

Abstract
Background
Delayed cerebral ischemia (DCI) is one of the main determinants of clinical outcome after aneurysmal subarachnoid hemorrhage (SAH). The classical description of risk for DCI over time is currently based on the outdated concept of angiographic vasospasm. The goal of this study was to assess the temporal risk profile of DCI, defined by extended clinical and radiological criteria, as well as the impact the time point of DCI onset has on clinical outcome.
Methods
All patients with aneurysmal SAH referred to a single tertiary care center between 2010 and 2018 were considered for inclusion. This study was designed as a retrospective cohort analysis and data were extracted from existing patient files. In conscious patients, DCI was diagnosed clinically, and in unconscious patients, diagnosis was based on perfusion computed tomography imaging and multimodal neuromonitoring. Extended Glasgow Outcome Scale scores were assessed after 12 months and compared between patients with early (< day 7) and late (≥ day 7) DCI onset.
Results
The median delay from day of the hemorrhage (day 0) until detection of the first DCI event was 7.0 days, with an interquartile range of 5 days. The probability of DCI development over time demonstrated a bimodal distribution with a peak risk on day 5 (0.084; confidence interval 0.05.5–0.122) and a second peak on day 9 (0.077; confidence interval 0.045–0.120). A total of 27 patients (15.6%) suffered dominant hemispheric or severe bilateral DCI-related infarctions, resulting in the withdrawal of technical life support. Of those, the majority (20 patients, 22.2%) presented with early DCI onset (vs. late onset: 7 patients, 8.4%; p = 0.013).
Conclusions
The risk profile of DCI over time mirrors the description of angiographic vasospasm; however, it comes with an added timely delay of 1 to 2 days. Early occurrence of DCI (before day 7) is associated with a higher infarct load and DCI-related mortality. Although the exact causal relationship remains to be determined, the time point of DCI onset may serve as an independent prognostic criterion in decision-making.
Keywords: Aneurysmal subarachnoid hemorrhage, Delayed cerebral ischemia, Timing, Outcome, Extended Glasgow Outcome Scale
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a severe type of hemorrhagic stroke involving relatively young patients. The rupture of an intracranial aneurysmal causes blood to enter the subarachnoid space under arterial pressure, leading to a sudden increase of intracranial pressure. The resulting transient drop in cerebral blood flow sets off an elusive cascade leading to endothelial cell damage, microglial activation, and edema formation. These pathophysiological processes beginning 24 to 48 h after aneurysm rupture are coined as early brain injury and can contribute to later morbidity and mortality [1]. Delayed cerebral ischemia (DCI) has been identified as one of the main contributors to poor clinical outcome after SAH because it may, if left untreated, lead to cerebral infarction [2]. Initially believed to be the result of angiographic vasospasm, DCI is now perceived as a multifactorial concept [3]. Disruption of the blood–brain barrier, microthrombosis, and loss of autoregulation have been identified as contributing factors [4]. Reactive side products of hemoglobin degradation can induce vasoconstriction in animal experiments; however, strategies to reduce cisternal blood load in patients have had limited success in improving outcome [5, 6]. The role blood degradation products play in inducing DCI in humans is being questioned. Apart from blood degradation, no other pathophysiological mechanism exists to explain the delayed nature of these insults.
The time frame during which angiographic narrowing of cerebral arteries occurs after SAH is well described. Angiographic vasospasm generally starts around day 3 to 4 after aneurysm rupture, peaks around day 7 to 10, and resolves by day 14 [7, 8]. However, the concept of angiographic vasospasm leading to cerebral infarction in a delayed fashion is now considered an oversimplification. Congruous to a better understanding and the conceptualization of DCI, diagnostic tools assessing functional changes within the brain, such as metabolism and perfusion, are more commonly used. Perfusion computed tomography (CT) imaging and multimodal monitoring have replaced angiography for DCI detection in unconscious patients. Therefore, we revisited the timeline of DCI onset and its daily probability after aneurysm rupture in a setting in which this functional definition of DCI is applied. Furthermore, we aimed at assessing whether the timing of DCI onset affects the severity of further disease progression and responsiveness to treatment. We hypothesize that earlier DCI onset might reflect more severe underlying pathophysiology and be more detrimental. Finally, we aimed to identify the rate of late (post day 14) DCI and associated factors when this functional definition is applied.
Methods
Participants and Study Design
This study was designed as an observational cohort analysis and entails a subgroup of data that were partially previously published [9, 10]. This study was registered (ClinicalTrials.gov identifier NCT02142166) and approved by the Ethics Committee of the Medical Faculty of RWTH Aachen University (EK 062/14). Patients 18 years of age or older with confirmed SAH (either in CT angiography or digital subtraction angiography) who were referred to a single university hospital between 2010 and 2018 were included. Data have been prospectively collected since 2014, and written informed consent was obtained for these patients. Data of patients before 2014 were retrospectively extracted from digital hospital records. Patients with a moribund initial presentation (dilated pupils, other signs of brain stem herniation, or severe dominant hemispheric damage) and anticipated early mortality, SAH due to mycotic or traumatic/pseudoaneurysms, and aneurysms associated with arteriovenous malformations were excluded.
SAH Treatment
After diagnosis, aneurysms were secured by either surgical clipping or endovascular occlusion within 48 h. The occlusion modality was based on interdisciplinary consensus. Each patient was observed in our neurointensive care unit for at least 14 days. The diagnostic and therapeutic treatment algorithm at our institution has been previously published in detail and is summarized below [11, 12].
In conscious patients, DCI was diagnosed clinically on the basis of the definition by Vergouwen et al. [13]. In unconscious patients not otherwise assessable, brain tissue oxygen probes (Neurovent PTO; Raumedic, Helmbrechts, Germany) and cerebral microdialysis catheters (71 High Cut-Off Brain Microdialysis Catheter; M dialysis, Stockholm, Sweden) were implanted in the watershed frontal region, ipsilateral to the ruptured aneurysm or highest blood load. Perfusion CT in conjunction with deterioration on multimodal neuromonitoring was used to define DCI in this cohort.
Induced hypertension (systolic blood pressure ≥ 180 mm Hg) via intravenous norepinephrine infusion was initiated in case of DCI-typical perfusion CT deficits (territorial cerebral blood flow/mean transit time mismatch) on the basis of neuroradiological consensus and/or oxygenation crises (brain tissue oxygen tension < 10 mm Hg) [14] or metabolic derangements (lactate/pyruvate ratio ≥ 40) [9, 15]. Onset of DCI was defined by the abovementioned extended criteria; however, for practical purposes, especially in respect to retrospectively collected data, the day of initiation of first tier treatment (induced hypertension) and the day of DCI diagnosis were equated. For all included patients, the binary outcome of DCI occurrence along its postictal day of onset was retrospectively reevaluated for each individual patient by two independent assessors (MV, MW), and discrepancies were discussed until consensus was reached.
In case perfusion deficits persisted under hypertensive treatment, endovascular rescue treatment consisting of transluminal balloon angioplasty or intraarterial spasmolysis was considered. For continuous local intraarterial nimodipine application, microcatheters (Excelsior SL-10; Stryker Neurovascular, Fremont, CA) were advanced into the internal carotid artery or vertebral artery and patients received weight-adapted intravenous tirofiban (Aggrastat; Correvia, Heathrow, UK). Treatment of DCI was reevaluated daily and deescalated as soon as possible. Tapering of induced hypertension was guided by imaging and multimodal monitoring.
Assessed Variables and Outcome Definition
The primary outcome was defined as the extended Glasgow Outcome Scale (GOSE) score 12 months after the initial hemorrhage and dichotomized as unfavorable (GOSE scores 1–4) and favorable (GOSE scores 5–8) outcome [16]. Most outcome data were collected prospectively during scheduled follow-ups, but missing information was either appended by analysis of medical records or a structured telephone interview by a blinded assessor [17]. In many cases included before 2014, the GOSE score at 12 months had to be reconstructed retrospectively by investigation of medical records or by contacting the patient, the next of kin, or the caregiver in a structured telephone interview by an assessor blinded for additional clinical information. Secondary outcome included the incidence of DCI -related infarction and DCI-related mortality. DCI-related mortality was defined as the withdrawal of technological life support due to severe DCI-induced dominant hemispheric or bilateral infarction.
Statistical Methods
Data are presented as the mean and standard deviation for continuous normally distributed variables. Nonnormal data are summarized as mean and interquartile range (quartile 1 to quartile 3). Categorical variables are provided as frequencies and proportions. After normality testing via the Shapiro–Wilk test, appropriate statistical tests were selected. Nominal data were tested with the χ2 test; for normally distributed continuous data, the unpaired t-test was used, and for nonnormally distributed data, the Mann–Whitney U-test was used. The Clopper–Pearson interval was used to calculate binomial confidence intervals (CIs) around daily DCI probability. All statistical analyses were performed using IBM SPSS Statistics 25 (SPSS Inc., Chicago, IL). Statistical significance was defined as a two-sided p value > 0.05.
Results
Patient Population
The recruitment process is depicted in Fig. 1. A total of 341 patients with SAH were treated at our institution between 2010 and 2018. Twenty-four patients were excluded because of loss of follow-up. DCI was diagnosed in 173 (50.7%) patients, of whom 124 (71.7%) were women and 49 (28.3%) were men. The mean age of patients developing DCI was 54.0 ± 12.7. The GOSE score after 1 year was available in all but 26 patients. Relevant baseline characteristics are presented in Table 1.
Fig. 1.
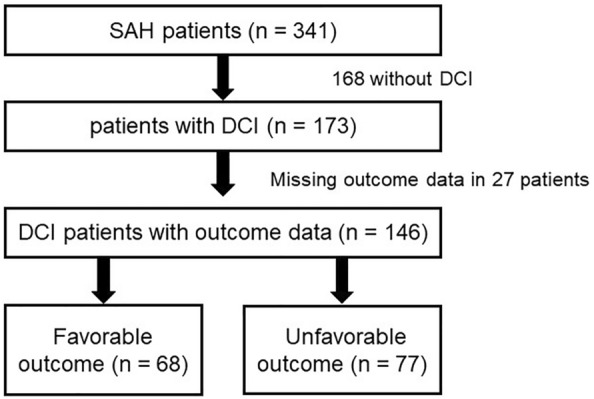
Inclusion flowchart. DCI delayed cerebral ischemia, SAH subarachnoid hemorrhage
Table 1.
Comparison of patient- and SAH-specific characteristics between early (< day 7) and late (≥ day 7) DCI onset
| DCI (n = 173) | Early DCI (n = 90) | Late DCI (n = 83) | p value | |
|---|---|---|---|---|
| Age (years), mean ± SD | 54.0 (12.7) | 52.5 ± 12.2 | 55.6 ± 13.1 | 0.106 |
| Sex (female/male), no. (%) | 124 (71.7)/49 (28.3) | 65 (72.2)/25 (27.8) | 59 (71.1)/24 (28.9) | 0.868 |
| Risk factors, no. (%) | ||||
| Hypertension | 69 (39.9) | 39 (43.3) | 30 (36.1) | 0.335 |
| Smoking | 59 (34.1) | 22 (24.4) | 37 (44.6) | 0.043 |
| Aneurysm location, no. (%) | 0.235 | |||
| Acomm | 60 (34.7) | 27 (30.0) | 33 (39.8) | |
| MCA | 49 (28.3) | 24 (26.7) | 25 (30.1) | |
| ICA | 27 (15.6) | 18 (20.0) | 9 (10.8) | |
| Others | 37 (21.4) | 21 (23.3) | 16 (19.3) | |
| Ant. circulation | 148 (85.5) | 78 (86.7) | 70 (84.3) | 0.663 |
| Post. circulation | 25 (14.5) | 12 (13.3) | 13 (15.7) | |
| Aneurysm size diameter maxium (mm), mean ± SD | 7.1 ± 3.7 | 6.9 ± 3.5 | 7.2 ± 4.0 | 0.574 |
| Hunt and Hess grade, no. (%) | 0.301 | |||
| Grade 1 | 19 (11.0) | 11 (12.2) | 8 (9.6) | |
| Grade 2 | 35 (20.2) | 14 (15.6) | 21 (25.3) | |
| Grade 3 | 56 (32.4) | 33 (36.7) | 23 (27.7) | |
| Grade 4 | 40 (23.1) | 18 (20.0) | 22 (26.5) | |
| Grade 5 | 23 (13.3) | 14 (15.6) | 9 (10.8) | |
| Intracerebral hemorrhage, no. (%) | 77 (44.5) | 39 (43.3) | 38 (45.8) | 0.746 |
| Acute hydrocephalus, no. (%) | 129 (74.6) | 65 (72.2) | 64 (77.1) | 0.461 |
| Modified Fisher scale, no. (%) | 0.269 | |||
| Grade 1 | 24 (13.9) | 10 (11.1) | 14 (16.9) | |
| Grade 2 | 17 (9.8) | 12 (13.3) | 5 (6.0) | |
| Grade 3 | 57 (32.9) | 32 (35.6) | 25 (30.1) | |
| Grade 4 | 74 (42.8) | 36 (40.0) | 39 (47.0) | |
| Aneurysm treatment, no. (%) | 0.976 | |||
| Clipping/endovascular | 79 (45.7)/94 (54.3) | 41 (45.6)/49 (54.4) | 38 (45.8)/45 (54.2) | |
| DCI surveillance | ||||
| INM, no. (%) | 0.735 | |||
| ptiO2 | 73 (42.2) | 39 (43.3) | 34 (41.0) | |
| CMD | 59 (34.1) | 30 (33.3) | 29 (34.9) | |
| Onset of first DCI event, days (IQR) | 6 (4–9) | 5 (3–5) | 9 (8–10) | < 0.001 |
Bold values indicate p < 0.05
Acomm Anterior communication aneurysm, CMD Cerebral microdialysis, DCI Delayed cerebral ischemia, ICA Internal carotid artery, INM Invasive neuromonitoring, IQR Interquartile range, MCA Middle cerebral artery, ptiO2 Brain tissue oxygen tension, SAH Subarachnoid hemorrhage
Risk of DCI Over Time
The median time until detection of the first DCI event was 7.0 days (interquartile range 4–9). The plotted prevalence of DCI per postictal day is skewed to the right and drops considerably after day 10 in Fig. 2a. For each postictal day, the probability of DCI development, with 95% CIs, was calculated and is plotted in Fig. 2b. Risk of DCI over time demonstrated a bimodal distribution with a peak risk on day 5 of 8.4% (CI 5.5–12.2%) and a second peak on day 9 of 7.7% (CI 4.5–12.0%). The risk of DCI rapidly decreased after day 10 and bottomed on day 14 (0.6%; CI 0.0–3.1%).
Fig. 2.

a, Prevalence distribution of DCI events over time in 173 patients. b, Probability distribution with confidence interval of DCI development per postictal day in 341 patients. DCI delayed cerebral ischemia, P(DCI), probability of DCI onset
Early Versus Late DCI
The median (day 7) was used as a dichotomization point, dividing patients into early (< day 7) and late (≥ day 7) DCI onset. A total of 90 (52.0%) patients developed early DCI, whereas 83 (48.0%) patients were diagnosed with late DCI. A higher rate of smoking was observed in the late DCI onset subgroup (44.6% vs. 24.4%; p = 0.043). No other relevant differences in patient-specific and SAH-relevant baseline characteristics were identified between early and late DCI. Especially, SAH severity (clinically, Hunt and Hess and radiologically, modified Fisher) had no effect on whether DCI occurred before or after postictal day 7 (Table 1).
Early DCI onset was not associated with longer length of stay or higher rates of intensive care unit (ICU)-related complications. A total of 27 patients (15.6%) suffered dominant hemispheric or severe bilateral DCI-related infarctions, resulting in the withdrawal of technical life support. Of those, the majority (20 patients, 11.6%) presented with early DCI onset (late DCI onset: 7 patients, 4.0%; p = 0.013). Overall mortality figures, however, were not affected by the time point of DCI onset. Moreover, rates of favorable outcome after 12 months were comparable between early DCI onset and late DCI onset (41.1% vs. 37.3%; p = 0.987) (Table 2).
Table 2.
Comparison of treatment characteristics and outcome between early (< day 7) and late (≥ day 7) DCI onset
| Early DCI (n = 90) | Late DCI (n = 83) | p value | |
|---|---|---|---|
| DCI detection, no. (%) | |||
| Clinical DCI | 37 (41.1) | 31 (37.3) | 0.613 |
| Metabolic DCI | 39 (59.1) | 35 (59.3) | 0.979 |
| Radiological DCI | 71 (81.6) | 73 (91.3) | 0.071 |
| Silent infarction, no. (%) | 24 (26.7) | 26 (31.3) | 0.499 |
| DCI treatment, no. (%) | |||
| iHT | 73 (81.1) | 64 (77.1) | 0.517 |
| ERT | 39 (43.3) | 40 (48.2) | 0.521 |
| Spasmolysis | 32 (35.6) | 37 (44.6) | 0.225 |
| Continuous spasmolysis | 17 (18.9) | 12 (14.5) | 0.361 |
| Clinical outcome | |||
| DCI-related infarction, no. (%) | 46 (51.1) | 43 (51.8) | 0.972 |
| Lag DCI-related infarction, mean ± SD | 4.5 ± 1.7 | 9.6 ± 2.6 | < 0.001 |
| Favorable outcome (GOSE scores 5–8), no. (%) | 37 (41.1) | 31 (37.3) | 0.987 |
| Unfavorable outcome (GOSE scores 1–4), no. (%) | 42 (46.7) | 35 (42.2) | |
| Overall mortality, no. (%) | 29 (32.2) | 21 (25.3) | 0.496 |
| DCI-related mortality, no. (%) | 20 (22.2) | 7 (8.4) | 0.013 |
| ICU LOS, days ± SD | 28.7 ± 17.5 | 30.9 ± 17.0 | 0.391 |
| ICU-related complications, no. (%) | |||
| Pneumonia | 55 (61.1) | 42 (50.6) | 0.164 |
| UTI | 76 (84.4) | 69 (83.1) | 0.684 |
| SIRS | 53 (58.9) | 42 (50.6) | 0.274 |
| Sepsis | 24 (26.7) | 15 (18.1) | 0.177 |
| Stunned myocardium | 11 (12.2) | 7 (8.4) | 0.415 |
| Cranial complications, no. (%) | |||
| ICP crisis | 33 (36.7) | 27 (32.5) | 0.517 |
| DHC | 24 (26.7) | 22 (26.5) | 0.981 |
Bold values indicate p < 0.05
DCI Delayed cerebral ischemia, DHC Decompressive hemicraniectomy, ERT Endovascular rescue treatment, GOSE Extended Glasgow Outcome Scale, ICP Intracranial pressure, ICU Intensive care unit, iHT Induced hypertension, LOS Length of stay, SIRS Systemic inflammatory response syndrome, UTI Urinary tract infection
DCI Beyond Day 14
DCI was diagnosed in two (1.2%) patients after day 14. Clinically silent infarction was the first sign of ongoing DCI in four (2.3%) patients. Demographic data did not differ for patients with DCI or DCI-related infarction after day 14 compared with the remainder of patients. Also, no differences were identified regarding aneurysm location, size, occlusion modality, or hemorrhage severity in patients with DCI after day 14. Very late DCI onset did not contribute to a longer length of stay at the ICU (Table 3).
Table 3.
Comparison of patient, SAH, and treatment characteristics alongside outcome between patients with very late DCI (> day 14) and the remainder of patients
| Ultralate DCI (n = 6) | Other patients with DCI (n = 167) | p value | |
|---|---|---|---|
| Sex (female/male), no. (%) | 4 (66.7)/2 (33.3) | 120 (71.9)/47 (28.1) | 0.782 |
| Risk factors | |||
| Age (years), mean ± SD | 56.5 ± 19.2 | 53.9 ± 12.5 | 0.621 |
| Hypertension, no. (%) | 4 (66.7) | 65 (38.9) | 0.173 |
| Smoking, no. (%) | 2 | 57 (34.1) | 0.968 |
| Aneurysm location, no. (%) | |||
| Acomm | 3 (50.0) | 57 (34.1) | 0.296 |
| MCA | 3 (50.0) | 46 (27.5) | |
| ICA | 0 (0) | 27 (16.2) | |
| Others | 0 (0) | 36 (21.6) | |
| Ant. circulation | 6 (100.0) | 142 (85.0) | 0.306 |
| Post. circulation | 0 (0) | 25 (15.0) | |
| Aneurysm size diameter maximum (mm), mean ± SD | 8.2 ± 5.6 | 7.0 ± 3.7 | 0.471 |
| Hunt and Hess grade, no. (%) | 0.299 | ||
| Grade 1 | 2 (33.3) | 17 (10.2) | |
| Grade 2 | 0 (0) | 35 (21.0) | |
| Grade 3 | 1 (16.7) | 55 (32.9) | |
| Grade 4 | 2 (33.3) | 38 (22.8) | |
| Grade 5 | 1 (16.7) | 22 (13.2) | |
| Modified Fisher scale, no. (%) | 0.571 | ||
| Grade 1 | 2 (33.3) | 22 (13.2) | |
| Grade 2 | 0 (0) | 17 (10.2) | |
| Grade 3 | 1 (16.7) | 56 (33.5) | |
| Grade 4 | 3 (50.0) | 71 (42.5) | |
| DCI detection, no. (%) | |||
| Clinical DCI | 3 (50.0) | 65 (38.9) | 0.585 |
| Technical DCI | 3 (50.0) | 102 (61.1) | |
| Silent infarction, no. (%) | 4 (66.7) | 46 (27.5) | 0.038 |
| DCI treatment, no. (%) | |||
| iHT | 4 (66.7) | 133 (79.6) | 0.442 |
| ERT | 2 (33.3) | 77 (46.1) | 0.537 |
| Spasmolysis | 2 (33.3) | 67 (40.1) | 0.709 |
| Continuous spasmolysis | 0 (0) | 29 (17.4) | 0.330 |
| Clinical outcome | |||
| DCI-related infarction, no. (%) | 4 (66.7) | 85 (50.9) | 0.448 |
| Lag DCI-related infarction, mean ± SD | 15.3 ± 3.4 | 6.8 ± 2.9 | < 0.001 |
| Favorable outcome (GOSE scores 5–8), no. (%) | 2 (33.3) | 65 (38.9) | 0.876 |
| Unfavorable outcome (GOSE scores 1–4), no. (%) | 2 (33.3) | 74 (44.3) | |
| Overall mortality, no. (%) | 1 (16.7) | 49 (29.3) | 0.354 |
| DCI-related mortality, no. (%) | 0 (0) | 27 (16.2) | 0.284 |
| ICU LOS, days ± SD | 41.7 ± 25.4 | 29.3 ± 16.9 | 0.085 |
| Length of ventilation | 23.8 ± 20.1 | 21.5 ± 16.2 | 0.289 |
| ICU-related complications, no. (%) | |||
| Pneumonia | 3 (50.0) | 94 (56.3) | 0.760 |
| UTI | 6 (100.0) | 139 (83.2) | 0.282 |
| SIRS | 3 (50.0) | 92 (55.1) | 0.806 |
| Sepsis | 1 (16.7) | 38 (22.8) | 0.726 |
| Stunned myocardium | 1 (16.7) | 17 (10.2) | 0.609 |
| SAH-specific complications, no. (%) | |||
| Hydrocephalus | 3 (50.0) | 126 (75.4) | 0.160 |
| ICP crisis | 0 (0) | 60 (35.9) | 0.184 |
| DHC | 0 (0) | 46 (27.5) | 0.132 |
Bold values indicate p < 0.05
Acomm, anterior communication aneurysm, DCI Delayed cerebral ischemia, DHC Decompressive hemicraniectomy, ERT Endovascular rescue treatment, GOSE Extended Glasgow Outcome Scale, ICA Internal carotid artery, ICP Intracranial pressure, ICU Intensive care unit, iHT Induced hypertension, LOS Length of stay, MCA Middle cerebral artery, SAH Subarachnoid hemorrhage, SD Standard deviation, SIRS Systemic inflammatory response syndrome, UTI Urinary tract infection
Discussion
DCI has been thoroughly reconceptualized as a multifactorial process in which macrovasospasm is only a contributing factor. New insights in DCI pathophysiology have broadened diagnostic criteria and made way for the application of additional diagnostic tools, i.e., perfusion CT imaging (typically triggered by transcranial Doppler), cerebral microdialysis, or brain oxygenation monitoring [9, 13]. In contemporary large SAH series in which DCI is assessed beyond angiographic vasospasm, the timeline of its onset is not addressed [2, 18].
We applied a combined clinical and functional DCI definition that included perfusion CT imaging. The risk of DCI presented as a bimodal distribution, with the highest risk on day 5 and a second peak on day 9 to 10. This mirrors the description of angiographic vasospasm but with an additional delay of 1 to 2 days. Earlier DCI onset was associated with a higher rate of DCI-related death because of more pronounced infarct load. It appears treatment in patients with early DCI proved not as effective in the prevention of severe infarction. This confirms the notion that earlier DCI may be an outing of more sever underlying pathophysiology. Early DCI onset presents a negative prognostic marker independent of clinical or radiological hemorrhage severity. DCI beyond day 14 was extremely rare and was observed only in 3.5% of cases. Of those, two thirds presented with already demarcated infarction, suggesting that the process of DCI started earlier. We were not able to identify any patient- or SAH-specific factors associated with very late DCI onset. Diagnosis of DCI after 14 days contributed to neither a longer ICU length of stay nor worse clinical outcome.
The classical understanding of the delayed nature of angiographic vasospasm is based on blood degradation and the time it takes for oxygen radicals to build up in the subarachnoid space. Hemolysis of red blood cells starts 16 to 32 h after aneurysm rupture and peaks around day 7 [19]. Although the exact spasmogenic agent has not been identified yet, the vasoconstrictive effect of blood degradation products has been well documented in animal SAH models [5]. In humans, efforts to increase the rate of subarachnoid blood clearance have had limited effect on outcome [6, 20].
In its most simplified form, DCI represents (still reversible) decompensation in oxygen/glucose supply and demand. Contribution factors can either cause decreased supply or increased demand. Angiographic macroscopic vasospasm causing decreased supply can largely be compensated as substantiated by the high rate of vasospasm remaining asymptomatic. Much more difficult to contemplate are microvascular changes within the neurovascular unit. Constriction of the microvasculature is consistently observed in experimental SAH models and remains irresponsive to induced hypertension [21]. A potassium surge drives inverted neurovascular coupling, creating a vicious circle in a brain where inflammatory repair mechanisms have already increased energy demand [22]. Local hypercoagulability and endothelial changes allow for formation of microthrombi, further reducing supply, and their presence has demonstrated spatial correlation with regions of neurol apoptosis [21, 23].
Neuroinflammation and the time required for the recruitment of circulating white blood cells, as well as buildup of a microglial response, offers an alternative explanation toward the delayed nature of DCI [24]. Inflammatory reactive changes in the central nervous system after SAH are incompletely understood but can be indirectly observed by a systemic increase of acute phase reactants, such as C-reactive protein and procalcitonin [25, 26]. Additionally, cerebrospinal fluid concentrations of proinflammatory cytokines, such as interleukin-1β, interleukin-6, interleukin-8, and tumor necrosis factor α, correlate with DCI and poor outcome [27–29]. Although immunosuppressive treatment, such as glucocorticoid use, has had no effect on DCI and outcome so far [30].
A possible explanation for milder ischemic injury in late DCI onset might be the longer recovery time the brain has after early brain injury before a second hit. After the initial insult, activated inflammatory cells become readily susceptible and primed for any subsequent cue. Consecutive stimuli can have a synergistic and potentiating effect on neuroinflammation, increasing tissue damage [31, 32]. Such a response is observed in traumatic brain injury, in which an early second hit can cause disproportional amounts of additional damage [33–35]. We speculate that this double-hit hypothesis can be an explanation for the increased mortality rate after early DCI, although future studies are needed to verify this concept.
An extended definition of DCI encompassing these changes, as well as allowing evaluation of unconscious patients, is urgently needed. Our compound definition of DCI highly relies on perfusion CT imaging. A relevant limitation thereof is the existing broad heterogeneity in perfusion CT definitions of DCI along with ubiquitous differences in image acquisition protocols and postprocessing algorithms. The definition of DCI in perfusion CT requires standardization before inclusion of this diagnostic modality in a newly phrased extended DCI definition, which is application for unconscious patients. Moreover, the lack of incorporating change over time of brain oxygenation and microdialysis parameter should be addressed as a limitation.
Despite rigorous documentation, the retrospective nature of this study allows for information bias to be introduced. Attrition bias due to loss to follow-up is suspected to have limited effect on results because loss to follow-up was equally distributed between early and late DCI. The accuracy of time specification for day 0 (day of hemorrhage) can be biased by aneurysm rupture earlier than the day of admittance. Whenever possible, the day of ictus was reconstructed on the basis of the patient’s history. Our applied functional DCI definition is heterogeneous and not yet widely accepted and most probably accounts for the high rate of DCI in our cohort. Outcome data were partially retrospectively assessed, creating potential observer and information bias.
Conclusions
The timeline of functional DCI mirrors that of angiographic vasospasm, with an additional delay of 1 to 2 days. Early onset of DCI is associated with a higher risk of severe cerebral infarction. Although the exact causal relationship remains to be determined, the time point of DCI onset may serve as an independent prognostic criterion in decision-making.
Author’s contributions
TPS and MV developed and designed the study. TPS, MW, AH, WA, and MV were involved in the acquisition of data. TPS and MV performed statistical analyses and drafted the manuscript. All authors were involved in the interpretation of data. The final manuscript was critically revised and approved by all authors.
Source of support
Open Access funding enabled and organized by Projekt DEAL. No funding bodies had any involvement in the preparation of this trial or in the decision to submit the article for publication.
Conflicts of Interest
The authors have no personal, financial, or institutional conflicts of interest to declare.
Ethical approval/informed consent
The trial was conducted in accordance with the recommendations of the Ethics Committee of the Medical Faculty of RWTH Aachen University (EK 062/14).
Clinical trial registration
The data collection process was part of a previously registered observational study (ClinicalTrials.gov identifier NCT02142166).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Research Treatment. 2013;2013:394036. doi: 10.1155/2013/394036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vergouwen MD, Etminan N, Ilodigwe D, Macdonald RL. Lower incidence of cerebral infarction correlates with improved functional outcome after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2011;31:1545–1553. doi: 10.1038/jcbfm.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10:44–58. doi: 10.1038/nrneurol.2013.246. [DOI] [PubMed] [Google Scholar]
- 4.Wan H, AlHarbi BM, Macdonald RL. Mechanisms, treatment and prevention of cellular injury and death from delayed events after aneurysmal subarachnoid hemorrhage. Expert Opin Pharmacother. 2014;15:231–243. doi: 10.1517/14656566.2014.865724. [DOI] [PubMed] [Google Scholar]
- 5.Megyesi JF, Vollrath B, Cook DA, Findlay JM. (2000) In vivo animal models of cerebral vasospasm: a review. Neurosurgery. 2000;46:448–460. doi: 10.1097/00006123-200002000-00035. [DOI] [PubMed] [Google Scholar]
- 6.Fistouris P, Scheiwe C, Grauvogel J, et al. (2022) Mitigation of Blood Load Impact in Patients with Subarachnoid Hemorrhage by Cisternal Lavage. Cerebrovascular diseases (Basel, Switzerland) 1–7. [DOI] [PubMed]
- 7.Lawton MT, Vates GE. Subarachnoid Hemorrhage. N Engl J Med. 2017;377:257–266. doi: 10.1056/NEJMcp1605827. [DOI] [PubMed] [Google Scholar]
- 8.Macdonald RL. Origins of the concept of Vasospasm. Stroke. 2016;47:e11–e15. doi: 10.1161/STROKEAHA.114.006498. [DOI] [PubMed] [Google Scholar]
- 9.Veldeman M, Albanna W, Weiss M, et al. Invasive neuromonitoring with an extended definition of delayed cerebral ischemia is associated with improved outcome after poor-grade subarachnoid hemorrhage. J Neurosurg. 2020;134(5):1527–1534. doi: 10.3171/2020.3.JNS20375. [DOI] [PubMed] [Google Scholar]
- 10.Veldeman M, Albanna W, Weiss M, et al. Treatment of delayed Cerebral Ischemia in Good-Grade Subarachnoid Hemorrhage: any Role for Invasive Neuromonitoring? Neurocrit Care. 2020;35(1):172–183. doi: 10.1007/s12028-020-01169-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albanna W, Weiss M, Muller M, et al. Endovascular rescue therapies for refractory vasospasm after subarachnoid hemorrhage: a prospective evaluation study using multimodal continuous event neuromonitoring. Neurosurgery. 2017;80:942–949. doi: 10.1093/neuros/nyw132. [DOI] [PubMed] [Google Scholar]
- 12.Weiss M, Conzen C, Mueller M, et al. Endovascular rescue treatment for delayed cerebral ischemia after Subarachnoid Hemorrhage Is Safe and effective. Front Neurol. 2019;10:136. doi: 10.3389/fneur.2019.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41:2391–2395. doi: 10.1161/STROKEAHA.110.589275. [DOI] [PubMed] [Google Scholar]
- 14.Maloney-Wilensky E, Gracias V, Itkin A, Hoffman K, Bloom S, Yang W, Christian S, LeRoux PDE, et al. Brain tissue oxygen and outcome after severe traumatic brain injury: a systematic review. Crit Care Med. 2009;37:2057–2063. doi: 10.1097/CCM.0b013e3181a009f8. [DOI] [PubMed] [Google Scholar]
- 15.Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, Boutelle MG, Chen JW, Claassen J, Dahyot-Fizelier C, Enblad P, et al. Consensus statement from 2014 the International Microdialysis Forum. Intensive Care Med. 2015;41:1517–1528. doi: 10.1007/s00134-015-3930-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jennett B, Snoek J, Bond MR, Brooks N. Disability after severe head injury: observations on the use of the Glasgow Outcome Scale. J Neurol Neurosurg Psychiatry. 1981;44:285–293. doi: 10.1136/jnnp.44.4.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson JT, Pettigrew LE, Teasdale GM. Structured interviews for the Glasgow Outcome Scale and the extended Glasgow Outcome Scale: guidelines for their use. J Neurotrauma. 1998;15:573–585. doi: 10.1089/neu.1998.15.573. [DOI] [PubMed] [Google Scholar]
- 18.Roos YB, de Haan RJ, Beenen LF, Groen RJ, Albrecht KW, Vermeulen M. Complications and outcome in patients with aneurysmal subarachnoid haemorrhage: a prospective hospital based cohort study in the Netherlands. J Neurol Neurosurg Psychiatry. 2000;68:337–341. doi: 10.1136/jnnp.68.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22:971–982. doi: 10.1161/01.STR.22.8.971. [DOI] [PubMed] [Google Scholar]
- 20.Veldeman M, Höllig A, Clusmann H, Stevanovic A, Rossaint R, Coburn M. Delayed cerebral ischaemia prevention and treatment after aneurysmal subarachnoid haemorrhage: a systematic review. Br J Anaesth. 2016;117:17–40. doi: 10.1093/bja/aew095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tso MK, Macdonald RL. Subarachnoid hemorrhage: a review of experimental studies on the microcirculation and the neurovascular unit. Transl Stroke Res. 2014;5:174–189. doi: 10.1007/s12975-014-0323-4. [DOI] [PubMed] [Google Scholar]
- 22.Koide M, Bonev AD, Nelson MT, Wellman GC. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc Natl Acad Sci USA. 2012;109:E1387–E1395. doi: 10.1073/pnas.1121359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabri M, Ai J, Lakovic K, D'Abbondanza J, Ilodigwe D, Macdonald RL. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience. 2012;224:26–37. doi: 10.1016/j.neuroscience.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 24.de Oliveira Manoel AL, Goffi A, Marotta TR, Schweizer TA, Abrahamson S, Macdonald RL. The critical care management of poor-grade subarachnoid haemorrhage. Crit Care. 2016;20:21. doi: 10.1186/s13054-016-1193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Höllig A, Stoffel-Wagner B, Clusmann H, Veldeman M, Schubert GA, Coburn M. Time courses of inflammatory markers after aneurysmal subarachnoid Hemorrhage and their possible relevance for future studies. Front Neurol. 2017;8:694. doi: 10.3389/fneur.2017.00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veldeman M, Lepore D, Höllig A, et al. Procalcitonin in the context of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Neurosurgery. 2020;135:29–37. doi: 10.3171/2020.5.JNS201337. [DOI] [PubMed] [Google Scholar]
- 27.Schoch B, Regel JP, Wichert M, Gasser T, Volbracht L, Stolke D. Analysis of intrathecal interleukin-6 as a potential predictive factor for vasospasm in subarachnoid hemorrhage. Neurosurgery. 2007;60:828–836. doi: 10.1227/01.NEU.0000255440.21495.80. [DOI] [PubMed] [Google Scholar]
- 28.Hendryk S, Jarzab B, Josko J. Increase of the IL-1 beta and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Neuro Endocrinol Lett. 2004;25:141–147. [PubMed] [Google Scholar]
- 29.Ahn SH, Savarraj JPJ, Parsha K, et al. Inflammation in delayed ischemia and functional outcomes after subarachnoid hemorrhage. J Neuroinflammation. 2019;16:213. doi: 10.1186/s12974-019-1578-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohney N, Williamson CA, Rothman E, et al. A propensity score analysis of the impact of dexamethasone use on delayed cerebral ischemia and poor functional outcomes after subarachnoid Hemorrhage. World neurosurgery. 2018;109:e655–e661. doi: 10.1016/j.wneu.2017.10.051. [DOI] [PubMed] [Google Scholar]
- 31.Jones H, Hopkins N, Bailey TG, Green DJ, Cable NT, Thijssen DH. Seven-day remote ischemic preconditioning improves local and systemic endothelial function and microcirculation in healthy humans. Am J Hypertens. 2014;27:918–925. doi: 10.1093/ajh/hpu004. [DOI] [PubMed] [Google Scholar]
- 32.Fiebich BL, Akter S, Akundi RS. The two-hit hypothesis for neuroinflammation: role of exogenous ATP in modulating inflammation in the brain. Front Cell Neurosci. 2014;8:260. doi: 10.3389/fncel.2014.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- 34.Schoettle RJ, Kochanek PM, Magargee MJ, Uhl MW, Nemoto EM. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats. J Neurotrauma. 1990;7:207–217. doi: 10.1089/neu.1990.7.207. [DOI] [PubMed] [Google Scholar]
- 35.Scholz M, Cinatl J, Schädel-Höpfner M, Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. 2007;27:401–416. doi: 10.1002/med.20064. [DOI] [PubMed] [Google Scholar]