshold predefined: [A], [B], [C] yes, according to manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: FDA EUA RT‐PCR assay (Abbott RealTime SARS‐CoV‐2 assay (Abbott Diagnostics Inc, Scarborough, ME), performed on the Abbott m2000 instrument, or the Simplex COVID‐19 Direct assay (DiaSorin Molecular LLC, Cypress CA), following manufacturer’s instructions Samples used: Nasopharyngeal swabs collected in either UTM or PBS Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: FDA EUA RT‐PCR assay (Abbott RealTime SARS‐CoV‐2 assay (Abbott Diagnostics Inc, Scarborough, ME), performed on the Abbott m2000 instrument, or the Simplex COVID‐19 Direct assay (DiaSorin Molecular LLC, Cypress CA), following manufacturer’s instructions) Samples used: Nasopharyngeal swabs collected in either UTM or PBS Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1] 1‐45 days overall (median: 9) post‐PCR+:
0‐6 days (median 5) post‐PCR+: 17/51
7‐13 days (median 9) post‐PCR+: 17/51
14+ days (median 18) post‐PCR+: 17/51
Combined samples were represented by the day farthest from the patient’s positive PCR test. All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: [C] For the purposes of analysis, equivocal results were treated as negative. 1 equivocal result for 0‐6 days post‐PCR+ Unit of analysis: Each sample represented a unique patient. |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. Publication status: Pre‐print (not peer‐reviewed) Source: medRxiv preprint Author COI: None declared. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Hogan 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Hogan 2020a [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Horber 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (186 samples from 58 patients) [2] Non‐COVID samples (n = 123) [2a] Pre‐pandemic samples collected before December 2019 (n = 88) [2b] Samples with potential cross‐reactive antibodies (n = 35) Recruitment: Not stated ([1] Routine blood samples of hospitalised COVID patients; [2a] Pre‐pandemic intensive care patients; [2b] Prospective or retrospective: [1] and [2b] Unclear [2a] Retrospective Sample size: 309 (186) samples of which 255 (132) were eligible for our review Further detail: [1] Hospitalised rt‐PCR‐confirmed COVID‐19 patients; [2a] Pre‐pandemic (before December 2019) intensive care patient; [2b] Not stated (samples with other acute viral and bacterial or fungal infections not suspicious of COVID‐19). |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: University Hospital Tübingen, Tübingen, Germany Country: Germany Dates: Not stated Symptoms and severity: All hospitalised COVID‐19 patients (most of the patients were critically ill and treated on the intensive care unit) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic, other diseases Source: [2a] Intensive care patients before December 2019 Characteristics: Intensive care patients (n = 88). Non‐Covid group 2: [2b] Cross‐reactivity Source: Patients with laboratory‐confirmed acute infections, time not stated. Possibly all from University Hospital Tübingen Characteristics: Potential cross‐reactive antibodies (n = 35): acute infections with influenza A virus (n = 5), human respiratory syncytial virus (n = 1); common cold coronaviruses (NL63: n = 1; HKU‐1 +NL63: n = 1; NL63 + 229E: n = 1) IgM antibodies against human cytomegalovirus (n = 5) and varicella zoster virus (n = 2), samples from patients with respiratory symptoms not suspicious of COVID‐19 disease (n = 11), samples containing antibodies (n = 6) against chlamydia pneumoniae (IgG, IgA and/or IgM) or candida albicans (IgG and/or IgA); samples positive for rheumatoid factor (n = 2) |
||
| Index tests | Test name: [A] SARS‐CoV‐2 Total (COV2T) [B] Elecsys anti‐SARS‐CoV‐2 [C] SARS‐CoV‐2‐ELISA (IgG) Manufacturer: [A] Siemens Healthineers [B] Roche Diagnostics [C] Euroimmun Antibody: [A] Total antibodies (IgG and IgM) [B] Antibodies (including IgG) [C] IgG Antigen target: [A] S1‐protein Receptor Binding domain (RBD) [B] N‐protein [C] S1 spike‐protein Evaluation setting: Lab tests performed in lab Test method: [A] CLIA [B] ECLIA [C] ELISA Run on fully automated platforms Timing of samples: Median time between positive PCR result and blood sample collection was 19 days (interquartile range: 12–29 days) 0‐6 days post‐PCR+: 23/186 7‐13 days post‐PCR+: 31/186 14+ days post‐PCR+: 132/186 Samples used: Plasma Test operator: Institute for Clinical Chemistry and Pathobiochemistry at the University Hospital Tübingen Definition of test positivity: [A] Cut‐off index (COI); < 1.0 negative; >= 1.0 positive; optimised: COI > 0.75 [B] Cut‐off index; < 1.0 negative; >= 1.0 positive; optimised: COI > 0.095 [C] Ratio; < 0.8: negative; 0.8– < 1.1: borderline; ≥ 1.1: positive; optimised: > 0.958 Blinding reported: Not stated Threshold predefined: Results of antibody measurements were evaluated according to the manufacturers’ cut‐off indices or ratios as positive or negative for the Roche and Siemens assays and as positive, borderline or negative for the Euroimmun assay. The study also used optimised cut‐offs (receiver operating characteristic (ROC) curve analysis and Youden index were used to identify optimised thresholds (cut‐off indices)). |
||
| Target condition and reference standard(s) | Reference standard: rt‐PCR, threshold not stated Samples used: Oro‐ and/or nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: [2a] Pre‐pandemic [2b] Not stated, none? Other laboratory confirmed acute infections Samples used: [2a] Pre‐pandemic [2b] Not stated, none? Timing of reference standard: [2a] Pre‐pandemic [2b] Not stated Blinded to index test: yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Median time between positive PCR result and blood sample collection was 19 days (interquartile range: 12–29 days). All patients received same reference standard: unclear/no for [2b] Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: yes (for test [C], 2/23 sampled 0‐6 days post‐PCR+ and 3/35 cross‐reaction samples borderline). For analyses, treated once as negative and once as positive Unit of analysis: [1] Samples [2] Patients |
||
| Comparative | |||
| Notes | Funding: None declared. Publication status: Published paper Source: Clinical Chemistry & Laboratory Medicine Author COI: Authors stated no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Horber 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Horber 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Hu 2020a.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity for detecting active or prior infection Confirmed COVID‐19 patients (211) Recruitment: NR; likely retrospective. Consecutive or otherwise NR | ||
| Patient characteristics and setting | Setting: inpatient Location: Chongqing Three Gorges Central Hospital, Chongqing Country: China Dates: 23 January‐3 March | ||
| Index tests | Test name: Magnetic Chemiluminescence Enzyme Immunoassay (MCLIA) kit Manufacturer: Bioscience Co., Ltd (Chongqing, China) Ab targets: IgM, IgG Antigens used: N and S (nucleoprotein and a peptide from the SARS‐CoV‐2 S‐protein) | ||
| Target condition and reference standard(s) | Reference standard for cases: Chinese CDC guidelines (Trial Version 6); included RT‐PCR Samples used: NR Timing of reference standard: unclear; appeared that repeat PCR undertaken during hospitalisation; 74/211 met discharge criteria during study period (normal temperature, significantly improving respiratory symptoms and chest radiology plus 2 repeat negative PCRs with ≥ 1‐day interval) Was it blind to index test: unclear | ||
| Flow and timing | Time interval between index and reference tests: NR Results presented by time period: yes All participants received the same reference standard: yes Missing data: none described; however text stated 993 samples but only 409 reported for IgM and 507 for IgG Uninterpretable results: none described | ||
| Comparative | |||
| Notes | Funding: funded by Chongqing Education Board “new coronavirus infection and prevention” emergency scientific research project (KYYJ202006YYJ202006). Chongqing Science and Technology Bureau “new crown pneumonia epidemic emergency science and technology special” the fourth batch of projects. Famous teacher project of Chongqing talent plan Publication status: preprint Source: medRxiv Study author COI: none declared | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Hu 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase Covid‐19 infection Design: Single or multi‐group study estimating sensitivity and specificity (design unclear), including: participants who visited Huangshi Central Hospital during specified time period, described as [1] PCR‐positive COVID‐19 group (n = 68) [2] Suspected Covid group (PCR‐negative but with fever and other respiratory symptoms) (n = 9) [3] Group with other diseases and negative PCR (n = 101) Study authors considered group [2] and [3] as disease‐negative. Recruitment: Unclear Prospective or retrospective: Unclear; presumed retrospective given time period from early in the pandemic Sample size: 178 (68) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear; no details Location: Huangshi Central Hospital, Hubei Province Country: China Dates: January and February 2020 Symptoms and severity: Unclear Demographics: Age: range 30 years to 90 years; no mean available Sex: 36/68 (53%) male Exposure history: Not stated Non‐Covid group 1: Suspected group Source: Characteristics: Age: range 2 months to 64 years; no mean available Sex: 7/9 (78%) male Non‐Covid group 2: Negative group Source: Characteristics: Age: range 2 years to 94 years; no mean available Sex: 48/101 (48%) male |
||
| Index tests | Test name: [A] YHLO SARS‐COV‐2 IgM [B] YHLO SARS‐COV‐2 IgG Manufacturer: [A] [B] Shenzhen YHLO Biotech Co. Ltd. Antibody: [A] IgM, [B] IgG Antigen target: [A] [B] Nucleocapsid protein, spike‐protein Evaluation setting: Laboratory Test method: Chemiluminescence immunoassay (CLIA) Timing of samples: < 7 days after symptom onset: 12/68 (18%) 7‐14 days after symptom onset: 25/66 (37%) > 14 days after symptom onset: 31/68 (46%) Samples used: Serum Test operator: Unclear Definition of test positivity: Cut‐off value for positive was 10 arbitrary units/mL Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR detecting open reading frame lab (ORFlab) and nucleocapsid protein (N) genes Samples used: Not stated Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No Definition of non‐COVID cases: As for cases; unclear if single or > 1 negative PCR result Samples used: Unclear Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No; possibility that serology may have influenced final diagnosis of 9 suspect cases |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: Nothing mentioned Uninterpretable results: Nothing mentioned Indeterminate results: Nothing mentioned Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article Publication status: Pre‐print (not peer reviewed) Source: medRxiv Author COI: The authors confirmed that there were no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Hu 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Hubbard 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection, current convalescent‐phase infection, prior infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (216 samples, 43 patients) [1a] Hospital inpatients (193 samples from 20 patients) [1b] Convalescent plasma donors (23 patients) [2] Non‐COVID samples (385 samples) [2a] Pre‐pandemic samples (170 samples) [2b] Hospital inpatients with negative COVID molecular diagnostic test (215 samples from 155 patients) Recruitment: [1a] and[2b] Remnant serum and lithium heparin plasma specimens collected for clinical purposes from hospitalised patients with and without confirmed SARS‐CoV‐2 infection [1b] Paired serum and lithium heparin plasma specimens collected were collected at least 14 days after resolution of symptoms. [2a] Remnant serum and lithium heparin plasma specimens collected for clinical purposes between September 2017 and June 2019 Prospective or retrospective: [2a] Retrospective [1a], [1b] and [2b] Unclear, prospective? (no longer than 3 weeks frozen storage of samples) Sample size: 601 (216) samples Further detail: [1a] Hospital inpatients with previously documented positive SARS‐CoV‐2 molecular diagnostic result [1b] at least 14 days after resolution of symptoms from individuals with a documented positive SARS‐CoV‐2 molecular diagnostic result [2a] Remnant serum and plasma specimens collected for clinical purposes between September 2017 and June 2019 [2b] Hospital inpatients with negative SARS‐CoV‐2 molecular diagnostic result on the same day or 1 day prior to serum or plasma specimen collection |
||
| Patient characteristics and setting | Setting: [1a] Hospital inpatients [1b] Convalescent plasma donors Location: Dartmouth‐Hitchcock Health System, Lebanon, NH Country: New Hampshire, USA Dates: Not stated Symptoms and severity: [1a] All hospitalised [1b] All convalescent (at least 14 days after resolution of symptoms) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic Source: Remnant serum and lithium heparin plasma specimens collected for clinical purposes between September 2017 and June 2019. Possibly all from the same hospital lab (Dartmouth‐Hitchcock Health System, Lebanon, NH)? Characteristics: Not stated Non‐Covid group 2: [2b] Current non‐COVID Source: Hospital inpatients with negative SARS‐CoV‐2 molecular diagnostic result; Dartmouth‐Hitchcock Health System, Lebanon, NH; time not stated Characteristics: Hospital inpatients so possibly other diseases |
||
| Index tests | Test name: [A] Abbott SARS‐CoV‐2 IgG assay [B] Roche Elecsys Anti–SARS‐CoV‐2 assay Manufacturer: [A] Abbott Laboratories Diagnostics Division [B] Roche Diagnostics Antibody: [A] IgG [B] Total antibodies Antigen target: [A] N‐protein [B] N‐protein Evaluation setting: Lab tests performed in lab Test method: [A] Chemiluminescence, on an Architect i1000 instrument [B] Chemiluminescence; on a Cobas e801 instrument Timing of samples: [1a] Not stated (could work out from Fig 1 and Tabl 1; 14+ days post‐PCR+: 10/193) [1b] Convalescent (14+ days symptom‐free) Samples used: Remnant serum and plasma samples, removed from refrigerated storage within 7 days of collection, aliquoted into sealed plastic tubes and frozen at ‐80C until further use no longer than 3 weeks in frozen storage for [1] and [2b], or at ‐20C for [2a] Test operator: Lab personnel Definition of test positivity: [A] Results were reported in a qualitative fashion with a signal/calibrator (S/C index) of < 1.4 interpreted as negative and >= 1.4 interpreted as positive. [B] cut‐off index (COI) of < 1.0 interpreted as nonreactive and >= 1.0 interpreted as reactive Blinding reported: Not stated Threshold predefined: Not stated, possibly yes |
||
| Target condition and reference standard(s) | Reference standard: [1a] All patients included in this study were tested for SARS‐CoV‐2 infection with one of 3 molecular diagnostic methods: the CDC format laboratory developed test, the Abbott RealTime SARS‐CoV‐2 m2000 assay, or the Diasorin Simplexa COVID‐19 assay. [1b] SARS‐CoV‐2 molecular diagnostic result (as [1a]?) Samples used: Not stated Timing of reference standard: [1a] ‐1 to 12 days pso [1b] Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: [2a] Pre‐pandemic [2b] All patients included in this study were tested for SARS‐CoV‐2 infection with one of 3 molecular diagnostic methods: the CDC format laboratory developed test, the Abbott RealTime SARS‐CoV‐2 m2000 assay, or the Diasorin Simplexa COVID‐19 assay. Samples used: [2a] Pre‐pandemic [2b] Not stated Timing of reference standard: [2a] Pre‐pandemic [2b] Not stated Blinded to index test: yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [2b] Reference standard on same day or one day before index test All others not stated (could get numbers for [1a] from Fig 1 and Tabl 1) All patients received same reference standard: no Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: NA as no intermediate range Unit of analysis: [1a] Samples [1b] Patients [2a] Not stated [2b] Patients |
||
| Comparative | |||
| Notes | Funding: No sponsor was declared. Publication status: Published paper Source: Journal of Applied Laboratory Medicine Author COI: Employment or Leadership: None declared. Consultant or Advisory Role: M.A. Cervinski, Roche Diagnostics. Stock Ownership: None declared. Honoraria: None declared. Research Funding: None declared. Expert Testimony: None declared. Patents: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Hubbard 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Imai 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase COVID‐19 Design: Two‐group study estimating sensitivity and specificity: [1] Patients with laboratory‐confirmed COVID‐19, including 74 symptomatic and 38 asymptomatic (total n = 112) [2] Pre‐pandemic samples from patients at Saitama Medical University Hospital (n = 48) Recruitment: Not reported Prospective or retrospective: Not reported Sample size: 160 (112) Further detail: NO further details |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Self‐Defense Forces Central Hospital and Saitama Medical University Hospital Country: Japan Dates: [1] February 11 to March 31, 2020 Symptoms and severity: 74, 66% symptomatic (fever, cough, nasal discharge, diarrhoea, malaise, dyspnoea, tachypnoea, peripheral capillary oxygen saturation < 93%, and need for oxygen therapy) Demographics: Median age (IQR) 67 y (45–74 y); 64 (57.1%) male Exposure history: Not reported Non‐Covid group 1: Pre‐pandemic samples Source: Saitama Medical University Hospital; April to October 2019 Characteristics: not stated |
||
| Index tests | Test name: One Step Novel Coronavirus (COVID‐19) IgM/IgG Antibody Test Manufacturer: Artron, Burnaby, Canada Antibody: IgM, IgG Antigen target: not stated Evaluation setting: POC; evaluation setting not reported (appeared to be lab‐based) Test method: LFA Timing of samples: day of admission and during hospitalisation (within 1 week (n = 90), 1–2 weeks (n = 25), and > 2 weeks after onset (n = 24) Samples used: serum Test operator: Not stated Definition of test positivity: the presence of both the control line and the IgM or IgG antibody line indicated a positive result for IgM or IgG antibody, respectively. Blinding reported: no Threshold predefined: Yes, according to manufacturer’s instructions. |
||
| Target condition and reference standard(s) | Reference standard: RT‐qPCR for SARS‐CoV2 Samples used: pharyngeal and nasopharyngeal swabs Timing of reference standard: [1] Of the 74 symptomatic patients, median time from onset to admission was 5 days (IQR, 2–7 days); [2] Of the 38 asymptomatic patients, median time from the first RT‐qPCR–positive day to admission was 5 days (IQR, 3–6 days). Blinded to index test: Yes Incorporated index test: no Definition of non‐COVID cases: Pre‐pandemic Samples used: serum Timing of reference standard: NA Blinded to index test: yes Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: unclear All patients received same reference standard: yes Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples; reported in two time periods |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or nonprofit sectors Publication status: Published Source: Journal of Clinical Virology Author COI: The authors declared that they had no conflicts of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Jaaskelainen 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase infection Design: Multi‐group study to assess sensitivity and specificity: [1] COVID‐19‐positive patients (n = 47) [2] Non‐COVID‐19 (n = 37 patients) [3] Probable COVID‐19 patients (according to WHO definition) who had tested negative for SARS‐CoV‐2 by NAT (n = 13) Recruitment: Unclear Prospective or retrospective: Restrospective Sample size: 97(47) but 84(37) could be used for our review Further detail: Inclusion: [1] Diagnosed with COVID‐19 by RT‐PCR [2] Patients diagnosed with seasonal human coronaviruses or other respiratory viruses or viral infections, either pre‐pandemic or RT‐PCR‐negative for SARS‐CoV‐2 Excluded: [1][2] Not stated |
||
| Patient characteristics and setting | Setting: Hospital Location: Helsinki University Hospital Country: Finland Dates: Not stated Symptoms and severity: Mild symptoms 9/37, moderate symptoms 15/37, severe symptoms 13/37 Demographics: [1] 23/40 (57.5%) males, median age 56 years (range: 24–77 years) Exposure history: Not stated Non‐Covid group 1: Non‐COVID‐19 patients Source: 2019‐2020, source not stated Characteristics: 15/37 (%) male, median age: 53 years (range: 5‐87 years). Patients diagnosed with seasonal human coronaviruses (OC43, NL63, 229E) or other respiratory viruses by nucleic acid tests (n = 11) and samples from patients who had been diagnosed as having adenovirus, enterovirus, influenza A, influenza B, parainfluenza, or respiratory syncytial (RSV) virus infections, through routine IgG antibody testing in 2019 (n = 26). Samples from 2019 were assumed to be from SARS‐CoV‐2 negative patients, while samples obtained in 2020 were from patients who had been tested for SARS‐CoV‐2 nucleic acid and found negative. |
||
| Index tests | Test name: SARS‐CoV‐2 IgG and IgA ELISA Manufacturer: Euroimmun, Lübeck, Germany Antibody: IgG, IgA Antigen target: S1‐protein Evaluation setting: Laboratory Test method: ELISA Timing of samples: 1‐23 days pso Samples used: Serum Test operator: Not stated Definition of test positivity: ratio < 0.8 was considered negative, ≥ 0.8 and < 1.1 inconclusive and ≥ 1.1 positive Blinding reported: Not stated Threshold predefined: Probably (commercial test), ratio < 0.8 was considered negative, ≥ 0.8 and < 1.1 inconclusive and ≥ 1.1 positive. |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: nasopharyngeal swabs Timing of reference standard: not stated Blinded to index test: yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic or RT‐PCR Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: 0‐14 days All patients received same reference standard: no Missing data: [1] One patient with a single sample taken before symptom onset was not further investigated. Uninterpretable results: not stated Indeterminate results: Indeterminate results classed as positive for analysis Unit of analysis: samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Rapid Communication Source: Euro Surveillance Author COI: COI declared: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Jin 2020.
| Study characteristics | |||
| Patient Sampling | 2‐group study recruiting patients estimating sensitivity and specificity [1] Laboratory‐confirmed COVID‐19 patients (n = 43); reported separately for 27 patients while still PCR‐positive and for 34 patients after becoming PCR‐negative (excluded from review) [2] Patients admitted with suspected SARS‐CoV‐2 infection, in whom the disease was eventually excluded in the hospital and who quarantined at home, were included as a control group (n = 33) Recruitment: unclear Sample size (virus/COVID cases): 76 (43) Inclusion and exclusion criteria: suspected SARS‐CoV‐2 infection (fever or any respiratory symptoms, especially in those with a history of travel to Wuhan or exposure to an infected case within 2 weeks) | ||
| Patient characteristics and setting | Setting: hospital inpatients
Location: Xixi Hospital of Hangzhou, Zhejiang Province
Country: China
Dates: January 2020‐4 March 2020
Symptoms and severity: [1] COVID‐19 patients: 27/43 (63%) fever; 26/43 (61%) cough; [2] non‐COVID‐19 patients: 24/43 (73%) fever; 15/33 (46%) cough. Sex: [1] COVID‐19 patients: 17/43 (40%) male; [2] Non‐COVID‐19 patients: 22/33 (67%) male. Age: [1] COVID‐19 patients: median age 47 (IQR 34–59) years; [2] non‐COVID‐19 patients: median age 31 (IQR 26–38) years. Exposure history: [1] NR; [2] NR |
||
| Index tests | Test name: The SARS‐CoV‐2 IgM and IgG CLIA kits
Manufacturer: Shenzhen YHLO Biotech Co., Ltd (China)
Ab targets: IgM, IgG
Antigens used: N‐protein, S‐protein
Test method: CLIA
Timing of samples: 1‐55 days pso whilst still in hospital
Samples used: serum
Test operators: laboratory
Definition of test positivity: > 10 AU/mL Blinded to reference standard: unclear Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard for cases: RT‐PCR testing at the Center for Disease Control of Hangzhou Samples used: oral swab or sputum Timing of reference standard: during patient care Blinded to index test: unclear Incorporated index test: no Reference standard for non‐cases: 2 consecutive negative RT‐PCR 24 h apart | ||
| Flow and timing | Time interval between index and reference tests: between 1 and 32 days Results presented by time period: days pso: 0‐5 6% (n = 6); 6‐10 12% (n = 12); 11‐15 15% (n = 15); 16‐20 22% (n = 22); 21‐25 22% (n = 22); 26‐30 15% (n = 15); 31‐55 8% (n = 8) All participants received the same reference standard: yes Missing data: review team excluded serology data for 34 participants after becoming PCR‐negative; no data reported for 16 participants while PCR‐positive Uninterpretable results: none mentioned Indeterminate results: none mentioned Unit of analysis: participants overall; samples by time period | ||
| Comparative | |||
| Notes | Funding: research Project on the Prevention and Treatment of COVID‐19 in Hangzhou (establishment of a clinical diagnosis and treatment system for COVID‐19 with treatment evaluation) Publication status: published paper Source: academic journal Study author COI: none mentioned | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Jung 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase infection Design: Multi‐group study to establish sensitivity and specificity. [1] Non‐COVID cases (57 samples) [1a] Non‐COVID cases with negative rt‐PCR test for SARS‐CoV‐2 (38 samples) for "clinical specificity", [1b] Non‐COVID cases with other diseases (cross‐reaction panel, 19 samples) for "analytical specificity". [2] confirmed COVID‐19 patients (n = 104 samples) for "clinical sensitivity". [2a] Only 42 inpatients for "seroconversion" study had days pso. Of these, only 19 samples had an eligible time split for our review. Recruitment: Not specified Prospective or retrospective: prospective (serum/plasma was not frozen but stored for up to 5 days at 4 °C until analysis) Sample size: 161 (104) samples of which 76 (19) samples were eligible for our review Further detail: Inclusion ‐ [2] COVID‐19 confirmed by PCR, [2a] Patients who were repeatedly assessed in our hospital, positive for SARS‐CoV‐2 by RT‐PCR, and had a known date of symptom onset [1a] negative for SARS‐CoV‐2 by RT‐PCR; [1b] patients with other confirmed viral infections; negative for SARS‐CoV‐2 by RT‐PCR or no known exposure, travel history, or symptoms of COVID‐19 No exclusion criteria defined |
||
| Patient characteristics and setting | Setting: [2] Not specified [2a] Hospital inpatients Location: [2] Texas Children’s Hospital or other in the Texas Medical Center (Baylor St. Luke’s and Ben Taub Hospitals), Houston, Texas [2a] Texas Children’s Hospital, Houston, Texas Country: Texas, USA Dates: Not specified (before 2020 August) Symptoms and severity: Not specified Demographics: Not specified Exposure history: Not stated Non‐Covid group 1: [1a] rt‐PCR‐negative samples (healthy volunteers?) Source: Not specified Characteristics: Not specified (negative SARS CoV‐2 RT‐PCR results) Non‐Covid group 2: [2b] Cross‐reaction panel Source: Concurrent, not stated Characteristics: Samples known to be positive for other viruses by molecular testing (including Influenza A, Influenza B, respiratory syncytial virus (RSV), adenovirus, rhinovirus), but negative for SARS‐CoV‐2 by RT‐PCR (3 samples did not have RT‐PCR result, but had no known exposure, travel history, or symptoms of COVID‐19) |
||
| Index tests | Test name: Ash Laboratories SARS‐CoV2 IgG and IgM ELISA Immunoassay Manufacturer: Ash Laboratories Antibody: IgG and IgM Antigen target: nucleocapsid (N) and spike (S) proteins. Evaluation setting: Laboratory Test method: ELISA (on Dynex‐DS2 automated immunoassay system) Timing of samples: [2a] < 6 days pso: n = 10, 6‐14 days pso: n = 9, > 14 days pso: n = 24 for [A] and n = 22 for [B]. Samples used: peripheral venous blood; plasma or serum stored for up to 5 days at 4 °C until analysis. Test operator: Not stated (different operators for [1a] and [2]) Definition of test positivity: > 12 AU/mL reactive; < 10 AU/mL non‐reactive, 10–12 AU/mL equivocal Blinding reported: Not stated Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: [2] COVID‐19 by RT‐PCR or TMA (Transcription‐mediated amplification) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (based on timing of tests) Incorporated index test: No Definition of non‐COVID cases: contemporaneous [1a] negative SARS CoV‐2 RT‐PCR results [1b] negative SARS CoV‐2 RT‐PCR results; 3 samples no known exposure, travel history, or symptoms of COVID‐19 Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (based on timing of tests) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no (rt‐PCR and TMA) [1] rt‐PCR [2a] rt‐PCR Remaining of [2] rt‐PCR and TMA Missing data: Yes, number of samples with IgM results lower than for IgG results (see Tables 2 and 3) Uninterpretable results: Not stated Indeterminate results: yes, but equivocal samples were considered positive Unit of analysis: [1a] Not stated [1b] Patients [2a] Samples |
||
| Comparative | |||
| Notes | Funding: EG and JJ were supported by the Ching Nan Ou Fellowship Endowment. Some of the validation kits used in this study were provided by Ansh Laboratories, but they did not participate in study design, validation, or data interpretation. Publication status: Published paper Source: Clinical Chimica Acta 510 (2020) 790–5 Author COI: Some of the validation kits used in this study were provided by Ansh Laboratories, but they did not participate in study design, validation, or data interpretation. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Kaltenbach 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase COVID‐19 infection Design: Three‐group study to estimate sensitivity and specificity: [1] Symptomatic and post‐symptomatic PCR‐confirmed Covid‐19 patients (n = 341) [2] PCR‐negative symptomatic patients (n = 115) [3] Pre‐pandemic blood donor controls (n = 150) Recruitment: Unclear; stated RT‐PCR‐positive 'committed' to participating (total positive at time of study period was 802), and RT‐PCR‐negative were randomly selected from 4509 negative results Prospective or retrospective: Prospective Sample size: 606 (341) Further detail: No more details available All RT‐PCR‐tested individuals were eligible for participation except when they were < 18 years of age, had a severely compromised immune system, were hospitalised at the time of sample collection, or were deceased. |
||
| Patient characteristics and setting | Setting: Community testing facility (hospitalised patients were excluded) Location: Basel‐Landschaft canton; 'Abklarungsstation COVID‐19' in Munchenstein Country: Switzerland Dates: 11th April 2020 to 22nd April 2020 Symptoms and severity: 35 (10%) bedridden during acute disease 62 (18%) required help for their daily activities 244 (72%) had no restrictions on daily activities Demographics: Sex: 177/349 (51%) male) Age: only available with the following breakdown: PCR‐positive <= 7 days (n = 31): median 45 years range 21‐80 years PCR‐positive > 7 days and <= 12 days (n = 46): median 51 years, range 20‐80 years PCR‐positive > 12 days (n = 272): median 51.5 years, range 17‐93 years [Numbers per group did not seem to correlate with accuracy data by time pso e.g. above added to 77 patients at <= 12 days, but Tabl 4 reported only 54 patients at <= 14 days] Exposure history: Not stated Non‐Covid group 1: PCR‐negative Source: Negative cohort from same source as positive patients Characteristics: Sex: 48/111 (43%) male Age: median 48 years, range 19‐87 years Non‐Covid group 2: Pre‐pandemic controls Source: Non‐renumerated blood donors from Swiss cantons of Thurgau, Basel, Bern, Waadt and Geneva, taken on 16th and 17th December 2016 Characteristics: Not stated |
||
| Index tests | Test name: [A] Anti‐SARS‐CoV‐2‐ELISA‐IgA (# EI 2606‐9601 A) [B] Anti‐SARS‐CoV‐2‐ELISA‐IgG (# EI 2606‐9601 G) [C] EDI Novel Coronavirus COVID‐19 IgM ELISA kit (# KT‐ 114 1033) [D] EDI Novel Coronavirus COVID‐19 IgG ELISA kit (# KT‐1032) Manufacturer: [A] Euroimmun AG, Lubeck, Germany [B] Euroimmun AG, Lubeck, Germany [C] Epitope Diagnostics, Inc., USA [D] Epitope Diagnostics, Inc., USA Antibody: [A] IgA [B] IgG [C] IgM [D] IgG Antigen target: Unclear Evaluation setting: Laboratory Test method: [A] ELISA [B] ELISA [C] ELISA [D] ELISA Timing of samples: <= 14 days: 54/345 (16%) 15‐20 days: 52/345 (15%) >= 12 days: 239/345 (69%) Samples used: [1] and [2] Serum [3] and [4] Serum and plasma Serum and plasma for all tests (some results in Suppl file) Test operator: Unclear Blood collection was performed by a medical assistant or nurse; samples either transferred to the diagnostic lab or directly processed on site in the make‐shift laboratory Definition of test positivity: [A] and [B] OD ≥ 1.1 xOD of the calibration sample [C and [D] "defined IgG‐ and IgM‐specific cut‐off values relative to the average OD of three negative controls (ODNC) as follows: OD sample ≥ (1.1+x)×ODNC is interpreted as positive" Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Unclear Timing of reference standard: Unclear Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: [2] PCR [3] Pre‐pandemic Samples used: Unclear Timing of reference standard: Unclear Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: Nothing mentioned 4 PCR‐negative found to be positive on both Euroimmun IgG and IgA and Epitope Diagnostics (EDI) IgG, considered FN PCR results were excluded. Study further reported variable numbers in each group in different parts of the paper. e.g. 341 patients in group [1] in the methods section, but 349 patients in Tabl 1 and 345 in Tabl 4 e.g. 115 PCR‐negative patients in group [2] in the methods section but 111 in Tabl 1 e.g. total sample size 606 in methods section but 605 in table 3 and 607 in figure 1 Uninterpretable results: None reported Indeterminate results: "All samples with uncertain result were considered negative for the analysis" [A] 27 (15 FN, 12 TN) uncertain results/345 (4%) [B] 14 (12 FN, 2 TN) uncertain results/345 (3%) [C] 37 (35 FN, 2 TN) uncertain results/345 (10%) [D] 23 (16 FN, 7 TN) uncertain results/345 (5%) Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This study was sponsored by Jurg Sommer, head of the "Amt fur Gesundheit".
FR is funded by the NCCR 'Molecular Systems Engineering'. Funding for JD from the two Cantons of Basel through project grant [X] granted by the ETH Zurich is acknowledged. Publication status: Pre‐print (not peer reviewed) Source: medRxiv Author COI: Nothing stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | No | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Kaltenbach 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kaltenbach 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kaneko 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity, [1] COVID‐19 patients (87 samples from 51 patients), [2] Pre‐pandemic controls (patients with other disease) (n = 100). Recruitment: Not stated Prospective or retrospective: [1] Prospective [2] Retrospective Sample size: 187 (87) samples Further detail: Inclusion ‐ [1] patients with acute COVID‐19 infection confirmed by RT‐PCR who were admitted to Musashino Red Cross Hospital and Tokyo Medical and Dental University Medical Hospital, between March and May 2020; [2] noninfected patients admitted to Musashino Red Cross Hospital and Tokyo Medical and Dental University Medical Hospital with other diseases in August and September 2019, before the spread of COVID‐19 infection. No exclusion criteria |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: Musashino Red Cross Hospital and Tokyo Medical and Dental University Medical Hospital Country: Japan Dates: March to May 2020 Symptoms and severity: All hospitalised All of the patients had clinical symptoms such as fever, cough, diarrhoea, malaise, and/or tachypnoea, no asymptomatic patients with COVID‐19. Demographics: median age 63 (25‐95) years, 37 (72.5%) male Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic controls Source: Musashino Red Cross Hospital and Tokyo Medical and Dental University Medical Hospital, August to September 2019 Characteristics: Admitted for other disease, such as hepatitis C virus infection Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: SARS‐Cov‐2 IgM/IgG Ab assay; 2019‐nCoV Ab Test Cassette (Colloidal Gold) Manufacturer: Innovita, Beijing, China Antibody: IgM/IgG Antigen target: antigen used not described Evaluation setting: POC test, unclear how it was used Test method: Lateral flow immunoassay (colloidal gold) (CGIA) Timing of samples: different time points 0‐4 days pso: 2/87 4‐7 days pso: 6/87 8‐14 days pso: 38/87 15‐28 days pso: 23/87 > 28 days pso: 18/87 Samples used: [1] Serum samples [2] serum samples stored at −80℃ Test operator: not stated Definition of test positivity: visible line The presence of only the control (C) line indicated a negative result; the presence of both the control line (C) and the IgM or IgG antibody (T) line indicated a positive result for IgM or IgG Ab, respectively. Blinding reported: No Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR for SARS‐CoV‐2 in accordance with the nationally recommended method in Japan. Samples used: Pharyngeal and nasopharyngeal swabs Timing of reference standard: not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: Pre‐pandemic, other disease Samples used: None (pre‐pandemic) Timing of reference standard: pre‐pandemic Blinded to index test: yes (presumed based on timing) Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: [1] Samples (87 samples from 51 patients) [2] Not stated (100 samples) |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Journal of Medical Virology Author COI: None |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Knauer 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase and convalescent infection Design: Two‐group to estimate sensitivity and specificity [1] Confirmed COVID cases (NAAT‐positive) [2] Suspected COVID, NAAT‐negative patients Recruitment: Not stated Prospective or retrospective: retrospective Sample size: 529 (529) samples from 366 NAAT‐tested individuals (unclear how many COVID cases, ranged from 71 to 206 NAAT‐positives per test) Eligible for our review were: [A] 204 (21) samples [B] 265 (60) samples [C] 228 (57) samples [D] 114 (11) samples [E] 261 (125) samples Further detail: Inclusion ‐ prior patients with nucleic acid amplification testing (NAAT) with confirmed or suspected COVID‐19, no exclusion criteria |
||
| Patient characteristics and setting | Setting: Not stated Location: Not stated Country: Canada Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Suspected, NAAT‐negative patients Source: Not stated (concurrent) Characteristics: Unclear Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: [A] DiaSorin SARS‐CoV‐2 S1/S2 IgG [B] EUROIMMUN Anti‐SARS‐CoV‐2 IgG [C] EUROIMMUN Anti‐SARS‐CoV‐2 IgA [D] Epitope Diagnostics Novel Coronavirus COVID‐19 IgM [E] Roche Elecsys Anti‐SARS‐CoV‐2 Total Assay Manufacturer: [A] DiaSorin [B] Euroimmun [C] Euroimmun [D] Epitope [E] Roche Antibody: [A] IgG [B] IgG [C] IgA [D] IgM [E] Total antibodies Antigen target: [A] S1/S2 from test name [B] ‐ [E] Not stated Evaluation setting: [A] ‐[E] Laboratory test (ELISA) performed in lab Test method: All ELISA [A] on Liaison XL [B} and [C] on the EUROIMMUN Analyzer‐1 [D] Manual [E] on the Cobas e801 Timing of samples: < 7 days after positive NAAT, 8‐14 days after positive NAAT, > 14 days after positive NAAT, 28 days post‐positive NAAT (n = 11 to 61). Samples used: Residual plasma samples, stored frozen at −20 °C Test operator: Not stated Definition of test positivity: All samples were tested in duplicate over the entire ELISA plate to evaluate any potential variability. Cut‐off not stated Blinding reported: Not specified Threshold predefined: Not specified, possibly yes |
||
| Target condition and reference standard(s) | Reference standard: Roche cobas SARS‐Cov‐2 NAAT, threshold not stated but included inconclusive results Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: Roche cobas SARS‐Cov‐2 NAAT, threshold not stated but included inconclusive results Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [2] Not stated [1] <= 7 to > 14 days post‐positive NAAT All patients received same reference standard: Yes Missing data: Not stated but not all samples measured with all tests Uninterpretable results: Not stated Indeterminate results: Borderline results in assays [B], [C] and [D] Borderline = result could not be clearly classified as positive or negative; borderline results were evaluated as positive for ELISA assays. Unit of analysis: [1] Serial samples for NAAT‐positive cohort [2] Not stated |
||
| Comparative | |||
| Notes | Funding: Academic Medical Organization of Southwestern Ontario (AMOSO) Publication status: Published letter Source: Clinical Biochemistry Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Knauer 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Knauer 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Knauer 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Knauer 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Ko 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent infection Design: Single‐group study to estimate sensitivity only [1] Confirmed COVID cases (51 samples from 29 patients) Recruitment: Not stated Prospective or retrospective: Unclear Sample size: 52 (52) Further detail: Patients with confirmed SARS‐CoV‐2 infections by RT‐PCR. Nothing else stated |
||
| Patient characteristics and setting | Setting: Tertiary care hospitals (8 inpatients) or life treatment centre (21 outpatients) Location: Not stated Country: Korea Dates: Not stated Symptoms and severity: 8 pneumonic COVID‐19 patients (hospitalised); 21 mild febrile without pneumonia. Demographics: 17 female, 12 male Age range 23‐80 years Mild febrile (n = 21): 8 (38.1%) male; mean age 32.2 years Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: Not stated Manufacturer: Wells Bio Inc., Seoul, Korea Antibody: IgG, IgM Antigen target: SARS‐CoV‐2 spike‐protein Evaluation setting: POCT, unclear how used Test method: Lateral flow immunoassay principle Timing of samples: Range 4‐56 days pso 4‐6 days pso: 3/52 7‐13 days pso: 6/52 14‐20 days pso: 6/52 21‐27 days pso: 10/52 28+ days pso: 27/52 Samples used: Serum (also used plasma and whole blood for evaluation of test performance according to the types of blood specimens; but this experiment was only done in 2 patients) Test operator: Not stated (test results were interpreted at the time of test and confirmed by the 2 investigators' agreement based on pictures) Definition of test positivity: 1) positive, if the IgG or IgM band showed similar intensity with the control band; 2) weakly positive, if the IgG or IgM band was clearly visible, but much fainter than the control band; 3) very weakly positive, if the IgG or IgM band was visible with very faint intensity; and 4) negative, if the IgG or IgM band was invisible, while the control band was visible. Test results were interpreted at the time of test, and finally confirmed by the two investigators’ agreement (J.‐H.K. and K.R.P) based on the pictures. Blinding reported: No (cases only) Threshold predefined: Yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: confirmed by RT‐PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Weakly positive and very weakly positive bands were classed as positive. Indeterminate results: Weakly positive and very weakly positive bands were classed as positive. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: None received
Wells Bio Inc., Seoul, Korea for donated pilot kits Publication status: Published paper (Short communication) Source: Journal of Microbiology, Immunology and Infection Author COI: The authors declared that they had no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Kohmer 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of multiple COVID‐19 diagnostic tests Design: Multi‐group study estimating both sensitivity and specificity Group [1] PCR‐confirmed COVID‐19 cases (n = 33) Group [2]: Other known infections (SARS, other coronaviruses, EBV, CMV) (n = 17) Recruitment: Unclear Prospective or retrospective: Not stated, but likely retrospective Sample size: 50 (33) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Both in‐ and outpatient (most cases were hospitalised) Location: University Hospital, Goethe University Frankfurt am Main, Frankfurt Country: Germany Dates: Not stated Symptoms and severity: Most cases were moderate to severe (numbers not available) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Group [2]: Other known infections (SARS, other coronaviruses, EBV, CMV) Source: Timing not specified, but 3 were SARS cases (2003) Characteristics: Not stated |
||
| Index tests | Test name: [A] Vircell COVID‐19 ELISA IgG [B] Euroimmun SARS‐CoV‐2 IgG ELISA [C] FaStep (COVID‐19 IgG/IgM) rapid test cassettes Manufacturer: [A] Vircell Spain S.L.U., Granada, Spain [B] EUROIMMUN AG, Germany [C] Assure Tech (Hangzhou) Co., Ltd, (China) Antibody: [A] IgG; [B] IgG; [C] IgG and IgM Antigen target: [A] S and N‐protein [B] S‐protein [C] not stated Evaluation setting: [A] and [B] Lab test; done in lab; [C] POC test; likely done in lab but unclear Test method: [A] and [B] Enzyme‐linked immunosorbent assay (ELISA); [C] Lateral flow immunoassay Timing of samples: Time since symptom onset not reported. For Group [1], time since PCR done: 17/33 (52%) collected 5‐9 days after PCR; 16/33 (48%) collected 10‐18 days after PCR Samples used: Serum Test operator: Not stated Definition of test positivity: [A] Index < 0.4 = negative, 0.4‐0.6 = equivocal, > 0.6 = positive; [B]: Ratio < 0.8 = negative, 0.8‐1.1 = equivocal, ≥ 1.1 = positive; [C]: Visual line Blinding reported: Not stated, but probably no Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: Group [1]: PCR (not further specified) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (done before index test) Incorporated index test: No Definition of non‐COVID cases: Group [2]: No testing Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Group [1]: index test done 5‐18 days after PCR Group [2]: NA All patients received same reference standard: No Missing data: Yes. Test [A] only had 13 negative cases in analysis, others had 24. Uninterpretable results: None Indeterminate results: None Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Published article Source: Academic journal Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Kohmer 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kohmer 2020a [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kohmer 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of clinical performance of multiple COVID‐19 tests Design: Multi‐group study estimating both sensitivity and specificity Group [1]: Symptomatic PCR‐confirmed COVID‐19 cases (n = 45) Group [2]: Other known infections (other coronaviruses, EBV, CMV) including some pre‐pandemic (n = 37); review team excluded 6 samples with serologically confirmed SARS‐COV‐2 infection based on PRNT) Recruitment: Unclear Prospective or retrospective: Not stated but likely retrospective Sample size: 82 (45) Further detail: No more details available |
||
| Patient characteristics and setting | |||
| Index tests | Test name: [A] SARS‐CoV‐2 IgG [B] Elecsys Anti‐SARS‐CoV‐2 [C] Liaison SARS‐CoV‐2 S1/S2 IgG [D] COVID‐19 VIRCLIA IgG MONOTEST [E] Anti‐SARS‐CoV‐2 ELISA (IgG) [F] Virotech SARS‐CoV‐2 ELISA IgG Manufacturer: [A] Abbott GbmH, Wiesbaden, Germany [B] Roche Diagnostics International AG, Rotkreuz, Switzerland [C] DiaSorin Deutschland GmbH, Dietzenbach, Germany [D] Vircell Spain S.L.U., Granada, Spain [E] Euroimmun, Lubeck, Germany [F] Virotech Diagnostics GmbH, Russelsheim, Germany Antibody: All tests except for [B]: IgG [B]: total antibody Antigen target: [A] N‐protein [B] N‐protein [C] S1 and S2‐protein [D] S1 and N‐protein [E] S1‐protein [F] N‐protein Evaluation setting: All lab tests, done in lab Test method: [A] Chemiluminescent microparticle assay (CMIA) [B] Electrochemiluminescence immunoassay (ECLIA) [C] Chemiluminescent immunoassay (CLIA) [D] Chemiluminescent immunoassay (CLIA) [E] Enzyme‐linked immunosorbent assay (ELISA) [F] Enzyme‐linked immunosorbent assay (ELISA) Timing of samples: No info regarding time since symptom onset. Group [1A]: collected 2‐49 days after PCR‐positive test. Not stated for the others. Samples used: Serum or plasma Test operator: Not stated Definition of test positivity: [A] positive if index S/C >= 1.4 [B] positive if signal sample/cut‐off >= 1.0 [C] positive if >= 15 AU/mL; equivocal if 12‐15 AU/mL [D] positive if AI >= 1.6; equivocal if AI 1.4‐1.6 [E] positive if ratio >= 1.1; equivocal if ratio 0.8‐1.1 [F] positive if Index > 1.1; equivocal if Index 0.9‐1.1 Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer. However, specified they allocated Euroimmun equivocal as negative (post hoc) |
||
| Target condition and reference standard(s) | Reference standard: Groups [1A] and [1B]: PCR (not further specified) Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: Group [2] and [3]: No testing Samples used: Group [2] and [3]: NA Timing of reference standard: Group [2] and [3]: NA Blinded to index test: Group [2] and [3]: NA Incorporated index test: Group [2] and [3]: NA |
||
| Flow and timing | Time interval between index and reference tests: Group [1A]: index tests done 2 to 49 days after PCR‐positivity (unknown for 4 cases) All patients received same reference standard: No Missing data: None (but not all index tests were used on all samples to assess specificity) Uninterpretable results: Yes, for [B]: 2 "equivocal" results, considered as negative Indeterminate results: None Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Published article Source: Academic journal Author COI: One author received speaker’s fee from Euroimmun (manufacturer of one of the index tests). |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Kohmer 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kohmer 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kohmer 2020b [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kohmer 2020b [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kohmer 2020b [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Korte 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Single‐group study to estimate sensitivity [1] Confirmed COVID patients (159 patients) Recruitment: [1] Potential participants were identified in the public health database and voluntary participation was based on the informed consent and documented positive SARS‐CoV‐2 PCR. Prospective or retrospective: Prospective Sample size: 159 (159) patients with 558 (558) samples Further detail: [1] Patients with a history of a positive SARS‐CoV‐2 PCR test |
||
| Patient characteristics and setting | Setting: Not stated (convalescent) Location: Public health database Country: Switzerland Dates: Not stated Symptoms and severity: Not stated Demographics: 52.2% females, 47.8% males Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] and [B] "anti‐spike protein IgG and IgA" [C] "anti‐nucleocapsid IgG" Names not stated Manufacturer: [A] and [B] Euroimmun, Lübeck, Germany; [C] Epitope Diagnostics, San Diego, USA Antibody: [A] IgG [B] IgA [C] IgG Antigen target: [A] Spike‐protein [B] Spike‐protein [C] Nucleocapsid‐protein Evaluation setting: [A]‐[C] Laboratory tests performed in lab Test method: [A]‐[C] ELISA Timing of samples: antibody tests were performed every week in the first month and then after another four weeks in the second month. Range: 2‐10 weeks after a positive SARS‐CoV‐2 PCR test Samples used: Not stated Test operator: Not stated Definition of test positivity: According the recommendations of the manufacturer. Blinding reported: Not stated but COVID cases only Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 PCR test Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior index test Incorporated index test: no Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: antibody tests were performed every week in the first month and then after another four weeks in the second month.
Range: 2‐10 weeks after a positive SARS‐CoV‐2 PCR test All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Funding came from the Center for Laboratory Medicine, the Swiss Federal Laboratories for Materials Science and Technology St. Gallen (Empa) and the Canton of St. Gallen Publication status: Published letter Source: Journal of Infection Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Korte 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Korte 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Kowitdamrong 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection, current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID cases (118 patients with 213 samples); [2] Non‐COVID controls (n = 171); [2a] COVID suspects with negative PCR (n = 49); [2b] Concurrent patients with other respiratory infections (n = 20); [2c] Healthy volunteers (pre‐pandemic) (n = 20); [2d] Pre‐pandemic healthy blood donors (n = 82). Recruitment: Not stated Prospective or retrospective: [1] Prospective [2a] and [2b] Prospective [2c] Unclear (possibly retrospective) [2d] Retrospective Sample size: 289 (118) participants with 384 (213) samples Further detail: Inclusion: [1] Confirmed COVID‐19 cases defined as those that tested positive for SARS‐CoV‐2 RNA using real‐time reverse transcription‐polymerase chain reaction (RT‐PCR) testing of combined nasopharyngeal and throat swab (NT) samples [2a] Plasma samples collected from May 1 to May 31, 2020, from patients under investigation (PUI) for COVID‐19 with RT‐PCR results that were negative for SARS‐CoV‐2 [2b] Serum specimens collected from May 1 to May 31, 2020 from patients with other infections (dengue, HBV, HCV, HIV, mumps, measles, rubella, EBV, CMV, VZV, HSV, and treponema) [2c] Plasma samples collected from healthy volunteers in the laboratory (prior to February 2020) [2d] Plasma samples leftover from healthy blood donors prior to February 2020 No exclusion criteria reported |
||
| Patient characteristics and setting | Setting: Not stated Location: Thai Red Cross Emerging Infectious Diseases Clinical Center (TRC‐EIDCC, King Chulalongkorn Memorial Hospital) and the Faculty of Medicine at Chulalongkorn University, Bangkok, Thailand Country: Thailand Dates: March 10 to May 31, 2020. Symptoms and severity: mild (upper respiratory symptoms) 59/118, moderate (pneumonia without hypoxia) 27/118, severe (pneumonia with hypoxia) 32/118. Demographics: Adult patients; median age of 38 years (IQR: 27–48); 47 (40%) male Exposure history: Not stated Non‐Covid group 1: [2a] Covid suspects with negative‐PCR Source: [2a] collected from May 1 to May 31, 2020, from patients under investigation (PUI) for COVID‐19 with RT‐PCR results that were negative for SARS‐CoV‐2 Characteristics: Median age 47 (IQR 28–65) years; 25 (51%) male Non‐Covid group 2: [2b] Concurrent patients with other diseases [2c] Pre‐pandemic healthy controls [2d] Pre‐pandemic healthy controls Source: [2b] Serum specimens collected from May 1 to May 31, 2020 from patients with other infections [2c] healthy volunteers in the laboratory prior to February 2020 [2d] healthy blood donors prior to February 2020 Characteristics: [2b] Patients with other infections (dengue, HBV, HCV, HIV, mumps, measles, rubella, EBV, CMV, VZV, HSV, and treponema). [2c] healthy volunteers in the laboratory [2d] healthy blood donors. |
||
| Index tests | Test name: [A] anti‐SARS‐CoV‐2 ELISA IgA kit [B] anti‐SARS‐CoV‐2 ELISA IgG kit Manufacturer: [A] and [B] EUROIMMUN Antibody: [A] IgA [B] IgG Antigen target: [A] and [B] S1‐protein Evaluation setting: [A] and [B] Lab test performed in lab Test method: [A] and [B] ELISA Timing of samples: 0‐3 days pso: 37/213 4‐7 days pso: 49/213 8‐14 days pso: 45/213 15‐28 days pso: 21/213 > 28 days pso: 61/213 Samples used: Plasma and serum were aliquoted and stored at ‐20˚C prior to serological testing. Test operator: Not stated Definition of test positivity: Semi‐quantitative results were evaluated by calculating the ratio of extinction at 450 nm of each sample over the calibrator. [A] A cut‐off ratio of 1.1 was used for SARS‐CoV‐2 IgA, as suggested by the package insert. [B] The borderline cut‐off ratio of 0.8 for SARS‐CoV‐2 IgG was assigned as positive. Blinding reported: Not stated Threshold predefined: Manufacturer's threshold but unclear why they used the borderline threshold for IgG |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 RNA using real‐time reverse transcription‐polymerase chain reaction (RT‐PCR) testing performed in the Department of Microbiology of the Faculty of Medicine at Chulalongkorn University. SARS‐CoV‐2 RNA was detected using the cobas1 SARS‐CoV‐2 kit (Roche Diagnostics, Basel, Switzerland) on a fully automated cobas1 6800 system (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s recommendations. Nucleic acid was automatically extracted from 400 μL of the NT specimens in viral transport medium (VTM) along with added internal control RNA (RNA IC). Subsequent real‐time RT‐PCR was performed automatically by the system, targeting ORF1a/b and E genes specific to SARS‐CoV‐2 and pan‐Sarbecovirus, respectively. Samples used: combined nasopharyngeal and throat swab (NT) samples. Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2a] RT‐PCR results that were negative for SARS‐CoV‐2 [2b] Not stated/ None? [2c] Pre‐pandemic (prior February 2020) [2d] Pre‐pandemic (prior February 2020) Samples used: [2a] Not stated (possibly as for cases) [2b] None [2c] Pre‐pandemic [2d] Pre‐pandemic Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Borderline results for IgG classed as positive Unit of analysis: [1] A total of 213 samples collected from 118 patients were tested for antibodies against SARS‐CoV‐2, with 36 patients having 1 sample, 69 patients having 2 samples, and 13 patients having 3 samples. [2] Patients |
||
| Comparative | |||
| Notes | Funding: This work was supported by funding to support Biobank from Ratchadapisek Sompoch Fund, Faculty of Medicine, Chulalongkorn University. Publication status: Published paper Source: PLOS One Author COI: The authors declared that no competing interests existed. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Kowitdamrong 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Krishnamurthy 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 cases (303 samples) [2] Non‐COVID samples (5262 samples) [2a] Pre‐pandemic healthy controls (n = 4502) [2b] Other disease controls including auto‐immune and infectious diseases (n = 464) [2c] SARS‐COV‐2 negative PCR (n = 296) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 5,565 (303) samples Further detail: Inclusion: [1] Individuals with SARS‐CoV2 microbiological confirmation from respiratory samples by PCR across multiple healthcare centres: 1. Gunnison Valley hospital, 2. Elite Medical Center, 3. commercial biospecimen laboratories [2a] Samples that were collected prior to the outbreak (January to April 2019) [2b] Disease controls including auto‐immune and infectious diseases from third‐party specimen providers [2c] Patients with SARS‐COV‐2 negative PCR results from respiratory samples by PCR across multiple healthcare centres: 1. Gunnison Valley hospital, 2. Elite Medical Center. Exclusions not stated |
||
| Patient characteristics and setting | Setting: Not stated Location: 1. Gunnison Valley hospital, 2. Elite Medical Center, Sunnyvale, CA, 3. commercial biospecimen laboratories. Country: USA Dates: Not stated Symptoms and severity: Not stated (the samples that were tested were ordered by physicians which would have inherent selection bias as to who was getting tested such as biased testing of individuals who were symptomatic) Demographics: Age: Mean 56 (range 17–87) years; 41% male, 59% female Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic healthy controls Source: Remnant samples, source not stated; January–April 2019 Characteristics: Age: mean 49 (range 17–90) years; 43% male, 57% female Non‐Covid group 2: [2b] Patients with other diseases [2c] COVID suspects with negative PCR Source: [2b] from third‐party specimen providers, time not stated [2c] 1. Gunnison Valley hospital, 2. Elite Medical Center, Sunnyvale, CA, Characteristics: [2b] Age and gender not stated for whole group Systemic lupus erythematosus (n = 26); Lyme disease (n = 20); CMV (n = 4); Hepatitis C (n = 20) Syphilis (n = 6); Celiac disease (n = 26); Rheumatoid arthritis (n = 26); ANA (Anti‐nuclear antibodies) (n = 79); HBV antibodies (n = 18); HCV antibodies (n = 14); Influenza A antibodies (n = 42) Influenza B antibodies (n = 26); Respiratory syncytial virus antibodies (n = 52); Common human coronavirus (n = 27); Adenovirus (n = 4); Coxsackie virus (n = 31); Echovirus (n = 28) Poliovirus (n = 11); Rhinovirus (n = 4) [2c] Age: Mean 51 (range 12–88) years; 45% male, 55% female |
||
| Index tests | Test name: Vibrant COVID‐19 Ab Manufacturer: Vibrant America Antibody: IgM, IgA and IgG Antigen target: S1 glycoprotein, Receptor binding domain (RBD), S2 glycoprotein, nucleoprotein Evaluation setting: Lab test performed in lab (test was only performed at Vibrant America) Test method: protein microarray technology; chemiluminescence Timing of samples: Not stated Samples used: Serum Test operator: Vibrant America Lab Definition of test positivity: The signal threshold was defined for each antigen by calculating the mean +/‐ SD of the signal intensity for the same antigen among the healthy controls collected prior to the infection outbreak. The raw data was converted into arbitrary chemiluminescent units (CU) based on each individual antigen cut‐off for further analysis. Blinding reported: no for [2a] as used to determine threshold Threshold predefined: no |
||
| Target condition and reference standard(s) | Reference standard: microbial RT‐PCR, threshold not stated Samples used: NP swab results Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2a] Pre‐pandemic (Jan‐April 2019) [2b] Unclear [2c] Negative PCR for SARS‐COV‐2 (unclear if at least 2 negative PCR tests) Samples used: [2a] Pre‐pandemic [2b] Unclear/none [2c] NP swab results Timing of reference standard: [2a] Pre‐pandemic [2b] Not stated [2c] Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients (see Table 2) |
||
| Comparative | |||
| Notes | Funding: Vibrant America provided funding for this study in the form of salaries for authors [HKK VJ KK TW KER KB].
Elite Medical Center provided support for this study in the form of salary for author IY.
The specific roles of these authors are articulated in the ‘author contributions’ section. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Publication status: Published paper Source: PLOS One Author COI: The authors have read the journal’s policy and the authors of this manuscript have the following competing interests: Authors HKK, VJ, KK, TW, KER, and KB are paid employees of Vibrant Sciences or Vibrant America which is a commercial lab and performs commercial antibody testing for the novel coronavirus. Author IY is a paid employee of Elite Medical Center, a commercial organisation. There were no patents, products in development or marketed products to declare. This did not alter our adherence to PLOS One policies on sharing data and materials. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lassauniere 2020 [A].
| Study characteristics | |||
| Patient Sampling | 2‐group design estimating sensitivity and specificity for 9 tests
Groups: [1] COVID‐19‐positive group (n = 30) admitted to ICU; [2] non‐COVID‐19 group (n = 82) including pre‐pandemic (2017) blood donors (n = 10); acute viral respiratory tract infections with other coronaviruses (n = 5) or non‐coronaviruses (n = 45); dengue virus (n = 9), CMV; n = 2 and Epstein Barr virus (n = 10). 1 additional patient positive for both CMV and Epstein Barr virus. Recruitment: [1] recruited consecutively (all cases in ICU on a single day); [2] unclear Sample size (virus/COVID cases): 112 (30) Inclusion and exclusion criteria: none stated |
||
| Patient characteristics and setting | Setting: [1] ICU; [2] biobank samples Location: [1] Hillerød Hospital Country: Denmark Dates: NR Symptoms and severity: NR Sex: 75% (24/32) male Age: median 67 years (IQR 52‐76) Exposure history: NR | ||
| Index tests | 9 tests evaluated, 3 ELISA and 6 LFIA; this entry ( Lassauniere 2020 [A] ), refers to test [A] in the list below:
[A] test name: Wantai SARS‐CoV‐2 Ab ELISA
Manufacturer: Beijing Wantai Biological Pharmacy Enterprise, Beijing, China; Cat # WS‐1096
Ab targets: total Ab
Antigens used: SARS‐CoV‐2 S‐protein RBD
Test method: ELISA
Timing of samples: not reported
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: calculated negative control value to 0.160
Blinded to reference standard: no
Threshold predefined: yes
[B] test name: Anti‐SARS‐CoV‐2 IgG ELISA
Manufacturer: Euroimmun Medizinische Labordiagnostika, Lübeck, Germany; Cat # EI 2668‐9601 G
Ab targets: IgG
Antigens used: SARS‐CoV‐2 S‐protein subunit 1 (S1)
Test method: ELISA
Timing of samples: not reported
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: ratio < 0.8 was considered negative, ≥ 0.8 and < 1.1 borderline, and ≥ 1.1 positive. For analysis 1.1, a more stringent cut‐off was used, and all values < 1.1 were considered negative.
Blinded to reference standard: no
Threshold predefined: yes
[C] test name: Anti‐SARS‐CoV‐2 IgA ELISA
Manufacturer: Euroimmun Medizinische Labordiagnostika, Lübeck, Germany; Cat # EI 2606‐9601 A
Ab targets: IgA
Antigens used: SARS‐CoV‐2 S‐protein subunit 1 (S1)
Test method: ELISA
Timing of samples: not reported
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: ratio < 0.8 was considered negative, ≥ 0.8 and < 1.1 borderline, and ≥ 1.1 positive. For analysis 1.1, a more stringent cut‐off was used, and all values < 1.1 were considered negative.
Blinded to reference standard: no
Threshold predefined: yes
[D] Test name: 2019‐nCOV IgG/IgM Rapid Test
Manufacturer: Dynamiker Biotechnology, Tianjin, China Cat # DNK‐1419‐1
Ab targets: IgM, IgG
Antigens used: NR
Test method: CGIA
Timing of samples: not reported
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: visible line
Blinded to reference standard: no
Threshold predefined: yes
[E] Test name: OnSiteTM COVID‐19 IgG/IgM Rapid Test
Manufacturer: CTK Biotech, Poway, CA, USA; Cat # R0180C
Ab targets: IgM, IgG
Antigens used: NR
Test method: CGIA
Timing of samples:
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: visible line
Blinded to reference standard: no
Threshold predefined: yes
[F] Test name: Anti‐SARS‐CoV‐2 Rapid Test
Manufacturer: AutoBio Diagnostics, Zhengzhou, China; Cat # RTA0204
Ab targets: IgM, IgG
Antigens used: NR
Test method: CGIA
Timing of samples: not reported
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: visible line
Blinded to reference standard: no
Threshold predefined: yes
[G] Test name: Coronavirus Diseases 2019 (COVID‐19) IgM/IgG Ab Test
Manufacturer: Artron Laboratories, Burnaby, Canada; Cat # A03‐51‐322
Ab targets: IgM, IgG
Antigens used: NR
Test method: CGIA
Timing of samples: not reported
Samples used: serum
Test operators: laboratory staff
Definition of test positivity: visible line
Blinded to reference standard: no
Threshold predefined: yes Insufficient samples available to report data by time pso for tests [H] and [I], therefore excluded from this iteration of the review [H] Test name: 2019‐nCoV IgG/IgM Rapid Test Cassette Manufacturer: Acro Biotech, Rancho Cucamonga, CA, USA; Cat # INCP‐402 Ab targets: IgM, IgG Antigens used: NR Test method: CGIA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: visible line Blinded to reference standard: no Threshold predefined: yes [I] Test name: 2019‐nCoV IgG/IgM Rapid Test Cassette Manufacturer: Hangzhou Alltest Biotech, Hangzhou, China; Cat # INCP‐402 Ab targets: IgM, IgG Antigens used: NR Test method: CGIA Timing of samples: not reported Samples used: serum Test operators: laboratory staff Definition of test positivity: visible line Blinded to reference standard: no Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard for cases (including threshold): viral nucleic acid detection (no further detail) in hospital patients Samples used: respiratory Timing of reference standard: during hospital stay Blinded to index test: yes Incorporated index test: no Reference standard for non‐cases: pre‐pandemic (2017) | ||
| Flow and timing | Time interval between index and reference tests: unclear Results presented by time period: days since onset: 7‐13 (n = 7); 14‐20 (n = 15); ≥ 21 (n = 8) All participants received the same reference standard: no Missing data: some participant samples were not tested with all assays. Only 32 of the 80 control participants were tested with POC assays. Unclear how the 32 were selected Uninterpretable results: not mentioned Indeterminate results: borderline results for [2] and [3] were considered test‐negative. For POC tests, weak signals for IgM and IgG were considered positive. Unit of analysis: participants | ||
| Comparative | |||
| Notes | Funding: Danish National Biobank resource, supported by the Novo Nordisk Foundation Publication status: preprint (not peer reviewed) Source: medRxiv Study author COI: none declared | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lassauniere 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Index tests | 9 tests evaluated, 3 ELISA and 6 LFIA; this entry ( Lassauniere 2020 [B] ) refers to test [B] [B] test name: Anti‐SARS‐CoV‐2 IgG ELISA Manufacturer: Euroimmun Medizinische Labordiagnostika, Lübeck, Germany; Cat # EI 2668‐9601 G Ab targets: IgG Antigens used: SARS‐CoV‐2 S protein subunit 1 (S1) Test method: ELISA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: ratio < 0.8 was considered negative, ≥ 0.8 and < 1.1 borderline, and ≥ 1.1 positive. For analysis 1.1, a more stringent cut‐off was used, and all values < 1.1 were considered negative. Blinded to reference standard: no Threshold predefined: yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
Lassauniere 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Index tests | Nine tests evaluated, 3 ELISA and six LFIA; this entry ( Lassauniere 2020 [C] ) refers to test [C] [C] test name: Anti‐SARS‐CoV‐2 IgA ELISA Manufacturer: Euroimmun Medizinische Labordiagnostika, Lübeck, Germany; Cat # EI 2606‐9601 A Ab targets: IgA Antigens used: SARS‐CoV‐2 S protein subunit 1 (S1) Test method: ELISA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: ratio < 0.8 was considered negative, ≥ 0.8 and < 1.1 borderline, and ≥ 1.1 positive. For analysis 1.1, a more stringent cut‐off was used, and all values < 1.1 were considered negative. Blinded to reference standard: no Threshold predefined: yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Comparative | |||
| Notes | |||
Lassauniere 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Index tests | 9 tests evaluated, 3 ELISA and 6 LFIA; this entry ( Lassauniere 2020 [D] ) refers to test [D] [D] Test name: 2019‐nCOV IgG/IgM Rapid Test Manufacturer: Dynamiker Biotechnology, Tianjin, China Cat # DNK‐1419‐1 Ab targets: IgM, IgG Antigens used: NR Test method: CGIA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: visible line Blinded to reference standard: no Threshold predefined: yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Comparative | |||
| Notes | |||
Lassauniere 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Index tests | 9 tests evaluated, 3 ELISA and 6 LFIA; this entry ( Lassauniere 2020 [E] ) refers to test [E] [E] Test name: OnSiteTM COVID‐19 IgG/IgM Rapid Test Manufacturer: CTK Biotech, Poway, CA, USA; Cat # R0180C Ab targets: IgM, IgG Antigens used: NR Test method: CGIA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: visible line Blinded to reference standard: no Threshold predefined: yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Comparative | |||
| Notes | |||
Lassauniere 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Index tests | 9 tests evaluated, 3 ELISA and 6 LFIA; this entry ( Lassauniere 2020 [F] ) refers to test [F] [F] Test name: Anti‐SARS‐CoV‐2 Rapid Test Manufacturer: AutoBio Diagnostics, Zhengzhou, China; Cat # RTA0204 Ab targets: IgM, IgG Antigens used: NR Test method: CGIA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: visible line Blinded to reference standard: no Threshold predefined: yes | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Comparative | |||
| Notes | |||
Lassauniere 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Index tests | 9 tests evaluated, 3 ELISA and 6 LFIA; this entry ( Lassauniere 2020 [G] ) refers to test [G] [G] Test name: Coronavirus Diseases 2019 (COVID‐19) IgM/IgG Ab Test Manufacturer: Artron Laboratories, Burnaby, Canada; Cat # A03‐51‐322 Ab targets: IgM, IgG. Antigens used: NR Test method: CGIA Timing of samples: Samples used: serum Test operators: laboratory staff Definition of test positivity: visible line Blinded to reference standard: no Threshold predefined: yes |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment ( Lassauniere 2020 [A] ) | ||
| Comparative | |||
| Notes | |||
Lau 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase Covid‐19 Design: [1] PCR‐confirmed Covid‐19 cases (n = 280 patients providing 415 samples) [2] Healthy heathcare worker controls (n = 597); 315 with annual southern hemisphere influenza vaccination 4 weeks prior [3] Antibody positive for different diseases: dengue (n = 74), hepatitis C (n = 3), hepatitis B (n = 12), syphilis (n = 1), antinuclear antibody (n = 16) double‐stranded DNA antibody (n = 4), rheumatoid factor (n = 7) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 994 (280) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear (hospital patients but unclear if inpatient or outpatient) Location: Changi General Hospital, Khoo Teck Puat Hospital, Sengkang General Hospital Country: Singapore Dates: April to June 2020 Symptoms and severity: Not stated Demographics: Unclear Not stated Exposure history: Unclear Not stated Non‐Covid group 1: Healthy healthcare workers Source: Volunteer staff in the same hospital, unclear if same time period as the cases or pre‐pandemic; described as 'coronavirus disease 2019 (COVID‐19)‐naive samples' Characteristics: No suspicion of Covid‐19 Non‐Covid group 2: Other disease serum samples Source: From ambulatory subjects with no suspicion for Covid‐19 or acute respiratory illness; unclear timing Characteristics: Not stated |
||
| Index tests | Test name: ELECSYS anti‐SARS‐CoV‐2 assay. Manufacturer: Roche Diagnostics, Switzerland Antibody: Unclear Not stated; appeared to be total Ab (not specified in IFU either) Antigen target: Biotinylated SARS CoV‐2 specific recombinant antigens and SARS‐CoV‐2 specific recombinant antigens labelled with ruthenium Evaluation setting: Laboratory Test method: CLIA (Sandwich immunoassay) Timing of samples: Timing reported was post‐PCR+ve 0‐7 days: 189/349 (54%) 7‐13 days: 90/349 (26%) 14‐20 days: 34/349 (10%) >= 21 days: 36/349 (10%) Samples used: Serum Test operator: Unclear Definition of test positivity: An anti‐SARS‐CoV‐2 index was derived with a reported cut‐off index (COI) of 1.0 for positivity. Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: PCR; no further details Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, as occurred before index test Incorporated index test: No Definition of non‐COVID cases: [2] None; unclear if pre‐pandemic [3] Unclear Samples used: Unclear Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: None reported; NB methods stated 415 samples included from 280 patients but results reported total of 419 samples Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Nothing stated Publication status: Pre‐print (not peer reviewed) Source: medRxiv Author COI: All authors had nothing to disclose. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Lau 2020b.
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to estimate sensitivity and specificity for diagnosis of acute and convalescent‐phase Covid Design: [1] PCR‐positive Covid‐19 cases (n = 338) of suspected or confirmed SARS‐CoV‐2 infection [2] Healthy healthcare workers (laboratory staff and frontline healthcare workers) with no suspicion for Covid‐19 (n = 294) [3] Samples positive for other antibodies including: dengue (n = 46), anti‐HCV (n = 3), HBsAg (n = 8), anti‐HBc IgM (n = 2), rheumatoid factor (n = 5). Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 696 (338) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear (includes hospital inpatients but unclear if outpatients also included) Location: Changi General Hospital Country: Singapore Dates: April to May 2020 Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Healthy healthcare workers Source: Volunteer staff in the same hospital, unclear if same time period as the cases or pre‐pandemic; described as 'coronavirus disease 2019 (COVID‐19)‐naive samples' Characteristics: No suspicion of Covid‐19 Non‐Covid group 2: Other disease serum samples Source: Unclear timing and source Characteristics: Not stated |
||
| Index tests | Test name: Architect SARS‐CoV‐2 IgG assay Manufacturer: Abbott Laboratories, USA Antibody: IgG Antigen target: Undisclosed epitope on the viral nucleocapsid Evaluation setting: Laboratory Test method: Qualitative chemiluminescent microparticle immunoassay (CLIA) Timing of samples: Timing was post‐PCR+ve: 0‐7 days: 155/266 (58%) 7‐14 days: 57/266 (21%) 14‐21 days: 22/266 (8%) >= 21 days: 32/266 (12%) Samples used: Serum Test operator: Unclear Definition of test positivity: The manufacturer cut‐off index (COI) of 1.4 was adopted to identify positivity Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: Duplex real‐time PCR targeting E and N gene Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, as occurred before index test Incorporated index test: No Definition of non‐COVID cases: [2] None; unclear if pre‐pandemic [3] Unclear Samples used: Unclear Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: Excluded 5/338 with unknown PCR status, 10 PCR‐negative, and 57 inpatients "not initially suspected of having COVID‐19 but subsequently tested positive for SARS‐CoV‐2 PCR" Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Patients Unclear; referred to 'cases' and samples |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Preprint Source: MedRxiv Author COI: All authors had nothing to disclose. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lau 2020c.
| Study characteristics | |||
| Patient Sampling | Purpose: To diagnose current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients, residual leftover sera (n = 353); [2] Non‐COVID Control ‐ [2a] Current healthy healthcare workers (HCWs) (n = 262); [2b] pre‐pandemic samples from our staff health screening (HS) programme in 2018 (n = 718); [2c] Cross‐reactivity panel (229/262 HCW volunteers from [2a] with recent influenza vaccination and 97 samples positive for dengue fever or other antibodies). Group [2a] and parts of [2c] were excluded from our review as they did not have an eligible reference standard. Recruitment: [1] Test samples ‐ Anonymised residual leftover sera (from other routine testing, e.g. renal panels, complete blood count) from subjects who had positive RT‐PCR at Changi General Hospital between April‐June 2020; unclear how recruited [2a] Excluded from review [2b] Stored samples from our staff health screening (HS) programme in 2018, unclear how recruited [2c] Samples of [2a] who had received a flu vaccination within 4 weeks of the antibody test plus samples that tested positive for dengue fever or other antibody‐positive subjects Prospective or retrospective: Both [1] test samples ‐ retrospective [2a] control group ‐ prospective [2b] pre‐pandemic healthy ‐ retrospective [2c] not stated for the 97 additional samples Sample size: 1430 (353) samples of which 1168 (353) were eligible for our review Further detail: Inclusion: [1] Subjects who had positive RT‐PCR at Changi General Hospital between April‐June 2020; [2a] Healthcare workers (HCWs) (laboratory staff, doctors, nurses, and housekeeping staff) volunteers at Changi General Hospital without symptoms of upper respiratory tract infection/fever and two serial antibody testing 14 days apart; [2b] Stored samples from staff health screening (HS) programme in 2018 (Changi General Hospital); [2c] HCW volunteers who had received the latest influenza vaccination (southern hemisphere) within four weeks of their first SARS‐CoV‐2 IgG test (see [2a]) or samples that tested positive for dengue fever or other antibodies [Anti‐HCV, Hepatitis B, anti‐nuclear antibody (ANA), double‐stranded DNA antibody (ds‐DNA), rheumatoid factor (RF), syphilis]. Exclusion: [1] Test group ‐ PCR‐negative samples [2] Not stated |
||
| Patient characteristics and setting | Setting: Hospital (Not stated if inpatients only or also outpatients) Location: Changi General Hospital, Singapore Country: Singapore Dates: April‐June 2020 Symptoms and severity: not mentioned (we did not have any data regarding symptom severity in our sensitivity cohort) Demographics: Sensitivity group (n = 279) Age: Mean 50.3 (SD 17.6) range 23 to 98; 234 (83.9%) males, 45 (16.1%) females Exposure history: not mentioned Non‐Covid group 1: [2c] Cross‐reactivity panel Source: [2c] 229/262 from [2a] not eligible for our review 97 additional samples, source and time for additional cross‐reactivity samples not stated Characteristics: [2c] 46 samples that tested positive for dengue fever, 51 other antibody‐positive subjects [Anti‐HCV – 4, Hepatitis B – 29, anti‐nuclear antibody (ANA) – 11, double‐stranded DNA antibody (ds‐DNA) – 1, rheumatoid factor (RF) – 5, syphilis – 1] Non‐Covid group 2: [2b] Pre‐pandemic healthy adults Source: stored samples from staff health screening (HS) programme in 2018 Characteristics: Age: Mean 44.2 (SD 13.4), range 20 to 85; 365 (50.8%) males, 353 (49.2%) females; healthy |
||
| Index tests | Test name: Abbott SARS‐CoV‐2‐IgG Manufacturer: Abbott Antibody: IgG Antigen target: Undisclosed epitope on the viral nucleocapsid Evaluation setting: laboratory Test method: qualitative chemiluminescent microparticle immunoassay Timing of samples: 0‐6 days post‐PCR+: 172/279 7‐13 days post‐PCR+: 47/279 14+ days post‐PCR+: 60/279 Samples used: Serum [1] Leftover sera (stored at 4 °C for 10 days) [2a] Serum [2b] Stored serum Test operator: not stated Definition of test positivity: Compared to the mean chemiluminescent signal of a calibrator, an IgG index is derived with a stated cut‐off index (COI) of 1.4 Blinding reported: not stated Threshold predefined: yes by the manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR‐ targets the N and E genes using a Qiagen EZ1 extraction system and Rotor Gene Q amplification system. Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, performed prior to index test (74 patients who were not initially suspected of having COVID‐19 but tested positive for SARS‐CoV‐2 RT‐PCR in their subsequent work‐up had samples for antibody test taken prior PCR+ test but these were excluded from analyses). Incorporated index test: No Definition of non‐COVID cases: [2a] NA as excluded from review; [2b] Pre‐pandemic (2018); [2c] Not stated for the additional 97 cross‐reactivity samples. Samples used: [2a] NA as excluded from review; [2b] Pre‐pandemic; [2c} Not stated. Timing of reference standard: [2a] NA as excluded from review; [2b] Pre‐pandemic; [2c] Not stated. Blinded to index test: yes, performed prior to index test Incorporated index test: No for [1], [2b] and eligible 97 samples from [2c] |
||
| Flow and timing | Time interval between index and reference tests: [1] 0‐6 days: 172/279; 7‐13 days: 47/279; 14+ days: 60/279; [2b] and remaining [2c] not stated All patients received same reference standard: No [1] rtPCR, [2b] pre‐pandemic, [2c] not stated. Missing data: Out of 353 RT‐PCR samples, 74 were excluded as these inpatients were not initially suspected of having COVID‐19 but tested positive for SARS‐CoV‐2 RT‐PCR in their subsequent work‐up. Of the remaining 279 samples, only 60 were eligible for our review (at least 14 days post‐PCR+). From our review, we also excluded group [2a] and 229 samples of group [2c]. Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: samples ([1] 279 samples from 160 individual SARS‐CoV‐2 RT‐PCR‐positive patients; [2c] patients; [2b] not stated) |
||
| Comparative | |||
| Notes | Funding:
We thank Temasek Holdings Pte Ltd and Abbott Diagnostics, Singapore, for sponsoring the test kits used in this study. Publication status: Published paper Source: Clinica Chimica Acta, Elsevier Author COI: None The authors declared that they had no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Lau 2020d.
| Study characteristics | |||
| Patient Sampling | Purpose: To diagnose Covid‐19 acute‐phase infection and convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Test group ‐ Confirmed COVID patients, residual leftover sera (n = 415) [2] Control group (n = 715): [2a] Non‐Covid control; current healthy healthcare workers (HCWs) (n = 597); [2b] Cross reactivity group‐ 315 HCWs from group [2a] who received their annual influenza vaccination 4 weeks prior to testing and 118 non‐Covid patients who had antibody positive samples [dengue, hepatitis C (HCV), hepatitis B (HBV), syphilis, antinuclear antibody (ANA), double‐stranded DNA antibody (anti ds‐DNA), rheumatoid factor (RF)] from ambulatory patients (n = 433) Recruitment: [1] Test samples: Residual serum samples from cases with suspected or confirmed infection from April to June 2020. Recruited from 3 institutions in Singapore: Changi General Hospital, Khoo Teck Puat Hospital, and Sengkang General Hospital, 415 excess serum samples (from 280 individual patients) [2a] Control group: 597 samples from consenting healthy (no self‐reported respiratory symptoms) healthcare workers (HCWs) were collected (laboratory staff, nurses, and housekeeping staff) [2b] Cross‐reactivity group: non‐Covid samples. Except for dengue, all other samples for cross‐reactivity analysis were from excess serum samples from before November 2019. Plus 315 from group [2a] who had their annual influenza jab 4 weeks prior to the antibody test. Prospective or retrospective: Both [1] test samples ‐ retrospective [2a] Healthy HCWs group ‐ prospective [2b] Cross‐reactivity ‐ retrospective, prospective for HCWs with influenza vaccination, unclear for dengue fever patients Sample size: 1130 (415) of which 785 (70) were eligible for our review (279 + 66 confirmed COVID cases without eligible time split excluded) Further detail: Inclusion: [1] Subjects who had positive RT‐PCR from April to June 2020, from 3 institutions in Singapore: Changi General Hospital, Khoo Teck Puat Hospital, and Sengkang General Hospital. [2a] Healthcare workers (HCWs) consenting healthy (no self‐reported respiratory symptoms) (laboratory staff, nurses, and housekeeping staff) [2b] HCWs with recent influenza vaccination, samples that tested positive for dengue fever or other antibodies [Anti‐HCV, Hepatitis B, anti‐nuclear antibody (ANA), double‐stranded DNA antibody (ds‐DNA), rheumatoid factor (RF), syphilis]. Except for dengue, all other samples for cross‐reactivity analysis were from excess serum samples from before November 2019. Exclusion: [1] Test group ‐ PCR‐negative samples [2] Not stated [3] Not stated |
||
| Patient characteristics and setting | Setting: Hospital (Not stated if inpatients only or also outpatients) Location: Changi General Hospital, Khoo Teck Puat Hospital, and Sengkang General Hospital Country: Singapore Dates: [1] Test samples ‐ from April to June 2020 Symptoms and severity: not mentioned Demographics: Data for 349 samples included in analyses: Age 49.8 (95% CI 47.7 to 51.8), range 23‐97 years; 282 males, 67 females Exposure history: Not mentioned Non‐Covid group 1: [2b] cross‐reactivity group Source: excess serum samples from before November 2019 (except for dengue) and samples from healthy HCWs who recently received influenza vaccination Characteristics: dengue N = 74, HCV N = 3, HBV N = 13, syphilis N = 1, ANA N = 16, anti‐ds‐DNA N = 4, and RF N = 7 315 healthy HCWs with recent influenza jab Non‐Covid group 2: [2a] current, healthy HCWs Source: Not mentioned, possibly HCWs (laboratory staff, nurses, and housekeeping staff) from the same 3 hospitals in Singapore Characteristics: 597 consenting healthy (no self‐reported respiratory symptoms) healthcare workers (HCWs) Characteristics only for [2a] and [2b] combined (n = 715): Age 40.4 (95% CI 38.9 to 41.9), range 19‐81 years; 126 males, 589 females |
||
| Index tests | Test name: Roche Elecsys anti‐SARS‐CoV‐2 assay Manufacturer: Roche Antibody: Total antibodies Antigen target: Undisclosed epitope Evaluation setting: Laboratory Test method: Sandwich immunoassay (where biotinylated SARS‐CoV‐2 specific recombinant antigens and SARS‐CoV‐2 specific recombinant antigens labelled with ruthenium form a sandwich complex with anti‐SARS‐CoV‐2). Electrochemiluminescent‐immunoassay Timing of samples: 0‐6 days: 189 7‐13 days: 90 >= 14 days: 70 Samples used: Serum [1] Leftover serum [2] Serum [3] Stored serum before 2019 or serum Test operator: Lab personnel from hospital laboratories (For serology, Changi and Sengkang hospitals employed the Roche Cobas e801 while Khoo Teck Puat used the Cobas e602 immunoassay analyser) Definition of test positivity: cut‐off index (COI) of 1.0 for a positive sample Blinding reported: Not stated Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: real‐time polymerase chain reaction (PCR) test systems that targeted at least 2 viral epitopes of SARS‐CoV‐2 Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes performed prior to index test Incorporated index test: No Definition of non‐COVID cases: [2a] Untested, no reported respiratory symptoms [2b] Pre‐pandemic, unclear for 74 dengue patients Samples used: [2a] Untested [2b] Pre‐pandemic or untested Timing of reference standard: [2a] Untested [2b] Pre‐pandemic or unclear Blinded to index test: yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1] 0–6 days ‐189, 7–13 days ‐ 90, 14+ days ‐ 70, 21+ days ‐36 [2] Untested [3] Pre‐pandemic or unclear All patients received same reference standard: No Missing data: A total of 415 excess serum samples (from 280 individual patients) that tested positive for SARSCoV‐2 by PCR were identified for sensitivity analysis. Of these, 66 were residual samples from inpatients not initially suspected of having COVID‐19 but who subsequently tested positive for SARS‐CoV‐2 PCR and were excluded from the sensitivity analysis. 279 Covid samples excluded from review as no eligible time split. Uninterpretable results: not stated Indeterminate results: No cases with indeterminate or missing results were used in our study. Unit of analysis: [1] Samples (349 samples from 205 individual patients) [2a] Unclear [2b] Unclear |
||
| Comparative | |||
| Notes | Funding: Research funding ‐ none declared; Temasek Holdings Pte Ltd sponsored the laboratory testing kits used in this study. Publication status: Published paper Source: JALM‐ Journal of Applied Laboratory Medicine Author COI: None declared, Honoraria: T.C. Aw, Abbott Diagnostics, Roche Diagnostics, Beckman‐Coulter |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Li 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase infections Design: Single‐group study estimating sensitivity alone: 49 NAT‐confirmed 2019‐nCoV infected patients (hospitalised patients) Recruitment: Not stated Prospective or retrospective: Not stated Sample size: 49 (49) Further detail: no more details available |
||
| Patient characteristics and setting | Setting: hospitalised patients Location: The Fifth Medical Centre of PLA General Hospital of China Country: China Dates: December 2019 to February 2020 Symptoms and severity: 12 (24%) severe; 37 (76%) mild illness; fever (41/49), cough (26/49), fatigue (11/49), dyspnoea (6/49), diarrhoea symptom (0/49); 17 had other systematic diseases (8 hypertension, 5 diabetes, 2 asthma, 1 AIDS, 1 tuberculosis, 1 hepatitis) Demographics: 30, 61% male; median age 43y (IQR: 3 to 79y) Exposure history: 35 patients had been to Wuhan before illness onset or lived in Wuhan city, others had never been to Wuhan recently. Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] SP‐based IgG/IgM ELISA; [B] N‐protein based IgG/IgM ELISA Manufacturer: [A] Hotgen Biotech (Beijing, China); [B] Livzon Group (Guangdong, China) Antigen target: [A] S (Spike); [B] N (Nucleocapsid) protein. Evaluation setting: Laboratory Test method: ELISA Timing of samples: Day 2 to 45 pso; 40 samples collected at < 10 days; up to 41 samples > 10 days Samples used: serum Test operator: not reported Definition of test positivity: the S/CO values ≥ 1 considered positive results, < 1 negative Blinding reported: Unclear Threshold predefined: not reported |
||
| Target condition and reference standard(s) | Reference standard: NAT; no further details Samples used: not reported Timing of reference standard: Not reported Blinded to index test: yes, reference standard done before index test Incorporated index test: no Definition of non‐COVID cases: NA Samples used: Timing of reference standard: Blinded to index test: Incorporated index test: |
||
| Flow and timing | Time interval between index and reference tests: not reported All patients received same reference standard: Yes; all NAT‐tested Missing data: none reported Uninterpretable results: none reported Indeterminate results: none reported Unit of analysis: 206 serum samples from 49 patients |
||
| Comparative | |||
| Notes | Funding: This work was supported by the Emergency Project for 2019‐nCoV of PLA General Hospital (20EP013). Publication status: Pre‐print Source: Lancet Infectious Diseases Author COI: authors reported no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Li 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Lippi 2020 [A].
| Study characteristics | |||
| Patient Sampling | 1‐group study recruiting patients estimating sensitivity and specificity [1] Suspected Covid‐19; subgroup of confirmed cases included Recruitment: consecutive patients Prospective or retrospective recruitment of cases: prospective Sample size (virus/Covid cases): 131 (NR); subgroup of 48 confirmed cases included Inclusion and exclusion criteria: suspected Covid‐19 patients hospitalised, in whom NP and OP swabs were collected along with blood samples during hospital stay, for purposes of COVID‐19 diagnosis and/or monitoring | ||
| Patient characteristics and setting | Setting: hospital inpatients Location: University Hospital of Verona Country: Italy Dates: NR Symptoms and severity: NR Sex: 60/131 (46%) male Age: mean 56 ± 21 years Exposure history: NR | ||
| Index tests | 2 tests were evaluated; this entry (Lippi 2020 [A]) refers to test [A] in the list below Test name: [A] MAGLUMI 2019‐nCoV IgG and IgM (2 indirect tests) [B] Anti‐SARS‐CoV‐2 IgA and IgG ELISA Manufacturer: [A] SNIBE – Shenzhen New Industries Biomedical Engineering Co., Ltd, Shenzhen, China [B] Euroimmun AG, Lübeck, Germany Ab targets: [A] IgM or IgG ; [B] IgA or IgG Antigens used: [A] CoV‐S (spike) and e CoV‐N (nucleocapsid); [B] NR Test method: [A] CLIA; [B] ELISAs Timing of samples: NR Samples used: blood, serum or plasma Test operators: NR Definition of test positivity: [A] ≥ 1.10 AU/mL [B] ≥ 1.1 (absorbance of patient sample/absorbance of calibrator) Blinded to reference standard: NR Threshold predefined: yes by manufacturer |
||
| Target condition and reference standard(s) | Reference standard for cases: RT‐PCR (commercial RT‐PCR method, Seegene AllplexTM2019‐nCoV Assay) Samples used: venous blood Timing of reference standard: during hospital stay Blinded to index test: NR Incorporated index test: no Reference standard for non‐cases: same reference standard, single‐group | ||
| Flow and timing | Time interval between index and reference tests: both during hospital stay Results presented by time period: no All participants received the same reference standard: yes Missing data: NR Uninterpretable results: NR Indeterminate results: 36 Inconclusive results Unit of analysis: per patient | ||
| Comparative | |||
| Notes | Funding: none declared Publication status: published letter Source: Clinical Chemistry and Laboratory Medicine Study author COI: study authors stated no conflict of interest. | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Lippi 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment (Lippi 2020 [A]) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment (Lippi 2020 [A]) | ||
| Index tests | 2 tests were evaluated; this entry (Lippi 2020 [B]) refers to test [B] in the list below Test name: [A] MAGLUMI 2019‐nCoV IgG and IgM (2 indirect tests) [B] Anti‐SARS‐CoV‐2 IgA and IgG ELISA Manufacturer: [A] SNIBE – Shenzhen New Industries Biomedical Engineering Co., Ltd, Shenzhen, China [B] Euroimmun AG, Lübeck, Germany Ab targets: [A] IgM or IgG ; [B] IgA or IgG Antigens used: [A] CoV‐S (spike) and e CoV‐N (nucleocapsid); [B] NR Test method: [A] CLIA (CLIAs); [B] ELISA Timing of samples: NR Samples used: blood, serum or plasma Test operators: NR Definition of test positivity: [A] ≥ 1.10 AU/mL [B] ≥ 1.1 (absorbance of patient sample/absorbance of calibrator) Blinded to reference standard: NR Threshold predefined: yes by manufacturer |
||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment (Lippi 2020 [A]) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment (Lippi 2020 [A]) | ||
| Comparative | |||
| Notes | |||
Liu 2020a.
| Study characteristics | |||
| Patient Sampling | 2‐group study estimating sensitivity and specificity for diagnosing active disease [1]. Consecutively‐recruited cohort of patients with confirmed or suspected Covid‐19 (n = 238; 153 PCR‐confirmed) [2]. Cohort of ordinary patients (n = 70); [3]. Cohort of randomly sampled healthy blood donors (n = 50) randomly sampled No further details | ||
| Patient characteristics and setting | [1]. Inpatients at General Hospital of Central Theater Command of People's Liberation Army (PLA), China (recruitment dates 6‐14 February 2020). Symptoms included fever (87%); dry cough (54%); fatigue (33%). 235/238 (99%) had CT ground glass opacity/patchy shadowing. Exposure history not described. Median age 55 [IQR 38.3‐65] years; 58% male [2]. Ordinary patients, characteristics not described. [3]. Healthy blood donors (n = 50), characteristics not described | ||
| Index tests | 2 Ab tests, blinding NR Both laboratory‐based a. ELISA kit (Lizhu, Zhuhai, China). Measured IgG and IgM detected using recombinant (rN) protein of SARS‐CoV‐2. Test threshold: NR, presumed as per manufacturer b. In‐house CLIA Timing: Serum samples acquired 17 (7%) day 0‐5; 41 (17%) day 6‐10; 21 (9%) day 11‐12; 48 (20%) day 13‐15; 111 (47%) day ≥ 16 | ||
| Target condition and reference standard(s) | 1. RT‐PCR (Daan Gene) targeting ORF1ab and N gene; Ct‐value ≤ 40 was defined as a positive test result. Pharyngeal swab specimens used Clinical diagnosis of highly‐suspected cases according to General Office of National Health Committee notice (General Office of National Health Committee. Office of State Administration of Traditional Chinese Medicine. Notice on the issuance of strategic guidelines for diagnosis and treatment of novel coronavirus (2019‐nCoV) infected pneumonia (Fifth edition draft) (2020‐02‐09) [EB/OL]) Timing: clinical diagnosis presumed on admission. RT‐PCR sampling ‐ 54 (23%) day 0‐5; 71 (30%) day 6‐10; 28 (12%) day 11‐12; 35 (15%) day 13‐15; 50 (21%) day ≥ 16 2. No reference standard described for 'ordinary' patients or healthy controls |
||
| Flow and timing | Time interval between index and reference NR, but within hospital stay. Data were disaggregated by time pso but different participants contributed samples at each time. No missing data, uninterpretable or indeterminate results described Basis for analysis: participants | ||
| Comparative | |||
| Notes | Funded by National Natural Science Foundation of China; National Key Research and Development Program of China; and the China Postdoctoral Science Foundation. Wuhan Institute of Virology of Chinese Academy of Sciences and Zhuhai Lizhu Diagnostics Inc. for providing assistance in ELISA detection Conflicts of interest: Zhuhai Lizhu Diagnostics Inc. acknowledged in Funding statement Preprint (not peer reviewed): medRxiv | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Liu 2020b [A].
| Study characteristics | |||
| Patient Sampling | 2‐group study to estimate sensitivity and specificity in acute and convalescent‐phase sera 1. RT‐PCR‐confirmed COVID‐19 cases (n = 214) 2. Healthy blood donors (n = 100) Retrospective design; recruitment method NR. No further detail | ||
| Patient characteristics and setting | [1] Inpatients at General Hospital of the Central Theater Command of the People’s Liberation Army (PLA), China (recruitment dates 18 January‐26 February). Exposure history and participant characteristics not described [2] Healthy blood donors; not further described | ||
| Index tests | 2 Ab tests, blinding NR; this entry (Liu 2020b [A]) refers to test [A] in the list below Laboratory‐based evaluations of ELISA assays measuring IgM and IgG using serum samples: A. rN‐based ELISA (Lizhu, Zhuhai, China), using recombinant N‐protein B. rS‐based ELISA (Hotgen, Beijing, China), using receptor‐binding domain of the recombinant S polypeptide (rS) Test thresholds: A. cut‐off calculated by summing 0.100 (IgM) or 0.130 (IgG) and the average A450 of negative control replicates. When A450 < cut‐off value, the test was considered negative, and when A450 was ≥ cut‐off value, the test was considered positive. B. cut‐off values (IgM and IgG) calculated by summing 0.250 and the average A450 of negative control replicates. When A450 < cut‐off value, the test was considered negative, and when A450 was ≥ cut‐off value, the test was considered positive. Samples acquired 0‐5 d 22, 10%; 6‐10 d 38, 18%; 11‐15 d 54, 25%; 16‐20 d 55, 26%; ≥ 21 d 45, 21% (32/45 were d 21‐30). Person applying the test not described | ||
| Target condition and reference standard(s) | [1] RT‐PCR (no further detail), using pharyngeal swabs samples. Positivity threshold NR. Samples acquired at a median of 15 d pso (range 0–55 days) 2. Healthy blood donors; no description of timing of serum sample collection | ||
| Flow and timing | Sampling for index and reference for cases was conducted within same time frame. No missing data, uninterpretable or indeterminate results described Basis for analysis: participants. Included a single sample per participant with results disaggregated by time pso, but different participants contributed data to each time period |
||
| Comparative | |||
| Notes | Supported by the National Natural Science Foundation, the China Postdoctoral Science Foundation (2019M664008), and the Wuhan Young and Middle‐aged Medical Backbone Talents Training Project (Wuweitong [2019] 87th266) Accepted manuscript (Journal of Clinical Microbiology) No conflicts of interest declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Liu 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment (Liu 2020b [A]) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment (Liu 2020b [A]) | ||
| Index tests | 2 Ab tests, blinding NR; this entry (Liu 2020b [B]) refers to test [B] in the list below Laboratory‐based evaluations of ELISA assays measuring IgM and IgG using serum samples A. rN‐based ELISA ( Lizhu, Zhuhai, China), using recombinant N protein B. rS‐based ELISA (Hotgen, Beijing, China), using receptor‐binding domain of the recombinant S polypeptide (rS) Test thresholds: A. cut‐off calculated by summing 0.100 (IgM) or 0.130 (IgG) and the average A450 of negative control replicates. When A450 < cut‐off value, the test was considered negative, and when A450 was ≥ cut‐off value, the test was considered positive. B. cut‐off values (IgM and IgG) calculated by summing 0.250 and the average A450 of negative control replicates. When A450 < cut‐off value, the test was considered negative, and when A450 was ≥ cut‐off value, the test was considered positive. Samples acquired 0‐5 d 22, 10%; 6‐10 d 38, 18%; 11‐15 d 54, 25%; 16‐20 d 55, 26%; ≥ 21 d 45, 21% (32/45 were d 21‐30). Person applying the test not described | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment (Liu 2020b [A]) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment (Liu 2020b [A]) | ||
| Comparative | |||
| Notes | |||
Liu 2020c.
| Study characteristics | |||
| Patient Sampling | Purpose: To diagnose Covid‐19 acute phase infection and convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Test group ‐ Confirmed COVID patients, serum from hospitalised patients (n = 206) [2] Control group (n = 270) – Non‐Covid pre‐pandemic healthy donors Recruitment: [1] Test samples ‐ Confirmed Covid patients, samples were collected from patients who were treated in the General Hospital of the Central Theatre Command of the People’s Liberation Army (PLA) between January 18 and April 4, 2020 [2] Control group (n = 270) – randomly collected from healthy blood donors who donated blood in May 2019, in Wuhan, China Prospective or retrospective: Both [1] Test samples – prospective [2] Pre‐pandemic healthy donors ‐ retrospective Sample size: 476 (206) Further detail: Inclusion: [1] Subjects who had positive RT‐PCR on pharyngeal swab specimens and were treated at the General Hospital of the Central Theatre Command of the People’s Liberation Army (PLA) between January 18 and April 4, 2020 [2] Healthy blood donors who donated blood in May 2019, in Wuhan, China. The healthy blood donors were healthy people without other infection and auto‐immune diseases. Exclusion: [1] Test group ‐ PCR‐negative samples [2] Those with other infections and auto‐immune diseases |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: General Hospital of the Central Theatre Command of the People’s Liberation Army (PLA), Wuhan, Hubei Country: China Dates: [1] between January 18 and April 4, 2020 Symptoms and severity: 54 patients were critical cases, 152 patients were non‐critical cases. Demographics: [1] Among the patients, 126 (61.1 %) were males, and 80 (38.8 %) were females. The median age of these patients was 57 years (IQR, 43–68 years), ranging from 17 to 91 years. Exposure history: Not stated (possibly all from Wuhan area) Non‐Covid group 1: [2] Healthy blood donors Source: They donated blood in May 2019, in Wuhan, China. Characteristics: The healthy blood donors were healthy people without other infection and auto‐immune diseases. The demographics (including age and gender) of patients and healthy donors were compared, with no significant differences. Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: Chemiluminescence Microparticle Immunoassays (CMIA). Test names not stated
[A] IgM‐CMIA
[B] Ab‐CMIA Manufacturer: [A] and [B] Xiamen InnoDx Biotech Co., Ltd., China (Xiamen, China) Antibody: [A] IgM [B] Ab (total antibodies) Antigen target: [A] and [B] RBD (receptor binding domain) of the SARS‐CoV‐2 spike‐protein, S‐protein of SARS‐CoV‐2 Evaluation setting: [A] and [B] Laboratory Test method: [A] chemiluminescence microparticle immunoassays (μ‐chain capture immunoassay) [B] chemiluminescence microparticle immunoassay (double‐antigens sandwich immunoassay) Timing of samples: Symptom onset 0‐7 days pso: 26/206 8‐14 days pso: 70/206 15‐21 days pso: 72/206 > 21 days pso: 38/206 Samples used: [1] Serum [2] Serum Test operator: Lab personnel from hospital laboratories Definition of test positivity: [A] and [B]: A test was determined as positive if the signal/cut‐off (S/CO) ratio > 1.0. Blinding reported: Not stated Threshold predefined: yes (the cut‐off value of IgM and total antibodies were calculated according to the manufacturer’s instructions) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR nucleic acid testing kit (Daan, Guangzhou, China) Samples used: [1] pharyngeal swab specimens Timing of reference standard: [1] During patient care, timing not stated Blinded to index test: yes, performed prior to index test Incorporated index test: No Definition of non‐COVID cases: [2] Untested, pre‐pandemic healthy blood donors who donated blood in May 2019 Samples used: [2] Untested, pre‐pandemic Timing of reference standard: [2] Untested, pre‐pandemic Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No ([1] PCR, [2] Pre‐pandemic samples) Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not mentioned (possibly none as test has no borderline range) Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding:
We thank Xiamen InnoDx Biotech Co., Ltd., China for providing assistance in CMIA detection. This work was supported by the
National Natural Science Foundation of China (81801984), the China Postdoctoral Science Foundation (2019M664008), and the Wuhan Young and Middle‐aged Medical Backbone Talents Training Project (Wuweitong [2019] 87th). Publication status: Published paper Source: Journal of Clinical Virology Elsevier Author COI: The authors declared that no conflict of interest existed. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Liu 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute or asymptomatic infection Design: [1] Confirmed COVID cases (n = 111) [1a] Symptomatic cases (n = 81) [1b] Asymptomatic cases (n = 30) [2] Non‐COVID patients (suspected COVID with multiple negative PCR tests) (n = 40) Recruitment: There were 111 patients with positive RT‐PCR test results at the time of admission and 40 suspected patients from Feb 3 to Mar 13 were enrolled. The suspected cases were based on clinical manifestation, chest radiography and epidemiology. All suspected patients were eventually "excluded from diagnosis" [and used as non‐COVID controls] based on clinical judgement as well as multiple negative RT‐PCR tests. Prospective or retrospective: Retrospective Sample size: 151 (111) patients, sample size unclear (65 COVID patients had a second blood sample and 54/62 discharged patients gave blood samples again in later check‐ups) 151 (111) samples seemed to be relevant for our review. Further detail: Inclusion: [1] rt‐PCR‐positive cases admitted to Union Jiangbei Hospital, Wuhan, China, from Feb 3 to Mar 13, 2020 [2] Suspected patients that were eventually excluded from diagnosis based on clinical judgement as well as multiple negative RT‐PCR tests |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Union Jiangbei Hospital, Wuhan, China Country: China Dates: Feb 3 to Mar 13 2020 Symptoms and severity: [1a] Symptomatic (n = 81); 17 (15.5%) severe, 42 (38.2%) common, 22 (20%) mild [1b] Asymptomatic (n = 30) Demographics: [1a] Age: Median 56 (range 23, 93) years; 48/81 (59.2%) male [1b] Age: Median 56.5 (range 20, 94) years; 22/30 (73.3%) Exposure history: Possibly all from Wuhan/Hubei province Non‐Covid group 1: [2] Suspected COVID cases with negative PCR Source: Union Jiangbei Hospital, Wuhan, China from Feb 3 to Mar 13, 2020 Characteristics: Age: Median 48.5 (range 23, 98) years; 23/40 (57.4%) male Non‐Covid group 2: NA |
||
| Index tests | Test name: "COVID‐19 IgG Detection Kits" Manufacturer: Hunan Yuanjing Biotechnology Co., Ltd. Antibody: IgM, IgG Antigen target: SARS‐CoV‐2 spike receptor‐binding domain (S‐RBD) and N spike‐protein as antigens Evaluation setting: Lab test performed in lab Test method: Magnetic Beads Chemiluminescent Immunoassay Timing of samples: [1a] First sample (n = 81): Median 7 days (range 4, 14) after symptom onset [1b] First sample (n = 30): Median 8 days (range 7, 9) after the positive RT‐PCR test detection [2] Median 9.5 (range 5, 12) day after symptom onset (n = 40) Samples used: Serum Test operator: Not stated Definition of test positivity: The test results in the sample were expressed in COI. Threshold not stated Blinding reported: no Threshold predefined: not stated |
||
| Target condition and reference standard(s) | Reference standard: real‐time RT‐PCR amplification of SARS‐CoV‐2 open reading frame 1ab (ORF1ab), nucleocapsid protein (NP) genes fragments using kits (Shanghai BioGerm Biotechnology Co., Ltd)
Conditions for amplification were 50 C for 10 min, 95 C for 5 min, followed by 40 cycles of 95 C for 10 s and 55 C for
40 s. The case would be considered to be laboratory confirmed when two targets (ORF1ab, NP) tested positive using specific real‐time RT‐PCR [19].
A cycle threshold value (Ct‐value) <= 38 was defined as a positive test, and a Ct‐value of > 38 was defined as a negative test. Samples used: nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: Yes, prior index test Incorporated index test: no Definition of non‐COVID cases: real‐time RT‐PCR amplification of SARS‐CoV‐2 open reading frame 1ab (ORF1ab), nucleocapsid protein (NP) genes fragments using kits (Shanghai BioGerm Biotechnology Co., Ltd) Conditions for amplification were 50 C for 10 min, 95 C for 5 min, followed by 40 cycles of 95 C for 10 s and 55 C for 40 s. The case would be considered to be laboratory confirmed when two targets (ORF1ab, NP) tested positive using specific real‐time RT‐PCR [19]. A cycle threshold value (Ct‐value) <= 38 was defined as a positive test, and a Ct‐value of > 38 was defined as a negative test. Classed as "Non‐COVID” control based on clinical judgement as well as multiple negative RT‐PCR tests Samples used: nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: Yes, prior index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1a] and [2] Not stated [1b] Median 8 days (range 7, 9) after the positive RT‐PCR test detection All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples but only 1 sample per time split |
||
| Comparative | |||
| Notes | Funding: This study was supported by the National Key R&D Programme of China [2019YFF0216303]. Publication status: Published paper Source: Annals of Medicine Author COI: No potential conflict of interest was reported by the author(s). |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Loconsole 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute phase infection Design: Prospective cohort study (n=819) Single‐group study to estimate sensitivity and specificity [1] PCR‐positive patients ‐ 148 [1a] < 7 days of symptoms ‐ 99 [1b] > 7 days of symptoms ‐ 44 [1c] Asymptomatic patients ‐ 5 [2] PCR‐negative patients ‐ 671 Recruitment: Consecutive (but convenient) Prospective or retrospective: Prospective Sample size: Total ‐ 819 PCR +ve ‐ 148 Further detail: Inclusion ‐ Consecutive patients presenting to the Emergency department between 23 March and 21 April 2020 [1] All patients with a positive PCR [1a] Patients with a positive PCR and symptoms < 7 days [1b] Patients with a positive PCR and symptoms > 7 days [1c] Patients with a positive PCR and no symptoms [2] PCR‐negative patients |
||
| Patient characteristics and setting | Setting: Emergency department Location: Policlinico Hospital of Bari, Italy Country: Italy Dates: 2020‐03‐23 to 2020‐04‐21 Symptoms and severity: 721/819 (88%) with respiratory symptoms (undefined). No indication of severity 98/819 (12.0%) no respiratory symptoms. Demographics: Median age ‐ 66 (IQR 52‐80) Male ‐ 454/819 (55.4%) Exposure history: Not stated Non‐Covid group 1: NA Source: NA Characteristics: NA Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: SARS‐CoV‐2 VivaDiagTM serological assay Manufacturer: Vivacheck Biotech, Hangzhou, China Antibody: IgM and/or IgG Antigen target: Not stated Evaluation setting: POC (All samples were analysed at the Laboratory of Molecular Epidemiology and Public Health of the Hygiene Unit of the Policlinico Hospital Bari, which is the Regional Reference Laboratory for surveillance and diagnosis of SARS‐CoV‐2) Test method: Lateral flow immunoassay (colloidal gold) (CGIA) Timing of samples: [1] PCR‐positive patients ‐ 148 [1a] < 7 days of symptoms ‐ 99 [1b] > 7 days of symptoms ‐ 44 [1c] Asymptomatic patients ‐ 5 Samples used: 10 micL of plasma or whole blood Test operator: Not stated Definition of test positivity: Visible line, read at 15 min If the quality control line “C” and the detection IgM and/or IgG lines were coloured, then the test was interpreted as positive for IgM and/or IgG anti‐SARS‐CoV‐2 antibodies. Blinding reported: Not stated (done at the same time as rt‐PCR so maybe yes as results were quicker) Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RNA was extracted using the Microlab Nimbus automated extraction system
(Seegene, Seoul, Republic of Korea), according to the manufacturer’s instructions. A commercial multiplex real‐time PCR kit (AllplexTM 2019‐nCoV Assay, Seegene, Seoul, Korea) was then used to detect the E, RdRP, and N genes of SARS‐CoV‐2. Results were considered positive when two or three genes were identified. The WHO Real‐time RT‐PCR protocol was used to confirm results when samples resulted positive for one gene. Samples used: Nasal and pharyngeal swabs Timing of reference standard: Prospective cohort study [1a] < 7 days of symptoms ‐ 99 [1b] > 7 days of symptoms ‐ 44 [1c] Asymptomatic patients ‐ 5 All patients ‐ on admission to ED Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: Contemporaneous [2] Negative SARS‐COV2 PCR RNA was extracted using the Microlab Nimbus automated extraction system (Seegene, Seoul, Republic of Korea), according to the manufacturer’s instructions. A commercial multiplex real‐time PCR kit (AllplexTM 2019‐nCoV Assay, Seegene, Seoul, Korea) was then used to detect the E, RdRP, and N genes of SARS‐CoV‐2. Results were considered positive when two or three genes were identified. The WHO Real‐time RT‐PCR protocol was used to confirm results when samples resulted positive for one gene. Samples used: Nasal and pharyngeal swabs Timing of reference standard: No respiratory symptoms ‐ 93 0‐7 days pso ‐ 415 > 7 days pso ‐ 52 Unknown time with symptoms ‐ 111 Performed on admission to ED Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated, possibly none (If the quality control line “C” was not coloured, the test was interpreted as invalid and repeated) Indeterminate results: None Unit of analysis: One sample per patient |
||
| Comparative | |||
| Notes | Funding: None
This research received no external funding. Publication status: Published paper Source: International Journal of Environmental Research & Public Health Author COI: None The authors declare no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Long 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Single‐group study to estimate sensitivity for diagnosing acute phase infection
Design:RT‐PCR‐positive confirmed cases (n = 285). No further detail of inclusion or exclusion criteria
Additional cohorts reported but not extracted included:
a. follow‐up cohort in RT‐PCR‐positive confirmed cases sampling every 3 days (n = 63 subset of cross‐sectional study); did not provide accuracy data
b. cohort of RT‐PCR‐negative suspects (n = 52); did not provide full accuracy data (specificity only could be extracted) c. cohort of asymptomatic contacts of 2 confirmed cases; only included 16 PCR+ |
||
| Patient characteristics and setting | Participants: Inpatients at 3 hospitals, Chongqing Three Gorges Central Hospital (TGH) (n = 158), Yongchuan Hospital Affiliated to Chongqing Medical University (YCH) (n = 75), and The Public Health Center of Chongqing (PHCC), China (n = 52), recruited 5 February 2020 Median age 47 years (IQR 34‐56 years); 55.4% male. 39/285 (14%) severe or critical in ICU. 103/285 (36%) patients had an history of exposure to transmission sources. | ||
| Index tests | One Ab test, blinding NR Laboratory‐based evaluated of magnetic CLIA kit (Bioscience (Chongqing) Co., Ltd), measuring IgM and IgG in serum samples, using recombinant antigen containing nucleoprotein and a peptide from S‐protein Test threshold not described; presume interpretation according to manufacturer's instructions Sample timing: 67/363 (18%) day 2‐7 from symptom onset; 149 (41%) day 8‐13; and 147 (40%) day 14+ | ||
| Target condition and reference standard(s) | RT‐PCR using nasal and pharyngeal swab specimens during hospital stay. No further detail. Theshold for positivity NR Single negative PCR for absence of infection Timing of reference standard sampling NR |
||
| Flow and timing | Time interval between index and reference NR. Data were disaggregated by time period but different participants contributed samples at each time pso. Missing data: 23 participants with no information on time pso were excluded leaving 363 samples from 262 participants. No uninterpretable or indeterminate results reported Basis for analysis: samples | ||
| Comparative | |||
| Notes | Funded by Emergency Project from the Science & Technology Commission of Chongqing; The Major National S&T programme grant from Science & Technology Commission of China No conflicts of interest declared; 1 study author from BioScience Co. Ltd, Chongqing, China Preprint paper (not peer reviewed) | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lou 2020 [A].
| Study characteristics | |||
| Patient Sampling | 2‐group study recruiting patients estimating sensitivity and specificity [1] n = 80 confirmed COVID cases [2] n = 300 healthy people enrolled from the community Recruitment: Prospective or retrospective recruitment of cases: Sample size (virus/COVID cases): 380 (80) Inclusion and exclusion criteria: willing to donate blood | ||
| Patient characteristics and setting | Setting: inpatient Location: First affiliated hospital of Zhejiang University Country: China Dates: 19 January‐9 February 2020 Symptoms and severity: n = 26. Critical case = any one of a) ARDS or oxygen saturation < 93% and needing mechanical ventilation invasively or non‐invasively; b) shock; c) complication of organ failure requiring ICU support N = 54 non‐critical case (not meeting criteria a) or b) or c) above) Sex: 38.7% female Age: 55 years (IQR 45‐64) Exposure history: for 45/80: incubation period (defined as interval between earliest date of SARS‐Cov‐2 exposure (unambiguous close contact with confirmed COVID‐19 case) and earliest date of symptom onset) range 0‐23 days, median 5 (IQR 2–10) | ||
| Index tests | 3 tests evaluated, data by time pso reported only for test [A]; tests [B] and [C] were excluded (B] Beijing Wantai ‐ SARS‐CoV‐2 IgG/IgM/Total Ab CGIA; [C] Xiamen InnoDx Biotech SARS‐CoV‐2 CLIA Test name: [A] SARS‐CoV‐2 IgG/IgM/Total Ab ELISA; Manufacturer: [A] Beijing Wantai; [C] Ab targets: Ab; IgM; IgG Antigens used: IgM and Ab: RBD of the SARS‐CoV‐2 S‐protein; IgG: indirect immunoassays using recombinant nucleoprotein of SARS‐CoV‐2 Test method: ELISA, CLIA; LFIA Timing of samples: between 0 and 29 days pso Samples used: serum Test operators: NR Definition of test positivity: NR Blinded to reference standard: unclear Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard for cases: confirmed case should meet 3 criteria: 1) fever and/or respiratory symptoms; 2) abnormal lung imaging findings; and 3) positive result of the nucleic acid of SARS‐CoV‐2
Samples used: deep sputum
Timing of reference standard: on admission
Blinded to index test: unclear
Incorporated index test: unclear Reference standard for non‐cases: NR |
||
| Flow and timing | Time interval between index and reference tests: NR
Results presented by time period: yes
All participants received the same reference standard: unclear
Missing data: [1] 36, 71 and 58/80 contributed to 0‐7, 8‐14 and 15‐29 days pso estimates of sensitivity for tests [A], [B] and [C] only [2] Not all control group participants were tested by all index tests (range 100‐300/300) Uninterpretable results: NR Indeterminate results: NR Unit of analysis: participant |
||
| Comparative | |||
| Notes | Funding: China National Mega‐Projects for Infectious Diseases and the Science and Technology Major Project of Xiamen Publication status: preprint Source:Pre‐print server (medRxiv) Study author COI: none declared | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lou 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Lynch 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: To report the evolution of antibody responses, and compare the magnitude of convalescent antibody responses to patients with critical and non‐critical COVID‐19 disease Design: Multi‐group study [1] COVID +ve patients [1a] ICU patients [1b] non‐ICU patients [1c] convalescent plasma donors (non‐ICU) [2] pre‐pandemic controls Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 613 (533) Further detail: Inclusion [1a] remnant serum or plasma samples from routine clinical laboratory testing [1b] and [1c] remnant serum or plasma samples from routine clinical laboratory testing at ZSFG hospital and COVID‐19 convalescent plasma donors No exclusion criteria [1c] Potential donors over 18 years of age with a self‐reported positive SARS‐CoV‐2 RT‐PCR test result were screened for allogeneic blood donation eligibility. |
||
| Patient characteristics and setting | Setting: Mixed
94 SARS‐CoV2 RT‐PCR‐positive patients, 62 (66%) admitted to the hospital and 32 outpatients Location: Zuckerberg San Francisco General Hospital Country: USA Dates: Not specified Symptoms and severity: ICU admission ‐ 26/94 (28%) Non‐ICU admission ‐ 36/94 (38%) Outpatient ‐ 32/94 (34%) Demographics: Male ‐ 64 (68%) Median age ‐ 49 (39‐58) Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name:
Pylon 3D automated immunoassay system Manufacturer: ET Healthcare, Palo Alto, CA using Pylon 3D automated immunoassay system Antibody: IgM and IgG Antigen target: nucleoprotein and a peptide from spike protein (N and S protein) Evaluation setting: Laboratory Test method: ELISA (Not stated) Timing of samples: 1‐70 days pso [1a] Week 2 [1b] Week 4 or later [1c] Two time periods ‐ 21‐40 days and 41‐70 days Samples used: Plasma or serum Test operator: Not stated Definition of test positivity: Mean plus 4 standard deviations (98.6% and 100% specificity for IgM and IgG) Blinding reported: Not stated Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes (based on timing) Incorporated index test: No (Based on timing) Definition of non‐COVID cases: Pre‐pandemic samples, prior to June 2018 Samples used: Blood samples Timing of reference standard: Pre‐pandemic Blinded to index test: Yes (based on timing) Incorporated index test: No (Based on timing) |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: 52/153 had more than 3 serial samples Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Funded by departmental discretionary funds. Reagents were donated by ET Healthcare. Publication status: Published Source: Clinical Infectious Disease Author COI: AHBW is on the scientific advisory board for ET Healthcare. Authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
MacMullan 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID samples (n = 123) [2] Current, PCR‐negative patients (n = 83) [3] Pre‐pandemic controls: serum samples collected prior to November 2019 (n = 76) Group [2] excluded from our review as test accuracy outcomes could not be read from Figure 1 Recruitment: [3] Purchased from Cureline (Brisbane, CA), and were collected before September 2019 from healthy adults in the USA Prospective or retrospective: [1] and [2] Prospective [3] Retrospective Sample size: 282 (123) of which 199 (123) were eligible for our review Further detail: Inclusion: [1] Samples from symptomatic participants collected more than 21 days post‐symptom onset, PCR‐positive [3] Collected before September 2019 from healthy adults in the USA Exclusions not stated |
||
| Patient characteristics and setting | Setting: Convalescent, setting not stated Location: Not stated (UCLA?) Country: USA Dates: Clinical serum samples collected between April and July 2020 Symptoms and severity: Symptomatic Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Current, PCR‐negative Source: Not stated (UCLA?) Characteristics: Not stated Non‐Covid group 2: [3] Pre‐pandemic healthy Source: Purchased from Cureline (Brisbane, CA), and were collected before September 2019 from healthy adults in the USA Characteristics: Healthy adults, USA |
||
| Index tests | Test name: [A] Gold Standard SARS‐CoV‐2 IgA ELISA (GSD01‐1029 IgA) [B] Gold Standard SARS‐CoV‐2 IgG ELISA (GSD01‐1028 IgG) [C] EuroImmun SARS‐CoV‐2 IgA ELISA (EI 2606‐9620 IgA) [D] EuroImmun SARS‐CoV‐2 IgG ELISA (EI 2606‐9620 IgG) Manufacturer: [A] Gold Standard Diagnostics, Davis, US [B] Gold Standard Diagnostics, Davis, US [C] EuroImmun, New Jersey, USA [D] EuroImmun, New Jersey, USA Antibody: [A] IgA [B] IgG [C] IgA [D] IgG Antigen target: [A] Nucleocapsid [B] Nucleocapsid [C] Spike [D] Spike Evaluation setting: [A] ‐ [D] Laboratory Test method: [A] ‐ [D] ELISA Timing of samples: > 21 days pso Samples used: Serum Test operator: Seemed to be scientists at Curative Inc (M.M and A.I. designed and ran experiments, analysed and interpreted data) Definition of test positivity: [A] and [B] Determination of sample positivity cut‐off value as an average of the calibrator values multiplied by a lot‐specific correction factor [C] and [D] Determination of sample absorbance ratio based on sample O.D. divided by the averaged O.D. of the calibrators Blinding reported: Not stated Threshold predefined: yes (based on cut‐off values for serum supplied by the manufacturers) |
||
| Target condition and reference standard(s) | Reference standard: [1] Curative’s oral fluid PCR test, positive for viral RNA was determined as below 35 cycle threshold (CT). Samples used: Oral fluid (participants coughed hard three times while shielding their cough via mask and/or coughing into the crook of their elbow. They then swabbed the inside of their cheeks, along the top and bottom gums, under the tongue, and finally on the tongue, to gather a sufficient amount of saliva. Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2] Curative’s oral fluid PCR test, positive for viral RNA was determined as below 35 cycle threshold (CT). [3] Pre‐pandemic Samples used: [2] oral fluid (participants coughed hard three times while shielding their cough via mask and/or coughing into the crook of their elbow. They then swabbed the inside of their cheeks, along the top and bottom gums, under the tongue, and finally on the tongue, to gather a sufficient amount of saliva). [3] Pre‐pandemic Timing of reference standard: [2] Not stated [3] Pre‐pandemic Blinded to index test: [2] and [3] yes, prior to index test Incorporated index test: [2] and [3] no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: yes (exclusion of group [2] from review) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Scientific Reports ‐ Nature Author COI: All authors are, or were at the time of research, employed by Curative Inc, a COVID‐19 diagnostics company. L.D., F.E.T. and V.S have partial ownership of Curative Inc. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
MacMullan 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
MacMullan 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
MacMullan 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Mairesse 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Independent validation ‐ evaluate the analytical and clinical performance of the iFlash® SARS‐CoV‐2 antibodies (IgM and IgG) chemiluminescence assay (CLIA) Design: Two groups [1] COVID‐19 confirmed patients ‐ n = 154 [2] Non‐SARS‐CoV‐2 sera ‐ n = 75 Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 253(178) Further detail: Inclusion ‐ Patients with RT‐PCR +ve and COVID symptoms Exclusion ‐ Not stated |
||
| Patient characteristics and setting | Setting: Not stated (specimens originated from two hospitals) Location: Saint Nikolaus Hospital, Eupen, Belgium; n = 66, and Clinique St‐Luc Bouge, Namur, Belgium; n = 112 Country: Belgium Dates: May 15 to 30, 2020 Symptoms and severity: Symptomatic patients, symptoms not described Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Non‐SARS‐CoV‐2 sera (n = 75) ‐ 38 sera from COVID‐19 negative healthy subjects and 37 sera from patients with a potential cross‐reaction Source: before the COVID‐19 pandemic and were stored at −20 °C Characteristics: Not stated Non‐Covid group 2: NA |
||
| Index tests | Test name: [A] iFlash® anti‐SARS‐CoV‐2 IgM [B] iFlash® anti‐SARS‐CoV‐2 IgG Manufacturer: [A] [B] YHLO biotechnology co., LTD, Shenzhen, China Antibody: [A] IgM [B] IgG Antigen target: [A] [B] S and N Evaluation setting: Laboratory Test method: Chemiluminescence Enzyme Immunoassay (CLIA) Timing of samples: 0‐6 days pso n = 45 7‐13 d pso ‐ n = 35 14‐20 d pso ‐n = 37 21‐27 d pso ‐ n = 29 > 28 d ‐ n = 32 Samples used: Blood (serum stored at ‐20 c) Test operator: Not stated Definition of test positivity: Two definitions 1 Manufacturer cut‐off (> 10 AU/mL) 2 ROC curve adapted cut‐offs (2.81 AU/mL for IgM; 4.86 AU/mL for IgG) Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: Confirmed RT‐PCR and with COVID‐19 symptoms Samples used: Serum Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Serum stored at ‐20 c Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Antibody kinetics since the onset of symptoms was evaluated in the full cohort of patients for which the information on the onset of symptoms was available ‐ total 154 but periodic results only for a small group Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published Source: Clinical Biochemistry Author COI: None Authors declared no known competing financial interests or personal relationships. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Mairesse 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Manalac 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Covid‐19 patients or healthcare workers with RT‐PCR‐confirmed and/or clinical assessment indicated SARS‐CoV‐2 infections (n = 97) [2] Non‐COVID samples (n = 1062) [2a] Concurrent, negative controls with no RT‐PCR results nor clinical assessment indicating SARS‐CoV‐2 infections (n = 137), [Excluded as no reference standard] [2b] Concurrent cross‐reactivity panel with positive serology test results of other infectious diseases or autoimmunity (n = 78) [2c] Pre‐pandemic samples with other diseases (n = 847) No relevant test accuracy results reported for group [2a] Recruitment: Not stated Prospective or retrospective: [1], [2a] and [2b] Unclear [2c] Retrospective Sample size: 1159 (97) of which 956 (31) were eligible for our review Further detail: [1] Specimens from patients or healthcare workers with RT‐PCR‐confirmed and/or clinical assessment indicated SARS‐CoV‐2 infections; [2a] Samples with no RT‐PCR results nor clinical assessment indicating SARS‐CoV‐2 infection; [2b] Samples with positive ANA (by ELISA), dsDNA, RF, cyclic‐citrullinated peptide IgG, RPR, and positive serology for HAV (IgG), HBV (HBV surface Ab, HBV core Ab), HCV, CMV, VZV, EBV, rubella, rubeola, mumps, HSV, and treponema pallidum, all of which were collected during the current COVID‐19 pandemic; [2c] Local patient populations seeking clinical care for rheumatoid diseases, thyroid cancer, and therapeutic drug monitoring. Remnant serum samples from rheumatoid disease screening (n = 643; 2011–2013), therapeutic drug monitoring (TDM) of lamotrigine, levetiracetam, testing for thyroglobulin (Tg), CA125, CA19‐9, CEA, AFP, and CA15‐3 (n = 94; before October 2019), and serum protein electrophoresis test (n = 110; 2012) |
||
| Patient characteristics and setting | Setting: Not stated (Covid‐19 patients or healthcare workers; our sample selection consisted of samples collected late in the disease course, mostly during follow up visits) Location: Not stated (Stanford Health Care?) Country: USA Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2b] Concurrent other diseases Source: Source not stated, collected during the current COVID‐19 pandemic Characteristics: positive ANA by ELISA (n = 5), dsDNA (n = 5), RF (n = 3), cyclic‐citrullinated peptide IgG (n = 2), and positive serology for HAV (n = 6), HBV (n = 11), HCV (n = 3), CMV (n = 2), VZV (n = 7), EBV (n = 6), rubella (n = 5), rubeola (n = 4), mumps (n = 2), HSV (n = 7), RPR (n = 5), and treponema pallidum (n = 5) Non‐Covid group 2: [2c] Pre‐pandemic, other diseases Source: Local patient populations seeking clinical care for rheumatoid diseases, thyroid cancer, and therapeutic drug monitoring Remnant serum samples from rheumatoid disease screening (n = 643; 2011–2013), therapeutic drug monitoring (TDM) of lamotrigine, levetiracetam, testing for thyroglobulin (Tg), CA125, CA19‐9, CEA, AFP, and CA15‐3 (n = 94; before October 2019), and serum protein electrophoresis test (n = 110; 2012) Characteristics: Samples were from patients ranged in age from 1 to 95 y with 67% female and 33% male A total of 165 samples were positive for one or more of ANA screening by ELISA or specific autoantibody results, with a positive rate of 25%. The samples with Tg results had 23% positive rate for the concurrent anti‐Tg autoantibodies. |
||
| Index tests | Test name: [A] Abbott Architect anti‐SARS‐CoV‐2 CMIA IgG [B] Euroimmun anti‐SARS‐CoV‐2 ELISA IgG assay Manufacturer: [A] Abbott; [B] Euroimmun Antibody: [A] IgG; [B] IgG Antigen target: [A] N‐protein; [B] S1 domain of viral spike‐protein Evaluation setting: Laboratory tests performed in lab Test method: [A] chemiluminescent microparticle immunoassay (CMIA); [B] ELISA Timing of samples: 14‐21 days pso: n = 4; > 21 days pso: n = 27; Unknown: n = 66 <= 10 days post‐PCR+: n = 8 > 10 days post‐PCR+: n = 48 Unknown: n = 41 Samples used: Abstract specified "Plasma" [2c] Remnant serum Test operator: Not stated Definition of test positivity: [A] The assay relies on an assay‐specific calibrator to report a ratio of specimen absorbance to calibrator absorbance. The interpretation of result is determined by an index (S/C) value, which is a ratio over the; threshold value. The Abbott IgG assay result is positive (index ≥ 1.4) or negative (index < 1.4). [B] The EI IgG or IgA assay result is positive (index ≥ 1.1), borderline (index ≥ 0.8 but < 1.1), or negative (index < 0.8). Blinding reported: Not stated Threshold predefined: [A]‐[C] by following manufacturer’s instructions |
||
| Target condition and reference standard(s) | Reference standard: [1] RT‐PCR‐confirmed and/or clinical assessment indicated SARS‐CoV‐2 infections, threshold not stated; clinical criteria not stated Samples used: [1] nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2b] Concurrent, not tested [2c] Pre‐pandemic Samples used: None Timing of reference standard: [2b] Untested [2c] Pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: yes, borderline results for [B] (see Table 3) [B] 35/847 controls borderline [1] No borderline result Unit of analysis: [1] Patients [2] Not stated, possibly patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Clinica Chimica Acta Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Manalac 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Marlet 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 hospital inpatients (63 samples) [2] Pre‐pandemic controls with other diseases (89 patients) The prospective study was not eligible for our review as < 25 COVID cases. 203 patients: COVID‐negative (n = 181), COVID‐positive (n = 22) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 152 (63) of which 102 (13) were eligible for our review Further detail: [1] Hospital inpatients with rt‐PCR‐confirmed COVID‐19 between 8th April and 11th May 2020 at Tours University Hospital [2] Patients from occupational medicine, emergency or pneumology departments or from patients tested positive by RT‐PCR for seasonal coronaviruses before the end of 2019 |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Tours University Hospital Country: France Dates: Plasma samples collected between April 8th and May 11th 2020 Symptoms and severity: Severe outcome 19/63 (30.2%) ICU 18/63 (28.6%) Death 3/63 (4.7%) Demographics: Age: Median 79 (IQR 67− 90); sex (F:M) 1.52 Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic, other diseases Source: Patients from occupational medicine (n = 30), emergency or pneumology departments (n = 26) or from patients tested positive by RT‐PCR (Allplex™ RP3, Seegene) for seasonal coronaviruses (n = 33, OC43, 229E or NL63) between 3–82 weeks before serology sampling before the end of 2019 Characteristics: Age: Median 30 (IQR 11− 54); sex (F:M) 1.17 |
||
| Index tests | Test name: [A] Euroimmun ELISA SARS‐CoV‐2 IgG, [B] Abbott SARS‐CoV‐2 IgG, [C] Wantai SARS‐CoV‐2 Ab ELISA [D] DiaPro COVID‐19 IgG Confirmation Manufacturer: [A] Euroimmun [B] Abbott [C] Wantai [D] DiaPro Antibody: [A] IgG [B] IgG [C] Total antibody [D] IgG Antigen target: [A] S1 [B] N [C] S (RBD) [D] S1, S2, N Evaluation setting: Laboratory Test method: [A] ELISA [B] CLIA (Alinity‐i) [C] ELISA [D] ELISA Timing of samples: 2–36 days after the onset of symptoms: 7‐13 days pso: 13 samples 14+ days pso: 45 samples Samples used: Plasma Test operator: Not stated Definition of test positivity: Not stated (performed according to the manufacturer’s recommendations) [A] and [C] Euroimmun IgG and Wantai Ab uninterpretable results were considered negative. [D] DiaPro IgG confirmation assay was considered positive when Ab against at least two targets (S1, S2 or nucleoprotein) were detected. Blinding reported: Not stated Threshold predefined: yes, performed according to the manufacturer’s recommendations |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 RT‐PCR were performed in respiratory samples using Allplex™ 2019‐nCOV assay (Seegene, Seoul, Republic of Korea), Abbott RealTime SARS‐CoV‐2 assay (Abbott Molecular, Illinois, USA) or
Bosphore 2019‐nCoV detection kit (Anatolia GeneWorks, Istanbul, Turkey) depending on reagents and systems availability.
Among the positive RT‐PCR results, inconclusive RT‐PCR results were defined as results positive only for one gene (E, ORF1ab or N). Samples used: Respiratory samples Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: [1] Sensitivity only reported for 13 + 45 = 58 samples but included in the study were 63 samples. They might have excluded samples taken before 7 days pso. Also exclusion of the prospective study Uninterpretable results: [A] and [C] Euroimmun IgG and Wantai Ab uninterpretable results were considered negative. Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. Publication status: Published paper Source: Journal of Clinical Virology Author COI: Dr. Marchand‐Adam reported financial relationships from Boehringer Ingelheim, Roche and Novartis outside the submitted work. Dr. Lemaignen reported financial relationships from Gilead, Pfizer and MSD outside the submitted work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Marlet 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Marlet 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Marlet 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Martinaud 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 161) [1a] Confirmed COVID patients for "clinical sensitivity" experiment (n = 101) [1b] Confirmed COVID patients for "analytical accuracy" experiment (n = 60) [2] Cross‐reactivity panel (n = 59) [3] Pre‐pandemic, healthy donors (n = 500) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 720 (161) of which 682 (123) were eligible for our review. Further detail: Inclusion: [1a] PCR‐confirmed COVID patients with a medical record of the date of symptomatic onset admitted to Military Medical Center, Percy [1b] Patients positive by RT‐PCR and more than 3 weeks after the symptoms onset [2] Sera obtained from patients positive for IgG and IgM against Dengue virus and Chikungunya virus, for HBsAg or anti−HCV, rheumatoid factor, monoclonal proteins, Abs against malaria, Abs against syphilis, IgG and IgM against EBV and IgG against CMV [3] Archived serum samples from healthy donors, obtained in March 2019 Exclusions not stated |
||
| Patient characteristics and setting | Setting: [1a] Hospital inpatients [1b] Not stated Location: [1a] Military Medical Center Percy, Clamart, France [1b] Unclear: Serum samples used in this study were obtained from the Medical Laboratory of the Military Medical Centers Percy (Clamart, France), Bégin (Saint‐Mandé, France) and Laveran (Marseille, France) and from the Military Biomedical Research Institute (Marseille, France). Country: [1] France Dates: [1] Not stated Symptoms and severity: [1a] 58/101 severe (= hospitalised, see results section); 43/101 non‐severe [1b] Not stated Demographics: [1] Not stated Exposure history: [1] Not stated Non‐Covid group 1: [2] Cross‐reactivity panel Source: Not stated Characteristics: Patients positive for IgG and IgM against Dengue virus (n = 5) and Chikungunya virus (n = 5), for HBsAg or anti−HCV (n = 5), rheumatoid factor (n = 5), monoclonal proteins (n = 10), Abs against malaria (n = 10), Abs against syphilis (n = 10), IgG and IgM against EBV (n = 4) and IgG against CMV (n = 5) Non‐Covid group 2: [3] Pre‐pandemic healthy Source: Healthy blood donors, obtained in March 2019 (possibly from the Blood Donation Screening Laboratory, French Military Blood Institute, Clamart, France) Characteristics: Healthy |
||
| Index tests | Test name: MosaiQ™ COVID‐19 antibody microarray Manufacturer: Quotient Antibody: IgM and IgG Antigen target: Spike S1‐protein Evaluation setting: Laboratory Test method: Solid‐phase photometric immunoassay Timing of samples: [1a] < 14 days pso: 38/101 14‐20 days pso: 33/101 > 20 days pso: 30/101 [1b] > 20 days pso: 60 samples Samples used: Serum Test operator: Not stated Definition of test positivity: Not stated Blinding reported: yes (as qualitative output) Threshold predefined: yes (qualitative output) |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 infection was confirmed by PCR in samples from the respiratory tract according to French guidelines, threshold not stated Samples used: samples from the respiratory tract (nasopharyngeal) Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2] Unclear [3] Pre‐pandemic Samples used: [2] Not stated [3] Pre‐pandemic Timing of reference standard: [2] Not stated [3] Pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: Yes (38 COVID samples < 14 days pso and 8 samples from "Analytical accuracy" experiment excluded from our review) Uninterpretable results: Not stated (in another experiment, there were 5 samples flagged with a data reduction error (DRE) making the result unavailable) Indeterminate results: 1 borderline result mentioned in Table 4. This sample was twice repeated and found negative on both occasions and finally concluded as negative. Unit of analysis: Unclear |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Journal of Clinical Virology Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
McAulay 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to estimate sensitivity and specificity for diagnosis of active disease and identification of previous disease. Design: [1] RT‐PCR‐positive COVID‐19 patients (predominantly hospitalised (n = 62 patients, 352 samples, Seattle cohort) [2] Specificity group: 74 pre‐pandemic clinical serum specimens and 31 “cross‐reactivity challenge” specimens (27 from individuals with a history of seasonal coronavirus infection within 3 years prior to collection and 4 specimens reactive for rheumatoid factor, HIV‐1 antibody, HAV total antibody, HBV core total antibody and surface antibody, HCV antibody and/or HSV2 antibody) (n = 105 people) Recruitment: [1] Samples were kindly shared by the Department of Laboratory Medicine at the University of Washington School of Medicine (Seattle, WA) [2] Unclear Prospective or retrospective: Retrospective Sample size: Samples: 457 (352). People: 167 (62) Further detail: [1] reverse‐transcription polymerase chain reaction (RT‐PCR)–confirmed COVID‐19 [2] Not stated |
||
| Patient characteristics and setting | Setting: "primarily hospitalised individuals with COVID‐19" (Supplementary Table S1) Location: Samples from Department of Laboratory Medicine at the University of Washington School of Medicine (Seattle, WA) Country: USA Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Specificity cohort (pre‐pandemic other disease or concurrent cross‐reactivity) Source: [2] 2 sources: 74 excess clinical serum specimens collected and stored in 2018, and 31 “cross‐reactivity challenge” specimens collected between March and April 2020 Characteristics: [2] 74 pre‐pandemic clinical samples: not stated; 31 "cross‐reactivity challenge" specimens: 27 from individuals with a history of seasonal coronavirus infection (as determined by a syndromic respiratory PCR test) within 3 years prior to collection (HKU1, n = 13; NL63, n = 6; OC43, n = 6; 229E, n = 2); 2 specimens reactive for rheumatoid factor; 1 reactive for HIV‐1 antibody, HAV total antibody, HBV core total antibody and surface antibody, and RPR; and 1 reactive for HCV antibody and HSV2 antibody |
||
| Index tests | Test name: [A] Rapid ResponseTM COVID‐19 Test Cassette (BTNX Inc.) Kit1 [B] SARS‐COV‐2 IgG/IgM Rapid Test (ACON Laboratories) [C]] Standard Q COVID‐19 IgM/IgG Duo (SD BIOSENSOR) [D] SARS‐CoV‐2 IgG immunoassay Manufacturer: [A] BTNX Inc. [B] ACON Laboratories [C] SD BIOSENSOR [D] Abbott Antibody: [A] IgM/IgG [B] IIgM/IgG [C] IgG (This kit was supplied as individual IgM and IgG cartridges; only the IgG cartridges were evaluated in this study) [D] IgG Antigen target: [A] Not stated [B] Not stated [C] N [D] N Evaluation setting: [A] POC, used in laboratory (Clinical Laboratory Improvement Amendments laboratory setting) [B] POC, used in laboratory (Clinical Laboratory Improvement Amendments laboratory setting) [C] POC, used in laboratory (Clinical Laboratory Improvement Amendments laboratory setting) [D] Lab test used in lab [Department of Laboratory Medicine at the University of Washington School of Medicine (Seattle, WA)] Test method: [A] Lateral Flow Immunoassay (LFIA) [B] Lateral Flow Immunoassay (LFIA) [C] Lateral Flow Immunoassay (LFIA) [D] CLIA Timing of samples: 1 to 31 days post‐symptom onset (Supplementary Table S1) < 7 days pso: 154/352 7‐13 days pso: 103/352 14‐31 days pso: 95/352 Samples used: [1] Mixed: 250 plasma, 77 serum, and 21 whole blood specimens (a further four unknown specimens were assumed to be either serum or plasma) received frozen; and underwent either 1 or 2 freeze–thaw cycles prior to testing. [2] Pre‐pandemic samples: 74 serum; Cross‐reactivity samples: not stated Test operator: [A]‐[D] Laboratory personnel Definition of test positivity: [A]‐[D] Visible lines Blinding reported: [A]‐[D] Yes Threshold predefined: As per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR ("RT‐PCR‐confirmed COVID‐19") Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic and “cross‐reactivity challenge” specimens determined by a syndromic respiratory PCR test Samples used: Pre‐pandemic and not stated Timing of reference standard: Pre‐pandemic and not stated Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated for [1] and pre‐pandemic samples from [2]
[2] Cross‐reactivity samples:
4 samples on the same day
27 samples: 1‐1159 days (within 3 years) All patients received same reference standard: No ‐ some pre‐pandemic Missing data: Yes (not all samples tested with tests [C], [D] and [E]: [C] only included 95 samples 14+ days pso; [D] only included 50 samples 14+ days pso; [E] 268/352 samples included in analyses) Uninterpretable results: 1 invalid result in specificity group excluded Indeterminate results: Not stated Unit of analysis: [1] Samples (Some patients had even several samples taken at the same day) [2] Patients |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors.
We would also like to thank the manufacturers for supplying some of the kits (ACON and BTNX kit 1). We also thank Safe Health Systems who supplied some kits (SD and BTNX kit 2) as part of a joint partnership with Mayo Clinic. Publication status: Pre‐print (not peer reviewed); now published Source: medRxiv preprint Journal (Diagnostic Microbiology and Infectious Disease) Author COI: TEG represents Mayo Clinic in a joint venture with Safe Health Systems and has shared intellectual property that may result in royalty sharing. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
McAulay 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
McAulay 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
McAulay 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Merrill 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐patients (54 specimens from 32 unique patients) [2] Suspected COVID cases and/or potential cross‐reactive with negative PCR (n = 35) [3] Pre‐pandemic samples (n = 139) Recruitment: Not stated Prospective or retrospective: [1]‐[3] Retrospective Sample size: 228 (54) of which 204‐210 (30‐36) were included in our review Further detail: Inclusion: [1] and [2] Remnant clinical specimens from individuals with SARS‐CoV‐2 PCR performed at our institution [3] Specimens collected prior to December 2019 for research and/or clinical assay validation studies Exclusions not stated |
||
| Patient characteristics and setting | Setting: Unclear (inpatients and outpatients?) Location: University of Iowa Hospitals and Clinics (UIHC), Iowa City, Iowa, USA Country: Iowa, USA Dates: Not stated Symptoms and severity: 13 asymptomatic; 41 symptomatic Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Current, PCR‐negative (COVID suspects or cross‐reactive) Source: University of Iowa Hospitals and Clinics (UIHC), Iowa City, Iowa, USA. Time not stated Characteristics: Asymptomatic n = 4; symptomatic n = 10 Other coronaviruses (229E, HKU1, NL63, OC43), n = 8 Respiratory pathogens: adenovirus n = 2, metapneumovirus n = 1, pneumocystis n = 1, rhinovirus/enterovirus n = 1, Or antibodies to other viruses: HAV n = 1, HBV/HCV n = 4, EBV/CMV n = 2, RF n = 1 Non‐Covid group 2: [3] Pre‐pandemic, healthy or other diseases Source: University of Iowa Hospitals and Clinics (UIHC), Iowa City, Iowa, USA. Before December 2019 Characteristics: HIV n = 12 No other diseases: n = 127 |
||
| Index tests | Test name: [A] DiaSorin Liaison SARS‐CoV‐2 S1/S2 IgG [B] Roche Diagnostics Elecsys Anti‐SARS‐COV‐2 assay Manufacturer: [A] DiaSorin [B] Roche Antibody: [A] IgG [B] total antibodies (IgG, IgM, IgA) Antigen target: [A] S1 and S2 domains of the spike (S)‐protein [B] Nucleocapsid (N)‐protein Evaluation setting: [A] and [B] Laboratory Test method: [A] chemiluminescent immunoassay [B] electrochemiluminescence immunoassay Timing of samples: < 7 days pso: 5/54 7‐13 days pso: 12/54 > 13 days pso: 12/54 Unknown: 12/54 Asymptomatic: 13/54 < 7 days post‐PCR+: 35/54 7‐13 days post‐PCR+: 13/54 > 13 days post‐PCR+: 6/54 Samples used: Plasma samples (lithium heparin and EDTA) Test operator: Not stated (possibly lab personnel at the Department of Pathology) Definition of test positivity: [A] signal of 15 AU/mL or higher indicating a positive result [B] cut‐off index (COI) of 1.0 or higher indicating a positive result Blinding reported: Not stated Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard: rt‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2] rt‐PCR, threshold not stated [3] Pre‐pandemic Samples used: [2] Not stated [3] Pre‐pandemic Timing of reference standard: [2] Not stated [3] Pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1] < 7 days post‐PCR+: 35/54 7‐13 days post‐PCR+: 13/54 > 13 days post‐PCR+: 6/54 [2] and [3] Not stated All patients received same reference standard: No Missing data: yes (our review excluded 12 samples that were > 13 days pso and 12 samples but included the group > 13 days post‐positive PCR. Unclear how the 6 samples > 13 days post‐positive PCR overlap with the other groups) Uninterpretable results: Not stated Indeterminate results: Possibly none as no borderline range Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: No sponsor was declared Publication status: Published paper Source: Journal of Applied Laboratory Medicine Author COI: No authors declared any potential conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Merrill 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Montesinos 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection using five tests for detection of SARS‐CoV‐2 IgG, IgM and IgA antibodies Design: Three‐group study to estimate sensitivity and specificity: [1] COVID‐19 patients confirmed by RT‐qPCR and CT‐ scans (n = 128) [B2 Negative controls. Stored sera from Jan 2018 to Aug 2019 (n = 62) included samples with a potential cross‐reaction to the SARS‐CoV‐2 immunoassays, namely, EBV infection (n = 5), CMV infection (n = 11), M. pneumoniae infection (n = 8), Parvovirus infection (n = 1), HBV infection (n = 1), Bartonella henselae infection (n = 1), Brucella spp infection (n = 1), auto‐immune pathologies (Anti‐DNA, n = 1; Anti‐PL12, n = 1; Anti Scl‐70, n = 1) and, [3] Sera from healthy volunteers (n = 10) obtained during the epidemic period (April 2020) Recruitment: Unclear. Prospective or retrospective: Retrospective Sample size: 200 (128) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear Location: Laboratoire Hospitalier Universitaire de Bruxelles ‐ Universitair Laboratorium Brussel (LHUB‐ULB) and the Microbiology Department of Cliniques Universitaires Saint Luc‐ UCLouvain (CUSL) in Brussels, Belgium. Country: Belgium Dates: Not stated Symptoms and severity: No information Demographics: No information Exposure history: No information Non‐Covid group 1: [2] Pre‐pandemic controls Source: Stored sera from Jan 2018 to Aug 2019 (n = 62) . Laboratoire Hospitalier Universitaire de Bruxelles ‐ Universitair Laboratorium Brussel (LHUB‐ULB) and the Microbiology Department of Cliniques Universitaires Saint Luc‐ UCLouvain (CUSL) in Brussels, Belgium Characteristics: No information Non‐Covid group 2: [3] Contemporaneous healthy Source: Sera from healthy volunteers (n = 10) obtained during the epidemic period (April 2020) Characteristics: No information |
||
| Index tests | Test name: [A] 2019‐nCov Antibody IgG/IgM [B] anti‐SARS‐COV‐2 IgA [C] anti‐SARS‐COV‐2 IgG [D] anti‐SARS‐COV‐2 IgA or IgG [E] rapid test cassette [F] MAGLUMI 2019‐nCoV IgG/IgM [G] QuickZen COVID‐19 IgM/IgG Manufacturer: [A] Avioq Bio‐Tech [B] EUROIMMUN [C] EUROIMMUN [D] EUROIMMUN [E] LaboOn Time [F] Snibe Diagnostic [G] ZenTech Antibody: [A] IgG, IgM, IgG or IgM [B] to [D] IgG, IgA, IgG or IgA Antigen target: [A] magnetic microbeads coated with SARS‐CoV‐2 recombinant antigen labelled with ABEI [B to D] recombinant S1 structural protein [C] to [G] SARS‐CoV‐2 antigen Evaluation setting: All laboratory‐evaluations Test method: [A, E, G] Lateral flow immunoassays; [B to D] Enzyme‐Linked Immunosorbent Assay (ELISA), [F] chemiluminescent immunoassay (CLIA) Timing of samples: Day 0 to > 15; no further details Samples used: All evaluated using serum; 10 μL serum used for LFAs Test operator: Not stated Definition of test positivity: [B to D] Ratio of the extinction of samples over the extinction of the calibrator calculated. The ratio interpretation was as follows: < 0.8 = negative, ≥ 0.8 to < 1.1 = borderline, ≥ 1.1 = positive. [F] The thresholds of positivity for these automated immunoassays were 1.0 AU/mL for IgM and IgG [A, E, G] Visible line ‐ read and interpreted 10 min after the test Blinding reported: Not stated Threshold predefined: as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR and CT scan
Two RT‐qPCR kits: RealStar® SARS‐CoV‐2 RT‐PCR kit 1.0 (Altona Diagnostics, Hambourg, Germany) at LHUB‐ULB; Genesig® Real‐Time PCR Coronavirus (COVID‐19) (Primerdesign Ltd, Chandlers Ford, United Kingdom) at CUSL
No further detail regarding how CT contributed to diagnosis Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, since it preceded it Incorporated index test: No Definition of non‐COVID cases: [2] Pre‐pandemic stored samples with known (non‐COVID) diagnoses [3] contemporaneous healthy; no reference standard reported to confirm absence of disease Samples used: Serum Timing of reference standard: NA Blinded to index test: Yes, since it preceded it Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Unclear; not stated whether CT used in all patients or whether +ve CT scan required to define positive Missing data: No; For ELISA and lateral flow tests all samples were reported in overall result but for CLIA, 2 cases were missing in IgG and IgM analyses, and 6 in IgG/IgM ‐ no reason given Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. Manufacturers offered the reagents for validation Publication status: Published paper Source: Journal of Clinical Virology Author COI: The authors declared that they had no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Montesinos 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Montesinos 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Montesinos 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Montesinos 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Montesinos 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Montesinos 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Muecksch 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Investigate performance of 4 SARS‐CoV‐2 serological assays in diagnosing prior infection Design: Single‐group study, sensitivity only [1] Prior RT‐PCR‐diagnosed SARS‐CoV‐2 positive (non‐hospitalised, relatively mildly symptomatic) Recruitment: Unclear, NHS Lothian Prospective or retrospective: Retrospective Sample size: 97 (97) Further detail: Inclusion criteria: Individuals with RT‐PCR diagnosed SARS‐CoV‐2 infection that did not require hospitalisation. Recruits surveyed to determine date of positive PCR test, date of onset of symptoms and if they required hospitalisation. 97 patients who were not hospitalised were included. Exclusion criteria: Not stated |
||
| Patient characteristics and setting | Setting: NHS outpatient clinics Location: Unclear, NHS Lothian (Abbott and Diasorin), NHS Lanarckshire (Roche), NHS Tayside (Siemens Atellica) NHS Lothian BioResource Country: United Kingdom (Scotland) Dates: Not stated Symptoms and severity: Mildy symptomatic, 70% of participants reported at least one of fever, cough or anosmia. Demographics: Mean age 44.2 years (21‐65 y), 70 female (72%) participants Exposure history: Not stated |
||
| Index tests | Test name: [A] Abbott SARS‐CoV‐2 IgG assay [B] DiaSorin SARS‐CoV‐2 IgG assay [C] Roche Anti‐SARS‐CoV total antibody assay [D] Siemens SARS‐CoV‐2 total antibody assay Manufacturer: [A] Abbott Laboratories, Illinois, USA [B] DiaSorin S.p.A., Saluggia, Italy [C] Roche Diagnostics, Rotkreuz, Switzerland [D] Siemens Healthcare Ltd, Surrey, United Kingdom Antibody: [A] IgG [B] IgG [C] total antibody [D] total antibody Antigen target: [A] N‐protein [B] S‐protein [C] N‐protein [D] RBD of S‐protein Evaluation setting: Laboratory Test method: [A] CMIA [B] CMIA [C] ECLIA [D] CLIA Timing of samples: Visit [1] (baseline) avg. 40.8 days post‐PCR +ve (range 24‐61 days), Visit [2] (two weeks post‐baseline) avg. 55.1 days post‐PCR +ve (range 40‐79 days), Visit [3] (four weeks post‐baseline) avg. 69.8 days post‐PCR +ve (range 55‐95 days), Visit [4] (8 weeks post‐baseline) avg. 98.4 days (85‐110 days) Samples used: Convalescent serum Test operator: Laboratory staff [NHS Lothian (Abbott and Diasorin), NHS Lanarckshire (Roche), NHS Tayside (Siemens Atellica)] Definition of test positivity: (All the assays generate a qualitative positive/negative result based on assay‐dependent signal thresholds. Each assay gives a qualitative positive or negative result, based on assay specific thresholds) [A] S/C [B] AU/mL [C] COI [D] AU Blinding reported: Unclear Threshold predefined: Not stated, possibly yes ("assay specific thresholds", unclear if this meant manufacturer recommended thresholds) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard:Unclear Blinded to index test: Yes Incorporated index test: no Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: 24‐110 days post‐PCR +ve All patients received same reference standard: Yes Missing data: Not stated (97 * 3 = 291 samples + 28 with a 4th sample = 319 samples, there seemed to be no samples missing but no flow diagram provided) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding:
This work was supported by the NHS and Grants from the National Institutes of Allergy and infectious Diseases R37AI640003 (to PDB) and R01AI078788 (to TH). There were no study sponsors. The funders played no role in the design, analysis or reporting of this research. Publication status: Pre‐print (not peer reviewed) Source: Pre‐print medRxiv Author COI: Authors declared no support from any organisation or financial relationships with any organisations that might have an interest in the submitted work in the previous three years, or no other relationships or activities that could appear to have influenced the submitted work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Muecksch 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Muecksch 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Muecksch 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Naaber 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Comparison of sensitivity of 7 commercial antibody tests to detect acute or convalescent‐phase SARS‐CoV‐2 infection Design: Multi‐group study to assess sensitivity and specificity of 7 commercial antibody tests [1] Confirmed COVID patients (n = 97) [1a] asymptomatic (n = 20) [1b] symptoms score 1‐6 (n = 43) [1c] symptoms score 7‐14 (n = 34) [2] Pre‐pandemic healthy controls (n = 100) Recruitment: [1] Recruited hospitalised and ambulatory patients, as well as healthy contacts of COVID‐19 patients selected randomly [2] Not stated Prospective or retrospective: [1] Unclear [2] Retrospective Sample size: 197 (97) samples of which 173 (73) were eligible for our review Further detail: [1] At least one‐week pso or post‐RT‐PCR +ve [2] anonymous serum samples collected before COVID‐19 pandemic and stored in SYNLAB Estonia from healthy persons for various health control laboratory tests Exclusions not stated Setting: Kurressaare Hospital inpatients, ambulatory patients and healthy contacts of COVID patients. Samples sent to SYNLAB Estonia central laboratory for testing Location: Kurressaare Hospital, Island of Saaremaa, Estonia Country: Estonia Dates: Serum samples collected between April 28 and May 07 2020 Symptoms and severity: Varied, 19.6% hospitalised, 44% recorded 1‐6 symptoms, 35% recorded 7+ symptoms, 20.6% asymptomatic. Demographics: Median age 59 years (21‐100 years), 32% male Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic, healthy persons Source: Anonymous serum samples collected before COVID‐19 pandemic (dates not stated), stored in SYNLAB Estonia Characteristics: Healthy, not screened for virus‐related antibodies |
||
| Patient characteristics and setting | Setting: Kurressaare Hospital inpatients, ambulatory patients and healthy contacts of COVID patients. Samples sent to SYNLAB Estonia central laboratory for testing Location: Kurressaare Hospital, Island of Saaremaa, Estonia Country: Estonia Dates: Serum samples collected between April 28 and May 07 2020 Symptoms and severity: Varied, 19.6% hospitalised, 44% recorded 1‐6 symptoms, 35% recorded 7+ symptoms, 20.6% asymptomatic Demographics: Median age 59 years (21‐100 years), 32% male Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic, healthy persons Source: Anonymous serum samples collected before COVID‐19 pandemic (dates not stated), stored in SYNLAB Estonia Characteristics: Healthy, not screened for virus‐related antibodies Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: [A] MAGLUMI 2019‐nCoV IgG, SNIBE [B] SARS‐CoV‐2 ELISA IgG, EUROIMMUN [C] SARS‐CoV‐2 IgG, Abbott [D] Elecsys Anti‐SARS‐CoV‐2, Roche [E] EDI Novel Coronavirus COVID‐19 IgG ELISA [F] LIAISON SARS‐CoV‐2 S1/S2 IgG [G] STANDARDTM Q COVID‐19 IgM/IgG Duo Test Manufacturer: [A] SNIBE (Shenzhen New Industries Biomedical Engineering Co) [B] EUROIMMUN AG [C] Abbott Laboratories [D] Roche Diagnostics GmbH [E] Epitope Diagnostics Inc [F] DiaSorin S.p.A [G] SD BioSensor Inc Antibody: [A] IgG [B] IgG [C] IgG [D] Total antibody [E] IgG [F] IgG [G] IgG Antigen target: [A] not specified [B] S1 [C] N‐protein [D] N‐protein [E] N and S‐protein [F] S1 and S2 [G] N‐protein Evaluation setting: [A], [B], [C], [D], [E], [F] laboratory tests [G] rapid IgG test (POCT) Test method: [A] CLIA [B] ELISA [C] CMIA [D] ECLIA [E] ELISA [F] CLIA [G] Rapid chromatographic immunoassay Timing of samples: At least one‐week pso or post‐PCR +ve Median 28 (range 7–57) days to test 7‐14 days, n = 20 15‐30 days, n = 35 31‐57 days, n = 42 Samples used: Serum [1] Serum was separated and aliquoted before storage. All aliquots were stored at– 30˚ C and analysed within one month applying one freezing/thawing cycle before testing Test operator: Staff at SYNLAB Estonia Central Laboratory Definition of test positivity: [A] > 1 pos [B] ≥ 0.8‐ < 1.1 borderline, ≥ 1.1 pos [C] ≥ 1.4 pos [D] ≥ 1 pos [E] neg 0.9 x (neg control + 0.10), borderline if > 0.9‐1.1x(neg control + 0.10)?, pos 1.1 x (neg control + 0.10) [F] ≥ 12‐ < 15 borderline, ≥ 15 pos [G] Positive: any line in test window Blinding reported: Unclear Threshold predefined: Yes (Commercial tests were performed and interpreted according to manufacturer instructions). |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard: Unclear Blinded to index test: Yes, performed prior to index test Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic healthy (date not stated) Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic controls, healthy persons Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No ([1] rt‐PCR [2] Pre‐pandemic) Missing data: Nothing stated Uninterpretable results: Nothing stated Indeterminate results: yes (text mentioned 2 borderline results for test [B] and 4 borderline results reported for test [E]) Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Funding acquired by Paul Naaber (first author), no more detail provided
The study was supported by Estonian Research Council grants PRG377 (LH, PR, PP) and IUT34‐19 (PN, ES). SYNLAB Estonia provided support in the form of salaries for authors (PN, KH, JH, IE) and research materials, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Publication status: Published paper Source: PLOS One Author COI: The authors have read the journal’s policy and have the following competing interests: PN, KH, JH, IE are employees of SYNLAB Estonia. There were no patents, products in development or marketed products associated with this research to declare. This does not alter our adherence to PLOS One policies on sharing data and materials. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Naaber 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Naaber 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Naaber 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Naaber 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Naaber 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Naaber 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nagasawa 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Detection of anti‐SARS‐CoV‐2 IgG and IgM antibodies in COVID‐19 patients, diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (45 samples from 26 patients) [1a] Moderate COVID patients (n = 19) [1b] Severe COVID patients (n = 7) [2] Controls, not eligible for our review Recruitment: Not stated. [1] Inpatients at Musashino Red Cross Hospital, Musashino City, Tokyo, Japan, admitted between April 12 and May 8 2020 Prospective or retrospective: Unclear Sample size: 57 (45) of which 45 (45) were eligible for our review Further detail: [1] Inclusion: Patients who were diagnosed as COVID‐19 from positive RT‐PCR test for SARS‐CoV‐2 by using naso‐pharynx swab specimens and admitted in our hospital between April 12 and May 8, 2020 Exclusion criteria not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Musashino Red Cross Hospital, Musashino City, Tokyo, Japan Country: Japan Dates: April 12 to May 8 2020 Symptoms and severity: [1a] Moderate COVID patients (n = 19) [1b] Severe COVID patients (n = 7) 26 confirmed pneumonia, 7 confirmed ventilator usage, 1 death. Demographics: 14 male, 12 female Age 19‐82 years, mean 52.6 (SD 6.3) Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] 2019‐nCoV Ab Test [B] COVID‐19 IgG/IgM Rapid Cassette Test [C] 2019‐nCoV IgG/IgM Rapid Test Casette Manufacturer: [A] INNOVITA Biological Technology Co., China [B] Zhejiang Orient Gene Biotech Co., China [C] Hangzhou AllTest Biotech Co., China Antibody: [A] IgG/IgM [B] IgG/IgM [C] IgG/IgM Antigen target: [A], [B], [C] Unclear Evaluation setting: Laboratory Test method: [A] [B] [C] Described in paper as ELISAs, but also as rapid tests; the test names matched available rapid tests and have been included in review as rapid tests. Timing of samples: 1‐29 days pso 1‐5 days pso: 1/45 6‐10 days pso: 10/45 11‐15 days pso: 19/45 16‐20 days pso: 9/45 21‐29 days pso: 6/45 Samples used: serum Test operator: Unclear, laboratory staff? Clinical staff? Result confirmed by at least two inspectors Definition of test positivity: Not stated. (Test was performed according to the protocol of each manufacturer. The result was confirmed by at least two inspectors and adopted only in case with unanimous decision) Blinding reported: Not stated Threshold predefined: yes, according to the protocol of each manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR performed at SRL laboratory (Tokyo, Japan); threshold not stated Samples used: naso‐pharynx swab specimens Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: Yes As in F1: samples missing for test [B] days 1‐5, test [A] days 11‐15, test [B] 11‐15 days IgG Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Test kits provided by SoftBank Cooperation (Tokyo, Japan) Publication status: Published paper Source: SN Comprehensive Clinical Medicine Author COI: Authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Nagasawa 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nagasawa 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nayak 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID‐19 patients, RT‐PCR +ve, n = 42 [2] Pre‐pandemic controls n = 22 Group [2] not eligible for our review as < 25 samples Recruitment: COVID‐19‐recovered individuals recruited from Shaheed Hasan Khan Mewati Government Medical College, Haryana, India, Super Specialty Pediatric Hospital and Post Graduate Teaching Institute, Noida and ICMR‐National Institute of Malaria Research, New Delhi Prospective or retrospective: [1] Unclear [2] Retrospective Sample size: 64 (42) of which 42 (42) were eligible for our review Further detail: [1] SARS‐CoV‐2 PCR‐positive at the time of initial diagnosis, and PCR‐negative when recruited for this study [2] Not stated Exclusions not reported |
||
| Patient characteristics and setting | Setting: Convalescent Location: Shaheed Hasan Khan Mewati Government Medical College, Haryana, India, Super Specialty Pediatric Hospital and Post Graduate Teaching Institute, Noida and ICMR‐National Institute of Malaria Research, New Delhi Country: India Dates: Not stated, 25‐84 days post‐PCR +ve Symptoms and severity: Not stated, all recovered Demographics: Mean age 39.4 years (range 15‐70); 38 males, 4 females Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: COVID‐Kavach ELISA test kit Manufacturer: Zydus diagnostics, Calida Healthcare Limited Antibody: IgG Antigen target: Not stated Evaluation setting: Laboratory Test method: ELISA Timing of samples: 25‐84 days post‐PCR +ve Samples used: Plasma Test operator: Not stated, laboratory staff? Definition of test positivity: >= 1.5 Blinding reported: Unclear Threshold predefined: Unclear, performed as per manufacturer instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR assay as the standard operating procedures established by Indian Council of Medical Research (ICMR)‐National Institute of Virology (NIV), Pune, India under the Government of India guidelines for COVID19 diagnosis (ICMR‐NIV, 2020)
Threshold not stated Samples used: Nasopharyngeal and throat swabs Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: 25‐84 days All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Supported in part by Indian Council of Medical Research VIR/COVID‐19/02/2020/ECD‐1. Individual authors supported through Dengue Translational Research Consortia National BioPharma Mission, DBT grant, DBT/Wellcome Trust India Alliance Early Career Fellowship grant Publication status: Published paper Source: Virology Author COI: Authors declared no known competing financial interests or personal relationships. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Ng 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection, and to evaluate immune response kinetics and seroprevalence Design: Multi‐group study estimating sensitivity and specificity (possible that [1] and [2] could be considered as a single group, but recruitment was not sufficiently clearly described) [1] RT‐PCR‐confirmed COVID‐19 cases (n = 43 patients) [2]: SARS‐CoV‐2 PCR‐negative UCSF patients (indication for PCR testing was not reported but implied COVID‐19 suspects?) (n = 163 patients for test [A] and 39 patients for test [B]) [3]: Pre‐pandemic controls collected by Abbott Laboratories (US blood donors) (n = 1013 for test [A], n = 1492 for test [B]) Two additional cohorts evaluated for seroprevalence survey not extracted for this review, including [4] patients hospitalised for indications other than COVID‐19 respiratory disease (March‐April 2020) (n = 387, and [5] contemporaneous blood donors (n = 1000) Recruitment: Unclear Prospective or retrospective: Not explicitly stated but likely retrospective Sample size: Covid suspects: 206 (43) for test A and 79 (42) for test B All patients: 1219 (43) for test [A]; 1574 (43) for test [B] Total samples: 1671 for test [A], 1877 for test [B] Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Mixed (outpatient and inpatient) Location: University of California, San Francisco (UCSF) Medical Center and the San Francisco Veterans Affairs (SFVA) Health Care System Country: United States Dates: March‐April 2020 Symptoms and severity: 2 (5%) asymptomatic; 38 (88%) >= 1 symptom (primarily cough, fever, shortness of breath); 3/43 (7%) info not available Severity: 15 (35%) reportedly admitted to ICU, however data by severity exceeded the total number of patients Demographics: 28 (65%) male; mean age 59 yrs (SD 18) Exposure history: Not stated Non‐Covid group 1: Group [2]: SARS‐CoV‐2 PCR‐negative UCSF patients Source: March‐April 2020 at UCSF Medical Center Characteristics: Not stated Non‐Covid group 2: Group [3]: Pre‐pandemic controls (US blood donors) Source: Samples collected by Abbott Labs before the COVID‐19 pandemic (no more details available) Characteristics: Not stated |
||
| Index tests | Test name: [A] Architect SARS‐CoV‐2 IgG assay [B] Architect SARS‐CoV‐2 IgM assay (reported as prototype; not currently commercially available) Manufacturer: Both Abbott Laboratories Antibody: [A] IgG [B] IgM Antigen target: [A] N‐protein [B] S‐protein Evaluation setting: Both Laboratory Test method: CLIA Timing of samples: day 1 to at least day 49 pso (Fig 2 D and E) [A] n samples by days pso: 41 (10%) day 1‐7 (from 16 patients); 106 (25%) day 8‐14 (from 24 patients); 113 (27%) day 15‐21 (from 21 patients); 163 (38%) day 22+ (up to 49) (from 18 patients) [B]: 26/346 (8%) day 1‐7 pso; 91/346 (26%) day 8‐14 pso; 83/346 (24%) day 15‐21 pso; 146/346 (42%) day 22+ pso Samples used: Serum, plasma Test operator: Not stated Definition of test positivity: Methods implied per manufacturer (i.e. IgG positive if Index S/C >= 1.4; IgM S/C >= 0.6) Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR test (no more details available) Samples used: NP and/or OP Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: Group [2]: RT‐PCR (no more details available) Group [3]: Pre‐pandemic Samples used: Group [2]: NP and/or OP Group [3]: NA Timing of reference standard: Not stated Blinded to index test: Yes (done earlier) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes (if we only included group [2], as we considered any RT‐PCR to be ok and 'the same', although that is a bit of a stretch); or, No if we included pre‐pandemic samples from group [3] Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Patients and samples [Fig 1 D and E gives per pt data for cases] |
||
| Comparative | |||
| Notes | Funding: This work was funded by multiple NIH grants and in part by Abbott Laboratories. Funders had no role in the study design, writing the manuscript, or decision to publish. However, employees from Abbott Labs contributed to sample collection, IgG and IgM testing, and data analysis. Publication status: Pre‐print article Source: Pre‐print server (medXriv) Author COI: One author is the director of the UCSF‐Abbott Viral Diagnostics and Discovery Center (VDDC) and receives research support funding from Abbott Laboratories. Five other authors are employees of Abbott Laboratories. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Ng 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nguyen 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent phase infection Design: Single‐group study to estimate sensitivity only [1] Confirmed COVID cases (hospitalised, ICU) (n = 99) Recruitment: Not stated Prospective or retrospective: Prospective Sample size: 99 (99) Further detail: Inclusion: [1] ICU patients presenting with severe SARS‐Cov‐2 infection confirmed by routine RT‐PCR methodology Exclusions not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients (ICU) Location: AP.HP. Centre Cochin university hospital, ICU department, Paris, France CMC Ambroise Paré, ICU department, Neuilly‐sur‐Seine, France Country: France Dates: Not stated Symptoms and severity: ICU patients presenting with severe SARS‐Cov‐2 infection Demographics: Age: mean 62.4 (SD 13.3) years; 34 (34.3%) women; BMI: 29.1 ± 5.9 kg/m2 Chronic immunosuppression: 9 (9.1%) Exposure history: Not stated Non‐Covid group 1: NA Source: NA Characteristics: NA Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: BIOSYNEX COVID‐19 BSS (IgG/IgM)® Manufacturer: Biosynex, Illkirch‐Graffenstaden, France Antibody: IgG/IgM Antigen target: Not stated Evaluation setting: POCT performed as POCT Test method: Lateral flow test (unspecified) Timing of samples: 17.9 ± 8.2 days since pso 0‐10 days: n = 18 11‐20 days: n = 45 21+ days: n = 35 1 sample unclear (only 98 samples in Fig. 1a) Samples used: Finger prick, with 10 μL of blood Test operator: Physicians Definition of test positivity: QC met and POCST showed no IgM and no IgG were considered negative. Positive: QC met and presence of IgM and/or IgG Blinding reported: no (only COVID cases, POCT) Threshold predefined: yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: positive for SARS‐Cov‐2 using routine RT‐PCR methodology, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: yes Missing data: yes, 1 sample seemed to be missing a result. Uninterpretable results: None. 2 (2.0%) in whom quality control was not met; hence, tests required to be performed twice. Indeterminate results: None (no indeterminate range) Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding
Biosynex which freely provided point‐of‐care serology tests Publication status: Published letter Source: Critical Care Author COI: All authors declared no conflict of interest regarding the content of this work. In particular, none had interests with BioSynex. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Nicol 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: The aim of the study was to assess the clinical performance of CE marked assays available in Europe to detect SARS‐CoV‐2 antibodies: two automated immunoassays (Euroimmun and Abbott assays) targeting two different proteins and also one lateral flow immunoassay (NG Biotech).
Multi‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: [1] patients with RT‐PCR‐confirmed SARS‐CoV‐2 infection (n = 82 patients, 141 samples) [2] patients with symptoms consistent with COVID‐19 but RT‐PCR‐negative (clinical diagnosis of pneumonia of unknown aetiology) (n = 52 patients, 57 samples) [3] Pre‐pandemic control group specimens (n = 50 samples) [4] Samples with pathogen potentially cross‐reactive with SARS‐CoV‐2 (n = 25 samples) [5] Samples from pregnant women (n = 10) [6] Samples from patients with positive rheumatoid factor (n = 10) Groups [4] to [6] combined Recruitment: Samples were collected in the virology laboratory of Angers University Hospital, France Prospective or retrospective: Retrospective Sample size: Individuals: 229 (82) Samples: 293 (141) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: [1] Not stated Location: [1] Virology laboratory of Angers University Hospital, France Country: [1] France Dates: Not stated Symptoms and severity: [1] Not stated Demographics: [1] median age: 67 years Exposure history: [1] Not stated Non‐Covid group 1: [2] Pneumonia of unknown aetiology, RT‐PCR‐negative Source: [2] Virology laboratory of Angers University Hospital, France Characteristics: [2] median age: 64 years Non‐Covid group 2: [3] Pre‐pandemic controls [4] Cross‐reactivity samples [5] Pregnant women [6] Patients with rheumatoid factor (RF) Source: [3] March 2019, Virology laboratory of Angers University Hospital, France [4]‐[6] Not stated Characteristics: [3] Not stated [4] Seasonal coronaviruses n = 2, influenza A virus n = 3, respiratory syncytial virus n = 3, rhinovirus n = 3, parainfluenzae virus n = 1, acute EBV infection (positive for EBV VCA IgM and EBV VCA IgG) n = 7, acute CMV infection (positive for CMV IgM) n = 1, M. pneumonia infection n = 2, acute Hepatitis A infection n = 1, acute hepatitis E infection n = 2 [5] Pregnant women [6] Rheumatoid factor |
||
| Index tests | Test name: [A] Abbott SARS‐CoV‐2 CLIA IgG assay [B] to [D] Euroimmun Anti‐SARS‐CoV‐2 ELISA IgG/IgA assays [E] LFIA NG‐Test® IgG‐IgM COVID‐19 Manufacturer: [A] Abbott Diagnostics, IL, USA [B] to [D] Euroimmun, Lübeck, Germany [E] NG Biotech Laboratoires, Guipry‐ Messac, France Antibody: [A] IgG [B] IgG [C] IgG or IgA [D] IgA [E] IgG, IgM Antigen target: [A] SARS‐CoV‐2 nucleoprotein (NP) [B] to [D] recombinant S1 structural protein ‐ assay detects antibodies against the viral spike‐protein [E] SARS‐CoV‐2 nucleoprotein Evaluation setting: [A] Laboratory, used in laboratory [B] to [D] Laboratory, used in laboratory [E] POC, used in laboratory Test method: [A] CLIA assay [B] to [D] ELISA [E] Lateral flow immunoassay (colloidal gold) (CGIA) Timing of samples: [A]‐[C] 0‐> 15 days after onset of symptoms 0‐7 days pso 32/141 8‐14 days pso 29/141 15+ days pso 80/141 [2] median time between symptom onset and sera: 9.5 days 0‐7 days pso 24/57 8‐14 days pso 15/57 15+ days pso 18/57 Samples used: [A]‐[C] Serum Test operator: [A]‐[C] Laboratory personnel Definition of test positivity: [A] cut‐off for positivity = ratio ≥ 1.4 [B] cut‐off for positivity = ratio ≥ 1.1; cut‐off for negativity = ratio < 0.8 [C] Visible lines Blinding reported: [A]‐[C] Unclear Threshold predefined: [A] Yes ‐ "performed according to the manufacturer’s instructions" [B] Yes ‐ "performed according to the manufacturer’s instructions" [C] Yes, visible lines |
||
| Target condition and reference standard(s) | Reference standard: [1] RT‐PCR Samples used: [1] Not reported Timing of reference standard: [1] Not reported Blinded to index test: [1] Yes, prior Incorporated index test: [1] No Definition of non‐COVID cases: [2] RT‐PCR for pneumonia PCR‐negative controls [3] pre‐pandemic [4]‐[6] None (no RT‐PCR detection performed) Samples used: [2] Not stated [3] Pre‐pandemic [4]‐[6] not tested Timing of reference standard: [2] Not stated [3] Pre‐pandemic [4]‐[6] not tested Blinded to index test: [2]‐[6] All prior to index test Incorporated index test: [2]‐[6] No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: To determine the specificity for IgG of the three assays, we excluded two specimens positive for serological assays but negative for RT‐PCR because the symptoms were strongly compatible with the COVID‐19 and RT‐PCR was performed 17–24 days after symptom onset. Uninterpretable results: Not stated Indeterminate results: [C] CLIA ‐ "Grey zone was considered positive for the statistical analyses." Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies. NG‐Test® IgG‐IgM COVID‐19 rapid test cassettes (NG Biotech Laboratoires) were kindly provided by the manufacturer. Publication status: Published paper Source: Journal of Clinical Virology Author COI: The authors declared that they had no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Nicol 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nicol 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nicol 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nicol 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Nilles 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: The study evaluated two commercial (Roche Diagnostics and Epitope Diagnostics IgM/IgG) and two non‐commercial (Simoa and Ragon/MGH IgG) immunoassays against 68 confirmed positive and 232 pre‐pandemic negative controls.
2‐group study to estimate sensitivity and specificity for diagnosis of active disease and identification of previous disease Design: [1] patients that had been hospitalised at the Brigham and Women’s Hospital testing positive by SARS‐CoV‐2 RT‐PCR (n = 28 patients, 68 samples) [2] Pre‐pandemic controls with and without recent respiratory infections (n = 232 patients/samples) Recruitment: Samples from Mass General Brigham Biobank (a biorepository that contains biological samples and linked demographic and clinical data from > 117,000 patients enrolled through the MGB network) Prospective or retrospective: Retrospective Sample size: Patients: 260 (28) Samples: 300 (68) Further detail: Cases = RT‐PCR‐positive To determine if recent respiratory infections may be associated with increased cross‐reactivity and false positives, we selected negative controls with and without recent respiratory infections. We only selected controls with both serum and plasma available from the same individual and time point. |
||
| Patient characteristics and setting | Setting: Hospital Inpatients Location: Brigham and Women’s Hospital (BWH). Samples from Mass General Brigham Biobank Country: USA Dates: March 30 to May 4, 2020 Symptoms and severity: 40/68 samples were from patients who required ICU. Symptoms included cough, fever, dyspnoea, myalgias, new loss of taste or smell, or sore throat. Demographics: Median age of patients was 57 years (range 32‐79) and 35/68 (51%) were female. Race: White 22/68 (32%); black 22/68 (32%); Asian or Pacific Islander 6/68 (9%); American Indian or Alaskan native 3/68 (4%); Other or not recorded 15/68 (22%) Ethnicity: Non‐Hispanic 53/68 (78%); Hispanic 9/68 (13%); other or not recorded 6/68 (9%) Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic controls Source: Samples from Mass General Brigham Biobank. August 28, 2017 to September 26, 2019 Characteristics: The median age was 55 years (range 20‐89) and 90/232 (39%) were female. Of the total 232 negative control samples, 100 were from individuals without recent respiratory illness; 31 from individuals with prior laboratory‐confirmed viral respiratory infections; 101 from individuals with a recent clinical diagnosis of respiratory infections including upper respiratory tract infection (n = 50) or viral (n = 11), bacterial (n = 20) or unspecified (n = 20) pneumonia based on diagnoses recorded in the electronic health record between 1 and 31 days prior to sample collection. Non‐Covid group 2: NA Source: NA Characteristics: NA |
||
| Index tests | Test name: [A] Elecsys Anti‐SARS‐CoV‐2 immunoassay [B] EDI New Coronavirus COVID‐19 IgG ELISAs [C] EDI Novel Coronavirus COVID‐19 IgM ELISA Manufacturer: [A] Roche Diagnostics, Indianapolis, USA [B] Epitope Diagnostics, USA [C] Epitope Diagnostics, USA Antibody: [A] IgG [B] IgG [C] IgM Antigen target: [A] nucleocapsid (NC) antigen (thought to include IgG, IgM, and IgA, although IgM and IgA were not specified in product information) [B] IgG against the NC antigen [C] IgM against an unspecified antigen Evaluation setting: [A] Laboratory, used in laboratory [B] Laboratory, used in laboratory [C] Laboratory, used in laboratory Test method: [A] Electro‐chemiluminescence immunoassay (ECLIA) [B] ELISA [C] ELISA Timing of samples: Samples were collected a mean of 10.5 days (standard deviation 6.0 days) post‐RT‐PCR confirmation and 16.1 days (standard deviation 5.4 days) post‐symptom onset (pso). 8‐14 days pso: 30/68 15‐21 days pso: 29/68 > 21 days pso: 9/68 Samples used: Serum or plasma (different requirements for different tests, but not specified) To ensure valid comparison between assays and given differences in plasma/sera requirements according to manufacturer/assay specifications, we only selected controls with both serum and plasma available from the same individual and time point. All samples were stored at ‐80°C following sample processing and none underwent thaw‐refreezing cycles prior to analysis. Test operator: [A] Brigham and Women’s Hospital laboratories [B] Brigham and Women’s Hospital laboratories [C] Brigham and Women's Hospital laboratories Definition of test positivity: [A], [B] and [C] Threshold cut‐offs for defining positive, negative or indeterminate/borderline test results were defined according to manufacturer specifications for commercial assays. Blinding reported: Yes ‐ "laboratories were blinded to sample group" Threshold predefined: [A] Threshold cut‐offs for defining positive, negative or indeterminate/borderline test results were defined according to manufacturer specifications for commercial assays. [B] and [C] Threshold cut‐offs for defining positive, negative or indeterminate/borderline test results were defined according to manufacturer specifications for commercial assays. |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard: Mean of 5.6 days after onset of symptoms Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated for pre‐pandemic samples
Samples for index test were collected a mean of 10.5 days (standard deviation 6.0 days) post‐RT‐PCR confirmation. All patients received same reference standard: No Missing data: Given limited negative control aliquots, the Epitope assays were tested against 230 samples, versus 232 for the remaining assays. Uninterpretable results: Not stated Indeterminate results: Indeterminate or borderline results were considered negative for all analyses. Unit of analysis: Samples The median number of samples per individual was two (range 1‐5) and the median interval between sample collection was three days (range 2‐6 days). |
||
| Comparative | |||
| Notes | Funding: This work was largely funded by Brigham Health. EN is supported by a CDC U01 GH002238. LB is supported by NIH UM1AI069412 and UL1TR001102. D.S. is supported by NIH K08 AR075850. Publication status: Pre‐print (not peer reviewed) Source: medRxiv preprint Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Nilles 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
NSAE 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Detection of prior infection with SARS‐CoV‐22 Design: Two‐group study to derive sensitivity and specificity [1] Samples from SARS‐CoV‐2 RT‐PCR‐positive participants including patients admitted to hospital or identified through surveillance of HCWs (n = 158) >= 18 years old at Oxford University Hospital NHS Foundation Trust and volunteer plasma donors (n = 378) (total n = 536) via NHS Blood and Transplant, across UK [2] Pre‐pandemic BioBank samples (n = 976) Recruitment: Not reported but appeared consecutive based on reporting of sample inclusion and PRISMA flow diagram Prospective or retrospective: Retrospective Sample size: 1512 (536) Further detail: All samples from individuals > 18 years old. (Four sources of known positives documented: Gastrointestinal illness sub‐study, ISARIC/WHO Clinical Characterisation Protocol for Severe Emerging Infections, Sepsis Immunomics project, and volunteer plasma donors) Inclusion: [1] Healthcare workers and patients >= 18 years old or plasma donors >= 18 years old with a previous positive SARS‐CoV‐2 RT‐PCR nose/throat swab, with blood samples taken ≥ 20 days post‐symptom onset [2] Healthy individuals 30‐50 years old, collected between 2015‐2018 Exclusion: Laboratory labelling mix up: n = 7; date of PCR or symptom onset not recorded: n = 3; samples missing: n = 2; insufficient sample to evaluate across all 4 platforms: n = 5 [1] De‐duplication by individual: n = 96; < 20 days post‐symptom onset/PCR‐positive test: n = 126 |
||
| Patient characteristics and setting | Setting: Mixed Location: Oxford University Hospital NHS Foundation Trust, Oxfordshire, UK Country: United Kingdom Dates: 1/2/20‐31/5/20 Symptoms and severity: [1a] Varied severity at the time of sampling; asymptomatic n = 13, mild n = 122, severe n = 16 and critical/death n = 7 [1b] All convalescent Demographics: Not reported, aged > 18 years Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic samples Source: Oxford BioBank ‐ samples collected between Sept 2014 and Oct 2016 Characteristics: Healthy individuals aged 30‐50 years old Non‐Covid group 2: NA |
||
| Index tests | Test name: [A]‐ SARS‐CoV‐2 IgG assay [B]‐ LIAISON SARS‐CoV‐2 S1/S2 IgG assay [C]‐ Elecsys Anti‐SARS‐CoV‐2 assay [D]‐ SARS‐CoV‐2 Total assay [Additional in‐house assay evaluated 'Oxford immunoassay'; not eligible for this review] Manufacturer: [A]‐ Abbott, Chicago, IL, USA [B]‐ DiaSorin, Saluggia, Italy [C]‐ Roche, Basel, Switzerland [D]‐ Siemens, Munich, Germany Antibody: A & B‐ IgG C & D‐ Total Ab Antigen target: A & C‐ Nucleocapsid B‐ Spike‐Protein S1/S2 D‐ Spike‐Protein S1 RBD Evaluation setting: Laboratory Test method: Not Stated Timing of samples: Of 158 admitted patients and HCWs median 36.5 d pso (IQR 28, 53; range 20 to 73) All 378 volunteer plasma donors were ≥ 28 days pso Samples used: Serum or plasma Test operator: Trained laboratory staff in UK Accreditation Service (UKAS) accredited laboratories: [A] [C] John Radcliffe Hospital Clinical Biochemistry and Microbiologylaboratories in Oxford [B] [D] PHE Porton Down Definition of test positivity: Table S2 appendix [A]‐ Positive: ≥ 1.4 [B]‐ Positive: ≥ 15.0 AU/mL [C]‐ Reactive: ≥ 1.0 [D]‐ Reactive: ≥ 1.0 Blinding reported: Yes Threshold predefined: Yes, as per manufacturers' instructions; alternative thresholds also explored, e.g. to optimise specificity |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; assays used not described Samples used: "Nose or throat swab" Timing of reference standard: Not stated Blinded to index test: Yes, on basis of timing and COVID‐19 group recruited on basis of positive RT‐PCR Incorporated index test: No. Definition of non‐COVID cases: Pre‐pandemic Samples used: NA Timing of reference standard: Pre‐pandemic controls Blinded to index test: Yes, as based on pre‐pandemic controls Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1a] Median 27 (range 3‐59) days (n = 105) [1b] Median 44 (range 32‐82) days All patients received same reference standard: No Missing data: None; stated 'No samples failed testing on any of the four commercial platforms.' Uninterpretable results: None reported Indeterminate results: Equivocal results reported separately; considered as index negative for purposes of this review Unit of analysis: Patients; stated "samples were de‐duplicated by individual, and the latest sample from each individual was analysed". |
||
| Comparative | |||
| Notes | Funding: Public Health England and UK National Institute for Health Research
This work was supported by the UK National Institute for Health Research (NIHR) Health Protection Research Unit in Healthcare Associated Infections and Antimicrobial Resistance at the University of Oxford, Oxford, UK in partnership with Public Health England, and the NIHR Oxford Biomedical Research Centre. Publication status: Published Paper Source: Lancet Infectious Diseases 2020 Author COI: None RC reported personal fees and reported acting as a co‐founder and consultant at MIROBIO, a University of Oxford spinout. The company targets immune inhibitory receptors as treatments for inflammation and auto‐immune disease. This work is unrelated to the serology work. DWE has received lecture fees from Gilead, outside of the submitted work. MGS reported grants from the UK Department of Health and Social Care, National Institute of Health Research UK, Medical Research Council UK, Health Protection Research Unit in Emerging and Zoonotic Infections, University of Liverpool, Liverpool, UK, during the conduct of the study; and acting as a member of the Infectious Disease Scientific Advisory Board to Integrum Scientific, Greensboro, NC, USA, outside of the submitted work. All other authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
NSAE 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
NSAE 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
NSAE 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Ong 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To evaluate flow immunochromatographic assays and an ELISA test for diagnosing COVID‐19. A small pilot study tested sensitivity and specificity of 6 rapid tests, after which the most sensitive was selected for evaluation in a larger cohort, alongside the ELISA test.
3‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: [1] COVID‐19‐positive patients presenting to a teaching hospital with respiratory symptoms that were suspected for respiratory tract infection (N = 99) [2] COVID‐19‐negative patients presenting to a teaching hospital with respiratory symptoms that were suspected for respiratory tract infection (N = 129) [3] randomly selected historical patient control sera (N = 50) Recruitment: consecutive patients presenting to a teaching hospital for prospective patients. No informed consent because tests were performed on samples that had been acquired for routine clinical care. Unclear for historical patient controls ("randomly selected") and retrospective cohort ("selected") Prospective or retrospective: Prospective (6th to 10th April 2020, n = 117) and retrospective (16th to 29th March 2020, n = 117, and September 2019, n = 50) Sample size: 278 (99) Further detail: had respiratory symptoms that were suspected for respiratory tract infection Unclear for historical patient controls ("adult patients in September 2019") |
||
| Patient characteristics and setting | Setting: Hospital A&E Location: A teaching hospital in the Netherlands Country: Netherlands Dates: 17 March 2020 to 10 April 2020 Symptoms and severity: 16/99 (16%) admitted to the ICU within 24 hours Total cohort (COVID +/‐) symptoms (from Supplementary materials): Coughing 68% Dyspnoea 59% Sore throat 17% Rhinorrhoea 15% Fever 48% Demographics: Not reported per group. Whole cohort (positive and negative, n = 228); median age of 61 years (interquartile range (IQR) 46‐74 years), 117 (52%) were male Exposure history: Not stated Non‐Covid group 1: [2] COVID suspects, reference standard‐negative Source: Same hospital as COVID cases, 17 March 2020 to 10 April 2020 Characteristics: Not reported per group. Whole cohort (positive and negative, n = 228); median age of 61 years (interquartile range (IQR) 46‐74 years), 117 (52%) were male Non‐Covid group 2: [3] Pre‐pandemic historic controls Source: September 2019 Characteristics: Adult patients |
||
| Index tests | Test name: [A] Orient Gene Biotech COVID‐19 IgG/IgM Rapid Test Cassette [B] Wantai SARS‐CoV‐2 Ab ELISA kit Manufacturer: [A] Orient Gene Biotech [B] Wantai Antibody: [A] IgG/IgM [B] Total antibody Antigen target: [A] Not stated [B] Not stated Evaluation setting: [A] POC, used in laboratory [B] Laboratory, used in laboratory Test method: [A] Lateral flow immunochromatographic assay [B] ELISA Timing of samples: At same time as nasopharyngeal samples. Median time from symptom onset to sample collection was 7 days (IQR 4‐14 days) for all 228 (positive and negative) patients. < 7 days: 39/99 cases 7+ days: 52/99 cases 14+ days: 14/99 cases 7‐13 days: 38/99 cases Unclear: 8/99 cases Timing of samples: At same time as nasopharyngeal samples. Median time from symptom onset to sample collection was 7 days (IQR 4‐14 days) for all 228 (positive and negative) patients. < 7 days: 39/99 cases 7+ days: 52/99 cases 14+ days: 14/99 cases 7‐13 days: 38/99 cases Unclear: 8/99 cases Samples used: [1] and [2] plasma samples [3] Serum Test operator: [A] Laboratory personnel [B] Laboratory personnel Definition of test positivity: [A] Any visible band for IgG, IgM or unspecified immunoglobulin was indicative for a positive result. [B] Not stated (interpreted according to the manufacturer's instructions) Blinding reported: Both clinical information and reference standard results were unavailable to the performers of LFAs and the ELISA. Threshold predefined: [A] Visual [B] interpreted according to the manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: PCR (referred to as PCR in Supplementary materials and as NAT in paper): Nucleic acid amplification tests performed according to the national reference method that was established after international collaboration, or by the CE‐IVD kit Gene‐ FinderTM COVID‐19 Plus RealAmp Kit using the Sample to Result Platform ELITe InGenius®. Samples used: Samples were taken from the oral cavity and subsequently from the nasal cavity using the same nasopharyngeal swab. In some cases, sputum samples were tested, because of persisting clinical suspicion of COVID‐19 despite a negative NAT on nasopharyngeal swabs. Timing of reference standard: Median time from symptom onset to sample collection was 7 days (IQR 4‐14 days) for all 228 (positive and negative) patients. < 7 days: 39/99 cases 7+ days: 52/99 cases 14+ days: 14/99 cases 7‐13 days: 38/99 cases Unclear: 8/99 cases Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: [2] COVID suspects = same nucleic test as COVID cases Nucleic acid amplification tests performed according to the national reference method that was established after international collaboration, or by the CE‐IVD kit Gene‐ FinderTM COVID‐19 Plus RealAmp Kit using the Sample to Result Platform ELITe InGenius®. [3] Historic controls = pre‐pandemic Samples used: [2] COVID suspects = nasopharyngeal swab Samples were taken from the oral cavity and subsequently from the nasal cavity using the same nasopharyngeal swab. In some cases, sputum samples were tested, because of persisting clinical suspicion of COVID‐19 despite a negative NAT on nasopharyngeal swabs. [3] Historic controls = pre‐pandemic Timing of reference standard: [2] COVID suspects = Median time from symptom onset to sample collection was 7 days (IQR 4‐14 days) for all 228 (positive and negative) patients. < 7 days: 40/129 cases 7+ days: 50/129 cases 14+ days: 32/129 cases 7‐13 days: 18/129 cases Unclear: 39/129 cases [3] Historic controls = pre‐pandemic Blinded to index test: [2] Not stated [3] Yes (pre‐pandemic) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1] and [2] none ‐ "plasma samples obtained upon hospital presentation, which corresponded to the dates of molecular testing." [3] Pre‐pandemic samples All patients received same reference standard: Yes (cohort), No (historic controls) Missing data: 5/228 samples were unavailable for ELISA. In some patients, time from symptom onset was undetermined or unavailable. Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding Publication status: Published paper Source: Clinical Microbiology and Infection Author COI: The authors declared no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ong 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Padoan 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Single‐group study recruiting patients estimating sensitivity Design: [1] Hospitalised patients with confirmed COVID‐19 Recruitment: cases with residual serum samples collected between 18 March‐26 March 2020 Prospective or retrospective recruitment of cases: retrospective Sample size (virus/COVID cases): 37 (37) Inclusion and exclusion criteria: Not stated | ||
| Patient characteristics and setting | Setting: inpatient Location: University Hospital of Padova Country: Italy Dates: 18 March‐26 March 2020 Symptoms and severity: NR Sex: NR Age: NR Exposure history: NR | ||
| Index tests | Test name: MAGLUMI 2000 Plus nCoV IgM and IgG Manufacturer: New Industries Biomedical Engineering Co., Ltd [Snibe], Shenzhen, China Ab targets: IgM; IgG Antigens used: NR Test method: CLIA Timing of samples: days since symptom onset: ≤ 5 days 4/37 (11%) 6‐7 days 6/37 (16%) 0‐7 days: 10/37 (27%) 8‐9 days 12/37 (32%) 10‐11 days 14/37 (38%) 12‐13 days 9/37 (24%) 8‐13 days: 35/37 (95%) > 13 days 25/37 (68%) Samples used: serum Test operators: NR Definition of test positivity: [A] IgM 1.0 AU/mL [B] IgG 1.1 AU/mL Blinded to reference standard: no Threshold predefined: yes | ||
| Target condition and reference standard(s) | Reference standard for cases: PCR Samples used: NP Timing of reference standard: NR Blinded to index test: yes Incorporated index test: no Reference standard for non‐cases: NA | ||
| Flow and timing | Time interval between index and reference tests: NR Results presented by time period: yes All participants received the same reference standard: yes Missing data: text described 87 samples from 37 participants but only 70 samples reported per time period and no per participant data were reported Uninterpretable results: NR Indeterminate results: NR Unit of analysis: sample | ||
| Comparative | |||
| Notes | Funding: none declared Publication status: published Source: Clinical Chemistry and Laboratory Medicine Study author COI: none declared | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Padoan 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase infection and antibody kinetics over time Design: Single‐group study to estimate sensitivity: [1] adult patients with PCR‐confirmed COVID‐19 (total N was not reported, 51 assessed for IgM and 19 assessed for IgA; any overlap between patient groups was not reported) The report contained two groups of COVID‐19 patients, one was assessed for IgM using CLIA and the other for IgA using ELISA. There was no non‐COVID‐19 or healthy control group. [1] Severely sick adult COVID‐19 (rRT‐PCR‐confirmed) patients longitudinally assessed for IgM using CLIA (n = 51) [2] Severely sick adult COVID‐19 (rRT‐PCR‐confirmed) patients longitudinally assessed for IgA using ELISA (n = 19) Recruitment: Unclear Prospective or retrospective: Not stated Sample size: Unclear; 51 reported for IgM and 19 for IgA; overlap not reported Further detail: No further details |
||
| Patient characteristics and setting | Setting: Not stated; 'patients', presumably inpatient as serial testing Location: University‐Hospital of Padova Country: Italy Dates: Not stated Symptoms and severity: Discussion described patients as 'severely sick' Demographics: n = 51; 37, 72.5% male; mean age, men 69.1 y (SD 13.5), range 22–89 y; women 62.6 y (SD 11.0), range 41–82 y; n = 19; 15, 79% male; mean age, men 65.4 y (SD 14.5), range 22–81 y; women 63.7 y (SD 7.8) range 53–70 y Exposure history: Not stated |
||
| Index tests | Test name: [A] MAGLUMI 2000 Plus [B] ELISA Manufacturer: [A] Not stated (manufacturer was SNIBE diagnostics) [B] Euroimmun Medizinische Laboradiagnostika, Luebeck, Germany Antibody: [A] IgM (IgG also measured, limited details in supplementary information) [B] IgA (IgG also measured, results not reported) Antigen target: [A] S‐antigen and N‐protein [B] S1‐specific IgA and IgG Evaluation setting: Laboratory Test method: [A] chemiluminescent (CLIA) assay [B] ELISA Timing of samples: from the onset of symptoms (fever) to 6 weeks after Samples used: Not stated Test operator: Not stated Definition of test positivity: 1. CLIA IgM cut‐off: 1.0 kAU/L 2. ELISA IgA cut‐off: ratio ≥ 1.1 Blinding reported: Not stated Threshold predefined: Yes, as previously published |
||
| Target condition and reference standard(s) | Reference standard: rRT‐PCR, no further details Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: Not stated |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Unclear; data for all reported patients seemed to be used to estimate mean titres over time. A subgroup of 18 patients with more than 3 serial measurements was also reported. The number of patients contributing data from day 0 to 23 in Tabl 1 was not reported, however, at each time point, results were presented for fewer (16‐58%) than the total 19 participants for the IgA ELISA and total 51 participants for IgM CLIA. Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients; however reported time periods were short (2‐day span), therefore, when results were combined to allow analysis per week post‐symptom onset, patients could contribute more than one sample per week. |
||
| Comparative | |||
| Notes | Funding: Not stated, except: "We acknowledge the support of Euroimmun Medizinische Laboradiagnostika, Luebeck, Germany for kindly supplying the reagents without any influence in study design and data analysis." Publication status: Published paper Source: International Journal of Clinical Chemistry Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Padoan 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Paiva 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Multi‐group study to estimate sensitivity and specificity for diagnosis of active disease Design: [1] RT‐PCR‐positive COVID‐19 cases (n = 71, 113 samples) [2] Healthy individuals (n = 126) [3] Samples positive for other viruses and pathogens (to test cross‐reaction of the assays) (n = 119) [4] serum or plasma samples collected before the pandemic started in the United States (n = 942) Recruitment: [1] Remnant/discarded serum or plasma samples were collected from the Clinical Immunology Lab at a major academic pathology department [2] Recruitment unclear ‐ random at pre‐employment screening (table 3) [3] Unclear ‐ probably also from the same laboratory as the COVID‐19 samples [4] Pre‐pandemic: 500 samples originally for reference range determination of a troponin assay, 371 prenatal samples for reference range determination of quadruple tests, 50 pre‐pandemic samples from transfusion service and 21 pre‐pandemic plasma segments from the Rhode Island Blood Center. Prospective or retrospective: Retrospective Sample size: 1300 (113) (samples) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Lifespan Health System (including Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island) Country: USA Dates: After 12 March 2020 Symptoms and severity: Highest level of treatment: Room air 26%; nasal cannula 45%; intubation 27%; extracorporeal membrane oxygenation 1% Demographics: 41% female; mean age 59.5 ± 1.9 White or Caucasian: 38/71 (53%) Hispanic or Latino: 22/71 (31%) African‐American: 10 (14%) Asian: 1/71 (1%) Exposure history: Not stated Non‐Covid group 1: [2] Current healthy controls Source: Pre‐employment screening before 12 March 2020 (early March 2020) Characteristics: Not stated Non‐Covid group 2: [3] Current, other viruses and pathogens [4] Pre‐pandemic healthy controls Source: [3] Collected from later March to early April 2020 [4] Collected before the pandemic started in the United States (before January 2020): Collected originally for: troponin study, prenatal plasma for quadruple test, transfusion service, Rhode Island Blood Center Characteristics: [3] The set included samples from patients testing positive for upper respiratory viruses and samples known to contain antibodies such as rheumatoid factor (RF), anti‐double stranded NA (ds‐DNA), antinuclear antibody (ANA), and paraprotein IgM and IgG Seasonal coronaviruses (n = 21): Other upper respiratory tract viruses (n = 27; influenza, metapneumovirus, rhinovirus/enterovirus, respiratory syncytial viruses and adenovirus) Samples with positive IgG or IgM against varicella zoster virus, rubella, Epstein‐Barr virus, cytomegalovirus (CMV) and hepatitis viruses (n = 71) [4] Not stated |
||
| Index tests | Test name: [A] SARS‐CoV‐2 Total Antibody Test [B] STANDARD Q COVID‐ 19 IgM/IgG Duo Test [C] SARS‐CoV‐2 IgG test Manufacturer: [A] Wondfo, Guangzhou, China [B] SD Biosensor, Gyeonggi‐do, Korea [C] Abbott Diagnostics, Lake Forest, IL Antibody: [A] Total antibody (IgM and IgG) [B] IgM/IgG [C] IgG Antigen target: [A] Spike‐protein [B] Not stated [C] Nucleocapsid protein Evaluation setting: [A] POC, used in laboratory [B] POC, used in laboratory [C] Laboratory test, used in laboratory Test method: [A] Lateral flow assay (no further detail) [B] Lateral flow assay (no further detail) [C] Chemiluminescent assay Timing of samples: [1] 1‐35 (38) days post‐symptom onset (mean 11.2) [2] NA [3] Unclear [4] NA Samples used: [A, B, C] serum (n = 16) or plasma (n = 97) for [1], serum for [2] and [3], plasma for [4] Test operator: [A] and [B] The reading was performed by three investigators (KJP, EWT, and SL) affiliated to the Department of Pathology and Laboratory Medicine, Warren Alpert Medical School of Brown University. Definition of test positivity: [A] and [B] positive result was indicated by a visible band in the designated area accompanied with an appropriate control band. The reading was performed by three investigators; consensus for any ambiguous results was obtained by at least two investigators. [3] Samples with signal‐to‐cut‐off (S/CO) ratio greater than or equal to 1.4 were considered positive. Blinding reported: Unclear Threshold predefined: Yes, by manufacturers |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR [ePlex® SARS‐CoV‐2 Test (GenMark, Carlsbad, CA) or Cobas® SARS‐CoV‐2 Test (Roche, Indianapolis, IN). The upper respiratory virus testing was performed on ePlex® Respiratory Pathogen Panel (GenMark)] Samples used: nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: [2] healthy controls ‐ not stated (pre‐employment screening in early March 2020; before the first COVID‐19 case was diagnosed in the Lifespan Health System) [3] Other viruses and pathogens ‐ viral respiratory pathogen nucleic acid test [the upper respiratory virus testing was performed on ePlex® Respiratory Pathogen Panel (GenMark)] [4] pre‐pandemic (before January 2020). [2] ‐ [4] The patients whose samples were reactive [positive result on index test] were followed by medical record review to ensure that they did not have COVID‐19. Samples used: Not stated Timing of reference standard: [2] and [3] Not stated (for index test positives, follow‐up history of 17‐38 days from medical records) [4] pre‐pandemic (before January 2020) Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not all the samples were available for the four tests (for the Abbott test, 3/1068 healthy samples, 1/105 COVID‐19 samples and 1/119 potential cross‐reactions missing; for SD IgM, 1/119 potential cross‐reactions missing; for SD IgG, 3/119 potential cross‐reactions missing) For sensitivity: 105 of 113 samples were selected to evaluate antibody‐positive rates every 2 days. 8 samples tested in the same 2 days were not used. Out of 8 patients with cross‐reactive results, 2 patients did not have follow‐up history in their medical records. Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding:The project was funded by Pathology Department of Lifespan Academic Center and Rhode Island Department of Health. Publication status: Pre‐print (not peer reviewed); now published Source: bioRxiv preprint doi: https://doi.org/10.1101/2020.05.29.124776; Journal of Medical Virology Author COI: The authors claimed no conflict of financial interest related to the project. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Paiva 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Paiva 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pan 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Single group of cases to estimate sensitivity in acute disease Design: SARS‐CoV‐2‐positive cases (n = 105, 134 samples) of which 67 cases (86 samples) confirmed by RT‐PCR, and 37 patients (39 samples) clinically diagnosed (RT‐PCR‐negative, radiography‐positive) Recruitment method: NR Exclusion criteria: NR | ||
| Patient characteristics and setting | Setting: Inpatients Location: Zhongnan hospital (Wuhan University) Country: China Dates: Testing 6 February‐23 February 2020, symptom onset 7 January‐18 February 2020 (for subgroup of 108) Participant characteristics: 48 male, 57 female, median age 58 years (range 20‐96) Symptoms and severity: NR Exposure status: NR |
||
| Index tests | Test name: Zhuhai Livzon Commercial Ab test
Manufacturer: Zhuhai Livzon Diagnositic Inc
Test method: LFA (conducted in laboratory setting). Colloidal gold‐based immunochromatographic strip assay Antibody target: IgM, IgG Antigen used: NR (as per manufacturer) Definition of positive: Presence of T line indicating positive Samples used: Serum or plasma samples used (included comparison with whole blood for subgroup; not extracted) Timing: No information on timing or who read the test results. |
||
| Target condition and reference standard(s) | Reference standard: 1. RT‐PCR following WHO guidelines for qRT‐PCR, using throat swabs (Chinese CDC recommended kit used, BioGerm, Shanghai, China) 2. clinically diagnosed as SARS‐CoV‐2 infection according to the 5th edition of guideline on diagnosis and treatment of the novel coronavirus pneumonia. Specifically, the clinical diagnosis means the suspected cases were negative to the real‐time RT‐PCR test but presented with viral pneumonia by radiography. Timing: Samples taken during inpatient stay but no details about timing or personnel for test interpretation | ||
| Flow and timing | Same reference standard: All participants received a reference standard, but there was differential verification with some patients confirmed by RT‐PCR and others RT‐PCR‐negative but confirmed by radiography. Subset who were RT‐PCR‐positive were reported separately. Timing of index tests and reference standard unclear Data reported only for those with symptom onset information; 26 samples excluded. No reporting of test failures or indeterminate results Unit of analysis: Per‐sample analysis; multiple samples (2 or 3) per participant disaggregated over time | ||
| Comparative | |||
| Notes | Funding: from the National Key Research and Development Program of China (2018YFE0204500) Author COI: Declared no conflict of interest Published in the Journal of Infection | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Pape 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to estimate sensitivity and specificity for diagnosis of active disease/identification of previous disease Design: [1] SARS‐ CoV‐2 positive sera were collected from PCR‐confirmed symptomatic COVID‐19 patients (n = 29, 57 samples, cohort C) [2] Pre‐pandemic negative control serum samples collected for various serological testing before the start of the SARS‐CoV‐2 outbreak (n = 218) ‐ healthy donors (n = 105, cohort B), patients that tested positive for common cold Corona viruses several months before the blood sample was taken (n = 34, all four types of ccCoV represented; cohort A), patients with diagnosed mycoplasma pneumoniae (n = 22; cohort Z), EBV or CMV infection (n = 57, cohort E) Recruitment: [1] SARS‐ CoV‐2 positive sera were collected from 29 PCR‐confirmed symptomatic COVID‐19 patients treated at the University Hospital Heidelberg. [2] Negative control serum samples were collected for various serological testing in the routine laboratory of the Center of Infectious Diseases, University Hospital Heidelberg between 2015 and 2019, before the start of the SARS‐CoV‐2 outbreak. Prospective or retrospective: [1] Unclear [2] Retrospective Sample size: People = 247 (29) ; samples = 275 (57) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: inpatients (n = 17) and outpatients (n = 12) Location: University Hospital Heidelberg Country: Germany Dates: Not stated Symptoms and severity: Not stated other than inpatients (n = 17) and outpatients (n = 12) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic controls Source: Negative control serum samples (n = 218) were collected for various serological testing procedures in the routine laboratory of the Center of Infectious Diseases, University Hospital Heidelberg between 2015 and 2019, before the start of the SARS‐CoV‐2 outbreak. Characteristics: Healthy 105/218 Positive for ccCoV several months before the blood sample was taken 34/218 Patients with diagnosed mycoplasma pneumoniae 22/218. Patients with EBV or CMV infection 57/218 |
||
| Index tests | Test name: [A] Euroimmun Anti‐SARS‐CoV‐2‐ELISA (IgA) [B] Euroimmun Anti‐SARS‐ CoV‐2‐ELISA (IgG) Manufacturer: [A] [B] Euroimmun, Lübeck, Germany Antibody: [A] IgA [B] IgG Antigen target: [A] [B] S1 domain of the viral spike‐protein Evaluation setting: Laboratory, used in laboratory Test method: ELISA Timing of samples: 5‐27 days post‐symptom onset 5‐11 days pso: 17/57 11‐14 days pso: 24/57 15‐27 days pso: 16/57 Samples used: Serum Test operator: Laboratory personnel Definition of test positivity: Optical densities (sample normalised to calibrator): values < 0.8 were classified as negative, 0.8‐1.1 as borderline, and values of 1.1 or higher as positive. For sensitivity or specificity calculations, borderline considered positive Blinding reported: Unclear Threshold predefined: As per manufacturer guidance |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA Timing of reference standard: Pre‐pandemic Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: For sensitivity or specificity calculations, borderline considered positive Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was in part supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and by the Deutsches Zentrum fuer Infektionsforschung. SB is supported by the Heisenberg programme and MLS is supported by the DFG. Open access funding enabled and organised by Projekt DEAL. Publication status: Pre‐print (not peer reviewed). Now published Source: bioRxiv preprint doi: https://doi.org/10.1101/2020.06.15.152587. Journal (BioEssays) Author COI: The authors declared they have no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Pape 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Patel 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of convalescent‐phase infection Design: Two‐group study to assess sensitivity and specificity for 5 commercially available serology assays [1] Covid‐19 convalescent plasma donors (n = 214 potential) [2] Pre‐pandemic samples from emergency department patients (n = 1099) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 1313 (214) Further detail: Inclusion: [1] Stored plasma specimens from a "convenience sample" of potential CCP donors that were recruited in the Baltimore, MD and Washington, DC areas from April 2020 to July 2020. Individuals were eligible for enrolment if they had a documented history of a positive molecular assay test result for SARS‐CoV‐2 infection and met standard self‐reported eligibility criteria for blood donation. [2] Stored serum specimens from an identity‐unlinked HIV serosurvey conducted in 2016 among adult patients attending the Johns Hopkins Hospital Emergency Department Exclusion: Not stated |
||
| Patient characteristics and setting | Setting: Community Location: Baltimore MD and Washington DC area Country: USA Dates: April 2020‐July 2020 Symptoms and severity: 16/214 required hospitalisation Demographics: Different samples used for different assays: [A]‐ n = 146, median age (IQR) = 41 (29‐53), male n (%) = 78 (53.4), white race n (%) = 112 (76.7), hospitalised n (%) = 12 (8.2), median days since PCR‐positive test (IQR) = 44 (39‐51) [B]‐ n = 146, median age (IQR) = 41 (29‐53), male n (%) = 78 (53.4), white race n (%) = 112 (76.7), hospitalised n (%) = 12 (8.2), median days since PCR‐positive test (IQR) = 44 (39‐51) [C]‐ n = 140, median age (IQR) = 40 (29‐53), male n (%) = 76 (54.3), white race n (%) = 108 (77.1), hospitalised n (%) = 12 (8.6), median days since PCR‐positive test (IQR) = 44 (38‐50) [D]‐ n = 146, median age (IQR) = 41 (29‐53), male n (%) = 78 (53.4), white race n (%) = 112 (76.7), hospitalised n (%) = 12 (8.2), median days since PCR‐positive test (IQR) = 44 (39‐51) [E]‐ n = 214, median age (IQR) = 44 (33‐56), male n (%) = 110 (51.4), white race n (%) = 165 (77.1), hospitalised n (%) = 16 (7.5), median days since PCR‐positive test (IQR) = 46 (39‐57) Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic controls Source: Stored serum specimens from an identity‐unlinked HIV serosurvey conducted in 2016 among adult patients attending the Johns Hopkins Hospital Emergency Department Characteristics: Different samples used for different assays: [A]‐ n = 561; median age (IQR) = 49 (32‐60); male n (%) = 247 (44.0); race/ethnicity: Non‐Hispanic white n (%) = 161 (28.7), Non‐Hispanic black n (%) = 345 (61.5), Hispanic n (%) = 19 (3.4), non‐Hispanic Asian n (%) = 10 (1.8), other n (%) = 26 (4.6); HIV Ab‐positive n (%) = 22 (3.9) [B]‐ n = 577; median age (IQR) = 48 (32‐60); male n (%) = 251 (43.5); race/ethnicity: Non‐Hispanic white n (%) = 166 (28.8), Non‐Hispanic black n (%) = 353 (61.2), Hispanic n (%) = 21 (3.6), non‐Hispanic Asian n (%) = 10 (1.7), other n (%) = 27 (4.7); HIV Ab‐positive n (%) = 26 (4.5) [C]‐ n = 306; median age (IQR) = 47 (31‐59); male n (%) = 135 (44.1); race/ethnicity: Non‐Hispanic white n (%) = 80 (26.1), Non‐Hispanic black n (%) = 191 (62.4), Hispanic n (%) = 12 (3.9), non‐Hispanic Asian n (%) = 7 (2.3), other n (%) = 16 (5.2); HIV Ab‐positive n (%) = 19 (6.1) [D]‐ n = 498; median age (IQR) = 45 (30‐59); male n (%) = 209 (42.0); race/ethnicity: Non‐Hispanic white n (%) = 130 (26.1), Non‐Hispanic black n (%) = 313 (62.9), Hispanic n (%) = 29 (5.8), non‐Hispanic Asian n (%) = 6 (1.2), other n (%) = 20 (4.0); HIV Ab‐positive n (%) = 19 (3.8) [E]‐ n = 498; median age (IQR) = 45 (30‐59); male n (%) = 209 (42.0); race/ethnicity: Non‐Hispanic white n (%) = 130 (26.1), Non‐Hispanic black n (%) = 313 (62.9), Hispanic n (%) = 29 (5.8), non‐Hispanic Asian n (%) = 6 (1.2), other n (%) = 20 (4.0); HIV Ab‐positive n (%) = 19 (3.8) |
||
| Index tests | Test name: [A]‐ Anti‐SARS‐CoV‐2 ELISA (IgG) [B]‐ EDI novel coronavirus COVID‐19 IgG ELISA kit [C]‐ SARS‐CoV‐2 NP IgG ELISA kit [D]‐ Abbott‐Architect SARS‐CoV‐2 IgG assay [E]‐ Elecsys anti‐SARS‐CoV‐2 Manufacturer: [A]‐ Euroimmun, Lubeck, Germany [B]‐ Epitope Diagnostics, Inc. (EDI), San Diego, CA, USA [C]‐ ImmunoDiagnostics Limited, Sha Tin, Hong Kong [D]‐ Abbott Laboratories Inc., Abbott Park, IL, USA [E]‐ Roche Diagnostics, Indianapolis, IN, USA Antibody: [A] to [D] ‐ IgG; [E]‐ Total Antibodies Antigen target: [A]‐ Spike 1‐Protein; [B] to [E] ‐ Nucleocapsid Protein Evaluation setting: Laboratory tests in laboratory Test method: [A] to [C] ‐ Manual ELISA [D]‐ Chemiluminescent microparticle immunoassay (CMIA) [E]‐ Electrochemiluminescence assay (ECLIA) Timing of samples: 38‐57 days post‐PCR + Samples used: Plasma/serum Test operator: Not stated Definition of test positivity: [A]‐ Negative, S/C ratio < 0.8; borderline, S/C ratio => 0.8 & < 1.1; positive, S/C ratio => 1.1 [B]‐ Negative, OD‐n =< 0.18; borderline, OD‐n > 0.18 & < 0.22; positive, OD‐n => 0.22 [C]‐ Negative, OD‐n < 0.15; borderline, OD‐n => 0.25 & =< 0.50; positive, OD‐n > 0.50 [D]‐ Negative, index (S/C) < 1.40; positive, index (S/C) => 1.40 [E]‐ Nonreactive, index < 1.0; reactive => 1.0 Borderline results considered seronegative Threshold predefined: Yes, manufacturer's cut‐off values used |
||
| Target condition and reference standard(s) | Reference standard: Positive molecular assay test Samples used: Not stated. Timing of reference standard: Not stated. Blinded to index test: Yes, based on timing of test (prior molecular confirmation of SARS‐CoV2 infection was required to be recruited to COVID‐19 group). Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic samples Samples used: NA Timing of reference standard: Pre‐pandemic controls Blinded to index test: Yes, as based on pre‐pandemic controls Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [A]‐ Median days since PCR+ test (IQR)‐ 44 (39‐51) [B]‐ Median days since PCR+ test (IQR)‐ 44 (39‐51) [C]‐ Median days since PCR+ test (IQR)‐ 44 (38‐50) [D]‐ Median days since PCR+ test (IQR)‐ 44 (39‐51) [E]‐ Median days since PCR+ test (IQR)‐ 46 (39‐57) All patients received same reference standard: No Missing data: All 214 Covid samples tested only on [E] Uninterpretable results: Not stated Indeterminate results: Borderline/indeterminate results treated as seronegative Unit of analysis: Patients. No individual contributed multiple specimens |
||
| Comparative | |||
| Notes | Funding: This work was supported in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), as well as by extramural support from NIAID and NIH Center of Excellence in Influenza Research and Surveillance; National Heart Lung and Blood Institute; National Institute of Drug Abuse; Bloomberg Philanthropies; and the Department of Defense. Publication status: Published paper Source: Journal of Clinical Microbiology Author COI: Authors declared no potential conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Patel 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Patel 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Patel 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Patel 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pere 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase infection and convalescent infection: analytically and clinically validate the Abbott SARS‐CoV‐2 IgG assay Design: Multi‐group study to establish sensitivity and specificity [1] Confirmed COVID‐19 patients [1a] COVID‐19 convalescent healthcare workers (n = 100) [1b] Hospitalised patients from COVID+ area (n = 63) [2] Non‐COVID patients [2a] Pre‐pandemic, other diseases (n = 117) [2b] Hospitalised patients from COVID‐free area (n = 96) Groups [1b] and [2b] were not eligible for our review. Recruitment: [1a] Cases ‐ not stated [2a] Controls ‐ October 2019 to January 2020; from patients randomly selected for whom serum samples were collected before the COVID‐19 epidemic Prospective or retrospective: Retrospective Sample size: 376 (163) of which 217 (100) were eligible for our review Further detail: Inclusion ‐ [1a] Hospital staff with a history of positive SARS‐CoV‐2 RT‐PCR at least 1 month before serology testing [2a] Leftover sera from pre‐epidemic period (collected from October 2019 to January 2020), available at the virology laboratory of Hôpital Européen Georges Pompidou No exclusion criteria defined |
||
| Patient characteristics and setting | Setting: [1a] Convalescent (specimens were collected by occupational medicine; hospital outpatients) Location: Hôpital Européen Georges Pompidou (HEGP), Assistance Publique Hôpitaux de Paris, Paris Country: France Dates: Not specified Health staff who had recovered from COVID‐19 Symptoms and severity: Pauci‐symptomatic (only 2 were hospitalised and 98 were not hospitalised and had only few symptoms) Demographics: Cases Median age (IQR) ‐ 34 (19.5) Male ‐ 31% Exposure history: Healthcare workers Non‐Covid group 1: [2a] Pre‐pandemic leftover sera Source: Virology laboratory of HEGP October 2019 to January 2020 Characteristics: Some of these sera came from patients with recent clinical history of viral respiratory infection including common coronaviruses (229E n = 2; NL63 n = 4; OC43 n = 1) as well as clinical history of malaria (n = 6). |
||
| Index tests | Test name: Abbott SARS‐CoV‐2 IgG assay Manufacturer: Abbott GmbH, Rungis, France Antibody: IgG Antigen target: SARS‐CoV‐2 nucleoprotein Evaluation setting: Laboratory Test method: Not stated (Abbott Architect™ i2000) Timing of samples: [1a] Cases ‐ 39.5 (median) days after PCR, at least 1 month after COVID diagnosis Samples used: Serum Test operator: Technicians at the Clinical Biochemistry department of Hôpital Européen Georges Pompidou Definition of test positivity: Index value threshold for positivity was 1.4. Blinding reported: Not stated Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: [1a] COVID‐19 by RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes (based on timing of tests) Incorporated index test: No Definition of non‐COVID cases: [2a] Pre‐pandemic Samples used: pre‐pandemic Timing of reference standard: pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1a] Cases ‐ Median interval between RT‐PCR and serology was 39.5 days (IQR = 9.25). [2a] Not stated All patients received same reference standard: No Missing data: Yes, groups [1b] and [2b] excluded from review Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None Publication status: Published paper Source: Journal of Clinical Virology Author COI: None |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Perez‐Garcia 2020(a).
| Study characteristics | |||
| Patient Sampling | Purpose: Study aimed to evaluate the diagnostic performance of a serologic rapid test in COVID‐19‐positive patients, COVID‐19‐negative patients with pneumonia, and pre‐pandemic patients.
Three‐group study to estimate sensitivity and specificity for diagnosis of active disease and identification of previous disease. Design: [1] randomly selected group of pre‐pandemic patients who had a serum sample taken for other serologic studies (n = 100) [2] patients admitted to the Emergency department with suspicion of COVID‐19 and PCR‐positive for SARS‐CoV‐2 (n = 90) [3] patients admitted for at least 5 days with a clinical and radiological diagnosis of pneumonia of unknown aetiology, PCR‐negative for SARS‐CoV‐2 (n = 61) Recruitment: [1] a randomly selected group of 100 pre‐pandemic serologic samples [2] patients admitted to the Emergency department with suspicion of COVID‐19 and PCR‐positive for SARS‐CoV‐2 [3] patients admitted for at least 5 days with pneumonia of unknown aetiology and a clinical diagnosis of COVID‐19 with negative PCR for SARS‐CoV‐2 (included as Perez‐Garcia 2020(b) Prospective or retrospective: [1] and [2] Retrospective ("Since the present study is retrospective, informed consent was not required.") [3] Prospective ("Fresh serum samples from these 61 patients were studied." "They were prospectively studied after the validation of the serologic test.") Sample size: 251 (151) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: [2] ED [14 (15.6 %) of them were discharged from ED, remaining 76 (84.4 %) patients were admitted to our hospital and 11 (14.5 %) required ICU admission] [3] inpatient Location: [2] and [3] Hospital Universitario Príncipe De Asturias, Madrid, Spain Country: [2] and [3] Spain Dates: [2] March 1 to April 6, 2020 [3] February 9 to April 2, 2020 Symptoms and severity: [2] Mild: 17/90 (18.9%) Non‐severe pneumonia: 47/90 (52.2%) Severe pneumonia: 20/90 (22.2%) Critical: 6/90 (6.7%) (3 ARDS and 3 with septic shock) [3] Mild: 0/61 (0.0%) Non‐severe pneumonia: 40/61 (65.6%) Severe pneumonia: 20/61 (32.8%) Critical (ARDS): 1/61 (1.6%) Demographics: [2] Age: median (IQR) 64 (55−79); 57.8% (52/90) male [3] Age: median (IQR) 67 (57‐73); 73.8% (45/61) male Exposure history: [2] and [3] Not stated Non‐Covid group 1: 1 Pre‐pandemic controls Source: patients who had a serum sample taken for other serologic studies, from September 1 to November 30, 2019 Characteristics: Age: median (IQR) 50 (33−65); 55% male |
||
| Index tests | Test name: AllTest COV‐19 IgG/IgM kit Manufacturer: AllTest Biotech, Hangzhou, China Antibody: IgG, IgM Antigen target: Unclear Evaluation setting: POC, performed in laboratory ("aliquots were previously obtained from samples sent to the laboratory to carry out other serologies") Test method: lateral flow immunoassay, LFA Timing of samples: [1] NA (pre‐pandemic) [2] median (IQR) days from symptom onset = 17 (9‐25) <= 7 days pso: 19/90 8‐14 days pso: 21/90 15‐21 days pso: 15/90 22‐28 days pso: 20/90 28 days pso: 15/90 [3] median (IQR) days from symptom onset = 17 (15‐20) <= 7 days pso: 0/61 8‐14 days pso: 15/61 15‐21 days pso: 31/61 22‐28 days pso: 14/61 28 days pso: 1/61 Samples used: Serum Test operator: Unclear Definition of test positivity: Visual Blinding reported: Unclear Threshold predefined:Yes, visual‐based. |
||
| Target condition and reference standard(s) | Reference standard: [2] and [3] RT‐PCR: VIASURE SARS‐CoV‐2 Real Time PCR Detection Kit (Certest Biotech, Zaragoza, Spain) and Allplex 2019‐nCoV assay (Seegene, Seoul, South Korea)
[3] Clinical diagnosis of COVID‐19 with negative PCR for SARS‐CoV‐2. Criteria for diagnosis not stated Samples used: [2] and [3] Unclear ‐ "clinical samples" Timing of reference standard: [2] and [3] Unclear Blinded to index test: [2] and [3] Yes, prior to index test Incorporated index test: [2] and [3] No Definition of non‐COVID cases: [1] Pre‐pandemic = not tested Samples used: [1] pre‐pandemic Timing of reference standard: [1] Pre‐pandemic samples Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors. Publication status: Published paper Source: Journal of Clinical Virology Author COI: The authors declared that they had no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Perez‐Garcia 2020(b).
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment (Perez‐Garcia 2020(a)) | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment (Perez‐Garcia 2020(a)) | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment (Perez‐Garcia 2020(a)) | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment (Perez‐Garcia 2020(a)) | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment (Perez‐Garcia 2020(a)) | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment (Perez‐Garcia 2020(a)) | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Perez‐Garcia 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute infection; compare the diagnostic performance of six serologic tests for the detection of antibodies against SARS‐CoV‐2 Design: Two‐group study, to assess sensitivity and specificity [1] Confirmed COVID‐19 (n = 80) [2] Pre‐pandemic control group (other diseases) (n = 60) Recruitment: [1] Unclear [2] Randomly selected group of patients with a sample taken for other serologic studies, from September 1 to November 30, 2019. Prospective or retrospective: Retrospective Sample size: 140 (80) Further detail: Inclusion: [1] Symptomatic patients admitted to the Emergency department between March 1 and April 28, 2020, with suspicion of COVID‐19 and confirmation by PCR [2] Patients with a sample taken for other serologic studies Exclusion ‐ not stated |
||
| Patient characteristics and setting | Setting: Emergency department Location: Hospital Universitario Príncipe de Asturias, Madrid Country: Spain Dates: Cases ‐ 2020‐03‐01 to 2020‐04‐28 Symptoms and severity: All cases were symptomatic. Severity not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: COVID‐negative Source: Pre‐pandemic stored serum samples 2019‐09‐01 to 2019‐11‐30 Characteristics: Sample taken for other serologic studies; 32 female, 28 male; mean age 48 years (median 44 years, range 18‐88 years) Rheumatoid arthritis: n = 5; psychiatric disorder: n = 3; psoriasis: n = 1; pregnancy: n = 6; mycosis fungoides: n = 1; multiple sclerosis: n = 1; lung cancer: n = 1; HIV infection: n = 2; haemodialysis: n = 5; HCV infection: n = 1; COPD, lung cancer: n = 1; chronic kidney disease: n = 1; breast cancer: n = 1; acute myeloid leukaemia: n = 1; acute lymphoid leukaemia: n = 1; no main underlying condition: n = 29 |
||
| Index tests | Test name:
[A] Hangzhou Alltest ‐ 2019‐nCoV IgG/IgM
[B] Innovita Biological ‐ 2019‐nCoV Ab test
[C] Epigentek SeroFlash IgM/IgG
[D] DiaPro COVID‐19 IgG Confirmation
[E] Roche ‐ Elecsys anti‐SARS‐CoV‐2 Ab
[F] Siemens Atellica Total‐Ab assay Manufacturer: [A] Hangzhou Alltest [B] Innovita Biological [C] Epigentek [D] DiaPro [E] Roche [F] Siemens Antibody: [A] to [D] IgG and IgM [E] [F]Total antibodies Antigen target: [A] N‐based, [B] N and S based, [C] N and S based, [D] N and S based, [E] N based, [F] Total antibodies Evaluation setting: [A] [B] [C] POCT performed retrospectively in lab [D] [E] [F] Laboratory Test method: LFA ‐ [A][B][C] ELISA ‐ [D] CLIA ‐ [E] [F] Timing of samples: 0‐7 days from onset of symptoms n = 18 8‐14 days from onset of symptoms n = 21 > 14 days from onset of symptoms n = 41 Samples used: Serum Test operator: Not stated Definition of test positivity: Positive serologic result was defined for LFA and ELISA tests for samples that resulted positive for either IgM or IgG antibodies. [A] [B] [C] visual‐based [D] [E] [F] Cut‐off not stated Blinding reported: Not stated Threshold predefined: [A] [B] [C] yes [D] [E] [F] Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, based on timing Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: None Timing of reference standard: Pre‐pandemic Blinded to index test: Yes, based on timing Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated
Study period and patient enrolment period almost identical All patients received same reference standard: No [1] PCR [2] Pre‐pandemic Missing data: yes, sensitivity evaluation of CLIA techniques could only be performed with 50 samples due to insufficient sample volume. 41 samples > 14 days pso not eligible for our review Uninterpretable results: Not stated Indeterminate results: Excluded from the analysis Two samples presented indeterminate result for IgG or IgM and were excluded from the analysis. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None Publication status: Published paper Source: Journal of Virological Methods Author COI: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Perez‐Garcia 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Perez‐Garcia 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Perez‐Garcia 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Perez‐Garcia 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Perez‐Garcia 2021 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pfluger 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Detection of current acute‐phase and current convalescent‐phase SARS‐CoV‐2 infection Design: Two‐group study to estimate sensitivity and specificity [1] Covid patients (n = 75) [2] Pre‐pandemic healthy controls (n = 320) Recruitment: [1] Not stated, hospital inpatients [2] Not stated, retained samples of a pre‐pandemic blood donor cohort Prospective or retrospective: [1] Prospective. First blood sample available after hospitalisation was used [2] Retrospective Sample size: 395 (75) Further detail: [1] COVID‐19 patients (positive RT‐PCR) in March and April of 2020 [2] Retained samples of a pre‐pandemic blood donor cohort collected 01.03.17–09.04.17 Exclusions Not stated |
||
| Patient characteristics and setting | Setting: Hospital, inpatient Location: University Medical Center Hamburg‐Eppendorf (UKE), Hamburg, Germany Country: Germany Dates: March and April 2020 Symptoms and severity: Mixed: based on WHO case definitions: critical, 31/75 (41.4%); severe 36/75 (48%); mild 7/75 (9.3%); asymptomatic 1/75 (1.3%) Demographics: Mean age 60.2 ± 15.4, range 16‐93 years; 33.3% female, 66.7% male Exposure history: Not stated. Non‐Covid group 1: Pre‐pandemic controls Source: Pre‐pandemic healthy blood donors, age 18‐70 years (equally distributed), male to female ratio 1:1, collected 01/03/17‐09/04/17 Characteristics: Healthy adults |
||
| Index tests | Test name: [A] Anti‐SARS‐CoV‐2 ELISA (IgG) [B] LIAISON SARS‐CoV‐2 S1/S2 IgG [C] Elecsys Anti‐SARS‐CoV‐2 [D] WANTAI SARS‐CoV‐2 Ab ELISA [E] Atellica IM SARS‐CoV‐2 Total (COV2T) Manufacturer: [A] EUROIMMUN AG, Lubeck, Germany [B] DiaSorin S.p.A, Saluggia, Italy [C] Roche Diagnostics Deutschland Gmbh, Mannheim, Germany [D] Beijing Wantai Biological Pharmacy Enterprise Co., Ltd., Beijing, China [E] Siemens Healthcare Gmbh, Erlangen, Germany Antibody: [A] IgG [B] IgG [C] Total Ab [D] Total Ab [E] Total Ab Antigen target: [A] S1‐domain, spike‐protein [B] S1 and S2‐protein [C] N‐protein [D] RBD [E] Spike‐protein Evaluation setting: Laboratory Test method: [A] ELISA [B] CLIA [C] ECLIA [D] ELISA [E] CLIA Timing of samples: Mean time pso was 11.4 days (± 6.6), range 1‐38 days 1‐10 days n = 37 11‐15 days n = 22 16‐38 days pso n = 16 Samples used: plasma/serum Test operator: Laboratory staff Definition of test positivity: [A] > 1.1 ratio (borderline 0.8‐1.1) [B] > 15 AU/mL (borderline 12‐15) [C] > 1 COI [D] > 1 A/C.O (borderline 0.9‐1.1) [E] > 1 Index Blinding reported: Not stated Threshold predefined: Yes (according to manufacturer's instructions) |
||
| Target condition and reference standard(s) | Reference standard: Positive RT‐PCR, either by modified E‐gene assay adapted as 'cobas Omni Utility Channel'‐protocol (Ct value < 34 positive in at least 2 independent samples) or by Roche SARS‐CoV‐2 IVD‐Test
9 samples received external ref standard PCR. Samples used: Naso‐pharyngeal swab Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA, pre‐pandemic Timing of reference standard: pre‐pandemic Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No [1] 3 RT‐PCR tests used. 66 samples tested in‐house using 'cobas Omni Utility Channel' or Roche SARS‐CoV‐2 IVD test. 9 samples external RT‐PCR test [2] Pre‐pandemic samples Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Some samples had borderline results, classed as positive Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Partially funded by the BGV (Behorde fur Gesundheit und Verbaucherschutz der Freien und Hansestadt Hamburg). Some authors funded by German Center for Infection Research (DZIF) and some by German Research Foundation (DGF, SFB841) Publication status: Published paper Source: Journal of Clinical Virology Author COI: Marc Lutgehetmann has received travel expenses and speakers' honoraria (Roche Diagnostics, DiaSorin,Biomerieux). Other authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Pfluger 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pfluger 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pfluger 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pfluger 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [A].
| Study characteristics | |||
| Patient Sampling | Design: Multi‐group study to assess sensitivity and specificity [1] Convalescent Covid patients (the total N samples used across all assay evaluations was not fully clear, however the overlap in samples between assays was provided. For each assay, numbers were made up to near 100 from the following sources: the Royal Free Hospital (RFH, n = 14), Basingstoke Hospital (n = 26) and the Porton Down laboratory (n = 4). [2] Non‐Covid patients (n = 499) [2a] historic negative samples (n = 399 per assay) [2b] cross‐reactive samples (n = 100 per assay unless otherwise stated) Recruitment: Not described; appeared to be convenience Prospective or retrospective: Retrospective Sample size: Total number unclear: [A] 592 (93) samples [B]‐[E] 599 (100) samples Further detail: [1] PCR‐confirmed Covid cases with sufficient volume of serum to cover multiple assays [2a] Historical serum samples collected before December 2019 [2b] Confounder serum samples collected before December 2019 Exclusion: [A] 7 sample results were removed post‐testing/post‐analysis as these were found to be PCR‐negative. No other exclusions stated |
||
| Patient characteristics and setting | Setting: Mainly community cases, very few admitted to hospital and those that were may have only been admitted for isolation during the containment phase Location: GPs in the community (FF 100 study, n = 82), Royal Free Hospital (RFH, n = 14), Basingstoke Hospital (n = 26) and the Porton Down laboratory (n = 4) Country: UK Dates: Date of evaluations: 5 April ‐ 14 July 2020 Samples collected before late April 2020 Symptoms and severity: Mostly mild disease (representative of the general population) [B] Samples were taken from patients with a range of disease severities Demographics: Not available for 14 positive samples from RFH Age range 10‐ > 64 years. 10‐24 years: 6‐8 samples 25‐34 years: 3‐6 samples 35‐44 years: 15‐17 samples 45‐54 years: 20‐22 samples 55‐64 years: 22‐27 samples > 64 years: 7‐28 samples Exposure history: Not stated. Non‐Covid group 1: [2a] Pre‐pandemic controls Source: Before December 2019, from existing reference panels at SEU, Manchester or Porton and Colindale. Characteristics: No confounder disease Non‐Covid group 2: [2b] Cross‐reactivity controls Source: Before December 2019, from SEU, Manchester or RIPL 2015 Lyme disease‐negative sample collection from Porton. Detail per assay: [A] 50 from SEU (12 RF, 6 CMV, 19 EBV, 13 VZV); 50 from RIPL 2015 Lyme disease‐negative sample collection; 399 pre‐pandemic [B] 351 samples CMV, EBV or VZV positive (no further details); 11 seasonal hCoV positive; 395 pre‐pandemic [C] 50 from SEU (12 RF, 6 CMV, 19 EBV, 13 VZV); 35 from RIPL 2015 Lyme disease‐negative sample collection; 387 pre‐pandemic [D] 49 from SEU (12 RF, 6 CMV, 19 EBV, 12 VZV); 50 from RIPL 2015 Lyme disease‐negative sample collection; 391 pre‐pandemic [E] 50 from SEU (12 RF, 6 CMV, 19 EBV, 13 VZV); 50 from RIPL 2015 Lyme disease‐negative sample collection; 399 pre‐pandemic (114 from PHE Immunoassay Group reference panel; 285 from SEU)* [F] to [H] appear to have used the same samples (all reported numbers were the same): 50 from SEU (12 RF, 6 CMV, 19 EBV, 13 VZV); 50 from RIPL 2015 Lyme disease negative sample collection; 399 pre‐pandemic (313 from PHE Immunoassay Group reference panel; 86 from SEU) |
||
| Index tests | [A] Euroimmun anti‐SARS‐CoV‐2 ELISA (IgG) serology assay (EI 2606‐9601 G) [B] Abbott SARS‐CoV‐2 IgG kit (Architect i2000SR system) (reagent batch number 16253FN00, exp date 16/07/2020) [C] Elecsys Anti‐SARS‐CoV‐2 assay (Lot 49025901, exp 31/05/20) [D] VITROS Immunodiagnostic Products anti‐SARS‐CoV‐2 IgG assay [E] Siemens Atellica‐IM SARS‐CoV‐2 Total (COV2T) serology assay (batch no. 11206711, exp 2021‐05‐12) [F] Ortho Clinical VITROS Anti‐SARS‐Cov‐2 Total Ab [G] LIAISON SARS‐CoV‐2 S1/S2 IgG serology assay [H] Beckman Coulter Access SARS‐CoV‐2 IgG Assay Manufacturer: [A] Euroimmun Medizinische Labordiagnostika AG [B] Abbott [C] Roche [D] Ortho Clinical Diagnostics [E] Siemens Healthcare GmbH [F] Ortho Clinical Diagnostics [G] DiaSorin S.p.A [H] Beckman Coulter Antibody: [A], [B], [D], [G], [H] IgG [C], [E], [F] Total antibody Antigen target: [A] S1‐protein [B] N‐protein [C] N‐protein [D] S‐based [E] Recombinant antigen [F] S1‐protein [G] S1 and S2‐protein [H] RBD of S1‐protein Evaluation setting: Laboratory used in laboratory Test method: [A] ELISA [B] CMIA [C] ECLIA [D] CLIA [E TO H] CLIA Timing of samples: variable; e.g. for the EUROIMMUN assay the interval pso was known for 79/93 samples; for 14/93 the interval was measured from when the patient was admitted to hospital to sample collection date (making the interval artificially low compared to actual time pso). Vast majority of samples across all evaluations was > 21 d pso, e.g. for EUROIMMUN, 75/93 were > 21 d [Data for <= 10 d was not included in the review because of lack of accurate sample timing] Samples used: Serum Test operator: Skilled research scientists in PHE Porton Down laboratory Definition of test positivity: [A] Ratio < 0.8 negative, >= 0.8 to < 1.1 borderline, >= 1.1 positive (borderline considered negative) [B] S/C < 1.4 negative, ≥ 1.4 positive [C] COI; signal sample/cut‐off < 1.0 negative, ≥ 1.0 positive [D] Signal for test sample/signal at cut‐off (cut‐off value) < 1.0 negative, ≥ 1.0 positive [E] < 1.0 index negative, >= 1.0 index positive [F] S/C < 1.0 negative; S/C >= 1.0 positive [G] < 12.0 AU/mL negative, 12.0 <= x < 15.0 AU/mL equivocal, >= 15.0 AU/mL positive. [H] <= 0.80 S/CO negative, > 0.80 to < 1.00 equivocal, >= 1.0 S/CO positive Blinding reported: Yes Threshold predefined: Yes, according to manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: swab sample Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA, pre‐pandemic Timing of reference standard: pre‐pandemic Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: yes, not all samples used for all test evaluations [E] 8 samples that did not yield results were excluded from the analysis. [A] [B] [C] [D] no exclusions Uninterpretable results: No results were excluded as uninterpretable [A] [B] [C] [D] no exclusions. [E] 8 samples that did not yield results were excluded from the analysis. Indeterminate results: [B] 3 equivocal results classed as negative for sensitivity [C] 6 equivocal results classed as negative for sensitivity [A] [D] [E] No equivocal range Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Asked to perform evaluation by Department of Health and Social Care. Funding not stated Publication status: Published report Source: Public Health England Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
PHE 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
PHE 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Phipps 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection; and assessment of analytical specificity (cross‐reactivity) Design: Multi‐group study, including: [1] Single group of suspected COVID‐19 cases with available prior or same‐day PCR swab test result (n = 173) Excluded from current review: additional groups included to assess analytical specificity: [2] Healthy blood donors (n = 656, 240 pre‐pandemic and 416 from 2020) [3] Patients with SLE (n = 29) [4] Patients with rheumatoid arthritis (n = 20) [5] Patients with previous positive respiratory viral PCR panel (n = 90) Recruitment: Unclear Prospective or retrospective: Retrospective (data collection based on chart review) Sample size: 173 (76) 795 additional non‐COVID‐19 samples excluded from current review Further detail: No more details available |
||
| Patient characteristics and setting | Setting: [1] Hospital inpatient Location: Not stated; author's institution University of Texas Southwestern Medical Center, Dallas Country: USA Dates: not stated Symptoms and severity: Unclear; both severe (requiring ICU) and mild/moderate cases included but n per group was not reported and data points reported in Figures did not sum to 76 cases Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: [A] SARS‐CoV‐2 IgG (Abbott 06R86) testing Second in‐house laboratory test reported (ineligible for this review) [B] SARS‐CoV‐2 IgM testing using a laboratory developed protein microarray Manufacturer: [A] Abbott Antibody: IgM or IgG Antigen target: SARS‐CoV‐2 nucleocapsid protein Evaluation setting: Laboratory Test method: [A] chemiluminescent microparticle immunoassay (CMIA) Timing of samples: Fig 3 showed samples collected between day 0 and day c45 Samples used: Plasma Test operator: not stated Definition of test positivity: [A] relative light units (RLU) positive at 1.4 or greater Blinding reported: Unclear; PCR same day or day before Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: [1] RT‐PCR; m2000 Abbott RealTime SARS Cov‐2 assay or [2] isothermal PCR; Abbott ID NOW COVID‐19 assay Samples used: nasopharyngeal swab Timing of reference standard: As for index test, samples collected between day 0 and day c45 Blinded to index test: not stated Incorporated index test: No Definition of non‐COVID cases: As above; single negative for absence of disease Samples used: not stated Timing of reference standard: not stated Blinded to index test: not stated Incorporated index test: unclear |
||
| Flow and timing | Time interval between index and reference tests: swab for PCR was same day or prior day All patients received same reference standard: No; isothermal or RT‐PCR (n not stated) Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: patients |
||
| Comparative | |||
| Notes | Funding: No external funding was received. Publication status: pre‐print Source: medRxiv Author COI: The authors have declared no competing interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Pickering 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to estimate sensitivity and specificity for diagnosis of active disease/identification of previous disease Design: [1] RT‐PCR‐positive COVID‐19 patients ‐ venous serum samples collected at St Thomas’ Hospital, London (N = 87 patients, 110 samples) [2] pre‐Covid‐19 pandemic control samples (n = 50 samples, 50 patients) Recruitment: [1] Surplus serum was retrieved from the routine biochemistry laboratory at point of discard. [2] Emergency admissions to St Thomas’ hospital in March 2019 Prospective or retrospective: Retrospective Sample size: Patients: 137 (87); samples 160 (110) Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Inpatients Location: St Thomas’ Hospital, London Country: UK Dates: 4 March‐21 April 2020 Symptoms and severity: Disease Severity: Level 0 N = 11 (12.6%); Level 1 N = 15 (17.2%); Level 2 N = 4 (4.6%); Level 3 N = 3 (3.4%); Level 4 N = 48 (55.2%); Level 5 N = 6 (6.9); Died N = 21 (24.1%). Level of Respiratory Support: No support N = 11 (12.6%); Supplemental oxygen N = 23 (26.4%); Non‐invasive ventilation N = 1 (1.1%); Mechanical ventilation N = 46 (52.8%); ECMO N = 6 (6.9%). Demographics: Mean age (years) 58.2 +/‐ 16.6; female 29 (33.3%) Exposure history: Not stated Non‐Covid group 1: pre‐Covid‐19 pandemic control samples Source: St Thomas’ Hospital, March 2019 Characteristics: Not stated |
||
| Index tests | Test name: [1] Accu‐Tell COVID‐19 IgG/IgM Cassette [2] COVID‐19 (SARS‐CoV‐2) Antibody Test Kit [3] SARS‐CoV‐2 IgM/IgG ANTIBODY TEST KIT [4] GenBody COVID‐19 IgM/IgG [5] COVID‐19 Spring IgM/IgG Rapid Test Cassette [6] COVID‐19 IgG/IgM Rapid Test Cassette [7] Rapid IgM‐IgG Combined Antibody Test Kit for SARS‐ CoV‐2 [8] SARS‐CoV‐2 Ab Diagnostic Test Kit [9] EUROIMMUN IgA (SARS‐CoV‐2 S1‐protein) [10] EUROIMMUN IgG (SARS‐CoV‐2 S1‐protein) Manufacturer: [1] AccuBiotech Co., Ltd. [2] Anhui DeepBlue Medical Technology Co., Ltd. [3] Biohit Healthcare Co., Ltd. [4] GenBody Inc. [5] Spring Healthcare Services AG [6] SureScreen Diagnostics Co., Ltd. [7] Jiangsu Medomics Medical Technology Co. Ltd. [8] Shenzen Watmind Medical Co., Ltd. [9] and [10] EUROIMMUN Medizinische Labordiagnostika AG Antibody: [1] ‐[7] IgG, IgM; [8] Total antibody (IgG, IgM, IgA); [9] IgA; [10] IgG Antigen target: [1]‐[7] Not reported; [8] total antibody against SARS‐CoV‐2; [9] SARS‐CoV‐2 S1‐protein; [10] SARS‐CoV‐2 S1‐protein Evaluation setting: All evaluated in laboratory setting Test method: [1]‐[7] colloidal‐gold‐based LFIA, [8] Chemiluminescence‐based Immunoassay, [9] and [10] Enzyme‐linked Immunosorbent Assay (ELISA) Timing of samples: 1 to 30 days after onset of self‐reported symptoms: < 10 days pso: 38/110 samples 10+ days pso: 72/110 samples < 14 days pso: 56/110 samples 14+ days pso: 54/110 samples 20+ days pso: 28/110 samples 10‐14 days pso: 18/110 samples 14‐20 days pso: 26/110 samples 10‐20 days pso: 44/110 samples Samples used: venous serum samples Test operator: Laboratory personnel Definition of test positivity: [1] ‐[7] Visible lines, on 4‐point scale (negative, borderline, positive, strong positive) for both IgM and IgG. Scoring was performed independently by two individuals. [8] Results equal to and below 1.0 AU (arbitrary units)/mL were negative, scores above 1.0 AU/mL were deemed positive. Scores > 10 AU/mL were deemed a strong positive. [9] and [10] Scores of < 0.8 were negative, >= 0.8 to < 1.1 were borderline, >= 1.2 to < 4 were positive, and >= 4 were strong positive. Blinding reported: Unclear Threshold predefined: [1]‐[7] Visible lines, according to manufacturer’s instructions, [8] according to manufacturer’s instructions, [9] and [10] Not reported. |
||
| Target condition and reference standard(s) | Reference standard: real‐time RT‐PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Pre‐pandemic Timing of reference standard: Pre‐pandemic (March 2019) Blinded to index test: Yes, prior to index test Incorporated index test: No, Pre‐pandemic |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No ‐ some pre‐pandemic Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: The research and the King’s College London Infectious Diseases Biobank were supported by the Department of Health via a National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s and St. Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. Development of SARS‐CoV‐2 reagents (RBD) was partially supported by the NIAID Centers of Excellence for Influenza Research and Surveillance (CEIRS). The work was supported by gifts from Peking University donors and Anhui Deep Blue company. The following donated test kits: the manufacturers of Spring, Biohit, Genbody, Medomics and Watmind. Authors supported by MRC‐KCL Doctoral Training Partnership in Biomedical Sciences, the Wellcome Trust, an MRC‐KCL Doctoral Training Partnership in Biomedical Sciences industrial Collaborative Award in Science & Engineering (iCASE) in partnership with Orchard Therapeutics, the Medical Research Council, King’s Together Rapid COVID‐19 Call awards, Fondation Dormeur, Vaduz, MRC Discovery Award Publication status: Published Source: Journal (PLOS Pathogens) Author COI: The authors have declared that no competing interests exist. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Pickering 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessmentSee main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pickering 2020 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Pollan 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to estimate sensitivity and specificity for diagnosis of acute Covid Design: [1] PCR‐confirmed Covid‐19 cases with serum samples (n = 82) [2] Pre‐pandemic serum samples for diagnosis of other pathogens (n = 42) [Study was reported as part of a wider seroprevalence survey; a second validation study of another immunoassay was also reported but not eligible for inclusion] Recruitment: Not stated Prospective or retrospective: Unclear; appeared to be retrospective Sample size: 124 (82); 66 (24) eligible for review Further detail: No further details reported |
||
| Patient characteristics and setting | Setting: Not stated Location: Not stated; validation study conducted by Spanish National Centre for Microbiology, Madrid Country: Spain Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic serum samples for diagnosis of other pathogens Source: Samples collected before December 8th 2019 Characteristics: Not stated |
||
| Index tests | Test name: SARS‐CoV‐2 IgG for use with ARCHITEC Manufacturer: Abbott Laboratories, IL, USA Antibody: IgG Antigen target: SARS‐CoV‐2 nucleoprotein Evaluation setting: Laboratory, used in laboratory Test method: Chemiluminescent microparticle immunoassay Timing of samples: All PCR+ were >= 10 days pso (n = 82), 58 (71%) >= 14 days pso Samples used: Serum samples Test operator: Not stated Definition of test positivity: Index S/C threshold of 1.4 Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Presumed yes as reference standard performed before index test Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA (pre‐pandemic) Timing of reference standard: NA (before December 8th 2019) Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Nothing mentioned Uninterpretable results: Nothing mentioned Indeterminate results: Nothing mentioned Unit of analysis: Not clear, did not state 1 sample per patient |
||
| Comparative | |||
| Notes | Funding: Spanish Ministry of Health and the Institute of Health Carlos III, in collaboration with the health services of the Spanish regions
The funders facilitated data acquisition but had no role in the design, analysis, interpretation, or writing. The
first three authors had full access to all the data. The first five authors and the senior author (RY) had final
responsibility for the decision to submit for publication. Publication status: Published paper Source: Lancet Author COI: Authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Prazuck 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: The study evaluated two Rapid Diagnostic Tests for the diagnosis of COVID‐19, compared with RT‐PCR
Single‐group study to estimate sensitivity and specificity for diagnosis of active disease. Design: [1] Suspected COVID‐19 patients who went to the hospital for a diagnostic consultation (n = 381) [1a] Patients with symptoms of COVID‐19 who went to the hospital for a diagnostic consultation with RT‐PCR‐positive for COVID‐19 (n = 238) [1b] Patients with symptoms of COVID‐19 who went to the hospital for a diagnostic consultation with RT‐PCR‐negative for COVID‐19 (n = 143) Recruitment: Patients with symptoms of COVID‐19 who went to the hospital for a diagnostic consultation Prospective or retrospective: Prospective (consent was obtained from each participant) Sample size: Patients = 381 (238); samples = 427 (284) Test [A] PRESTO: 222 (150) samples Test [B] DUO: 205 (134) samples (24 samples tested with both tests) Further detail: Patients with symptoms of COVID‐19 who went to the hospital for a diagnostic consultation. Adult patients visiting the infectious disease department (Centre Hospitalier Regional Orle´ans, France) from March, 18th, 2020 to April 10th, 2020. This department receives patients whose symptoms, such as headache, fatigue, fever or respiratory signs suggest a COVID infection, and for whom a diagnosis is requested. |
||
| Patient characteristics and setting | Setting: Inpatient and outpatient Location: Centre Hospitalier Regional Orléans, France Country: France Dates: March 18, 2020 to April 10, 2020 Symptoms and severity: Not stated Demographics: mean age of patients was 53.68 years ± 20.18 (median 54; range 19‐96). Exposure history: Not stated Non‐Covid group 1: RT‐PCR‐negative Source: Centre Hospitalier Regional Orléans, France ‐ March 18, 2020 to April 10, 2020 Characteristics: 48.20 years (SD: 17.00; range 19‐72) , median 46 |
||
| Index tests | Test name: [A] COVID‐PRESTO [B] COVID‐DUO Manufacturer: [A] AAZ‐LMB [B] AAZ‐LMB Antibody: [A] and [B] IgM, IgG Antigen target: [A] and [B] recombinant COVID‐19 antigens labelled with colloidal gold Evaluation setting: [A] and [B] POC used at PC ("at the site by clinical staff, physicians or nurses") Test method: [A] and [B] lateral flow immune‐chromatographic assay (recombinant COVID‐19 antigens labelled with colloidal gold) Timing of samples: For [1a] 0‐ > 15 days post‐onset [A] 0‐5 days pso: 20/150 6‐10 days pso: 43/150 11‐15 days pso: 39/150 15‐31 days pso: 48/150 [B] 0‐5 days pso: 14/134 6‐10 days pso: 42/134 11‐15 days pso: 44/134 15‐31 days pso: 34/134 For [1b] 24 hours to 8 days from onset of symptoms (median 2 days; range 1–8 days) Samples used: Capillary whole blood samples taken at the fingertip Test operator: Conducted at the site by clinical staff, physicians or nurses. Health workers involved in the study received a two‐hours training session for each type of test prior to the beginning of the study. Result read within 10 minutes by two independent operators Definition of test positivity: Visual interpretation of coloured bands according to manufacturer's instructions Blinding reported: Unclear Threshold predefined: Visual interpretation of coloured bands according to manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: Real‐time RT‐PCR assays for the detection of SARS‐CoV‐2 Samples used: Nasopharyngeal (NP) swab specimens Timing of reference standard: At first consultation ‐ timing unclear Blinded to index test: Yes, prior to index Incorporated index test: No Definition of non‐COVID cases: Real‐time RT‐PCR assays for the detection of SARS‐CoV‐2 Samples used: Nasopharyngeal (NP) swab specimens Timing of reference standard: At first consultation ‐ timing unclear Blinded to index test: Yes, prior to index Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Rapid Diagnostic Tests were provided free of charge by AAZ‐LMB.
The study was funded by CHR Orleans (Orle´ans Regional Hospital Centre), a public hospital with no‐profit status, of which all authors are employees. Publication status: Pre‐print (not peer reviewed), now published Source: medRxiv preprint, PLOS One Author COI: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Prazuck 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Qian 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Detect current acute or convalescent Covid infection Design: Multi‐group study to estimate sensitivity and specificity [1] Covid patients, n = 565 [1a] PCR+ COVID patients, n = 513 [1b] Suspected COVID patients with typical epidemiological history, clinical symptoms and featured chest CT images, n = 52 (54?) [2] Controls, n = 1558 [2a] Hospitalised patients (concurrent, other diseases, PCR‐ for SARS‐COV‐2), n = 972 [2b] Normal population (untested), n = 586 Group [1b] has no time pso reported so was not eligible for our review. Recruitment: [1] Recruited individuals from 10 hospitals, 4 in Hubei province, 6 from other provinces in China [2a] Hospitalised patients with diseases other than COVID‐19 from the four hospitals in the outbreak Hubei province, and the six hospitals in other provinces in China [2b] Recruited from a physical examination centre in a hospital in Shenzhen Prospective or retrospective: Unclear possibly prospective Sample size: 2123 (565) of which 2071 (513) were eligible for our review Further detail: [1a] Included confirmed RT‐PCR‐positive COVID inpatients, all ages (aged 1 month to 92 years) [1b] recruited following guidelines of diagnosis and treatment of COVID‐19, including typical epidemiological history, clinical symptoms and featured chest CT image [2] No epidemiological history and clinical symptoms of COVID19, and excluded for SARS‐CoV‐2 infection by a negative nucleic acid test with RT‐PCR [2b] from a physical examination centre in a hospital in Shenzhen Exclusion not stated |
||
| Patient characteristics and setting | Setting: [1] Hospital inpatient, 10 hospitals Location: [1] 4 hospitals in Hubei province, China, 6 hospitals in other provinces in China (possibly the 10 hospitals from the author affiliations) Country: China Dates: Not stated Symptoms and severity: Not stated, hospitalised Demographics: [1a] Age range 1 month to 92 years, average age 53 years; no gender/sex specified Exposure history: [1a] 296/513 from Hubei province, 217/513 from other provinces. Non‐Covid group 1: [2a] Hospitalised controls, other disease Source: 317 from 4 hospitals in Hubei province, 655 from 6 other hospitals Time not stated (concurrent) Characteristics: RT‐PCR‐negative, no epidemiological history or symptoms of COVID‐19 Age range 1 to 90 years, average 48 years 3 to 9 samples positive for IgM and/or IgG for the four common human respiratory coronaviruses (229E, NL63, OC43, and HKU1), influenza A and B viruses, seasonal influenza virus (H1N1, H5N1, H3N2, and H7N9), legionella pneumophila, mycoplasma pneumoniae, chlamydia pneumoniae, adenovirus, respiratory syncytial virus, measles virus, mumps virus, rhinovirus, enterovirus, Epstein‐Barr virus, CMV, and rotavirus, autoantibodies to rheumatoid factors and some major anti‐nucleic antibodies (dsDNA, Sm, SS‐A, SS‐B, Jo‐1, Ro‐52) Non‐Covid group 2: [2b] Normal control population Source: Physical examination centre in a hospital in Shenzhen. No time stated (concurrent) Characteristics: Not tested with RT‐PCR but assumed COVID‐negative Age range 18 to 35 years, average 25 years |
||
| Index tests | Test name: Name not stated. SARS‐CoV‐2 IgM and IgG immunoassay in development. Manufacturer: Shenzhen YHLO Biotech Co., Ltd Antibody: IgM and IgG Antigen target: SARS‐CoV‐2 nucleocapsid protein and spike‐protein Evaluation setting: Laboratory Test method: CLIA Timing of samples: [1a] < 7 days pso, n = 63 7‐14 days pso, n = 99 > 14 days pso, n = 351 Samples used: serum Test operator: Laboratory staff Definition of test positivity: >= 10 kAU/L Blinding reported: Not stated Threshold predefined: No |
||
| Target condition and reference standard(s) | Reference standard: [1a] RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated. Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: [2a] 972 hospitalised controls tested negative by RT‐PCR [2b] 586 normal population controls received no reference standard Samples used: [2a] Not stated [2b] untested Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated. All patients received same reference standard: Yes, all Covid patients received same ref standard. Not all control patients received ref standard. Missing data: Not stated. Uninterpretable results: Not stated. Indeterminate results: Not stated. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: B.F. Liu, Fundamental Research Funds for the Central Universities. Publication status: Published paper. Source: Clinical Chemistry & Laboratory Medicine Author COI: Authors stated no conflict of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Qiu 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Detection of acute or convalescent‐phase Covid infection Design: Two‐group study to estimate sensitivity and specificity: [1] Confirmed Covid cases n = 475 [2] Non‐Covid controls, concurrent non‐COVID patients n = 389 Recruitment: [1] and [2] Individuals enrolled from four medical institutions in Hubei Province between January 20 2020 and March 12 2020 Prospective or retrospective: Retrospective Sample size: 864 (475) Further detail: Included adults >= 18 years age Excluded pregnant women |
||
| Patient characteristics and setting | Setting: Hospital inpatients. Location: Four medical institutions, Hubei province: Zhongnan Hospital of Wuhan University, Wuhan Third Hospital‐Tongren Hospital of Wuhan University, Huang Gang Central Hospital, Hebi City Centre for Disease Control and Prevention Country: China Dates: Between January 20 2020 and March 12 2020 Symptoms and severity: Hospital inpatients, symptoms recorded but data not shown Demographics: Of 409 cases used for Ab testing: 217 males, 192 females, median age 60 years (IQR, 49‐69) Exposure history: Not stated Non‐Covid group 1: Non‐Covid controls Source: Hospital inpatients Four medical institutions between January 20 2020 and March 12 2020, Hubei province: Zhongnan Hospital of Wuhan University, Wuhan Third Hospital‐Tongren Hospital of Wuhan University, Huang Gang Central Hospital, Hebi City Centre for Disease Control and Prevention Characteristics: 224 males, 165 females; median age 45 years (IQR, 29‐61) |
||
| Index tests | Test name: SARS‐CoV‐2 IgG and IgM CLIA Microparticle detection kit Manufacturer: Autobio Diagnostics Co., Ltd. (Henan, China) Antibody: IgG or IgM Antigen target: S‐protein Evaluation setting: Laboratory Test method: CLIA Timing of samples: 1 to 87 days pso 1‐10 days pso: 66/409 11‐20 days pso: 70/409 21+ days pso: 273/409 Samples used: Serum Test operator: Laboratory staff Definition of test positivity: S/CO >= 1 Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐qPCR, the target genes included the open reading frame 1ab (ORF1ab) gene, and the nucleocapsid protein (N) gene of SARS‐CoV2, analysed according to manufacturer’s protocol. Samples used: Throat swabs Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: RT‐qPCR, the target genes included the open reading frame 1ab (ORF1ab) gene, and the nucleocapsid protein (N) gene of SARS‐CoV2, analysed according to manufacturer’s protocol. Samples used: throat swabs Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not clear, time between symptom onset and collection of serum samples for index test ranged from 1 to 87 days. All patients received same reference standard: Yes Missing data: 475 Covid patients recruited, results only available for 409 cases Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Work supported by Hubei Province Health and Family Planning Scientific Research Project Publication status: Published paper Source: Emerging Microbes & Infections Author COI: No COI reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ragnesola 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients, convalescent plasma donor samples (n = 63) [2] Pre‐pandemic samples (n = 10) Group [2] has < 25 samples and was excluded from our review. Recruitment: [1] Convalescent donor plasma was collected by the New York Blood Center (NYBC). Recruitment not stated. [2] Not stated Prospective or retrospective: [1] Prospective [2] Retrospective Sample size: 73 (63) of which 63 (63) were eligible for our review Further detail: Inclusion: [1] All donors had self‐reported documented COVID‐19 disease by positive SARS‐CoV‐2 RT‐PCR test (manufacturer and documentation not provided from referring institution of CP donors), had complete resolution of symptoms at least 14 days prior to donation, and otherwise met all criteria for donating blood consistent with FDA’s policy on the Collection of COVID‐19 Convalescent Plasma. [2] Frozen plasma was used that was collected prior to the beginning of the epidemic. Exclusions not reported |
||
| Patient characteristics and setting | Setting: Convalescent plasma donors Location: New York Blood Center Lindsley F. Kimball Research Institute, 310 E 67th Street, New York, NY 10065, USA Country: New York, USA Dates: Not stated Symptoms and severity: Convalescent, at least 14 days since symptom resolution Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Clungene® SARS‐CoV‐2 IgG/IgM Rapid Test Cassettes Manufacturer: Hangzhou Clongene Biotech Co., Ltd., Hangzhou, China Antibody: IgM / IgG Antigen target: receptor‐binding domain (RBD) of the spike and nucleocapsid protein Evaluation setting: POCT performed in lab Test method: Lateral flow test (no details) Timing of samples: Symptom‐free for at least 14 days so at least 14 days post‐PCR+ Samples used: Plasma Test operator: four independently trained operators Definition of test positivity: Positive and negative IgG/IgM band determinations were made by visual inspection with accordance to manufacturer instructions. Blinding reported: Not stated Threshold predefined: yes (visual‐based) |
||
| Target condition and reference standard(s) | Reference standard: Self‐reported documented COVID‐19 disease by positive SARS‐CoV‐2 RT‐PCR test (manufacturer and documentation not provided from referring institution of CP donors), threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes (unclear as self‐reported) Missing data: yes, group [2] excluded from review Uninterpretable results: All samples yielded an interpretable result with no invalid result. Indeterminate results: No intermediate range Unit of analysis: [1] Not quite clear, possibly yes |
||
| Comparative | |||
| Notes | Funding: The LFD used in the testing were provided by CL/BioSolutions services LLC. Publication status: Published paper Source: BMC Research Notes Author COI: CL worked with the LFD manufacturer on the Emergency Use Authorization submission to the US FDA. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Renard 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute or convalescent SARS‐CoV‐2 infection Design: Multi‐group study estimating sensitivity and specificity: [1] Covid‐positive, N = 405 samples, n = 142 patients [2] Covid‐negative controls, N = 989 patients, pre‐pandemic healthy donors [3] Serum cross‐reactivity pre‐pandemic samples, n = 276 Recruitment: [1] RT‐PCR‐positive, symptomatic patients from three hospitals: Centre Hospitalier Saint Joseph Saint Luc, Lyon, France; Centre de Ressources Biologiques (CRB) des Hospices Civils de Lyon, CRB Nord and CRB Sud, Lyon, France [2] Pre‐pandemic adult donors, before September 2019. Healthy donors. Collected at Etablissement Francais du Sang (EFS), France and Clinilabs, Inc., United States [3] Frozen pre‐pandemic sera from patients with other potentially interfering infections or medical conditions (bioMerieux, Centre Hospitalier Grenoble‐Alpes and Saint Joseph Saint Luc Lyon collections) Prospective or retrospective: [1] Unclear [2] [3] Retrospective Sample size: 1670 (405) Further detail: [1] Inclusion: Symptomatic patients from three hospitals (inpatient and outpatients). Exclusion: Asymptomatic patients. [2] Inclusion: Healthy adult blood donors. Exclusion: Not stated [3] Inclusion: Patients with potentially interfering infections or medical conditions, including 5 pregnant women. Exclusion: Not stated. |
||
| Patient characteristics and setting | Setting: Hospital inpatients and hospital outpatients Location: Centre Hospitalier Saint Joseph Saint Luc, Lyon, France; Centre de Resources Biologiques (CRB) des Hospices Civils de Lyon, CRB Nord and CRB Sud, Lyon, France Country: France Dates: March 31 to June 2, 2020 Symptoms and severity: Symptomatic, severity not stated. Data for 130 patients (time post‐PCR+ analyses) Hospitalised (n = 54) and non‐hospitalised (n = 61), 15 missing data Data for 63 patients (time pso analyses) 48 (76.2%) hospitalised 15 (23.8%) missing Demographics: Data for 130 patients (time post‐PCR+ analyses) 61 non‐hospitalised: missing data on age, 69 other patients: median 70 (range 27–96) years; 47 male, 22 female, 61 missing Data for 63 patients (time pso analyses) Age: median 70 (range 27–96) years; 45 (71.4%) male Exposure history: Not stated. Non‐Covid group 1: [2] pre‐pandemic healthy donors Source: Etablissement Francais du Sang (EFS), France and Clinilabs, Inc., United States. Collected before September 2019 Characteristics: Healthy donors Non‐Covid group 2: [3] Cross‐reactivity sera, pre‐pandemic Source: Cross‐reactivity sera from bioMerieux, Centre Hospitalier Grenoble‐Alpes and Saint Joseph Saint Luc Lyon collections Time not stated Characteristics: Patients with potentially interfering infections or medical conditions: Pregnant women 5 Antinuclear antibody (ANA)a 47 Rheumatoid factor 19 Human anti‐mouse antibody (HAMA) 5 Borrelia burgdorferib 10 Haemophilus influenzae B 5 Plasmodium falciparum 3 Toxoplasma gondiib 10 Treponema pallidum 3 Trypanosoma cruzi 5 Hepatitis A virus (HAV) 3 Hepatitis B virus (HBV) 5 Hepatitis C virus (HCV) 5 Hepatitis E virus (HEV)b 7 Herpes simplex virus (HSV)b 6 Human immunodeficiency virus (HIV) 5 Cytomegalovirus (CMV) 4 Measles virus (MV) 4 Mumps virus (MuV) 1 Rubella virus (RuV)b 10 Dengue virus (DENV) 3 West Nile virus (WNV) 4 Yellow fever virus (YFV) 4 Zika virus (ZIKV)b 5 Adenovirus (AdV) 2 Metapneumovirus (MPV) 4 Rhinovirus/enterovirus (RV/EnteroV)c 20 Influenza A and B virus (IAV/IBV) 30 Parainfluenza viruses 1/2/3 (PIV‐1/2/3) 11 Respiratory syncytial virus A or B (RSV A or B) 13 Coronavirus NL63/HKU1 (CoV‐NL63/HKU1)d 9 Coronavirus 229E (CoV‐229E) 7 Coronavirus OC43 (CoV‐OC43) 2 |
||
| Index tests | Test name: [A] Vidas SARS‐CoV‐2 IgM (423833) [B] Vidas SARS‐CoV‐2 IgG (423834) Manufacturer: [A] [B] bioMerieux, France Antibody: [A] IgM [B] IgG Antigen target: [A] [B] RBD of spike‐protein Evaluation setting: [A] [B] Laboratory Test method: [A] [B] two‐step enzyme immunoassay combined with an enzyme‐linked fluorescent assay (ELFA) detection technique Timing of samples: 0 to 32+ 1‐65 days pso (n = 105), 0‐7 days: n = 22 8‐15 days: n = 29 16‐23 days: n = 26 24‐31 days: n = 18 >= 32 days: n = 10 0‐65 days post‐PCR +ve (n = 232) 0‐7 days: n = 110 8‐15 days: n = 60 16‐23 days: n = 38 24‐31 days: n = 13 >= 32 days: n = 11 Samples used: Serum or plasma Test operator: Laboratory staff Definition of test positivity: [A] [B] Positive COI >= 1.00, negative < 1.00 Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated. Blinded to index test: Yes Incorporated index test: No Definition of non‐COVID cases: [2] Pre‐pandemic (before September 2019) [3] Pre‐pandemic (timing unclear) Samples used: [2] [3] Pre‐pandemic Timing of reference standard: [2] [3] Pre‐pandemic Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Index tests conducted 0‐65 days post‐reference test
0‐7 days: n = 110
8‐15 days: n = 60
16‐23 days: n = 38
24‐31 days: n = 13
>= 32 days: n = 11 All patients received same reference standard: No Missing data: [1] 173 samples excluded from time split post‐PCR+ analyses: 2 missing date of positive PCR, 171 as IgM test or IgG test not done, multiple measurements per patient in one time frame, or missing paired test 300 samples excluded from time split post‐symptom onset analyses: 194 Missing date of symptom onset, 106 as IgM test or IgG test not done, multiple measurements per patient in one time frame, or missing paired test [2] [3] Not all samples tested with IgG test [B] Uninterpretable results: Not stated. Indeterminate results: No borderline range Unit of analysis: Samples. To avoid a statistical bias, only one patient’s measurement per time period was included in the analysis. |
||
| Comparative | |||
| Notes | Funding: Work was supported by bioMerieux.
J.L received research funding from bioMerieux for this study. Publication status: Published paper. Source: Journal of Clinical Microbiology. Author COI: M.P declared a consulting contract with bioMerieux. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Renard 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rijkers 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: To compare the antibody response in patients with severe (hospitalised) and mild (non‐hospitalised) COVID‐19 and determine sensitivity for diagnosis of current acute infection and current convalescent infection Design: Single‐group study to estimate sensitivity only [1] Confirmed COVID patients (n = 62) [1a] Severe Covid‐19 group (n = 38) [1b] Mild Covid‐19 group (n = 24) Recruitment: [1a] Consecutive hospital Covid patients admitted to the Admiral de Ruyter Hospital in Goes, The Netherlands, in the period March 2020–May 2020 [1b] Not stated Prospective or retrospective: [1a] Prospective [1b] Not stated (possibly prospective) Sample size: 62 (62) patients, number of samples unclear (serial sampling from week 1 to week 4 post‐symptom onset) Further detail: Inclusion: [1a] Subjects who had positive RT‐PCR and were hospitalised, both ICU and non‐ICU (admitted to the Admiral de Ruyter Hospital in Goes, The Netherlands) [1b] Hospital personnel (both from clinical departments as well as laboratory departments) who developed fever, coughing, and/or dyspnoea and had positive RT‐PCR and were non‐hospitalised with mild disease Exclusion: [1] Test group ‐ PCR‐negative samples |
||
| Patient characteristics and setting | Setting: [1a] Hospital inpatients [1b] non‐hospitalised patients (home isolation, under control of GP) Location: [1] Admiral de Ruyter Hospital in Goes Country: The Netherlands Dates: [1a] March 2020–May 2020 [1b] Not stated Symptoms and severity: [1a] The criteria for hospital admission were severity and/or progression of clinical symptoms, as assessed by the referring general practitioner. The presenting clinical symptoms included fever (n = 17), cough (n = 18), dyspnoea (n = 11), dizziness and/or confusion (n = 4), and general malaise (n = 6). The clinical criteria for admission of hospitalised patients to the ICU primarily were respiratory insufficiency, haemodynamic instability, and/or multiorgan failure. ICU 15/38 non‐ICU 23/38 6/38 died [1b] Mild symptoms (fever, coughing, and/or dyspnoea), non‐hospitalised Demographics: [1a] Age (years) ‐ median 70 (range 38‐87) Male gender ‐ 26 (68%) Any comorbidities ‐ 26 (68%) Diabetes mellitus ‐ 4 (11%) Hypertension ‐ 13 (34%) Coronary heart disease ‐ 8 (21%) COPD ‐ 10 (26%) Body Mass Index ‐ median 27 (range 19‐41) [1b] Median age 42 years (range, 21–66 years) Exposure history: [1a] Not stated [1b] Hospital personnel |
||
| Index tests | Test name: The Wantai SARS‐CoV‐2 total antibody ELISA (catalog number WS1096) Manufacturer: Beijing Wantai Biological Pharmacy Enterprise, Beijing, China Antibody: Total antibodies Antigen target: receptor binding domain antigen of SARS‐CoV‐2 Evaluation setting: Hospital laboratory Test method: sandwich ELISA Timing of samples: Serial blood sampling (3 times per week) was started a median of 2 days (range, 1–7 days) after positive RT‐PCR 1‐7 days pso 8‐14 days pso 15‐21 days pso 22‐28 days pso Samples used: Serum Test operator: Lab personnel from hospital laboratories Definition of test positivity: [A] and [B] Optical density (OD) was measured at 450 nm and the antibody titer for each sample was calculated as the ratio of the reading of that sample to the reading of a calibrator (included in the kit):OD ratio. Threshold not stated Blinding reported: Not stated (no as only COVID cases included) Threshold predefined: yes (according to the manufacturer’s instructions) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: [1a] Nasopharyngeal swabs [1b] Not stated Timing of reference standard: [1a] On the first hospital day [1b] Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: [1a] Serial blood sampling (3 times per week) was started at a median of 2 days (range, 1–7 days) after positive RT‐PCR. [1b] Not stated All patients received same reference standard: Yes Missing data: yes (no sensitivity data for 24 non‐hospitalised patients [1b] for test [B], no sensitivity data for time points 1‐7 days, 8‐14 days and 15‐21 days pso reported for both groups) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Multiple samples per patient but only 1 sample per patient included per time split |
||
| Comparative | |||
| Notes | Funding: None stated Publication status: Published paper Source: Journal of Infectious Diseases Author COI: All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors considered relevant to the content of the manuscript have been disclosed. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Rode 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection Design: Single‐group study to estimate sensitivity [1] Confirmed COVID patients (n = 21, 60 samples) Recruitment: Randomly selected hospitalised adult patients (consecutive sera analysed) Prospective or retrospective: Prospective Sample size: 60 (60) samples Further detail: Inclusion: Subjects who had positive RT‐PCR and were hospitalised Exclusion: Test group ‐ PCR‐negative samples |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: University Hospital for Infectious Diseases “Dr. Fran Mihaljević”, Mirogojska 8, 10000 Zagreb. Country: Croatia Dates: Not stated Symptoms and severity: The most common symptoms were cough (95.2%), fever (90.5%), fatigue (42.9%) and shortness of breath (42.9%). Pulmonary opacities showed in 76.2% of patients. Severity ‐ mild, moderate and severe; Mild disease 5 (23.8%) Moderate disease 10 (47.6%) Severe disease 6 (28.6%) Demographics: Age median (range), years 56 (26–81); male/female 13 (61.9%)/8 (38.1%); comorbidity 10 (47.6%) Exposure history: Not stated |
||
| Index tests | Test name: [A] Anti‐SARS‐CoV‐2 IgA ELISA [B] Anti‐SARS‐CoV‐2 IgG ELISA [C] SARS‐CoV‐2 IgM/IgG Antibody Assay Kit Manufacturer: [A,B] Euroimmun, Germany [C] Maccura Biotechnology Co., Ltd. Antibody: [A] IgA, [B] IgG [C] IgM and IgG Antigen target: [A,B] S1 antigen [C] N/S antigen Evaluation setting: [A,B] Laboratory [C] POCT performed in lab Test method: [A,B] ELISA and [C] Collodial Gold Timing of samples: Range 0‐22 days post‐symptom onset: 0‐3 days pso: n = 11, 4‐7 days pso: n = 17, 8‐11 days pso: n = 18, >= 12 days from onset of illness: n = 14 Samples used: Serum Test operator: Laboratory personnel Definition of test positivity: [A,B] The antibody levels were determined by calculating the extinction ratio of the patient samples (S) over the cut‐off calibrator value (CO; S/CO). Cut‐off not stated [C] A clearly visible coloured quality control band and detection line, either IgG or IgM, were deemed positive for anti‐SARS‐CoV‐2 antibodies. The final results were always read by two independent investigators. Blinding reported: Not stated (no as only COVID cases included) Threshold predefined: yes by the manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐qPCR Roche Total Nucleic Acid Isolation kit on a Roche MagnaPure LC 2.0 (Roche, Germany) According to the WHO‐recommended Charité protocol, utilising the E and RdRP gene targets on an Applied Biosystems 7500 real‐time thermocycler (Applied Biosystems, USA), 5 μL of RNA was used for the detection of SARS‐CoV‐2.
Threshold not stated Samples used: Combined nasopharyngeal and oropharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples (Eleven patients had 2 consecutive sera, 6 had 3 sera, and 3 had 4 sera and for one patient, 8 samples were tested) |
||
| Comparative | |||
| Notes | Funding: None stated Publication status: Published paper Source: European Journal of Clinical Microbiology & Infectious Diseases Author COI: The authors declared that they had no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Rode 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rode 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection or current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID samples (n = 366) [2] Non‐COVID samples [2a] Blood donor samples from influenza seasons 2016/17 and 2017/18 (n = 500) [2b] Samples which tested PCR‐negative for SARS‐CoV‐2 (n = 110) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 976 (366) Further detail: Inclusion: [1] Serum of our previously described positive (SERO‐BL‐positive) cohort of study participants testing PCR‐positive for SARS‐CoV‐2 during the initial wave of COVID‐19 infections in the canton of Basel‐Landschaft Switzerland [2a] Blood donor cohort composed of donations from December 2016, February 2017, and February 2018 [2b] Serum of our previously described negative (SERO‐BL‐negative) cohort of study participants testing PCR‐negative for SARS‐CoV‐2 during the initial wave of COVID‐19 infections in the canton of Basel‐Landschaft,2 Switzerland Exclusions not reported. |
||
| Patient characteristics and setting | Setting: Convalescent study participants of SERO‐BL‐COVID‐19 Location: Biobank of the Canton Basel‐Landschaft Country: Switzerland Dates: During the first wave of the pandemic in Switzerland Symptoms and severity: Wide range of disease severity; these samples were representative for symptomatic and oligosymptomatic cases. Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic healthy Source: Blood donor cohort composed of donations from December 2016, February 2017, and February 2018 Characteristics: Blood donor samples from previous flu seasons Non‐Covid group 2: [2a] Current, PCR‐negative Source: Study participants of "COVID‐19 in Baselland Investigation and Validation of Serological Diagnostic Assays and Epidemiological Study of Sars‐CoV‐2 specific Antibody Responses (SERO‐BL‐COVID‐19)", during the first wave of the pandemic in Switzerland Characteristics: PCR‐negative |
||
| Index tests | Test name: [A] OnSite™ COVID‐19 IgG/IgM Rapid Test (LOT F0507R1C00) [B] SARS‐CoV‐2 IgM/IgG Antibody Rapid Test (LOT COV1252006A) [C] SimtomaX® Corona Check (LOT GGM20089W) [D] SARS‐CoV‐2 Antibody Lateral Flow Test (LOT 20200428) [E] NTBIO One Step Rapid Test ‐ COVID‐19 IgG/IgM Antibody Test (LOT V02009201) [F] QuickTestCorona™ COVID‐19 IgG/IgM (LOT MC0000102) [G] SARS‐Cov‐2 IgG/IgM Rapid Qualitative Test (LOT X2003602) [H] BIOZEK COVID‐19 IgG/IgM Rapid Test Cassette (LOT BNCP40200080) [I] MEDsan COVID‐19 IgM/IgG Rapid Test (LOT 20200325) [J] SARS‐CoV‐2 IgM/IgG Ab Rapid Test (LOT COV1252003C) [K] The RightSignTM COVID‐19 IgG/IgM Rapid Test Cassette / Lumiratek (LOT COV20040013) Manufacturer: [A] CTK Biotech, Inc. (US) [B] Sure Bio‐tech (USA) Co., Ltd (US) [C] Augurix SA (CH) [D TAmiRNA GmbH (AT) [E] NTBIO® Diagnostics Inc. (CA) [F] MEXACARE GmbH (DE) [G] Xiamen Biotime Biotechnology Co., Ltd. (CN) [H] Inzek International Trading B.V. (NL) [I] MPC International S.A. (LU) [J] Qingdao HIGHTOP Biotech Co., Ltd. (CN) [K] Hangzhou Biotest Biotech Co Ltd (CN) Antibody: [D] IgM/IgG (single band) All other tests: separate lines for IgM and IgG Antigen target: [A] Spike [B] S1, S2, RBD [C] RBD, N‐protein [D] S1 [E] Not stated [F] RBD, N‐protein [G] Not stated [H] Not stated [I] Not stated [J] Spike, N‐protein [K] Spike Evaluation setting: All POCT performed in lab Test method: All lateral flow tests (no details) Timing of samples: wide range of days post‐symptom onset <= 14 days pso 15‐21 days pso > 21 days pso Numbers differed between tests. Samples used: We assayed the Hightop test using whole blood, serum and plasma, while all other tests were assayed using serum and plasma. Test operator: The Hightop [J] and MEDSan [I] assays were characterised at the SwissTPH using the identical biobank and experimental setup as outlined previously. Eight tests were characterised simultaneously at the KUSPO Munchenstein and the Biotime [G} at the FHNW, Muttenz. Definition of test positivity: Presence of bands was visually inspected, and each test was imaged with a digital camera (different models) under standardised lightning conditions. We considered a test valid if its control band was present, and we considered a valid test positive for the respective antibody if the SARS‐CoV‐2 specific IgM, IgG or IgM/IgG band was detected in the sample. Blinding reported: Unclear Threshold predefined: Yes, visual‐based |
||
| Target condition and reference standard(s) | Reference standard: [1] PCR‐positive for SARS‐CoV‐2 Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: [2a] Pre‐pandemic [2b] PCR‐negative for SARS‐CoV‐2 Samples used: [2a] Pre‐pandemic [2b] Not stated Timing of reference standard: [2a] Pre‐pandemic [2b] Not stated Blinded to index test: Yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: Yes (not all samples tested with all index tests; time split <= 14 days pso not eligible for our review) Uninterpretable results: Not stated Indeterminate results: No intermediate range Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | Funding: The Swiss Red Cross financed all the used LFA except for the Hightop and Biotime assays. The Hightop
was purchased by the canton Basel‐Landschaft and the Biotime was provided by the Swiss importer.
FR is funded by the NCCR ’Molecular Systems Engineering’. Publication status: Pre‐print (not peer reviewed) Source: medRxiv preprint Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Rudolf 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Rudolf 2020 [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Ruetalo 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients [1a] Non‐hospitalised COVID‐patients (n = 49), 46 PCR+, 3 symptomatic close contacts [1b] one hospitalised, convalescent COVID patient (2 samples) [2] Healthy donors (n = 4); Group [2] excluded from our review as < 25 samples Group [1b] not included as no information on time pso or time post‐PCR+ [1a] 3 symptomatic close contacts excluded as not PCR‐confirmed Recruitment: Not stated Prospective or retrospective: Not stated Sample size: 55 (49) samples of which 46 (46) rt‐PCR positive COVID patients were eligible for our review Further detail: Inclusions [1a] Potential blood donors for convalescent plasma therapy after written consent at the Clinical Transfusion Medicine, Tübingen between April 04 and May 12, 2020 Older than 18 years old with a PCR‐confirmed diagnosis of SARS‐CoV‐2 (n = 46) or symptomatic and close contacts to positively diagnosed COVID‐19 patients (partners tested positive) [1b] Hospitalised, convalescent COVID patient [2] Healthy donors Exclusions not reported |
||
| Patient characteristics and setting | Setting: Convalescent (potential convalescent plasma donors) Location: Clinical Transfusion Medicine, Tübingen Country: Germany Dates: between April 04 and May 12, 2020 Symptoms and severity: non‐hospitalised, asymptomatic to a mild course of disease, cough (69%), fever (59%), limb pain and headache (35%), diarrhoea (10%), and loss of taste (10%). Now all convalescent Demographics: Age ranged from 19‐66 years (median 40 years); 24 male, 25 female Exposure history: Not stated |
||
| Index tests | Test name: [A] Euroimmun SARS‐CoV‐2‐ELISA (IgG) [B] S1 RBD SARS‐CoV‐2 (IgG, IgA, IgM) (test name not stated) [C] Elecsys anti‐SARS‐CoV‐2 *Additional assay (SARS‐COV‐2 DigiWest assay) excluded as not commercially available Manufacturer: [A] Euroimmun, Lübeck, Germany [B] Mediagnost [C] Roche Antibody: [A] IgG [B] IgG, IgA, IgM[C] Total antibody Antigen target: [A] S1‐based [B] [C] N‐protein Evaluation setting: All laboratory tests Test method: [A] ELISA [B] ELISA [C] ECLIA Timing of samples: The time from positive SARS‐CoV‐2 test to blood sampling was 14‐64 days (median 45 days). Samples used: Serum samples were stored at ‐80°C. Test operator: [A] Institute for Transfusion Medicine, University Hospital Tübingen, Tübingen, Germany [B] Mediagnost GmbH, Reutlingen, Germany [C] Institute for Medical Virology and Epidemiology of Viral Diseases, University Hospital Tübingen, Tübingen, Germany Definition of test positivity: [A] Ratios were classified as negative (< 0.8), borderline (≥ 0.8– < 1.1) and positive (≥ 1.1) [B] Ratios were classified as: negative (< 0.42), borderline (≥ 0.42‐0.7) and positive (≥ 0.7) for IgG; negative (< 0.33), borderline (≥ 0.33‐0.7) and positive (≥ 0.7) for IgA; negative (< 0.87), borderline (≥ 0.87‐1.47) and positive (≥ 1.47) for IgM [C] If the numeric COI result was ≥ 1.0, the serum was diagnosed as reactive, COI < 1.0 were attributed as non‐reactive. Blinding reported: Not stated Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard: PCR‐confirmed diagnosis of SARS‐CoV‐2 (n = 46) and three were symptomatic and close contacts to positively diagnosed COVID‐19 patients (partners tested positive), PCR threshold not stated.
3 COVID patients without PCR test not included in review Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: The time from positive SARS‐CoV‐2 test to blood sampling was 14‐64 days (median 45 days). All patients received same reference standard: yes Missing data: yes (exclusion of groups [1b] and [2] and 3 patients from group [1a] from our review) Uninterpretable results: Not stated Indeterminate results: yes [A] n = 1 [B] n = 6 [C] n = 15 Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This work was supported by grants to MS from the Baden‐Württemberg foundation (BW Stiftung), the Deutsche Forschungsgemeinschaft, the MWK Baden‐Würtemberg as well as by basic funding provided to MS by the University Hospital Tübingen and TÜFF Gleichstellungsförderung to K.A. (2563‐0‐0). Publication status: Pre‐print (not peer reviewed) Source: medRxiv pre‐print Author COI: The authors reported no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Ruetalo 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Ruetalo 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Schnurra 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Single‐group study to estimate sensitivity only [1] Confirmed COVID patients (73 sera from 57 patients) Recruitment: Not stated Prospective or retrospective: Prospective Sample size: 73 (73) samples from 57 (57) patients Further detail: Inclusion: Adult individuals with positive SARS CoV‐2 RNA test after informed consent Exclusion: Ex post, four viral RNA‐positive participants that were asymptomatic and were tested as part of routine screening for healthcare workers or before surgery without contact with infected persons were excluded. |
||
| Patient characteristics and setting | Setting: Not stated (none required hospital care, all convalescent) Location: University Clinics and Medical Faculty, University of Leipzig, Leipzig, Germany Country: Germany Dates: Not stated Symptoms and severity: with mild to moderate disease or asymptomatic infection; none was seriously ill or required hospital care. 3 asymptomatic 25 mild (e.g. fatigue, sore throat, headache) 28 moderate (e.g. fever, myalgia, no or mild pneumonia) 1 severe disease (e.g. with dyspnoea, hypoxia, or > 50 percent lung involvement on imaging within 24 to 48 hours) Demographics: Adults Exposure history: Not stated |
||
| Index tests | Test name: [A] Roche Elecsys Anti‐SARS‐CoV‐2 [B] Abbott Architect SARS‐CoV‐2 IgG [C] Novatec Novalisa SARS‐CoV‐2 IgG ELISA [D] Virotech SARS‐CoV‐2 IgG ELISA [E] Euroimmun Anti‐SARS‐CoV‐2‐ELISA (IgG) [F] Mediagnost AntiSARS CoV‐2 ELISA [G] Siemens Atellica IM COV2T Manufacturer: [A] Roche [B] Abbott [C] Novatec [D] Virotech [E] Euroimmun [F] Mediagnost [G] Siemens Antibody: [A] IgM, IgG and other Ig antibody bridging [B] IgG [C] IgG [D] IgG [E] IgG [F] IgG [G] IgM, IgG and other Ig antibody bridging Antigen target: [A] N‐protein [B] N‐protein [C] N‐protein [D] N‐protein [E] S1 glycoprotein [F] RBD of S1 glycoprotein [G] RBD of S1 glycoprotein Evaluation setting: All laboratory tests Test method: [A] ECLIA [B] CMIA [C] ELISA [D] ELISA [E] ELISA [F] ELISA [G] Microparticle immunoassay (chemiluminescence?) Timing of samples: Between 2 and 10 weeks after symptom onset or viral RNA testing (additional data provided by author show range from 14 to 70 days post‐PCR test): 2‐3 weeks after PCR+: n = 25 4‐10 weeks after PCR+: n = 48 Sera from symptomatic participants fell into the same groups when classified according to the day of symptom onset. Samples used: Sera were frozen at −20 °C until testing. During the study, serum samples were thawed 1–4 times. Test operator: The tests were performed in three diagnostic routine laboratories and a research laboratory according to the instructions of the manufacturers. Definition of test positivity: [A] Positive ≥ 1 COI, negative < 1 COI [B] Positive ≥ 1.4 index (S/C), negative < 1.4 index (S/C) [C] Positive > 11 NTU, borderline 9–11 NTU, negative < 9 NTU [D] Positive > 11 VE, borderline 9–11 VE, negative < 9 VE [E] Positive ≥ 1.1, borderline ≥ 0.8 to < 1.1, negative < 0.8 [F] Positive > 5 x OD negative control, borderline 3–5 x OD negative control, negative < 3 x OD negative control [G] Positive ≥ 1 index, negative < 1 index Blinding reported: No as only COVID cases included Threshold predefined: yes (all tests performed according to the manufacturer's instructions) |
||
| Target condition and reference standard(s) | Reference standard: Positive SARS CoV‐2 RNA test, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: 2−3 weeks (N = 25) or > 4 weeks (N = 48) after symptom onset and viral RNA test All patients received same reference standard: yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: For calculations, borderline results were considered negative. [C] 8 borderline results [D] 5 borderline results [E] 4 borderline results [F] 15 borderline results Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: We are also grateful to ... Novatec Immundiagnostica GmbH, Virotech Diagnostics GmbH and Mediagnost
GmbH for making their test kits available. The Siemens COV2T tests were provided by Siemens Healthineers and performed by Labor alphaomega, Leipzig. The research did not receive any specific grant from funding agencies. Publication status: Published paper Source: Journal of Clinical Virology Author COI: None. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | No | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Schnurra 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Schnurra 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Schnurra 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Schnurra 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Schnurra 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Schnurra 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] SARS‐CoV‐2‐infected inpatients (126 samples from 89 patients) [2] Pre‐pandemic controls (36 samples) [2a] Healthy (n = 25) and [2b] HIV and other viral diseases (n = 11) Recruitment: [1] Not stated [2] Matched samples were selected based on COVID‐19 patients’ sex and age Prospective or retrospective: [1] Prospective [2] Retrospective Sample size: 162 (126) samples Further detail: Inclusion: [1] Patients living in the Minho region of Portugal who were inpatients at Senhora da Oliveira Hospital (Guimarães) or Braga Hospital, admitted with COVID‐19 (diagnosed by RT‐qPCR at a reference laboratory; at least 2 positive RT‐qPCR results were obtained from each patient) [2] SARS‐COV‐2 non‐infected controls were selected from banked human plasma samples from 2 pre‐COVID‐19 pandemic studies conducted by the study authors (the first COVID‐19 case in Portugal was reported on 2 March 2020): [2a] a study with healthy individuals > 55 years old (samples collected between April 2019 and January 2020); [2b] a study with HIV‐infected patients on antiretroviral therapy (54–60 months; samples collected between January 2016 and August 2018). Matched samples were selected based on COVID‐19 patients’ sex and age. |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Senhora da Oliveira Hospital (Guimarães, Portugal) or Braga Hospital (Braga, Portugal) Country: Portugal Dates: Not stated Symptoms and severity: Severe 32/89; non‐severe 57/89 Demographics: Age: median 71 [range 30;96] years; female 51/89 (57.3%) None of the COVID‐19 patients were HIV‐positive or had a history of organ transplantation. Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic healthy Source: Banked human plasma samples from a pre‐COVID‐19 pandemic study with healthy individuals > 55 years old, samples collected between April 2019 and January 2020 Characteristics: Age: Median 71 [range 59 to 80] years; 13/25 (52.0%) female Non‐Covid group 2: [2b] Pre‐pandemic, other diseases Source: Banked human plasma samples from a pre‐COVID‐19 pandemic study with HIV‐infected patients on antiretroviral therapy, samples collected between January 2016 and August 2018 Characteristics: Age: Median 57 [range 33;72] years; 3/11 (27.3%) female |
||
| Index tests | Test name: [A] Abbott Architect anti‐SARS‐CoV‐2 IgG (no. 06R86) [B] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG (no. EI 2606‐9601 G) [C] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA (no. EI 2606‐9601 A) [D] Snibe Diagnostic ‐ MAGLUMI 2019‐nCoV IgG/IgM (no. 130219016M) [E] Cellex qSARS‐CoV‐ 2 IgG/IgM (no. WI5513C) [F] Getein One Step Test (no. CG2057) [G] Innovita Biological ‐ 2019‐nCoV Ab test [H] Liming Bio StrongStep1 IgM/IgG [I] Leccurate ‐ SARS‐CoV‐2 [J] Jiangsu Medomics Combined Ab [K] Render COVID‐19 IgM/IgG (no. K‐20‐RC‐CoV‐2) [L] SD Biosensor IgM/IgG Duo (no. Q‐NCOV‐01D) Manufacturer: [A] Abbott Architect [B] EUROIMMUN [C] EUROIMMUN [D] Snibe Diagnostic [E] Cellex [F] Getein [G] Innovita Biological [H] Liming Bio [I] Leccurate [J] Jiangsu Medomics [K] Render [L] SD Biosensor Antibody: [A] IgG [B] IgG [C] IgA [D] IgM/IgG [E] IgM/IgG [F] Total antibodies [G] IgM/IgG [H] IgM/IgG [I] IgM/IgG [J] IgM/IgG [K] IgM/IgG [L] IgM/IgG Antigen target: [A] N‐protein [B] S1‐protein [C] S1‐protein [D] S antigen and N‐protein [E] N and S‐proteins [F] N and S‐proteins [G] N and S‐proteins [H] Not specified [I] Not specified [J] Not specified [K] Not specified [L] N‐protein Evaluation setting: [A] ‐ [D] Laboratory tests used in lab [E] ‐ [L] POCT performed in lab (frozen plasma) Test method: [A] CLIA [B] ELISA [C] ELISA [D] CLIA [E] LFIA [F] LFIA [G] LFIA [H] LFIA [I] LFIA [J] LFIA [K] LFIA [L] LFIA Timing of samples: Days since symptom onset: Numbers varied per test: < 10 days: 12‐24 samples; 10–15 days: 12‐33 samples; 16–21 days: 20‐34 samples; > 21 days: 16‐35 samples Samples used: Plasma frozen at ‐80 degrees Celsius Test operator: C.S‐M., C.N., S.R., J.C‐G., C.S.S., N.V., P.B‐S. and P.A‐P. performed the experiments. Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal and ICVS/3B’s‐PT Government Associate Laboratory, Braga/Guimarães, Portugal. Definition of test positivity: Threshold not stated for [A] ‐ [E] (see Fig 2; according to the manufacturer's instructions) [A] Index (S/C) [B] Ratio (between 0.8 and 1.1 borderline) [C] Ratio (between 0.8 and 1.1 borderline) [D] Arbitrary units/mL [E]‐[L] Visual‐based Blinding reported: Not stated Threshold predefined: yes, tested according to the manufacturer’s instructions ([E] ‐ [L] visual‐based) |
||
| Target condition and reference standard(s) | Reference standard: [1] Diagnosed by RT‐qPCR at a reference laboratory; at least 2 positive RT‐qPCR results were obtained from each patient); threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2] Pre‐pandemic Samples used: [2] Pre‐pandemic Timing of reference standard: [2] Pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: yes (not all patients tested with all tests, only 1 sample per time split used per patient) Uninterpretable results: Not stated Indeterminate results: Not stated (according to Fig 2, test [B] could have borderline results) Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was funded by National funds, through the Foundation for Science and Technology (FCT) R4COVID (
596694995), POCI‐01‐0145‐FEDER‐016428, UIDB/50026/2020 and UIDP/50026/2020; and by the projects NORTE‐01‐0145‐
FEDER‐000013 and NORTE‐01‐0145‐FEDER‐000023, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). CN, SR and NV are junior researchers under the scope of the FCT Transitional Rule DL57/2016. JC‐G is supported by an FCT PhD grant, in the context the Doctoral Program in Aging and Chronic Diseases (PhDOC; PD/ BD/137433/2018); CSS is supported by an FCT PhD grant, in the context of the Doctoral Program in Applied Health Sciences(PD/BDE/142976/2018). Publication status: Published paper Source: International Journal of Infectious Diseases Author COI: Getein kits were provided free of charge by the manufacturer. No other conflicts of interest reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Serre‐Miranda 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Serre‐Miranda 2021 [L].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Shamsollahi 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to evaluate the sensitivity and specificity of a rapid serological test for diagnosis of active or previous COVID‐19 using serum samples Design: [1] 114 RT PCR‐confirmed COVID‐19 patients in hospitals affiliated to Tehran University of Medical Sciences in 2020 [2] 198 frozen serum specimens taken from healthy people in summer and autumn 2019 (pre‐COVID‐19) From group [1], time split 0‐19 days pso was excluded from our review (n = 31). Recruitment: COVID‐19 cases were PCR‐confirmed patients in hospitals affiliated to Tehran University of Medical Sciences in 2020; test‐negative controls were a random sample of frozen serum specimens from healthy people participating in a Tehran University of Medical Sciences Employees COHORT study, taken in summer and autumn 2019 (months before reporting the first case of COVID‐19 by China) Prospective or retrospective: Unclear Sample size: 312 (114) of which 312 (83) were eligible for our review Further detail: No more details available |
||
| Patient characteristics and setting | Setting: RT‐PCR‐confirmed COVID‐19 cases in several hospitals ‐ unclear whether inpatient or outpatient Location: Several hospitals affiliated to Tehran University of Medical Sciences Country: Iran Dates: Unclear Symptoms and severity: Not stated Demographics: Average age: 44.0 (± 12.1) years Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic negative controls Source: healthy people participating in a Tehran University of Medical Sciences Employees COHORT study, taken in summer and autumn 2019 Characteristics: Average age: 39.2 (± 8.0) years |
||
| Index tests | Test name: “VivaDiag” COVID‐19 IgM/IgG Manufacturer: VivaChek Inc., China Antibody: IgM, IgG Antigen target: Not stated Evaluation setting: POC test; unclear where testing was done for cases; as negative controls were frozen samples, these must have been done in a laboratory Test method: Not stated (Seemed to be colloidal gold from website) Timing of samples: 5‐53 days (mean: 27.9) pso 0‐19 days pso: 31/114 (27.2%) 20‐39 days pso 65/114 (57.0%) 40+ days pso: 18/114 (15.8%) Samples used: 10 μL (whole) blood for cases or frozen serum for pre‐pandemic samples Test operator: Not stated Definition of test positivity: Not stated ; "Based on kit instructions" Visual‐based Blinding reported: No; tests appeared to have been conducted separately for known positives and for known negatives (which had to be thawed before testing) Threshold predefined: Based on kit instructions (visual‐based) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR ‐ no threshold reported Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes. For cases, samples were already PT PCR‐confirmed when index test used. Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic blood samples. No report of these being tested by RT‐PCR Samples used: 10 μL blood Timing of reference standard: Pre‐pandemic blood samples. Blinded to index test: Yes. For controls, blood samples were drawn months before the study. Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated for cases; pre‐pandemic controls All patients received same reference standard: No (PCR test for cases, pre‐pandemic samples for controls) Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: 1 sample per patient. |
||
| Comparative | |||
| Notes | Funding: Not stated; VivaDiag kit donated to Tehran University of Medical Sciences (TUMS) by the Ministry of Health and Medical Education (MOHME) Publication status: Preprint, now published Source: Preprint server ‐ medRxiv Journal (Archives of Iranian Medicine) Author COI: The authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Shen 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: Single‐group study to estimate sensitivity and specificity: Patients with fever or respiratory symptoms suspected as having COVID‐19 based on China CDC guideline (v5) (including 97 RT‐PCR confirmed) [A separate cohort of 26 healthy blood donors were tested ‐ not included in main analysis] Recruitment: Not reported; could be consecutive Prospective or retrospective: Prospective Sample size: 150 (97) Further detail: Participants met the suspected COVID‐19 case definition according to the Diagnosis and Treatment Guideline (trial version 5) of China. A suspected COVID‐19 case was defined as a pneumonia that had related epidemiological history (likely exposure) and fulfilled two of the three criteria: fever and/or respiratory symptoms; imaging manifestations of pneumonia; low or normal white‐cell count or low lymphocyte count. |
||
| Patient characteristics and setting | Setting: Unclear; Public Health Medical Center (all quarantined for 2 weeks) Location: Taizhou Hospital of Zhejiang Province, China Country: China Dates: January 20, 2020 to February 2, 2020 Symptoms and severity: Clinical severity ‐ ordinary = 76/97 = (78%) Clinical severity ‐ severe = 21/97 (22%) Fever = 71/97 (73%) Cough = 19/97 (20%) Fatigue = 3/97 (3%) Dizziness = 3/97 (3%) Chest tightness = 3/97 (3%) Diarrhoea = 2/97 (2%) Demographics: 59/97 (60.8%) male; median age (IQR) = 46 (38‐56) [NB age data in Tab 1 were incorrectly printed and implausible. These figures in review text are from the results text] Exposure history: 75/97 (77.3%) with Wuhan exposure Non‐Covid group 1: PCR‐negative Source: Public Health Medical Center, Taizhou Hospital of Zhejiang Province, China; January 20, 2020 to February 2, 2020 Characteristics: 30/53 (56.6%) male; median age (IQR) = 32 (20‐42.5) Fever = 30/53 (57%) Cough = 23/53 (43%) Fatigue = 3/53 (6%) Dizziness = 2/53 (4%) Chest tightness = 6/53 (11%) Diarrhoea = 1/53 (2%) 25/53 (47.2%) with Wuhan exposure |
||
| Index tests | Test name: colloidal gold immunochromatography assay for SARS‐Cov‐2 IgM/IgG (LOT: 20200101) Manufacturer: Shanghai Outdo Biotech Co. Ltd, China Antibody: SARS‐Cov‐2 IgM/IgG Antigen target: synthetic antigens of the S, M, and N‐proteins of COVID‐19 Evaluation setting: Intended for POC use. For the study "sera were incubated at 56°C for 30 minutes to heat‐inactivate viruses before serological analysis", so study use was laboratory‐dependent. Test method: Lateral flow immunoassay (colloidal gold immunochromatography assay) Timing of samples: At time of consultation. Time since symptom onset for COVID 19‐positive cases: 0‐7 days = 40/97 (41.2%) 8‐14 days = 33/97 (34.0%) ≥ 15 days = 24/97 (24.7%) Since symptom onset for COVID 19‐negative: 0‐7 days = 50/53 (94.3%) 8‐14 days = 3/53 (5.7%) Samples used: serum (3 mL of peripheral venous blood collected) Test operator: Not stated Definition of test positivity: Visible line on immunochromatography antibody detection kit Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR at referral laboratory; not further described
PCR‐positive = Ct threshold < 37
PCR‐negative = Ct threshold > 40
Those with results between 37 to 40 Ct were resampled and PCR performed by CDC.
Absence of COVID‐19 required at least 2 RT‐PCR‐negative results; authors still considered 34/53 PCR‐negative as 'inconclusive' Covid‐19 due to lack of other 'identified condition or infection' or recovery after treatment. Samples used: Nasopharyngeal and oropharyngeal swab samples Timing of reference standard: At same time as serology samples taken Blinded to index test: Not stated Incorporated index test: No as PCR was done using nose/throat swabs and not blood; antibody tests were not part of guideline definitions at this time. Definition of non‐COVID cases: As above Samples used: As above Timing of reference standard: As above Blinded to index test: As above Incorporated index test: As above |
||
| Flow and timing | Time interval between index and reference tests: Samples (blood and swabs) taken at same time All patients received same reference standard: yes Missing data: No losses to follow‐up. "The clinical record for each patient was complete." Uninterpretable results: None Indeterminate results: Not stated Unit of analysis: patient |
||
| Comparative | |||
| Notes | Funding: Financially supported by National Natural Science Foundation of China (Grant NO. 81672086 and 81903417) Publication status: Published paper Source: American Journal of Translational Research Author COI: Authors reported no conflicts of interest present. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Shen 2020b.
| Study characteristics | |||
| Patient Sampling | Purpose: To investigate the utility of the IgM‐based gold immunochromatographic assay as a candidate clinical diagnostics assay in COVID‐19 patients
Multi‐group study to estimate sensitivity and specificity for the diagnosis of active disease/identification of previous disease [1] COVID‐19 patients (n = 58) in time‐based analysis [2] COVID‐19 patients (n = 70, incl 45 PCR‐positive); acute phase only (4‐14 days pso); not eligible for inclusion [3] patients with non‐coronaviral respiratory illness (n = 10) (2 confirmed for influenza A virus, 3 confirmed for influenza B virus, 3 confirmed for respiratory syncytial virus and 2 confirmed for adenovirus) [4] negative control, consisted of 50 sera samples collected from 50 healthy people assessed by physical examination (n = 50) Recruitment: patients at the Xiangyang Central Hospital ‐ no further information Prospective or retrospective: unclear. Sample size: 118 (58) patients eligible for inclusion Further detail: Not stated |
||
| Patient characteristics and setting | Setting: patients at the Xiangyang Central Hospital ‐ appeared to be mixed settings Location: Xiangyang Central Hospital, Affiliated Hospital of Hubei University of Arts and Science, Xiangyang Hubei Province 441021, People’s Republic of China Country: China Dates: Not stated Symptoms and severity: mild (n = 50) and severe (n = 8) Demographics: Age: Median (IQR) 52 (36–61); range 8‐81 years; 55% (32/58) female Exposure history: Not stated |
||
| Index tests | Test name: SARS‐CoV‐2 IgM GICA kit Manufacturer: Shanghai Outdo Biotech Co., China Antibody: IgM Antigen target: immobilised SARS‐CoV‐2 antigen (N and S recombinant proteins forming an antibody–antigen complex) Evaluation setting: POC, performed in laboratory Test method: colloidal gold immunochromatographic assay (GICA) Timing of samples: 0 to 31 days after symptom onset < 4 days pso: 41/155 (26.5%) 4‐7 days pso: 31/155 (20.0%) 8‐14 days pso: 48/155 (31.0%) 15‐21 days pso: 23/155 (14.8%) > 21 days pso: 12/155 (7.7%) Samples used: Serum Test operator: Not stated Definition of test positivity: The serum was considered positive if bands could be visualised on both the test and control lines. Each sample was repeated in triplicate. Blinding reported: Unclear Threshold predefined: As per manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by the Doctoral Fund of Xiangyang Central Hospital (RC202001), the One Belt and One Road major project for infectious diseases (2018ZX10101004‐ 003). Gary Wong is supported by a G4 grant from IP, FMX and CAS. Publication status: Published paper Source: Emerging Microbes & Infections Author COI: No potential conflict of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Soleimani 2021.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (176 samples obtained from 125 patients) [2] Non‐COVID (100 samples) [2a] Pre‐pandemic healthy (n = 40) [2b Pre‐pandemic, other diseases (n = 40) [2c] Asymptomatic subjects in March 2020 (n = 20) (excluded as no reference standard) Recruitment: [1] Randomly collected from 25 February to 10 March 2020 [2a] and [2b] Randomly selected among stored sera collected between October 2018 and February 2019 [2c] Not stated Prospective or retrospective: [1] Prospective [2a] and [2b] Retrospective (residual samples) [2c] Unclear Sample size: 276 (176) samples Further detail: Inclusion criteria: [1] Symptomatic and hospitalised patients >= 18 years with positive RT‐qPCR tests on nasopharyngeal swab samples and characteristic radiological lung patterns such as ground glass opacity and/or bilateral involvement [2a] Residual samples obtained from COVID‐19 negative subjects with no known confounding factors [2b] Residual samples obtained from COVID‐19 negative subjects with supposedly confounding factors known to interfere with serological assays such as auto‐immune Ab and infectious diseases Ab [2c] Asymptomatic subjects during the overlapping period of Flu epidemic and COVID‐19 outbreak in March 2020 Exclusion criteria: [1] < 18 years; immunocompromised patients were not excluded from the study. [2] Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Cliniques Universitaires Saint‐Luc, Brussels, Belgium Country: Belgium Dates: 25 February to 10 March 2020 Symptoms and severity: All symptomatic with viral pneumonia and hospitalised Symptoms on admission: Fever (> 38‐38.5°C) 125/125 Cough 119/125 Dyspnoea 120/125 Myalgia and or fatigue 48/125 Rhinorrhea and sore throat 16/125 Diarrhoea 14/125 Nausea, vomiting and abdominal pain 14/125 Other symptoms (including headache, confusion, unconsciousness, anosmia and dysgeusia) 55/125 Final outcomes: 17/125 death 101/125 recovered 7/125 remained in hospital by 30 April 2020 Demographics: 58/125 female; mean age 65.2 (95% CI 62.3‐68.1) years Exposure history: Not stated Non‐Covid group 1: [2] Non‐COVID samples Source: [2a] and [2b] stored sera collected between October 2018 and February 2019 [2c] Asymptomatic subjects during the overlapping period of Flu epidemic and COVID‐19 outbreak in March 2020 Characteristics: 60/100 females; mean age = 37.2 years 20 asymptomatic; 40 no confounding factors; 40 other diseases: 23 infectious diseases Ab including:
17 auto‐immune diseases Ab including:
|
||
| Index tests | Test name: [A] Snibe MAGLUMI 2019‐Novel Coronavirus (nCoV) Kit Manufacturer: [A] Shenzhen New Industries Biomedical Engineering [Snibe] Co., Ltd., Shenzhen, China Antibody: [A] IgG and/or IgM Antigen target: [A] nucleocapsid and Spike‐proteins Evaluation setting: [A] Laboratory test Test method: [A] CLIA Timing of samples: 0 to 4 days pso n = 21, 5 to 9 days pso n = 50, 10 to 14 days pso n = 61, 15 to 25 days pso n = 44. Samples used: Serum Test operator: Lab technicians Definition of test positivity: A level greater than 1.00 AU/mL was interpreted as positive for both Ab. Blinding reported: Not stated Threshold predefined: yes, seropositivity cut‐off value claimed by the manufacturer (1 AU/mL) |
||
| Target condition and reference standard(s) | Reference standard: COVID‐19 was confirmed by positive RT‐qPCR on nasopharyngeal swab and by radiographic criteria (bilateral chest involvement and/or ground‐glass opacity [GGO] identified by X‐ray or computed tomography [CT] scan) Samples used: nasopharyngeal swab samples Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2a] and [2b] Pre‐pandemic [2c] Current asymptomatic, untested Samples used: [2a] and [2b] Pre‐pandemic [2c] untested Timing of reference standard: [2a] and [2b] Pre‐pandemic [2c] untested Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: no Missing data: yes (data for Euroimmun ELISA test not included in review as no eligible time split) Uninterpretable results: Not stated Indeterminate results: No borderline range Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Journal of Medical Virology Author COI: The authors declared that there were no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Sterlin 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To evaluate immune response in individuals with SARS‐CoV‐2 infection
1‐group study to estimate sensitivity for diagnosis of active disease and identification of previous disease Design: Group [1]: PCR‐confirmed adult COVID‐19 cases (n = 135) Group [2]: Age‐ and sex‐matched healthy donors (n = 20) Group [3]: 10 cases with CT scan displaying features suggesting a COVID‐19 infection and tested positive for the presence of serum anti‐SARSCoV‐2 antibodies Group [2] was excluded from the review as < 25 controls. Group [3] was excluded as < 10 cases and no test accuracy outcomes. Recruitment: [1] Consecutive [2] Age‐ and sex‐matched [3] Not stated Prospective or retrospective: Prospective (patients gave informed consent and samples were immediately collected) Sample size: 155 (135) of which only 135 (135) cases/214 (214) samples were eligible for our review Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: Department of Internal Medicine 2, Pitié‐Salpêtrière Hospital, Paris Country: France Dates: March 22 to April 24, 2020 Symptoms and severity: All symptomatic and hospitalised 39/135 (29%) admitted to ICU (severe/critical) Pneumonia 123 (91%)
Acute respiratory distress syndrome 13 (10%) Heart failure 5 (4%) Acute renal injury 15 (11%) Demographics: Age, median (IQR: 61.3 y (49.7‐72.0); sex: 55/135 (41%) female Exposure history: Not stated |
||
| Index tests | Test name: Maverick SARS‐CoV‐2 Multi‐Antigen Serology Panel Manufacturer: Genalyte Inc., USA Antibody: IgA, IgM, IgG Antigen target: N, S1 RBD, S1/S2, S2 and S1 (multiplex format based on photonic ring resonance technology) Evaluation setting: Lab test, done in lab Test method: Photonic ring immunoassay Timing of samples: Multiple samples obtained from each patient (214 samples from 135 patients): 48 samples collected 1‐7 days pso; 8‐14 days pso: 81/214 15‐21 days pso: 39/214 22‐28 days pso: 20/214 > 28 days pso: 26/214 Samples used: Serum Test operator: Not stated Definition of test positivity: 20 sera collected before December 2019 were analysed to calculate cut‐off values. Positivity was defined as results above the 99th percentile. Blinding reported: Not stated Threshold predefined: Yes, 20 sera collected before December 2019 (independent samples) were analysed to calculate cut‐off values. Positivity was defined as results above the 99th percentile. |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR assay (no more details available) Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Unclear, but likely done earlier Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Unclear (PCR test used not mentioned ‐ perhaps different tests used for different patients) Missing data: Unclear (numbers not provided in the text, figures hard to interpret because of overlapping circles) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by Fondation de France, Tous unis contre le virus framework Alliance (Fondation de France, AP‐HP, Institut Pasteur) in collaboration with Agence Nationale de la Recherche (ANR Flash COVID19 programme), and by the SARS‐CoV‐2 Program of the Faculty of Medicine from Sorbonne University ICOViD programs, PI: G.G.).
One author received a Pasteur/APHP interface fellowship for this study. Publication status: Pre‐print paper Source: Pre‐print server (medXriv) Author COI: One author received consulting fees from Genalyte Inc. 3 years ago. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Sterlin 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Suhandynata 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase COVID‐19 Design: Multiple‐group study to estimate sensitivity and specificity: [1] laboratory confirmed COVID‐19 patients (n = 54); [2] patients PCR‐positive on a respiratory panel nucleic acid (RPNA) test for other infections (n = 21), [3] patients with positive for antinuclear antibodies (ANA) or anti‐double stranded DNA (dsDNA) (n = 24) [4] HIV positive patients (n = 10), [5] apparently healthy subjects (no respiratory symptoms per self‐report) (n = 78), [6] pre‐pandemic samples (n = 102) Recruitment: Unclear Prospective or retrospective: Not stated; presume retrospective Sample size: 289 (54) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Mixed; primarily inpatient Location: University of California San Diego Health (UCSD) Country: USA Dates: Not stated Symptoms and severity: Discussion reported 50 (93%) inpatient, and 30/54 (56%) not intubated 'at the time of writing' Demographics: Cases only (calculated from Tab 3): median age 54.5 y (IQR 41, 70.5 y; range 20 to 91 y); 35 (65%) male Exposure history: not stated Non‐Covid group 1: [2] to [4] other conditions or respiratory pathogens (n = 55) Source: Not stated; presume same medical centre Characteristics: not stated Non‐Covid group 2: [5] contemporaneous healthy, [6] pre‐pandemic healthy Source: [5] Not stated, [6] 2018 Characteristics: No further details |
||
| Index tests | Test name: [A] DZ‐LITE 2019‐nCoV IgG (CLIA) Assay Kit (Cat # 130219015M) and [B] DZ‐LITE 2019‐ nCoV‐2 IgM (CLIA) Assay Kit (Cat # 130219016M) Manufacturer: Diazyme Antibody: IgM, IgG, IgM or IgG Antigen target: SARS‐CoV‐2 recombinant nucleocapsid (N) and spike (S) proteins Evaluation setting: Laboratory Test method: CLIA Timing of samples: Unclear, data by time were in relation to first positive PCR result Samples used: Serum or plasma, collected in BD Vacutainer collection tubes (K‐EDTA, lithium‐Heparin plasma separator tubes, and/or serum separator tubes) Test operator: not stated Definition of test positivity: ≥ 1.00 AU/mL considered reactive Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: Not stated, EUA NAT that had been clinically validated in the laboratory Samples used: not stated Timing of reference standard: First positive PCR was median of 5 days pso (IQR 2.25, 7.75; range 0 to 22 days); data calculated from Tabl 3 Blinded to index test: Not stated; probably Yes Incorporated index test: No Definition of non‐COVID cases: Not stated for group [2] to [5]; group [6] was pre‐pandemic Samples used: Serum or plasma Timing of reference standard: not stated Blinded to index test: Unclear for [2] to [5]; Yes for [6] Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Serum sampling for cases reported by days post‐PCR (Suppl Tabl 3): Day 0 to 7 ‐ 36 (67%), day 8 to 14 ‐ 22 (41%), day >= 15 ‐ 18 (33%) (reported in paper, 19 reported in Table) All patients received same reference standard: No Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: patients |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published paper Source: Journal of Applied Laboratory Medicine Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Suhandynata 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (n = 60) [2] Non‐COVID subjects (n = 179) [2a] Current, other diseases (n = 22) [2b] Current, positive for other antibodies, DNA or IgM/IgG (n = 27) [2c] Current, apparently healthy subjects (n = 20) [2d] Pre‐pandemic samples (n = 110) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 239 (60) patients with 339 (160) samples of which 204 (25) were eligible for our review Further detail: Inclusion: [1] Patients which tested PCR‐positive for SARS‐CoV‐2 [2a] Patients which tested PCR‐positive on a respiratory panel nucleic acid (RPNA) test infections other than SARS‐CoV‐2 [2b] Patients which tested positive for antinuclear antibodies (ANA) or anti‐double stranded DNA (dsDNA) or patients with clinically elevated levels of IgM/IgG [2c] Apparently healthy subjects (no respiratory symptoms per self‐report) [2d] Patient samples that had been stored frozen (‐20 degrees C) since 2018 Exclusions not reported |
||
| Patient characteristics and setting | Setting: Not stated Location: UC San Diego Health clinical laboratories, California Country: California, USA Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2a] and [2b] Cross‐reactivity panel Source: UC San Diego Health clinical laboratories, current (time not stated) Characteristics: Human metapneumovirus n = 4 Influenza A H1‐2009 PCR n = 1 Mycoplasma pneumoniae n = 1 Non‐COVID coronavirus n = 7 Parainfluenza 4 PCR n = 1 Respiratory syncytial virus A n = 2 Respiratory syncytial virus B n = 2 Rhinovirus/enterovirus n = 4 Anti‐dsDNA (> 100 IU/mL) n = 4 Antinuclear antibodies n = 20 Elevated IgG/IgM n = 3 Non‐Covid group 2: [2c] Current, healthy (untested) [2d] Pre‐pandemic Source: [2c] Source not stated, current [2d] UC San Diego Health clinical laboratories, patient samples that had been stored frozen (‐20 degrees C) since 2018 Characteristics: [2c] Apparently healthy subjects (no respiratory symptoms per self‐report) [2d] Not stated |
||
| Index tests | Test name: [A] Diazyme DZ‐LITE 2019‐nCoV IgG, IgM (CLIA) Assay Kits (Cat # 130219015M; Cat # 130219016M) [B] Roche Elecsys Anti‐SARS‐CoV‐2 total Ig (Ref # 09203079190) [C] Abbott SARS‐CoV‐2 IgG (Ref # 06R8620) reagent kit Manufacturer: [A] Diazyme [B] Roche [C] Abbott Antibody: [A] IgG, IgM [B] Total antibodies [C] IgG Antigen target: Not stated Evaluation setting: All laboratory tests Test method: [A] CLIA [B] CLIA [C] CMIA Timing of samples: ≤ 7 days post‐PCR+ (n = 43) 8–14 days post‐PCR+ (n = 31) ≥ 15 days post‐PCR+ (n = 25) Samples used: Plasma (Li‐Heparin or K‐EDTA) and serum samples Test operator: Department of Pathology UC San Diego Health Definition of test positivity: [A] Absorbance units per mL (AU/mL), values ≥ 1.00 AU/mL were considered reactive. [B] A cut‐off index (COI; signal of sample/cut‐off); values ≥ 1.00 COI were considered reactive. [C] Index value (S/C); Index values ≥ 1.4 S/C were considered positive. Blinding reported: Not stated Threshold predefined: yes (analysed in a manner consistent with the package inserts) |
||
| Target condition and reference standard(s) | Reference standard: Positive for COVID‐19 by a nucleic acid amplification test that had been clinically validated in our laboratory and had an emergency use authorisation (EUA) listing with the US Food and Drug Administration
Threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no Definition of non‐COVID cases: [2a], [2b] To identify patient specimens containing other PCR‐confirmed microbes, the respiratory pathogen nucleic acid (RPNA) test was performed on the GenMark ePlex. This panel detects Adenovirus (A‐F), coronavirus (229E, HKU1, NL63, OC42), human metapneumovirus, human rhinovirus/enterovirus, influenza A, B and C, influenza 2009 H1N1, parainfluenza (1‐4), respiratory syncytial virus (A and B), chlamydia pneumoniae and mycoplasma pneumoniae. [2c] Untested (no respiratory symptoms per self‐report) [2d] Pre‐pandemic Samples used: [2a] and [2b] Not stated or untested [2c] Untested [2d] Pre‐pandemic Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: ≤ 7 days post‐PCR+ (n = 43), 8–14 days post‐PCR+ (n = 31), ≥ 15 days post‐PCR+ (n = 25) All patients received same reference standard: no Missing data: 74 COVID samples < 15 days post‐positive PCR not included in review; only 1 sample used per patient per time split (160‐99 = 61 samples excluded from analyses) Uninterpretable results: Not stated Indeterminate results: No borderline range Unit of analysis: [1] Samples but only one sample from each PCR‐positive patient used per specified time frame [2] Patients |
||
| Comparative | |||
| Notes | Funding: Research Funding: R.T. Suhandynata, Waters Corporation; M.A. Hoffman, Roche Diagnostics
The funding organisations played no role in the design of study, choice of enrolled patients, review and interpretation of data, preparation of manuscript, or final approval of manuscript Publication status: Published paper Source: Journal of Applied Laboratory Medicine Author COI: Employment or leadership: None declared Consultant or advisory role: None declared Stock ownership: None declared. Honoraria: None declared Research Funding: R.T. Suhandynata, Waters Corporation; M.A. Hoffman, Roche Diagnostics Expert testimony: None declared Patents: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Suhandynata 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Suhandynata 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Sun 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection and current convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity [1] Confirmed COVID patients (209 samples from 35 patients) [2] Healthy close contacts (n = 21) Group [2] excluded from review as < 25 samples Recruitment: [1] From 23 January to 27 February 2020, 38 hospitalised COVID‐19 cases were consecutively recruited. Prospective or retrospective: Prospective Sample size: 56 (35) patients with 230 (209) samples of which 209 (209) samples were eligible for our review Sensitivity results reported for 70 (70) samples Further detail: Inclusion: Hospitalised COVID‐19 cases in two designated hospitals for COVID‐19 between 23 January and 27 February 2020 Exclusion: [1] One mild and two severe cases were transferred to other hospitals after hospitalisation in these two hospitals and were excluded from this study. |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Two designated hospitals for COVID‐19, the Guangdong Seconded Provincial General Hospital and the First Hospital of Foshan in Guangdong, China Country: China Dates: 23 January to 27 February 2020 Symptoms and severity: 28 mild and 7 severe cases Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: [A] Cat. no. IEQ‐CoVS1RBD‐IgG [B] Cat. No. IE‐CoVS1RBD‐IgA [C] Cat. No. IE‐CoVS1RBD‐IgM Manufacturer: [A] ‐ [C] RayBiotech, GA, USA Antibody: [A] IgG [B] IgA [C] IgM Antigen target: [A] ‐ [C] RBD (from cat. No.) Evaluation setting: [A] ‐ [C] Laboratory tests performed in lab Test method: [A] ELISA [B] ELISA [C] ELISA Timing of samples: Serum samples were collected prospectively from cases every 3 days from hospitalisation until the date of discharge from hospital. Samples used: Serum Test operator: L.C., Z.L., H.L., R.Y., Z.P., H.X., X.Q., P.J., C.F., K.B., S.J., L.Z. and L.J. carried out the investigations. All from Guangdong Provincial Institute of Public Health, Guangdong Provincial Centre for Disease Control and Prevention, Guangzhou, China Definition of test positivity: According to the manufacturer's instructions, threshold not stated Blinding reported: Not stated Threshold predefined: yes, according to the manufacturer's instructions |
||
| Target condition and reference standard(s) | Reference standard: The laboratory‐confirmed case was defined as a case with respiratory specimens that tested positive for the SARS‐CoV‐2 by at least one of the following three methods: isolation of virus, positive results of real time reverse transcription polymerase chain reaction (rRT‐PCR) assay or a genome sequence that matched SARS‐CoV‐2.
A commercial rRT‐PCR kit targeting the ORF1ab and N genes was used to detect SARS‐CoV‐2 RNA (DaAn Gene, Guangzhou, China. Cat.No.DA0931).
Amplification was performed on an Applied Biosystems™ 7500 machine (ThermoFisher Scientific, USA). Specimens were considered positive for SARS‐CoV‐2 RNA if both ORF1ab and N gene target amplification curves were generated within 40 cycles. Samples used: Respiratory specimens Timing of reference standard: Not stated Blinded to index test: yes, prior to index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: yes Missing data: yes, group [2] excluded from review. No sensitivity data reported for test [C]; sensitivity results not available for all time points Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by grants from the Guangdong Provincial Novel Coronavirus Scientific and Technological Project (2020111107001) and Guangzhou Novel Coronavirus Scientific and Technological Project (202008040004). Publication status: Published paper Source: Clinical Microbiology and Infection Author COI: All authors reported no conflicts of interest relevant to this article. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Sweeney 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study to estimate sensitivity and specificity for diagnosis of acute and previous Covid‐19 Design: [1] PCR‐confirmed SARS‐CoV‐2 positive individuals (n = 301) [2] Pre‐pandemic stored serum samples (n = 200) [3] Pre‐pandemic stored acute and convalescent confounder samples from individuals with a range of viral, bacterial and fungal pathogens (n = 100) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 601 (301) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Unclear Location: Guy's and St Thomas' NHS Foundation Trust, London Country: UK Dates: Unclear Symptoms and severity: Unclear Demographics: Unclear Exposure history: Unclear Non‐Covid group 1: Pre‐pandemic stored samples Source: 43525 Characteristics: Unclear Non‐Covid group 2: Pre‐pandemic confounder samples Source: Not stated Characteristics: Cytomegalovirus (n = 8), Epstein‐Barr virus (EBV) (n = 10), hepatitis A virus (n = 8), hepatitis B virus (n = 7), hepatitis C virus (n = 5), human immunodeficiency virus (HIV) (n = 9), Kaposi's sarcoma herpesvirus 1/2 (n = 5), measles virus (n = 6), mumps (n = 9), mycobacterium (n = 1), parvovirus (n = 7), pneumocystis pneumonia (n = 4), rubella virus (n = 5), syphilis virus (n = 4), toxoplasma gondii (n = 7), varicella zoster virus (n = 5) |
||
| Index tests | Test name: SureScreen LFIA Manufacturer: Surescreen Diagnostics, UK Antibody: IgM/IgG Antigen target: "detecting antibodies to SARS‐CoV‐2 spike proteins" Evaluation setting: POC, used in the laboratory Test method: Lateral flow immunoassay Timing of samples: [1] 14+ days post‐onset of symptoms: 301/301 (100%), of which: 14‐19 days post‐onset of symptoms: 97/301 (32%) 20+ days post‐onset of symptoms: 204/301 (68%) Samples used: Serum Test operator: Laboratory staff Definition of test positivity: 2 independent operators evaluating the result. A detectable band of either IgM or IgG (or both) was reported to the clinician as “antibodies detected". Blinding reported: Unclear Threshold predefined: yes, visual‐based test |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (AusDiagnostics); threshold not stated (reference PHE 2020 rapid assessment) Samples used: Unclear Timing of reference standard: Not stated Blinded to index test: Yes, occurred before Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: None Timing of reference standard: NA Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: Nothing mentioned Uninterpretable results: Nothing mentioned Indeterminate results: Nothing mentioned Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: King’s Together Rapid COVID‐19 Call awards to KJD, SJDN and RMN. MRC Discovery Award MC/PC/15068 to SJDN, KJD and MHM. National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London, programme of Infection and Immunity to MHM and JE. AWS and CG were supported by the MRC‐KCL Doctoral Training Partnership in Biomedical Sciences. GB was supported by the Wellcome Trust. SA was supported by an MRC‐KCL Doctoral Training Partnership in Biomedical Sciences industrial Collaborative Award in Science & Engineering (iCASE) in partnership with Orchard Therapeutics. NK was supported by the Medical Research Council. SP, HDW and SJDN were supported by a Wellcome Trust Senior Fellowship. Fondation Dormeur, Vaduz for funding equipment (KJD). Development of SARS‐CoV‐2 reagents (RBD) was partially supported by the NIAID Centers of Excellence for Influenza Research and Surveillance (CEIRS) Publication status: Pre‐print (not peer reviewed) Source: medRxiv Author COI: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf and declared: no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Tan 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute or convalescent‐phase infection Design: [1] Covid patients (n = 170) [2] Non‐Covid patients (n = 163) [2a] Pre‐pandemic healthy controls (n = 60) [2b] Pre‐pandemic, cross‐reactivity group (n = 103) Recruitment: [1] Prospectively‐selected samples between 30 March‐15 May 2020 from COVID patients with at least one positive RT‐PCR respiratory sample. [2] Not stated Prospective or retrospective: [1] Prospective [2] Retrospective Sample size: 333 (170) Further detail: Inclusion: [1] Inpatients with >= 1 RT‐PCR‐positive result [2] Archived negative controls were utilised with samples taken from patients prior to December 2019. These included patients with and without other positive serological tests. Exclusion: [1] Asymptomatic cases [2] Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: National University Hospital, Singapore Country: Singapore Dates: Samples from patients collected between 30 March 2020 and 15 May 2020 Symptoms and severity: Symptomatic. Severity unclear Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic controls Source: Hospital patients (National University Hospital) pre‐December 2019 Characteristics: [2a] Healthy (n = 60), [2b] Sero‐positive viruses or auto‐immune disorders (n = 103):
|
||
| Index tests | Test name: [A] Roche Elecsys Anti‐SARS‐CoV‐2 assay [B] Abbott Architect Anti‐SARS‐CoV‐2 assay Manufacturer: [A] Roche Diagnostics, Rotkruez, Switzerland [B] Abbott Diagnostics, Chicago, USA Antibody: [A] Total Antibodies (IgG and IgM) [B] IgG Antigen target: [A] Nucleocapsid protein [B] Nucleocapsid protein Evaluation setting: Laboratory Test method: [A] and [B] CLIA Timing of samples: < 7 days pso (n = 80) 7‐13 days pso (n = 37) 14‐20 days pso (n = 21) >= 21 days pso (n = 32) Samples used: Serum Test operator: Not stated Definition of test positivity: [A] signal cut‐off index (COI) of >= 1.0 was positive for Roche [A], < 1.0 was negative. [B} signal cut‐off index (S/C) ratio of >= 1.4 was positive for Abbott [B], < 1.4 was negative. Blinding reported: Not stated Threshold predefined: Yes, according to manufacturers' instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, at least one positive on the Cobas 6800 SARS‐CoV‐2 assay (Roche Diagnostics, Rotkruez, Switzerland), with the cycle threshold value being lower than cut‐off (not stated) Samples used: Respiratory samples Timing of reference standard: Not stated Blinded to index test: Yes, previous Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic. Samples used: None for reference standard, pre‐pandemic Timing of reference standard: Pre‐pandemic controls Blinded to index test: Yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: No indeterminate threshold Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: We would like to thank Temasek Holdings Pte Ltd for sponsoring the laboratory testing kits used in this study.
Dr Tambyah has received grants paid to the National University Hospital from Roche, Johnson & Johnson, Sanofi Pasteur, GlaxoSmithKline, and Shionogi. Publication status: Published paper Source: Archives of Pathology & Laboratory Medicine Author COI: Dr Tambyah has received grants paid to the National University Hospital from Roche, Johnson & Johnson, Sanofi Pasteur, GlaxoSmithKline, and Shionogi. Other authors have no relevant financial interest in the products or companies described in this article. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Tan 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tang 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection using three commercial SARS‐CoV‐2 IgG assays Design: Multiple‐group study estimating sensitivity and specificity: [1] residual serum samples from patients with laboratory‐confirmed COVID‐19 infection and physician ordered completed blood count (n = 48, providing 103 samples) [2] PCR‐negative COVID‐19 suspects (n = 80); [3] pre‐pandemic serum (n = 50) [4] PCR‐negative, with other confirmed coronavirus (HKU1, NL63, and 229E) (n = 5) or influenza A or B (n = 4) [5] serum from patients with potentially interfering antibodies (n = 14; CMV IgG (n = 5), EBV VCA IgG (n = 3) or IgM (n = 3) or both (n = 2), RF+ (n = 1)) Recruitment: Not stated Prospective or retrospective: Retrospective Sample size: 256 (103) Further detail: No further details |
||
| Patient characteristics and setting | Setting: Unclear; 'a majority of our patient population (were) hospitalised' Location: Barnes Jewish Hospital, St. Louis, MO Country: USA Dates: No information Symptoms and severity: No information; 'majority' hospitalised Demographics: No information Exposure history: No information Non‐Covid group 1: Presumed negative controls Source: Source unclear Characteristics: No information |
||
| Index tests | Test name: [A] Abbott SARS‐CoV‐2 IgG assay [B] EUROIMMUN SARS‐CoV‐2 IgG assay [C] Roche Elecsys Anti‐SARS‐CoV‐2 Manufacturer: [A] Abbott diagnostics [B] EUROIMMUN [C] Roche Antibody: [A] and [B] IgG [C] total Ab Antigen target: [A] undisclosed epitope of the SARS‐CoV‐2 nucleocapsid protein [B] S1 domain of viral spike‐protein [C] nucleocapsid protein from SARS‐CoV‐2 Evaluation setting: Laboratory‐based assays Test method: [A] CLIA [B] ELISA [C] CLIA Timing of samples: Day 0 to >= 14 days pso Timing of samples: Day 0 to >= 14 days pso Samples used: Discussion stated plasma; PCR+ samples collected in EDTA Vacutainer tubes; controls were either stored or recent specimens (source unclear). Test operator: Not stated Definition of test positivity: [1] ratio ≥ 1.4 [B] positive = ratio ≥ 1.1 ; borderline = ratio < 1.1 to ≥ 0.8; results extracted considering borderline results +ve or ‐ve [C] ratio of specimen electrochemiluminescent signal to calibrator; cut‐off index (ratio) ≥ 1.0. Blinding reported: Not stated Threshold predefined: as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR using one of three platforms due to reagent shortages:
[1] Quidel Lyra RT‐PCR assay (majority)
[2] Xpert Xpress SARS‐CoV‐2 molecular assay (Cepheid)
[3] Simplexa COVID‐19 Direct Assay using a LIAISON MDX (Diasorin) Samples used: nasopharyngeal (NP) swabs, oropharyngeal (OP) swabs, or lower respiratory tract specimens (only latter used with Diasorin Simplexa) Timing of reference standard: varying times from symptom onset Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: RT‐PCR for COVID‐19 suspects (n = 80) and for other infection samples (5 with other CoV); Unclear reference for other interfering antibody samples (n = 14); Pre‐pandemic for remaining 50 Samples used: Serum Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Reported as 0 to >= 14 days after positive PCR All patients received same reference standard: Yes; all RT‐PCR (different kits) Missing data: None reported Uninterpretable results: None reported Indeterminate results: For EUROIMMUN ELISA, borderline results were initially considered positive (main text) and reported as negative in Supplementary Information. Unit of analysis: Samples; patients per week (all 48 reported at >= 14 days pso) |
||
| Comparative | |||
| Notes | Funding: None declared Publication status: Accepted manuscript and subsequently research letter Source: Clinical Chemistry Author COI: Employment or leadership: A.M. Gronowski, Clinical Chemistry, AACC. Consultant or Advisory Role: N.W. Anderson, Diasorin Molecular |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Tang 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tang 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Theel 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To evaluate four high throughput serologic tests for detection of anti‐SARS‐CoV‐2 IgG antibodies
Multi‐group study to estimate sensitivity and specificity for diagnosis of active disease/identification of previous disease Design: [1] serum samples from patients with confirmed COVID‐19 (n = 56, 224 samples) [2] healthy donor sera from 2018 (n = 149 samples) [3] cross‐reactivity serum panel collected in early 2020 (n = 105 samples, see comments) In group [1], 11 samples from outpatients would be excluded from our review as taken 0‐7 days post‐positive PCR. Recruitment: [1] Serum samples were collected as available throughout the hospital stay for the inpatient group until discharge, whereas prospective collection of acute and convalescent sera was completed for outpatients. [2] Samples collected in 2018, prior to the SARS‐CoV‐2 outbreak [3] Samples submitted for testing as part of routine clinical care in January and early February 2020 Prospective or retrospective: Mixed (as above) Sample size: 478 (224 samples from 56 patients) of which 476 (213 samples from 56 patients were eligible for our review). Further detail: Not stated |
||
| Patient characteristics and setting | Setting: Inpatients and outpatients Location: Division of Clinical Microbiology, Department of Laboratory Medicine, Mayo Clinic, Rochester, MN Country: USA Dates: [1] COVID cases March and April 2020 Symptoms and severity: 33 were hospitalised (inpatient group) and 23 were treated as outpatients (outpatient group) Demographics: Median age of the 33 inpatients was 61 years (range: 24 to 90 years) and 61% (20/33) were male. Among the 23 outpatients, the median age was 37 years (range: 21 to 64 years) and 43% (10/23) were male. Exposure history: Not stated Non‐Covid group 1: Healthy donors Source: Pre‐pandemic, 2018 Characteristics: Not stated Non‐Covid group 2: Cross‐reactivity Source: January and early February 2020 Characteristics: Not stated |
||
| Index tests | Test name: [A] Euroimmun Anti‐SARS‐CoV‐2 IgG ELISA [B] Epitope Novel Coronavirus COVID‐19 IgG ELISA [C] Abbott Laboratories SARS‐CoV‐2 IgG Chemiluminescent Microparticle Immunoassay [D] VITROS Anti‐SARS‐CoV‐2 IgG Chemiluminescent Immunoassay Manufacturer: [A] Euroimmun, Lübeck, Germany [B] Epitope Diagnostics Inc., San Diego, CA [C] Abbott Laboratories, Abbott Park, IL [D] Ortho‐Clinical Diagnostics, Rochester, NY Antibody: [A] IgG [B] IgG [C] IgG [D] IgG Antigen target: [A] S1‐protein from the SARS‐CoV‐2 spike‐protein [B] nucleocapsid protein from SARS‐CoV‐2 [C] SARS‐CoV‐2 nucleocapsid antigen [D] SARS‐CoV‐2 spike antigen Evaluation setting: [A] Laboratory, used in laboratory [B] Laboratory, used in laboratory [C] Laboratory, used in laboratory [D] Laboratory, used in laboratory Test method: [A] ELISA [B] ELISA [C] Chemiluminescent Microparticle Immunoassay (CMIA) [D] Chemiluminescent Immunoassay (CLIA) Timing of samples: Inpatients: 0 to 26 days post‐symptom onset Outpatients: 11 patients had both baseline and convalescent serum samples collected at 3 to 7 days and 20 to 31 days post‐initial positive SARS‐CoV‐2 RT‐PCR result, respectively, and the remaining 12 outpatients only had a convalescent sample collected. 33 inpatients (190 samples) 0‐7 days pso: 38 8‐14 days pso: 91 15‐26 days pso: 61 23 outpatients (34 samples): 0‐7 days post‐PCR+: 11 (excluded from review) 20‐31 days post‐PCR+: 23 Samples used: [1] Serum [2] Serum [3] Serum [4] Serum Test operator: Laboratory personnel Definition of test positivity: [A] Index values (signal to cut‐off [S/Co] ratios) of < 0.8, ≥ 0.8 to < 1.1, and ≥ 1.1 were interpreted as negative, indeterminate, and positive, respectively, per the instructions for use. [B] The qualitative index value (S/Co) cut‐off thresholds used for negative, indeterminate and positive results were < 1.01, ≥ 1.01 to < 1.21, and ≥ 1.21, respectively. [C] The patient sample signal was divided by the calibrator signal, with calculated signal to cut‐off (S/Co) values of < 1.4 and ≥ 1.4 reported as negative and positive, respectively. [D] The patient sample signal was divided by the calibrator signal, with calculated signal to cut‐off (S/Co) values of < 1.00 and ≥ 1.00 reported as negative and positive, respectively. Blinding reported: Not stated Threshold predefined: [A] Yes, per the instructions for use [B] No, laboratory‐determined cut‐off threshold. Modified to optimise assay specificity [C] Yes, per the instructions for use [D] Yes, per the instructions for use |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 RT‐PCR assay (laboratory‐developed or commercially available FDA EUA) Samples used: nasopharyngeal swab Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: [2] Pre‐pandemic [3] Not stated Samples used: [2] Pre‐pandemic [3] Not stated Timing of reference standard: [2] Pre‐pandemic [3] Not stated Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Inpatients: Not stated Outpatients: 11 patients had both baseline and convalescent serum samples collected at 3 to 7 days and 20 to 31 days post‐initial positive SARS‐CoV‐2 RT‐PCR result, respectively, and the remaining 12 outpatients only had a convalescent sample collected. All patients received same reference standard: No Missing data: Not stated Uninterpretable results: None reported Indeterminate results: For statistical analysis, indeterminate results by the Euroimmun and Epitope anti‐SARS‐CoV‐2 IgG ELISAs were considered ‘negative’. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Accepted Manuscript Source: Journal of Clinical Microbiology, doi:10.1128/JCM.01243‐20 Author COI: Not stated |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Theel 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Theel 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Theel 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Thijsen 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: Assessment of cell‐mediated and humoral immune response in COVID‐19 cases according to disease severity Design: Two‐group study estimating both sensitivity and specificity Group [1]: PCR confirmed COVID‐19 cases (n = 27) Group [2]: Healthy controls (n = 16) Recruitment: Unclear Prospective or retrospective: Not stated Sample size: 43 (27) |
||
| Patient characteristics and setting | Setting: Inpatient services (ICU and pulmonary ward) Location: Diakonessenhuis Utrecht Country: Netherlands Dates: Not stated Symptoms and severity: Severity: 18/27 (67%) severe/critical (ICU); 9/27 (33%) moderate/severe (pulmonary ward) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Healthy controls Source: Not stated Characteristics: Not stated |
||
| Index tests | Test name: [A] Euroimmun Anti‐SARS‐CoV‐2 ELISA IgG [B] Euroimmun Anti‐SARS‐CoV‐2 ELISA IgA Manufacturer: [A], [B]: EUROIMMUN AG, Germany Antibody: [A] IgG [B] IgA Antigen target: [A], [B]: S1 domain of the spike‐protein Evaluation setting: [A], [B]: Lab test, done in lab Test method: [A], [B]: Enzyme‐linked immunosorbent assay (ELISA) Timing of samples: 6‐32 days post‐symptom onset Samples used: Not stated (likely serum or plasma) Test operator: Not stated Definition of test positivity: Not stated (likely as per manufacturer; plot showed threshold of OD ratio approximately 1.1 which is consistent with manufacturer recommended threshold) Blinding reported: Not stated, but probably no Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (no more details available) Samples used: Not stated Timing of reference standard: Unclear Blinded to index test: Yes (done earlier) Incorporated index test: No Definition of non‐COVID cases: Not stated, but likely no testing Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated. Only stated that the index test was done 6‐32 days post‐symptom onset All patients received same reference standard: No (Group [2] received no testing and patients from Group [1] were likely tested with various RT‐PCR assays. Missing data: Not stated, but the total sample size did not appear to match what was reported in the figures for tests [A] and [B] ‐ Unclear whether some patients were tested only with one test Data by time period (suppl Fig 3) did not sum to total shown for full time period (Fig 1C and suppl Fig 2), nor to total reportedly included. Uninterpretable results: No Indeterminate results: No Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Published letter Source: Academic journal Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Trabaud 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Single‐group study to estimate sensitivity [1] COVID patients (N = 68, 82 samples) [1a] infected hospitalised patients (N = 40) [1b] infected non‐hospitalised healthcare workers (N = 28) Recruitment: Recruited infected hospitalised patients and non‐hospitalised infected healthcare workers Prospective or retrospective: Retrospective Sample size: 82 (82) samples from 68 (68) patients of which 66 (66) samples were eligible for our review Further detail: Inclusion: hospitalised patients or non‐hospitalised healthcare workers with RT‐PCR confirmed COVID positive |
||
| Patient characteristics and setting | Setting: Inpatients and outpatients Location: Hopital de la Croix‐Rousse, Hospices Civils de Lyon, Lyon Country: France Dates: Not stated Symptoms and severity: 40 hospitalised with 25 in intensive care units 28 non‐hospitalised. All symptomatic (symptoms not stated) Demographics: [1] Age range 7‐81 years (median = 51) [1a] Age range 7‐81 years (median = 64), 11/40 female (27.5%) [1b] Age range 25‐59 years (median = 36), 22/28 female (78.6%) Exposure history: [1a] Not stated [1b] Healthcare workers (HCW) (including physicians, nurses, and lab staff) |
||
| Index tests | Test name: [A] Diasorin Liaison [B] bioMeriuex Vidas [C] Siemens Atellica [D] Wantai [E] Abbott Architect [F] Roche Elecsys [G] BioRad Platelia [H] Epitope Diagnostics EDI Manufacturer: [A] Diasorin S.p.A. [B] bioMerieux diagnostics [C] Siemens Healthcare GmbH [D] Beijing Wantai Biological Pharmacy [E] Abbott Diagnostics [F] Roche Diagnostics [G] Bio‐Rad Laboratories, Inc. [H] Epitope Diagnostics Inc. Antibody: [A] IgG [B] IgG [C] Total antibody [D] Total antibody [E] IgG [F] Total antibody [G] Total antibody [H] IgG Antigen target: [A] S1 and S2 [B] S1 and peptide [C] RBD [D] RBD [E] N‐protein [F] N‐protein [G] N‐protein [H] N‐protein Evaluation setting: Laboratory Test method: [A] indirect CLIA [B] Enzyme Linked Fluorescent Assay (ELFA) [C] CLIA [D] ELISA [E] CMIA [F] ECLIA [G] ELISA [H] ELISA Timing of samples: Range 4 to 52 days post‐symptom onset: <= 15 days pso (n = 16) 16‐20 days pso (n = 21) > 20 days pso (n = 45) Samples used: All serum/plasma Test operator: Technicians from the laboratory Definition of test positivity: [A] AU/mL; 12, > 12‐ < 15 borderline [B] ratio; 1 [C] ratio; 1 [D] ratio; > 1.1; >= 0.9‐ <= 1.1 borderline [E] ratio; 1.4 [F] ratio; 1 [G] ratio; 1; >= 0.8‐ < 1 borderline [H] >= 1.1x (NC + 0.18); ≥ 0.9x (NC + 0.18) < 1.1x (NC + 0.18) borderline Blinding reported: not stated (but only COVID cases included in study) Threshold predefined: yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: Yes as 16 samples 4‐15 days pso excluded from review as interval too wide Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: 82 samples from 68 patients |
||
| Comparative | |||
| Notes | Funding: This work did not receive any specific grant from funding agencies, in the public, commercial or not‐for‐profit sectors. The assay kits were provided by the manufacturers. Publication status: Published paper Source: Journal of Clinical Virology Author COI: Authors declared no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Trabaud 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Trabaud 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Trabaud 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Trabaud 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Trabaud 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Trabaud 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Trabaud 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Traugott 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase COVID‐19 Design: Multi‐group study estimating sensitivity and specificity [1] Symptomatic patients with acute PCR‐confirmed COVID‐19 infection (n = 77) [2] Symptomatic patients with negative PCR results (n = 30) [3] Healthy volunteers with negative PCR (n = 30) [4] Stored samples from individuals with previous PCR‐confirmed coronavirus OC43 infection (n = 10); interval from infection to sampling of 4 to 1452 days [5] Pre‐pandemic samples from patients with pneumonia (n = 30) Recruitment: Not stated Prospective or retrospective: Unclear; all recruitment appeared to be retrospective Sample size: 177 (77) |
||
| Patient characteristics and setting | Setting: Mixed; majority were inpatients at time of sample collection Location: 4th Medical Department, Department of Infectious Diseases and Tropical Medicine, Kaiser‐Franz‐Josef Hospital, Vienna Country: Austria Dates: 27th February to 30th March 2020 Symptoms and severity: 59 (75%) hospitalised due to moderate/severe illness, 17 (22%) 'dismissed to home care', 1 sample from HCW Demographics: Median age 63, range 15‐92; 248/77 (62%) male Exposure history: Not stated Non‐Covid group 1: [2] Symptomatic COVID‐19 suspects Source: [2] 27th February to 30th March 2020 Characteristics: [2] No further details per group; overall 40% male, median age 49 y (2–93 y) [3] Healthy volunteers [4] Other coronavirus [5] Pre‐pandemic pneumonia Source: [3] Contemporaneous [4] Unclear; 'stored' [5] Before December 2019 Characteristics: [3] No further details per group [4] previous PCR‐confirmed coronavirus OC43 infections [5] No further details per group |
||
| Index tests | Test name: [A] Euroimmun SARS‐CoV‐2 IgA ELISA [B] Euroimmun SARS‐CoV‐2 IgG ELISA [C] Wantai SARS‐CoV‐2 IgM ELISA [D] Wantai SARS‐CoV‐2 total antibody ELISA [E] Wantai SARS‐CoV‐2 Ab Rapid Test [F] Hangzhou Alltest 2019‐nCoV IgG/IgM Rapid Test Manufacturer: [A], [B] Euroimmun, Germany [C] to [E] Beijing Wantai Biological Pharmacy, China [F] Hangzhou AllTest Biotech, China Antibody: [A] IgA [B] IgG [C] IgM [D] total antibody [E] Total antibody [F] IgG/IgM Antigen target: [A], [B] S1 domain of the spike‐protein [C], [D] Spike‐protein receptor binding domain [E], [F] Not stated Evaluation setting: [A] to [D] Laboratory [E], [F] Laboratory, but intended as a POC Test method: [A] to [D] ELISA [E], [F] Lateral flow assay Timing of samples: PCR+ cases only: 1‐5 days post‐symptom onset: 30 (39%) 6‐10 days post‐symptom onset: 25 (32%) 11‐29 days post‐symptom onset: 22 (29%) Timing of samples: PCR+ cases only: 1‐5 days post‐symptom onset: 30 (39%) 6‐10 days post‐symptom onset: 25 (32%) 11‐29 days post‐symptom onset: 22 (29%) Samples used: Serum or plasma Test operator: Laboratory staff Definition of test positivity: [A] to [D] Positive when antibody ratio was > 1.1 [E],[F] All tests with (still) visible bands [to the naked eye] were considered positive. Blinding reported: Unclear Threshold predefined: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR with WHO‐recommended primers and probe located in the E‐gene Samples used: Nasopharyngeal swab/respiratory secretion samples Timing of reference standard: Not stated Blinded to index test: Unclear; but likely conducted first Incorporated index test: No Definition of non‐COVID cases: [2] RT‐PCR [3] RT‐PCR [4] Unclear ('stored') [5] Pre‐pandemic Samples used: [2], [3] Nasopharyngeal swab/respiratory secretion samples Timing of reference standard: [2], [3] Unclear, but contemporaneous [4] Unclear [5] Pre‐pandemic Blinded to index test: [2], [3] Unclear ‐ likely conducted first [4] Yes, it seems these stored samples were from before the observational period [5] Yes, pre‐pandemic Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: None reported Uninterpretable results: None reported Indeterminate results: No All indeterminate results (0.8 to 1.1) on ELISA were counted as index‐negative; weakly positive rapid test results counted as positive Unit of analysis: Patients; only included one sample per patient |
||
| Comparative | |||
| Notes | Funding: Medical Scientific Fund of the Mayor of the City of Vienna Publication status: Published paper Source: Journal of Infectious Diseases Author COI: Authors reported no conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Traugott 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Traugott 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Traugott 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Traugott 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Traugott 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tre‐Hardy 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Retrospective two‐group analysis to estimate sensitivity and specificity (n = 125) [1] Covid patients (n = 44) [2] Non‐Covid pre‐pandemic patients (n = 81) [2a] Cross‐reactivity panel (n = 75) [2b] Healthy subjects (n = 6) Recruitment: [1][2] All sera originated from blood samples taken during previous clinical requests for diagnostic purposes. [1] Blood samples positive for COVID‐19 were collected from patients with mild, severe or critical infection. Prospective or retrospective: Retrospective Sample size: 125 (44) Further detail: [1] Blood samples positive for COVID‐19 were collected from patients with mild, severe or critical infection. Patients were considered positive according to the results of the RT‐qPCR. [2a] Patients with other viral, bacterial, parasitic or auto‐immune pathologies that could be considered as confounding factors or to another strain of coronavirus, collected in 2019 [2b] No history of known auto‐immune pathologies and without any acute infection of viral or bacterial origin, collected in 2019 |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Iris Sud Hospitals (laboratory serum biobank), Brussels, Belgium Country: Belgium Dates: April 16 to 20, 2020 Symptoms and severity: Mild, severe or critical infection based on the extent of anomalies observed on CT scans: moderate (10%–25%), extensive (25%–50%), severe (> 50%) or critical > 75% and on clinical symptoms (headache, fever, fatigue, cough and sore throat, myalgia, shortness of breath or digestive signs) Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: [2] Non‐Covid patients Source: 2019 prior to the pandemic Characteristics: [2a] Sera positive for the following viral, bacterial and infection from parasite origin were included to assess the possible cross‐reactivity: HBsAg (n = 7), HAV IgM (n = 3), adenovirus (n = 1), HSV IgM and CMV IgM (n = 1), IgM CMV (n = 8), IgM parvovirus B19 (n = 5), HIV (n = 1), ASLO (antistreptolysin O) (n = 4), anti‐treponema pallidum antibody (n = 1), IgG borrelia (n = 1), IgM mycoplasma pneumoniae (n = 10), toxoplasma gondii IgM (n = 16) The cross‐reactivity of the following auto‐immune pathologies was also assessed: rheumatoid factor (n = 1), anti‐TPO antibody (n = 7), irregular antibodies (n = 4), direct coombs (n = 1). Two sera from COVID‐19‐negative patients but positive to another strain of coronavirus Finally, one serum with a high level of total IgM (9.01 g/L) (normal range: 0.40–2.30 g/L), one serum with high total IgA (4.47 g/L) (normal range: 0.70–4.00 g/L) [2b] six sera from COVID‐19‐negative healthy subjects with no history of known auto‐immune pathologies and without any acute infection of viral or bacterial origin |
||
| Index tests | Test name: [A] LIAISON SARS‐CoV‐2 IgG [B] anti‐SARS‐CoV‐2 ELISA IgG Manufacturer: [A] Diasorin, Saluggia, Italy [B] Euroimmun, Medizinische Labordiagnostika, Lubeck, Germany Antibody: [A] IgG [B] IgG Antigen target: [A] S1 and S2 subunits [B] S1 subunit Evaluation setting: [A] and [B] Laboratory Test method: [A] CLIA [B] ELISA Timing of samples: >= 14 days post‐PCR + Samples used: Serum stored in the laboratory serum biobank at ≤− 20 °C Test operator: Clinical laboratory staff Definition of test positivity: Manufacturer’s cut‐off: [A] >= 15.0 AU/mL is positive, < 12.0 AU/mL is negative, in between is doubtful. [B] Ratio >= 1.1 is positive, < 0.8 is negative, in between is doubtful. ROC curve analyses cut‐off: [A] > 6.1 AU/mL [B] > 0.708 Blinding reported: Not stated Threshold predefined: Yes, using the cut‐off provided by the manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐qPCR, threshold not stated. Samples used: Respiratory samples. Timing of reference standard: Delay between first symptom onset and RT‐qPCR test was estimated at 4 days (± 1 days). Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA as pre‐pandemic Timing of reference standard: NA as pre‐pandemic. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: >= 14 days All patients received same reference standard: No [1] PCR [2] Pre‐pandemic Missing data: not stated Uninterpretable results: not stated Indeterminate results: Thresholds for 'doubtful' results but no results recorded in this category [A] For the doubtful sample with the LIAISON®SARS‐CoV‐2 IgG kit, the sample must be retested in duplicate. If at least two of three results were doubtful, the sample was considered positive. If two of the results/three are < 12.0 AU/mL, the sample was negative. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None declared Publication status: Published paper Source: De Gruyter Clinical Chemistry & Laboratory Medicine Author COI: Authors stated no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Tre‐Hardy 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity, including: [1] Hospitalised patients with PCR‐proven or suspected COVID‐19 Infection (PCR‐negative were excluded), n = 38 samples [2] Pre‐pandemic controls (samples collected in 2017‐2018 from patients care in the Department of Infectious Diseases), n = 20 Recruitment: Consecutive cases Prospective or retrospective: Prospective Sample size: 58 (38) Further detail: Inclusion criteria: Patients care at the Montpellier University Hospital suspected of a COVID‐19 infection |
||
| Patient characteristics and setting | Setting: Inpatient Location: University Hospital, Montpellier Country: France Dates: From 18 March 2020 (ongoing) Symptoms and severity: Severe cases 26/38 (68%); 4/9 day 1 to 6; 9/14 day 7 to 14; 13/15 day >= 15 Demographics: Age reported subgroup (mean, SD): Day 1 to 6 72 y (55‐90y); Day 7 to 14 65 y (39‐86y); Day >= 15 66 y (51‐83y). Sex: 22/38 cases were male (58%). Exposure history: Not stated Non‐Covid group 1: Control Source: 2017‐2018 (pre‐pandemic) Characteristics: Age (mean, SD): 41 (17‐72); sex: 10/20 (50%) Non‐Covid group 2: NA |
||
| Index tests | Test name: [A] Zhuhai Livzon Pharmaceutical Group ‐ 2019‐nCoV IgM/IgG [B] UNscience Biotechnology ‐ COVID‐19 IgG/IgM [C] Chongqing iSIA BIO‐Technology ‐ 2019‐nCoV IgM/IgG kit [D] Guangdong Hecin Biotech ‐ 2019‐nCoV IgM kit [E] AccuBiotech ‐ Accu‐Tell COVID‐19 IgG/IgM [F] Acro Biotech ‐ 2019‐nCoV IgM/IgG [G] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA [H] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgG [I] EUROIMMUN ‐ anti‐SARS‐COV‐2 IgA or IgG [J] ID.Vet ‐ ID Screen SARS‐CoV‐2‐N IgG Indirect ELISA Manufacturer: [A] Zhuhai Livzon Pharmaceutical Group [B] UNscience Biotechnology [C] Chongqing iSIA BIO‐Technology [D] Guangdong Hecin Biotech [E] AccuBiotech [F] Acro Biotech [G] EUROIMMUN [H] EUROIMMUN [I] EUROIMMUN [J] ID.Vet Antibody: [A] [B] [C] IgG and IgM, [C] IgM, [D] [E] [F] IgG and IgM,[G] IgA, [H] IgG, [I] IgA and IgG, [J] IgG Antigen target: [A] [B] [C] [D] [E] [F] unclear, [G] [H] [I] S1,[J] N Evaluation setting: [A] to [F] POC tests [G] to [J] Laboratory Test method: [A] CGIA, [B] CGIA, [C] LFA, [D] CGIA, [E] CGIA, [F] LFA, [G] ELISA, [H] ELISA, [I] ELISA, [J] ELISA Timing of samples: [1] 1‐6 days (n = 9), 7‐14 days (n = 14), ≥ 15 days (n = 15) from the onset of symptoms Samples used: Plasma (as per Material and Methods, 1st paragraph) Test operator: Not stated Definition of test positivity: [A] to [F] any band, even weakly visible: positive [G] ≥ 1.1 positive [H] cut‐off value for a positive ≥ 70% Blinding reported: Not stated Threshold predefined: Yes, as per manufacturer’s instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; no details Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: Not stated Timing of reference standard: Pre‐pandemic controls (2017‐2018) Blinded to index test: Not applicable (NA) Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: PCR‐negatives excluded Uninterpretable results: None reported Indeterminate results: EUROIMMUN borderline results considered negative Unit of analysis: samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by Grants from Montpellier University Hospital and Montpellier University (MUSE). Publication status: pre‐print Source: medRxiv Author COI: The authors have declared no competing interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Tuaillon 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Tuaillon 2020 [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Valdivia 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Two‐group study to assess sensitivity and specificity: [1] Laboratory‐confirmed SARS‐CoV‐2 infection (n = 90) [2] Pre‐pandemic controls (n = 20) Recruitment: Non‐consecutive Prospective or retrospective: Retrospective Sample size: 110 (90) Further detail: Inclusion: [1] Laboratory‐confirmed SARS‐CoV‐2 infection by RT‐PCR with leftover sera obtained for routine SARS‐CoV‐2 serological testing [2] Sera collected from healthy individuals in 2019, 10 of which had prior endemic coronavirus infection Exclusion: [1] [2] Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: Hospital Clínico Universitario of Valencia Country: Spain Dates: March 5 and April 30, 2020 Symptoms and severity: All 51 patients presented with pneumonia and imaging or laboratory findings compatible with COVID‐19 and were hospitalised in either the pneumology ward (n = 27) or the intensive care unit (ICU; n = 24) Demographics: Male/female ‐ 32/19, mean age ‐ 53, median hospitalisation days ‐ 17, number with other comorbidities ‐ 35 Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic controls Source: 20 pre‐pandemic sera from healthy individuals collected within 2019, of which 10 belonged to patients with prior endemic coronavirus infections Characteristics: (n = 10) healthy no disease, HCoV‐229E (n = 8); HCoV NL63 (n = 1); HCoVHKU (n = 1) |
||
| Index tests | Test name: [A] LIAISON SARS‐CoV‐2 S1/S2 [B] Euroimmun SARS‐CoV‐2 IgG ELISA [C] MAGLUMI 2019‐nCoV IgG [D] COVID‐19 ELISA IgG Manufacturer: [A] DiaSorin S.p.A., Saluggia, Italy [B] Euroimmun, Lübeck, Germany [C] Shenzhen New Industries Biomedical Engineering Co., Ltd., Shenzhen, China [D] Vircell Spain, S.L.U., Granada, Spain Antibody: [A][B][C][D] IgG Antigen target: [A] S protein [B] S1 domain [C] N protein [D] S1 and N protein Evaluation setting: Laboratory Test method: [A] Chemiluminescent immunoassay [B] ELISA [C] CLIA [D] ELISA Timing of samples: Samples were stored for a maximum 1 month from point of collection. Samples used: Serum Test operator: Not stated Definition of test positivity: [A] > 15 AU/mL positive, 12.0‐15.0 AU/mL indeterminate [B] >= 1.1 positive COI, 0.8‐1.09 indeterminate [C] >= 1.10 AU/mL positive [D] > 1.6 AI positive, 1.4‐1.6 AI indeterminate Blinding reported: Not stated Threshold predefined: As per manufacturers specifications |
||
| Target condition and reference standard(s) | Reference standard: SARS‐COV2‐RT PCR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA Timing of reference standard: NA Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Indeterminate results to be classed as positive Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Valencian Government grant IDIFEDER/2018/056 to JRD and Covid_19‐SCI to RG Publication status: Published paper Source: European Journal of Clinical Microbiology & Infectious Diseases Author COI: Declared none |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Valdivia 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Valdivia 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Valdivia 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To evaluate diagnostic performance of 8 antibody tests for COVID Design: Multiple‐group design with separate estimates of sensitivity and specificity [1] Symptomatic PCR‐confirmed COVID‐19 cases (n = 94) [2] Pre‐pandemic patients with a respiratory infection who had a PCR test for respiratory pathogens (n = 49) [3] Pre‐pandemic other infections (patients with confirmed non‐SARS‐CoV‐2 coronavirus infection) (n = 14) [4] Pre‐pandemic other infections (patients with antigens against other pathogens (e.g. CMV, EBV, HIV) from routine serology testing) (n = 40) [Suppl file described all controls as 'pre‐pandemic' so I've changed throughout] Recruitment: Unclear Prospective or retrospective: Unclear; described requirement for residual samples so likely retrospective Sample size: 197 (94) Further detail: Inclusion: only patients for whom residual samples were available were included. Exclusion: two cases excluded due to treatment with rituximab for B‐cell malignancy |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: University Hospitals Leuven, Leuven Country: Belgium Dates: March and April 2020 Symptoms and severity: 29 (35%) of patients were critical (required mechanical ventilation/died). Demographics: age: median 67.6 years, range 23‐90 years; sex: 66/94 (70%) male Exposure history: Unknown Non‐Covid group 1: [2] Pre‐pandemic respiratory infections Source: [2] September to November 2019 Characteristics: [2] Consecutive patients with a respiratory infection who had PCR test for respiratory pathogens Non‐Covid group 2: [3] Other infection (non‐SARS‐CoV‐2 coronavirus) [4] Other infections (patients with antibodies against other pathogens) Source: [3] Not stated; 'pre‐COVID‐19' [4] Not stated; 'pre‐COVID‐19' Characteristics: [3] PCR‐positive for a different coronavirus [4] Patients with antibodies against other pathogens e.g. cytomegalovirus (CMV) Epstein‐Barr virus (EBV), human immunodeficiency virus (HIV) from routine serology testing |
||
| Index tests | Test name: [A] Clungene COVID‐19 IgG/IgM Rapid test cassette [B] OrientGene COVID‐19 IgG/IgM Rapid test cassette [C] VivaDiag COVID‐19 IgM/IgG Rapid test [D] StrongStep SARS‐CoV‐2 IgM/IgG Antibody Rapid test [E] Dynamiker 2019‐nCOV IgG/IgM Rapid test [F] Multi‐G MGA 2019‐nCoV IgG/IgM Rapid test cassette [G] Prima COVID‐19 IgG/IgM Rapid test [H] Euroimmun Anti‐SARS‐Cov‐2 IgG/IgA ELISA Manufacturer: [A] Clungene Biotech, China [B] Zheijang OrientGene Biotech, China [C] VivaCheck Biotech, China [D] Liming Bio‐Products, China [E] Dynamiker Biotechnology, China [F] Multi‐G, Belgium [G] Prima Lab SA, Switzerland [H] Euroimmun, Germany Antibody: [A] IgG/IgM [B] IgG/IgM [C] IgG/IgM [D] IgG/IgM [E] IgG/IgM [F] IgG/IgM [G] IgG/IgM [H] IgG (specificity data only available for the IgA version; data not included) Antigen target: [A] Recombinant envelope antigens [B] Recombinant antigens [C] Recombinant antigen [D] Recombinant antigen [E] Nucleocapsid protein [F] Nucleocapsid protein [G] COVID‐19 antigen [H] S1‐protein Evaluation setting: [A] to [G]: designed to be POC but unclear whether used at POC or in laboratory [H] laboratory Test method: [A] to [G]: lateral flow assay [H] ELISA Timing of samples: Day 0‐6 post‐symptom onset: 37 (24%) Day 7‐13 post‐symptom onset: 78 (51%) Day 14‐25 post‐symptom onset: 38 (25%) Samples used: Serum or plasma (according to suppl file) Test operator: Unclear Definition of test positivity: [A] to [G]: pink/red test line indicating a positive result [H]: according to manufacturer's instruction, but borderline results (0.8‐1.1) were considered positive for further analysis Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; described as 'in‐house method complying with the WHO guidelines' Samples used: Nasopharygeal swabs in UTM Timing of reference standard: During hospital stay Blinded to index test: Unclear Incorporated index test: No Definition of non‐COVID cases: [2] Pre‐pandemic [3] PCR [4] Antibody test Samples used: [2] Unclear [3] Unclear [4] "Serology" Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: No Uninterpretable results: OrientGene LFA was the only kit with more than one device failure (8 failures). Indeterminate results: No; indeterminate results on EUROIMMUN were considered positive. Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: The research did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors. Publication status: Published paper Source: Clinical Microbiology and Infection Author COI: PV reported personal fees from Roche, outside the submitted work, and is a senior clinical investigator of the FWO‐Vlaanderen. KL reported personal fees and non‐financial support from Pfizer, personal fees and non‐financial support from MSD, personal fees from SMB Laboratoires, personal fees from Gilead, and personal fees from FUJIFILM Wako, outside the submitted work. The other authors stated no conflicts of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Van Elslande 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020a [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Multi‐group study to assess sensitivity and specificity [1] Covid‐positive (n = 233 samples, 114 patients) [2] Covid‐negative, pre‐pandemic (n = 113) [2a] Pre‐pandemic respiratory infection (n = 49) [2b] Pre‐pandemic coronavirus (n = 24) [2c] Pre‐pandemic other infections (n = 40) Recruitment: [1] Patients PCR‐positive for COVID‐19 [2a] Pre‐pandemic serum samples from consecutive patients with a respiratory infection who had a PCR test for respiratory pathogens between September and November 2019 [2b] Pre‐pandemic patients with a confirmed non‐SARS‐CoV‐2 coronavirus infection collected 12‐42 days after the positive PCR, not stated [2c] Pre‐pandemic patients with antibodies against other pathogens (e.g. cytomegalovirus, Epstein Barr virus, human immunodeficiency virus) from routine serology testing Prospective or retrospective: Retrospective Sample size: 346 (233) Further detail: Inclusion [1] PCR‐confirmed SARS‐CoV‐2 infection [2a] Respiratory infection [2b] Non‐SARS‐CoV‐2 coronavirus infection [2c] Antibodies against other pathogens Exclusion: [1] Immunocompromised patients (e.g. acute leukaemia, treatment with azathioprine) excluded [2a] [2b][2c] Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: University Hospitals Leuven, Leuven Country: Belgium Dates: Not stated Symptoms and severity: All symptomatic, 36/114 patients were classified as critical (needed mechanical ventilation or fatal infection), 78 non‐critical (moderate) Demographics: 81 male, 33 female; median age 66.5 years (rage 23‐90 years) Exposure history: Not stated Non‐Covid group 1: [2a] Respiratory infections, pre‐pandemic Source: September to November 2019, University Hospitals Leuven Characteristics: Confirmed respiratory infection: HSV n = 19, CMV n = 13, entero/rhinovirus n = 8, S. pneumoniae n = 7, RSV n = 3, parainfluenza virus n = 2, HMPV n = 1, P. jirovecii n = 1, bocavirus n = 1, L. pneumophila n = 1 Non‐Covid group 2: [2b] Other human coronaviruses, pre‐pandemic [2c] Antibodies against various viruses, pre‐pandemic Source: [2b] [2c] Pre‐pandemic (before January 2020), University Hospitals Leuven Characteristics: [2b] Non‐SARS‐CoV‐2 coronavirus infection: a‐Cov HCoV‐229E n = 7, a‐Cov HCoV‐NL63 n = 6, B‐Cov HCoV‐OC43 n = 7, B‐CoV HCoV‐HKU1 n = 4 [2c] Antibodies against other pathogens: CMV n = 21, EBV n = 15, VZV IgG n = 10, HIV‐1 n = 8, HSV IgG n = 7, HAV/HBV/HCV n = 14 |
||
| Index tests | Test name: [A] Roche Ig anti‐N [B] Abbott IgG anti‐N [C] Euro NCP IgG anti‐N [D] Mikrogen IgG anti‐N [E] Maglumi IgG anti‐N/S [F] Diasorin IgG anti‐S [G] Euro S1 IgG anti‐S Manufacturer: [A] Roche Diagnostics, Basel, Switzerland [B] Abbott Diagnostics, Lake Forest, Illinois [C] Euroimmun, Lubeck, Germany [D] Mikrogen, Neuried, Germany [E] Snibe, Shenzen, China [F] Diasorin, Saluggia, Italy [G] Euroimmun, Lubeck, Germany Antibody: [A] Total Ig antibodies [B]‐[G] IgG Antigen target: [A] N‐protein [B] N‐protein [C] N‐protein [D] N‐protein [E] N and S‐protein [F] S‐protein (S1 and S2) [G] S1‐protein Evaluation setting: Laboratory performed in laboratory Test method: [A] CLIA [B] CLIA [C] ELISA [D] ELISA [E] CLIA [F] CLIA [G] ELISA Timing of samples: 0‐6 days pso, n = 43 7‐13 days pso, n = 98 14‐17 days pso, n = 42 18‐21 days pso, n = 16 22‐27 days pso, n = 13 28‐37 days pso, n = 11 Samples used: Serum Test operator: Staff at University Hospitals Leuven (technical assistants) Definition of test positivity: Cut‐off [A] >= 1.0 [B] >= 1.4 [C] >= 0.8 positive, equivocal zone 0.8/1.1 [D] >= 20 positive, equivocal zone 20/24 [E] >= 1.0 [F] >= 12 positive, equivocal zone 12/15 [G] >= 0.8 positive, equivocal zone 0.8/1.1 Blinding reported: Not stated Threshold predefined: Yes, according to manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; described as 'in‐house method complying with the WHO guidelines', threshold not stated Samples used: Nasopharyngeal swabs (UTM, Copan, Italy) Timing of reference standard: 83.3% of patients were admitted the day of the first PCR‐positive result. Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA pre‐pandemic Timing of reference standard: Pre‐pandemic Blinded to index test: yes, prior to index test Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: 83.3% of patients were admitted the day of the first PCR‐positive result.
The median time between onset of symptoms and admission to the hospital was 7 days.
0‐6 days pso, n = 43
7‐13 days pso, n = 98
14‐17 days pso, n = 42
18‐21 days pso, n = 16
22‐27 days pso, n = 13
28‐37 days pso, n = 11 All patients received same reference standard: No Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Equivocal results [C][D][F][G] treated as positive Unit of analysis: Samples, only one sample included per patient per time frame |
||
| Comparative | |||
| Notes | Funding: Pieter Vermeersch reported personal fees from Roche, outside the submitted work. Katrien Lagrou reported personal fees and non‐financial support from Pfizer, personal fees and non‐financial support from MSD, personal fees from SMB Laboratoires, personal fees from Gilead, and personal fees from FUJIFILM Wako, outside the submitted work.
The research did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors. Publication status: Published paper Source: Clinical and Microbiology and Infection Author COI: Pieter Vermeersch reported personal fees from Roche, outside the submitted work. Katrien Lagrou reported personal fees and nonfinancial support from Pfizer, personal fees and non‐financial support from MSD, personal fees from SMB Laboratoires, personal fees from Gilead, and personal fees from FUJIFILM Wako, outside the submitted work. The other authors stated no conflicts of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Van Elslande 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020b [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020b [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020b [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Van Elslande 2020b [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Velay 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection of COVID‐19 Design: Multi‐group analysis to estimate sensitivity and specificity (n = 325) [1a] PCR‐confirmed hospital patients (n = 55) [1b] PCR‐confirmed healthcare workers (n =143) [2a] Pre‐pandemic controls (n = 100) [2b] Cross‐reactivity negative controls (n = 27) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 325 (198) Further detail: Inclusion: [1a] Hospitalised PCR‐positive Covid patients (n = 55) [1b] PCR‐positive healthcare workers (n = 143) [2a] Pre‐pandemic healthy blood donors [2b] Pre‐pandemic non‐SARS‐CoV‐2 infection: (n = 20) anti‐hCoV positive, (n = 2) anti‐influenza A virus positive, (n = 1) anti‐rhinovirus positive, (n = 2) rheumatoid factor positive, (n = 2) antinuclear antibodies positive Exclusion: Not stated |
||
| Patient characteristics and setting | Setting: [1a] Hospital inpatients (n = 55) [1b] Outpatients Location: Strasbourg University Hospital (Strasbourg, France) Country: France Dates: 2020 April Symptoms and severity: [1a] 23 were admitted to ICU [1b] Not stated Demographics: Patient group ‐ median age 68, male/female = 17/38. Healthcare workers ‐ median age ‐ 32, male/female ‐ 96/47. Total ‐ median age ‐ 43, male/female ‐ 113/85 Exposure history: Not stated Non‐Covid group 1: [2a] Pre‐pandemic controls Source: March to November 2019 Characteristics: Serum samples from 40 patients and plasma samples from 60 healthy blood donors collected before the COVID‐19 pandemic onset Non‐Covid group 2: [2b] Controls for cross‐reactivity Source: 27 serum samples collected before the COVID‐19 pandemic onset were used to study cross‐reactivity. Characteristics: Previous human coronavirus infections ‐ HCoV‐229E, HCoV‐HKU1, HCoV‐NL63, and HCoV‐OC43), 2 from patients previously infected with influenza A virus, 1 from a patient previously infected with human rhinovirus, 2 containing rheumatoid factor, and 2 positive for antinuclear antibodies |
||
| Index tests | Test name: [A] Biosynex COVID‐19 BSS [B] COVID‐19 Sign IgM/IgG [C] ELISA anti–SARS‐CoV‐2 IgA and IgG [D] EDI™ novel coronavirus COVID‐19 IgM and IgG Manufacturer: [A] Biosynex, Switzerland, Fribourg [B] Servibio/VEDALAB, France, Alençon [C] Euroimmun, Lübeck, Germany [D] Epitope Diagnostics, San Diego, California Antibody: [A] IgM and IgG [B] IgM and IgG [C] IgA and IgG [D] IgM and IgG Antigen target: [A] N‐protein [B] S1‐protein Evaluation setting: [A][B] POC [C][D] Laboratory All performed in laboratory Test method: [A] [B] Lateral flow assay [C] [D] ELISA Timing of samples: [1a] Serum samples were collected at a median of 7 days pso (range, 0–31 days pso). [1b] 24 days pso (range, 15–39 days pso) Samples used: Serum and plasma Test operator: Unclear Definition of test positivity: [A] [B] Visible line [C] >= 1.1 positive [D] Values greater than the cut‐off positive Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR testing of nasopharyngeal swab specimens according to current guidelines (Institut Pasteur, Paris, France; WHO technical guidance). This assay targets 2 regions of the viral RNA‐dependent RNA polymerase (RdRp) gene, with a threshold limit of detection of 10 copies per reaction. Samples used: Nasopharyngeal Timing of reference standard: Total median time since symptom onset ‐ 2 days Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA Timing of reference standard: NA Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Median time difference 20 days ‐ median time to PCR 2 days and median time to serum collection ‐ 22 days All patients received same reference standard: Yes Missing data: Not stated Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Study was supported by the Strasbourg University Hospital (COVID‐HUS study) Publication status: Published paper Source: Diagnostic Microbiology and Infectious Disease Author COI: None declared |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Unclear | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Velay 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Velay 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Velay 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Veyrenche 2021 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute‐phase infection Design: Two‐group study to assess sensitivity and specificity [1] Covid‐19 cases (n = 45) [2] Non‐Covid controls (n = 20) Group [2] not eligible for our review as < 25 samples leaving a single‐group study to estimate sensitivity only Recruitment: [1] Patients admitted in Montpellier University Hospitals between 14 March and 11 April 2020 who tested positive for SARS‐CoV‐2 RNA [2] Samples collected in the pre‐COVID‐19 period (2017‐2018) in patients Prospective or retrospective: [1] Prospective [2] Retrospective Sample size: 65 (45) but 45 (45) included in our study Further detail: [1] Inclusion: Hospital inpatients with RT‐PCR confirmed SARS‐CoV‐2 infection. Any disease severity Exclusion: Not stated [2] Inclusion: Samples from patients collected pre‐pandemic (2017‐2018) Exclusion: Not stated |
||
| Patient characteristics and setting | Setting: Hospital inpatients Location: Montpellier University hospitals (Centre Hospitalier Universitaire de Montpellier, Montpellier) Country: France Dates: 14 March to 11 April 2020 Symptoms and severity: 26/45, 58% cases 'severe' according to WHO guideline (similar numbers per Ct subgroup) All hospitalised Demographics: 32/45, 71% male Exposure history: Not stated |
||
| Index tests | Test name: [A] SARS‐CoV‐2 IgG immunoassay (Alinity) [B] ELISA COVID‐19 THERA02 IgM assay [C] SureScreen COVID‐19 IgM/IgG Rapid Test [D] Syzbio SARS‐CoV‐2 IgM/IgG Antibody Assay Kit Manufacturer: [A] Abbott Diagnostics, Illinois, USA [B] Theradiag, Marne la Vallee, France [C] SureScreen Diagnostics Ltd, Derby, UK [D]Syzbio Biotech Joint Stock Co., Ltd, Wuhan, China Antibody: [A] IgG [B] IgM [C] IgM and/or IgG [D] IgM and/or IgG Antigen target: [A] N‐protein [B] S‐protein [C] not stated [D] not stated Evaluation setting: [A] [B] Laboratory test [C] [D] POCT performed in lab Test method: [A] CMIA [B] ELISA [C] [D] lateral flow Timing of samples: Day 1‐20 pso. 1‐7 days (n = 22) 7‐14 days (n = 14) 14 ‐ 20 days (n = 9) Samples used: [A] [B] [C] [D] Plasma Test operator: Nicolas Veyrenche, Karine Bolloré and Amandine Pisoni have performed experiments (Pathogenesis and Control of Chronic Infections, INSERM, Etablissement Français du Sang, CHU Montpellier, Université de Montpellier, Montpellier, France). All tests were performed in the laboratory of Virology. Definition of test positivity: [A] ratio (S/C) >= 1.4 is positive, < 1.4 negative. [B] positive cut‐off is ratio >= 1. [C] [D] any signal visible, even weak, at 15 mins on the test line is positive. Blinding reported: Not stated. Threshold predefined: Yes, according to manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Allplex™ 2019‐nCoV Assay (Seegene, Seoul, South Korea);
COVID‐19 confirmed‐subjects were grouped according to the average value of the cycle threshold (Ct), Ct ≤ 25, 25 < Ct < 35 and Ct ≥ 35. Samples used: Nasopharyngeal Timing of reference standard: Hospital admission ranged from 1‐20 days pso; PCR performed prospectively on admission within a few hours after collection Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests:
[1] Same day All patients received same reference standard: Yes for [1], group [2] excluded from review Missing data: yes, group [2] excluded from review Uninterpretable results: none reported Indeterminate results: no indeterminate threshold Unit of analysis: patients |
||
| Comparative | |||
| Notes | Funding: This work was funded by the Montpellier University Hospital, Muse I‐SITE Program Grant, University of Montpellier. Publication status: Published paper Source: Journal of Medical Virology Author COI: The authors declared that there were no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Veyrenche 2021 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Veyrenche 2021 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Veyrenche 2021 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wang 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: Single‐group study to estimate sensitivity and specificity in: [1] patients who visited the hospital with respiratory complaints during January to March 2020 (n = 375) Recruitment: Consecutive (all patients in a time period) Prospective or retrospective: Retrospective Sample size: 375 (141) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: First People’s Hospital of Jingmen, Hubei Province Country: China Dates: 25th January to 16th March 2020 Symptoms and severity: Not reported Demographics: 65 (46%) male, median age 58 years, range 21 to 95 years Exposure history: Unclear |
||
| Index tests | Test name: Xiamen Biotime IgG/IgM Manufacturer: Xiamen Wantai Kairui Biological Tecnhology Co. Ltd, China Antibody: Total antibody Antigen target: Recombinant antigens containing the receptor binding domain (RBD) Evaluation setting: Laboratory Test method: CLIA Timing of samples: Day 0 to > 20 days pso; 0‐10 days after symptom onset: 61 (43%) 11‐20 days after symptom onset: 72 (51%) 21+ days after symptom onset: 8 (6%) Samples used: Serum Test operator: Unclear Definition of test positivity: Signal‐to‐cut off ratio >= 1 represented antibody positivity. Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: New Coronavirus Pneumonia Prevention and Control Program (7th edition) definition; specifically:
[1] RT‐PCR (Applied Biosystems ViiA7 Dx (Applied Biosystems, Singapore) and RT‐PCR reagent BioGerm (Shanghai BioGerm Medical Technology Co., Ltd.); threshold > 40 Ct defined negative, or
[2] RT‐PCR‐negative with characteristic CT changes of the lungs Samples used: Throat swabs Timing of reference standard: Of 1415 cases: 39.7% positive day 0‐3 62.4% positive by day 5 86.7% positive by day 7 92.2% positive by day 10 or more 11 patients remained PCR‐negative Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Varied, as reference tests were repeated up to 5 times until positive, and index tests were performed on discharge Missing data: None reported Uninterpretable results: None reported Indeterminate results:None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Published paper Source: Journal of Virological Methods Author COI: The author declared that there was no conflict of interest related to this article content. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Weidner 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Sensitivity for identification of previous disease Design: Single‐group study estimating sensitivity Used serum samples from convalescent plasma donors with Nucleic Acid Test (NAT)‐confirmed COVID‐19 (n = 100) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 100 (100) |
||
| Patient characteristics and setting | Setting: Convalescent plasma donors Location: Austrian Red Cross, Blood Service for Vienna, Lower Austria and Burgenland, Vienna Country: Austria Dates: Not stated Symptoms and severity: Severity: 93/100 (93%): mild or no symptoms (WHO class 1‐2); 6/100 (6%): moderate‐severe symptoms (WHO class 3‐6); no details on 1 individual. Reported symptoms: 63% fever, 48% headache, 44% body aches, 43% loss of taste and smell, 40% cough, 31% fatigue, 23% gastrointestinal symptoms, 29% sore throat Demographics: Age range: 18‐66 y; age, median (SD): 47 y (12.7); sex: 61/100 (61%) male Exposure history: Not stated |
||
| Index tests | Test name: [A] Euroimmun SARS‐CoV‐2 IgG ELISA [B] Wantai SARS‐CoV‐2 Ab ELISA [C] Roche Elecsys Anti‐SARS‐CoV‐2 [D] LIAISON® SARS‐CoV‐2 S1/S2 IgG [E] MEDsan COVID‐19 IgM/IgG Rapid Test [F] Wantai SARS‐CoV‐2 Ab Rapid Test Manufacturer: [A] Euroimmun, Lübeck, Germany [B] Wantai Biological Pharmacy, Beijing, China [C] Roche Diagnostics, Rotkreuz, Switzerland [D] DiaSorin S.p.A., Saluggia, Italy [E] MPC International S.A., Luxemburg [F] Wantai Biological Pharmacy, Beijing, China Antibody: [A] IgG [B] IgM [C] Total antibodies [D] IgG [E] IgM, IgG [F] Total antibodies Antigen target: [A] S1 domain of the spike‐protein [B] Not stated [C] N‐protein [D] S1 and S2 domains of the spike‐protein [E] Not stated [F] Not stated Evaluation setting: [A]‐[E]: Lab test, done in lab [F]: POC test, unclear if used as POC Test method: [A] [B]: Enzyme‐linked immunosorbent assay (ELISA) [C]: Electrochemilumescence sandwich assay (ECLIA) [D]: Chemiluminescence immunoassay (CLIA) [E], [F]: Lateral flow assay Timing of samples: Samples collected between 26 and 61 days pso (median 47 days, standard deviation 6.6 days) Samples used: Serum, plasma Test operator: Not stated ELISA tests performed at the Center for Virology, Medical University of Vienna; CLIA test performed by Department for Blood Group Serology and Transfusion Medicine, Medical University Graz. Sounded like lab personal for [A]‐[E] Definition of test positivity: [A]: Positive if ratio >= 1.1; borderline if ratio 0.8‐1.09; negative if ratio < 0 [B]: Positive if ratio > 1.0 (the cut‐off is calculated as the mean of three negative controls (minimum 0.03) plus 0.16). [C]: Positive if COI >= 1 [D]: Positive if >= 15 AU/mL; equivocal if 12‐14.9 AU/mL; negative if < 12 AU/mL [E], [F]: Visual‐based (read after 15 min and classified according to their strength, from 0 to 4+. 0 is negative and 4+ corresponds to an intensity equivalent to the control line. A picture card was used to standardise interpretation of the result) Blinding reported: Not stated Threshold predefined: [A]‐[F]: Yes, as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: positive PCR test for COVID‐19 Samples used: Nasopharyngeal swabs or pharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Unclear ‐ multiple assays were likely used Missing data: Yes: 2 for test [A], 1 for test [C], 1 for test [E], 2 for test [F] Uninterpretable results: No Indeterminate results: yes (classed as TPs) [A] 2 borderline or equivocal results (ID) [D] 5 ID Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: None reported
(This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors) Publication status: Published paper Source: Academic journal (Journal of Clinical Virology) Author COI: Two authors are employees of Baxter AG, a Takeda company and have Takeda stock interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Weidner 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Weidner 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Weidner 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Weidner 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Weidner 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wellinghausen 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Single‐group analysis to assess sensitivity [1] Covid patients (n = 67 samples from 58 patients) [1a] Covid outpatients (n = 60 samples from 51 patients) [1b] Asymptomatic Covid patients (n = 7 samples from 7 patients) Recruitment: [1a] Patients with clinical symptoms and confirmed‐PCR, ambulatory treated SARS‐CoV‐2 infection [1b] Asymptomatic persons with a positive SARS‐CoV‐2 PCR in the past who were contact persons to PCR‐confirmed COVID‐19 patients Recruitment unclear. Prospective or retrospective: Retrospective Sample size: 67 (67) samples from 58 (58) patients of which 58 (58) samples are used for sensitivity estimation Further detail: Inclusion: [1a] PCR‐positive for SARS‐CoV‐2 in a nasopharyngeal swab (at least 7 days before serum collection) in our laboratory information system (LIS), with clinical symptoms, ambulatory treated patients fulfilling the clinical diagnostic criteria of the Robert‐Koch‐Institut [1b] Asymptomatic Covid contacts with a positive SARS‐CoV‐2‐PCR in the past Exclusion: Not stated |
||
| Patient characteristics and setting | Setting: [1a] Outpatients or [1b] community Location: MVZ Labor Ravensburg Country: Germany Dates: March 24th to May 6th 2020 Symptoms and severity: [1a] Symptomatic, ambulatory treated [1b] Asymptomatic Demographics: Not stated Exposure history: [1a] Not stated [1b] Contacts of Covid patients |
||
| Index tests | Test name: [A] Anti‐SARS‐CoV‐2 ELISA IgG [B] EDI Novel Coronavirus COVID‐19 IgG ELISA [C] Liaison SARS‐CoV‐2 S1/S2 IgG [D] SARS‐CoV‐2 IgG [E] Elecsys Anti‐SARS‐CoV‐2 (IgM/IgA/IgG) Manufacturer: [A] Euroimmun, Luebeck, Germany [B] Epitope Diagnostics, San Diego (CA) [C] Diasorin, Dietzenbach, Germany [D] Abbott Diagnostics, Wetzlar, Germany [E] Roche Diagnostics, Mannheim, Germany Antibody: [A] IgG [B] IgG [C] IgG [D] IgG [E] Total Ab Antigen target: [A] S1‐protein [B] N‐protein [C] S1 and S2‐protein [D] N‐protein [E] N‐protein Evaluation setting: Laboratory Test method: [A] ELISA [B] ELISA [C] ELISA [D] CLIA [E] CLIA Timing of samples: [1a] 10 to 54 days post‐symptom onset (median 24 days) day 10‐20 (n = 11), day 21 to 54 (n = 40), plus 3 patients with three follow‐up serum samples each. [1b] 9‐56 days post‐PCR+ Samples used: serum Test operator: staff at MVZ Labor Ravensburg Definition of test positivity: [A] Ratio < 0.8 negative, 0.8‐1.09 equivocal, >= 1.1 positive [B] Ratio < 0.9 negative, 0.9‐1.09 equivocal, >= 1.1 positive [C] < 12 AU/mL negative, 12.0‐14.5 AU/mL equivocal, >= 15 AU/mL positive [D] COI < 1.4 negative, COI >= 1.4 positive [E] COI < 1.0 negative, COI >= 1.0 positive Blinding reported: Not stated (but study only included COVID cases) Threshold predefined: Yes, manufacturers |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR with the Cobas SARS‐CoV‐2 assay, the AmpliGnost SARS‐CoV‐2 E‐Gen qPCR and the AmpliGnost SARS‐CoV‐2 E‐Gen PCR(PIIM) and AmpliGnost SARS‐CoV‐2 N‐Gen PCR (PIIM), threshold not stated Samples used: nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior. Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: Yes Missing data: yes, 9 follow‐up samples not included in review Uninterpretable results: Not stated Indeterminate results: Equivocal results were counted as negative; (n = 2) Euroimmun and Liaison, (n = 4) EDI Unit of analysis: Samples, 58 patients, 3 patients with 4 samples each For review, only 58 samples from 58 patients were included. |
||
| Comparative | |||
| Notes | Funding: None stated. Publication status: Published paper Source: GMS Infectious Diseases Author COI: The authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Wellinghausen 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wellinghausen 2020a [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wellinghausen 2020a [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wellinghausen 2020a [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wellinghausen 2020b.
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current convalescent‐phase infection Design: Single‐group study to assess sensitivity [1] Covid patients (n = 137) [1a] Symptomatic outpatients (n = 111) [1b] Asymptomatic, PCR‐confirmed contacts (n = 26) Recruitment: [1] All serum samples sent to our laboratory for SARS‐CoV‐2‐IgG determination between March 24th and May 6th 2020 from outpatients with a positive result of SARS−COV‐2‐RT‐PCR in a nasopharyngeal swab (at least 7 days before serum collection) were considered for analysis (n = 158). Patients with past hospital treatment for COVID‐19 (n = 11) and patients in whom clinical information could not be obtained (n = 10) have been excluded from analysis. Prospective or retrospective: Retrospective Sample size: 137 (137) but 126 (126) included in our review Further detail: Inclusion: [1a] PCR‐positive for SARS‐CoV‐2 in a nasopharyngeal swab (at least 7 days before serum collection) in our laboratory information system (LIS), with clinical symptoms, ambulatory treated patients fulfilling the clinical diagnostic criteria of the Robert‐Koch‐Institute [1b] Asymptomatic Covid contacts with a positive SARS‐CoV‐2‐PCR at least 7 days before serum collection Exclusion: Patients with past hospital treatment for COVID‐19 (n = 11); patients in whom clinical information could not be obtained (n = 10) |
||
| Patient characteristics and setting | Setting: [1a] Outpatients or [1b] community All had recovered at the time point of blood collection. Location: MVZ Labor Ravensburg, Ravensburg (private laboratory serving a large number of private practices and hospitals in Southwest Germany as well as most coronavirus test centres in the region) Country: Germany Dates: March 24th to May 6th 2020 Symptoms and severity: [1a] Symptomatic, ambulatory treated [1b] Asymptomatic Demographics: Not stated Exposure history: [1a] Not stated [1b] Contacts of Covid patients Non‐Covid group 1: NA |
||
| Index tests | Test name: Anti‐SARS‐CoV‐2‐ELISA IgG Manufacturer: Euroimmun, Luebeck, Germany Antibody: IgG Antigen target: S1‐protein Evaluation setting: Laboratory used in lab Test method: ELISA Timing of samples: All had recovered at the time point of blood collection. [1a] Day 10‐20 pso, n = 11; day 21‐68 pso, n = 100 [1b] Day 9‐20 post‐PCR+, n = 10; day 21‐56 post‐PCR+, n = 16; day 28‐56 post‐PCR+, n = 14 Samples used: Serum Test operator: Lab staff at MVZ Labor Ravensberg Definition of test positivity: Not stated, according to the manufacturer’s instructions Blinding reported: No, no negative group Threshold predefined: Yes, according to manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR with the Cobas SARS‐CoV‐2 assay, the AmpliGnost SARS‐CoV‐2 E‐Gen qPCR and the AmpliGnost SARS‐CoV‐2 E‐Gen PCR (PIIM) and AmpliGnost SARS‐CoV‐2 N‐Gen PCR (PIIM), threshold not stated Samples used: Nasopharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: NA Samples used: NA Timing of reference standard: NA Blinded to index test: NA Incorporated index test: NA |
||
| Flow and timing | Time interval between index and reference tests: [1a] Not stated [1b] 9‐56 days All patients received same reference standard: Yes Missing data: yes, 11 samples from [1a] with time split 10‐20 days pso excluded from review Uninterpretable results: Not stated Indeterminate results: Equivocal results counted as negative [1a] 10‐20 days pso, one equivocal result. 21‐68 days, three equivocal results Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Journal of Clinical Virology Author COI: No conflicts of interest by all authors |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Whitman 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute or convalescent‐phase infection Design: Multi‐group study to assess sensitivity and specificity [1] Covid patients (n = 128 samples from 79 patients) [2] Non‐Covid patients (n = 159) [2a] Pre‐pandemic (n = 108) [2b] Cross‐reactivity, concurrent (n = 41) [2c] Concurrent, SARS‐COV‐2 PCR‐negative, no other viruses detected (n = 10) Recruitment: [1] Patients diagnosed at University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital. Not admitted, admitted or ICU [2a] Blood donors before 2019 [2b] Patients with other respiratory pathogen testing at University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital [2c] Patients at University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital Recruitment method not stated. Prospective or retrospective: Retrospective Sample size: 287 (128) Further detail: [1] Inclusion: In or outpatients at University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital with symptomatic infection and positive SARS‐CoV‐2 RT–PCR testing of nasopharyngeal or oropharyngeal swabs and remnant plasma and serum samples in associated laboratories Exclusion: If an individual had more than one specimen for a given time interval, only the later specimen was included. [2a] Inclusion: Blood donors before July 2018. Exclusion: Not stated [2b] Inclusion: Concurrent patients from 2020 with detection of other respiratory viruses. Exclusion: Not stated [2c] Inclusion: Concurrent patients from 2020, RT‐PCR‐negative for SARS‐CoV‐2 by RT–PCR. Exclusion: Not stated Exclusions: Data that did not fit our study design were excluded after the fact. This included all data and statistics derived from specimens from individuals who were mis‐assigned to a data analysis group (including time interval of days from symptom onset, or RT‐PCR status), duplicate patient specimens not originally identified prior to obtaining results or data from confirmatory spot testing described in the manuscript. |
||
| Patient characteristics and setting | Setting: Hospital inpatients (82%) and outpatients (ambulatory) (18%) Location: University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital, San Francisco, CA Country: USA Dates: Not stated Symptoms and severity: 18% (14/79) not admitted, 46% (36/79) inpatients without ICU care, 37% (29/79) required ICU care Demographics: Age range 22‐90 years, mean 52.9 (SD 15) years 68% Hispanic/Latino 9% Asian 9% White 8% Black 6% Other/not reported Male sex 54/79 (68%) Exposure history: Not stated. Non‐Covid group 1: [2a] Pre‐pandemic Source: Blood donors before July 2019. From University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital Characteristics: Healthy Non‐Covid group 2: [2b] Cross‐reactivity [2c] RT‐PCR‐negative, no other respiratory viruses detected Source: [2b] [2c] University of California, San Francisco (UCSF) hospital system or Zuckerberg San Francisco General (ZSFG) Hospital, in 2020 Characteristics: [2b] Influenza A (n = 2), human rhinovirus/enterovirus (n = 17), human metapneumovirus (n = 54), respiratory syncytial virus (n = 9), parainfluenza (n = 3), adenovirus (n = 2), other coronaviruses (n = 4) [2c] Not other respiratory viruses |
||
| Index tests | Test name: [A] COVID‐19 IgM‐IgG Rapid Test (51‐002‐20) [B] Perfect POC Novel Corona Virus (SARS‐CoV‐2) IgM/IgG Rapid Test Kit (SC30201 W) [C] Novel Coronavirus (SARS‐CoV‐2) IgM/IgG Combo Rapid Test‐Cassette [D] COVID‐19 (SARS‐CoV‐2) IgG/IgM Antibody Test Kit (Colloidal Gold) [E] Novel Coronavirus (2019‐nCoV) Ab Test (Colloidal Gold) IgM [F] COVID‐19 IgG/IgM Rapid Test Cassette (INGMMC42S) (RightSign assay from Hangzhou Biotest) [G] SARS‐CoV‐2 IgM/IgG Antibody Rapid Test (VC01210 3) [H] Coronavirus IgG/IgM Antibody (COVID‐19) Test Cassette (U‐CoV102) [I] VivaDiag SARS‐CoV‐2 IgM/IgG Rapid Test (VID35‐08‐011) [J] SARS‐CoV‐2 Antibody Test (W195) [K] EDI Novel Coronavirus COVID‐19 IgM or IgG ELISA (KT‐1033; KT‐1032) [Addional in‐House ELISA reported; not included in review] Manufacturer: [A] BioMedomics Inc, Morrisville, NC, USA [B] Bioperfectus Technologies Co Ltd, Jiangsu, China [C] DecomBio Biotechnology Co Ltd, Beijing, China [D] DeepBlue Medical Technology Co Ltd, Anhui, China [E] Innovita Biological Technology Co Ltd, Qian'an, China [F] Premier Biotech, Minneapolis, MN, USA (RightSign assay from Hangzhou Biotest, marketed by Premier Biotech under a different name) [G] Sure Biotech, New York, USA; Wan Chai, Hong Kong [H] UCP Biosciences, San Jose, CA, USA [I] VivaChek Biotech Co, Hangzhou, China [J] Wondfo Biotech Co Ltd, Guagzhou, China [K] Epitope Diagnostics, San Diego, USA Antibody: [A]‐[I] IgM and/or IgG [J] Total Ab [K] IgM, or IgG Antigen target: [A] RBD [B] N and S [C] Not stated [D] Not stated [E] N and S [F] Not stated [G] N and S [H] Not stated [I] Not stated [J] Not stated [K] N Evaluation setting: [A]‐[J] POCT, performed in lab [K] Laboratory Test method: [C] [F]‐[J] Lateral flow assays [D] [E] Colloidal gold [K] ELISA Timing of samples: 0‐ > 20 days pso 1‐5 days pso: n = 28 6‐10 days pso: n = 36 11‐15 days pso: n = 34 16‐20 days pso: n = 19 > 20 days pso: n = 11 Samples used: Plasma or serum Test operator: Laboratory staff Definition of test positivity: [A]‐[J] All visual‐based tests, each cartridge was assigned an integer score (0 for negative, 1–6 for positive) for test line intensity by two independent readers blinded to specimen status and to each other’s scores. [K] IgM positive cut‐off = 1.1 × ((average of negative control readings) + 0.10). Values less than or equal to the positive cut‐off were interpreted as negative; IgG positive cut‐off = 1.1 × ((average of negative control readings) + 0.18). Values less than or equal to the positive cut‐off were interpreted as negative. Blinding reported: [A]‐[J] two independent readers blinded to specimen status and to each other’s scores [K] Not stated Threshold predefined: [A]‐[L] Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, threshold not stated Samples used: Nasopharyngeal or oropharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: [2a] Pre‐pandemic [2b] RT‐PCR‐negative or none [2c] RT‐PCR‐negative Samples used: [2a] NA, pre‐pandemic [2b] Nasopharyngeal or oropharyngeal swabs or none [2c] Nasopharyngeal or oropharyngeal swabs Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: Some specimens were exhausted during the analysis and were not included in all tests. One test result missing for test [E], IgM, 11‐15 days Uninterpretable results: Not stated Indeterminate results: Not stated. Unit of analysis: Samples but only 1 sample per patient per time‐split included. If an individual had more than one specimen for a given time interval, only the later specimen was included. |
||
| Comparative | |||
| Notes | Funding: This work was supported by gifts from Anthem Blue Cross Blue Shield, the Chan Zuckerberg Biohub and anonymous philanthropy.
We thank the following sources for donation of test kits: the manufacturers of Bioperfectus, DecomBio, Sure Biotech, UCP Biosciences; D. Friedberg, J. Hering and H. Schein.
The Wilson lab has received support from the Rachleff Family Foundation.
The Hsu lab has received support from S. Altman, V. and N. Khosla, D. and S. Deb, the Curci Foundation and Emergent Ventures.
The Marson lab has received gifts from J. Aronov, G. Hoskin, K. Jordan, B. Bakar, the Caufield family and funds from the Innovative Genomics Institute, the Northern California JDRF Center of Excellence and the Parker Institute for Cancer Immunotherapy.
We thank the National Institutes of Health for its support (to J.D.W., R38HL143581; to A.E.G., F30AI150061; to D.N.N., L40 AI140341; to S.P.B., NHLBI R38HL143581, to G.M.G., NHLBI R38HL143581; to T.A.M., 1F30HD093116; to D.W., 1F31NS106868‐01; to J.G.C., R01 AI40098; to E.T.R. and R.C.C., CDC U01CK000490; MSTP students were supported by T32GM007618). R.Y. was supported by an AP Giannini Postdoctoral Fellowship. J.A.S. was supported by the Larry L. Hillblom
Foundation (2019‐D‐006‐FEL). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub and is a recipient of the Cancer Research Institute Lloyd J. Old STAR grant.
C.Y.C. is the director of the UCSF‐Abbott Viral Diagnostics and Discovery Center, receives research support funding from Abbott Laboratories and is on the scientific advisory board of Mammoth Biosciences. C.J.Y. is co‐founder of DropPrint Genomics and serves as an advisor to them. M.S.A. holds stock in Medtronic and Merck. P.D.H. is a co‐founder of Spotlight
Therapeutics and serves on the board of directors and scientific advisory board and is an advisor to Serotiny. P.D.H. holds stock in Spotlight Therapeutics and Editas Medicine. A.M. is a co‐founder of Spotlight Therapeutics and Arsenal Biosciences and serves on their board of directors and scientific advisory board. A.M. has served as an advisor to Juno Therapeutics, was a member of the scientific advisory board at PACT Pharma and was an advisor to Trizell. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. R.Y. owns stock in AbbVie, Bluebird Bio, Bristol‐Myers Squibb, Cara
Therapeutics, Editas Medicine, Esperion and Gilead Sciences. Unrelated to this current work, the Marson lab has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and GlaxoSmithKline and a gift from Gilead. Publication status: Published paper Source: Nature Biotechnology Author COI: C.Y.C. is the director of the UCSF‐Abbott Viral Diagnostics and Discovery Center, receives research support funding from Abbott Laboratories and is on the scientific advisory board of Mammoth Biosciences. C.J.Y. is co‐founder of DropPrint Genomics and serves as an advisor to them. M.S.A. holds stock in Medtronic and Merck. P.D.H. is a co‐founder of Spotlight Therapeutics and serves on the board of directors and scientific advisory board and is an advisor to Serotiny. P.D.H. holds stock in Spotlight Therapeutics and Editas Medicine. A.M. is a co‐founder of Spotlight Therapeutics and Arsenal Biosciences and serves on their board of directors and scientific advisory board. A.M. has served as an advisor to Juno Therapeutics, was a member of the scientific advisory board at PACT Pharma and was an advisor to Trizell. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. R.Y. owns stock in AbbVie, Bluebird Bio, Bristol‐Myers Squibb, Cara Therapeutics, Editas Medicine, Esperion and Gilead Sciences. Unrelated to this current work, the Marson lab has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and GlaxoSmithKline and a gift from Gilead. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Whitman 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [G].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [H].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [I].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [J].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020a [K].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute‐phase infection Design: Two‐group study estimating sensitivity and specificity [1] SARS‐CoV‐2 RT‐PCR‐positive (n = 44) [2] pre‐pandemic asymptomatic adults (n = 30) [3] pre‐pandemic other infection controls with febrile and/or respiratory illness (n = 30) Recruitment: Not reported Prospective or retrospective: Not reported Sample size: 104 (44) Further detail: No further details |
||
| Patient characteristics and setting | Setting: Inpatient Location: Massachusetts General Hospital Country: USA Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated Non‐Covid group 1: Pre‐pandemic, healthy Source: No further details Characteristics: Non‐Covid group 2: Pre‐pandemic, other infection controls Source: No further details Characteristics: No further details |
||
| Index tests | Test name: See below Manufacturer: [A] SD Biosensor ‐ Standard Q COVID‐19 IgM/IgG Duo (KT1032; lot P630C) [B] Biolidics ‐ 2019‐nCoV IgG/IgM antibody detection kit (CBB‐F015016‐V; V2020 0330) [C] Biomedomics ‐ COVID‐19 IgM and IgG Rapid Test (51‐002‐20; lot 20200, 22702, 20200, 32103) Antibody: IgG, IgM Antigen target: [A] N‐based [B] N‐ and S‐based [C] S‐based Evaluation setting: POC or laboratory Test method: LFA Timing of samples: Not stated Samples used: serum/plasma Test operator: Not stated Definition of test positivity: Not stated; assume as per manufacturer Blinding reported: Not reported Threshold predefined: Not stated; assume as per manufacturer |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; threshold NR Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes; prior to index Incorporated index test: No Definition of non‐COVID cases: [2] + [3] Samples used: Not stated Timing of reference standard: [2] + [3] pre‐pandemic Blinded to index test: yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not reported; presume as per main study reported in Whitman 2020 (A) Publication status: pre‐print Source: medRxiv Author COI: Not reported; presume as per main study reported in Whitman 2020 (A) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Whitman 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Whitman 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wolff 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity [1] Confirmed Covid patients (n = 111) [1a] Symptomatic Covid (n = 87) [1b] Asymptomatic Covid (n = 24) [2] Pre‐pandemic, non‐Covid (n = 96) Recruitment: Not stated. Prospective or retrospective: [1] Unclear [2] Retrospective Sample size: 207 (111) Further detail: [1] Included symptomatic (mild to moderate or severe) cases and asymptomatic cases confirmed by qRT‐PCR [1b] Asymptomatic patients were defined as individuals without any symptoms who were screened positive for SARS‐CoV‐2 nucleic acid due to close contacts with COVID‐19 patients. [2] Residual serum samples non‐SARS‐CoV‐2 collected before the pandemic COVID‐19 from January to February 2019 Exclusion criteria not stated |
||
| Patient characteristics and setting | Setting:
Not stated (seems to be mixed) Location: Laboratoire Hospitalier Universitaire de Bruxelles, Université Libre de Bruxelles, Brussels, Belgium Country: Belgium Dates: Not stated Symptoms and severity: Mild to moderate (n = 47): fever, headache, cough, myalgia Severe (n = 40): need for oxygen supplementation, respiratory failure requiring mechanical ventilation, admission to ICU or death Asymptomatic (n = 24) Demographics: [1a] median age 60 years, range 21‐88 years, 36 women, 51 men [1b] median age 61 years, range 20‐85 years, 11 women, 13 men Exposure history: [1a] not stated [1b] close contacts of Covid cases Non‐Covid group 1: Pre‐pandemic, non‐Covid patients (n = 96) Source: Residual samples collected between January to February 2019. source not stated Characteristics: Median age 38, range 0 ‐87 years, 62 women, 38 men |
||
| Index tests | Test name: [A] Elecsys Anti‐SARS CoV‐2 [B] Liaison SARS‐CoV‐2 S1/S2 IgG [C] Euroimmun Anti‐SARS CoV‐2 IgG ELISA [D] Euroimmun Anti‐SARS CoV‐2 IgA ELISA [E] VIDAS Anti‐SARS CoV‐2 IgG [F] VIDAS Anti‐SARS CoV‐2 IgM Manufacturer: [A] Roche Diagnostics, Vilvoorde, Belgium [B] Diasorin, Saluggia, Italy [C] [D] Euroimmun, Luebeck, Germany [E] [F] BioMerieux, Marcy‐l'Etoile, France Antibody: [A] IgM/IgG (total antibodies including IgG) [B] IgG [C] IgG [D] IgA [E] IgG [F] IgM Antigen target: [A] N‐protein [B] S1/S2‐protein [C] [D] S1‐protein [E] [F] S‐protein Evaluation setting: Laboratory Test method: [A] CLIA [B] CLIA [C] [D] ELISA [E] [F] enzyme linked fluorescence assay (ELFA) Timing of samples: [1a] 0‐54 days pso [1b] 0‐15 days post‐PCR + 0–7 days post‐symptoms or post + PCR: n = 35 8–14 days post‐symptoms or post + PCR: n = 31 > 15 days post‐symptoms or post + PCR: n = 45 Samples used: Serum Test operator: Laboratory staff Definition of test positivity: [A] negative COI < 1, positive COI >= 1 [B] negative < 12 AU/mL, borderline >= 12 to < 15 AU/mL, positive >= 15 AU/mL [C] [D] negative < 0.8, borderline >= 0.8 to < 1.1, positive >= 1.1 [E] [F] negative (index < 1) or positive (index ≥ 1) Blinding reported: Unclear Threshold predefined: Yes, according to manufacturer |
||
| Target condition and reference standard(s) | Reference standard: qRT‐PCR using the RealStar SARS‐CoV‐2 RT‐PCR kit 1.0, threshold not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Yes, prior Incorporated index test: No Definition of non‐COVID cases: Pre‐pandemic Samples used: NA, pre‐pandemic Timing of reference standard: NA, pre‐pandemic Blinded to index test: yes, prior Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: [1a] Not stated [1b] 0‐15 days post‐PCR + (n = 24) All patients received same reference standard: No, [2] pre‐pandemic Missing data: 45 samples 16‐54 days pso not included in review Uninterpretable results: Not stated Indeterminate results: Borderline data were found for [B] four samples analysed using the Liaison IgG, two samples using the [C] Euroimmun IgG and [D] IgA. Borderline data were considered positive for the statistical analyses. Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated Publication status: Published paper Source: Diagnostic Microbiology and Infectious Disease Author COI: No declaration of competing interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Wolff 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wolff 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wolff 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wolff 2020 [E].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wolff 2020 [F].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wu 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: Two‐group study to estimate sensitivity and specificity, however it appeared that all participants met Taiwanese reporting criteria for COVID‐19: [1] PCR‐confirmed symptomatic and hospitalised Covid‐19 patients (n = 16) [2] Inpatients with respiratory tract infection or fever but 2 negative PCR results for SARS‐CoV‐2 (n = 58) (All patients meeting criteria for testing were simultaneously evaluated for SARS‐CoV‐2 and influenza A/B; if both PCR results were negative, an additional SARS‐CoV‐2 test was performed using a second sample from the suspected COVID patient) Recruitment: All admitted cases between January 23 and April 25 2020 Prospective or retrospective: Retrospective Sample size: 74 (16) Further detail: No more details available |
||
| Patient characteristics and setting | Setting: Hospital inpatient Location: National Taiwan University Hospital Country: Taiwan Dates: January 23rd to April 25, 2020 Symptoms and severity: 12/16 (75%) with lower respiratory tract symptoms 10/16 (63%) with upper airway symptoms 8/16 (50%) with body temperature > 38 C 5/16 (31%) with headache or myalgia 3/16 (19%) with gastrointestinal symptoms 3/16 (19%) required intensive care, 1/16 (6%) of which received extracorporeal membrane oxygenation support Demographics: Age: mean 45.6 years, SD 15.5; sex: 9/16 (56%) male) Exposure history: Unclear Non‐Covid group 1: Control group Source: January 23rd to April 25, 2020 Characteristics: Patients hospitalised with respiratory tract infection or fever but with two negative RT‐PCR results for SARS‐CoV‐2 |
||
| Index tests | Test name: [A] ALLTEST 2019‐nCoV IgG/IgM Rapid Test [B] Dynamiker 2019‐nCoV IgG/IgM Rapid Test [C] ASK COVID‐19 IgG/IgM Rapid Test [D] Wondfo SARS‐CoV‐2 Antibody Test Manufacturer: [A] Hangzhou ALLTEST Biotech Co., Ltd., China [B] Dynamiker Biotechnology (Tianjin) Co., Ltd., China [C] TONYAR Biotech Inc., Taiwan [D] Guangzhou Wondfo Biotech Co., Ltd., China Antibody: [A] IgG/IgM [B] IgG/IgM [C] IgG/IgM [D] Total antibody (Separate results were plotted for IgG and for IgM alone, however insufficient data were available to construct 2 x 2 tables) Antigen target: [A] Nucleocapsid [B] Nucleocapsid [C] Spike [D] Not described Evaluation setting: Designed POC, unclear use Test method: [A]‐[D] Lateral flow assays (no further details) Timing of samples: [1] Day 1‐14 post‐symptom onset: 46/99 (46%) Day 15‐21 post‐symptom onset: 23/99 (23%) > Day 21 post‐symptom onset: 30/99 (30%) [2] Day 1‐14 post‐symptom onset: 37/58 (64%) Day 15‐21 post‐symptom onset: 11/58 (19%) > Day 21 post‐symptom onset: 10/58 (17%) Samples used: Serum Test operator: Unclear Definition of test positivity: Considered as positive according to the manufacturers’ instructions Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: rRT‐PCR targeting envelope, nucleocapsid and RNA‐dependent RNA polymerase genes Samples used: Throat or lower respiratory specimens (OP, NP, sputum, gargling) Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No Definition of non‐COVID cases: rRT‐PCR targeting envelope, nucleocapsid and RNA‐dependent RNA polymerase genes (at least two negative results) Samples used: Throat or lower respiratory specimens Timing of reference standard: Unclear Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported Indeterminate results: None reported Unit of analysis: Samples; 99 samples from 16 patients, disaggregated by time period, but certainly multiple examples of multiple samples from the same patient within each time period. If multiple samples per day then only one sample used for rapid antibody testing |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published Source: Journal of Infection Author COI: The authors declared no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Wu 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wu 2020 [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Wu 2020 [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Xiang 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group study recruiting patients estimating sensitivity and specificity Design: PCR conducted for patients presenting with a history of travel to or residence in Wuhan or local endemic areas; [1] 85 RT‐PCR‐confirmed cases [2] 24 suspected cases with ≥ 2 negative RT‐PCR and none positive (and protocol is to retest RT‐PCR‐negatives every 1‐2 days) [3] 60 healthy blood donors (control group) (hospital staff) or from patients with other lung diseases in the same hospital (all PCR‐negative) Recruitment: NR Prospective or retrospective recruitment of cases: unclear Sample size (virus/COVID cases): 169 (109; data for 66 lab‐confirmed and 24 suspected cases extracted as D+ group) Inclusion and exclusion criteria: unclear |
||
| Patient characteristics and setting | Setting: hospital inpatients Location: Wuhan Country: China Dates: 19 January‐2 March 2020 Symptoms and severity: [1] severe 18/85 (21%) [2] 2/24 (8%) severe Sex: [1] female 54/85 (64%) [2] female 12/24 (50%) [3] 35/60 (58%) female Age: [1] median 51 (IQR 32‐65) [2] median 44 (IQR 36‐61) [3] median 34 (IQR 29‐51) Exposure history: NR | ||
| Index tests | Test name: Zhuhai Livzon SARS‐CoV‐2 ELISA Manufacturer: ELISA kits, Livzon Inc, Zhuhai, P.R.China, lot number of IgM: 20200308, IgG: 20200308 Ab targets: IgG IgM Antigens used: N‐protein Test method: ELISA Timing of samples: NR Samples used: serum Test operators: NR Definition of test positivity: unclear ‐ "The optical density of each well was determined by a microplate reader set to 450 nm within 30 min. The ratio of optical density to the cut‐off value (optical density of the blank well + 0.1) was reported as the Ab concentration. For detection of IgG, the dilution factor was changed (1:20) and the cut off value was modified (optical density of the blank well + 0.13)." Blinded to reference standard: no Threshold predefined: unclear | ||
| Target condition and reference standard(s) | Reference standard for cases: [1] RT‐PCR [2] Symptoms and PCR‐negative (no guideline cited but criteria clearly elaborated) Samples used: NP and/or OP swabs Timing of reference standard: NR Blinded to index test: yes Incorporated index test: no Reference standard for non‐cases: (no exposure or symptoms) and RT‐PCR‐negative | ||
| Flow and timing | Time interval between index and reference tests: NR Results presented by time period: no All participants received the same reference standard: Missing data: data per sample were provided for the 85 confirmed cases, however per participant data were available only for 66/85 confirmed cases plus 24/24 suspected cases (total number of cases reported = 90) Uninterpretable results: NR Indeterminate results: NR Unit of analysis: reported both samples and participants | ||
| Comparative | |||
| Notes | Funding: this work is funded by National Natural Science Foundation of China (No. 81973990, 91643101), and Science Foundation of Huazhong University of Science and Technology (No. 2020kfyXGYJ100). Publication status: published in journal Source: Infectious Disease Society of America Study author COI: declared that they have none | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Xiao 2020a.
| Study characteristics | |||
| Patient Sampling | Purpose: Single‐group study to estimate sensitivity for diagnosing active or prior infection
Design: Confirmed cases of COVID‐19 (n = 34) according to the diagnosis and treatment guideline for SARS‐CoV‐2 from Chinese National Health Committee (Version 5) and the interim guidance from Centers for Disease Control and Prevention
Recruitment method: not reported Sample size: 34 (34) |
||
| Patient characteristics and setting | Setting: Inpatients Location: Tongji Hospital, Wuhan Country: China Dates: 1‐29 February 2020; final follow‐up date 3 March 2020 Exposure history: NR Patient characteristics: 12 female, 22 male. Median age (review team estimated) 49 years (range 26‐87), 22 (65%) male |
||
| Index tests | Test name: Manufacturer: Shenzhen Yahuilong Biotechnology Co. Ltd. Antibody: IgM and IgG Antigen target: Not described Evaluation setting: laboratory test Test method: CLIA Timing of samples: Samples acquired ≥ 2 weeks after symptoms onset for 32/34 participants; and on day 2 and day 3 for remaining 2 participants Samples used: Plasma Test operator: not reported Definition of test positivity: not reported |
||
| Target condition and reference standard(s) | Reference standard: COVID‐19 according to diagnosis and treatment guideline for SARS‐CoV‐2 from Chinese National Health Committee (Version 5) Timing of reference standard: not described Blinded to index test: Not described Incorporated index test: |
||
| Flow and timing | Time interval between index and reference standard: Not described Timing: Not stated Missing data: None Uninterpretable results: None Indeterminate results: None |
||
| Comparative | |||
| Notes | Funding: No funding sources declared Author COI: No conflicts of interest declared Source: Pre‐proof paper accepted for publication (Journal of Infection) | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Xiao 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: screening and diagnosing asymptomatic carriers; comparing asymptomatic, pre‐symptomatic and symptomatic cases
Diagnosis of current or prior infection Design: Three‐group study to estimate sensitivity according to symptomatic status: (1) 23 asymptomatic cases, (2) 33 pre‐symptomatic cases, (3) 19 age‐matched symptomatic cases Recruitment: unclear Participants selected from consecutive series of 449 COVID‐19 patients observed at single hospital: a. 77 asymptomatic on admission. Excluded: 21 due to severe disease (n = 2), inpatients (n = 5) or having undetectable RNA and IgM (n = 14), leaving 56 discharged patients for inclusion: 1) 23 who remained asymptomatic and 2) 33 who became symptomatic after admission (pre‐symptomatic group) b. 372 symptomatic on admission; random sample of 19 age‐matched cases selected (group 3) Prospective or retrospective: retrospective analysis Sample size: 75 (75) |
||
| Patient characteristics and setting | Setting: hospital inpatients Location: Shenzhen Third People's Hospital Country: China Dates: January 23, 2020‐April 1, 2020 Symptoms and severity: Pre‐symptomatic* ‐ fever 11 (13%), cough 22 (67%), chest tightness 2 (6%); Symptomatic ‐ fever 13 (68%), cough 13 (68%), chest tightness 1 (5%) *2/77 asymptomatic on admission were excluded due to disease severity and 5/77 excluded as remained as inpatients Demographics: Asymptomatic: Age: median (IQR): 30 (41.8), gender, n (%): male 5 (21.7), female 18 (78.3); pre‐symptomatic: Age: median (IQR): 45 (30.5), gender, n (%): male 18 (54.6), female 15 (45.5); symptomatic: Age: median (IQR): 25 (36.0), gender, n (%): male 9 (47.4), female 10 (52.6) Exposure history: not clearly reported; asymptomatic on admission (n = 77) identified through active surveillance and contact tracing Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] Wantai Biological Pharmacy Enterprise CLIA Total‐Ab assay [B] Wantai Biological Pharmacy Enterprise SARS‐CoV‐2 IgG ELISA [C] Wantai Biological Pharmacy Enterprise SARS‐CoV‐2 IgM ELISA [D] Wantai Biological Pharmacy Enterprise ELISA IgA assay Manufacturer: [A] Wantai Biological Pharmacy Enterprise [B] Wantai Biological Pharmacy Enterprise [C] Wantai Biological Pharmacy Enterprise [D] Wantai Biological Pharmacy Enterprise Antibody: [A] total antibody (Ab), [B] IgG, [C] IgM, and [D] IgA Antigen target: [A] RBD [B] [C] [D] S based Evaluation setting: laboratory test Test method: [A] CLIA [B] [C] [D] ELISA Timing of samples: day 0 to 65 post‐symptom onset (or post‐admission for asymptomatic group). Number of patients with samples obtained per week varied (total (asymptomatic/pre‐symptomatic/symptomatic)): day 1‐7 48/75 (17/23/8); day 8‐14 38/75 (9/17/22); day 15‐30 48/75 (10/21/19); day 31‐65 64/75 (17/28/19) Samples used: plasma Test operator: not reported Definition of test positivity: The relative fluorescence of sample to control (COI) was used to estimate the result. Positive: COI > 1 Blinding reported: Not stated Threshold predefined: not reported; presumably as per manufacturer instructions |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (GeneoDX Co., Ltd., Shanghai, China on an ABI 7500 thermo cycler) or antibody tests for SARS‐CoV‐2 (not described), as per Chinese NHC guidelines (version 6)
RT‐PCR‐positive if Ct < 40.0, and negative if the viral load was undetectable. Samples with Ct > 37 were retested. Samples used: respiratory specimens for COVID‐19 confirmation; anal swabs also obtained (appeared from Figure that fewer anal swabs obtained compared to respiratory) Timing of reference standard: not reported; appeared to be on admission for majority (64/75) and repeated over time. Tabl 1 reported no obvious difference in the calculated initial Ct value of NP samples between groups: mean (SD) 29.9 (4.8) (n = 19 asymptomatic; 29.1 (6.8) (n = 30 pre‐symptomatic); 29.2 (5.7) (n = 15 symptomatic) Blinded to index test: yes, as only confirmed cases were included Incorporated index test: No Definition of non‐COVID cases: NA Samples used: Timing of reference standard: Blinded to index test: Incorporated index test: |
||
| Flow and timing | Time interval between index and reference tests: not reported, but ref standard was performed before index test as only COVID‐19 confirmed cases were included All patients received same reference standard: not reported, but unlikely, as it was reported that RT‐PCR tests or antibody tests for SARS‐CoV‐2 used to confirm diagnosis Missing data: Unclear; data were not reported for all participants per week since onset and only 32/33 pre‐symptomatic were reported in supplementary tables Uninterpretable results: none reported Indeterminate results: none reported Unit of analysis: Patients; multiple samples obtained per participant (total 324; 77 asymptomatic, 142 pre‐symptomatic, 105 symptomatic), however data per week and overall were reported on a per patient basis (not every patient contributed samples to each week of data post‐onset) |
||
| Comparative | |||
| Notes | Funding: This work was supported by Shenzhen Bay Laboratory Open Fund (SZBL202002271001). Publication status: Pre‐print Source: medRxiv Author COI: Authors declared no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Unclear | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Xiao 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Xiao 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Xiao 2020b [D].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Yang 2020 [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of acute and convalescent‐phase infection Design: Multi‐group study to estimate sensitivity and specificity: [1] patients presenting to ED and displaying signs and symptoms suspicious for COVID 19 (n = 87, only 42 PCR‐confirmed cases providing 120 samples could be included in the review) [2] Pre‐pandemic ED patients (sample number = 320; unclear patient number) [3] Pre‐pandemic healthy blood donors (n = 256) Groups [2] and [3] used for different assays Additional cohorts reported but not extracted for the purposes of this review: [4] convalescent patients who were PCR‐positive or had Covid‐19‐like illness but were not tested, and had been symptom‐free for at least 14 days (n = 145) [5] Cross‐reactivity panel, including: patients treated for recent non‐Covid‐19 respiratory infections (n = 30); patients with antibodies to known microbial agents or with autoantigens (n = 78); patients who tested positive for one of the respiratory viruses in the Respiratory Pathogen PCR Panel (n = 16) Recruitment: Unclear Prospective or retrospective: Retrospective Sample size: 696 (120) Further detail: Not further described |
||
| Patient characteristics and setting | Setting: Accident and Emergency; hospital inpatient Location: Wells Cornell Medicine, New York Country: United States Dates: 6th March to 4th April 2020 Symptoms and severity: 14/42 (33%) discharged from ED 28/42 (67%) inpatients 23/42 (55%) required ICU care 24/42 (57%) required intubation Demographics: age: mean 56.5 years, SD 16.0; sex: 33/42 male (79%) Exposure history: Not stated Non‐Covid group 1: [2] Pre‐pandemic ED patients Source: [2] Pre‐pandemic (July 2019) Characteristics: [2] Not stated Non‐Covid group 2: [3] Pre‐pandemic healthy blood donors Source: [3] Pre‐pandemic (before 2019) Characteristics: [3] Not stated |
||
| Index tests | Test name: [A] Pylon COVID‐19 IgM and IgG assays; [B] New York SARS‐CoV‐2 MIA Manufacturer: [A] ET Healthcare, Palo Alto, CA, USA [B] Luminex Corporation, Austin, TX, USA (uses recombinant antigen produced at the Wadsworth Center/NYSDOH coupled with a cDNA copy of the N gene of SARS‐CoV; coupling carried out using a purchased kit from Luminex) Antibody: [A] IgG, IgM, IgG or and IgM [B] Total antibody Antigen target: [A] S‐receptor binding domain and recombinant nucleocapsid protein [B] recombinant nucleocapsid protein Evaluation setting: Laboratory Test method: [A] cyclic enhanced fluorescence assay (CEFA) [B] microsphere immunoassay (MIA) Timing of samples: 0 to > 32 days pso, of the 120 samples from 42 PCR+ cases: 8, 7% day 0‐3 33, 28% day 4‐7 42, 35% day 8‐14 15, 13% day 15‐20 21, 18%, day 21‐32 1, 0.8% day > 32 Samples used: Serum Test operator: Not stated Definition of test positivity: [A] Samples with an index value ≥ 1 were designated as positive [B] Samples with an index value ≥ 1.78 were designated as positive Blinding reported: Unclear Threshold predefined: Yes |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (RealStar SARS CoV‐2 RT‐PCR kit 1.0; Altona Diagnostics USA, Inc) Samples used: Nasopharyngeal swabs Timing of reference standard: Unclear; on presentation at ED Blinded to index test: Unclear; probably yes Incorporated index test: No Definition of non‐COVID cases: [2] and [3] Pre‐pandemic controls [4] [5] unclear [6] PCR+ for other infection Samples used: NA Timing of reference standard: NR Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: Yes, MIA results reported for only 114/120 samples from PCR+ cases; no a‐b data for 45 PCR‐ COVID suspects Uninterpretable results: NR Indeterminate results: NR Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Unclear Publication status: Published paper Source: Clinica Chimica Acta Author COI: ZZ received seed instruments and sponsored travel from ET Healthcare. The manufacturers did not review the article and had no input on data analysis prior to the manuscript submission. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Yang 2020 [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Yongchen 2020.
| Study characteristics | |||
| Patient Sampling | Purpose: one‐group study recruiting patients estimating sensitivity
Design: [1] 11 non‐severe COVID‐19 patients [2] 5 severe COVID‐19 patients [3] 5 asymptomatic carriers Recruitment: retrospective Sample size (virus/COVID cases): 21 (21) Inclusion and exclusion criteria: no more details available |
||
| Patient characteristics and setting | Setting: Hospital Location: 2 medical centres ‐ Second Hospital of Nanjing and the Affiliated Hospital of Xuzhou Medical University in Jiangsu Province Country: China Dates: 25 January‐18 March 2020 Symptoms and severity: 5 severe, 11 non‐severe and 5 asymptomatic cases. Asymptomatic carriers were defined as individuals who were positive for COVID‐19 nucleic acid but without any symptoms during screening of close contacts. Sex: 13/21 (62%) male; age: median (range) = 37 (10‐73) Exposure history: NR | ||
| Index tests | Test name: no commercial name stated Manufacturer: Innovita Co., Ltd, China Ab targets: IgG and IgM Antigens used: SARS‐CoV‐2 S‐protein and N‐protein Test method: GICA Timing of samples: IPD presented in Fig 1; 1 sample included per patient per time slot Samples used: serum Test operators: NR Definition of test positivity: NR Blinded to reference standard: NR and no assumptions made based on timing of the test Threshold predefined: NR | ||
| Target condition and reference standard(s) | Reference standard for cases: RT‐PCR ‐ confirmed after 2 sequential positive respiratory tract sample results Samples used: throat swabs Timing of reference standard: throat swab samples collected every 1‐2 days Blinded to index test: yes (serum samples for serological evaluation were stored for later evaluation) Incorporated index test: no Reference standard for non‐cases: NA | ||
| Flow and timing | Time interval between index and reference tests: NR
Results presented by time period: yes All participants received the same reference standard: yes Missing data: NR Uninterpretable results: NR Indeterminate results: NR Unit of analysis: participant |
||
| Comparative | |||
| Notes | Funding: supported by the National Natural Science Foundation of China, Jiangsu Provincial Medical Talent, Six talent peaks project of Jiangsu Province, Advanced health talent of six‐one project of Jiangsu Province, Nanjing Medical Science and Technique Development Foundation Publication status: published paper Source: Emerging Microbes & Infections Study author COI: none was declared | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Zhang 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Diagnosis of current acute and convalescent‐phase infection Design: Multi‐group study to assess sensitivity and specificity [1] Covid cases (572 samples) [1a] confirmed hospitalised cases (338 samples from 164 patients) [1b] Follow‐up cases (234 samples from 234 patients) [2] Non‐Covid cases (n = 996) [2a] Healthy controls (n = 600) [2b] With other diseases (n = 396) [3] Suspected COVID patients (162 samples from 154 patients) Recruitment: Samples obtained between December 2019 and March 2020 from Wuhan Recruitment method not stated. Prospective or retrospective: Retrospective Sample size: 1730 (574) samples Further detail: [1] Inclusion: Hospitalised clinically confirmed Covid patients. Exclusion: Not stated [2a] Inclusion: Healthy. Exclusion: Not stated [2b] Inclusion: Other diseases, respiratory disease (n = 57), orthopaedic disease (n = 8), hepatobiliary disease (n = 48), gynaecological disease (n = 50), auto‐immune disease (n = 10), endocrine disease (n = 41), dermal disease (n = 18), nervous system disease (n = 13), kidney disease (n = 32), digestive disease (n = 64), cardiovascular disease (n = 24), blood disease (n = 21), other disease (n = 10). Exclusion: Not stated. [3] Suspected COVID cases, close contact with COVID patients |
||
| Patient characteristics and setting | Setting: [1a] Hospitalised [1b] Outpatients/community (follow‐up patients) [3] close contacts with COVID patients (screening) Location: Wuhan Huoshenshan Hospital, Wuhan, First Hospital of Changsha, Changsha, and Chinese PLA General Hospital, Beijing Country: China Dates: December 2019 to March 2020 Symptoms and severity: [1a] Ordinary cases (n = 141), severe cases (n = 23) based on the diagnosis and treatment of novel coronavirus pneumonia (trial version 6) [1b] Not stated [3] 153/154 asymptomatic (no fever, no abnormalities in CT image) 1/154 first asymptomatic; later developed fever Demographics: [1a] Male (n = 92), female (n = 72), age range 25‐91 years, median age 62 years [1b] male (n = 115), female (n = 119), age range 1‐84 years, median age 49 years [3] Not stated Exposure history: [1] Not stated [3] Close contacts of confirmed COVID patients. Non‐Covid group 1: [2a] Healthy controls Source: [2a] December 2019 to March 2020, Wuhan Huoshenshan Hospital, First Hospital of Changsha and Chinese PLA General Hospital? Characteristics: Healthy, male, n = 313, female, n = 287; age range: 9–74, median age: 45 years Non‐Covid group 2: [2b] With other diseases Source: December 2019 to March 2020, Wuhan Huoshenshan Hospital, First Hospital of Changsha and Chinese PLA General Hospital? Characteristics: male, n = 185, female, n = 211; age range: 1–94, median age: 50 years, respiratory disease (n = 57), orthopaedic disease (n = 8), hepatobiliary disease (n = 48), gynaecological disease (n = 50), auto‐immune disease (n = 10), endocrine diseases (n = 41), dermal disease (n = 18), nervous system diseases (n = 13), kidney disease (n = 32), digestive disease (n = 64), cardiovascular disease (n = 24), blood diseases (n = 21), other diseases (neonatal diseases, oral diseases) (n = 10) |
||
| Index tests | Test name: [A] 2019‐nCoV IgM Antibody Determination Kit [B] 2019‐nCoV IgG Antibody Determination Kit Manufacturer: [A] [B] Beijing Diagreat Biotechnology Co., Ltd., Beijing, People’s Republic of China Antibody: [A] IgM [B] IgG Antigen target: [A] [B] S1 and N‐protein Evaluation setting: POCT, unclear setting Test method: [A] [B] Fluorescence‐based lateral flow assay Timing of samples: [1] 0‐70 days of onset of fever [1a] < 15 days pso: n = 9 15‐21 days pso (n = 38) > 21 days pso (n = 291) [1b] > 21 days pso: n = 234 [3] Asymptomatic Samples used: whole blood Test operator: Lab staff Definition of test positivity: The 95% percentile of the T/C ratio (the ratio between the fluorescence intensity in test area [T] and the fluorescence intensity in control area [C] on test strip card) was defined as 1 U/L, and this was set as the cut‐off value. Blinding reported: Not stated Threshold predefined: No, in the present primary experiment, 200 samples obtained from healthy controls were detected to determine the cut‐off value. |
||
| Target condition and reference standard(s) | Reference standard: [1] Clinically defined, criteria not described Possibly RNA test and CT image [3] RNA test and characteristic CT image Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: [1] Yes, prior [3] Not stated Incorporated index test: No Definition of non‐COVID cases: [2] Not stated, none Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: [2] yes, prior [3] Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated All patients received same reference standard: No Missing data: yes (9 samples collected in first two weeks not included in review) Uninterpretable results: Not stated Indeterminate results: Not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: This work was supported by the Beijing Science and Technology Planning Project. Publication status: Published paper Source: Emerging Microbes and Infections Author COI: XXL and ZJP are employees of Beijing Diagreat Biotechnology, the commercial manufacturer of the test strips. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | No | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Zhang 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Zhang 2020b [A].
| Study characteristics | |||
| Patient Sampling | Purpose: To investigate the potential relationships between immune antibodies and disease progression
Diagnosis of acute and convalescent‐phase infection Design: Single‐group study to estimate sensitivity only: [1] COVID‐19 patients (all RT‐PCR‐positive) Recruitment: Not stated Prospective or retrospective: retrospective Sample size: 112 (112) |
||
| Patient characteristics and setting | Setting: Department of Neurology, Inpatient (all admitted) Location: Renmin Hospital of Wuhan University, Hubei Country: China Dates: February 1 to February 29, 2020 Symptoms and severity: Severity: all described as mild, none sent to ICU Symptoms: 10 (8.9%) asymptomatic; 61 (54%) fever, 52 (46%) cough, 29 (26%) fatigue, 15 (13%) pharyngeal pain, (< 10%) diarrhoea, vomiting, myalgia, headache, and eye discomfort Demographics: 33 (29.5%) male; median age 38.6 SD 14.9) y, range 25‐78 y Exposure history: Not stated Non‐Covid group 1: NA |
||
| Index tests | Test name: [A] YHLO SARS‐CoV‐2 iFlash IgM assay [B] YHLO SARS‐CoV‐2 iFlash IgG assay [C] YHLO SARS‐CoV‐2 iFlash IgG/IgM assay Manufacturer: [A] [B] [C] Shenzhen YHLO (Yahuilong Biotechnology, Shenzhen, China) Antibody: [A] IgM, [B] IgG, [C] IgG and IgM Antigen target: [A] [B] [C] N and S based Evaluation setting: laboratory Test method: [A] [B] [C] CLIA Timing of samples: range < 10 to 49 d post‐symptom onset; data presented by time period Serological antibody tests were performed at different times post–disease onset: < 10 days, 10‐20 days, 20‐30 days, 30‐40 days, 40‐50 days Samples used: Not stated; presume serum from introduction/discussion Test operator: Not stated Definition of test positivity: > 10 AU/mL was considered as positive. Blinding reported: Not stated Threshold predefined: Yes (as per manufacturer instructions) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (SARS‐CoV‐2 open reading frame 1ab (ORF1ab)/nucleocapsid protein (N) gene); BioGerm, Shanghai, China Samples used: Nasopharynx and oropharynx swabs Timing of reference standard: Not stated Blinded to index test: Unclear Incorporated index test: No Definition of non‐COVID cases: NA |
||
| Flow and timing | Time interval between index and reference tests: unclear All patients received same reference standard: yes Missing data: Unclear Uninterpretable results: None reported Indeterminate results: no Unit of analysis: patients |
||
| Comparative | |||
| Notes | Funding: This work was supported by grants from National Natural Science Foundation of China (No.81822016 and 81771382) to Z. Zhang. Publication status: Accepted manuscript (NB Journal of Infectious Diseases® 2020;XX:1–6 as major article) Source: Journal of Infectious Diseases Author COI: The authors declared no conflict of interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Zhang 2020b [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Zhang 2020b [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Zhao 2020a [A].
| Study characteristics | |||
| Patient Sampling | Purpose: Two‐group design estimating sensitivity and specificity in acute‐phase sera
Design: [1] Confirmed COVID‐19 cases (n = 173) with positive RT‐PCR testing for COVID‐19 (providing 535 plasma samples) [2] Controls ‐ pre‐pandemic healthy individuals (n = 213) Recruitment: Not stated Sample size: 386 (173) |
||
| Patient characteristics and setting | Setting: Hospital Location: Shenzhen Third People’s Hospital Country: China Dates: 11 January to 9 February, 2020 Symptoms and severity: 32/173 (18%) considered critical Demographics: Median (IQR) age 48 (35‐61). 84/173 (49%) male Exposure history: 26/173 (73%) clear exposure identified Non‐Covid group 1: No information given |
||
| Index tests | Test name: IgM and IgG antibody detection kit Manufacturer: Beijing Wantai Antibody: [A] Total Ab, [B] IgM, [C] IgG Antigen target: [A] RBD [B] RBD [C] N‐protein Evaluation setting: laboratory Test method: All ELISA assays Timing of samples: Median 7 days pso [IQR 5 ‐ 10 d) Samples used: Plasma Definition of test positivity: Not stated Blinding reported: Not stated Threshold predefined: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Samples used: Respiratory tract samples Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No Definition of non‐COVID cases:Reference standard based on being pre‐pandemic samples |
||
| Flow and timing | Time interval between index and reference tests: Unclear All patients received same reference standard: No Missing data: Inadequate plasma samples for 2 IgM tests and 1 IgG test Uninterpretable results: None reported Indeterminate results: not stated Unit of analysis: Samples |
||
| Comparative | |||
| Notes | Funding: Supported by Bill & Melinda Gates Foundation Author COI: No conflicts of interest noted Publication status: Report from a preprint (not peer reviewed) | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (All tests) | |||
| DOMAIN 2: Index Test (Antibody tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | No | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| The reference standard does not incorporate the index test | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Zhao 2020a [B].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
Zhao 2020a [C].
| Study characteristics | |||
| Patient Sampling | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Patient characteristics and setting | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Index tests | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Target condition and reference standard(s) | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Flow and timing | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
| Comparative | |||
| Notes | See main entry for this study for characteristics and QUADAS‐2 assessment | ||
A&E: Accident and Emergency Department Ab: antibody ABEI: (4‐aminobutyl)‐N‐ethylisoluminol AdV: adenovirus AFI: acute febrile illness ANA: antinuclear antibody ARDS: acute respiratory distress syndrome ARI: acute respiratory infection ASLO: antistreptolysin O antibody AU: arbitrary unit BMI: body mass index CDC: Center for Disease Control CE: Conformité Européene CLIA: chemiluminescent immunoassay CMIA: chemiluminescent microparticle immunoassay CMV: cytomegalovirus CT: computed tomography CGIA: colloidal gold immunoassay CHIKV: chikungunya virus CLIA: chemiluminescence immunoassay COI: conflict of interest CRU: cardiorespiratory unit CU: chemiluminescent units D‐: disease negative D+: disease positive DABA: double antigen binding assay DENV: dengue virus DNA: deoxyribonucleic acid DRE: digital rectal exam E: envelope EBV: Epstein‐Barr virus ECDC: European Centre for Disease Prevention and Control ECLIA: electrochemiluminescent immunoassay ECMO: Extracorporeal membrane oxygenation ED: emergency department DTA: ethylenediamine tetraacetic acid EIA: enzyme immunoassay ELFA: enzyme‐linked fluorescent assay ELISA: enzyme‐linked immunosorbent assay ER: emergency room EUA: emergency use authorisation Flu: fluorescence intensity FMN: flavin mononucleotide GGO: ground‐glass opacity GI: gastrointestinal GICA: gold immunochromatography assay GP: general practitioner H: hour HAMA: human anti‐mouse antibodies HAV: hepatitis A virus HBV: hepatitis B virus HBcAb: hepatitis B core antibody HCV: hepatitis C virus HCW: healthcare worker HepB: hepatitis B HEV: hepatitis E virus HIV: Human immunodeficiency virus HMPV: Human metapnuemovirus HS: Hidradenitis suppurativa HTLV: Human T‐lymphotropic virus IAV: influenza A virus IBV: Infectious bronchitis virus IC: intensive care ICU: intensive care unit ID: immunodiagnostics IgA: immunoglobulin A IgG: immunoglobulin G IgM: immunoglobulin M IFU: instructions for use IIFT: indirect Immunofluorescence test IQR: interquartile range LFA: lateral flow assay LFIA: lateral flow immunoassay LIPS: luciferase immunoprecipitation system LIS: laboratory information system LRTI: lower respiratory tract infection MCLIA: magnetic chemiluminescent immunoassay MERS: middle east respiratory syndrome MFI: multiplex fluorescent immunoassay MPV: mean platelet volume MuV: mumps virus MV: measles virus N‐protein: nucleocapsid protein NA: not applicable NAAT: nucleic acid amplification test NAT: nucleic acid testing NB: nota bene NC: negative control NHS: National Health Service NIH: National Institues of Health NIHR: National Institute for Health Research NP: nasopharyngeal NR: not reported NTU: NovaTec‐Units OD: optical density OP: oropharyngeal PBS: phosphate buffered saline PCR: polymerase chain reaction PHE: Public Health England PIV: parainfluenza PLHA: people living with HIV/AIDS P/N: positive/negative ratio POC: point‐of‐care POCT: point‐of‐care test PRNT: plaque reduction neutralization test pso: post‐symptom onset PUI: person under investigation QUADAS‐2: quality assessment tool for diagnostic accuracy studies 2 RBD: receptor binding domain RdRp: RNA‐dependent RNA polymerase RDT: rapid diagnostic test RF: rheumatoid factor RLU: relative light unit rN: recombinant RNA: ribonucleic acid rpm: revolutions per minute RPNA: reverse phase protein microarray RPP: respiratory pathogen panel ROC: receiver operating characteristic rS: recombinant spike RSVA/B: respiratory syncytial virus A/B RT‐PCR: reverse transcriptase polymerase chain reaction RT‐qPCR: reverse transcriptase quantitative polymerase chain reaction RuV: rubella virus RV: rhinovirus S1: spike 1 SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2 S/C: signal/calibrator S/CO: signal/cutoff SD: standard deviation SE: standard error S‐flow: flow‐cytometry based test SLE: systemic lupus erythematosus SP: spike‐protein TB: tuberculosis T/C: ratio between the fluorescence intensity in test area [T] and the fluorescence intensity in control area [C] on test strip card TDM: therapeutic drug monitoring Tg: thyroglobulin TMA: transcription‐mediated amplification TN: true negative TP: true positive TPHA: treponema pallidum haemagglutination TPO: thyroid peroxidase UTM: universal transport medium VE: Virotech units VIDRL: Victorian infectious diseases research laboratory VZV: varicella‐zoster virus WHO: World Health Organization WNV: west Nile virus YFV: yellow fever virus y: years ZIKV: Zika virus
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abravanel 2020 | Index test ‐ no eligible time split |
| Adams 2020b | Index test ‐ assays could not be identified |
| Alger 2020 | Population ‐ no data for sensitivity |
| Amanat 2020 | Accuracy data cannot be extracted |
| Amrun 2020 | Index test ‐ no eligible time split |
| Antoine‐Reid 2020 | Index test ‐ no eligible time split |
| Arumugam 2020 | Ineligible study design |
| Ayouba 2020 | Index test ‐ no eligible time split |
| Barallat 2020 | Study design ‐ not test accuracy |
| Baron 2020 | Accuracy data cannot be extracted |
| Batra 2020 | Index test ‐ no eligible time split |
| Becker 2020 | Index test ‐ inhouse assay |
| Bendavid 2020 | Index test ‐ cannot identify assay |
| Black 2020 | Index test ‐ no eligible time split |
| Bortz 2020 | Index test ‐ inhouse assay |
| Brandstetter 2020 | Index test ‐ no eligible time split |
| Brantley 2020 | Index test ‐ assay not identified |
| Brecher 2020 | Population ‐ specificity only |
| Bruni 2020 | Index test ‐ no eligible time split |
| Bryan 2020b | Index test ‐ no eligible time split |
| Buntinx 2020 | Index test ‐ no eligible time split |
| Burbelo 2020 | Index test ‐ inhouse assay |
| Byrnes 2020 | Index test ‐ inhouse assay |
| Cai 2020 | Index test ‐ inhouse assay |
| Cassaniti 2020 | Index test ‐ no eligible time split |
| Chatzidimitriou 2020 | Index test ‐ no eligible time split |
| Chen 2020a | Index test ‐ inhouse assay |
| Choe 2020 | Index test ‐ no eligible time split |
| Chughtai 2020 | Index test ‐ assay not identified |
| Colavita 2020 | Index test ‐ no eligible time split |
| Comar 2020 | Ineligible reference standard |
| Dahlke 2020 | Inadequate sample size |
| Das 2020 | Inadequate sample size |
| Demey 2020 | Inadequate sample size |
| Di Lorenzo 2020 | Index test ‐ no eligible time split |
| Dittadi 2020 | Index test ‐ no eligible time split |
| Dobaño 2020 | Index test ‐ inhouse assay |
| Dohla 2020 | Index test ‐ cannot identify assay |
| Du 2020 | Inadequate sample size |
| Edouard 2020 | Index test ‐ inhouse assay |
| Erikstrup 2020 | Index test ‐ no eligible time split |
| Espino 2020 | Index test ‐ no eligible time split |
| Fong 2020 | Index test ‐ inhouse assay |
| Freeman 2020 | Index test ‐ inhouse assay |
| Garcia‐Basteiro 2020 | Index test ‐ no eligible time split |
| Garcia Garmendia 2020 | Inadequate sample size |
| Grzelak 2020 | Index test ‐ inhouse assay |
| Guo 2020a | Index test ‐ inhouse assay |
| Guo 2020c | Ineligible population |
| Guthmiller 2020 | Index test ‐ inhouse assay |
| He 2020 | Index test ‐ no eligible time split |
| He 2020a | Index test ‐ no eligible time split |
| Hou 2020 | Ineligible study design ‐ not test accuracy |
| Huang 2020a | Accuracy data cannot be extracted |
| Huang 2020b | Index test ‐ inhouse assay |
| Huang 2020c | Accuracy data cannot be extracted |
| Hung 2020 | Index test ‐ no eligible time split |
| Imam 2020 | Accuracy data cannot be extracted |
| Infantino 2020 | Population pre‐selected ‐ all sero‐positive in week 1 |
| Jia 2020 | Index test ‐ no eligible time split |
| Jiang 2020b | Accuracy data cannot be extracted |
| Karp 2020 | Index test ‐ no eligible time split |
| Karp 2020a | Index test ‐ inhouse assay |
| Klumpp‐Thomas 2020 | Index test ‐ inhouse assay |
| Kruttgen 2020 | Index test ‐ no eligible time split |
| Kushemererwa 2020 | Index test ‐ assay not identified |
| Lahner 2020 | Index test ‐ no eligible time split |
| Lapuente 2020 | Index test ‐ no eligible time split |
| Lee 2020 | Inadequate sample size |
| Lei 2020 | Index test ‐ no eligible time split |
| Li 2020a | Index test ‐ no eligible time split |
| Li 2020b | Index test ‐ no eligible time split |
| Li 2020c | Index test ‐ no eligible time split |
| Li 2020d | Accuracy data cannot be extracted |
| Li 2020e | Index test ‐ no eligible time split |
| Lin 2020 | Index test ‐ no eligible time split |
| Linares 2020 | Paper withdrawn by authors; https://www.biorxiv.org/content/10.1101/2020.07.01.182618v2 |
| Lippi 2020 | Ineligible reference standard ‐ used EUROIMMUN ELISA |
| Liu 2020d | Index test ‐ cannot identify assay |
| Liu 2020e | Index test ‐ no eligible time split |
| Liu 2020f | Index test ‐ no eligible time split |
| Lopez de la Iglesias 2020 | Index test ‐ no eligible time split |
| Ma 2020a | Index test ‐ inhouse assay |
| McAndrews 2020 | Index test ‐ inhouse assay |
| Morley 2020 | Index test ‐ inhouse assay |
| Munitz 2020 | Index test ‐ inhouse assay |
| Mutnal 2020 | Inadequate sample size |
| Nath 2020 | Population ‐ specificity only |
| Nguyen 2020a | Inadequate sample size |
| Nie 2020 | Ineligible study design |
| Norman 2020 | Index test ‐ inhouse assay |
| Nuccetelli 2020 | Index test ‐ no eligible time split |
| Okba 2020a | Ineligible study design ‐ not intended as test accuracy |
| Olivares 2020 | Inadequate sample size |
| Ossareh 2020 | Ineligible study design ‐ not test accuracy |
| Ozturk 2020 | Inadequate sample size |
| Paradiso 2020a | Inadequate sample size |
| Paradiso 2020b | Accuracy data cannot be extracted |
| Patel 2020 | Ineligible reference standard ‐ no reference standard |
| Pellanda 2020 | Index test ‐ no eligible time split |
| Perkmann 2020 | Index test ‐ no eligible time split |
| Phan 2020 | Index test ‐ inhouse assay |
| Plebani 2020 | Index test ‐ no eligible time split |
| Prince 2020 | Ineligible reference standard ‐ serological consensus‐based |
| Qian 2020 | Index test ‐ no eligible time split |
| Qu 2020 | Index test ‐ no eligible time split |
| Rabets 2020a | Index test ‐ inhouse assay |
| Randad 2020 | Index test ‐ inhouse assay |
| Robledo Gomez 2020 | Index test ‐ no eligible time split |
| Rosado 2020 | Index test ‐ inhouse assay |
| Rosendal 2020 | Inadequate sample size |
| Rushworth 2020 | Index test ‐ inhouse assay |
| Santos 2020 | Index test ‐ no eligible time split |
| Serrano 2020 | Index test ‐ no eligible time split |
| Shaw 2020 | Index test ‐ inhouse assay |
| Solodky 2020 | Index test ‐ no eligible time split |
| Song 2020 | Inadequate sample size |
| Spicuzza 2020 | Index test ‐ no eligible time split |
| Staines 2020 | Accuracy data cannot be extracted |
| Steiner 2020 | Index test ‐ inhouse assay |
| Strömer 2020 | Index test ‐ no eligible time split |
| Sun 2020a | Accuracy data cannot be extracted |
| Tan 2020 | Index test ‐ no eligible time split |
| Tan 2020a | Ineligible study design |
| Teng 2020 | Population ‐ specificity only |
| Thevis 2020 | Index test ‐ no eligible time split |
| To 2020a | Index test ‐ inhouse assay |
| Tre‐Hardy 2020 | Inadequate sample size |
| Valenti 2020 | Inadequate sample size |
| Van Praet 2021 | Inadequate sample size |
| Varadhachary 2020 | Index test ‐ no eligible time split |
| Vasarhelyi 2020 | Index test ‐ no eligible time split |
| Vidal‐Anzardo 2020 | Inadequate sample size |
| Villarreal 2020 | Ineligible study design ‐ development phase; may be commercial assay |
| Wajnberg 2020 | Inadequate reference standard |
| Wan 2020 | Index test ‐ no eligible time split |
| Wang 2020b | Inadequate sample size |
| Wang 2020c | Accuracy data cannot be extracted |
| Wang 2020d | Accuracy data cannot be extracted |
| Wang 2020e | Accuracy data cannot be extracted |
| Wechselberger 2020 | Ineligible reference standard ‐ composite based on 5 commercially available assays |
| Weiss 2020 | Accuracy data cannot be extracted |
| Wen 2020 | Index test ‐ inhouse assay |
| Wheeler 2020 | Inadequate sample size |
| Woelfel 2020 | Ineligible reference standard |
| Wu 2020a | Accuracy data cannot be extracted |
| Xiang 2020b | Index test ‐ no eligible time split |
| Xie 2020a | Accuracy data cannot be extracted |
| Xu 2020a | Index test ‐ inhouse assay |
| Xu 2020b | Index test ‐ no eligible time split |
| Xu 2020c | Inadequate sample size |
| Xue 2020 | Inadequate sample size |
| Yamaoka 2021 | Index test ‐ inhouse assay |
| Yan 2021 | Accuracy data cannot be extracted |
| Yildirim 2020 | Ineligible population ‐ all seropositive |
| Yokoyama 2020 | Index test ‐ no eligible time split |
| Yu 2020 | Index test ‐ inhouse assay |
| Yue 2020 | Index test ‐ no eligible time split |
| Zeng 2020a | Ineligible stdy design |
| Zeng 2020b | Accuracy data cannot be extracted |
| Zhang 2020c | Index test ‐ inhouse assay |
| Zhang 2020d | Index test ‐ inhouse assay |
| Zhang 2020e | Index test ‐ no eligible time split |
| Zhang 2020f | Inadequate sample size |
| Zhao 2020b | Index test ‐ inhouse assay |
| Zhong 2020 | Index test ‐ inhouse assay |
| Zhou 2020 | Index test ‐ no eligible time split |
ELISA: enzyme‐linked immunoabsorbent assay
Differences between protocol and review
We planned to check the following websites for eligible index tests, however, these did not prove to be very accessible or easy to use and, after initial review, were not further considered:
National Institute for Health Research (NIHR) Innovation Observatory (www.io.nihr.ac.uk/)
Meta‐evidence (meta-evidence.co.uk/the-role-of-evidence-synthesis-in-covid19/)
QUADAS‐2 (Whiting 2011), item "Was there an appropriate interval between index test(s) and reference standard?" was dropped from assessment because, for antibody tests, the body's immune response to SARS‐CoV‐2 infection tends to increase over time such that the time between confirmation of the presence of SARS‐CoV‐2 and the index test is less relevant than the time from symptom onset to the application of the index test.
We intended for two authors to independently perform data extraction, however, one review author extracted study characteristics, and a second author checked them. Contingency table data were extracted independently by two review authors as planned.
We did not undertake planned sensitivity analyses because we did not include any unpublished studies, company documents, and no study used spiked samples.
Differences between the original review and this review update
As the evidence base evolves over the course of the pandemic, we have made some adjustments to our original approach with the following changes between earlier versions of the review and this update:
Review inclusion criteria amended to only include studies evaluating commercially developed tests and to only include studies reporting sensitivity in predefined time periods.
Search sources included in the protocol and the previous version of this review, the Cochrane COVID‐19 Study Register and the CDC Database of COVID‐19 Research Articles, were not included in this version as the single source from the University of Bern living search database did not involve manual effort to de‐duplicate and, therefore, proved more efficient to process. The exceptionally large numbers of COVID‐19 studies available only as preprints also contributed to this decision as preprints were not covered by the Cochrane COVID‐19 Study Register at that time.
We checked for published versions of studies identified only as preprints in the electronic searches such that some studies have study IDs reflecting a 2021 publication date, despite the study having been identified prior to the 30 September 2020 search cut‐off.
We increased the minimum number of samples or participants required for a study to be included to 25. In the previous version of this review we excluded studies with fewer than 10 samples or participants.
We made further efforts to separate studies that evaluated the test in patients who were symptomatic (with active infection) from those who had recovered from their symptoms (convalescent), however, differences in reporting between studies (some by week after onset of symptoms and some in longer time periods) meant that we were still not able to fully separate these groups. Our stratification of results according to time since onset of symptoms will better reflect these categorisations compared to the previous review iteration, however.
We did not conduct planned heterogeneity investigations by reference standard for COVID‐19 cases because the majority used RT‐PCR alone.
We investigated differences in reference standards used for non‐COVID‐19 cases and time after onset of symptoms as part of the primary analyses.
Contributions of authors
JDi was the contact person with the editorial base. TF and JDi co‐ordinated contributions from the co‐authors and wrote the final draft of the review. JDi, CD, YT, JJD, JG, STP, GS, JB screened papers against eligibility criteria. RS conducted the literature searches TF, JG, JDi, JB, GS, DH, YM, PW, DW, BB, HB, KP, YS, JJD, YT, CD, STP appraised the quality of papers. TF, JG, JDi, JB, GS, DH, YM, PW, DW, BB, HB, KP, YS, JJD, YT, CD, STP extracted data for the review
TF, JG, JDi sought additional information about papers from study authors. TF and JDi entered data into Review Manager 5.4.1. KS, TF and JDi analysed and interpreted data. TF, JDi, JJD, YT, CD, STP, RS, ML, MM, LH, AVB, DE, SD, JC, JV worked on the methods sections.
All authors reviewed, edited, contributed to, and approved this review. JDi is the guarantor of the update.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK
University of Birmingham, UK
External sources
-
Foreign, Commonwealth and Development Office (FCDO), UK
Project number: 300342‐104
National Institute for Health Research (NIHR), UK
NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham, UK
Declarations of interest
Tilly Fox: none known
Julia Geppert: none known
Jacqueline Dinnes: FIND (grant/contract); Cochrane DTA editor
Katie Scandrett: none known
Jacob Bigio: none known
Giorgia Sulis: none known
Dineshani Hettiarachchi: none known
Yasith Mathangasinghe: none known
Praveen Weeratunga: none known
Dakshitha Wickramasinghe: none known
Hanna Bergman: none known
Brian S Buckley: none known
Katrin Probyn: none known
Yanina Sguassero: Cochrane Response (employment); editor assistant of CDPLPG and Cochrane Clinical Answers.
Jane Cunningham: no relevant interests; affiliated to WHO, which produces guidance on use of SARS‐CoV‐2 rapid tests.
Sabine Dittrich: FIND (employment), the global alliance for diagnostic.
Devy Emperador: no relevant interests; employed by FIND with funding from FCDO and KFW. FIND is a global non‐for profit product development partnership and WHO Diagnostic Collaboration Centre. It is FIND’s role to accelerate access to high quality diagnostic tools for low‐resource settings and this is achieved by supporting both R&D and access activities for a wide range of diseases, including COVID‐19. FIND has several clinical research projects to evaluate multiple new diagnostic tests against published Target Product Profiles that have been defined through consensus processes. These studies are for diagnostic products developed by private sector companies who provide access to know how, equipment/reagents, and contribute through unrestricted donations as per FIND policy and external SAC review.
Lotty Hooft: no relevant interests; DTA Editorial Team; PMG implementation team.
Mariska MG Leeflang: no relevant interests; Diagnostic Test Accuracy Editorial Team member.
Matthew DF McInnes: no relevant interests; works as a health professional at the Ottawa Hospital.
René Spijker: none known
Thomas Struyf: none known
Ann Van den Bruel: none known
Jan Verbakel: none known
Clare Davenport: no relevant interests; Contact Editor for the Cochrane DTA Editorial Team; not involved in the editorial process for this review update because of this conflict of interest.
Yemisi Takwoingi: no relevant interests; Member, Cochrane Editorial Board; Editor, Cochrane Infectious Diseases Group; Statistical Editor, Cochrane BJMT Group; Cochrane DTA Editor.
Sian Taylor‐Phillips: finddx (grant/contract) ‐ funding to Warwick University from Birmingham University to fund a staff member time (approx 6 months part time) working on this review. No funding to individuals (funds originally came from FIND diagnostics, a charity); National Institute for Health Research (grant/contract) ‐ NIHR Career Development Fellowship NIHR‐CDF‐2016‐018 for methods of evaluating screening tests. Money to institution (University of Warwick); involved with the EDSAB‐HOME study, Public Health England, as co‐author but did not receive any funds for participation.
Jonathan J Deeks: no relevant interests; Eight podcasts, including Talk Evidence (BMJ), More‐or‐Less (Radio 4), Inside Science (Radio 4), The Newscast (Radio 4). Five opinion pieces in Guardian, unHerd and the BMJ. Numerous television, radio and mainstream media interviews giving substantial coverage of scientific issues related to test evaluation for COVID‐19. Presented evidence to the House of Lords Select Committee, and the All Parliamentary Party Investigation on COVID‐19. Two invited editorials on COVID‐19 for the BMJ; Cochrane DTA editor.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Adams 2020 {published data only}
- Adams ER, Augustin Y, Byrne RL, Clark DJ, Cocozza M, Cubas-Atienzar AI, et al. Rapid development of COVID-19 rapid diagnostics for low resource settings: accelerating delivery through transparency, responsiveness, and open collaboration. medRxiv [Preprint] 2020. [DOI: ]
Alvim 2020 {published data only}
- Alvim RGF, Lima TM, Rodrigues DAS, Marsili FF, Bozza VBT, Higa LM, et al. An affordable anti-SARS-CoV-2 spike ELISA test for early detection of IgG seroconversion suited for large-scale surveillance studies in low-income countries. medRxiv [Preprint] 2020. [DOI: ]
Andrey 2020a [A] {published data only}
- Andrey DO, Cohen P, Meyer B, Torriani G, Yerly S, Mazza L, et al. Diagnostic accuracy of Augurix COVID-19 IgG serology rapid test. European Journal of Clinical Investigation 2020;50(10):e13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andrey 2020a [B] {published data only}
- See first entry for publication details for this study. Andrey 2020a [A].
Andrey 2020b [A] {published data only}
- Andrey DO, Cohen P, Meyer B, Torriani G, Yerly S, Mazza L, et al. Head-to-head accuracy comparison of three commercial COVID-19 IgM/IgG serology rapid tests. Journal of Clinical Medicine 2020;9(8):2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andrey 2020b [B] {published data only}
- See first entry for publication details for this study. Andrey 2020b [A].
Andrey 2020b [C] {published data only}
- See first entry for publication details for this study. Andrey 2020b [A].
Bartolini 2020 [A] {published data only}
- Bartolini A, Scapaticci M, Bioli M, Lazzarotto T, Re MC, Mancini R. Immunochromatographic assays for COVID-19 epidemiological screening: our experience. medRxiv [Preprint] 2020. [DOI: ]
Bartolini 2020 [B] {published data only}
- See first entry for publication details for this study. Bartolini 2020 [A].
Beavis 2020 {published data only}
- Beavis KG, Matushek SM, Abeleda APF, Bethel C, Hunt C, Gillen S, et al. Evaluation of the EUROIMMUN Anti-SARS-CoV-2 ELISA assay for detection of IgA and IgG antibodies. Journal of Clinical Virology 2020;129:104468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bernasconi 2020 {published data only}
- Bernasconi L, Oberle M, Gisler V, Ottiger C, Fankhauser H, Schuetz P, et al. Diagnostic performance of a SARS-CoV-2 IgG/IgM lateral flow immunochromatography assay in symptomatic patients presenting to the emergency department. Clinical Chemistry and Laboratory Medicine 2020;58(9):e159-61. [DOI] [PubMed] [Google Scholar]
Bettencourt 2020 {published data only}
- Bettencourt P, Fernandes C, Gil A, Almeida A, Alvelos M. Qualitative serology in patients recovered from SARS CoV-2 infection. Journal of Infection 2020;81(2):e120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bond 2020 {published data only}
- Bond K, Nicholson S, Lim S, Karapanagiotidis T, Williams E, Johnston D, et al. Evaluation of serological tests for SARS-CoV-2: Implications for serology testing in a low-prevalence setting. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Bond K, Nicholson S, Lim SM, Karapanagiotidis T, Williams E, Johnson D, et al. Evaluation of serological tests for SARS-CoV-2: implications for serology testing in a low-prevalence setting. Journal of Infectious Diseases 2020;222(8):1280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boukli 2020 [A] {published data only}
- Boukli N, Le Mene M, Schnuriger A, Cuervo NS, Laroche C, Morand-Joubert L, et al. High incidence of false-positive results in patients with acute infections other than COVID-19 by the Liaison SARS-CoV-2 commercial chemiluminescent microparticle immunoassay for detection of IgG Anti-SARS-CoV-2 antibodies. Journal of Clinical Microbiology 2020;58(11):e01352-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boulki 2020 [B] {published data only}
- See first entry for publication details for this study. Boukli 2020 [A].
Brochot 2020 [A] {published data only}
- Brochot E, Demey B, Handala L, Francois C, Duverlie G, Castelain S. Comparison of different serological assays for SARS-CoV-2 in real life. Journal of Clinical Virology 2020;130:104569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brochot 2020 [B] {published data only}
- See first entry for publication details for this study. Brochot 2020 [A].
Brochot 2020 [C] {published data only}
- See first entry for publication details for this study. Brochot 2020 [A].
Brochot 2020 [D] {published data only}
- See first entry for publication details for this study. Brochot 2020 [A].
Brochot 2020 [E] {published data only}
- See first entry for publication details for this study. Brochot 2020 [A].
Bryan 2020a {published data only}
- Bryan A, Fink SL, Gattuso MA, Pepper G, Chaudhary A, Wener MH, et al. Anti-SARS-CoV-2 IgG antibodies are associated with reduced viral load. medRxiv [Preprint] 2020. [DOI: ]
- Bryan A, Pepper G, Wener MH, Fink SL, Morishima C, Chaudhary A, et al. Performance characteristics of the Abbott Architect SARS-CoV-2 IgG assay and seroprevalence in Boise, Idaho. Journal of Clinical Microbiology 2020;58(8):941. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bundschuh 2020 {published data only}
- Bundschuh C, Egger M, Wiesinger K, Gabriel C, Clodi M, Mueller T, et al. Evaluation of the EDI enzyme linked immunosorbent assays for the detection of SARS-CoV-2 IgM and IgG antibodies in human plasma. Clinica Chimica Acta 2020;509:79-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Butterfield 2021 [A] {published data only}
- Butterfield TR, Bruce-Mowatt A, Phillips YZR, Brown N, Francis K, Brown J, et al. Assessment of commercial SARS-CoV-2 antibody assays, Jamaica. International Journal of Infectious Diseases 2021;105:333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield TR, Bruce-Mowatt A, Phillips YZR, Brown N, Francis K, Brown J, et al. Assessment of commercial SARS-CoV-2 antibody assays, Jamaica. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Butterfield 2021 [B] {published data only}
- See first entry for publication details for this study. Butterfield 2021 [A].
Butterfield 2021 [C] {published data only}
- See first entry for publication details for this study. Butterfield 2021 [A].
Butterfield 2021 [D] {published data only}
- See first entry for publication details for this study. Butterfield 2021 [A].
Butterfield 2021 [E] {published data only}
- See first entry for publication details for this study. Butterfield 2021 [A].
Butterfield 2021 [F] {published data only}
- See first entry for publication details for this study. Butterfield 2021 [A].
Candel 2020 {published data only}
- Candel FJ, Vinuela-Prieto JM, Gonzalez Del Castillo J, Barreiro Garcia P, Fragiel Saavedra M, Hernandez Piriz A, et al. Utility of lateral flow tests in SARS-CoV-2 infection monitorization. Revista Española de Quimioterapia 2020;33(4):258-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Carozzi 2020 [A] {published data only}
- Carozzi FM, Cusi MG, Pistello M, Galli L, Bartoloni A, Anichini G, et al. Detection of asymptomatic SARS-CoV-2 infections among healthcare workers: results from a large-scale screening program based on rapid serological testing. medRxiv [Preprint] 2020. [DOI: ]
Carozzi 2020 [B] {published data only}
- See first entry for publication details for this study. Carozzi 2020 [A].
Carta 2020 {published data only}
- Carta M, Bragagnolo L, Tramarin A, Barzon E, Cappelletti A, Pascarella M, et al. Anti SARS-CoV-2 antibodies monitoring in a group of residents in a long term care facility during COVID-19 pandemic peak. Diagnosis 2020;7(4):395-400. [DOI] [PubMed] [Google Scholar]
Case 2020 [A] {published data only}
- Case JB, Rothlauf PW, Chen RE, Liu Z, Zhao H, Kim AS, et al. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Case JB, Rothlauf PW, Chen RE, Liu Z, Zhao H, Kim AS, et al. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host & Microbe 2020;28(3):475-85.e5. [DOI: 10.1016/j.chom.2020.06.021] [DOI] [PMC free article] [PubMed] [Google Scholar]
Case 2020 [B] {published data only}
- See first entry for publication details for this study. Case 2020 [A].
Caturegli 2020 {published data only}
- Caturegli G, Materi J, Howard BM, Caturegli P. Clinical validity of serum antibodies to SARS-CoV-2: a case-control study. Annals of Internal Medicine 2020;173(8):614-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cervia 2020 {published data only}
- Cervia C, Nilsson J, Zurbuchen Y, Valaperti A, Schreiner J, Wolfensberger A, et al. Systemic and mucosal antibody secretion specific to SARS-CoV-2 during mild versus severe COVID-19. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Chan 2020a {published data only}
- Chan CW, Parker K, Tesic V, Baldwin A, Tang NY, Wijk XMR, et al. Analytical and clinical evaluation of the automated Elecsys anti-SARS-CoV-2 antibody assay on the Roche cobas e602 Analyzer. American Journal of Clinical Pathology 2020;154(5):620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Charlton 2020 [A] {published data only}
- Charlton CL, Kanji JN, Johal K, Bailey A, Plitt SS, MacDonald C, et al. Evaluation of six commercial mid- to high-volume antibody and six point-of-care lateral flow assays for detection of SARS-CoV-2 antibodies. Journal of Clinical Microbiology 2020;58(10):e01361-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Charlton 2020 [B] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [C] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [D] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [E] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [F] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [G] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [H] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [I] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [J] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [K] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charlton 2020 [L] {published data only}
- See first entry for publication details for this study. Charlton 2020 [A].
Charpentier 2020 [A] {published data only}
- Charpentier C, Ichou H, Damond F, Bouvet E, Chaix ML, Ferre V, et al. Performance evaluation of two SARS-CoV-2 IgG/IgM rapid tests (Covid-Presto and NG-Test) and one IgG automated immunoassay (Abbott). Journal of Clinical Virology 2020;132:104618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Charpentier 2020 [B] {published data only}
- See first entry for publication details for this study. Charpentier 2020 [A].
Charpentier 2020 [C] {published data only}
- See first entry for publication details for this study. Charpentier 2020 [A].
Chaudhuri 2020 [A] {published data only}
- Chaudhuri S, Thiruvengadam R, Chattopadhyay S, Mehdi F, Kshetrapal P, Shrivastava T, et al. Comparative evaluation of SARS-CoV-2 IgG assays in India. Journal of Clinical Virology 2020;131:104609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chaudhuri 2020 [B] {published data only}
- See first entry for publication details for this study. Chaudhuri 2020 [A].
Chen 2020 [A] {published data only}
- Chen SY, Lee YL, Lin YC, Lee NY, Liao CH, Hung YP, et al. Multicenter evaluation of two chemiluminescence and three lateral flow immunoassays for the diagnosis of COVID-19 and assessment of antibody dynamic responses to SARS-CoV-2 in Taiwan. Emerging Microbes & Infections 2020;9(1):2157-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020 [B] {published data only}
- See first entry for publication details for this study. Chen 2020 [A].
Chen 2020 [C] {published data only}
- See first entry for publication details for this study. Chen 2020 [A].
Chen 2020 [D] {published data only}
- See first entry for publication details for this study. Chen 2020 [A].
Chen 2020 [E] {published data only}
- See first entry for publication details for this study. Chen 2020 [A].
Chew 2020 {published data only}
- Chew KL, Tan SS, Saw S, Pajarillaga A, Zaine S, Khoo C, et al. Clinical evaluation of serological IgG antibody response on the Abbott Architect for established SARS-CoV-2 infection. Clinical Microbiology and Infection 2020;26(9):1256.e9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chong 2021 {published data only}
- Chong Y, Ikematsu H, Tani N, Arimizu Y, Watanabe H, Fukamachi Y, et al. Clinical significance of SARS-CoV-2-specific IgG detection with a rapid antibody kit for COVID-19 patients. Influenza and Other Respiratory Viruses 2021;15(1):13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Clarke 2020 {published data only}
- Clarke C, Prendecki M, Dhutia A, Ali MA, Sajjad H, Shivakumar O, et al. High prevalence of asymptomatic COVID-19 infection in hemodialysis patients detected using serologic screening. Journal of the American Society of Nephrology 2020;31(9):1969-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Conklin 2020 [A] {published data only}
- Conklin SE, Martin K, Manabe YC, Schmidt HA, Keruly M, Klock E, et al. Evaluation of serological SARS-CoV-2 lateral flow assays for rapid point of care testing. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Conklin 2020 [B] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [C] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [D] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [E] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [F] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [G] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [H] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [I] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [J] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [K] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [L] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [M] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [N] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [O] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Conklin 2020 [P] {published data only}
- See first entry for publication details for this study. Conklin 2020 [A].
Costa 2020 {published data only}
- Costa SF, Buss L, Espinoza EPS, Vieira JM Jr, Oliveira da Silva LC, Souza RM, et al. Performance of a qualitative rapid chromatographic immunoassay to diagnose COVID-19 in patients in a middle-income country. Journal of Clinical Virology 2020;131:104592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coste 2021 [A] {published data only}
- Coste AT, Jaton K, Papadimitriou-Olivgeris M, Greub G, Croxatto A. Comparison of SARS-CoV-2 serological tests with different antigen targets. Journal of Clinical Virology 2021;134:104690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste AT, Jaton K, Papadimitriou-Olivgeris M, Greub G, Croxatto A. Comparison of SARS-CoV-2 serological tests with different antigen targets. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Coste 2021 [B] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [C] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [D] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [E] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [F] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [G] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [H] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [I] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [J] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [K] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [L] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [M] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [N] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Coste 2021 [O] {published data only}
- See first entry for publication details for this study. Coste 2021 [A].
Criscuolo 2020 [A] {published data only}
- Criscuolo E, Diotti RA, Strollo M, Rolla S, Ambrosi A, Locatelli M, et al. Poor correlation between antibody titers and neutralizing activity in sera from SARS-CoV-2 infected subjects. medRxiv [Preprint] 2020. [https://doi.org/10.1101/2020.07.10.20150375] [DOI] [PMC free article] [PubMed]
Criscuolo 2020 [B] {published data only}
- See first entry for publication details for this study. Criscuolo 2020 [A].
Dave 2020 {published data only}
- Dave M, Poswal L, Bedi V, Regar L, Vijayvargiya R, Sharma M, et al. Study of antibody-based rapid card test in COVID-19 patients admitted in a tertiary care COVID hospital in Southern Rajasthan. Journal, Indian Academy of Clinical Medicine 2020;21(1-2):7-11. [Google Scholar]
Decru 2020 [A] {published data only}
- Decru B, Van Elslande J, Weemaes M, Houben E, Empsen I, Andre E, et al. Comparison of the diagnostic performance with whole blood and plasma of four rapid antibody tests for SARS-CoV-2. Clinical Chemistry and Laboratory Medicine 2020;58(10):e197-9. [DOI] [PubMed] [Google Scholar]
Decru 2020 [B] {published data only}
- See first entry for publication details for this study. Decru 2020 [A].
Decru 2020 [C] {published data only}
- See first entry for publication details for this study. Decru 2020 [A].
Decru 2020 [D] {published data only}
- See first entry for publication details for this study. Decru 2020 [A].
Delliere 2020 [A] {published data only}
- Delliere S, Salmona M, Minier M, Gabassi A, Alanio A, Le Goff J, et al. Evaluation of the COVID-19 IgG/IgM rapid test from Orient Gene Biotech. Journal of Clinical Microbiology 2020;58(8):e01233-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Delliere 2020 [B] {published data only}
- See first entry for publication details for this study. Delliere 2020 [A].
Doherty Institute 2020 [A] {published data only}
- Doherty Institute. Post-market validation of a further three serological assays for COVID-19 (10th Aug 2020). Report prepared for Office of Health Protection, Commonwealth Government of Australia; The Therapeutics Goods Administration (TGA) of Australia. https://www.health.gov.au/resources/publications/post-market-validation-of-a-further-three-serological-assays-for-covid-19-updated-report.
- Doherty Institute. Post-market validation of serological assays for COVID-19 - Updated Report (2 June 2020). Report prepared for: Office of Health Protection, Commonwealth Government of Australia; The Therapeutics Goods Administration (TGA) of Australia. https://www.health.gov.au/resources/publications/post-market-validation-of-serological-assays-for-covid-19-updated-report.
- Doherty Institute. Post-market validation of three serological assays for COVID-19 (29th April 2020). Report prepared for: Office of Health Protection, Commonwealth Government of Australia; The Therapeutics Goods Administration (TGA) of Australia. https://www.health.gov.au/resources/publications/post-market-validation-of-three-serological-assays-for-covid-19-final-report.
Doherty Institute 2020 [B] {published data only}
- See first entry for publication details for this study. Doherty Institute 2020 [A].
Doherty Institute 2020 [C] {published data only}
- See first entry for publication details for this study. Doherty Institute 2020 [A].
Doherty Institute 2020 [D] {published data only}
- See first entry for publication details for this study. Doherty Institute 2020 [A].
Doherty Institute 2020 [E] {published data only}
- See first entry for publication details for this study. Doherty Institute 2020 [A].
Doherty Institute 2020 [F] {published data only}
- See first entry for publication details for this study. Doherty Institute 2020 [A].
Doherty Institute 2020 [G] {published data only}
- See first entry for publication details for this study. Doherty Institute 2020 [A].
DomBourian 2020 [A] {published data only}
- DomBourian MG, Annen K, Huey L, Andersen G, Merkel PA, Jung S, et al. Analysis of COVID-19 convalescent plasma for SARS-CoV-2 IgG using two commercial immunoassays. Journal of Immunological Methods 2020;486:112837. [DOI] [PMC free article] [PubMed] [Google Scholar]
DomBourian 2020 [B] {published data only}
- See first entry for publication details for this study. DomBourian 2020 [A].
Dora 2020 {published data only}
- Dora AV, Winnett A, Fulcher JA, Sohn L, Calub F, Lee-Chang I, et al. Using serologic testing to assess the effectiveness of outbreak control efforts, serial PCR testing, and cohorting of positive SARS-CoV-2 patients in a skilled nursing facility. Clinical Infectious Diseases 2021;73:545-48. [DOI: 10.1093/cid/ciaa1286] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dortet 2020 {published data only}
- Dortet L, Emeraud C, Vauloup-Fellous C, Khecharem M, Ronat J-B, Fortineau N, et al. Rapid determination of SARS-CoV-2 antibodies using a bedside, point-of-care, serological test. Emerging Microbes & Infections 2020;9(1):2212-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dortet L, Emeraud C, Vauloup-Fellous C, Khecharem M, Ronat J-B, Fortineau N, et al. Rapid determination of SARS-CoV-2 antibodies using a bedside, point-of-care, serological test. SSRN [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Dortet 2021 [A] {published data only}
- Dortet L, Ronat JB, Vauloup-Fellous C, Langendorf C, Mendels DA, Emeraud C, et al. Evaluating 10 commercially available sars-cov-2 rapid serological tests by use of the STARD (Standards for reporting of diagnostic accuracy studies) method. Journal of Clinical Microbiology 2021;59(2):e02342-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dortet L, Ronat J-B, Vauloup-Fellous C, Langendorf C, Mendels D-A, Emeraud C, et al. Evaluating ten commercially-available SARS-CoV-2 rapid serological tests using the STARD (Standards for Reporting of Diagnostic Accuracy Studies) method. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Dortet 2021 [B] {published data only}
- See first entry for publication details for this study. Dortet 2021 [A].
Du 2021 {published data only}
- Du J, Chu E, Zhang D, Lu CM, Zhang A, Sha MY. A high-throughput Anti-SARS-CoV-2 IgG testing platform for COVID-19. Journal of Virological Methods 2021;287:114009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Chu E, Zhang D, Lu CM, Zhang A, Sha MY. A high-throughput Anti-SARS-CoV-2 IgG testing platform for COVID-19. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Egger 2020 [A] {published data only}
- Egger M, Bundschuh C, Wiesinger K, Gabriel C, Clodi M, Mueller T, et al. Comparison of the Elecsys(R) Anti-SARS-CoV-2 immunoassay with the EDI enzyme linked immunosorbent assays for the detection of SARS-CoV-2 antibodies in human plasma. Clinica Chimica Acta 2020;509:18-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 2020 [B] {published data only}
- See first entry for publication details for this study. Egger 2020 [A].
Fafi‐Kremer 2020 {published data only}
- Fafi-Kremer S, Bruel T, Madec Y, Grant R, Tondeur L, Grzelak L, et al. Serologic responses to SARS-CoV-2 infection among hospital staff with mild disease in eastern France. EBioMedicine 2020;59:102915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fafi-Kremer S, Bruel T, Madec Y, Grant R, Tondeur L, Grzelak L, et al. Serologic responses to SARS-CoV-2 infection among hospital staff with mild disease in eastern France. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Favresse 2020a {published data only}
- Favresse J, Eucher C, Elsen M, Tre-Hardy M, Dogne JM, Douxfils J. Clinical performance of the Elecsys electrochemiluminescent immunoassay for the detection of SARS-CoV-2 total antibodies. Clinical Chemistry 2020;66(8):1104-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Favresse 2020b {published data only}
- Favresse J, Eucher C, Elsen M, Laffineur K, Dogne JM, Douxfils J. Response of anti-SARS-CoV-2 total antibodies to nucleocapsid antigen in COVID-19 patients: a longitudinal study. Clinical Chemistry and Laboratory Medicine 2020;58(10):e193-6. [DOI] [PubMed] [Google Scholar]
Fenwick 2021 [A] {published data only}
- Fenwick C, Croxatto A, Coste AT, Pojer F, André C, Pellaton C, et al. Changes in SARS-CoV-2 spike versus nucleoprotein antibody responses impact the estimates of infections in population-based seroprevalence studies. Journal of Virology 2021;95(3):e01828-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick C, Croxatto A, Coste AT, Pojer F, André C, Pellaton C, et al. Qualitative changes in the SARS-CoV-2 antibody response in the post-infection phase impact the estimates of infections in population-based seroprevalence studies. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Fenwick 2021 [B] {published data only}
- See first entry for publication details for this study. Fenwick 2021 [A].
Fenwick 2021 [C] {published data only}
- See first entry for publication details for this study. Fenwick 2021 [A].
Fenwick 2021 [D] {published data only}
- See first entry for publication details for this study. Fenwick 2021 [A].
Fenwick 2021 [E] {published data only}
- See first entry for publication details for this study. Fenwick 2021 [A].
Flinck 2021 [A] {published data only}
- Flinck H, Rauhio A, Luukinen B, Lehtimaki T, Haapala AM, Seiskari T, et al. Comparison of 2 fully automated tests detecting antibodies against nucleocapsid N and spike S1/S2 proteins in COVID-19. Diagnostic Microbiology and Infectious Disease 2021;99(1):115197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flinck 2021 [B] {published data only}
- See first entry for publication details for this study. Flinck 2021 [A].
Flower 2020 [A] {published data only}
- Flower B, Brown JC, Simmons B, Moshe M, Frise R, Penn R, et al. Clinical and laboratory evaluation of SARS-CoV-2 lateral flow assays for use in a national COVID-19 seroprevalence survey. Thorax 2020;75(12):1082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flower 2020 [B] {published data only}
- Flower B, Brown JC, Simmons B, Moshe M, Frise R, Penn R, et al. Clinical and laboratory evaluation of SARS-CoV-2 lateral flow assays for use in a national COVID-19 seroprevalence survey. Thorax 2020;75(12):1082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flower 2020 [C] {published data only}
- Flower B, Brown JC, Simmons B, Moshe M, Frise R, Penn R, et al. Clinical and laboratory evaluation of SARS-CoV-2 lateral flow assays for use in a national COVID-19 seroprevalence survey. Thorax 2020;75(12):1082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flower 2020 [D] {published data only}
- Flower B, Brown JC, Simmons B, Moshe M, Frise R, Penn R, et al. Clinical and laboratory evaluation of SARS-CoV-2 lateral flow assays for use in a national COVID-19 seroprevalence survey. Thorax 2020;75(12):1082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flower 2020 [E] {published data only}
- Flower B, Brown JC, Simmons B, Moshe M, Frise R, Penn R, et al. Clinical and laboratory evaluation of SARS-CoV-2 lateral flow assays for use in a national COVID-19 seroprevalence survey. Thorax 2020;75(12):1082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fragkou 2020 {published data only}
- Fragkou PC, Papaevangelou V, Antoniadou A, Kavvatha D, Ploussi A, Pantazis N, et al. Preliminary data of a quantitative point of care test for SARS-CoV-2 antibodies from Greece. In Vivo 2020;34(5):3039-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fujigaki 2020 [A] {published data only}
- Fujigaki H, Takemura M, Osawa M, Sakurai A, Nakamoto K, Seto K, et al. Reliability of serological tests for COVID-19: comparison of three immunochromatography test kits for SARS-CoV-2 antibodies. Heliyon 2020;6(9):e04929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujigaki H, Takemura M, Osawa M, Sakurai A, Nakamoto K, Seto K, et al. Reliability of serological tests for COVID-19: Comparison of three immunochromatography test kits for SARS-CoV-2 antibodies. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Fujigaki 2020 [B] {published data only}
- See first entry for publication details for this study. Fujigaki 2020 [A].
Fujigaki 2020 [C] {published data only}
- See first entry for publication details for this study. Fujigaki 2020 [A].
Gao 2020a {published data only}
- Gao Y, Yuan Y, Li TT, Wang WX, Li YX, Li A, et al. Evaluation of the auxiliary diagnosis value of antibodies assays for the detection of novel coronavirus (SARS-Cov-2). medRxiv [Preprint] 2020. [DOI: ]
- Gao Y, Yuan Y, Li TT, Wang WX, Li YX, Li A, et al. Evaluation of the auxiliary diagnostic value of antibody assays for the detection of novel coronavirus (SARS-CoV-2). Journal of Medical Virology 2020;133(12):1975-79. [DOI: 10.1002/jmv.25919] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gao 2020b [A] {published data only}
- Gao HX, Li YN, Xu ZG, Wang YL, Wang HB, Cao JF, et al. Detection of serum immunoglobulin M and immunoglobulin G antibodies in 2019-novel coronavirus infected cases from different stages. Chinese Medical Journal 2020;133(12):1479-80. [DOI: 10.1097/CM9.0000000000000820] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gao 2020b [B] {published data only}
- See first entry for publication details for this study. Gao 2020b [A].
Gao 2020b [C] {published data only}
- See first entry for publication details for this study. Gao 2020b [A].
Garnett 2020 {published data only}
- Garnett E, Jung J, Tam E, Rajapakshe D, Cheney S, Brown C, et al. Clinical validation and performance evaluation of the automated vitros total anti-SARS-CoV-2 Antibodies assay for screening of serostatus in COVID-19. American Journal of Clinical Pathology 2020;154(6):742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
GeurtsvanKessel 2020 [A] {published data only}
- GeurtsvanKessel CH, Okba NMA, Igloi Z, Bogers S, Embregts CWE, Laksono BM, et al. An evaluation of COVID-19 serological assays informs future diagnostics and exposure assessment. Nature Communications 2020;11(1):3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
GeurtsvanKessel 2020 [B] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
GeurtsvanKessel 2020 [C] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
GeurtsvanKessel 2020 [D] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
GeurtsvanKessel 2020 [E] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
GeurtsvanKessel 2020 [F] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
GeurtsvanKessel 2020 [G] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
GeurtsvanKessel 2020 [H] {published data only}
- See first entry for publication details for this study. GeurtsvanKessel 2020 [A].
Graham 2021 {published data only}
- Graham NSN, Junghans J, McLaren R, Randell P, Lang N, Ladhani SN, et al. High rates of SARS-CoV-2 seropositivity in nursing home residents. Journal of Infection 2021;82(2):282-327. [DOI: 10.1016/j.jinf.2020.08.040] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gudbjartsson 2020 [A] {published data only}
- Gudbjartsson DF, Norddahl GL, Melsted P, Gunnarsdottir K, Holm H, Eythorsson E, et al. Humoral immune response to SARS-CoV-2 in Iceland. New England Journal of Medicine 2020;383(18):1724-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gudbjartsson 2020 [B] {published data only}
- See first entry for publication details for this study. Gudbjartsson 2020 [A].
Gudbjartsson 2020 [C] {published data only}
- See first entry for publication details for this study. Gudbjartsson 2020 [A].
Guedez‐Lopez 2020 [A] {published data only}
- Guedez-Lopez GV, Alguacil-Guillen M, Gonzalez-Donapetry P, Bloise I, Tornero-Marin C, Gonzalez-Garcia J, et al. Evaluation of three immunochromatographic tests for rapid detection of antibodies against SARS-CoV-2. European Journal of Clinical Microbiology and Infectious Diseases 2020;39(12):2289-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guedez‐Lopez 2020 [B] {published data only}
- See first entry for publication details for this study. Guedez-Lopez 2020 [A].
Guedez‐Lopez 2020 [C] {published data only}
- See first entry for publication details for this study. Guedez-Lopez 2020 [A].
Haljasmagi 2020 {published data only}
- Haljasmagi L, Remm A, Rumm AP, Krassohhina E, Sein H, Tamm A, et al. LIPS method for the detection of SARS-CoV-2 antibodies to spike and nucleocapsid proteins. European Journal of Immunology 2020;50(8):1234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamilton 2020 {published data only}
- Hamilton F, Muir P, Attwood M, Vipond ANB, Hopes R, Moran E, et al. Kinetics and performance of the Abbott architect SARS-CoV-2 IgG antibody assay. Journal of Infection 2020;81(6):e7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harritshoej 2021 [A] {published data only}
- Harritshøj LH, Gybel-Bask M, Afzal S, Kamstrup PR, Jørgensen CS, Thomsen MK, et al. Comparison of 16 serological SARS-CoV-2 immunoassays in 16 clinical laboratories. Journal of Clinical Microbiology 2021;59(5):e02596-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harritshøj LH, Gybel-Brask M, Afzal S, Kamstrup PR, Jørgensen CS, Thomsen MK, et al. Comparison of sixteen serological SARS-CoV-2 immunoassays in sixteen clinical laboratories. medRxiv [Preprint] 2020. [DOI: ]
Harritshoej 2021 [B] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [C] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [D] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [E] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [F] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [G] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [H] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [I] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [J] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [K] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [L] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Harritshoej 2021 [M] {published data only}
- See first entry for publication details for this study. Harritshoej 2021 [A].
Haselmann 2020 [A] {published data only}
- Haselmann V, Kittel M, Gerhards C, Thiaucourt M, Eichner R, Costina V, et al. Comparison of test performance of commercial anti-SARS-CoV-2 immunoassays in serum and plasma samples. Clinica Chimica Acta 2020;510:73-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Haselmann 2020 [B] {published data only}
- See first entry for publication details for this study. Haselmann 2020 [A].
Haselmann 2020 [C] {published data only}
- See first entry for publication details for this study. Haselmann 2020 [A].
Herroelen 2020 [A] {published data only}
- Herroelen PH, Martens GA, De Smet D, Swaerts K, Decavele AS. Humoral immune response to SARS-CoV-2. American Journal of Clinical Pathology 2020;154(5):610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herroelen PH, Martens GA, De Smet D, Swaerts K, Decavele A-S. Kinetics of the humoral immune response to SARS-CoV-2: comparative analytical performance of seven commercial serology tests. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Herroelen 2020 [B] {published data only}
- See first entry for publication details for this study. Herroelen 2020 [A].
Herroelen 2020 [C] {published data only}
- See first entry for publication details for this study. Herroelen 2020 [A].
Herroelen 2020 [D] {published data only}
- See first entry for publication details for this study. Herroelen 2020 [A].
Herroelen 2020 [E] {published data only}
- See first entry for publication details for this study. Herroelen 2020 [A].
Herroelen 2020 [F] {published data only}
- See first entry for publication details for this study. Herroelen 2020 [A].
Herroelen 2020 [G] {published data only}
- See first entry for publication details for this study. Herroelen 2020 [A].
Hoffman 2020 {published data only}
- Hoffman T, Nissen K, Krambrich J, Ronnberg B, Akaberi D, Esmaeilzadeh M, et al. Evaluation of a COVID-19 IgM and IgG rapid test; an efficient tool for assessment of past exposure to SARS-CoV-2. Infection Ecology & Epidemiology 2020;10(1):1754538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hogan 2020a [A] {published data only}
- Hogan KO, Klippel D, Plapp FV, Liesman RM. Comparative Evaluation of Three Serologic Assays for the Identification of SARS-CoV-2 Antibodies. medRxiv [Preprint] 2020. [DOI: ]
Hogan 2020a [B] {published data only}
- See first entry for publication details for this study. Hogan 2020a [A].
Hogan 2020a [C] {published data only}
- See first entry for publication details for this study. Hogan 2020a [A].
Horber 2020 [A] {published data only}
- Horber S, Soldo J, Relker L, Jurgens S, Guther J, Peter S, et al. Evaluation of three fully-automated SARS-CoV-2 antibody assays. Clinical Chemistry and Laboratory Medicine 2020;58(12):2113-20. [DOI] [PubMed] [Google Scholar]
Horber 2020 [B] {published data only}
- See first entry for publication details for this study. Horber 2020 [A].
Horber 2020 [C] {published data only}
- See first entry for publication details for this study. Horber 2020 [A].
Hu 2020a {published data only}
- Hu Q, Cui X, Liu X, Peng B, Jiang J, Wang X, et al. The production of antibodies for SARS-CoV-2 and its clinical implication. medRxiv [Preprint] 2020. [DOI: ]
Hu 2020b [A] {published data only}
- Hu F, Shang X, Chen M, Zhang C. Joint detection of serum IgM/IgG antibody is an important key to clinical diagnosis of SARS-CoV-2 infection. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Hu 2020b [B] {published data only}
- See first entry for publication details for this study. Hu 2020b [A].
Hubbard 2021 [A] {published data only}
- Hubbard JA, Geno KA, Khan J, Szczepiorkowski ZM, Gijsel D, Ovalle AA, et al. Comparison of two automated immunoassays for the detection of SARS-CoV-2 nucleocapsid antibodies. Journal of Applied Laboratory Medicine 2021;6(2):429-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hubbard 2021 [B] {published data only}
- See first entry for publication details for this study. Hubbard 2021 [A].
Imai 2020 {published data only}
- Imai K, Tabata S, Ikeda M, Noguchi S, Kitagawa Y, Matuoka M, et al. Clinical evaluation of an immunochromatographic IgM/IgG antibody assay and chest computed tomography for the diagnosis of COVID-19. Journal of Clinical Virology 2020;128:104393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaaskelainen 2020 {published data only}
- Jaaskelainen AJ, Kekalainen E, Kallio-Kokko H, Mannonen L, Kortela E, Vapalahti O, et al. Evaluation of commercial and automated SARS-CoV-2 IgG and IgA ELISAs using coronavirus disease (COVID-19) patient samples. Eurosurveillance 2020;25(18):2000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jin 2020 {published data only}
- Jin Y, Wang M, Zuo Z, Fan C, Ye F, Cai Z, et al. Diagnostic value and dynamic variance of serum antibody in coronavirus disease 2019. International Journal of Infectious Diseases 2020;94:49-52. [DOI: 10.1016/j.ijid.2020.03.065] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jung 2020a {published data only}
- Jung J, Garnett E, Jariwala P, Pham H, Huang R, Benzi E, et al. Clinical performance of a semi-quantitative assay for SARS-CoV2 IgG and SARS-CoV2 IgM antibodies. Clinica Chimica Acta 2020;510:790-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kaltenbach 2020 [A] {published data only}
- Kaltenbach H-M, Rudolf F, Linnik J, Deichmann J, Ruf T, Altamura R, et al. Initial characterisation of ELISA assays and the immune response of the clinically correlated SARS-CoV-2 biobank SERO-BL-COVID-19 collected during the pandemic onset in Switzerland. medRxiv [Preprint] 2020. [https://doi.org/10.1101/2020.07.05.20145888]
Kaltenbach 2020 [B] {published data only}
- See first entry for publication details for this study. Kaltenbach 2020 [A].
Kaltenbach 2020 [C] {published data only}
- See first entry for publication details for this study. Kaltenbach 2020 [A].
Kaneko 2021 {published data only}
- Kaneko S, Nukui Y, Arashiro T, Aiso Y, Sugii M, Hadano Y, et al. Clinical validation of an immunochromatographic SARS-Cov-2 IgM/IgG antibody assay with Japanese cohort. Journal of Medical Virology 2021;93(1):569-72. [DOI] [PubMed] [Google Scholar]
Knauer 2020 [A] {published data only}
- Knauer MJ, Hedley BD, Bhayana V, Payne M, Chin-Yee I, Delport J. Interim analysis of the clinical performance of five SARS-Cov-2 serology assays. Clinical Biochemistry 2020;86:28-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Knauer 2020 [B] {published data only}
- See first entry for publication details for this study. Knauer 2020 [A].
Knauer 2020 [C] {published data only}
- See first entry for publication details for this study. Knauer 2020 [A].
Knauer 2020 [D] {published data only}
- See first entry for publication details for this study. Knauer 2020 [A].
Knauer 2020 [E] {published data only}
- See first entry for publication details for this study. Knauer 2020 [A].
Ko 2021 {published data only}
- Ko JH, Joo EJ, Kim SH, Kim YJ, Huh K, Cho SY, et al. Clinical application of rapid diagnostic test kit for SARS-CoV-2 antibodies into the field of patient care. Journal of Microbiology, Immunology, and Infection 2021;54(1):97-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kohmer 2020a [A] {published data only}
- Kohmer N, Westhaus S, Ruhl C, Ciesek S, Rabenau HF. Brief clinical evaluation of six high-throughput SARS-CoV-2 IgG antibody assays. Journal of Clinical Virology 2020;129:104480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kohmer 2020a [B] {published data only}
- See first entry for publication details for this study. Kohmer 2020a [A].
Kohmer 2020a [C] {published data only}
- See first entry for publication details for this study. Kohmer 2020a [A].
Kohmer 2020b [A] {published data only}
- Kohmer N, Westhaus S, Ruhl C, Ciesek S, Rabenau HF. Clinical performance of different SARS-CoV-2 IgG antibody tests. Journal of Medical Virology 2020;92(10):2243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kohmer 2020b [B] {published data only}
- See first entry for publication details for this study. Kohmer 2020b [A].
Kohmer 2020b [C] {published data only}
- See first entry for publication details for this study. Kohmer 2020b [A].
Kohmer 2020b [D] {published data only}
- See first entry for publication details for this study. Kohmer 2020b [A].
Kohmer 2020b [E] {published data only}
- See first entry for publication details for this study. Kohmer 2020b [A].
Kohmer 2020b [F] {published data only}
- See first entry for publication details for this study. Kohmer 2020b [A].
Korte 2021 [A] {published data only}
- Korte W, Buljan M, Rosslein M, Wick P, Golubov V, Jentsch J, et al. SARS-CoV-2 IgG and IgA antibody response is gender dependent; and IgG antibodies rapidly decline early on. Journal of Infection 2021;82(1):e11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Korte 2021 [B] {published data only}
- See first entry for publication details for this study. Korte 2021 [A].
Korte 2021 [C] {published data only}
- See first entry for publication details for this study. Korte 2021 [A].
Kowitdamrong 2020 [A] {published data only}
- Kowitdamrong E, Puthanakit T, Jantarabenjakul W, Prompetchara E, Suchartlikitwong P, Putcharoen O, et al. Antibody responses to SARS-CoV-2 in coronavirus diseases 2019 patients with different severity. medRxiv [Preprint] 2020. [https://doi.org/10.1101/2020.09.06.20189480] [DOI] [PMC free article] [PubMed]
- Kowitdamrong E, Puthanakit T, Jantarabenjakul W, Prompetchara E, Suchartlikitwong P, Putcharoen O, et al. Antibody responses to SARS-CoV-2 in patients with differing severities of coronavirus disease 2019. PLoS One 2020;15(10):e0240502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kowitdamrong 2020 [B] {published data only}
- See first entry for publication details for this study. Kowitdamrong 2020 [A].
Krishnamurthy 2020 {published data only}
- Krishnamurthy HK, Jayaraman V, Krishna K, Rajasekaran KE, Wang T, Bei K, et al. Antibody profiling and prevalence in the US population during the SARS-CoV-2 pandemic. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Krishnamurthy HK, Jayaraman V, Krishna K, Rajasekaran KE, Wang T, Bei K, et al. Antibody profiling and prevalence in US patients during the SARS-CoV2 pandemic. PLoS One 2020;15(11):e0242655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lassauniere 2020 [A] {published data only}
- Lassauniere R, Frische A, Harboe ZB, Nielsen AC, Fomsgaard A, Krogfelt KA, et al. Evaluation of nine commercial SARS-CoV-2 immunoassays. medRxiv [Preprint] 2020. [DOI: ]
Lassauniere 2020 [B] {published data only}
- See first entry for publication details for this study. Lassauniere 2020 [A].
Lassauniere 2020 [C] {published data only}
- See first entry for publication details for this study. Lassauniere 2020 [C].
Lassauniere 2020 [D] {published data only}
- See first entry for publication details for this study. Lassauniere 2020 [A].
Lassauniere 2020 [E] {published data only}
- See first entry for publication details for this study. Lassauniere 2020 [A].
Lassauniere 2020 [F] {published data only}
- See first entry for publication details for this study. Lassauniere 2020 [A].
Lassauniere 2020 [G] {published data only}
- See first entry for publication details for this study. Lassauniere 2020 [A].
Lau 2020a {published data only}
- Lau CS, Hoo SP, Yew SF, Ong SK, Lum LT, Heng PY, et al. Evaluation of the Roche Elecsys anti-SARS-CoV-2 assay. medRxiv [Preprint] 2020. [https://doi.org/10.1101/2020.06.28.20142232]
Lau 2020b {published data only}
- Lau CS, Hoo SP, Liang YL, Aw TC. Evaluation of the Abbott SARS-CoV-2 Ig-G assay. medRxiv [Preprint] 2020. [DOI: ]
Lau 2020c {published data only}
- Lau CS, Oh HML, Hoo SP, Liang YL, Phua SK, Aw TC. Performance of an automated chemiluminescence SARS-CoV-2 IG-G assay. Clinica Chimica Acta 2020;510:760-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lau 2020d {published data only}
- Lau CS, Hoo SP, Yew SF, Ong SK, Lum LT, Heng PY, et al. Evaluation of an electrochemiluminescent SARS-CoV-2 antibody assay. Journal of Applied Laboratory Medicine 2020;5(6):1313-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020 [A] {published data only}
- Li B, Feng F, Yang G, Liu A, Yang N, Jiang Q, et al. Immunoglobulin G/M and cytokines detections in continuous sera from patients with novel coronaviruses (2019-nCoV) infection. SSRN [Preprint] 2020. [DOI: ]
Li 2020 [B] {published data only}
- See first entry for publication details for this study. Li 2020 [A].
Lippi 2020 [A] {published data only}
- Lippi G, Salvagno GL, Pegoraro M, Militello V, Caloi C, Peretti A, et al. Assessment of immune response to SARS-CoV-2 with fully automated MAGLUMI 2019-nCoV IgG and IgM chemiluminescence immunoassays. Clinical Chemistry and Laboratory Medicine 2020;58(7):1156-9. [DOI: 10.1515/cclm-2020-0473] [DOI] [PubMed] [Google Scholar]
Lippi 2020 [B] {published data only}
- See first entry for publication details for this study. Lippi 2020 [A].
Liu 2020a {published data only}
- Liu L, Liu W, Wang S, Zheng S. A preliminary study on serological assay for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 238 admitted hospital patients. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Liu L, Liu W, Zheng Y, Jiang X, Kou G, Ding J, et al. A preliminary study on serological assay for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 238 admitted hospital patients. Microbes and Infection 2020;22(4-5):206-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2020b [A] {published data only}
- Liu W, Liu L, Kou G, Zheng Y, Ding Y, Ni W, et al. Evaluation of nucleocapsid and spike protein-based ELISAs for detecting antibodies against SARS-CoV-2. Journal of Clinical Microbiology 2020;58(6):e00461-20. [DOI: 10.1128/JCM.00461-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu L, Kou G, Zheng Y, Ding Y, Ni W, et al. Evaluation of nucleocapsid and spike protein-based ELISAs for detecting antibodies against SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Liu 2020b [B] {published data only}
- See first entry for publication details for this study. Liu 2020b [A].
Liu 2020c {published data only}
- Liu W, Kou G, Dong Y, Zheng Y, Ding Y, Ni W, et al. Clinical application of Chemiluminescence Microparticle Immunoassay for SARS-CoV-2 infection diagnosis. Journal of Clinical Virology 2020;130:104576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2021 {published data only}
- Liu J, Lian R, Zhang G, Hou B, Wang C, Dong J, et al. Changes in serum virus-specific IgM/IgG antibody in asymptomatic and discharged patients with reoccurring positive COVID-19 nucleic acid test (RPNAT). Annals of Medicine 2021;53(1):34-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Loconsole 2020 {published data only}
- Loconsole D, Centrone F, Morcavallo C, Campanella S, Sallustio A, Quarto M, et al. The light and shadow of rapid serological tests for SARS-CoV-2 infection: results from a study in a large emergency department. International Journal of Environmental Research and Public Health 2020;17(18):6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Long 2020 {published data only}
- Long Q, Deng H, Chen J, Hu J, Liu B, Liao P, et al. Antibody responses to SARS-CoV-2 in COVID-19 patients: the perspective application of serological tests in clinical practice. medRxiv [Preprint] 2020. [DOI: ]
- Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nature Medicine 2020;26:845-8. [DOI] [PubMed] [Google Scholar]
Lou 2020 [A] {published data only}
- Lou B, Li T, Zheng S, Su Y, Li Z, Liu W, et al. Serology characteristics of SARS-CoV-2 infection since the exposure and post symptoms onset. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Lou B, Li TD, Zheng SF, Su YY, Li ZY, Liu W, et al. Serology characteristics of SARS-CoV-2 infection after exposure and post-symptom onset. European Respiratory Journal 2020;56(2):2000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lou 2020 [B] {published data only}
- See first entry for publication details for this study. Lou 2020 [A].
Lynch 2021 {published data only}
- Lynch KL, Whitman JD, Lacanienta NP, Beckerdite EW, Kastner SA, Shy BR, et al. Magnitude and kinetics of anti-SARS-CoV-2 antibody responses and their relationship to disease severity. Clinical Infectious Diseases 2021;72(2):301-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KL, Whitman JD, Lacanienta NP, Beckerdite EW, Kastner SA, Shy BR, et al. Magnitude and kinetics of anti-SARS-CoV-2 antibody responses and their relationship to disease severity. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
MacMullan 2020 [A] {published data only}
- MacMullan MA, Ibrayeva A, Trettner K, Deming L, Das S, Tran F, et al. ELISA detection of SARS-CoV-2 antibodies in saliva. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- MacMullan MA, Ibrayeva A, Trettner K, Deming L, Das S, Tran F, et al. ELISA detection of SARS-CoV-2 antibodies in saliva. Scientific Reports 2020;10(1):20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
MacMullan 2020 [B] {published data only}
- See first entry for publication details for this study. MacMullan 2020 [A].
MacMullan 2020 [C] {published data only}
- See first entry for publication details for this study. MacMullan 2020 [A].
MacMullan 2020 [D] {published data only}
- See first entry for publication details for this study. MacMullan 2020 [A].
Mairesse 2020 [A] {published data only}
- Mairesse A, Favresse J, Eucher C, Elsen M, Tre-Hardy M, Haventith C, et al. High clinical performance and quantitative assessment of antibody kinetics using a dual recognition assay for the detection of SARS-CoV-2 IgM and IgG antibodies. Clinical Biochemistry 2020;86:23-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mairesse 2020 [B] {published data only}
- See first entry for publication details for this study. Mairesse 2020 [A].
Manalac 2020 [A] {published data only}
- Manalac J, Yee J, Calayag K, Nguyen L, Patel PM, Zhou D, et al. Evaluation of Abbott anti-SARS-CoV-2 CMIA IgG and Euroimmun ELISA IgG/IgA assays in a clinical lab. Clinica Chimica Acta 2020;510:687-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Manalac 2020 [B] {published data only}
- See first entry for publication details for this study. Manalac 2020 [A].
Marlet 2020 [A] {published data only}
- Marlet J, Petillon C, Ragot E, Abou El Fattah Y, Guillon A, Marchand Adam S, et al. Clinical performance of four immunoassays for antibodies to SARS-CoV-2, including a prospective analysis for the diagnosis of COVID-19 in a real-life routine care setting. Journal of Clinical Virology 2020;132:104633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marlet 2020 [B] {published data only}
- See first entry for publication details for this study. Marlet 2020 [A].
Marlet 2020 [C] {published data only}
- See first entry for publication details for this study. Marlet 2020 [A].
Marlet 2020 [D] {published data only}
- See first entry for publication details for this study. Marlet 2020 [A].
Martinaud 2020 {published data only}
- Martinaud C, Hejl C, Igert A, Bigaillon C, Bonnet C, Merens A, et al. Evaluation of the Quotient(R) MosaiQ COVID-19 antibody microarray for the detection of IgG and IgM antibodies to SARS-CoV-2 virus in humans. Journal of Clinical Virology 2020;130:104571. [DOI] [PMC free article] [PubMed] [Google Scholar]
McAulay 2020 [A] {published data only}
- McAulay K, Bryan A, Greninger AL, Grill F, Lake D, Kaleta EJ, et al. Retrospective clinical evaluation of 4 lateral flow assays for the detection of SARS-CoV-2 IgG. Diagnostic Microbiology and Infectious Disease 2020;98(3):115161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAulay K, Bryan A, Greninger AL, Grill F, Lake D, Kaleta EJ, et al. Retrospective clinical evaluation of four lateral flow assays for the detection of SARS-CoV-2 antibodies. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
McAulay 2020 [B] {published data only}
- See first entry for publication details for this study. McAulay 2020 [A].
McAulay 2020 [C] {published data only}
- See first entry for publication details for this study. McAulay 2020 [A].
McAulay 2020 [D] {published data only}
- See first entry for publication details for this study. McAulay 2020 [A].
Merrill 2020 [A] {published data only}
- Merrill AE, Jackson JB, Ehlers A, Voss D, Krasowski MD. Head-to-head comparison of two SARS-CoV-2 serology assays. Journal of Applied Laboratory Medicine 2020;5(6):1351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Merrill 2020 [B] {published data only}
- See first entry for publication details for this study. Merrill 2020 [A].
Montesinos 2020 [A] {published data only}
- Montesinos I, Gruson D, Kabamba B, Dahma H, Van den Wijngaert S, Reza S, et al. Evaluation of two automated and three rapid lateral flow immunoassays for the detection of anti-SARS-CoV-2 antibodies. Journal of Clinical Virology 2020;128:104413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Montesinos 2020 [B] {published data only}
- See first entry for publication details for this study. Montesinos 2020 [A].
Montesinos 2020 [C] {published data only}
- See first entry for publication details for this study. Montesinos 2020 [A].
Montesinos 2020 [D] {published data only}
- See first entry for publication details for this study. Montesinos 2020 [A].
Montesinos 2020 [E] {published data only}
- See first entry for publication details for this study. Montesinos 2020 [A].
Montesinos 2020 [F] {published data only}
- See first entry for publication details for this study. Montesinos 2020 [A].
Montesinos 2020 [G] {published data only}
- See first entry for publication details for this study. Montesinos 2020 [A].
Muecksch 2020 [A] {published data only}
- Muecksch F, Wise H, Batchelor B, Squires M, Semple E, Richardson C, et al. Longitudinal analysis of clinical serology assay performance and neutralising antibody levels in COVID19 convalescents. medRxiv [Preprint] 2020. [DOI: ]
Muecksch 2020 [B] {published data only}
- See first entry for publication details for this study. Muecksch 2020 [A].
Muecksch 2020 [C] {published data only}
- See first entry for publication details for this study. Muecksch 2020 [A].
Muecksch 2020 [D] {published data only}
- See first entry for publication details for this study. Muecksch 2020 [A].
Naaber 2020 [A] {published data only}
- Naaber P, Hunt K, Pesukova J, Haljasmagi L, Rumm P, Peterson P, et al. Evaluation of SARS-CoV-2 IgG antibody response in PCR positive patients: Comparison of nine tests in relation to clinical data. PLoS One 2020;15(10):e0237548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naaber P, Hunt K, Pesukova J, Haljasmagi L, Rumm P, Peterson P, et al. Evaluation of SARS-CoV-2 IgG antibody response in PCR positive patients: comparison of nine tests in relation with clinical data. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Naaber 2020 [B] {published data only}
- See first entry for publication details for this study. Naaber 2020 [A].
Naaber 2020 [C] {published data only}
- See first entry for publication details for this study. Naaber 2020 [A].
Naaber 2020 [D] {published data only}
- See first entry for publication details for this study. Naaber 2020 [A].
Naaber 2020 [E] {published data only}
- See first entry for publication details for this study. Naaber 2020 [A].
Naaber 2020 [F] {published data only}
- See first entry for publication details for this study. Naaber 2020 [A].
Naaber 2020 [G] {published data only}
- See first entry for publication details for this study. Naaber 2020 [A].
Nagasawa 2020 [A] {published data only}
- Nagasawa M, Yamaguchi Y, Furuya M, Takahashi Y, Taki R, Nagata K, et al. Investigation of anti-SARS-CoV-2 IgG and IgM antibodies in the patients with COVID-19 by three different ELISA test kits. SN Comprehensive Clinical Medicine 2020;2(9):1323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nagasawa 2020 [B] {published data only}
- See first entry for publication details for this study. Nagasawa 2020 [A].
Nagasawa 2020 [C] {published data only}
- See first entry for publication details for this study. Nagasawa 2020 [A].
Nayak 2021 {published data only}
- Nayak K, Gottimukkala K, Kumar S, Reddy ES, Edara VV, Kauffman R, et al. Characterization of neutralizing versus binding antibodies and memory B cells in COVID-19 recovered individuals from India. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Nayak K, Gottimukkala K, Kumar S, Reddy ES, Edara VV, Kauffman R, et al. Characterization of neutralizing versus binding antibodies and memory B cells in COVID-19 recovered individuals from India. Virology 2021;558:13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ng 2020 [A] {published data only}
- Ng DL, Goldgof GM, Shy BR, Levine AG, Balcerek J, Bapat SP, et al. Sars-cov-2 seroprevalence and neutralizing activity in donor and patient blood. Nature Communications 2020;11(1):4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DL, Goldgof GM, Shy BR, Levine AG, Balcerek J, Bapat SP, et al. SARS-CoV-2 seroprevalence and neutralizing activity in donor and patient blood from the San Francisco Bay Area. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Ng 2020 [B] {published data only}
- See first entry for publication details for this study. Ng 2020 [A].
Nguyen 2020 {published data only}
- Nguyen LS, Laghlam D, De Gonfreville E, Pene F, Rozenberg F, Mira JP. Sensitivity of point-of-care IgM and IgG test in critically ill patients with SARS-Cov-2. Critical Care 2020;24(1):573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nicol 2020 [A] {published data only}
- Nicol T, Lefeuvre C, Serri O, Pivert A, Joubaud F, Dubee V, et al. Assessment of SARS-CoV-2 serological tests for the diagnosis of COVID-19 through the evaluation of three immunoassays: Two automated immunoassays (Euroimmun and Abbott) and one rapid lateral flow immunoassay (NG Biotech). Journal of Clinical Virology 2020;129:104511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nicol 2020 [B] {published data only}
- See first entry for publication details for this study. Nicol 2020 [A].
Nicol 2020 [C] {published data only}
- See first entry for publication details for this study. Nicol 2020 [A].
Nicol 2020 [D] {published data only}
- See first entry for publication details for this study. Nicol 2020 [A].
Nicol 2020 [E] {published data only}
- See first entry for publication details for this study. Nicol 2020 [A].
Nilles 2020 [A] {published data only}
- Nilles EJ, Karlson EW, Norman M, Gilboa T, Fischinger S, Atyeo C, et al. Evaluation of two commercial and two non-commercial immunoassays for the detection of prior infection to SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Nilles 2020 [B] {published data only}
- See first entry for publication details for this study. Nilles 2020 [A].
NSAE 2020 [A] {published data only}
- National SARS-CoV-2 Serology Assay Evaluation Group. Performance characteristics of five immunoassays for SARS-CoV-2: a head-to-head benchmark comparison. Lancet Infectious Diseases 2020;20(12):1390-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Public Health England. Evaluation of sensitivity and specificity of four commercially available SARS-CoV-2 antibody immunoassays (July 2020). https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/898437/Evaluation__of_sensitivity_and_specificity_of_4_commercially_available_SARS-CoV-2_antibody_immunoassays.pdf.
NSAE 2020 [B] {published data only}
- See first entry for publication details for this study. NSAE 2020 [A].
NSAE 2020 [C] {published data only}
- See first entry for publication details for this study. NSAE 2020 [A].
NSAE 2020 [D] {published data only}
- See first entry for publication details for this study. NSAE 2020 [A].
Ong 2020 [A] {published data only}
- Ong DSY, Man SJ, Lindeboom FA, Koeleman JGM. Comparison of diagnostic accuracies of rapid serological tests and ELISA to molecular diagnostics in patients with suspected coronavirus disease 2019 presenting to the hospital. Clinical Microbiology and Infection 2020;26(8):1094.e7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ong 2020 [B] {published data only}
- See first entry for publication details for this study. Ong 2020 [A].
Padoan 2020a {published data only}
- Padoan A, Cosma C, Sciacovelli L, Faggian D, Plebani M. Analytical performances of a chemiluminescence immunoassay for SARS-CoV-2 IgM/IgG and antibody kinetics. Clinical Chemistry and Laboratory Medicine 2020;58(7):1081-8. [DOI: ] [DOI] [PubMed] [Google Scholar]
Padoan 2020b [A] {published data only}
- Padoan A, Sciacovelli L, Basso D, Negrini D, Zuin S, Cosma C, et al. IgA-Ab response to spike glycoprotein of SARS-CoV-2 in patients with COVID-19: A longitudinal study. Clinica Chimica Acta 2020;507:164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Padoan 2020b [B] {published data only}
- See first entry for publication details for this study. Padoan 2020b [A].
Paiva 2021 [A] {published data only}
- Paiva KJ, Grisson RD, Chan PA, Huard RC, Caliendo AM, Lonks JR, et al. Validation and performance comparison of three SARS-CoV-2 antibody assays. Journal of Medical Virology 2021;93(2):916-23. [DOI] [PubMed] [Google Scholar]
Paiva 2021 [B] {published data only}
- See first entry for publication details for this study. Paiva 2021 [A].
Paiva 2021 [C] {published data only}
- See first entry for publication details for this study. Paiva 2021 [A].
Pan 2020a {published data only}
- Pan Y, Li X, Yang G, Fan J, Tang Y, Zhao J, et al. Serological immunochromatographic approach in diagnosis with SARS-CoV-2 infected COVID-19 patients. Journal of Infection 2020;81(1):e28-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Li X, Yang G, Fan J, Tang Y, Zhao J, et al. Serological immunochromatographic approach in diagnosis with SARS-CoV-2 infected COVID-19 patients. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Pape 2021 [A] {published data only}
- Pape C, Remme R, Wolny A, Olberg S, Wolf S, Cerrone L, et al. Microscopy-based assay for semi-quantitative detection of SARS-CoV-2 specific antibodies in human sera: A semi-quantitative, high throughput, microscopy-based assay expands existing approaches to measure SARS-CoV-2 specific antibody levels in human sera. Bioessays 2021;43(3):e2000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape C, Remme R, Wolny A, Olberg S, Wolf S, Cerrone L, et al. Microscopy-based assay for semi-quantitative detection of SARS-CoV-2 specific antibodies in human sera. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Pape 2021 [B] {published data only}
- See first entry for publication details for this study. Pape 2021 [A].
Patel 2021 [A] {published data only}
- Patel EU, Bloch EM, Clarke W, Hsieh YH, Boon D, Eby Y, et al. Comparative performance of five commercially available serologic assays to detect antibodies to SARS-CoV-2 and identify individuals with high neutralizing titers. Journal of Clinical Microbiology 2021;59(2):e02257-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel EU, Bloch EM, Clarke W, Hsieh YH, Boon D, Eby Y, et al. Comparative performance of five commercially available serologic assays to detect antibodies to SARS-CoV-2 and identify individuals with high neutralizing titers. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Patel 2021 [B] {published data only}
- See first entry for publication details for this study. Patel 2021 [A].
Patel 2021 [C] {published data only}
- See first entry for publication details for this study. Patel 2021 [A].
Patel 2021 [D] {published data only}
- See first entry for publication details for this study. Patel 2021 [A].
Patel 2021 [E] {published data only}
- See first entry for publication details for this study. Patel 2021 [A].
Pere 2020 {published data only}
- Pere H, Vedie B, Vernet R, Demory N, Kassis N, Mirault T, et al. Unexpected diagnosis of COVID-19-associated disorders by SARS-CoV-2-specific serology. Journal of Clinical Virology 2020;132:104568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Perez‐Garcia 2020(a) {published data only}
- Pérez-García F, Pérez-Tanoira R, Romanyk J, Arroyo T, Gómez-Herruz P, Cuadros-González J. Rapid diagnosis of SARS-CoV-2 infection by detecting IgG and IgM antibodies with an immunochromatographic device: a prospective single-center study. medRxiv [Preprint] 2020. [DOI: ]
- Perez-Garcia F, Perez-Tanoira R, Romanyk J, Arroyo T, Gomez-Herruz P, Cuadros-Gonzalez J. Alltest rapid lateral flow immunoassays is reliable in diagnosing SARS-CoV-2 infection from 14 days after symptom onset: A prospective single-center study. Journal of Clinical Virology 2020;129:104473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Perez‐Garcia 2020(b) {published data only}
- Pérez-García F, Pérez-Tanoira R, Romanyk J, Arroyo T, Gómez-Herruz P, Cuadros-González J. Rapid diagnosis of SARS-CoV-2 infection by detecting IgG and IgM antibodies with an immunochromatographic device: a prospective single-center study. medRxiv [Preprint] 2020. [DOI: ]
- Perez-Garcia F, Perez-Tanoira R, Romanyk J, Arroyo T, Gomez-Herruz P, Cuadros-Gonzalez J. Alltest rapid lateral flow immunoassays is reliable in diagnosing SARS-CoV-2 infection from 14 days after symptom onset: A prospective single-center study. Journal of Clinical Virology 2020;129:104473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Perez‐Garcia 2021 [A] {published data only}
- Perez-Garcia F, Perez-Tanoira R, Iglesias ME, Romanyk J, Arroyo T, Gomez-Herruz P, et al. Comparative evaluation of six immunoassays for the detection of antibodies against SARS-CoV-2. Journal of Virological Methods 2021;289:114047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia F, Perez-Tanoira R, Iglesias ME, Romanyk J, Arroyo T, Gomez-Herruz P, et al. Comparative evaluation of six immunoassays for the detection of antibodies against SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Perez‐Garcia 2021 [B] {published data only}
- See first entry for publication details for this study. Perez-Garcia 2021 [A].
Perez‐Garcia 2021 [C] {published data only}
- See first entry for publication details for this study. Perez-Garcia 2021 [A].
Perez‐Garcia 2021 [D] {published data only}
- See first entry for publication details for this study. Perez-Garcia 2021 [A].
Perez‐Garcia 2021 [E] {published data only}
- See first entry for publication details for this study. Perez-Garcia 2021 [A].
Perez‐Garcia 2021 [F] {published data only}
- See first entry for publication details for this study. Perez-Garcia 2021 [A].
Pfluger 2020 [A] {published data only}
- Pfluger LS, Bannasch JH, Brehm TT, Pfefferle S, Hoffmann A, Norz D, et al. Clinical evaluation of five different automated SARS-CoV-2 serology assays in a cohort of hospitalized COVID-19 patients. Journal of Clinical Virology 2020;130:104549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pfluger 2020 [B] {published data only}
- See first entry for publication details for this study. Pfluger 2020 [A].
Pfluger 2020 [C] {published data only}
- See first entry for publication details for this study. Pfluger 2020 [A].
Pfluger 2020 [D] {published data only}
- See first entry for publication details for this study. Pfluger 2020 [A].
Pfluger 2020 [E] {published data only}
- See first entry for publication details for this study. Pfluger 2020 [A].
PHE 2020 [A] {published data only}
- Public Health England. Evaluation of DiaSorin LIAISON SARSCoV-2 S1/S2 IgG serology assay for the detection of anti-SARS-CoV-2 antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/893435/Evaluation_of_Diasorin_Liaison_anti_SARS_CoV_2.pdf.
- Public Health England. Evaluation of Roche Elecsys Anti-SARS-CoV-2 serology assay for the detection of anti-SARS-CoV-2 antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/989460/Evaluation_of_Roche_Elecsys_anti_SARS_CoV_2_S_assay_PHE.pdf.
- Public Health England. Evaluation of Siemens Atellica-IM Total (COV2T) SARS-CoV-2 serology assay for the detection of anti-SARS-CoV-2 total antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/894176/Evaluation_of_Siemens_Atellica-IM_anti_SARS_CoV_2_Total.pdf.
- Public Health England. Evaluation of the Abbott SARS-CoV-2 IgG for the detection of anti-SARS-CoV-2 antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/890566/Evaluation_of_Abbott_SARS_CoV_2_IgG_PHE.pdf.
- Public Health England. Evaluation of the Beckman Coulter Access Anti-SARS-CoV-2 IgG assay for the detection of anti-SARS-CoV-2 antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/913571/Evaluation_Beckman_Coulter_Access_Anti-SARS_CoV2_August-2020_FINAL.pdf.
- Public Health England. Evaluation of the Euroimmun Anti-SARS-CoV-2 ELISA (IgG) serology assay for the detection of anti-SARS-CoV-2 antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/893433/Evaluation_of_Euroimmun_SARS_CoV_2_ELISA_IgG__1_.pdf.
- Public Health England. Evaluation of the Ortho Clinical Diagnostics Vitros Immunodiagnostic Products Anti-SARS-CoV-2 IgG serology assay for the detection of anti-SARS-CoV-2 antibodies. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/894173/Evaluation_of_OCD_Vitros_Immunodiagnostic_Anti-SARS_CoV2_total_antibody_serology_assay.pdf.
- Public Health England. Supporting information for the PHE commercial serology assay evaluations. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/970010/Supporting_information_for_the_PHE_commercial_serology_assay_evaluations.pdf.
PHE 2020 [B] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
PHE 2020 [C] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
PHE 2020 [D] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
PHE 2020 [E] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
PHE 2020 [F] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
PHE 2020 [G] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
PHE 2020 [H] {published data only}
- See first entry for publication details for this study. PHE 2020 [A].
Phipps 2020 {published data only}
- Phipps WS, SoRelle JA, Li QZ, Mahimainathan L, Araj E, Markantonis J, et al. SARS-CoV-2 antibody responses do not predict COVID-19 disease severity. American Journal of Clinical Pathology 2020;154(4):459-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps WS, SoRelle JA, Li QZ, Mahimainathan L, Araj E, Markantonis J, et al. SARS-CoV-2 antibody responses do not predict COVID-19 disease severity. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Pickering 2020 [A] {published data only}
- Pickering S, Betancor G, Galao RP, Merrick B, Signell AW, Wilson HD, et al. Comparative assessment of multiple COVID-19 serological technologies supports continued evaluation of point-of-care lateral flow assays in hospital and community healthcare settings. PLoS Pathogens 2020;16(9):e1008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering S, Betancor G, Pedro Galao R, Merrick B, Signell AW, Wilson HD, et al. Comparative assessment of multiple COVID-19 serological technologies supports continued evaluation of point-of-care lateral flow assays in hospital and community healthcare settings. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Pickering 2020 [B] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [C] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [D] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [E] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [F] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [G] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [H] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [I] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pickering 2020 [J] {published data only}
- See first entry for publication details for this study. Pickering 2020 [A].
Pollan 2020 {published data only}
- Pollan M, Perez-Gomez B, Pastor-Barriuso R, Oteo J, Hernan MA, Perez-Olmeda M, et al. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): a nationwide, population-based seroepidemiological study. Lancet 2020;396(10250):535-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prazuck 2020 [A] {published data only}
- Prazuck T, Colin M, Giachè S, Gubavu C, Seve A, Rzepecki V, et al. Evaluation of performance of two SARS-CoV-2 Rapid whole-blood finger-stick IgM-IgG combined antibody tests. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Prazuck T, Colin M, Giache S, Gubavu C, Seve A, Rzepecki V, et al. Evaluation of performance of two SARS-CoV-2 Rapid IgM-IgG combined antibody tests on capillary whole blood samples from the fingertip. PLoS One 2020;15(9):e0237694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prazuck 2020 [B] {published data only}
- See first entry for publication details for this study. Prazuck 2020 [A].
Qian 2020a {published data only}
- Qian C, Zhou M, Cheng F, Lin X, Gong Y, Xie X, et al. Development and multicenter performance evaluation of fully automated SARS-CoV-2 IgM and IgG immunoassays. Clinical Chemistry and Laboratory Medicine 2020;58(9):1601-7. [DOI] [PubMed] [Google Scholar]
Qiu 2020 {published data only}
- Qiu X, Xiang Y, Sun J, Wang X, Chen G, Xu X, et al. Dynamic changes of throat swabs RNA and serum antibodies for SARS-CoV-2 and their diagnostic performances in patients with COVID-19. Emerging Microbes & Infections 2020;9(1):1974-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ragnesola 2020 {published data only}
- Ragnesola B, Jin D, Lamb CC, Shaz BH, Hillyer CD, Luchsinger LL. COVID19 antibody detection using lateral flow assay tests in a cohort of convalescent plasma donors. BMC Research Notes 2020;13(1):372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Renard 2021 [A] {published data only}
- Renard N, Daniel S, Cayet N, Pecquet M, Raymond F, Pons S, et al. Performance characteristics of the VIDAS® SARS-COV-2 IgM and IgG serological assays. medRxiv [Preprint] 2020. [https://doi.org/10.1101/2020.09.28.20196030] [DOI] [PMC free article] [PubMed]
- Renard N, Daniel S, Cayet N, Pecquet M, Raymond F, Pons S, et al. Performance characteristics of the VIDAS SARS-CoV-2 IgM and IgG serological assays. Journal of Clinical Microbiology 2021;59(4):e02292-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Renard 2021 [B] {published data only}
- See first entry for publication details for this study. Renard 2020 [A].
Rijkers 2020 {published data only}
- Rijkers G, Murk JL, Wintermans B, Looy B, den Berge M, Veenemans J, et al. Differences in antibody kinetics and functionality between severe and mild severe acute respiratory syndrome coronavirus 2 infections. Journal of Infectious Diseases 2020;222(8):1265-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rode 2021 [A] {published data only}
- Rode OD, Kurolt IC, Puljiz I, Civljak R, Balent NC, Laskaj R, et al. Antibody response and the clinical presentation of patients with COVID-19 in Croatia: the importance of a two-step testing approach. European Journal of Clinical Microbiology and Infectious Diseases 2021;40(2):261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rode 2021 [B] {published data only}
- See first entry for publication details for this study. Rode 2021 [A].
Rode 2021 [C] {published data only}
- See first entry for publication details for this study. Rode 2021 [A].
Rudolf 2020 [A] {published data only}
- Rudolf F, Kaltenbach H-M, Linnik J, Ruf M-T, Niederhauser C, Nickel B, et al. Clinical characterisation of eleven lateral flow assays for detection of COVID-19 antibodies in a population. medRxiv [Preprint] 2020. [DOI: ]
Rudolf 2020 [B] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [C] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [D] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [E] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [F] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [G] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [H] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [I] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [J] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Rudolf 2020 [K] {published data only}
- See first entry for publication details for this study. Rudolf 2020 [A].
Ruetalo 2020 [A] {published data only}
- Ruetalo N, Businger R, Althaus K, Fink S, Ruoff F, Hamprecht K, et al. Neutralizing antibody response in non-hospitalized SARS-CoV-2 patients. medRxiv [Preprint] 2020. [DOI: ]
Ruetalo 2020 [B] {published data only}
- See first entry for publication details for this study. Ruetalo 2020 [A].
Ruetalo 2020 [C] {published data only}
- See first entry for publication details for this study. Ruetalo 2020 [A].
Schnurra 2020 [A] {published data only}
- Schnurra C, Reiners N, Biemann R, Kaiser T, Trawinski H, Jassoy C. Comparison of the diagnostic sensitivity of SARS-CoV-2 nucleoprotein and glycoprotein-based antibody tests. Journal of Clinical Virology 2020;129:104544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schnurra 2020 [B] {published data only}
- See first entry for publication details for this study. Schnurra 2020 [A].
Schnurra 2020 [C] {published data only}
- See first entry for publication details for this study. Schnurra 2020 [A].
Schnurra 2020 [D] {published data only}
- See first entry for publication details for this study. Schnurra 2020 [A].
Schnurra 2020 [E] {published data only}
- See first entry for publication details for this study. Schnurra 2020 [A].
Schnurra 2020 [F] {published data only}
- See first entry for publication details for this study. Schnurra 2020 [A].
Schnurra 2020 [G] {published data only}
- See first entry for publication details for this study. Schnurra 2020 [A].
Serre‐Miranda 2021 [A] {published data only}
- Serre-Miranda C, Nobrega C, Roque S, Canto-Gomes J, Silva CS, Vieira N, et al. Performance assessment of 11 commercial serological tests for SARS-CoV-2 on hospitalised COVID-19 patients. International Journal of Infectious Diseases 2021;104:661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre-Miranda C, Nobrega C, Roque S, Canto-Gomes J, Silva CS, Vieira N, et al. Performance assessment of 11 commercial serological tests for SARS-CoV-2 on hospitalized COVID-19 patients. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Serre‐Miranda 2021 [B] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [C] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [D] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [E] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [F] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [G] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [H] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [I] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [J] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [K] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Serre‐Miranda 2021 [L] {published data only}
- See first entry for publication details for this study. Serre-Miranda 2021 [A].
Shamsollahi 2020 {published data only}
- Shamsollahi HR, Amini M, Alizadeh S, Nedjat S, Akbari-Sari A, Rezaei M, et al. Assessment of a serological diagnostic kit of sars-cov-2 available in Iran. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PubMed]
- Shamsollahi HR, Amini M, Alizadeh S, Nedjat S, Akbari-Sari A, Rezaei M, et al. Validity of a serological diagnostic kit of SARS-CoV-2 available in Iran. Archives of Iranian Medicine 2020;23(9):629-32. [DOI] [PubMed] [Google Scholar]
Shen 2020a {published data only}
- Shen B, Zheng Y, Zhang X, Zhang W, Wang D, Jin J, et al. Clinical evaluation of a rapid colloidal gold immunochromatography assay for SARS-Cov-2 IgM/IgG. American Journal of Translational Research 2020;12(4):1348-54. [PMC free article] [PubMed] [Google Scholar]
Shen 2020b {published data only}
- Shen L, Wang C, Zhao J, Tang X, Shen Y, Lu M et al. Delayed specific IgM antibody responses observed among COVID-19 patients with severe progression. Emerging Microbes & Infections 2020;9(1):1096-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Soleimani 2021 {published data only}
- Soleimani R, Khourssaji M, Gruson D, Rodriguez-Villalobos H, Berghmans M, Belkhir L, et al. Clinical usefulness of fully automated chemiluminescent immunoassay for quantitative antibody measurements in COVID-19 patients. Journal of Medical Virology 2021;93(3):1465-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterlin 2021 [A] {published data only}
- Sterlin D, Mathian A, Miyara M, Mohr A, Anna F, Claer L, et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
- Sterlin D, Mathian A, Miyara M, Mohr A, Anna F, Claer L, et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Science Translational Medicine 2021;13:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterlin 2021 [B] {published data only}
- See first entry for publication details for this study. Sterlin 2021 [A].
Suhandynata 2020a {published data only}
- Suhandynata RT, Hoffman MA, Kelner MJ, McLawhon RW, Reed SL, Fitzgerald RL. Longitudinal monitoring of SARS-CoV-2 IgM and IgG seropositivity to detect COVID-19. Journal of Applied Laboratory Medicine 2020;5(5):908-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Suhandynata 2020b [A] {published data only}
- Suhandynata RT, Hoffman MA, Kelner MJ, McLawhon RW, Reed SL, Fitzgerald RL. Multi-platform comparison of SARS-CoV-2 serology assays for the detection of COVID-19. Journal of Applied Laboratory Medicine 2020;5(6):1324-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Suhandynata 2020b [B] {published data only}
- See first entry for publication details for this study. Suhandynata 2020b [A].
Suhandynata 2020b [C] {published data only}
- See first entry for publication details for this study. Suhandynata 2020b [A].
Sun 2020 {published data only}
- Sun J, Tang X, Bai R, Liang C, Zeng L, Lin H, et al. The kinetics of viral load and antibodies to SARS-CoV-2. Clinical Microbiology and Infection 2020;26(12):1690.e1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sweeney 2020 {published data only}
- Sweeney N, Merrick B, Galão RP, Pickering S, Botros A, Wilson H, et al. Clinical utility of targeted SARS-CoV-2 serology testing to aid the diagnosis and management of suspected missed, late or post-COVID-19 infection syndromes: results from a pilot service. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Tan 2020 [A] {published data only}
- Tan SS, Saw S, Chew KL, Wang C, Pajarillaga A, Khoo C, et al. Comparative clinical evaluation of the Roche Elecsys and Abbott SARS-CoV-2 serology assays for COVID-19. Archives of Pathology and Laboratory Medicine 2020;145:32-8. [DOI] [PubMed] [Google Scholar]
Tan 2020 [B] {published data only}
- See first entry for publication details for this study. Tan 2020 [A].
Tang 2020 [A] {published data only}
- Tang MS, Hock KG, Logsdon NM, Hayes JE, Gronowski AM, Anderson NW, et al. Clinical performance of two SARS-CoV-2 serologic assays. Clinical Chemistry 2020;66(8):1055-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tang 2020 [B] {published data only}
- See first entry for publication details for this study. Tang 2020 [A].
Tang 2020 [C] {published data only}
- See first entry for publication details for this study. Tang 2020 [A].
Theel 2020 [A] {published data only}
- Theel ES, Harring J, Hilgart H, Granger D. Performance characteristics of four high-throughput immunoassays for detection of IgG antibodies against SARS-CoV-2. Journal of Clinical Microbiology 2020;58(8):e01243-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Theel 2020 [B] {published data only}
- See first entry for publication details for this study. Theel 2020 [A].
Theel 2020 [C] {published data only}
- See first entry for publication details for this study. Theel 2020 [A].
Theel 2020 [D] {published data only}
- See first entry for publication details for this study. Theel 2020 [A].
Thijsen 2020 {published data only}
- Thijsen S, Heron M, Gremmels H, Kieft R, Reusken C, Kremer K, et al. Elevated nucleoprotein-induced interferon-gamma release in COVID-19 patients detected in a SARS-CoV-2 enzyme-linked immunosorbent spot assay. Journal of Infection 2020;81(3):452-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Trabaud 2020 [A] {published data only}
- Trabaud MA, Icard V, Milon MP, Bal A, Lina B, Escuret V. Comparison of eight commercial, high-throughput, automated or ELISA assays detecting SARS-CoV-2 IgG or total antibody. Journal of Clinical Virology 2020;132:104613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Trabaud 2020 [B] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Trabaud 2020 [C] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Trabaud 2020 [D] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Trabaud 2020 [E] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Trabaud 2020 [F] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Trabaud 2020 [G] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Trabaud 2020 [H] {published data only}
- See first entry for publication details for this study. Trabaud 2020 [A].
Traugott 2020 [A] {published data only}
- Traugott M, Aberle SW, Aberle JH, Griebler H, Karolyi M, Pawelka E, et al. Performance of severe acute respiratory syndrome coronavirus 2 antibody assays in different stages of infection: comparison of commercial enzyme-linked immunosorbent assays and rapid Tests. Journal of Infectious Diseases 2020;222(3):362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Traugott 2020 [B] {published data only}
- See first entry for publication details for this study. Tragoutt 2020 [A].
Traugott 2020 [C] {published data only}
- See first entry for publication details for this study. Tragoutt 2020 [A].
Traugott 2020 [D] {published data only}
- See first entry for publication details for this study. Tragoutt 2020 [A].
Traugott 2020 [E] {published data only}
- See first entry for publication details for this study. Tragoutt 2020 [A].
Traugott 2020 [F] {published data only}
- See first entry for publication details for this study. Tragoutt 2020 [A].
Tre‐Hardy 2021 [A] {published data only}
- Tre-Hardy M, Wilmet A, Beukinga I, Favresse J, Dogne JM, Douxfils J, et al. Analytical and clinical validation of an ELISA for specific SARS-CoV-2 IgG, IgA, and IgM antibodies. Journal of Medical Virology 2021;93(2):803-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tre‐Hardy 2021 [B] {published data only}
- See first entry for publication details for this study. Tre-Hardy 2021 [A].
Tuaillon 2020 [A] {published data only}
- Tuaillon E, Bollore K, Pisoni A, Debiesse S, Renault C, Marie S, et al. Detection of SARS-CoV-2 antibodies using commercial assays and seroconversion patterns in hospitalized patients. Journal of Infection 2020;81(2):e39-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tuaillon 2020 [B] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [C] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [D] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [E] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [F] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [G] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [H] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [I] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Tuaillon 2020 [J] {published data only}
- See first entry for publication details for this study. Tuallion 2020 [A].
Valdivia 2020 [A] {published data only}
- Valdivia A, Torres I, Latorre V, Francés-Gómez C, Albert E, Gozalbo-Rovira R, et al. Inference of SARS-CoV-2 spike-binding neutralizing antibody titers in sera from hospitalized COVID-19 patients by using commercial enzyme and chemiluminescent immunoassays. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Valdivia 2020 [B] {published data only}
- See first entry for publication details for this study. Valdivia 2020 [A].
Valdivia 2020 [C] {published data only}
- See first entry for publication details for this study. Valdivia 2020 [A].
Valdivia 2020 [D] {published data only}
- See first entry for publication details for this study. Valdivia 2020 [A].
Van Elslande 2020a [A] {published data only}
- Van Elslande J, Houben E, Depypere M, Brackenier A, Desmet S, Andre E, et al. Diagnostic performance of seven rapid IgG/IgM antibody tests and the Euroimmun IgA/IgG ELISA in COVID-19 patients. Clinical Microbiology and Infection 2020;26(8):1082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Elslande 2020a [B] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020a [C] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020a [D] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020a [E] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020a [F] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020a [G] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020a [H] {published data only}
- See first entry for publication details for this study. Van Elslande 2020a [A].
Van Elslande 2020b [A] {published data only}
- Van Elslande J, Decru B, Jonckheere S, Van Wijngaerden E, Houben E, Vandecandelaere P, et al. Antibody response against SARS-CoV-2 spike protein and nucleoprotein evaluated by four automated immunoassays and three ELISAs. Clinical Microbiology and Infection 2020;26(11):1557.e1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Elslande 2020b [B] {published data only}
- See first entry for publication details for this study. Van Elslande 2020b [A].
Van Elslande 2020b [C] {published data only}
- See first entry for publication details for this study. Van Elslande 2020b [A].
Van Elslande 2020b [D] {published data only}
- See first entry for publication details for this study. Van Elslande 2020b [A].
Van Elslande 2020b [E] {published data only}
- See first entry for publication details for this study. Van Elslande 2020b [A].
Van Elslande 2020b [F] {published data only}
- See first entry for publication details for this study. Van Elslande 2020b [A].
Van Elslande 2020b [G] {published data only}
- See first entry for publication details for this study. Van Elslande 2020b [A].
Velay 2020 [A] {published data only}
- Velay A, Gallais F, Benotmane I, Wendling MJ, Danion F, Collange O, et al. Evaluation of the performance of SARS-CoV-2 serological tools and their positioning in COVID-19 diagnostic strategies. Diagnostic Microbiology and Infectious Disease 2020;98(4):115181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Velay 2020 [B] {published data only}
- See first entry for publication details for this study. Velay 2020 [A].
Velay 2020 [C] {published data only}
- See first entry for publication details for this study. Velay 2020 [A].
Velay 2020 [D] {published data only}
- See first entry for publication details for this study. Velay 2020 [A].
Veyrenche 2021 [A] {published data only}
- Veyrenche N, Bolloré K, Pisoni A, Bedin A-S, Mondain A-M, Ducos J, et al. Diagnosis value of SARS-CoV-2 antigen/antibody combined testing using rapid diagnostic tests at hospital admission. Journal of Medical Virology 2021;93(5):3069-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrenche N, Bolloré K, Pisoni A, Bedin A-S, Mondain A-M, Ducos J, et al. Diagnosis value of SARS-CoV-2 antigen/antibody combined testing using rapid diagnostic tests at hospital admission. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Veyrenche 2021 [B] {published data only}
- See first entry for publication details for this study. Veyrenche 2021 [A].
Veyrenche 2021 [C] {published data only}
- See first entry for publication details for this study. Veyrenche 2021 [A].
Veyrenche 2021 [D] {published data only}
- See first entry for publication details for this study. Veyrenche 2021 [A].
Wang 2020a {published data only}
- Wang P. Combination of serological total antibody and RT-PCR test for detection of SARS-COV-2 infections. Journal of Virological Methods 2020;283:113919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weidner 2020 [A] {published data only}
- Weidner L, Gansdorfer S, Unterweger S, Weseslindtner L, Drexler C, Farcet M, et al. Quantification of SARS-CoV-2 antibodies with eight commercially available immunoassays. Journal of Clinical Virology 2020;129:104540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weidner 2020 [B] {published data only}
- See first entry for publication details for this study. Weidner 2020 [A].
Weidner 2020 [C] {published data only}
- See first entry for publication details for this study. Weidner 2020 [A].
Weidner 2020 [D] {published data only}
- See first entry for publication details for this study. Weidner 2020 [A].
Weidner 2020 [E] {published data only}
- See first entry for publication details for this study. Weidner 2020 [A].
Weidner 2020 [F] {published data only}
- See first entry for publication details for this study. Weidner 2020 [A].
Wellinghausen 2020a [A] {published data only}
- Wellinghausen N, Voss M, Ivanova R, Deininger S. Evaluation of the SARS-CoV-2-IgG response in outpatients by five commercial immunoassays. GMS Infectious Diseases 2020;8:Doc22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wellinghausen 2020a [B] {published data only}
- See first entry for publication details for this study. Wellinghausen 2020a [A].
Wellinghausen 2020a [C] {published data only}
- See first entry for publication details for this study. Wellinghausen 2020a [A].
Wellinghausen 2020a [D] {published data only}
- See first entry for publication details for this study. Wellinghausen 2020a [A].
Wellinghausen 2020a [E] {published data only}
- See first entry for publication details for this study. Wellinghausen 2020a [A].
Wellinghausen 2020b {published data only}
- Wellinghausen N, Plonne D, Voss M, Ivanova R, Frodl R, Deininger S. SARS-CoV-2-IgG response is different in COVID-19 outpatients and asymptomatic contact persons. Journal of Clinical Virology 2020;130:104542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Whitman 2020a [A] {published data only}
- Whitman JD, Hiatt J, Mowery CT, Shy BR, Yu R, Yamamoto TN, et al. Evaluation of SARS-CoV-2 serology assays reveals a range of test performance. Nature Biotechnology 2020;38(10):1174-83. [DOI: 10.1038/s41587-020-0659-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Whitman 2020a [B] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [C] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [D] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [E] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [F] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [G] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [H] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [I] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [J] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020a [K] {published data only}
- See first entry for publication details for this study. Whitman 2020a [A].
Whitman 2020b [A] {published data only}
- Whitman JD, Hiatt J, Mowery CT, Shy BR, Yu R, Yamamoto TN, et al. Test performance evaluation of SARS-CoV-2 serological assays. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.25.20074856] [DOI]
Whitman 2020b [B] {published data only}
- See first entry for publication details for this study. Whitman 2020b [A].
Whitman 2020b [C] {published data only}
- See first entry for publication details for this study. Whitman 2020b [A].
Wolff 2020 [A] {published data only}
- Wolff F, Dahma H, Duterme C, Van den Wijngaert S, Vandenberg O, Cotton F, et al. Monitoring antibody response following SARS-CoV-2 infection: diagnostic efficiency of 4 automated immunoassays. Diagnostic Microbiology and Infectious Disease 2020;98(3):115140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolff 2020 [B] {published data only}
- See first entry for publication details for this study. Wolff 2020 [A].
Wolff 2020 [C] {published data only}
- See first entry for publication details for this study. Wolff 2020 [A].
Wolff 2020 [D] {published data only}
- See first entry for publication details for this study. Wolff 2020 [A].
Wolff 2020 [E] {published data only}
- See first entry for publication details for this study. Wolff 2020 [A].
Wolff 2020 [F] {published data only}
- See first entry for publication details for this study. Wolff 2020 [A].
Wu 2020 [A] {published data only}
- Wu JL, Tseng WP, Lin CH, Lee TF, Chung MY, Huang CH, et al. Four point-of-care lateral flow immunoassays for diagnosis of COVID-19 and for assessing dynamics of antibody responses to SARS-CoV-2. Journal of Infection 2020;81(3):435-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2020 [B] {published data only}
- See first entry for publication details for this study. Wu 2020 [A].
Wu 2020 [C] {published data only}
- See first entry for publication details for this study. Wu 2020 [A].
Wu 2020 [D] {published data only}
- See first entry for publication details for this study. Wu 2020 [A].
Xiang 2020a {published data only}
- Xiang F, Wang X, He X, Peng Z, Yang B, Zhang J, et al. Antibody detection and dynamic characteristics in patients with COVID-19. Clinical Infectious Diseases 2020;71(8):1930-4. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xiao 2020a {published data only}
- Xiao DA, Gao DC, Zhang DS. Profile of specific antibodies to SARS-CoV-2: the first report. Journal of Infection 2020;81(1):147-78. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xiao 2020b [A] {published data only}
- Xiao T, Wang Y, Yuan J, Ye H, Wei L, Liao X, et al. Early viral clearance and antibody kinetics of COVID-19 among asymptomatic carriers. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Xiao 2020b [B] {published data only}
- See first entry for publication details for this study. Xiao 2020b [A].
Xiao 2020b [C] {published data only}
- See first entry for publication details for this study. Xiao 2020b [A].
Xiao 2020b [D] {published data only}
- See first entry for publication details for this study. Xiao 2020b [A].
Yang 2020 [A] {published data only}
- Yang HS, Racine-Brzostek SE, Lee WT, Hunt D, Yee J, Chen Z, et al. SARS-CoV-2 antibody characterization in emergency department, hospitalized and convalescent patients by two semi-quantitative immunoassays. Clinica Chimica Acta 2020;509:117-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2020 [B] {published data only}
- See first entry for publication details for this study. Yang 2020 [A].
Yongchen 2020 {published data only}
- Yongchen Z, Shen H, Wang X, Shi X, Li Y, Yan J, et al. Different longitudinal patterns of nucleic acid and serology testing results based on disease severity of COVID-19 patients. Emerging Microbes and Infections 2020;9(1):833-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020a [A] {published data only}
- Zhang G, Nie S, Zhang Z, Zhang Z. Longitudinal change of severe acute respiratory syndrome coronavirus 2 antibodies in patients with coronavirus disease 2019. Journal of Infectious Diseases 2020;222(2):183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020a [B] {published data only}
- See first entry for publication details for this study. Zhang 2020a [A].
Zhang 2020b [A] {published data only}
- Zhang C, Zhou L, Liu H, Zhang S, Tian Y, Huo J, et al. Establishing a high sensitivity detection method for SARS-CoV-2 IgM/IgG and developing a clinical application of this method. Emerging Microbes & Infections 2020;9(1):2020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020b [B] {published data only}
- See first entry for publication details for this study. Zhang 2020b [A].
Zhang 2020b [C] {published data only}
- See first entry for publication details for this study. Zhang 2020b [A].
Zhao 2020a [A] {published data only}
- Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clinical Infectious Diseases 2020;71:2027-34. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Zhao 2020a [B] {published data only}
- See first entry for publication details for this study. Zhao 2020a [A].
Zhao 2020a [C] {published data only}
- See first entry for publication details for this study. Zhao 2020a [A].
References to studies excluded from this review
Abravanel 2020 {published data only}
- Abravanel F, Miedouge M, Chapuy-Regaud S, Mansuy JM, Izopet J. Clinical performance of a rapid test compared to a microplate test to detect total anti SARS-CoV-2 antibodies directed to the spike protein. Journal of Clinical Virology 2020;130:104528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2020b {published data only}
- Adams ER, Anand R, Andersson MI, Auckland K, Baillie JK, Barnes E, et al. Evaluation of antibody testing for SARS-Cov-2 using ELISA and lateral flow immunoassays. medRxiv [Preprint] 2020. [www.medrxiv.org/content/10.1101/2020.04.15.20066407v1.full.pdf]
Alger 2020 {published data only}
- Alger J, Cafferata ML, Alvarado T, Ciganda A, Corrales A, Desale H, et al. Using prenatal blood samples to evaluate COVID-19 rapid serologic tests specificity. Maternal and Child Health Journal 2020;24(9):1099-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Amanat 2020 {published data only}
- Amanat F, Nguyen T, Chromikova V, Strohmeier S, Stadlbauer D, Javier A, et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.17.20037713] [DOI] [PMC free article] [PubMed]
Amrun 2020 {published data only}
- Amrun SN, Lee CY, Lee B, Fong SW, Young BE, Chee RS, et al. Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS-CoV-2 exposure and disease severity. EBioMedicine 2020;58:102911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Antoine‐Reid 2020 {published data only}
- Antoine-Reid T, Malone J, Maris AS, White-Abell J, Schmitz JE. Cross-comparison of a chemiluminescent platform and a commercial receptor binding domain-based ELISA assay for detecting SARS-CoV-2 IgG. Journal of Applied Laboratory Medicine 2020;5(6):1416-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Arumugam 2020 {published data only}
- Arumugam A, Wong SS. The potential use of unprocessed sample for RT-qPCR detection of COVID-19 without an RNA extraction step. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.06.028811] [DOI]
Ayouba 2020 {published data only}
- Ayouba A, Thaurignac G, Morquin D, Tuaillon E, Raulino R, Nkuba A, et al. Multiplex detection and dynamics of IgG antibodies to SARS-CoV2 and the highly pathogenic human coronaviruses SARS-CoV and MERS-CoV. Journal of Clinical Virology 2020;129:104521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barallat 2020 {published data only}
- Barallat J, Fernández-Rivas G, Quirant-Sánchez B, González V, Doladé M, Martinez-Caceres E, et al. Seroprevalence of SARS-CoV-2 IgG Specific Antibodies among Healthcare Workers in the Northern Metropolitan Area of Barcelona, Spain, after the first pandemic wave. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Baron 2020 {published data only}
- Baron RC, Risch L, Weber M, Thiel S, Grossmann K, Wohlwend N, et al. Frequency of serological non-responders and false-negative RT-PCR results in SARS-CoV-2 testing: a population-based study. Clinical Chemistry and Laboratory Medicine 2020;58(12):2131-40. [DOI] [PubMed] [Google Scholar]
Batra 2020 {published data only}
- Batra R, Olivieri LG, Rubin D, Vallari A, Pearce S, Olivo A, et al. A comparative evaluation between the Abbott Panbio COVID-19 IgG/IgM rapid test device and Abbott Architect SARS CoV-2 IgG assay. Journal of Clinical Virology 2020;132:104645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Becker 2020 {published data only}
- Becker M, Strengert M, Junker D, Kerrinnes T, Kaiser PD, Traenkle B, et al. Going beyond clinical routine in SARS-CoV-2 antibody testing - A multiplex corona virus antibody test for the evaluation of cross-reactivity to endemic coronavirus antigens. medRxiv [Preprint] 2020. [DOI: ]
Bendavid 2020 {published data only}
- Bendavid E, Mulaney B, Sood N, Shah S, Ling E, Bromley-Dulfano R, et al. COVID-19 antibody seroprevalence in Santa Clara County, California. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Black 2020 {published data only}
- Black MA, Shen G, Feng X, Garcia B, Feng Y, Vasudevaraja V, et al. Analytical performance of lateral flow immunoassay for SARS-CoV-2 exposure screening on venous and capillary blood samples. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Bortz 2020 {published data only}
- Bortz RH, Florez C, Laudermilch E, Wirchnianski AS, Lasso G, Malonis RJ, et al. Development, clinical translation, and utility of a COVID-19 antibody test with qualitative and quantitative readouts. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Brandstetter 2020 {published data only}
- Brandstetter S, Roth S, Harner S, Buntrock-Dopke H, Toncheva AA, Borchers N, et al. Symptoms and immunoglobulin development in hospital staff exposed to a SARS-CoV-2 outbreak. Pediatric Allergy and Immunology 2020;31(7):841-7. [DOI] [PubMed] [Google Scholar]
Brantley 2020 {published data only}
- Brantley HL, Yoo RM, Jones GI, Stock MA, Park PJ, Sheils NE, et al. Variation across population subgroups of COVID-19 antibody testing performance. medRxiv [Preprint] 2020. [DOI: ]
Brecher 2020 {published data only}
- Brecher SM, Dryjowicz-Burek J, Yu H, Campbell S, Ratcliffe N, Gupta K. Patients with common cold coronaviruses tested negative for IgG antibody to SARS-CoV-2. Journal of Clinical Microbiology 2020;58(8):e01029-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bruni 2020 {published data only}
- Bruni M, Cecatiello V, Diaz-Basabe A, Lattanzi G, Mileti E, Monzani S, et al. Persistence of anti-SARS-CoV-2 antibodies in non-hospitalized COVID-19 convalescent health care workers. Journal of Clinical Medicine 2020;9(10):3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bryan 2020b {published data only}
- Bryan A, Pepper G, Wener MH, Fink SL, Morishima C, Chaudhary A, et al. Performance characteristics of the Abbott Architect SARS-CoV-2 IgG assay and seroprevalence in Boise, Idaho. Journal of Clinical Microbiology 2020;58(8):e00941-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Buntinx 2020 {published data only}
- Buntinx F, Claes P, Gulikers M, Verbakel JY, De Lepeleire J, Van der Elst M, et al. Early experiences with antibody testing in a Flemish nursing home during an acute COVID-19 outbreak: a retrospective cohort study. medRxiv [Preprint] 2020. [DOI: ]
Burbelo 2020 {published data only}
- Burbelo PD, Riedo FX, Morishima C, Rawlings S, Smith D, Das S, et al. Detection of nucleocapsid antibody to SARS-CoV-2 is more sensitive than antibody to spike protein in COVID-19 patients. MedRxiv [Preprint] 2020. [DOI: ]
Byrnes 2020 {published data only}
- Byrnes JR, Zhou XX, Lui I, Elledge SK, Glasgow JE, Lim SA, et al. Competitive SARS-CoV-2 serology reveals most antibodies targeting the spike receptor-binding domain compete for ACE2 binding. mSphere 2020;5(5):e00802-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cai 2020 {published data only}
- Cai XF, Chen J, Hu JL, Long QX, Deng HJ, Fan K, et al. A peptide-based magnetic chemiluminescence enzyme immunoassay for serological diagnosis of Coronavirus Disease 2019 (COVID-19). Journal of Infectious Diseases 2020;222:189-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cassaniti 2020 {published data only}
- Cassaniti I, Novazzi F, Giardina F, Salivaro F, Sachs M, Perlini S, et al. Performance of VivaDiagTM COVID-19 IgM/IgG Rapid Test is inadequate for diagnosis of COVID-19 in acute patients referring to emergency room department. Journal of Medical Virology 2020;92(10):1724-7. [DOI: 10.1002/jmv.25800] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chatzidimitriou 2020 {published data only}
- Chatzidimitriou M, Chatzopoulou F, Gavriilaki E, Chatzivasileiou P, Rousis D, Meletis G, et al. Repeated negative serological testing in otherwise healthy patients with COVID-19. Journal of Infectious Diseases 2020;223:924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020a {published data only}
- Chen Z, Zhang Z, Zhai X, Li Y, Lin L, Zhao H, et al. Rapid and sensitive detection of anti-SARS-CoV-2 IgG using lanthanide-doped nanoparticles-based lateral flow immunoassay. Analytical Chemistry 2020;92(10):7226-31. [DOI] [PubMed] [Google Scholar]
Choe 2020 {published data only}
- Choe JY, Kim JW, Kwon HH, Hong HL, Jung CY, Jeon CH, et al. Diagnostic performance of immunochromatography assay for rapid detection of IgM and IgG in coronavirus disease 2019. Journal of Medical Virology 2020;92(11):2567-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chughtai 2020 {published data only}
- Chughtai OR, Batool H, Khan MD, Ashraf S. Seroconversion in newly diagnosed cases of coronavirus disease. Journal of the College of Physicians and Surgeons--Pakistan 2020;30(8):801-4. [DOI] [PubMed] [Google Scholar]
Colavita 2020 {published data only}
- Colavita F, Brogi A, Lapa D, Bordi L, Matusali G, Meschi S, et al. Evaluation of ELISA tests for the qualitative determination of IgG, IgM and IgA to SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ]
Comar 2020 {published data only}
- Comar M, Brumat M, Concas MP, Argentini G, Bianco A, Bicego L, et al. COVID-19 experience: first Italian survey on healthcare staff members from a mother-child research hospital using combined molecular and rapid immunoassays test. MedRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.19.20071563] [DOI]
Dahlke 2020 {published data only}
- Dahlke C, Heidepriem J, Kobbe R, Santer R, Koch T, Fathi A. Distinct early IgA profile may determine severity of COVID-19 symptoms: an immunological case series. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.14.20059733] [DOI]
Das 2020 {published data only}
- Das MK, Chaudhary A, Bryan A, Wener MH, Fink SL, Morishima C. Rapid screening evaluation of SARS-CoV-2 IgG assays using z-scores to standardize results. Emerging Infectious Diseases 2020;26(10):2501-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Demey 2020 {published data only}
- Demey B, Daher N, Francois C, Lanoix JP, Duverlie G, Castelain S, et al. Dynamic profile for the detection of anti-SARS-CoV-2 antibodies using four immunochromatographic assays. Journal of Infection 2020;81:e6-e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Di Lorenzo 2020 {published data only}
- Di Lorenzo G, Toniolo P, Lurani C, Foresti L, Carrisi C. Evaluating the adequacy of Prima Covid-19 IgG/IgM Rapid Test for the assessment of exposure to SARS-CoV-2 virus. medRxiv [Preprint] 2020. [DOI: ]
Dittadi 2020 {published data only}
- Dittadi R, Afshar H, Carraro P. The early antibody response to SARS-Cov-2 infection. Clinical Chemistry and Laboratory Medicine 2020;58(10):e201-3. [DOI] [PubMed] [Google Scholar]
Dobaño 2020 {published data only}
- Dobaño C, Vidal M, Santano R, Jiménez A, Chi J, Barrios D, et al. Highly sensitive and specific multiplex antibody assays to quantify immunoglobulins M, A and G against SARS-CoV-2 antigens. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Dohla 2020 {published data only}
- Dohla M, Boesecke C, Schulte B, Diegmann C, Sib E, Richter E, et al. Rapid point-of-care testing for SARS-CoV-2 in a community screening setting shows low sensitivity. Public Health 2020;182:170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Du 2020 {published data only}
- Du Z, Zhu F, Guo F, Yang B, Wang T. Detection of antibodies against SARS-CoV-2 in patients with COVID-19. Journal of Medical Virology 2020;92(10):1735-8. [DOI: 10.1002/jmv.25820] [DOI] [PMC free article] [PubMed] [Google Scholar]
Edouard 2020 {published data only}
- Edouard S, Colson P, Melenotte C, De Pinto F, Thomas L, La Scola B, et al. Evaluating the serological status of COVID-19 patients using an indirect immunofluorescent assay, France. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Erikstrup 2020 {published data only}
- Erikstrup C, Hother CE, Vestager Pedersen OB, Mølbak K, Skov RL, Holm DK, et al. Estimation of SARS-CoV-2 infection fatality rate by real-time antibody screening of blood donors. medRxiv [Preprint] 2020. [https://doi.org/10.1101/2020.04.24.20075291] [DOI] [PMC free article] [PubMed]
Espino 2020 {published data only}
- Espino AM, Pantoja P, Sariol CA. Validation and performance of a quantitative IgG assay for the screening of SARS-CoV-2 antibodies. bioRxiv [Preprint] 2020. [DOI: ]
Fong 2020 {published data only}
- Fong CH, Cai JP, Dissanayake TK, Chen LL, Choi CY, Wong LH, et al. Improved detection of antibodies against SARS-CoV-2 by microsphere-based antibody assay. International Journal of Molecular Sciences 2020;21:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Freeman 2020 {published data only}
- Freeman B, Lester S, Mills L, Rasheed MA, Moye S, Abiona O, et al. Validation of a SARS-CoV-2 spike ELISA for use in contact investigations and serosurveillance. bioRxiv [Preprint] 2020. [DOI: ]
Garcia‐Basteiro 2020 {published data only}
- Garcia-Basteiro AL, Moncunill G, Tortajada M, Vidal M, Guinovart C, Jiménez A, et al. Seroprevalence of antibodies against SARS-CoV-2 among health care workers in a large Spanish reference hospital. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Garcia Garmendia 2020 {published data only}
- Garcia Garmendia JL, Ramirez Arcos M, Barrero Almodovar AE, Chavez Caballero M, Jorge Amigo V, Serrano Martino MC. Viral detection and serological response in critically ill patients with SARS-CoV-2. Implications for isolation withdrawal [Detección viral y respuesta serológica en pacientes críticos intubados con SARS-CoV-2. Implicaciones para retirada de aislamiento]. Medicina Intensiva 2020;44(9):586-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grzelak 2020 {published data only}
- Grzelak L, Temmam S, Planchais C, Demeret C, Huon C, Guivel F, et al. SARS-CoV-2 serological analysis of COVID-19 hospitalized patients, pauci-symptomatic individuals and blood donors. medRxiv [Preprint] 2020. [DOI: ]
Guo 2020a {published data only}
- Guo L, Ren L, Yang S, Xiao M, Chang, Yang F, et al. Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clinical Infectious Diseases 2020;71:778-85. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guo 2020c {published data only}
- Guo X, Guo Z, Duan C, Chen Z, Wang G, Lu Y, et al. Long- term persistence of IgG antibodies in SARS-CoV infected healthcare workers. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.02.12.20021386] [DOI]
Guthmiller 2020 {published data only}
- Guthmiller JJ, Stovicek O, Wang J, Changrob S, Li L, Halfmann P, et al. SARS-CoV-2 infection severity is linked to superior humoral immunity against the spike. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
He 2020 {published data only}
- He J, Hu P, Gao Y, Zheng S, Xu C, Liu R, et al. Comparison and application of different immunoassay methods for the detection of SARS-CoV-2. Journal of Medical Virology 2020;92(11):2777-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
He 2020a {published data only}
- He Y, Luo J, Yang J, Song J, Wei L, Ma W. Value of viral nucleic acid in sputum and feces and specific IgM/IgG in serum for the diagnosis of coronavirus disease 2019. Frontiers in Cellular and Infection Microbiology 2020;10:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hou 2020 {published data only}
- Hou H, Wang T, Zhang B, Luo Y, Mao L, Wang F, et al. Detection of IgM and IgG antibodies in patients with coronavirus disease 2019. Clinical & Translational Immunology 2020;9(5):e01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2020a {published data only}
- Huang M, Lu QB, Zhao H, Zhang Y, Sui Z, Fang L, et al. Temporal antibody responses to SARS-CoV-2 in patients of coronavirus disease 2019. Cell Discovery 2020;6:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2020b {published data only}
- Huang Z, Chen H, Xue M, Huang H, Zheng P, Luo W, et al. Characteristics and roles of severe acute respiratory syndrome coronavirus 2-specific antibodies in patients with different severities of coronavirus 19. Clinical and Experimental Immunology 2020;202(2):210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2020c {published data only}
- Huang J, Mao T, Li S, Wu L, Xu X, Li H, et al. Long period dynamics of viral load and antibodies for SARS-CoV-2 infection: an observational cohort study. medRxiv [Preprint] 2020. [DOI: ]
Hung 2020 {published data only}
- Hung IF-N, Cheng VC-C, Li X, Tam AR, Hung DL-L, Chiu KH-Y, et al. SARS-CoV-2 shedding and seroconversion among passengers quarantined after disembarking a cruise ship: a case series. Lancet Infectious Diseases 2020;20(9):1051-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Imam 2020 {published data only}
- Imam M, Khawaja S, Naz A, Siddiqui A, Nafees TS, Younus A, et al. Performance assessment of first-generation anti-SARS-CoV-2 serological assays. medRxiv [Preprint] 2020. [DOI: ]
Infantino 2020 {published data only}
- Infantino M, Grossi V, Lari B, Bambi R, Perri A, Manneschi M, et al. Diagnostic accuracy of an automated chemiluminescent immunoassay for anti-SARS-CoV-2 IgM and IgG antibodies: an Italian experience. Journal of Medical Virology 2020;92:1671-75. [DOI: 10.1002/jmv.25932] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jia 2020 {published data only}
- Jia X, Zhang P, Tian Y, Wang J, Zeng H, Wang J, et al. Clinical significance of IgM and IgG test for diagnosis of highly suspected COVID-19 infection. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Jiang 2020b {published data only}
- Jiang HW, Li Y, Zhang HN, Wang W, Men D, Yang X, et al. Global profiling of SARS-CoV-2 specific IgG/ IgM responses of convalescents using a proteome microarray. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.20.20039495] [DOI] [PMC free article] [PubMed]
Karp 2020 {published data only}
- Karp DG, Cuda D, Tandel D, Danh K, Robinson PV, Seftel D, et al. Sensitive and specific detection of SARS-CoV-2 antibodies using a high-throughput, fully automated liquid-handling robotic system. SLAS Technology 2020;25(6):545-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karp 2020a {published data only}
- Karp DG, Danh K, Seftel D, Robinson P, Tsai CT. A serological assay to detect SARS-CoV-2 antibodies in at-home collected finger-prick dried blood spots. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Klumpp‐Thomas 2020 {published data only}
- Klumpp-Thomas C, Kalish H, Drew M, Hunsberger S, Snead K, Fay MP, et al. Standardization of enzyme-linked immunosorbent assays for serosurveys of the SARS-CoV-2 pandemic using clinical and at-home blood sampling. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Kruttgen 2020 {published data only}
- Kruttgen A, Cornelissen CG, Dreher M, Hornef M, Imohl M, Kleines M. Comparison of four new commercial serologic assays for determination of SARS-CoV-2 IgG. Journal of Clinical Virology 2020;128:104394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kushemererwa 2020 {published data only}
- Kushemererwa GE, Kayongo I, Semanda P, Nansumba H, Tadeo I, Namulindwa C, et al. Combination of antibody based rapid diagnostic tests used in an algorithm may improve their performance in SARS CoV-2 diagnosis. medRxiv [Preprint] 2020. [DOI: ]
Lahner 2020 {published data only}
- Lahner E, Dilaghi E, Prestigiacomo C, Alessio G, Marcellini L, Simmaco M, et al. Prevalence of SARS-Cov-2 infection in health workers (HWs) and diagnostic test performance: the experience of a teaching hospital in central Italy. International Journal of Environmental Research and Public Health 2020;17(12):4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lapuente 2020 {published data only}
- Lapuente D, Maier C, Irrgang P, Huebner J, Peter SA, Hoffmann M, et al. Rapid response flow cytometric assay for the detection of antibody responses to sars-cov-2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Lee 2020 {published data only}
- Lee YL, Liao CH, Liu PY, Cheng CY, Chung MY, Liu CE, et al. Dynamics of anti-SARS-Cov-2 IgM and IgG antibodies among COVID-19 patients. Journal of Infection 2020;81(2):e55-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lei 2020 {published data only}
- Lei Q, Li Y, Hou H, Wang F, Ouyang Z, Zhang Y, et al. Antibody dynamics to SARS-CoV-2 in asymptomatic and mild COVID-19 patients. medRxiv [Preprint] 2020. [DOI: ]
Li 2020a {published data only}
- Li Z, Yi Y, Luo X, Xiong N, Liu Y, Li S, et al. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. Journal of Medical Virology 2020;92:1518-24. [DOI: 10.1002/jmv.25727] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020b {published data only}
- Li K, Wu M, Huang B, Zhong A, Li L, Cai Y, et al. The dynamic changes of antibodies against SARS-CoV-2 during the infection and recovery of COVID-19. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Li 2020c {published data only}
- Li T, Wang L, Wang H, Li X, Zhang S, Xu Y, et al. Serum SARS-COV-2 nucleocapsid protein: a sensitivity and specificity early diagnostic marker for SARS-COV-2 infection. Frontiers in Cellular and Infection Microbiology 2020;10:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020d {published data only}
- Li Y, He Q, Yu R, Jiang H, Wang W, Feng D, et al. Highlighted prospects of an IgM/IgG antibodies test in identifying individuals with asymptomatic SARS CoV-2 infection. Archives of Pathology and Laboratory Medicine 2020;145:39-45. [DOI] [PubMed] [Google Scholar]
Li 2020e {published data only}
- Li H, Liu Z, He Y, Qi Y, Chen J, Ma Y, et al. A new and rapid approach for detecting COVID-19 based on S1 protein fragments. Clinical and Translational Medicine 2020;10(2):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2020 {published data only}
- Lin D, Liu L, Zhang M, Hu Y, Yang Q, Guo J, et al. Evaluations of the serological test in the diagnosis of 2019 novel coronavirus (SARS-CoV-2) infections during the COVID-19 outbreak. European Journal of Clinical Microbiology and Infectious Diseases 2020;17:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Linares 2020 {published data only}
Lippi 2020 {published data only}
- Lippi G, Salvagno GL, Pegoraro M, Militello V, Caloi C, Peretti A, et al. Preliminary evaluation of Roche Cobas Elecsys Anti-SARS-CoV-2 chemiluminescence immunoassay. Clinical Chemistry and Laboratory Medicine 2020;58:e251-3. [DOI] [PubMed] [Google Scholar]
Liu 2020d {published data only}
- Liu Y, Liu Y, Diao B, Ren F, Wang Y, Ding J, et al. Diagnostic indexes of a rapid IgG/IgM combined antibody test for SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ]
Liu 2020e {published data only}
- Liu R, Liu X, Yuan L, Han H, Shereen MA, Zhen J, et al. Analysis of adjunctive serological detection to nucleic acid test for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection diagnosis. International Immunopharmacology 2020;86:106746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2020f {published data only}
- Liu R, Liu X, Han H, Shereen MA, Niu Z, Li D, et al. The comparative superiority of IgM-IgG antibody test to real-time reverse transcriptase PCR detection for SARS-CoV-2 infection diagnosis. medRxiv [Preprint] 2020. [DOI: ]
Lopez de la Iglesias 2020 {published data only}
- Lopez de la Iglesia J, Fernandez-Villa T, Rivero A, Carvajal A, Bay Simon E, Martinez Martinez M, et al. Predictive factors of COVID-19 in patients with negative RT-qPCR. Semergen 2020;46(Suppl 1):6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ma 2020a {published data only}
- Ma H, Zeng W, He H, Zhao D, Jiang D, Zhou P, et al. Serum IgA, IgM, and IgG responses in COVID-19. Cellular & Molecular Immunology 2020;17(7):773-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
McAndrews 2020 {published data only}
- McAndrews KM, Dowlatshahi DP, Hensel J, Ostrosky-Zeichner LL, Papanna R, LeBleu VS, et al. Identification of IgG antibody response to SARS-CoV-2 spike protein and its receptor binding domain does not predict rapid recovery from COVID-19. medRxiv [Preprint] 2020. [DOI: ]
Morley 2020 {published data only}
- Morley GL, Taylor S, Jossi S, Perez-Toledo M, Faustini SE, Marcial-Juarez E, et al. Sensitive detection of SARS-CoV-2-specific antibodies in dried blood spot samples. Emerging Infectious Diseases 2020;26(12):2970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Munitz 2020 {published data only}
- Munitz A, Edry-Botzer L, Itan M, Tur-Kaspa R, Dicker D, Markovitch D, et al. SARS-CoV-2 serological testing using electrochemiluminescence reveals arapid onset of seroconversion in severe COVID-19 patients. medRxiv [Preprint] 2020. [DOI: ]
Mutnal 2020 {published data only}
- Mutnal MB, Mohammad AA, Arroliga AC, Hua Y, Wang L, Koss W, et al. Role of Anti-SARS-CoV-2 antibodies in different cohorts: Can they provide clues for appropriate patient triaging? bioRxiv [Preprint] 2020. [DOI: ]
Nath 2020 {published data only}
- Nath H, Mallick A, Roy S, Sukla S, Basu K, De A, et al. Dengue antibodies can cross-react with SARS-CoV-2 and vice versa-Antibody detection kits can give false-positive results for both viruses in regions where both COVID-19 and Dengue co-exist. medRxiv [Preprint] 2020. [DOI: ]
Nguyen 2020a {published data only}
- Nguyen NN, Mutnal MB, Gomez RR, Pham HN, Nguyen LT, Koss W, et al. Correlation of ELISA based with random access serologic immunoassays for identifying adaptive immune response to SARS-CoV-2. medRxiv [Preprint] 2020;10. [DOI: ]
Nie 2020 {published data only}
- Nie J, Li Q, Wu J, Zhao C, Hao H, Liu H, et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerging Microbes and Infections 2020;9(1):680-6. [DOI: 10.1080/22221751.2020.1743767 10.1080/22221751.2020.1743767.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Norman 2020 {published data only}
- Norman M, Gilboa T, Ogata AF, Maley AM, Cohen L, Busch EL, et al. Ultrasensitive high-resolution profiling of early seroconversion in patients with COVID-19. Nature Biomedical Engineering 2020;4(12):1180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nuccetelli 2020 {published data only}
- Nuccetelli M, Pieri M, Grelli S, Ciotti M, Miano R, Andreoni M, et al. SARS-CoV-2 infection serology: a useful tool to overcome lockdown? Cell Death Discovery 2020;6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Okba 2020a {published data only}
- Okba N, Muller MA, Li W, Wang C, GeurtsvanKessel CH, Corman VM, et al. SARS-CoV-2 specific antibody responses in COVID-19 patients. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.18.20038059] [DOI]
- Okba NM, Muller MA, Li W, Wang C, GeurtsvanKessel CH, Corman VM, et al. Severe acute respiratory syndrome coronavirus 2-specific antibody responses in coronavirus disease 2019 patients. Emerging Infectious Diseases 2020;26(7):1478-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Muller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020;581:465-9. [DOI] [PubMed] [Google Scholar]
Olivares 2020 {published data only}
Ossareh 2020 {published data only}
- Ossareh S, Bagheri M, Abbasi M, Abolfathi S, Bohlooli A. Role of screening for COVID-19 in hemodialysis wards, results of a single center study. Iranian Journal of Kidney Diseases 2020;14(5):389-98. [PubMed] [Google Scholar]
Ozturk 2020 {published data only}
- Ozturk T, Howell C, Benameur K, Ramonell RP, Cashman K, Pirmohammed S, et al. Cross-sectional IgM and IgG profiles in SARS-CoV-2 infection. medRxiv [Preprint] 2020. [DOI: ]
Paradiso 2020a {published data only}
- Paradiso AV, De Summa S, Loconsole D, Procacci V, Sallustio A, Centrone F, et al. Clinical meanings of rapid serological assay in patients tested for SARS-Co2 RT-PCR. medRxiv [Preprint] 2020. [DOI: ]
Paradiso 2020b {published data only}
- Paradiso AV, De Summa S, Silvestris N, Tommasi S, Tufaro A, De Palma G, et al. Rapid serological tests have a role in asymptomatic health workers COVID-19 screening. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.15.20057786] [DOI]
Patel 2020 {published data only}
- Patel MM, Thornburg NJ, Stubblefield WB, Talbot HK, Coughlin MM, Feldstein LR, et al. Change in antibodies to SARS-CoV-2 over 60 days among health care personnel in Nashville, Tennessee. JAMA 2020;17:1781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pellanda 2020 {published data only}
- Pellanda LC, da Ros Wendland EM, McBride AJA, Tovo-Rodrigues L, Ferreira MRA, Dellagostin OA, et al. Sensitivity and specificity of a rapid test for assessment of exposure to SARS-CoV-2 in a community-based setting in Brazil. medRxiv [Preprint] 2020. [DOI: ]
Perkmann 2020 {published data only}
- Perkmann T, Perkmann-Nagele N, Breyer MK, Breyer-Kohansal R, Burghuber OC, Hartl S, et al. Side-by-side comparison of three fully automated SARS-CoV-2 antibody assays with a focus on specificity. Clinical Chemistry 2020;66(11):1405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Phan 2020 {published data only}
- Phan IQ, Subramanian S, Kim D, Carter L, King N, Anishchenko I, et al. In silico detection of SARS-CoV-2 specific B-cell epitopes and validation in ELISA for serological diagnosis of COVID-19. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Plebani 2020 {published data only}
- Plebani M, Padoan A, Negrini D, Carpinteri B, Sciacovelli L. Diagnostic performances and thresholds: The key to harmonization in serological SARS-CoV-2 assays? Clinica Chimica Acta 2020;509:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prince 2020 {published data only}
- Prince HE, Givens TS, Lape-Nixon M, Clarke NJ, Schwab DA, Batterman HJ, et al. Detection of SARS-CoV-2 IgG targeting nucleocapsid or spike protein by four high-throughput immunoassays authorized for emergency use. Journal of Clinical Microbiology 2020;58(11):e01742-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Qian 2020 {published data only}
Qu 2020 {published data only}
- Qu J, Wu C, Li X, Zhang G, Jiang Z, Li X, et al. Profile of immunoglobulin G and IgM antibodies against Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clinical Infectious Diseases 2020;71(16):2255-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rabets 2020a {published data only}
- Rabets A, Bila G, Grytsko R, Samborsky M, Rebets Y, Vari S, Pagneux Q, et al. Development of antibodies to pan-coronavirus spike peptides in convalescent COVID-19 patients. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Randad 2020 {published data only}
- Randad PR, Pisanic N, Kruczynski K, Manabe YC, Thomas D, Pekosz A, et al. COVID-19 serology at population scale: SARS-CoV-2-specific antibody responses in saliva. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Robledo Gomez 2020 {published data only}
- Robledo Gomez AY, Hunt NR, Chambliss AB, Pham HP, Emerson JF, Marin MJ. An initial evaluation of the agreement between two SARS-CoV-2 serologic assays. Journal of Applied Laboratory Medicine 2020;5(5):1139-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rosado 2020 {published data only}
- Rosado J, Pelleau S, Cockram C, Hélène Merkling S, Nekkab N, Demeret C, et al. Serological signatures of SARS-CoV-2 infection: Implications for antibody-based diagnostics. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Rosendal 2020 {published data only}
- Rosendal E, Wigren J, Groening R, Yongdae K, Nilsson E, Sharma A, et al. Detection of asymptomatic SARS-CoV-2 exposed individuals by a sensitive S-based ELISA. medRxiv [Preprint] 2020. [DOI: ]
Rushworth 2020 {published data only}
- Rushworth SA, Johnson BB, Ashurst K, Davidson R, Paddy P, Mistry JJ, et al. Performance and health economic evaluation of the Mount Sinai COVID-19 serological assay identifies modification of thresholding as necessary to maximise specificity of the assay. medRxiv [Preprint] 2020. [DOI: ]
Santos 2020 {published data only}
- Santos VAD, Rafael MM, Sabino EC, Duarte A. Sensitivity of the Wondfo One Step COVID-19 test using serum samples. Clinics 2020;75:e2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Serrano 2020 {published data only}
- Serrano MM, Rodriguez DN, Palop NT, Arenas RO, Cordoba MM, Mochon MDO, et al. Comparison of commercial lateral flow immunoassays and ELISA for SARS-CoV-2 antibody detection. Journal of Clinical Virology 2020;129:104529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shaw 2020 {published data only}
- Shaw AM, Hyde C, Merrick B, James-Pemberton P, Squires BK, Olkhov RV, et al. Real-world evaluation of a novel technology for quantitative simultaneous antibody detection against multiple SARS-CoV-2 antigens in a cohort of patients presenting with COVID-19 syndrome. Analyst 2020;145(16):5638-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Solodky 2020 {published data only}
- Solodky ML, Galvez C, Russias B, Detourbet P, N'Guyen-Bonin V, Herr AL, et al. Lower detection rates of SARS-COV2 antibodies in cancer patients versus health care workers after symptomatic COVID-19. Annals of Oncology 2020;31(8):1087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Song 2020 {published data only}
- Song KH, Kim DM, Lee H, Ham SY, Oh SM, Jeong H, et al. Dynamics of viral load and anti-SARS-CoV-2 antibodies in patients with positive RT-PCR results after recovery from COVID-19. Korean Journal of Internal Medicine 2020;36:11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spicuzza 2020 {published data only}
- Spicuzza L, Montineri A, Manuele R, Crimi C, Pistorio MP, Campisi R, et al. Reliability and usefulness of a rapid IgM-IgG antibody test for the diagnosis of SARS-CoV-2 infection: A preliminary report. Journal of Infection 2020;81(2):e53-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Staines 2020 {published data only}
- Staines HM, Kirwan DE, Clark DJ, Adams ER, Augustin Y, Byrne RL, et al. Dynamics of IgG seroconversion and pathophysiology of COVID-19 infections. medRxiv [Preprint] 2020. [DOI: ]
Steiner 2020 {published data only}
- Steiner DJ, Cognetti JS, Luta EP, Klose AM, Bucukovski J, Bryan MR, et al. Array-based analysis of SARS-CoV-2, other coronaviruses, and influenza antibodies in convalescent COVID-19 patients. Biosensors and Bioelectronics 2020;169:112643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Strömer 2020 {published data only}
- Strömer A, Grobe O, Rose R, Fickenscher H, Lorentz T, Krumbholz A. Diagnostic accuracy of six commercial SARS-CoV-2 IgG/total antibody assays and identification of SARS-CoV-2 neutralizing antibodies in convalescent sera. medRxiv [Preprint] 2020. [DOI: ]
Sun 2020a {published data only}
- Sun B, Feng Y, Mo X, Zheng P, Wang Q, Li P, et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerging Microbes & Infections 2020;9(1):940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tan 2020 {published data only}
- Tan X, Krel M, Dolgov E, Park S, Li X, Wu W, et al. Rapid and quantitative detection of SARS-CoV-2 specific IgG for convalescent serum evaluation. Biosensors and Bioelectronics 2020;169:112572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tan 2020a {published data only}
- Tan W, Lu Y, Zhang J, Wang J, Dan Y, Tan Z, et al. Viral kinetics and antibody responses in patients with COVID-19. medRxiv [Preprint] 2020. [DOI: ]
Teng 2020 {published data only}
- Teng J, Dai J, Su Y, Zhou Z, Chi H, Wan L, et al. Detection of IgM and IgG antibodies against SARS-CoV-2 in patients with autoimmune diseases. Lancet Rheumatology 2020;2(7):e384-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thevis 2020 {published data only}
- Thevis M, Knoop A, Schaefer MS, Dufaux B, Schrader Y, Thomas A, et al. Can dried blood spots (DBS) contribute to conducting comprehensive SARS-CoV-2 antibody tests? Drug Testing and Analysis 2020;12(7):994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
To 2020a {published data only}
- To KK, Tsang OT, Leung WS, Tam AR, Wu TC, Lung DC, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infectious Diseases 2020;20(5):565-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tre‐Hardy 2020 {published data only}
- Tre-Hardy M, Blairon L, Wilmet A, Beukinga I, Malonne H, Dogne JM, et al. The role of serology for COVID-19 control: Population, kinetics and test performance do matter. Journal of Infection 2020;81(2):e91-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Valenti 2020 {published data only}
- Valenti L, Bergna A, Pelusi S, Facciotti F, Lai A, Tarkowski M, et al. SARS-CoV-2 seroprevalence trends in healthy blood donors during the COVID-19 Milan outbreak. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Van Praet 2021 {published data only}
- Van Praet JT, Coene AS, Van De Moortele K, Descheemaeker P, Reynders M. Comparison of four commercial SARS-CoV-2 IgG immuno-assays in RT-PCR negative patients with suspect CT findings. Infection 2021;49(1):145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Varadhachary 2020 {published data only}
- Varadhachary A, Chatterjee D, Garza J, Garr RP, Foley C, Letkeman AF, et al. Salivary anti-SARS-CoV-2 IgA as an accessible biomarker of mucosal immunity against COVID-19. medRxiv [Preprint] 2020. [DOI: ]
Vasarhelyi 2020 {published data only}
- Vasarhelyi B, Kristof K, Ostorhazi E, Szabo D, Prohaszka Z, Merkely B. The diagnostic value of rapid anti IgM and IgG detecting tests in the identification of patients with SARS CoV-2 virus infection. Orvosi Hetilap 2020;161(20):807-12. [DOI] [PubMed] [Google Scholar]
Vidal‐Anzardo 2020 {published data only}
- Vidal-Anzardo M, Solis G, Solari L, Minaya G, Ayala-Quintanilla B, Astete-Cornejo J, et al. Evaluation of a rapid serological test for detection of IgM and igG antibodies against SARS-CoV-2 under field conditions. Revista Peruana de Medicina Experimental y Salud Publica 2020;37(2):203-9. [DOI] [PubMed] [Google Scholar]
Villarreal 2020 {published data only}
- Villarreal A, Rangel G, Zhang X, Wong D, Britton G, Fernandez PL, et al. Performance of a point of care test for detecting IgM and IgG antibodies against SARS-CoV-2 and seroprevalence in blood donors and health care workers in Panama. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Wajnberg 2020 {published data only}
- Wajnberg A, Mansour M, Leven E, Bouvier NM, Patel G, Firpo A, et al. Humoral immune response and prolonged PCR positivity in a cohort of 1343 SARS-CoV 2 patients in the New York City region. medRxiv [Preprint] 2020. [DOI: ]
Wan 2020 {published data only}
- Wan Y, Li Z, Wang K, Li T, Liao P. Performance verification of anti-SARS-CoV-2-specific antibody detection by using four chemiluminescence immunoassay systems. Annals of Clinical Biochemistry 2020;57(6):429-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020b {published data only}
- Wang Q, Du Q, Guo B, Guo Y, Fang L, X Guo. Performance of urea-mediated dissociation in reducing false-positive of 2019-nCoV IgM test. Chinese Journal of Laboratory Medicine 2020;43:889-93. [DOI: 10.3760/cma.j.cn114452-20200219-00091] [DOI] [Google Scholar]
- Wang Q, Du Q, Guo B, Mu D, Lu X, Ma Q, et al. A method to prevent SARS-CoV-2 IgM false positives in gold immunochromatography and enzyme-linked immunosorbent assays. Journal of Clinical Microbiology 2020;58(6):e00375-20. [DOI: 10.1128/JCM.00375-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020c {published data only}
- Wang Z, Li H, Li J, Yang C, Guo X, Hu Z, et al. Elevated serum IgM levels indicate poor outcome in patients with coronavirus disease 2019 pneumonia: a retrospective case-control study. medRxiv [Preprint] 2020. [DOI: ]
Wang 2020d {published data only}
- Wang X, Guo X, Xin Q, Chu Y, Li J, Pan Y, et al. Neutralizing antibodies responses to SARS-CoV-2 in COVID-19 inpatients and convalescent patients. medRxiv [Preprint] 2020. [DOI: ]
Wang 2020e {published data only}
- Wang B, Wang L, Kong X, Geng J, Xiao D, Ma C. Long-term coexistence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with antibody response in non-severe coronavirus disease 2019 (COVID-19) patients. medRxiv [Preprint] 2020. [DOI: ]
Wechselberger 2020 {published data only}
- Wechselberger C, Sussner S, Doppler S, Bernhard D. Performance evaluation of serological assays to determine the immunoglobulin status in SARS-CoV-2 infected patients. Journal of Clinical Virology 2020;131:104589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weiss 2020 {published data only}
- Weiss S, Klingler J, Hioe C, Amanat F, Baine I, Kojic EM, et al. A high through-put assay for circulating antibodies directed against the S protein of severe acute respiratory syndrome corona virus 2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Wen 2020 {published data only}
- Wen T, Huang C, Shi FJ, Zeng XY, Lu T, Ding SN, et al. Development of a lateral flow immunoassay strip for rapid detection of IgG antibody against SARS-CoV-2 virus. Analyst 2020;145(15):5345-52. [DOI] [PubMed] [Google Scholar]
Wheeler 2020 {published data only}
- Wheeler SE, Shurin GV, Keetch C, Mitchell G, Kattel G, McBreen J, et al. Evaluation of SARS-CoV-2 prototype serologic test in hospitalized patients. Clinical Biochemistry 2020;86:8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Woelfel 2020 {published data only}
- Woelfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Mueller MA, et al. Clinical presentation and virological assessment of hospitalized cases of coronavirus disease 2019 in a travel-associated transmission cluster. medRxiv [Preprint] 2020. [DOI: ]
Wu 2020a {published data only}
- Wu X, Fu B, Chen L, Feng Y. Serological tests facilitate identification of asymptomatic SARS-CoV-2 infection in Wuhan, China. Journal of Medical Virology 2020;92:1795-6. [DOI: 10.1002/jmv.25904] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xiang 2020b {published data only}
- Xiang J, Yan M, Li H, Liu T, Lin C, Huang S, et al. Evaluation of enzyme-linked immunoassay and colloidal gold- immunochromatographic assay kit for detection of novel coronavirus (SARS-Cov-2) causing an outbreak of pneumonia (COVID-19). medRxiv [Preprint] 2020. [DOI: ]
Xie 2020a {published data only}
- Xie J, Ding C, Li J, Wang Y, Guo H, Lu Z, et al. Characteristics of patients with coronavirus disease (COVID-19) confirmed using an IgM-IgG antibody test. Journal of Medical Virology 2020;92:2004-10. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xu 2020a {published data only}
- Xu Y. Dynamic profile of severe or critical COVID-19 cases. medRxiv [Preprint] 2020. [DOI: ]
Xu 2020b {published data only}
- Xu G, Emanuel AJ, Nadig S, Mehrotra S, Caddell BA, Curry SR, et al. Evaluation of orthogonal testing algorithm for detection of SARS-CoV-2 IgG antibodies. Clinical Chemistry 2020;66:1531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xu 2020c {published data only}
- Xu Y, Xiao M, Liu X, Xu S, Du T, Xu J, et al. Significance of serology testing to assist timely diagnosis of SARS-CoV-2 infections: implication from a family cluster. Emerging Microbes and Infection 2020;9:924-7. [DOI: 10.1080/22221751.2020.1752610] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xue 2020 {published data only}
- Xue X, Zhu C, Huang S, Pan L, Xu J, Li W. Effect of heat inactivation of blood samples on the efficacy of three detection methods of SARS-CoV-2 antibodies [血液样本灭活处理对三种SARS-CoV-2抗体检测方法结果的影响]. Journal of Southern Medical University 2020;40(3):316-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yamaoka 2021 {published data only}
- Yamaoka Y, Jeremiah SS, Miyakawa K, Saji R, Nishii M, Takeuchi I, et al. Whole nucleocapsid protein of severe acute respiratory syndrome coronavirus 2 may cause false-positive results in serological assays. Clinical Infectious Diseases 2021;72(7):1291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yan 2021 {published data only}
- Yan M, Zheng Y, Sun Y, Wang L, Luan L, Liu J, et al. Analysis of the diagnostic value of serum specific antibody testing for coronavirus disease 2019. Journal of Medical Virology 2021;93(1):441-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yildirim 2020 {published data only}
- Yildirim F, Yildiz Gulhan P, Diken OE, Capraz A, Simsek M, Botan Yildirim B, et al. Alternative or complementary role of serological rapid antibody test in the management of possible COVID-19 cases. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Yokoyama 2020 {published data only}
- Yokoyama R, Kurano M, Morita Y, Shimura T, Nakano Y, Qian C, et al. Validation of a new automated chemiluminescent anti-SARS-CoV-2 IgM and IgG antibody assay system detecting both N and S proteins in Japan. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Yu 2020 {published data only}
- Yu HQ, Sun BQ, Fang ZF, Zhao JC, Liu XY, Li YM, et al. Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. European Respiratory Journal 2020;56(2):2001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yue 2020 {published data only}
- Yue H, Nowak RP, Overwijn D, Payne NC, Fischinger S, Atyeo C, et al. Rapid 'mix and read' assay for scalable detection of SARS-CoV-2 antibodies in patient plasma. medRxiv [Preprint] 2020. [DOI: ]
Zeng 2020a {published data only}
- Zeng Z, Chen L, Pan Y, Deng Q, Ye G, Li Y, et al. Profile of specific antibodies to SARS-CoV-2: the first report. Journal of Infection 2020;81(1):e80-1. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2020b {published data only}
- Zeng F, Dai C, Cai P, Wang J, Xu L, Li J, et al. A comparison study of SARS-CoV-2 IgG antibody between male and female COVID-19 patients: a possible reason underlying different outcome between gender. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Zhang 2020c {published data only}
- Zhang W, Du RH, Li B, Zheng XS, Yang X L, Hu B, et al. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerging Microbes and Infections 2020;9(1):386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020d {published data only}
- Zhang P, Gao Q, Wang T, Ke Y, Mo F, Jia R, et al. Evaluation of recombinant nucleocapsid and spike proteins for serological diagnosis of novel coronavirus disease 2019 (COVID-19). medRxiv [Preprint] 2020. [DOI: ]
Zhang 2020e {published data only}
- Zhang B, Zhou X, Zhu C, Song Y, Feng F, Qiu Y, et al. Immune phenotyping based on the neutrophil-to-lymphocyte ratio and IgG level predicts disease severity and outcome for patients with COVID-19. Frontiers in Molecular Biosciences 2020;7:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020f {published data only}
- Zhang J, Liu J, Li N, Liu Y, Ye R, Qin X, et al. Serological detection of 2019-nCoV respond to the epidemic: a useful complement to nucleic acid testing. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed]
Zhao 2020b {published data only}
- Zhao R, Li M, Song H, Chen J, Ren W, Feng Y, et al. Serological diagnostic kit of SARS-CoV-2 antibodies using CHO-expressed full-length SARS-CoV-2 S1 proteins. medRxiv [Preprint] 2020. [DOI: ]
Zhong 2020 {published data only}
- Zhong L, Chuan J, Gong BO, Shuai P, Zhou Y, Zhang Y, et al. Detection of serum IgM and IgG for COVID-19 diagnosis. Science China Life Sciences 2020;63(5):777-80. [DOI: 10.1007/s11427-020-1688-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhou 2020 {published data only}
- Zhou Q, Zhu D, Yan H, Quan J, Kuang Z, Zhang W, et al. A preliminary study on analytical performance of serological assay for SARS-CoV-2 IgM/IgG and application in clinical practice. medRxiv [Preprint] 2020. [DOI: ]
Additional references
Agarwal 2020
- Agarwal A, Rochwerg B, Lamontagne F, Siemieniuk RA, Agoritsas T, Askie L, et al. A living WHO guideline on drugs for covid-19 (last update Mar 3 2022). BMJ 2020;370:m3379. [DOI] [PubMed] [Google Scholar]
Arevalo‐Rodriguez 2020a
- Arevalo-Rodriguez I, Buitrago-Garcia D, Simancas-Racines D, Zambrano-Achig P, Del Campo R, Ciapponi A, et al. False-negative results of initial RT-PCR assays for COVID-19: A systematic review. PLoS One 2020;15(12):e0242958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bonanni 2021
- Bonanni P, Cantón R, Gill D, Halfon P, Liebert UG, Crespo KAN, et al. The role of serology testing to strengthen vaccination initiatives and policies for COVID-19 in Europe. COVID 2021;1(1):20-38. [Google Scholar]
Bossuyt 2015
- Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig L, et al. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. BMJ 2015;351:h5527. [DOI: 10.1136/bmj.h5527] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2020a
- Centers for Disease Control and Prevention (CDC). Multisystem Inflammatory Syndrome in Adults (MIS-A) Case definition information for healthcare providers (last reviewed November 13, 2020). Available from: cdc.gov/mis/mis-a (accessed 31 March 2021).
CDC 2021a
- Centers for Disease Control and Prevention (CDC). Information for Healthcare Providers about Multisystem Inflammatory Syndrome in Children (MIS-C) (last reviewed May 20, 2021). Available from: cdc.gov/mis/mis-c/hcp/index.html (accessed 31 March 2021).
CDC 2021b
- Centers for Disease Control and Prevention (CDC). Interim Guidelines for COVID-19 Antibody Testing (updated September 21, 2021). Available from: www.cdc.gov/coronavirus/2019-ncov/lab/resources/antibody-tests-guidelines.html (accessed 9 December 2021).
CDC China 2020
- National Health Commission of the People's Republic of China. Release of 7th edition of case definitions. www.nhc.gov.cn/yzygj/s7653p/202003/46c9294a7dfe4cef80dc7f5912eb1989.shtml (accessed prior to 23 June 2020).
Chua 2021
- Chua CRR, los Santos EDE, Escasa KVH, Estolas RLG, Feliciano J, Ortega SAE, et al. Diagnostic accuracy of COVID-19 antibody tests authorized by FDA Philippines: a systematic review and meta-analysis. SciMedicine Journal 2021;3(4):283-301. [Google Scholar]
Corman 2020
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu D, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020;25(3):2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
COVID‐19 Open Access Project 2021
- COVID-19 Open Access Project. Living Evidence on COVID-19. Available at ispmbern.github.io/covid-19/living-review/.
Covidence [Computer program]
- Covidence. Version accessed 27 April 2020. Melbourne, Australia: Veritas Health Innovation. Available at covidence.org.
De Carvalho 2021
- Carvalho CC, Cardozo MM, Gonelli R, Regueira SL, Souza AB, Ramos IB, et al. Diagnostic accuracy of serological tests for COVID-19: a systematic review and meta-analysis of cohort studies. La Rivista Italiana della Medicina di Laboratorio 2021;17(4):227-36. [DOI: 10.23736/S1825-859X.21.00122-5] [DOI] [Google Scholar]
Dinnes 2021
- Dinnes J, Deeks JJ, Berhane S, Taylor M, Adriano A, Davenport C, et al. Rapid, point-of-care antigen and molecular-based tests for diagnosis of SARS-CoV-2 infection. Cochrane Database of Systematic Reviews 2021, Issue 3. Art. No: CD013705. [DOI: 10.1002/14651858.CD013705.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Doust 2021
- Doust JA, Bell KJL, Leeflang MMG, Dinnes J, Lord SJ, Mallett S, et al. Guidance for the design and reporting of studies evaluating the clinical performance of tests for present or past SARS-CoV-2 infection. BMJ 2021;372:n568. [DOI] [PubMed] [Google Scholar]
ECDC 2021
- European Centre for Disease Prevention and Control. The use of antibody tests for SARS-COV-2 in the context of Digital Green Certificates; 10 May 2021. Available at https://www.ecdc.europa.eu/sites/default/files/documents/Use-of-antibody-tests-for-SARS-COV-2-in-the-context-of-Digital-Green-Certificates.pdf.
FDA 2021a
- FDA. SARS-CoV-2 viral mutations: impact on COVID-19 tests. https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests (accessed 31 March 2022).
FDA 2022
- FDA. Coronavirus (COVID-19) Update: FDA authorizes new monoclonal antibody for treatment of COVID-19 that retains activity against omicron variant (Feb 11 2022). Available at www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-new-monoclonal-antibody-treatment-covid-19-retains.
Gracienta 2022
- Gracienta TJ, Herardi R, Santosa F, Pasiak TF, Tjang YS. Diagnostic accuracy of antibody-based rapid diagnostic test in detecting coronavirus disease 2019: systematic review. Archives of Medical Science 2022;18(4):949-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Greaney 2022
- Greaney AJ, Starr TN, Eguia RT, Loes AN, Khan K, Karim F, et al. A SARS-CoV-2 variant elicits an antibody response with a shifted immunodominance hierarchy. PLoS Pathogens 2022;18(2):e1010248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Healy 2021
- Healy B, Khan A, Metezai H, Blyth I, Asad H. The impact of false positive COVID-19 results in an area of low prevalence. Clinical Medicine Journal 2021;21(1):e54-6. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
HIQA 2021
- Health Information and Quality Authority. Duration of protective immunity (protection from reinfection) following SARS-CoV-2 infection; updated on 18 Nov 2021. https://www.hiqa.ie/reports-and-publications/health-technology-assessment/duration-protective-immunity-protection.
Islam 2021
- Islam N, Ebrahimzadeh S, Salameh JP, Kazi S, Fabiano N, Treanor L, et al. Thoracic imaging tests for the diagnosis of COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 3. Art. No: CD013639. [DOI: 10.1002/14651858.CD013639.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Junker 2022
- Junker D, Becker M, Wagner TR, Kaiser PD, Maier S, Grimm TM, et al. Antibody binding and ACE2 binding inhibition is significantly reduced for the Omicron variant compared to all other variants of concern. medRxiv [Preprint] 2021. [DOI: ]
Kucirka 2020
- Kucirka LM, Lauer SA, Laeyendecker O, Boon D, Lessler J. Variation in false-negative rate of reverse transcriptase polymerase chain reaction-based SARS-CoV-2 tests by time since exposure. Annals of Internal Medicine 2020;173(4):262-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kumar 2021
- Kumar M, Swarnim S, Pallavi P. Clinical characteristics of multisystem inflammatory syndrome in children and young adults with COVID-19: a rapid systematic review. Journal of Pediatrics Review 2022;10:367-88. [Google Scholar]
Leeflang 2021
- Leeflang MMG, Bell K, Deeks JJ, Dinnes J, Doust J, Korevaar DA, et al. Electronic and animal noses for detecting SARS-CoV-2 infection. Cochrane Database of Systematic Reviews 2021, Issue 6. Art. No: CD015013. [DOI: 10.1002/14651858.CD015013] [DOI] [Google Scholar]
Macaskill 2010
- Macaskill P, Gatsonis C, Deeks JJ, Harbord RM, Takwoingi Y. Chapter 10: analysing and presenting results. In: Deeks JJ, Bossuyt PM, Gatsonis C editor(s). Cochrane Handbook for Systematic Reviews of Diagnostic Test Accuracy Version 1.0. The Cochrane Collaboration, 2010. Available from methods.cochrane.org/sdt/handbook-dta-reviews.
Macedo 2022
- Macedo ACL, Prestes GdS, Colonetti T, Candido ACR, Uggioni MLR, Gomes AC, et al. A systematic review and meta-analysis of the accuracy of SARS-COV-2 IGM and IGG tests in individuals with COVID-19. Journal of Clinical Virology 2022;148:105121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Makoah 2022
- Makoah NA, Tipih T, Litabe MM, Brink M, Sempa JB, Goedhals D, et al. A systematic review and meta-analysis of the sensitivity of antibody tests for the laboratory confirmation of COVID-19. Future Virology 2022;17(2):119-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mayers 2020
- Mayers C, Baker K, Government Office for Science. Impact of false-positives and false-negatives in the UK’s COVID-19 RT-PCR testing programme; 3 June 2020. Available at assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/895843/S0519_Impact_of_false_positives_and_negatives.pdf.
McInnes 2018
- McInnes MDF, Moher D, Thombs BD, McGrath TA, Bossuyt PM, the PRISMA-DTA Group. Preferred reporting items for a systematic review and meta-analysis of diagnostic test accuracy studies: The PRISMA-DTA Statement. JAMA 2018;319(4):388-96. [DOI: 10.1001/jama.2017.19163] [PMID: ] [DOI] [PubMed] [Google Scholar]
McInnes 2020
- McInnes M, Leeflang MM, Salameh J-P, McGrath T, Van der Pol CB, Frank RA, et al. Imaging tests for the diagnosis of COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 4. Art. No: CD013639. [DOI: 10.1002/14651858.CD013639] [DOI] [PubMed] [Google Scholar]
MHRA 2021a
- Medicines and Healthcare Products Regulatory Authority. Target product profile: Antibody tests to help determine if people have immunity to SARS-CoV-2 (v2; updated 14 February 2022). Available at www.gov.uk/government/publications/how-tests-and-testing-kits-for-coronavirus-covid-19-work/target-product-profile-antibody-tests-to-help-determine-if-people-have-recent-infection-to-sars-cov-2-version-2.
MHRA 2021b
- Medicines and Healthcare Products Regulatory Authority. Target product profile: Enzyme immunoassay (EIA) antibody tests to help determine if people have antibodies to SARS-CoV-2 (v2; updated 14 February 2022). Available at www.gov.uk/government/publications/how-tests-and-testing-kits-for-coronavirus-covid-19-work/target-product-profile-enzyme-immunoassay-eia-antibody-tests-to-help-determine-if-people-have-antibodies-to-sars-cov-2.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. PLoS Medicine 2009;6(7):1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
ONS 2020
- Office for National Statistics. Coronavirus (COVID-19) Infection Survey, UK: 21 August 2020. Available at ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/coronaviruscovid19infectionsurveypilot/englandandwales21august2020.
Patel 2021
- Patel P, DeCuir J, Abrams J, Campbell AP, Godfred-Cato S, Belay ED. Clinical characteristics of multisystem inflammatory syndrome in adults: a systematic review. JAMA Network Open 2021;4(9):e2126456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Post 2021
- Post N, Eddy D, Huntley C, Schalkwyk MCI, Shrotri M, Leeman D, et al. Antibody response to SARS-CoV-2 infection in humans: A systematic review. PLoS One 2021;15(12):e0244126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Riley 2020
- Riley S, Ainslie KE, Eales O, Walters CE, Wang H, Atchison C, et al, REACT Study Investigators. Transient dynamics of SARS-CoV-2 as England exited national lockdown. medRxiv [Preprint] 2020;.. [DOI: 10.1101/2020.08.05.20169078] [DOI: ] [DOI]
RSS 2021
- Royal Statistical Society. Royal Statistical Society Diagnostic Tests Working Group Report; June 2021. Available at https://rss.org.uk/RSS/media/File-library/Policy/2021/RSS-Diagnostic-tests-report-FINAL.pdf.
Stata [Computer program]
- Stata. Version 17. College Station, TX, USA: StataCorp, 2021. Available at www.stata.com.
Stegeman 2020
- Stegeman I, Ochodo EA, Guleid F, Holtman G, Yang B, Cunningham J, et al. Routine laboratory testing to determine if a patient has COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 11. Art. No: CD013787. [DOI: 10.1002/14651858.CD013787] [DOI] [PMC free article] [PubMed] [Google Scholar]
Struyf 2021
- Struyf T, Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 2. Art. No: CD013665. [DOI: 10.1002/14651858.CD013665.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Takwoingi 2017
- Takwoingi Y, Guo B, Riley RD, Deeks JJ. Performance of methods for meta-analysis of diagnostic test accuracy with few studies or sparse data. Statistical Methods in Medical Research 2017;26(4):1896-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Veroniki 2022
- Veroniki AA, Tsokani S, Agarwal R, Pagkalidou E, Rücker G, Mavridis D, et al. Diagnostic test accuracy network meta-analysis methods: A scoping review and empirical assessment. Journal of Clinical Epidemiology 2022;146:86-96. [DOI] [PubMed] [Google Scholar]
Ward 2022
- Ward H, Whitaker M, Flower B, Tang SN, Atchison C, Darzi A, et al. Population antibody responses following COVID-19 vaccination in 212,102 individuals. Nature Communications 2022;13(1):907. [DOI] [PMC free article] [PubMed] [Google Scholar]
West 2021
- West RM, Kobokovich A, Connell N, Gronvall GK. Antibody (serology) tests for COVID-19: a case study. mSphere 2021;6(3):e00201-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Whiting 2011
- Whiting PF, Rutjes AW, Westwood ME, Mallett S, Deeks JJ, Reitsma JB, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Annals of Internal Medicine 2011;155(8):529-36. [DOI] [PubMed] [Google Scholar]
WHO 2020
- World Health Organization (WHO). Global surveillance for COVID-19 caused by human infection with COVID-19 virus Interim guidance. www.who.int/publications/i/item/global-surveillance-for-human-infection-with-novel-coronavirus-(2019-ncov) (accessed 23 June 2020). [https://www.who.int/publications/i/item/global-surveillance-for-human-infection-with-novel-coronavirus-(2019-ncov)]
Yadav 2022
- Yadav PD, Sapkal GN, Sahay RR, Potdar VA, Deshpande GR, Patil DY, et al. Substantial immune response in Omicron infected breakthrough and unvaccinated individuals against SARS-CoV-2 variants of concern. Journal of Infection 2022;84(5):e80-1. [DOI: 10.1016/j.jinf.2022.02.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2020a
- Yang Y, Yang M, Yuan J, Wang F, Wang Z, Li J, et al. Laboratory diagnosis and monitoring the viral shedding of SARS-CoV-2 infection. Innovation 2020;1(3):100061. [DOI: 10.1016/j.xinn.2020.100061] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2021
- Yang B, Mallett S, Takwoingi Y, Davenport CF, Hyde CJ, Whiting PF, et al. QUADAS-C: a tool for assessing risk of bias in comparative diagnostic accuracy studies. Annals of Internal Medicine 2021;174(11):1592-9. [DOI] [PubMed] [Google Scholar]
Zhao 2020
- Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clinical Infectious Diseases 2020;71(16):2027-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Deeks 2020a
- Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Spijker R, Taylor-Phillips S, et al. Antibody tests for identification of current and past infection with SARS-CoV-2. Cochrane Database of Systematic Reviews 2020, Issue 6. Art. No: CD013652. [DOI: 10.1002/14651858.CD013652] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2020b
- Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, Spijker R, et al. Diagnosis of SARS-CoV-2 infection and COVID-19: accuracy of signs and symptoms; molecular, antigen, and antibody tests; and routine laboratory markers. Cochrane Database of Systematic Reviews 2020, Issue 4. Art. No: CD013596. [DOI: 10.1002/14651858.CD013596] [DOI] [Google Scholar]