

Abstract
Background
PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitors are important therapeutic options for reducing cardiovascular disease risk; however, questions remain regarding potential differences in the neuropsychiatric impact of long‐term PCSK9 inhibition between men and women.
Methods and Results
Using PCSK9 gene single‐nucleotide polymorphisms from European ancestry–based genome‐wide association studies of low‐density lipoprotein cholesterol (N=1 320 016), circulating PCSK9 protein levels (N=10 186), tissue‐specific PCSK9 gene expression, sex‐specific genome‐wide association studies of anxiety, depression, cognition, insomnia, and dementia (ranging from 54 321 to 194 174), we used drug‐target inverse variance–weighted Mendelian randomization (MR) and complementary MR methods (MR Egger, weighted median, and weighted mode) to investigate potential neuropsychiatric consequences of genetically proxied PCSK9 inhibition in men and women. We failed to find evidence surpassing correction for multiple comparisons of relationships between genetically proxied PCSK9 inhibition and the risk for the 12 neuropsychiatric end points in either men or women. Drug‐target analyses were generally well‐powered to detect effect estimates at several hypothesized thresholds for both combined‐sex and sex‐specific end points, especially analyses using PCSK9 instruments derived from protein and expression quantitative trait loci. Further, MR estimates across complementary MR methods and additional models using genetic instruments derived from circulating PCSK9 protein levels and tissue‐specific PCSK9 expression were in alignment, strengthening causal inference.
Conclusions
Genetically proxied PCSK9 inhibition showed a neutral neuropsychiatric side effect profile with no major sex‐specific differences. Given statistical power considerations, replication with larger samples, as well as data from other ancestral populations, are necessary. These findings may have important clinical implications for lipid‐lowering drug‐prescribing practices and side effect monitoring of approved and future PCSK9 therapies.
Keywords: Alzheimer disease, cholesterol, cognition, dementia, depression, low‐density lipoprotein, Mendelian randomization, PCSK9
Subject Categories: Epigenetics, Atherosclerosis, Genetics, Mental Health
Nonstandard Abbreviations and Acronyms
- CRISPR
clustered regularly interspaced short palindromic repeats
- eQTL
expression quantitative trait loci
- GLGC
Global Lipids Genetics Consortium
- GTEx
Genotype‐Tissue Expression
- InSIDE
Instrument Strength Independent of Direct Effect
- IV
instrumental variable
- IVW
inverse variance–weighted
- MR
Mendelian randomization
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PCSK9i
proprotein convertase subtilisin/kexin type 9 inhibitor
- pQTL
protein quantitative trait loci
- TPM
transcripts per million
Clinical Perspective.
What Is New?
PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitors exhibit a safe neuropsychiatric outcome profile, which is not significantly different in male and female samples.
What Are the Clinical Implications?
Genetic evidence suggests that PCSK9 inhibitors do not pose a significant risk for adverse cognitive outcomes reported in some studies of statins, and concern regarding cognitive side effects contributing to underuse of PCSK9 inhibitors may be unfounded.
Hyperlipidemia is a disease marked by a high concentration of lipids in the blood and has been known for some time to play a role in the pathogenesis of diseases of the cardiovascular and neurological system, including myocardial infarction, stroke, and sudden cardiac death. 1 , 2 , 3 , 4 , 5 , 6 While hyperlipidemia includes cases of elevated blood triglycerides, hypercholesterolemia is marked by elevated blood low‐density lipoprotein cholesterol (LDL‐C). Hyperlipidemia and hypercholesterolemia are among the leading causes of cardiovascular disease, the leading cause of death worldwide, through their role in the formation of atherosclerotic plaques. 7 , 8 , 9 , 10 , 11
Pharmacologic modification of atherogenic lipoprotein levels by lipid‐lowering therapies such as statins 12 and PCSK9 (proprotein convertase subtilisin/kexin type 9) (monoclonal antibody inhibitors (alirocumab and evolocumab) 13 are considered effective approaches to reducing cardiovascular disease risk. 14 New pharmaceutical approaches to PCSK9 inhibition (PCSK9i) are also being developed. Inclisiran, a small inhibitory RNA molecule which inhibits PCSK9 via the RNA interference (RNAi) pathway 15 has recently been approved by the European Commission and US Food and Drug Administration to be used in combination with statins and diet to lower LDL‐C levels in adults with hypercholesterolemia or atherosclerosis, 15 , 16 , 17 , 18 and recent preclinical work using CRISPR gene editing to induce a PCSK9 knockout genotype reported long‐term cholesterol reduction in nonhuman primates. 19 , 20
While short‐term clinical trials investigating the neuropsychiatric impact of PCSK9 monoclonal antibodies reported no major adverse neuropsychiatric events among study participants, 13 , 21 , 22 , 23 there remains concern about the potential adverse neuropsychiatric impact of PCSK9i therapy, owing in part to potential cognitive effects that have been observed in some studies of statins, 24 , 25 and in vitro and in vivo studies implicating PCSK9 in a range of neural processes. 26 , 27 For example, PCSK9 has been shown to be involved in neuronal differentiation, apoptosis, astrocytes, and glial cell activation; neuronal PCSK9 expression has been shown to be upregulated in adult brains during disease states, including Alzheimer disease (AD), alcohol use disorder, ischemic stroke, and mood disorders 26 , 27 , 28 ; and Mendelian randomization (MR) analyses have suggested that genetically proxied long‐term PCSK9i is associated with increased risk for depression in individuals of European ancestry. 29
As new PCSK9i therapeutics become approved for clinical use, it is important to examine and understand their long‐term efficacy and side effect profiles in populations representative of the patients who will use the therapeutics, including any potential differences between those of men and women. 29 , 30 Neuropsychiatric disorder prevalence and risk profiles are known to differ between men and women. 31 , 32 Women are twice as likely as men to be diagnosed with depression, 31 are more likely to present with most major subtypes of depression, and report greater symptom severity. 33 , 34 , 35 , 36 Given sex‐related differences in mood disorders and dementia risk, it is important to investigate any potential sex‐specific effects of PCSK9i, and while previous short‐term clinical trials and genetics‐based studies have failed to find evidence of large‐scale neuropsychiatric effects related to PCSK9i, 37 , 38 , 39 , 40 any potential differences in risk between men and women have not been investigated. Although randomized controlled trials (RCTs) remain the gold standard for assessing causal relationships between risk factors and disease outcomes, 41 , 42 an RCT investigating the long‐term and potential sex‐specific impact of PCSK9i on neuropsychiatric disorders would be challenging because of the recency of PCSK9i approval, and consequently long‐term neuropsychiatric data from RCTs are not yet available. In addition, despite improved sex equity in RCTs over the past 20 years, there remains sex bias within clinical trials that may not capture important differences between men and women. 30
Therefore, the present study sought to determine whether there is genetic evidence of a sex‐specific impact of PCSK9i therapy on mood disorders, cognition, or dementia. We employed drug‐target MR, a recent extension of MR using single‐nucleotide polymorphisms (SNPs) associated with druggable gene targets (ie, SNPs within or near the PCSK9 gene locus), 43 to genetically proxy and evaluate the lifelong impact on outcomes of interest caused by pharmacological modulation of the gene target (ie, inhibition of PCSK9), 43 and summary‐level genome‐wide association study (GWAS) data in men, women, and in combined‐sex samples. In addition to instrumenting genetically proxied PCSK9i in LDL‐C levels (the primary physiological response to pharmacological PCSK9i), we also leveraged recently released GWAS data on circulating PCSK9 protein levels and cerebral cortex– and liver‐specific PCSK9 gene expression data in order to better genetically model the mechanisms of action for the anti‐PCSK9 monoclonal antibodies 44 and inclisiran, 15 respectively.
Methods
Code Availability
Code is available from the authors upon reasonable request. The current study used the TwoSampleMR R package (https://mrcieu.github.io/TwoSampleMR/). Figures 1 and 2 were created using BioRender.com. The mRnd Shiny App used for power calculations is available at https://shiny.cnsgenomics.com/mRnd/.
Figure 1. Overview of study methods and procedures.
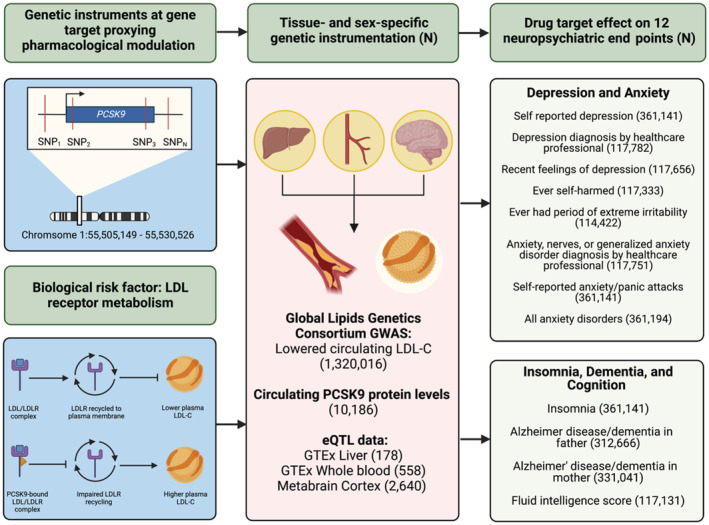
All summary‐level genetic associations were derived from genome‐wide association studies (GWAS) of European ancestry. Additional information regarding the GWAS data (consortium, study cohort, and author information of the GWAS for the exposure and neuropsychiatric outcomes) are located in Table S1. We performed cis‐instrumentation of genetically predisposed PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibition in several complementary data sets. First, single‐nucleotide polymorphisms (SNPs) ±100 kilobases of the PCSK9 gene locus were extracted from the Global Lipids Genetics Consortium (GLGC) 2021 meta‐analysis on circulating low‐density lipoprotein (LDL) cholesterol (LDL‐C) levels surpassing conventional genome‐wide significance (P<5×10−8). We also proxied PCSK9 inhibition using circulating levels of the PCSK9 protein and tissue‐specific gene expression of PCSK9 in the liver, whole blood, and brain (cortex). These PCSK9 SNPs were then extracted from selected neuropsychiatric end points spanning mood disorders, insomnia, dementia, and cognition from UK Biobank data that combined men and women, as well as male‐only and female‐only GWASs. Finally, we performed drug‐target Mendelian randomization to evaluate the neuropsychiatric impact of genetically predisposed PCSK9 inhibition across men and women (see Methods section). eQTL indicates expression quantitative trait loci; GTEx Genotype‐Tissue Expression; and LDLR, low‐density lipoprotein receptor.
Figure 2. Inverse variance–weighted Mendelian randomization results of genetically proxied PCSK9 (proprotein convertase subtilisin/kexin type 9) in circulating low‐density lipoprotein cholesterol (LDL‐C) and circulating protein levels on neuropsychiatric outcomes for men and women.
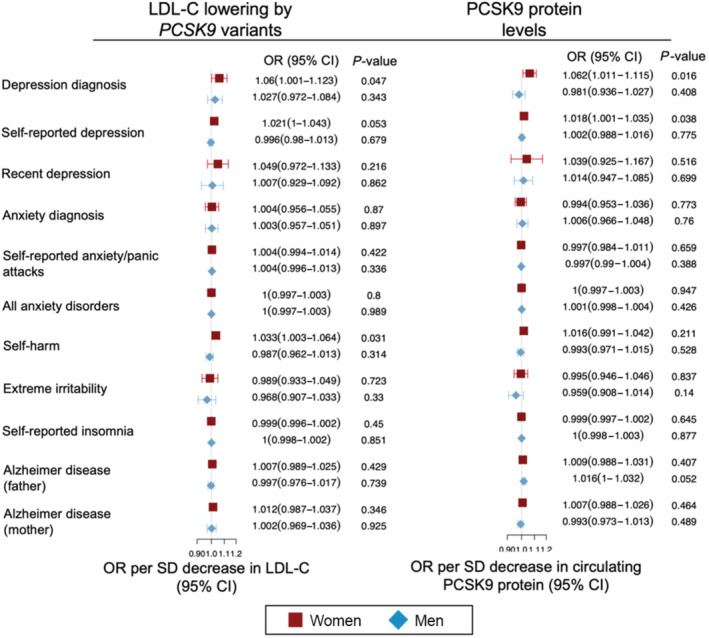
Estimates for the LDL‐C–lowering impact of PCSK9 inhibition are reported as odds ratios (ORs) corresponding to a change in the risk for the neuropsychiatric end point per 1‐SD reduction of genetically determined circulating LDL‐C levels (ie, the primary physiological response of pharmacologic PCSK9 inhibition). For the analyses using circulating PCSK9 protein levels, the ORs correspond to a change in genetically determined normalized circulating PCSK9 protein levels (ie, the primary physiological target of monoclonal PCSK9 inhibitors). Because fluid intelligence is a continuous variable, it was not included in the Forest plot but is discussed in the Results section. Full results, including combined‐sex results, are presented in Table S4 and Table S5.
iData Availability
All exposure instruments required to replicate the analyses are located in the Supplemental Tables. The current study used publicly available GWAS summary statistics. The neuropsychiatric endpoint data for men, women, and combined‐sex samples are available from the Neale Lab repository (http://www.nealelab.is/uk‐biobank). The Global Lipids Genetics Consortium (GLGC) LDL‐C data are available from the GLGC downloads page (http://csg.sph.umich.edu/willer/public/glgc‐lipids2021/). GWAS results of circulating PCSK9 protein levels are available at https://zenodo.org/record/5643551. Genotype‐Tissue Expression (GTEx) expression quantitative trait loci (eQTL) data are available from its respective downloads page (https://gtexportal.org/home/datasets). MetaBrain eQTL data are available at: https://metabrain.nl/.
Approval and Data Sources
Figure 1 provides a study overview. The present study used publicly available, summary‐level genome‐wide association study (GWAS) data. The data sources used (UK Biobank [https://www.ukbiobank.ac.uk] and GLGC [http://lipidgenetics.org/]) have existing approvals from their respective institutional review boards. All participants provided written informed consent. Full information and references for data sources can be found in Table S1. The current study is reported in accordance with the MR STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines (Data S1).
MR Assumptions
MR uses SNPs as instrumental variables (IVs) to identify associations between the genetic liability for an exposure trait and an outcome. 43 , 45 , 46 MR has 3 main assumptions (Figure S1): (1) the IV itself must be associated with the exposure (the relevance assumption); (2) there must not exist any causes of the IV that also affect the outcomes through mechanisms other than the exposure of interest (the exchangeability assumption); and (3) the IV must not affect the outcome through a mechanism independent of the exposure, nor affect another trait with a downstream effect on the outcome of interest (the exclusion restriction assumption). 45 , 46
The current study used drug‐target MR, a recent extension of MR, leveraging variants located within the genomic region of a druggable gene (cis‐instrumentation) to evaluate whether modulation of a specific drug target (ie, PCSK9) will have an impact on the outcomes of interest (ie, neuropsychiatric end points). 43 This interpretation is different than conventional biomarker MR analyses, which investigates the causal impact of the biomarker (eg, circulating LDL‐C levels) on the outcomes of interest. 43 Drug‐target MR using cis‐instruments are less prone to bias because of horizontal pleiotropy. 43 Nevertheless, we included additional MR methods used to assess the sensitivity of our results to different patterns of violations of IV assumptions, which we describe in the Statistical Analysis section.
PCSK9 Instruments
To proxy therapeutic inhibition of PCSK9, we included several separate genetic models incorporating data at the biomarker, protein, and gene expression levels. A brief summary of the PCSK9 instruments used in the present study can be found in Table 1; detailed instrument descriptions are presented in Table S2. First, because the primary physiological response of pharmacological PCSK9 inhibition is lowering circulating levels of LDL‐C, 44 as has been done in previous MR studies of PCSK9 and other lipid‐lowering target genes, 39 , 47 , 48 we extracted SNPs located within 100 kb of the PCSK9 gene locus (chromosome 1:55505221–55 530 525 GRCh37/hg19) associated with LDL‐C levels from participants of European ancestry in the 2021 GLGC LDL‐C European meta‐analysis (N≤1 320 016) 49 at conventional genome‐wide significance P<5×10−8. 49 We clumped SNPs at linkage disequilibrium R 2=0.001, leaving 6 independent SNPs comprising the LDL‐C–based PCSK9 instrument. Because only 3 of the 6 independent PCSK9 variants in the LDL‐C instrument were found in the PCSK9 protein level data, 50 we also created a PCSK9–LDL‐C instrument comprising only variants associated with both LDL‐C (P<5×10−8) and PCSK9 protein levels (P<5×10−6), 50 as a biologically conservative instrument and sensitivity analysis to assess the MR exclusion restriction assumption. We included an LDL‐C–based PCSK9 instrument with 3 independent SNPs derived from the earlier 2013 GLGC GWAS of circulating LDL‐C levels (N≤173 082) as an additional sensitivity analysis. 51 Circulating LDL‐C levels are reported in SD units.
Table 1.
Summary of PCSK9 Instruments Included in the Current Study
| PCSK9 instruments | No. of genetic variants (SNPs) | Average sample size (minimum, maximum) | Average F statistic (minimum, maximum) | Total R 2 |
|---|---|---|---|---|
| Circulating LDL‐C levels | ||||
| PCSK9 SNPs in LDL‐C (GLGC 2021 49 ) | 6 | 1 196 479 (1 094 709, 1 228 324) | 484.95 (83.35, 913.76) | 0.0024 |
| PCSK9 SNPs in LDL‐C (GLGC 2013 51 ) | 3 | 81 322 (77 417, 86 399) | 308.17 (21.66, 762.37) | 0.0118 |
| PCSK9 SNPs in LDL‐C with variants also associated with PCSK9 protein | 2 | 1 229 493 (1 227 744, 1 231 241) | 1206.53 (798.60, 1614.53) | 0.00196 |
| Circulating PCSK9 protein levels (pQTL) | ||||
| PCSK9 in whole blood | 2 | 9905 (9623, 10 186) | 153.42 (92.24, 207.20) | 0.03001 |
| Tissue‐specific PCSK9 expression (eQTLs) | ||||
| PCSK9 in liver | 1 | 178 (NA) | 26.83 (NA) | 0.1323 |
| PCSK9 in whole blood | 3 | 558 (NA) | 44.21 (24.87, 58.54) | 0.1374 |
| PCSK9 in cortex | 1 | 2640 (NA) | 96.96 (NA) | 0.0355 |
#Genetic variants were the number of PCSK9 (proprotein convertase subtilisin/kexin type 9) single‐nucleotide polymorphisms (SNPs) within ±100 kilobases of the PCSK9 locus included in the instrument before harmonization with the neuropsychiatric outcome data. Average sample size reports the average genome‐wide association studies (GWAS) summary statistics sample size for each PCSK9 instrument SNP. Average F statistics for each PCSK9 variant in the instrument. Total R 2 is the variance explained in the GWAS summary statistics by all of the PCSK9 SNPs for each instrument. PCSK9 quantitative trait loci data were obtained from the Pott et al meta‐analysis of circulating PCSK9 protein, Genotype‐Tissue Expression (GTEx) project portal, and MetaBrain Consortia (links provided in Table S2). Additional PCSK9 instrument information is presented in Table S2. eQTL indicates expression quantitative trait loci; GLGC, Global Lipid Genetics Consortium; LDL‐C, low‐density lipoprotein cholesterol; and pQTL, protein expression quantitative trait loci.
Next, because instrumentation of PCSK9 in LDL‐C does not measure changes in PCSK9 levels directly, and because current anti‐PCSK9 monoclonal antibodies target the PCSK9 protein, 44 we supplemented the PCSK9 instrument derived from LDL‐C data with SNPs associated with circulating PCSK9 protein levels using protein quantitative loci (pQTL) data from 10 186 individuals of European ancestry. 50 We extracted and clumped SNPs as above, leaving 2 independent SNPs within the PCSK9 locus. PCSK9 protein levels were measured in normalized protein units. 50 Given the liver‐specific mechanism of action of recently approved small interfering RNA inhibitor, inclisiran, 15 we extracted eQTL from GTEx version 8 52 PCSK9 data–derived liver tissue (N=178). We cis‐instrumented the liver PCSK9 instrument (clumped at linkage disequilibrium R 2=0.001), leaving one variant. Finally, we supplemented these eQTL analyses with additional PCSK9 instruments from PCSK9 data derived from whole blood (N=558), and the brain tissue from the MetaBrain analysis (N=2640). 53 The whole blood eQTL PCSK9 instrument had 2 independent SNPs and the eQTL PCSK9 instrument from cortex had 1 independent SNP (Table S2). eQTL data are measured in transcripts per million (TPM). 53 , 54
To test the MR relevance assumption and because MR analyses may be biased by the inclusion of weak instrument SNPs, which may occur when the variants comprising the MR instrument explain only a small proportion of the exposure, resulting in reduced statistical power to reject the null hypothesis, 55 we tested the strength of each PCSK9 instrument by calculating the variance explained by the instrument (ie, the R 2) and the corresponding F statistics. 56 By convention, SNP F statistics >10 provide evidence that the instruments are unlikely to be subject to weak instrument bias. 56 Every PCSK9 SNP used in this study had estimated F statistics exceeding 20 (Table S2). F statistics for PCSK9 SNPs within the 2021 LDL‐C data ranged from 72.52 to 913.71 (average F statistic=484.49). F statistics for pQTL and eQTL PCSK9 instruments were similarly strong (minimum, maximum pQTL: 92.24, 207.20; whole blood eQTL: 24.47, 58.54; liver eQTL: 26.83). These F statistics indicate minimal bias from weak instruments in the MR analyses. 55
Circulating Lipid Levels
LDL‐C levels have been implicated in neuropsychiatric disorders, 57 , 58 , 59 and because LDL‐C is the primary biomarker measured with PCSK9i, 44 we also evaluated the relationships of LDL‐C levels and neuropsychiatric outcomes using a polygenic LDL‐C instrument. To proxy LDL‐C levels, we extracted 400 independent (linkage disequilibrium R 2 < 0.001) variants associated with LDL‐C at conventional genome‐wide significance, irrespective of their genomic position, from the GLGC meta‐analysis (N≤1 320 016). 49 A full list of variants used for the polygenic LDL‐C instrument is available in Table S3. Instrument SNPs had strong F statistics (average F statistic, 206.32; range, 29.73–4799.60).
Neuropsychiatric Outcomes From the UK Biobank
We obtained summary‐level GWAS data from end points related to depression, anxiety, cognition, and dementia from the UK Biobank for the sex‐specific analysis (http://www.nealelab.is/uk‐biobank). 60 Additional information for all end points are available in Figure 2 and Table S1. These UK Biobank data sets were derived from participants of European ancestry (ages 40–69 years at the start of data collection). For depression and anxiety, we included self‐reported depression (men: 7156 cases/159 832 controls; women: 13 492 cases/180 661 controls); whether the participant reported ever having depression diagnosed by a healthcare professional (men: 8166 cases/43 675 controls; women: 16 921 cases/49 020 controls); whether the participant reported ever having self‐harmed (men: 1594/50 262; women: 3505/62 372); whether the participant reported ever having a period of extreme irritability (men: 12 626 cases/37 987 controls; women: 17 121 cases/46 778 controls); whether the participant reported ever having anxiety, nerves, or generalized anxiety disorder diagnosed by a professional healthcare worker (men: 5649 cases/46 176 controls; women: 11 081 cases/54 845 controls); and self‐reported anxiety/panic attacks (men: 1813 cases/165 175 controls; women: 3148 cases/191 005 controls).
Given the average age of UK Biobank participants, there are few cases of AD among UK Biobank participants. Therefore, we leveraged the high heritability of AD, which implies that AD case status for offspring can be, to some extent, inferred by parental AD case status (ie, offspring of parents with AD may have higher genetic risk for AD) 60 and used a phenotype‐by‐proxy approach with GWAS data on parental AD among participants in the UK Biobank. We used AD status of both fathers (men: 6617 cases/135 280 controls; women: 8405 cases/162 364 controls) and mothers (men: 12 324 cases/136 664 controls; women: 16 183 cases/165 870 controls). Finally, we used a continuous measure of fluid intelligence (men=54 321; women=62 810). For all end points, we analyzed the male, female, and combined GWAS data (Figure 2 and Table S1). We extracted PCSK9 and polygenic LDL‐C SNPs for each instrument from each of the outcome GWAS and then harmonized the exposure and outcome data (ie, aligned effect alleles, β coefficients). All PCSK9 instrument SNPs were found in the outcome GWAS data.
Power Calculations
We performed MR power calculations for the primary analyses based on available outcome sample sizes and the variance explained (R 2) of the PCSK9 and polygenic LDL instruments using the mRnd Shiny App. 61 We used an α level of 0.05 and calculated the statistical power to detect odds ratios (ORs) at 3 separate true effect sizes (0.50, 0.80, and 0.90) and considered analyses sufficiently powered where calculated power exceeded 80%.
Sample Independence
The 2021 GLGC meta‐analysis incorporated LDL‐C from 440 546 UK Biobank participants. 49 For neuropsychiatric outcomes included in this study, the Neale Lab release of the UK Biobank had samples ranging from N=114 422 to N=361 114. Therefore, for analyses with the LDL‐C instruments, there is an up to 27% overlap for these combined‐sex analyses. However, the maximum overlap for male and female analyses is 12.7% and 14.7%, respectively. While sample overlap in summary‐level GWAS data used to estimate genetic associations between exposure and outcome 2‐sample MR may potentially bias results, 62 , 63 any bias would likely be minimal, 62 , 63 and it has also been shown that 2‐sample MR may be safely used in single samples when the data are derived from large biobanks, such as the UK Biobank. 64 We report no sample overlap for the 2013 GLGC LDL‐C, PCSK9 pQTL, and GTEx eQTL analyses.
Statistical Analysis
We performed all analysis in R version 4.0.2 using the TwoSampleMR R package. 41 For instruments with 2+ SNPs (ie, PCSK9 variants in LDL‐C, PCSK9 pQTL, PCSK9 eQTL in whole blood, and the polygenic LDL‐C instrument), we used the inverse variance–weighted (IVW) analyses as the primary MR method. For analyses with instruments comprised of a single SNP (PCSK9 liver and cortex eQTLs), we used the Wald ratio method as the primary method. 65 For analyses with >2 SNPs (PCSK9 variants associated with LDL‐C levels and the polygenic LDL‐C analyses), MR Egger, weighted median, and weighted mode analyses are presented as sensitivity analyses to assess the robustness of the MR IVW results and evaluate the exclusion restriction MR assumption. It is assumed that MR IVW gives consistent estimates when all genetic variants are valid IVs. 41 , 66 Compared with the MR IVW method, MR Egger uses a relaxed assumption rather than the strict MR assumption of no pleiotropy (the Instrument Strength Independent of Direct Effect [InSIDE] assumption). 66 , 67 MR Egger extends MR IVW by not setting the linear regression intercept to zero, allowing the average horizontal pleiotropic MR estimate across all SNPs to be unbalanced or directional, ie, some variants may be acting on the neuropsychiatric outcomes via ≥1 pathways other than through the PCSK9 or LDL‐C exposures. 66 , 67 The weighted median method uses the median association of all available instrument SNPs. 45 Therefore, only half of the SNPs need to be valid instruments, ie, variants with no horizontal pleiotropy, no associations with confounders, and robust associations with the exposure, to return an unbiased MR estimate. 68 The weighted mode method weights the contribution of each instrument SNP to the clustering by the inverse variance of its association with each neuropsychiatric outcome. Assuming the most common MR instrument is consistent, the overall MR estimate will be unbiased, even if all other SNPs within the instrument are invalid. 45
While weighted median estimates generally are nearly as precise as MR IVW estimates, both are substantially more precise than MR Egger estimates, with MR Egger estimates particularly imprecise if all IVs have similar exposure effect sizes. 68 Complementary MR methods help evaluate the sensitivity of the results to different patterns of violations of IV assumptions, including horizontal pleiotropy, 45 and consistency of MR estimates across all methods suggests an unbiased MR estimate, 41 , 42 , 45 strengthening causal inference. 41 In addition, for the PCSK9 target instrumented in LDL‐C and polygenic LDL‐C instrument, we also used the MR Egger intercept test, 69 Cochran Q heterogeneity test, 70 MR Lasso test, 71 and MR Steiger test 56 assessing causal direction between hypothesized circulating LDL‐C and depression, anxiety, cognition, and dementia outcomes.
Because the primary physiological response of pharmacological PCSK9 inhibition is lowered LDL‐C levels, we validated our pQTL and eQTL instruments by investigating their impact on LDL‐C. To simulate the pharmacological impact of PCSK9 inhibitors, the reported drug‐target MR estimates were transformed to correspond to a decrease of LDL‐C in units of SD, circulating PCSK9 protein levels, and lower PCSK9 gene expression. For instruments derived from LDL‐C levels, this corresponds to a change in the likelihood of reporting a positive neuropsychiatric end point per a 1‐SD decrease in circulating LDL‐C levels. For the PCSK9 protein and gene expression analyses, this instead corresponds to a change in the likelihood per a 1‐SD change in normalized PCSK9 protein levels and TPM, respectively. We report 95% CIs estimates for the MR analyses. The strength of evidence was indexed against the null hypotheses (no association) by the exact P value before and after correction for multiple testing. To account for multiple testing bias, a Bonferroni correction was used, where the adjusted P value threshold was 4.17×10−3 (0.05/12 end points tested). For any nominally significant findings (P<0.05) in the sex‐specific end points, a post hoc hypothesis test was performed to evaluate whether the MR estimates between the male and female end point were significantly different (P value <0.05) from each other.
Results
Impact of LDL‐C Lowering by PCSK9 on Neuropsychiatric Risk
Drug‐target MR analyses proxying genetic PCSK9 inhibition in LDL‐C levels failed to find any IVW MR estimates among either men or women surpassing Bonferroni correction for multiple comparisons (Figure 2). Full results are presented in Table S4 and power calculations in Table S5. MR estimates (β) for fluid intelligence were 0.0246 (P=0.877) and − 0.0048 (P=0.644) for men and women, respectively. For women, genetically predisposed PCSK9 inhibition was associated with a nominally significant increase in the risk of self‐harm (OR, 1.003; P=0.031). Genetically predisposed PCSK9 inhibition was also associated with depression diagnosis (OR, 1.06; P=0.047). MR estimates for men for these end points were null (self‐harm OR, 0.99; P=0.313; depression OR, 1.023; P=0.343); however, the post hoc hypothesis test revealed no statistically significant difference between the male and female estimates (P=0.091) (Table S4).
Results using the PCSK9 instrument derived from 2013 GLGC LDL‐C data were generally aligned (Table S6); however, associations with self‐harm in women (OR, 1.025; P=0.155) were not statistically significant. Overall, these results were consistent across MR methods. MR Egger intercept analysis suggested no evidence of horizontal pleiotropy and the Cochran Q test did not indicate heterogeneity, which improves the causal inference of the MR estimates. 45
Further, analysis of circulating LDL‐C levels proxied by instruments comprised of SNPs throughout the genome did not yield evidence for any association with depression risk in women or men (Table S7). We did observe one end point surpassing correction for multiple comparisons: in the combined‐sex analysis, increased LDL‐C levels were associated with decreased risk for paternal AD risk (OR, 0.995; P=0.0013) but not maternal AD (P=0.029).
Assessing the Neuropsychiatric Impact of PCSK9 QTL Instruments
MR testing validity of the QTL PCSK9 instruments by evaluating their causal impact on circulating LDL‐C levels showed that increased genetically proxied PCSK9 inhibition was associated with lowered LDL‐C (Table S8). Our pQTL and tissue‐specific eQTL results aligned with the LDL‐C–based PCSK9 analyses failing to find evidence of a neuropsychiatric impact of genetically proxied PCSK9 inhibition and, as before, we observed nominally significant increased risk for self‐reported depression in women (Table S9), ie, an OR of 1.018 (P=0.038) for reduced circulating PCSK9 protein (Figure 2), an OR of 1.0094 (P=0.0064) for liver PCSK9 expression, and an OR of 1.024 (P=0.036) for whole blood PCSK9 expression (Figure 3). Genetically proxied circulating PCSK9 protein inhibition was associated with an increased risk for depression diagnosis in women (OR, 1.062; P=0.016). Post hoc hypothesis testing found a statistical difference between men and women in self‐reported depression for liver PCSK9 expression (P=0.035) but not for whole blood genetic PCSK9 expression (P=0.18) or circulating genetic PCSK9 protein (P=0.12).
Figure 3. Inverse variance–weighted and Wald ratio Mendelian randomization results of genetically proxied lowering of tissue‐specific PCSK9 (proprotein convertase subtilisin/kexin type 9) gene expression on neuropsychiatric outcomes for men and women.
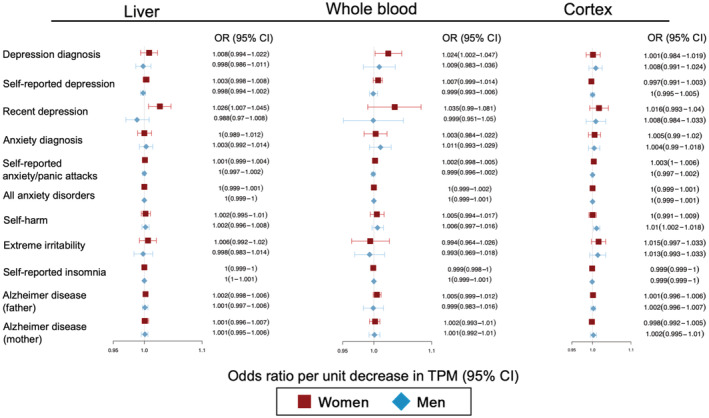
Estimates are reported odds ratios (ORs) and 95% CIs corresponding to a change in the risk for the neuropsychiatric end point for a change in genetically determined liver, whole blood, and brain PCSK9 gene expression (measured in transcripts per million [TPM]). Full results, including the combined‐sex results, are presented in Table S9.
Power Calculations
Power calculations showed that the eQTL and pQTL analyses were generally sufficiently powered (power exceeding 80%) to detect the presence of ORs of ≥1.2 at a type 1 error rate (α level) of 0.05 for the combined‐sex and sex‐specific analyses for all neuropsychiatric outcomes except for self‐reported anxiety/panic attacks (Table S9). Broadly, PCSK9 instruments using eQTL and pQTL data were better powered than the PCSK9 instruments derived from circulation LDL data because of the increased variance explained by the instruments (eg, PCSK9 liver eQTL variants explained 13.74% of the variance in PCSK9 liver expression compared with 0.242% of the variance of LDL‐C levels [Table S9]). As expected, the estimated power to detect ORs of ≤1.1 was reduced; however, the eQTL‐ and pQTL‐based analyses remained well powered for several end points, including irritability, depression, cognition, and AD (Table S5).
Discussion
The present study found that the neuropsychiatric impact of genetically proxied inhibition of PCSK9 was both generally neutral and similar between men and women, adding to the growing body of literature suggesting a safe neuropsychiatric profile for PCSK9 inhibitors and other lipid‐lowering therapies. 13 , 21 , 22 , 23 , 38 , 72 , 73 Our results provide an important sex‐specific comparison of potential neuropsychiatric adverse side effects related to genetically proxied PCSK9i, which has not been heretofore evaluated despite the call for more studies stratifying analyses by sex 30 and reported differences in prevalence of these disorders between men and women. 31 , 32 , 33 , 34 , 35 , 36 Overall, these results may help mitigate ongoing fears of adverse neuropsychiatric side effects related to PCSK9i that have contributed, in part, to their underutilization. 74
We did observe some associations between genetically proxied PCSK9 inhibition and depression, which adds to the growing body of MR literature suggesting a causal relationship between depression and cardiovascular disease. 75 , 76 , 77 Notably, our corresponding null estimates on depression risk from polygenic LDL‐C instruments using variants across the genome suggest that the relationship is not mediated by the primary physiological response to PCSK9 inhibition. Further, while some instruments suggested a nominal statistical increase in the risk of depression, other instruments suggested potential beneficial effects of PCSK9 inhibition (eg, the biologically conservative PCSK9 instrument). Further, causal inference requires triangulating study designs, 78 and an impact by PCSK9i on depression has not been observed in RCTs or observational data, 13 , 21 , 22 , 23 , 38 , 72 suggesting more work is needed to further elucidate the direction of these relationships.
The present study made use of the ability of drug target MR to estimate the causal relationships between a modifiable exposure (PCSK9 levels) and neuropsychiatric outcomes of interest by examining the association between exposure‐associated genetic variants, as IVs, and the outcome of interest. Analyses of participant data from short‐term RCTs have used data from patients on concurrent statin therapy, 13 , 22 potentially complicating evaluation of the neuropsychiatric impact specifically related to PCSK9i; however, the MR framework can estimate these relationships while minimizing the impact of confounding variables, 41 , 42 allowing use of population‐based observational data to strengthen causal inference regarding the long‐term neuropsychiatric effects of PCSK9i. We also leveraged new data sources measuring the genetic component of PCSK9 gene expression and protein levels in addition to liver‐specific PCSK9 gene expression, 50 which, in addition to providing multiple PCSK9 instruments to evaluate potential neuropsychiatric effects, may also yield important information regarding specific PCSK9 drug classes (ie, approved anti‐PCSK9 monoclonal antibodies that target circulating PCSK9 protein levels, 44 while inclisiran targets liver‐specific PCSK9 expression 15 ). Further, the use of PCSK9 instruments from gene expression, protein levels, and biomarkers reduces the risk of reverse causation and can provide important complementary evidence in drug‐target MR studies. 43
We note several limitations and important factors for interpretation. These results represent the analysis of only summary statistics taken from large, public GWAS data sets. As such, no clinical data or data from RCTs were included in this analysis. While MR is a powerful method for assessing genetic relationships, it is not a substitute for RCTs, which remain the gold standard for assessing causal biological relationships. Additionally, causal inference in MR depends on several assumptions that cannot be verified (eg, the exchangeability and exclusion restriction assumptions), 45 and while cis‐instrument MR is less prone to horizontal pleiotropy, 43 , 48 and sensitivity analyses yielded consistent MR estimates across all methods employed in the study, the possibility of bias caused by confounding or pleiotropy cannot be completely disregarded. Further, our cis‐instrument made use only of those SNPs in the PCSK9 gene associated with LDL‐C, and therefore did not assess for any effects of PCSK9 inhibition through alternative pathways. Additional research in real‐world settings, including long‐term studies of postmarketing data, are needed to address the remaining uncertainties regarding PCSK9 inhibition. Relatedly, drug‐target MR cannot itself proxy any potential off‐target effects of specific drug classes, such as adverse effects of the RNAi delivery system used for treatment with inclisiran. Moreover, MR estimates assess preexisting, permanent genetic variants with permanent biological effects beginning in early development. In contrast, PCSK9 inhibitors are generally prescribed to adults; the temporal difference in the effective developmental stage of the 2 populations represents a limitation of drug‐target MR methods for completely proxying pharmacological outcome profiles. 41 , 45 Therefore, we urge caution regarding the interpretation of these results for clinical decision‐making, pending replication and further investigation. 79 In addition, the current study outcomes were based on self‐reported data, including self‐reported diagnoses. Self‐reported outcomes in the behavioral and healthcare literature are subject to response bias, either due to underreporting or overreporting, 80 which might also impact the analyses.
There are additional limitations related to statistical power and sample overlap. While power calculations suggested the analyses were generally sufficiently powered to detect the evidence of the potential neuropsychiatric impact of genetically predisposed PCSK9 inhibition, some analyses using the PCSK9 variants instrumented in the LDL‐C data, despite strong F statistics, may be underpowered. Nevertheless, power calculations of analyses of these same end points using pQTL and eQTL instruments were well‐powered, which improves the causal inference of the null MR estimates. In addition, while there was no sample overlap between the 2013 LDL‐C, pQTL, and eQTL analyses, the 2021 GLGC data included UK Biobank participants, 49 which may bias these estimates. 63 Recent work has shown that 2‐sample MR may safely be used in single samples (ie, 100% sample overlap) provided the data are derived from large biobanks, such as the UK Biobank, 64 so any sample overlap bias is likely minimal. Finally, because our analyses were limited to participants of European ancestry, including outcomes drawn exclusively from the UK Biobank, which has been shown to be more educated and generally healthier than the general UK population, 81 caution is warranted before generalizing our findings to other populations; replication is needed in populations of other ancestries when such data become available.
Conclusions
Our drug‐target MR analysis of genetically proxied PCSK9 inhibition suggests a similar and neutral neuropsychiatric profile for PCSK9i therapy in men and women. While it is possible that the lack of statistically significant findings for certain end points may be attributable to insufficient power, our findings generally aligned across the neuropsychiatric end points using PCSK9 instruments derived from gene expression data, circulating PCSK9 protein levels, and PCSK9 locus‐lowering of LDL‐C. Future studies with larger, more diverse GWAS data, and additional long‐term and postmarketing research will continue to further our understanding of the possible neuropsychiatric impact of PCSK9 inhibition.
Sources of Funding
This research was facilitated by the Medical Research Council Integrative Epidemiology Unit (MRC‐IEU, University of Bristol, UK), especially the developers of the MRC‐IEU UK Biobank GWAS Pipeline. We gratefully acknowledge their contributing studies and the participants and investigators in those studies without whom this effort would not be possible. This work was supported by the National Institutes of Health intramural funding (ZIA‐AA000242 to F.W.L); Division of Intramural Clinical and Biological Research of the National Institute on Alcohol Abuse and Alcoholism.
Disclosures
None.
Supporting information
Data S1
Figure S1
Table S1–S9
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.026122
For Sources of Funding and Disclosures, see page 11.
Contributor Information
Andrew S. Bell, Email: andy.bell@nih.gov.
Falk W. Lohoff, Email: falk.lohoff@nih.gov.
References
- 1. Ahn JM, Lee KH, Yoo SY, Cho YR, Suh J, Shin ES, Lee JH, Shin DI, Kim SH, Baek SH, et al. Prognosis of variant angina manifesting as aborted sudden cardiac death. J Am Coll Cardiol. 2016;68:137–145. doi: 10.1016/j.jacc.2016.04.050 [DOI] [PubMed] [Google Scholar]
- 2. Eaton CB. Hyperlipidemia. Primary care: clinics in office practice. 2005;32:1027–1055. doi: 10.1016/j.pop.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 3. Goldstein JL, Hazzard WR, Schrott HG, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease I. Lipid levels in 500 survivors of myocardial infarction. J Clin Investig. 1973;52:1533–1543. doi: 10.1172/JCI107331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Menet R, Bernard M, ElAli A. Hyperlipidemia in stroke pathobiology and therapy. Insights Perspect. 2018;9. doi: 10.3389/fphys.2018.00488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer JS, Rogers RL, Mortel KF, Judd BW. Hyperlipidemia is a risk factor for decreased cerebral perfusion and stroke. Arch Neurol. 1987;44:418–422. doi: 10.1001/archneur.1987.00520160052014 [DOI] [PubMed] [Google Scholar]
- 6. Ross R, Harker L. Hyperlipidemia and atherosclerosis. Science. 1976;193:1094–1100. doi: 10.1126/science.822515 [DOI] [PubMed] [Google Scholar]
- 7. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, Ferranti Sd, Després JP, Fullerton HJ, et al. Heart disease and stroke statistics—2016 update. Circulation 2016;133:e38‐e360. doi: 10.1161/CIR.0000000000000350 [DOI] [PubMed] [Google Scholar]
- 8. Karr S. Epidemiology and management of hyperlipidemia. Am J Manag Care. 2017;23:S139–S148. [PubMed] [Google Scholar]
- 9. Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, Ali S, Banerjee P, Chan KC, Gipe DA, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711–720. doi: 10.1056/NEJMoa2004215 [DOI] [PubMed] [Google Scholar]
- 10. Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–1217. doi: 10.1038/nm1102-1211 [DOI] [PubMed] [Google Scholar]
- 11. Stewart J, McCallin T, Martinez J, Chacko S, Yusuf S. Hyperlipidemia. Pediatr Rev. 2020;41:393–402. doi: 10.1542/pir.2019-0053 [DOI] [PubMed] [Google Scholar]
- 12. Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, Blumenthal R, Danesh J, Smith GD, DeMets D, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388:2532–2561. doi: 10.1016/s0140-6736(16)31357-5 [DOI] [PubMed] [Google Scholar]
- 13. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664 [DOI] [PubMed] [Google Scholar]
- 14. Holmes MV, Smith GD. Dyslipidaemia: revealing the effect of CETP inhibition in cardiovascular disease. Nat Rev Cardiol. 2017;14:635–636. doi: 10.1038/nrcardio.2017.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, Bisch JA, Richardson T, Jaros M, Wijngaard PLJ, et al. Two phase 3 trials of Inclisiran in patients with elevated LDL cholesterol. N Engl J Med. 2020;382:1507–1519. doi: 10.1056/NEJMoa1912387 [DOI] [PubMed] [Google Scholar]
- 16. Administration UFD. FDA Approves Add‐on Therapy to Lower Cholesterol among Certain High‐Risk Adults. https://www.fda.gov/drugs/news‐events‐human‐drugs/fda‐approves‐add‐therapy‐lower‐cholesterol‐among‐certain‐high‐risk‐adults. 2021. Accessed March 27. [Google Scholar]
- 17. Leiter LA, Teoh H, Kallend D, Wright RS, Landmesser U, Wijngaard PLJ, Kastelein JJ, Ray KK. Inclisiran lowers LDL‐C and PCSK9 irrespective of diabetes status: the ORION‐1 randomized clinical trial. Diabetes Care. 2019;42:173–176. doi: 10.2337/dc18-1491 [DOI] [PubMed] [Google Scholar]
- 18. Raal FJ, Kallend D, Ray KK, Turner T, Koenig W, Wright RS, Wijngaard PLJ, Curcio D, Jaros MJ, Leiter LA, et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N Engl J Med. 2020;382:1520–1530. doi: 10.1056/NEJMoa1913805 [DOI] [PubMed] [Google Scholar]
- 19. Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, Wang K, Iyer S, Dutta C, Clendaniel V, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y [DOI] [PubMed] [Google Scholar]
- 20. Rothgangl T, Dennis MK, Lin PJC, Oka R, Witzigmann D, Villiger L, Qi W, Hruzova M, Kissling L, Lenggenhager D, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guedeney P, Giustino G, Sorrentino S, Claessen BE, Camaj A, Kalkman DN, Vogel B, Sartori S, De Rosa S, Baber U, et al. Efficacy and safety of alirocumab and evolocumab: a systematic review and meta‐analysis of randomized controlled trials. Eur Heart J. 2019;43:e17–e25. doi: 10.1093/eurheartj/ehz430 [DOI] [PubMed] [Google Scholar]
- 22. Giugliano RP, Mach F, Zavitz K, Kurtz C, Schneider J, Wang H, Keech A, Pedersen TR, Sabatine MS, Sever PS, et al. Design and rationale of the EBBINGHAUS trial: a phase 3, double‐blind, placebo‐controlled, multicenter study to assess the effect of evolocumab on cognitive function in patients with clinically evident cardiovascular disease and receiving statin background lipid‐lowering therapy‐a cognitive study of patients enrolled in the FOURIER trial. Clin Cardiol. 2017;40:59–65. doi: 10.1002/clc.22678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–2107. doi: 10.1056/NEJMoa1801174 [DOI] [PubMed] [Google Scholar]
- 24. Evans MA, Golomb BA. Statin‐associated adverse cognitive effects: survey results from 171 patients. J Hum Pharmacol Drug Ther. 2009;29:800–811. doi: 10.1592/phco.29.7.800 [DOI] [PubMed] [Google Scholar]
- 25. Parker BA, Polk DM, Rabdiya V, Meda SA, Anderson K, Hawkins KA, Pearlson GD, Thompson PD. Changes in memory function and neuronal activation associated with atorvastatin therapy. Pharmacol Drug Ther. 2010;30:625. doi: 10.1592/phco.30.6.625 [DOI] [Google Scholar]
- 26. O'Connell EM, Lohoff FW. Proprotein convertase subtilisin/Kexin type 9 (PCSK9) in the brain and relevance for neuropsychiatric disorders. Front Neurosci. 2020;14:609. doi: 10.3389/fnins.2020.00609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lohoff FW, Sorcher JL, Rosen AD, Mauro KL, Fanelli RR, Momenan R, Hodgkinson CA, Vendruscolo LF, Koob GF, Schwandt M, et al. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol Psychiatry. 2018;23:1900–1910. doi: 10.1038/mp.2017.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bell AS, O'Connell EM, Lohoff FW. Chapter 28 ‐ cholesterol and alcohol. In: Bukiya AN, Dopico AM, eds Cholesterol. Academic Press; 2022:747–767. doi: 10.1016/B978-0-323-85857-1.00036-5 [DOI] [Google Scholar]
- 29. Ziajka P. Gender differences in response to PCSK9 inhibitor therapy. J Clin Lipidol. 2019;13:e42. doi: 10.1016/j.jacl.2019.04.071 [DOI] [Google Scholar]
- 30. Steinberg JR, Turner BE, Weeks BT, Magnani CJ, Wong BO, Rodriguez F, Yee LM, Cullen MR. Analysis of female enrollment and participant sex by burden of disease in US clinical trials between 2000 and 2020. JAMA Network Open. 2021;4:e2113749. doi: 10.1001/jamanetworkopen.2021.13749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riecher‐Rössler A. Sex and gender differences in mental disorders. Lancet Psychiatry. 2017;4:8–9. doi: 10.1016/S2215-0366(16)30348-0 [DOI] [PubMed] [Google Scholar]
- 32. Beam CR, Kaneshiro C, Jang JY, Reynolds CA, Pedersen NL, Gatz M. Differences between women and men in incidence rates of dementia and Alzheimer's disease. J Alzheimers Dis. 2018;64:1077–1083. doi: 10.3233/JAD-180141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bogren M, Brådvik L, Holmstrand C, Nöbbelin L, Mattisson C. Gender differences in subtypes of depression by first incidence and age of onset: a follow‐up of the Lundby population. Eur Arch Psychiatry Clin Neurosci. 2018;268:179–189. doi: 10.1007/s00406-017-0778-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eid RS, Gobinath AR, Galea LA. Sex differences in depression: insights from clinical and preclinical studies. Prog Neurobiol. 2019;176:86–102. doi: 10.1016/j.pneurobio.2019.01.006 [DOI] [PubMed] [Google Scholar]
- 35. Marcus SM, Kerber KB, Rush AJ, Wisniewski SR, Nierenberg A, Balasubramani GK, Ritz L, Kornstein S, Young EA, Trivedi MH. Sex differences in depression symptoms in treatment‐seeking adults: confirmatory analyses from the sequenced treatment alternatives to relieve depression study. Comprehensive Psychiatry. 2008;49:238–246. doi: 10.1016/j.comppsych.2007.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Helton SG, Lohoff FW. Serotonin pathway polymorphisms and the treatment of major depressive disorder and anxiety disorders. Pharmacogenomics. 2015;16:541–553. doi: 10.2217/pgs.15.15 [DOI] [PubMed] [Google Scholar]
- 37. Robinson JG, Rosenson RS, Farnier M, Chaudhari U, Sasiela WJ, Merlet L, Miller K, Kastelein JJ. Safety of very low low‐density lipoprotein cholesterol levels with alirocumab: pooled data from randomized trials. J Am Coll Cardiol. 2017;69:471–482. doi: 10.1016/j.jacc.2016.11.037 [DOI] [PubMed] [Google Scholar]
- 38. Lyall DM, Ward J, Banach M, Smith GD, Gill JG, Pell JP, Holmes MV, Sattar N. PCSK9 genetic variants and cognitive abilities: a large‐scale Mendelian randomization study. Arch Med Sci. 2021;17:241–244. doi: 10.5114/aoms/127226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Williams DM, Finan C, Schmidt AF, Burgess S, Hingorani AD. Lipid lowering and Alzheimer disease risk: a Mendelian randomization study. Ann Neurol. 2020;87:30–39. doi: 10.1002/ana.25642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alghamdi J, Matou‐Nasri S, Alghamdi F, Alghamdi S, Alfadhel M, Padmanabhan S. Risk of neuropsychiatric adverse effects of lipid‐lowering drugs: a Mendelian randomization study. Int J Neuropsychopharmacol. 2018;21:1067–1075. doi: 10.1093/ijnp/pyy060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR‐base platform supports systematic causal inference across the human phenome. Elife. 2018;7. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–R98. doi: 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt AF, Finan C, Gordillo‐Marañón M, Asselbergs FW, Freitag DF, Patel RS, Tyl B, Chopade S, Faraway R, Zwierzyna M, et al. Genetic drug target validation using Mendelian randomisation. Nat Commun. 2020;11:3255. doi: 10.1038/s41467-020-16969-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol. 2019;16:155–165. doi: 10.1038/s41569-018-0107-8 [DOI] [PubMed] [Google Scholar]
- 45. Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, Palmer T, Schooling CM, Wallace C, Zhao Q, et al. Mendelian randomization. Nat Rev Methods Primers. 2022;2:6–21. doi: 10.1038/s43586-021-00092-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res. 2007;16:309–330. doi: 10.1177/0962280206077743 [DOI] [PubMed] [Google Scholar]
- 47. Yarmolinsky J, Bull CJ, Vincent EE, Robinson J, Walther A, Smith GD, Lewis SJ, Relton CL, Martin RM. Association between genetically proxied inhibition of HMG‐CoA reductase and epithelial ovarian cancer. JAMA. 2020;323:646–655. doi: 10.1001/jama.2020.0150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Holmes MV, Richardson TG, Ference BA, Davies NM, Davey SG. Integrating genomics with biomarkers and therapeutic targets to invigorate cardiovascular drug development. Nat Rev Cardiol. 2021;18:435–453. doi: 10.1038/s41569-020-00493-1 [DOI] [PubMed] [Google Scholar]
- 49. Graham SE, Clarke SL, Wu KH, Kanoni S, Zajac GJM, Ramdas S, Surakka I, Ntalla I, Vedantam S, Winkler TW, et al. The power of genetic diversity in genome‐wide association studies of lipids. Nature. 2021;600:675–679. doi: 10.1038/s41586-021-04064-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pott J, Gådin JR, Theusch E, Kleber ME, Delgado GE, Kirsten H, Hauck SM, Burkhardt R, Scharnagl H, Krauss RM, et al. Meta‐GWAS of PCSK9 levels detects two novel loci at APOB and TM6SF2. Hum Mol Genet. 2022;31:999–1011. doi: 10.1093/hmg/ddab279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Consortium GT. The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Klein N, Tsai EA, Vochteloo M, Baird D, Huang Y, Chen CY, van Dam S, Deelen P, Bakker OB, Garwany OE, et al. Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain‐related diseases. bioRxiv 2021:2021.2003.2001.433439. doi: 10.1101/2021.03.01.433439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Consortium G. Human genomics. The genotype‐tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burgess S, Thompson SG, Collaboration CC. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755–764. doi: 10.1093/ije/dyr036 [DOI] [PubMed] [Google Scholar]
- 56. Hemani G, Tilling K, Davey SG. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13:e1007081. doi: 10.1371/journal.pgen.1007081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wagner CJ, Musenbichler C, Böhm L, Färber K, Fischer AI, von Nippold F, Winkelmann M, Richter‐Schmidinger T, Mühle C, Kornhuber J, et al. LDL cholesterol relates to depression, its severity, and the prospective course. Prog Neuropsychopharmacol Biol Psychiatry. 2019;92:405–411. doi: 10.1016/j.pnpbp.2019.01.010 [DOI] [PubMed] [Google Scholar]
- 58. Hu X, Wang T, Luo J, Liang S, Li W, Wu X, Jin F, Wang L. Age‐dependent effect of high cholesterol diets on anxiety‐like behavior in elevated plus maze test in rats. Behav Brain Funct. 2014;10:30. doi: 10.1186/1744-9081-10-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mefford MT, Chen L, Lewis CE, Muntner P, Sidney S, Launer LJ, Monda KL, Ruzza A, Kassahun H, Rosenson RS, et al. Long‐term levels of LDL‐C and cognitive function: the CARDIA study. J Int Neuropsychol Soc. 2021;27:1048–1057. doi: 10.1017/s1355617721000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Neale‐Lab . UK Biobank GWAS. Available at: http://www.nealelab.is/uk‐biobank/. 2018. Accessed June.
- 61. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42:1497–1501. doi: 10.1093/ije/dyt179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181:251–260. doi: 10.1093/aje/kwu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two‐sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608. doi: 10.1002/gepi.21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Minelli C, Fabiola Del Greco M, van der Plaat DA, Bowden J, Sheehan NA, Thompson J. The use of two‐sample methods for Mendelian randomization analyses on single large datasets. bioRxiv 2020:2020.2005.2007.082206. doi: 10.1101/2020.05.07.082206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hemani G, Bowden J, Davey SG. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. 2018;27:R195–R208. doi: 10.1093/hmg/ddy163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bowden J, Smith GD, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Davey Smith G, Bowden J, Del Greco MF, Minelli C, Thompson JR, Sheehan NA. Assessing the suitability of summary data for two‐sample Mendelian randomization analyses using MR‐egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45:1961–1974. doi: 10.1093/ije/dyw220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bowden J, Del Greco MF, Minelli C, Smith GD, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two‐sample summary data Mendelian randomization. Stat Med. 2017;36:1783–1802. doi: 10.1002/sim.7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bowden J, Del Greco MF, Minelli C, Zhao Q, Lawlor DA, Sheehan NA, Thompson J, Davey SG. Improving the accuracy of two‐sample summary‐data Mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48:728–742. doi: 10.1093/ije/dyy258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. doi: 10.1093/ije/dyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhou Z, Ryan J, Ernst Michael E, Zoungas S, Tonkin Andrew M, Woods Robyn L, McNeil John J, Reid Christopher M, Curtis Andrea J, Wolfe R, et al. Effect of statin therapy on cognitive decline and incident dementia in older adults. J Am Coll Cardiol. 2021;77:3145–3156. doi: 10.1016/j.jacc.2021.04.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rosoff Daniel B, Bell Andrew S, Jung J, Wagner J, Mavromatis Lucas A, Lohoff FW. Mendelian randomization study of PCSK9 and HMG‐CoA reductase inhibition and cognitive function. J Am Coll Cardiol. 2022;80:653–662. doi: 10.1016/j.jacc.2022.05.041 [DOI] [PubMed] [Google Scholar]
- 74. Ahmed HM, Nissen SE. Nonstatin therapy for dyslipidemia. Circ Res. 2018;123:1036–1038. doi: 10.1161/CIRCRESAHA.118.313829 [DOI] [PubMed] [Google Scholar]
- 75. Khandaker GM, Zuber V, Rees JM, Carvalho L, Mason AM, Foley CN, Gkatzionis A, Jones PB, Burgess S. Shared mechanisms between coronary heart disease and depression: findings from a large UK general population‐based cohort. Mol Psychiatry. 2020;25:1477–1486. doi: 10.1038/s41380-019-0395-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tang B, Yuan S, Xiong Y, He Q, Larsson SC. Major depressive disorder and cardiometabolic diseases: a bidirectional Mendelian randomisation study. Diabetologia. 2020;63:1305–1311. doi: 10.1007/s00125-020-05131-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu Y, Wang Z, Georgakis MK, Lin H, Zheng L. Genetic liability to depression and risk of coronary artery disease, myocardial infarction, and other cardiovascular outcomes. J Am Heart Assoc. 2021;10:e017986. doi: 10.1161/JAHA.120.017986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hammerton G, Munafò MR. Causal inference with observational data: the need for triangulation of evidence. Psychol Med. 2021;51:563–578. doi: 10.1017/s0033291720005127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Narasimhan S, Lohoff FW. Pharmacogenetics of antidepressant drugs: current clinical practice and future directions. Pharmacogenomics. 2012;13:441–464. doi: 10.2217/pgs.12.1 [DOI] [PubMed] [Google Scholar]
- 80. Rosenman R, Tennekoon V, Hill LG. Measuring bias in self‐reported data. Int J Behav Healthc Res. 2011;2:320–332. doi: 10.1504/IJBHR.2011.043414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, Collins R, Allen NE. Comparison of sociodemographic and health‐related characteristics of UK biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–1034. doi: 10.1093/aje/kwx246 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figure S1
Table S1–S9