

Abstract
Objective.
To investigate whether Ccr2 inactivation in aggrecan-expressing cells induced before PTOA onset or during progression, improves joint structures, synovial thickness and pain.
Design.
We induced a Ccr2 deletion in aggrecan-expressing cells (CCR2-AggKO) in skeletally mature mice using a tamoxifen-inducible Ccr2 inactivation.. We stimulated PTOA changes (DMM) in CCR2-AggKO and CCR2+/+ mice, inducing recombination before DMM or 4wks after DMM (early- vs late-inactivation). Joint damage was evaluated 2, 4, 8, 12wks post-DMM using multiple scores: articular-cartilage structure (ACS), Safranin-O, histomorphometry, osteophyte size/maturity, subchondral bone thickness and synovial hyperplasia. Spontaneous (incapacitance meter) and evoked pain (von-Frey filaments) were assessed up to 20wks.
Results.
Early aggrecan-Ccr2 inactivation in CCR2-AggKO mice (N=8) resulted in improved ACS score (8–12wk, p=0.002), AC area (4–12wk, p<0.05) and Saf-O score (2wks p=0.004, 4wks p=0.02, 8–12wks p=0.002) compared to CCR2+/+. Increased subchondral bone thickness was delayed only at 2wks and exclusively following early recombination. Osteophyte size was not affected, but osteophyte maturation (cartilage-to-bone) was delayed (4wks p=0.04; 8wks p=0.03). Although late aggrecan-Ccr2 deletion led to some cartilage improvement, most data did not reach statistical significance; osteophyte maturity was delayed at 12wks. Synovial thickness did not change in all experiments. Early aggrecan-Ccr2 deletion led to improved pain measures of weight bearing compared to CCR2+/+ mice (N=9, 12wks diff 0.13 [0.01, 0.26], 16wks diff 0.15[0.05, 0.26], 20wks diff 0.23 [0.14, 0.31]). Improved mechanosensitivity in evoked pain, although less noticeable, was detected.
Conclusions.
We demonstrated that deletion of Ccr2 in aggrecan expressing cells reduces the initiation but not progression of OA.
Keywords: Chemokines, Osteoarthritis, Joint disease, Animal Model, Cartilage, Bone
Introduction
Increased expression of chemokines in joint tissues are believed to be linked to osteoarthritis (OA) progression[1]. This work focuses on the specific targeting of CC-chemokine Receptor-2 (CCR2) during post-traumatic OA (PTOA); our interest stems from previous mouse developmental studies where we found that abnormal CCR2 signal in the forming joint leads to Collagen-2 dysregulation and affects proper limb ossification, suggesting that similar compartment-specific regulation might be related to OA progression, involving both chondrocytes and osteoblasts[2]. CCR2 is expressed in several tissues, including chondrocytes and osteoblasts[3, 4] and has been recognized as an important potential target in OA; CCR2 and its ligands CCL2 (a.k.a. MCP1) and CCL12 (a.k.a. MCP-5) are significantly increased in both rodent OA models and in humans with OA and have been shown to mediate OA pain[5–11]. In previous clinical studies, we determined that increasing CCL2 serum levels at baseline were associated with radiographic knee OA progression and joint space narrowing at 5-year follow up[7]. Previous animal studies, including ours, have demonstrated that systemic pharmacological inactivation of CCR2 successfully prevented OA progression and pain[5, 8]. However, our previous studies using the destabilization of medial meniscus (DMM) post-traumatic OA (PTOA) model, demonstrated that the efficacy of CCR2 systemic targeting was stage-specific and dependent on treatment length. Early and transient CCR2 inhibition improved cartilage and bone structures, while continuous CCR2 inhibition initiated at joint injury, as well as delayed treatments, did not improve PTOA outcomes, but alleviated pain. These results suggest that joint tissues might respond in a temporal specific manner to CCR2 stimulation[8]. We also showed that during DMM, protein levels of the CCR2 ligand CCL12, are detectable in articular cartilage (AC) in OA lesions, increasing with OA severity, but undetectable in controls[8]. Furthermore, our in-vitro studies in human chondrocytes highlighted how a certain CCL2 balance was critical for selective activation of cartilage degradation markers, suggesting an important role of CCR2 in cartilage degeneration[12].
Therefore, we decided to specifically target CCR2 in cartilage and determine both its preventive and therapeutic effects on the whole joint degeneration. To this scope, we used an inducible aggrecan-specific Ccr2 inactivation, inducing recombination before DMM (preventive) or during OA progression (therapeutic), and followed disease modifications in cartilage, bone and synovium. Although the beneficial systemic targeting of CCR2 on pain management has been demonstrated, we are the first to report the direct contribution of cartilage levels of CCR2 with joint structure and its correlation to pain perception[8, 9, 13]. This information is critical to the future development of CCR2 as a target for both pain and structure in PTOA.
Method
Animals
Animal protocols follow ARRIVE guidelines and were approved by UNC Animal Care and Use Committee. Aggrecan-CreERT2 mice have an inducible Cre recombinase integrated in the 3’-UTR of the endogenous aggrecan gene[14]. In the CCR2-floxed mice (CCR2flx/flxeGFP), the exon 3 of the Ccr2 allele is flanked by loxP sites, followed by an eGFP cassette[15]. Mice were on a C57BL/6 background.
We crossed AggCreERT2 with CCR2flx/flx-eGFP mice to obtain CCR2flx/flx-eGFP/AggCreERT2-positive and CCR2flx/flx-eGFP/AggCreERT2-negative. We induced Cre recombination in CCR2flx/flx-eGFP/AggCreERT2-positive by tamoxifen injection to obtain a mouse with Ccr2 inactivation in aggrecan-expressing cells (referred as CCR2-AggKO). Injections were performed either 2-weeks (wks) before surgery (to achieve an early CCR2 inactivation) or 4wks after surgery (for late CCR2 inactvation). Mice were euthanized for OA assessment at 2, 4, 8 and 12wks post-surgery for the early CCR2 inactivation group or 8 and 12wks for the late CCR2 inactivation. Tamoxifen-injected CCR2flx/flx-eGFP/AggCreERT2-negative mice, where the Cre is not expressed, were used as controls (CCR2+/+) (details in Supplemental Methods).
Induction of OA
DMM/Sham surgery was induced in the right leg of sixteen-week-old CCR2-AggKO or CCR2+/+ male mice by transecting the menisco-tibial ligament, as previously described[16–18]. In the Sham, the ligament is visualized and left untouched (Supplemental Methods). Mice subjected to DMM/Sham were assessed for pain behaviors up to 20wks post-surgery. In a parallel set of experiments, dissected knees from mice at 2, 4, 8 and 12wks post-surgery were fixed and prepared for histology.
Behavioral pain assessment
Spontaneous hindlimb weight distribution was measured with an incapacitance meter as previously reported[19];the static weight distribution on the two hind paws was measured as the force exerted by each limb on a plate over a given time period. We used a ratio between the two paws (Left/un-operated vs Right/operated) as measure of pain. Higher ratios resulting from decreased weight bearing on the operated knee indicate increased pain. Evoked pain was assessed by application of von Frey filaments to the plantar surface of the hindpaw to determine a mechanosensitivity threshold. A mouse responding to a filament of less force (grams) was more sensitive (Supplemental Methods).
Histopathologic assessment of arthritis
Dissected knees were prepared and assessed. For OA grading, we used the AC Structure score (ACS, scale 0–12) and the Safranin-O staining score (Saf-O, scale 0–12), to provide in-depth information regarding changes within the lesions, for both AC structure and extracellular matrix integrity[20] (Supplemental Methods). Results were expressed as the average of scores in all quadrants in all sections.
Histomorphometric analyses using Image-J were performed to quantify area of AC and subchondral bone[21], from mice used for ACS/Saf-O semiquantitative assessments[8] (Supplemental Methods).
Osteophyte assessment.
We used the scoring system developed to assess osteophyte size and maturity, with the latter reflecting the osteophyte tissue composition[22] (Supplemental Methods). Histomorphometric analysis were performed to quantify area of cartilage in the osteophytes and expressed as percentage of the total osteophyte areas (Image-J, Supplemental Methods[21].
Because osteophytes in the DMM are predominately localized on the medial-side, only sections from this region were graded.
Immunohistochemistry (IHC) studies
Sections adjacent to those used for histopathologic assessment were used for IHC staining for CCR2, Col10 and GFP (Supplemental Methods). As control, representative sections were incubated without primary antibody, to exclude non-specific binding.
Images were taken with an Olympus BX51 microscope and a DP71 camera.
Assessment of synovial thickness
One H&E stained section/mouse from the posterior joint compartment was scored for the synovial hyperplasia based on the scoring system described by Rowe et al. scale of 0–3)[23] (Supplemental Methods). Joint medial and lateral compartments were scored separately.
All scores/measures were acquired in a blinded manner by three independent investigators, and results were expressed as an average.
Statistical analysis
Statistical analyses were performed using SAS v 9.4 (SAS Institute) and based on a 0.05 significance level. Shapiro-Wilk and Levene’s tests were used for assessing normality of data and homogeneity of variance. Due to the violation of normality assumption in most outcomes, which did not improved after the logarithmic or square root transformation, non-parametric Wilcoxon rank sum tests following Benjamini and Hochberg adjustment for p-values in multiple comparisons were performed to examine differences in mean ACS, Saf-O, histomorphometry among groups. Analyses were performed separately at 2, 4, 8, and 12wks.
For osteophyte size/maturity and synovial hyperplasia Wilcoxon tests were used.
For longitudinal von Frey and incapacitance measures, linear mixed effects models were fit with unstructured covariance structure to account for repeated measures over time. Fixed-effect terms included indicators for group (DMM or Sham, for each genotype, and left/right for von Frey measurements), time in weeks and their interaction. To account for von Frey measurements on both knees collected for each mouse, mouse random effect was included in the model. Model assumption of normality of residuals was checked visually with Q-Q plot. Equal variance of residuals was checked with a plot of residuals vs. fitted values. For von Frey, a square root transformation was used to achieve normality. Mice missing follow-up measurements were still included in the model under a ‘missing at random’ paradigm. Proc plm was used to estimate comparisons of interest by specifying differences between means. Plots were generated using GraphPad Prism Software (9.1.0).
Results
CCR2 inactivation in aggreacan-expressingjoint tissues.
We successfully generated inducible CCR2-AggKO mice, by injecting tamoxifen in CCR2flx/flx-eGFP/AggCreERT2-positive mice, where Cre recombination leads to Gfp expression. Injected CCR2flx/flx-eGFP/AggCreERT2-negative mice were used as Controls (CCR2+/+). Figure-1 shows the presence of GFP protein staining in the AC and meniscus of CCR2-AggKO 2wks after first injection, while it was undetectable in the CCR2-AggKO. To confirm the CCR2 inactivation, we assessed protein expression levels of CCR2 in the AC and meniscus and, as expected, we found CCR2 staining in both compartments of CCR2+/+ mice but not in CCR2-AggKO (Fig.-1 A–C). To understand the contribution from non-articular cartilage CCR2-expressing tissues within the joint, we also assessed protein levels of CCR2 and GFP in ligaments, bone and synovial capsule. We found no detectable expression of GFP in ligaments (Supplemental Fig. S1), bone (Supplemental Fig. S2), or in the lining cells of the synovium (Supplemental Fig. S3) as confirmed by the positive staining of CCR2 in both CCR2+/+ and CCR2-AggKO mice.
Figure 1. Protein levels of CCR2 and GFP in the AC of CCR2-AggKO and CCR2+/+ mice following Tam injections.
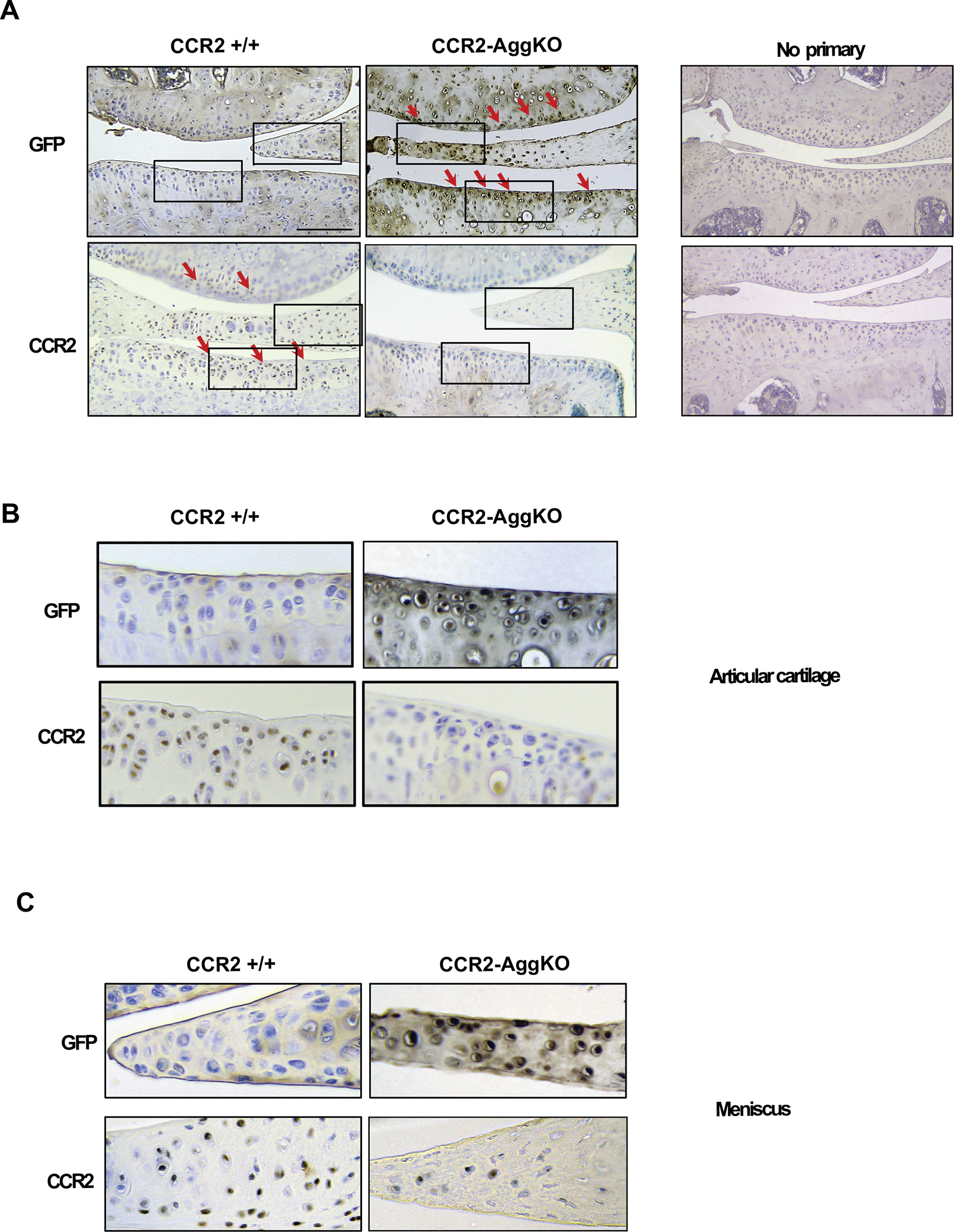
(A) Paraffin embedded knee joint sections are immunostained for CCR2 and GFP, two weeks after the first Tam injection. Positive staining is detected as brown precipitate (red arrows). CCR2 staining is detected in the AC of CCR2+/+ mice, while is absent in CCR2-AggKO. Conversely, GFP staining is visible in CCR2-AggKO (red arrows) but undetected in CCR2+/+ mice. (B) Images represent a magnification of the rectangle in the articular cartilage defined in panel A. (C) Images represent a magnification of the rectangle in the meniscus defined in panel A. Images are representative of 6 different mice for each of the experimental groups described, ranging between 14 and 18 weeks of age. Scale bars are 100 μm.
Early -CCR2 inactivation in aggrecan-expressing cells decreases PTOA cartilage damage.
We induced Cre recombination in CCR2flx/flx-eGFP/AggCreERT2-positive mice before DMM to obtain CCR2-AggKO and followed PTOA damage in cartilage, bone and synovium at mild (2, 4wks), moderate (8wks) and severe (12wks) stages. To provide a comprehensive characterization of the cartilage lesions, we used both the ACS and Saf-O semiquantitative scores[20]. In Figure-2A–B, early Ccr2 inactivation leads to lower ACS score at moderate/severe OA stages compared to the CCR2+/+; although a decrease in ACS score is detected also at mild stages (Table-1), results were not statistically significant. Differently from ACS score, changes in Saf-O in CCR2+/+ DMM mice are detected as early as 2wks post-surgery (median=2.00), compared to Sham controls (median=0.38), indicating a rapid deterioration of the extracellular matrix (Fig.-2C–D). Interestingly, DMM CCR2-AggKO mice showed improvement of the extracellular matrix at all stages, compared to DMM CCR2+/+ (Fig. 2C–D, Table-1). We also performed AC histomorphometric analysis to quantify cartilage loss. Starting 4wks post-surgery, CCR2-AggKO DMM mice showed increased AC area compared to CCR2+/+ DMM (Fig.2E, Table-1).
Figure 2. Histopathological evaluation of CCR2-AggKO and CCR2+/+ mouse knees, following early CCR2 inactivation.
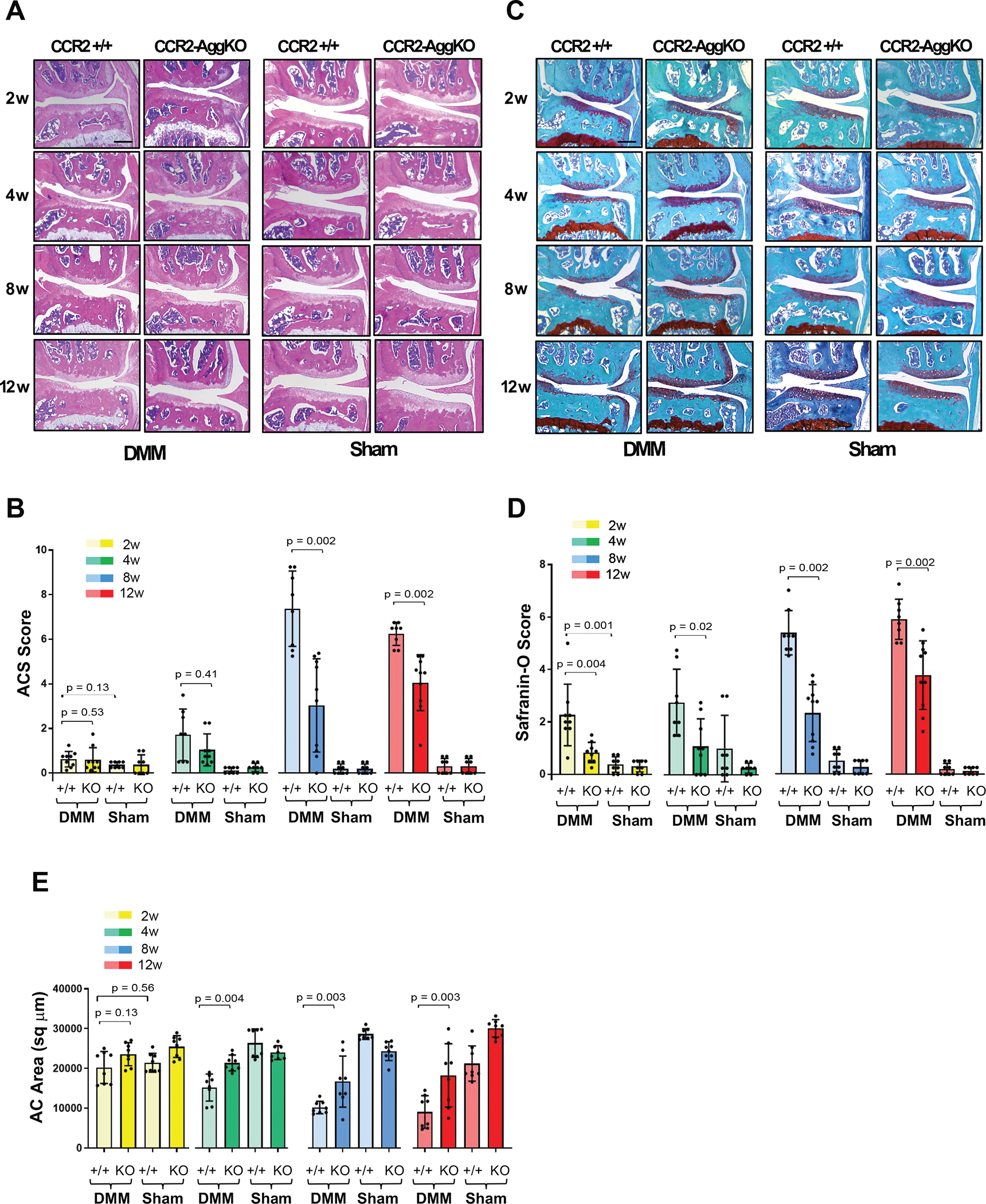
(A) H&E staining of the knee medial compartment from CCR2-AggKO and CCR2+/+ mice after DMM/Sham (time indicated); images are representative of N=8 for each time point. (B) ACS score (0–12 scale) of DMM/Sham knees at the time point indicated. Results are expressed as average of 4 quadrants (medial and lateral tibial plateau, medial and lateral femoral condyles); N=8 mice for each experimental point. (C) Safranin-O/Fast green staining of the knee medial compartment after DMM/Sham (time indicated); images are representative of N=8 for each time points. (D) Safranin-O staining Score (0–12 scale) of DMM/Sham knees at the time point indicated. Results are expressed as average of 4 quadrants as described above; N=8 mice for each experimental point. (E) Quantification of the AC area (sq μm) of the medial plateau of DMM/Sham knees by histomorphometric analysis at the time point indicated; N=8 mice for each experimental point. The graphs represents the mean ± standard deviation Indicated p-values were determined by Wilcoxon rank sum tests at each time point, following adjustment for multiple comparisons. Scale bars of the images are 100 μm.
Table 1.
Effect of early CCR2 inactivation (before DMM) on OA assessment and pain in mouse CCR2-AggKO and CCR2+/+ knee joints
| OA parameters | 2 weeks | 4 weeks | 8 weeks | 12 weeks | |||||
| Z (pvalue*) | median | Z (pvalue) | median | Z (pvalue) | median | Z (pvalue) | median | ||
| ACS | DMM CCR2+/+ | 0.63 (0.53) | 0.75 | 1.03 (0.41) | 1.75 | 3.38 (0.002) | 7.63 | 3.52 (0.002) | 6.50 |
| DMM CCR2-AggKO | 0.5 | 0.75 | 3.50 | 4.50 | |||||
| Saf-o | DMM CCR2+/+ | 3.01 (0.004) | 2.00 | 2.42 (0.02) | 2.38 | 3.52 (0.002) | 5.25 | 3.21 (0.002) | 5.88 |
| DMM CCR2-AggKO | 0.89 | 1.00 | 2.75 | 4.00 | |||||
| Cartilage Quantification | DMM CCR2+/+ | −1.52 (0.13) | 20927 | −3.20 (0.004) | 16083 | −2.26 (0.03) | 9712 | −3.20 (0.03) | 8811 |
| DMM CCR2-AggKO | 23874 | 21128 | 15589 | 19264 | |||||
| Osteophyte size | DMM CCR2+/+ | 0.15 (0.88) | 0.63 | 2.02 (0.16) | 1.13 | −1.03 (0.60) | 1.00 | 0.25 (0.88) | 1.13 |
| DMM CCR2-AggKO | 0.63 | 0.88 | 1.25 | 1.00 | |||||
| Osteophyte maturity | DMM CCR2+/+ | 0.77 (0.44) | 0.50 | 3.04 (0.04) | 1.25 | 2.65 (0.03) | 1.25 | 0.98 (0.44) | 1.00 |
| DMM CCR2-AggKO | 0.50 | 0.75 | 1.00 | 0.86 | |||||
| Subchondral bone thickness | DMM CCR2+/+ | 2.47 (0.02) | 3740 | 0.05 (0.96) | 3544 | 1.84 (0.09) | 6356 | 2.47 (0.02) | 6547 |
| DMM CCR2-AggKO | 3127 | 3421 | 4789 | 4705 | |||||
| Osteophyte Cartilage Quantification | DMM CCR2+/+ | 0.00 (1.00) | 48.00 | −3.33 (0.004) | 19.5 | −2.89 (0.008) | 15.50 | −0.05 (1.00) | 16.00 |
| DMM CCR2-AggKO | 46.05 | 38.90 | 41.00 | 17.00 | |||||
| Pain | Incapacitance Meter | Von-Frey Filaments* | |||||||
| Time PO/Stage | DMM CCR2+/+ vs. DMM CCR2-AggKO | DMM CCR2+/+ vs. DMM CCR2-AggKO | DMM CCR2+/+ vs. Contralateral CCR2+/+ | DMM CCR2-AggKO vs. Contralateral -CCR2-AggKO | |||||
| 4 wks/mild | 0.10 (0.00, 0.19) | 0.21 (−0.22,0.64) | 0.62 (0.13,1.11) | 0.2 (−0.21,0.61) | |||||
| 8 wks/moderate | 0.10 (−0.02, 0.23) | 0.11 (−0.32,0.55) | 0.38 (−0.11,0.87) | 0.37 (−0.09,0.83) | |||||
| 12 wks/severe | 0.13 (0.01, 0.26) | −0.25 (−0.61,0.11) | 0.55 (0.19,0.91) | 0.28 (−0.1,0.66) | |||||
| 16 wks/severe | 0.15 (0.05, 0.26) | 0.26 (−0.37,0.89) | 0.68 (0.3,1.07) | 0.67 (0.28,1.06) | |||||
| 20 wks/severe | 0.23 (0.14, 0.31) | −0.42 (−0.81,−0.04) | 1.00 (0.58,1.41) | 0.67 (0.19,1.15) | |||||
OA parameters: Data presented show median for each group at separate time points. Indicated z- and p-values were determined by Wilcoxon rank sum tests between DMM CCR+/+ and dMm CCR-AggKO groups separately at each time point, following adjustment for multiple comparisons (Benjamini-Hochberg). For more complete information, see Supplementary Table X.
Pain: Results are least-squares mean differences (and corresponding 95% CIs) from separate linear mixed effects models.
Early CCR2 inactivation in aggrecan-expressing cells before injury partially affects bone damage..
We evaluated changes in osteophyte formation and subchondral bone thickness in both genotypes. We rarely observe osteophytes in Shams at all time-points (Fig.-3A), therefore they were excluded from the analysis. Early Ccr2 deletion does not affect osteophyte size during OA progression (Fig.-3B, Table-1). Changes in osteophyte tissue composition (maturity score and osteophyte cartilage quantification) were detected at 4wks and 8wks, indicating a persistence of cartilaginous tissue in the forming osteophyte; however, such differences are not relevant at 10wks, where bone tissue seems to be predominant in both genotypes (Fig.-3C–D). This result is validated by IHC, where increased expression of Collagen-10 is visible Fig.-3E in the osteophytes of CCR2-AggKO mice at 4 and 8wks, compared to CCR2+/+ mice. This outcome indicates a persistence of hypertrophic chondrocytes in the forming osteophytes of CCR2-AggKO mice, that is not seen in the CCR2+/+. When evaluating subchondral bone thickness, we found that CCR2 ablation in aggrecan-expressing cells was delaying subchondral changes at 2wks, but no relevant differences were noticed as OA progressed (Fig.-3F). No changes in subchondral bone thickness were detected in shams (data not shown), therefore they were not included in the analysis.
Figure 3. Osteophyte assessment in mouse knee joints of CCR2-AggKO and CCR2+/+, following early CCR2 inactivation.
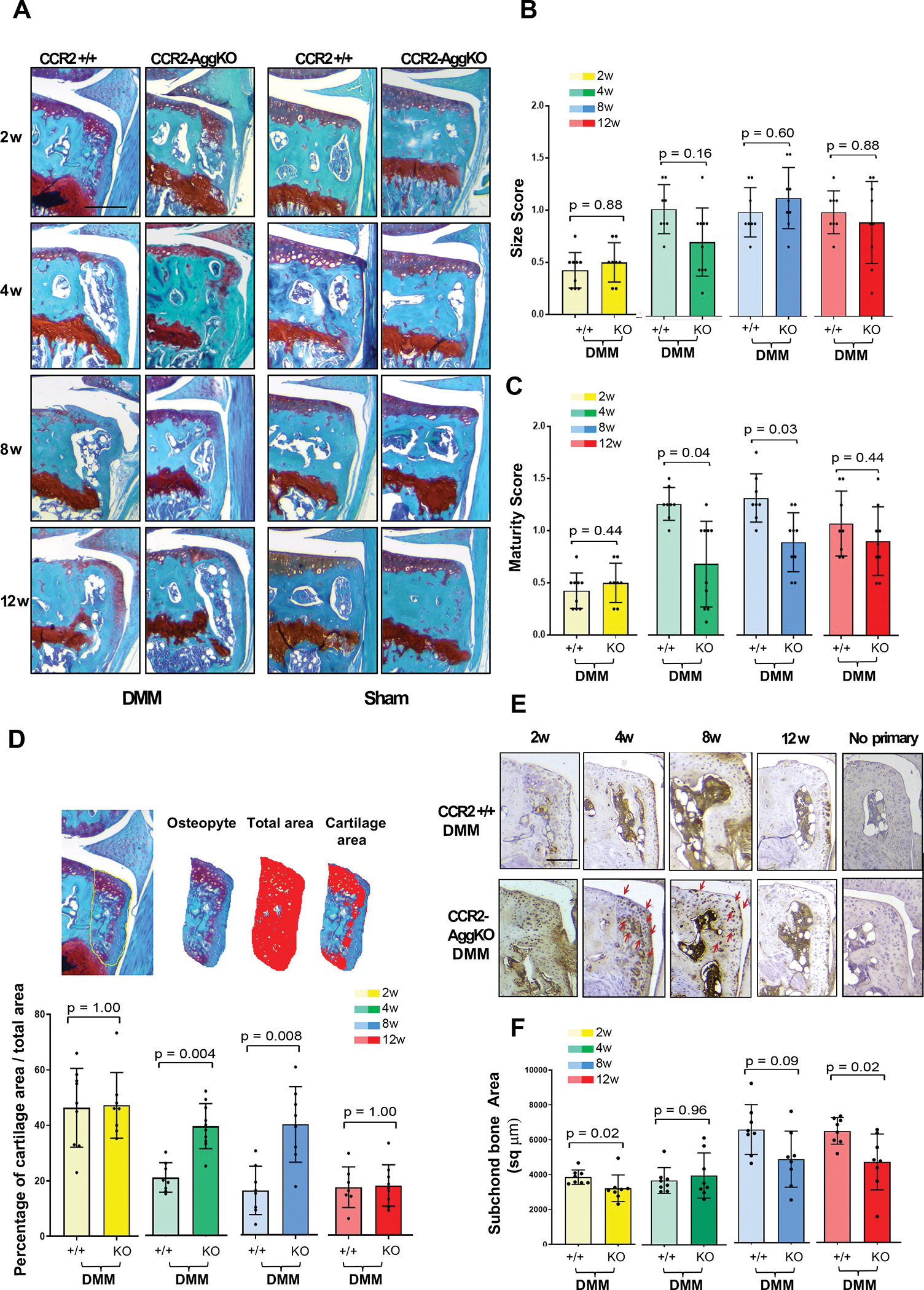
(A) Safranin-O/Fast green staining of the medial compartment of CCR2-AggKO and CCR2+/+ mouse knees at 2, 4, 8 and 12 weeks after DMM surgery. Images are representative of N=8 for each of the experimental points described. (B) Osteophyte size and (C) osteophyte maturity scores (scale 0–3) of CCR2-AggKO and CCR2+/+ mouse knees at the time point indicated and described in panel A. Results are expressed as average of 4 quadrants (medial and lateral tibial plateau, medial and lateral femoral condyles); N=8 mice for each experimental point. The graphs represent the mean ± standard deviation; indicated p-values were determined by Wilcoxon rank sum tests at each time point, following adjustment for multiple comparisons. (D) Paraffin embedded knee joint sections are immunostained for Collagen 10 (Col10) at the time point indicated. Positive staining is detected as brown precipitate. Images are representative of 6 different mice for each of the experimental groups described. Scale bars of the images are 100 μm.
Early CCR2 inactivation in aggrecan-expressing cells does not affect PTOA synovial hyperplasia.
As a chemokine receptor, CCR2 has a role in macrophage recruitment and inflammation[1, 11], therefore, we analyzed PTOA synovial hyperplasia in both genotypes. Although some decrease in lining cell thickness was observed in a few samples, the high variability among replicates did not produce significant changes between genotypes (Supplemental Figure-S4, Supplemental Table-S1). Synovial thickness is rarely present in shams, therefore they were not included.
Late CCR2 inactivation in aggrecan-expressing cells does not significantly prevent PTOA cartilage damage.
We induced Cre recombination in CCR2flx/flx-eGFP/AggCreERT2-positive mice during PTOA, following joint degeneration in CCR2-AggKO and CCR2+/+ mice at moderate and severe stages (8–12wks). Late Ccr2 inactivation was not able to consistently prevent or slow cartilage ACS score at both stages, and differences among DMM groups were not significant (Fig.-4A–B, Table-2). The extracellular matrix composition showed a slightly better outcome, with score improvement on moderate PTOA (8wks), but not on severe OA (Fig. 4C–D). Histomorphometric analysis confirmed the loss of efficacy in preventing AC damage following late Ccr2 inactivation and no changes in area quantification were detected at any OA stages (Fig. 4E).
Figure 4. Histopathological evaluation of CCR2-AggKO and CCR2+/+ mouse knees, following late CCR2 inactivation.
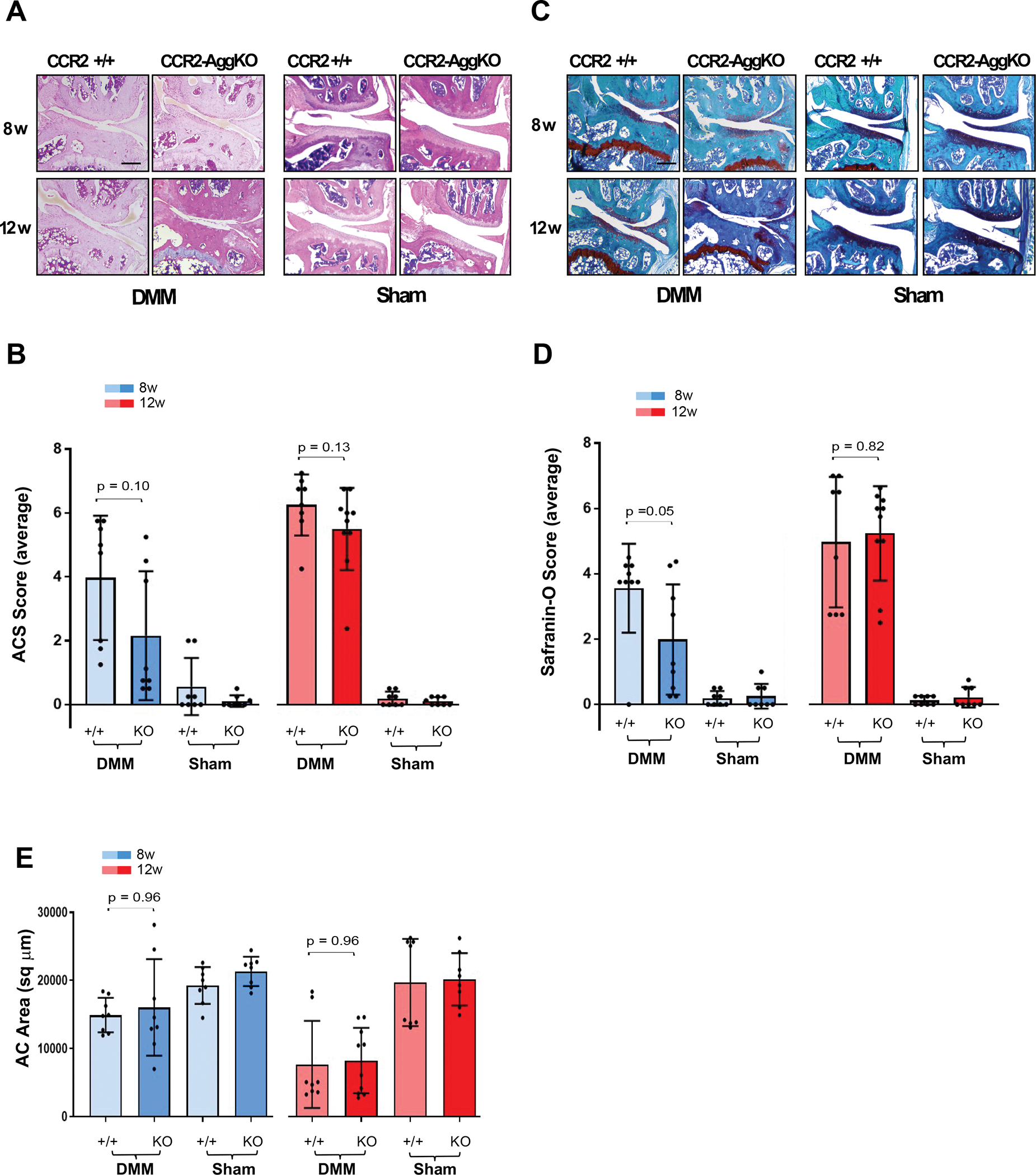
(A) H&E staining of the knee medial compartment from CCR2-AggKO and CCR2+/+ mice after DMM/Sham (time indicated); images are representative of N=8 for each time point. (B) ACS score (0–12 scale) of DMM/Sham knees at the time point indicated. Results are expressed as average of 4 quadrants (medial and lateral tibial plateau, medial and lateral femoral condyles); N=8 mice for each experimental point. (C) Safranin-O/Fast green staining of the knee medial compartment after DMM/Sham (time indicated); images are representative of N=8 for each time points. (D) Safranin-O staining Score (0–12 scale) of DMM/Sham knees at the time point indicated. Results are expressed as average of 4 quadrants as described above; N=8 mice for each experimental point. (E) Quantification of the AC area (sq μm) of the medial plateau of DMM/Sham knees by histomorphometric analysis at the time point indicated; N=8 mice for each experimental point. The graphs represents the mean ± standard deviation; indicated p-values were determined by Wilcoxon rank sum tests at each time point, following adjustment for multiple comparisons. Scale bars of the images are 100 μm.
Table 2.
Effect of late CCR2 inactivation (4wks post-DMM) on OA assessment (N=8) in mouse CCR2-AggKO and CCR2+/+ knee joints.
| OA PARAMETERS | Week 8 | Week 12 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Z(pvalue) | median | mean (SD) | n | Z (pvalue) | Median | mean (SD) | ||
| ACS | CCR2+/+ | 8 | 2.16 (0.10) | 4.88 | 3.97 (1.95) | 8 | 1.52 (0.13) | 6.50 | 6.25 (0.95) |
| CCR2-AggKO | 8 | 0.94 | 2.16 (2.02) | 10 | 5.88 | 5.50 (1.29) | |||
| Saf-o | CCR2+/+ | 9 | 1.60 (0.05) | 3.75 | 3.56 (1.36) | 8 | 0.22 (0.82) | 5.50 | 4.97 (2.00) |
| CCR2-AggKO | 9 | 1.25 | 1.99 (1.69) | 10 | 5.86 | 5.24 (1.45) | |||
| Cartilage Quantification | CCR2+/+ | 8 | 0.05 (0.96) | 14849 | 14907 (2538) | 8 | −0.04 (0.96) | 4860 | 7655 (6398) |
| CCR2-AggKO | 8 | 13854 | 16038 (7093) | 10 | 8269 | 8221 (4809) | |||
| Osteophyte size | CCR2+/+ | 9 | −0.90 (0.41) | 0.63 | 0.49 (0.28) | 8 | −0.82 (0.41) | 1.13 | 1.10 (0.51) |
| CCR2-AggKO | 9 | 0.63 | 0.72 (0.35) | 10 | 1.25 | 1.27 (0.23) | |||
| Osteophyte maturity | CCR2+/+ | 9 | 0.00 (1.00) | 0.63 | 0.54 (0.28) | 8 | 2.48 (0.02) | 1.35 | 1.30 (0.27) |
| CCR2-AggKO | 9 | 0.50 | 0.57 (0.24) | 10 | 1.00 | 0.95 (0.26) | |||
| Subchondral bone thickness | CCR2+/+ | 8 | 1.97 (0.10) | 4850 | 4869 (509) | 8 | 0.00 (1.00) | 7387 | 7224 (1713) |
| CCR2-AggKO | 9 | 4125 | 4191 (598) | 10 | 7189 | 7298 (1417) | |||
| Osteophyte Cartilage quantification | CCR2+/+ | 6 | −0.58 (0.56) | 16.25 | 15.77 (5.57) | 6 | −2.89 (0.008) | 12.10 | 12.42 (6.79) |
| CCR2- AggKO | 8 | 18.8 | 20.58 (10.49) | 9 | 28.7 | 35.44 (15.03) | |||
Data presented show median, mean and standard deviation (SD), and number of animals in each group at 8 and 12 weeks. Indicated z- and p-values were determined by Wilcoxon rank sum tests between DMM CCR+/+ and DMM CCR-AggKO groups separately at each time point, following adjustment for multiple comparisons (Benjamini-Hochberg).
Late CCR2 inactivation in aggrecan-expressing cells does not affect bone damage but delays their maturation from cartilage to bone.
Ccr2 deletion during OA progression did not affect osteophyte size at any disease stages (Fig.-5A–B and Table-2) in both genotypes, while a decrease in osteophyte maturity was detected in CCR2-AggKO mice at 12wks, compared to CCR2+/+, confirming an action of Ccr2 on osteophyte tissue composition (Fig.-5C–D). Figure 5E confirmed an increased expression of Col10 in the CCR2-AggKO osteophytes at 12wks, compared to CCR2+/+ mice. Late CCR2 inactivation did not change the DMM-induced increase in subchondral thickness (Fig.-5F). As above, shams were excluded from the analysis (Fig.-5A)
Figure 5. Osteophyte assessment in mouse knee joints of CCR2-AggKO and CCR2+/+, following late CCR2 inactivation.

(A) Safranin-O/Fast green staining of the medial compartment of CCR2-AggKO and CCR2+/+ mouse knees at 8 and 12 weeks after DMM surgery. Images are representative of N=8 for each of the experimental points described. (B) Osteophyte size and (C) osteophyte maturity scores (scale 0–3) of CCR2-AggKO and CCR2+/+ mouse knees at the time point indicated and described in panel A. Results are expressed as average of 4 quadrants (medial and lateral tibial plateau, medial and lateral femoral condyles); N=8 mice for each experimental point. The graphs represent the mean ± standard deviation; indicated p-values were determined by Wilcoxon rank sum tests at each time point, following adjustment for multiple comparisons (D). Paraffin embedded knee joint sections are immunostained for Collagen 10 (Col10) at the time point indicated. Positive staining is detected as brown precipitate. Images are representative of 6 different mice for each of the experimental groups described. Scale bars of the images are 100 μm.
Late CCR2 inactivation in aggrecan-expressing cells does not affect synovial hyperplasia.
No changes in synovial hyperplasia scores were detected between CCR2-AggKO and CCR2+/+ mice at the time points observed (8 and 12wks), with high variability among replicates (Supplemental Figure-S5 and Supplemental Table-S2).
Early CCR2 inactivation in aggrecan-expressing cells reduces pain on severe PTOA.
We previously reported that systemic CCR2 targeting decreased DMM-induced static weight bearing pain measures[8]. In this study we analyzed whether the CCR2-AggKO improved cartilage structure was translated to improved spontaneous pain behavior compared to CCR2+/+ mice, measured by hindlimb weight distribution. Table-1 and Supplementary Table-S3 include estimated between-group differences (corresponding 95% CIs) at each time point. Early Ccr2 deletion resulted in decreased pain responses during PTOA progression (4wks: mean difference, 0.10; 95%CI, [0.00, 0.19]; 8wks: mean difference, 0.10; 95%CI, [−0.02, 0.23];12wks: mean difference, 0.13; 95%CI, [0.01, 0.26]; 16wks: mean difference, 0.15; 95%CI, [0.05, 0.26]; 20wks: mean difference, 0.23; 95%CI, [0.14, 0.31]) (Fig.-6A, CCR2+/+ vs CCR2-AggKO). As OA progressed up to 20wks, we found no increase in pain perception in the CCR2-AggKO mice, with values that are not different from the sham (4wks: mean difference, 0.01; 95%CI, [−0.09, 0.11]; 8wks: mean difference, 0.01; 95%CI, [−0.12, 0.15];12wks: mean difference, 0.02; 95%CI, [−0.11, 0.15]; 16wks: mean difference, 0.02; 95%CI, [−0.08, 0.13]; 20wks: mean difference, 0.01; 95%CI, [−0.08, 0.10]). In contrast, CCR2+/+ mice progressed with higher static pain measures, as compared to CCR2-AggKO mice (Fig.-6A).
Figure 6. Pain assessment of CCR2-AggKO and CCR2+/+ mice, following early CCR2 inactivation.
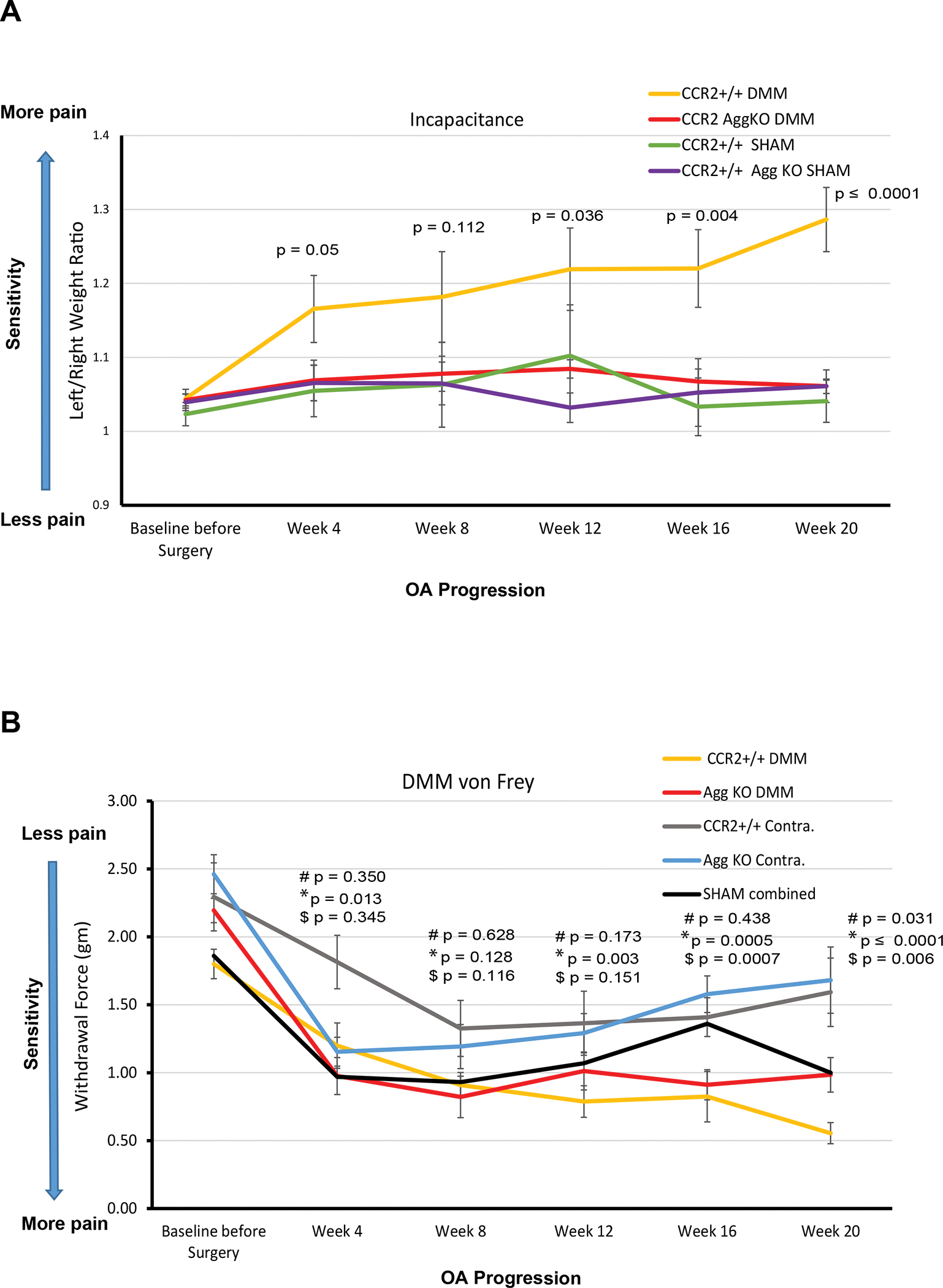
(A) Weight bearing longitudinal measures of CCR2-AggKO and CCR2+/+ mice were assessed at the indicated time points after DMM or Sham (N=9). Baseline measures were obtained before DMM/Sham and before CCR2 recombination. The graph represents observed mean scores by group and time point; error bars indicate the SEM by group and time point. Indicated p-values were determined for differences between corresponding groups by linear mixed effects model (LMM) as described in Statistical Analysis section. Numerical summaries of LMM are provided in details in Table-1 and Supplemental Table-S3. (B) Evoked pain (von Frey) was longitudinally assessed in CCR2-AggKO and CCR2+/+ mice in both the operated (DMM leg) and the un-operated limb (Contralateral), at the indicated time points after DMM (N=9). Baseline measures were obtained before DMM/Sham and before CCR2 recombination. Shams measures have been combined at each time point and the average is indicated by a black line (N=12). The graph represents observed mean scores by group and time point; error bars indicate the SEM. Indicated p-values were determined for differences between corresponding groups by LMM as described in Statistical Analysis section. Numerical summaries of LMM are provided in details in Table-1 and Supplemental Table-S4 : # indicates differences in values between DMM CCR2+/+ and DMM CCR2-AggKO mice (yellow line vs red line); *represents differences in values between the DMM and its Contralateral leg in CCR2+/+ mice (yellow line vs gray line, respectively), at the time point indicated; $ represents differences in values between the DMM and its Contralateral leg in CCR2-AggKO mice (red line vs blue line, respectively), at the time point indicated.
We also evaluated changes in evoked pain caused by DMM using von Frey filaments (Fig.-6B, Table-1 and Supplemental Table-S4). We found that pain values between the DMM leg vs its contralateral were only relevant starting at 16–20wks, in both genotypes(); when comparing DMM legs between genotypes, no differences were detected at any time point, although a trend was observed by 20wks (mean difference, −0.42; 95%CI, [−0.81, −0.04]). No differences between shams of different genotypes were found, and no difference were found when comparing non-OA legs (shams, contralateral of sham and contralateral of DMM) for both genotypes (Supplemental Fig.--S6 and Supplemental Table-S4, therefore sham values have been grouped together at each time point and the average is shown as black line (Fig.-6B).
Discussion
Although several studies indicate that CCR2 inhibition can reduce pain in pre-clinical OA models, evidence that it provides a benefit for structural OA damage has varied depending on the model system studied[5, 8–11]. Studies using the DMM model in global Ccr2-null mice demonstrated a key role for CCR2 in establishing OA pain but observed a modest reduction in chondropathy, evident only at the most severe stages[9–11]; we believe the lack of more significant reduction in cartilage lesions might be due to the limitations in using Ccr2 global germ-line deletion as a model system for OA, such as the reported pre- and post-natal growth plate defects[24]. We also showed that CCR2 blockade caused impaired limb ossification during embryogenesis[2] and found evidence post-natally of disorganized proliferative columns in the growth plate of Ccr2-null mice (P10, Supplemental Fig.-S7). Therefore, altered bone remodeling may mask amelioration of OA.
Mounting data have highlighted the importance of CCLs/CCR2 axis in knee injuries and OA, in both clinical[7, 25–28] and animal studies[5, 8, 29]. More importantly, in accordance with other investigators[5], we previously showed that pharmacological inactivation of CCR2 successfully prevented PTOA progression[8]; while this evidence recognizes CCR2 as a promising therapeutic target, past studies in arthritis have led to contrasting results using systemic approaches[30, 31]. Studies in inflammatory arthritis using Ccr2-null mice have led to the identification of CCR2+ T cell subpopulation that down-modulates the inflammatory response during disease progression[30]; therefore, a systemic approach to CCR2 targeting during OA might not be ideal, because it could enhance inflammation at certain disease stages and impact its therapeutic application. In line with this hypothesis, our previous DMM data demonstrated a temporal-specific effect of CCR2 targeting on cartilage and bone structural changes, with treatment efficacy limited at the early and moderate OA stages, highlighting the need to address the tissue-specific action of Ccr2 for therapeutic purposes[8, 32].
Given the critical role of Ccr2 in cartilage metabolism and OA[12, 33–35], we focused on cartilaginous joint tissues, using aggrecan-Cre to drive Ccr2 recombination. Aggrecan is a major component of the extracellular matrix, necessary for the hydration of cartilage and its weight-bearing mechanical demands, therefore it is widely used for deleting genes in cartilage and the inner meniscus in preclinical OA models. We modulated Ccr2 deletion after skeletal maturity at different times during PTOA to determine the contribution of cartilage levels of CCR2 to the whole joint pathology and pain. To provide more in-depth information of changes within the lesions, we dissected the contribution of CCR2 to the integrity of the basal lamina (ACS), and to changes in the cell compartment or the extracellular matrix[20]. We noticed that extracellular matrix composition was the first parameter to improve by Ccr2 inactivation (Fig.-2D), as early as 2wks post-surgery, followed by AC thinning (Fig.-2E, 4wks) and AC structure (Fig.-2B, 8wks). Such time-dependency in different cartilage properties might reflect a stage-specific activation of distinctive cartilage degradation markers by CCR2, as corroborated by our previous human in vitro data demonstrating a differential regulation of signaling pathways involved in cartilage degradation in normal chondrocytes vs OA samples in response to CCL2/CCR2 stimulation[12].
We next determined whether a delayed cartilage-specific targeting of the CCR2 after disease onset could improve cartilage changes at later stages. Delayed Ccr2 targeting drastically reduced the beneficial effect on cartilage damage, with transitory improvements only limited to the extracellular matrix(Fig.-4D). In accordance with our previous DMM studies[8], these data suggest the need of early intervention on CCR2 targeting for potential therapeutic approaches on PTOA cartilage damage.
Bone involvement is a hallmark of OA[36–38] and CCR2 has been shown to modulate skeletal repair and bone remodeling[39]. In the DMM, chondro/osteophytes and increased bone thickness are early PTOA features[8]; we demonstrated that early systemic pharmacological CCR2 targeting led to amelioration of such bone parameters[8]. Since Ccr2 was not ablated in bone tissues in the present study (Supplemental Fig. S2), we were able to analyze how cartilage and meniscus levels of CCR2 indirectly contributed to bone lesions. In contrast with pharmacological systemic targeting[8], subchondral thickness and osteophyte size were not changed following Agg-specific CCR2 inactivation, either with early (Fig.-3B, 5F) or late recombination (Fig.-5B, 5F), although a delay in subchondral changes was noticed at early stages, following early CCR2 ablation (2wks, Fig.-3F). When analyzing osteophyte tissue composition, a persistence of cartilage matrix was observed with both early and late CCR2 inactivation, although with some variability in the timeline (4wks/8wks vs 12wks respectively, Fig.-3C-D-E; Fig.-5C-D-E). These differences suggest that Ccr2 expression in other cell compartments might have a role in DMM-induced bone changes and that CCR2 levels in cartilage might only partially mediate their ossification. Studies are in progress in our lab to understand the contribution of osteoblast-Ccr2 expression to the whole PTOA pathology.
Synovitis has been reported after 4wks in DMM mice compared to shams [40] therefore, macrophage activation by CCR2 may contribute to synovial damage during DMM-induced OA. However, cartilage CCR2 levels don’t seem to affect the increase in synovial thickness induced by DMM, either with early or late receptor inactivation, suggesting that the CCR2-mediated synovial hyperplasia might be directly correlated to Ccr2 expression in other cell compartments, such as macrophages or other inflammatory cells. Further studies are needed to understand the contribution of CCR2 to the whole inflammatory response during PTOA.
Sensory innervations are abundant in the joint and pain can originate from many articular tissues[41]. CCR2 is expressed in the dorsal root ganglia (DRG) and its role in pain perception has been confirmed by different investigators[8–10, 13]. In a study in Ccr2 null mice, Abbadie et al. compared nociceptive responses in both inflammatory and neuropathic pain models describing differences in the contribution of CCR2 to chronic and acute pain[13]. Several studies report a correlation between specific structural changes in a defined joint tissue with pain[42, 43]. For example, joint space narrowing was more strongly associated with pain than was the presence of osteophytes[43]. We previously showed that early and transient systemic CCR2 targeting decreased PTOA pain, but we could not exclude an involvement of DRG in this perception, since CCR2 mRNA, protein, and signaling activity are up-regulated in the DRG following DMM surgery[9]. Here, we establish a link between Ccr2 levels in aggrecan-expressing cells and cartilage degeneration post-trauma, and the contribution of such structural changes to PTOA pain. We demonstrated that amelioration of joint structure by Ccr2 early ablation was able to decrease static measures of pain at severe OA stages (12wks), with benefits increasing as OA progressed (up to 20wks, Fig.-6A). Evoked pain, induced by mechanostimulation, was also affected by Ccr2 deletion in the DMM leg compared to the contralateral, however between- group differences were smaller and did not reach statistical significance. These discrepancies might be attributed to the activation of different pain pathways and progression of chronic pain. A human OA study by Parks et al. corroborates the hypothesis of distinct mechanisms in evoked vs spontaneous knee pain[44]. Mechanical pressure-evoked pain ratings were minimally different between OA and healthy subjects, while spontaneous OA knee pain induced brain activity like chronic back pain. Furthermore, when using OA animal models to understand pain-related outcomes, differences in pain measures might also reflect, to some extent, the limitations of approaches and available techniques[45].
Aggrecan expression has been found in brain tissue, where it is thought to restrict certain developmental processes during neural maturation[46]. When comparing evoked mechanosensitivity values in absence of OA degeneration (Von Frey, Shams and Contralateral legs of both CCR2-AggKO and CCR2+/+), our results show no differences from baseline up to 20ks post-surgery, with CCR2-AggKO similar to CCR2+/+ (Supplemental Fig.-S7 and Supplemental Table-S4). These data confirm that both genotypes show comparable sensitivity to evoked stimulation, suggesting that the Ccr2 ablation in aggrecan-expressing cells did not affect mechanosensitivity in the absence of joint tissue degeneration.
Several strategies to target cartilage and bone tissues independently are currently being developed[47–49]. There is also a move in the field to better phenotype OA by assessing specific joint tissues affected in a given individual to optimize therapies. In this respect, understanding the role of CCR2 in cartilage and its associated pain, will help to define a critical window for PTOA intervention and determine the value of tissue specific-targeting for CCR2-mediated therapies. Finally, our studies shed light on how reducing damage in one tissue might benefit other joint tissues.
Limitations of the study
As discussed, besides articular cartilage, aggrecan is also expressed in other joint tissues, such as the inner zone of the meniscus, that also express CCR2. Although we did not detect any recombination in other joint tissues, such as ligaments and synovium, we cannot exclude that some recombination might occur in other joint compartments below the level of detection of the IHC technique. Therefore, it’s likely that CCR2 expression in joint tissues other than AC, might contribute to the PTOA and pain that we assessed in our model. Further studies are needed to determine the specific contribution of CCR2 in each of these tissues to the whole joint pathology and pain.
The exclusive use of young male mice might constitute a limitation of the study, since it is known that age can exacerbate DMM-induced damage and estrogen can delay OA progression and alter pain responses[50]. Further studies in our lab are planned to understand the role of CCR2 in aging, as well as the impact of estrogen in PTOA.
Supplementary Material
Acknowledgements
We thank the Animal Histopathology Core at the University of North Carolina at Chapel Hill for their technical assistance.
We thank Dr. Manolis Pasparakis, University of Cologne, Germany, for providing the CCR2flx/flxeGFP mice.
We thank Dr. Peng Liu, School of Medicine, UNC, for providing the global CCR2-null mice.
The Aggrecan-CreERT2 mice are originally produced by Benoit de Crombrugghe, University of Texas MD Anderson Cancer Center, Houston, TX.
We thank Amanda Le and Amaya Esterellas for their technical assistance in the behavioral experiments and Jose’ Valdes-Fernandez for technical assistance in image acquisition.
In addition, we would like to thank Dr. Richard Loeser, Thurston Arthritis Research Center, UNC, Chapel Hill, for his constant advice and support.
Role of the funding source
This work has been sponsored by the National Institute of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR070821 to Dr. Longobardi; P30 AR072580 to Dr. Callahan, UNC Core Center for Clinical Research, for Liubov Arbeeva support).
Sponsors had no involvement in the study design, collection, analyses, interpretation of data and in the manuscript writing.
Footnotes
Conflict of interest
Authors do not have conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 2011; 7(1):33–42. [DOI] [PubMed] [Google Scholar]
- 2.Longobardi L, Li T, Myers TJ, O’Rear L, Ozkan H, Li Y, et al. A: TGF-beta type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Dev Cell 2012; 23(1):71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charo IF. CCR2: from cloning to the creation of knockout mice. Chem Immunol 1999; 72:30–41. [DOI] [PubMed] [Google Scholar]
- 4.Muller K, Ehlers S, Solbach W, Laskay T. Novel multi-probe RNase protection assay (RPA) sets for the detection of murine chemokine gene expression. J Immunol Methods 2001; 249(1–2):155–165. [DOI] [PubMed] [Google Scholar]
- 5.Appleton CT, Usmani SE, Pest MA, Pitelka V, Mort JS, Beier F. Reduction in disease progression by inhibition of transforming growth factor alpha-CCL2 signaling in experimental posttraumatic osteoarthritis. Arthritis Rheumatol 2015; 67(10):2691–2701. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Jiang BE. Serum and synovial fluid chemokine ligand 2/monocyte chemoattractant protein 1 concentrations correlates with symptomatic severity in patients with knee osteoarthritis. Ann Clin Biochem 2015; 52(Pt 2):276–282. [DOI] [PubMed] [Google Scholar]
- 7.Longobardi L, Jordan JM, Shi XA, Renner JB, Schwartz TA, Nelson AE, et al. Associations between the chemokine biomarker CCL2 and knee osteoarthritis outcomes: the Johnston County Osteoarthritis Project. Osteoarthritis Cartilage 2018; 26(9):1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longobardi L, Temple JD, Tagliafierro L, Willcockson H, Esposito A, D’Onofrio N, et al. Role of the C-C chemokine receptor-2 in a murine model of injury-induced osteoarthritis. Osteoarthritis Cartilage 2017; 25(6):914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A 2012; 109(50):20602–20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM, et al. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage 2017; 25(3):406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F, et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis 2017; 76(5):914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willcockson HOH, Chubinskaya S, Loeser RF, and Longobardi L CCL2 Induces Articular Chondrocyte MMP expression through ERK and p38 Signaling Pathways. Osteoarthritis and Cartilage Open 2021; 3 (2021) 100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A 2003; 100(13):7947–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 2009; 47(12):805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood; 120(3):613–625. [DOI] [PubMed] [Google Scholar]
- 16.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005; 434(7033):644–648. [DOI] [PubMed] [Google Scholar]
- 17.Glasson SS, Askew R, Sheppard B, Carito BA, Blanchet T, Ma HL, et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis and rheumatism 2004; 50(8):2547–2558. [DOI] [PubMed] [Google Scholar]
- 18.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007; 15(9):1061–1069. [DOI] [PubMed] [Google Scholar]
- 19.Bove SE, Calcaterra SL, Brooker RM, Huber CM, Guzman RE, Juneau PL, et al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society 2003; 11(11):821–830. [DOI] [PubMed] [Google Scholar]
- 20.McNulty MA, Loeser RF, Davey C, Callahan MF, Ferguson CM, Carlson CS. A Comprehensive Histological Assessment of Osteoarthritis Lesions in Mice. Cartilage 2011; 2(4):354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagira K, Ikuta Y, Shinohara M, Sanada Y, Omoto T, Kanaya H, et al. Histological scoring system for subchondral bone changes in murine models of joint aging and osteoarthritis. Sci Rep 2020; 10(1):10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 2009; 60(12):3723–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowe MA, Harper LR, McNulty MA, Lau AG, Carlson CS, Leng L, et al. Reduced Osteoarthritis Severity in Aged Mice With Deletion of Macrophage Migration Inhibitory Factor. Arthritis Rheumatol 2017; 69(2):352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binder NB, Niederreiter B, Hoffmann O, Stange R, Pap T, Stulnig TM, et al. Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat Med 2009l; 15(4):417–424. [DOI] [PubMed] [Google Scholar]
- 25.Arkestal K, Mints M, Enocson A, Linton L, Marits P, Glise H, et al. CCR2 upregulated on peripheral T cells in osteoarthritis but not in bone marrow. Scand J Immunol 2018; 88(6):e12722. [DOI] [PubMed] [Google Scholar]
- 26.Garriga C, Goff M, Paterson E, Hrusecka R, Hamid B, Alderson J, et al. Clinical and molecular associations with outcomes at 2 years after acute knee injury: a longitudinal study in the Knee Injury Cohort at the Kennedy (KICK). Lancet Rheumatol 2021; 3(9):e648–e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lisee C, Spang JT, Loeser R, Longobardi L, Lalush D, Nissman D, et al. Tibiofemoral articular cartilage composition differs based on serum biochemical profiles following anterior cruciate ligament reconstruction. Osteoarthritis Cartilage 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watt FE, Paterson E, Freidin A, Kenny M, Judge A, Saklatvala J, et al. Acute Molecular Changes in Synovial Fluid Following Human Knee Injury: Association With Early Clinical Outcomes. Arthritis Rheumatol 2016; 68(9):2129–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ismail HM, Yamamoto K, Vincent TL, Nagase H, Troeberg L, Saklatvala J. Interleukin-1 Acts via the JNK-2 Signaling Pathway to Induce Aggrecan Degradation by Human Chondrocytes. Arthritis Rheumatol 2015; 67(7):1826–1836. [DOI] [PubMed] [Google Scholar]
- 30.Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol 2004; 172(2):890–898. [DOI] [PubMed] [Google Scholar]
- 31.Quinones MP, Ahuja SK, Jimenez F, Schaefer J, Garavito E, Rao A, et al. Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. J Clin Invest 2004; 113(6):856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller RE, Malfait AM; Can we target CCR2 to treat osteoarthritis? The trick is in the timing! Osteoarthritis Cartilage 2017; 25(6):799–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favero M, Belluzzi E, Trisolino G, Goldring MB, Goldring SR, Cigolotti A, et al. Inflammatory molecules produced by meniscus and synovium in early and end-stage osteoarthritis: a coculture study. J Cell Physiol 2019; 234(7):11176–11187. [DOI] [PubMed] [Google Scholar]
- 34.Goldring MB, Otero M, Plumb DA, Dragomir C, Favero M, El Hachem K, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater 2011; 21:202–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scanzello CR. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J Orthop Res 2017; 35(4):735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burr DB. Increased biological activity of subchondral mineralized tissues underlies the progressive deterioration of articular cartilage in osteoarthritis. J Rheumatol 2005; 32(6):1156–1158; discussion 1158–1159. [PubMed] [Google Scholar]
- 37.Kouri JB, Aguilera JM, Reyes J, Lozoya KA, Gonzalez S. Apoptotic chondrocytes from osteoarthrotic human articular cartilage and abnormal calcification of subchondral bone. J Rheumatol 2000; 27(4):1005–1019. [PubMed] [Google Scholar]
- 38.Zamli Z, Robson Brown K, Tarlton JF, Adams MA, Torlot GE, Cartwright C, et al. Subchondral bone plate thickening precedes chondrocyte apoptosis and cartilage degradation in spontaneous animal models of osteoarthritis. Biomed Res Int 2014; 2014:606870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu AC, Morrison NA, Kelly WL, Forwood MR. MCP-1 expression is specifically regulated during activation of skeletal repair and remodeling. Calcif Tissue Int 2013; 92(6):566–575. [DOI] [PubMed] [Google Scholar]
- 40.Jackson MT, Moradi B, Zaki S, Smith MM, McCracken S, Smith SM, et al. Depletion of protease-activated receptor 2 but not protease-activated receptor 1 may confer protection against osteoarthritis in mice through extracartilaginous mechanisms. Arthritis Rheumatol 2014; 66(12):3337–3348. [DOI] [PubMed] [Google Scholar]
- 41.Malfait AM, Schnitzer TJ. Towards a mechanism-based approach to pain management in osteoarthritis. Nature reviews Rheumatology 2013; 9(11):654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncan R, Peat G, Thomas E, Hay E, McCall I, Croft P. Symptoms and radiographic osteoarthritis: not as discordant as they are made out to be? Ann Rheum Dis 2007; 66(1):86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neogi T, Felson D, Niu J, Nevitt M, Lewis CE, Aliabadi P, et al. Association between radiographic features of knee osteoarthritis and pain: results from two cohort studies. BMJ 2009; 339:b2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parks EL, Geha PY, Baliki MN, Katz J, Schnitzer TJ, Apkarian AV. Brain activity for chronic knee osteoarthritis: dissociating evoked pain from spontaneous pain. Eur J Pain 2011; 15(8):843 e841–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller RE, Malfait AM. Osteoarthritis pain: What are we learning from animal models? Best Pract Res Clin Rheumatol 2017; 31(5):676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morawski M, Bruckner G, Arendt T, Matthews RT. Aggrecan: Beyond cartilage and into the brain. Int J Biochem Cell Biol 2012; 44(5):690–693. [DOI] [PubMed] [Google Scholar]
- 47.Laroui H, Grossin L, Leonard M, Stoltz JF, Gillet P, Netter P, et al. Hyaluronate-covered nanoparticles for the therapeutic targeting of cartilage. Biomacromolecules 2007; 8(12):3879–3885. [DOI] [PubMed] [Google Scholar]
- 48.Sandker MJ, Petit A, Redout EM, Siebelt M, Muller B, Bruin P, et al. In situ forming acyl-capped PCLA-PEG-PCLA triblock copolymer based hydrogels. Biomaterials 2013; 34(32):8002–8011. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Wei L, Miron RJ, Shi B, Bian Z. Anabolic bone formation via a site-specific bone-targeting delivery system by interfering with semaphorin 4D expression. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 2015; 30(2):286–296. [DOI] [PubMed] [Google Scholar]
- 50.Hwang HS, Park IY, Hong JI, Kim JR, Kim HA. Comparison of joint degeneration and pain in male and female mice in DMM model of osteoarthritis. Osteoarthritis Cartilage 2021; 29(5):728–738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.