

Abstract
Aims
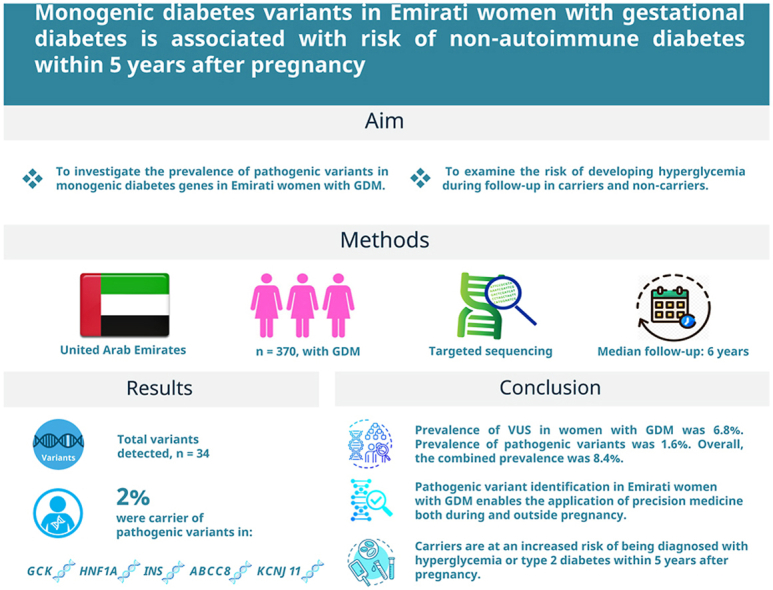
To investigate the prevalence of pathogenic variants in monogenic diabetes genes in Emirati women with gestational diabetes (GDM) and examine the risk of developing hyperglycemia during follow-up in carriers and non-carriers.
Methods
Female patients with GDM (n = 370) were identified. Selected monogenic diabetes genes, GCK, HNF1A, HNF4A, HNF1B, INS, ABCC8 and KCNJ1I, were examined by sequencing and identified variants were classified. Anthropometrics and subsequent diagnosis of diabetes were extracted from hospital records. Median follow-up time was 6-years.
Results
A total of 34 variants were detected. Seven women (2%) were carriers of pathogenic variants in GCK, HNF1A, INS, ABCC8 or KCNJ11. A significantly larger fraction of women carrying pathogenic variants were diagnosed with any form of hyperglycemia or diabetes postpartum (risk ratio = 1.8 (1.1–2.9), p = 0.02) or 2.5 (1.3–4.8; p = 0.009), respectively) and they had a shorter disease-free period after GDM compared to women without such variants. There were no significant associations between carrying pathogenic variants and anthropometric measures or C-peptide.
Conclusions
Pathogenic variants were found in known monogenic diabetes genes in two percent of Emirati women with GDM, allowing for precision medicine utilisation in these women both during and outside pregnancy. Carriers were at an increased risk of being diagnosed with hyperglycemia or type 2 diabetes mellitus within 5 years after pregnancy.
Keywords: Gestational diabetes, Maturity onset diabetes of the young, Monogenic, Type 2 diabetes mellitus, Gene
Graphical abstract
Abbreviations
- ABCC8
ATP Binding Cassette Subfamily C Member 8
- ACMG
American College of Medical Genetics and Genomics
- GAD
Glutamic acid decarboxylase
- GCK
Glucokinase
- GDM
Gestational diabetes
- HNF1A
Hepatocyte nuclear factor-1 alpha
- HNF1B
Hepatocyte nuclear factor-1 beta
- HNF4A
Hepatocyte nuclear factor-4 alpha
- HG
Hyperglycemia
- IA2
Islet antigen 2
- INS
Insulin
- IQR
Interquartile range
- KCNJ11
Potassium Inwardly Rectifying Channel Subfamily J Member 11
- MODY
Maturity-onset diabetes of the young
- RR
Risk ratio
- T2DM
Type 2 diabetes mellitus
- VUS:
Variants of uncertain significance
1. Introduction
In 2019, the prevalence of diabetes in the United Arab Emirates (UAE) was reported by the International Diabetes Federation (IDF) to be 16.3% for the 20–79 years age group [1]. The number of people with diabetes in the Middle Eastern region is projected to double by 2035 [2]. Women in the Middle East and North Africa have the highest risk globally of developing metabolic diseases [3] and a prevalence of diabetes of 8.6% has been found in a study of Emirati female students [4]. Risk factors like obesity, urbanization, changes in dietary habits, lack of physical activity and genetic factors are likely to play an important role [2].
Little is known about the genetic background of the Emirati population; although, a recent study has shed some light on the genomic architecture of the Emirati population [5]. Furthermore, a study performed whole exome sequencing in two Emirati nationals showed extensive genetic admixture with genetic contribution from the Middle East, Sub-Saharan and North Africa, Central and South Asia as well as Europe and Oceania [6]. In addition, the effect of 101 known type 2 diabetes mellitus (T2DM) loci was examined in Emiratis (422 patients with T2DM and 455 controls) and common SNPs in HHEX, BCL2, ADAMTS9, SLC22A3 and MTNR1B associated with T2DM were detected [7]. However, the prevalence of rare variants, detected only by sequencing, has not been examined among Emirati patients with diabetes.
The most common form of monogenic diabetes is maturity-onset diabetes of the young (MODY), characterised by autosomal dominant inherited non-autoimmune diabetes, with early-onset of diabetes (before the age of 25 years in at least one family member) [8]. Rare variants in fourteen genes are so far known to cause MODY and precision medicine-based intervention (involving precise molecular diagnosis and precise path to medical care for an individual) can be applied for the most common of these genetic etiologies [9]. The presence of such variants has been discovered in different populations with diabetes including women with gestational diabetes (GDM) [[9], [10], [11], [12], [13]]. This is of great importance as the identification of such variants in the mother can guide treatment before and during the pregnancy, fetus growth risks, complications during delivery, possible implications for risk of developing diabetes later in life and counselling [12,13]. The aim of the present study was therefore 1) to investigate if pathogenic variants in GCK, HNF1A, HNF4A, HNF1B, INS, ABCC8 and KCNJ1I are present in Emirati women with GDM, 2) to study the associated clinical characteristics of carriers of such variants and 3) to examine if such variants associate with risk of developing hyperglycemia (HG) postpartum.
2. Materials and methods
2.1. Population
Female patients were recruited from Imperial College London Diabetes Centre, Abu Dhabi, UAE. Inclusion criteria were; diagnosis with diabetes for the first time during pregnancy and before the age of 45. Women were excluded if either glutamic acid decarboxylase (GAD) or islet antigen 2 (IA2) autoantibodies were detected. Clinical diagnosis was based on the electronic medical record. The diagnosis of gestational diabetes was based on a ADA guidelines using either the one-step or the two-step diagnostic criteria [14]. The average time from diagnosis with GDM until the study ended in February 2019 was 71 months (approximately 6 years) ranging from 2 to 385 months. In total, 370 women were included in the present study (Supplementary Table S1).
Informed consent was obtained from all participants. The study design was in accordance with the ethical scientific principles of the Helsinki Declaration II and approved by Imperial College London Diabetes Centre Research Ethics Committee (reference number: IREC032).
2.2. Anthropometric and biochemical measurements
Weight (kg) was measured to the nearest 1.0 kg with the participant wearing light clothes and no shoes. Change in weight was calculated as the difference between post-pregnancy weight and pre-pregnancy weight.
GAD and IA2 antibodies were analyzed by Enzyme-Linked Immunosorbent Assay (ELISA) tests using anti-GAD and anti-IA2 commercial kits (EUROIMMUN AG, Germany). C-peptide was measured at the time of recruitment by the electrochemiluminescence immunoassay (ECLIA) method using a Cobas 6000 (Roche, Switzerland).
The hospital records available did not include date of giving birth, thus the time from the registration of GDM to a subsequent registration of either overt T2DM or any form of HG (if either pre-diabetes, impaired glucose tolerance (IGT), impaired fasting glucose (IFG) or T2DM was recorded in hospital records) was defined as being a minimum of 8 months, in order to ensure that the subsequent diagnosis of diabetes was indeed postpartum, meaning that if follow-up is stated as 2 months, it is 10 months after the first diagnosis of GDM was recorded.
2.3. Targeted sequencing
Targeted regions including the coding regions and exon/intron boundaries of the GCK, HNF1A, HNF4A, HNF1B, INS, ABCC8 and KCNJ11 genes, were captured and sequenced using the Illumina HiSeq2000 Analyzer as previously described [15]. Sequencing data were analyzed for variants using the Annovar software [16] as per the transcripts (GCK: NM_000162, HNF1A: NM_000545.5, HNF4A: NM_175914, HNF1B: NM_000458.2, NM_001304286 and NM_001304286, INS: NM_000207.2, ABCC8: NM_000352 and KCNJ11: NM_000525).
2.4. Pathogenicity of variants
Variants were classified as being either pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign or benign, as per American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines [17]. This classification was based on: 1) the location and function of variants; 2) minor allele frequency (MAF) in both the Gnomad database [18] and a Middle Eastern reference population [19]; 3) previous information of variants being involved in MODY [[20], [21], [22], [23]]; 4) CADD score (http://cadd.gs.washington.edu/info); 5) ClinVar classification (if available) [24] and 6) information of the prevalence of variants in the in house database of approximately 6000 Danish population-based individuals without diabetes (the Inter99 cohort) [25].
In the remainder of the manuscript, variants classified as either likely pathogenic or pathogenic will be denoted pathogenic, variants classified as either likely benign or benign will be denoted benign and variants classified as being of uncertain significance will be denoted VUS.
2.5. Statistical analysis
For all analyses below, we will refer to the non-carrier group – often working as a reference group – which includes individuals with no or only benign variants. The differences in quantitative traits (age at examination and at diagnosis, C-peptide, pre- and post-pregnancy weight, change in weight and age-of-diagnosis) between carriers and non-carriers of pathogenic variants and/or VUS in the investigated genes, were examined using standard linear regression with carrier-status as the explanatory variable and quantitative trait as outcome. All investigated traits (approximately) followed a normal distribution except C-peptide which was log-transformed.
The risk ratio (RR) was calculated as the ratio between the risk of developing the disease (overt T2DM or any form of HG) after GDM according to carrier-status and was tested for associations between exposure (carrier-status) and disease (developing T2DM or HG).
Median follow-up time was calculated as 1) the time period from the GDM diagnosis noted in the hospital records to the first date of any form of HG (noted in the records as either IFG, IGT or T2DM) or exclusively as T2DM (overt T2DM) or 2) for patients remaining disease free, from the time period from the GDM diagnosis noted in the hospital records until end of study (February 2019). The median follow-up for overt T2DM was 3.7 years (IQR: 2.0–6.7 years) and for any form of HG 3.4 years (IQR: 1.9–6.3 years).
The Kaplan-Meier log-rank test was used to compare the estimated disease-free ratio of women who remained free of either any form of HG or T2DM in carriers of, respectively, VUS or pathogenic variants compared to non-carriers.
Statistical analyses were performed using RStudio software version 3.6.1 and 4.0.2 (R Foundation for Statistical Computing, Boston, MA, USA). A p-value < 0.05 was considered statistically significant.
3. Results
3.1. Prevalence of identified variants
We identified 34 different variants in the studied genes in 370 women with GDM (Table 1). Eleven of the variants were common (MAF >1%) (HNF1A: I27L, A98V and S487 N; HNF4A: T117I and I441V; ABCC8: A1370S and V1573I; KCNJ11: K23E, L270V, V337I and S385C), six were low frequency variants (MAF between 1% and 0.1%) (GCK: H318P, HNF1A: A239V, P291A, and S574G; HNF4A:V147I, and HNF1B: F443L) and the remaining 17 were rare (minor allele frequency <0.1%) (GCK: Q19H, R64H and T207 M; HNF1A: G52A, L254R, S328 N and L389V; HNF1B: V25L, S75F, S342F and R435H, INS: R55H; ABCC8: R285Q, A355T, E612D, and A958V; KCNJ11: R365H) (Table 1).
Table 1.
Variants identified in 370 Emirati women with GDM.
| Variant | Position hg19/rs-number | No. carriers: WT/HE/HO (MAF) | Gnomad (all) [18] | GME [19] | Pathogenicity classification [17] |
|---|---|---|---|---|---|
| GCK (NM_033507) | |||||
| Q19H | 44193054/NA | 369/1/0 (0.1%) | 0% | 0% | VUS |
| R64H | 44192920/rs746444094 | 369/1/0a (0.1%) | 0.001% | 0% | Pathogenic |
| T207 M | 44189421/NA | 368/2/0 (0.3%) | 0.0004% | 0% | Pathogenic |
| H318P | 44186131/NA | 369/1/0 (0.1%) | 0% | 0.3% | VUS |
| HNF1A (NM_000545) | |||||
| I27L | 121416650/rs1169288 | 101/185/64 (48%) | 36% | 43% | Benign |
| G52A | 121416726/rs142318174 | 369/1/0 (0.1%) | 0.02% | 0% | VUS |
| A98V | 121416864/rs1800574 | 335/33/2 (5%) | 3% | 4% | Benign |
| A239V | 121431969/rs587778397 | 368/2/0 (0.3%) | 0.02% | 0.1% | VUS |
| L254R | 121432014/NA | 369/1/0 (0.1%) | 0% | 0% | Pathogenic |
| P291A | 121432124/rs151256267 | 369/1/0 (0.1%) | 0.0004% | 0.1% | VUS |
| S328 N | 121434092/NA | 369/1/0 (0.1%) | 0% | 0% | VUS |
| L389V | 121434401/rs115080759 | 367/3/0 (0.4%) | 0.04% | 0% | VUS |
| S487 N | 121435427/rs2464196 | 92/173/105 (52%) | 33% | 42% | Benign |
| S574G | 121437382/rs1169305 | 369/1/0 (0.1%) | 0.3% | 0.5% | Benign |
| HNF4A (NM_175914) | |||||
| T117I | 43042364/rs1800961 | 362/8/0 (1%) | 3% | 0.9% | Benign |
| V147I | 43043159/rs142204928 | 367/3/0 (0.4%) | 0.2% | 0.3% | VUS |
| I441V | 43058267/rs147638455 | 362/8/0 (1%) | 0.03% | 2% | Benign |
| HNF1B (NM_000458) | |||||
| V25L | 36104803/rs139107479 | 364/5/1b (1%) | 0.002% | 0% | Benign |
| S75F | 36104652/NA | 369/1/0 (0.1%) | 0.0004% | 0% | VUS |
| S342F | 36091606/rs780035561 | 363/7/0 (1%) | 0.003% | 0% | VUS |
| R435H | 36047353/rs200421746 | 369/1/0a (0.1%) | 0.005% | 0% | VUS |
| F443L | 36047328/rs8068014 | 366/4/0 (0.5%) | 0.7% | 0% | Benign |
| INS (NM_000207.2) | |||||
| R55H | 2182038/NA | 369/1/0b (0.1%) | 0.0008% | 0% | Pathogenic |
| ABCC8 (NM_001287174) | |||||
| R285Q | 17482192/rs199616008 | 369/1/0 (0.1%) | 0.01% | 0% | VUS |
| A355T | 17474779/rs145136257 | 369/1/0 (0.1%) | 0.05% | 0% | VUS |
| E612D | 17450199/rs764753690 | 369/1/0 (0.1%) | 0.0008% | 0% | Pathogenic |
| A958V | 17428948/rs772574110 | 369/1/0 (0.1%) | 0.0004% | 0% | VUS |
| A1370S | 17418477/rs757110 | 32/143/195 (72%) | 64% | 73% | Benign |
| V1573I | 17414570/rs8192690 | 303/65/2 (9%) | 5% | 8.8% | Benign |
| KCNJ11 (NM_000525) | |||||
| K23E | 17409572/rs5219 | 28/145/197 (73%) | 65% | 73% | Benign |
| L270V | 17408831/rs1800467 | 343/27/0 (4%) | 4% | 4% | Benign |
| V337I | 17408630/rs5215 | 29/146/195 (72%) | 65% | 73% | Benign |
| R365H | 17408545/rs750689750 | 369/1/0 (0.1%) | 0.003% | 0% | Pathogenic |
| S385C | 17408485/rs41282930 | 344/25/1 (3.6%) | 0.5% | 3% | Benign |
Legend. GME: Greater Middle East.
Carrier of both the GCK R64H and the HNF1B R435H variant.
Carrier of both the INS R55H and the HNF1B V25L variant.
Prevalence of identified variants was compared with available references including a Middle Eastern reference [18,19] and three novel mutations were found (Q19H in GCK; L254R and S328 N in HNF1A) (Table 1). The remaining 31 variants have previously been described [18,19].
3.2. Pathogenicity of variants
The variants were classified according to pathogenicity, resulting in the identification of six pathogenic variants, 14 VUS and 14 benign variants. All of the common variants were benign.
VUS were present in GCK, HNF1A, HNF4A, HNF1B and ABCC8 in 25 carriers, resulting in a prevalence of 6.8% (95% CI: 4.6–9.8%) in our samples (Table 2). Seven women were carriers of pathogenic variants located in GCK, HNF1A, INS, ABCC8 and KNJ11, resulting in a prevalence of 1.9% (0.9–3.9%) (Table 2). It should be noted that two patients carried two rare variants each. One carried the combination of a pathogenic and a benign variant and the other a pathogenic and a VUS. Thus, the combined prevalence was 8.4% (6.0–11.7%).
Table 2.
Number of carriers of VUS and pathogenic variants among 370 women with GDM.
| GDM (n = 370) | NC or benign, n | Carriers of VUS, n | Carriers of pathogenic variants, n | Prevalence of carriers of VUS, % (95% CI) | Prevalence of carriers of pathogenic variants, % (95% CI) |
|---|---|---|---|---|---|
| GCK | 365 | 2 | 3 | 0.5 (0.2–2.0) | 0.8 (0.3–2.4) |
| HNF1A | 361 | 8 | 1 | 2.2 (1.1–4.2) | 0.3 (0.05–1.5) |
| HNF4A | 367 | 3 | 0 | 0.8 (0.3–2.4) | 0 |
| HNF1B | 361 | 9 | 0 | 2.4 (1.3–4.6) | 0 |
| INS | 369 | 0 | 1 | 0 | 0.3 (0.05–1.5) |
| ABCC8 | 366 | 3 | 1 | 0.8 (0.3–2.4) | 0.3 (0.05–1.5) |
| KCNJ11 | 369 | 0 | 1 | 0 | 0.3 (0.05–1.5) |
| Total, specific | 340 | 25 | 7 | 6.8 (4.6–9.8) | 1.9 (0.9–3.9) |
| Total, combined | 339a | 31a | 8.4% (6.0–11.7) | ||
Legend. NC: Non-carrier. VUS: Variant of uncertain clinical significance. CI: Confidence interval.
One individual carried a pathogenic and a VUS and one carried a pathogenic and a benign variant.
3.3. Phenotypic characteristics
We did not observe any significant associations between age of examination, age of diagnosis, pre- and post-pregnancy weight or change in weight and carrying either VUS or pathogenic variants (Table 3).
Table 3.
Phenotypical characteristics among women with GDM with and without pathogenic variants in GCK, HNF1A, HNF4A, HNF1B, INS, ABCC8 and KCNJ11.
| GDM (n = 370) | N | NC or benign (n = 339) | Carriers of VUS (n = 24a) | Carriers of pathogenic variants (n = 7) | p-value (non-carriers vs. carriers of VUS) | p-value (non-carriers vs. carriers of pathogenic variants) |
|---|---|---|---|---|---|---|
| Age at examination (years) | 370 | 39.0 (34.5; 44.0) | 36.5 (34.5; 4050) | 39.0 (33.0; 41.00) | 0.3 | 0.5 |
| Age of diagnosis (years) | 370 | 33.0 (30.0; 38.0) | 32.5 (30.0; 35.0) | 34.0 (26.0; 35.0) | 0.3 | 0.5 |
| C-peptide (nmol/l) | 367 | 1.0 (0.75; 1.34) | 0.89 (0.72; 1.20) | 1.16 (0.99; 1.48) | 0.2 | 0.5 |
| Pre-pregnancy weight (kg) | 139 | 74.9 (65.0; 85.4) | 79.2 (66.4; 93.0) | 74.2 (65.8; 83.7) | 0.6 | 0.8 |
| Post-pregnancy weight (kg) | 218 | 78.3 (66.6; 91.0) | 77.8 (62.9; 84.5) | 71.8 (63.2; 77.9) | 0.6 | 0.3 |
| Change in weight (kg) | 116 | 1.5 (−1.68; 5.25) | 6.2 (4.3; 6.6) | −0.4 (−2.2; 1.9) | 0.3 | 0.7 |
Legend. Data is presented as median (interquartile range (IQR)). GDM: Gestational diabetes. NC: Non-carrier. VUS: variant of uncertain significance.
Carrier with both a VUS and a pathogenic variant is included among carriers of pathogenic variants.
3.4. Risk of developing HG or T2DM
Within the study period a total of 89 out of 370 women developed overt T2DM (24%; 95% CI: 20–29%). Among carriers of pathogenic variants, 57% (25–84%; n = 4) developed T2DM following GDM, 24% (12–43%; n = 6) of the carriers of VUS and 23% (19–28%; n = 79) of the non-carriers (Table 4).
Table 4.
Number of women with and without post-partum development of T2DM or HG.
| Without T2DM |
With T2DM |
Proportion T2DM (95% CI) |
RR vs. reference (non-carriers) | |
|---|---|---|---|---|
| Non-carriers (n = 339) | 260 | 79 | 23% (19–28%) | |
| Carriers of VUS (n = 24a) | 19 | 6 | 25% (12–45%) | 1.0 (0.5–2.1), p = 0.9 |
| Carriers of pathogenic variants (n = 7) |
3 |
4 |
57% (25–84%) |
2.5 (1.3–4.8), p = 0.009 |
| Without HG | With HG | Proportion with HG (95% CI) | ||
| Non-carriers (n = 339) |
202 |
137 |
40% (35–46%) |
|
| Carriers of VUS (n = 24a) | 17 | 8 | 33% (18–53%) | 0.8 (0.45–1.4), p = 0.4 |
| Carriers of pathogenic variants (n = 7) | 2 | 5 | 71% (36–92%) | 1.8 (1.1–2.9), p = 0.02 |
Legend. HG: Hyperglycemia. RR: Risk ratio. T2DM: type 2 diabetes mellitus. VUS: variant of uncertain significance.
Carrier with both a VUS and a pathogenic variant is included among carriers of pathogenic variants.
Hence, women with pathogenic variants had more than a 2-fold risk of developing overt postpartum T2DM compared to non-carriers, risk ratio (RR) = 2.5 (1.3–4.8; p = 0.009) (Table 4). There were no significant difference in the fraction of women with VUS who developed T2DM when compared to non-carriers (Table 4).
The total number of women who developed any form of HG within the study period was 147 (40%; 35–45%). Among carriers of pathogenic variants 71% (36–92%; n = 5) developed any form of HG compared to 32% (17–52%; n = 8) of the carriers of VUS and 40% (35–45%; n = 134) of the non-carriers. The RR of developing HG among carriers of pathogenic variant was 1.8 (1.1–2.9; p = 0.01) compared to non-carriers (Table 4). The RR was not significant when comparing non-carriers and carriers of VUS (Table 4).
3.5. Length of time from GDM to subsequent development of overt T2DM or any form of HG
The median time from GDM to development of T2DM was 41.2 months (IQR: 28.6; 49.5, n = 4) among carriers of pathogenic variants, 62.1 months (IQR: 53.0; 87.7, n = 6) among carriers of VUS and 43.0 months (IQR: 19.7; 77.3, n = 79) among non-carriers.
The median time from GDM to development of any form of HG was 28.9 months (IQR: 10.8; 35.2, n = 5) among carriers of pathogenic variants, 43.6 months (IQR: 37.3; 75.2, n = 8) among carriers of VUS and 39.3 months (IQR: 17.6; 77.4, n = 134) among non-carriers. The overall follow-up time for all the women was comparable between women who developed either any form of HG or overt T2DM versus those women who remained disease free until end of study according to carrier-status (Supplementary Table S2).
Kaplan-Meier analysis was used to compare time from GDM to development of T2DM or any form of HG between non-carriers vs. carriers of VUS and pathogenic variants. This analysis showed that women with pathogenic variants had a shorter disease-free period until either development of T2DM or HG compared to non-carriers, p = 0.005 and p = 0.0004, respectively (Fig. 1) with all of the women diagnosed with HG within 5 years after GDM (Fig. 1 and Supplementary Table S2). There were no significant differences in the proportion of women who remained disease-free between carriers of VUS and non-carriers (Fig. 1).
Fig. 1.

Kaplan-Meier curves of the time from GDM to development of overt T2DM (A) or any form of HG (B) for non-carriers and carriers of VUS and pathogenic variants.
Legend. HG: Hyperglycemia. T2DM: Type 2 diabetes mellitus. VUS: Variants of uncertain clinical significance.
4. Discussion
Seven carriers of pathogenic variants in GCK, HNF1A, INS, ABCC8 and KCNJ11 were found in a cohort of 370 Emirati women with GDM, corresponding to a prevalence of two percent. Carrying such variants predisposed the women to develop HG within 5 years after GDM. Moreover, identification of pathogenic variants in selected MODY genes in the Emirati population may be important as precision intervention can be applied. For example, women carrying variants in the GCK gene generally do not require treatment outside of the pregnancy [26], however, in women carrying a child who did not inherit the GCK mutation, the fetus will increase insulin secretion in response to the slight HG in the mother. This in turn increases the risk of macrosomia. As such, current recommendations state that a mother carrying pathogenic GCK variants is followed closely during pregnancy and that C-section is performed at signs of macrosomia [12].
Women carrying pathogenic variants in the transcription factor HNF1A as well as the two genes (ABCC8 and KCNJ11) encoding a beta-cell K-ATP channel are sensitive to sulphonylurea treatment [[26], [27], [28]]. Yet, due to the risk of trans-placental transfer of sulphonylurea, carriers of pathogenic HNF1A, ABCC8 or KCNJ11 variants are recommended to be transferred onto insulin before the third trimester in order to reduce the risk of fetal hyperinsulinism and macrosomia [12,13].
Women with a pathogenic variant in INS gene often require insulin treatment regardless of pregnancy as their insulin production is significantly reduced [29].
In our cohort of women with GDM, carriers of pathogenic variants had a higher risk of developing any form of HG and T2DM after delivery. A previous study on Danish women with a history of GDM, found that 71% of the women with pathogenic MODY variants had glucose levels corresponding to diabetes at follow-up after approximately 10 years [10]. This is in line with the current study, stating that 71% of women carrying pathogenic variants developed HG postpartum with a median follow-up time of 6 years. Thus, in addition to tailoring treatment during and post-pregnancy, identification of women with pathogenic MODY mutations can aid the identification of women in need of more frequent control visits postpartum.
Previous studies in women from other ethnic groups with GDM, have found prevalence of pathogenic GCK variants between 2 and 6% [10,11,30,31] and pathogenic HNF1A variants between 1 and 2% [10,11,30], thus, slightly higher than seen in our study. This could be due to ethnic differences or a more strict pathogenicity classification of identified variants in recent studies. More extensive variant information is now available, including prevalence of variants in individuals without diabetes, resulting in fewer variants classified as pathogenic.
One carrier of an INS variant was identified in a previous study [10], but none of the previous studies investigated the prevalence of rare ABCC8 and KCNJ11 pathogenic variants in patients with GDM. A systematic review investigated the effect of a polymorphism (rs5219) in KCNJ11, and found a slight association with elevated risk of GDM [32].
In the current study we identified three novel variants in the Emirati population (GCK: Q19H and HNF1A: L254R; S328 N). Two HNF1B variants (V25L and S342F) found to be rare in other populations, had a MAF of 1% in the Emirati population [18,19]. However, in order to validate whether these variants are more frequent in the Emirati population, a local reference genome is needed. The remaining variants were found in allele frequencies comparable to the ones published online [18,19].
One limitation of the study is the small sample size due to which we were not able to extrapolate the estimated prevalence in the overall population. Additionally, the number of patients developing HG subsequent to GDM is connected to the length of follow-up and it can be expected that a larger number of women would develop HG with increased follow-up time. Yet, follow-up time was longer for non-carriers and carriers of VUS compared to carriers of pathogenic variants, thus the lower number of events among non-carriers and carriers of VUS is likely not a consequence of follow-up time. However, median follow-up time for carriers of pathogenic variants without any events was shorter than the median follow-up for the pathogenic events group. This could perhaps indicate that even more events would have been observed, if follow-up times were lengthened. Finally, a limitation of the study is the lack of a healthy control group to which results could have been compared. Nevertheless, our study does show that pathogenic variants in MODY genes are present in the Emirati population and may be the cause of diabetes in women with GDM. In addition, the majority of carriers have developed HG within a few years after their pregnancy. Thus, accurate genetic diagnosis enables better treatment, early detection of complication related to fetal growth and a better follow-up after delivery.
5. Conclusions
In the present study, two percent of Emirati women with GDM carry pathogenic variants in genes known to be involved in monogenic diabetes. Correct diagnosis of women with GDM due to the presence of pathogenic variant in MODY genes, enables the application of precision medicine both during and outside of pregnancy and also allows for screening of family members, with early detection leading to earlier diagnosis and adequate treatment.
Data availability
The data supporting the results of this research paper is included within this article and its additional supplementary files.
Funding
The study was partially performed at the Novo Nordisk Foundation Center for Basic Metabolic Research, an independent research centre at the University of Copenhagen, partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk). The study was also partially performed at the Research Institute at Imperial College London Diabetes Centre (ICLDC) and partially funded by ICLDC (www.icldc.ae). The study was also supported by Augustinus Foundation (16–2922) and the Novo Nordisk Foundation through the Challenge Program (NNFOC0033950) and the Steno Collaborative Grants (NNF17OC0028328). Funders did not have any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CRediT authorship contribution statement
Hinda Daggag: were responsible for, Conceptualization, were responsible for design of the study, were involved in, Funding acquisition, critically revised the manuscript and contributed to the discussion, The final version of the paper was read and approved by all authors. Anette P. Gjesing: were responsible for, Conceptualization, were responsible for design of the study, took part in the, Formal analysis, were involved in, Funding acquisition, Writing – original draft, the article, critically revised the manuscript and contributed to the discussion, The final version of the paper was read and approved by all authors. Alshafi Mohammad: was involved in, Writing – review & editing, electronic medical records and, Data curation, critically revised the manuscript and contributed to the discussion, The final version of the paper was read and approved by all authors. Lars Ängquist: took part in the, Formal analysis, critically revised the manuscript and contributed to the discussion, The final version of the paper was read and approved by all authors. Bindu Shobi: were responsible for DNA extraction and, Data curation, collection. Suma Antony: were responsible for DNA extraction and, Data curation, collection. Dalia Haj: was responsible for recruitment. Alia Al Tikriti: was involved in extracting phenotypic, Data curation. Adam Buckley: critically revised the manuscript and contributed to the discussion, The final version of the paper was read and approved by all authors. Torben Hansen: were responsible for conception, were responsible for design of the study, took part in the, Formal analysis, were involved in, Funding acquisition, Writing – original draft, the article, critically revised the manuscript and contributed to the discussion, The final version of the paper was read and approved by all authors. Maha T. Barakat: were responsible for, Conceptualization, were involved in, Funding acquisition, critically revised the manuscript and contributed to the discussion. The final version of the paper was read and approved by all authors.
Declaration of competing interest
None.
Acknowledgement
From Novo Nordisk Foundation Centre for Basic Metabolic Research, University of Copenhagen, Denmark, we wish to thank A. Forman, T. H. Lorentzen and G. J. Klavsen for laboratory assistance, P. Sandbeck for data management, and C. Verdich and L. Ryborg for grant management. From ICLDC Research Institute, we wish to thank H. Al-Karbi for patient recruitment and T. Ghulam for laboratory assistance. Authors have no relevant conflicts of interest to disclose.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.metop.2022.100213.
Contributor Information
Hinda Daggag, Email: hindadaggag@gmail.com.
Anette P. Gjesing, Email: anette.gjesing@sund.ku.dk.
Alshafi Mohammad, Email: alshafimd@yahoo.com.
Lars Ängquist, Email: lars.angquist@sund.ku.dk.
Bindu Shobi, Email: bshobi@icldc.ae.
Suma Antony, Email: santony@icldc.ae.
Dalia Haj, Email: dhaj@icldc.ae.
Alia Al Tikriti, Email: aaltikriti@icldc.ae.
Adam Buckley, Email: abuckley@icldc.ae.
Torben Hansen, Email: torben.hansen@sund.ku.dk.
Maha T. Barakat, Email: mbarakat@icldc.ae.
Appendix A. Supplementary data
The following is/are the supplementary data to this article:
References
- 1.Federation I.D. ninth ed. 2019. IDF diabetes atlas.https://www.diabetesatlas.org/en/resources 2019. [Google Scholar]
- 2.Abuyassin B., Laher I. Diabetes epidemic sweeping the Arab world. World J Diabetes. 2016;7:165–174. doi: 10.4239/wjd.v7.i8.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azizi F., Hadaegh F., Hosseinpanah F., Mirmiran P., Amouzegar A., Abdi H., Asghari G., Parizadeh D., Montazeri S.A., Lotfaliany M., et al. Metabolic health in the Middle East and north Africa. Lancet Diabetes Endocrinol. 2019;7:866–879. doi: 10.1016/S2213-8587(19)30179-2. [DOI] [PubMed] [Google Scholar]
- 4.Mohamad M.N., Ismail L.C., Stojanovska L., Apostolopoulos V., Feehan J., Jarrar A.H., Al Dhaheri A.S. The prevalence of diabetes amongst young Emirati female adults in the United Arab Emirates: a cross-sectional study. PLoS One. 2021;16 doi: 10.1371/journal.pone.0252884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.AlSafar H.S., Al-Ali M., Elbait G.D., Al-Maini M.H., Ruta D., Peramo B., Henschel A., Tay G.K. Introducing the first whole genomes of nationals from the United Arab Emirates. Sci Rep. 2019;9 doi: 10.1038/s41598-019-50876-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osman W., Hassoun A., Jelinek H.F., Almahmeed W., Afandi B., Tay G.K., Alsafar H. Genetics of type 2 diabetes and coronary artery disease and their associations with twelve cardiometabolic traits in the United Arab Emirates population. Gene. 2020;750 doi: 10.1016/j.gene.2020.144722. [DOI] [PubMed] [Google Scholar]
- 7.Murphy R., Ellard S., Hattersley A.T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metabol. 2008;4:200–213. doi: 10.1038/ncpendmet0778.ncpendmet0778. [pii] [DOI] [PubMed] [Google Scholar]
- 8.Anik A., Catli G., Abaci A., Bober E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab : JPEM (J Pediatr Endocrinol Metab) 2015;28:251–263. doi: 10.1515/jpem-2014-0384. [DOI] [PubMed] [Google Scholar]
- 9.Monsonego S., Clark H., Karovitch A., O'Meara P., Shaw T., Malcolm J. Management and outcomes of maturity-onset diabetes of the young in pregnancy. Can J Diabetes. 2019;43:647–654. doi: 10.1016/j.jcjd.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Gjesing A.P., Rui G., Lauenborg J., Have C.T., Hollensted M., Andersson E., Grarup N., Sun J., Quan S., Brandslund I., et al. High prevalence of diabetes-predisposing variants in MODY genes among Danish women with gestational diabetes mellitus. Journal of the Endocrine Society. 2017;1:681–690. doi: 10.1210/js.2017-00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weng J., Ekelund M., Lehto M., Li H., Ekberg G., Frid A., Aberg A., Groop L.C., Berntorp K. Screening for MODY mutations, GAD antibodies, and type 1 diabetes--associated HLA genotypes in women with gestational diabetes mellitus. Diabetes Care. 2002;25:68–71. doi: 10.2337/diacare.25.1.68. [DOI] [PubMed] [Google Scholar]
- 12.Dickens L.T., Naylor R.N. Clinical management of women with monogenic diabetes during pregnancy. Curr Diabetes Rep. 2018;18:12. doi: 10.1007/s11892-018-0982-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shepherd M., Brook A.J., Chakera A.J., Hattersley A.T. Management of sulfonylurea-treated monogenic diabetes in pregnancy: implications of placental glibenclamide transfer. Diabet Med : a journal of the British Diabetic Association. 2017 doi: 10.1111/dme.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.American Diabetes A. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care. 2021;44:S15–S33. doi: 10.2337/dc21-S002. [DOI] [PubMed] [Google Scholar]
- 15.Gao R., Liu Y., Gjesing A.P., Hollensted M., Wan X., He S., Pedersen O., Yi X., Wang J., Hansen T. Evaluation of a target region capture sequencing platform using monogenic diabetes as a study-model. BMC Genet. 2014;15:13. doi: 10.1186/1471-2156-15-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet Med : official journal of the American College of Medical Genetics. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.project G.V. 2020. The greater Middle East (GME) variome project. [Google Scholar]
- 20.De Franco E., Saint-Martin C., Brusgaard K., Knight Johnson A.E., Aguilar-Bryan L., Bowman P., Arnoux J.B., Larsen A.R., May S., Greeley S.A.W., et al. Update of variants identified in the pancreatic beta-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat. 2020;41:884–905. doi: 10.1002/humu.23995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osbak K.K., Colclough K., Saint-Martin C., Beer N.L., Bellanne-Chantelot C., Ellard S., Gloyn A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- 22.Colclough K., Bellanne-Chantelot C., Saint-Martin C., Flanagan S.E., Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013;34:669–685. doi: 10.1002/humu.22279. [DOI] [PubMed] [Google Scholar]
- 23.Bonnefond A., Boissel M., Bolze A., Durand E., Toussaint B., Vaillant E., Gaget S., Graeve F., Dechaume A., Allegaert F., et al. Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat Metab. 2020;2:1126–1134. doi: 10.1038/s42255-020-00294-3. [DOI] [PubMed] [Google Scholar]
- 24.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glumer C., Jorgensen T., Borch-Johnsen K., Inter s. Prevalences of diabetes and impaired glucose regulation in a Danish population: the Inter99 study. Diabetes Care. 2003;26:2335–2340. doi: 10.2337/diacare.26.8.2335. [DOI] [PubMed] [Google Scholar]
- 26.Steele A.M., Shields B.M., Wensley K.J., Colclough K., Ellard S., Hattersley A.T. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311:279–286. doi: 10.1001/jama.2013.283980. [DOI] [PubMed] [Google Scholar]
- 27.Pearson E.R., Starkey B.J., Powell R.J., Gribble F.M., Clark P.M., Hattersley A.T. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275–1281. doi: 10.1016/S0140-6736(03)14571-0. [DOI] [PubMed] [Google Scholar]
- 28.Hansen T., Eiberg H., Rouard M., Vaxillaire M., Moller A.M., Rasmussen S.K., Fridberg M., Urhammer S.A., Holst J.J., Almind K., et al. Novel MODY3 mutations in the hepatocyte nuclear factor-1alpha gene: evidence for a hyperexcitability of pancreatic beta-cells to intravenous secretagogues in a glucose-tolerant carrier of a P447L mutation. Diabetes. 1997;46:726–730. doi: 10.2337/diab.46.4.726. [DOI] [PubMed] [Google Scholar]
- 29.Harris A.G., Letourneau L.R., Greeley S.A.W. Monogenic diabetes: the impact of making the right diagnosis. Curr Opin Pediatr. 2018;30:558–567. doi: 10.1097/MOP.0000000000000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zurawek M., Wender-Ozegowska E., Januszkiewicz-Lewandowska D., Zawiejska A., Nowak J. GCK and HNF1alpha mutations and polymorphisms in Polish women with gestational diabetes. Diabetes Res Clin Pract. 2007;76:157–158. doi: 10.1016/j.diabres.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Stoffel M., Bell K.L., Blackburn C.L., Powell K.L., Seo T.S., Takeda J., Vionnet N., Xiang K.S., Gidh-Jain M., Pilkis S.J., et al. Identification of glucokinase mutations in subjects with gestational diabetes mellitus. Diabetes. 1993;42:937–940. doi: 10.2337/diab.42.6.937. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C., Bao W., Rong Y., Yang H., Bowers K., Yeung E., Kiely M. Genetic variants and the risk of gestational diabetes mellitus: a systematic review. Hum Reprod Update. 2013;19:376–390. doi: 10.1093/humupd/dmt013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the results of this research paper is included within this article and its additional supplementary files.