

Abstract
Mitochondrial DNA (mtDNA) mutations are among the most common genetic events in all tumors and directly impact metabolic homeostasis. Despite the central role mitochondria play in energy metabolism and cellular physiology, the role of mutations in the mitochondrial genomes of tumors has been contentious. Until recently, genomic and functional studies of mtDNA variants were impeded by a lack of adequate tumor mtDNA sequencing data. These barriers, and a conceptual fog surrounding the functional impact of mtDNA mutations in tumors has begun to lift, revealing a path to understanding the role of this essential metabolic genome in cancer initiation and progression. Here we discuss the history, recent developments, and challenges that remain for this emerging field as the impact of a major new class of cancer-associated mutations is unveiled.
Keywords: Mitochondrial DNA, cancer, mutation selection, genome editing
Introduction
The mitochondrial genome (mtDNA) is among the most mutated regions of the cancer genome, undergoing somatic mutation in approximately 50% of all tumors [1]. Mutations to mtDNA affect the small repertoire of protein-coding genes, non-protein-coding transfer and ribosomal RNAs, and noncoding DNA that are essential to oxygen-dependent mitochondrial respiration (Box 1). That mtDNA mutations occur so commonly, and affect genetic loci, which are often under immense purifying selection in the human germline, reflects a basic underlying paradox: how can mtDNA mutations confer a selective advantage to cancer cells while simultaneously impairing a central node for ATP generation and maintenance of redox balance? This question sits at the center of a historically controversial debate around the selection and function of mtDNA mutations in cancer [1–3].
Box 1. A Primer on Mitochondrial Genetics and Human Disease.
Mitochondria are double membrane-bound organelles found in eukaryotic cells. These organelles evolved from a proteobacterial ancestor [12] and retain a vestigial genome known as mitochondrial DNA (mtDNA). Most of the genes in mtDNA have been lost or transferred to the nuclear genome through evolution, resulting in a small, circular 16.5 kb genome, coding for 11 mRNAs, 22 tRNAs and 2 rRNAs in humans [13,14]. The 11 mRNAs are translated into 13 essential subunits of complex I, III and IV of the electron transport chain and ATP synthase, which are critical to oxygen-dependent cellular metabolism [13,14]. Mitochondrial DNA is genetically compact, with very few non-coding regions and no redundancy in the encoded components, and intact expression of all genes is necessary for a functional mitochondrial respiratory chain [15].
Mitochondrial DNA is a multi-copy genome, typically found in the range of 100–10,000 molecules per cell, depending on the cell type [16]. Consequently, when mutations arise in mtDNA, they do not affect every copy in a cell and instead produce a mixed population of mutant and wild-type genomes, a phenomenon known as heteroplasmy [17]. Mutations of mtDNA have been extensively studied in the context of rare metabolic diseases caused by hereditary transmission of deleterious mtDNA alleles, affecting approximately 1 per 4,000 people [18]. The clinical severity of these primary mitochondrial disorders critically depends on heteroplasmy levels. Mutation heteroplasmies of ~60–80% are mostly associated with relatively mild juvenile or adult-onset pathologies, demonstrating limited mitochondrial dysfunction. However, mutation heteroplasmies of >90% are generally associated with profound mitochondrial dysfunction that accompanies severe, multi-system disorders with significant morbidity and mortality [19]. Such phenotypic thresholds in heteroplasmy levels are highly variable and dependent on numerous factors, including the identity of the mutant allele and the tissue in which it arises [20], as well as the presence of nuclear-DNA-encoded gene variants modifying penetrance of mtDNA alleles [21]. Such observations have, by default, drawn the field to a view that mutation heteroplasmies beneath clinical thresholds or < 60% are not relevant to immediate disease processes and do not exert a significant effect on mitochondrial function, as evidenced by the lack of impact on oxygen consumption [17,22,23]. However, given the emergence of mtDNA mutations with a broader range of heteroplasmies in cancer (which we describe below), we contend that this historical perspective from the primary mitochondrial disease literature likely needs to evolve to reconcile recent data from the cancer genomics field.
Distilling somatic mutations that endow cells with a selective advantage (“driver mutations”, see Glossary) from the vast reservoir of somatic mutations that do not (“passenger mutations”) and delineating the mechanism by which driver mutations confer a selective advantage, remains a cornerstone of basic and translational cancer research. While an obvious place to search for driver mutations is in genes or regions with high somatic mutation rates, such regions are often as likely to be passengers (e.g. mutations to the extremely long gene TTN) as they are to be drivers (KRAS) [4,5]. Historically, a combination of forward and reverse genetics has been required to determine the function and fitness-modifying properties of potential drivers in nuclear DNA. This advanced was achieved through production of model systems where specific, cancer-relevant modifications to DNA are identified, inserted, and studied. The hunt for nuclear drivers of cancer co-evolved with, and in some instances directly advanced the capacity for, genetic engineering of nuclear DNA. Developments in engineering mtDNA have been comparatively glacial because of a double-membrane barrier and the high copy number of the mitochondrial genome [6,7]. However, recent years have seen the emergence of key genome manipulation technologies, optimized for use in mitochondria, that are poised to make an impact [8–11].
In this review, we summarize our current understanding of the incidence, selection, and function of mtDNA variants in human cancers, and discuss how emerging technologies are positioned to resolve open questions about the functional significance of mtDNA mutagenesis. We also explore single cell sequencing and mtDNA editing systems as advanced, highly scalable platforms to determine dosage and threshold effects in transcriptional phenotypes to experimentally identify cancer genes of mtDNA.
The Landscape of mtDNA Mutations Across Cancers
In 1956, Otto Warburg proposed that dysfunctional mitochondria are responsible for the fermentative catabolism of glucose to lactate and lack of respiration in aerobic conditions in tumor cells [24,25]. While decades of research have led to the discovery of numerous mechanistic causes for aerobic glycolysis not mediated by mtDNA mutations [26], including somatic mutation of metabolic regulators and selective expression/regulation of enzymes favoring lactate production [27–31], these discoveries have gone hand in hand with the observation that apparently pathogenic mtDNA mutations are abundantly common in tumors [32]. One of the earliest descriptions of mtDNA mutations in tumors came from Polyak and colleagues, who reported the presence of seemingly homoplasmic mutations in a small number of colorectal cancer cell lines [33]. Since then, numerous studies have described an abundance of somatic and predominantly heteroplasmic mutations in mtDNA (reviewed comprehensively in [2]). The most comprehensive study analyzed whole genome sequencing from ICGC/PCAWG [32], whole genome sequencing of childhood tumors from St. Jude’s Childrens Hospital [34], and exome sequencing of diverse adult cancers from TCGA [35]. The key findings of these analyses converged on several discoveries.
First, these studies independently confirmed that most tumors (~50%) have readily detectable somatic mutations in their mtDNA. These findings were largely reproducible across sequencing cohorts with distinct disease compositions and thresholds for variant calling (from 5% in the TCGA [35] to as low as 1% in PCAWG [32]). Most of these mutations likely originate from the relatively high mtDNA background mutation rate, about 10- to 17-fold higher than nuclear DNA [36]. These studies reported that somatic mtDNA mutations in tumors largely obeyed a unique strand-specific pattern of mutations (preferring C>T mutations on the heavy strand, and T>C mutations on the light strand) [1]. This mutational pattern was initially attributed to oxidative damage via generation of reactive oxygen species (ROS) by the physically proximal electron transport chain (ETC) [37,38]. Emerging evidence [39,40], however, suggested that mutation rates do not necessarily correlate with ROS levels, and concurrent genetic ablation of mitochondrial base excision repair pathways alone or in combination with mitochondrial superoxide dismutase in mice did not lead to elevated mutation rates [39]. A competing theory suggests that the accumulation of replicative errors from differing modes of mtDNA strand replication are a likely source of mutations found in cancer (for a review see [41]) [42–44]. Accordingly, it has been proposed [41] that because mtDNA replication is uncoupled from the cell cycle [45,46] or continually replicates over time in the context of a proliferating cancer cell, this can lead to a considerable build-up of low level replicative errors [43] despite the mitochondrial replicative polymerase γ (Polγ) having one of the highest processive fidelities across all domains of life [47].
Second, the analyses indicated that mtDNA mutation burden was highly lineage-specific and that certain cancer types were subject to a significantly higher burden of mtDNA mutations than others [1,32,35] (Figure 1A). This lineage-specificity manifests both in the total burden of mutations in some diseases as well as the functional effect of the mutation (i.e. in the preference for/against protein-truncating variants). Analysis of both PCAWG and TCGA data suggested that tumors of the colorectal tract, thyroid, and certain histological subtypes of kidney cancers, such as chromophobe and papillary renal cell carcinoma, are enriched specifically for protein-truncating variants, whereas hematological malignancies such as acute myeloid leukemia have qualitatively low burdens of all somatic mitochondrial mutations [32,35]. A recent analysis of mutant heteroplasmy in patients with the m.A3243G (known to underlie the mitochondrial genetic condition MELAS) indicated that T cells undergo purifying selection against the mutant allele [48], consistent with the apparent purifying selection in human AML tumors.
Figure 1. Hierarchy of variation in mtDNA genotypes.
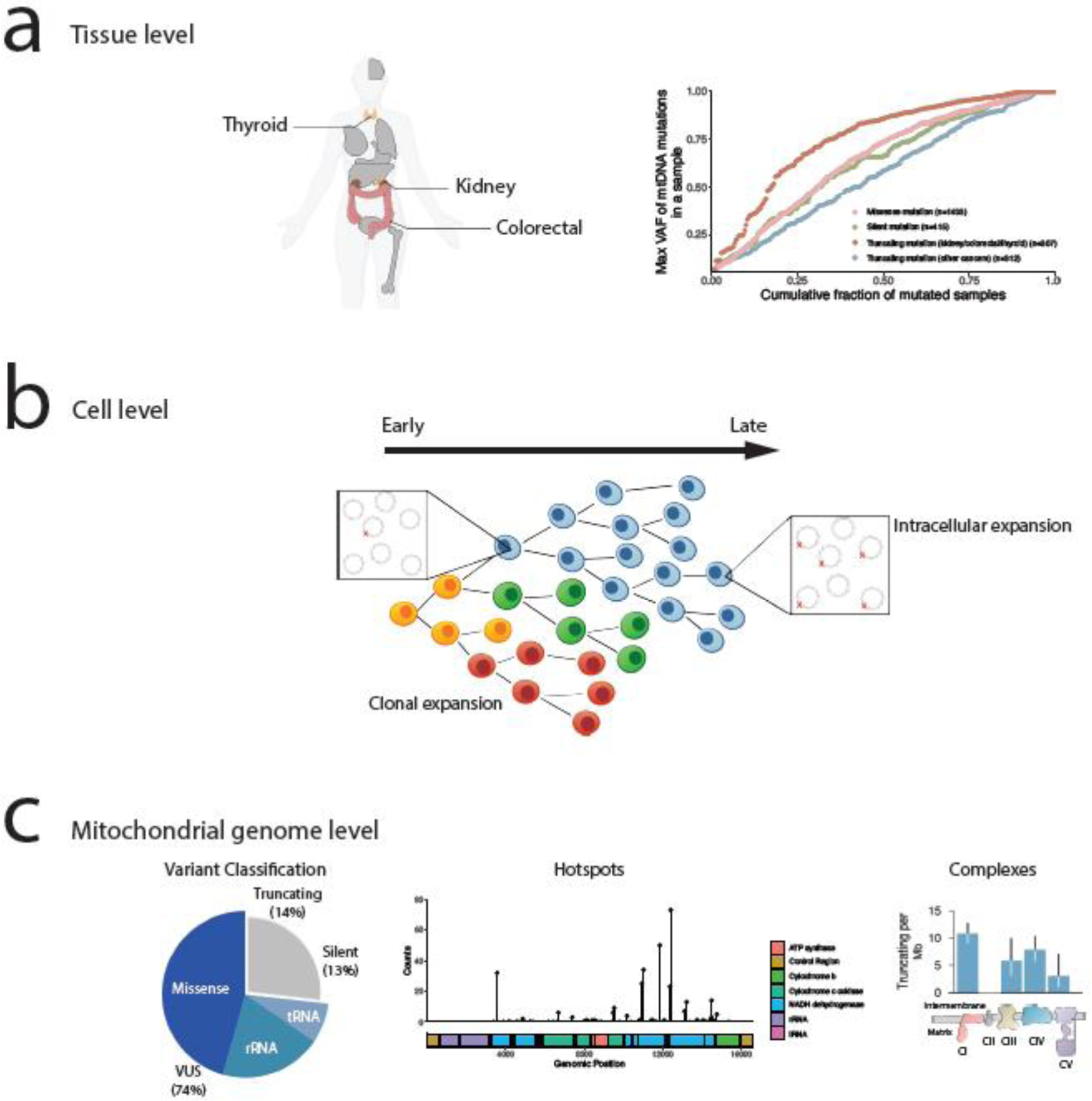
(A) At the resolution of tissues, renal, thyroid, and colorectal cancer types have enrichment of truncating mutations with higher heteroplasmy levels, suggestive of positive selection in those tumor types. The right figure is a reproduction from [32]. (B) At the resolution of individual cells, tumors are composed of genetically diverse cell populations with distinct mtDNA genotypes. Changes in bulk mtDNA heteroplasmy over time thus reflect both changes in clonal composition, as intracellular shifts in heteroplasmy. Both processes may be subject to evolutionary selection. (C) At the resolution of mtDNA genotypes, mutations non-randomly accumulate in specific regions of the genome. Protein-truncating mutations, which have been the primary subject of interest in genomic studies, account for only 27% of variants observed in genomes. Such truncating mutations preferentially accumulate in complex I subunits, suggesting that the function of truncating mutations is shaped by their impact on respiratory function. The figures are a reproduction from [35].
More recent large-scale studies have found that the vast majority of somatic mtDNA mutations in cancer are heteroplasmic, reaching allele frequencies well below 100% [1,32]. The prevalence of heteroplasmy suggests that most mtDNA-mutant tumors retain some spare mitochondrial respiratory capacity in the form of wild-type mtDNA. Experimental data from a mouse model with a heteroplasmic mtDNA variant in tRNAAla supports this hypothesis. There, an increase in mtDNA copy number rescued the COX deficiency observed in cells with high heteroplasmy level [49]. This finding suggests that the absolute copy number of wild type mtDNA is an important determinant of the pathological impact of these variants, and that at sufficiently high heteroplasmies, negative selection (potentially acting via compensation in wild-type mtDNA copy number) emerges to resist further expansion of the mutant mtDNA pool. However, understanding these selective pressures in the context of heterogeneous cell populations with potentially diverse mtDNA genotypes requires a detailed quantification of heteroplasmy at the level of single cells.
Single cell sequencing of mtDNA unmasks latent diversity in mtDNA heteroplasmy
Tumors are heterogeneous compositions of malignant, immune, and stromal cells with potentially distinct mtDNA genotypes. Consequently, quantitative determination of heteroplasmy levels in tumors using conventional bulk sequencing methods is confounded by the abundance of malignant and non-malignant cell populations, and by cell-to-cell variation in heteroplasmy levels within each cell population. Recently, the development of new sequencing approaches that directly assay the genotype of individual cells (“single cell sequencing” technologies) has enabled a direct interrogation of mtDNA genotype and parallel measurement of cellular phenotypes (e.g. transcriptional profiles via RNA sequencing, or DNA accessibility profiles via ATAC profiling) (Figure 1B,C). Thus, single cell sequencing technologies, together with computational methods for identifying mtDNA variants [50] and experimental methods for recovering low heteroplasmic mtDNA variants with targeted deep sequencing of mtDNA [51], allow for the precise measurement of both per-cell heteroplasmy level and corresponding changes in transcriptional phenotype.
The earliest single cell studies on mtDNA used the high copy number of mitochondrial genomes and the high mutation rate of mtDNA as an endogenous barcode; paired with the stable propagation of mtDNA variants to daughter cells, this allowed for lineage tracing and subclonal reconstruction of distinct clones in hematopoietic cells (Figure 2A,B) [52]. Subsequent work using diverse single cell RNA- and DNA-sequencing technologies demonstrated the capability to assess heteroplasmy levels across cell types [48], and define the evolutionary and phylogenetic relationships between clones in primary tumors and patient-derived xenografts. These studies collectively highlighted that even genetically related clones often acquired individual mtDNA variants and clone-specific heteroplasmy levels (suggestive of founder effects) that tracked with phylogenies derived from expression data [52] and/or chromatin accessibility profiles [53,54].
Figure 2. Single cell sequencing of mtDNA.
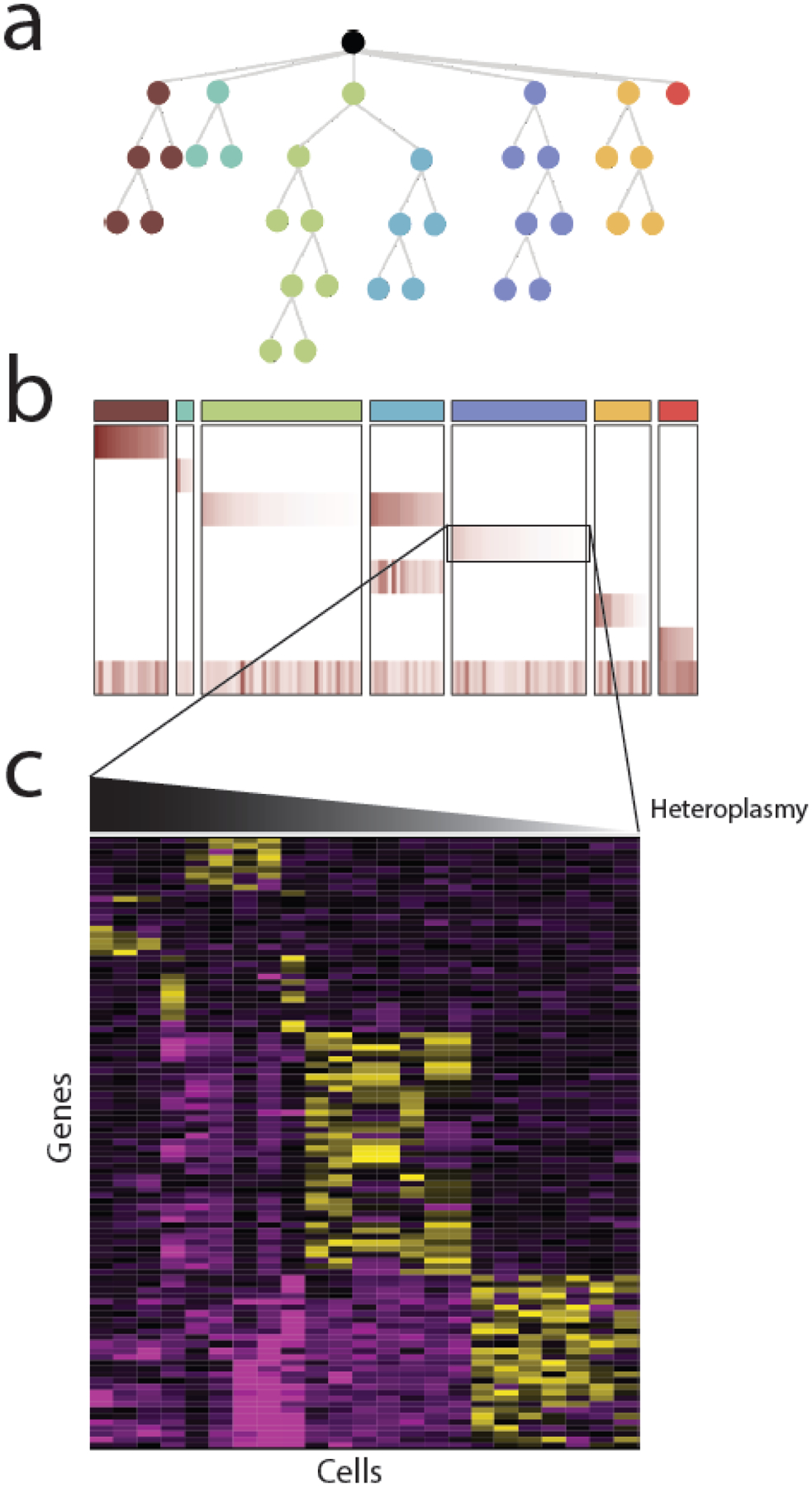
Most single cell analysis of mtDNA variants to date has been for the purposes of reconstructing evolutionary phylogenies, using mtDNA variants as cell-endogenous barcodes. (A) As cancer cells divide, they accumulate new somatic changes to nuclear and mitochondrial DNA that reflect their evolutionary phylogeny. (B) Evolutionarily related cells can be clustered into clones using information about the presence and heteroplasmy of heteroplasmic mtDNA variants. In htis example, a heatmap of the heteroplasmy levels of mtDNA variants reflects the evolutionary structure shown above in (A) The columns indicate each cell and rows indicate mtDNA variants. (C) Some mtDNA variants may not be silent, but rather may be associated with a particular phenotype. In this case, examination of nuclear gene expression patterns (e.g. derived from in single cell RNA-seq) may identify phenotypes associated with heteroplasmic dosage.
Cell-to-cell variation in mtDNA heteroplasmy also has significant implications for the functional interpretation of mtDNA variants in non-cancer settings. When studying the heteroplasmy level of m.A8344G, a known pathogenic variant associated with Myclonic epilepsy with red ragged fibers (MERRF), in a lymphoblastoid cell line [55], it was reported that while the bulk heteroplasmy level (44%) and the median per-cell heteroplasmy (38%) were comparable, the per-cell heteroplasmy level varied extensively from total absence to complete homoplasmy. This finding suggests that variants with low heteroplasmy based on bulk sequencing could attain high heteroplasmy in rare subsets of cells, and in turn might show exceptionally high fitness in certain conditions, such as under therapeutic treatment.
Selective pressure for cancer-associated mtDNA mutations
There is ongoing controversy as to whether somatic mtDNA variants in cancer constitute bona fide driver mutations, contributing to the fitness of the tumor, or if instead they simply represent passenger alterations with neutral or non-fitness-modifying effects on tumor physiology [2,33,35,41]. Below, we attempt to disentangle this controversy by discussing (i) the evidence for and against selective pressure for somatic mtDNA mutations, and (ii) the evidence for the functional significance of mtDNA mutations in cancer.
Selective pressure for somatic mtDNA mutations
Prior studies have largely focused on three complementary pieces of evidence when assessing the extent of positive and negative selective pressure for somatic mutations in cancers affecting protein-coding genes in mtDNA: (i) the ratio of non-synonymous to synonymous mutations (“dN/dS”), (ii) the allele frequency of non-synonymous and truncating mutations, and (iii) the identification of recurrent mutations or “hotspots”. At least three independent analyses have examined dN/dS, a quantitative measure of selection whose value exceeds 1 in instances of positive selective pressure and is below 1 in instances of negative selective pressure [56–58]. The apparent dN/dS of protein-coding mtDNA mutations was initially determined to be quite high (~4), suggesting a strong and positive selective pressure for mutations [34]. However, another study cautioned that because mtDNA has faced a strong C>T/T>C mutational bias over an extended evolutionary period, the number of admissible silent mutations have been systematically eliminated [1]. Only a weak positive selection for missense mutations (95% CI 1.015–1.434), and neutral selection for nonsense mutations (95% CI 0.699–1.443) was found when controlling for this background mutation signature. Conversely, a subsequent analysis that utilized a Bayesian approach to simulate the expected dN/dS in order to account for the strong C>T/T>C mutational bias found the dN/dS of the mtDNA mutations in adult and pediatric malignancies to be significantly higher than expected by chance [34]. In contrast, normal healthy tissues derived from blood demonstrated a significantly lower dN/dS than expected by chance. These results highlight the ongoing uncertainty surrounding the selective pressure for mtDNA mutations in cancer and emphasize that the background mutational process is an important confounder in directly assessing selective pressure with dN/dS.
In addition to dN/dS, variant allele frequency of non-synonymous or protein-truncating mutations have been used as a surrogate measure of selection [1,32,42]. The distribution of heteroplasmies of a given class of mutations (often nonsense/frameshift mutations or missense mutations) were compared to a reference class under putatively neutral selection (i.e. silent mutations). The findings of these analyses have proven reproducible and illuminating. Frameshift indels and nonsense mutations demonstrate consistently lower heteroplasmies than silent mutations, whereas missense mutations show a largely indistinguishable heteroplasmy distribution compared to silent mutations [1]. These data collectively suggest that highly disruptive mutations to mtDNA are under a distinct form of dosage-dependent negative selection that prevents their expansion to higher dosages. Subsequent analysis revealed that heteroplasmy levels of truncating mutations are dependent on cancer tissue lineage [35]. Tissues from the colorectal tract, the kidney, and the thyroid are not only distinguished by an enrichment of protein truncating variants, but also by high heteroplasmy levels of these truncating mutations. Truncating mutations in these three tumor lineages affect 20–30% of all samples, rendering them as common as mutation to canonical tumor suppressors in these diseases. Similarly connecting elevated heteroplasmy with increased selection, a subset of somatic mtDNA mutations were identified in normal tissue adjacent to tumors but absent in normal blood [58]. Non-synonymous substitutions increased in bulk heteroplasmy to a median allele frequency of 58.8% in the tumor compared to 18.8% for synonymous substitutions when focusing on heteroplasmic expansions in variants present at very low allele frequency in normal adjacent tissues (<5%) and elevated heteroplasmy in tumors. These findings suggest that positive selection may act on pre-existing somatic mtDNA mutations by promoting the expansion of otherwise low-level heteroplasmic variants.
Further supporting a role for positive selection in mtDNA is the appearance of recurrent mutations in specific genomic loci and respiratory complexes, which likely reflects alteration in cellular fitness and preferential retention in the population [59] (Figure 1C). Truncating mutations in the kidney, colorectal tract, and thyroid were not randomly distributed across the mtDNA, but preferentially affected subunits of Complex I, NADH dehydrogenase (in contrast to missense variants, which typically occur in Complex III, and silent variants, which show no preference) [35]. Conversely, truncating mutations affecting Complex V were depleted and showed characteristically lower heteroplasmies. The kidney, thyroid, and adrenal glands develop oncocytomas, metaplasia characterized by a high cytoplasmic density of mitochondria. Such oncocytomas possess high-heteroplasmy (or even homoplasmy) truncating mutations in mtDNA that preferentially affect Complex I [60–62].
Truncating mtDNA mutations demonstrate patterns of recurrence not only within specific respiratory complexes, but also at specific genetic loci. Certain mutant mtDNA alleles, especially those associated with truncating variants, seem to recurrently arise at the same genomic position, akin to cancer-associated hotspot somatic mutations in nuclear-DNA encoded genes commonly referred to as hotspots [63]. Such hotspot alleles in nuclear-encoded genes can be associated with either gain-of-function (more common, e.g. KRAS G12D) or loss-of-function events (less-common, e.g. TP53 and APC alleles). Early studies of hotspot alleles in mtDNA were, given the extent of recurrence, comparatively underpowered to detect them and to determine whether their recurrence exceeded what would be expected by chance. 6 hotspot alleles, all arising in Complex I subunits, have been recently identified as using a suitably powered cohort of ~6,000 tumors and a background statistical model of mutation incidence [35]. These hotspots are represented throughout most cancer types with variable and occasionally high frequency. For example, truncating hotspots in mtDNA are represented in 24% of all colorectal tumors, though these recurrent mutations are not detectable in prostate cancers. In total, these observations suggest that there is differential selection for mutations across the mtDNA that is shaped by a combination of tissue lineage, gene function, and mutant allele outcome.
Unresolved functional significance and dark matter in mtDNA mutations
What is the functional consequence of somatic mtDNA mutations once they emerge? Relative to the abundant data on the patterns of mtDNA mutations, data on the functional consequences of these same mutant alleles is comparatively scarce.
With the increased use of cytoplasmic hybrid (cybrid) cells, clonal chimeras resulting from fusion of mtDNA-free ρo cells with cytoplasm containing mutant mtDNA, many studies have tried to experimentally validate the function of mtDNA mutations in cancer. For example, pathogenic mutations in MT-ATP6 gene of mtDNA generated ROS and enhanced tumor growth [64]. mtDNA mutations were also found to result in increased ROS levels and metastatic potential. However, the enhanced growth rate of primary tumors did not correlate with high metastatic potential, suggesting that metastatic potential was regulated indirectly through increased ROS levels [65,66]. Increased ROS levels and tumorigenicity because of mtDNA mutation was further confirmed when an increase in cell survival under stress resulted in enhanced tumorigenicity with heteroplasmic mutation in MT-ND5 [67]. However, the authors also highlighted the importance of heteroplasmy levels in the function of these mutations since homoplasmic MT-ND5 mutation on the same locus resulted in increased rate of apoptosis and inhibited tumorigenicity. This finding was attributed to limited ATP production, which resulted in increased apoptotic potency. However, this is likely dependent on several factors such as mutation type, tissue distribution, and tissue type [17]. For example, homoplasmic mtDNA mutations are a common occurrence in renal, thyroid and other oncocytomas, linking mitochondrial dysfunction with increases in mitochondrial biogenesis [61,62,68,69], a compensatory phenomenon which is observed in mitochondrial disease [19]. A link between mtDNA mutations, in this case homoplasmic mutation of MT-CYTB, and altered TCA cycle fluxes favoring reductive carboxylation of glutamine in support of lipid synthesis and cell proliferation, was also demonstrated for the first time using cybrid cells [70]. A related and more recent study demonstrated that complete loss of Complex III impaired tumor growth, but that this phenotype could be rescued through introduction of ectopic expression of an enzyme capable of oxidizing ubiquinol [71]. Finally, the presence of increased mtDNA mutational burden (produced through mutation of the mtDNA polymerase Polg) enhances tumor growth through upregulation of the de novo serine biosynthesis pathway in mouse models of colorectal carcinogenesis [72].
There have also been efforts to associate the presence of mtDNA mutations in tumors with clinical outcomes. In prostate cancer, mutations in the HV1 segment in the control region are associated with better patient outcome, adjusting for age, pre-treatment PSA, T-category, and GS levels [73]. In contrast, a seemingly more important regulatory region of H-strand origin of replication (OHR) and the essential protein coding gene of ATP8 is associated with inferior patient outcome. Another study directly measuring bioenergetics of mitochondrial mutant prostate tumors associated missense mutations in Complex I with increases in succinate oxidation and inferior patient outcome [74]. Similarly, the presence of truncating mutations was associated with a lineage-agnostic transcriptional phenotype characterized by upregulation of OXPHOS genes and the downregulation of numerous innate immune pathways [35]. However, these analyses fail to delineate the mechanistic link connecting the presence of mtDNA mutations to their (potential) functional outcomes.
While limited progress has been made in understanding the function of a small number of select mutant alleles, the vast majority of somatic mtDNA mutations in cancer are variants of entirely unknown functional significance. Adding to the complexity of interpreting these variants is that nearly 25% of them occur in non-protein-coding regions including tRNAs (8%) and the highly conserved ribosomal RNA (16%) [35]. Mutations to tRNA and rRNA represent particularly exotic mutations that, because of the broad impact a disruptive variant would have on all translated genes, are uniquely tolerated in the mitochondrial genome, where the effects of their mutation are likely limited to the 13 protein-coding genes in mtDNA. Mutations to tRNAs are a well-known and common cause of inherited mitochondrial disease, and similar mutations, including both m.3243A>G and the adjacent m.3244G>A, are observed in human tumors as recurrent hotspots [68,69]. Data suggests that cancer-associated mutations to tRNAs may arise at specific secondary structure elements [35]. rRNA mutations in the germline are not common causes of mitochondrial disease, with the exception of two partially penetrant variants known to produce deafness in patients with MT-RNR1, m.1555A>G and m.1494C>T [75]. Understanding the functional outcome of such “dark matter” rRNA and tRNA mutations, which likely have broad effects on all OXPHOS complexes, is of significant biological interest.
Existing and emerging opportunities for functional interrogation of mtDNA mutations through mitochondrial genome engineering
A key limitation to studying of mitochondrial genetics has been a lack of reliable and robust mtDNA editing tools [6]. Much of our understanding of nuclear oncogenes and tumor suppressors in the modern era derives from the ability to perform both forward and reverse genetics to determine the function of a given cancer-associated gene. While targeting some nuclear genome-editing tools, such as CRISPR, to mtDNA have proven unsuccessful due to differences in mitochondrial mechanisms of RNA import and mtDNA repair [6], the past five years have yielded an mtDNA-editing toolkit of engineered enzymes that can shift heteroplasmies or induce mutagenesis both in vitro and in vivo [7,8,76–79]. Whilst manipulation of any mtDNA sequence at will remains elusive, these tools hold great potential for enhancing our knowledge of mtDNA in cancer (Figure 3)
Figure 3. Current approaches for editing mtDNA in animals.

(A) Random mutators of mtDNA, based on the defective mtDNA polymerase (PolgAmut) or the mitochondrially targeted monomeric cytosine deaminase APOBEC1 [88,89] allow generation of abundant, low heteroplasmy mutations of mtDNA resulting in non-physiological forms of mitochondrial dysfunction. Heteroplasmy shifting exploits the tendency for mitochondria to rapidly degrade genomes containing DNA double strand breaks. If allele-specific double strand breaks can be introduced in the context of a cell bearing a pre-existing mtDNA mutation, the heteroplasmy of that mutation can be amplified or diminished. Several approaches to heteroplasmy shifting, with robust effects in vitro and in vivo, have been developed, including but not limited to mitochondrially targeted: restriction enzymes [81,91,92], zinc-finger nucleases [8,77] and transcription activator-like effector nucleases [76,78]. Base editing, at present consisting exclusively of engineered, dimeric DddAtox cytosine deaminase base editor (DdCBE) domains fused to TALE DNA binding domains, permit a degree of selective mutation introduction into mtDNA, in situ, for the first time. These chimeric enzymes are limited in terms of targetable sites (based on TALE DNA binding constraints, need for 5’TC’3 context, immediate off-target considerations) and the mutations they can produce (C>T or G>A only, no indels), but allow for production of many previously unobtainable genotypes at relevant levels of heteroplasmy. (B) Depiction of the impact of cumulative heteroplasmies shown in A on mitochondrial function.
Enzymes designed to shift heteroplasmy began as a curiosity in a field with no better options. These were initially a small selection of mitochondrially-targeted restriction enzymes ([9,10,76,80–83] (mtREs), able to target only canonical RE target sites. While such enzymes were profoundly limited in their scope, the approach inspired further developments in heteroplasmy shifting with engineerable DNA target specificities, yielding mitochondrially-targeted zinc-finger nucleases (mtZFNs), mitochondrially-targeted transcription activator-like effector nucleases (mitoTALENs), and meganucleases [9,10,76,80–83]. These chimeric enzymes worked by binding to a target site and inducing a double-strand break. Linearised mtDNAs would then be rapidly degraded through combined exonucleolytic processing by Mgme1 and the replicative mitochondrial polymerase gamma [84–86]. The remaining mtDNA molecules, a majority of which will be wild-type, replicated to recover steady-state copy number, thereby shifting heteroplasmy. Although limited by their capacity to manipulate existing heteroplasmy in situ, shifting heteroplasmy in pre-existing model systems of mitochondrial mutation has proven useful in the study of cancer metabolism. Indeed, mtZFNs have been used to alter the mutant load in m.8993T>G osteosarcoma cells [9], producing isogenic cell lines with different levels of mutant heteroplasmy, termed mTUNE [87]. This model system allowed the identification of a cellular metabolic strategy employed by cells with high levels of mitochondrial dysfunction, whereby cytosolic reductive carboxylation of glutamine acts through malate dehydrogenase to enable NAD+ recycling and support glycolytic flux, enhancing cell proliferation and migration [87]. Thus, manipulating mtDNA illuminated how mitochondrial dysfunction ultimately altered growth and movement of cancer cells.
A major limitation of heteroplasmy shifting methods is their capacity to manipulate only pre-existing heteroplasmies. Methods for ab initio introduction of mtDNA alleles are still required to allow a thorough assessment of mitochondrial genetics in cancer. In recent years, the cytosine deaminases mitoAPOBEC1 and DddA-derived cytosine base editors (DdCBEs) have been employed to address this issue. mitoAPOBEC1 is a mitochondrially targeted form of rat APOBEC1, a potent cytosine deaminase capable of introducing C:G>T:A transitions within mtDNA [88]. Expression of mitoAPOBEC1 induced point mutations randomly, with no significant impact on mtDNA copy number, and these mutations negatively impacted organismal fitness in Drosophila due to mitochondrial dysfunction. As such, mitoAPOBEC1 holds the potential for use to study the effects of mtDNA point mutations in diseases, particularly cancer, as it produces the same type of mutations that are overrepresented across tumors [42]. However, mitoAPOBEC1 is a random mutator, like other model systems such as the error prone mitochondrial DNA polymerase [89], and would be limited in forward screening approaches.
Recently the first reliable, targeted approach to mtDNA mutagenesis was reported [90]. This system utilizes DdCBEs, which are composed of a split interbacterial toxin DddAtox, TALE DNA-binding domains and a uracil glycosylase inhibitor (UGI). Owing to substrate preference of the DddAtox enzyme, DdCBEs can induce C>T point mutations in a sequence-specific context where a T must be 5’ of the target C. In a proof-of-concept study, m.11922G>A mutations were induced in MT-ND4 to mimic the mutations seen in Complex I of rare tumors of the kidney and thyroid. These cells with high mutational load showed a reduction in complex I assembly and in oxidative phosphorylation, due to reduced activity. DdCBEs have also been applied in vivo where the mutations m.12539C>T and m.12542G>A were separately introduced into Complex I to create mouse embryos with a silent mutation [7]. The embryos maintained these mutations up to 50-days post-birth and were transferable to offspring. However, the same transmission for deleterious mutations is yet to be shown. DdCBEs have also recently been used to edit mtDNA in target organs via AAV delivery [7]. While being of major utility, DdCBEs are limited by a number of key constraints, including i.) the efficiency with which they can bind target DNA via TALE domains; ii.) sequence-specific mutational biases, and iii.) off-target mutagenesis, both within the target site and throughout the mitochondrial genome, although these limitations may be addressed through subsequent evolution and optimization [11]. Nevertheless, as the sole platform capable of inducing targeted mtDNA mutations, they can begin to be applied to create model systems that may shed light on the role of mtDNA mutations in cancer.
Concluding remarks
The field of mitochondrial oncogenetics today stands at a precarious crossroad: while the case for a functional role for mtDNA mutations found in most cancers has strengthened in the past decade (Figure 4), it primarily rests on patient genomics and clinical data. To tease apart potential mechanisms that underlie patterns of recurrence and selection observed at a population level, these increasingly rich observational data need to be complemented with rigorous experimental approaches. The experimental support for a functional role of mitochondrial DNA mutations in cancer (reviewed previously (see [41]) has not kept pace with the genomic developments of the past decade, still suffering from a lack of well-controlled model systems. Some recent experimental reports that suggest altered cancer cell characteristics in the context of mtDNA mutations are encouraging [72,74], though they remain a distance from determining relevant mechanisms or vulnerabilities. The synergy between two evolving technologies, one for the direct editing of mtDNA and the other for high-resolution phenotypic interrogation of heteroplasmic mtDNA alleles at single cell level, will herald the dawn of a new era in mitochondrial oncogenetics as the varied impacts of mitochondrial mutational processes are determined and a world of untapped therapeutic opportunities are revealed (see Outstanding Questions).
Figure 4. A model for selection of deleterious heteroplasmies in cancer cells.
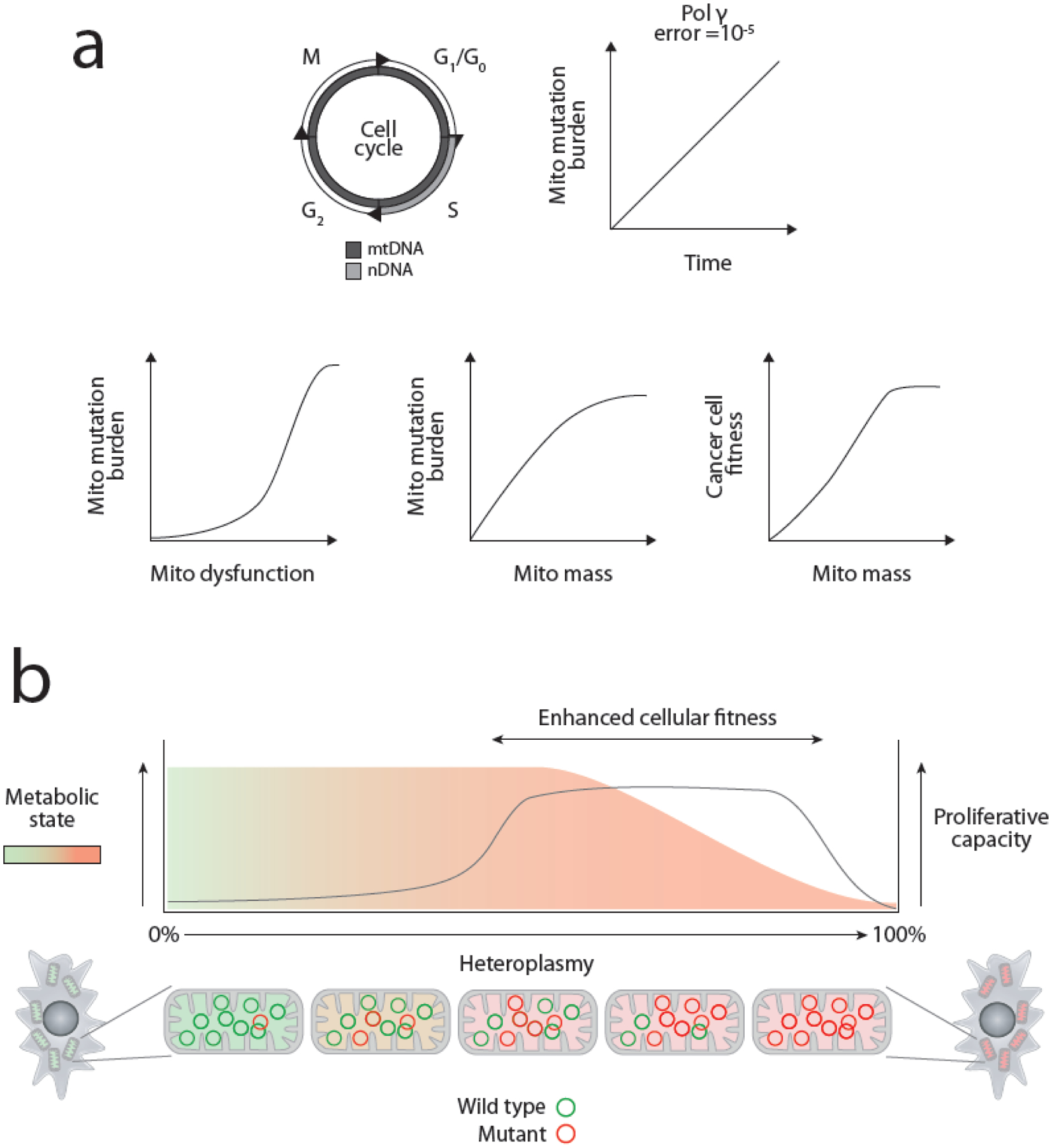
(A) Unlike nuclear DNA, mtDNA is replicated both during S phase and throughout all stages of the cell cycle. While the misincorporation rate of the mitochondrial replicative polymerase (Pol γ) is considered very low, the impact of continuous replication is such that mitochondrial mutational heterogeneity is seeded throughout the body in a time-dependent fashion, in agreement with both the strand asymmetric distribution of mutations (due to mode of mtDNA replication) and type of mutations found in cancers. Cells bearing mtDNA mutations in proliferative neoplasms acquire a fitness advantage due to increased capacity for anabolism and proliferation due to enhanced mitochondrial mass, a common response to mitochondrial dysfunction. The coupling of mtDNA mutations to increases in mitochondrial mass and consequent cellular fitness provides a rational framework for the consistent selection of mitochondrial mutations by cancer cells. (B) A visual representation of the proposed framework for selection of mtDNA mutations in cancer.
Outstanding Questions.
How does tissue lineage modulate the phenotype of a given mtDNA allele?
How does heteroplasmic dosage mediate the phenotype of a mutation?
What are the pan-cancer and lineage specific impacts of mtDNA mutations on tumor formation?
What is the full spectrum of non-metabolic phenotypes produced by mtDNA mutations?
Highlights.
Somatic mutations to mtDNA in cancers are abundant, but their selection is highly gene- and context-dependent
Truncating and tRNA mutations to mtDNA are drivers in certain diseases, but the function of the vast majority of somatic mtDNA variants is uncharacterized
Heteroplasmic dosage is likely a critical determinant of the phenotype produced by somatic mtDNA mutations
New techniques for single cell profiling technologies and mitochondrial genome editing overcome the key obstacles to delineating the function of mtDNA mutations in cancer
Glossary
- Clone
Cluster of cells derived from a common ancestor with a similarly inherited nuclear genome.
- dN/dS
the ratio of non-synonymous to synonymous mutations, in which a value greater than 1 suggests positive selective pressure and a value less than 1 suggests negative selective pressure.
- Control region
Approximately 1122-bp long non-coding region within the mitochondrial genome that includes one of the origins of replication, promoter sequences, and the D-loop. It is a region with the highest rate of polymorphisms observed in the mitochondrial genome.
- Cybrids
Cells engineered by fusing mtDNAs from a donor cell to a different, recipient cell of the same nuclear background.
- DdCBE
DddA-derived cytosine base editors are capable of inducing targeted C>T mtDNA point mutations in a sequence-specific manner.
- Driver mutation
a term used to describe a mutation that is advantageous to the fitness of cancer cells and can undergo positive selection
- Heteroplasmy
The fraction of the mutant mitochondrial genomes in the total mtDNA pool of a given tissue specimen.
- Heteroplasmy threshold effect
a critical threshold in mutational load beyond which a deleterious variant can trigger mitochondrial and cellular dysfunctions that ultimately lead to emergence of diseases. Passenger mutation: a term used to describe a mutation that does not alter the fitness of cancer cells and undergoes neutral selection.
- Meganuclease
Small, monomeric enzyme that recognizes 22bp sequences within the DNA to induce a double-stranded break.
- mitoAPOBEC1
can induce random C>T point mutations across mtDNA without inducing significant indels or depletions of mtDNA copy number.
- mitoTALEN
mitochondrially-targeted transcription activator-like effector nucleases are engineered to bind to specific regions of mtDNA via their TALE domains to induce changes in heteroplasmy levels.
- mtRE
Mitochondrially-targeted restriction enzymes are bacterially-derived restriction enzymes that can robustly induce changes in heteroplasmy.
- mtZFN
mitochondrially-targeted zinc-finger nucleases are engineered to bind to specific regions of mtDNA via their zinc-finger domain to induce changes in heteroplasmy levels.
- Selective pressure
Force that provides growth advantage to cells with genetic variation over other cells. It can be negative, in which the variant is disadvantageous (decreases the fitness of the cell) or positive, in which the variant is advantageous (increases the fitness of the cell).
- Sequencing depth
the number of sequencing reads covering the position.
- Truncating mutations
mutations that introduce a stop codon within a protein sequence producing a truncated variant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife. 2014;3. doi: 10.7554/eLife.02935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hertweck KL, Dasgupta S. The Landscape of mtDNA Modifications in Cancer: A Tale of Two Cities. Frontiers in Oncology. 2017. doi: 10.3389/fonc.2017.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhidkov I, Livneh EA, Rubin E, Mishmar D. MtDNA mutation pattern in tumors and human evolution are shaped by similar selective constraints. Genome Res. 2009;19: 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hess JM, Bernards A, Kim J, Miller M, Taylor-Weiner A, Haradhvala NJ, et al. Passenger Hotspot Mutations in Cancer. Cancer Cell. 2019;36: 288–301.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell. 2018;173: 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gammage PA, Moraes CT, Minczuk M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends in Genetics. 2018. pp. 101–110. doi: 10.1016/j.tig.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva-Pinheiro P, Nash PA, Van Haute L, Mutti CD, Turner K, Minczuk M. In vivo mitochondrial base editing via adeno-associated viral delivery to mouse post-mitotic tissue. Nat Commun. 2022;13: 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gammage PA, Viscomi C, Simard M-L, Costa ASH, Gaude E, Powell CA, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med. 2018;24: 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gammage PA, Gaude E, Van Haute L, Rebelo-Guiomar P, Jackson CB, Rorbach J, et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016;44: 7804–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Wu H, Kang X, Liang Y, Lan T, Li T, et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell. 2018;9: 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mok BY, Kotrys AV, Raguram A, Huang TP, Mootha VK, Liu DR. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat Biotechnol. 2022. doi: 10.1038/s41587-022-01256-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martijn J, Vosseberg J, Guy L, Offre P, Ettema TJG. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature. 2018;557: 101–105. [DOI] [PubMed] [Google Scholar]
- 13.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290: 457–465. [DOI] [PubMed] [Google Scholar]
- 14.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23: 147. [DOI] [PubMed] [Google Scholar]
- 15.Kotrys AV, Szczesny RJ. Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology. Cells. 2019;9: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clay Montier LL, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics. 2009;36: 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nature Reviews Genetics. 2015. pp. 530–542. doi: 10.1038/nrg3966 [DOI] [PubMed] [Google Scholar]
- 18.Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77: 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2: 16080. [DOI] [PubMed] [Google Scholar]
- 20.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat J-P, Letellier T. Mitochondrial threshold effects. Biochemical Journal. 2003. pp. 751–762. doi: 10.1042/bj20021594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kullar PJ, Gomez-Duran A, Gammage PA, Garone C, Minczuk M, Golder Z, et al. Heterozygous SSBP1 start loss mutation co-segregates with hearing loss and the m.1555A>G mtDNA variant in a large multigenerational family. Brain. 2018. pp. 55–62. doi: 10.1093/brain/awx295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boulet L, Karpati G, Shoubridge EA. Distribution and threshold expression of the tRNA(Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1992;51: 1187–1200. [PMC free article] [PubMed] [Google Scholar]
- 23.Masucci JP, Davidson M, Koga Y, Schon EA, King MP. In vitro analysis of mutations causing myoclonus epilepsy with ragged-red fibers in the mitochondrial tRNA(Lys)gene: two genotypes produce similar phenotypes. Mol Cell Biol. 1995;15: 2872–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warburg O On the origin of cancer cells. Science. 1956;123: 309–314. [DOI] [PubMed] [Google Scholar]
- 25.Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell. 2016;166: 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganapathy-Kanniappan S Molecular intricacies of aerobic glycolysis in cancer: current insights into the classic metabolic phenotype. Crit Rev Biochem Mol Biol. 2018;53: 667–682. [DOI] [PubMed] [Google Scholar]
- 27.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metabolism. 2016. pp. 27–47. doi: 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel PH, Chadalavada RSV, Chaganti RSK, Motzer RJ. Targeting von Hippel-Lindau Pathway in Renal Cell Carcinoma: Fig. 1. Clinical Cancer Research. 2006. pp. 7215–7220. doi: 10.1158/1078-0432.ccr-06-2254 [DOI] [PubMed] [Google Scholar]
- 29.Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277: 23111–23115. [DOI] [PubMed] [Google Scholar]
- 30.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4: 891–899. [DOI] [PubMed] [Google Scholar]
- 31.Harris AL. Hypoxia — a key regulatory factor in tumour growth. Nature Reviews Cancer. 2002. pp. 38–47. doi: 10.1038/nrc704 [DOI] [PubMed] [Google Scholar]
- 32.Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet. 2020;52: 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20: 291–293. [DOI] [PubMed] [Google Scholar]
- 34.Triska P, Kaneva K, Merkurjev D, Sohail N, Falk MJ, Triche TJ Jr, et al. Landscape of Germline and Somatic Mitochondrial DNA Mutations in Pediatric Malignancies. Cancer Res. 2019;79: 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorelick AN, Kim M, Chatila WK, La K, Hakimi AA, Berger MF, et al. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nature Metabolism. 2021;3: 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parsons TJ, Muniec DS, Sullivan K, Woodyatt N, Alliston-Greiner R, Wilson MR, et al. A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet. 1997;15: 363–368. [DOI] [PubMed] [Google Scholar]
- 37.Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radical Biology and Medicine. 1999. pp. 463–471. doi: 10.1016/s0891-5849(98)00216-0 [DOI] [PubMed] [Google Scholar]
- 38.Richter C Oxidative damage to mitochondrial DNA and its relationship to ageing. The International Journal of Biochemistry & Cell Biology. 1995. pp. 647–653. doi: 10.1016/1357-2725(95)00025-k [DOI] [PubMed] [Google Scholar]
- 39.Kauppila JHK, Bonekamp NA, Mourier A, Isokallio MA, Just A, Kauppila TES, et al. Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Research. 2018. pp. 6642–6669. doi: 10.1093/nar/gky456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9: e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gammage PA, Frezza C. Mitochondrial DNA: the overlooked oncogenome? BMC Biology. 2019. doi: 10.1186/s12915-019-0668-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart JB, Alaei-Mahabadi B, Sabarinathan R, Samuelsson T, Gorodkin J, Gustafsson CM, et al. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLOS Genetics. 2015. p. e1005333. doi: 10.1371/journal.pgen.1005333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, et al. Oxidative Stress Is Not a Major Contributor to Somatic Mitochondrial DNA Mutations. PLoS Genetics. 2014. p. e1003974. doi: 10.1371/journal.pgen.1003974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otten ABC, Stassen APM, Adriaens M, Gerards M, Dohmen RGJ, Timmer AJ, et al. Replication Errors Made During Oogenesis Lead to Detectable De Novo mtDNA Mutations in Zebrafish Oocytes with a Low mtDNA Copy Number. Genetics. 2016;204: 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falkenberg M, Larsson N-G, Gustafsson CM. DNA Replication and Transcription in Mammalian Mitochondria. Annual Review of Biochemistry. 2007. pp. 679–699. doi: 10.1146/annurev.biochem.76.060305.152028 [DOI] [PubMed] [Google Scholar]
- 46.Bogenhagen D, Clayton DA. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell. 1977;11: 719–727. [DOI] [PubMed] [Google Scholar]
- 47.Lynch M The Lower Bound to the Evolution of Mutation Rates. Genome Biology and Evolution. 2011. pp. 1107–1118. doi: 10.1093/gbe/evr066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker MA, Lareau CA, Ludwig LS, Karaa A, Sankaran VG, Regev A, et al. Purifying Selection against Pathogenic Mitochondrial DNA in Human T Cells. New England Journal of Medicine. 2020. pp. 1556–1563. doi: 10.1056/nejmoa2001265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filograna R, Koolmeister C, Upadhyay M, Pajak A, Clemente P, Wibom R, et al. Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci Adv. 2019;5: eaav9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwok AWC, Qiao C, Huang R, Sham M-H, Ho JWK, Huang Y. MQuad enables clonal substructure discovery using single cell mitochondrial variants. Nat Commun. 2022;13: 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller TE, Lareau CA, Verga JA, DePasquale EAK, Liu V, Ssozi D, et al. Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat Biotechnol. 2022. doi: 10.1038/s41587-022-01210-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell. 2019;176: 1325–1339.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu J, Nuno K, Litzenburger UM, Qi Y, Corces MR, Majeti R, et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. Elife. 2019;8. doi: 10.7554/eLife.45105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Penter L, Gohil SH, Lareau C, Ludwig LS, Parry EM, Huang T, et al. Longitudinal Single-Cell Dynamics of Chromatin Accessibility and Mitochondrial Mutations in Chronic Lymphocytic Leukemia Mirror Disease History. Cancer Discov. 2021;11: 3048–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lareau CA, Ludwig LS, Muus C, Gohil SH, Zhao T, Chiang Z, et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol. 2021;39: 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grandhi S, Bosworth C, Maddox W, Sensiba C, Akhavanfard S, Ni Y, et al. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum Mol Genet. 2017;26: 2912–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nielsen R Molecular Signatures of Natural Selection. Annual Review of Genetics. 2005. pp. 197–218. doi: 10.1146/annurev.genet.39.073003.112420 [DOI] [PubMed] [Google Scholar]
- 60.Simonnet H, Demont J, Pfeiffer K, Guenaneche L, Bouvier R, Brandt U, et al. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis. 2003;24: 1461–1466. [DOI] [PubMed] [Google Scholar]
- 61.Gasparre G, Hervouet E, de Laplanche E, Demont J, Pennisi LF, Colombel M, et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum Mol Genet. 2008;17: 986–995. [DOI] [PubMed] [Google Scholar]
- 62.Mayr JA, Meierhofer D, Zimmermann F, Feichtinger R, Kögler C, Ratschek M, et al. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res. 2008;14: 2270–2275. [DOI] [PubMed] [Google Scholar]
- 63.Larman TC, DePalma SR, Hadjipanayis AG, Cancer Genome Atlas Research Network, Protopopov A, Zhang J, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci U S A. 2012;109: 14087–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65: 1655–1663. [DOI] [PubMed] [Google Scholar]
- 65.Ishikawa K, Koshikawa N, Takenaga K, Nakada K, Hayashi J-I. Reversible regulation of metastasis by ROS-generating mtDNA mutations. Mitochondrion. 2008;8: 339–344. [DOI] [PubMed] [Google Scholar]
- 66.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320: 661–664. [DOI] [PubMed] [Google Scholar]
- 67.Park JS, Sharma LK, Li H, Xiang R, Holstein D, Wu J, et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet. 2009;18: 1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gopal RK, Kübler K, Calvo SE, Polak P, Livitz D, Rosebrock D, et al. Widespread Chromosomal Losses and Mitochondrial DNA Alterations as Genetic Drivers in Hürthle Cell Carcinoma. Cancer Cell. 2018;34: 242–255.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ganly I, Makarov V, Deraje S, Dong Y, Reznik E, Seshan V, et al. Integrated Genomic Analysis of Hürthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes. Cancer Cell. 2018;34: 256–270.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mullen AR, Wheaton WW, Jin ES, Chen P-H, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martínez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature. 2020;585: 288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith ALM, Whitehall JC, Bradshaw C, Gay D, Robertson F, Blain AP, et al. Author Correction: Age-associated mitochondrial DNA mutations cause metabolic remodeling that contributes to accelerated intestinal tumorigenesis. Nat Cancer. 2021;2: 129. [DOI] [PubMed] [Google Scholar]
- 73.Hopkins JF, Sabelnykova VY, Weischenfeldt J, Simon R, Aguiar JA, Alkallas R, et al. Mitochondrial mutations drive prostate cancer aggression. Nat Commun. 2017;8: 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schöpf B, Weissensteiner H, Schäfer G, Fazzini F, Charoentong P, Naschberger A, et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat Commun. 2020;11: 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao Z, Yuan Y-S. Screening for mitochondrial 12S rRNA C1494T mutation in 655 patients with non-syndromic hearing loss: An observational study. Medicine. 2020;99: e19373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med. 2013;19: 1111–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M. Mitochondrially targeted ZFN s for selective degradation of pathogenic mitochondrial genomes bearing large‐ scale deletions or point mutations. EMBO Molecular Medicine. 2014. pp. 458–466. doi: 10.1002/emmm.201303672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bacman SR, Kauppila JHK, Pereira CV, Nissanka N, Miranda M, Pinto M, et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nature Medicine. 2018. pp. 1696–1700. doi: 10.1038/s41591-018-0166-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mok S, Stokes BH, Gnädig NF, Ross LS, Yeo T, Amaratunga C, et al. Artemisinin-resistant K13 mutations rewire Plasmodium falciparum’s intra-erythrocytic metabolic program to enhance survival. Nature Communications. 2021. doi: 10.1038/s41467-020-20805-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gammage PA, Van Haute L, Minczuk M. Engineered mtZFNs for Manipulation of Human Mitochondrial DNA Heteroplasmy. Methods in Molecular Biology. 2016. pp. 145–162. doi: 10.1007/978-1-4939-3040-1_11 [DOI] [PubMed] [Google Scholar]
- 81.Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet. 2001;10: 3093–3099. [DOI] [PubMed] [Google Scholar]
- 82.Tanaka M, Borgeld H-J, Zhang J, Muramatsu S-I, Gong J-S, Yoneda M, et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci. 2002;9: 534–541. [DOI] [PubMed] [Google Scholar]
- 83.Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet. 1996;14: 146–151. [DOI] [PubMed] [Google Scholar]
- 84.Peeva V, Blei D, Trombly G, Corsi S, Szukszto MJ, Rebelo-Guiomar P, et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat Commun. 2018;9: 1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nicholls TJ, Zsurka G, Peeva V, Schöler S, Szczesny RJ, Cysewski D, et al. Linear mtDNA fragments and unusual mtDNA rearrangements associated with pathological deficiency of MGME1 exonuclease. Hum Mol Genet. 2014;23: 6147–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao L Mitochondrial DNA degradation: A quality control measure for mitochondrial genome maintenance and stress response. Enzymes. 2019;45: 311–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gaude E, Schmidt C, Gammage PA, Dugourd A, Blacker T, Chew SP, et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol Cell. 2018;69: 581–593.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andreazza S, Samstag CL, Sanchez-Martinez A, Fernandez-Vizarra E, Gomez-Duran A, Lee JJ, et al. Mitochondrially-targeted APOBEC1 is a potent mtDNA mutator affecting mitochondrial function and organismal fitness in Drosophila. Nat Commun. 2019;10: 3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429: 417–423. [DOI] [PubMed] [Google Scholar]
- 90.Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tanaka M, Borgeld H-J, Zhang J, Muramatsu S-I, Gong J-S, Yoneda M, et al. Gene therapy for mitochondrial disease by delivering restriction endonucleaseSmaI into mitochondria. Journal of Biomedical Science. 2002. pp. 534–541. doi: 10.1007/bf02254980 [DOI] [PubMed] [Google Scholar]
- 92.Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci U S A. 2005;102: 14392–14397. [DOI] [PMC free article] [PubMed] [Google Scholar]