

Abstract
Extrachromosomal DNA (ecDNA) amplification is an important driver alteration in cancer. It has been observed in most cancer types and is associated with worse patient outcome. The functional impact of ecDNA has been linked to its unique properties, such as its circular structure that is associated with altered chromatinization and epigenetic regulatory landscape, as well as its ability to randomly segregate during cell division, which fuels inter-cellular copy number heterogeneity. Recent investigations suggest that ecDNA is structurally more complex than previously anticipated and that it localizes to specialized nuclear bodies (hubs) and can act in trans as enhancers for genes on other ecDNAs or chromosomes. In this Review, we synthesize what is currently known about how ecDNA is generated and how its genetic and epigenetic architecture affects proto-oncogene deregulation in cancer. We discuss how recently identified ecDNA functions may on the one hand impact oncogenesis, but also serve as new therapeutic vulnerabilities in cancer.
Introduction
Aberrantly high expression of proto-oncogenes is a hallmark of oncogenesis and can be caused by structural or numerical chromosome anomalies1. Somatic copy-number alterations are the most common drivers of variations in oncogene expression2. Extrachromosomal DNA (ecDNA) is a type of circular DNA element that has emerged as one of the most important forms of somatic copy-number amplification3–5. Since its discovery in 1965 in neuroblastoma cell lines6, re-evaluation of ecDNA in large-scale DNA sequencing data sets has revealed that it is present in a subset of tumours across most cancer types (Figure 1a)7–10. Patients with tumours containing ecDNA have worse clinical outcomes compared to those with other types of focal amplification, corroborating the functional importance of ecDNA elements9,11. Two commonly accepted features make ecDNA a unique and extraordinarily potent vehicle of proto-oncogene amplification. First, their circular structure has been associated with elevated transcription, resulting in increased proto-oncogene expression compared to linear amplifications12. Second, relieved from chromosomal positional constraints and lacking centromeres, ecDNAs are unequally segregated into daughter cells, which can quickly increase ecDNA copy number and drive intratumoral heterogeneity (Figure 1b)4,13,14. Thus, ecDNA represents an important vehicle for oncogene amplification in cancer with distinct molecular features, most of which are currently not fully understood.
Figure 1. Oncogene amplification on ecDNA is a frequent event in cancer and promotes tumour heterogeneity.
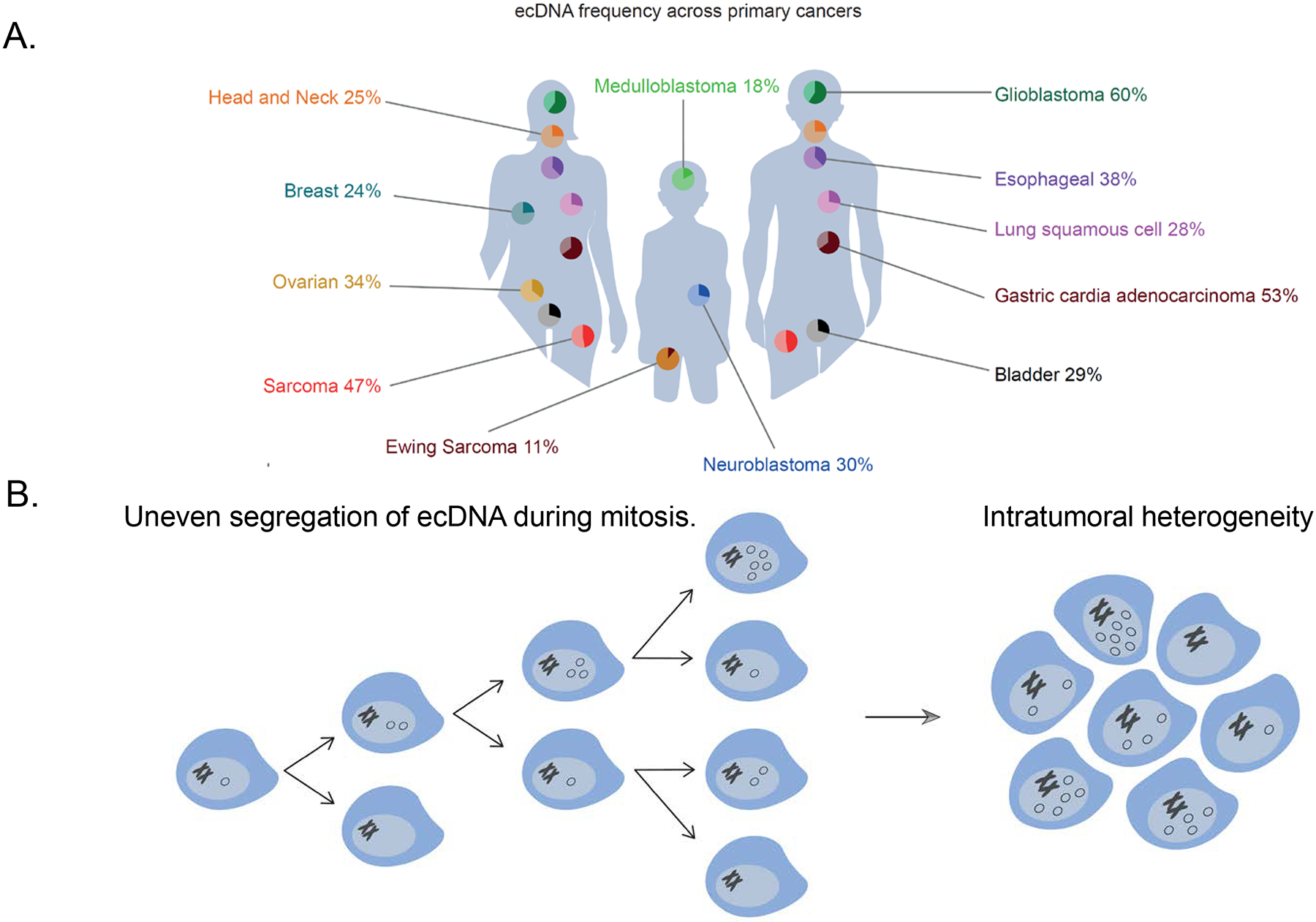
a | The frequency of extrachromosomal DNA (ecDNA) amplification across newly diagnosed cancer7–10. b | EcDNA elements are replicated in S-phase and due to the absence of centromeres, segregate unevenly to daughter cells during mitosis. Clonal selection of tumour cells with an ecDNA-endowed proliferative advantage enables rapid intercellular diversification of ecDNA copy number and increases intratumoral heterogeneity.
Other types of extrachromosomal DNAs have been described15–19. These include a class of extrachromosomal circular DNA elements often referred to as eccDNA and which are present in similar low copy numbers in non-neoplastic and cancer cells (2–3 copies of each small extrachromosomal circular DNA element per nucleus on average)18,20. By contrast, dozens of ecDNA copies can accumulate in cancer cells9,12. EccDNAs are mostly shorter than 1kb, compared to the much larger size of ecDNAs (50kb–5Mb size range for ecDNAs based on the latest analyses of patient tumour samples)7–10. Although we refer here to eccDNAs as small circular elements, we note that nomenclature for circular DNA elements is not fully standardized, hence ‘eccDNA’ has also been used more broadly to describe Eukaryotic covalently closed circular DNAs of any size29. The majority of small extrachromosomal circular DNAs do not contain full-length genes or regulatory elements. The role of eccDNAs in cellular homeostasis will not be discussed in this Review.
When cancer cells contain ecDNAs, these ecDNAs can drive dosage-dependent increases of cargo gene expression. However, recent reports have described unique aspects of ecDNA biology, which challenge the view that ecDNA only affects oncogene expression levels through increased copy number dosage. EcDNA chromatinization, the co-amplification and resulting structural proximity of enhancer elements, and ecDNA clustering in specialized nuclear bodies (ecDNA hubs) jointly create conditions for increased cargo gene transcription efficiency. Moreover, ecDNAs and ecDNA hubs may serve a double role as ectopic enhancers in trans for genes on other ecDNAs or chromosomes13,21–23. These and other discoveries of the unique properties of ecDNAs are resulting in the development of new methods to detect, sequence and track ecDNA in cancer cells, and are fuelling a new revolution in cancer genetics.
In this Review, we discuss our current understanding of how ecDNA is structured and how it contributes to oncogene expression, including properties related to ecDNA chromatin organization and nuclear positioning13,21,23. We describe the development of new tools for the detection and sequencing of ecDNA (Box 1) and their use for the characterization of the structure and abundance of ecDNAs in cancers. We discuss the altered chromatin organization of ecDNA and its impact on oncogene regulation, the spatial organization and movement of ecDNA in the nucleus and its clustering in specialized hubs. We also discuss the ability of ecDNAs to interact with other chromosomes, and the implications for genome-wide transcriptional regulation. We argue that unanticipated oncogenic functions may arise from these new ecDNA properties which may provide opportunities for therapeutic intervention, and conclude by reflecting on the most pressing open biological questions related to ecDNA biology.
Box 1. Methods for ecDNA detection.
Accurate and sensitive methods for extrachromosomal DNA (ecDNA) characterization are imperative to understand ecDNA biology. Traditionally, cytogenetics methods have been used for ecDNA detection, which include DAPI (4’,6-diamidino-2-phenylindole) staining, which reveals the presence of an ecDNA element, and fluorescence in situ hybridization (FISH), a highly targeted approach detecting and quantifying ecDNA elements of interest4,5. As ecDNAs are present at high copy-number, this facilitates detection through sequencing-based approaches, as the number of copies scales linearly with the number of derived sequencing reads. Sequencing and cytogenetic methods rely on several unique properties of ecDNAs: circularity12; high average number of ecDNA elements, often greater than ten copies per cell across a specimen; and high level of variability between individual cells.
Whole-genome sequencing can be used to assemble ecDNA structures in silico9,27. However, it remains a major computational challenge to accurately distinguish chromosomal breakage-fusion-bridge (BFB) structures from ecDNAs, co-existing homogeneously staining regions (HSRs) and HSRs that have circularized, and ecDNAs in samples where some ecDNAs have reinserted into the genome. Nonetheless, software tools such as AmpliconArchitect47, AmpliconReconstructor83, Circle_Finder and circMap84 are used to infer ecDNA structures from whole-genome sequencing data (see the table). These methods start by identifying regions of the genome with elevated copy number, and use those loci as a seed to construct a circular graph. If circularity can be achieved, this supports the presence of an ecDNA while enabling resolution of the structure and gene or regulatory element cargo content. The specificity of sequencing-based ecDNA detection is over 80% when validated using cytogenetics, although quantifying sensitivity is challenging owing to the lack of golden truth data4,5,47,83–85.
New and ecDNA-specific developments for ecDNA characterization include Circle-Seq, a sequencing library enrichment method which uses exonuclease digestion of linear DNA to isolate circular DNA prior to sequencing and, combined with short-read or long-read sequencing, provides a powerful platform for comprehensive ecDNA characterization10,20. Another powerful method for targeted profiling of ecDNA (CRISPR-CATCH), uses CRISPR–Cas9-mediated enrichment of ecDNA fragments followed by sequencing, which allows sequence reconstructions at base-pair resolution86. Chromatin accessibility and chromosomal conformation sequencing approaches have demonstrated unique ecDNA patterns and hold promise in combination with single-cell methods11,12,21,87. Tagging ecDNA-specific sequences using guide RNAs combined with fluorescent tags, such as the ecTag method, enables ecDNA visualization in live cells13.
The interpretation of ecDNA composition from sequencing data should account for allele specificity and the status of the chromosomal segments from which the ecDNA arose, and whether there are confounding factors when deletions are hemizygous. Long-read sequencing holds promise as a method for phasing as well as ecDNA characterization, for example in combination with ecDNA library enrichment methods23,86.
Table for box 1.
Extrachromosomal DNA detection and characterization methods.
| Method | Input data | Output | Refs |
|---|---|---|---|
| AmpliconArchitect | Whole-genome sequencing | Unsupervised | 47 |
| AmpliconReconstructor | Optical mapping with whole-genome sequencing | Unsupervised | 83 |
| ecSeg | DAPI stain or FISH images | Unsupervised (DAPI), targeted (FISH) | 88 |
| circMap | Whole-genome sequencing | Unsupervised | 84 |
| CRISPR-CATCH | Circular DNA sequencing | Targeted | 86 |
| CircleSeq | Circular DNA sequencing | Unsupervised | 20 |
| Circle_Finder | ATAC-seq | Unsupervised | 87 |
| ecTag | Confocal microscopy of cells transduced with guide RNAs specific for ecDNA breakpoint sequences | Targeted | 13 |
ATAC-seq, assay for transposase-accessible chromatin sequencing.
Structural ecDNA properties
Composition and amplification.
Focal DNA amplifications in cancer are observable in linear intrachromosomal or circular extrachromosomal forms, cytogenetically appearing as homogeneously staining regions (HSRs) and double minutes, respectively6,24. The term ‘double minutes’ was derived from their appearance in doublets during metaphase6. Comprehensive genomic characterization across cancer types and cancer models9,25–27 have since provided extensive catalogues of chromosomal rearrangements and have demonstrated that there is substantial diversity in focal chromosomal and extrachromosomal amplifications between and within tumour types. Focal DNA amplifications vary greatly in amplification structure, genomic content, amplicon size and copy number level. These genomic analyses are consistent with the earlier cytogenetic studies in that, in broad terms, amplifications can be classified in two groups: linear intrachromosomal increases in DNA copies (HSRs) and ecDNA.
When the resulting chromosomal abnormalities contain a proto-oncogene and/or an oncogenic regulatory element that provides the cell with a proliferative or survival advantage, clonal selection occurs. At least 70 genomic regions have been reported as recurrently amplified in cancer, including loci containing proto-oncogenes such as EGFR, MYC, MYCN and CCND228. The majority of genes that are focally amplified at high copy number levels in cancer are extrachromosomally amplified in a subset of tumours, with ecDNAs detected in 14% of all newly diagnosed and untreated cancers9 (Figure 1a). EcDNAs frequently harbour functional enhancer elements juxtaposed to cargo genes, i.e. enhancer hijacking, which contributes to the very high level of expression observed for ecDNA-residing genes. Some ecDNAs contain only enhancers and no genes, suggesting that selection of ecDNA-containing tumour cells can be based on roles of ecDNA beyond activating cargo oncogene expression11,21.
Focal amplifications are likely to arise as a result of DNA damage at random locations across the genome followed by erroneous repair17,29,30. In its simplest form, an ecDNA element consists of a single chromosomal DNA segment created by two DNA double-strand breaks that have been circularized through end-to-end ligation (Figure 2). Such simple amplicons can be observed, for example, in leukaemias31. More commonly, ecDNAs consist of tens to hundreds geographically separated DNA segments from different chromosomes, recombined into a single element4,12,23 (Figure 2). Focal DNA copy number amplifications are created in cells in vitro during, for example, methotrexate treatment32–37. Resistant cells outgrow sensitive cells through focal amplification of the DFHR gene, which drives resistance to methotrexate. The resulting amplicon structure ranges from single-segment linear or extrachromosomal amplifications to arrangements consisting of many DNA segments from different chromosomes with variable transcriptional orientations38. The diversity of amplification observed under controlled circumstances suggests that amplicon formation is subject to different directives. Complex ecDNA structure formation has been linked to at least two mechanisms: first, chromosome shattering (chromothripsis), creating clustered rearrangements and leading to ecDNA formation, as evidenced by footprints of chromothripsis in approximately 36% of ecDNAs detected across primary cancers9; and second, breakage-fusion-bridge cycles (BFBs), as evidenced by head-to-head fold-back inversions found on some ecDNAs (Figure 2)39. BFBs result in arrays of genomic segments. The existence of near-identical amplicons existing in parallel as either an HSR or as an ecDNA indicates that some BFB amplicons may circularize to terminate the cyclic process, resulting in ecDNAs39,40. The presence of multiple co-existing amplicons in the same specimen may also suggest a multi-step process whereby simple ecDNAs evolve into complex multi-fragment structures over time, or structural evolution of ecDNAs through subsequent rounds of chromothripsis and incorporation of damaged DNA38,41–44. These dynamic processes may however present a chicken-and-egg problem as ecDNAs have been observed to re-insert into the genome, especially when under selective pressure and when DNA damage repair occurs10,27,38,45–47. Following re-insertion, ecDNAs may subsequently reform when the selective pressure is removed, reflecting that ecDNAs are not static structures but undergo evolution (Figure 2)38,45.
Figure 2. EcDNA life cycle.
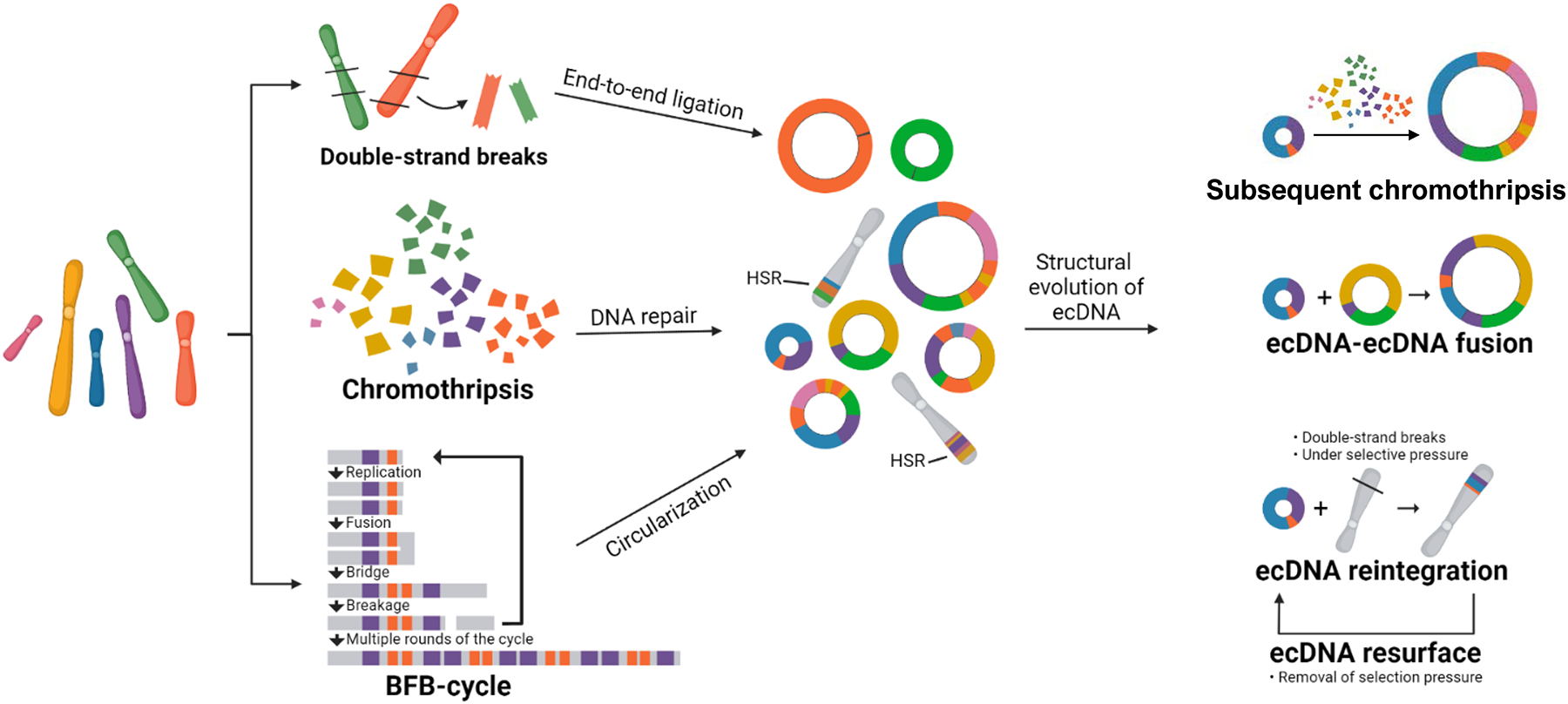
Following DNA breakage, extrachromosomal DNA (ecDNA) structures are formed through end-to-end ligation. Chromothripsis creates massive rearrangements and, like breakage-fusion-bridge (BFB) cycles, can result in complex ecDNA structures. EcDNA structures may evolve by acquiring new genome segments following additional breakage events, including through merging of co-existing ecDNAs. Selective pressure and linear DNA damage may cause ecDNA molecules to reintegrate into the linear genome and generate homogeneously staining regions (HSRs). Upon removal of selection pressure or changing circumstances, reintegrated ecDNAs may re-emerge.
More evidence of ecDNA sequence evolution was derived from recent discoveries of kataegis (clusters of mutations), which was detected on 30% of ecDNAs in primary tumours48. In this pan-cancer analysis, ecDNA-associated kataegis — named kyklonas — occurred relatively distant from ecDNA breakpoints implicating that kyklonas probably occurred after ecDNA formation and as part of an adaptive process. Kyklonas carried features of mutagenesis by activation-induced deaminase (AID) and APOBEC3 deaminase, which are enzymes that are part of the endogenous response to viral DNA in the nucleus. Whether this response mechanism actively targets ecDNA, deriving mutation clusters as a result, or whether this is a passive process requires additional research. By contrast, APOBEC-mediated kataegis was found to occur near breakpoints in neuroblastoma ecDNAs, suggesting these mutations were induced during ecDNA formation49, and implicating that kyklonas can be derived through different mechanisms.
We do not yet fully understand the rules that govern clonal selection and spur the evolutionary process to an equilibrium of ecDNA element frequencies across the tumour. We speculate that the ecDNA cell-to-cell copy number distribution is determined by the cost of reproducing and maintaining the copy number levels of single or parallel HSRs and ecDNAs, as well as by the growth advantages provided by the increased oncogenic signals created by the amplification.
Distribution.
The number of ecDNA molecules varies from cell to cell, which implies uneven segregation of ecDNAs during mitosis (Figure 1b)3–5. The absence of centromeres in ecDNAs prevents even mitotic distribution through spindle-complex forces during metaphase–anaphase stages of the cell cycle50. If ecDNA elements are not included in a daughter cell’s nucleus after mitosis, however, they become entrapped in micronuclei51–54. Thus, mechanisms ensuring mitotic ecDNA distribution need to exist. EcDNAs replicate early in S-phase, a property often associated with active transcription55,56 (Figure 3). EcDNA molecules relocate from the nuclear periphery into the centre of the nucleus upon initiation of DNA replication, which may imply the existence ecDNA-specific replication machinery57. In M-phase and during segregation, ecDNAs appear to ‘hitchhike’ by binding preferentially to the telomeric regions of linear chromosomes50,54,58. Sister ecDNA molecules migrate into the same daughter cell during mitosis, which may indicate that physical post-replication bonds such as ecDNA concatenation exist50,58.
Figure 3. EcDNA movement and location during the cell cycle.
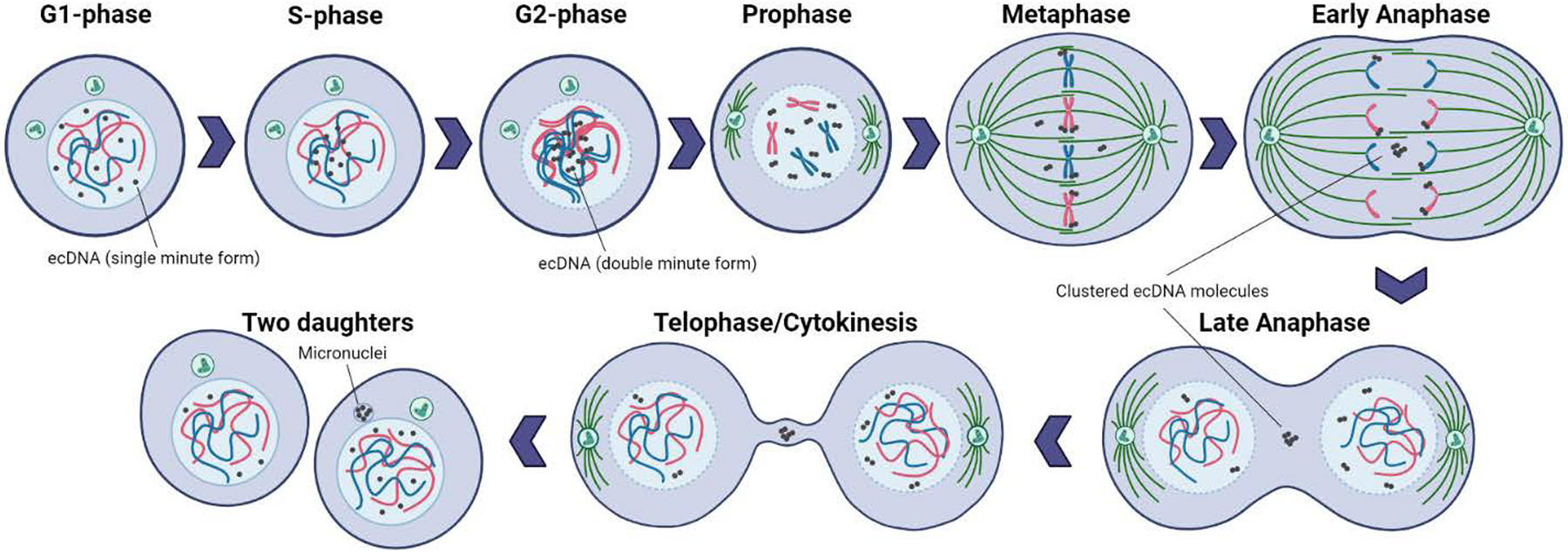
During S-phase, extrachromosomal DNA (ecDNA) molecules relocate from the periphery to the centre of the nucleus. Replication occurs, resulting in double-minute shaped sister ecDNA chromatids, prior to the initiation of mitosis. During mitosis, ecDNAs randomly bind to chromosome ends and divide unequally to daughter cells. During anaphase, ecDNAs that are not tethered to chromosomal ends are clustered, are not carried into the newly forming daughter nuclei and remain in one daughter cell in micronuclei.
The frequency of ecDNA molecules across cells can fluctuate rapidly and in response to changing circumstances13,22,57,59. Uneven segregation in combination with the competitive advantage provided by oncogene overexpression can result in accelerated expansion of ecDNA-containing clones and the sometimes hundreds of ecDNA copies observed inside a single nucleus4,5,40,45. Adaptive responses have been observed in patient tumours, where ecDNA-containing subclones rapidly shrink under targeted treatment, but re-emerge when the therapeutic stress is removed45,60. The dynamic ability to decrease and increase ecDNA levels may be particularly effective under stress conditions, which may include hypoxia and high acidity encountered in tumour microenvironments46,61. Epigenetic states have been associated with responses to stress and can promote transient site-specific copy number gains of the EGFR locus, specifically when extrachromosomal62,63. EcDNA may provide less advantage under stable circumstances, such as cell culture where ecDNAs are often lost44. Lastly, ecDNAs are more prone to acquiring activating mutations relative to chromosomal regions64, which further aids positive selection. Thus, even though ecDNA replicates once per cell cycle, it does not follow most rules of mitotic inheritance, enabling tumours to rapidly evolve and maintain high genetic intra-tumour heterogeneity.
Transcriptional regulation of ecDNA
Chromatin organization.
Until recently, increased proto-oncogene copy number was believed to be the predominant mechanism through which DNA amplification altered proto-oncogene expression2. However, several recent reports demonstrating a crucial role for chromatin organization, and the presence of regulatory sequences for oncogene expression on ecDNA, indicate key roles for factors other than dosage. Human chromosomes are composed of DNA wound around nucleosomes that tightly control access of transcriptional regulators to the DNA65. This arrangement regulates associations between genes and proteins and their response to intra- and extracellular regulatory stimuli, and provides a control mechanism that prevents incorrect interactions. Changes in chromatin organization are associated with cancer66.
Recent evidence suggests that ecDNA harbours increased chromatin accessibility and less compact nucleosomal organization compared to chromosomal DNA (Figure 4a)7,11,12,21. A significantly higher signal from assay for transposase-accessible chromatin sequencing (ATAC-seq) was observed in extrachromosomal circular amplicons compared to linear amplicons across tumour samples and after normalizing for DNA copy number12. A similar contrast was observed in isogenic cell lines, in which the ecDNA amplicon displays an increased chromatin accessibility compared to the identical locus amplified chromosomally as an HSR12. This increased density of ATAC-seq signal within the circularized and amplified loci was confirmed in medulloblastoma tumours7. Using a recently developed method, ‘sequencing of enzyme-accessible chromatin in circular DNA’ (CCDA-seq) that combines the use of methylases for soft-labelling of open chromatin without DNA fragmentation, and exonuclease digestion to enrich for ecDNA, Chen and colleagues were able to use long-read nanopore sequencing to assess chromatin state on ecDNA at single-molecule resolution67. They observed that chromatin on ecDNA is on average two-fold more accessible compared to chromatin of homologous linear DNA, and 80% of ecDNA areas were highly accessible. These highly accessible areas not only included genic regions but also, and mostly, introns and intergenic regions. Overall, genic regions on ecDNA were 62% more accessible than their linear counterparts67. The increase in chromatin accessibility contributed to the increase in expression levels of ecDNA cargo genes, which were significantly higher than expected based on ecDNA copy number9,12. The mechanisms driving increased chromatin accessibility on ecDNA are currently unknown, as is whether accessibility precedes ecDNA formation or is a consequence thereof.
Figure 4. Chromatin organization on ecDNA.
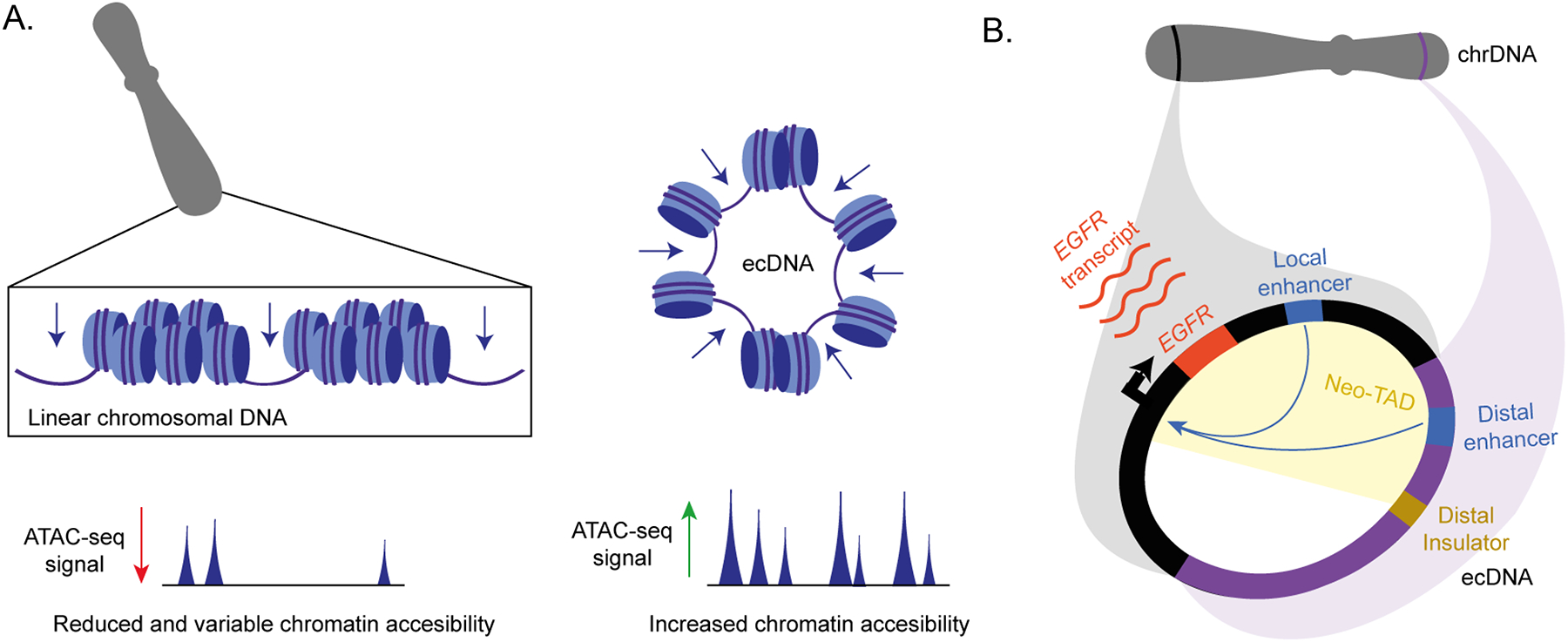
a Extrachromosomal DNA (ecDNA) contains more accessible chromatin compared to linear chromosomal DNA, reflected by the increased read density and number of peaks in assay for transposase-accessible chromatin sequencing (ATAC-seq) profiles. b | The re-arrangement of distal chromosomal fragments into ecDNAs results in three-dimensional reorientation of chromatin elements and enables the ‘hijacking’ of distal enhancers.
Enhancer co-amplification.
The circular structure of ecDNA results in three-dimensional re-orientation of chromatin elements, which enables local and distal enhancer–gene contacts (Figure 4b)11,12,21,62,68. In addition to past reports suggesting that amplicon borders were shaped by DNA fragile sites69, recent studies have provided evidence that co-amplified neighbouring enhancer elements active in the cancer cell type of origin play a major role in shaping amplicon borders11,68. Many of the co-amplified enhancers represent lineage-specific super-enhancers, characterized by large domains of high-intensity peaks of H3K27 acetylation (H3K27ac) and H3K4 methylation (H3K4me1)21. In neuroblastoma, a prototypical ecDNA-harbouring cancer type in which the MYCN oncogene is commonly extrachromosomally amplified, two distinct classes of amplicons were described based on the presence or absence of local enhancers68. Class I amplicons incorporated local enhancer elements, were simple in structure and only rarely incorporated additional distal DNA segments from other chromosomes. The rare subset of Class I cases where both local and distal enhancers can be found on the same ecDNA was also observed in glioblastoma11; in these cases, local and distal enhancers for EGFR were preserved on the ecDNA, thereby expanding 3D contacts within the boundaries of the canonical topologically associating domains (TADs) regulating EGFR expression. By contrast, Class II amplicons lacked local enhancer co-amplification, but compensated through ligation of DNA segments from distant sites, such as from other chromosomes. Co-amplified distant DNA segments harboured lineage-specific enhancer elements as well as insulators, forming highly complex, multi-fragment amplicons with new spatially interacting regulatory neighbourhoods (neo-TADs) and resulting in enhancer hijacking68. Enhancer hijacking on ecDNA has also been detected in medulloblastoma7,11. Co-amplified regulatory elements and their contacts with the oncogene promoter are not just preserved on ecDNA, but actively contribute to oncogene expression and cancer cell fitness11. Thus, multiple layers of ecDNA chromatin are altered: nucleosomal organization and compaction; enhancer–oncogene contacts, including enhancer hijacking; and 3D conformation, such as through changes to TAD structures. All of these chromatin features contribute to high oncogene expression from ecDNAs.
Together, these recent reports raise the fascinating possibility that oncogene expression from ecDNA is only partly determined by increased oncogene dosage on ecDNA. It is tempting to integrate these new insights into a unifying model of ecDNA-driven oncogene deregulation determined by the law of mass action. The model would comprise an interplay between local and distant enhancer strengths, the extent of chromatin compaction and accessibility, transcription factor affinity at enhancer binding sites, and DNA copy number, that together determine the transcriptional output from ecDNA.
EcDNA hubs.
The smaller size and circular nature of ecDNA enables functional properties that are physically restrictive to linear chromosomes22,50,54,56–58. For example, ecDNA molecules were reported to cluster in hubs during interphase51,57 and mitosis54. Moreover, enabled by the development of sequencing and CRISPR-mediated live-cell ecDNA imaging technologies (Box 1), ecDNA hubs have been established as transcriptional hot spots in interphase13,23. EcDNA clustering is a dynamic process and does not happen continuously. A not-yet peer reviewed preprint study70 suggests that hubs do not form in all cell types, suggesting that yet unknown factors may be required for hub formation. Detection of ecDNA hub formation requires extended imaging windows13, and shorter exposure times may be an alternative explanation for the observed absence of ecDNA clustering70.
The nucleus during interphase is spatially organized into a compartmentalized structure with congregation of transcriptionally active sites towards the nuclear interior71–73. Nuclear compartments that are enriched in RNA polymerase II (RNAPII) serve as transcription factories73. The transcriptionally activated state of ecDNAs may contribute to the formation of ecDNA clusters and suggests that ecDNAs leverage similar principles to form nuclear aggregates and may share transcriptional machineries with other ecDNAs (Figure 5a).
Figure 5. The role of ecDNA and ecDNA hubs in transcriptional regulation.
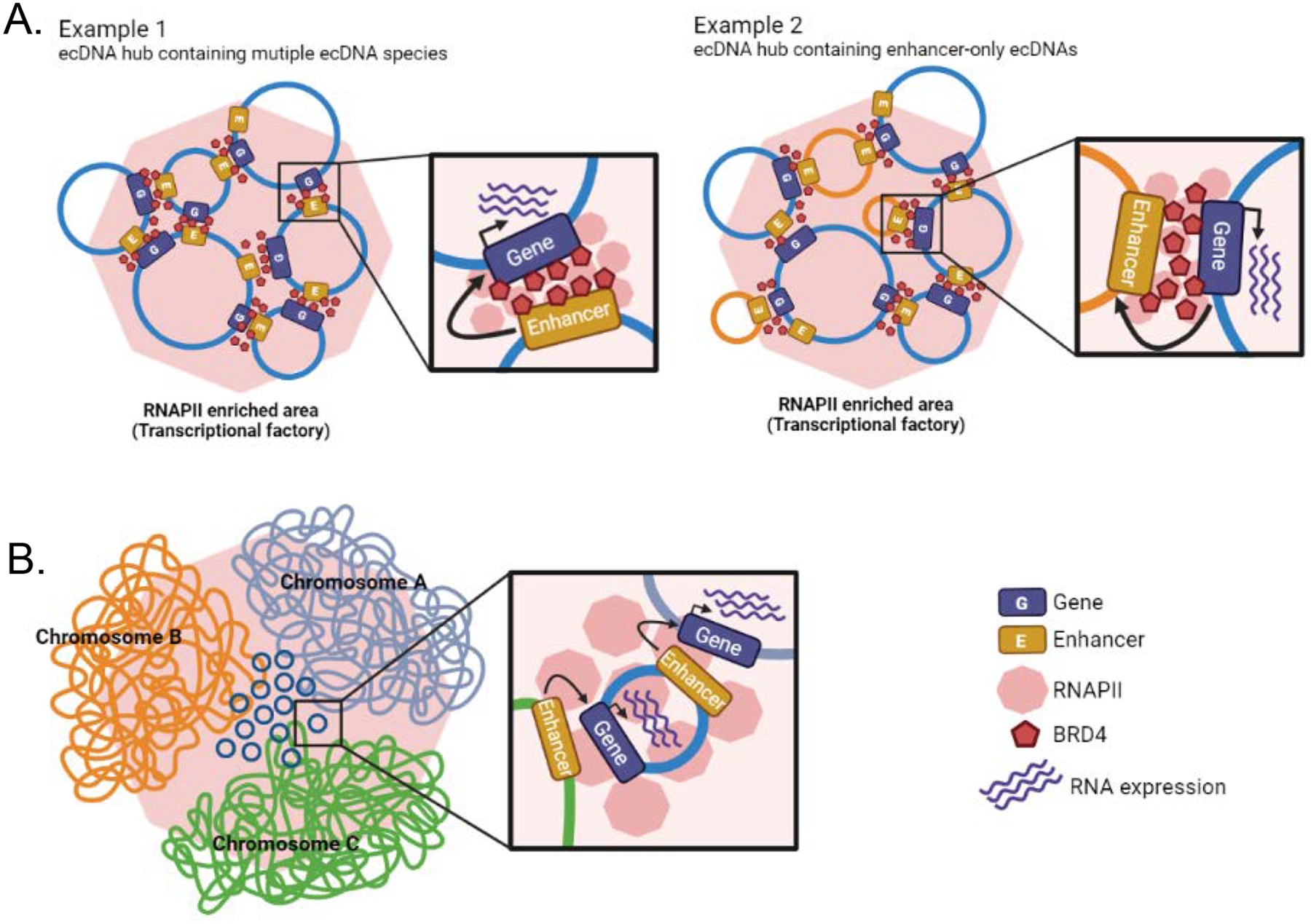
a | Representative examples of extrachromosomal DNA (ecDNA) hubs. Example 1: an ecDNA hub comprising different ecDNA species containing cargo oncogene and cargo enhancers. Example 2: an ecDNA hub including some enhancer-only ecDNAs without cargo genes. EcDNAs are glued together by BRD4 and recruit RNA polymerase II (RNAPII). Co-amplified enhancer elements regulate gene transcription of ecDNA hub partners. b | EcDNAs that are in contact with chromosomes recruit RNAPII. Co-amplified enhancers regulate transcription of linear chromosome genes in proximity to contact regions.
The bromodomain and extraterminal domain 4 (BRD4) protein, which accumulates on transcriptionally active regulatory elements to promote gene expression, plays an important role in ecDNA hub formation23. BRD4 inhibition is being explored as a therapeutic strategy in cancers dependent on the oncogene MYC, a gene frequently extrachromosomally amplified, as BRD4 and MYC are often found to be co-bound on active promoters74. Intriguingly, BRD4 inhibition in ecDNA-positive cells led to dispersion of both MYC-harbouring ecDNA and hubs with ecDNAs encoding for other genes, as well as reduced cargo gene expression23. Although both single ecDNA molecules and ecDNA hubs are preferentially associated with RNAPII relative to the linear genome, RNAPII molecules are relatively more abundant in ecDNA hubs, further substantiating the role of ecDNA hubs in transcriptional regulation13,21. Inhibition of RNAPII does not disrupt ecDNA hubs, indicating that RNAPII may be recruited into ecDNA hubs after their formation23. Cancer cells in which ecDNAs are aggregated into hubs transcribe increased levels of the cargo oncogene compared to cancer cells in which ecDNAs do not form hubs, to an extent where ecDNA hubs determine the level of oncogene transcription over the number of ecDNA copies (Figure 5a)13. Taken together, these results suggest that ecDNA hubs are biologically relevant nuclear bodies with roles in transcriptional regulation of oncogenes, and hence are likely to influence the maintenance and progression of cancer.
Transcriptional regulation in trans.
The detachment of ecDNAs from their chromosome of origin enables ecDNA elements to move around the nucleus with fewer constraints, and provides opportunity for physical interactions with other ecDNAs and linear chromosomes. Although chromosomal tethering of ecDNA during cell division has been reported, the extent and biological function of the ecDNA–chromosome interactions has remained unclear50,54,58. Genome-wide profiling studies using chromosome conformation capture approaches (such as Hi-C) and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) have demonstrated that ecDNAs make contacts with other ecDNAs at high frequencies that are not seen for any other chromosomal loci11,12,21. This high interaction frequency may reflect ecDNA hubs, as well as connections made between individual ecDNAs outside of such hubs.
EcDNAs are also in physical contact with the linear genome, and in particular with chromosomal regions that are transcriptionally active, reflected by enrichment of RNAPII and H3K27 acetylation signals in ecDNA–chromosome interactomes21. Accordingly, there is an increase in gene expression at regions with high ecDNA–chromosome contact frequency, in comparison to those not tethered to ecDNA (Figure 5b). Thus, the regulatory function of co-amplified ecDNA enhancers to regulate oncogenes on the same ecDNA may extend to chromosomal genes and oncogenes in particular. This intriguing emerging function of ecDNAs as mobile enhancers creates a synthetic aneuploidy effect of transcription, comparable to how copy number gains of whole chromosomes or chromosome arms increase expression levels of all resident genes. It also provides additional explanation for why ecDNAs often contain non-coding chromosomal DNA and may explain the trans-regulatory function of ecDNAs that only contain regulatory elements and no genes11,68. Moreover, this trans-activator model provides a secondary but potentially important mechanism by which ecDNA may rapidly promote intratumour heterogeneity at the level of chromosomal transcription. However, the affinity of ecDNA tethering to specific chromosomal regions and the detailed processes of ecDNA–chromosome interactome formation remain to be discovered.
Conclusions and perspectives
Our understanding of the role of extrachromosomal DNA elements in cancer is rapidly improving, aided by technological advances such as sequencing for ecDNA characterization and CRISPR for live-cell studies and targeted analysis (Box 1). We now know that ecDNA can play a dominant role in driving intratumoral heterogeneity and tumour evolution, and that unique regulatory mechanisms exist that govern the transcription of resident oncogenes. Recent discoveries uncovering regulatory functions of ecDNA in chromosomal gene transcription further expand on the unique biology provided by this class of cancer-associated chromosomal rearrangement.
Opportunities for therapeutic targeting.
Many cancer-driving oncogenes are activated by amplifications, including on ecDNAs exclusively found in cancer cells. A major therapeutic strategy is to directly inhibit the protein products of these amplified oncogenes. This approach has been particularly successful for oncogenes encoding protein kinases. Examples include the anti-HER2 monoclonal antibody trastuzumab used to treat HER2-positive breast cancers, and the small molecule afatinib which is approved for treatment of non-small cell lung cancers carrying EGFR amplifications. Beyond these examples, as of 2021 there were 62 small-molecule protein kinase inhibitors approved by the US Food and Drug Administration (FDA)75 that provide substantial survival benefits to patients with cancer. However, most oncogenes activated through focal amplification, including on ecDNA, are not kinases28. Targeting frequently amplified genes such as MYC, TERT and MCL1 has been long considered out of reach. Perturbing ecDNA maintenance, propagation, structures or function may provide an orthogonal opportunity to inhibit oncogene activation and may even prevent ecDNA-mediated targeted therapy resistance45,60. The excitement over therapeutic strategies that target ecDNA is derived from the concept of blocking the vehicle of oncogene amplification rather than the oncogene product. In an ideal scenario, a single drug would have the potential to improve outcomes across many cancer types and regardless of the amplified oncogene as long as the structure is extrachromosomal.
The unique features of ecDNA — their replication, segregation, clustering into hubs, genesis, and expulsion in micronuclei — provide at least five potential strategies for therapy, as discussed next and outlined in Figure 6.
Figure 6. Opportunities for development of ecDNA targeting therapeutic strategies.
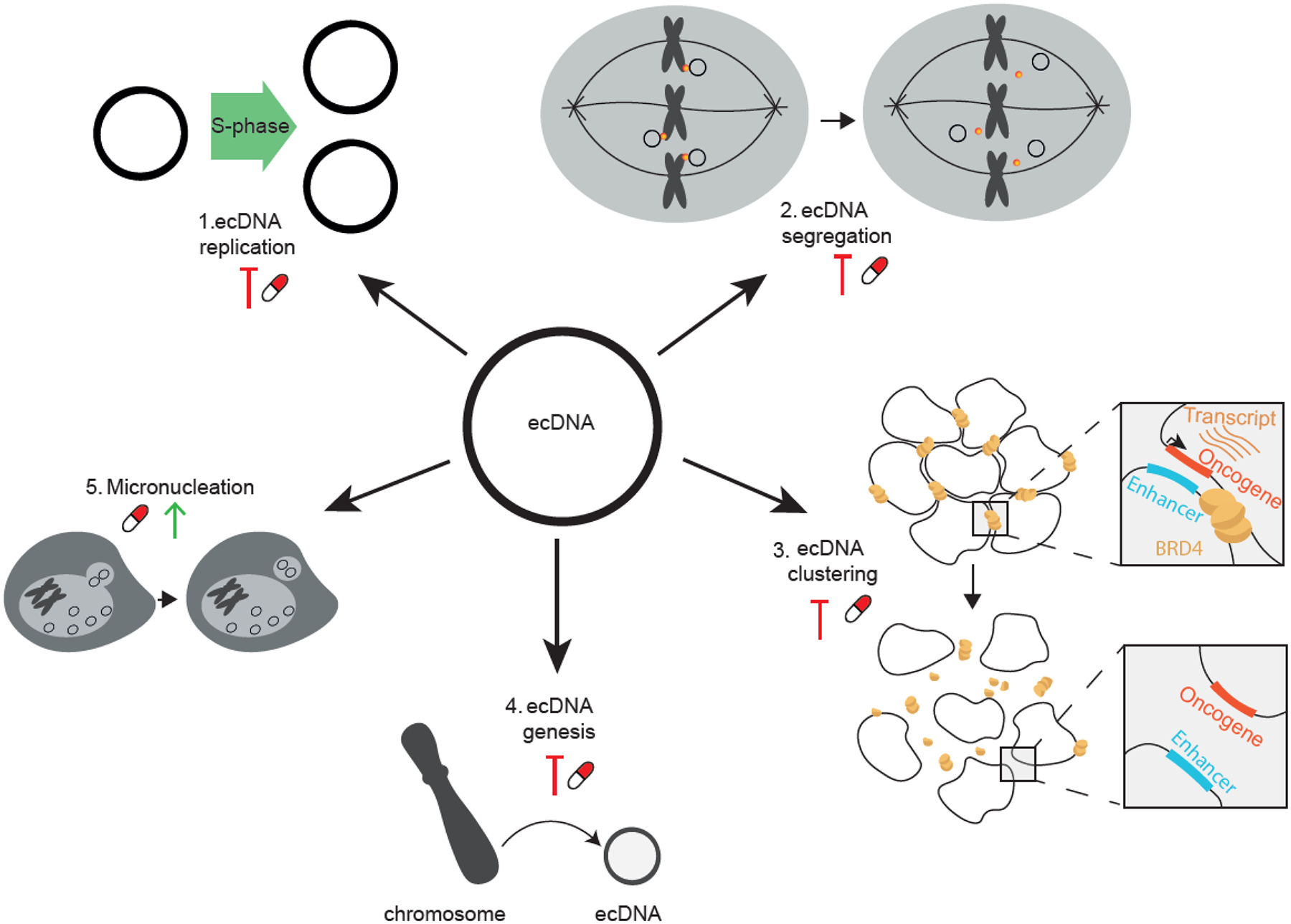
New therapeutic approaches may be designed to target the amplification vehicle rather than amplified cargo genes, for improved outcomes of patients with extrachromosomal DNA (ecDNA)-containing cancer. At least five potential strategies for therapy are under consideration, including: blocking ecDNA replication (1); perturbing ecDNA segregation (2); disrupting ecDNA clustering into hubs (3); preventing new ecDNA formation (4); and promoting ecDNA expulsion in micronuclei (5).
Strategy 1: targeting ecDNA replication.
DNA replication starts with the unwinding of the double helix by helicases. There are 95 human DNA helicases, each with DNA or RNA structure specificity76. EcDNA replication may be subject to unique helicase activity, as occurs for mitochondrial DNA replication by specific helicases77. Another option includes interfering with de novo synthesis of the deoxyribonucleotide triphosphate DNA building blocks, to limit nucleotide production. Ribonucleotide reductase is the rate limiting enzyme for chromosomal DNA replication and can be targeted using for example gemcitabine or hydroxyurea37,78. Whether ecDNA-specific replication enzymes exist is currently unknown.
Strategy 2: targeting ecDNA segregation.
Following replication, ecDNAs may segregate to daughter cells through hitchhiking on chromosomal DNA54. It is unclear what enables ecDNA to attach to chromosomes and what molecular glue may exist. A better understanding of ecDNA segregation may present opportunities to modulate the process.
Strategy 3: targeting ecDNA clustering into hubs.
Specific ecDNA functions such as ecDNA hub formation may be pharmacologically perturbed and thereby decrease the transcription and activity of the ecDNA cargo, for example by targeting the BET protein BRD4 which may be critical in ecDNA tethering13,23.
Strategy 4: targeting ecDNA genesis.
DNA repair following chromothripsis and other DNA breakage events results in ecDNA formation. Perturbation of DNA damage repair processes such as homologous recombination (by PARP inhibitors), and non-homologous end joining (by DNA-PKcs inhibitors) may disadvantage tumours that benefit from ecDNA amplifications38 and may be most effective in combination with other DNA damaging treatments, such as radiotherapy.
Strategy 5: targeting ecDNA micronucleation.
Finally, through yet unknown molecular mechanisms, ecDNA is more prone to micronuclear expulsion and elimination than chromosomal DNA, a process also facilitated by hydroxyurea37,79. Micronucleation may prevent DNA repair of entrapped ecDNAs, thus rendering ecDNA vulnerable to DNA damaging strategies through for example ionizing radiation. An in-depth analysis of the molecular pathways involved in micronuclear expulsion may reveal new therapeutic targets.
Challenges of therapeutic ecDNA targeting.
Despite these promising opportunities for ecDNA-directed drug development, several complicating factors remain. First, it is clear that the term ‘ecDNA’ represents a diverse group of focal amplifications, which vary in various properties such as size and structural complexity9,27. This molecular heterogeneity may mean that different strategies are needed for targeting distinct subclasses of ecDNA. A second potentially complicating factor is that we have limited data on the clonality of ecDNA. Like tyrosine kinase inhibitors, targeting a subclonal ecDNA may create a growth advantage of cells lacking ecDNA and drive rapid clonal selection and treatment resistance. Thirdly, we currently lack understanding of the ability of ecDNAs to re-integrate into the genome39. Integration may occur randomly in locations of DNA damage38, and this provides another trajectory that may limit treatment efficacy, as ecDNAs might re-integrate as a mechanism of drug resistance and then re-emerge extrachromosomally once treatment is removed45. Finally, targeting a molecule that resides in the nucleus requires an ability to pass through multiple cellular membranes and endosome escape, adding additional chemical biology complexity80–82. Filling these knowledge gaps will be paramount for maximizing the chances of achieving successful anti-ecDNA therapy. As multiple mechanisms are likely to contribute to ecDNA generation, maintenance, and evolution, targeting a single mechanism amongst those discussed in this Review may not be enough to successfully treat ecDNA-harbouring cancer. Moreover, given the heterogeneity of cancer, it is likely that a one-size-fits-all approach will not work, and that subcategories of ecDNA-driven cancers exist with differential sensitivity to the therapeutic approaches outlined above. However, we believe that lowering ecDNA frequency, which could reduce the risk of ecDNA-driven therapy resistance, would improve therapy efficacy, and eventually provide prolonged patient survival.
Outlook.
As pointed out in this Review, important progress has been made in characterizing the unique sequence and chromatin composition of ecDNAs. Many questions regarding these features still remain unanswered. For example, the molecular mechanisms leading to altered chromatin compaction on ecDNA remain unknown, so does how ecDNA interactions with other ecDNAs (hubs) and chromosomal DNA are formed. Future investigations into these mechanisms not only have the potential to reveal new biology, but may also uncover factors that may serve as therapeutic targets. Furthermore, forthcoming studies using new single-cell-based methods are expected to yield important new insights into ecDNA intercellular heterogeneity and structure–function relationships. A seemingly basic challenge for future research may lie in the quantification of ecDNA properties in such datasets. Lastly, many other organisms such as yeast and others also contain circular DNAs, which may in part resemble ecDNA with regards to properties such as maintenance, chromatin regulation and replication, suggesting that some of the outstanding questions about ecDNA in cancer may only be solvable through interdisciplinary research approaches in multiple model systems. Thus, to fully deliver the potential of ecDNAs to improve the treatment and diagnosis of patients suffering from ecDNA-driven cancer, sustained academic and industrial investigations in ecDNA-related research will be needed.
Acknowledgements
We thank Kevin Seburn for providing constructive feedback while writing the manuscript (The Jackson Laboratory (JAX), Research Program Development). We thank Chia-lin Wei (JAX) and Richard Koche (Memorial Sloan–Kettering Cancer Center) for helpful discussions. This work was supported by the following grants from the US National Institutes of Health (NIH): R01 CA237208, R21 NS114873, R21 CA256575, R33 CA236681 and Cancer Center Support Grant P30 CA034196 (R.G.W.V.); a grant from the Brain Tumour Charity (R.G.W.V.). E.Y. is supported by a basic research fellowship from the American Brain Tumor Association (BRF1800014). A.G.H. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 398299703 and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 949172). R.C.G. is supported by a fellowship from the ‘la Caixa’ Foundation (ID 100010434). The fellowship code is LCF/BQ/EU20/11810051.
Glossary
- Proto-oncogenes
Genes involved in normal cell growth that, when abnormally activated, lead or contribute to cancer development
- Focal amplification
A DNA region that only spans a subchromosomal-arm proportion of the chromosome and is amplified at high level, i.e. more than eight copies
- Intratumour heterogeneity
The differences among cancer cells within the same tumour
- Cargo gene
Any gene (or genes) harboured on the sequence of an extrachromosomal DNA (ecDNA) element
- Homogeneously staining regions
(HSRs). Chromosomal regions with DNA amplification presenting a uniformed staining pattern with Giemsa nucleic acid stain
- Enhancer hijacking
A process in which a somatic structural genomic rearrangement brings an enhancer into physical proximity of a gene it does not normally interact with, and activates it ectopically
- Chromothripsis
A massive chromosomal rearrangement resulting from a chromosome shattering event, characterized by more than 20 DNA fragments stitched together in an abnormal order
- Breakage-fusion-bridge cycles
(BFBs). A mechanism of chromosomal instability caused by a cycle of telomere breaks and dicentric chromosome formation
- Mitosis
A cellular process in which replicated genetic information in a single cell is divided into two identical nuclei
- Micronuclei
The small nuclear structures that reside in the cytoplasm and contain damaged DNA fragments which were not incorporated into the main nucleus after mitosis
- EcDNA concatenation
A structure in which two or more closed circular DNAs are interlinked
- Chromatin accessibility
The extent to which proteins are able to interact with chromatinized DNA, which is regulated through nucleosome occupancy and other factors occluding access to DNA
- Transcription factories
Molecular complexes consisting of extrachromosomal DNAs (ecDNAs) and transcription machinery components, with high transcriptional activity of ecDNA sequences
- Synthetic aneuploidy effect
Increased transcriptional activity of regions of the genome where extrachromosomal DNAs (ecDNAs) make a physical connection, similar to the effect of aneuploidy
- Hemizygous
The type of zygosity in which only one allele contains a gene or mutation
- Phasing
The process of inferring haplotype information of a sequence from genomic data
Footnotes
Competing interests
R.G.W.V. is a cofounder and/or advisor of Boundless Bio and Stellanova Therapeutics. There is no commercial interest or intellectual property associated with this work. The remaining authors declare no competing interests.
References
- 1.Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Group, P.T.C. et al. Genomic basis for RNA alterations in cancer. Nature 578, 129–136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RGW, Bafna V & Mischel PS Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer 19, 283–288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.deCarvalho AC et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet 50, 708–717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; deCarvalho et al. (2018) and Turner et al. (2017) demonstrated that ecDNA is highly frequently observed in brain tumours, providing early suggestions that ecDNA incidence in cancer is much higher than previously thought.
- 5.Turner KM et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; deCarvalho et al. (2018) and Turner et al. (2017) demonstrated that ecDNA is highly frequently observed in brain tumours, providing early suggestions that ecDNA incidence in cancer is much higher than previously thought.
- 6.Cox D, Yuncken C & Spriggs AI Minute Chromatin Bodies in Malignant Tumours of Childhood. Lancet 1, 55–8 (1965). [DOI] [PubMed] [Google Scholar]; The initial discovery of extrachromosomal DNA in the nuclei of the neoplastic cells.
- 7.Chapman OS et al. The landscape of extrachromosomal circular DNA in medulloblastoma. bioRxiv, 2021.10.18.464907 (2021). [Google Scholar]
- 8.Zhao XK et al. Focal amplifications are associated with chromothripsis events and diverse prognoses in gastric cardia adenocarcinoma. Nat Commun 12, 6489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim H et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 52, 891–897 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first whole-genome sequencing based pan-cancer analysis of extrachromosomal DNA amplifications across newly diagnosed tumours and cancer types.
- 10.Koche RP et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet 52, 29–34 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this work, the authors used whole-genome sequencing and, for the first time, circular DNA sequencing in neuroblastoma cancer samples to demonstrate how ecDNA impacts genome organization.
- 11.Morton AR et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 179, 1330–1341 e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that co-amplification of enhancers and oncogenes determines the genomic breakpoints that define ecDNA boundaries.
- 12.Wu S et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi E et al. Live-Cell Imaging Shows Uneven Segregation of Extrachromosomal DNA Elements and Transcriptionally Active Extrachromosomal DNA Hubs in Cancer. Cancer Discov 12, 468–483 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; Yi et al. (2022) reported the discovery that ecDNA molecules form clusters in which cargo gene transcription is strongly increased.
- 14.Weller M et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 18, 1373–1385 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Liehr T, Claussen U & Starke H Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res 107, 55–67 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Moller HD, Parsons L, Jorgensen TS, Botstein D & Regenberg B Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci U S A 112, E3114–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen T, Kumar P, Koseoglu MM & Dutta A Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet 34, 270–278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata Y et al. Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science 336, 82–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This landmark study reported on the presence of small circular DNA elements in human cells.
- 19.Shoura MJ et al. Intricate and Cell Type-Specific Populations of Endogenous Circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3 (Bethesda) 7, 3295–3303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moller HD et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun 9, 1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 39, 694–707 e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanda T, Sullivan KF & Wahl GM Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8, 377–85 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Hung KL et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 600, 731–736 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; Hung et al. (Nature, 2021) reported the discovery that ecDNA molecules form clusters in which cargo gene transcription is strongly increased.
- 24.Balaban-Malenbaum G & Gilbert F Double minute chromosomes and the homogeneously staining regions in chromosomes of a human neuroblastoma cell line. Science 198, 739–41 (1977). [DOI] [PubMed] [Google Scholar]
- 25.Nik-Zainal S et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y et al. Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadi K et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 183, 197–210 e32 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scully R, Panday A, Elango R & Willis NA DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20, 698–714 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noer JB, Horsdal OK, Xiang X, Luo Y & Regenberg B Extrachromosomal circular DNA in cancer: history, current knowledge, and methods. Trends Genet (2022). [DOI] [PubMed] [Google Scholar]
- 31.L’Abbate A et al. MYC-containing amplicons in acute myeloid leukemia: genomic structures, evolution, and transcriptional consequences. Leukemia 32, 2152–2166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alt FW, Kellems RE, Bertino JR & Schimke RT Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem 253, 1357–70 (1978). [PubMed] [Google Scholar]
- 33.Haber DA, Beverley SM, Kiely ML & Schimke RT Properties of an altered dihydrofolate reductase encoded by amplified genes in cultured mouse fibroblasts. J Biol Chem 256, 9501–10 (1981). [PubMed] [Google Scholar]
- 34.Haber DA & Schimke RT Unstable amplification of an altered dihydrofolate reductase gene associated with double-minute chromosomes. Cell 26, 355–62 (1981). [DOI] [PubMed] [Google Scholar]
- 35.Beverley SM, Coderre JA, Santi DV & Schimke RT Unstable DNA amplifications in methotrexate-resistant Leishmania consist of extrachromosomal circles which relocalize during stabilization. Cell 38, 431–9 (1984). [DOI] [PubMed] [Google Scholar]
- 36.Schimke RT Gene amplification in cultured animal cells. Cell 37, 705–13 (1984). [DOI] [PubMed] [Google Scholar]
- 37.Von Hoff DD et al. Hydroxyurea accelerates loss of extrachromosomally amplified genes from tumor cells. Cancer Res 51, 6273–9 (1991). [PubMed] [Google Scholar]
- 38.Shoshani O et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Storlazzi CT et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res 20, 1198–206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohl NE et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35, 359–67 (1983). [DOI] [PubMed] [Google Scholar]; The identification of oncogene sequences on extrachromosomal DNA amplifications.
- 41.L’Abbate A et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res 42, 9131–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nonet GH, Carroll SM, DeRose ML & Wahl GM Molecular dissection of an extrachromosomal amplicon reveals a circular structure consisting of an imperfect inverted duplication. Genomics 15, 543–58 (1993). [DOI] [PubMed] [Google Scholar]
- 43.Carroll SM et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol 8, 1525–33 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stark GR, Debatisse M, Giulotto E & Wahl GM Recent progress in understanding mechanisms of mammalian DNA amplification. Cell 57, 901–8 (1989). [DOI] [PubMed] [Google Scholar]
- 45.Nathanson DA et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; The identification of ecDNA modulation as a mechanism of resistance to targeted drug treatment.
- 46.Song K et al. Plasticity of Extrachromosomal and Intrachromosomal BRAF Amplifications in Overcoming Targeted Therapy Dosage Challenges. Cancer Discov 12, 1046–1069 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deshpande V et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 10, 392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bergstrom EN et al. Mapping clustered mutations in cancer reveals APOBEC3 mutagenesis of ecDNA. Nature 602, 510–517 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosswog C et al. Chromothripsis followed by circular recombination drives oncogene amplification in human cancer. Nat Genet 53, 1673–1685 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Levan A & Levan G Have double minutes functioning centromeres? Hereditas 88, 81–92 (1978). [DOI] [PubMed] [Google Scholar]
- 51.Shimizu N, Itoh N, Utiyama H & Wahl GM Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol 140, 1307–20 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimizu N, Kanda T & Wahl GM Selective capture of acentric fragments by micronuclei provides a rapid method for purifying extrachromosomally amplified DNA. Nat Genet 12, 65–71 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Tanaka T & Shimizu N Induced detachment of acentric chromatin from mitotic chromosomes leads to their cytoplasmic localization at G(1) and the micronucleation by lamin reorganization at S phase. J Cell Sci 113 (Pt 4), 697–707 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Kanda T, Otter M & Wahl GM Mitotic segregation of viral and cellular acentric extrachromosomal molecules by chromosome tethering. J Cell Sci 114, 49–58 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Pope BD et al. Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barker PE, Drwinga HL, Hittelman WN & Maddox AM Double minutes replicate once during S phase of the cell cycle. Exp Cell Res 130, 353–60 (1980). [DOI] [PubMed] [Google Scholar]
- 57.Itoh N & Shimizu N DNA replication-dependent intranuclear relocation of double minute chromatin. J Cell Sci 111 (Pt 22), 3275–85 (1998). [DOI] [PubMed] [Google Scholar]
- 58.Barker PE & Hsu TC Are double minutes chromosomes? Exp Cell Res 113, 456–8 (1978). [DOI] [PubMed] [Google Scholar]
- 59.Lundberg G et al. Binomial mitotic segregation of MYCN-carrying double minutes in neuroblastoma illustrates the role of randomness in oncogene amplification. PLoS One 3, e3099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xue Y et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med 23, 929–937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lange JT et al. Principles of ecDNA random inheritance drive rapid genome change and therapy resistance in human cancers. bioRxiv, 2021.06.11.447968 (2021). [Google Scholar]
- 62.Clarke TL et al. Histone Lysine Methylation Dynamics Control EGFR DNA Copy-Number Amplification. Cancer Discov 10, 306–325 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson KC et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat Genet 53, 1456–1468 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nikolaev S et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun 5, 5690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Res 21, 381–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spielmann M, Lupianez DG & Mundlos S Structural variation in the 3D genome. Nat Rev Genet 19, 453–467 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Chen W et al. Sequencing of methylase-accessible regions in integral circular extrachromosomal DNA reveals differences in chromatin structure. Epigenetics Chromatin 14, 40 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helmsauer K et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat Commun 11, 5823 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blumrich A et al. The FRA2C common fragile site maps to the borders of MYCN amplicons in neuroblastoma and is associated with gross chromosomal rearrangements in different cancers. Hum Mol Genet 20, 1488–501 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Purshouse K et al. Oncogene expression from extrachromosomal DNA is driven by copy number amplification and does not require spatial clustering. bioRxiv, 2022.01.29.478046 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meldi L & Brickner JH Compartmentalization of the nucleus. Trends Cell Biol 21, 701–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chuang CH et al. Long-range directional movement of an interphase chromosome site. Curr Biol 16, 825–31 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Osborne CS et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36, 1065–71 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Delmore JE et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol Res 165, 105463 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Umate P, Tuteja N & Tuteja R Genome-wide comprehensive analysis of human helicases. Commun Integr Biol 4, 118–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szczesny RJ et al. Yeast and human mitochondrial helicases. Biochim Biophys Acta 1829, 842–53 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Yu L et al. Gemcitabine eliminates double minute chromosomes from human ovarian cancer cells. PLoS One 8, e71988 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Von Hoff DD et al. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci U S A 89, 8165–9 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rajendran L, Knolker HJ & Simons K Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov 9, 29–42 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Petros RA & DeSimone JM Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov 9, 615–27 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Pouton CW, Wagstaff KM, Roth DM, Moseley GW & Jans DA Targeted delivery to the nucleus. Adv Drug Deliv Rev 59, 698–717 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Luebeck J et al. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nat Commun 11, 4374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prada-Luengo I, Krogh A, Maretty L & Regenberg B Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads. BMC Bioinformatics 20, 663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng S et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev 27, 1462–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hung KL et al. Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH. bioRxiv, 2021.11.28.470285 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar P et al. ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci Adv 6, eaba2489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rajkumar U et al. EcSeg: Semantic Segmentation of Metaphase Images Containing Extrachromosomal DNA. iScience 21, 428–435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]