

Abstract
Introduction
We examined longitudinal cerebrospinal fluid (CSF) Alzheimer's disease (AD) biomarker changes among cognitively normal individuals with 10.7 years follow‐up, on average.
Methods
Analyses included 278 participants (M age = 57.5 years); 94 have progressed from normal cognition to mild cognitive impairment (MCI). Amyloid beta (Aβ)42/Aβ40, phosphorylated tau181 (p‐tau181), and total tau (t‐tau) were measured using automated electrochemiluminescence assays.
Results
Apolipoprotein E (APOE) ε4 carriers had lower baseline Aβ42/Aβ40, but longitudinal Aβ42/Aβ40 decreases did not differ by APOE ε4 after accounting for Aβ42/Aβ40 positivity. Lower baseline Aβ42/Aβ40 was associated with greater increases in tau (more strongly in males), and APOE ε4 genotype was associated with greater tau increases after reaching Aβ42/Aβ40 positivity. Participants who progressed to MCI had more abnormal biomarker levels and greater tau increases prior to MCI symptom onset. Biomarkers were more abnormal among older adults, but unrelated to sex or education.
Discussion
Our results confirm accelerated biomarker changes during preclinical AD and highlight the important role of amyloid levels in tau accelerations.
Keywords: amyloid, apolipoprotein E genotype, biomarkers, cerebrospinal fluid, preclinical Alzheimer's disease, tau
1. BACKGROUND
The preclinical phase of Alzheimer's disease (AD) is characterized by the accumulation of amyloid plaques and neurofibrillary tangles years to decades before clinical symptoms emerge. 1 Despite significant advances in techniques for measuring these pathologies in vivo, current hypothetical models of biomarker trajectories are based largely on cross‐sectional studies across the clinical disease spectrum or short‐term evaluations (typically 2 to 5 years) of biomarker changes in older adults (baseline age = 70 years). 2 This limits understanding of longitudinal within‐person biomarker changes during preclinical AD, especially from midlife. Addressing this gap is important, given clinical trials are increasingly targeting the earliest disease phases and using biomarkers for trial enrollment and outcomes.
RESEARCH IN CONTEXT
Systematic review: Traditional sources (e.g., PubMed) were used to review prior literature. Although hypothetical biomarker models of Alzheimer's disease (AD) describe biomarker changes during the preclinical phase of AD, these models are based primarily on cross‐sectional and short‐term longitudinal studies in older adults.
Interpretation: Our findings suggest that among cognitively normal, largely middle‐aged individuals, lower levels of amyloid are associated with increases in tau (as measured in cerebrospinal fluid), while accelerations in tau are more closely linked to mild cognitive impairment symptom onset. They also suggest apolipoprotein E ε4 carriers have an earlier age of amyloid onset and higher rates of amyloid positivity, but no differences in amyloid rates of change over time.
Future directions: Future studies are needed to further address possible sex differences in AD biomarker trajectories. Additional longitudinal studies among more diverse groups of cognitively normal, middle‐aged participants are needed to confirm the generalizability of these findings.
This study's aim was to characterize longitudinal AD biomarker trajectories spanning an average of 10.7 years (max = 23 years) in 278 cognitively normal, primarily middle‐aged individuals (M baseline age = 57.5 years). AD biomarkers were measured by cerebrospinal fluid (CSF) amyloid beta (Aβ)42/Aβ40, phosphorylated tau181 (p‐tau181), and total tau (t‐tau). We examined the impact of demographics (age, sex, education) and apolipoprotein E (APOE) ε4 genotype on biomarker trajectories. We also examined associations among the biomarker measures, and tested the hypothesis that APOE ε4‐related differences in biomarker trajectories are due to differences in amyloid positivity between APOE ε4 carriers and non‐carriers, given APOE ε4 carriers accumulate amyloid earlier than non‐carriers. 3 , 4 , 5 Furthermore, we explored interactions with sex, because prior studies have reported sex‐related differences in AD biomarkers 6 , 7 , 8 and clinical outcomes. 9 , 10 , 11 , 12 Finally, given the sizeable number of individuals who progressed to mild cognitive impairment (MCI) or dementia (n = 94) during follow‐up, we examined whether CSF biomarker trajectories differed with respect to clinical outcomes. The focus on middle‐aged individuals is particularly important because AD pathology begins to develop in midlife, 5 , 13 and abnormal midlife levels of CSF amyloid and tau are associated with poorer cognitive outcomes. 11 , 14 , 15
2. METHODS
2.1. Study design and participant selection
Data were derived from the BIOCARD study, which was designed to identify variables among cognitively normal individuals that predict subsequent development of cognitive decline and dementia. As described previously, 16 the study was initiated in 1995 at the National Institutes of Health (NIH). Between 1995 and 2005, 349 cognitively normal, primarily middle‐aged (M = 57.3 years, standard deviation = 10.4) participants were enrolled after providing written informed consent. By design, 75% of the cohort had a first‐degree relative with AD dementia. While at the NIH, participants completed comprehensive clinical and cognitive assessments annually; CSF and other biomarkers were obtained approximately every 2 years. The study was stopped in 2005 and re‐established in 2009 at Johns Hopkins University (JHU), and annual clinical and cognitive assessments and collection of blood specimens were reinitiated. The biennial collection of CSF was reinitiated in 2015. Participants have undergone repeated visits over a long period of time and longitudinal data collection is ongoing (see supporting information Text S1 for additional details, including a study timeline). This study was approved by the JHU Institutional Review Board.
These analyses included 278 participants who were cognitively normal at their “baseline” CSF measure (i.e., first available). Participants were excluded for the following reasons: (1) 26 had not yet re‐enrolled or had withdrawn, (2) 21 had an estimated age of MCI clinical symptom onset at or prior to their first CSF measure, and (3) 24 did not have CSF collected.
2.2. Clinical assessments
Annual assessments include physical and neurological examinations, record of medication use, behavioral and mood assessments, family history of dementia, history of symptom onset, a neuropsychological test battery, and the Clinical Dementia Rating (CDR 17 ) based on a semi‐structured interview. Consensus diagnoses used in study analyses (see Albert et al. 16 and Text S1) involved first establishing a syndromic diagnosis (normal, MCI, dementia, impaired‐not‐MCI) based on (1) clinical data (medical, neurological, and psychiatric status), (2) reports of changes in cognition (based on the CDR), and (3) decline in cognitive test performance. Second, for participants with cognitive impairment, the likely syndromic etiology (/etiologies) was determined based on the neurologic, medical, and psychiatric information collected (without knowledge of biomarker measures). These consensus diagnoses follow the recommendations of the National Institute on Aging/Alzheimer's Association working group reports for the diagnosis of MCI 18 and AD dementia. 19 The estimated age of MCI clinical symptom onset was established separately, based primarily on the CDR interview.
For these analyses, we created a dichotomous indicator variable for follow‐up diagnosis, reflecting each participant's last (i.e., most recent) diagnosis (remain normal = 0, progress from normal cognition to MCI or dementia = 1).
2.3. Cerebrospinal fluid biomarker measures
CSF samples used in these analyses were collected over time while the study was at the NIH (1995‐2005) and JHU (since 2015; see Text S2 in supporting information for CSF measures across sites). CSF was collected via lumbar puncture after an overnight fast. Samples were aliquoted into polypropylene cryotubes that were kept on dry ice and immediately transferred to a –80°C freezer for long‐term storage. Samples were thawed for the first time since collection to measure Aβ40, Aβ42, p‐tau181, and t‐tau using fully automated electrochemiluminescence assays (Lumipulse G1200 platform; Fujirebio Diagnostics, Inc.; for coefficients of variation, see Greenberg et al. 11 ). The present analyses used the Aβ42/Aβ40 ratio (vs. Aβ42 alone) to account for individual differences in total Aβ production (as measured by Aβ40) and reduce the impact of pre‐analytic variables. 20 , 21 , 22 , 23 The three main outcome variables were Aβ42/Aβ40, p‐tau181, and t‐tau. See Text S3 in supporting information for the p‐tau181/(Aβ42/Aβ40) and t‐tau/(Aβ42/Aβ40) ratio results.
2.4. APOE genotype
APOE genotypes were determined by restriction endonuclease digestion of polymerase chain reaction amplified genomic DNA (performed by Athena Diagnostics, Worcester, MA). Genotypes were coded dichotomously (APOE ε4 non‐carriers = 0, carriers = 1).
2.5. Statistical analyses
Group differences in descriptive statistics were compared using t tests or chi‐square tests, as appropriate. Rates of change in the CSF biomarkers over time were analyzed with li mixed effects models that included random intercepts and slopes with unstructured covariance. CSF measures were standardized (z‐scored) separately for CSF collected at NIH versus JHU using the first available measure at each site, so that model coefficients reflected z‐scores while simultaneously accounting for potential differences due to collection site. Time was modeled in the unit of years (since baseline).
The primary analyses evaluated the relationship of demographics and APOE ε4 genotype to levels and rates of change in CSF biomarkers. Model predictors included: baseline age (centered), sex, years of education (centered), APOE ε4 genotype, time, the interaction (cross‐product) of each predictor with time, and time2 for evaluating non‐linear (quadratic) trajectories. Models also included a “collection site” indicator, reflecting the location of CSF collection (NIH vs. JHU) to control for potential systematic site differences. To examine the robustness of our primary findings, models using chronological age (in years) as the timescale are also presented (excluding terms for baseline age).
The first set of follow‐up models evaluated associations between (1a) baseline tau biomarkers (p‐tau181 or t‐tau) and change in Aβ42/Aβ40 (additional model terms: baseline tau and baseline tau x time), and (1b) baseline Aβ42/Aβ40 and change in tau (additional model terms: baseline Aβ42/Aβ40 and baseline Aβ42/Aβ40 x time). The second set of follow‐up models evaluated whether accounting for Aβ42/Aβ40 positivity affected the relationship between APOE ε4 status and biomarker change (additional model terms: Aβ42/Aβ40 positivity and Aβ42/Aβ40 positivity x time, and the interactions of these terms with APOE ε4). Aβ42/Aβ40 positivity was calculated as a time‐varying variable reflecting dichotomous Aβ42/Aβ40 negative versus positive status at each time point using clinically derived cut‐points (Aβ42/Aβ40 positive = 1 if Aβ42/Aβ40 ≤ 0.075, otherwise 0; see Text S5 in supporting information for the cut‐point derivation). The third set of follow‐up models evaluated differences in CSF biomarker trajectories by follow‐up diagnosis (additional model terms: follow‐up diagnosis and follow‐up diagnosis x time). The fourth set of follow‐up models explored sex differences in CSF biomarker changes by including interactions between sex and baseline age (4a), or sex and APOE ε4 genotype (4b), as well as the interactions of these terms with time. Last, we examined whether sex modified the relationship between baseline tau and change in Aβ42/Aβ40 (4c), and between baseline Aβ42/Aβ40 and change in tau (4d) by including additional terms for the respective baseline biomarkers, interactions with sex, and their interactions with time.
Analyses were run in Stata (v17.0). Estimates and 95% confidence intervals are reported with p‐values ≤0.05 considered significant.
3. RESULTS
Participants (N = 278; M age = 57.5 years) were cognitively normal at baseline and had 3.7 CSF measures over 10.7 years, on average (86% had ≥2 CSF measures; Table 1). Those who subsequently progressed to MCI or dementia (n = 94; 34%) were older, had shorter follow‐up, and more abnormal biomarkers (i.e., lower Aβ42/Aβ40, higher tau).
TABLE 1.
Baseline characteristics for participants included in the analyses, for the full sample and stratified by follow‐up diagnosis
| All participants | Remain normal | Progress to MCI/dementia^ | |
|---|---|---|---|
| N | 278 | 184 | 94 |
| Age, M (SD) [range] | 57.5 (10.29) [20.4–92.5] | 55.2 (10.06) [20.4–92.5] | 61.9 (9.30) [38.7–84.8]* |
| Female sex, n (%) | 165 (59%) | 114 (62%) | 51 (54%) |
| Years of education, M (SD) | 17.0 (2.37) | 17.2 (2.33) | 16.8 (2.43) |
| APOE ε4 carriers, n (%) | 96 (35%) | 61 (33%) | 35 (37%) |
| White race, n (%) | 271 (98%) | 181 (98%) | 90 (96%) |
| Number of CSF measures over time, M (SD) [range] | 3.7 (1.9) [1‐9] | 3.8 (2.0) [1‐8] | 3.5 (1.79) [1‐9] |
| Years between baseline and last CSF measure, M (SD) [range] | 10.7 (7.3) [0‐23] | 11.4 (7.5) [0‐23] | 9.2 (6.8) [0‐22]* |
| Years between baseline CSF measure and last (i.e., most recent) diagnosis, M (SD) [range] | 13.9 (5.7) [0–23.6] | 15.4 (5.3) [0–23.6] | 10.8 (5.0) [1–22.8]** |
| Years between baseline CSF measure and MCI clinical symptom onset, M (SD) [range] | — | — | 7.6 (4.5) [0.8–22.4] |
| CSF Aβ42/Aβ40, M (SD) | 0.09 (0.02) | 0.09 (0.02) | 0.08 (0.02)** |
| CSF p‐tau181 (pg/ml), M (SD) | 34.60 (17.45) | 31.30 (13.69) | 41.07 (21.77)** |
| CSF t‐tau (pg/ml), M (SD) | 259.61 (138.51) | 236.05 (111.53) | 305.73 (171.52)** |
| Participants with ≥2 CSF measures over time, n (%) | 239 (86%) | 158 (86%) | 81 (86%) |
| Number of CSF measures over time for participants with ≥2 CSF measures, M (SD) | 4.1 (1.7) | 4.2 (1.8) | 4.0 (1.6) |
| Years between baseline and last CSF measure for participants with ≥2 CSF measures, M (SD) | 12.4 (6.4) | 13.3 (6.3) | 10.7 (6.2)* |
* p < 0.05 and ** p < 0.001 for significant differences between remain normal versus progress to MCI/dementia.
^ Of the 94 participants who progressed from normal cognition to MCI/dementia, n = 86 and n = 8 had a diagnosis of MCI and dementia, respectively, at their last (i.e., most recent) visit.
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; p‐tau, phosphorylated tau; SD, standard deviation; t‐tau, total tau.
3.1. CSF biomarker changes and impact of demographics, APOE ε4 genotype
In the primary analyses (Table 2, Figure 1), with time since baseline as the timescale, there was a significant main effect of APOE ε4 genotype for Aβ42/Aβ40, indicating lower Aβ42/Aβ40 levels among APOE ε4 carriers. Additionally, there were significant APOE ε4 x time interactions for all CSF biomarkers, indicating greater Aβ42/Aβ40 decreases and p‐tau181 and t‐tau increases, among APOE ε4 carriers. There were also significant main effects of age (for all biomarkers) and significant age x time interactions (for all biomarkers except p‐tau181, p = 0.093). Older participants had more abnormal CSF biomarker levels and greater rates of biomarker change. In contrast, CSF biomarker levels and rates of change were unrelated to sex and years of education. Using chronological age as the timescale, the patterns of results were similar, except that the quadratic effect of time (i.e., age2) was significant for Aβ42/Aβ40 (p = 0.006; Table 2). Similar effects were found for the tau/(Aβ42/Aβ40) ratios (Text S3), and in models excluding extreme participant age groups (i.e., <40 and >85 years; Text S4 in supporting information).
TABLE 2.
Results of the longitudinal mixed effects regression models examining CSF biomarker trajectories
| Timescale: Years from baseline | ||||||
|---|---|---|---|---|---|---|
| Outcome: Aβ42/Aβ40 | Outcome: p‐tau181 | Outcome: t‐tau | ||||
| Model predictor | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value |
| Time | 0.017 (–0.003, 0.038) | 0.100 | 0.030 (0.009, 0.051) | 0.005 | 0.043 (0.020, 0.065) | < 0.001 |
| Time x time | –0.001 (–0.002, 0.000) | 0.093 | 0.002 (0.001, 0.003) | < 0.001 | 0.001 (0.000, 0.002) | 0.044 |
| Baseline age | –0.033 (–0.043, –0.023) | < 0.001 | 0.035 (0.024, 0.046) | < 0.001 | 0.035 (0.024, 0.046) | < 0.001 |
| Baseline age x time | –0.001 (–0.002, –0.000) | 0.025 | 0.001 (‐0.000, 0.002) | 0.093 | 0.002 (0.001, 0.003) | 0.001 |
| Sex, female | 0.005 (–0.205, 0.215) | 0.962 | 0.013 (–0.214, 0.240) | 0.910 | –0.004 (–0.231, 0.224) | 0.973 |
| Sex x time | –0.001 (–0.016, 0.013) | 0.841 | 0.003 (–0.017, 0.023) | 0.797 | 0.009 (–0.008, 0.026) | 0.318 |
| Education | 0.015 (–0.029, 0.058) | 0.511 | –0.017 (–0.064, 0.031) | 0.487 | –0.044 (–0.091, 0.003) | 0.068 |
| Education x time | 0.002 (–0.001, 0.005) | 0.278 | –0.001 (–0.005, 0.003) | 0.670 | 0.000 (–0.004, 0.004) | 0.998 |
| APOE ε4 | –0.508 (–0.721, –0.294) | < 0.001 | 0.106 (–0.126, 0.337) | 0.371 | 0.062 (–0.170, 0.293) | 0.602 |
| APOE ε4 x time | –0.045 (‐0.059, –0.030) | < 0.001 | 0.044 (0.024, 0.064) | < 0.001 | 0.039 (0.022, 0.056) | < 0.001 |
| Timescale: Chronological age | ||||||
|---|---|---|---|---|---|---|
| Outcome: Aβ42/Aβ40 | Outcome: p‐tau181 | Outcome: t‐tau | ||||
| Model predictor | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value |
| Age | –0.010 (–0.023, 0.003) | 0.148 | 0.034 (0.020, 0.048) | < 0.001 | 0.033 (0.020, 0.047) | < 0.001 |
| Age x age | –0.001 (–0.001, –0.000) | 0.006 | 0.001 (0.000, 0.001) | < 0.001 | 0.001 (0.001, 0.001) | < 0.001 |
| Sex, female | 0.041 (–0.147, 0.230) | 0.667 | 0.039 (–0.227, 0.305) | 0.773 | 0.074 (–0.164, 0.313) | 0.540 |
| Sex x age | –0.003 (–0.016, 0.011) | 0.690 | 0.010 (–0.005, 0.024) | 0.195 | 0.01 (–0.004, 0.023) | 0.167 |
| Education | 0.003 (–0.036, 0.042) | 0.895 | –0.001 (–0.056, 0.055) | 0.983 | –0.020 (–0.070, 0.029) | 0.426 |
| Education x age | 0.002 (‐0.001, 0.005) | 0.156 | –0.001 (–0.004, 0.002) | 0.690 | 0.000 (–0.003, 0.003) | 0.830 |
| APOE ε4 | –0.688 (–0.879, –0.497) | < 0.001 | 0.177 (–0.092, 0.446) | 0.198 | 0.119 (–0.122, 0.360) | 0.332 |
| APOE ε4 x age | –0.044 (‐0.058, –0.030) | < 0.001 | 0.044 (0.029, 0.059) | < 0.001 | 0.038 (0.024, 0.052) | < 0.001 |
Note: Different models were estimated for each CSF measure. Models were also adjusted for collection site (National Institutes of Health vs. Johns Hopkins University).
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; CI, confidence interval; CSF, cerebrospinal fluid; p‐tau, phosphorylated tau; t‐tau, total tau.
FIGURE 1.
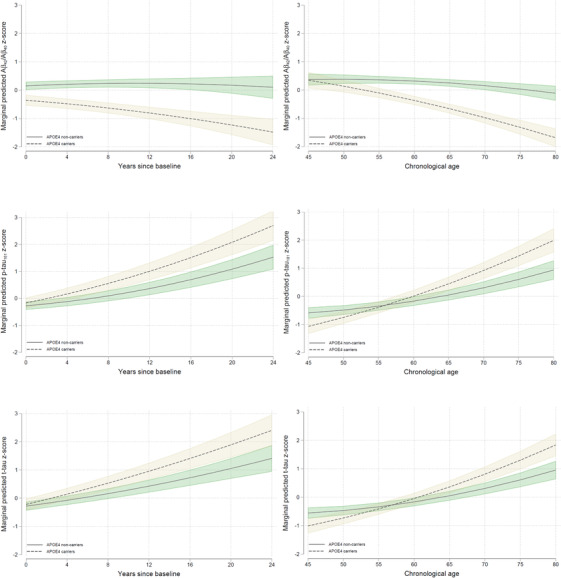
Adjusted estimates (95% confidence interval) of longitudinal trajectories of z‐scored cerebrospinal fluid (CSF) amyloid beta (Aβ)42/Aβ40 (top), phosphorylated tau (p‐tau181; middle) and total tau (t‐tau; bottom) biomarkers over time by, separately for apolipoprotein E (APOE) ε4 carriers (dashed tan lines) and non‐carriers (solid green lines). Results are shown using years since baseline (left) and chronological age (right) as the timescale
3.2. Associations between CSF biomarkers
For the models that examined baseline levels of p‐tau181 or t‐tau in relationship to change in Aβ42/Aβ40, there were main effects of tau but no tau x time interactions (Table 3). Higher baseline p‐tau181 and t‐tau were each associated with lower Aβ42/Aβ40 levels, but not with change in Aβ42/Aβ40 over time. In the models that examined baseline Aβ42/Aβ40 in relationship to change in tau, both the main effects of Aβ42/Aβ40 and the Aβ42/Aβ40 x time interaction terms were significant. Lower baseline Aβ42/Aβ40 was associated with higher levels of p‐tau181 and t‐tau, and with greater increases in p‐tau181 and t‐tau over time.
TABLE 3.
Results of the longitudinal mixed effects regression models examining the relationship of baseline biomarker levels to CSF biomarker trajectories
| Outcome: Aβ42/Aβ40 | Outcome: Aβ42/Aβ40 | Outcome: p‐tau181 | Outcome: t‐tau | |||||
|---|---|---|---|---|---|---|---|---|
| Model predictor | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value |
| Time | 0.017 (–0.003, 0.038) | 0.098 | 0.017 (–0.003, 0.038) | 0.101 | 0.034 (0.014, 0.054) | 0.001 | 0.045 (0.023, 0.067) | < 0.001 |
| Time x time | –0.001 (–0.002, 0.000) | 0.078 | –0.001 (–0.002, 0.000) | 0.087 | 0.002 (0.001, 0.003) | < 0.001 | 0.001 (0.000,0.002) | 0.049 |
| APOE ε4 | –0.490 (–0.692, –0.288) | < 0.001 | –0.449 (–0.636, –0.261) | < 0.001 | –0.086 (–0.298, 0.126) | 0.427 | –0.073 (–0.298, 0.152) | 0.525 |
| APOE ε4 x time | –0.045 (–0.059, –0.030) | < 0.001 | –0.043 (–0.058, –0.029) | < 0.001 | 0.030 (0.011, 0.050) | 0.002 | 0.029 (0.012, 0.046) | 0.001 |
| Baseline t‐tau | –0.295 (–0.397, –0.193) | < 0.001 | — | — | — | — | — | — |
| Baseline t‐tau x time | –0.009 (–0.018, 0.001) | 0.066 | — | — | — | — | — | — |
| Baseline p‐tau181 | — | — | –0.438 (–0.534, –0.343) | < 0.001 | — | — | — | — |
| Baseline p‐tau181 x time | — | — | –0.006 (–0.016, 0.003) | 0.208 | — | — | — | — |
| Baseline Aβ42/Aβ40 | — | — | — | — | –0.455 (–0.562, –0.348) | < 0.001 | –0.320 (–0.434, –0.206) | < 0.001 |
| Baseline Aβ42/Aβ40 x time | — | — | — | — | –0.031 (–0.043, –0.020) | < 0.001 | –0.022 (–0.033, –0.011) | < 0.001 |
Note: Different models were estimated for each CSF measure. Models were also adjusted for all variables indicated in Table 2.
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; CI, confidence interval; CSF, cerebrospinal fluid; p‐tau, phosphorylated tau; t‐tau, total tau.
3.3. CSF biomarker changes: impact of APOE ε4 when co‐varying Aβ42/Aβ40 positivity
With Aβ42/Aβ40 as the outcome, there was a main effect of Aβ42/Aβ40 positivity and an Aβ42/Aβ40 positivity x time interaction (both P ≤ 0.009; Text S5), indicating lower Aβ42/Aβ40 levels and greater Aβ42/Aβ40 decreases over time among Aβ42/Aβ40‐positive individuals. There was a main effect of APOE ε4 (p < 0.001) but no APOE ε4 x time or APOE ε4 x Aβ42/Aβ40 positivity x time interactions (both p > 0.59): APOE ε4 carriers had lower Aβ42/Aβ40 levels but rates of Aβ42/Aβ40 change over time did not differ by APOE ε4 genetic status after accounting for Aβ42/Aβ40 positivity.
For p‐tau181 and t‐tau as outcomes, Aβ42/Aβ40 positivity was associated with higher levels of p‐tau181 and t‐tau (both P ≤ 0.003). There were significant Aβ42/Aβ40 positivity x APOE ε4 and Aβ42/Aβ40 positivity x APOE ε4 x time interactions (all p < 0.02): relative to APOE ε4 non‐carriers, APOE ε4 carriers had lower tau levels when Aβ42/Aβ40 positive, but greater increases in p‐tau181 and t‐tau after reaching Aβ42/Aβ40 positivity.
3.4. CSF biomarker changes and follow‐up diagnosis
There were significant main effects of follow‐up diagnosis for Aβ42/Aβ40, p‐tau181, and t‐tau, indicating more abnormal baseline CSF biomarker levels among those who progressed to MCI/dementia over time. Additionally, there were follow‐up diagnosis x time interactions for p‐tau181 and t‐tau, indicating greater increases in tau among participants who progressed to MCI/dementia versus remained normal (Table 4; Figure 2). Further exploratory analyses examining CSF biomarker change before versus after MCI symptom onset indicated greater increases in tau prior to MCI clinical symptom onset, compared to those who remained cognitively normal (Text S6).
TABLE 4.
Results of the longitudinal mixed effects regression models examining CSF biomarker trajectories by follow‐up diagnosis (remain normal vs. progress to MCI/dementia)
| Outcome: Aβ42/Aβ40 | Outcome: p‐tau181 | Outcome: t‐tau | ||||
|---|---|---|---|---|---|---|
| Model predictor | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value | Estimate (95% CI) | p‐value |
| Time | 0.018 (–0.004, 0.040) | 0.101 | 0.015 (–0.007, 0.038) | 0.176 | 0.034 (0.010, 0.057) | 0.005 |
| Time x time | –0.001 (–0.002, 0.000) | 0.092 | 0.002 (0.001, 0.003) | < 0.001 | 0.001 (0.000, 0.002) | 0.037 |
| APOE ε4 | –0.492 (–0.701, –0.284) | < 0.001 | 0.094 (–0.134, 0.322) | 0.420 | 0.051 (–0.177, 0.280) | 0.659 |
| APOE ε4 x time | –0.045 (–0.059, –0.030) | < 0.001 | 0.042 (0.022, 0.062) | < 0.001 | 0.037 (0.020, 0.054) | < 0.001 |
| Follow‐up diagnosis (progressed vs. remained normal) | –0.415 (–0.637, –0.193) | < 0.001 | 0.365 (0.122, 0.607) | 0.003 | 0.318 (0.074, 0.561) | 0.011 |
| Follow‐up diagnosis x time | –0.001 (–0.017, 0.015) | 0.883 | 0.038 (0.016, 0.060) | 0.001 | 0.023 (0.004, 0.042) | 0.019 |
Note: Different models were estimated for each CSF measure. Models were also adjusted for all variables indicated in Table 2.
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; CI, confidence interval; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; p‐tau, phosphorylated tau; t‐tau, total tau.
FIGURE 2.
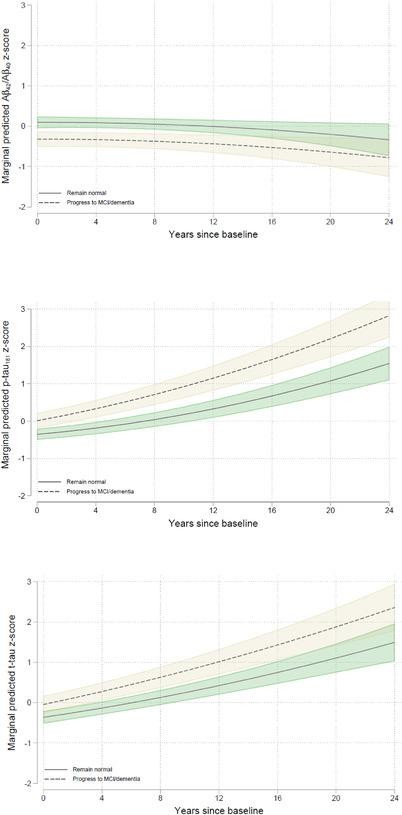
Adjusted estimates (95% confidence interval) of longitudinal trajectories of z‐scored cerebrospinal fluid (CSF) amyloid beta (Aβ)42/Aβ40 (top), phosphorylated tau (p‐tau181; middle) and total tau (t‐tau; bottom) biomarkers over time, separately for those who remained normal (solid green lines) versus progressed to mild cognitive impairment dementia (dashed tan lines). Results are modeled using years since baseline as the timescale
3.5. CSF biomarker changes: interactions among sex and age, APOE, and baseline biomarker levels
There were no significant interactions between sex and age or APOE ε4 genotype with respect to change in Aβ42/Aβ40, p‐tau181, or t‐tau over time (all p > 0.11; data not shown).
Sex did not modify the association between baseline p‐tau181 or t‐tau and Aβ42/Aβ40 (all p > 0.06). However, sex influenced the association between baseline Aβ42/Aβ40 and rate of change in p‐tau181. Significant sex x baseline Aβ42/Aβ40 (estimate = 0.295, 95% confidence interval [CI] = [0.090, 0.500], p = 0.005) and sex x baseline Aβ42/Aβ40 x time (estimate = 0.027, 95% CI = [0.005, 0.050], p = 0.017) interactions indicated that the association between baseline Aβ42/Aβ40 and both levels and rate of p‐tau181 change was weaker in females versus males.
4. DISCUSSION
This study examined CSF biomarker trajectories, measured over 10.7 years of follow‐up on average, among individuals who were initially cognitively normal and largely middle‐aged at baseline. There are several important findings. First, lower CSF Aβ42/Aβ40 levels were associated with increases in tau, and accelerations in CSF tau were closely linked to MCI symptom onset. Second, APOE ε4 genotype was unrelated to Aβ42/Aβ40 rates of change after accounting for Aβ42/Aβ40 positivity, but APOE ε4 was associated with greater increases in p‐tau181 and t‐tau after reaching Aβ42/Aβ40 positivity. Finally, AD biomarker trajectories did not differ by sex or years of education, though there was some evidence that sex may modify the relationship between Aβ42/Aβ40 and subsequent change in p‐tau181. These findings are consistent with hypothetical AD biomarker models, 2 proposing accelerated biomarker changes during preclinical AD, with these changes occurring over long periods of time beginning as early as midlife.
Compared to those who remained normal, those who progressed to MCI/dementia had greater rates of change in p‐tau181 and t‐tau (and tau/[Aβ42/Aβ40] ratios; see also Moghekar et al. 14 ), with exploratory analyses suggesting changes in tau occurred prior to MCI clinical symptom onset. This suggests accelerations in tau are more closely linked to MCI symptom onset than changes in amyloid. This is in line with recent findings from our group, 11 which reported that baseline levels of CSF tau were associated with proximal progression to MCI clinical symptoms (within 7 years, with 7 years being the mean time from baseline to MCI clinical symptom onset), whereas baseline Aβ42/Aβ40 was associated with more distal progression (>7 years). It is also consistent with findings showing strong relationships between tau and cognition among cognitively normal individuals, particularly in the presence of amyloid, 8 , 15 , 24 , 25 , 26 , 27 and with short‐term longitudinal CSF findings in autosomal dominant AD (ADAD), suggesting increases in CSF tau are most evident prior to an individual's expected age of symptom onset. 28
In contrast, the rate of change in CSF Aβ42/Aβ40 did not differ between those who remained normal versus progressed to MCI/dementia. Given there were baseline differences between these groups, it is likely that the greatest Aβ42/Aβ40 change occurred prior to the observation period (particularly for individuals who progressed to MCI more proximally to baseline), which in turn initiated increases in tau prior to symptom onset. For example, it has been suggested that CSF Aβ42 plateaus at least 9 years before dementia onset, 29 with studies in ADAD reporting similar CSF Aβ42 plateaus relative to the estimated age of symptom onset. 30 Additionally, it may be that some participants who have remained cognitively normal to date are undergoing amyloidosis associated with preclinical AD, reducing our ability to detect diagnostic group differences. Our findings differ from Roe et al., 31 which reported that participants who progressed to MCI/dementia had greater declines in CSF Aβ42 compared to those who remained cognitively normal, but no difference in p‐tau181 and t‐tau rates of change. Reasons for these different findings might be related to the measure of Aβ used (Aβ42/Aβ40 vs. Aβ42), cohort characteristics (e.g., AD family history; baseline age), or mean number of biomarker assessments (3.7 vs. 1.8). Of note, our results (and Roe et al. 31 ) indicate that CSF AD biomarker changes occur very early in the disease process. This aligns with autopsy findings that have identified neurofibrillary tangles and neuritic plaques in middle‐aged and younger individuals. 5 , 13
We also found that lower baseline Aβ42/Aβ40 was associated with longitudinal increases in p‐tau181 and t‐tau, but baseline tau was not associated with change in Aβ42/Aβ40. Although prior studies among cognitively normal individuals have reported associations between amyloid measures and longitudinal increases in tau, these studies were conducted in older (M age >70 years) cohorts with shorter follow‐up. 6 , 8 , 24 , 25 , 32 Together, these findings support the view that early amyloid changes may initiate or accelerate tau accumulation and neuronal injury, 2 , 33 providing an important therapeutic window for slowing AD pathogenesis prior to symptom onset.
Notably, APOE ε4 carriers had lower levels of Aβ42/Aβ40 (but not tau), but no differences in Aβ42/Aβ40 declines after accounting for APOE ε4‐related differences in Aβ42/Aβ40 positivity over time. These findings are in accordance with the known role for APOE ε4 in amyloid aggregation and clearance, and consistent with an earlier age of amyloid and AD dementia onset among APOE ε4 carriers. 3 , 34 , 35 Interestingly, prior studies on the relationship of APOE ε4 to amyloid change have been mixed. Some studies among cognitively normal individuals have reported increased rates of amyloid accumulation among APOE ε4 carriers, 36 , 37 , 38 whereas others have reported faster changes only among APOE ε4 carriers with low amyloid burden (Burnham et al. 4 and Lim and Mormino; 39 see also Schindler et al. 40 ), or no difference in amyloid accumulation by APOE ε4 genotype. 4 , 41 , 42 Our findings suggest that observed differences in Aβ42/Aβ40 trajectories between APOE ε4 carriers and non‐carriers reflect higher rates and earlier ages of amyloid positivity among APOE ε4 carriers (resulting in a more advanced preclinical disease stage).
APOE ε4 genotype was associated with greater rates of change in CSF p‐tau181 and t‐tau after reaching Aβ42/Aβ40 positivity. We hypothesize much of this effect is driven by associations between APOE ε4 and Aβ42/Aβ40: because APOE ε4 carriers are more likely to develop amyloid positivity at earlier ages, 3 they may also be more likely to show amyloid‐related increases in tau during the observation period. Consistent with this, a prior study estimated that the effect of APOE ε4 genotype on neurofibrillary tangle pathology is mainly mediated by amyloid pathology. 43 However, APOE ε4 genotype was associated with changes in p‐tau181 and t‐tau independent of baseline Aβ42/Aβ40 levels (Table 3), which may suggest some amyloid‐independent effects of APOE ε4 on tau accumulation. 44 , 45 Few comparable studies have evaluated the relationship of APOE ε4 to longitudinal changes in tau. Consistent with our results, one study reported a trend toward greater CSF p‐tau181 and t‐tau accumulation among older cognitively normal APOE ε4 carriers with lower baseline Aβ42. 6 Our results also appear consistent with a prior study showing that rates of change in p‐tau181 and t‐tau over ~7 years of follow‐up occurred earlier in cognitively normal, middle‐aged APOE ε4 carriers compared to non‐carriers. 38 In contrast, other studies of older cognitively normal individuals, or with shorter biomarker follow‐up, have found no direct association between APOE ε4 genotype and tau biomarker change (CSF: 6 , 31 , 46 ; PET: 8 ). Inconsistencies may be due to differences in length of biomarker follow‐up and participant age, as biomarker studies capture only a snapshot of neurobiological changes that occur over decades.
Our results also suggest that there are no sex‐related differences in CSF AD biomarker trajectories; however, there was some evidence of weaker coupling between Aβ42/Aβ40 and change in p‐tau181 among females compared to males. This is consistent with recent findings from our group, whereby Aβ42/Aβ40 and p‐tau181 had weaker associations with risk of MCI symptom onset in females than males at younger baseline ages, but the differences between sexes diminished with increasing baseline age. 11 However, sex differences in amyloid biomarkers are controversial, 47 and the lack of relationship between sex and tau differs from recent findings in various diagnostic groups, which have reported higher tau levels 7 , 9 , 48 (but see 49 ) and greater short‐term tau accumulation 6 , 8 in APOE ε4‐carrying or amyloid‐positive females. Reasons for these mixed findings are not clear, warranting additional studies.
Finally, the CSF biomarkers were unrelated to years of education. Although additional studies with other measures of intellectual/cognitive experiences are needed, this suggests education‐related differences in MCI/AD dementia risk are not due to differences in the accumulation of AD pathology (as measured by these biomarkers) during the earliest disease phases, in line with a recent review reporting limited evidence of a relationship between AD biomarkers and intellectual experiences. 50
This study has limitations. First, generalizability is limited because this volunteer sample was predominantly White, highly educated, and enriched for a family history of AD dementia. Although the mean baseline age was 57.5 years, the age range was large (20–93 years). Second, despite the moderate sample size, we may have been underpowered to detect three‐way interactions. Third, while we took several approaches to adjust for pre‐analytic factors related to Aβ42 (see Sections 2.3 and 2.5), these may have been insufficient and resulted in an underestimation of Aβ42/Aβ40 effects. Fourth, despite the long duration of follow‐up, CSF collection among those who progressed to MCI/dementia was more limited, particularly after MCI symptom onset, impacting our ability to examine biomarker changes during the earliest symptomatic disease phases. We were also unable to examine whether biomarker trajectories differed for those who progressed to MCI versus dementia, or separately for APOE ε2/ε4, APOE ε3/ε4, and APOE ε4/ε4, although these are important questions for future work.
To our knowledge, no prior study among initially cognitively normal, primarily middle‐aged individuals has described CSF AD biomarker changes over this duration of follow‐up. These findings advance our understanding of early AD‐related biomarker changes by further highlighting the important role of amyloid in the acceleration of tau, as well as the importance of studying longitudinal AD biomarker changes during middle age.
CONFLICTS OF INTEREST
CP, AS, JW, and MCW have no disclosures. Dr. Greenberg is Editor‐in‐Chief of the other open access journal of the Alzheimer's Association: Alzheimer's & Dementia—Translational Research and Clinical Interventions. Dr. Albert is an advisor to Eli Lilly. Dr. Moghekar receives research support to JHU (but no direct or indirect salary support) from Fujirebio Diagnostics to analyze CSF preanalytics.
Supporting information
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant numbers U19‐AG033655, P30‐AG066507). The authors are grateful to the members of the BIOCARD Scientific Advisory Board who provide continued oversight and guidance regarding the conduct of the study including: Drs David Holtzman, William Jagust, David Knopman, Walter Kukull, and Kevin Grimm, and Drs John Hsiao and Laurie Ryan, who provide oversight on behalf of the National Institute on Aging. The authors thank the members of the BIOCARD Resource Allocation Committee who provide ongoing guidance regarding the use of the biospecimens collected as part of the study, including: Drs Constantine Lyketsos, Carlos Pardo, Gerard Schellenberg, Leslie Shaw, Madhav Thambisetty, and John Trojanowski. The authors acknowledge the contributions of the Geriatric Psychiatry Branch of the intramural program of NIMH, who initiated the study (Principal investigator: Dr. Trey Sunderland). The authors are indebted to Dr. Karen Putnam, who provided documentation of the Geriatric Psychiatry Branch study procedures and the data files received from NIMH.
The BIOCARD Study consists of 7 Cores and 2 Projects with the following members: (1) the Administrative Core (Marilyn Albert, Rostislav Brichko); (2) the Clinical Core (Marilyn Albert, Anja Soldan, Corinne Pettigrew, Greg Pontone, Leonie Farrington, Jules Gilles, Nicole Johnson, Maura Grega, Gay Rudow, Scott Rudow); (3) the Imaging Core (Michael Miller, Susumu Mori, Tilak Ratnanather, Andrea Faria, Anthony Kolasny, Kenichi Oishi, Laurent Younes, Hanzhang Lu); (4) the Biospecimen Core (Abhay Moghekar, Jacqueline Darrow, Alexandria Lewis); (5) the Informatics Core (Ann Ervin, David Shade, Jennifer Jones, Hamadou Coulibaly, Kathy Moser); (6) the Biostatistics Core (Mei‐Cheng Wang, Yuxin [Daisy] Zhu, Jiangxia Wang); (7) the Neuropathology Core (Juan Troncoso, Javier Redding, Roberta Knox); (8) Project 1 (Paul Worley, Jeremy Walston), and (9) Project 2 (Mei‐Cheng Wang, Sufei Sun).
Pettigrew C, Soldan A, Wang J, et al. Longitudinal CSF Alzheimer's disease biomarker changes from middle age to late adulthood. Alzheimer's Dement. 2022;14:e12374. 10.1002/dad2.12374
REFERENCES
- 1. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Jr., Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Betthauser TJ, Bilgel M, Koscik RL, et al. Multi‐method investigation of factors influencing amyloid onset and impairment in three cohorts. Brain. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burnham SC, Laws SM, Budgeon CA, et al. Impact of APOE‐epsilon4 carriage on the onset and rates of neocortical Abeta‐amyloid deposition. Neurobiol Aging. 2020;95:46‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pletnikova O, Kageyama Y, Rudow G, et al. The spectrum of preclinical Alzheimer's disease pathology and its modulation by ApoE genotype. Neurobiol Aging. 2018;71:72‐80. [DOI] [PubMed] [Google Scholar]
- 6. Buckley RF, Mormino EC, Chhatwal J, et al. Associations between baseline amyloid, sex, and APOE on subsequent tau accumulation in cerebrospinal fluid. Neurobiol Aging. 2019;78:178‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hohman TJ, Dumitrescu L, Barnes LL, et al. Sex‐specific association of apolipoprotein E with cerebrospinal fluid levels of tau. JAMA Neurol. 2018;75:989‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jack CR, Wiste HJ, Weigand SD, et al. Predicting future rates of tau accumulation on PET. Brain. 2020;143:3136‐3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Altmann A, Tian L, Henderson VW, Greicius MD, Alzheimer's disease neuroimaging initiative I. Sex modifies the APOE‐related risk of developing Alzheimer disease. Ann Neurol. 2014;75:563‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 11. Greenberg BD, Pettigrew C, Soldan A, et al. CSF Alzheimer's disease biomarkers: Time‐varying relationships with MCI symptom onset, and associations with age, sex and ApoE4. Neurology. 2022;99:e1640‐e1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neu SC, Pa J, Kukull W, et al. Apolipoprotein E genotype and sex risk factors for Alzheimer disease: a meta‐analysis. JAMA Neurol. 2017;74:1178‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960‐969. [DOI] [PubMed] [Google Scholar]
- 14. Moghekar A, Li S, Lu Y, et al. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology. 2013;81:1753‐1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soldan A, Pettigrew C, Cai Q, et al. Hypothetical preclinical Alzheimer disease groups and longitudinal cognitive change. JAMA Neurol. 2016;73:698‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Albert M, Soldan A, Gottesman R, et al. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Curr Alzheimer Res. 2014;11:773‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. [DOI] [PubMed] [Google Scholar]
- 18. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gervaise‐Henry C, Watfa G, Albuisson E, et al. Cerebrospinal fluid Abeta42/Abeta40 as a means to limiting tube‐ and storage‐dependent pre‐analytical variability in clinical setting. J Alzheimers Dis. 2017;57:437‐445. [DOI] [PubMed] [Google Scholar]
- 21. Lewczuk P, Matzen A, Blennow K, et al. Cerebrospinal fluid Abeta42/40 corresponds better than Abeta42 to amyloid PET in Alzheimer's disease. J Alzheimers Dis. 2017;55:813‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toombs J, Foiani MS, Wellington H, et al. Amyloid beta peptides are differentially vulnerable to preanalytical surface exposure, an effect incompletely mitigated by the use of ratios. Alzheimers Dement (Amst). 2018;10:311‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willemse E, van Uffelen K, Brix B, Engelborghs S, Vanderstichele H, Teunissen C. How to handle adsorption of cerebrospinal fluid amyloid beta (1‐42) in laboratory practice? Identifying problematic handlings and resolving the issue by use of the Abeta42/Abeta40 ratio. Alzheimers Dement. 2017;13:885‐892. [DOI] [PubMed] [Google Scholar]
- 24. Gomar JJ, Conejero‐Goldberg C, Davies P, Goldberg TE, Alzheimer's disease neuroimaging I. anti‐correlated cerebrospinal fluid biomarker trajectories in preclinical Alzheimer's disease. J Alzheimers Dis. 2016;51:1085‐1097. [DOI] [PubMed] [Google Scholar]
- 25. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76:915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sperling RA, Mormino EC, Schultz AP, et al. The impact of amyloid‐beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85:181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Llibre‐Guerra JJ, Li Y, Schindler SE, et al. Association of longitudinal changes in cerebrospinal fluid total tau and phosphorylated tau 181 and brain atrophy with disease progression in patients with Alzheimer disease. JAMA Netw Open. 2019;2:e1917126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stomrud E, Minthon L, Zetterberg H, Blennow K, Hansson O. Longitudinal cerebrospinal fluid biomarker measurements in preclinical sporadic Alzheimer's disease: A prospective 9‐year study. Alzheimers Dement (Amst). 2015;1:403‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schindler SE, Fagan AM. Autosomal dominant alzheimer disease: a unique resource to study CSF biomarker changes in preclinical AD. Front Neurol. 2015;6:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roe CM, Ances BM, Head D, et al. Incident cognitive impairment: longitudinal changes in molecular, structural and cognitive biomarkers. Brain. 2018;141:3233‐3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sutphen CL, McCue L, Herries EM, et al. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer's disease. Alzheimers Dement. 2018;14:869‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353‐356. [DOI] [PubMed] [Google Scholar]
- 34. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bilgel M, An Y, Zhou Y, et al. Individual estimates of age at detectable amyloid onset for risk factor assessment. Alzheimers Dement. 2016;12:373‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mishra S, Blazey TM, Holtzman DM, et al. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE epsilon4 genotype. Brain. 2018;141:1828‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stomrud E, Hansson O, Zetterberg H, Blennow K, Minthon L, Londos E. Correlation of longitudinal cerebrospinal fluid biomarkers with cognitive decline in healthy older adults. Arch Neurol. 2010;67:217‐223. [DOI] [PubMed] [Google Scholar]
- 38. Sutphen CL, Jasielec MS, Shah AR, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 2015;72:1029‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lim YY, Mormino EC, Alzheimer's Disease Neuroimaging I. APOE genotype and early beta‐amyloid accumulation in older adults without dementia. Neurology. 2017;89:1028‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schindler SE, Li Y, Buckles VD, et al. Predicting symptom onset in sporadic Alzheimer disease with amyloid PET. Neurology. 2021;97:e1823‐e1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Resnick SM, Bilgel M, Moghekar A, et al. Changes in Abeta biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiol Aging. 2015;36:2333‐2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vlassenko AG, Mintun MA, Xiong C, et al. Amyloid‐beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data. Ann Neurol. 2011;70:857‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mungas D, Tractenberg R, Schneider JA, Crane PK, Bennett DA. A 2‐process model for neuropathology of Alzheimer's disease. Neurobiol Aging. 2014;35:301‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid‐beta‐independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21:21‐35. [DOI] [PubMed] [Google Scholar]
- 45. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lleo A, Alcolea D, Martinez‐Lage P, et al. Longitudinal cerebrospinal fluid biomarker trajectories along the Alzheimer's disease continuum in the BIOMARKAPD study. Alzheimers Dement. 2019;15:742‐753. [DOI] [PubMed] [Google Scholar]
- 47. Ferretti MT, Iulita MF, Cavedo E, et al. Sex differences in Alzheimer disease ‐ the gateway to precision medicine. Nat Rev Neurol. 2018;14:457‐469. [DOI] [PubMed] [Google Scholar]
- 48. Damoiseaux JS, Seeley WW, Zhou J, et al. Gender modulates the APOE epsilon4 effect in healthy older adults: convergent evidence from functional brain connectivity and spinal fluid tau levels. J Neurosci. 2012;32:8254‐8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Toledo JB, Zetterberg H, van Harten AC, et al. Alzheimer's disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain. 2015;138:2701‐2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soldan A, Pettigrew C, Albert M. Cognitive reserve from the perspective of preclinical Alzheimer disease: 2020 Update. Clin Geriatr Med. 2020;36:247‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION