

Abstract
Background
A beneficial role for prostanoids in hypertension is suggested by clinical studies showing nonsteroidal anti‐inflammatory drugs, which block the production of all prostanoids, cause sodium retention and exacerbate hypertension. Among prostanoids, prostaglandin E2 and its E‐prostanoid receptor 4 receptor (EP4R) have been implicated in blood pressure control. Our previous study found that conditional deletion of EP4R from all tissues in adult mice exacerbates angiotensin II‐dependent hypertension, suggesting a powerful effect of EP4R to resist blood pressure elevation. We also found that elimination of EP4R from vascular smooth muscle cells did not affect the severity of hypertension, suggesting nonvascular targets of prostaglandin E mediate this antihypertensive effect.
Methods and Results
Here we generated mice with cell‐specific deletion of EP4R from macrophage‐specific EP4 receptor knockouts or kidney epithelial cells (KEKO) to assess the contributions of EP4R in these cells to hypertension pathogenesis. Macrophage‐specific EP4 receptor knockouts showed similar blood pressure responses to alterations in dietary sodium or chronic angiotensin II infusion as Controls. By contrast, angiotensin II‐dependent hypertension was significantly augmented in KEKOs (mean arterial pressure: 146±3 mm Hg) compared with Controls (137±4 mm Hg; P=0.02), which was accompanied by impaired natriuresis in KEKOs. Because EP4R expression in the kidney is enriched in the collecting duct, we compared responses to amiloride in angiotensin II‐infused KEKOs and Controls. Blockade of the epithelial sodium channel with amiloride caused exaggerated natriuresis in KEKOs compared with Controls (0.21±0.01 versus 0.15±0.02 mmol/24 hour per 20 g; P=0.015).
Conclusions
Our data suggest EP4R in kidney epithelia attenuates hypertension. This antihypertension effect of EP4R may be mediated by reducing the activity of the epithelial sodium channel, thereby promoting natriuresis.
Keywords: EP4 receptor, hypertension, kidney epithelial cells, prostaglandin E2
Subject Categories: Hypertension, Animal Models of Human Disease, Basic Science Research, Nephrology and Kidney, Physiology
Nonstandard Abbreviations and Acronyms
- Ang II‐HTN
angiotensin II‐dependent hypertension
- CD
collecting duct
- EF
effect size
- EP4R
EP4 receptor
- KEKO
kidney epithelial cell‐specific EP4 receptor knockout
- MϕKO
macrophage‐specific EP4 receptor knockout
Clinical Perspective.
What Is New?
We demonstrated for the first time that the E‐prostanoid receptor 4 in kidney epithelium plays a pivotal role in maintaining sodium balance in angiotensin II‐dependent hypertension.
We described in vivo effects of kidney epithelial E‐prostanoid receptor 4 to control the epithelial sodium channel activity.
We identified a specific molecular pathway by which nonsteroidal anti‐inflammatory drugs can cause sodium retention.
What Are the Clinical Implications?
Our study demonstrated that kidney tubular epithelia are critical targets of prostaglandin E2 acting via E‐prostanoid receptor 4 to oppose the development of hypertension.
Our findings may suggest a specific strategy for treatment for hypertension that develops with the administration of drugs that inhibit the synthesis or actions of prostaglandin E2, such as nonsteroidal anti‐inflammatory drugs or novel E‐prostanoid receptor 4 antagonists.
Uncontrolled hypertension remains a significant unmet medical challenge leading to severe complications such as stroke, kidney disease, and heart failure, resulting in substantial mortality. 1 , 2 Although it is the most common chronic human disease worldwide, the factors that cause essential hypertension are poorly understood. Furthermore, >50% of patients fail to achieve blood pressure (BP) targets with conventional treatments 3 ; thus, identifying new therapeutic targets for hypertension remains a high priority. Historically, physiological pathways causing increased BP, such as the renin‐angiotensin system, have been significant areas of research focus. 4 However, naturally occurring mechanisms counteracting BP elevation are also crucial for determining the severity of hypertension. Although most hypertension therapies target systems that increase BP, preserving and/or augmenting pathways with the potential to lower BP can be complementary approaches for improving BP control.
Prostanoids, produced by the cyclooxygenase (COX) metabolism of arachidonic acid, have significant roles in various biological processes from inflammation to cardiovascular disease. 5 A role for prostanoids in attenuating hypertension has been suggested by clinical studies showing that nonsteroidal anti‐inflammatory drugs, which inhibit COX enzymes and block the production of all prostanoids, are associated with increased risk for hypertension. 6 , 7 Prostaglandin E2 (PGE2) is a highly abundant prostanoid with diverse biological functions mediated by a family of 4 E‐prostanoid (EP) receptors. 8 , 9 Among these receptors, the EP4 receptor (EP4R) has actions potentially affecting BP, including regulating vascular tone, 9 , 10 renin release, 11 , 12 and inflammation. 13 Furthermore, we showed that deletion of EP4R from all tissues in adult mice caused dramatic exacerbated angiotensin II‐dependent hypertension (Ang II‐HTN), 10 demonstrating powerful actions of EP4R to resist BP elevation. However, the cellular targets mediating these antihypertensive effects of EP4R are unclear.
PGE2 is a potent vasodilator, and EP4 has been classically considered a “vaso‐relaxant” receptor 9 based on its preferential coupling to Gs enhancing cAMP formation. 14 Moreover, it has been suggested that compensatory vasodilation might be responsible for the effects of PGE2 to attenuate hypertension. 15 Yet, in previous studies, we found that cell‐specific elimination of EP4R from vascular smooth muscle cells did not affect the severity of Ang II‐HTN. 10 Alternatively, the immune system plays a critical role in hypertension pathogenesis 16 and PGE2 is a potent immune modulator. 17 For example, we have previously demonstrated actions of EP4R to inhibit T cell and macrophage functions. 18 Indeed, EP4R deficiency in macrophages has been suggested to promote BP increases with high‐salt feeding. 19 Finally, BP is critically regulated by the kidney through precise control of sodium excretion, and potent actions of PGE2 to induce natriuresis are well documented. 20 Although recent single‐cell RNA sequencing studies have shown that EP4R is enriched explicitly in principal and intercalated cells of the collecting duct (CD), 21 , 22 and previous studies have suggested a role for EP4R in influencing urinary concentration, 23 , 24 effects of EP4R to affect sodium handling by the kidney have not been documented. Our studies in mice lacking EP4R in all tissues demonstrated potent actions of EP4R to resist the development of hypertension. 10 Based on our work and published studies from others, we hypothesized that the antihypertensive effects of EP4 receptors are mediated by the actions in macrophages or kidney epithelial cells. We tested this hypothesis using transgenic mice with EP4R specifically deleted from macrophages or kidney epithelial cells in the studies described here.
Methods
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Experimental Animals
Mouse lines were bred in AAALAC‐accredited animal facilities at the Durham Veterans' Affairs Medical Center. All studies were approved by Duke University and Durham Veterans' Affairs Medical Center Institutional Animal Care and Use Committees and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were housed in temperature‐controlled rooms with 12‐hour light/dark cycles and had free access to standard rodent chow and water unless specified.
The macrophage‐specific EP4 deletion mice (MϕKO) were generated by crossing the Ptger flox/flox mice 10 , 25 with a mouse line harbor nuclear‐localized Cre recombinase inserted into the first coding ATG of the lysozyme 2 gene (LysM‐Cre). 26 The littermate LysM‐Cre − Ptger4 flox/flox mice served as Controls.
Conditional deletion of EP4R in the renal tubular epithelium was achieved by crossing the Ptger4 flox/flox mouse line with Pax8‐rtTA and TetO‐Cre mouse lines. The Pax8‐rtTA mouse line has a Tet‐On tool that allows the expression of genes in renal tubular epithelial cells (kidney tubular epithelial cells). TetO‐Cre mice express Cre bacterial recombinase under the control of the tetracycline‐responsive element provided by Pax8‐rtTA. 27 The generated Pax8‐rtTA + TetO‐Cre + Ptger4 flox/flox mice were fed doxycycline hydrochloride (2 mg/mL with 5% sucrose) in drinking water to induce Cre expression 27 , 28 in kidney tubular epithelial cells and lead to deletion of EP4R specifically in kidney epithelial cells (KEKO). The littermate Pax8‐rtTA − TetO‐Cre + Ptger4 flox/flox mice served as Controls. To validate the cell‐specific expression of Pax8‐rtTA‐TetO‐Cre, we crossed the Pax8‐rtTA + TetO‐Cre + mice with the membrane‐targeted tdTomato/ EGFP (mTmG) reporter mice 29 and mapped the TetO‐Cre + expression in kidney.
All experimental mice are on a C57BL/6 background. We used only male mice because the antihypertension effects of EP4R we demonstrated in our previous work 10 were observed in male mice.
Primary Peritoneal Macrophage
Mice were anesthetized with isoflurane, and the abdominal skin was cut and pulled up to expose the intact peritoneum. Using a 27 g needle 5 mL ice‐cold PBS was injected into the peritoneal cavity. The fluid in the peritoneal cavity was then collected through a 25 g needle after a gentle massage on the peritoneum for ~1 minute. The peritoneal cavity cells were then collected by centrifuge at 4 °C (300g, 7 minutes) and seeded on 6‐well‐plate in a DMEM cell culture medium containing 4.5 g/L glucose and 10% fetal bovine serum. After 1.5 hours, the cells were washed, and any unattached cells were removed. The attached macrophages were cultured in DMEM containing 20% fetal bovine serum overnight and harvested for RNA extraction.
Kidney Tubule Segments Enrichment
To verify the deletion of EP4R in kidney tubule epithelial cells, we isolated kidney tubule segments by sieve. The kidneys were harvested, and the kidney cortex was quickly removed. The remaining kidney tissue was gently pressed down through a 180 μm sieve. The tissue suspension was collected and passed through a 70 μm cell culture filter to remove the remaining glomeruli. The tissue suspension was then passed through a 40 μm cell culture filter to remove single cells. The tissue that did not pass the 40 μm filter was collected for RNA extraction.
Reverse Transcription Quantitative Polymerase Chain Reaction
Reverse transcription quantitative polymerase chain reaction was used to assess mRNA expression levels. Isoflurane anesthetized mice were perfused with ice‐cold PBS, and kidney tissues were snap frozen in liquid nitrogen and stored at −80 °C for RNA isolation. The macrophages and kidney tubule segments were prepared as described. Total RNA was extracted using DirectZol Kit (Zymo, CA). Reverse transcription was performed using qScript cDNA Supermix (Quanta Biosciences, Gaithersburg, MD), and quantitative polymerase chain reaction was carried out using the SYBR Green PCR Master Mix (BioRad, Hercules, CA). The amount of target gene relative to an endogenous control (GAPDH) was determined by the ΔCT method. The primer sequences are provided in Table 1.
Table 1.
Sequences of Primers for RT‐PCR
| Variable | Forward Primers (5′ → 3′) | Reverse Primers (5′ → 3′) |
|---|---|---|
| EP4 | TCTCTGGTGGTGCTCATCTG | TGCAAATCTGGGTTTCTGCT |
| Renin‐1 | ATGAAGGGGGTGTCTGTGGGGTC | ATGCGGGGAGGGTGGGCACCTG |
| GAPDH | TCACCACCATGGAGAAGGC | GCTAAGCAGTTGGTGGTGCA |
BP Measurements and Ang II Infusion
BP was measured continuously in conscious mice using radiotelemetry as described previously. 10 After telemetry implantation, mice were allowed to recover for 7 days before BP recording started. The BP was collected at baseline for 7 days when mice were fed with regular chow containing 0.4% NaCl, followed by low‐salt (<0.01% NaCl, Envigo TD 90228) and high‐salt (6% NaCl, Envigo TD 30230) diets for 7 days respectively. Then after another 5 days of feeding with regular chow, Ang II (Sigma Aldrich, Saint Louis, MO) was infused subcutaneously using an osmotic mini‐pump (Alzet 2004, Cupertino, CA) at 1000 ng/kg per min as described previously. 10 BP was continuously monitored for 3 (in MϕKO) or 4 (in KEKO) weeks after Ang II infusion was initiated.
Metabolic Balance Study
Separate cohorts of Control (n=8) and KEKO (n=7) mice were individually placed in the metabolic cages 29 (Hatteras Instruments, Cary, NC) for 2 days for the mice to familiarize themselves with the environment. The 24‐hour urine was collected under baseline for 3 days and during Ang II infusion (1000 ng/kg per min) for 12 days. The mice were fed with 10 mg per day gel diet containing nutrients, water, and 0.1% w/w sodium (Nutra‐Gel; Bio‐Serv, Frenchtown, NJ). Urinary sodium content was measured daily using an IL943 Automatic Flame photometer (Instrumentation Laboratory, Lexington, MA). Sodium balance was determined by subtracting the total sodium ingested daily by the total amount of sodium excreted in the urine over 24 hours.
Sodium Excretion Following Amiloride
Mice (n=4/group) were placed in the metabolic cage on day 9 of Ang II infusion. After 2 days of calibration, 24 hours of urine was collected as the baseline. The mice then received an intraperitoneal amiloride injection (4 mg/kg), and 24 hours of urine was collected again. The urine sodium excretion was tested as described. The change in urine sodium excretion from baseline to post‐amiloride was used to calculate the amiloride‐sensitive sodium excretion.
Semiquantitative Immunoblotting
The mouse kidneys were harvested at the end of the metabolic balance study and frozen in liquid nitrogen to measure the sodium transporters' pool sizes. One kidney was used for semiquantitative immunoblotting, as previously described. 30 In brief, whole kidneys were thawed in cold PBS and minced on ice, homogenized in 1 mL of ice‐cold isolation buffer (5% sorbitol buffer, 0.5 mmol/L disodium EDTA, and 5 mmol/L histidine‐imidazole buffer, pH=7.5, with the addition of 0.2 mmol/L phenylmethylsulfonyl fluoride, 9 μg/mL aprotinin, and 5 μL/mL of a phosphatase inhibitor cocktail; Sigma‐Aldrich, MO) for 5 minutes at the lowest setting with an Ultra‐Turrax T25 (IKA‐Labortechnik). Homogenates were centrifuged at 2000g for 10 minutes at 4 °C to remove unbroken cells. Supernatants were retained, and the pellets were rehomogenized in another 1 mL isolation buffer, centrifuged again, and supernatants pooled with the first. Samples were divided into single‐use aliquots, quick‐frozen in liquid nitrogen, and stored at −80°, pending assay. Protein concentrations were determined by BCA assay (Thermo Fisher Scientific, IL). Samples were denatured in Laemmli sample buffer (20 minutes, 60 °C). Uniform protein loading was ascertained by resolving a constant amount of protein from each sample by SDS‐PAGE, gel stained with Coomassie blue, and multiple random bands quantified and assessed by densitometry. For blots, 1X and ½X amounts of protein were assayed to verify the linearity of the detection system, as previously described, 30 , 31 and illustrated in Figure S1. The quantity of protein assayed, antibody resources, dilutions, and incubation times are listed in Table 2. Because each sample was run twice (at 1X and ½X amounts), the normalized values were averaged and mean values compiled for statistical analysis. Signals were detected and quantified with the Odyssey Infrared Imaging System and accompanying software (LI‐COR).
Table 2.
Mouse Immunoblot Details
| Antibody target | ~kDa | μg/lane cortex | Primary ab supplier | Ab host | Dilution | Time | Secondary ab supplier | Host and target | Dilution | Time |
|---|---|---|---|---|---|---|---|---|---|---|
|
(Pro)renin receptor Full‐length cleaved |
~37 ~28 |
40 | Sigma | Rb | 1:1000 | O/N | Invitrogen | GAR 680 | 1:5000 | 1 h |
|
ENaC‐α Full‐length cleaved |
~80 ~20 |
40, 20 |
Loffing (Zurich) |
Rb | 1:5000 | 1 h | Invitrogen | GAR 680 | 1:5000 | 1 h |
| ENaC‐β | ~90 | 40, 20 |
Loffing (Zurich) |
Rb | 1:1500 | O/N | Invitrogen | GAR 680 | 1:5000 | 1 h |
|
ENaC‐γ Full‐length cleaved |
~80 ~60 |
40, 20 |
Loffing (Zurich) |
Rb | 1:1000 | O/N | Invitrogen | GAR 680 | 1:5000 | 1 h |
| NHE3 | ~83 | 40, 20 | McDonough | Rb | 1:2000 | O/N | Invitrogen | GAR 680 | 1:5000 | 1 h |
| NHE3pS552 | ~83 | 5, 2.5 |
Santa Cruz (sc‐53 962) |
Mu | 1:1000 | 2 h | Invitrogen | GAM 680 | 1:5000 | 1 h |
| NKCC2 | 160 | 20, 10 |
DSHB (Iowa) |
Mu | 1:6000 | O/N | LI‐COR | GAM 800 | 1:5000 | 1 h |
| NKCC2p‐ S87 | 160 | 40, 20 |
DSTT (Dundee) |
Sh | 1:2500 | 2 h | Invitrogen | DAS 680 | 1:5000 | 1 h |
Ab indicates antibody; ENAC, epithelial sodium channel; Mu, mouse; NHE3, sodium‐hydrogen antiporter 3; NKCC2, Na‐K‐Cl cotransporter 2; O/N, overnight; Rb, rabbit; and Sh, sheep.
PRR ((Pro)renin receptor) protein levels were measured after 4 weeks of Ang II infusion and BP measurement. 30 , 32 In brief, the kidney medulla will be dissected on ice and homogenized in lysis buffer containing 20 mmol/L HEPES (pH 7.4), 2 mmoL/L EDTA, 0.3 mol/L sucrose, 0.1% SDS, 10 μL/mL Ipegal CA‐630, 10 μL/mL protease inhibitor cocktail, and 10 μL/mL phenylmethylsulfonyl fluoride (all from Sigma). Samples were centrifuged at 12 000g for 5 minutes at 4 °C, and protein content in the supernatant was determined by BCA assay (Thermo Fisher Scientific). Fifty μg of protein was denatured, separated on a 4% to 12% NuPAGE gel (Life Technologies), and transferred to a polyvinylidene difluoride membrane (Thermo Fisher Scientific). The membrane was incubated in blocking buffer (50 mmoL/L Tris, 500 mmoL/L NaCl, 5% nonfat dried milk, and 0.1% Tween 20) for 30 minutes and then with a rabbit anti‐PRR polyclonal antibody (Sigma, Table 2) in blocking buffer overnight at 4°C. After washing, the membrane was incubated with a secondary antibody (Thermo Fisher Scientific). Reaction products were exposed by SuperSignal West Pico Chemiluminescence Substrate kit (Thermo Fisher Scientific), and the signal was detected and analyzed using Bio‐rad Touch Gel Imaging System.
Statistical Analysis
GraphPad v9.0 (GraphPad Software, San Diego, CA) was used for statistical analysis. Comparisons between 2 groups were assessed using the Mann–Whitney test, and data are expressed as median with interquartile range. Multiple comparisons between groups and treatments or between groups and time points were assessed by 2‐way ANOVA followed by Sidak post hoc test, and data are expressed as mean±SEM. The probability value for significance was defined as P<0.05.
Results
BP Responses to High‐Salt Feeding and Chronic Ang II Infusion Are Unaffected in Mice Lacking EP4R in Macrophages
A recent study suggested that eliminating COX2 or EP4R from macrophages promoted salt‐sensitive hypertension, accompanied by increased phosphorylation of the thiazide‐sensitive sodium‐chloride cotransporter. 19 Accordingly, to evaluate whether EP4R in macrophages also contributes to Ang II‐HTN, we generated mice lacking EP4R in macrophages (LysM‐Cre + EP4 flox/flox , MϕKO). Expression of EP4 mRNA in primary peritoneal macrophages was reduced by more than 95% in MϕKos, as shown in Figure S2 (P=0.008; n=5; effect size [EF]: large), confirming the efficient elimination of EP4R expression in macrophages.
To assess the impact of EP4R in macrophages on BP regulation, we measured BP in MϕKOs and Controls using radiotelemetry. As shown in Figure 1A, there were no differences in mean arterial pressures (MAP) between Control and MϕKO mice at baseline (111.8±2 versus 110.2±1.4 mm Hg; P=0.89), during low‐salt (112.5±2.2 versus 110.8±1.4 mm Hg; P=0.9) or high‐salt feeding (114.7±2.1 versus 114.5±1.8 mm Hg; P=1; n=9 and 10/group). Although the MAP change from low‐salt feeding to high‐salt feeding period tends to be higher in MϕKO mice, the differences between the 2 groups are not statistically significant (2.2±0.2 versus 3.7±0.7 mm Hg; P=0.1; n=9 & 10/group; Figure 1B). After a washout period of 5 days with a regular diet, we examined the impact of EP4R‐deficiency in macrophages on BP response to chronic Ang II infusion. As shown in Figure 1C, Control and MϕKO mice had similar MAP during Ang II infusion (average of week 2–3 of Ang II infusion: 139±5 versus 131±3 mm Hg; P=0.23; n=9 and 10). Control and MϕKO mice also showed similar MAP elevation from baseline (washout period) to Ang II infusion (averaged BP of week 2–3 of Ang II infusion) period (24 [10] versus 19 [11] mm Hg; P=0.19; Figure 1D). Additionally, the magnitude of cardiac hypertrophy after Ang II‐HTN was virtually identical in Control and MϕKO mice (7.2 [1.8] versus 6.9 [2.1] mg/g heart‐to‐body weight ratio; P=0.63; Figure 1E), consistent with similar levels of BP elevation in the 2 groups. Thus, our findings suggest that EP4R actions in macrophages do not contribute to the enhanced hypertensive phenotype we observed in whole‐body EP4R deletion mice. 10
Figure 1. Mice lack EP4R in macrophages maintained normal salt sensitivity and BP response to Ang II.
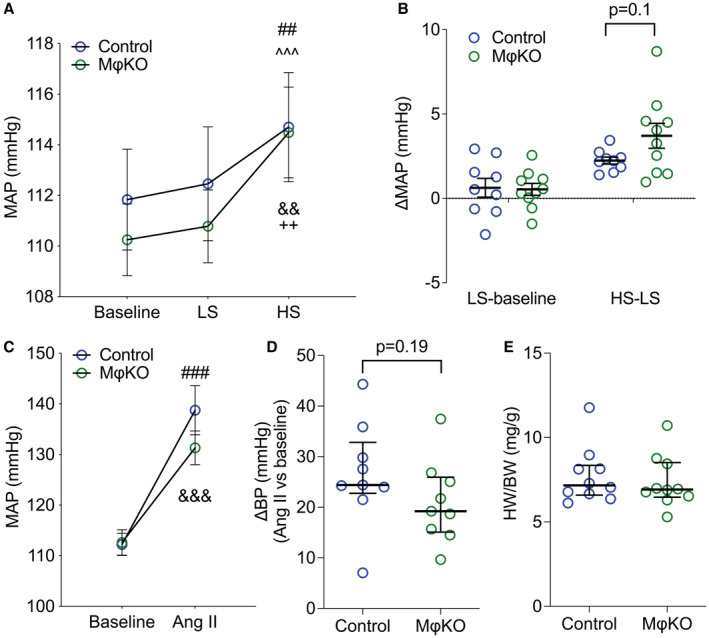
(A) The MϕKOs have similar MAP measured by telemetry at baseline and during low‐ or high‐salt feeding (1 week respectively) compared with Controls. (B) The BP changes from normal to low‐salt feeding and low‐ to high‐salt feeding periods were also similar between genotypes. (C) The mice received Ang II infusion for 3 weeks, and the averaged MAP during the week 2 to 3 of the Ang II infusion period was similar between Control and MϕKO mice. (D) The Ang II‐induced MAP increase tends to be higher in MϕKOs than in Control mice; however, the difference was not statistically significant. (E) The heart weight/body weight ratio was at the same level in Controls and MϕKOs following Ang II infusion. Data in (A through C) are expressed as mean±SEM and analyzed by 2‐way ANOVA (factors: genotype and treatment) with Sidak multiple comparisons tests; data in (D and E) are expressed as median and interquartile range and analyzed by Mann–Whitney test; n=9 to 10/group; ## P<0.01, ### P<0.001 vs Control baseline; ^^^ P<0.001 vs Control LS; && P<0.01, &&& P<0.001 vs KEKO baseline; ++ P<0.01 vs KEKO LS; effect size: trivial for (A), large for (C). Ang II indicates angiotensin II; BP, blood pressure; HS, high salt; HW/BW, heart weight/body weight ratio; KEKO, kidney epithelial cell‐specific EP4 receptor knockout; LS, low salt; MAP, mean arterial pressure; and MϕKO, macrophage‐specific EP4 receptor knockout.
A prior study had reported that deletion of EP4R in macrophages caused exaggerated responses to high‐salt feeding. 19 We also tested the effects of more prolonged exposure to a high‐salt diet (6% NaCl) for 3 weeks on BP in MϕKO and Control mice. As shown in Figure S3, BP response to prolonged high‐salt feeding was similarly unaffected in EP4R MϕKO mice.
Generation of a Mouse Line with Conditional Deletion of EP4R in Renal Tubular Epithelial Cells
Based on our findings of a negligible contribution of EP4R in macrophages to BP regulation, we considered the possibility that EP4R in the kidney may play a role in the well‐established contributions of altered kidney sodium handling to the development of hypertension. 33 Accordingly, we generated a mouse line with conditional EP4R deletion from kidney epithelia across the nephron (Pax8‐rtTA + TetO‐Cre + Ptger4 flox/flox , KEKO). The specificity of inducible Cre expression in kidney epithelia was confirmed using mTmG reporter mice (Figure S4A). To document the degree of EP4R excision in the kidney, EP4R mRNA expression was quantified by real‐time polymerase chain reaction in the kidney cortex and medulla from KEKO and Control mice (Pax8‐rtTA − TetO‐Cre + Ptger4 flox/flox ). After 2 weeks of doxycycline hydrochloride treatment, only ≈ 12% reduction of EP4 expression was seen in the cortex (P=0.48; EF: medium; Figure S4B). As the expression of Pax8‐rtTA + is limited to tubular epithelia, 27 we presume that the failure to observe more substantial levels of EP4R deletion in the cortex is related to high levels of EP4R expression in glomeruli. 21 By contrast, EP4 mRNA levels were significantly reduced in the kidney medulla by ≈50% (P=0.04; n=6 & 4; EF: large; Figure S4C).
To further validate the EP4 deletion from kidney epithelia in KEKOs, we measured the EP4 mRNA expression in enriched kidney tubule segments and found that KEKOs showed ≈ 65% reduction in EP4 expression (P=0.01; n=3–4; EF: large; Figure S4D).
Enhanced Ang II‐HTN in KEKO Mice
We carried out a series of studies to examine the impact of EP4R in kidney epithelia on BP control using KEKO and Control mice. As shown in Figure 2A, BP measured by radiotelemetry were similar in KEKO and Control mice at baseline on a standard (0.4% NaCl) diet (113.4±1.9 versus 113.1±2.2 mm Hg; n=12 & 10). During feeding of low‐salt (<0.01% NaCl) chow, there was an equivalent, minimal reduction in MAP in both groups (Figure 2A and 2B). Although the high‐salt feeding increased MAPs in both Control and KEKOs (EF: trivial), the average MAPs during the high‐salt feeding period (115.5±1.8 versus 116.4±2.4 mm Hg) and the BP change from low‐salt to high‐salt feeding period (3.3±0.6 versus 4.1±1 mm Hg) were similar in Control and KEKO mice (Figure 2A and 2B). Therefore, our data suggest that the elimination of EP4R from kidney tubular epithelia does not affect salt sensitivity.
Figure 2. Mice lacking EP4R in kidney tubular epithelial cells maintain normal baseline BP and salt sensitivity.
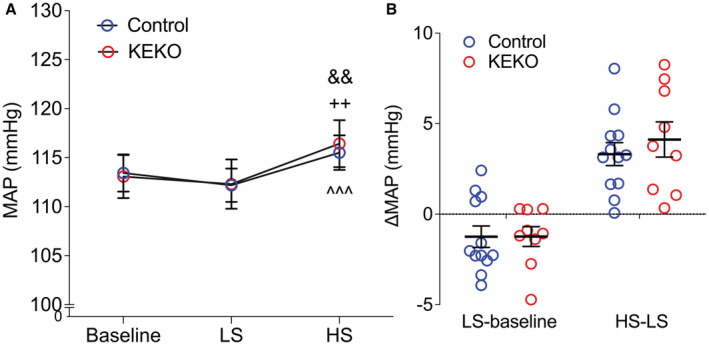
The KEKOs have similar MAP measured by telemetry at baseline and during low‐ or high‐salt feeding (1 week respectively) compared with Controls (A). The BP changes from normal to low‐salt feeding and low‐ to high‐salt feeding periods were also similar between genotypes (B). Data are expressed as mean±SEM and analyzed by 2‐way ANOVA (factors: genotype and treatment) with Sidak multiple comparisons tests; n=9‐12/group; ^^^ P<0.001 vs Control LS (effect size: trivial); && P<0.01 vs KEKO baseline (effect size: trivial); ++ P<0.01 vs KEKO LS (effect size: trivial); BP indicates blood pressure; EP4R, E‐prostanoid receptor 4; HS, high salt; KEKO, kidney epithelial cell‐specific EP4 receptor knockout; LS, low salt; MAP, mean arterial pressure; and NS, normal salt.
We next examined the consequences of eliminating EP4R from kidney epithelium on the development of Ang II‐HTN. As shown in Figure 3A, Ang II caused similar rapid increases in BP in both groups. Yet, after 14 days of Ang II infusion, the BP curves separated, and BPs in KEKOs were consistently higher through the remainder of the Ang II infusion (EF: medium for day 14 and 23, large for other days; Figure 3A). As such, MAP during weeks 3 to 4 of the Ang II infusion period was significantly higher in KEKOs (146±3 mm Hg; n=8) than in Controls (137±4 mm Hg; P=0.02; n=9; EF: large; Figure 3B). Likewise, the change in MAP from baseline averaged over the last 3 weeks of Ang II infusion was also significantly higher in KEKOs (+33 [7] mm Hg) than in Controls (+18 [14] mm Hg; P=0.02; EF: large; Figure 3C). Additionally, cardiac hypertrophy measured as heart‐to‐body‐weight ratio was also significantly higher in the KEKOs (8.5 [1.7] mg/g; n=10) compared with Controls (7.2 [1.7] mg/g; P=0.02; EF: large; n=12; Figure 3D), consistent with a higher level of chronic pressure load in the KEKO mice. 34 Thus, elimination of EP4R from kidney epithelia increases the severity of Ang II‐HTN.
Figure 3. Eliminating EP4R from kidney tubular epithelial cells leads to exaggerated BP response to Ang II.
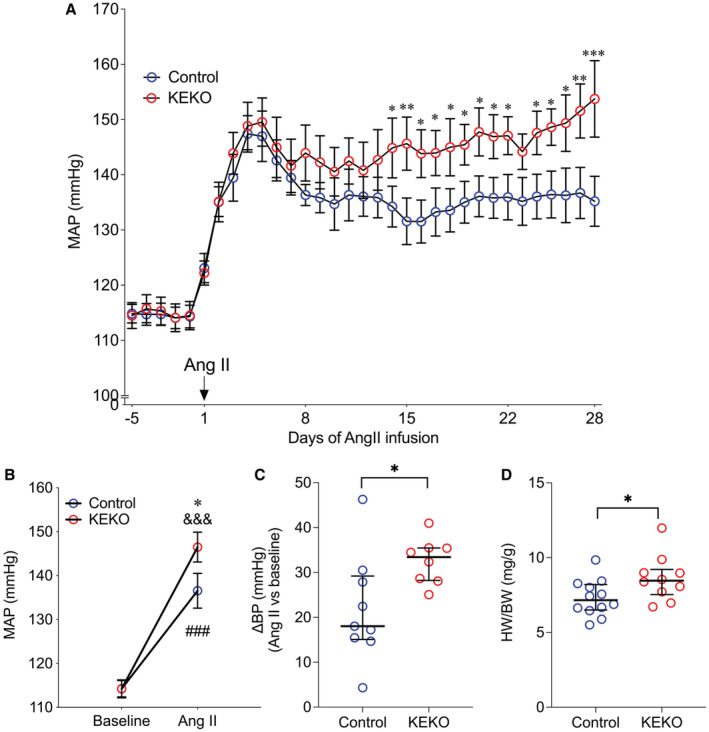
(A) KEKO mice have significantly higher MAP after day 14 of Ang II infusion than Controls. (B) The averaged MAP of week 3 to 4 of Ang II infusion was significantly higher in KEKO mice than Controls. (C) The Ang II‐induced MAP increase was exacerbated significantly in KEKO mice. (D) The heart weight/body weight ratio was significantly higher in KEKO mice than Controls following Ang II infusion. Data in (A and B) are expressed as mean±SEM and analyzed by 2‐way ANOVA (factors: genotype and days for A; genotype and treatment for B) with Sidak multiple comparisons tests; data in (C and D) are expressed as median and interquartile range and analyzed by Mann–Whitney test; *P<0.05, **P<0.01, ***P<0.001 vs Control at the same time point or the same treatment (effect size: medium—large for (A), large for (Bfollowing Ang II infusion through D); ### P<0.001 vs Control baseline (effect size: large); &&& P<0.001 vs KEKO baseline (effect size: large); n=9 to 12/group in (A and B); n=8 to 10/group in (C and D); Ang II indicates angiotensin II; BP, blood pressure; EP4R, E‐prostanoid receptor 4; HW/BW, heart weight/body weight ratio; KEKO, kidney epithelial cell‐specific EP4 receptor knockout; and MAP, mean arterial pressure.
Impaired Natriuresis in KEKOs During Chronic Ang II Infusion
To determine whether alterations in renal sodium handling contributed to augmented Ang II‐HTN in KEKOs, we measured sodium balance during the first 12 days of Ang II infusion by clamping sodium intake and measuring daily urinary sodium excretion as described previously. 29 Notably, the urine production and urine sodium excretion were significantly reduced during the Ang II infusion period compared with baseline in both Control and KEKO mice. Yet, no differences in these parameters were observed at any time points between the groups (Figure S5A and S5B). Additionally, Ang II caused significantly reduced sodium consumption compared with baseline in all the mice. Although KEKO mice tend to ingest more sodium during days 7 to 12 of Ang II infusion than Controls, which was accompanied by moderate‐higher body weight during these days, these differences were not statistically significant (Control versus KEKO during Ang II infusion days 7 to 12: P=0.25 for sodium consumption, EF: large; P=0.85 for body weight, EF: small; Figure S5C and S5D). We then calculated the sodium balance (subtract sodium excretion in urine from the sodium consumption) in the mice. There were no differences in sodium balance between the groups averaged over the first 6 days of Ang II infusion. However, from days 7 to 12 of Ang II infusion, the time when the BP levels first diverged between the 2 groups, sodium retention was significantly higher in KEKOs (+0.2±0.01 mmol/24 hour; n=7) compared with Controls (+0.16±0.01 mmol/24 hour; P=0.02; n=8; EF: large; Figure 4). Thus, enhanced Ang II‐HTN in KEKOs is associated with an impaired pressure‐natriuresis response and sodium retention, indicating that EP4R in kidney epithelia promotes sodium excretion in Ang II‐HTN.
Figure 4. Kidney epithelial cells‐specific EP4R contributes to the regulation of sodium balance during Ang II infusion.
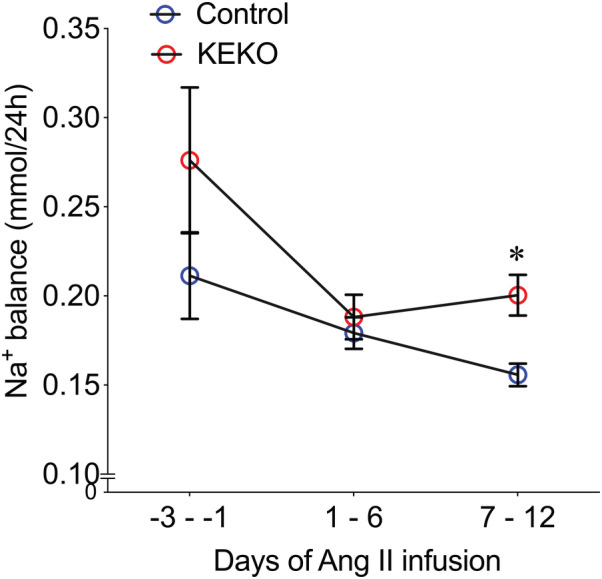
The sodium balance was calculated in Control and KEKO mice before and during the first 12 days of Ang II infusion. From day 7 to 12 of Ang II infusion, sodium balance was significantly higher in KEKOs. Data are expressed as mean±SEM and analyzed by 2‐way ANOVA (factors: genotype and days) with Sidak multiple comparisons tests; *P<0.05 vs Control at the same time point (effect size: large); n=7 to 8/group. Ang II indicates angiotensin II; EP4R, E‐prostanoid receptor 4; and KEKO, kidney epithelial cell‐specific EP4 receptor knockout.
EP4R in Kidney Tubular Epithelia Promote Pressure‐Natriuresis by Inhibiting Epithelial Sodium Channel Function
EP4R expression in the kidney is enriched in CD. 21 , 22 In CD, the major sodium transporter is the epithelial sodium channel (ENaC), which plays a key role in regulating sodium balance and BP. Therefore, we considered the possibility that enhanced ENaC activity might contribute to the exacerbated hypertension in KEKOs. Therefore, we compared the natriuretic actions of the diuretic amiloride, which specifically blocks sodium reabsorption by ENaC, in KEKOs and Controls during Ang II infusion. As shown in Figures 5A and 5B, amiloride caused a significant increase in urine sodium excretion in both Control (~40% increase, P=0.01 versus baseline; EF: large) and KEKO (~60% increase, P=0.0002 versus baseline; EF: large) mice. However, total 24‐hour urinary sodium excretion following amiloride treatment was significantly higher in KEKOs (0.21±0.01 mmol/24 hour per 20 g) compared with Controls (0.15±0.02 mmol/24 hour per 20 g; P=0.015; n=4/group; EF: large). The increase of 24‐hour sodium excretion in response to amiloride compared with baseline was also significantly enhanced in KEKOs (0.08 [0.01] mmol/24 hour/20 g) compared with Controls (0.04 [0.046] mmol/24 hour per 20 g; P=0.013; EF: large; Figure 5C). These data suggest that in chronic Ang II infusion, EP4R signaling in kidney epithelia inhibits ENaC activity thereby promoting natriuresis.
Figure 5. Kidney epithelial cells‐specific EP4 deletion leads to augmented natriuretic response to amiloride during Ang II‐HTN.
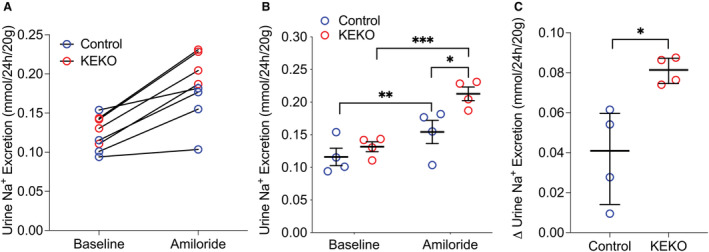
Mice received Ang II infusion at 1000 ng/kg per min. On day 11 of Ang II infusion, 24 hours of urine was collected, and on day 12 of Ang II infusion, mice were given amiloride 4 mg/kg (intraperitoneal.) and 24 hours of urine was collected immediately after the amiloride injection. Following amiloride treatment, the urine sodium excretion significantly increased in both Control and KEKO mice (A and B). However, the total urine sodium excretion (A and B) and the change in urine sodium excretion compared with baseline (C) were significantly higher in KEKOs. Data are expressed as raw value (A), mean±SEM (B), or median with interquartile range (C); (B) is analyzed by 2‐way ANOVA (factors: genotype and treatment) with Sidak multiple comparisons tests; (C) is analyzed by Mann–Whitney test; *P<0.05, **P<0.01; ***P<0.001; effect size: medium for (B) Control baseline vs KEKO baseline, large for all other significant comparisons; n=4/group. Ang II‐HTN indicates angiotensin II‐dependent hypertension; BP, blood pressure; EP4, E‐prostanoid receptor 4; and KEKO, kidney epithelial cell‐specific EP4 receptor knockout.
Sodium Transporter Profiles in Ang II‐HTN
Based on our observation of impaired natriuresis in KEKOs during days 7 to 12 of Ang II infusion, we analyzed key kidney sodium transporters by semiquantitative immunoblotting. 30 Kidneys were harvested for these measurements on day 14 of Ang II infusion, corresponding with the timing of observed differences in BP, sodium handling, and responses to amiloride. There were no differences in protein levels between KEKOs and Controls for most of the major transporters, including NKCC2 (Na‐K‐Cl cotransporter 2) and NKCC2p (Figure S6). Notably, despite the significant differences in amiloride responses, levels of full‐length and cleaved subunits of the epithelial sodium channel—ENaCα, ENaCβ, and ENaCγ—were similar in Controls and KEKOs (Figure 6A through 6F). By contrast, total NHE3 (sodium‐hydrogen antiporter 3) and NHE3pS552, a phosphorylated form of NHE3 sequestered at the base of microvilli in an inactive state, 30 , 35 , 36 are significantly lower ~20% in KEKOs compared with Controls (P=0.009 respectively; n=6/group; EF: large; Figure 6G through 6I). Furthermore, the similar levels of cleaved‐ENaC subunit proteins in KEKOs and Controls suggest that EP4R is likely inhibiting the channel activity.
Figure 6. The protein abundance of ENaC and NHE3 during Ang II‐HTN.
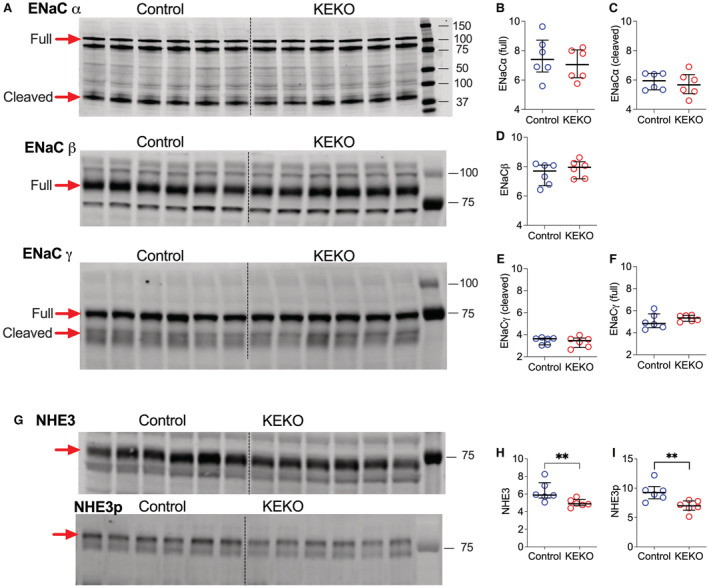
The proteins were extracted after 14 days of Ang II infusion. (A) Gel images of Western blotting for ENaC subtypes. (B through F) Quantification of the western blotting for ENaC subtypes. The full length or cleaved ENaCα, ENaCβ, or ENaCγ proteins in the kidney were similar in Control and KEKO mice. (G) Gel images of western blotting for NHE3 and phosphorylated NHE3 (NHE3pS552). Both NHE3 (H) and NHE3pS552 (I) protein levels were significantly reduced in KEKOs. Data are expressed as median with interquartile range and analyzed by Mann–Whitney test; **P<0.05 (effect size: large); n=6/group; ENaC indicates epithelial sodium channel; KEKO, kidney epithelial cell‐specific EP4 receptor knockout; and NHE3, sodium‐hydrogen antiporter 3.
Reduced PRR Levels in EP4R‐KEKOs
Regulation of renin expression and release represents another potential pathway for BP control by EP4R. 11 For example, our group has previously demonstrated direct effects of EP4R to stimulate renin expression at the juxtaglomerular apparatus. 10 , 11 Therefore, we tested whether kidney epithelial EP4R affected renin expression after 4 weeks of Ang II infusion and found no differences in renin mRNA levels in the kidney cortex (1 [0.9] versus 0.9 [0.6]; P>0.99; n=4) and medulla (1 [0.3] versus 1 [1.3]; P=0.3; n=4) between KEKOs and Controls, respectively. EP4R has also been implicated in regulating the PRR expression in renal medulla. 37 Thus, we measured PRR levels in the kidney medulla of Ang II infused KEKO and Controls by immunoblotting. It has been suggested that increased levels of PRR may contribute to BP elevation during Ang II infusion. 37 Yet, both full‐length and soluble PRR protein levels were significantly lower in KEKOs than in Controls (P=0.009 and 0.004, respectively; EF: large; Figure S7). Therefore, our data suggest that kidney epithelial EP4R is required for the PRR protein expression during Ang II infusion. However, the actions of EP4R to attenuate hypertension do not seem to be mediated by dysregulation of PRR or renin.
Discussion
Prostanoids are lipid mediators with wide‐ranging biological effects generated by COX metabolism of arachidonic acid. 5 Several lines of evidence support a role for prostanoids in human hypertension, including demonstrations of increased risk for hypertension in individuals taking nonsteroidal anti‐inflammatory drugs, 6 , 7 which act by specifically inhibiting COX isoforms and reducing the formation of all prostanoid species. 38 In this regard, a protective role for PGE2 in hypertension has been well documented. 17 , 39 The actions of PGE2 are mediated by 4 distinct EP receptors. 8 , 9 We have previously shown that inducible deletion of the EP4 receptor isoform from all tissues causes dramatic exaggeration of Ang II‐HTN. 10 Because of its preferential coupling to Gs‐proteins linked to enhanced formation of cAMP and relaxation of bronchial and vascular smooth muscle, 40 it seemed likely that the actions of EP4R to mitigate hypertension might depend upon compensatory vasodilation. However, although cell‐specific elimination of EP4R from vascular smooth muscle cells abrogated acute PGE2‐dependent vasodilation, we found no impact on the development of Ang II‐HTN hypertension. 10
Immune and inflammatory cells also make critical contributions to the pathogenesis of hypertension and hypertension‐induced target organ damage. 16 , 41 , 42 , 43 , 44 In this regard, EP4R is the primary EP receptor isoform expressed in myeloid cells, and previous studies from our laboratory have shown that EP4R mediates potent inhibition of macrophage functions. 18 Moreover, macrophages play a role in controlling the deposition of sodium in the skin and salt‐dependent BP homeostasis. 45 , 46 A recent study suggested that deletion of EP4R from macrophages promotes salt‐sensitive hypertension. 19 Accordingly, to understand the contribution of this pathway to Ang II‐HTN, we generated mice with cell‐specific deletion of EP4R from macrophages (MϕKO). Despite efficient (>95%) deletion of EP4R from macrophages, BPs at baseline and with low‐salt or high‐salt feeding were similar in MϕKO and Control mice (Figures 1 and Figure S3). Thus, we find negligible contributions of EP4R in macrophages to BP responses to the high‐salt diet. These results differ from the work of Zhang et al., which suggested a significant impact of macrophage EP4R on salt‐sensitive BP changes. 19 Some differences in experimental protocols between the 2 studies might contribute to the divergent outcomes, including the Cre transgenic lines used to eliminate EP4R from macrophages. We used the LysM‐Cre line, 26 whereas Zhang et al. 19 used the CD11b‐Cre mice, yet both lines achieved efficient deletion of EP4R from macrophages. There were also differences in the salt feeding protocol. We administered a diet with high‐salt content (6% NaCl) for 3 weeks, whereas the diet used by Zhang et al. had an even higher concentration of NaCl (8%) fed for a more extended period of 4 to 6 weeks. 19 Although we are not aware of previous studies addressing the impact of macrophage EP4R in Ang II‐HTN, we did not observe increased severity of hypertension during chronic Ang II infusion in MϕKO (Figure 1).
Because we found no evidence for mitigation of hypertension by EP4R in macrophages, we turned to the kidney as a potentially relevant target tissue. A central role for the kidney in BP control has been well documented, 33 , 47 whereby the kidney controls BP by regulating salt balance and the pressure‐natriuresis response. 48 , 49 , 50 PGE2 is the most abundant prostanoid synthesized in the kidney, 51 , 52 and infusions of PGE2 have been long known to cause natriuresis. 20 Nonetheless, cellular targets of PGE2 in the kidney mediating this natriuretic response are not clear. We generated mice lacking EP4R in kidney epithelium to explore this issue using the well‐characterized Pax8‐Cre line, which expresses broadly in epithelial cell populations across the nephron. 27 Elimination of EP4R from kidney epithelia did not affect baseline BPs or responses to altered dietary sodium content (Figure 2). However, KEKO mice showed significantly exaggerated BP elevation with chronic Ang II infusion and increased cardiac hypertrophy (Figure 3), recapitulating the phenotype we observed in mice lacking EP4R in all tissues. 10 In addition, the more severe hypertension was accompanied by an impaired natriuretic response temporally coinciding with the development of elevated BPs in KEKOs. We found that the KEKO mice showed moderate higher sodium consumption during days 7 to 12 of Ang II infusion, which is not statistically significant compared with Controls (Figure S5). Although the absolute levels of sodium excretion were similar between the 2 groups (Figure S5), the overall sodium balance (consumption of sodium from food subtracted by excreted sodium in the urine) was significantly higher in KEKO mice (Figure 4), suggesting increased sodium retention in the KEKO mice. Thus, our data indicate that kidney epithelium is a significant target for PGE2 acting via EP4R to oppose the development of Ang II‐HTN, and the mechanism of this effect is by promoting enhanced natriuresis. 10
Recent studies using single‐cell sequencing demonstrated that expression of EP4R and PGE synthase‐1 (mPGES1), which synthesizes PGE2 from COX‐derived precursors, 53 are enriched explicitly in kidney CD epithelia. 21 , 22 In addition, we previously found that activation of Ang II receptor type 1 in CD triggers an antihypertensive pathway involving upregulation of COX‐2 in intercalated cells, causing increased production of PGE2 and enhanced natriuresis. 54 ENaC is the major sodium transporter in the CD, where it plays a crucial role in BP control and the development of hypertension. 55 , 56 , 57 , 58 , 59 Furthermore, in vitro studies have suggested a potential for PGE2 to affect ENaC function via autocrine and paracrine mechanisms. 55 , 60 , 61 Therefore, we considered that enhanced ENaC activity might contribute to exaggerated hypertension in KEKOs. To test this point, we compared the functional activity of ENaC in vivo in KEKOs and Controls by measuring responses to amiloride, a diuretic that specifically blocks sodium reabsorption by ENaC. 62 Indeed, natriuretic response to amiloride was more significant by 29% in KEKOs, indicating higher sodium reabsorption by ENaC (Figure 5). Our findings suggest that EP4R promotes natriuresis by inhibiting ENaC activity. These studies are consistent with a local pathway in CD epithelia whereby activation of Ang II receptor type 1 receptors in CD triggers COX2 expression and generation of PGE2, which may act via EP4R to promote natriuresis by inhibiting ENaC and attenuating hypertension. 54 Yet, our study design cannot rule out the possibility that the enhanced natriuresis response to amiloride in KEKO mice was driven by the exacerbated sodium retention but not dependent on ENaC. Further studies using diuretics that are independent of ENaC regulation may be beneficial to verify the mechanisms underlying the amiloride‐induced natriuresis response in KEKO mice.
To further explore this mechanism, we examined the profile of sodium transporter abundance in kidneys during Ang II infusion using well‐established immunoblotting techniques. 30 However, we found no differences between KEKOs and Controls in the quantity of full‐length or cleaved (activated) ENaC subunits α, β, and γ (Figure 6), suggesting that EP4R in kidney epithelia inhibits channel activity without altering the abundance of activated ENaC protein subunits. Indeed, the ENaC function is regulated by very complex mechanisms, including extrinsic and intrinsic factors that affect expression, conductance properties, and intracellular trafficking of the channel. 57 Investigating these mechanisms will involve advanced technical approaches, for example, performing electrophysiology in isolated CD 63 , 64 , 65 or isolating proteins from cell membranes and mitochondria. 66 Although we did not include these studies in the current work because of the technical limitations, it would be interesting for future studies to explore the alternative mechanisms that may contribute to the ENaC regulation by EP4R. We also found no differences in the abundance of other essential sodium transporters, including NKCC2, and NKCC2p in KEKOs (Figure S6), but levels of total NHE3 and phosphorylated NHE3 (NHE3pS552) protein were ≈20% lower in KEKOs (Figure 6). Previous studies have suggested that proportionate, compensatory reductions in NHE3 levels are typically observed as BP increases. 67 , 68 , 69 This smaller NHE3 pool size in KEKO versus Controls may reflect an effect of EP4R KO to lower baseline abundance of NHE3 proteins (independent of Ang II infusion), or greater suppression of NHE3 owing to the higher BP in the KEKO versus Controls during Ang II‐HTN, as part of the pressure‐natriuresis response.
Previous studies using EP4R antagonists and agonists suggested that EP4R regulates the expression of the PRR in the kidney medulla. Medullary PRR is upregulated with Ang II infusion. 37 A series of studies have shown that elimination of PRR from kidney epithelia broadly or specifically in principal cells of the CD attenuates Ang II‐HTN, indicating that increased expression of PRR may promote hypertension in this setting. 70 , 71 , 72 , 73 Moreover, the mechanism of action of elevation of PRR levels to promote hypertension involves increased expression and activity of ENaC. 72 Accordingly, we examined the impact of EP4R deficiency in kidney epithelia on PRR expression. During Ang II infusion, we found that levels of both full‐length and soluble PRR were reduced in KEKO mice (Figure S7), indicating that the effects of EP4R on BP and ENaC activity cannot be explained by effects on PRR expression, as diminished PRR expression in KEKOs would be expected to lower BP.
Conclusions
Taken together, the findings of our study demonstrate that the renal tubular epithelia are critical targets of PGE2 acting via EP4R to oppose the BP elevation. EP4R may resist the development of hypertension through actions in the CD to reduce sodium reabsorption via ENaC.
Our study identifies critical cell targets for PGE2 signaling via the EP4 receptor in kidney epithelium to actively attenuate angiotensin II‐dependent hypertension. Although EP4R in macrophages have been previously implicated in the modulation of BP, they are not involved in this response despite high levels of EP4R expression. Instead, EP4R in kidney epithelia temper the rise in BP during chronic Ang II infusion by promoting natriuresis. Our studies suggest that this enhanced natriuresis is the result of diminished activity of ENaC. Inhibition of this pathway may contribute to sodium retention and BP elevation commonly observed in patients receiving nonsteroidal anti‐inflammatory drugs or novel EP4R antagonists, which are being developed to treat inflammatory disorders. Our findings also suggest that amiloride might help treat hypertension in these settings.
Sources of Funding
This work was supported by the American Society of Nephrology and Kidney Cure Carl W. Gottschalk Research Scholar Award (T.Y.), Duke University Medical Center Department of Medicine Chair Research Award (T.Y.), National Institutes of Health Grant DK105049 (T.M.C.), Clapp Distinguished Professorship Fund (T.M.C.), and the Career Development Award from the Biomedical Laboratory Research and Development Service of the Department of Veterans Affairs Office of Research and Development (M.A.S.).
Disclosures
None.
Supporting information
Figure S1–S7
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.026581
For Sources of Funding and Disclosures, see page 13.
References
- 1. Chobanian A. The hypertension paradox: more uncontrolled disease despite improving therapy. New Engl J Med. 2009;361:991–994. doi: 10.1056/NEJMsa0903829 [DOI] [PubMed] [Google Scholar]
- 2. Lawes C, Vander Hoorn S, Rodgers A, Hypertension ISo . Global burden of blood pressure related disease. Lancet. 2008;371:1513–1518. doi: 10.1016/S0140-6736(08)60655-8 [DOI] [PubMed] [Google Scholar]
- 3. Burnier M, Egan BM. Adherence in hypertension. Circ Res. 2019;124:1124–1140. doi: 10.1161/CIRCRESAHA.118.313220 [DOI] [PubMed] [Google Scholar]
- 4. Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin‐angiotensin system in kidney physiology. Compr Physiol. 2014;4:1201–1228. doi: 10.1002/cphy.c130040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Suppl):S423–S428. doi: 10.1194/jlr.R800094-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Solomon DH, Schneeweiss S, Levin R, Avorn J. Relationship between COX‐2 specific inhibitors and hypertension. Hypertension. 2004;44:140–145. doi: 10.1161/01.HYP.0000136134.31846.83 [DOI] [PubMed] [Google Scholar]
- 7. White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49:408–418. doi: 10.1161/01.HYP.0000258106.74139.25 [DOI] [PubMed] [Google Scholar]
- 8. Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 9. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193 [DOI] [PubMed] [Google Scholar]
- 10. Herrera M, Yang T, Sparks MA, Manning MW, Koller BH, Coffman TM. Complex role for E‐Prostanoid 4 receptors in hypertension. J Am Heart Assoc. 2019;8:e010745. doi: 10.1161/JAHA.118.010745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Facemire CS, Nguyen M, Jania L, Beierwaltes WH, Kim HS, Koller BH, Coffman TM. A major role for the EP4 receptor in regulation of renin. Am J Physiol Renal Physiol. 2011;301:F1035–F1041. doi: 10.1152/ajprenal.00054.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Poschke A, Kern N, Maruyama T, Pavenstadt H, Narumiya S, Jensen BL, Nusing RM. The PGE(2)‐EP4 receptor is necessary for stimulation of the renin‐angiotensin‐aldosterone system in response to low dietary salt intake in vivo. Am J Physiol Renal Physiol. 2012;303:F1435–F1442. doi: 10.1152/ajprenal.00512.2011 [DOI] [PubMed] [Google Scholar]
- 13. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Breyer MD, Breyer RM. G protein‐coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001;63:579–605. doi: 10.1146/annurev.physiol.63.1.579 [DOI] [PubMed] [Google Scholar]
- 15. Yang T, Du Y. Distinct roles of central and peripheral prostaglandin E2 and EP subtypes in blood pressure regulation. Am J Hypertens. 2012;25:1042–1049. doi: 10.1038/ajh.2012.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrison DG, Coffman TM, Wilcox CS. Pathophysiology of hypertension: the mosaic theory and beyond. Circ Res. 2021;128:847–863. doi: 10.1161/CIRCRESAHA.121.318082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23. doi: 10.1172/JCI13416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI13640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase‐2 in hematopoietic cells results in salt‐sensitive hypertension. J Clin Invest. 2015;125:4281–4294. doi: 10.1172/JCI81550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haas JA, Hammond TG, Granger JP, Blaine EH, Knox FG. Mechanism of natriuresis during intrarenal infusion of prostaglandins. Am J Physiol. 1984;247:F475–F479. doi: 10.1152/ajprenal.1984.247.3.F475 [DOI] [PubMed] [Google Scholar]
- 21. Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Susztak K. Single‐cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360:758–763. doi: 10.1126/science.aar2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen L, Chou CL, Knepper MA. A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol. 2021;32:897–912. doi: 10.1681/ASN.2020101406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao M, Cao R, Du S, Jia X, Zheng S, Huang S, Han Q, Liu J, Zhang X, Miao Y, et al. Disruption of prostaglandin E2 receptor EP4 impairs urinary concentration via decreasing aquaporin 2 in renal collecting ducts. Proc Natl Acad Sci USA. 2015;112:8397–8402. doi: 10.1073/pnas.1509565112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang F, Lu X, Peng K, Fang H, Zhou L, Su J, Nau A, Yang KT, Ichihara A, Lu A, et al. Antidiuretic action of collecting duct (pro)renin receptor downstream of vasopressin and PGE2 receptor EP4. J Am Soc Nephrol. 2016;27:3022–3034. doi: 10.1681/ASN.2015050592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gruzdev A, Nguyen M, Kovarova M, Koller BH. PGE2 through the EP4 receptor controls smooth muscle gene expression patterns in the ductus arteriosus critical for remodeling at birth. Prostaglandins Other Lipid Mediat. 2012;97:109–119. doi: 10.1016/j.prostaglandins.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960 [DOI] [PubMed] [Google Scholar]
- 27. Traykova‐Brauch M, Schonig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med. 2008;14:979–984. doi: 10.1038/nm.1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schonig K, Schwenk F, Rajewsky K, Bujard H. Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res 2002;30:e134, 134e, 1134, DOI: 10.1093/nar/gnf134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol. 2015;26:2953–2962. doi: 10.1681/ASN.2014080816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veiras LC, McFarlin BE, Ralph DL, Buncha V, Prescott J, Shirvani BS, McDonough JC, Ha D, Giani J, Gurley SB, et al. Electrolyte and transporter responses to angiotensin II induced hypertension in female and male rats and mice. Acta Physiol. 2020;229:e13448. doi: 10.1111/apha.13448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McDonough AA, Veiras LC, Minas JN, Ralph DL. Considerations when quantitating protein abundance by immunoblot. Am J Physiol Cell Physiol. 2015;308:C426–C433. doi: 10.1152/ajpcell.00400.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen D, Stegbauer J, Sparks MA, Kohan D, Griffiths R, Herrera M, Gurley SB, Coffman TM. Impact of angiotensin type 1A receptors in principal cells of the collecting duct on blood pressure and hypertension. Hypertension. 2016;67:1291–1297. doi: 10.1161/HYPERTENSIONAHA.115.06987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest. 2014;124:2341–2347. doi: 10.1172/JCI72274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kocinsky HS, Dynia DW, Wang T, Aronson PS. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am J Physiol Renal Physiol. 2007;293:F212–F218. doi: 10.1152/ajprenal.00042.2007 [DOI] [PubMed] [Google Scholar]
- 36. Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo‐Branco RC, Pastor‐Soler N, Arranz CT, Yu ASL, et al. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol. 2017;28:3504–3517. doi: 10.1681/ASN.2017030295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A, Yang T. Prostaglandin E‐prostanoid4 receptor mediates angiotensin II‐induced (pro)renin receptor expression in the rat renal medulla. Hypertension. 2014;64:369–377. doi: 10.1161/HYPERTENSIONAHA.114.03654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Francois H, Coffman TM. Prostanoids and blood pressure: which way is up? J Clin Invest. 2004;114:757–759. doi: 10.1172/JCI22929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Facemire CS, Griffiths R, Audoly LP, Koller BH, Coffman TM. The impact of microsomal prostaglandin e synthase 1 on blood pressure is determined by genetic background. Hypertension. 2010;55:531–538. doi: 10.1161/HYPERTENSIONAHA.109.145631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010–1052. doi: 10.1124/pr.112.007195 [DOI] [PubMed] [Google Scholar]
- 41. Schiffrin EL. The immune system: role in hypertension. Can J Cardiol. 2013;29:543–548. doi: 10.1016/j.cjca.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 42. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, et al. Interleukin‐17A regulates renal sodium transporters and renal injury in angiotensin II‐induced hypertension. Hypertension. 2016;68:167–174. doi: 10.1161/HYPERTENSIONAHA.116.07493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end‐organ damage. Circ Res. 2015;116:1022–1033. doi: 10.1161/CIRCRESAHA.116.303697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Muller DN, Derer W, et al. Macrophages regulate salt‐dependent volume and blood pressure by a vascular endothelial growth factor‐C‐dependent buffering mechanism. Nat Med. 2009;15:545–552. doi: 10.1038/nm.1960 [DOI] [PubMed] [Google Scholar]
- 46. Wiig H, Schroder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, Boschmann M, Goss J, Bry M, Rakova N, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. 2013;123:2803–2815. doi: 10.1172/JCI60113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, Stegbauer J, Jin H, Gomez JA, Buckley AF, et al. Type 1 angiotensin receptors on macrophages ameliorate IL‐1 receptor‐mediated kidney fibrosis. J Clin Invest. 2014;124:2198–2203. doi: 10.1172/JCI61368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guyton AC. Blood pressure control—Special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193 [DOI] [PubMed] [Google Scholar]
- 49. Aperia AC, Broberger CG, Soderlund S. Relationship between renal artery perfusion pressure and tubular sodium reabsorption. Am J Physiol. 1971;220:1205–1212. doi: 10.1152/ajplegacy.1971.220.5.1205 [DOI] [PubMed] [Google Scholar]
- 50. Coffman TM, Crowley SD. Kidney in hypertension: guyton redux. Hypertension. 2008;51:811–816. doi: 10.1161/HYPERTENSIONAHA.105.063636 [DOI] [PubMed] [Google Scholar]
- 51. Jensen BL, Stubbe J, Hansen PB, Andreasen D, Skott O. Localization of prostaglandin E(2) EP2 and EP4 receptors in the rat kidney. Am J Physiol Renal Physiol. 2001;280:F1001–F1009. doi: 10.1152/ajprenal.2001.280.6.F1001 [DOI] [PubMed] [Google Scholar]
- 52. Bonvalet JP, Pradelles P, Farman N. Segmental synthesis and actions of prostaglandins along the nephron. Am J Physiol. 1987;253:F377–F387. doi: 10.1152/ajprenal.1987.253.3.F377 [DOI] [PubMed] [Google Scholar]
- 53. Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione‐dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stegbauer J, Chen D, Herrera M, Sparks MA, Yang T, Konigshausen E, Gurley SB, Coffman TM. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight. 2017;2:e92720. doi: 10.1172/jci.insight.92720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol. 2015;10:135–146. doi: 10.2215/CJN.05760513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. 2005;16:3154–3159. doi: 10.1681/ASN.2005050460 [DOI] [PubMed] [Google Scholar]
- 57. Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol. 2008;19:1845–1854. doi: 10.1681/ASN.2008020225 [DOI] [PubMed] [Google Scholar]
- 58. Shimkets RA, Warnock DG, Bositis CM, Nelson‐Williams C, Hansson JH, Schambelan M, Gill JR Jr, Ulick S, Milora RV, Findling JW, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x [DOI] [PubMed] [Google Scholar]
- 59. Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson‐Williams C, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248 [DOI] [PubMed] [Google Scholar]
- 60. Gueutin V, Vallet M, Jayat M, Peti‐Peterdi J, Corniere N, Leviel F, Sohet F, Wagner CA, Eladari D, Chambrey R. Renal beta‐intercalated cells maintain body fluid and electrolyte balance. J Clin Invest. 2013;123:4219–4231. doi: 10.1172/JCI63492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mansley MK, Niklas C, Nacken R, Mandery K, Glaeser H, Fromm MF, Korbmacher C, Bertog M. Prostaglandin E2 stimulates the epithelial sodium channel (ENaC) in cultured mouse cortical collecting duct cells in an autocrine manner. J Gen Physiol. 2020;152. doi: 10.1085/jgp.201912525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone‐dependent and ‐independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol. 2012;303:F1289–F1299. doi: 10.1152/ajprenal.00247.2012 [DOI] [PubMed] [Google Scholar]
- 63. Prieto MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, McLellan M, Gentile O, Jensen VB, Ichihara A, et al. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II‐induced hypertensive mice. Am J Physiol Renal Physiol. 2017;313:F1243–F1253. doi: 10.1152/ajprenal.00152.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mamenko M, Zaika O, Tomilin V, Jensen VB, Pochynyuk O. Compromised regulation of the collecting duct ENaC activity in mice lacking AT1a receptor. J Cell Physiol. 2018;233:7217–7225. doi: 10.1002/jcp.26552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mamenko M, Zaika O, Prieto MC, Jensen VB, Doris PA, Navar LG, Pochynyuk O. Chronic angiotensin II infusion drives extensive aldosterone‐independent epithelial Na+ channel activation. Hypertension. 2013;62:1111–1122. doi: 10.1161/HYPERTENSIONAHA.113.01797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ware AW, Rasulov SR, Cheung TT, Lott JS, McDonald FJ. Membrane trafficking pathways regulating the epithelial Na(+) channel. Am J Physiol Renal Physiol. 2020;318:F1–F13. doi: 10.1152/ajprenal.00277.2019 [DOI] [PubMed] [Google Scholar]
- 67. Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II‐dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol. 2013;305:F510–F519. doi: 10.1152/ajprenal.00183.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sparks MA, Dilmen E, Ralph DL, Rianto F, Hoang TA, Hollis A, Diaz EJ, Adhikari R, Chew G, Petretto EG, et al. Vascular control of kidney epithelial transporters. Am J Physiol Renal Physiol. 2021;320:F1080–F1092. doi: 10.1152/ajprenal.00084.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McDonough AA, Nguyen MT. Maintaining balance under pressure: integrated regulation of renal transporters during hypertension. Hypertension. 2015;66:450–455. doi: 10.1161/HYPERTENSIONAHA.115.04593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chan SH, Chan JY. (pro)renin receptor as a therapeutic target for the treatment of hypertension? Hypertension. 2015;65:278–279. doi: 10.1161/HYPERTENSIONAHA.114.04532 [DOI] [PubMed] [Google Scholar]
- 71. Wang F, Lu X, Liu M, Feng Y, Zhou SF, Yang T. Renal medullary (pro)renin receptor contributes to angiotensin II‐induced hypertension in rats via activation of the local renin‐angiotensin system. BMC Med. 2015;13:278. doi: 10.1186/s12916-015-0514-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II‐induced hypertension. Am J Physiol Renal Physiol. 2017;312:F245–F253. doi: 10.1152/ajprenal.00178.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ramkumar N, Kohan DE. The (pro)renin receptor: an emerging player in hypertension and metabolic syndrome. Kidney Int. 2019;95:1041–1052. doi: 10.1016/j.kint.2018.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1–S7