

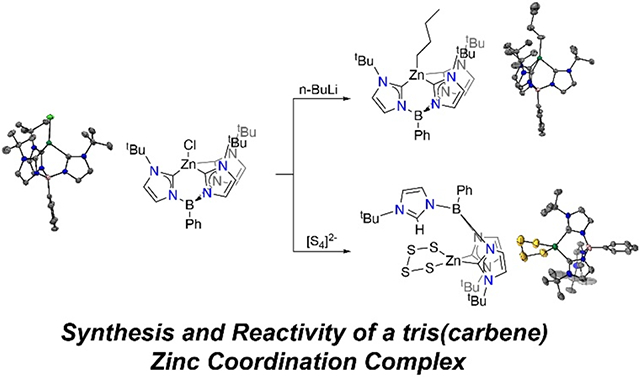
Abstract
The PhB(tBuIm)31− ligand has gained increased attention since it was first reported in 2006 due to its ability to stabilize highly reactive first row transition metal complexes. In this work, we explore the coordination chemistry of this ligand with redox-inert zinc to understand how a zinc metal center behaves in such a strong coordinating environment. The PhB(tBuIm)3ZnCl (1) complex can be formed via deprotonation of [PhB(tBuIm)3][OTf]2 followed by the addition of ZnCl2. Salt metathesis reaction with nucleophilic n-BuLi yields the highly carbon-rich zinc coordination complex PhB(tBuIm)3BuIm)3ZnBu (2) with three carbene atom donors and one carbanion donor. In contrast, reaction of complex 1 with a less nucleophilic polysulfide reagent, [K.18-C-6]2[S4], leads to the formation of a tetrahedral zinc tetrasulfido complex via protonation of one carbene donor to form PhB(tBuIm)2(tBuImH)Zn(κ2-S4) (3).
Graphical Abstract
Introduction
Studies of zinc coordination chemistry generally focus on the implications of its role as a redox-inert, abundant, and bioavailable metal. Zinc is an attractive metal for biological systems because of its predictable coordination chemistry, which favors a four-coordinate tetrahedral geometry. Furthermore, the ability to ligate both larger polarizable as well as smaller non-polarizable atoms such as S, and O, respectively, allows Nature to utilize zinc in a wide array of functions. These range from structural motifs in zinc finger proteins1 to Lewis acidic sites in the enzyme carbonic anhydrase that activate water molecules for CO2 hydration.2 These characteristics have allowed synthetic chemists to investigate the coordination chemistry of zinc and discover a wide array of reactivity, including small molecule activation,3 secondary coordination sphere effects,4-6 catalysis,7, 8 and metal-ligand cooperative reactivity.9-11
One area of zinc coordination chemistry that has gained increased attention involves its reactivity with reactive sulfur species (RSS) due to the common coordination of thiolate ligands to zinc in biology. Tsui and coworkers have utilized the redox-inert properties of zinc to investigate the redox chemistry of ligand-based thiolates with RSS. Reaction of a bis(carboxamide)pyridine zinc complex featuring two flanking thiolate chelates with elemental sulfur demonstrated sulfane sulfur insertion into the Zn-S bond yielding an unusual tetrasulfinado zinc complex, and which was shown to be reversible in the presence of 3 equiv of a PPh3 reductant.12 The coordination chemistry of anions such as thionitrite (SNO−) and perthionitrite (SSNO−), which are subject to rapid reactivity in the presence of redox-active reagents, have also taken advantage of the redox-inert properties of zinc to form the first structurally characterized transition metal complexes of SNO− and SSNO−.13
In this work, we investigate the coordination chemistry of zinc using a highly donating tris(carbene) ligand (phenyltris(3-tert-butylimidazol-2-ylidene)borato, [PhB(tBuIm)3]1−). The [PhB(tBuIm)3]1− ligand platform has been employed by others to synthesize and stabilize highly reactive molecules including some with unusual geometries. For example, Smith and coworkers synthesized the first FeV nitride and noted that the highly σ-donating carbene ligands interacts with iron dxz, dyz and dz2 orbitals, which are the same orbitals involved in multiple bonding of the nitride ligand.14 Using an adamantyl rather than tert-butyl version of this ligand, Anderson and coworkers synthesized a rare Co-oxo species15 and demonstrated the biomimetic C─H activation properties of Co-oxo complexes using an adamantyl derivative.16 Nickel complexes featuring the [PhB(tBuIm)3]1− ligand were also used to stabilize a bridging peroxo complex that was synthesized via O2 activation, which further demonstrates the impacts of the strongly donating ligand platform towards small molecule reactivity.17 In our own work, we found that the PhB(tBuIm)3NiCl can react with RSS to form square planar bis(hydrosulfide) or disulfide complexes with [NBu4][SH] or [K.18-C-6]2[S4] reagents, respectively.18
Here we incorporate the [PhB(tBuIm)3]1− ligand into Zn coordination complex to investigate the reactivity of this electron-rich zinc complex in ligand substitution reactions. Carbene based complexes of zinc have gained popularity in recent years, however the incorporation of multiple carbene based ligands is more uncommon and to the best of our knowledge zinc complexes with three carbene based ligands have not been reported. We demonstrate that treatment of ZnCl2 with [PhB(tBuIm)31− forms a PhB(tBuIm)3ZnCl complex, and that salt metatheses reactions result in [PhB(tBuIm)3)]1− ligand protonation, which is consistent with work with other metals.
Results and Discussion
Complex 1 was synthesized by deprotonation of [PhB(tBuIm)3][OTf]2 with 3 equiv. of lithium diisopropyl amide (LDA) generated in situ in THF, to generate the PhB(tBuIm)3Li intermediate, followed by salt metathesis with ZnCl2 to provide PhB(tBuIm)3ZnCl (1) as a white powder in 37% yield. The formation of 1 was confirmed from analysis of the 1H NMR spectrum of the product, which showed a single peak at 1.61 ppm corresponding to the tBu peaks of the PhB(tBuIm)3 ligand indicating a single C3 symmetric product. Furthermore, the absence of the H-Im peak of the free ligand as well as the shifted resonances compared to the PhB(tBuIm)3Li intermediate provided strong evidence for the formation of 1. In addition, the 13C carbene resonance shifts significantly from 135.3 ppm in PhB(tBuIm)3Li19 to 186.70 ppm in 1. The structure of 1 was confirmed by single crystal X-ray diffraction studies using colorless single crystals grown from a concentrated THF solution layered with hexanes and placed in the −25 °C freezer overnight. The molecular geometry about Zn in complex 1 is best described as pseudo-tetrahedral with three carbon donor atoms from the tris(carbene) borate ligand and one chloride ligand. Zn-CIm bond lengths in 1 vary between 2.045(2) and 2.086(2) Å, which is in the standard range of other Zn-carbene species.20 When compared to other metal centers bearing the same [PhB(tBuIm)3]1− and chloride ligands, the Zn-CIm distances are longer than expected despite Zn having the smallest atomic radius. The average M-CIm bond lengths where for M = Fe, Co, Ni and Zn are 2.093(3) Å,21 2.035(2) Å,19 2.003(2) Å,22 and 2.069(2) Å, respectively. We initially expected that that the Zn-CIm distances would decrease with respect to atomic radii, however the observed elongation may be due to the decreased covalent nature of the Zn-CIm bond when compared to more covalency of M-CIm bonds due to the partially filled d-orbitals of pseudo tetrahedral Fe2+, Co2+ or Ni2+.
Addition of n-BuLi to 1 resulted in formation of Cl exchange to form alkyl analog PhB(tBuIm)3ZnBu (2). Treatment of a solution of 1 in THF with 2.0 equiv. of a 1.6 M n-BuLi solution in hexanes afforded 2 as a white powder in 70% yield. We chose to use 2.0 equiv. of n-BuLi because we noticed that when only 1 equiv. of n-BuLi was added that the reaction proceeded very slowly to completion. The 1H NMR spectrum of 2 showed four new resonances at 2.31, 2.02, 1.43 and 0.93 ppm corresponding to the butyl ligand in addition to minor shifts of the [PhB(tBuIm)3]1− ligand. The structure of 2 was confirmed by single crystal X-ray diffraction studies using colorless single crystals grown from a concentrated Et2O solution that was placed in the −25 °C freezer overnight. The molecular geometry about Zn in complex 2 is best described as pseudo-tetrahedral with three carbon donor atoms from the tris(carbene) borate ligand and one alkyl butyl ligand. Most notable changes in the metal ligand bond distances include elongation of the average Zn-CIm distances from 2.069(2) Å in 1 to 2.138(2) Å in 2. As expected, the strong σ-donating alkyl ligand has a Zn-C bond length of 2.050(3) Å, significantly shorter compared to the Zn-Cl bond length of 2.2492(5) Å observed in 1. This change agrees with the elongation of Zn-CIm bond distances observed in 2 due to the greater electron density residing on the Zn center. Complex 2 is comprised of a Zn center with four Zn-C bonds, which to the best of our knowledge is the first Zn complex with three carbene and one alkyl-based ligands. We exclusively handled 2 under an inert atmosphere and did not observe any significant stability issues despite the electron rich nature of this complex.
To expand the coordination chemistry of 1, we also investigated the reaction with electron rich RSS. Stirring 1 with 1 equiv. of [K.18-C-6]2[S4] in THF led to a color change from a colorless to light-yellow over the course of 16 h and formation of the tetrasulfido complex PhB(tBuIm)2(tBuImH)Zn(κ2-S4) (3). Compound 3 was isolated in 54% yield, which was not improved by varying the [K.18-C-6]2[S4] stoichiometry. Complex 3 is best described as tetrahedral and features two C donor atoms from the PhB(tBuIm)2(ttBuImH) ligand and two S donor atoms from the tetrasulfide ligand. The most notable difference in 3 in comparison to the molecular structures of 1 and 2 is that it contains one protonated imidazolylidene that is no longer coordinated to the metal center. The observation of the protonated imidazolylidene is supported by changes in the 1H NMR spectrum. We observe a reduction in symmetry that is consistent with formation of a two-fold symmetric species, with the tBu resonances at 1.72 and 1.62 ppm integrating with a 2:1 stoichiometry, respectively. Furthermore, the Im-H proton is observed at 7.55 ppm. We have previously observed similar protonation reactivity of one carbene of the PhB(tBuIm)31− ligand in prior work when PhB(tBuIm)3NiCl was treated with [K.18-C-6]2[S4] or [NBu4][SH] to form PhB(tBuIm)2(tBuImH)NiS2 and PhB(tBuIm)2(tBuImH)Ni(SH)2, respectively.18 Mechanistic studies revealed that the most likely source of this advantageous proton was from unidentified species reacting with either the [NBu4]+ or [K.18-C-6]+.
Complex 3 is just one of three other zinc tetrasulfido complexes that has been structurally characterized, and emphasizes the utility of the [K.18-C-6]2[S4] reagent as an effective method for the delivery of tetrasulfides. Other methods for zinc tetrasulfido synthesis rely on the unpredictable decomposition of zinc polysulfides23, 24 or by the reduction of larger polysulfides with PPh3.25 The S-S bond distances in 3 range between 2.012(4) Å and 2.100(5) Å and are consistent with other ZnS4 motifs.25 We have recently commented on the stabilizing factors of C─H…S interactions and upon further inspection of the structure of complex 3 we find multiple short C─H…S contacts that meet the definition of a hydrogen bond.26 Multiple contacts between the C─H groups of the flanking tBu of the PhB(tBuIm)2(tBuImH) ligand point directly at the sulfur atoms of the S42− chain with some contacts as short as 2.730 Å with a C─H…S bond angle near 160°, which is significantly shorter than the sum of the van der Waals radii of the H and S atoms (3.0 Å). In addition to crystallographic evidence for C─H…S interactions in complex 3, we also performed variable temperature 1H NMR experiments (233 – 323 K) and observed a downfield shift of the flanking tBu and imidazolium hydrogens at lower temperatures, and an upfield shift of these resonances at elevated temperatures (see SI Figures S7 and S8). Both observations further support H-bonding to the nearby S atoms.27
Conclusions
We have demonstrated the reactivity and coordination chemistry of a zinc complex with a tris(carbene) ligand scaffold. The chloride analog, complex 1, performs a salt metathesis reaction with nucleophilic n-BuLi yielding a highly carbon rich zinc coordination complex 2 with three carbene atom donors and one carbanion donor. In contrast, reaction of complex 1 with a less nucleophilic polysulfide reagent, [K.18-C-6]2[S4], leads to the formation of a tetrahedral zinc tetrasulfido complex via protonation of one carbene donor to form complex 3. Taken together this work demonstrates the reactivity of the donating PhB(tBuIm)31− ligand when coordinated to a redox-inert metal such as zinc.
Experimental Section
Materials and Methods
All manipulations were performed under an inert atmosphere using an Innovative Atmospheres N2-filled glove box unless otherwise noted. [PhB(tBuIm)3][OTf]2,19 and [K18-C-6]2[S4]27 were synthesized from established procedures. C6D6 was degassed with N2 and stored in an inert atmosphere glove box over 4 Å molecular sieves. d8-THF was degassed with N2, distilled from Na/benzophenone, and stored in an inert atmosphere glove box over 4 Å molecular sieves. All commercially available chemicals were used as received and purchased from Strem or Sigma Aldrich. C6D6 and d8-THF were purchased from Cambridge Isotope Laboratories. UV/Vis spectra were acquired on an Agilent Cary 60 UV/Vis spectrophotometer equipped with a Quantum Northwest TC-1 temperature controller set at 25.0 ± 0.05 °C. NMR spectra were acquired on a Varian 500 MHz or a Bruker 600 MHz spectrometer. Chemical shifts are reported in parts per million (δ) and are referenced to an internal standard. We did not observe the 13C resonance corresponding to the carbon atom bound to boron in compounds 1-3. We attribute this absence to the quadrupolar nuclear spin of boron and expected significant signal broadened of the carbon atom. Mass spectrometric measurements were performed by the University of Illinois, Urbana Champaign MS facility. IR spectra were acquired on a Nicolet 6700 IR Spectrometer as ATR or KBr pellet samples.
X-Ray Crystallography
Diffraction intensities for 1, 2 and 3 were collected at 173 K (1 and 2) and 213 K (3) on a Bruker Apex2 CCD diffractometer using a Incoatec Cu IμS source, CuKα radiation, 1.54178 Å. Space groups were determined based on systematic absences. Absorption corrections were applied by SADABS.28 Structures were solved by direct methods and Fourier techniques and refined on F2 using full matrix least-squares procedures. H atoms were refined in calculated positions in a rigid group model. The terminal ─CH2CH2CH2CH3 group in 2 is disordered over three positions. Refinement of occupation factors shown that occupation factors for these positions are close to 0.45; 0.35 and 0.20. The final refinement was completed for a model where this terminal group is disordered over three positions and occupation factors for these positions have been fixed at the mentioned values. The C atoms in this group were refined with isotropic thermal parameters. This group was also refined with restrictions on its geometry; the standard C─C distances were used in the refinement as the targets for the corresponding C─C bonds. In the crystal structure of 3 there are three tetrahydrofuran solvent molecules filling empty spaces in the packing. Two THF molecules were found from the diffraction data and refined although there are significant elongations of thermal parameters for atoms in these solvent molecules. The third molecule is highly disordered and was treated by SQUEEZE.29 Correction of the X-ray diffraction was 148 electron/cell; the required value is 160 electron/cell for four THF molecules in the full unit cell. X-ray diffraction from crystals of 3 at high angles was very weak due to the small sizes of crystals and the high disorder inside the structure. Even by using a strong Incoatec Cu IμS source it was only possible to collect diffraction up to 2θmax = 99.62°. Some atoms in the structure of 3 were refined with isotropic thermal parameters. The data collected provide the appropriate number of measured reflections per refined parameters, 7645 per 740. The structure of 3 has not been precisely determined but provides clear evidence for the chemical results. All calculations were performed by the Bruker SHELXL-2014/7 package.30
Synthesis of PhB(tBuIm)3ZnCl (1).
A 20 mL scintillation vial containing solution of diisopropyl amine (0.251 g, 2.40 mmol) in 5 mL THF was cooled in a cold well at −78 °C in the glovebox. To this solution was added 1.55 mL (2.48 mmol) of a 1.6 M n-BuLi solution in hexanes to generate lithium diisopropylamide (LDA). The cooled LDA solution was added to a separate, cooled, 20 mL scintillation vial containing a slurry of [PhB(tBuIm)3][OTf]2 (0.567 g, 0.74 mmol) in 5 mL THF and let stir for one hour and allowed to warm to room temperature. Solid ZnCl2 (0.10 g, 0.74 mmol) was added to the triply-deprotonated ligand solution. No immediate color change was observed, and the solution was left stirring for 1 hour. After 1 hour the THF solvent was removed under reduced pressure. The slightly yellow residue was extracted in benzene (3 mL), filtered over Celite, and the benzene was removed under reduced pressure. The residue was then heated under vacuum for 1 hour at 60 °C to remove THF coordinated to the LiOTf byproduct. The residue was then redissolved in benzene, and the purification process was repeated to remove the LiOTf(THF)x byproduct until the 1H NMR indicated that no more THF (in the form of LiOTf(THF)x) was present. The fine white powder product was isolated in 37% yield (155 mg). Single crystals suitable for X-ray diffraction were grown from a concentrated THF solution layered with hexanes and placed in the −25 °C freezer overnight to yield colorless crystals of PhB(tBuIm)3ZnCl. 1H NMR (500 MHz, C6D6) δ: 8.10 (d, J = 6.9 Hz, 2H), 7.42 (m, 2H), 7.36 (t, J = 7.2 Hz, 1H), 6.97 (d, J = 1.7 Hz, 3H), 6.39 (d, J = 1.7 Hz, 3H), 1.61 (s, 27H). 13C NMR (126 MHz, C6D6) δ: 186.70, 135.91, 135.85, 122.47, 114.68, 114.54, 57.03, 31.27.
Synthesis of PhB(tBuIm)3ZnBu (2)
In a 20 mL scintillation vial, PhB(tBuIm)3ZnCl (0.0114 g, 0.0203 mmol) was dissolved in THF and placed in a cold well at −78 °C in the glovebox. To this solution was added 0.025 mL (0.0400 mmol) of a 1.6 M n-BuLi solution in hexanes, and the solution was allowed to warm to room temperature while stirring for one hour. The solvent was removed under reduced pressure to afford a white residue that was extracted with benzene to remove salts and the filtered over a pad of Celite. The benzene was then removed under reduced pressure leaving the product in 70% yield (8.0 mg). Single crystals suitable for X-ray diffraction were grown from a concentrated Et2O solution and placed in the −25 °C freezer overnight to yield colorless crystals of PhB(tBuIm)3ZnBu. 1H NMR (600 MHz, C6D6) δ: 8.19 (d, J = 7.3 Hz, 2H), 7.43 (t, J = 7.4 Hz, 2H), 7.36 (t, J = 7.2 Hz, 1H), 7.04 (s, 3H), 6.44 (s, 3H), 2.31 (dt, J = 16.5, 7.4 Hz, 2H), 2.02 (h, J = 7.2 Hz, 2H) 1.52 (s, 27H), 1.43 (t, J = 7.3 Hz, 3H) 0.93 (m, 2H). 13C NMR (151 MHz, C6D6) δ: 191.85, 136.17, 128.59, 127.43, 122.27, 113.99, 56.07, 34.25, 32.32, 31.34, 14.98, 14.73.
Synthesis of PhB(tBuIm)2(tBuImH)Zn(κ2-S4) (3)
In a 20 mL scintillation vial, PhB(tBuIm)3ZnCl (0.019 g, 0.035 mmol) was dissolved in THF. Solid [K.18-C-6]2[S4] (0.028 g, 0.038 mmol) was added to this solution, and it was allowed to stir at room temperature for 16 h. As the sparingly soluble orange [K1.8-C-6]2[S4] was consumed, the reaction mixture changed from colorless to a light-yellow solution. The solvent was removed under reduced pressure and the solution was triturated with hexanes (20 mL) to remove the crown ether byproduct. The residue was then extracted in benzene and filtered over a pad of Celite to remove any salts. The benzene was removed under reduced pressure to provide a light-yellow residue that was recrystallized from a concentrated THF solution layered with hexanes and placed in the −25 °C freezer overnight to yield yellow crystals of PhB(tBuIm)2(tBuImH)Zn(κ2-S4) (15 mg, 54% yield). 1H NMR (600 MHz, THF-d8) δ: 7.55 (t, J = 1.7 Hz, 1H), 7.34 (m, 3H), 7.24 (d, J = 1.8 Hz, H), 7.11 (m, 2H), 6.79 (t, J = 1.5 Hz, 1H), 6.61 (d, J = 1.8 Hz, 1H), 6.50 (t, J = 1.7 Hz, 1H), 1.72 (s, 18H), 1.62 (s, 9H). 13C NMR (151 MHz, THF) δ: 185.84, 134.35, 134.28, 129.20, 128.77, 125.07, 124.80, 121.35, 117.76, 59.62, 59.43, 32.69, 29.73. HRMS m/z [M + H]+ calcd. For [C27H40BN6S4Zn]+ 651.1582; found 651.1587.
Supplementary Material
Figure 1.

Solid-state structure of 1. Ellipsoids are shown at 50% probability levels. Pink, gray, blue, neon green, and forest green ellipsoids represent B, C, N, Cl, and Zn atoms, respectively. Hydrogen atoms are omitted for clarity.
Figure 2.

Solid-state structure of 2. Ellipsoids are shown at 50% probability levels. Pink, gray, blue, and forest green ellipsoids represent B, C, N, and Zn atoms, respectively. Hydrogen atoms are omitted for clarity. The terminal butyl group in 2 is disordered over three positions, and the C atoms in this group were refined with isotropic thermal parameters.
Figure 3.

Side (left) and top (right) views of solid-state structure of 3. Ellipsoids are shown at 50% probability levels. White, pink, gray, blue, yellow, and forest green ellipsoids represent H, B, C, N, S and Zn atoms, respectively. Hydrogen atoms, except Im-H, are omitted for clarity.
Scheme 1.

Synthesis of complexes 1, 2, and 3.
Table 1.
Selected bond distances and angles for 1, 2, and 3. For 1, X = Cl; for 2, X = C, and for 3, X = S.
| 1 | 2 | 3 | |
|---|---|---|---|
| d(Zn-CIm1) (Å) | 2.045(2) | 2.160(2) | 2.064(10) |
| d(Zn-CIm2) (Å) | 2.075(2) | 2.160(2) | 2.083(9) |
| d(Zn-CIm3) (Å) | 2.086(2) | 2.093(2) | N/A |
| d(Zn–X) (Å) | 2.2492(5) | 2.050(3) | 2.366(3), 2.360(3) |
In complex 3 there are two independent molecules in the asymmetric unit, and the values shown are the average of these two values.
Acknowledgements
We thank the NIH (F32-GM139372 to T.J.S.), the NSF (CHE-2004150 to M.D.P.), an NSF training grant (DGE-2022168 to KL) for support of this research.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: X-ray, NMR, and UV-vis data. See DOI: 10.1039/x0xx00000x. CCDC deposition numbers 2179150-2179152 contain the supplementary crystallographic data for this paper.
References
- 1.Laitaoja M, Valjakka J and Jänis J, Inorg. Chem, 2013, 52, 10983–10991. [DOI] [PubMed] [Google Scholar]
- 2.Christianson DW and Fierke CA, Acc. Chem. Res, 1996, 29, 331–339. [Google Scholar]
- 3.Jochmann P and Stephan DW, Organometallics, 2013, 32, 7503–7508. [Google Scholar]
- 4.Dahl EW, Kiernicki JJ, Zeller M and Szymczak NK, J. Am. Chem. Soc, 2018, 140, 10075–10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiernicki JJ, Norwine EE, Lovasz MA, Zeller M and Szymczak NK, Chem. Commun, 2020, 56, 13105–13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiernicki JJ, Norwine EE, Zeller M and Szymczak NK, Inorg. Chem, 2021, 60, 13806–13810. [DOI] [PubMed] [Google Scholar]
- 7.Cronin SP, Strain JM, Mashuta MS, Spurgeon JM, Buchanan RM and Grapperhaus CA, Inorg. Chem, 2020, 59, 4835–4841. [DOI] [PubMed] [Google Scholar]
- 8.Procter RJ, Uzelac M, Cid J, Rushworth PJ and Ingleson MJ, ACS Catal., 2019, 9, 5760–5771. [Google Scholar]
- 9.Ward MB, Scheitler A, Yu M, Senft L, Zillmann AS, Gorden JD, Schwartz DD, Ivanović-Burmazović I and Goldsmith CR, Nat. Chem, 2018, 10, 1207–1212. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee P, Company A, Weyhermüller T, Bill E and Hess CR, Inorg. Chem, 2009, 48, 2944–2955. [DOI] [PubMed] [Google Scholar]
- 11.Myers TW, Sherbow TJ, Fettinger JC and Berben LA, Dalton Trans., 2016, 45, 5989–5998. [DOI] [PubMed] [Google Scholar]
- 12.Ballesteros M and Tsui EY, Inorg. Chem, 2019, 58, 10501–10507. [DOI] [PubMed] [Google Scholar]
- 13.Hosseininasab V, Bertke JA and Warren TH, Angew. Chem. Int. Ed, 2021, 60, 21184–21188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scepaniak JJ, Vogel CS, Khusniyarov MM, Heinemann FW, Meyer K and Smith JM, Science, 2011, 331, 1049–1052. [DOI] [PubMed] [Google Scholar]
- 15.Goetz MK, Hill EA, Filatov AS and Anderson JS, J. Am. Chem. Soc, 2018, 140, 13176–13180. [DOI] [PubMed] [Google Scholar]
- 16.Goetz MK, Schneider JE, Filatov AS, Jesse KA and Anderson JS, J. Am. Chem. Soc, 2021, 143, 20849–20862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao N, Filatov AS, Xie J, Hill EA, Rogachev AY and Anderson JS, J. Am. Chem. Soc, 2020, 142, 21634–21639. [DOI] [PubMed] [Google Scholar]
- 18.Sherbow TJ, Zakharov L and Pluth MD, Inorg. Chem, 2021, 60, 8135–8142. [DOI] [PubMed] [Google Scholar]
- 19.Cowley RE, Bontchev RP, Duesler EN and Smith JM, Inorg. Chem, 2006, 45, 9771–9779. [DOI] [PubMed] [Google Scholar]
- 20.Roberts AJ, Clegg W, Kennedy AR, Probert MR, Robertson SD and Hevia E, Dalton Trans., 2015, 44, 8169–8177. [DOI] [PubMed] [Google Scholar]
- 21.Scepaniak JJ, Fulton MD, Bontchev RP, Duesler EN, Kirk ML and Smith JM, J. Am. Chem. Soc, 2008, 130, 10515–10517. [DOI] [PubMed] [Google Scholar]
- 22.Hill EA, Zhao N, Filatov AS and Anderson JS, Chem. Commun, 2020, 56, 7861–7864. [DOI] [PubMed] [Google Scholar]
- 23.Pafford RJ and Rauchfuss TB, Inorg. Chem, 1998, 37, 1974–1980. [Google Scholar]
- 24.Pafford RJ, Chou J-H and Rauchfuss TB, Inorg. Chem, 1999, 38, 3779–3786. [Google Scholar]
- 25.Coucouvanis D, Patil PR, Kanatzidis MG, Detering B and Baenziger NC, Inorg. Chem, 1985, 24, 24–31. [Google Scholar]
- 26.Fargher HA, Sherbow TJ, Haley MM, Johnson DW and Pluth MD, Chem. Soc. Rev, 2022, 51, 1454–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smiles DE, Wu G and Hayton TW, Inorg. Chem, 2014, 53, 12683–12685. [DOI] [PubMed] [Google Scholar]
- 28.Sheldrick G, Journal, 1998. [Google Scholar]
- 29.van der Sluis P and Spek AL, Acta Crystallogr. A, 1990, 46, 194–201. [Google Scholar]
- 30.Sheldrick G, Acta Crystallogr A, 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.