

Abstract
Background and objectives:
We sought to determine clinical significance of neuronal septin autoimmunity and evaluate for potential IgG effects.
Methods:
Septin-IgGs were detected by indirect immunofluorescence assays (IFA, mouse tissue and cell based) or western blot. IgG binding to (and internalization of) extracellular septin epitopes were evaluated for by live rat hippocampal neuron assay. The impact of purified patient IgGs on murine cortical neuron function was determined by recording extracellular field potentials in a multielectrode array platform.
Results:
Septin-IgGs were identified in 23 patients. All 8 patients with septin-5-IgG detected had cerebellar ataxia, 7 with prominent eye movement disorders. One of 2 with coexisting septin-7-IgG had additional psychiatric phenotype (apathy, emotional blunting and poor insight). Fifteen patients had septin-7 autoimmunity, without septin-5-IgG detected. Disorders included encephalopathy (11; 2 with accompanying myelopathy, 2 were relapsing), myelopathy (3) and episodic ataxia (1). Psychiatric symptoms (≥1 of agitation, apathy, catatonia, disorganized thinking, and paranoia) were prominent in 6/11 with encephalopathic symptoms. Eight of 10 with data available (from 23 total) improved after immunotherapy, a further 2 spontaneously. Staining of plasma membranes of live hippocampal neurons produced by patient IgGs (subclasses 1 and 2) colocalized with pre- and post-synaptic markers. Decreased spiking and bursting behavior in mixed cultures of murine glutamatergic and GABAergic cortical neurons produced by patient IgGs were attributable to neither antigenic crosslinking and internalization nor complement activation.
Interpretation:
Septin-IgGs are predictive of distinct treatment-responsive autoimmune CNS disorders. Live neuron binding and induced electrophysiologic effects by patient IgGs may support septin-specific pathophysiology.
Keywords: septin-5, septin-7, ataxia, encephalitis, psychosis
Summary for social media
Septin autoimmunity has been reported as a cause of cerebellar ataxia in a limited number of patients. This current study purpose is to evaluate for phenotypic associations of neural septin IgGs (septin-5 and septin-7) and to establish if septin-IgGs had pathogenic potential. Septin-5-IgG is associated with ataxia with prominent hyperkinetic eye movement disorders and septin-7-IgG is associated with encephalopathy with prominent neuropsychiatric features; both disorders are often immune therapy responsiveness. Consistent with septin-IgGs having pathogenic potential, CSF IgGs from both septin autoimmune patient groups bound to the surface of live rodent neurons and colocalized with polyclonal septin antibodies. In addition, IgGs purified from serum of septin autoimmune patients decreased spiking and bursting behavior in mixed cultures of glutamatergic and GABAergic cortical neurons. In clinical practice, septin autoimmunity should be considered in the differential diagnosis of immune therapy-responsive potentially IgG mediated autoimmune CNS diseases.
Introduction
We previously reported septin-5-immunoglobulin-G (IgG) autoantibody as an autoimmune CNS disease biomarker in 4 patients with ataxia, an association confirmed by others.1, 2 We have since determined an additional form of septin neurological autoimmunity for which septin-7 is the principal target. The clinical spectrum of neuronal septin autoimmunity requires elucidation. Differences in brain regional distributions and functions of septins (regulation of neurotransmitter exocytosis by septin-5, and dendritic branching by septin-7) raises the question of distinct septin autoimmune neurological phenotypes.
Septins are pan-eukaryotic guanosine-5’-triphosphate (GTP)-binding proteins originally observed in the septa of yeast.3 In mammals, including humans, neuronal septins-5 and -7 have structural and functional diversity, existing as monomers and in complexes with septins-6 and −11, among others.4 They have diverse specialized biological functions determined by their location and tertiary structure. In general, septins are known to regulate cytoplasmic processes that are dependent on microtubules (mitosis and meiosis) or actin (cellular morphogenesis, cell migration and host defense), and also serve as diffusion barriers.3 It is also possible that extracellular domains of neuropil-facing, membrane-bound septins might be targets of pathogenic IgGs.5
Herein, we report our neuronal septin autoimmune clinical and serological experience to date. We demonstrate 2 distinct CNS phenotypic groups, septin-5-IgG with cerebellar ataxia, and septin-7-IgG with encephalopathy predominant. Both disorders have strong propensity for immune therapy response. Though septins are abundant in cell cytoplasm, polyclonal septin antibodies are known to bind to extracellular epitopes on live hippocampal neurons.6 Therefore, we hypothesized that patient septin-IgGs would also bind to live rodent neurons, and that there might be occurrence of functional effects of purified patient IgGs on spiking and bursting behavior of murine cortical neuronal cultures.
Methods
The Mayo Clinic Institutional Review Board (IRB) approved human specimen acquisition and retrospective review of patients’ histories (IRB#, 16–009814).
Patients and septin antibody testing
Over the course of January 1st 1997 until December 31st 2021, 651694 serums and 266507 CSF specimens from patients with diverse subacute neurological illnesses (encephalopathies, movement disorders, myelopathies, neuropathies, paraneoplastic disorders) were evaluated for neural IgG antibodies. Of those, 30 specimens (18 serums [0.003%] and 12 CSF [0.004%]) from 23 patients fulfilled mouse tissue indirect immunofluorescence assay (IFA) ‘septin pattern’ criteria (IgG staining of diffuse neural synapses [cerebrum- and cerebellar molecular layer-predominant] and renal glomeruli), as previously described.1 Septin specificities (5 and 7) were confirmed molecularly by fixed cell-based indirect immunofluorescence assays (CBAs) in all serum and CSF samples, and IgG subclasses 1–4 in a subset of samples (5 serums, 2 CSFs). The CBAs utilized human embryonic kidney (HEK)293 cells transfected with full-length recombinant histidine-tagged septin-5 or septin-7 cDNAs (isoform 1 for both). Control cells were transfected with empty vector. Cells were grown on glass coverslips, fixed with ice-cold acetone, and prepared as millimeter-sized biochip fragments on microscope slides as a mosaic of septin-expressing and control cells (Euroimmun AG, Lübeck, Germany). Secondary anti-human IgG antibodies were utilized undiluted (Euroimmun kit secondary pan-IgG) or diluted 1:200 (IgG1 [Invitrogen] and IgG2–4 [Southern Biotech]). Serum from the initial 11 patients evaluated were also tested for other neural septin-IgGs (6 and −11) by recombinant protein western blot.
Hippocampal neuron preparation
Rat hippocampi were dissected from embryonic (embryonic [E] day 18–20) animals, and meninges were removed. Tissue was minced in Hanks Balanced Salt Solution (HBSS). Minced tissue was incubated in 4.5 mL HBSS with 0.5 mL 0.25% trypsin in a 37°C water bath for 15 minutes with gentle inversion every 5 minutes. Tissue was washed with HBSS +10% fetal bovine serum with DNAse at 37°C for 5 minutes. After washing 2 more times in HBSS at 37°C, tissue was triturated in HBSS. After larger tissue chunks settled, supernatants were transferred to new tubes. Trituration was repeated, and supernatants were combined. Cells were counted and plated at 5×104 in Neural Basal Plus media with B27 supplement (Gibco, Gaithersburg, MD) in poly-L-lysine coated chamber slides (Corning) at 3×104 cells/well and held at 37°C in a humidified atmosphere of 5% CO2. Fresh medium was exchanged for half the spent medium volume every 4 days.
Live hippocampal neuron immunofluorescence
Live cell immunostaining was performed at 4°C. After washing with PBS, neurons were exposed for 30 minutes to undiluted patient CSF samples and, where applicable, mouse monoclonal antibody against human septin-5 (1:400; Invitrogen). Six patient CSF specimens were evaluated for neuronal cell surface IgG binding in live rat hippocampal neuronal cultures. The 2 control CSF specimens were from patients with anti-N-methyl-D-aspartate receptor (NMDA-R) encephalitis or normal pressure hydrocephalus (NPH). Cells were washed in PBS and exposed for 30 minutes to fluoroscein isothiocyanate (FITC)-conjugated goat anti-human IgG (Southern Biotech), and where applicable, Alexa fluor 647 goat anti-mouse IgG (heavy [H] and light [L] chain-specific, 1:1000, Invitrogen) secondary antibody diluted 1:200 in 10% normal goat serum (NGS). Cells were washed in PBS and exposed to 4% paraformaldehyde at room temperature for 15 minutes. To label neurons post-fixation, cells were permeabilized in 0.2% Triton X-100 for 5 minutes, washed in PBS, then blocked in 10% NGS for 30 minutes. Cells were subsequently exposed to primary commercial antibodies; mouse anti-acetylated tubulin IgG (1:1000; Sigma-Aldrich [St. Louis, MO]) or anti-PSD95 (1:400; Invitrogen), and rabbit anti-synaptophysin (1:400; Invitrogen). Immunostaining of septins-5 and -7 post-fixation was accomplished using commercial antibodies (1:400 dilution) against human septins: monoclonal anti-septin-5 (Invitrogen) raised in mouse, and polyclonal anti-septin-5 (Sigma-Aldrich) and anti-septin-7 (Bethyl Laboratories), both raised in rabbit. For comparison, binding assay was also done in green fluorescent protein (GFP)-tagged septin-5 transfected HEK293 cells (fixed and live) using mouse anti-septin-5.1 Cells were washed, probed with TRITC-conjugated goat anti-rabbit IgG and Alexa fluor 647 goat anti-mouse IgG (H+L) (1:1000, Invitrogen), washed in PBS, and mounted in Prolong Gold with DAPI (Invitrogen, Carlsbad, CA). Confocal images were captured using a Zeiss LSM710 confocal microscope with ×40 water immersion lens or an Olympus Fluoview FV1000 confocal microscope with ×60 water immersion lens.
Septin modulation (antigen internalization) assay
To test modulating activity of the patient septin antibodies, live neurons were exposed to patient CSF (1:2) or serum (1:10) diluted in Neural Basal Plus media with B27 supplement (Gibco, Gaithersburg, MD). Cells were incubated 6.5 hours at 37°C in a humidified atmosphere of 5% CO2. Cells were placed on ice, washed with cold PBS and exposed for 30 minutes to FITC-conjugated goat anti-human IgG (1:200; Southern Biotech). After washing with PBS, cells were fixed in 4% PFA for 15 minutes at ambient temperature, exposed for 5 minutes to 0.2% Triton X-100 and blocked in 10% NGS for 30 minutes. Cells were incubated overnight at 4oC with primary commercial antibodies, washed in PBS and probed after 1 hour at ambient temperature with secondary antibodies.
Cortical neuron preparation
Murine cortices were dissected from E15 fetuses and meninges were removed as described previously.7 Tissue was enzymatically-digested in 2 mg/mL papain (Worthington Biochemical, # LS003126) in HBSS (Gibco, 14025076) for 10 minutes at 37°C followed by quenching of enzymatic digestion in ‘Plate Media’ composed of high glucose Dulbecco’s Modified Eagle Medium (DMEM, [ThermoFisher, #11965092]) with 10% fetal bovine serum and 10% F12 supplement (Invitrogen 31765–035). Cells were mechanically triturated to dissociate cortices 20 times with a P1000 tip and 20 times with a fire-polished glass Pasteur pipet (VWR, #14672–380). Cells were pelleted at 250g for 5 minutes before repeated trituration with a P1000 and glass-fired Pasteur pipet tip in Plate Media. Axion MEA plates (Axion Biosystems, # M768-tMEA-48B) were coated for 4 hours with 0.1% sterile polyethylenimine (Sigma Aldrich, #408727) before being washed 4 times with sterile water and allowed to air dry overnight. Before the addition of cortical neurons, plates were coated with 20 ug/mL laminin (Sigma Aldrich L2020) for 45 min at 37°C. Laminin was removed from each well immediately prior to plating 105 cells in 50 uL Plate Media. After 2 hours at 37°C/5%CO2 450 uL/well of ‘Feed Media’ was added consisting of Neurobasal media (Invitrogen 21103–049) with 2% B27 supplement (Invitrogen, #17504–044), 1% penicillin-streptomycin (Life Technologies, #10378–016), and 0.5 mM Glutamax (ThermoFisher, #35050–061) supplemented with BDNF (10 ng/mL, PeproTech, #450–02) and IGF-1 (10 ng/mL, BD Biosciences, #354037). Half-media changes were performed every other day.
Live cortical neuron electrophysiology
IgGs were purified from 4 individual autoimmune septin patient sera using Protein G columns (HP SpinTrap, GE healthcare, #GE-289031–34) and then dialyzed against artificial CSF (136 mM Na, 145.1 mM Cl, 1.3 mM Mg, 2 mM Ca, 10 mM HEPES) using 0.025um dialysis membranes (MF-Millipore, #VSWP01300) before co-incubation with neuronal cultures. IgG titers for the 4 septin patient serums used were: septin-5-IgG, 1:7680 and 1:30720; septin-7-IgG, 1:3840 and 1:7680 (normal, ≤ 240). Realtime electrophysiological data were obtained from mixed murine glutamatergic and GABAergic cortical neurons on day 14 in vitro using Axion Cytoview 48-well culture plates with extracellular electrodes (Axion Biosystems, #M768-tMEA-48B) on the Axion Maestro platform.7 Spontaneous extracellular potentials were recorded for 10 minutes and wells with ≤ 2 active electrodes were excluded from the assay design. Active wells were divided into tiers according to overall spike frequency and then proportionately assigned to experimental conditions. The volume of media was reduced to 37.5 uL/well and spontaneous firing was recorded for three 10-minute epochs at 37°C/5%CO2. Septin autoimmune patient and control specimens derived from individual patients (12.5μl of each; 25% final concentration by volume) were added to condition-matched wells. Controls were artificial CSF (1), CSF from myelin oligodendrocyte glycoprotein (MOG)-IgG positive patients (2), and CSF from a non-autoimmune disease patient (1). Extracellular field potential recordings were collected every 10 minutes with a 10-minute rest between recordings over 6 hours. Filtered extracellular field potentials (Butterworth high pass at 200 Hz; lowpass at 3000 Hz) with unipolar peaks that exceeded 6 standard deviations (STD) above baseline as assessed by adaptive thresholding for each individual electrode were defined as spikes8,9. Bursts were defined as ≥ 5 spikes on a single electrode with a maximum inter-spike interval of 100 msec and network bursts were defined as ≥ 50 spikes detected on ≥ 35% of electrodes/well with a maximum inter-spike interval of 100 msec. Data were analyzed in GraphPad Prism Software v9.1.0 using 2-way ANOVA with a mixed-effects model for repeated measures analysis. Correction for multiple comparisons was performed using Dunnett’s method with artificial CSF as the control condition and alpha ≤0.05 for significance. Bicinchoninic acid analysis was performed on dialyzed CSF samples to ascertain protein concentration, and values were compared to bursts at 300 minutes (logistic regression analysis).
Results
Diagnostic serology findings
For the 23 included patients, a septin-specific IgG pattern of staining was identified by tissue IFA in all 12 CSF samples and 17/18 sera (1 patient was positive in CSF only). Median IgG value for CSF was 1:256 (range, 4–2048; normal, ≤2) and, for serum, 1:15360 (range, 480–61440; normal ≤240). Tissue staining characteristics included prominent staining of synaptic elements (neuropil) of the mouse cerebrum and cerebellum (Figure 1). The molecular layer of the cerebellum and the thalamus demonstrated stronger immunoreactivity than the midbrain, hippocampus, cortex, and basal ganglia. The myenteric plexus of the gastric mucosa and renal glomeruli were also reactive in all specimens. By tissue IFA, additional interstitial staining of non-neural gastric mucosal and kidney tubular elements was observed only in those specimens that were septin-7-IgG CBA positive (Figure 1). Septin-5- and -7 CBAs yielded IgGs reactive with septin-7 (15 patients), septin-5 (6), or both (2).
Figure 1. Distinct patterns of septin-5- and septin-7-IgG staining by mouse tissue-based indirect immunofluorescence assay.
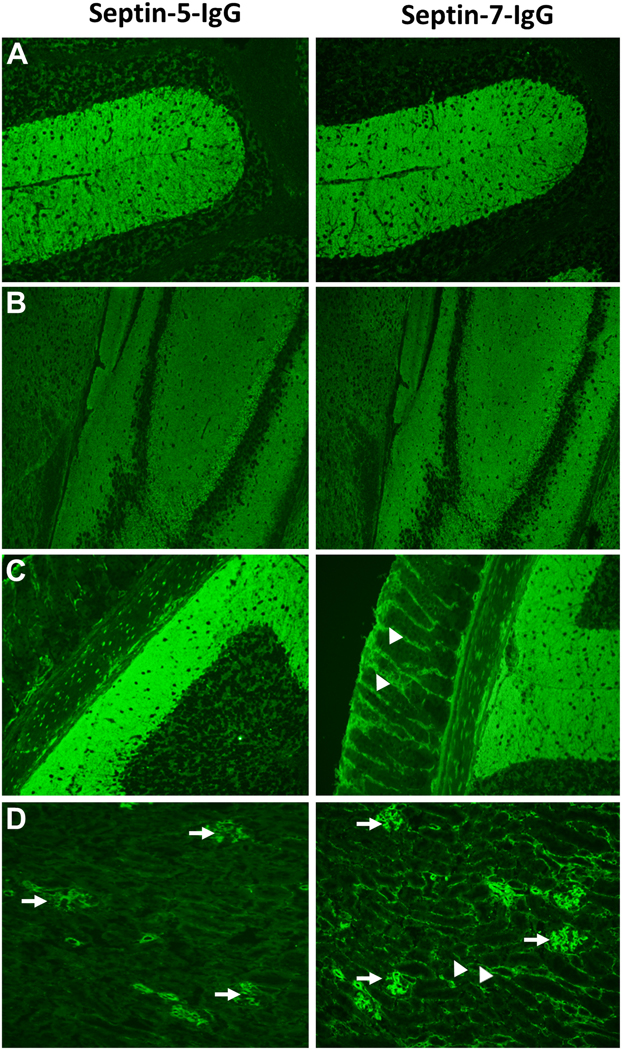
Both septin-5 and -7-IgGs produce staining of synaptic elements (within the cerebellum [A, molecular layer predominant], hippocampus and cerebral cortex [B], myenteric plexus and gastric mucosal neurites [C]) and renal glomerular podocytes (D, arrows). In addition, septin-7 IgG also produces staining of gastric mucosal and renal interstitial elements (C & D, right panels, arrow heads).
IgG subclasses detected were IgG1 (all 7 samples tested) and IgG2 (2 tested, both with septin-7 autoimmunity). IgG3 and IgG4 subclasses were not detected in any sample. One of 4 septin-5-IgG positive patients tested, and 6 of 7 septin-7-IgG positive patients tested had coexisting septin-6- and −11-IgGs detected by recombinant protein western blot. Two patients (7 and 22) had coexisting non-septin IgGs detected in CSF (GTPase regulator associated with Focal Adhesion Kinase [GRAF] and neurofilament heavy chain, 1 each).
Patient demographic, clinical & test findings
Fourteen of 23 patients were men (61%). Median symptom onset age was 63 years (range, 40–85), Supplementary tables 1 and 2. Septin-IgGs were identified in both serum and CSF (8), serum only (9), or CSF only (6). These differences were mostly related to sample availability, though 1 patient had positive CSF with negative serum.
Eight patients were septin-5-IgG seropositive by IFA and CBA (Supplementary table 1). All had pancerebellar ataxia, 7 with prominent eye movement disorders. The eye movement and speech exam for patient 6 is demonstrated in the Video. None had neoplasms reported to indicate a paraneoplastic cause; one had symptom onset 1 month after a mild SARS-CoV-2 respiratory illness. Two had coexisting septin-7-IgG detected, one of whom developed apathy, emotional blunting and poor insight after ataxia onset.
Video.
Examination from patient 6 is demonstrated. The video demonstrates coarse saccadic eye movements and nystagmus, ataxia dysarthria and dysmetria.
Fifteen patients were septin-7-IgG seropositive by IFA and CBA (Supplementary table 2) and were septin-5-IgG negative. Neurological phenotypes were encephalopathy (9, relapsing in 2), encephalomyelopathy (2), myelopathy (2), painful myelopolyradiculopathy (1) and episodic ataxia with normal exam (1). Psychiatric symptoms (≥1 of agitation, apathy, catatonia, disorganized thinking, and paranoia) were prominent in 6 of 11 patients with encephalopathic symptoms. Two patients had preexisting poorly defined neuropsychiatric problems before subacute encephalopathic decline. Four had ≥1 concurrent neoplasm (breast adenocarcinoma, 2; non-Hodgkin lymphoma, 1; carcinoid, 1; myelodysplastic syndrome, 1). Another 2 patients developed neurological symptoms post-herpes simplex virus (HSV)-1 encephalitis.
Imaging findings in 7 septin-5 autoimmune cases (including both with septin-7-IgG coexisting) with data available were normal MRI (4), focal cerebellar atrophy on MRI (2, Figure 2) and focal cerebellar FDG-hypermetabolism on PET-CT scan (1), Supplementary table 1. Findings in 9 septin-7 cases with MRI data available were normal or non-specific (3), bilateral hippocampal T2 signal abnormality (2, superimposed on typical HSV-1 type T2 hyperintense temporal neocortical changes in 1), generalized cerebral atrophy (2), pre-frontal T2 hyperintensity and progressive frontal lobe atrophy (1, Figure 2) and temporal lobe atrophy (post-HSV encephalitis, 1), Supplementary table 2.
Figure 2. MRI images from septin-5 and -7-IgG positive patients.

MRI images from patient 4 and patient 18. A, Patient 4 had cerebellar atrophy (T1 sagittal). B, Patient 18 (early imaging, left; follow-up imaging, right) carried a diagnosis of PLS and had increased T2 signal in unilateral followed later by bilateral superior precentral gyri (arrow heads), and had progressive frontal lobe atrophy over 4 years (all images axial T2 FLAIR).
Of 23 total patients, 15 had CSF data available, 11 of whom had inflammatory changes (at least one of protein > 100mg/dL, lymphocytic white blood cell count elevation, elevated IgG synthesis rate or CSF-exclusive oligoclonal bands), Supplementary tables.
Patient treatments & Outcomes
Outcomes available in 6 septin-5-IgG ataxia cases were: improvements with immunotherapy (4), rapid spontaneous improvement before treatment could be initiated (1) and death (1), Supplementary table 1. Immune treatment responses, available in 5 of the septin-7-IgG encephalopathy cases were rapid improvements in 4, and death in 1 (from likely corticosteroid-related bowel perforation). For 3 other patients, 1 with HSV-1 encephalitis recovered after acyclovir treatment, 1 had rapid spontaneous improvement before treatment could be initiated, and another untreated worsened, Supplementary table 2.
Live hippocampal neuron binding
All 6 patient CSF specimens tested (3 septin-5-IgG positive only, 3 septin-7-IgG positive only) were reactive with plasma membrane surfaces of live hippocampal neurons (Figures 3 and 4). Staining was punctate and more restricted in comparison to that produced by a control CSF from a patient with anti-NMDA-R encephalitis (Figure 3). Both commercial septin-5 antibodies (mouse and rabbit) produced an identical diffuse pattern of staining of permeabilized neurons (Figure 4A), though only the mouse monoclonal antibody produced staining of live neurons (Supplementary Figure 1). The mouse monoclonal septin-5 antibody produced punctate membranous staining of live neurons (Figure 4B), but not live septin-5 transfected HEK293 cells (Supplementary Figure 2). This staining by mouse anti-septin-5 of live neurons colocalized with staining produced by septin-5 autoimmune patient CSF (Figure 4B). A presynaptic localization for septins-5 and -7 was supported by colocalization with anti-synaptophysin staining (Figure 4C and 4D). A post-synaptic localization for septin-7 was supported by colocalization with anti-PSD-95 (Figure 4E). After incubating the live neurons with patient serum or CSF at 37°C for 6.5 hours, septin-5 and -7-IgG staining was preserved, suggesting antigenic modulation (crosslinking and internalization) had not occurred.
Figure 3. Septin patient IgG binds to living hippocampal neurons.

Live immunostaining of control and patient CSF. NMDAR-IgG (green, upper left) serves as a positive control. NPH-IgG (lower left) lacks specific binding. Septin-5 patient IgG (2 patients shown, upper middle and right) binds in a punctate linear pattern. Septin-7 patient IgG (2 patients shown, lower middle and right) binds in a punctate pattern, more diffuse than seen for septin-5-IgG. DAPI, blue. Scale bar, 50 μm.
Figure 4. Septin patient IgG colocalizes with synaptic proteins.
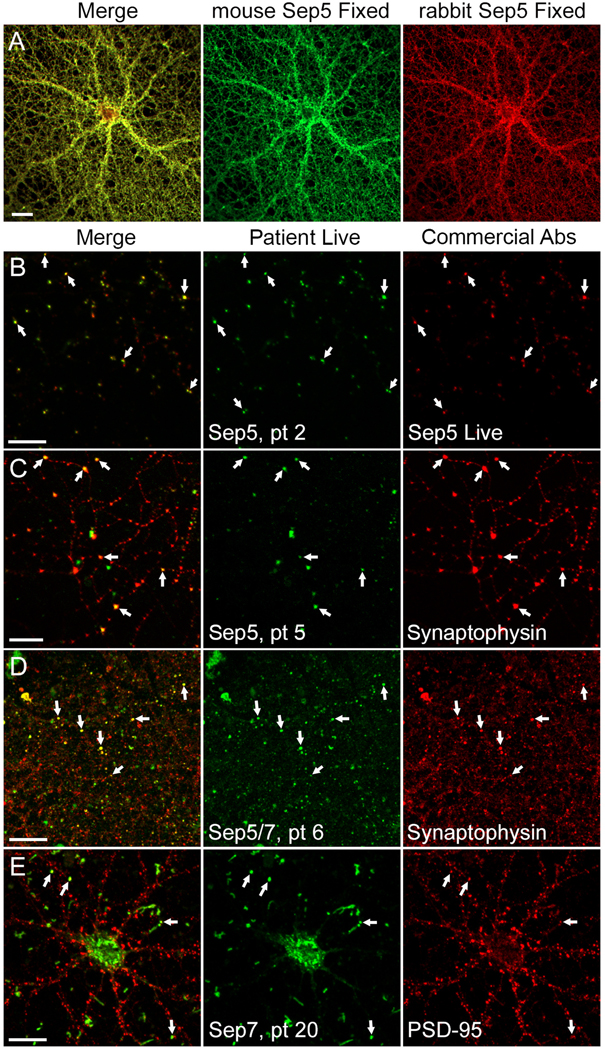
Live and fixed immunostaining of hippocampal neurons. A) Assessment of anti-septin-5 reagents: dual septin-5 immunostaining (mouse monoclonal, green; rabbit polyclonal, red) in fixed and permeabilized neuron cultures. Co-localization (yellow in merge) of antibody signals is consistent with septin-5 specificity. B) Live immunostaining of septin-5 autoimmune patient CSF (green) colocalizes with mouse monoclonal septin-5 antibody (red) at punctate foci on plasma membranes (arrows). C) Live immunostaining of septin-5 patient CSF (green) colocalizes with synaptophysin (red) post-fixation (arrows; yellow in merged). D) Live immunostaining of septin-5/7 patient CSF (green) colocalizes with synaptophysin (red) post-fixation (arrows; yellow in merged). E) Live binding of septin-7 autoimmune patient CSF (green) partially colocalizes with post-fixation with anti PSD-95 (red). Shown as white in merged panel. Scale bars, 20 μm.
Effects of patient septin-IgGs on murine cortical neurons
The impact of septin IgGs on murine cortical neuron function was determined by recording extracellular field potentials in a multielectrode array platform. Mixed cultures of glutamatergic and GABAergic neurons were matured for 14 days in vitro, and were then exposed to: individual patient serum-derived purified IgGs diluted in artificial CSF (septin 5-IgG, n=2 patients; septin-7-IgG, n=2 patients), MOG-IgG positive CSF (n=2), CSF from a patient with no detectable autoantibodies (non-autoimmune; n=1 patient) or artificial CSF (n=1). To control for interwell-variation in spontaneous neuronal activity, spike, bursts, and network burst parameters were normalized to the within-well baseline recorded prior to IgG addition. Notably, neuronal activity did not significantly differ across groups at baseline. This analysis revealed that both IgGs from septin-5 and septin-7 autoimmune patients acutely inhibited the electrophysiologic parameters examined. Normalized spikes (activity recorded on a single electrode) were shown to be inhibited starting at 200 minutes post-treatment by septin-7 patient IgGs and at 260 minutes post treatment by septin-5 patient IgGs, Figure 5A. Bursting activity (repeated firing on a single electrode; Figure 5B) and network bursting activity (activity detected across multiple electrodes; Figure 5C) were also inhibited by 220 minutes after treatment. Notably, the number of extracellular field potentials comprising both bursts and network bursts was decreased by 40–100 minutes following addition of purified septin-IgGs (Figure 5, D and E). This response was not correlated with total protein content in the CSF used for stimulation (R2=0.038) but was highly correlated with septin-IgG seropositivity (Pearson r = 0.65). Taken together, these results indicate addition of IgGs from septin autoimmune patients resulted in an acute decrease of extracellular field potentials in synaptically-mature circuits comprised of glutamatergic and GABAergic neurons.
Figure 5. Effects of septin IgGs on extracellular field potentials in cultures of glutamatergic and GABAergic murine cortical neurons.

A-C) The number of spikes (A), bursts (B), and network bursts (C) per condition were recorded in 10-minute epochs for 6 hrs and normalized to percent of individual baseline counts. D-E) Normalized percent baseline of spikes per burst (D) and spikes per network bursts (E) were also recorded. Data are normalized within each well to the baseline firing activity to control for inter-well variability in spontaneous activity. Across-well averages are shown for each condition with error bars representing 95% confidence intervals. n=5–6 wells/patient sample. * indicates p≤0.05
Discussion
We report novel forms of immune therapy-responsive autoimmune disorders targeting neuronal septins whereby clinical presentations are distinct depending on the IgG profile. Patients with septin-5 autoimmunity (with or without septin-7-IgG coexisting) had ataxia with prominent hyperkinetic eye movement disorders1, 2 In keeping with this, septin-5 expression is particularly abundant in cerebellum & basal ganglia, and most especially enriched in the oculomotor control-relevant cerebellar dentate nuclei.1, 10 CNS phenotypes were more diverse among septin-7-IgG positive patients. Encephalopathy with prominent neuropsychiatric features was most common. In some septin-7 autoimmune cases, chronic psychiatric illness preceded subacute cognitive decline. Immune-therapy responsiveness was common to both patient groups. Furthermore, the only patient with progressive symptoms who was untreated had a poor outcome.
Evaluation of serum and CSF permitted detection of similar (but distinct) tissue IFA patterns for septin-5- and -7-IgGs, confirmed by septin-specific CBAs. We have also previously demonstrated that patient septin-IgG staining is specific by confocal IFA with rabbit septin antibody, and elimination of IFA staining after preabsorption of patient serum with recombinant septin protein.1 CSF from both septin autoimmune patient groups in this study bound to the surface of live rodent neurons, and colocalized with polyclonal septin antibodies, in a similar pattern to that previously published.6 In a small study (4 cases, 4 controls), IgGs purified from serum of septin autoimmune patients decreased spiking and bursting behavior in mixed cultures of glutamatergic and GABAergic cortical neurons. Further studies are needed to confirm septin-specificity and determine neurochemical and immunologic underpinnings of these observations.
Both septin-5-IgG and septin-7-IgG positive CSF specimens produced sparse, punctate patterns of staining on plasma membranes of live rat hippocampal neurons, that co-localized with established anti-septin reagents, with adjacency to presynaptic (anti-synaptophysin) and postsynaptic (anti-PSD95) markers. These observations are consistent with that previously reported for septin-5 and -7 rabbit polyclonal antibodies (septin-5 around presynaptic vesicles, septin-7 just beneath axon terminal membranes).6, 11 This staining was not observed in septin-5 transfected live HEK293 cells, which we speculate is due to biological differences between HEK293 cells and rodent neurons. Abrogation of septin-IgG live neuron staining was not observed under physiologic conditions, implying classical cross-linking and internalization (modulation) is not mechanistically pertinent. Though not formally assessed, complement cascade activation by IgG should not be the principal mechanism either (the electrophysiologic multielectrode array system utilized purified IgG rather than complement component containing serum). Future studies should explore potential antagonist effects of septin-IgGs on septins and disruption of interactions between septins and physically linked molecules12.
Septins have highly conserved GTPase and polybasic domain regions, but there is divergence in their N- and C-termini.13 14, 15 The polybasic domain binds directly to phosphoinositides on plasma membranes.16 They associate with each other to form diverse higher order hetero-oligomeric complexes, filament polymers and rings.17, 18 Different structures impart differentiated functions including cell membrane structure, and cytoplasmic scaffold and diffusion barrier capabilities.19 They also coordinate highly regulated processes such as cytokinesis (including maintenance of cell polarity and membrane remodelling), ciliogenesis and phagocytosis.20 Septin-5 and -7 are expressed predominantly in neurons of the mammalian brain, and are abundant in hippocampal synaptic vesicle fractions.21 They form heterooligomeric complexes with other septins, including −6, and −11. Coexisting septin-6- and 11-IgGs occurred among most septin-7-IgG patients, which implies polyclonal autoimmunity targeting septin complexes in those patients. Other coexisting antibodies were uncommon but were also pertinent to the neurological phenotype (1 patient each with GRAF1 and ataxia, or NF-H and encephalopathy).22, 23
At a neurochemical level, mechanistic possibilities include upregulation of presynaptic inhibitory GABA transmission (as an outcome of septin-5 autoimmunity) and downregulation of glutamatergic transmission (as an outcome of septin-7 autoimmunity). Electron microscopy studies have been informative for some differentiation between septin-5 and -7 expression.24 Septin-5 is abundant in inhibitory presynaptic terminals and associated with GABAergic vesicles in the thalamus, globus pallidus, and cerebellar nuclei. Septin-5 is considered a negative regulator of synaptic vesicle release at inhibitory presynaptic terminals by competing with α-soluble N-ethylmaleimide-sensitive factor attachment protein (α-SNAP) for binding SNAP receptor protein (SNARE), a vesicle docking-regulating protein complex.25 Septin-5 also interacts with syntaxin-1, resulting in decreased vesicle exocytosis.25–27 Thus, septin-5 appears to serve as a negative regulator of GABA vesicle release.24, 26 In our mixed cultures of glutamatergic and GABAergic cortical neurons, IgGs from septin-5 autoimmune patients resulted in attenuation of spiking and bursting parameters consistent with diminution of excitatory neurotransmission due to increased GABAergic inhibitory tone. In keeping with the purported role for septin-5 as a ‘brake’ on GABA vesicle release, our findings support dysregulation of GABA release as being mechanistically relevant but requiring further exploration. Our findings cannot directly address the impact of septin-5-IgG on GABAergic signaling in cerebellar circuitry where dysregulated GABA transmission may be directly neurotoxic, as proposed as a mechanism in alcohol-induced cerebellar degeneration.28, 29
Patients with septin-7 autoimmunity, without septin-5-IgG positivity, had diverse phenotypic associations, but were dominated by encephalopathy with neuropsychiatric clinical presentations. Septin-7 is thought to be vital to all septin complex formation, having a widespread distribution across CNS regions and nerve roots, along, or just beneath axon membranes, but not colocalizing with synaptic vesicles.24 In particular, septin-7 is enriched at the bases of dendritic protrusions and branch points.30 Overexpression of septin-7 increases dendrite branching and dendritic protrusion density, whereas septin-7 knockdown interferes with dendritic branching and maturation, and axon growth.31 In schizophrenia patients, spine density on the basilar dendrites of pyramidal neurons is reduced in layer 3 (but not layers 5 and 6) of the dorsolateral prefrontal cortex, as compared to controls. SEPT7 mRNA levels have been shown to parallel these interlayer differences in dendritic spine density.32 In our cortical neuron cultures, IgGs from septin-7 autoimmune patients functionally exhibited earlier inhibition of spiking activity in mixed glutamatergic and GABAergic cortical circuits than IgGs from septin-5 patients. This is perhaps not surprising given the clinical presentation of septin-7 autoimmunity being illustrative of disruption of cortical rather than cerebellar circuity.
We cannot conclude whether the functional impact of patient IgG was mediated by presynaptic modulation of vesicular release or altered post-synaptic dendrite branching. The hyperacute increase in electrophysiologic activity in our cultures immediately observed upon addition of CSF across all conditions is interpreted to arise as a volumetric effect. Despite this initial increase, the acute nature of septin-IgG-mediated electrophysiologic effects (as early as 40 minutes when examining spikes per burst) is consistent with an antibody-mediated alteration of synaptic vesicles or other presynaptic elements.33, 34
One of our patients with septin-5- and septin-7-IgGs coexisting developed personality change characterized by apathy, blunted emotion and loss of insight, after onset and treatment of autoimmune ataxia. These psychiatric symptoms might be attributable to autoimmune targeting of septin-5, -7, or both. In addition to the importance of SEPT7 in thinking and behavior, SEPT5 knockout mice are also known to have decreased anxiety-related behavior and increased prepulse inhibition.35 Also, patient sera which bind more than one septin might recognize epitopes which are homologous between the different septins (septin 5 and 7 have 56% sequence identity). The observed psychological phenotype in septin autoimmune patients (which contrasts with that of GAD65 autoimmunity, where anxiety is a common feature) may serve as a diagnostic clue to septin autoimmunity.36 Patient 18, diagnosed with progressive lateral sclerosis (PLS), was assumed to have a neurodegenerative disorder, and was not treated with immune therapy. A clue to an alternative autoimmune diagnosis was markedly elevated phosphorylated neurofilament heavy chain protein level in CSF. PLS tends to be a slowly progressive disorder. Neurofilament protein levels in CSF are generally highest at disease onset in patients with conditions characterized by rapid axon neuronal loss, including autoimmune disease.23, 37
Potential etiologic factors were evident in 40% of septin-7-IgG cases, and just 1 of 8 septin-5 autoimmune cases. For septin-7-IgG positive patients, either cancers (paraneoplastic, though no one specific systemic cancer type) or parainfectious (post-HSV-1 encephalitis) causes were possible. A biphasic clinical course post HSV-1 encephalitis was encountered (infection first, autoimmune second), identical to the trajectory reported in NMDA-receptor encephalitis post-HSV-1.38 The importance of SARS-CoV-2 infection occurring 1 month prior to septin-5 ataxia in one patient was unclear, and may have been coincidental. Occasional cases of post-infectious autoimmune encephalitis have been reported post-SARS-CoV-2 infection, usually seronegative, though NMDA-R-IgG positivity has been reported.39, 40
In summary, differentiated septin-IgG profiles are predictive of distinct immune therapy responsive neurological syndromes (septin-5-IgG for cerebellar ataxia; septin-7-IgG for an encephalopathy predominant disorder). Live neuron studies support IgG binding and pathogenic effects as revealed by realtime, acute dampening of electrophysiologic activity in cortical neuron networks. Further study is needed to assess septins as mechanistically relevant targets of IgGs in the pathogenesis of these disorders.
Supplementary Material
Abbreviations:
- α-SNAP
α-soluble N-ethylmaleimide-sensitive factor attachment protein
- BDNF
brain derived neurotrophic factor
- CBA
cell-based assay
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle Medium
- FITC
fluoroscein isothiocyanate
- GABA
gamma amino butyric acid
- GFP
green fluorescent protein
- GTP
Guanosine-5’-triphosphate
- GRAF
GTPase Regulator Associated with Focal adhesion kinas
- IFA
indirect immunofluorescence
- IGF-1
insulin-like growth factor-1
- H
heavy chain
- HEK
human embryonic kidney
- HBSS
Hanks balanced salt solution
- HSV
herpes simplex virus
- IgG
immunoglobulin G
- L
light chain
- MOG
myelin oligodendrocyte glycoprotein
- NGS
normal goat serum
- NMDA
n-methyl-D-aspartate
- NPH
normal pressure hydrocephalus
- PSD-95
postsynaptic density 95
- R
receptor
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SNARE
SNAP receptor
- STD
standard deviation
- TRITC
Tetramethylrhodamine-isothiocyanate
Footnotes
Potential conflicts of interest
Drs Honorat, Miske, Sharf, Pittock and McKeon have patent applications pending for septin-5 and -7-IgGs as biomarkers of autoimmune neurologic disease.
References
- 1.Honorat JA, Lopez-Chiriboga AS, Kryzer TJ, et al. Autoimmune septin-5 cerebellar ataxia. Neurol Neuroimmunol Neuroinflamm. 2018. Sep;5(5):e474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrero San Martin A, Amarante Cuadrado C, Gonzalez Arbizu M, et al. Autoimmune Septin-5 Disease Presenting as Spinocerebellar Ataxia and Nystagmus. Neurology. 2021. Aug 10;97(6):291–2. [DOI] [PubMed] [Google Scholar]
- 3.Spiliotis ET, Nakos K. Cellular functions of actin- and microtubule-associated septins. Curr Biol. 2021. May 24;31(10):R651–R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung KY, Dai L, Trimble WS. Cell and molecular biology of septins. Int Rev Cell Mol Biol. 2014;310:289–339. [DOI] [PubMed] [Google Scholar]
- 5.Ageta-Ishihara N, Kinoshita M. Developmental and postdevelopmental roles of septins in the brain. Neurosci Res. 2020. Nov 4. [DOI] [PubMed] [Google Scholar]
- 6.Fujishima K, Kiyonari H, Kurisu J, Hirano T, Kengaku M. Targeted disruption of Sept3, a heteromeric assembly partner of Sept5 and Sept7 in axons, has no effect on developing CNS neurons. J Neurochem. 2007. Jul;102(1):77–92. [DOI] [PubMed] [Google Scholar]
- 7.Clarkson BDS, Kahoud RJ, McCarthy CB, Howe CL. Inflammatory cytokine-induced changes in neural network activity measured by waveform analysis of high-content calcium imaging in murine cortical neurons. Sci Rep. 2017. Aug 22;7(1):9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam Y, Wheeler BC. In vitro microelectrode array technology and neural recordings. Crit Rev Biomed Eng. 2011;39(1):45–61. [DOI] [PubMed] [Google Scholar]
- 9.Bradley JA, Luithardt HH, Metea MR, Strock CJ. In Vitro Screening for Seizure Liability Using Microelectrode Array Technology. Toxicol Sci. 2018. May 1;163(1):240–53. [DOI] [PubMed] [Google Scholar]
- 10.Prevosto V, Graf W, Ugolini G. The control of eye movements by the cerebellar nuclei: polysynaptic projections from the fastigial, interpositus posterior and dentate nuclei to lateral rectus motoneurons in primates. Eur J Neurosci. 2017. Jun;45(12):1538–52. [DOI] [PubMed] [Google Scholar]
- 11.Xue J, Tsang CW, Gai WP, et al. Septin 3 (G-septin) is a developmentally regulated phosphoprotein enriched in presynaptic nerve terminals. J Neurochem. 2004. Nov;91(3):579–90. [DOI] [PubMed] [Google Scholar]
- 12.Planaguma J, Haselmann H, Mannara F, et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann Neurol. 2016. Sep;80(3):388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson EA, Petty EM. Conquering the complex world of human septins: implications for health and disease. Clin Genet. 2010. Jun;77(6):511–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beise N, Trimble W. Septins at a glance. J Cell Sci. 2011. Dec 15;124(Pt 24):4141–6. [DOI] [PubMed] [Google Scholar]
- 15.Low C, Macara IG. Structural analysis of septin 2, 6, and 7 complexes. J Biol Chem. 2006. Oct 13;281(41):30697–706. [DOI] [PubMed] [Google Scholar]
- 16.Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. 2012. Feb 8;13(3):183–94. [DOI] [PubMed] [Google Scholar]
- 17.Estey MP, Kim MS, Trimble WS. Septins. Curr Biol. 2011. May 24;21(10):R384–7. [DOI] [PubMed] [Google Scholar]
- 18.Dolat L, Hu Q, Spiliotis ET. Septin functions in organ system physiology and pathology. Biol Chem. 2014. Feb;395(2):123–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neubauer K, Zieger B. The Mammalian Septin Interactome. Front Cell Dev Biol. 2017;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bridges AA, Gladfelter AS. Septin Form and Function at the Cell Cortex. J Biol Chem. 2015. Jul 10;290(28):17173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsang CW, Estey MP, DiCiccio JE, Xie H, Patterson D, Trimble WS. Characterization of presynaptic septin complexes in mammalian hippocampal neurons. Biol Chem. 2011. Aug;392(8–9):739–49. [DOI] [PubMed] [Google Scholar]
- 22.Jarius S, Martinez-Garcia P, Hernandez AL, et al. Two new cases of anti-Ca (anti-ARHGAP26/GRAF) autoantibody-associated cerebellar ataxia. J Neuroinflammation. 2013. Jan 15;10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKeon A, Shelly S, Zivelonghi C, et al. Neuronal intermediate filament IgGs in CSF: Autoimmune Axonopathy Biomarkers. Ann Clin Transl Neurol. 2021. Feb;8(2):425–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinoshita A, Noda M, Kinoshita M. Differential localization of septins in the mouse brain. J Comp Neurol. 2000. Dec 11;428(2):223–39. [DOI] [PubMed] [Google Scholar]
- 25.Beites CL, Campbell KA, Trimble WS. The septin Sept5/CDCrel-1 competes with alpha-SNAP for binding to the SNARE complex. Biochem J. 2005. Jan 15;385(Pt 2):347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beites CL, Xie H, Bowser R, Trimble WS. The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat Neurosci. 1999. May;2(5):434–9. [DOI] [PubMed] [Google Scholar]
- 27.Asada A, Takahashi J, Taniguchi M, et al. Neuronal expression of two isoforms of mouse Septin 5. J Neurosci Res. 2010. May 1;88(6):1309–16. [DOI] [PubMed] [Google Scholar]
- 28.Luo J. Effects of Ethanol on the Cerebellum: Advances and Prospects. Cerebellum. 2015. Aug;14(4):383–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaatinen P, Rintala J. Mechanisms of ethanol-induced degeneration in the developing, mature, and aging cerebellum. Cerebellum. 2008;7(3):332–47. [DOI] [PubMed] [Google Scholar]
- 30.Tada T, Simonetta A, Batterton M, Kinoshita M, Edbauer D, Sheng M. Role of Septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr Biol. 2007. Oct 23;17(20):1752–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y, Vessey JP, Konecna A, Dahm R, Macchi P, Kiebler MA. The GTP-binding protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr Biol. 2007. Oct 23;17(20):1746–51. [DOI] [PubMed] [Google Scholar]
- 32.Ide M, Lewis DA. Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 2010. Jul 1;68(1):25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fewou SN, Rupp A, Nickolay LE, et al. Anti-ganglioside antibody internalization attenuates motor nerve terminal injury in a mouse model of acute motor axonal neuropathy. J Clin Invest. 2012. Mar;122(3):1037–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YI, Neher E. IgG from patients with Lambert-Eaton syndrome blocks voltage-dependent calcium channels. Science. 1988. Jan 22;239(4838):405–8. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki G, Harper KM, Hiramoto T, et al. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum Mol Genet. 2009. May 1;18(9):1652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Black JL, Barth EM, Williams DE, Tinsley JA. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998. Jan-Feb;39(1):38–44. [DOI] [PubMed] [Google Scholar]
- 37.Falzone YM, Domi T, Agosta F, et al. Serum phosphorylated neurofilament heavy-chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J Neurol. 2020. Aug;267(8):2272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armangue T, Moris G, Cantarin-Extremera V, et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology. 2015. Oct 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilotto A, Masciocchi S, Volonghi I, et al. SARS-CoV-2 encephalitis is a cytokine release syndrome: evidences from cerebrospinal fluid analyses. Clin Infect Dis. 2021. Jan 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McHattie AW, Coebergh J, Khan F, Morgante F. Palilalia as a prominent feature of anti-NMDA receptor encephalitis in a woman with COVID-19. J Neurol. 2021. Apr 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.