

Abstract
We identified three types of monosynaptic cholinergic inputs spatially arranged onto medial substantia nigra dopaminergic neurons in male and female mice: cotransmitted acetylcholine (ACh)/GABA, GABA-only, and ACh only. There was a predominant GABA-only conductance along lateral dendrites and soma-centered ACh/GABA cotransmission. In response to repeated stimulation, the GABA conductance found on lateral dendrites decremented less than the proximally located GABA conductance, and was more effective at inhibiting action potentials. While soma-localized ACh/GABA cotransmission showed depression of the GABA component with repeated stimulation, ACh-mediated nicotinic responses were largely maintained. We investigated whether this differential change in inhibitory/excitatory inputs leads to altered neuronal excitability. We found that a depolarizing current or glutamate preceded by cotransmitted ACh/GABA was more effective in eliciting an action potential compared with current, glutamate, or ACh/GABA alone. This enhanced excitability was abolished with nicotinic receptor inhibitors, and modulated by T- and L-type calcium channels, thus establishing that activity of multiple classes of ion channels integrates to shape neuronal excitability.
SIGNIFICANCE STATEMENT Our laboratory has previously discovered a population of substantia nigra dopaminegic neurons (DA) that receive cotransmitted ACh and GABA. This study used subcellular optogenetic stimulation of cholinergic presynaptic terminals to map the functional ACh and GABA synaptic inputs across the somatodendritic extent of substantia nigra DA neurons. We determined spatially clustered GABA-only inputs on the lateral dendrites while cotransmitted ACh and GABA clustered close to the soma. We have shown that the action of GABA and ACh in cotransmission spatially clustered near the soma play a critical role in enhancing glutamate-mediated neuronal excitability through the activation of T- and L-type voltage-gated calcium channels.
Keywords: cholinergic, GABA, nicotinic receptors, optogenetics, substantia nigra, synaptic transmission, cotransmission
Introduction
Since the seminal computational work of Wilfrid Rall (1962), it has become clear that dendritic morphology critically influences neuronal excitability by differentially filtering local time-dependent changes in excitatory and inhibitory currents (Rall, 1977, 1995; Bekkers and Stevens, 1996; Stuart and Spruston, 1998; Gentet et al., 2000; Roth and Häusser, 2001). Consequently, an appreciation of network functionality requires both an understanding of the synaptic properties between defined neuron populations and knowledge of the precise location of synaptic inputs since those synaptic inputs spatiotemporally summate and interact with intrinsic neuronal conductances to shape the neurons' activity. Previous approaches used to study synapse distribution on the dendrites of CNS neurons have typically used light or electron microscopy (EM) with immunohistochemical techniques (Caruncho et al., 1996; Nusser et al., 1998; Arroyo-Jiménez et al., 1999; Fabian-Fine et al., 2001; Unal et al., 2015; Fitzgerald et al., 2019), which cannot probe the functional consequences of synaptic inputs with regards to their location on the neuron's dendrites. Furthermore, the anatomic overlap of axonal projections with dendritic branches as a classical predictor of input strength and/or connectivity between different neuronal populations has not been corroborated by recent experiments (Callaway, 2002; Callaway and Yuste, 2002; White, 2002; Petreanu et al., 2009; Mao et al., 2011). Therefore, since an understanding of network activity requires an accurate physiological probing of circuit connectivity, there is a need for research to implement a functional approach to investigate spatial profiles of synaptic inputs to understand how such profiles shape the multisynaptic integrative properties of neurons that define their firing patterns. The need for functional and anatomic characterization of synaptic inputs is more critical in systems in which neurotransmitter corelease/cotransmission adds another layer of integrative complexity (Lee et al., 2010; Shabel et al., 2014; Saunders et al., 2015; Granger et al., 2016; Sethuramanujam et al., 2016; Case et al., 2017; Estakhr et al., 2017; Takács et al., 2018; Obermayer et al., 2019).
Paired recordings have been applied to probe CNS neural networks and detect functionally connected neurons; however, those methods often rely on fully intact presynaptic and postsynaptic neuron populations and are thus mainly limited to local circuits that can be retained in a brain slice preparation (Luo et al., 2008). Neurotransmitter uncaging has also been used, but the spillover effect produced with uncaging can obscure local input contributions (Schubert et al., 2001; Anastasiades et al., 2018). With the discovery of the light-activated cation channel channelrhodopsin-2 (ChR2) from the algae Chlamydomonas reinhaardtii (Nagel et al., 2003), optogenetics has become a common methodology to selectively photostimulate presynaptic terminals to evoke postsynaptic responses (Boyden et al., 2005; X. Li et al., 2005; Nagel et al., 2005; Lin, 2011). It has also permitted the development of fine scale optical stimulation techniques to investigate neuronal circuits by probing the functional connectivity between target nuclei without requiring the preservation of the cell body of the presynaptic neurons as one can effectively photostimulate presynaptic axonal terminals even when severed from their somata (Petreanu et al., 2007; Wang et al., 2007). The study of Petreanu et al. (2007) was the first to effectively map out monosynaptic connections between neurons using optical stimulation. Since then, many studies have used optogenetics to map neuronal connectivity in the mammalian brain (Wang et al., 2007; Petreanu et al., 2009; Mao et al., 2011; Bhatia et al., 2019; D'Souza et al., 2019). However, to date, there has been no fine resolution mapping of functional synaptic inputs onto substantia nigra (SN) dopaminergic (DA) neurons. Our laboratory has previously discovered cotransmitted ACh and GABA onto medial SN DA neurons (Estakhr et al., 2017). Here, we have applied a fine-scale ChR2-based input mapping technique that permits a true optical following of the dendritic tree with subcellular resolution (∼15 μm) in conjunction with voltage-gated channel blockade to probe the spatial profile of monosynaptic ACh and GABA release onto medial SN DA neurons receiving cotransmitted inputs from cholinergic brainstem nuclei and to investigate how the spatial distribution of excitatory and inhibitory inputs from cholinergic axons shapes the activity of SN DA neurons.
Materials and Methods
Animal care and breeding
All experiments were conducted in accordance with animal protocols approved by the University of Victoria Animal Care Committee following guidelines for the care and use of animals set by the Canadian Council on Animal Care. Mice were housed at the University of Victoria Animal Care Unit (University of Victoria ACU) under a 12 h light/dark cycle and ad libitum access to food and water.
ChATcre::ChR2(H134R)-eYFP mice were produced by crossing ChATcre mice (JAX stock #006410) (Rossi et al., 2011), a knock-in mouse strain expressing cre-recombinase driven by the choline acetyltransferase (ChAT) promoter, with ChR2(H134R)-eYFP mice (JAX stock #012569) (Madisen et al., 2012), a knock-in mouse strain in which a cre-dependent (loxP-flanked stop cassette) ChR2 with the H13R mutation and tagged with yellow fluorescent protein (eYFP) inserted in the ROSA26 locus. Mice were bred to homozygosity for both genes.
We also used an Ai9-tdTomato (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J) mouse strain (JAX, stock #007909) (Madisen et al., 2010) that conditionally expresses a red fluorescent protein variant (tdTomato) in cre-recombinase-expressing neurons. Homozygous ChATcre(neodel) mice were mated with homozygous Ai9-tdTomato mice to generate a ChATcre(neodel)::Ai9-tdTomato reporter line heterozygous for both genes to express tdTomato in cholinergic neurons (Nasirova et al., 2020). P35 ChATcre(neodel)::Ai9-tdTomato mice were used for immunohistochemistry experiments while 4- to 5-week-old offspring (P25-P35) from ChATcre::ChR2(H134R)-eYFP-expressing lines were used for electrophysiology. We have previously validated the ChATcre::ChR2(H134R)-eYFP mouse line showing that ChR2-eYFP was correctly localized in only choline acetylcholine transferase-positive immunolabeled neurons in several cholinergic nuclei in the brain (Estakhr et al., 2017).
Brain slice preparation for electrophysiology
Acute brain slices were prepared from P25- to P35-day-old mice of either sex. Mice were deeply anesthetized with isoflurane (Fresenius Kabi, product #CP0406V2) and intracardially perfused with 10 ml of 2°C-4°C carbogen-bubbled (95% O2/5% CO2) N-methyl-D-glucamine (NMDG) aCSF protective cutting solution (92 mm NMDG, 2.5 mm KCl, 1.25 mm NaH2PO4, 30 mm NaHCO3, 20 mm HEPES, 25 mm D-glucose, 5 mm Na-ascorbate, 3 mm Na-pyruvate, 0.5 mm CaCl2, and 10 mm MgCl2, pH 7.3). At the end of the perfusion, the mouse was decapitated, and the brain was extracted and immersed in 2°C-4°C NMDG aCSF for a minute. The brain was then blocked in 3% agar-A (CAS #9002-18-0, Bio Basic Canada) caudal side up and placed on the slicing platform of the vibratome (Leica VT 1000S) with the ventral side facing the blade. The block was immersed in 2°C-4°C bubbled NMDG aCSF, and the brain was sectioned coronally at 320 μm thickness. Sections that included the SNc were transferred to a prewarmed (32°C-34°C) NMDG aCSF solution for an initial protective recovery period of 10 min. After the initial recovery period, slices were washed with room temperature (22°C-24°C) bubbled recording aCSF (118 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 24 mm NaHCO3, 12.5 mm D-glucose, 10 mm D-mannitol, 3 mm Na-ascorbate, 1.5 mm Na-pyruvate, 2 mm CaCl2, and 2 mm MgCl2, 308-310 mOsm) before transferring to a final holding chamber containing room temperature (22°C-24°C) bubbled recording aCSF for a period of 45 min before use for electrophysiology. The recording aCSF solution was prepared fresh on the day of the experiment.
Electrophysiological recordings
Brain slices were transferred to a recording chamber perfused with carbogenated aCSF heated to 31°C with a dual-channel temperature controller (catalog #TC-344C, Warner Instruments). Neurons were imaged using differential interference microscopy on an upright Nikon FN1 microscope system equipped with a CFI APO 40× W NIR objective (water, 0.8 NA, 3.5 mm working distance). The medial SNc was identified as the region of the SNc medial to the oculomotor nerve that passes through the SN lateral to the medial lemniscus as defined previously by our laboratory (Estakhr et al., 2017). DA neurons in the medial SN were imaged with a Thorlabs Kiralux CMOS camera (CS505MU) under infra-red differential interference contrast illumination microscopy. Whole-cell patch-clamp recordings were performed using 4-6 mΩ borosilicate glass electrodes (catalog #1B150F-4, World Precision Instruments) filled with one of two internal solutions for voltage-clamp experiments. All voltage-clamp experiments in the paper used internal Solution 1 except for Figures 1 and 3B1–B3, which used internal Solution 2. Internal Solution 1 is a modified Cs gluconate-based solution to optimize the voltage clamp for the fine spatial profiling of nicotinic and GABA-mediated postsynaptic currents (PSCs) onto medial SN DA neurons (125 mm Cs gluconate, 2 mm TEABr, 2 mm MgCl2, 10 mm HEPES, 0.5 mm CaCl2, 5 mm EGTA, 5 mm phosphocreatine Tris, 3 mm Mg-ATP, 0.2 mm GTP Tris, 5 mm QX314Br, and 2 mm L-glutathione with 80 μm Alexa-555/568 hydrazide titrated to pH 7.4, 295 mOsm, resulting in a Goldman-Hodgkin-Katz theoretically calculated membrane potential [GHK] = −30 mV and ECl = −55 mV). Internal Solution 2 was only used for the experiment in Figures 1 and 3B1–B3. Internal Solution 2 is a K gluconate-based solution was used for the large-scale spatial profiling of nicotinic and GABA-mediated PSCs onto medial SN DA neurons (130 mm K gluconate, 1.3 mm EGTA, 10 mm HEPES, 2 mm MgCl2, 0.5 mm CaCl2, 5 mm phosphocreatine Tris, 3 mm Mg-ATP, 0.2 mm GTP Tris, 2 mm QX314Cl, and 2 mm L-glutathione with 80 μm Alexa-555 titrated to pH 7.4, 289 mOsm, ECl = −80 mV, EK = −102 mV, GHK = −70 mV). In voltage-clamp experiments, cells were held close to their GHK at −30 mV with Solution 1 and at −60 mV with Solution 2 before stepping to various membrane potentials. We used cells with holding currents not exceeding ±100 pA. For current-clamp recordings, the patch pipettes were filled with a K gluconate internal solution (280 mOsm, pH 7.4, ECl = −65 mV, EK = −103 mV) containing the following: 125 mm K gluconate, 5 mm KCl, 1.3 mm EGTA, 10 mm HEPES, 2 mm MgCl2, 0.5 mm CaCl2, 5 mm phosphocreatine Tris, 3 mm Mg-ATP, 0.2 mm GTP Tris, and 2 mm L-glutathione with 80 μm Alexa-555/568 hydrazide (catalog #A20501MP/catalog #A10437, Thermo Fisher Scientific). In all experiments except if otherwise explicitly mentioned CNQX (10 μm) and D-APV (20 μm) were included in the bath to inhibit AMPA/kainate and NMDA receptors, respectively. Nicotinic (dihydro-β-erythroidine [DHβE], 1 μm; methyllycaconitine [MLA], 10 nm; mecamylamine [MEC], 10 μm) and GABAergic (SR 95531, 10 μm; or bicuculline, 10 μm) antagonists were added sequentially in cotransmission experiments to confirm the identity of transmitters generating optically evoked EPSCs/IPSCs. For the targeted spatial input mapping experiments, TTX (1 μm) and 4-AP (500 μm) were used to eliminate action potential (AP)-dependent release and block K+ channels necessary for repolarization (Petreanu et al., 2007, 2009; Mao et al., 2011). Finally, caged MNI-glutamate (200 μm), an L-type Ca2+ channel agonist (Bay K 8644, 10 μm) and blocker (nifedipine, 1 μm), and a T-type Ca2+ channel blocker (TTA-P2, 8 μm) were used during voltage or current-clamp experiments to investigate the role of ACh/GABA transmission on SN DA excitability and its modulation by Ca2+ channels.
Figure 1.
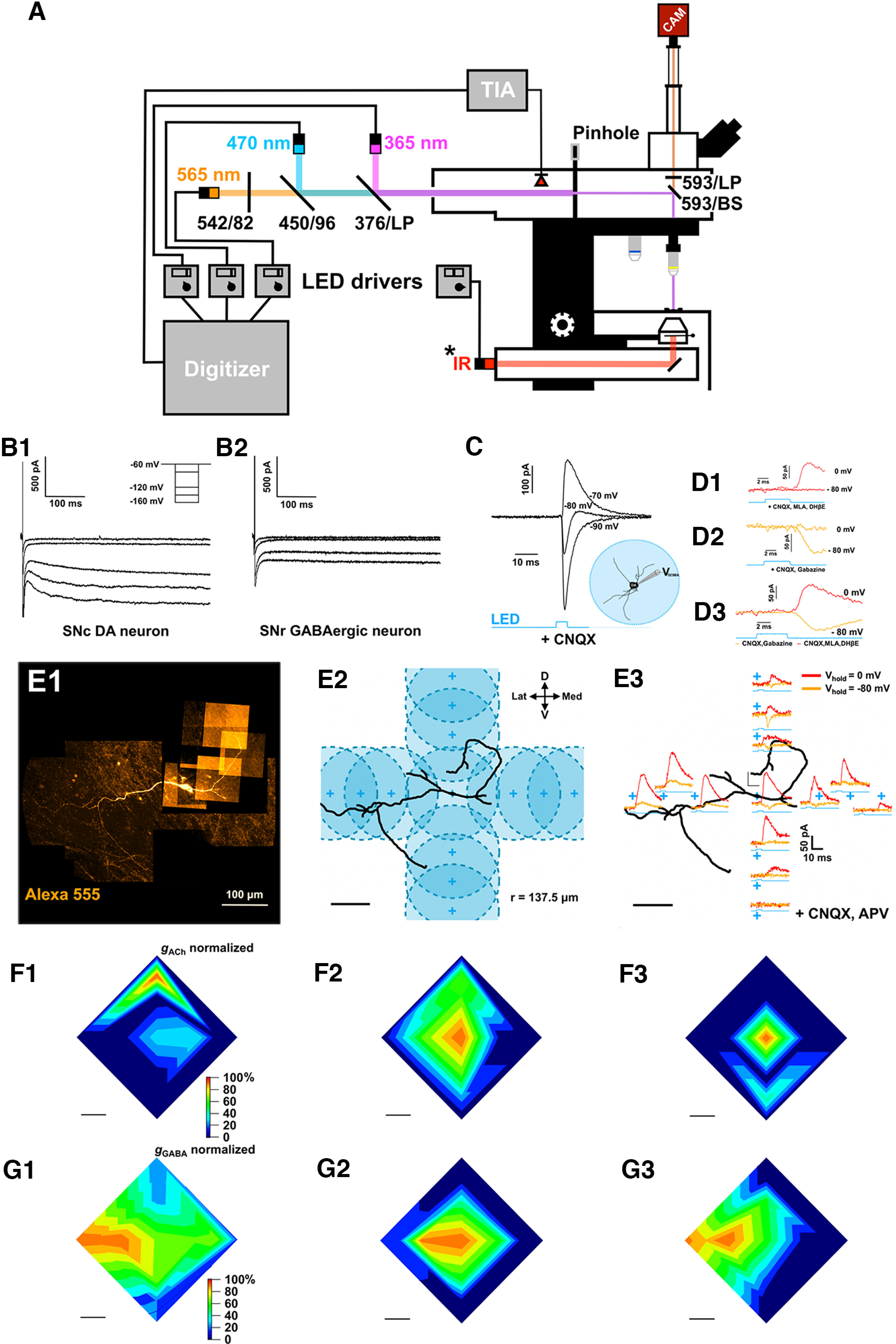
Setup and coarse functional mapping of cholinergic ACh/GABA cotransmitted synaptic inputs. A, Schematic diagram of the electrophysiological and optical stimulation setup. The microscope is equipped with transmitted light and fluorescent LEDs, a photodiode, and a camera (CAM) for one-photon imaging. Light paths (fluorescence: orange, blue, violet; transmitted: red) and filters are indicated. The camera is controlled by a computer running μManager that is independent from the computer used for electrophysiology, fluorescent LED control, and photodiode measurements, run by pClamp. Fluorescent LED intensity and duration are measured online by a photodiode placed in the light path and connected to a transimpedance amplifier that converts the photodiode current signal into a voltage step that is recorded by pClamp. Finally, a custom-made pinhole controls the optical stimulation field diameter that is projected onto the slice (from 550 to 30 μm). B1, B2, Whole-cell voltage-clamp recordings of an SNc DA cell and an SNR GABAergic cell over a range of hyperpolarizing potentials. Note the large HCN-mediated current in the SNc DA cell and, conversely, the lack of HCN-mediated current in the GABAergic neuron. C, Whole-cell recordings in voltage-clamp mode of full-field light-evoked ChAT-DA transmission at different holding potentials under bath application of CNQX shows a biphasic response, indicating cotransmission of ACh and GABA from cholinergic axons onto the recorded DA cell. D1–D3, Pharmacology confirms the identity of the light-evoked PSCs from cholinergic fibers onto a DA neuron receiving putative ACh/GABA cotransmitted PSCs and defines their respective reversals. D1, Bath application of AMPAR inhibitor (CNQX, 10 μm) and nAChR inhibitors (DHβE, 1 μm; MLA, 10 nm) pharmacologically isolates a GABA component that reverses at −80 mV. D2, Washout of the nAChR inhibitors and bath application of a GABAARs inhibitor (gabazine, 10 μm) show an isolated ACh component that rectifies at 0 mV. D3, Superimposition of pharmacologically isolated GABA (c1) and ACh (c2) PSCs shows an identical onset corroborating cotransmission. E1, Example DA neuron showing light-evoked, cotransmitted ACh/GABA PSCs from ChR2+ cholinergic axons under full-field 470 nm LED stimulation. Alexa-555 hydrazide (80 μm) was included in the patch pipette to image the neuron during electrophysiological recordings. The slice was subsequently fixed with 4% PFA and imaged with confocal microscopy to reconstruct neuronal morphology. E2, Experimental design to identify the distribution of ACh and GABA currents across DA dendrites. The stimulation field (137.5 μm radius) was moved in all four orthogonal vectors to locally sample ACh and GABA-mediated PSCs in the dorsal, ventral, medial, and lateral axes. Each vector covered a range of 320 μm from the origin point set on the cell body of the neuron. Three zones per vector were sampled with an initial distance step of 160 μm from the origin, and two subsequent steps of 80 μm (see 1, 2, and 3 on the panel) to cover the 320 μm range. E3, Light-evoked, voltage-isolated ACh (orange) and GABA (red) PSCs for each stimulation area sampled as per the experimental design outlined in E2. F1–F3, Spatial mapping of voltage-isolated light-evoked ACh (recorded at −80 mV) conductances for three recorded SN DA cells normalized to the maximum ACh conductance. G1–G3, Spatial mapping of voltage-isolated light-evoked GABA (recorded at 0 mV) conductances for all three recorded SN DA cells normalized to the maximum GABA conductance. F, G, Scale bars, 100 μm.
Figure 3.
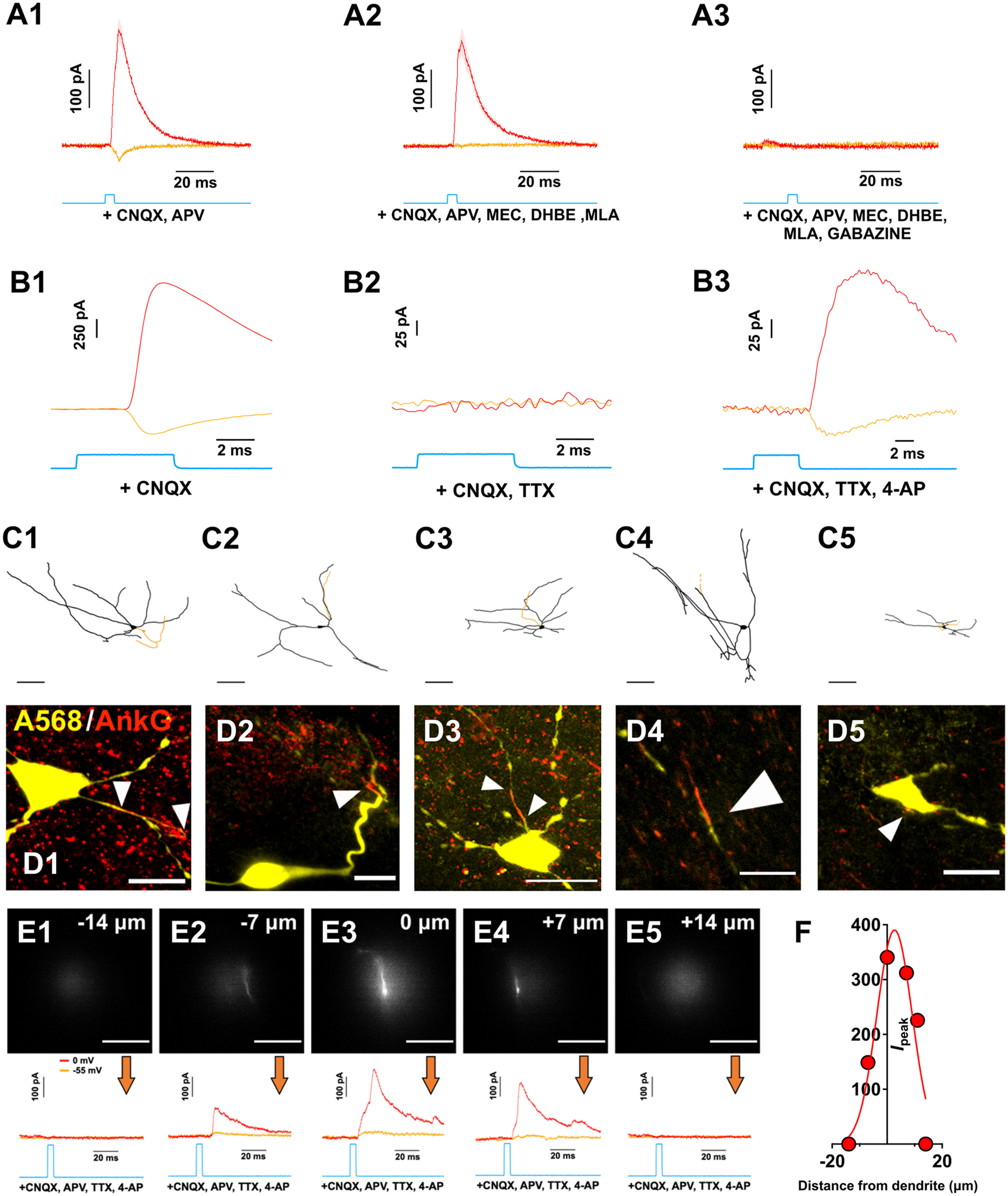
Control experiments showing pharmacological inhibition of ACh and GABA responses, pharmacological isolation of monosynaptic synaptic inputs with TTX and 4-AP, ankyrin staining of dye-filled neurons, and quantification of the effective resolution of the functional synaptic mapping technique. A1–A3, Example of light-evoked pharmacologically isolated ACh/GABA PSCs from a medial SN DA neuron. The neuron was voltage-clamped at −55 mV (orange) to isolate EPSCs and at 0 mV (red) to isolate IPSCs. All PSCs were recorded in the presence of glutamate receptor antagonists CNQX and D-APV (baseline). ACh PSCs were subsequently suppressed using a cocktail of nicotinic receptor antagonists (MLA, DHβE, MEC) and GABA PSCs were suppressed with gabazine (from left to right). n = 1 cell from 1 mouse. B1–B3, Example light-evoked PSCs from a medial SN DA neuron voltage-clamped at −90 and 0 mV in the presence of CNQX (baseline) and following subsequent bath application of (left to right) TTX and 4-AP. This indicates that the evoked nicotinic EPSCs and evoked GABA IPSCs are AP dependent and monosynaptic. n = 1 cell from 1 mouse. C1–C5, Reconstructed medial SN neurons immunostained for ankyrin-G to label the axon initial segment. Scale bar, 15 μm. n = 5 cells from 5 mice. D1–D4, Confocal images of recorded medial SN neurons filled will Alexa-568 dye and immunostained for ankyrin-G (red) to indicate the localization of the axon initial segments (white arrowheads). E1–E5, Measurements of the postsynaptic GABA currents evoked by an illumination spot positioned lateral to the dendrite of the recorded neuron. Optical illumination spot (∼30 μm diameter) was moved perpendicular to the dendrite in the X-Y plane in 7 μm increments (dendrite visualized with 80 μm Alexa-568 hydrazide). Scale bar, all images: 20 μm. Associated voltage-clamp recordings (orange = −55 mV; red = 0 mV) in the presence of CNQX, D-APV, TTX, and 4-AP. F, Responses showed in E were fitted with a Gaussian function. Width at half maximum was found to be 15 μm. E, F, n = 1 cell from 1 mouse.
All recordings were amplified and low-pass filtered at 4 kHz using a MultiClamp 700B amplifier (Molecular Devices), digitized at a sampling rate of 10-20 kHz with a Digidata 1440A A/D board (Molecular Devices) using pCLAMP 10.2 acquisition software (Molecular Devices). The holding voltage in voltage clamp was corrected for the liquid junction potential (−13 mV), and the series resistance was corrected 40%. In current-clamp mode, bridge balance and capacitance neutralization of the pipette were applied. Cells with access resistance RA between 10 and 20 mΩ were kept for analysis while cells with RA > 25 mΩ or with an ΔRA >20% over the course of the recording session were excluded. Electrophysiological responses were analyzed in Igor Pro 8 (Wavemetrics) using the Neuromatic plugin (version 3.0; http://www.neuromatic.thinkrandom.com).
DA cell phenotype was confirmed by measuring hyperpolarization-activated cyclic nucleotide-gated currents (Ih) in SN neurons in cells recorded with K gluconate-based internal solution and by also performing post hoc immunohistochemistry against TH in brain slices containing dye-filled SN neurons as described (Estakhr et al., 2017). SN DA neurons were readily distinguished by their large Ih currents compared with GABAergic neurons as well as by their colocalization of the Alexa-555/568 dye with the TH antibody (Figs. 1, 2).
Figure 2.
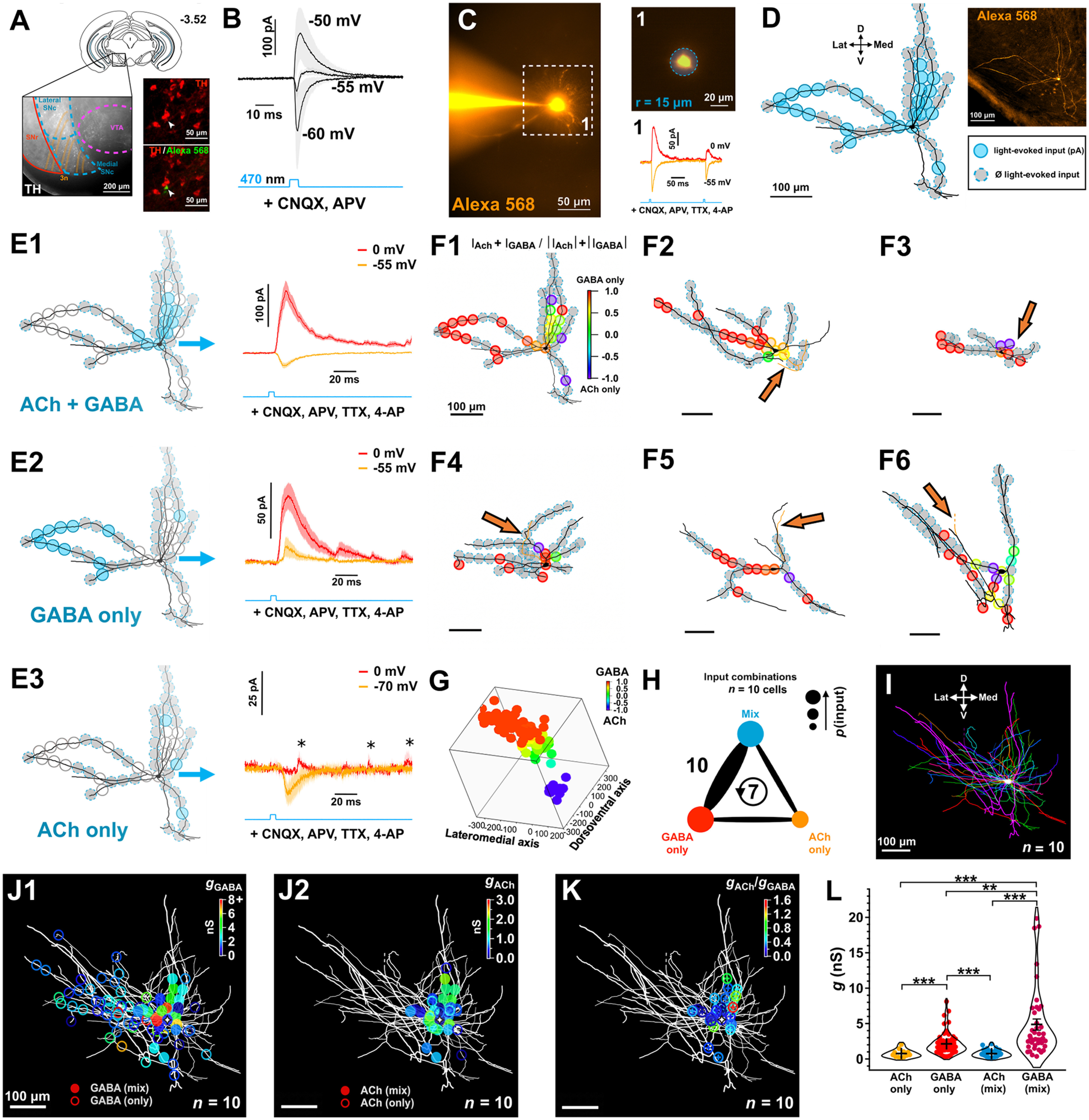
Subcellular mapping of functional ACh and GABA cholinergic synaptic inputs. A, Diagram of a coronal slice (bregma −3.52 mm) showing the substantia nigra (medial portion) region where we studied ChAT-DA connections using optogenetic stimulation of ChAT+ axons and whole-cell recordings of DA neurons. Branches of the oculomotor nerve (3n) were used as a landmark to target the medial SN. Patched neurons were dye-filled to confirm DA phenotype post hoc via TH immunolabeling. B, Whole-cell voltage-clamp recordings of light-evoked ChAT-DA transmission at different holding potentials in the presence of CNQX and D-AP5 (n = 3 cells from 3 mice). Note the biphasic component indicating putative ACh/GABA cotransmission. C, Example of monosynaptic light-evoked ACh (hold = −55 mV) and GABA (hold = 0 mV) PSCs from a 30-μm-diameter optical stimulation on a medial SN DA neuron dye-filled with Alexa-568 (80 μm) following bath application of CNQX, D-AP5, TTX, and 4-AP. D, Input map overlaid on the recorded neuron following post hoc neuronal tracing. Inputs where PSCs were evoked are color-coded. Recorded neurons in this and subsequent experiments were orthogonally aligned to the midline position. E1–E3, Focal whole-cell recordings of monosynaptic light-evoked PSCs across inputs show differential subcellular distribution of ACh and GABA release from ChAT:ChR2 axons. PSCs were organized by input type (mean ± SEM is plotted). F1–F6, Examples of input maps overlaid on reconstructed neurons. The relative contributions of ACh and GABA to the total recorded current across the two voltage potentials (−55 and 0 mV) are color-coded for each local optical stimulation. Ankyrin-G staining on DA cells (n = 5 cells from 5 mice) before neuronal tracing reveals variation in location of the axon initial segment relative to the soma (orange arrows). G, 3D superimposition of input maps as a function of the relative contributions of ACh and GABA to each local input site shows a differential distribution of input types across orthogonal axes. H, Summary of input type pairs observed on mapped DA neurons. Thickness of the connecting line indicates the frequency with which a pair was observed. Values are number of cells per pair. Circular arrow represents cells where all three input types were observed. I, Superimposition of mapped DA neurons aligned to the midline and cell-body centered. Note the asymmetry of dendritic fields. J1-J2, ACh and GABA input maps overlaid on superimposed neurons. ACh and GABA conductances were derived for individual local inputs and organized by input type. K, Absolute ratios of ACh and GABA conductances for recorded ACh/GABA inputs. F–K, n = 10 cells from 10 mice. L, ACh and GABA conductances per local input as a function of the input type. *p < 0.05; **p < 0.01; ***p < 0.001; Aligned Rank Transformed (ART) ANOVA with post hoc pairwise contrast tests for main effects with Tukey correction. Data are mean ± SEM.
LED light stimulation and imaging
Glutamate uncaging, ChR2 stimulation, and Alexa dye visualization during brain slice physiology were done with ultrafast collimated LEDs mounted on a microscope for illumination through a CFI APO 40× W NIR objective (water, 0.8 NA, 3.5 mm working distance), with peak wavelengths at 365, 470, and 565 nm (Thorlabs, M365LP1, M470L3-C5, M565L3). To prevent the cross-stimulation of channelrhodopsin, Alexa dyes, and caged glutamate compounds, we restricted the emission of LED spectra with bandpass filters (Semrock, Thorlabs): 365 nm LED with a 376 nm long-pass dichroic filter, 470 nm LED with a 473 ± 23 nm filter, and 565 nm LED with a 562 ± 20 nm filter (Fig. 1A). Light power was controlled through LED drivers (Thorlabs LEDD1B or custom-made) using analog voltage pulses. Illumination power was measured with a photometer (PM100USB, Thorlabs) at the front of the objective. Photon fluxes were set to 40 mW/mm2 at 365 nm, 20 mW/mm2 at 470 nm, and 5 mW/mm2 at 565 nm. The LED illumination duration and onset were recorded with a photodiode system integrated into the light path, and the signal was directed to the Digidata 1440A A/D converter (Fig. 1A). The illumination field was controlled with custom-made pinholes that projected circular light spots with diameters ranging from 30 to 560 μm in the FOV (Fig. 1A). Pinholes were calibrated before the experiments with different objectives using a stage micrometer and appropriate fluorescent slides.
In Figure 2, to achieve high-resolution functional mapping of synaptic inputs, 30-μm-diameter LED stimulation was used. However, optical stimulation at 30 μm field illumination resulted in significant rates of synaptic transmission failures because of the probabilistic properties of neurotransmitter vesicular fusion (data not shown). Therefore, when examining short-term plasticity and induction of APs (see Figs. 7–12), we wanted to recruit more synaptic terminals by using a larger illumination area (80 μm diameter) to lessen the contribution of synaptic failures to our overall response to achieve smoother changes in ACh or GABA conductances with repeated stimulation and to have a more robust effect in induction of APs.
Figure 7.
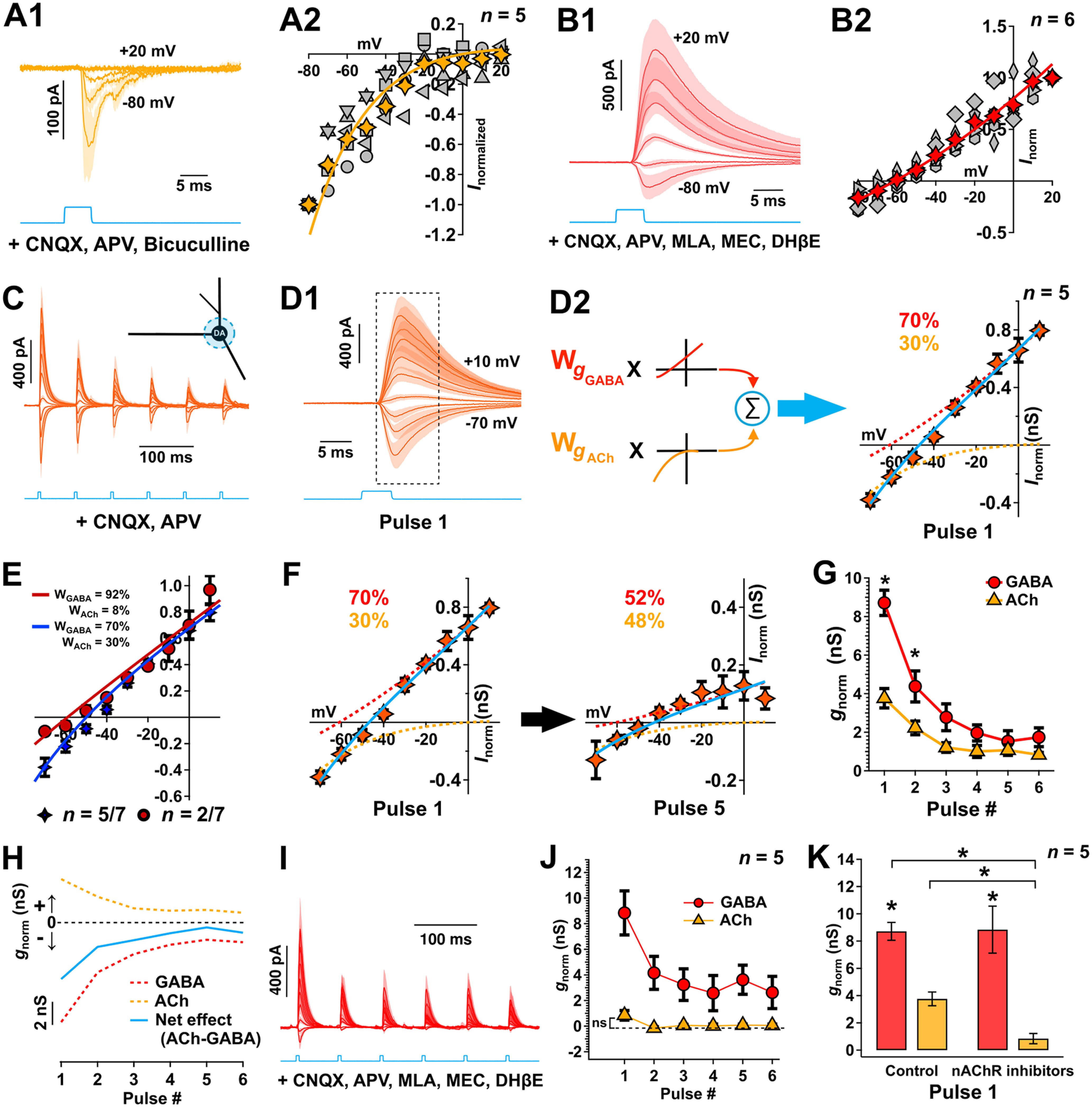
Soma-centered ACh/GABA cotransmission shows different short-term plasticity and validation of methodology of calculation of ACh/GABA weighting using nicotinic inhibitors. A1, Whole-cell voltage-clamp recordings in DA neurons of light-evoked nAChR currents measured at a series of holding potentials (as indicated) in the presence of CNQX, D-APV, and bicuculline. n = 5 cells from 5 mice. Data are mean ± SEM. A2, The normalized I–V relationship computed from the nAChR current averaged over a 10 ms period from –1 ms to onset. Each gray marker represents an individual cell. Yellow marker represents average. The average I–V was fit by a derivation of the Woodhull equation (see Materials and Methods). B1, B2, Similar to A1, A2, GABAR currents were measured following pharmacological isolation (CNQX, D-APV, MEC, DHβE, MLA), and the I–V relationship was fit by an exponential function. n = 6 cells from 6 mice. Data are mean ± SEM. C, Whole-cell voltage-clamp recordings of local (80-μm-diameter; blue light) soma-centered mix nAChR- and GABAR-mediated currents with repeated ChR2 stimulation (6 pulses, 15 Hz) over a range of holding potentials (−70 to 10 mV, 10 mV increments). Responses are measured in the presence of CNQX and D-APV. n = 5 cells from 5 mice. Data are mean ± SEM. D1, D2, The I–V relationship computed from the current in C averaged over a 10 ms period from time to onset (rounded to the nearest 0.5 ms), which corresponds to the boxed region in D1. The average current was then modeled, as displayed in D2 by a weighted linear sum of the basis fit functions of each receptor type as estimated in A and B (solid blue line, see Materials and Methods). The computed fractional conductances (gACh,gGABA) at −60 mV were obtained following deconvolution (see text) and are indicated on the graph for the first ChR2 pulse. Responses were initially dominated by GABARs (dashed red line). E, The I–V relationships computed from the currents from the population data (n = 7 cells from 7 mice) and modeled by their respective weighted linear sum of the receptor basis functions as estimated in A and B; 30% of the cells (2 of 7, burgundy) showed little to no ACh contribution on the first pulse compared with 70% of the cells (5 of 7, blue) that showed a 30% contribution of ACh on the first pulse and which were subsequently used to study the dynamics of ACh/GABA transmission in (G). F, The average I–V relationship between the first and fifth pulse. The estimated GABAR contribution (dashed red line) decreases with repeated stimulation to levels equal to that of the nAChR component of the response (dashed orange line). G, Normalized estimated ACh and GABA conductances as a function of the pulse number. Nonparametric F2-LD-F2 factorial test followed by post hoc paired Wilcoxon rank sum tests with continuity correction. H, Temporal evolution of nAChR (orange) and GABAR (red) conductance estimated at −60 mV in the same DA neurons using deconvolution. Subtracting the GABA from the nAChR conductance yields an estimate of the net excitation/inhibition (blue). C, D, F–H, n = 5 cells from 5 mice. *p < 0.05. **p < 0.01. ***p < 0.001. Data are mean ± SEM. I–K, Control experiments to validate the methodology of calculation of ACh/GABA weighting using nicotinic inhibitors. I, Whole-cell voltage-clamp recordings of local (80 μm) soma-centered mix nAChR- and GABAR-mediated currents with repeated ChR2 stimulation (6 pulses, 15 Hz) over a range of holding potentials (−70 to 10 mV, 10 mV increments). Responses are measured in the presence of CNQX, D-APV, MEC, MLA, and DHβE to inhibit AMPA/kainate/NMDARs and nAChRs. n = 5 cells from 5 mice. Data are mean ± SEM. J, Normalized estimated ACh and GABA conductances as a function of the pulse number. The estimated gACh across pulses is not significantly different from 0. Data are mean ± SEM. K, Normalized estimated ACh (orange) and GABA (red) conductances for the first pulse with and without nicotinic receptor inhibitors. *p < 0.05; **p < 0.01; ***p < 0.001; paired Wilcoxon rank sum tests. Data are mean ± SEM.
Figure 12.
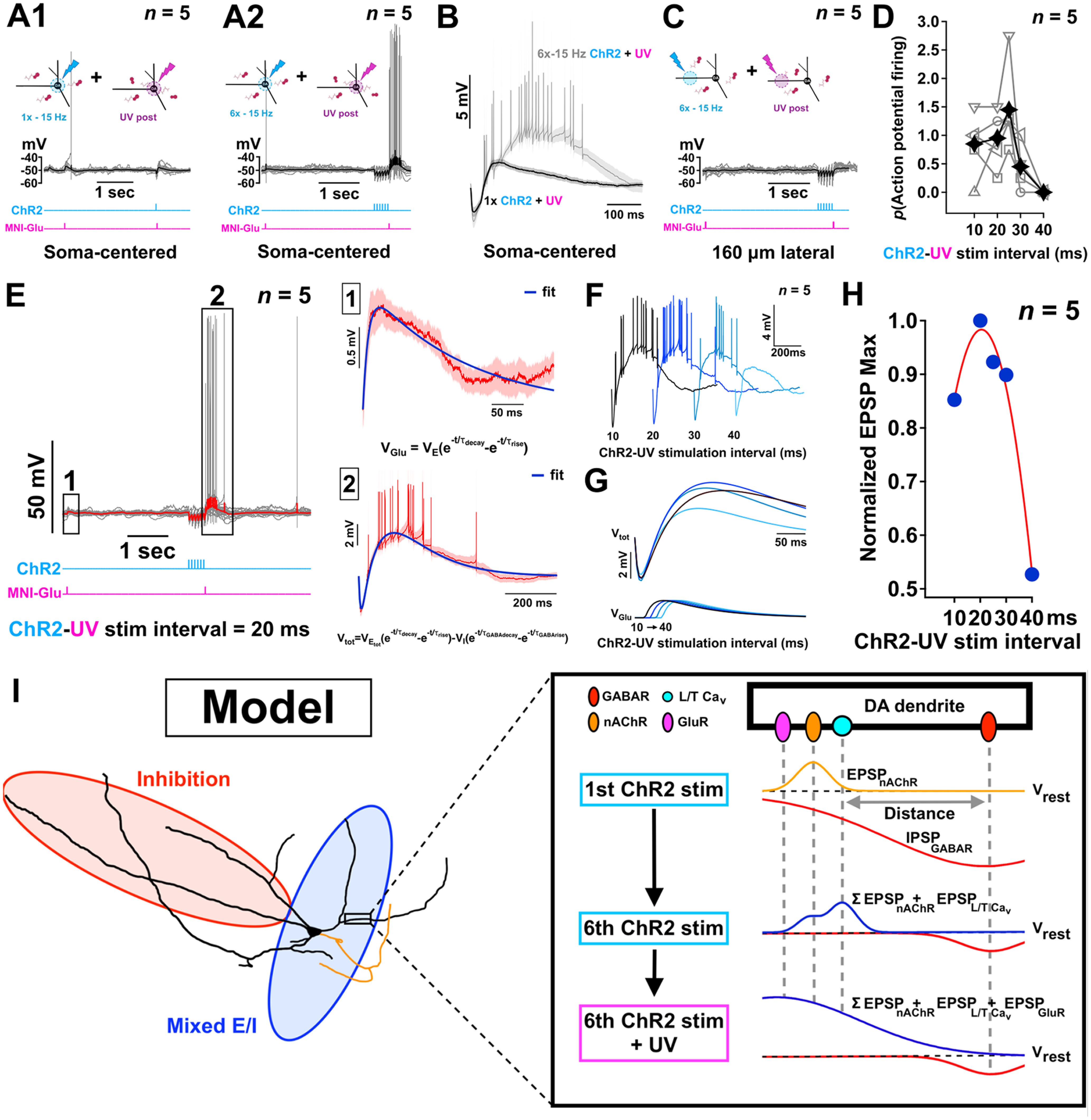
Mechanistic explanation of how combined cotransmitted ACh/GABA followed by glutamate increases neuronal excitability. A1, A2, Soma-centered subthreshold glutamate photolysis following a single optical stimulation of ACh/GABA transmission in medial SN DA neurons current-clamped ∼5 mV below their firing threshold confirms the initial dominant inhibitory conductance (see Fig. 7) and shunts the glutamate-mediated PSP to an amplitude that is lower than the PSP generated before ACh/GABA transmission, while repeated stimulation of ACh/GABA transmission and subsequent recruitment of L- and T-type Cavs (Figs. 10, 11) contribute to a positive E/I balance shift that permits optimal summation of the glutamate-mediated PSP and neuronal spiking. Data are mean with individual traces superimposed in gray. B, Superimposition of current-clamp traces of the last ChR2 stimulation and the following subthreshold glutamate photolysis from either a single optical stimulation of ACh/GABA transmission (black trace) or the 15 Hz stimulation train (gray trace) reveals the augmented and sustained neuronal depolarization generated because of the shift in ACh/GABA conductances and Cav recruitment. Traces are shown as mean ± SEM. C, Subthreshold glutamate photolysis following 15 Hz optical stimulation of the lateralized GABA transmission 160 μm lateral to the soma of medial SN DA neurons current-clamped ∼5 mV below their firing threshold does not induce AP firing. A–C, n = 5 cells from 5 mice. D, Probability of AP generation with varied interpulse intervals between the 6th 470 nm pulse and the following 365 nm pulse (soma centered) identifies a peak integration window of ∼25 ms between ACh/GABA transmission and UV-mediated photolysis of MNI-glutamate. n = 5 cells from 5 mice. E, Whole-cell current-clamp recordings of local soma-centered optical stimulation of ACh/GABA transmission in medial SN DA neurons current-clamped ∼5 mV below their firing threshold combined with subthreshold glutamate photolysis with a 20 ms interpulse interval were fitted (blue lines, 1 and 2) to model the kinetics and amplitude of both the glutamate-mediated PSP alone and the spatiotemporal summation of ACh/GABA-, Cav-, and glutamate-mediated PSPs following the optical stimulation train, as displayed in Box 1 and Box 2, respectively. The equations of the fits are shown. Red traces represent the mean ± SEM with individual traces superimposed in gray. F, G, Average somatic voltage traces for increasing interpulse intervals between the last 470 nm and 365 nm pulses reveal a supralinear mode of summation that further supports the synergistic role of ACh/GABA transmission with dendritic L- and T-type Cavs that cannot be explained by the amplitude and kinetics of the summed Glu-mediated PSP alone as shown in G by the fits of the modeled kinetics and amplitudes of the voltage summation and its underlying Glu-mediated PSP contribution. H, Summary data of the normalized EPSP maximum amplitudes for the fits of each 470-365 nm interpulse interval EPSP responses reveals the temporal integrative window for supralinear summation to occur. F–H, n = 5 cells from 5 mice. I, Summary model of cholinergic and glutamatergic transmission in a medial SN dopaminergic neuron. Cholinergic projections spatially distribute onto the dendritic fields of medial SN dopaminergic neurons to preferentially provide strong synaptic GABA-mediated inhibition laterally while differentially modulating the E/I balance somatically via ACh/GABA cotransmission and recruitment of intrinsic dendritic L- and T-type Cavs. As evidenced by our immunohistochemical findings, the nature of soma-centered ACh/GABA transmission arises from ACh and GABA release onto spatially distinct dendritic microdomains that minimize their respective shunting of conductances and serves a critical role in the activation of Cavs to prime the temporal summation of subsequent excitatory glutamate-mediated PSPs. During repeated release of ACh and GABA, the initial dominant GABA-mediated hyperpolarization serves to deinactivate L- and T-type Cavs that can be subsequently recruited by the smaller ACh-mediated PSPs and in turn activate L-type Cavs as the GABA contribution decreases with subsequent cholinergic release. This generates an increased time window of synaptic integration for other excitatory inputs (i.e., glutamate) to positively modulate DA excitability.
Access resistance RA was monitored throughout. One, three, four, or five repetitions of optical stimulation were obtained per trial depending on the experiment. All experiments included a 20-30 s period in the dark between repetitions to allow for complete channelrhodopsin recovery and/or replenishment of caged compound in the illuminated region.
Expression of ChR2(H134R)-eYFP was confirmed by brief visual inspection of the oculomotor nerve and surrounding fluorescence levels in the SN region with the 470 nm LED (Thorlabs). For spatial mapping experiments, dye-filled dopaminergic (DA) neurons were imaged between recording trials with Nikon CFI APO 40× W NIR objective (water, 0.8 NA, 3.5 mm working distance) using Thorlabs Kiralux CS505MU under identical illumination parameters throughout: 565 nm LED (Thorlabs) at 5 mW/mm2 with the 562 ± 20 nm filter.
LED light optogenetic stimulation for coarse spatial mapping of ACh and GABA currents
To generate the conductance maps in Figure 1, light-evoked synaptic currents were recorded at the pharmacologically defined ECl (Vhold = −80 mV) and ECat (Vhold = 0 mV) with bath application of CNQX and D-APV at each optical stimulation coordinate to voltage-isolate the individual ACh and GABA currents. The peak of light-evoked ACh and GABA currents at each coordinate was computed offline with peaks at or below noise level (5 pA) assigned a value of 0. Peak values (pA) for ACh and GABA PSCs were subsequently divided by their driving force (80 mV) to derive the respective ACh and GABA-mediated conductances (nS). Conductance values and their assigned spatial coordinates were organized into XYZ triplets in Igor Pro 8 to generate a Delauney triangulation of the data and create the conductance maps via XYZ contouring. Contouring levels (n = 11) were automatically assigned, and the interpolation was set as linear in light of the resolution of optical stimulation (circular beam diameter, 275 μm).
LED light optogenetic stimulation for synaptically localized spatial mapping of ACh and GABA currents
To generate the fine-scale inputs maps in Figure 2, light-evoked synaptic currents were recorded at −55 mV (ECl) and 0 mV (ECat) to voltage-isolate ACh and GABA PSCs, respectively, under bath application of AMPA and NMDA receptor inhibitors (CNQX, D-APV). The stimulation field was reduced to a 30-μm-diameter area, and recordings were made in the presence of TTX and 4-AP to eliminate AP-dependent release and evoke voltage-isolated ACh and GABA monosynaptics by limiting excitation at the presynaptic terminals (Petreanu et al., 2007). The presence of ACh or GABA inputs for each individual stimulation field was verified by repeated light stimulation (3 pulses, 5 ms pulse duration, 5 Hz), and the input nature of each field was assigned based on PSC repeatability (i.e., the local inputs were assigned as ACh or GABA or mix ACh/GABA if at least 2 of 3 PSCs were above noise level, 5 pA, at each voltage potential hold). Stimuli were restricted to fluorescently labeled dendrites and delivered sequentially following manual refocusing to sample the entire dendritic field. Stimulation coordinates were recorded during electrophysiological recordings, and inputs maps were aligned onto the reconstructed DA neurons after physiology. Peak conductance values were calculated based on the peak current measurement of the first pulse for each local field divided by their respective nAChR (Ecat = 0, driving force = 55 mV for mix ACh/GABA fields or driving force = 70 mV for primary ACh fields) and GABAR driving forces (∼55 mV, which approximates the driving force in light of theoretical ECl = −55 mV and experimental ECl = −61 mV).
Immunohistochemistry
Mice were deeply anesthetized with isoflurane (Fresenius Kabi, product #CP0406V2) and intracardially perfused with PBS, followed by PFA via a peristaltic pump (Masterflex Easy Load, Cole-Parmer, catalog #EW-07518-00) at a rate of 6 ml/min. All solutions were prepared 1 d before the intracardial perfusion and left at 4°C overnight. The perfusion began with 25 ml of PBS, pH 7.6, followed by 25 ml of 4% PFA (in PBS, pH 7.6, Electron Microscopy Sciences, catalog #15710), and finally 20 ml of PBS (pH 7.6). After perfusion, the mouse was decapitated, the brain extracted, and placed in 4% PFA in PBS, pH 7.6. After 2 d in the dark at 4°C, the cerebellum was removed and the remainder of the brain was oriented rostral side up in a weight-boat and submerged in 1.5% Agar A (Bio Basic, catalog #4583); 80-µm-thick coronal sections were cut using a vibratome (Pelco 101, 1000 Series) and mounted on lysine-coated slides (NewSilane, Newcomer Supply, catalog #5070, or Superfrost Plus Gold, Fisher Scientific, catalog #15-188-48).
Brain sections on slides were rinsed three times with PBS, pH 7.6, for 10 min and then permeabilized with 0.25% Triton X-100 for 10 min. The sections were rinsed 3 times for 10 min with PBS and then blocked with 10% donkey serum (diluted in PBS; Jackson ImmunoResearch Laboratories, catalog #017-000-121) for 30 min. The primary antibody (TH, Abcam, catalog #AB76442, host: chicken; vesicular ACh transporter [VAChT], Millipore, catalog #ABN100, host: goat; vesicular GABA transporter [VGAT], Millipore, catalog #AB5062P, host: rabbit) was diluted in 3% donkey serum (diluted in PBS) at 1:250 and incubated at room temperature overnight. Sections were washed with PBS 3 times for 10 min. The secondary antibody (AlexaFluor-405 IgG secondary antibody, Invitrogen, catalog #A-31556; AlexaFluor-488 IgG secondary antibody; AlexaFluor-647 IgG secondary antibody, Invitrogen, catalog #A-21447; Cy3-conjugated IgG secondary antibody, Jackson ImmunoResearch Laboratories, catalog #711-165-152) was diluted in 3% donkey serum, at 1:300 concentration and incubated overnight at room temperature. Brain sections were washed with PBS 3 times for 10 min, then washed 3 times at 1 h intervals and mounted with 80 µl Immu-Mount (Thermo Fisher Scientific, catalog #9990402). Following coverslipping, slides were left at 4°C overnight before imaging.
For cell type confirmation and cellular reconstruction of recorded DA neurons filled with Alexa-555/568 hydrazide dye, brain slices were fixed in 4% PFA, pH 7.6, diluted in PBS for 1 h on a shaker and stored at 4°C overnight before immunolabeling similarly described by Káradóttir and Attwell (2006). Slices were incubated overnight on a shaker at room temperature with either a primary antibody against TH (Millipore, catalog #AB1542(CH), host: sheep) or ankyrin G (Invitrogen, catalog #33-8800, host: mouse) diluted 1:250 in 3% donkey serum in PBS. Secondary antibodies (Cy3-conjugated IgG secondary antibody, Jackson ImmunoResearch Laboratories, catalog #711-165-152; AlexaFluor-647 IgG secondary antibody, catalog #A-31 571) were diluted in 3% donkey serum, at 1:300 concentration and incubated overnight on shaker at room temperature. Finally, sections were washed 3 times at 1 h intervals, mounted, and coverslipped.
Confocal microscopy
Images were acquired using a Nikon C1si spectral confocal microscope. For neuronal reconstruction and AnkyrinG imaging, sections were imaged with a 40× air Plan Fluor DIC objective (0.75 NA, 0.66 mm working distance) and a 60× oil-immersion Plan Apo VC objective (1.4 NA, 0.12 mm working distance), respectively. For neuronal reconstruction, AlexaFluor-555 and -568 hydrazide were excited using a 561 nm laser line at 10%-25% maximum transmission of a 10 mW diode pumped solid state laser while AlexaFluor-647 was excited using a 640 nm laser line at 1% maximum transmission of a 100 mW laser. Images were collected at 5.52 μs pixel dwell time and averaged over three scans through a 60-μm-diameter pinhole with a spectral detector gain set at 220. Settings were optimized to ensure that grayscale intensity values fell below saturating values (<4095 for 12-bit grayscale) and kept consistent across all images and experiments. Image z stacks comprised a 200 × 200 μm2 field with a 0.5 μm step size over a 20-30 μm depth across a spectral range of 660-750 nm (A647), or 570-590/670/730 nm (A555/A568).
For vesicular transporter immunostaining, sections were imaged with a 40× oil-immersed Plan Apo VC objective (1.3 NA, 0.24 mm working distance). AlexaFluor-647 was excited using a 640 nm laser line at 1% maximum transmission, tdTomato was excited using a 561 nm laser line at 8% maximum transmission, AlexaFluor-488 was excited using a 488 nm laser line at 2% maximum transmission of a 40 mW Argon laser, and Alexa-CF 430 was excited using a 405 nm laser line at 3.2% maximum transmission of a 36 mW laser. Images were collected with a 5.52 μs pixel dwell time and averaged over 2 scans through a 60-μm-diameter pinhole with a spectral detector gain of 220 (A647, tdTomato, CF430) or 175 (A488). Settings were optimized to ensure that grayscale intensity values fell below saturating value and kept consistent across all images and experiments; 150 × 150 μm2 z stack images with a 0.5 μm step size over a ∼20 μm depth across a spectral range of 660-750 nm (A647), 580-640 nm (tdTomato), 493-543 nm (A488), and 430-480 nm (CF430).
Analysis of confocal microscopy images
Analysis of vesicular transporters in cholinergic axons using confocal microscopy images was performed in Fiji (ImageJ, version 2.1.0/1.53c) with the 3D imageJ suite (https://imagej.net/3D_ImageJ_Suite). A 4.0 pixel median filter was applied to all images of the stacks followed by subsequent background subtraction (rolling ball radius: 50.0 pixels). Automated thresholding with the RenyiEntropy method was applied to VAChT and VGAT substacks that successfully selected puncta while maximally excluding noise. Similarly, automated thresholding with the Triangle method was applied to ChAT and TH substacks that successfully selected axons and dendrites, respectively. Corresponding thresholded VAChT and VGAT slices were combined with thresholded ChAT slices using the Image calculator command with an “AND” operator to analyze only colocalized VAChT-ChAT and VGAT-ChAT puncta. For the lateral stacks, to restrict VAChT-ChAT and VGAT-ChAT puncta analysis to the vicinity of TH dendrites, colocalized VAChT-ChAT and VGAT-ChAT slices were combined with thresholded TH slices that had been 3D dilated by 1 μm (maximum 3D filter application on thresholded TH slices, x = 7 pixels, y = 7 pixels, z = 3.5 pixels). Puncta analyses were performed using the 3D Object Counter plugin (voxel limit: 10/500) and the 3D distances plugin (from the 3D ImageJ suite) with parameters set for closest neighbor computation and border distances.
Tissue preparation for EM
Mice were anesthetized with isoflurane and transcardially perfused with 100 ml of heparinized PBS followed by fixation with 100 ml of 2% PFA and 2% glutaraldehyde in 0.15 m sodium cacodylate. The brain was extracted and fixed overnight at 4°C in the same fixative, and then sectioned at 200 µm thickness with a vibratome. Small regions (1 × 1 mm) of the substantia nigra pars compacta were microdissected using the oculomotor nerve, medial lemniscus, and VTA as landmarks. Tissue blocks were then postfixed in 1% osmium tetroxide and 1% potassium ferrocyanide in 0.15 m sodium cacodylate (4 h at room temperature), washed with dH2O (3 × 10 min each), followed by en bloc staining with 2% uranyl acetate (aq) overnight. Blocks were then dehydrated in an ascending series of ethanols and embedded in Spurr's resin and polymerized overnight at 70°C. Ultrathin sections (40-60 nm thick) were cut using a diamond knife on a Leica Ultracut E ultramicrotome and collected onto copper grids. Sections were observed on a JEOL JEM-1400 transmission electron microscope operated at 80 kV and images acquired with a Gatan SC-1000 digital camera. All supplies for tissue processing were purchased from Electron Microscopy Sciences.
Double and triple postembedding immunogold EM
Immunogold cytochemistry was performed as previously described (Puente et al., 2019). Small rectangular fixed pieces measuring 0.5 × 0.5 × 1 mm from the medial substantia nigra pars compacta region were rinsed in PB (4°C, overnight), cryoprotected in glycerol (10%, 20%, and 30% in PB), and rapidly frozen in liquid propane in a cryofixation unit (KF80; Reichert). They were then freeze-substituted with methanol and 0.5% uranyl acetate, and subsequently embedded in Lowicryl HM20 (Lowi).
Ultrathin sections (80 nm) were collected on 300 mesh nickel grids. Then, they were washed in TBS with Triton X-100 (TBST: 50 mm Tris-HCl, pH 7.4; 0.15 m NaCl; 0.1% Triton X-100; 0.02% NaN3) containing 0.1% NaBH4 and 50 mm glycine for 10 min, and rinsed 3 times for 1 min in TBST. Tissue sections were preincubated in blocking solution: 3% (w/v) human serum albumin (HSA) in TBST for at least 10 min. They were subsequently incubated overnight with different affinity-purified primary antisera, diluted in TBST with 2% HSA. Primary antibodies included rabbit anti-VAChT (20 μg/ml, Frontier Institute, catalog #VAChT-Rb, RRID:AB_2571850), mouse anti-VGAT at 1:100 (Synaptic Systems, catalog #131011, RRID:AB_887872), and guinea pig anti-tdTomato (20 μg/ml, Frontier Institute, catalog #tdTomato-GP-Af430, RRID:AB_2631185). Tissue processing was followed by thorough washes in TBST, preincubated with the blocking solution for 10 min, and then incubated with a mixture of secondary antibodies coupled to colloidal gold particles of different sizes diluted 1:20 in TBST containing 2% HSA and 0.5% polyethylene glycol. Secondary antibodies included goat anti-rabbit IgG F(ab)′2 antibody, 10 nm gold conjugated (1:20, BBI Solutions catalog #EM.GFAR10), goat anti-mouse IgG 30 nm gold antibody (BBI Solutions catalog #EM.GAM30), goat anti-mouse IgG 20 nm gold antibody (BBI Solutions catalog #EM.GAM20), goat anti-guinea pig IgG 20 nm gold antibody (BBI Solutions catalog #EM.GAG20), and goat anti-guinea pig IgG 10 nm gold antibody (BBI Solutions catalog #EM.GAG10). Finally, the grids were rinsed several times in double-distilled water, dried, counterstained with 2% uranyl acetate (90s) and 2.5% lead citrate (90s), and examined in a JEOL JEM 1400 Plus electron microscope. Tissue samples were photographed by using a digital camera coupled to the electron microscope (Morada, Olympus; sCMOS). Figures were created with Adobe Photoshop (CS3, Adobe Systems; RRID:SCR_014199).
Algebraic deconvolution of nAChR and GABAR conductances
Light-evoked ACh/GABA currents were recorded at membrane potentials between ∼−70 and ∼10 mV stepped in 10 mV increments under repeated stimulation (15 Hz, 6 pulses). The average current response was measured over a time window (10 ms following current onset, with the onset rounded to the nearest 0.5 ms) to increase signal-to-noise ratio. Responses were consequently modeled as the sum of their respective nAChR- and GABAR-mediated currents (least-squares fit) as per (Manookin et al., 2010) as follows:
where ITOT (V) is the total current at the membrane voltage V, ECl the chloride reversal = −61 mV, and Ecat the cationic reversal = 0 mV. gGABA and gACh are the respective basis conductance functions of GABARs and nAChRs, respectively, normalized to 1 at −60 mV (see below). WGABA and WACh denote the conductance weight at −60 mV and represented the reported values for GABA and ACh conductances.
Based on previous experiments (Manookin et al., 2010), the basis function for GABA (gGABA) was defined as an exponential equation:
where a = 4.43851e−5 and b = −0.986892 are constant experimentally derived from the fit of light-evoked GABAR current–voltage (I–V) relationships measured from DA cells (5 cells from 5 mice, see Fig. 7) under pharmacological inhibition of nAChRs and AMPA/NMDARs. gGABA was finally multiplied by a factor to normalize the conductance at –60 mV to 1 (i.e., multiplying by the inverse of gGABA at –60 mV): 0.9567.
The basis ACh function was well described by a simplification of the Woodhull equation (Haghighi and Cooper, 1998) to account for the voltage-dependent inward rectification of nAChRs:
where a = 426.773 and b = −0.0236513 are constants experimentally derived from the fit of light-evoked nAChR I–V relationships measured from DA cells (5 cells from 5 mice, see Fig. 7) under pharmacological inhibition of GABARs and AMPA/NMDARs. gACh was consequently multiplied by a factor to normalize the conductance at −60 mV to 1 (i.e., multiplying by the inverse of gACh at −60 mV): 1.045.
Statistical analyses
Values are reported as mean ± SEM and were computed using R (version 4.0.3, 2020) (R Core Team, 2018). Data were graphed using Igor Pro software. When applicable, assumptions of normality and equal variances were tested using a Shapiro–Wilk normality test and an F test, respectively. Nonparametric F2-LD-F2 or F1-LD-F1 longitudinal data factorial tests (Noguchi et al., 2012) followed by post hoc paired Wilcoxon rank sum tests with continuity correction were used to evaluate longitudinal data when parametric assumptions were not met. A linear regression mixed model (Type III ANOVA with Satterthwaite's method) followed by post hoc pairwise contrast tests for main effects with Tukey correction was used when parametric assumptions were not met for 2-factorial nonlongitudinal data. Either Friedman's test followed by pairwise Wilcoxon rank-sum test with Bonferroni correction or Dunn's test with Bonferroni correction was used to evaluate mean group differences for repeated measurements when parametric assumptions were not met. Wilcoxon rank-sum tests were used to evaluate mean group differences when parametric assumptions were not met. If both samples had normal distribution but unequal variances, a Welch's t test was used to examine differences between the two means. Statistical significance was determined at the α level of 0.05 in all analyses. For detailed statistical results, see Table 1.
Table 1.
| Figure | Sample size (n) | Statistical test | Values |
|---|---|---|---|
| 2 L | n = 149 inputs from 10 neurons, 10 mice | Aligned Rank Transformed (ART) ANOVA with post hoc pairwise contrast tests for main effects with Tukey correction | ART ANOVA F = 31.16, p = 1.319e-15, df = 3, 145 Pairwise contrasts: gACh–gGABA, df = 145, t.ratio = −4.53, p = 0.0001 gACh–gAChMIX, df = 145, t.ratio = −0.84, p = 0.835 gACh–gGABAMIX, df = 145, t.ratio = −6.68, p < 0.0001 gGABA–gAChMIX, df = 145, t.ratio = 5.65, p < 0.0001 gGABA–gGABAMIX, df = 145, t.ratio = −3.74, p = 0.0015 gAChMIX–gGABAMIX, df = 145, t.ratio = −8.54, p < 0.0001 |
| 4 C | n = 4 mice, n = 6 z stack images per mouse; each dot represents one z stack; n = 24 z stacks per group (VGAT+, VAChT+, VGAT+:VAChT+); n = 48 medial z stacks; n = 24 lateral z stacks | Linear regression mixed model (Type III ANOVA with Satterthwaite's method) followed by post hoc pairwise contrast tests for main effects with Tukey correction | Type III ANOVA with Satterthwaite's method: Type: F = 62.996, p = 4.973e-16, df = 2, 66 Location: F = 0, p = 1, df = 1, 66 Type:location: F = 12.582, p = 2.348e-05, df = 2, 66 Pairwise contrasts (interaction = true): VGAT–VACHT/MED–LAT, df = 44, t.ratio = −5.007, p < 0.0001 VGAT–MIX/MED–LAT, df = 44, t.ratio = −2.232, p = 0.0308 VACHT–MIX/MED–LAT, df = 44, t.ratio = 2.775, p = 0.0081 df method: Kenward–Roger Pairwise contrasts: VGAT MED-VACHT MED, df = 44, t.ratio = 1.799, p = 0.477 VGAT MED-MIX MED, df = 44, t.ratio = 7.66, p < 0.0001 VGAT MED-VGAT LAT, df = 66, t.ratio = −3.41, p = 0.0135 VGAT MED-VACHT LAT, df = 66, t.ratio = 5.137, p < 0.0001 VGAT MED-MIX LAT, df = 66, t.ratio = 6.002, p < 0.0001 VACHT MED-MIX MED, df = 44, t.ratio = 5.866, p < 0.0001 VACHT MED-VGAT LAT, df = 66, t.ratio = −4.88, p < 0.0001 VACHT MED-VACHT LAT, df = 66, t.ratio = 3.67, p = 0.0063 VACHT MED-MIX LAT, df = 66, t.ratio = 4.53, p = 0.0003 MIX MED-VGAT LAT, df = 66, t.ratio = −9.67, p < 0.0001 MIX MED-VACHT LAT, df = 66, t.ratio = −1.122, p = 0.87 MIX MED-MIX LAT, df = 66, t.ratio = −0.256, p = 0.9998 VGAT LAT-VACHT LAT, df = 44, t.ratio = 7.404, p < 0.0001 VGAT LAT-MIX LAT, df = 44, t.ratio = 8.15, p < 0.0001 VACHT LAT-MIX LAT, df = 44, t.ratio = 0.75, p = 0.974 df method: Kenward–Roger |
| 4 D | Type III ANOVA with Satterthwaite's method: Type: F = 18.87, p = 1.207e-06, df = 2, 44 Location: F = 11.73, p = 0.00242, df = 1, 22 Type:location: F = 4.456, p = 0.0173, df = 2, 44 Pairwise contrasts (interaction = true): VGAT–VACHT MED–LAT, df = 44, t.ratio = −0.220, p = 0.827 VGAT–MIX MED–LAT, df = 44, t.ratio = −2.468, p = 0.0175 VACHT–MIX MED–LAT, df = 44, t.ratio = 2.688, p = 0.0101 df method: Kenward–Roger Pairwise contrasts: VGAT MED-VACHT MED, df = 44, t.ratio = 2.53, p = 0.137 VGAT MED-MIX MED, df = 44, t.ratio = 7.46, p < 0.0001 VGAT MED-VGAT LAT, df = 44.2, t.ratio = 3.54, p = 0.0114 VGAT MED-VACHT LAT, df = 44.2, t.ratio = 5.23, p < 0.0001 VGAT MED-MIX LAT, df = 44.2, t.ratio = 5.39, p < 0.0001 VACHT MED-MIX MED, df = 44, t.ratio = 4.92, p = 0.0002 VACHT MED-VGAT LAT, df = 44.2, t.ratio = 2.075, p = 0.319 VACHT MED-VACHT LAT, df = 44.2, t.ratio = 3.76, p = 0.0061 VACHT MED-MIX LAT, df = 44.2, t.ratio = 3.920, p = 0.0061 MIX MED-VGAT LAT, df = 44.2, t.ratio = −0.779, p = 0.97 MIX MED-VACHT LAT, df = 44.2, t.ratio = 0.909, p = 0.942 MIX MED-MIX LAT, df = 44.2, t.ratio = 1.065, p = 0.892 VGAT LAT-VACHT LAT, df = 44, t.ratio = 2.060, p = 0.326 VGAT LAT-MIX LAT, df = 44, t.ratio = 2.250, p = 0.236 VACHT LAT-MIX LAT, df = 44, t.ratio = 0.190, p = 1.000 df method: Kenward–Roger |
||
| 4 E | n = 4512 puncta, n = 24 z stacks, n = 4 mice, n = 6 z stack images per mouse | Two-sided Welch's t test | F = 31.61, p = 1.997e-08, df = 1, 4410.7 |
| 4 F | n = 4512 puncta, n = 24 z stacks, n = 4 mice, n = 6 z stack images per mouse | Linear regression model followed by post hoc pairwise contrast tests for main effects with Tukey correction | ANOVA: Type: F = 30.299, p = 3.91e-08, df = 1, 4508 Location: F = 18.16, p = 2.073e-05, df = 1, 4508 Type:location: F = 4.08, p = 0.043, df = 1, 4508 Pairwise contrasts (interaction = true): VACHT-VGAT MED-LAT, df = 4508, t.ratio = −2.02, p = 0.043 Pairwise contrasts: VACHT MED-VGAT, df = 4508, t.ratio = −5.55, p < 0.0001 VACHT MED-VACHT, df = 44.2, t.ratio = −4.16, p = 0.0002 VACHT MED-VGAT LAT, df = 44.2, t.ratio = −5.68, p < 0.0001 VGAT MED-VACHT LAT, df = 44.2, t.ratio = −1.50, p = 0.436 VGAT MED-VGAT LAT, df = 44.2, t.ratio = −2.21, p = 0.12 VACHT LAT-VGAT LAT, df = 44.2, t.ratio = −0.14, p = 0.999 |
| 5 G | n = 16 z stack images, 4 z stack images from each of 4 mice medial SN | Paired Wilcoxon rank sum tests | Wilcoxon rank sum test, paired = true, alternative = two-sided Medial: M1-M190°, V = 136, p = 0.00048 M2-M290°, V = 136, p = 0.00048 |
| 5 H | n = 8 z stack images, 2 z stack images from each of 4 mice lateral SN | Paired Wilcoxon rank sum tests | Wilcoxon rank sum test, paired = true, alternative = two-sided Lateral: M1-M190°, V = 21, p = 0.036 M2-M290°, V = 21, p = 0.036 |
| 7 G | n = 60 conductance estimates, n = 5 longitudinal repeated conductance estimates for treatments = 2 (gACh, gGABA) across 5 cells from 5 mice | Nonparametric F2-LD-F2 factorial test followed by post hoc paired Wilcoxon rank sum tests | F2-LD-F2 with Wald test: gWeight (ACh, GABA): df = 1, z = 6.76, p = 9.31e-03 Pulse number: df = 5, z = 332.681305, p = 9.35e-70 gWeight:pulse number: df = 5, z = 8.82, p = 1.16e-01 Wilcoxon rank sum test, paired = true, alternative = less AChP1-GABAP1, V = 0, p = 0.031 AChP2-GABAP2, V = 0, p = 0.031 AChP3-GABAP3, V = 1, p = 0.0625 AChP4-GABAP4, V = 2, p = 0.094 AChP5-GABAP5, V = 6, p = 0.41 AChP6-GABAP6, V = 1, p = 0.062 |
| 7 K |
n = 20 conductance estimates, n = 2 treatments (-/+ nAChR inhibitors), n = 5 cells from 5 mice |
Paired Wilcoxon rank sum tests | Wilcoxon rank sum test, paired = true, alternative = less ACh-GABA, V = 0, p = 0.03125 ACh+NicInhib-GABA+NicInhib, V = 0, p = 0.031 Wilcoxon rank sum test, paired = false, alternative = less AChNicInhib-GABA, V = 0, p = 0.031 AChNicInhib-ACh, V = 0, p = 0.031 Wilcoxon rank sum test, paired = true, alternative = two.sided GABANicInhib-GABA, V = 7, p = 1 |
| 8 E | n = 66 conductance estimates, n = 5 longitudinal repeated conductance estimates for conditions = 2 (gGABAMED, gGABALAT) across 4 (gGABALAT) and 7 (gGABAMED) cells from 7 mice | Nonparametric F1-LD-F1 factorial test | F1-LD-F1 with Wald test: Location: df = 1, z = 6.188, p = 1.287e-02 Pulse number: df = 5, z = 138.28, p = 4.298e28 Location:pulse number: df = 5, z = 21.928, p = 5.42e-04 F1-LD-F1 with ANOVA test: Location: df = 1, z = 6.187809, p = 1.286333e-02 Pulse number: df = 1.840753, z = 20.985173, p = 1.597254e-07 Location:pulse number: df = 1.840753, z = 3.108420, p = 4.892182e-02 |
| 8 G | n = 54 average frequency estimates from treatments = 3 (pre, stim, post) and conditions = 2 (soma-centered, 160 μm lateral), n = 9 cells from 9 mice | Dunn's test with Bonferroni correction | Soma-centered: Kruskal−Wallis χ2 = 5.09, df = 2, p = 0.08 pre-stim: 1.259, p = 0.312 post-stim: −2.25, p = 0.0365 pre-post: −0.992, p = 0.481 160 μm lateral: Kruskal−Wallis χ2 = 21.092, df = 2, p = 0 pre-stim: 2.708, p = 0.0102* post-stim: −4.566, p = 0.0000* pre-post: −1.858, p = 0.095 alpha = 0.05 Reject Ho if p < alpha/2 |
| 10 F | n = 28 spike probability estimates from conditions = 4 (control pre, control post, bay K, nicotinic inhibitors), n = 5 cells from 5 mice for all four conditions, n = 7 cells from 7 mice for 3 of 4 conditions (control pre, control post, nicotinic inhibitors) | Friedman rank sum test followed by pairwise Wilcoxon with or without Bonferroni correction | Friedman χ2 = 14.12, df = 3, p = 0.0027 Pairwise comparisons using Wilcoxon rank sum test without Bonferroni correction: pre-post, p = 0.0017 pre-bay k, p = 0.0048 pre-nic inhib, p = 0.253 post-bay K, p = 0.042 post-nic inhib, p = 0.0016 bay K-nic inhib, p = 0.0046 Pairwise comparisons using Wilcoxon rank sum test with Bonferroni correction: pre-post, p = 0.0100 pre-bay k, p = 0.0288 pre-nic inhib, p = 1.0000 post-bay K, p = 0.255 post-nic inhib, p = 0.0096 bay K-nic inhib, p = 0.027 |
| 10 H | n = 6 cells from 6 mice | Friedman rank sum test followed by pairwise Wilcoxon with Bonferroni correction | Friedman χ2 = 17.773, df = 4, p = 0.001367 Pairwise comparisons using Wilcoxon rank sum test with Bonferroni correction: pre-post, p = 0.0105 post-nifedipine, p = 0.0105 nic inhib-wash, p = 0.0279 |
| 11 E | n = 10 δ time-to-peak estimates for conditions = 2 (control, TTA-P2), n = 5 cells from 5 mice | Friedman rank sum test | Friedman χ2 = 5, df = 1, p = 0.025 |
Results
Functional cotransmitted ACh and GABA from cholinergic axons onto mouse SN dopaminergic neurons show a differential spatial distribution
Prior studies have shown that cholinergic neurons from select brainstem nuclei project onto DA neurons of the VTA and SN and can release ACh and GABA in a cotransmitted manner and modulate motor behaviors (Estakhr et al., 2017; Li and Spitzer, 2020). To assess the nature and physiological consequences of this cotransmission of excitatory and inhibitory neurotransmitters onto DA neurons in the medial SN, we combined local optogenetic stimulation of cholinergic axons with whole-cell patch-clamp recordings of Alexa-555 or -568 dye-filled DA neurons in ex vivo brain slices from ChATcre::ChR2(H134R)-eYFP mice. ChATcre::ChR2(H134R)-eYFP mice express cre recombinase under the transcriptional control of the ChAT promoter, enabling selective expression of the opsins in cholinergic neurons (Rossi et al., 2011; Estakhr et al., 2017). The medial SN region is separated from the VTA by the dorsal medial lemniscus and the lateral SN by the tracts of the oculomotor nerve (Fig. 2A). Whole-cell patch-clamped DA neurons were identified by their large Ih currents, and the identification was confirmed via post hoc immunolabeling (Figs. 1B1–B2, 2A). Experiments were done on DA cells showing ACh/GABA cotransmission. Photostimulation of cholinergic axons was performed in voltage-clamp mode across a range of stepped membrane potentials to enable the simultaneous recording of synaptic ACh and GABA currents (Figs. 1C, 2B). Individual components were pharmacologically isolated using either a cocktail of nAChR antagonists (MLA, DHβE, and MEC) or a GABAAR antagonist (gabazine) to confirm that components displayed identical latencies and thus were cotransmitted (Fig. 1D1–D3). We initially investigated the spatial profiles of ACh and GABA release onto DA neurons displaying full-field light-evoked ACh/GABA cotransmitted PSCs to assess whether the distribution of cotransmission was similar across the dendritic arbor (Fig. 1E1–E3). Interestingly, we found a differential spatial profile of ACh versus GABA transmission wherein the ACh conductance extended primarily ventrodorsally from the soma, while the GABA conductance was more uniformly distributed with a slightly stronger conductance in the lateromedial axis (Figs. 1F1–F3,G1-G3).
We then applied a finer-scale ChR2-based input mapping strategy to understand the nature and strength of inputs from cholinergic axons onto medial SN DA neurons across their dendritic arbor. For these experiments, the patch pipette contained a cesium gluconate-based internal solution with bromide forms of QX314 and TEA to block Na2+, K+, and Ca2+ channels and improve the quality of the voltage clamp at depolarized potentials while preserving membrane properties (Zhang et al., 1994; Talbot and Sayer, 1996; Kaczorowski et al., 2007). This pipette solution also shifted the effective chloride reversal to less hyperpolarized potentials (from −80 to −55 mV) (Bormann et al., 1987; Petreanu et al., 2007). Voltage-clamp recordings confirmed the voltage shift of the biphasic component of ACh/GABA cotransmission (Fig. 2B), which was verified by pharmacology (Fig. 3A). The illumination field was subsequently reduced to a 30-μm-diameter area, and recordings were made in the presence of TTX and 4-AP to evoke voltage-isolated ACh and GABA monosynaptic responses by preventing initiation of AP propagation along axons of passage, thus limiting excitation to the presynaptic terminals (Petreanu et al., 2009). Under these conditions, brief 470 nm LED pulses (5 ms) depolarized ChR2-expressing presynaptic boutons, to trigger release of ACh and GABA (Figs. 2C, 3B1–B3). Stimuli were restricted to fluorescently labeled dendrites and somata and delivered following sequential manual movement and refocusing of the optical stimulus to eventually sample the entire somatodendritic field of each neuron. Measurements of the functional synaptic mapping point spread function indicated that our methodology maps specific types of inputs with high subcellular spatial resolution (15 μm) (Fig. 3E,F). Aligning the input maps with the reconstructed DA neurons after physiology revealed the dendritic locations of synaptic contacts with ChR2-positive cholinergic axons (Fig. 2D). Furthermore, voltage-clamping at −55 and 0 mV while locally stimulating the individual 30 μm fields revealed three different types of monosynaptic transmitter release from cholinergic axons: cotransmitted ACh/GABA, GABA-only, and ACh only (Fig. 2E1–E3). The distribution of inputs was sparse across the DA dendrites, suggesting a small number of synaptic contacts (Fig. 2D–F,J1,J2,K). However, alignment of all the input maps revealed a somatodendritic patterning of input distribution consistent with the predominant GABA-only conductance located on dendrites lateral to the soma and a soma-centered ACh/GABA cotransmission (Fig. 2F,G). This is regardless of the location of the axon initial segment (Fig. 2F, arrows; Fig. 3C,D). All cells (10 of 10 cells) showed both ACh/GABA cotransmission and GABA-only input types, and most cells (7 of 10 cells) showed all three, including ACh only synapses (Fig. 2H). Superimposition of all reconstructed cells with their conductance maps suggested that ACh/GABA inputs were found close to the cell body, while GABA-only and ACh only inputs extended toward the lateral and dorsoventral axes, respectively (Fig. 2I,J1,J2). Finally, we found a larger GABA conductance for the proximally located mixed ACh/GABA inputs compared with other GABA-only input types located predominantly along the lateral dendrites (Fig. 2L). However, the lateral dendrites expressed a greater density/number of GABA-only inputs than the proximally located mixed ACh/GABA inputs (Fig. 2L). This suggests that there might be a difference in either the density of synaptic contacts between cholinergic axons and medial SN DA neurons as a function of the neurotransmitter released, or a different probability of release, or both (Estakhr et al., 2017; Li and Spitzer, 2020).
Anatomical evidence for differential subcellular localization of ACh and GABA presynaptic release sites along the somatodendritic extent of SN DA neurons
SN DA neurons are innervated by long-range cholinergic projections from the laterodorsal tegmental and pedunculopontine tegmental nuclei (PPN) (Clarke et al., 1987; Cornwall et al., 1990; Futami et al., 1995; Takakusaki et al., 1996; Lokwan et al., 1999; Xiao et al., 2016; Estakhr et al., 2017; Li and Spitzer, 2020). To further validate the differential nature of ACh and GABA synaptic release from cholinergic axons onto medial SN DA neurons, we used ChATcre(neo-del)+/−::Ai9-tdTomato+/− mice that show selective labeling of cholinergic neurons and their axonal processes (Nasirova et al., 2020). Triple labeling the SN for TH, VAChT, and VGAT identified proportional differences in the number of VAChT and VGAT puncta found in tdTomato+ putative cholinergic terminals as a function of the DA somatic or lateral dendritic regions (Fig. 4A,B). Similar to Estakhr et al. (2017), we identified VAChT-VGAT puncta that were either fully colocalized or ≤1 μm distance (both criteria we referred to as “mix” VAChT/VGAT puncta); however, mix puncta represented the minority of presynaptic cholinergic terminals in contrast with individual VGAT+ only and VAChT+ only puncta that overlapped with identified tdTomato+ cholinergic axons (Figs. 4B,C, 5A–H). Moreover, both our own data and previous research confirmed that subpopulations of cholinergic neurons in those nuclei, in particular the PPN, possess the cellular machinery for acetylcholine and GABA transmitter release in their presynaptic terminals found in the SN (Fig. 5I) (Estakhr et al., 2017; Li and Spitzer, 2020). Expanding on the findings of Li and Spitzer (2020) and consistent with our electrophysiological results, a greater proportion of VGAT+ puncta (69%) were found within a 1 μm vicinity of the TH+ dendrites 300 μm lateral to the soma of medial SN DA neurons while similar amounts of VGAT+ (49%) and VAChT+ (40%) puncta were found in proximity of the cell bodies (Fig. 4C). Meanwhile, the lower absolute number of lateralized puncta is likely reflective of the lower density of lateral TH+ dendrites 200-400 μm away from the somata of medial SN DA cells (Fig. 4D,E). Furthermore, differences in VGAT+ and VAChT+ puncta sizes in identified tdTomato+ putative terminals suggest a greater density of GABA transmitter packaging across cholinergic terminals compared with ACh packaging, which in addition to differences in release probabilities outlined in Estakhr et al. (2017), further strengthens the differences in ACh and GABA conductances observed with soma-centered ACh/GABA transmission (Fig. 4F).
Figure 4.
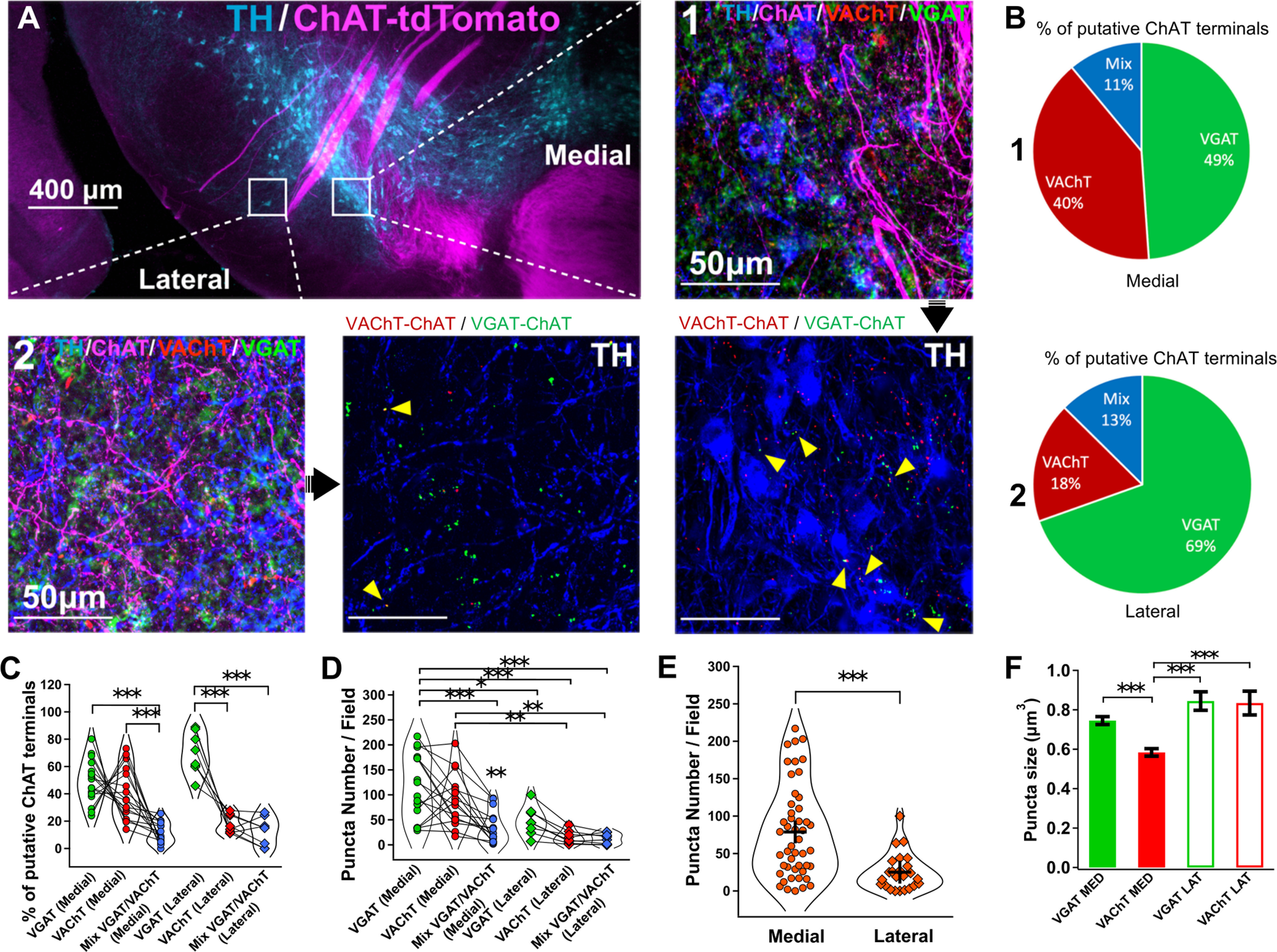
Immunohistochemical mapping of ACh and GABA cholinergic synaptic inputs. A, Immunohistochemistry to identify the distribution of ACh and GABA transporter expression of cholinergic axons in the SN. A ChATcre(neodel)+/−:tdTomato+/− mouse cross was used to label the cholinergic innervation of the SN. Coronal sections containing the SN were triple labeled for TH, VGAT, and VAChT. Two locations were bilaterally sampled (two z stack images centered on the cell bodies of medial DA neurons, one lateral z stack image) to enable a comparison in transporter expression for putative cholinergic terminals between the somatic and lateral dendritic regions of DA cells. Analyses were performed on VAChT+:tdTomato+ and VGAT+:tdTomato+ puncta. For the lateral field, puncta at distances ≥ 1 μm away from TH+ dendrites were excluded from analysis. n = 4 mice, n = 6 z stack images per mouse. B, Summary of the percentage of VAChT+ and VGAT+ terminals in cholinergic axons projecting to either the somata of DA cells or in the vicinity of lateralized TH+ dendrites. C, Percentage of VAChT+, VGAT+, and mixed VGAT+:VAChT+ (≤1 μm away) in putative cholinergic terminals in the SN as a function of their location. D, VAChT+, VGAT+, and mixed VGAT+:VAChT+ puncta numbers in putative cholinergic terminals in the SN as a function of their location. C, D, Linear regression mixed model (Type III ANOVA with Satterthwaite's method) followed by post hoc pairwise contrast tests for main effects with Tukey correction. E, Absolute number of puncta in putative cholinergic terminals in the SN as a function of their location. Two-sided Welch's t test. C–E, n = 4 mice, n = 6 z stack images per mouse. Each dot represents one z stack. n = 24 z stack per group (VGAT+, VAChT+, VGAT+:VAChT+). n = 48 medial z stack, n = 24 lateral z stack. F, VAChT+:tdTomato+ and VGAT+:tdTomato+ puncta sizes as a function of their location. Linear regression model followed by post hoc pairwise contrast tests for main effects with Tukey correction.
Figure 5.

Immunohistochemistry to quantify colocalization of VAChT and VGAT containing presynaptic boutons in the SN and validate of PPN cholinergic neurons coexpressing ChAT and GABA. A–C, Immunolabeling of VAChT (A) and VGAT (B) and overlap (C) representing their presynaptic terminals in the medial SN. DA neurons were labeled with TH (C). D–F, Similar to A–C, but for the lateral SN. G, Quantification analysis of the colocalization of VAChT and VGAT in the medial SN by calculating the Mander's coefficients (M1 and M2). M1 and M2 were greater for the original images versus when one image was rotated 90 degrees. H, Quantification analysis of the colocalization of VAChT and VGAT in the lateral SN by calculating the Mander's coefficients (M1 and M2). M1 and M2 were greater for the original images versus when one image was rotated 90 degrees. G, H, Pairwise Wilcoxon tests. n = 4 mice, n = 16 medial stacks, n = 8 lateral stacks. *p < 0.05. **p < 0.01. ***p < 0.001. Data are mean ± SEM. I, Experimental design to identify the distribution of transmitter expression in cholinergic cells in the PPN. A ChATcre(neodel)+/−:tdTomato+/− mouse was used to label the cholinergic cell population in the PPN. Coronal sections containing the PPN were labeled for GABA. z stack sampling in the PPN shows a subpopulation of cholinergic cells that are positive for GABA among primary cholinergic and GABAergic cell populations.
Finally, using EM (Fig. 6A1–A3), we observed that dendrites of DA neurons from the medial SN received synaptic inputs containing mainly spherically shaped excitatory vesicles (putative cholinergic inputs) (Fig. 6A2), while we also found presynaptic boutons that had a mixed population of oblong (putative GABA) and round vesicles (putative ACh) synapsing onto DA dendrites (Fig. 6A3). To confirm the three different cholinergic synapse types at an ultrastructural level, postembedding immunogold staining in the medial SN of ChATcre(neo-del)+/−::Ai9-tdTomato+/− mice was performed (Fig. 6B1–B3). We found presynaptic boutons synapsing onto dendrites of DA neurons that immunolabelled for tdTomato and the ACh transport machinery VAChT (Fig. 6B1); presynaptic boutons expressing tdTomato and VGAT (Fig. 6B2); and presynaptic boutons expressing tdTomato, VAChT, and VGAT (Fig. 6B3). To our knowledge, these results provide the first ultrastructural evidence of ACh/GABA cotransmission in the medial SN.
Figure 6.

Ultrastructure of ACh and GABA cholinergic presynaptic inputs onto SN DA neurons. Low (A1) and high (A2,A3) magnification views of a dopamine neuron and surrounding synapses in the substantia nigra pars compacta. A large dopamine neuron (shaded blue) is surrounded by neuropil filled with dendrites (Den, shaded blue), myelinated axons (Ax), glial cells, and synaptic contacts on dendrites (boxed regions shown at high magnification in A2 and A3). Two distinct types of synaptic terminals are seen in this region. A putative cholinergic terminal (A2, shaded red) on a dendrite is filled with large uniform-sized round vesicles ∼50-60 nm (*) in diameter (shown at high magnification in inset). Another type of terminal (A3, shaded yellow) exhibits a mixture (**) of small oblong vesicles ∼20 × 80 nm in size (arrowheads in inset) corresponding to putative GABA-type vesicles and large putative cholinergic-type round vesicles (arrows in inset). B1–B3, Immunogold EM of brain sections of the medial SN of a ChATcre(neo-del)+/−::tdTomato+/− mouse. B1, A cholinergic terminal (*, shaded red) synapsing onto a dopaminergic dendrite (Den) showing colabeling of VAChT (10 nm; white arrows) and tdTomato (20 nm; black arrows). B2, Colabeling of tdTomato (10 nm; black arrow) and VGAT (20 nm; arrowhead) in a presynaptic bouton (shaded green) indicating GABA vesicles within a cholinergic terminal. B3, Triple labeling of a mixed terminal (**, shaded yellow) containing VAChT (10 nm; arrowheads), tdTomato (20 nm; black arrow), and VGAT (30 nm; white arrow). Mi, Mitochondria.
Generating the nAChR and GABAR basis functions for deconvolution of cotransmitted ACh and GABA
Cholinergic PPN neurons in rodents fire APs in a frequency range between 3 and 16 Hz (Takakusaki et al., 1997). Recent optogenetic experiments have shown a dichotomy in the plasticity of ACh and GABA release following activation of cholinergic terminals that are because of differences in release probabilities with low stimulation frequencies (5 Hz) supporting stronger inhibitory neurotransmission while high stimulation frequencies (15 Hz) would suggest a shift toward excitatory neurotransmission (Estakhr et al., 2017). However, this study optogenetically stimulated in full-field illumination mode an area with a circular diameter of 550 μm and, therefore, did not account for the localized subcellular diversity of distribution of ACh and GABA inputs and the higher proportion of “cholinergic terminals” containing only ACh or GABA compared with colocalized terminals. To investigate aspects of soma-centered ACh and GABA transmission, we restricted our stimulation field to 80 μm focused on the cell body to measure the contribution of ACh/GABA transmission without recruiting the lateralized population of primarily GABA-only inputs. We then used a deconvolution method developed by Manookin et al. (2010) whereby I–V plots obtained following repeated optogenetic stimulation can be deconvolved into the weighted sum of their individual GABAR and nAChR conductances. The conductance deconvolution method is advantageous to understand the physiological nature of ACh versus GABA transmission in that it can estimate the relative strengths of their respective receptor contributions without pharmacological application of drugs that could disturb the local network properties (Manookin et al., 2010; Sethuramanujam et al., 2016).
We first measured the full-field isolated voltage-dependent properties of nAChRs and GABARs using a cocktail of receptor antagonists in medial SN DA cells to obtain accurate parameters to apply to the deconvolution procedure. Cholinergic synaptic currents were evoked under bath application of glutamate and GABA receptor antagonists (CNQX + D-APV + bicuculline) and showed an inward rectification with a cationic reversal (Ecat) at 0 mV (Haghighi and Cooper, 1998) that was well described by a derivation of the Woodhull equation (Fig. 7A1,A2; n = 5 cells, 5 mice). Similarly, pharmacologically isolated GABAR-mediated currents (CNQX + D-APV + MLA + DHβE + MEC) showed a slight outward rectification with a chloride reversal (ECl) at −61 mV, and the I–V behavior was fit by a single exponential function as previously described (Manookin et al., 2010) (Fig. 7B1,B2; n = 6 cells, 6 mice).
nAChRs and GABARs show differential short-term plasticity in ACh/GABA neurotransmission
To estimate the relative contributions of the different receptors to the total current response with repeated local stimulations on the cell body, PSCs were evoked over a range of step potentials (Fig. 7C). The mix I–V relationship was then modeled as a weighted linear sum of the basis functions for nAChRs and GABARs, which on deconvolution, yielded an estimate of the fractional conductance for each receptor type at −60 mV (Fig. 7D1,D2). Interestingly, a dominant GABAergic component in the initial stimulation that was consistently observed across 71% of the recorded cells (n = 5/7 cells, 7 mice, 1 cell/mouse) (gGABA = 70.2 ± 2.3%, gACh = 29.7 ± 2.3%; where gGABA and gACh indicate the respective GABA and nAChR conductance weights), while 29% of medial SN DA neurons (n = 2/7 cells) had very little nAChR contribution (gGABA = 92.1 ± 2.8%, gACh = 7.9 ± 2.8%) (Fig. 7D2,E). Focusing on the majority of medial SN DA neurons that reliably display ACh/GABA transmission on initial soma-centered local optogenetic stimulation, we found that the weight of the GABAergic contribution showed a greater decrement compared with the ACh component with repeated stimulation, resulting an initial dominant inhibition followed by a subsequent equalization of excitatory and inhibitory conductances (Fig. 7F–H). To validate the analysis using the deconvolution model, we tested its performance under bath application of nAChR antagonists. In this case, the ACh-mediated inward current component seen at hyperpolarized holding voltages and the associated nAChR conductance were suppressed to levels that were not significantly greater than zero (Fig. 7I–K).
The isolated GABAR conductance is more sustained on the distal lateral dendrite than the proximal soma
In light of the robust performance of the deconvolution procedure, we performed similar experiments to investigate the plasticity of the isolated GABA conductance between the populations of mix ACh/GABA inputs proximally and the population of GABA-only inputs found on the lateral dendrites 160 μm away from the cell body (Fig. 8A–C). We pharmacologically isolated GABAergic responses by inhibiting glutamatergic responses (CNQX and APV) and nicotinic receptor responses (MLA, MEC, and DHβE). The I–V relationships between distal and proximal GABA inputs were overlapping on the first pulse across the range of voltage potentials, suggesting that the voltage clamp data did not suffer from space-clamp issues and were of adequate quality for the deconvolution procedure (Fig. 8D). Interestingly, the lateral GABA component was more sustained across repeated stimulations compared with the proximal GABA conductances (Fig. 8E). Along with research indicating DA neurons are electrotonically compact (Häusser et al., 1995; Hage and Khaliq, 2015; Moubarak et al., 2019), this suggests a differential contribution to excitatory-inhibitory balance by spatially distributed populations of ACh and GABA inputs from cholinergic axons onto the dendrites of medial SN DA neurons.
Figure 8.
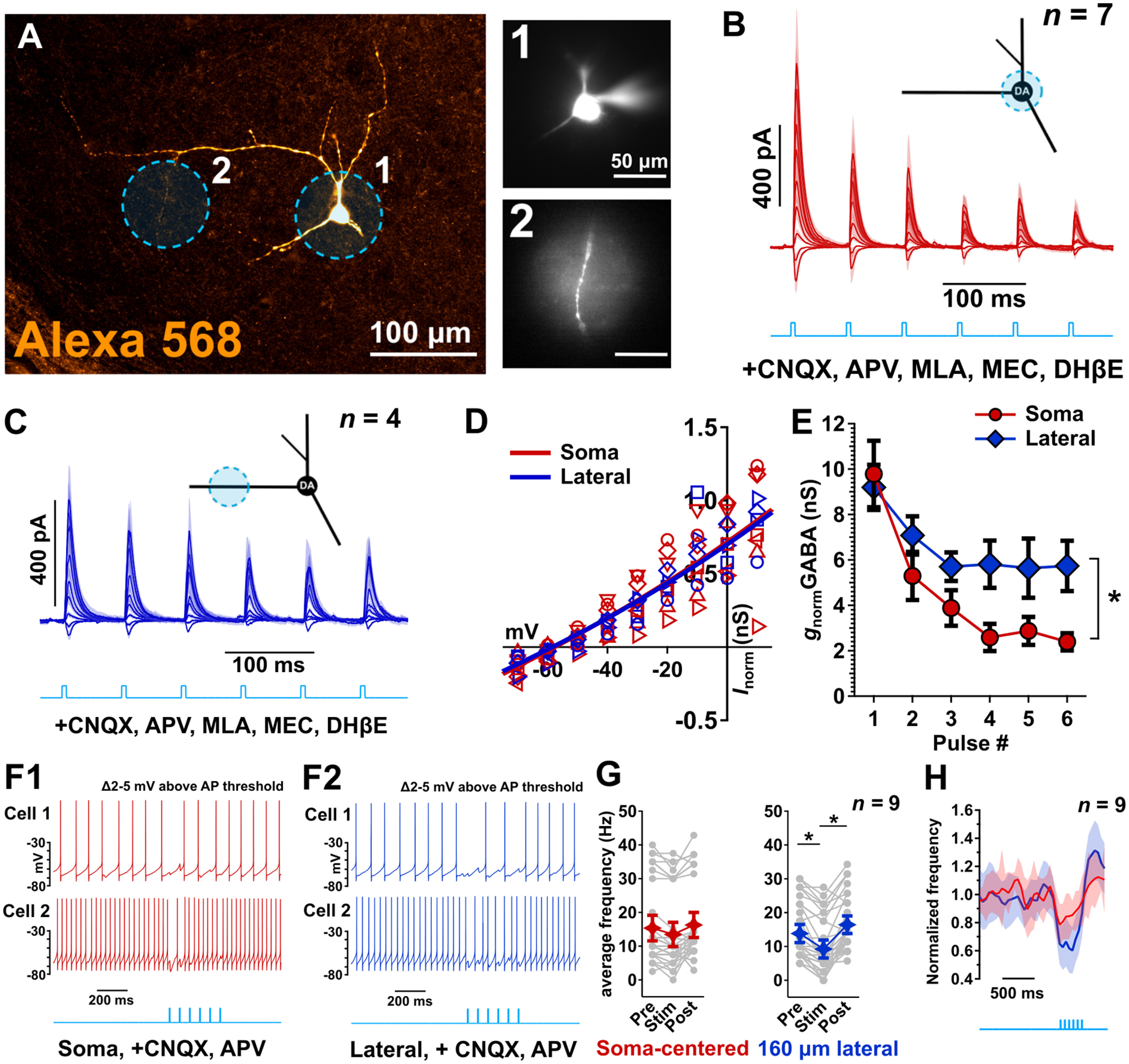
Soma-centered versus lateral dendrite located GABA synaptic transmission shows different short-term plasticity and effects on neuronal excitability. A, Experimental design to investigate the isolated GABAR conductance between the mix nAChR/GABAR conductance proximally and the lateral primary GABAR conductance. Stimulation fields were restricted to 80 μm. An example DA neuron used in the experiment is shown. B, Whole-cell voltage-clamp recordings of local (80-μm-diameter; blue light) soma-centered GABAR-mediated currents in conditions similar to that of Figure 7. Responses are measured in the presence of CNQX, D-APV, MEC, DHβE, and MLA. Data are mean ± SEM. C, Similar to B, GABAR currents were measured on a lateral dendrite 160 μm away from the cell body. B, C, n = 7 cells from 7 mice, n = 7 cells for the proximal focal stimulation, n = 4 cells for the lateral focal stimulation (repeated measurements). D, The I–V relationships computed from the currents in B and C and modeled by their respective weighted linear sum of the receptor basis functions as estimated in Figure 7 (thick blue and red lines). The high degree of overlap between fitted I–V shows that the voltage-clamp data were of adequate quality for the deconvolution procedure. E, Normalized estimated medial and lateral GABAR conductances as a function of the pulse number. The lateral GABAR contribution shows less decrease with repeated stimulation compared with the medial GABAR response. Nonparametric F1-LD-F1 factorial test. F1, F2, High current-driven firing (∼15 Hz) in medial DA cells was differentially diminished following local (80 μm) soma-centered versus lateral optical stimulation (15 Hz, 6 pulses). G, Average frequencies prior, during, and after optical stimulation reveal a stronger distal lateral inhibition of AP generation compared with soma-centered optical stimulation. Dunn's test with Bonferroni correction. H, Firing frequencies of medial SN DA cells normalized to baseline confirm the transient inhibitory potential of soma-centered optical stimulation (red line) compared with the more sustained lateral inhibition (blue line). F–H, n = 9 cells from 9 mice (F, examples; G,H, population data). *p < 0.05. **p < 0.01. ***p < 0.001. Data are mean ± SEM.
The GABAR conductance on the distal lateral dendrite is more effective in inhibiting APs than GABA in the proximal soma
Considering the differential short-term plasticity of GABA release that we revealed at the single DA neuron level as a function of their spatial distribution on the somatodendritic surface, we investigated how the observed GABA and mix ACh/GABA conductances would affect firing during a state of tonic medial SN DA pacemaking. We recorded medial SN DA neurons in whole-cell current-clamp mode while maintaining them at a relatively high rate of tonic pacemaker firing (by injecting slow continuous steady current) and then exciting ChR2 (6 pulses at 15 Hz) to highlight the effects of the cholinergic-mediated GABAergic inhibition as a function of their spatial distribution (Fig. 8F1,F2). Interestingly, we revealed that the local lateralized GABAergic conductance was more sustained and inhibited DA pacemaking to a greater extent, despite dendritic filtering, compared with soma-centered ACh/GABA release (Fig. 8G,H). Furthermore, the transient inhibition of AP generation during the first 2-3 pulses in the optogenetic train for soma-centered optogenetic stimulation confirmed the conductance results outlined in Figure 7 (see Fig. 8H).
Repeated cotransmission in the proximal soma summed with proximal glutamate or somatic current injection is more likely to fire APs than glutamate or current alone
Focusing on soma-centered ACh/GABA transmission, we sought to understand its role in modulating SN DA excitability. Since the data on conductance temporal dynamics did not reveal a dramatic switch from inhibition to excitation, a plausible mechanism could involve an interplay between the equalization of net excitatory and inhibitory conductances and the intrinsic voltage-gated properties of DA neurons. This could in turn favor an “up” state of DA excitability that would promote EPSP summation and consequent AP firing. To test these hypotheses, we recorded from medial SN DA neurons (n = 10 cells, 10 mice) in the presence of glutamate receptor inhibitors CNQX and APV and maintained the cells 5 mV below their firing threshold under current clamp (by injecting slow continuous and steady hyperpolarizing current) while optically stimulating ACh/GABA release locally on the soma (15 Hz, 6 pulses; Fig. 9A,B). Concurrent with the conductance results derived from the population data in Figure 7, only a minority of DA cells (2 of 10 cells) repeatedly fired APs with post-optogenetic stimulation while the majority of DA cells (8 of 10 cells) remained silent (Fig. 9A,B). This confirmed that, while repeated ACh/GABA transmission could be sufficient to transition from an inhibitory to an excitatory state for some DA neurons, this was not its preferred physiological mechanism to modulate SN DA excitability in the absence of glutamatergic neurotransmission and that a switch to promote multisynaptic spatiotemporal integration by including glutamatergic neurotransmission was more likely. To this point, we implemented an experimental design that would allow for a combination of optogenetics and UV one-photon uncaging of glutamate (Fig. 10A–H). We achieved reliable soma-targeted photolysis of MNI-glutamate within an 80 μm field at different UV intensities (Fig. 10A). However, extensive research describes the variable antagonistic character of MNI-glutamate toward GABAA receptors with IC50 ranging from 105 to 500 μm (Fino et al., 2009; Palma-Cerda et al., 2012; Olson et al., 2013). In our case, we verified that bath application of 200 μm caged MNI-glutamate for 15 min did not induce a decrease in the amount nor alter the short-term plasticity of optogenetically evoked local GABA currents (Fig. 10B), and we consequently used MNI-glutamate to induce subthreshold depolarizations in medial SN DA cells (Fig. 10C). We compared the kinetics of the evoked EPSC elicited by UV uncaging of MNI-glutamate to that of sEPSCs. For the largest AMPA-mediated evoked EPSC elicited by UV MNI-glutamate uncaging in Figure 10A, the 10%-90% rise time was 6.4 ms and the 1/e decay time constant was 23.2 ms. In the same cell, the average of 13 individual sEPSCs had more rapid kinetics with a 10%-90% rise time of 1.6 ± 0.2 ms and the 1/e decay time constant of 3.5 ± 1.6 ms. This is consistent to previous reports of the kinetic properties of sEPSCs in the SN (Pearlstein et al., 2015) and EPSCs evoked by UV uncaging with MNI-glutamate (Passlick and Ellis-Davies, 2018).
Figure 9.

ChR2 stimulation on its own without glutamatergic neurotransmission induces AP firing in a minority of neurons. A, Soma-centered optical stimulation of ACh/GABA transmission (15 Hz, 6 pulses, 80 μm field stimulation) in medial SN DA neurons (in the presence of glutamate receptor inhibitors CNQX and APV) with current held ∼5 mV below their firing threshold further strengthens the shift toward net excitation with repeated local ACh/GABA transmission as seen in Figure 7 with a depolarization rebound that is sufficient to generate APs in 20% of the sampled DA neurons (2 of 10 cells). B, However, 80% of medial SN DA cells (8 of 10 cells) do not spike following repeated soma-centered stimulation under the same conditions. n = 10 cells from 10 mice.
Figure 10.
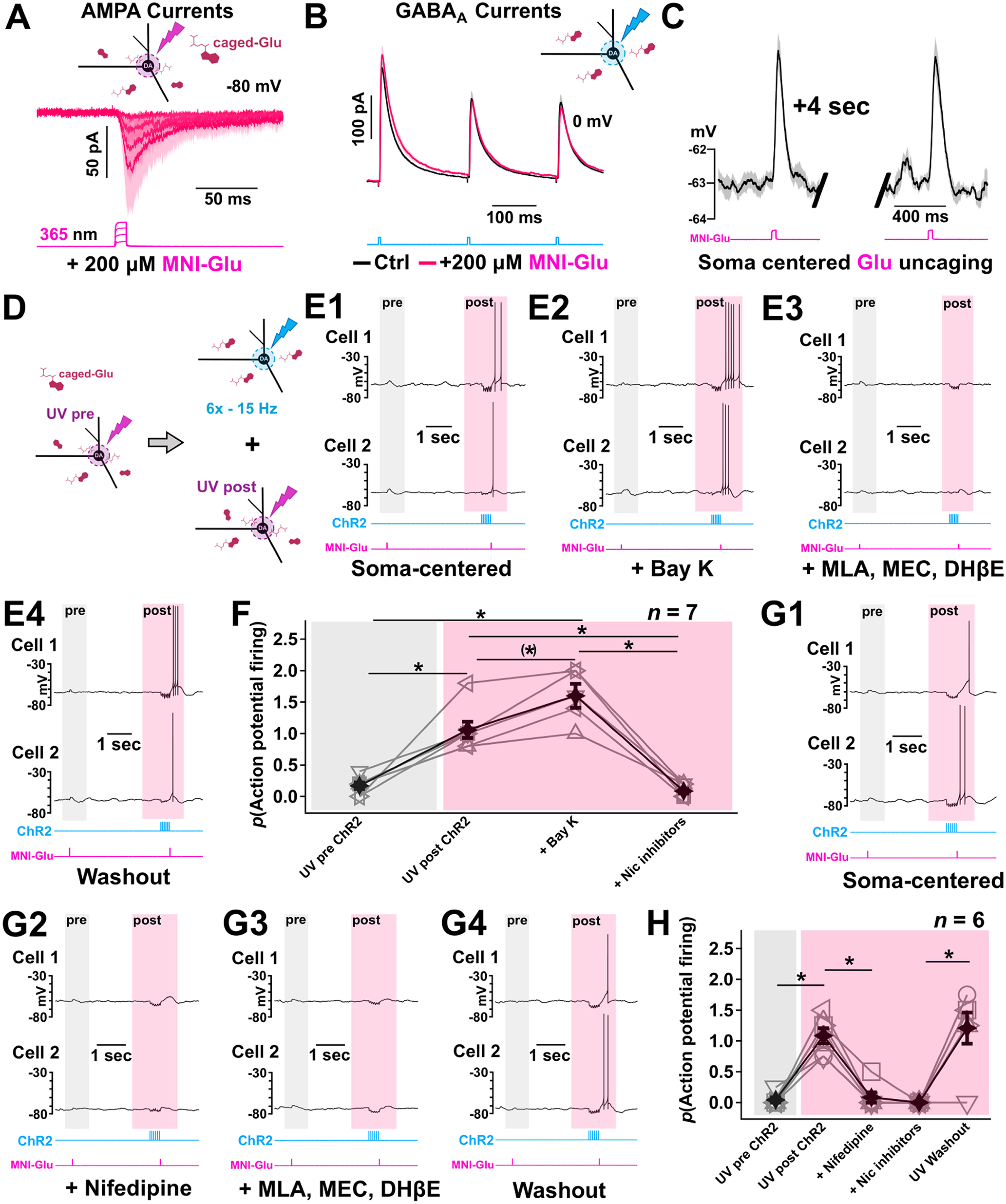
A train of ACh/GABA cotransmission is sufficient to enhance the action of glutamate to increase neuronal excitability through activation of L-type Ca2+ channels. A, Whole-cell recordings in voltage-clamp mode of medial SN DA cells reveal somatic glutamate-mediated inward currents induced by local photolysis of caged MNI-glutamate (200 μm) at increasing UV intensities. Optical stimulation fields were restricted to 80 μm centered on DA cell bodies. n = 3 cells from 3 mice. B, Local light-evoked soma-centered IPSCs from a medial SN DA neuron suggest that the concentration of caged MNI-glutamate used does not interfere with synaptic activation of GABA receptors. Voltage-clamp recordings of PSCs at 0 mV show no initial amplitude nor plasticity difference following a 15 min bath application of MNI-glutamate (200 μm, pink trace) compared with baseline (black trace). Data are mean ± SEM. C, Example glutamate-induced PSPs from a medial SN DA neuron current-clamped ∼5 mV below its firing threshold. Local soma-centered photolysis of caged glutamate can produce reliable subthreshold EPSPs 4 s apart with similar amplitudes. Data are mean ± SEM. D, Experimental design to investigate the role of soma-centered ACh/GABA transmission and its potential with regard to multisynaptic integration with glutamate in medial SN DA neurons. Local MNI-glutamate photolysis is used to generate an initial local subthreshold EPSP. Similar to C, UV intensities and duration parameters are kept similar for a second photolysis event that is preceded by repeated light-evoked ACh/GABA transmission (15 Hz, 6 pulses). Optical stimulation fields were restricted to 80 μm centered on DA cell bodies and kept consistent for both MNI-glutamate and ChR2 stimulation. E1–E4, Example glutamate-induced PSPs from two medial SN DA neurons current-clamped ∼5 mV below their firing threshold with and without bath application of a L-type Ca2+ channel agonist and nicotinic receptor inhibitors reveal a nicotinic enhancement of SN DA excitability that is modulated by L-type Ca2+ channels and favors somatic glutamate-mediated PSP summation. Responses are measured in the presence of caged MNI-glutamate and under subsequent bath applications of Bay K 8644 (10 μm) and nicotinic inhibitors (MEC, DHβE, MLA). Washout of pharmacological agents is shown. The second photolysis event is delivered ∼25 ms after the end of the last 470 nm pulse. F, Probability of AP generation following subthreshold soma-centered photolysis of MNI-glutamate with and without a preceding light-evoked repeated ACh/GABA transmission and in the presence of Bay K 8644 and nicotinic inhibitors. Friedman test followed by post hoc Wilcoxon pairwise comparisons with Bonferroni correction. E, F, n = 7 cells from 7 mice (E, examples; F, population data). n = 7 cells from 7 mice. n = 2 of 7 cells did not receive BAY K but did receive nicotinic inhibitors (symbols but no lines connecting the symbols). n = 5 of 7 cells received all conditions, including BAY K and nicotinic inhibitors (symbols connected with lines). Filled black symbols with black lines represent averages. G1–G4, Example glutamate-induced PSPs from two medial SN DA neurons current-clamped ∼5 mV below their firing threshold with and without bath application of an L-type Ca2+ channel antagonist and nicotinic receptor inhibitors reveal a nicotinic enhancement of SN DA excitability that is positively modulated by L-type Ca2+ channels and favors somatic glutamate-mediated PSP summation. G1, Responses are measured in the presence of caged MNI-glutamate and under subsequent bath applications of nifedipine (1 μm) (G2) and nicotinic inhibitors (MEC 20 μm, DHβE 10 μm, MLA 10 nm) (G3). Washout of pharmacological agents is shown (G4). The second photolysis event is delivered ∼25 ms after the end of the last 470 nm pulse. H, Summary graph showing probability of AP induction following subthreshold soma-centered photolysis of MNI-glutamate with and without a preceding light-evoked repeated ACh/GABA transmission and in the presence of the L-type Ca2+ channel inhibitor nifedipine and nicotinic inhibitors (n = 6 cells from 6 mice). *p < 0.05 before and after Bonferroni correction (if corrected). (*)p < 0.05 before, but not after, correction. **p < 0.01. ***p < 0.001. Data are mean ± SEM.
We then proceeded with our paradigm to investigate the effect of repeated soma-centered ACh/GABA cotransmission combined with subthreshold glutamate-mediated depolarizations (Fig. 10D). We found that ACh/GABA transmission preceding subthreshold glutamate-mediated depolarization was more effective in generating spikes in medial SN DA cells compared with glutamate alone (Fig. 10E1). Bath application of the L-type Ca2+ channel agonist Bay K 8644 (10 μm) dramatically increased the number of APs elicited with the combined stimulation of local ACh/GABA transmission and glutamate-mediated depolarization (Fig. 10E1,E2,F) and was suppressed by either bath application of the selective L-type Ca2+ channel blocker nifedipine (1 μm) or nAChR inhibitors (Fig. 10E3,F,G1-G4,H). Finally, washing out the pharmacological agents restored the ACh/GABA-mediated enhancement of excitability (Fig. 10E4,G4). This suggested a local recruitment of L-type Ca2+ channels by nicotinic receptors following ACh/GABA release that can help promote spatiotemporal summation of glutamate-mediated inputs (Fig. 10F,H).
In addition, replacing caged-glutamate by somatic current injections replicated the phenomenon of enhanced dopaminergic excitability mediated by nicotinic receptors following soma-centered ACh/GABA transmission (Fig. 11A). Furthermore, suprathreshold somatic current injections preceded by optogenetic stimulation of ACh/GABA release revealed an additional depolarizing contribution because of the recruitment of T-type Ca2+ channels (Fig. 11B–E). We found that T-type calcium channels decreased the latency of triggering an AP with channelrhodopsin-mediated ACh/GABA release and current injection compared with when T-type calcium channels were blocked by TTA-P2 (8 μm) (Fig. 11D,E). We observed that, following the final (6th) ChR2-mediated pulse of the stimulation train, there was a subsequent repolarization that was dampened with T-type channel blocker and more severely reduced with the nicotinic receptor inhibitor cocktail (Fig. 11C).
Figure 11.
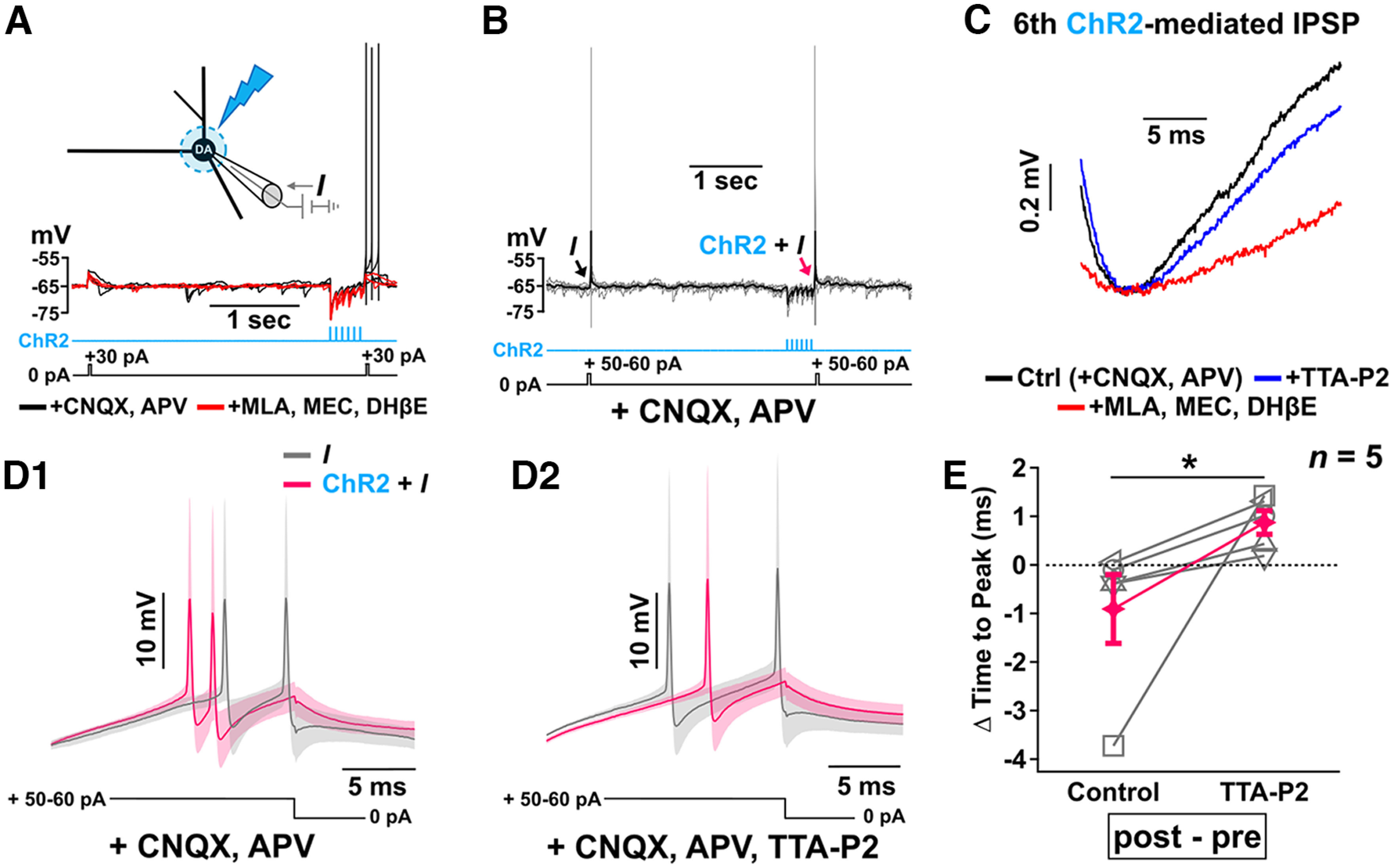
A train of ACh/GABA cotransmission is sufficient to enhance the action of membrane depolarizing current to increase neuronal excitability and speed to onset through T-type Ca2+ channels. A, Subthreshold depolarizations from somatic current injections in a medial SN DA neuron clamped ∼5 mV below its firing threshold replicates the phenomenon seen in Figure 10 of induction of AP stimulation when the current injection follows a train of light-evoked ACh/GABA transmission (15 Hz, 6 pulses) (80-μm-diameter; black trace). Induction of APs is abolished by bath application of nicotinic inhibitors (red trace). Current-clamp recordings were obtained in the presence of CNQX and APV and following subsequent application of MEC, DHβE, and MLA. B, D1, D2, Suprathreshold somatic depolarization reveals a faster AP onset after light-evoked soma-centered ACh/GABA cotransmission and depolarizing current (red arrow in B; red traces in D1) compared with the depolarizing current injection alone before the light-evoked stimulation (black arrow in B; gray traces in D1) that is delayed by bath application of a T-type Ca2+ channel antagonist (D2). C, In the presence of the T-type Ca2+ channel antagonist (blue trace), there was a slower membrane repolarization following the 6th and final pulse of the optical stimulation train of ACh/GABA cotransmission compared with controls with only CNQX and APV (black trace). There was a further dampening of the repolarization following the 6th optical stimulation when the nicotinic receptor inhibitors were subsequently applied (red trace). Current-clamp recordings were obtained in the presence of CNQX and APV and following subsequent application of the T-type Ca2+ channel antagonist TTA-P2 (8 μm) and then the nicotinic receptor inhibitors MLA, MEC, and DHβE. E, Differences in time to AP peak (post-pre) induced by suprathreshold depolarizing current injection before and following repeated soma-centered ACh/GABA cotransmission confirms an additional modulation of the nicotinic-mediated enhancement of SN DA excitability by T-type Ca2+ channels as inhibition of T-type Ca2+ channels significantly delays the onset of APs. Friedman test. B–E, n = 5 cells from 5 mice (B,C,D1,D2, examples; E, population data). *p = 0.025. Data are mean ± SEM.
Mechanism of how temporal integration of synaptic inputs and voltage-gated channels increases neuronal excitability
We sought to explain mechanistically how repeated ACh/GABA cotransmission followed by glutamate was able to induce AP firing. We noticed that a single stimulation pulse of ACh/GABA followed by glutamate uncaging (25 ms later) produced a depolarization that was insufficient to trigger an AP (Fig. 12A1). Only when there was a train (15 Hz, 6 pulses) of ChR2 stimulation followed by glutamate uncaging (25 ms later) was there a robust induction of APs (Fig. 12A2). It was clear that the membrane depolarization produced by glutamate uncaging following the train of ChR2 stimulation was much greater and more prolonged (gray trace) than that of the glutamate uncaging following a single pulse of ChR2 stimulation (black trace) (Fig. 12B). When examining the timing between ChR2 train and UV uncaging, we found that the probability of AP induction was high when glutamate uncaging followed the final ChR2 train pulse by 10 ms, but firing probability increased further when the ChR2 to UV-uncaging interval increased to 20 ms and then peaked at 25 ms (Fig. 12D). The probability of firing dropped precipitously when the ChR2-UV interval was increased to 30 ms and reached zero probability of firing when the interval reached 40 ms (Fig. 12D). We surmised that the probability of AP may be influenced by the magnitude and kinetics of the underlying initial hyperpolarization and subsequent depolarization following the ChR2 train and UV glutamate uncaging. So we fitted the glutamate uncaging-alone response of 5 averaged cells (red trace) (Fig. 12E1) and the glutamate uncaging following the train of ChR2 stimulation (red trace) (Fig. 12E2), each with their own multiexponential equations so that we can estimate and measure more readily the amplitude of the underlying depolarization. To show the temporal relationship of ChR2-UV uncaging interval to membrane depolarization, we show the raw traces of post-ChR2-UV uncaging at ChR2-UV intervals of 10, 20, 30, and 40 ms (Fig. 12F) and their respective multiexponential fits of the underlying membrane depolarization (top traces) and the temporally aligned glutamate uncaging response (bottom traces) (Fig. 12G). The ChR2-UV uncaging stimulation combination regardless of the temporal interval had much larger membrane depolarizations and longer time courses than the temporally aligned glutamate-mediated PSP alone (Fig. 12G). Second, the 20-25 ms ChR2-UV uncaging intervals had the largest membrane depolarizations of the various intervals tested (Fig. 12G,H). This prolonged membrane depolarization is likely the result of the integration of nicotinic receptors, AMPA glutamate receptors, and L- and T-type Cav channels following the recovery of the hyperpolarizing potential of the final pulse of the train of ACh/GABA cotransmission (Fig. 12I).
Discussion
The present study investigated the spatial distribution and physiological effects of ACh/GABA cotransmitting synapses onto medial SN DA neurons to understand their role in tuning the excitatory/inhibitory balance of these neurons. We confirmed results from our previous study (Estakhr et al., 2017) that medial SN DA neurons receive cotransmitted ACh/GABA monosynaptic inputs. We identified three different types of monosynaptic connections from cholinergic axons onto DA cells: cotransmitted ACh/GABA, GABA-only, and ACh only. There was a heterogeneous somatodendritic patterning of cholinergic input distribution onto DA cells with a predominant GABA-only conductance along the lateral dendrites and a soma-centered ACh/GABA cotransmission. Immunofluorescent labeling of neurotransmitter vesicular transporters on cholinergic terminals revealed that cotransmission represents a minority of the presynaptic mode of release from cholinergic axons onto medial SN DA neurons. GABA cotransmission onto proximal soma depressed more rapidly than GABA-only on the lateral dendrites. The lateral GABA-only release was more sustained in inhibiting AP firing. In the proximal soma, nAChR-mediated conductances evoked by localized ACh release were initially lower than the competing cotransmitted GABA responses, but equalized with subsequent illuminations as the GABA responses decayed more rapidly while nAChR responses were substantially maintained. This resulted in greater neuronal excitability when either MNI-glutamate uncaging or depolarizing current injection was applied following the train of ACh and GABA cotransmission. This increased excitability was positively modulated by L-type calcium channel activator Bay K and T-type calcium channels but was completely abolished with nAChR inhibitors.
Our immunohistochemical data showed mostly a differential spatial organization of excitatory and inhibitory synaptic inputs labeled by VAChT and VGAT antibodies that overlapped with cholinergic terminals (87%-89%) and relatively few colocalized terminals (11%-13%) (Fig. 4B,C). One question would be: what physiological advantage would cholinergic cotransmitted ACh-containing and GABA-containing vesicles have in being segregated in separate presynaptic boutons? Distinct spatial organization of synaptic inputs is a well-known mechanism for overcoming the sublinear summation of PSPs associated with spatiotemporally colocalized signals (Mel, 1993; Koch, 1998; Poirazi et al., 2003; Stuart et al., 2016). Theoretical modeling predicts that GABAergic inputs are more effective in inhibiting excitatory inputs when GABA inputs are spatially separated and localized between the excitatory inputs and the soma, and ineffective when located distal to the excitatory inputs (Rall, 1967; Koch et al., 1983; Koch, 1998; Hao et al., 2009; Stuart et al., 2016). A similar physiological advantage could be addressed in the differential spatial distribution of cholinergic VGAT- and VAChT-containing terminals. Subpopulations of cholinergic neurons in the PPN are known to possess the cellular machinery for acetylcholine and GABA transmitter release and extend their presynaptic terminals in the SN (Fig. 5) (Estakhr et al., 2017; Li and Spitzer, 2020). Since we found the neuronal source of these cholinergic inputs in the PPN contain many neurons that colocalize ChAT and GABA (Fig. 5), these neurons may sort their respective neurotransmitter containing vesicles in mostly distinct presynaptic boutons, although a minority of these ACh/GABA-containing vesicles do appear in the same presynaptic boutons (Fig. 4).
Surprisingly, the histologic data also revealed that a small proportion of mix ACh/GABA puncta and primary ACh puncta were found ∼300 μm lateral to the soma of medial SN DA neurons, while electrophysiological experiments did not detect ACh conductance laterally. However, there are a few reasons why such immunohistochemical findings were not evidenced functionally: (1) ACh and mix ACh/GABA puncta representing a disynaptic or trisynaptic complex with primary presynaptic modulatory effects which could not be discerned anatomically from a postsynaptic role. Finally, (2) the low proportion of ACh puncta laterally can be easily reconciled based on prior findings observing a heterogeneous cholinergic innervation across the medial-lateral extent of SN DA neurons with lateral SN DA neurons primarily receiving ACh-only-mediated inputs compared with the medial SN DA neuron populations, which mostly receive cotransmitted ACh/GABA (Estakhr et al., 2017).
The 15 Hz stimulation in the somatic region produced an initial dominant GABAergic component of ACh/GABA transmission (Estakhr et al., 2017); however, we found that, with repeated stimulation, the GABAergic contribution diminished more rapidly than the ACh component, resulting in an initial dominant inhibition followed by a subsequent equalization of excitatory and inhibitory conductances (Fig. 7). However, there were only a minority of cells (2 of 10) in which this resulted in increased neuronal firing (Fig. 9). Therefore, we hypothesized that this equalization of GABA and ACh conductances would tip the balance toward increased neuronal excitation when another incoming excitatory neurotransmitter is present. Under conditions where uncaging glutamate alone was insufficient to induce increased AP firing, eliciting cotransmitted ACh and GABA with glutamate uncaging was sufficient (Fig. 10E1). This increased excitability was nAChR-mediated as this effect was abolished with nAChR antagonists (Fig. 10E3). Furthermore, we discovered that the depolarization provided by nAChR activation enhanced activity of L- and T-type calcium channels to positively modulate AP firing (Figs. 10E-H, 11C–E), while inhibition of L-type calcium channels with nifedipine almost completely abolished AP firing (Fig. 10G2,H). L-type channel activation caused a prolonged plateau depolarization with an increased number of APs (Fig. 10E2,F), while T-type channels reduced the latency to AP onset while not affecting the number of APs elicited (Fig. 11B–E). If these actions are presumably contributed by the depolarization of the membrane potentials via nAChRs, then what would be the rationale of cotransmitted GABA? We propose that the strong initial GABA hyperpolarization may play a role in de-inactivating L- and T-type calcium channels as well as voltage-gated sodium channels. Thus, GABA may “prime” these voltage-gated channels to more strongly depolarize the membrane potential to initiate AP firing. We also determined that the release of glutamate had to be precisely timed to the 15 Hz train of ACh/GABA in that there was a 20-25 ms window after the sixth and final pulse of the 15 Hz train in which glutamate release was optimal to elicit APs (Fig. 12D–H). This brief time window shortly after the ACh/GABA release argues against a metabotropic effect of ACh release as a contributor to increased excitability; furthermore, inhibitors of the ionotropic nAChRs completely abolished the increased excitability (Fig. 10E3,F,G3,H). Having said this, we also cannot rule out the contribution of disynaptic GABA inputs in addition to cotransmitted ACh/GABA in modulating neuronal excitability. We have previously shown evidence of disynaptic GABA mediated by ACh release in dopaminergic neurons of the medial SN (Estakhr et al., 2017).
The fact that cotransmitted GABA release depressed more rapidly than ACh release on repeated stimulation may reflect that GABA-containing vesicles have an initially higher probability of release than ACh-containing vesicles. This could be because of a variety of mechanisms. The synaptotagmin of the GABA-containing vesicles may be more closely situated to the calcium source than the ACh-containing vesicles. The two vesicle populations may have different subtypes of synaptotagmins with differing calcium sensitivity of phospholipid binding (C. Li et al., 1995). Having ACh and GABA release sites spatially segregated in different presynaptic boutons may allow the possibility that these distinct sites have different concentrations of calcium buffer proteins (Smith and Zucker, 1980; Rozov et al., 2001; Jackman and Regehr, 2017), different voltage-gated calcium channel subtypes with distinct gating kinetics and voltage sensitivities (Dolphin, 2021) or differential localization of metabotropic receptors on the presynaptic terminals, which can have profound effects on modulating neurotransmitter release (Burke et al., 2018). Another potential explanation of why repetitive GABA responses depress more quickly than ACh responses could be that ACh-containing vesicles may replenish more quickly than GABA-containing vesicles. Vesicle replenishment has been shown to occur at high rates (10s of milliseconds) and therefore can profoundly affect short-term synaptic plasticity (Crowley et al., 2007; Butola et al., 2017; Bornschein et al., 2020). We also recognize that there are challenges at studying short-term plasticity of release dynamics using optogenetic approaches. Schoenenberger et al. (2011) showed that stimulation of hippocampal CA3 to CA1 synapses resulted in paired-pulse inhibition of glutamate release with channelrhodopsin stimulation while electrical stimulation resulted in paired-pulse facilitation with electrical stimulation. Optogenetic stimulation may exaggerate release probability since ChR2 itself is permeable to calcium.
We also observed that the lateral cholinergic responses are GABA-only and display significantly less depression with repeated illumination than the cotransmitted GABA responses at the soma (Figs. 2, 8), suggesting a lower GABA release probability onto the lateral dendrite versus the soma. Interestingly, the lateral GABA release resulted in a stronger and more maintained inhibition of AP firing than the cotransmitted GABA and ACh which showed a transient, weaker inhibition of AP firing. This result goes against the prevailing theory that GABAergic inputs are more effective in inhibiting excitatory inputs when they are spatially localized at more proximally between the soma and the excitatory input, and ineffective when located distal to the excitatory inputs (Rall, 1967; Koch et al., 1983; Koch, 1998; Hao et al., 2009). Our results support that dopaminergic neurons are electrotonically compact as previously shown by others (Häusser et al., 1995; Hage and Khaliq, 2015; Moubarak et al., 2019; Otomo et al., 2020). The strong lateralized GABA-only inputs and the proximal localized ACh/GABA cotransmitted inputs may play another physiological role. Dopaminergic neurons are known to release DA locally in the SN through dendritic release of DA (Geffen et al., 1976; Mendez et al., 2011). GABA inputs could shunt back-propagating APs to inhibit dendritic DA release in the SN and modifying local network activity, while proximally localized ACh/GABA cotransmitted inputs could potentially enhance or inhibit dendritic DA release depending on the train frequency of local cholinergic inputs.
Thus, these results suggest that precise spatial and temporal patterning of synaptic inputs of ACh, GABA, and glutamate can integrate and recruit a variety of voltage-gated cation channe ls to shape neuronal excitability of DA SN neurons. We have, for the first time, mapped in spatial detail the functional synaptic inputs of cholinergic originating GABA and ACh release onto DA cells. Future work will need to map out the functional glutamatergic inputs onto these cells. In addition, we need to deduce how rapid successive stimulation of transmitter release at different sites along the somatodendritic extent of the neuron can impact synaptic integration and affect neuronal excitability. The specificity of the localization and timing of synaptic inputs and their integration of innate voltage-gated channels along the somatodendritic extent greatly augment the computational power of neurons.
Footnotes
This work was supported by Natural Sciences and Engineering Research Council of Canada Discovery Grant RGPIN4581-2018 and Canadian Institutes of Health Research Project Grant PJT-159548 to R.N.; Natural Sciences and Engineering Research Council of Canada Discovery Grant RGPIN 2019-06871 to K.R.D.; Basque Government Grant IT1230-19 and Ministry of Science and Innovation Grant PID2019-107548RB-I00 to P.G.; and Canadian Institutes of Health Research–Frederick Banting and Charles Best Canada Graduate Scholarship to K.L.L.G. We thank Drs. Santhosh Sethuramanujam, Craig Brown, and Gautam Awatramani for helpful discussions; Prof. Niels Christian Danbolt and Shreyas B. Rao for the Lowicryl HM20 embedding (Department of Molecular Medicine, Laboratory of Molecular Neuroscience, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway); and the animal care staff at the University of Victoria for providing excellent mouse husbandry.
The authors declare no competing financial interests.
References
- Anastasiades PG, Marques–Smith A, Butt SJ (2018) Studies of cortical connectivity using optical circuit mapping methods. J Physiol 596:145–162. 10.1113/JP273463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo-Jiménez MM, Bourgeois JP, Marubio LM, Le Sourd AM, Ottersen OP, Rinvik E, Fairén A, Changeux JP (1999) Ultrastructural localization of the alpha4-subunit of the neuronal acetylcholine nicotinic receptor in the rat substantia nigra. J Neurosci 19:6475–6487. 10.1523/JNEUROSCI.19-15-06475.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1996) Cable properties of cultured hippocampal neurons determined from sucrose-evoked miniature EPSCs. J Neurophysiol 75:1250–1255. 10.1152/jn.1996.75.3.1250 [DOI] [PubMed] [Google Scholar]
- Bhatia A, Moza S, Bhalla US (2019) Precise excitation-inhibition balance controls gain and timing in the hippocampus. Elife 8:e43415. 10.7554/eLife.43415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B (1987) Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol 385:243–286. 10.1113/jphysiol.1987.sp016493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornschein G, Brachtendorf S, Schmidt H (2020) Developmental increase of neocortical presynaptic efficacy via maturation of vesicle replenishment. Front Synaptic Neurosci 11:36. 10.3389/fnsyn.2019.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8:1263–1268. 10.1038/nn1525 [DOI] [PubMed] [Google Scholar]
- Burke KJ, Keeshen CM, Bender KJ (2018) Two forms of synaptic depression produced by differential neuromodulation of presynaptic calcium channels. Neuron 99:969–984.e5. 10.1016/j.neuron.2018.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butola T, Wichmann C, Moser T (2017) Piccolo promotes vesicle replenishment at a fast central auditory synapse. Front Synaptic Neurosci 9:14. 10.3389/fnsyn.2017.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway EM (2002) Cell type specificity of local cortical connections. J Neurocytol 31:231–237. 10.1023/a:1024165824469 [DOI] [PubMed] [Google Scholar]
- Callaway EM, Yuste R (2002) Stimulating neurons with light. Curr Opin Neurobiol 12:587–592. 10.1016/S0959-4388(02)00364-1 [DOI] [PubMed] [Google Scholar]
- Caruncho HJ, Liste I, Labandeira-García J (1996) GABAA receptor α1-subunit-immunopositive neurons in the rat striatum. Brain Res 722:185–189. 10.1016/0006-8993(96)00197-7 [DOI] [PubMed] [Google Scholar]
- Case DT, Burton SD, Gedeon JY, Williams SP, Urban NN, Seal RP (2017) Layer- and cell type-selective cotransmission by a basal forebrain cholinergic projection to the olfactory bulb. Nat Commun 8:652. 10.1038/s41467-017-00765-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PB, Hommer DW, Pert A, Skirboll LR (1987) Innervation of substantia nigra neurons by cholinergic afferents from pedunculopontine nucleus in the rat: neuroanatomical and electrophysiological evidence. Neuroscience 23:1011–1019. 10.1016/0306-4522(87)90176-X [DOI] [PubMed] [Google Scholar]
- Cornwall J, Cooper JD, Phillipson OT (1990) Afferent and efferent connections of the laterodorsal tegmental nucleus in the rat. Brain Res Bull 25:271–284. 10.1016/0361-9230(90)90072-8 [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Carter AG, Regehr WG (2007) Fast vesicle replenishment and rapid recovery from desensitization at a single synaptic release site. J Neurosci 27:5448–5460. 10.1523/JNEUROSCI.1186-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza RD, Bista P, Meier AM, Ji W, Burkhalter A (2019) Spatial clustering of inhibition in mouse primary visual cortex. Neuron 104:588–600.e5. 10.1016/j.neuron.2019.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2021) Functions of presynaptic voltage-gated calcium channels. Function (Oxf) 2:zqaa027. 10.1093/function/zqaa027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estakhr J, Abazari D, Frisby K, McIntosh JM, Nashmi R (2017) Differential control of dopaminergic excitability and locomotion by cholinergic inputs in mouse substantia nigra. Curr Biol 27:1900–1914.e4. 10.1016/j.cub.2017.05.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A (2001) Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci 21:7993–8003. 10.1523/JNEUROSCI.21-20-07993.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Araya R, Peterka DS, Salierno M, Etchenique R, Yuste R (2009) RuBi-glutamate: two-photon and visible-light photoactivation of neurons and dendritic spines. Front Neural Circuits 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald ML, Mackie K, Pickel VM (2019) Ultrastructural localization of cannabinoid CB1 and mGluR5 receptors in the prefrontal cortex and amygdala. J Comp Neurol 527:2730–2741. 10.1002/cne.24704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futami T, Takakusaki K, Kitai ST (1995) Glutamatergic and cholinergic inputs from the pedunculopontine tegmental nucleus to dopamine neurons in the substantia nigra pars compacta. Neurosci Res 21:331–342. 10.1016/0168-0102(94)00869-H [DOI] [PubMed] [Google Scholar]
- Geffen LB, Jessell TM, Cuello AC, Iversen LL (1976) Release of dopamine from dendrites in rat substantia nigra. Nature 260:258–260. 10.1038/260258a0 [DOI] [PubMed] [Google Scholar]
- Gentet LJ, Stuart GJ, Clements JD (2000) Direct measurement of specific membrane capacitance in neurons. Biophys J 79:314–320. 10.1016/S0006-3495(00)76293-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Mulder N, Saunders A, Sabatini BL (2016) Cotransmission of acetylcholine and GABA. Neuropharmacology 100:40–46. 10.1016/j.neuropharm.2015.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage TA, Khaliq ZM (2015) Tonic firing rate controls dendritic Ca2+ signaling and synaptic gain in substantia nigra dopamine neurons. J Neurosci 35:5823–5836. 10.1523/JNEUROSCI.3904-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, Cooper E (1998) Neuronal nicotinic acetylcholine receptors are blocked by intracellular spermine in a voltage-dependent manner. J Neurosci 18:4050–4062. 10.1523/JNEUROSCI.18-11-04050.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Wang X, Dan Y, Poo M, Zhang X (2009) An arithmetic rule for spatial summation of excitatory and inhibitory inputs in pyramidal neurons. Proc Natl Acad Sci USA 106:21906–21911. 10.1073/pnas.0912022106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Stuart G, Racca C, Sakmann B (1995) Axonal initiation and active dendritic propagation of action potentials in substantia nigra neurons. Neuron 15:637–647. 10.1016/0896-6273(95)90152-3 [DOI] [PubMed] [Google Scholar]
- Jackman SL, Regehr WG (2017) The mechanisms and functions of synaptic facilitation. Neuron 94:447–464. 10.1016/j.neuron.2017.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft J, Spruston N (2007) Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. J Physiol 578:799–818. 10.1113/jphysiol.2006.124586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Attwell D (2006) Combining patch-clamping of cells in brain slices with immunocytochemical labeling to define cell type and developmental stage. Nat Protoc 1:1977–1986. 10.1038/nprot.2006.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C (1998) Biophysics of computation: information processing in single neurons. New York: Oxford UP. [Google Scholar]
- Koch C, Poggio T, Torre V (1983) Nonlinear interactions in a dendritic tree: localization, timing, and role in information processing. Proc Natl Acad Sci USA 80:2799–2802. 10.1073/pnas.80.9.2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim K, Zhou ZJ (2010) Role of ACh-GABA cotransmission in detecting image motion and motion direction. Neuron 68:1159–1172. 10.1016/j.neuron.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Davletov BA, Südhof TC (1995) Distinct Ca2+ and Sr2+ binding properties of synaptotagmins: definition of candidate Ca2+ sensors for the fast and slow components of neurotransmitter release. J Biol Chem 270:24898–24902. 10.1074/jbc.270.42.24898 [DOI] [PubMed] [Google Scholar]
- Li HQ, Spitzer NC (2020) Exercise enhances motor skill learning by neurotransmitter switching in the adult midbrain. Nat Commun 11:2195. 10.1038/s41467-020-16053-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gutierrez DV, Hanson MG, Han J, Mark MD, Chiel H, Hegemann P, Landmesser LT, Herlitze S (2005) Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci USA 102:17816–17821. 10.1073/pnas.0509030102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY (2011) A user's guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol 96:19–25. 10.1113/expphysiol.2009.051961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokwan SJ, Overton PG, Berry MS, Clark D (1999) Stimulation of the pedunculopontine tegmental nucleus in the rat produces burst firing in A9 dopaminergic neurons. Neuroscience 92:245–254. 10.1016/S0306-4522(98)00748-9 [DOI] [PubMed] [Google Scholar]
- Luo L, Callaway EM, Svoboda K (2008) Genetic dissection of neural circuits. Neuron 57:634–660. 10.1016/j.neuron.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, et al. (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, et al. (2012) A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15:793–802. 10.1038/nn.3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manookin MB, Weick M, Stafford BK, Demb JB (2010) NMDA receptor contributions to visual contrast coding. Neuron 67:280–293. 10.1016/j.neuron.2010.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, Kusefoglu D, Hooks BM, Huber D, Petreanu L, Svoboda K (2011) Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron 72:111–123. 10.1016/j.neuron.2011.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mel BW (1993) Synaptic integration in an excitable dendritic tree. J Neurophysiol 70:1086–1101. 10.1152/jn.1993.70.3.1086 [DOI] [PubMed] [Google Scholar]
- Mendez JA, Bourque MJ, Fasano C, Kortleven C, Trudeau LE (2011) Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J Biol Chem 286:23928–23937. 10.1074/jbc.M111.218032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moubarak E, Engel D, Dufour MA, Tapia M, Tell F, Goaillard JM (2019) Robustness to axon initial segment variation is explained by somatodendritic excitability in rat substantia nigra dopaminergic neurons. J Neurosci 39:5044–5063. 10.1523/JNEUROSCI.2781-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E (2003) Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA 100:13940–13945. 10.1073/pnas.1936192100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A (2005) Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol 15:2279–2284. 10.1016/j.cub.2005.11.032 [DOI] [PubMed] [Google Scholar]
- Nasirova N, Quina LA, Agosto-Marlin IM, Ramirez JM, Lambe EK, Turner EE (2020) Dual recombinase fate mapping reveals a transient cholinergic phenotype in multiple populations of developing glutamatergic neurons. J Comp Neurol 528:283–307. 10.1002/cne.24753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi K, Gel YR, Brunner E, Konietschke F (2012) nparLD: an R software package for the nonparametric analysis of longitudinal data in factorial experiments. J Stat Softw 50:1–23. 10.18637/jss.v050.i1225317082 [DOI] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P (1998) Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci 18:1693–1703. 10.1523/JNEUROSCI.18-05-01693.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermayer J, et al. (2019) Prefrontal cortical ChAT-VIP interneurons provide local excitation by cholinergic synaptic transmission and control attention. Nat Commun 10:5280. 10.1038/s41467-019-13244-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JP, Kwon HB, Takasaki KT, Chiu CQ, Higley MJ, Sabatini BL, Ellis-Davies GC (2013) Optically selective two-photon uncaging of glutamate at 900 nm. J Am Chem Soc 135:5954–5957. 10.1021/ja4019379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo K, Perkins J, Kulkarni A, Stojanovic S, Roeper J, Paladini CA (2020) In vivo patch-clamp recordings reveal distinct subthreshold signatures and threshold dynamics of midbrain dopamine neurons. Nat Commun 11:6286. 10.1038/s41467-020-20041-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma-Cerda F, et al. (2012) New caged neurotransmitter analogs selective for glutamate receptor sub-types based on methoxynitroindoline and nitrophenylethoxycarbonyl caging groups. Neuropharmacology 63:624–634. 10.1016/j.neuropharm.2012.05.010 [DOI] [PubMed] [Google Scholar]
- Passlick S, Ellis-Davies GC (2018) Comparative one- and two-photon uncaging of MNI-glutamate and MNI-kainate on hippocampal CA1 neurons. J Neurosci Methods 293:321–328. 10.1016/j.jneumeth.2017.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlstein E, Gouty-Colomer LA, Michel FJ, Cloarec R, Hammond C (2015) Glutamatergic synaptic currents of nigral dopaminergic neurons follow a postnatal developmental sequence. Front Cell Neurosci 9:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Huber D, Sobczyk A, Svoboda K (2007) Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci 10:663–668. 10.1038/nn1891 [DOI] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K (2009) The subcellular organization of neocortical excitatory connections. Nature 457:1142–1145. 10.1038/nature07709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirazi P, Brannon T, Mel BW (2003) Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron 37:977–987. 10.1016/S0896-6273(03)00148-X [DOI] [PubMed] [Google Scholar]
- Puente N, Río IB, Achicallende S, Nahirney PC, Grandes P (2019) High-resolution immunoelectron microscopy techniques for revealing distinct subcellular type 1 cannabinoid receptor domains in brain. Bio Protoc 9:e3145. 10.21769/BioProtoc.3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing. [Google Scholar]
- Rall W (1962) Theory of physiological properties of dendrites. Ann NY Acad Sci 96:1071–1092. 10.1111/j.1749-6632.1962.tb54120.x [DOI] [PubMed] [Google Scholar]
- Rall W (1967) Distinguishing theoretical synaptic potentials computed for different soma-dendritic distributions of synaptic input. J Neurophysiol 30:1138–1168. 10.1152/jn.1967.30.5.1138 [DOI] [PubMed] [Google Scholar]
- Rall W (1977) Core conductor theory and cable properties of neurons. In Handbook of physiology: the nervous system (Poeter R, ed), pp 39–97. Bethesda, MD: American Physiological Society. [Google Scholar]
- Rall W (1995) Theoretical significance of dendritic trees for neuronal input-output relations. In: The theoretical foundation of dendritic function (Segev I, Rinzel J, Shepherd GM, eds), pp 122–146. Cambridge, MA: Massachusetts Institute of Technology. [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK (2011) Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13:195–204. 10.1016/j.cmet.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Häusser M (2001) Compartmental models of rat cerebellar Purkinje cells based on simultaneous somatic and dendritic patch-clamp recordings. J Physiol 535:445–472. 10.1111/j.1469-7793.2001.00445.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E (2001) Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol 531:807–826. 10.1111/j.1469-7793.2001.0807h.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Granger AJ, Sabatini BL (2015) Corelease of acetylcholine and GABA from cholinergic forebrain neurons. Elife 4:e06412. 10.7554/eLife.06412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenenberger P, Schärer YP, Oertner TG (2011) Channelrhodopsin as a tool to investigate synaptic transmission and plasticity. Exp Physiol 96:34–39. 10.1113/expphysiol.2009.051219 [DOI] [PubMed] [Google Scholar]
- Schubert D, Staiger JF, Cho N, Kötter R, Zilles K, Luhmann HJ (2001) Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J Neurosci 21:3580–3592. 10.1523/JNEUROSCI.21-10-03580.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethuramanujam S, McLaughlin AJ, deRosenroll G, Hoggarth A, Schwab DJ, Awatramani GB (2016) A central role for mixed acetylcholine/GABA transmission in direction coding in the retina. Neuron 90:1243–1256. 10.1016/j.neuron.2016.04.041 [DOI] [PubMed] [Google Scholar]
- Shabel SJ, Proulx CD, Piriz J, Malinow R (2014) Mood regulation: GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science 345:1494–1498. 10.1126/science.1250469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Zucker RS (1980) Aequorin response facilitation and intracellular calcium accumulation in molluscan neurones. J Physiol 300:167–196. 10.1113/jphysiol.1980.sp013157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N (1998) Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci 18:3501–3510. 10.1523/JNEUROSCI.18-10-03501.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Häusser M (2016) Dendrites. Oxford: Oxford UP. [Google Scholar]
- Takács VT, et al. (2018) Cotransmission of acetylcholine and GABA regulates hippocampal states. Nat Commun 9:2848. 10.1038/s41467-018-05136-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakusaki K, Shiroyama T, Kitai ST (1997) Two types of cholinergic neurons in the rat tegmental pedunculopontine nucleus: electrophysiological and morphological characterization. Neuroscience 79:1089–1109. 10.1016/S0306-4522(97)00019-5 [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Shiroyama T, Yamamoto T, Kitai ST (1996) Cholinergic and noncholinergic tegmental pedunculopontine projection neurons in rats revealed by intracellular labeling. J Comp Neurol 371:345–361. [DOI] [PubMed] [Google Scholar]
- Talbot MJ, Sayer RJ (1996) Intracellular QX-314 inhibits calcium currents in hippocampal CA1 pyramidal neurons. J Neurophysiol 76:2120–2124. 10.1152/jn.1996.76.3.2120 [DOI] [PubMed] [Google Scholar]
- Unal G, Joshi A, Viney TJ, Kis V, Somogyi P (2015) Synaptic targets of medial septal projections in the hippocampus and extrahippocampal cortices of the mouse. J Neurosci 35:15812–15826. 10.1523/JNEUROSCI.2639-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. (2007) High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci USA 104:8143–8148. 10.1073/pnas.0700384104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EL (2002) Specificity of cortical synaptic connectivity: emphasis on perspectives gained from quantitative electron microscopy. J Neurocytol 31:195–202. [DOI] [PubMed] [Google Scholar]
- Xiao C, Cho JR, Zhou C, Treweek JB, Chan K, McKinney SL, Yang B, Gradinaru V (2016) Cholinergic mesopontine signals govern locomotion and reward through dissociable midbrain pathways. Neuron 90:333–347. 10.1016/j.neuron.2016.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Weiner JL, Valiante TA, Velumian AA, Watson PL, Jahromi SS, Schertzer S, Pennefather P, Carlen PL (1994) Whole-cell recording of the Ca(2+)-dependent slow afterhyperpolarization in hippocampal neurones: effects of internally applied anions. Pflugers Arch 426:247–253. 10.1007/BF00374778 [DOI] [PubMed] [Google Scholar]