

Abstract
In 2019, the European Society of Cardiology/European Atherosclerosis Society stated that apolipoprotein B (apoB) was a more accurate marker of cardiovascular risk than low‐density lipoprotein cholesterol (LDL‐C) and non–high‐density lipoprotein cholesterol. Since then, the evidence has continued to mount in favor of apoB. This review explicates the physiological mechanisms responsible for the superiority of apoB as a marker of the cardiovascular risk attributable to the atherogenic apoB lipoprotein particles chylomicron remnants, very low‐density lipoprotein, and low‐density lipoprotein particles. First, the nature and relative numbers of these different apoB particles will be outlined. This will make clear why low‐density lipoprotein particles are almost always the major determinants of cardiovascular risk and why the concentrations of triglycerides and LDL‐C may obscure this relation. Next, the mechanisms that govern the number of very low‐density lipoprotein and low‐density lipoprotein particles will be outlined because, except for dysbetalipoproteinemia, the total number of apoB particles determines cardiovascular risk, Then, the mechanisms that govern the cholesterol mass within very low‐density lipoprotein and low‐density lipoprotein particles will be reviewed because these are responsible for the discordance between the mass of cholesterol within apoB particles, measured either as LDL‐C or non–high‐density lipoprotein cholesterol, and the number of apoB particles measured as apoB, which creates the superior predictive power of apoB over LDL‐C and non–high‐density lipoprotein cholesterol. Finally, the major apoB dyslipoproteinemias will be briefly outlined. Our objective is to provide a physiological framework for health care givers to understand why apoB is a more accurate marker of cardiovascular risk than LDL‐C or non–high‐density lipoprotein cholesterol.
Keywords: apoB, apolipoprotein B, cardiovascular disease prevention, cardiovascular risk
Subject Categories: Basic Science Research, Lipids and Cholesterol, Cardiovascular Disease, Epidemiology
Nonstandard Abbreviations and Acronyms
- ANGPTL3
angiopoietin like protein 3
- CE
cholesteryl ester
- CETP
cholesteryl ester transfer protein
- FH
familial hypercholesterolemia
- Lp(a)
lipoprotein(a)
- PCSK9
proprotein convertase subtilisin kexin 9
Deposition of cholesterol within the arterial wall caused by trapping of apolipoprotein B (apoB)‐containing lipoprotein particles initiates and drives the evolution of atherosclerosis, from the beginning to the end, from the appearance of the first fatty streaks, to the complex, calcified atherosclerotic plaques that cause the clinical events. 1 , 2 However, cholesterol only circulates in plasma within lipoprotein particles, and deposition of cholesterol within the arterial wall only occurs with trapping of apoB particles within the arterial wall. Based on evidence from prospective epidemiological studies, randomized clinical trials, and Mendelian randomization analyses, the 2019 European Society of Cardiology/European Atherosclerosis Society Guidelines concluded that apoB, which measures the number of apoB particles in plasma, is a more accurate marker of the cardiovascular risk attributable to the apoB lipoproteins than low‐density lipoprotein cholesterol (LDL‐C) or non–high‐density lipoprotein cholesterol (non–HDL‐C) and a more accurate index of the adequacy of lipid lowering therapy than LDL‐C or non–HDL‐C. 3 The evidence has grown even stronger since. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 Moreover, apoB can be measured more accurately by clinical laboratories than LDL‐C or non–HDL‐C, but just as rapidly, and almost as inexpensively, using standardized automated technologies that are available in any modern clinical chemistry laboratory. 12 , 13
Because cholesterol is a major component of all apoB particles, apoB, LDL‐C, and non–HDL‐C are highly correlated. As values of 1 increase, so do values of the 2 others, and so does cardiovascular risk. Conversely, as values of 1 decrease, so do values of the 2 others, and so does cardiovascular risk. However, LDL‐C and non–HDL‐C are not always accurate equivalents of apoB, because the mass of cholesterol per apoB particle varies. 14 , 15 Therefore, at any level of LDL‐C or non–HDL‐C, there is considerable variance in the value of apoB. Similarly, at any level of apoB, there is considerable variance in the levels of LDL‐C and non–HDL‐C. 16
The risk of cardiovascular disease is driven by the number of apoB particles, which are trapped within the arterial wall, and cardiovascular risk relates more directly to the number of apoB particles in plasma than to the mass of cholesterol within them. 1 , 2 Therefore, the variance or discordance between the levels of apoB on the one hand, and the levels of LDL‐C and non–HDL‐C on the other, underlie why apoB is a more accurate marker of cardiovascular risk in individuals than either LDL‐C or non–HDL‐C.
LDL‐C has traditionally been the primary marker and target in clinical care of the risk of cardiovascular disease caused by apoB lipoprotein particles, a decision that was based on masses of published evidence, which have resulted in wide familiarity and acceptance of its value. However, this is changing as health care providers become more aware of the evidence supporting apoB as a more accurate marker of cardiovascular risk. That a new marker may work better than the old marker does not negate the benefits that the old marker brought. apoB does not contradict LDL‐C and non–HDL‐C. apoB extends LDL‐C and non–HDL‐C. However, there is no recent summary to outline the pathophysiological bases for the superiority of apoB over LDL‐C and non–HDL‐C. Accordingly, this essay will review the mechanisms that regulate the number of apoB particles in plasma and their lipid composition.
Based on these concepts, care givers will understand why, except for dysbetalipoproteinemia, apoB includes all the information about cardiovascular risk from LDL‐C, non–HDL‐C, and triglycerides, and therefore, how apoB could improve diagnosis and simplify therapy. We will explain why LDL particles are more important drivers of cardiovascular risk than very low‐density lipoprotein (VLDL) particles, even in those with hypertriglyceridemia, and why not all individuals with hypertriglyceridemia have an elevated apoB, and therefore, why not all those with high triglycerides have the same cardiovascular risk. We will also demonstrate how the number of LDL apoB particles, and therefore cardiovascular risk, can be elevated when LDL‐C and/or non–HDL‐C are normal. Finally, we will illustrate the most significant proatherogenic apoB lipoprotein phenotypes to help reframe the conventional disorders of lipid metabolism as disorders of apoB lipoprotein particle metabolism.
apoB Particles and apoB Particle Number
The different classes of the apoB lipoproteins are illustrated schematically in Figure 1. apo B48 is 2143 amino acids in length and 264 kDa in size, and is a truncated, lighter version of apo B100, which is 4136 amino acids in length and 550 kDa in size. 17 , 18 , 19 , 20 apo B48 encircles chylomicron particles secreted by the intestine, providing an exoskeleton for these particles; apo B48 does not contain the low‐density lipoprotein (LDL) receptor binding region of apoB, which is excised by a distinctive intestine‐specific RNA editing mechanism. 20
Figure 1. apoB lipoprotein particles.
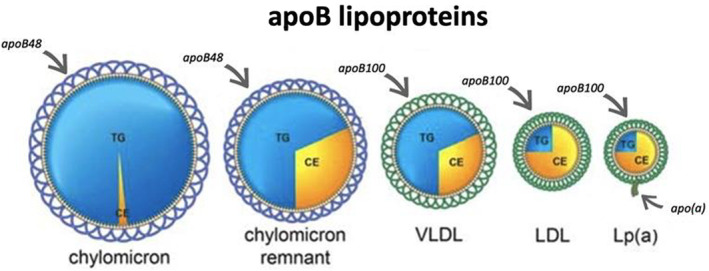
One molecule of apo B48 encircles each chylomicron and chylomicron remnant particle. The chylomicron remnant particle contains less TG but the same amount of cholesterol as the intact chylomicron particle. The difference in TG mass represents the mass of TG delivered to adipose tissue and skeletal muscle. One molecule of apo B100 encircles VLDL, LDL, and Lp(a) particles. Lp(a) particles are an LDL particle to which a molecule of apo(a) has been attached. One molecule of apo B48 encircles a chylomicron or chylomicron remnant particle. apoB indicates apolipoprotein B; CE, cholesteryl ester; LDL, low‐density lipoprotein; Lp(a), lipoprotein(a); TG, triglycerides; and VLDL, very low‐density lipoprotein.
Intact chylomicrons transport dietary triglycerides to adipose tissue and skeletal muscle. Chylomicron remnant particles are released back into plasma from these tissues after removal of most of their triglycerides, but with the original mass of cholesterol still intact. These chylomicron remnant particles deliver the dietary cholesterol and the triglycerides that remain in their core to the liver and, in normal individuals, these are rapidly removed from plasma. 17 , 21
Each VLDL particle secreted by the liver (Figure 1) is encircled by 1 molecule of apo B100, which provides structural integrity. 22 , 23 A molecule of apo B100 also contains a specific binding site for the LDL receptor, and once bound to an LDL receptor, that apo B100 particle, along with its cholesterol cargo, is rapidly removed from plasma. In contrast to the multiple other apolipoproteins, which can be present on its surface, particularly on VLDL and chylomicron particles, apo B100 and apo B48 are essential structural components of apoB particles and are present throughout their lifetime in plasma. Because there is 1 molecule of apoB per particle, 22 , 23 total plasma apoB is an exact measure of the total number of apoB particles in plasma.
Other apolipoproteins, such as apoE, apo CII, and apo CIII, play important roles in regulating the metabolism of the chylomicron and VLDL particles. However, we will not deal with these here, because these other markers have not yet been shown to add clinically relevant information beyond that provided by apoB. That is not the case for lipoprotein(a) (Lp[a]) particles. Lp(a) is an LDL particle to which a molecule of apo(a) has been disulfide bonded to apo B100. 24 Lp(a) appears to increase the risk of arteriosclerotic cardiovascular disease independently of LDL‐C or total apoB. 21 Therefore, to assess the atherogenic risk attributable to the apoB lipoproteins, Lp(a) must be measured as well as apoB. Lp(a) also plays a causal role in the genesis of calcific aortic stenosis. 25 Lp(a) particles generally account for only a small minority of total apoB particles in plasma. Occasionally, however, they may contribute significantly to total apoB and LDL‐C. 26 Newer assays that report Lp(a) in nanomoles per liter rather than mass should solve this problem, because the proportion of total apoB attributable to Lp(a) will be calculable. 27 The levels of Lp(a) are determined genetically, and there is little variance over our lifetimes. Medications to lower Lp(a) with the goal to reduce cardiovascular risk and the likelihood of aortic stenosis are being developed, but the results of randomized controlled trials of cardiovascular outcomes are not yet available. 28 Therefore, we will not deal further with Lp(a).
apoB Particle Number
The relative numbers of the different apoB particles in the plasma of a normal person are illustrated in Figure 2. Chylomicron particles are the largest, 75 to 1200 nm in diameter, and the fewest; they are normally almost all removed from plasma within 3 to 4 hours of eating. 29 Chylomicron particles are too large to enter the arterial wall and therefore are not atherogenic. However, they are not entirely innocuous. Pancreatitis can occur secondary to a marked increase in chylomicron particle numbers and residence time. However, pathogenic chylomicronemia is relatively rare and, although important, will not be dealt with here. All the other apoB particles, including chylomicron remnant particles, can enter the arterial wall and therefore are atherogenic. These are the particles on which we will focus.
Figure 2. Relative numbers of apoB particles.
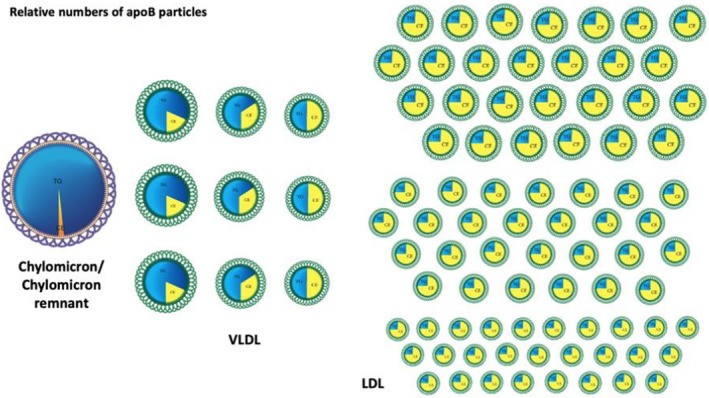
LDL particles (on the right) are by far the most numerous, whereas chylomicron particles (the large particle on the left) are by far the least numerous. There are, on average, 9 times the number of VLDL particles as the sum of chylomicron and chylomicron particles in the postprandial period but 9 times the number of LDL particles as VLDL particles. Note VLDL and LDL particles can differ in size based on the relative amounts of triglycerides and cholesterol, respectively. LDL particles differ in the mass of cholesterol they contain, and therefore their size, resulting in the subclasses of LDL particles: LDL1, LDL2, and LDL3. In this figure, 1 chylomicron and 1 chylomicron remnant particle are combined, so that the proper relative number of apoB particles in the postprandial period can be illustrated. apoB indicates apolipoprotein B; LDL, low‐density lipoprotein; and VLDL, very low‐density lipoprotein.
At peak postprandial periods, there are 9 to 10 times as many VLDL apo B100 particles as apo B48 particles, 30 , 31 , 32 and there are 9 times as many LDL apoB particles as VLDL apoB particles 30 , 31 (Figure 2). All clinically available immunoassays to measure apoB recognize apo B48 as well as apo B100. Yet, because there are so few apo B48 particles relative to the number of apo B100 particles, 29 , 30 , 33 , 34 there is no significant difference between fasting and postprandial apoB level. Consequently, fasting samples are not required to measure apoB.
In normotriglyceridemic subjects, 5% to 10% of total apoB particles are VLDL particles; the rest are LDL particles. 30 , 31 As plasma triglycerides increase, this proportion may increase to 15% to 20%. 35 Nevertheless, even in severe hypertriglyceridemia, except for dysbetalipoproteinemia (formerly called type III hyperlipoproteinemia, a rare dyslipidemia characterized by abnormal accumulation of remnant particles [see below]), LDL particles make up the great majority of apoB particles in plasma. 32 , 36 The great excess of LDL particles over VLDL particles explains why LDL, and by extension LDL‐C, is such a strong risk factor for atherosclerotic cardiovascular disease (ASCVD), whereas triglycerides are not, and why, even in patients who are hypertriglyceridemic, LDL particles are the major drivers of risk.
VLDL particles differ in size; the larger ones contain considerably more triglycerides than the smaller ones. Accordingly, plasma triglycerides are not a measure of the number of VLDL particles. The mass of cholesterol per LDL particle also varies, although less so than the variance of triglycerides within VLDL particles. Subclasses of LDL particles have been defined; LDL1 particles have the greatest mass of cholesterol per particle, LDL2 particles have an average mass of cholesterol per particle, and LDL3 particles have the least mass of cholesterol per particle. 15 The overall ratio of cholesterol per LDL particle will depend on which of these species predominates. This variance in composition explains why LDL‐C is an inexact, and therefore clinically unreliable, measure of the number of LDL particles.
Pathophysiological Determinants of the Number of apoB Particles in Plasma
Because apo B48 contributes so little to total apoB, plasma apoB is effectively determined by the total number of VLDL, and especially LDL, particles. The number of VLDL and LDL particles can increase either because their production increases, their clearance decreases, or by a combination of increased production and decreased clearance (Figure 3). Thus, a high number of VLDL particles could be because of (1) increased production of VLDL particles by the liver, (2) impaired clearance of VLDL particles by the liver, (3) impaired conversion of VLDL to LDL particles, or (4) any combination of these factors. Similarly, an elevated number of LDL particles could be because of (1) impaired clearance of LDL particles by the LDL receptor pathway, (2) to increased production of LDL particles from VLDL particles, or (3) to a combination of decreased clearance and increased production. Accordingly, the pathophysiology of every apoB dyslipoproteinemia can be characterized by whether the primary defect is in clearance or production of apoB particles or whether there are significant defects in both.
Figure 3. VLDL‐LDL particle metabolism.
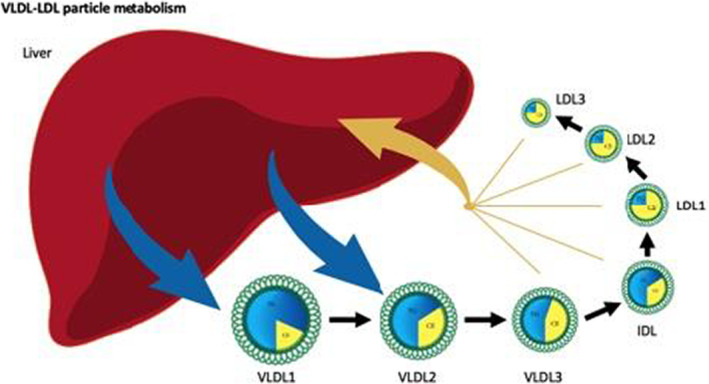
This figure illustrates schematically the regulation of VLDL and LDL particle number in plasma. VLDL particles are secreted by the liver into the plasma compartment, from which they are either removed directly by the liver after much of their TG have been removed or converted to LDL particles. At steady state, the rate at which VLDL particles are secreted from the liver is equal to the rate at which they are removed from plasma. Once steady state is achieved, either an increase in the rate of production or a decrease in the rate of removal will produce an increased VLDL particle number. Similarly, the rate at which LDL particles are produced at steady state is equal to the rate at which they are removed from plasma. LDL particles are produced by conversion of VLDL to LDL particles and are removed either by a specific clearance pathway or by multiple nonspecific pathways. Almost all LDL particles are cleared from plasma by the liver. Only a small minority are removed by peripheral cells. IDL indicates intermediate‐density lipoprotein; LDL indicates low‐density lipoprotein; TG, triglycerides; and VLDL, very low‐density lipoprotein.
Determinants of VLDL Particle Number
VLDL apoB particles are secreted by the liver, and after much of their triglycerides are removed and deposited in adipose tissue or skeletal or cardiac muscle, they are either cleared from plasma by the liver or converted to LDL apoB particles (Figure 4). At steady state, the rate at which VLDL particles are secreted by the liver into plasma equals the rate at which VLDL particles are removed from circulation by the liver plus the rate at which VLDL particles are converted to LDL particles (Figure 3 and 4). Our knowledge of the determinants of VLDL production and secretion by the liver is incomplete, although genetic studies are providing new leads. It has been established that the rate at which apoB molecules are synthesized by the liver varies little, whereas the rate at which newly synthesized apoB molecules are incorporated into nascent VLDL particles varies substantially. 37 , 38 Most evidence indicates that regulation of secretion of apoB particles occurs at this step or in the final assembly of VLDL particles within the endoplasmic reticulum. 39 Increased rates of triglycerides and cholesterol ester (CE) synthesis correlate with increased rates of secretion of VLDL particles, but whether these are associated or controlling events has not been determined with any certainty. 40 , 41 , 42 VLDL secretion rates are increased in patients with hypertriglyceridemia, 43 abdominal obesity, insulin resistance, and diabetes, 44 , 45 and in patients with familial combined hyperlipidemia, 46 , 47 , 48 the most common atherogenic dyslipoproteinemia associated with premature coronary artery disease. 49 , 50 , 51 The liver secretes at least 2 different VLDL particles at rates that can differ substantially. One is larger, with more triglycerides than the other, which explains why the relationship between the secretion rate of triglycerides within VLDL particles and the secretion rate of VLDL particles can differ so significantly. 52
Figure 4. Regulation of VLDL and LDL apoB particle number.
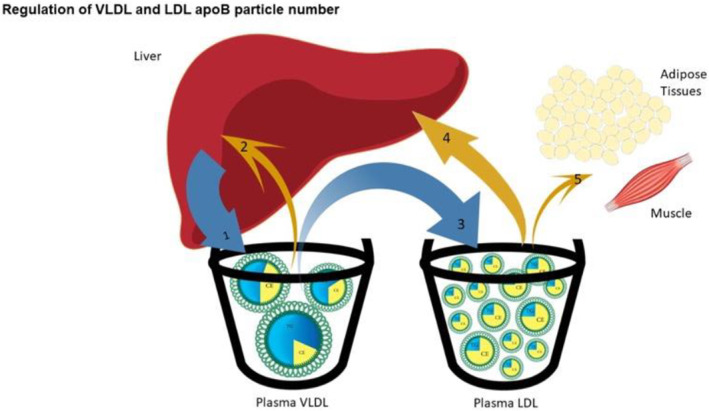
This figure illustrates the metabolic relations among the various VLDL and LDL particles. The liver secretes VLDL1 and VLDL2 particles. VLDL1 particles are the most TG rich and can be converted to VLDL2 particles by hydrolysis of core TG in peripheral tissues. Similarly, VLDL2 particles can be converted to VLDL3 particles, which can be converted to IDL particles or removed by the liver. IDL particles can be converted to LDL1 particles, which, following core lipid exchanges as illustrated in Figure 3, can be converted successively to LDL1, LDL2, and LDL3 particles. VLDL1 are the most TG‐rich particles secreted by the liver; VLDL2 contain an intermediate mass of TG and are also secreted by the liver. VLDL1 and VLDL2 particles can be sequentially converted to VLDL3 particles, which can be converted to IDL particles, which can be converted successively to LDL1, LDL2, and LDL3 particles. apoB indicates apolipoprotein B; CE, cholesteryl ester; IDL, intermediate density lipoprotein; LDL, low‐density lipoprotein; TG, triglycerides; and VLDL, very low‐density lipoprotein.
Just as with VLDL secretion, little is known as to what determines whether VLDL particles are removed from plasma or converted to LDL particles. apoE likely plays an important role 53 and perhaps so also does apo CIII. 54 As well, we suspect, so does the mass of cholesterol relative to triglycerides within the particle. The processes that catabolize the smaller VLDL particles, from which much of the triglycerides have been removed, are also not well defined. Specific receptors for VLDL have been postulated, but none have been rigorously demonstrated. On the other hand, there is good evidence that binding to heparan sulfate proteoglycans may be a critical step in the removal process. 55 Whether the LDL receptor pathway is involved at all is an open question, particularly because turnover studies suggest statin treatment increases VLDL clearance from plasma, and VLDL particles all contain a molecule of apo B100 and many apoE as well, which are both ligands for the LDL receptor. However, the fact that triglyceride levels are characteristically low in patients with familial hypercholesterolemia (FH), in whom the LDL receptor pathway is genetically defective, argues against a critical role for the LDL receptor in this process.
Determinants of LDL Particle Number
LDL particles are produced when triglycerides are removed from VLDL particles. Thus, LDL particles are produced indirectly by the liver, because they are products of metabolized VLDL particles. In general, the rate of production of VLDL particles is the major determinant of the rate of production of LDL particles. 56 Therefore, to the extent that the determinants of production of VLDL particles remain to be fully clarified, so do the determinants of production of LDL particles. A second mechanism that can affect production of LDL particles is variance in the proportion of VLDL particles converted to LDL particles. In FH, an increased proportion of VLDL particles are converted to LDL particles, perhaps because of decreased reuptake of newly secreted VLDL particles by the liver, 57 whereas in dysbetalipoproteinemia, a decreased proportion is converted, perhaps because of impaired interaction with the liver. 58 The APOB p.Arg3480Pro variant is an unusual but apt and vivid example of a critically sited mutation producing hypobetalipoproteinemia because of reduced conversion of VLDL to LDL. 59 Understanding the metabolic determinants of this process should be a high priority, because it appears to be an important target for drug development; the inhibitors of ANGPTL3 (angiopoietin like protein type 3), for example, might act at this site. 60
As with VLDL particles, the number of LDL particles in plasma is a function of the rate at which they are produced and the rate at which they are removed or cleared from plasma (Figure 3). The plasma half‐life of a VLDL particle is 3 to 4 hours, whereas the plasma half‐life of an LDL particle is 3 to 4 days. 43 , 61 Thus, although each LDL particle was derived from a VLDL particle, this dramatic difference in clearance rates results in the dominance of LDL particles versus VLDL particles. Almost all attention has focused on clearance as the primary determinant of LDL particle number, based, in large part, on the brilliant studies by Brown and Goldstein and their colleagues, who not only described the LDL receptor pathway as the physiological route to remove LDL particles from plasma, but also demonstrated that the highest levels of LDL occur in individuals in whom the LDL receptor pathway is completely defective. 62
The LDL receptor pathway operates as follows: the apoB on the LDL particle binds to the LDL receptor, the receptor‐LDL particle complex is rapidly internalized, the apoB is hydrolyzed within a lysosome, and the cholesterol that is released initiates an intricate sequence of metabolic responses within the cell that results in reduced synthesis of LDL receptors and cholesterol. The liberated receptor is recycled to the cell surface, a process that occurs up to 100 times over its lifespan until it is finally degraded by PCSK9 (proprotein convertase subtilisin kexin 9). In this model, the LDL receptor pathway meets the cell's needs by delivering cholesterol to the interior of the cell, thereby reducing the need for synthesis or uptake of more cholesterol. Accordingly, the metabolism of cholesterol within LDL particles is part of a tightly integrated homeostatic system.
However, does the LDL receptor pathway fulfill this function in the intact organism? Not obviously. 63 The great majority of LDL particles are removed from plasma by the liver. 64 Only a small number are removed by other tissues. 65 Except for the adrenal glands, LDL particles play only a minor physiological role in the transport of cholesterol to peripheral tissues. Peripheral cells synthesize all the cholesterol they need. Not only is the physiological role of the LDL receptor pathway to deliver cholesterol to cells not clear, its capacity as a transport system to remove LDL particles from plasma is limited. The LDL receptor binds apo B100 with high affinity, but is saturated at relatively low concentrations of LDL. Are other mechanisms required to clear LDL particles from plasma? Obviously yes, otherwise LDL particles would continue to accumulate in plasma indefinitely.
These undefined receptors appear to bind LDL particles nonspecifically, but without limit. 66 Accordingly, these nonspecific pathways clear a constant fraction of LDL particles in plasma regardless of their number. Importantly, recent large‐scale genetic studies examining the relation between causal variants of significant dysfunction in the LDL receptor pathway and levels of LDL‐C in plasma demonstrate that only a small minority of individuals with marked hypercholesterolemia have demonstrable single gene defects in this pathway, whereas a surprisingly large number of individuals with mostly imputed causal variants have normal or only moderately elevated levels of LDL‐C. These observations demonstrate that factors other than the LDL receptor pathway must play critical roles in determining the concentration of LDL, and therefore apoB, in plasma. 63
Cholesterol Fluxes and Removal of LDL Particles From Plasma
The liver stands at the center of all the major cholesterol fluxes within the organism, and uptake of cholesterol within LDL particles via the LDL receptor pathway is only one of the multiple sources of cholesterol for the liver. Chylomicron remnant particles deliver dietary cholesterol to the liver. Hepatocytes actively synthesize cholesterol using 3‐hydroxy‐3‐methyl glutaryl‐(HMG) CoA reductase. High‐density lipoprotein (HDL) particles deliver cholesterol from peripheral cells to the liver. So also do VLDL particles taken up by the liver. Given that the liver is the only tissue that can break down and secrete cholesterol in meaningful quantities, and that all cells synthesize cholesterol, all routes must lead to the liver to maintain cholesterol homeostasis within the organism.
In species, such as humans, in which CETP (cholesteryl ester transfer protein) is present, VLDL and LDL particles also participate in reverse cholesterol transport from peripheral tissues. In these species, CE is transferred to VLDL particles from HDL particles and can be returned to the liver either within VLDL or LDL particles. There is no evidence that this improves reverse cholesterol transport; merely, that the route cholesterol may take to return to the liver is more complex than it might otherwise have been. In this accounting, the LDL receptor pathway is but one of the multiple routes by which cholesterol is returned to the liver to maintain cholesterol homeostasis in the organism.
What physiological process is served by the cholesterol that was exported from the liver within VLDL particles and returned as VLDL or LDL particles? Is this just a futile cycle of cholesterol or could it be that cholesterol and CE are essential elements to form VLDL particles? Alternatively, could it be that at least in certain circumstances VLDL particles also export excess cholesterol from the liver? These are questions without answers at present.
Clearance Versus Production as Determinants of LDL Particle Number
Whatever the uncertainties as to its physiological usefulness or its final importance in clearance, the LDL receptor pathway is a major determinant of LDL particle number in plasma. LDL particles can be cleared from plasma by the LDL receptor pathway or by nonspecific pathways. These latter pathways are also principally situated in the liver, and although they bind LDL particles with low affinity, they have an unlimited capacity to transport LDL particles. 67 In patients with homozygous FH, in whom the LDL receptor pathway does not function because of biallelic pathogenic variants in the LDLR, APOB, PCSK9, or LDLRAP1 genes, clearance of LDL particles must presumably occur, principally or exclusively, via nonspecific pathways. apoB turnover studies in patients with FH and Watanabe rabbits with a pathogenic LDLR variant causing a phenotype analogous to human FH, as well as studies using LDL containing a binding defective variant of apo B100, have all demonstrated that the nonspecific pathways clear a constant fraction, ≈0.17 pools of LDL per day, of the LDL particles in plasma. 64 , 68 , 69 , 70 , 71 Although the actual mass of cholesterol delivered to the liver is greater than normal, these nonspecific pathways have no clinical benefit in homozygous FH, because this increased delivery of cholesterol is achieved at the cost of a disproportionately increased LDL particle number and therefore vastly increased cardiovascular risk.
Thus, there are 2 routes to clear LDL from plasma: the specific LDL receptor pathway and the nonspecific pathways. The former is highly effective but limited in capacity. The latter is much less effective, but unlimited in capacity. Dietschy and his colleagues worked out in detail the relationships among production of LDL‐C and clearance of LDL particles by the specific LDL receptor pathway versus the nonspecific pathways. 71 Based on their work and corresponding apoB turnover studies, we have calculated the impact of varying clearance or production or both on the number of LDL particles, LDL apoB. 72
Tables 1 through 3 quantify the effect on LDL apoB that occurs with changes in clearance of LDL particles, production of LDL particles, or both. Table 1 lists the effects on LDL apoB of serial reduction in the activity of the LDL receptor pathway in a 70‐kg hypothetical individual with a normal production rate of LDL particles. 72 The fractional clearance rate of LDL particles by the nonspecific pathway is constant (ie, 0.17 pools of LDL per day). The fractional clearance rate of removal by the specific pathway varies from 0.28 pools of LDL per day, when its activity is normal, to 0 when the LDL receptor pathway is totally dysfunctional, as in homozygous biallelic FH. Thus, the fractional clearance rate decreases progressively from 0.45 to 0.17 pools of LDL per day as the activity of the LDL removal pathway is reduced from normal to 0. The net effect is that LDL apoB would increase progressively from 45 to 118 mg/dL, a substantial but not extraordinary increase. This is not consistent with the levels of LDL‐C in patients with homozygous FH, which are much higher. This discordance between what is calculated and what has been measured was noted by Bilheimer and colleagues and explained by the fact that production of LDL was increased in FH as well as its clearance decreased, 68 a finding that has been replicated in other studies. 67 , 73 , 74
Table 1.
Effect on Plasma LDL apoB of Decreased Activity of the LDL Clearance Pathway With Normal Production of LDL Particle
| Production rate of LDL apoB | FCR LDL apoB, pools/d | LDL apoB, mg/dL |
|---|---|---|
| 700 mg/d | 0.45 | 45 |
| 700 mg/d | 0.32 | 63 |
| 700 mg/d | 0.17 | 118 |
apoB indicates apolipoprotein B; FCR, fractional clearance rate; and LDL, low‐density lipoprotein.
Table 3.
Effect on Plasma LDL apoB of Increased Production of LDL With Moderately Reduced Clearance (FCR 0.33)
| Production rate of LDL apoB | FCR LDL apoB, pools/d | LDL apoB, mg/dL |
|---|---|---|
| 700 mg/d | 0.29 | 69 |
| 1000 mg/d | 0.24 | 119 |
| 1300 mg/d | 0.22 | 170 |
apoB indicates apolipoprotein B; FCR, fractional clearance rate; and LDL, low‐density lipoprotein.
Table 2 quantifies the effect on LDL apoB that occurs in another hypothetical individual with increased production of LDL particles with persistent normal activity of the LDL receptor pathway. Note the calculated fractional clearance rate decreases as production increases, although the LDL receptor pathway activity is normal. This is because of the increase in the plasma pool of LDL apoB. A larger pool means a smaller fraction must be removed to achieve metabolic equilibrium. The plasma LDL apoB pool increased substantially because production increased, and the transport capacity of the LDL receptor pathway, although normal, was saturated. Therefore, more and more clearance of LDL particles must occur through nonspecific pathways. Because the nonspecific pathways have low affinity for LDL particles, there is a dramatic rise in the concentrations of LDL apoB particles as their production increases. Note that the impact of overproduction on increasing LDL apoB is more dramatic than the effect of reducing clearance, a critical insight made by Meddings and Dietschy. 66
Table 2.
Effect on Plasma LDL apoB of Increased Production of LDL With Normal Activity of the LDL Clearance Pathway
| Production rate of LDL apoB | FCR LDL apoB, pools/d | LDL apoB, mg/dL |
|---|---|---|
| 700 mg/d | 0.45 | 45 |
| 1000 mg/d | 0.32 | 95 |
| 1300 mg/d | 0.17 | 145 |
apoB indicates apolipoprotein B; FCR, fractional clearance rate; and LDL, low‐density lipoprotein.
Finally, Table 3 quantifies the effect on LDL apoB of a simultaneous increase in production of LDL apoB with partial, but constant, decrease in activity of the LDL receptor pathway. This indicates that a dual defect, increased production with impaired clearance, produces the most dramatic increases in LDL apoB. 66
Summary
Except for dysbetalipoproteinemia, even in patients with severe hypertriglyceridemia, LDL particles are much more numerous than VLDL particles, just as VLDL particles are much more numerous than chylomicrons and remnant particles. Therefore, LDL apoB is the predominant determinant of apoB. VLDL apoB or LDL apoB levels can increase because production increases, clearance decreases, or both. Total apoB reflects the total number of apoB particles, and except for dysbetalipoproteinemia, the cardiovascular risk is attributable to the apoB lipoproteins, no matter the pathophysiological mechanism responsible for the elevation in apoB. This is how apoB integrates information and simplifies care.
Variance in the Composition of apoB Particles
Triglycerides are the major lipid component of VLDL particles, but the amount of triglycerides per VLDL particle is highly variable (Figure 1B). Therefore, the correlations between plasma triglycerides and VLDL apoB or total apoB are low. 35 The mass of cholesterol within LDL particles also varies, although not to the same extent (Figure 1B). Therefore, the correlation between apoB and LDL‐C or non–HDL‐C is relatively high. 35 Nevertheless, 3 classes of LDL particles can be distinguished: LDL1 particles contain the greatest mass of cholesterol, LDL2 particles contain an average mass of cholesterol, and LDL3 particles contain the least cholesterol. 75 The overall LDL‐C/apoB ratio is determined by the relative amounts of these 3 subclasses of LDL particles. The variance in their proportions creates variance in the average mass of cholesterol per apoB particle. This variance is sufficiently great and sufficiently common that neither LDL‐C nor non–HDL‐C are accurate estimates of LDL apoB or total apoB in many individual patients. 4
Because VLDL particles are large, 30 to 80 nm in diameter, the mass of triglycerides per particle is high. Thus, a few VLDL particles can transport large masses of triglycerides through plasma. By contrast, because LDL particles are much smaller than VLDL particles, 20 to 25 nm in diameter, the mass of cholesterol per LDL particle is much less than the mass of triglycerides per VLDL particle. The volume of the average VLDL particle is ≈10 times the volume of the average LDL particle. Therefore, many more LDL particles are required to transport the same mass of cholesterol as the mass of triglycerides carried by VLDL particles. The conventional measurements of plasma lipids do not reflect these physical realities. The absolute concentration of triglycerides in plasma measured as milligrams per deciliter or millimole per liter can often be close to, or greater than, the absolute concentration of LDL‐C. A mole of triglycerides has a greater mass than a mole of cholesterol. This creates a misleading impression of how many VLDL particles there are compared with how many LDL particles there are.
Pathophysiology of Discordance Between LDL‐C/Non–HDL‐C and apoB
If the mass of cholesterol per apoB particle were invariant, LDL‐C, non–HDL‐C, and apoB would be identical predictors of the risk of cardiovascular disease. If so, there would be no reason to measure atherogenic particle number rather than LDL‐C or non–HDL‐C. However, the cholesterol mass per apoB can vary substantially. The result is that at any level of apoB, there is significant variance in the mass of cholesterol per apoB particle. The result is that LDL‐C and non–HDL‐C are imperfect surrogates of apoB.
Exchange of core lipids is the major mechanism responsible for the variance in the cholesterol mass in apoB particles 76 (Figure 5). The lipids within the core of lipoprotein particles, CE and triglycerides, can be exchanged among the apoB and HDL particles by CETP. If a CE molecule is exchanged for another CE molecule between an apoB particle and an HDL particle, or a triglyceride molecule is exchanged for another triglyceride molecule, there is no change in the composition of these particles. However, if a CE molecule is transferred from an LDL or HDL particle to a VLDL or a chylomicron particle, and in return a triglyceride molecule is transferred from that VLDL or chylomicron particle to that LDL or HDL particle, the composition of both particles has been changed. The VLDL particle has become enriched in CE whereas the LDL or HDL particle has become enriched in triglycerides. The LDL particle has not changed significantly in size because the partial specific volumes of triglycerides and CE are comparable. If the triglyceride is hydrolyzed, the mass of lipid in the particle core has been diminished and the LDL particle has become smaller. These 2 steps in succession explain the pathogenesis of smaller, denser, cholesterol‐depleted LDL particles. As noted above, the average mass of cholesterol per apoB particle, which can be estimated from the LDL‐C/apoB ratio, is determined by the relative amounts of the LDL subfractions. Although the scheme of core lipid exchanges we have outlined is well known and well accepted, it is not well appreciated that triglycerides are not the only physiological determinants of this process, and that small, dense cholesterol‐depleted apoB particles can be present at any level of triglycerides. 77 , 78
Figure 5. Lipid exchanges amongst plasma lipoprotein particles.
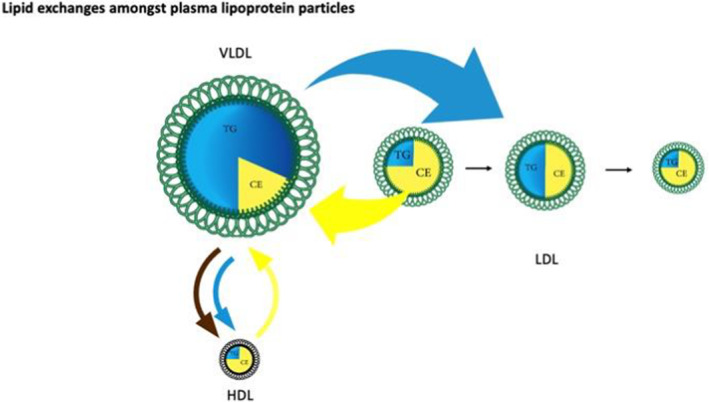
This figure illustrates the exchange of the core lipids, cholesterol ester (CE) and triglycerides (TG) mediated by CETP among VLDL, LDL, and HDL. If a CE molecule from LDL or HDL is exchanged (yellow arrow) for a TG molecule from VLDL (turquoise arrow), the LDL or HDL particle becomes enriched in TG, but depleted in CE, whereas the VLDL particle loses TG but gains CE. In the case of LDL, the TG is subsequently hydrolyzed, producing a smaller, CE‐depleted LDL particle. Illustrated also is the transfer of free cholesterol from a VLDL particle to HDL as it is being hydrolyzed, where it can subsequently be esterified to form CE. CE indicates cholesterol ester; CETP, cholesterol ester transfer protein; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; TG, triglycerides; and VLDL, very low‐density lipoprotein.
The physiological process that produces larger, cholesterol‐enriched LDL particles is not as clear. No process resulting in net transfer of CE from 1 lipoprotein particle to another without exchange of triglycerides has been described. Nevertheless, VLDL particles secreted by the liver might be enriched in CE under some circumstances such as FH. Alternatively, an increased LDL‐C/apoB ratio could reflect reduced amounts of cholesterol‐depleted LDL particles in patients. Conversely, if VLDL particles contain fewer triglycerides, LDL particles, on average, will contain more because there will be less exchange of triglycerides from VLDL for CE from LDL.
Finally, as their triglycerides content is removed in peripheral tissues, chylomicrons and VLDL particles become smaller and release cholesterol and phospholipids from their external surface. This cholesterol can also be transferred to HDL particles. This cholesterol can then be esterified, trapping the cholesterol within the core of the HDL particles as CE, which results in net transfer of cholesterol from one lipoprotein particle to another (Figure 5). Thus, a significant portion of the CE within HDL particles can originate from the intestine and the liver; not all come from peripheral cells.
Summary
The average mass of triglycerides within VLDL particles is determined by the relative rates of the production of the different VLDL particles, the rate at which the triglycerides within the VLDL particles is hydrolyzed, and CETP‐mediated exchanges of triglycerides and CE between VLDL, on the one hand, and HDL and LDL, on the other. Correlation describes how closely changes in 1 marker are associated with changes in another. Concordance describes the variance in 1 marker for a given value of another. VLDL triglycerides and VLDL apoB are only moderately correlated and poorly concordant. 35 The net result is that triglycerides are a poor estimate of VLDL particle number. The average mass of cholesterol within LDL particles is determined by the numbers of the different size LDL particles. This is a product of triglyceride concentration within VLDL particles, CETP activity, and the number of VLDL, LDL, and HDL particles as well as multiple other factors. Thus, the relation between plasma triglycerides and the cholesterol‐depleted LDL particles is more complex and uncertain than is generally appreciated. 77 , 78 The net result is that LDL‐C and non–HDL‐C are highly correlated with apoB, but only moderately concordant. Thus, neither LDL‐C or non–HDL‐C are reliable estimates of LDL apoB or total apoB for an individual patient.
apoB Dyslipoproteinemic Phenotypes
Based on the rates at which VLDL and LDL particles are produced and cleared, and the changes in their composition that are produced by exchange of core lipids, distinctive patterns of dyslipoproteinemia can be characterized. This concept of specific dyslipoproteinemias related to differences in pathophysiology is similar to that introduced by Fredrickson and his colleagues to differentiate the different lipid phenotypes. 79 , 80 , 81 , 82 , 83 The principal difference between then and now is that particle number as determined by apoB is included as well as lipoprotein lipids. Given that the atherogenic lipids circulate within apoB lipoprotein particles, and that trapping of apoB particles within the arterial wall is the fundamental cause of atherosclerosis, this new approach is more physiologically based than the original although it is conceptually similar. 2
The Fredrickson classification of dyslipidemia follows from whether plasma triglycerides are normal or elevated and whether LDL‐C is normal or elevated. This system was useful in the past, but we feel it has outlived its usefulness. 84 Triglycerides were the measure of VLDL and LDL‐C the measure of LDL, and the labor‐intensive laboratory method of ultracentrifugation was required for accurate phenotyping of these variables. However, as we have demonstrated, neither are acceptable surrogates for VLDL apoB and LDL apoB. In patients with hypertriglyceridemia, attributable to cholesterol‐depleted LDL particles, LDL‐C may be normal or only moderately elevated, but LDL apoB, and therefore total apoB, may be markedly elevated. A normal LDL‐C does not imply a normal LDL particle number. Conversely, an elevated LDL‐C does not imply an abnormal LDL particle number. This was not appreciated at the time, although it is now accepted.
Nevertheless, as clinicians came to see triglycerides and LDL‐C as independent variables, and as statin use surged, classification of dyslipidemic phenotypes fell out of favor. However, as demonstrated above, VLDL and LDL are not independent variables. LDL particles are the product of the metabolism of VLDL particles, and core lipid exchanges between VLDL and LDL alter their composition without affecting their number. The levels of triglycerides and LDL‐C reflect the outcome of the processes, which regulate the composition of apoB particles as well as their number.
The production and clearance rates of VLDL and LDL apoB particles have been determined in normal individuals and in the various dyslipoproteinemias. Thus, the metabolic causes of each dyslipoproteinemia can be specified in terms of alterations of apoB metabolism. Moreover, by adding apoB to the conventional lipid panel, the application of a simple algorithm allows all the major apoB dyslipoproteinemias to be differentiated. 85 This algorithm classifies an elevated apoB as >120 mg/dL (>1.2 g/L), which is the 90th percentile of the American population. However, as the evidence demonstrating that apoB is a more accurate marker of cardiovascular risk than LDL‐C and non–HDL‐C has mounted, we believe the appropriate clinical cut point to designate high apoB should now be 105 mg/dL (1.05 g/L), the 75th percentile of the American population, which is the equivalent decisional level for LDL‐C and non–HDL‐C. 86
The pathophysiological characteristics of the 4 major apoB dyslipoproteinemic phenotypes: hypertriglyceridemic normal apoB (hyperTG normoapoB), hypertriglyceridemic hyperapoB (hyperTG hyperapoB), normotriglyceridemia and hyper apolipoprotein B (normoTG hyperapoB), and dysbetalipoproteinemia will be summarized below.
Pathophysiological Characteristics of the 4 Major apoB Dyslipoproteinemic Phenotypes
Normal
Normal (Figure 6A) is defined as a normal production and clearance rate of VLDL apoB particles and a normal production and clearance rate of LDL particles, which results in a normal level of triglycerides, total cholesterol, and apoB. A normal level of triglycerides is defined as a value <1.5 mmol/L (130 mg/dL). This level was selected because it is the level above which cholesterol‐depleted LDL particles become more common. 77 , 78 As explained above, a normal number of VLDL and LDL apoB particles is defined as an apoB <105 mg/dL. The triglycerides/apoB ratio (millimoles per gram per liter) must be <10/1 and the TC/apoB (millimoles per gram per liter) ratio will be <6.2. 17 , 85 Importantly, in some individuals, the LDL‐C will be elevated (≥3.6 mmol/L, >135 mg/dL [ie, ≥75th percentile]), but the total apoB will be <105 mg/dL (ie, <75th percentile). In these cases, the LDL particles are cholesterol enriched, but the total atherogenic particle number is not increased. Therefore, cardiovascular risk attributable to the apoB lipoproteins is not as increased in these individuals as the elevated LDL‐C suggests. The degree of risk attributable to the apoB lipoproteins is determined by the plasma apoB. 4 Such individuals should be reassured and informed that this is a normal variant.
Figure 6. Pathophysiological characteristics of the 4 major apoB dyslipoproteinemic phenotypes.
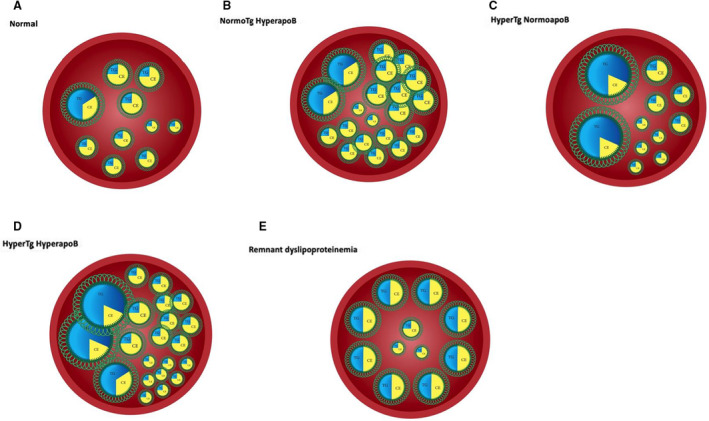
A, Normal number of VLDL and LDL particles within an arterial lumen. B, Increased number of VLDL particles with an increased number of cholesterol‐rich LDL particles. Familial hypercholesterolemia is the prototypic disorder that represents the extreme manifestations of this phenotype. C, Relative number of VLDL and LDL particles within an arterial lumen in a patient with hyperTG normoapoB. The VLDL particles are enriched in triglycerides or increased in number, but LDL particle number is normal. The net result is that apoB is normal. Familial hypertriglyceridemia is present when this is the dominant phenotype within a family. D, Increased number of VLDL and LDL particles within the arterial lumen of a patient with hyperTG hyperapoB. This occurs because of increased production of VLDL and LDL particles in hyperTG hyperapoB. Familial combined hyperlipidemia represents the expression of this disorder within a family. Levels of apoB are >120 mg/d (>90th percentile) in subjects with familial combined hyperlipidemia. E, Markedly increased number of cholesterol‐enriched VLDL and chylomicron remnants with a normal number of LDL particles in a patient with dysbetalipoproteinemia (type III hyperlipoproteinemia). apoB indicates apolipoprotein B; CE, cholesterol ester; hyperTG hyperapoB, hypertriglyceridemic hyperapoB; hyperTG normoapoB, hypertriglyceridemic normal apoB; LDL, low‐density lipoprotein; TG, triglycerides; and VLDL, very low‐density lipoprotein.
NormoTG HyperapoB
NormoTG hyperapoB is defined as plasma triglycerides <1.5 mmol/L (<130 mg/dL) and apoB <105 mg/dL. 85 LDL‐C may be normal or elevated (< or > 135 mg/dL, 3.6 mmol/L). Extreme elevations of either apoB (>150 mg/dL) or LDL‐C (>190 mg/dL, 5.0 mmol/L), both >95th percentile of the population, raise the possibility of FH. Figure 6B illustrates the consequences for the artery; the combination of markedly reduced clearance and moderately increased production in FH produces massively increased numbers of apoB particles within the arterial lumen and consequently a massively increased risk of a cardiovascular event. Lowering apoB is the objective of therapy. With moderate elevations of apoB, statins often suffice, although addition of ezetimibe may be necessary. With more extreme elevations of apoB, addition of PCSK9 inhibitors may be required, and in the extreme case of homozygous FH, LDL apheresis is the foundation of care, 87 and the anti‐ANGPTL3 monoclonal antibody evinacumab has been recently approved for this condition by the US Food and Drug Administration.
HyperTG NormoapoB
HyperTG normoapoB is defined as plasma triglycerides >1.5 mmol/L (>130 mg/dL) and plasma apoB <105 mg/dL. Plasma triglycerides/apoB ratio (millimoles per gram) is <10:1 and total cholesterol (TC)/plasma apoB ratio is <6.2:1. 85 LDL‐C is usually normal (ie, <135 mg/dL, 3.6 mmol/L) and HDL‐C is typically low. The cardiovascular risk attributable to the apoB lipoproteins is related more closely to apoB than to triglycerides or LDL‐C. As illustrated in Figure 6C, the number of apoB particles (ie, the sum of VLDL and LDL particles) is not increased. The hypertriglyceridemia is attributable to a normal number of triglyceride‐enriched VLDL particles. LDL particle number is also normal.
HyperTG normoapoB is the characteristic phenotype seen in mild‐to‐moderate hypertriglyceridemia, which is primarily a non‐Mendelian polygenic phenotype that corresponds to former type IV hyperlipoproteinemia. 88 This contrasts with hyperTG hyperapoB, which is the phenotype of familial combined hyperlipidemia, also primarily a non‐Mendelian polygenic phenotype that corresponds to the former type IIB hyperlipoproteinemia, 89 in which plasma triglycerides and apoB are both increased. 90 In combined hyperlipidemia, hypertriglyceridemia is attributable increased secretion of VLDL particles, whereas, in mild‐to‐moderate hypertriglyceridemia, hypertriglyceridemia is attributable to secretion of triglyceride‐enriched VLDL particles at a normal rate. 90 , 91 The difference in apoB correlates with the difference in cardiovascular risk, which is substantially higher in hypertriglyceridemic patients with combined hyperlipidemia than in patients with mild‐to‐moderate hypertriglyceridemia. 92 , 93
The elevation in plasma triglycerides in these patients is primarily because of VLDL particles, not chylomicrons, and therefore, these patients are not at risk for pancreatitis. However, for reasons still not well understood, plasma triglycerides in these patients may suddenly rise substantially because of a failure in chylomicron clearance and, if this occurs, pancreatitis is possible. Cardiovascular risk attributable to the apoB lipoproteins in these patients is proportional to the number of apoB particles. Typically, apoB is low. Sometimes, however, it may be high, pointing to concurrent elevation of VLDL and LDL apoB particles. 35 , 85 In all instances, other causes of cardiovascular risk may be present, such as diabetes or hypertension, and these must be considered in determining the overall level of cardiovascular risk. When lipid‐lowering therapy is required, statins are the preferred therapy, not fibrates, because the target of therapy is apoB, not triglycerides. Fibrates substantially reduce triglycerides and VLDL apoB, but only modestly reduce LDL apoB. 35 Because LDL apoB accounts for the great majority of total apoB, the effect of fibrates on apoB is modest. This modest effect explains why fibrates were successful in some randomized trials but not in most. By contrast, statins reduce VLDL apoB moderately, LDL apoB markedly, and therefore total apoB markedly, and this explains why statins, not fibrates, were virtually uniformly successful in randomized clinical trials of cardiovascular outcomes. 35
HyperTG HyperapoB
HyperTG hyperapoB (Figure 6D) is defined as an elevated apoB primarily because of increased secretion of VLDL particles with increased production of LDL particles. 43 , 46 , 47 , 48 The diagnostic criteria are a plasma triglycerides >1.5 mmol/L (>130 mg/dL) and apoB >105 mg/dL. The triglycerides/apoB ratio will be <10:1 (triglycerides, millimoles per liter/apoB, grams per liter) and TC/apoB ratio will be <6.2 (TC, millimoles per liter/apoB grams per liter). 85 LDL‐C may be normal or elevated (>3.6 mmol/L; 135 mg/dL), and HDL‐C is typically low.
Affected patients may be hypertriglyceridemic, hypercholesterolemic, hypertriglyceridemic and hypercholesterolemic, or occasionally, even normolipidemic with borderline triglyceride levels. All affected individuals with this phenotype have increased numbers of apoB particles, which on average are smaller and more cholesterol depleted than normal. 94 , 95 It is the variance in the number of apoB particles that explains why so many different lipid phenotypes could be created with the same basic building blocks. Just as with FH, atherogenic particle number is markedly increased, and therefore so is the rate of entry and trapping of apoB particles within the arterial wall.
HyperTG hyperapoB is the most common atherogenic dyslipoproteinemia in people with abdominal obesity, type 2 diabetes, 96 , 97 and in individuals with premature coronary artery disease. HyperTG hyperapoB is the hallmark dyslipoproteinemia in familial combined hyperlipidemia. 51 , 94 Familial combined hyperlipidemia is the most common familial disorder associated with premature coronary artery disease. 98 , 99 As mentioned, familial combined hyperlipidemia is a polygenic condition 100 with no major gene(s) as in FH, despite >4 decades of intensive searching by geneticists.
HyperTG hyperapoB is attributable to increased production of VLDL and LDL particles, hence the elevated triglycerides and the elevated apoB. 43 , 46 , 47 , 48 Because the rate at which VLDL particles can be cleared is limited, VLDL apoB particle numbers increase. Because conversion of VLDL to LDL particles continues, production of LDL particles is increased. Because the capacity of the high‐affinity LDL receptor pathway to clear LDL particles is limited, LDL particle numbers in plasma are increased. The cholesterol‐depleted LDL particles, so characteristic in this disorder, bind less well to the LDL receptor than cholesterol‐replete ones, which further diminishes the effectiveness of LDL clearance in the absence of any fault in the LDL receptor pathway itself. 101
Statins are the frontline therapy for this disorder, and apoB is the target of therapy. Addition of ezetimibe may be required. In high‐risk individuals, PCSK9 inhibitors may be required as well.
Dysbetalipoproteinemia
Dysbetalipoproteinemia (Figure 6E) is characterized by markedly increased numbers of cholesterol‐enriched chylomicron and VLDL remnant particles. By contrast, the number of LDL particles is low. Total apoB is <120 mg/dL, triglycerides are elevated (>1.5 mmol/L), the triglycerides/apoB ratio is <10:1, and the TC apoB ratio is >6.2:1. 85 Dysbetalipoproteinemia is a highly atherogenic dyslipoproteinemia, less common than heterozygous FH, but often much more easily treatable. The pathophysiology is complex. The great majority of those affected are homozygous for the apoE E2 isoform. However, this is only a precondition, not a sufficient requirement. 32 , 102 Obesity, diabetes, and exogenous hormones are among the factors that may trigger the expression of the abnormal accumulation of remnant particles, but the precise mechanisms remain unknown. In patients with dysbetalipoproteinemia, in contrast to all the other phenotypes, apoB is not the primary target of therapy. The targets of therapy are the cholesterol‐enriched chylomicron VLDL and remnant particles, but statins, once again, are still be the preferred initial therapy, although fibrates and even niacin are effective in many patients. 103
SUMMARY
Our objective has been to present a physiological framework in which the determinants of apoB particle number and the composition of apoB particles could be understood by caregivers. Cardiovascular risk relates directly and powerfully to the level of apoB in plasma. This level is determined by the rates at which VLDL and LDL particles are produced and are cleared from plasma. The LDL receptor pathway, the specific pathway to effectively clear LDL particles from plasma, has a limited transport capacity. Any decrease in its effectiveness means even more LDL particles must be cleared by the nonspecific pathways, with the result that levels of apoB rise sharply. Similarly, increased production of LDL particles because of increased production of VLDL particles overwhelms the clearance capacity of the LDL receptor pathway, also producing markedly increased levels of apoB.
Neither triglycerides nor LDL‐C are accurate markers of VLDL or LDL particle numbers. VLDL and LDL particles are atherogenic, but there is no way to integrate their risk using triglycerides and LDL‐C. apoB simplifies clinical care because it provides an accurate summary estimate of the atherogenic risk attributable to all apoB lipoprotein particles. Except for dysbetalipoproteinemia, triglycerides, LDL‐C, and non–HDL‐C add no significant information about cardiovascular risk to apoB, whereas apoB adds significant information to triglycerides, LDL‐C, and non–HDL‐C. At the same time, by integrating the information from lipids and apoB, detailed and accurate discrimination of the various dyslipoproteinemias becomes possible. Adding apoB to a conventional lipid panel converts the disorders of lipid metabolism to the disorders of lipoprotein metabolism.
Our argument is not to abandon the familiar traditional lipid profile. Our argument is to move from lipids to lipoproteins, evolving toward a more physiological construct. apoB is not the final step in the characterization of the dyslipoproteinemias. apoB is only an intermediate, but essential, step in this process. All cholesterol within an atheroma was deposited there from within an apoB particle. The disease process itself, atherosclerosis, reflects a myriad of responses to the entrapment of apoB particles within the arterial wall. The number of apoB particles circulating within the lumen of our arteries is the primary determinant of the number of apoB particles that will enter and become trapped within the arterial wall.
However, the apoB particle story does not end there. Some apoB particles may be more atherogenic than others. The abnormal, cholesterol‐enriched remnant apoB particles in dysbetalipoproteinemia do appear to be particularly atherogenic. Whether certain triglyceride‐rich apoB particles or smaller LDL particles are also particularly atherogenic is not as clear, but this remains a legitimate question for investigation. An essential objective must be to increase our understanding of the processes that regulate plasma apoB concentrations, the processes that regulate the production and clearance of these particles, because the number of apoB particles in plasma is the primary determinant of the cardiovascular risk. However, apoB particle number is not the only issue of interest. We must learn much more about the processes that alter the trapping of apoB particles within the arterial wall, because trapping of apoB particles within the arterial wall is the fundamental cause of atherosclerosis. We are not at the end of our understanding of the apoB lipoproteins and atherogenesis. We are only at the end of one phase and the beginning of another.
Sources of Funding
This work was supported by an unrestricted grant from the Doggone Foundation.
Disclosures
Dr Thanassoulis reported receiving personal fees from Amgen, Sanofi/Regeneron Pharmaceuticals, and Boehringer Ingelheim and grants from Ionis Pharmaceuticals and Servier Laboratories outside the submitted work. Dr Couture has received funding in the past 5 years from the Canadian Institutes for Health Research, Agriculture and Agri‐Food Canada (Growing Forward program supported by the Dairy Farmers of Canada, Canola Council of Canada, Flax Council of Canada, Dow Agrosciences), Dairy Research Institute, Dairy Australia, Danone Institute, Merck, Pfizer, Atrium Innovations, and Kaneka. Dr Couture reports consulting and/or speaker honoraria from Acasti, Aegerion, Akcea/Ionis, Amgen, Arrowhead, HLS Therapeutics, Novartis, Pfizer, Regeneron, and Sanofi. Dr Hegele is supported by the Jacob J. Wolfe Distinguished Medical Research Chair, the Edith Schulich Vinet Canada Research Chair in Human Genetics, the Martha G. Blackburn Chair in Cardiovascular Research, and operating grants from the Canadian Institutes of Health Research (Foundation Grant) and the Heart and Stroke Foundation of Ontario (G‐21‐0031455) and the Academic Medical Association of Southwestern Ontario (INN21‐011). The remaining authors have no disclosures to report.
For Sources of Funding and Disclosures, see page 14 and 15.
References
- 1. Borén J, Williams KJ. The central role of arterial retention of cholesterol‐rich apolipoprotein‐B‐containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–483. doi: 10.1097/MOL.0000000000000330 [DOI] [PubMed] [Google Scholar]
- 2. Sniderman AD, Thanassoulis G, Glavinovic T, Navar AM, Pencina M, Catapano A, Ference BA. Apolipoprotein B particles and cardiovascular disease: a narrative review. JAMA Cardiol. 2019;4:1287–1295. doi: 10.1001/jamacardio.2019.3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, Ference BA, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. doi: 10.1093/eurheartj/ehz455 [DOI] [PubMed] [Google Scholar]
- 4. Sniderman AD, Navar AM, Thanassoulis G. Apolipoprotein B vs low‐density lipoprotein cholesterol and non‐high‐density lipoprotein cholesterol as the primary measure of apolipoprotein B lipoprotein‐related risk: the debate is over. JAMA Cardiol. 2022;7:257–258. doi: 10.1001/jamacardio.2021.5080 [DOI] [PubMed] [Google Scholar]
- 5. Richardson TG, Sanderson E, Palmer TM, Ala‐Korpela M, Ference BA, Davey Smith G, Holmes MV Rader DJ, ed. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis, PLoS Med. 2020;17:e1003062, doi: 10.1371/journal.pmed.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zuber V, Gill D, Ala‐Korpela M, Langenberg C, Butterworth A, Bottolo L, Burgess S. High‐throughput multivariable Mendelian randomization analysis prioritizes apolipoprotein B as key lipid risk factor for coronary artery disease. Int J Epidemiol. 2021;50:893–901. doi: 10.1093/ije/dyaa216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yuan S, Tang B, Zheng J, Larsson SC. Circulating lipoprotein lipids, apolipoproteins and ischemic stroke. Ann Neurol. 2020;88:1229–1236. doi: 10.1002/ana.25916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levin MG, Zuber V, Walker VM, Klarin D, Lynch J, Malik R, Aday AW, Bottolo L, Pradhan AD, Dichgans M, et al. Prioritizing the role of major lipoproteins and subfractions as risk factors for peripheral artery disease. Circulation. 2021;144:353–364. doi: 10.1161/CIRCULATIONAHA.121.053797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richardson TG, Wang Q, Sanderson E, Mahajan A, McCarthy MI, Frayling TM, Ala‐Korpela M, Sniderman A, Smith GD, Holmes MV. Effects of apolipoprotein B on lifespan and risks of major diseases including type 2 diabetes: a mendelian randomisation analysis using outcomes in first‐degree relatives. Lancet Healthy Longev. 2021;2:e317–e326. doi: 10.1016/S2666-7568(21)00086-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johannesen CD, Mortensen MB, Langsted A, Nordestgaard B. Apolipoprotein B and Non‐HDL‐C better reflect residual risk than LDL cholesterol in statin‐treated patients with atherosclerosis. J Am Coll Cardiol. 2021;77:1439–1450. doi: 10.1016/j.jacc.2021.01.027 [DOI] [PubMed] [Google Scholar]
- 11. Marston NA, Giugliano RP, Melloni G, Park J‐G, Morrill V, Blazing MA, Ference GA, Stein E, Stroes E, Braunwald E, et al. Association of apolipoprotein‐B‐containing lipoproteins and risk of myocardial infarction in individuals with and without atherosclerosis: distinguishing between particle concentration, Type and Content. JAMA Cardiol. 2022;7:250–256. doi: 10.1001/jamacardio.2021.5083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Langlois MR, Nordestgaard BG, Langsted A, Chapman MJ, Aakre KM, Baum H, Borén J, Bruckert E, Catapano A, Cobbaert C, et al. European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Joint Consensus Initiative. Quantifying atherogenic lipoproteins for lipid‐lowering strategies: consensus‐based recommendations from EAS and EFLM. Clin Chem Lab Med. 2020;58:496–517. doi: 10.1515/cclm-2019-1253 [DOI] [PubMed] [Google Scholar]
- 13. Kohli‐Lynch CN, Thanassoulis G, Moran AE, Sniderman AD. The clinical utility of apoB versus LDL‐C/non‐HDL‐C. Clin Chim Acta. 2020;508:103–108. doi: 10.1016/j.cca.2020.05.001 [DOI] [PubMed] [Google Scholar]
- 14. Fisher WR. Heterogeneity of plasma low density lipoproteins manifestations of the physiologic phenomenon in man. Metab Clin Exp. 1983;32:283–291. doi: 10.1016/0026-0495(83)90194-4 [DOI] [PubMed] [Google Scholar]
- 15. Krauss RM, Burke DJ. Identification of multiple subclasses of plasma low density lipoproteins in normal humans. J Lipid Res. 1982;23:97–104. doi: 10.1016/S0022-2275(20)38178-5 [DOI] [PubMed] [Google Scholar]
- 16. Sniderman AD, St‐Pierre AC, Cantin B, Dagenais GR, Després J‐P, Lamarche B. Concordance/discordance between plasma apolipoprotein B levels and the cholesterol indexes of atherosclerotic risk. Am J Cardiol. 2003;91:1173–1177. doi: 10.1016/S0002-9149(03)00262-5 [DOI] [PubMed] [Google Scholar]
- 17. De Graaf J, Couture P, Sniderman AD. ApoB in Clinical Care. Bohn Stafleu van Loghum; 2015. doi: 10.1007/978-90-368-0980-1 [DOI] [Google Scholar]
- 18. Kane JP, Havel RJ. In: Scriver C, ed The Metabolic Basis of Inherited Disease. 6th ed. McGraw‐Hill; 1139. –1164. [Google Scholar]
- 19. Kane JP, Hardman DA, Paulus HE. Heterogeneity of apolipoprotein B: isolation of a new species from human chylomicrons. Proc Natl Acad Sci USA. 1980;77:2465–2469. doi: 10.1073/pnas.77.5.2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591 [DOI] [PubMed] [Google Scholar]
- 21. Afshar M, Rong J, Zhan Y, Chen HY, Engert JC, Sniderman AD, Larson MG, Vasan RS, Thanassoulis G. Risks of incident cardiovascular disease associated with concomitant elevations in lipoprotein(a) and low‐density lipoprotein cholesterol‐The Framingham Heart Study. J Am Heart Assoc. 2020;9:e014711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elovson J, Chatterton JE, Bell GT, Schumaker VN, Reuben MA, Puppione DL, Reeve JR, Young NL. Plasma very low density lipoproteins contain a single molecule of apolipoprotein B. J Lipid Res. 1988;29:1461–1473. doi: 10.1016/S0022-2275(20)38425-X [DOI] [PubMed] [Google Scholar]
- 23. Phillips ML, Pullinger C, Kroes I, Kroes J, Hardman DA, Chen G, Curtiss LK, Gutierrez MM, Kane JP, Schumaker VN. A single copy of apolipoprotein B‐48 is present on the human chylomicron remnant. J Lipid Res. 1997;38:1170–1177. doi: 10.1016/S0022-2275(20)37199-6 [DOI] [PubMed] [Google Scholar]
- 24. Utermann G. The Metabolic and Molecular Bases of Inherited Disease. In: Scriver C, Beaudet AL, Sly WS, Valle D, eds. Lipoprotein(a). 8th ed. McGraw‐Hill; 2001:2753–2787. [Google Scholar]
- 25. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fatica EM, Meeusen JW, Vasile VC, Jaffe AS, Donato LJ. Measuring the contribution of Lp(a) cholesterol towards LDL‐C interpretation. Clin Biochem. 2020;86:45–51. doi: 10.1016/j.clinbiochem.2020.09.007 [DOI] [PubMed] [Google Scholar]
- 27. Cegla J, France M, Marcovina SM, Neely RDG. Lp(a): When and how to measure it. Ann Clin Biochem. 2021;58:16–21. doi: 10.1177/0004563220968473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Afshar M, Yazdan‐Ashoori S, Engert JC, Thanassoulis G. Drugs for prevention and treatment of aortic stenosis: how close are we? Can J Cardiol. 2021;37:1016–1026. doi: 10.1016/j.cjca.2021.02.017 [DOI] [PubMed] [Google Scholar]
- 29. Park CY, Park J‐Y, Choi J, Kim DJ, Park KS, Yoon K‐H, Lee M‐K, Park SW. Increased postprandial apolipoprotein B‐48 level after a test meal in diabetic patients: a multicenter, cross‐sectional study. Metab Clin Exp. 2016;65:843–851. doi: 10.1016/j.metabol.2016.02.008 [DOI] [PubMed] [Google Scholar]
- 30. Durrington PN, Bolton CH, Hartog M. Serum and lipoprotein apolipoprotein B levels in normal subjects and patients with hyperlipoproteinaemia. Clin Chim Acta. 1978;82:151–160. doi: 10.1016/0009-8981(78)90038-4 [DOI] [PubMed] [Google Scholar]
- 31. Sniderman A, Vu H, Cianflone K. Effect of moderate hypertriglyceridemia on the relation of plasma total and LDL apo B levels. Atherosclerosis. 1991;89:109–116. doi: 10.1016/0021-9150(91)90050-D [DOI] [PubMed] [Google Scholar]
- 32. Sniderman AD, De Graaf J, Thanassoulis G, Tremblay AJ, Martin SS, Couture P. The spectrum of type III hyperlipoproteinemia. J Clin Lipidol. 2018;12:1383–1389. doi: 10.1016/j.jacl.2018.09.006 [DOI] [PubMed] [Google Scholar]
- 33. Xiao C, Dash S, Morgantini C, Lewis GF. Intravenous glucose acutely stimulates intestinal lipoprotein secretion in healthy humans. Arterioscler Thromb Vasc Biol. 2016;36:1457–1463. doi: 10.1161/ATVBAHA.115.307044 [DOI] [PubMed] [Google Scholar]
- 34. Mori K, Ishida T, Yasuda T, Monguchi T, Sasaki M, Kondo K, Hasokawa M, Nakajima H, Haraguchi Y, Sun L, et al. Fasting serum concentration of apolipoprotein B48 represents residual risks in patients with new‐onset and chronic coronary artery disease. Clin Chim Acta. 2013;421:51–56. doi: 10.1016/j.cca.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 35. Sniderman AD, Couture P, Martin SS, DeGraaf J, Lawler PR, Cromwell WC, Wilkins JT, Thanassoulis G. Hypertriglyceridemia and cardiovascular risk: a cautionary note about metabolic confounding. J Lipid Res. 2018;59:1266–1275. doi: 10.1194/jlr.R082271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marais D. Dysbetalipoproteinemia: an extreme disorder of remnant metabolism. Curr Opin Lipidol. 2015;26:292–297. doi: 10.1097/MOL.0000000000000192 [DOI] [PubMed] [Google Scholar]
- 37. Olofsson S‐O, Borén J. Apolipoprotein B secretory regulation by degradation. Arterioscler Thromb Vasc Biol. 2012;32:1334–1338. doi: 10.1161/ATVBAHA.112.251116 [DOI] [PubMed] [Google Scholar]
- 38. Ginsberg HN, Fisher EA. The ever‐expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50:S162–S166. doi: 10.1194/jlr.R800090-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olofsson SO, Asp L, Boren J. The assembly and secretion of apolipoprotein B‐containing lipoproteins. Curr Opin Lipidol. 1999;10:341–346. doi: 10.1097/00041433-199908000-00008 [DOI] [PubMed] [Google Scholar]
- 40. Sniderman AD, Cianflone K. Substrate delivery as a determinant of hepatic apoB secretion. Arterioscler Thromb. 1993;13:629–636. doi: 10.1161/01.ATV.13.5.629 [DOI] [PubMed] [Google Scholar]
- 41. Sniderman AD, Cianflone K, Arner P, Summers LK, Frayn KN. The adipocyte, fatty acid trapping, and atherogenesis. Arterioscler Thromb Vasc Biol. 1998;18:147–151. doi: 10.1161/01.ATV.18.2.147 [DOI] [PubMed] [Google Scholar]
- 42. Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro‐Paavonen A, Westerbacka J, Vehkavaara S, Häkkinen A, Olofsson SO, Yki‐Järvinen H, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49:755–765. doi: 10.1007/s00125-005-0125-z [DOI] [PubMed] [Google Scholar]
- 43. Teng B, Sniderman AD, Soutar AK, Thompson GR. Metabolic basis of hyperapobetalipoproteinemia. Turnover of apolipoprotein B in low density lipoprotein and its precursors and subfractions compared with normal and familial hypercholesterolemia. J Clin Invest. 1986;77:663–672. doi: 10.1172/JCI112360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ying Q, Chan DC, Barrett PHR, Watts GF. Unravelling lipoprotein metabolism with stable isotopes: tracing the flow. Metab Clin Exp. 2021;124:154887. doi: 10.1016/j.metabol.2021.154887 [DOI] [PubMed] [Google Scholar]
- 45. Adiels M, Borén J, Caslake MJ, Stewart P, Soro A, Westerbacka J, Wennberg B, Olofsson S‐O, Packard C, Taskinen M‐R. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2005;25:1697–1703. doi: 10.1161/01.ATV.0000172689.53992.25 [DOI] [PubMed] [Google Scholar]
- 46. Chait A, Foster DM, Albers JJ, Failor RA, Brunzell JD. Low density lipoprotein metabolism in familial combined hyperlipidemia and familial hypercholesterolemia: kinetic analysis using an integrated model. Metab Clin Exp. 1986;35:697–704. doi: 10.1016/0026-0495(86)90236-2 [DOI] [PubMed] [Google Scholar]
- 47. Kissebah AH, Alfarsi S, Adams PW. Integrated regulation of very low density lipoprotein triglyceride and apolipoprotein‐B kinetics in man: normolipemic subjects, familial hypertriglyceridemia and familial combined hyperlipidemia. Metab Clin Exp. 1981;30:856–868. doi: 10.1016/0026-0495(81)90064-0 [DOI] [PubMed] [Google Scholar]
- 48. Cortner JA, Coates PM, Bennett MJ, Cryer DR, Le NA. Familial combined hyperlipidaemia: use of stable isotopes to demonstrate overproduction of very low‐density lipoprotein apolipoprotein B by the liver. J Inherit Metab Dis. 1991;14:915–922. doi: 10.1007/BF01800473 [DOI] [PubMed] [Google Scholar]
- 49. Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–1568. doi: 10.1172/JCI107332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Graaf J, van der Vleuten G, Stalenhoef AFH. Diagnostic criteria in relation to the pathogenesis of familial combined hyperlipidemia. Semin Vasc Med. 2004;4:229–240. doi: 10.1055/s-2004-861490 [DOI] [PubMed] [Google Scholar]
- 51. Sniderman AD, Castro Cabezas M, Ribalta J, Carmena R, de Bruin TWA, de Graaf J, Erkelens DW, Humphries SE, Masana L, Real JT, et al. A proposal to redefine familial combined hyperlipidaemia ‐‐ third workshop on FCHL held in Barcelona from 3 to 5 May 2001, during the scientific sessions of the European Society for Clinical Investigation. 2002; 32:71–73. [DOI] [PubMed] [Google Scholar]
- 52. Adiels M, Packard C, Caslake MJ, Stewart P, Soro A, Westerbacka J, Wennberg B, Olofsson S‐O, Taskinen M‐R, Borén J. A new combined multicompartmental model for apolipoprotein B‐100 and triglyceride metabolism in VLDL subfractions. J Lipid Res. 2005;46:58–67. doi: 10.1194/jlr.M400108-JLR200 [DOI] [PubMed] [Google Scholar]
- 53. Khalil YA, Rabès J‐P, Boileau C, Varret M. APOE gene variants in primary dyslipidemia. Atherosclerosis. 2021;328:11–22. doi: 10.1016/j.atherosclerosis.2021.05.007 [DOI] [PubMed] [Google Scholar]
- 54. Borén J, Packard CJ, Taskinen M‐R. The roles of ApoC‐III on the metabolism of triglyceride‐rich lipoproteins in humans. Front Endocrinol (Lausanne). 2020;11:474. doi: 10.3389/fendo.2020.00474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Williams KJ. Molecular processes that handle — and mishandle — dietary lipids. J Clin Invest. 2008;118:3247–3259. doi: 10.1172/JCI35206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sniderman AD, Zhang XJ, Cianflone K. Governance of the concentration of plasma LDL: a reevaluation of the LDL receptor paradigm. Atherosclerosis. 2000;148:215–229. doi: 10.1016/S0021-9150(99)00282-8 [DOI] [PubMed] [Google Scholar]
- 57. Williams KJ, Brocia RW, Fisher EA. The unstirred water layer as a site of control of apolipoprotein B secretion. J Biol Chem. 1990;265:16741–16744. doi: 10.1016/S0021-9258(17)44822-8 [DOI] [PubMed] [Google Scholar]
- 58. Chait A, Hazzard WR, Albers JJ, Kushwaha RP, Brunzell JD. Impaired very low density lipoprotein and triglyceride removal in broad beta disease: comparison with endogenous hypertriglyceridemia. Metab Clin Exp. 1978;27:1055–1066. doi: 10.1016/0026-0495(78)90151-8 [DOI] [PubMed] [Google Scholar]
- 59. Benn M, Nordestgaard BG, Jensen JS, Nilausen K, Meinertz H, Tybjaerg‐Hansen A. Mutation in apolipoprotein B associated with hypobetalipoproteinemia despite decreased binding to the low density lipoprotein receptor. J Biol Chem. 2005;280:21052–21060. doi: 10.1074/jbc.M413877200 [DOI] [PubMed] [Google Scholar]
- 60. Reeskamp LF, Millar JS, Wu L, Jansen H, van Harskamp D, Schierbeek H, Gipe DA, Rader DJ, Dallinga‐Thie GM, Hovingh GK, et al. ANGPTL3 inhibition with evinacumab results in faster clearance of IDL and LDL apoB in patients with homozygous familial hypercholesterolemia‐brief report. Arterioscler Thromb Vasc Biol. 2021;41:1753–1759. doi: 10.1161/ATVBAHA.120.315204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marsh JB, Welty FK, Lichtenstein AH, Lamon‐Fava S, Schaefer EJ. Apolipoprotein B metabolism in humans: studies with stable isotope‐labeled amino acid precursors. Atherosclerosis. 2002;162:227–244. doi: 10.1016/S0021-9150(01)00709-2 [DOI] [PubMed] [Google Scholar]
- 62. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sniderman AD, Glavinovic T, Thanassoulis G. Key questions about familial hypercholesterolemia. J Am Coll Cardiol. 2022;79:1023–1031. doi: 10.1016/j.jacc.2022.01.010 [DOI] [PubMed] [Google Scholar]
- 64. Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34:1637–1659. doi: 10.1016/S0022-2275(20)35728-X [DOI] [PubMed] [Google Scholar]
- 65. Dietschy JM, Spady DK, Stange EF. Quantitative importance of different organs for cholesterol synthesis and low‐density‐lipoprotein degradation. Biochem Soc Trans. 1983;11:639–641. doi: 10.1042/bst0110639 [DOI] [PubMed] [Google Scholar]
- 66. Meddings JB, Dietschy JM. Regulation of plasma levels of low‐density lipoprotein cholesterol: interpretation of data on low‐density lipoprotein turnover in man. Circulation. 1986;74:805–814. doi: 10.1161/01.CIR.74.4.805 [DOI] [PubMed] [Google Scholar]
- 67. Zulewski H, Ninnis R, Miserez AR, Baumstark MW, Keller U. VLDL and IDL apolipoprotein B‐100 kinetics in familial hypercholesterolemia due to impaired LDL receptor function or to defective apolipoprotein B‐100. J Lipid Res. 1998;39:380–387. doi: 10.1016/S0022-2275(20)33899-2 [DOI] [PubMed] [Google Scholar]
- 68. Bilheimer DW, Stone NJ, Grundy SM. Metabolic studies in familial hypercholesterolemia. Evidence for a gene‐dosage effect in vivo. J Clin Invest. 1979;64:524–533. doi: 10.1172/JCI109490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pittman RC, Carew TE, Attie AD, Witztum JL, Watanabe Y, Steinberg D. Receptor‐dependent and receptor‐independent degradation of low density lipoprotein in normal rabbits and in receptor‐deficient mutant rabbits. J Biol Chem. 1982;257:7994–8000. doi: 10.1016/S0021-9258(18)34287-X [DOI] [PubMed] [Google Scholar]
- 70. Carew TE, Pittman RC, Steinberg D. Tissue sites of degradation of native and reductively methylated [14C]sucrose‐labeled low density lipoprotein in rats. Contribution of receptor‐dependent and receptor‐independent pathways. J Biol Chem. 1982;257:8001–8008. doi: 10.1016/S0021-9258(18)34288-1 [DOI] [PubMed] [Google Scholar]
- 71. Meddings JB, Spady DK, Dietschy JM. Kinetic characteristics and mechanisms of regulation of receptor‐dependent and receptor‐independent LDL transport in the liver of different animal species and humans. Am Heart J. 1987;113:475–481. doi: 10.1016/0002-8703(87)90617-X [DOI] [PubMed] [Google Scholar]
- 72. Sniderman AD, De Graaf J, Couture P, Williams K, Kiss RS, Watts GF. Regulation of plasma LDL: the apoB paradigm. Clin Sci (Lond). 2009;118:333–339. doi: 10.1042/CS20090402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Millar JS, Maugeais C, Ikewaki K, Kolansky DM, Barrett PHR, Budreck EC, Boston RC, Tada N, Mochizuki S, Defesche JC, et al. Complete deficiency of the low‐density lipoprotein receptor is associated with increased apolipoprotein B‐100 production. Arterioscler Thromb Vasc Biol. 2005;25:560–565. doi: 10.1161/01.ATV.0000155323.18856.a2 [DOI] [PubMed] [Google Scholar]
- 74. Tremblay AJ, Lamarche B, Ruel IL, Hogue J‐C, Bergeron J, Gagné C, Couture P. Increased production of VLDL apoB‐100 in subjects with familial hypercholesterolemia carrying the same null LDL receptor gene mutation. J Lipid Res. 2004;45:866–872. doi: 10.1194/jlr.M300448-JLR200 [DOI] [PubMed] [Google Scholar]
- 75. Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43:1363–1379. doi: 10.1194/jlr.R200004-JLR200 [DOI] [PubMed] [Google Scholar]
- 76. Nurmohamed NS, Ditmarsch M, Kastelein JJP. CETP‐inhibitors: from HDL‐C to LDL‐C lowering agents? Cardiovasc Res 2021. Epub ahead of print. PMID: 34849601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Griffin BA, Freeman DJ, Tait GW, Thomson J, Caslake MJ, Packard CJ, Shepherd J. Role of plasma triglyceride in the regulation of plasma low density lipoprotein (LDL) subfractions: relative contribution of small, dense LDL to coronary heart disease risk. Atherosclerosis. 1994;106:241–253. doi: 10.1016/0021-9150(94)90129-5 [DOI] [PubMed] [Google Scholar]
- 78. Watson TD, Caslake MJ, Freeman DJ, Griffin BA, Hinnie J, Packard CJ, Shepherd J. Determinants of LDL subfraction distribution and concentrations in young normolipidemic subjects. Arterioscler Thromb. 1994;14:902–910. doi: 10.1161/01.ATV.14.6.902 [DOI] [PubMed] [Google Scholar]
- 79. Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins‐‐an integrated approach to mechanisms and disorders. N Engl J Med 1967;276:34–42contd. [DOI] [PubMed] [Google Scholar]
- 80. Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins‐‐an integrated approach to mechanisms and disorders. N Engl J Med 1967;276: 94– 103contd. [DOI] [PubMed] [Google Scholar]
- 81. Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins‐‐an integrated approach to mechanisms and disorders. N Engl J Med 1967;276:148–156contd. [DOI] [PubMed] [Google Scholar]
- 82. Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins‐‐an integrated approach to mechanisms and disorders. N Engl J Med 1967;276:215– 225contd. [DOI] [PubMed] [Google Scholar]
- 83. Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins–an integrated approach to mechanisms and disorders. N Engl J Med 1967;276:273–281contd. [DOI] [PubMed] [Google Scholar]
- 84. Berberich AJ, Hegele RA. A modern approach to dyslipidemia. Endocr Rev: 2021. Epub ahead of print. PMID: 34676866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sniderman A, Couture P, De Graaf J. Diagnosis and treatment of apolipoprotein B dyslipoproteinemias. Nat Rev Endocrinol. 2010;6:335–346. doi: 10.1038/nrendo.2010.50 [DOI] [PubMed] [Google Scholar]
- 86. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella‐Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2019;139:e1082–e1143. doi: 10.1161/CIR.0000000000000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Raal FJ, Hovingh GK, Catapano AL. Familial hypercholesterolemia treatments: guidelines and new therapies. Atherosclerosis. 2018;277:483–492. doi: 10.1016/j.atherosclerosis.2018.06.859 [DOI] [PubMed] [Google Scholar]
- 88. Dron JS, Wang J, McIntyre AD, Cao H, Hegele RA. The polygenic nature of mild‐to‐moderate hypertriglyceridemia. J Clin Lipidol. 2020;14:28–34.e2. doi: 10.1016/j.jacl.2020.01.003 [DOI] [PubMed] [Google Scholar]
- 89. Gill PK, Hegele RA. Familial combined hyperlipidemia is a polygenic trait. Curr Opin Lipidol. 2022;33:126–132. doi: 10.1097/MOL.0000000000000796 [DOI] [PubMed] [Google Scholar]
- 90. Brunzell JD, Albers JJ, Chait A, Grundy SM, Groszek E, McDonald GB. Plasma lipoproteins in familial combined hyperlipidemia and monogenic familial hypertriglyceridemia. J Lipid Res. 1983;24:147–155. doi: 10.1016/S0022-2275(20)38008-1 [DOI] [PubMed] [Google Scholar]
- 91. Chait A, Albers JJ, Brunzell JD. Very low density lipoprotein overproduction in genetic forms of hypertriglyceridaemia. Eur J Clin Invest. 1980;10:17–22. doi: 10.1111/j.1365-2362.1980.tb00004.x [DOI] [PubMed] [Google Scholar]
- 92. Brunzell JD, Schrott HG, Motulsky AG, Bierman EL. Myocardial infarction in the familial forms of hypertriglyceridemia. Metab Clin Exp. 1976;25:313–320. doi: 10.1016/0026-0495(76)90089-5 [DOI] [PubMed] [Google Scholar]
- 93. Austin MA, McKnight B, Edwards KL, Bradley CM, McNeely MJ, Psaty BM, Brunzell JD, Motulsky AG. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: a 20‐year prospective study. Circulation. 2000;101:2777–2782. doi: 10.1161/01.CIR.101.24.2777 [DOI] [PubMed] [Google Scholar]
- 94. Veerkamp MJ, De Graaf J, Hendriks JCM, Demacker PNM, Stalenhoef AFH. Nomogram to diagnose familial combined hyperlipidemia on the basis of results of a 5‐year follow‐up study. Circulation. 2004;109:2980–2985. doi: 10.1161/01.CIR.0000130646.93255.86 [DOI] [PubMed] [Google Scholar]
- 95. Ayyobi AF, McGladdery SH, McNeely MJ, Austin MA, Motulsky AG, Brunzell JD. Small, dense LDL and elevated apolipoprotein B are the common characteristics for the three major lipid phenotypes of familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2003;23:1289–1294. doi: 10.1161/01.ATV.0000077220.44620.9B [DOI] [PubMed] [Google Scholar]
- 96. Brouwers MCGJ, De Graaf J, Simons N, Meex S, Doeschate Ten S, van Heertum S, Heidemann B, Luijten J, de Boer D, Schaper N, et al. Incidence of type 2 diabetes in familial combined hyperlipidemia. BMJ Open Diabetes Res Care. 2020;8:e001107. doi: 10.1136/bmjdrc-2019-001107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sniderman AD, Scantlebury T, Cianflone K. Hypertriglyceridemic hyperapob: the unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus. Ann Intern Med. 2001;135:447–459. doi: 10.7326/0003-4819-135-6-200109180-00014 [DOI] [PubMed] [Google Scholar]
- 98. Sagris M, Antonopoulos AS, Theofilis P, Oikonomou E, Siasos G, Tsalamandris S, Antoniades C, Brilakis ES, Kaski JC, Tousoulis D. Risk factors profile of young and older patients with Myocardial Infarction. Cardiovasc Res 2021; Epub ahead of print. PMID: 34358302. [DOI] [PubMed] [Google Scholar]
- 99. Wiesbauer F, Blessberger H, Azar D, Goliasch G, Wagner O, Gerhold L, Huber K, Widhalm K, Abdolvahab F, Sodeck G, et al. Familial‐combined hyperlipidaemia in very young myocardial infarction survivors (< or =40 years of age). Eur Heart J. 2009;30:1073–1079. doi: 10.1093/eurheartj/ehp051 [DOI] [PubMed] [Google Scholar]
- 100. Gill PK, Dron JS, Berberich AJ, Wang J, McIntyre AD, Cao H, Hegele RA. Combined hyperlipidemia is genetically similar to isolated hypertriglyceridemia. J Clin Lipidol. 2021;15:79–87. doi: 10.1016/j.jacl.2020.11.006 [DOI] [PubMed] [Google Scholar]
- 101. Teng B, Sniderman A, Krauss RM, Kwiterovich PO, Milne RW, Marcel YL. Modulation of apolipoprotein B antigenic determinants in human low density lipoprotein subclasses. J Biol Chem. 1985;260:5067–5072. doi: 10.1016/S0021-9258(18)89180-3 [DOI] [PubMed] [Google Scholar]
- 102. Koopal C, Marais AD, Visseren FLJ. Familial dysbetalipoproteinemia: an underdiagnosed lipid disorder. Curr Opin Endocrinol Diabetes Obes. 2017;24:133–139. doi: 10.1097/MED.0000000000000316 [DOI] [PubMed] [Google Scholar]
- 103. Blom DJ, Marais AD, Retterstøl K, Stein EA, Ycas J, Gu Z, Miller E. Rosuvastatin reduces non‐high‐density lipoprotein cholesterol and lipoprotein remnants in patients with dysbetalipoproteinemia (Fredrickson type III hyperlipoproteinemia). J Clin Lipidol. 2008;2:418–425. doi: 10.1016/j.jacl.2008.10.002 [DOI] [PubMed] [Google Scholar]