

Abstract
Background
Atrial fibrillation (AF) is the most common and progressive tachyarrhythmia. Diabetes is a common risk factor for AF. Recent research findings revealed that microtubule network disruption underlies AF. The microtubule network mediates the contact between sarcoplasmic reticulum and mitochondria, 2 essential organelles for normal cardiomyocyte function. Therefore, disruption of the microtubule network may impair sarcoplasmic reticulum and mitochondrial contacts (SRMCs) and subsequently cardiomyocyte function. The current study aims to determine whether microtubule‐mediated SRMCs disruption underlies diabetes‐associated AF.
Methods and Results
Tachypacing (mimicking AF) and high glucose (mimicking diabetes) significantly impaired contractile function in HL‐1 cardiomyocytes (loss of calcium transient) and Drosophila (reduced heart rate and increased arrhythmia), both of which were prevented by microtubule stabilizers. Furthermore, both tachypacing and high glucose significantly reduced SRMCs and the key SRMC tether protein mitofusin 2 (MFN2) and resulted in consequent mitochondrial dysfunction, all of which were prevented by microtubule stabilizers. In line with pharmacological interventions with microtubule stabilizers, cardiac‐specific knockdown of MFN2 induced arrhythmia in Drosophila and overexpression of MFN2 prevented tachypacing‐ and high glucose–induced contractile dysfunction in HL‐1 cardiomyocytes and/or Drosophila. Consistently, SRMCs/MFN2 levels were significantly reduced in right atrial appendages of patients with persistent AF compared with control patients, which was aggravated in patients with diabetes.
Conclusions
SRMCs may play a critical role in clinical AF, especially diabetes‐related AF. Furthermore, SRMCs can be regulated by microtubules and MFN2, which represent novel potential therapeutic targets for AF.
Keywords: atrial fibrillation, diabetes, Drosophila, microtubules, mitosusin 2, sarcoplasmic reticulum‐mitochondrial contacts
Subject Categories: Basic Science Research, Atrial Fibrillation, Arrhythmias
Nonstandard Abbreviations and Acronyms
- βOHB
β‐hydroxybutyrate
- CaT
calcium transients
- ER
endoplasmic reticulum
- HG
high glucose
- MAM
mitochondrial‐associated membranes
- NG
normal glucose
- PeAF
persistent AF
- RAA
right atrial appendages
- RT
room temperature
- SR
sarcoplasmic reticulum
- SRMCs
SR and mitochondrial contacts
Clinical Perspective.
What Is New?
We identified a significant loss of sarcoplasmic reticulum and mitochondrial contacts (SRMCs) and the tether protein mitofusin 2 in experimental and clinical atrial fibrillation (AF).
SRMCs and the tether protein mitofusin 2 are reduced in the experimental models for diabetes.
Microtubule stabilizers protect against loss of SRMCs in the experimental models for AF and diabetes.
Pharmacological and genetic preservation of mitofusin 2 protects against cardiomyocyte dysfunction in experimental models of AF and diabetes.
What Are the Clinical Implications?
SRMCs and its tether protein mitofusin 2, are novel druggable targets for AF, especially diabetes‐related AF.
The ketone body β‐hydroxybutyrate can prevent loss of mitofusin 2 and SRMCs in experimental models of AF and diabetes and may be of interest to test in a clinical trial.
Atrial fibrillation (AF) is the most common progressive cardiac tachyarrhythmia in clinical practice with increased cardiovascular morbidity and mortality worldwide, despite some innovative improvements in clinical management. 1 Because of the aging population, AF shows a steep increase in incidence. Its prevalence in the developed world is estimated to be 1.5% to 2% of the general population, and steadily rises in individuals between 75 and 85 years of age. 2 Around 12 million people are estimated to have this condition in the United States by 2050 and 17.9 million people in Europe by 2060. 3 The health care costs of AF treatment are approaching epidemic proportions. Unfortunately, no effective curative therapy exists for the majority of patients with AF. This is because the exact processes underlying AF remain unclear. Therefore, research directed at uncovering mechanisms driving AF is urgently needed.
Well‐known risk factors for AF include aging, hypertension, valvular heart disease, heart failure, and diabetes. Because of the increasing population with obesity, more attention has recently been drawn to diabetes as a risk factor for AF, 1 , 4 , 5 which is a strong and independent risk factor for AF occurrence. 6 Patients diagnosed with diabetes have ≈40% greater risk of developing AF, compared with the nondiabetic population. 7 Moreover, AF in patients with diabetes is associated with a 61% greater risk of all‐cause mortality and higher risks of cardiovascular death, stroke, and heart failure. 8 In addition, hypertensive patients who develop new onset of diabetes have a significantly higher AF incidence and a higher risk of developing persistent AF, suggesting that diabetes may contribute to the onset and progression of AF. 9 However, the precise pathophysiological mechanisms underlying diabetes‐related AF persistence have not been deciphered.
It has been recognized that AF persistence is rooted in the derailment in cardiomyocyte proteostasis (protein expression, function, and clearance). 10 , 11 An important contributing factor for proteostasis derailment is tachypacing‐induced microtubule disruption, which results in contractile dysfunction and increased arrhythmogenesis in experimental models for AF and patients with AF. 12 Previously, we showed that microtubule disruption in AF is mainly attributed to the upstream activation of the deacetylase histone deacetylase 6. 12 However, the downstream mechanisms by which disrupted microtubules regulate cardiomyocyte contractions are still unclear. Sarcoplasmic reticulum (SR) and mitochondria are the 2 central organelles for normal cardiac contractions by controlling Ca2+ and energy (ATP) homeostasis. 13 , 14 , 15 The crosstalk between SR and mitochondria is mediated via SR‐mitochondria contacts (SRMCs), which are critical for normal mitochondrial and cardiac function. 16 Mitofusin 2 (MFN2), a physical tether between mitochondria and SR, is essential for the structure and function of SRMCs. 17 Besides MFN2, functional SRMCs are also highly dependent on an intact microtubule network. 13 , 18 Moreover, microtubules are also associated with dysregulation of mitochondria in experimental and human studies on diabetes and obesity. 19 , 20 However, whether microtubules regulate SRMCs changes in atrial cardiomyocytes and thereby drive (diabetes‐related) AF is unknown. Therefore, this study aims to investigate whether microtubule disruption has a detrimental effect on SRMCs and the function of SRMCs in experimental AF model systems and clinical AF.
METHODS
Availability of Data and Materials
All the data used in this study are available within the article and the supplemental material. The original data and algorithm for quantification are available from the corresponding authors upon reasonable request.
HL‐1 Cardiomyocyte Culture, Drug Treatment, and Calcium Transient Measurements
HL‐1 atrial cardiomyocytes, obtained from the Claycomb Lab (Louisiana State University, New Orleans) and purchased from Sigma (SCC065, Sigma), were seeded in 0.02% gelatin (Sigma) coated cell culture plates, and were cultured in Claycomb medium (Sigma) supplemented with 10% FBS (PAA cell culture company), 100 μg/mL penicillin/streptomycin (Lonza BioWhittaker), 0.1 mmol/L norepinephrine (Sigma), and 2 mmol/L l‐glutamine (Lonza BioWhittaker).
To mimic AF, HL‐1 cardiomyocytes were tachypaced (5 Hz, 40 V, and 20‐ms pulse duration) in 4‐well plates (Thermo Scientific) with a C‐pace100 culture pacer (IonOptix) for 8 hours. Before tachypacing, HL‐1 cardiomyocytes were treated for 12 hours in medium with either vesicle or with microtubule stabilizers including 5 nmol/L taxol (Sigma), 21 10 mmol/L β‐hydroxybutyrate (βOHB; Sigma), 22 , 23 , 24 or 1 μmol/L tubacin (Sigma) 12 to study the role of microtubules on SRMCs and contractile function. For genetic manipulation, cells were co‐transfected using lipofectamine 3000 (Thermo Fisher) with plasmid RFP (Addgene) and either control plasmid pcDNA3.1 (Addgene) or MFN2‐MYC (Addgene) for 48 hours to study the specific role of MFN2. Calcium transients (CaT) were previously found to correlate well with cellular contractility/shortening in HL‐1 cardiomyocytes 25 and are therefore used as a proxy for HL‐1 cellular contractility/shortening. CaT were imaged using a high‐speed confocal microscope (Nikon A1R) as previously described. 26 Briefly, HL‐1 cardiomyocytes were loaded with 2 μmol/L of the Ca2+ sensitive Fluo‐4‐AM dye (Invitrogen), followed by a washing and imaging with excitation at 488 nm and emission at 500 to 550 nm under normal pacing (40 V, 1 Hz, and 20‐ms pulse duration). CaT measurements were performed in a blinded manner by selection of normal‐shaped cardiomyocytes using bright field settings, followed by switching to the fluorescence filter to determine the CaT.
Drosophila Stocks, Tachypacing, and Heart Contraction Assays
The wild‐type Drosophila melanogaster strain w1118 was used for all drug exposure experiments. Male and female adult flies were crossed in tubes with standard food (containing 26 g yeast, 17 g agar, 54 g sucrose, and 1.3 g nipagin per L), and flies were removed from the embryos‐containing tubes after 3 days, and either drug (50 nmol/L taxol or 100 mmol/L βOHB) or the same amount of vehicle (DMSO) were added to the food. Drosophila were incubated at 25 °C for 48 hours, with larvae consuming the drug or vehicle before entering the prepupa stage. The Drosophila prepupae were collected and subjected to tachypacing for 20 minutes (20 V, 4 Hz, 10‐ms pulse duration) and heart wall functions (heart rate and arrhythmia index) were measured as previously described. 26 Contractile dysfunction is defined as a significant reduction in heart rate and increase in arrhythmia.
To create cardiac overexpression or suppression of MFN2 in Drosophila, the GAL4‐UAS system was used. For overexpression, the UAS‐MFN2 UAS Drosophila line (BDSC_59044; Bloomington Drosophila Stock Center) was utilized; for suppression, the MFN2 UAS‐RNAi Drosophila line (VDRC, ID:40478; Vienna Drosophila RNAi Center) was utilized. Both UAS Drosophila lines were crossed with a Hand‐GAL4 driver strain (kind gift of Prof. Dr Achim Paululat, University of Osnabrück, Germany) to manipulate gene expression specifically in the heart. 26 As control, wild‐type files w1118 were crossed with Hand‐GAL4 driver flies. Prepupae of the F1 progeny were used.
High Glucose Treatment of HL‐1 Cardiomyocytes and Drosophila
For the cardiomyocyte high glucose model (HG, mimicking diabetes), HL‐1 cardiomyocytes were incubated with 25 mmol/L glucose d‐(+)‐glucose sugar (Sigma‐Aldrich) in complete Claycomb medium for 48 hours or 72 hours, and then CaT measurement was performed with control (0 mmol/L glucose) and HG‐exposed HL‐1 cardiomyocytes.
For Drosophila, 96 g or 246 g d‐(+)‐glucose (Sigma‐Aldrich) per L were additionally added to standard food to make 150 g/L HG food and 300 g/L HG food, respectively. High sugar diet has been shown to induce insulin resistance that mimics the pathophysiology of type 2 diabetes in humans. 27
Male and female adult flies were crossed in tubes with different doses of HG, and adult flies were removed from the embryos‐containing tubes after 4 days. The embryos were incubated at 25 °C for at least 3 days until developing into the prepupa stage. The prepupae were collected and heart wall functions were measured as previously described. 26
Right Atrial Tissue Samples From Patients
Before surgery, patient characteristics were assessed (Table1) as previously described. 28 Tissue samples from right atrial appendages (RAA) were obtained from patients with no history of AF (sinus rhythm, SiRh) or patients with persistent AF (PeAF) with coronary artery and/or valvular heart disease undergoing open‐heart surgery. After excision, atrial appendage tissue was immediately snap‐frozen in liquid nitrogen and stored at −80 °C. Tissue was used to perform biochemical and immunofluorescent staining assays. Unfortunately, the number of patients for each condition is limited; however, at least 4 samples per group were used for these assays, as indicated in the figure legends. The study conformed to the principles of the Declaration of Helsinki and complied with all relevant ethical regulations. The institutional medical ethical committee approved the study (MEC‐2014‐393) and patients gave written informed consent before inclusion.
Table 1.
Characteristics of Patients for Western Blot and Imaging Analysis (N=30)
| SiRh | SiRh+Diabetes | PeAF | PeAF+Diabetes | |
|---|---|---|---|---|
| RAA (N) | 8 | 7 | 9 | 6 |
| Age, y, mean±SD | 60.2±21.1 | 65.5±8.1 | 67.3±6.5 | 72.9±5.6 |
| BMI, kg/m2, mean±SD | 25.5±3.1 | 31.4±5.4* | 28.1±5.2 | 28.7±4.8 |
| Hypertension (N, %) | 2 (25.0) | 7 (100.0) | 4 (44.4) | 5 (83.3) |
| Male (N, %) | 6 (75.0) | 4 (57.1) | 8 (88.9) | 4 (66.7) |
| Underlying heart disease/surgical procedure (N, %) | ||||
| CHD | 3 (37.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| AVD | 1 (12.5) | 0 (0.0) | 3 (33.3) | 0 (0.0) |
| AVD+CAD | 1 (12.5) | 1 (14.3) | 0 (0,0) | 1 (16.7) |
| MVD | 1 (12.5) | 0 (0.0) | 4 (44.4) | 3 (50.0) |
| CAD | 2 (25) | 6 (85.7) | 1 (11.1) | 1 (16.7) |
| TVD | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) |
| MAZE | 0 (0.0) | 0 (0.0) | 1 (11.1) | 0 (0.0) |
| Medication (N, %) | ||||
| ACE inhibitor | 4 (50.0) | 4 (57.1) | 5 (55.6) | 6 (100.0) |
| Statin | 3 (37.5) | 6 (85.7) | 3 (33.3) | 4 (67.7) |
| Type I AAD | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Type II AAD | 3 (37.5) | 5 (71.4) | 5 (55.6) | 6 (100.0) |
| Type III AAD | 0 (0.0) | 0 (0.0) | 2 (22.2) | 0 (0.0) |
| Type IV AAD | 0 (0.0) | 1 (14.3) | 2 (22.2) | 0 (0.0) |
| Digoxin | 0 (0.0) | 0 (0.0) | 2 (22.2) | 2 (33.3) |
AAD indicates anti‐arrhythmic drug; ACE, angiotensin‐converting enzyme; AVD, aortic valve disease; BMI, body mass index; CAD, coronary artery disease; CHD, congenital heart disease; MAZE, atrial arrhythmia surgery; MVD, mitral valve disease; PeAF, persistent atrial fibrillation; SiRh, sinus rhythm; TVD, tricuspid valve disease.
P<0.05 vs SiRh. Two‐tailed Student t test.
Protein Isolation and Western Blot Analysis
RAA tissue samples or HL‐1 cardiomyocytes were lysed on ice by the radioimmunoprecipitation assay buffer supplemented with protease inhibitor cocktail (Roche), sodium orthovanadate (Sigma), sodium fluoride (Sigma), and β‐mercaptoethanol (Bio‐Rad), followed by shearing of the DNA with a 26G needle and measuring protein concentration with Bradford assay (Bio‐Rad). Next, protein homogenates were separated by 4% to 20% TGX gels (Bio‐Rad), transferred to nitrocellulose membranes. Subsequently, immunodetection was done by incubation with primary antibodies: anti‐MFN2 (1:1000, Cell Signaling, 9482), anti‐MFN1 (1:500, Abcam, ab104274), anti‐SERCA2 (1:1000, Cell Signaling, 4388), anti‐VDAC1 (1:1000, Abcam, ab15895), anti‐TOM20 (1:1000, Santa Cruz, sc17764), and anti‐GAPDH (1:25000, Fitzgerald, 10R‐G109A) overnight at 4 °C in a cold room, followed by a 3‐time wash with 1% Tween 20 in TRIS‐buffered saline solution, and incubation with horseradish peroxidase–conjugated anti‐mouse or anti‐rabbit secondary antibodies (Dako, 1:2000) for 1 hour at room temperature (RT). After washing with 1x TRIS‐buffered saline solution, the intensity of the bands was quantified by using ImageQuant TL (GE Healthcare).
Mitochondrial‐Associated Membranes Isolation
Mitochondrial‐associated membranes (MAM) were isolated according to the protocol from the Wieckowski lab. 29 Briefly, to isolate crude mitochondria from HL‐1 cardiomyocytes, homogenized cells were centrifuged twice at 600g for 5 minutes at 4 °C to remove unbroken cells and nuclei. To obtain a mitochondrial‐enriched pellet, the supernatant was collected and centrifuged at 7000g for 10 minutes at 4 °C. To obtain a MAM fraction, the pellet was resuspended and separated in 8 mL Percoll gradient medium (Sigma‐Aldrich) and centrifuged at 950 00g for 30 minutes at 4 °C. The MAM fraction was collected from the Percoll gradient, resuspended, and centrifuged at 100 000g for 1 hour at 4 °C to get the MAM fraction. See Method S1 for detailed information.
Immunofluorescent Staining and Co‐Localization Quantification
HL‐1 cardiomyocytes were washed with PBS, incubated with 250 nmol/L Mitotracker Deep Red (Invitrogen) for 30 minutes at 37 °C, and washed with PBS. Then HL‐1 cardiomyocytes or RAA tissue samples of patients with SR or AF were fixed with 4% paraformaldehyde (Sigma) at RT for 15 minutes, washed 3 times with 1% Tween 20 in PBS (1x PBS‐T), blocked with 10% goat serum in PBS at RT for 60 minutes, incubated overnight at 4 °C with anti‐SEC61β (1:250, Thermo Fisher, PA3‐015), anti‐acetylated α‐tubulin (1:200, Sigma, T7451), anti‐SERCA2 (1:100, Cell signaling, 4388) or anti‐TOM20 (1:100, Santa Cruz, SC17764, only used for patient samples) primary antibodies diluted in in PBS with 1% BSA (1% PBSA), washed 3 times with 1× PBS‐T, incubated with donkey anti‐mouse (Alexa 488, Invitrogen) and anti‐rabbit (Alexa 555, Invitrogen) in 1% PBSA at RT in the dark for 1 to 2 hours, washed 3 times with 1× PBS‐T at RT, incubated with WGA Alexa 647 (1:50, Invitrogen) in PBS at RT 30 minutes, washed 3 times with 1× PBS‐T, incubated with DAPI (1:1000 in PBS, Invitrogen) at RT for 5 minutes, washed twice with 1× PBS‐T, then washed with PBS. The coverslips were mounted with DABCO (15 μL/sample, Invitrogen), and stored at 4 °C before microscopic analysis. The images were taken with a confocal microscope (Leica TCS SP8 STED). The protein co‐localization was calculated by using Mander's coefficient, analyzed in Image J by using COLOC 2 plugin with a COSTES threshold. 30 , 31
Determination of Oxygen Consumption Rate by Seahorse XF96 Analyzer
HL‐1 cardiomyocytes were cultured to ≈80% confluency, trypsinized, and counted. Forty thousand cells were reseeded per well (coated with 0.02% gelatin) on a Seahorse XF96 96‐well culture plate (Seahorse Bioscience), and then incubated for 16 to 24 hours at 37 °C and 5% CO2. One hour before the measurement, medium was removed and replaced by DMEM (Sigma) containing 25 mmol/L glucose (Sigma), 1 mmol/L sodium pyruvate (Lonza), and 2 mmol/L l‐glutamine (Life Technologies), followed by incubation for 1 hour in a non‐CO2 incubator at 37 °C. The mitochondrial oxygen consumption rate was measured using the Seahorse Bioscience XF96 Extracellular Flux Analyzer (Seahorse Bioscience) as follows: (1) basal levels (no additives); (2) leak respiration (15 μmol/L oligomycin final concentration); (3) maximal respiration (10 μmol/L FCCP), and (4) residual respiration (5 μmol/L antimycin A and 5 μmol/L rotenone). Experiments were performed with 10 wells per condition. The results were plotted and calculated by Seahorse software.
NADH Measurement in HL‐1 Cardiomyocytes
HL‐1 cardiomyocytes were washed with cold PBS, and 2×105 HL‐1 cardiomyocytes were pelleted per condition for each assay. The level of NADH was measured with the NAD/NADH Quantification Kit (Sigma, MAK037) according to the instructions of the manufacturer.
Statistical Analysis
Results are expressed as mean± SEM. Biochemical analyses were performed at least in duplicate. The 2‐tailed Student t test was evaluated within 2 individual group mean differences. One‐way ANOVA and post hoc Tukey test were used to assess the differences among more than 2 groups. All P values were 2 sided. Values of P<0.05 were considered significant. GraphPad Prism 8.2.1 was used for all the statistical evaluations and figures.
RESULTS
Tachypacing Induces Loss of Microtubule‐SRMCs Interaction and Contractile Dysfunction in HL‐1 Cardiomyocytes
Previously, we showed that the microtubule stabilizer tubacin prevented tachypacing‐induced contractile dysfunction. 12 To test whether additional microtubule stabilizers, taxol and βOHB, 21 , 22 , 23 , 24 could also prevent tachypacing‐induced contractile dysfunction, HL‐1 cardiomyocytes were pretreated with taxol (5 nmol/L) or βOHB (10 mmol/L) for 12 hours, and then subjected to tachypacing for 8 hours, followed by measurement of the CaT amplitude. Similar to tubacin, taxol as well as βOHB attenuated tachypacing‐induced loss of CaT in HL‐1 cardiomyocytes, compared with untreated controls (Figure 1A and 1B). Moreover, microtubule stabilizers tubacin, taxol, and βOHB attenuated the tachypacing‐induced reduction in the levels of both ɑ‐tubulin and acetylated ɑ‐tubulin in HL‐1 cardiomyocytes (Figure 1C through 1E). To determine whether taxol and βOHB affected SRMCs in HL‐1 cardiomyocytes, we isolated MAM from HL‐1 cardiomyocytes. We found that tachypacing induced a significant reduction in SRMCs, as the SRMC tether protein MFN2 was significantly reduced (Figure 1F and 1G), and the SR membrane protein SERCA2 that is enriched in SRMCs 17 , 32 was also reduced in MAM (Figure 1F and 1H), compared with nonpaced controls. Interestingly, tachypacing‐induced SRMC reduction was prevented by the microtubule stabilizers tubacin, taxol, as well as βOHB (Figure 1F through 1H). Consistently, immunofluorescent staining of markers for stable microtubules (acetylated ɑ‐tubulin), mitochondria (Mitotracker), and ER/SR (SEC61β) showed that tachypacing induced a significant reduction in SRMCs, and in contact sites between acetyl microtubules and SR/mitochondria (Figure 2A through 2G). The reductions in SRMCs and in contact between microtubules and SR/mitochondria were prevented by tubacin, taxol, or βOHB (Figure 2A through 2G). Furthermore, the microtubule stabilizers tubacin, taxol, or βOHB had no significant side effect on the interaction among ER/SR, mitochondria, and microtubules in control nonpaced HL‐1 cardiomyocytes (Figure S1A through S1F). Taken together, these findings indicate that tachypacing induced disruption of the microtubule network, reduction of the SRMC tether protein MFN2, and consequent downstream loss of SRMCs, which mediates contractile dysfunction as indicated by CaT loss in the HL‐1 cardiomyocyte model for AF.
Figure 1. Tachypacing‐Induced Loss of SR and Mitochondria Contacts (SRMCs) in HL‐1 Cardiomyocytes Is Prevented by Microtubule Stabilizing Compounds.
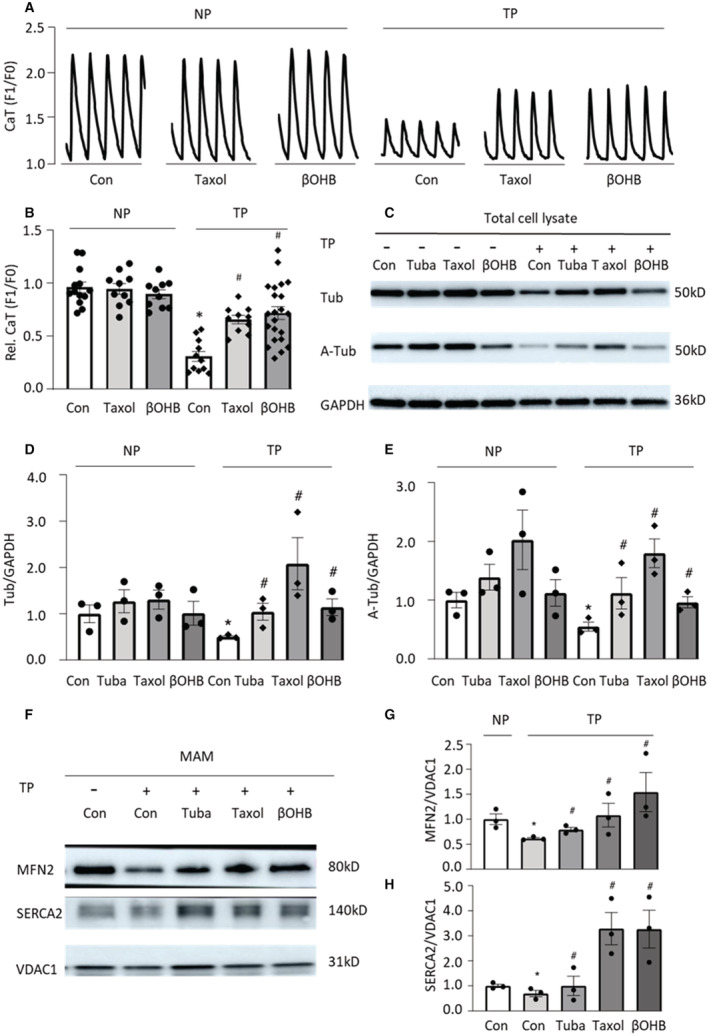
A and B, HL‐1 cardiomyocytes were pretreated with taxol or β‐hydroxybutyrate (βOHB) for 12 hours, followed by 8 hours tachypacing at 5 Hz (TP) or nonpacing (NP). Calcium transients (CaT) were reduced after TP, compared with NP. Taxol and βOHB counteracted TP‐induced loss of CaT in HL‐1 cardiomyocytes. C through E, Tachypacing reduced the levels of acetylated ɑ‐tubulin and ɑ‐tubulin in HL‐1 cardiomyocytes, and tubacin (Tuba), taxol, and βOHB attenuated the TP‐induced reduction of microtubule proteins analyzed by Western blot. F through H, Western blot analysis of mitochondrial‐associated membranes (MAM) isolated from HL‐1 cardiomyocytes after experimental treatments. Tubacin, taxol, and βOHB inhibited the TP‐induced loss of SRMCs, as indicated by the fraction of ER/SR protein (SERCA2) in MAM and by the SRMC tether protein MFN2 in HL‐1 cardiomyocytes. Each experiment was repeated at least 3 times. Data are presented as the mean±SEM. N=independent experiments, 2‐tailed Student t test was used for statistical significance. *P<0.05 vs control (Con) NP, # P<0.05 vs Con tachypacing (TP).
Figure 2. Tachypacing‐Induced Reduction in the SR and Mitochondria Contacts (SRMCs) and Microtubule‐SRMC Contacts Is Prevented by Microtubule‐Stabilizing Compounds.
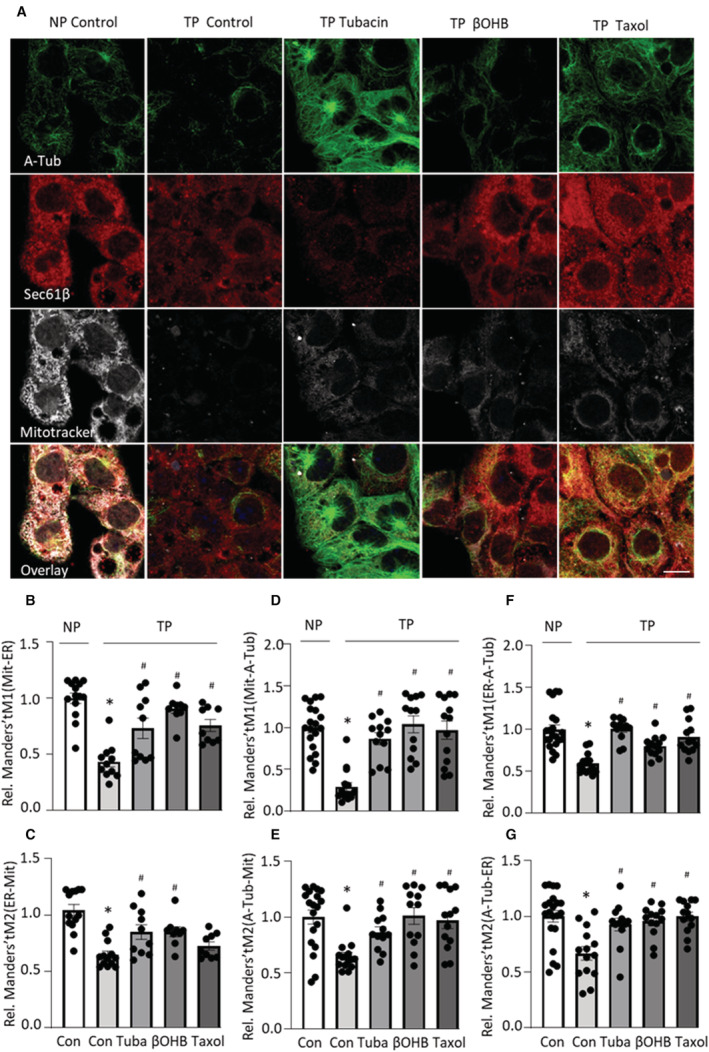
A, Representative images showing mitochondria (Mitotracker deep red; Mit), ER/SR (SEC61β), acetylated microtubule (acetylated ɑ‐tubulin; A‐Tub), and the co‐localization (overlay) of mitochondria, ER, and acetylated microtubule in HL‐1 cardiomyocytes. Scale bar = 10 μm. B and C, Quantified data showing that tachypacing significantly reduced co‐localization between mitochondria and ER/SR, which was prevented by microtubule stabilizers tubacin, β‐hydroxybutyrate (βOHB), and taxol. Manders' tM1: the fraction of mitochondria co‐localized with ER. Manders' tM2: the fraction of ER colocalized with mitochondria. D and E, Quantified data showing tachypacing significantly reduced co‐localization between mitochondria and acetyl microtubule, which was prevented by microtubule stabilizers tubacin, βOHB, and taxol. Manders' tM1: the fraction of mitochondria co‐localized with microtubule. Manders' tM2: the fraction of microtubule co‐localized with mitochondria. F and G, Quantified data showing tachypacing significantly reduced co‐localization between ER/SR and microtubule, which was prevented by microtubule stabilizers tubacin, βOHB, and taxol. Manders' tM1: the fraction of ER colocalized with microtubule. Manders' tM2: the fraction of microtubule co‐localized with ER. Each experiment was performed 2 times, and for each condition 10 to 15 images of ≈50 to 100 cardiomyocytes were selected for analysis. Statistical analysis was performed by using 1‐way ANOVA followed by Tukey's multiple comparisons test. *P<0.05 vs normal pacing (NP Control), # P<0.05 vs tachypacing (TP Control).
Microtubule Stabilizers Prevent Tachypacing‐Induced Contractile Dysfunction in Drosophila
To further confirm findings from HL‐1 cardiomyocytes, similar experiments were performed in the previously well‐established in vivo Drosophila model for AF. 33 For this purpose, microtubule stabilizers (taxol and βOHB) or vehicle were added to Drosophila food, and after at least 48 hours, heart wall contraction of Drosophila prepupae was monitored before and after tachypacing. As expected, the microtubule stabilizer taxol prevented tachypacing‐induced contractile dysfunction, which was defined as a significant reduction in heart rate and increase in arrhythmia in Drosophila prepupae (Figure 3A through 3C). Similarly, βOHB also prevented tachypacing‐induced contractile dysfunction (Figure 3D through 3F). Since both taxol and βOHB prevented tachypacing‐induced loss of microtubule‐SRMCs contacts in HL‐1 cardiomyocytes, these findings suggest that microtubule‐SRMCs contacts may mediate contractile dysfunction in the Drosophila model for AF.
Figure 3. Microtubule Stabilizers Prevent Tachypacing‐Induced Contractile Dysfunction in Drosophila.
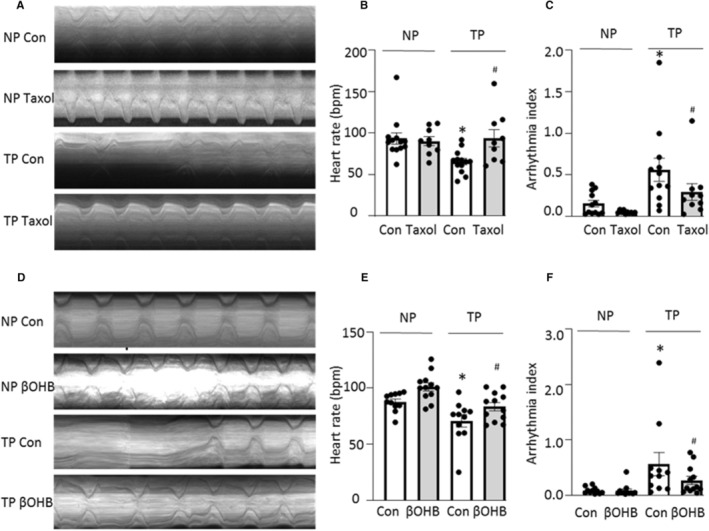
A, Representative heart wall motions (during 5 s). B and C, Taxol protected against tachypacing (TP)‐induced reduction in heart rate and against increase in arrhythmia. D, Representative heart wall motions (during 5 s). E and F, β‐Hydroxybutyrate (βOHB) protected against TP‐induced reduction in heart rate and against increase in arrhythmia. bpm, beats per minute. Each experiment was performed 3 times, and N=10 to 15 Drosophila prepupae for each condition. One‐way ANOVA followed by Tukey's multiple comparisons test were used for statistical significance. *P<0.05 vs control before tachypacing (NP Con), # P<0.05 vs Taxol group before tachypacing (NP Taxol).
Preservation of Microtubule‐SRMC Contacts Attenuates Tachypacing‐Induced Mitochondrial Dysfunction in HL‐1 Cardiomyocytes
SRMCs are crucial for mitochondrial function in cardiomyocytes by regulating calcium‐mediated NADH production and ATP production in the mitochondria. 15 , 17 , 34 Because tachypacing resulted in reduced SRMCs and contractile dysfunction in HL‐1 cardiomyocytes, which was prevented by microtubule stabilizers, we set out to determine whether microtubule stabilizers also prevented tachypacing‐induced mitochondrial dysfunction in HL‐1 cardiomyocytes. Therefore, mitochondrial oxygen consumption rate was determined as a key measurement for mitochondrial function. Basal respiration, ATP production rate, and maximal respiration were significantly reduced in tachypaced HL‐1 cardiomyocytes, suggesting mitochondrial dysfunction in this tachypaced HL‐1 cardiomyocyte model for AF (Figure 4A through 4D). As expected, tubacin, taxol, and βOHB counteracted tachypacing‐induced mitochondrial dysfunction, indicating that preservation of the microtubule–SRMC interactions ameliorates mitochondrial dysfunction in this model (Figure 4A through 4D). In agreement, supplementation with βOHB attenuated the tachypacing‐induced reduction in NADH levels in HL‐1 cardiomyocytes (Figure 4D). Collectively, we showed that microtubule stabilizers, which inhibited loss of SRMCs, also attenuated tachypacing‐induced mitochondrial dysfunction.
Figure 4. Preservation of Microtubule‐SRMC Contacts With Microtubule Stabilizers Attenuated Tachypacing‐Induced Mitochondrial Dysfunction in HL‐1 Cardiomyocytes.
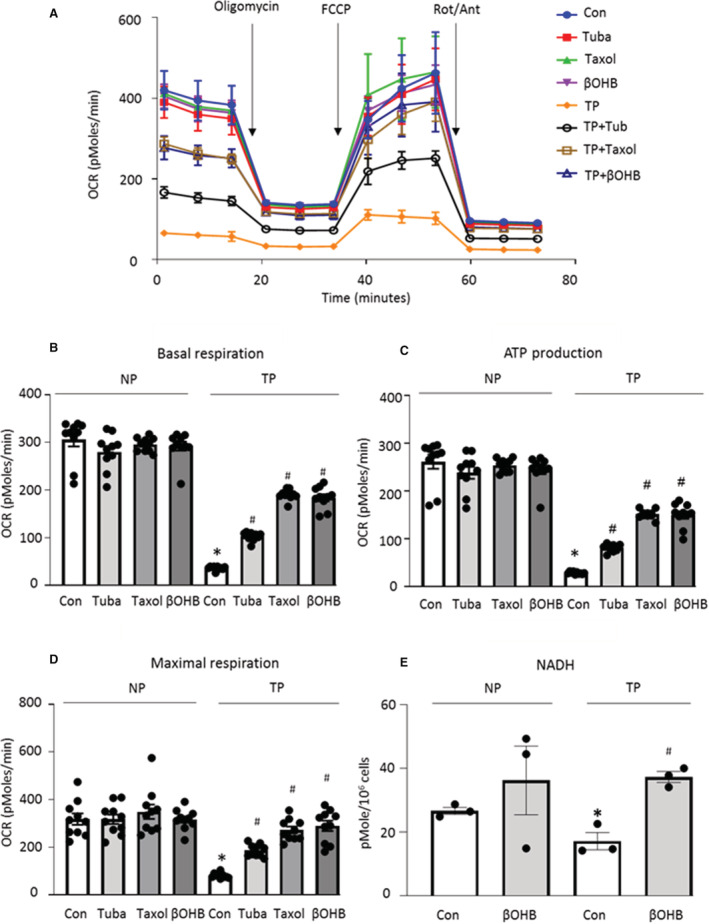
A through D, We determined whether upstream and downstream preservation of microtubule‐SRMC (sarcoplasmic reticulum and mitochondrial contacts) contacts protected against tachypacing (TP)‐induced mitochondrial dysfunction. Before 8 h tachypacing, HL‐1 cardiomyocytes were pretreated with tubacin, taxol or β‐hydroxybutyrate (βOHB) for 12 h. Mitochondrial oxygen consumption rate (OCR) was determined using Seahorse XF96 Flux analysis as a key measurement of mitochondrial function in HL‐1 cardiomyocytes. Tubacin, taxol, and βOHB improved TP‐induced basal respiration, ATP production, and maximal respiration. E, Biochemical analysis of NADH levels. βOHB prevented the TP‐induced reduction in the level of NADH of HL‐1 cardiomyocytes. Each experiment was performed 3 times. Statistical significance was determined using 1‐way ANOVA followed by Tukey's multiple comparisons test. *P<0.05 vs Control (Con) NP, # P<0.05 vs Con TP.
MFN2‐Mediated Loss of SRMCs Underlies HG‐Induced Contractile Dysfunction in HL‐1 Cardiomyocytes and Drosophila
Microtubule‐dependent mitochondrial regulation is also affected by diabetes, a strong and independent risk factor for AF incidence. 9 , 19 , 20 Therefore, we explored whether microtubule–SRMC contacts are involved in diabetes‐induced contractile dysfunction. To this end, HL‐1 cardiomyocytes were grown in 25 mmol/L high glucose (HG, mimicking diabetes) for increasing time periods, followed by measurement of CaT. After 72 hours of HG treatment, CaT of HL‐1 cardiomyocytes was significantly reduced compared with controls, suggesting that long‐term HG treatment induced contractile dysfunction in HL‐1 cardiomyocytes (Figure S2A and S2B). Since βOHB offered the best protection against tachypacing‐induced SRMCs reduction and contractile dysfunction in HL‐1 cardiomyocytes and Drosophila prepupae (Figure 1, 2, 3, 4) without affecting SRMCs in control nonpaced conditions (Figure S1), we examined βOHB in the HG model. Importantly, βOHB prevented HG‐induced CaT loss in HL‐1 cardiomyocytes (Figure 5A and 5B). MAM isolation showed that HG induced a significant reduction of the SRMCs, as represented by a reduction in SR (measured by SERCA2 levels as proxy) in MAM as well as a reduction of the tether protein MFN2, which was prevented by βOHB (Figure 5C through 5E). Moreover, immunofluorescent staining of mitochondria and ER/SR showed that HG significantly reduced co‐localization of mitochondria with ER/SR, further confirming that HG induced loss of SRMCs in HL‐1 cardiomyocytes (Figure 5F through 5H). Again, this was prevented by βOHB (Figure 5F through 5H). HG, however, had no significant effect on the levels of ɑ‐tubulin and acetylated ɑ‐tubulin in HL‐1 cardiomyocytes, and no effect of βOHB on the levels of ɑ‐tubulin and acetylated ɑ‐tubulin was seen (Figure S2C). Consistently, HG had no effect on the contacts between microtubules and ER/SR or mitochondria (Figure 5I through 5L). Taken together, this suggests that βOHB stabilizes SRMCs in the HG model, independently of its role in stabilizing microtubules, but dependent on its direct effect on the SRMC tether protein MFN2 (Figure 5C through 5L).
Figure 5. High Glucose Induces Loss of CaT and reduces SR and Mitochondria Contacts (SRMCs) and Its Tether Protein MFN2, Which Is Prevented by β‐Hydroxybutyrate (βOHB) in HL‐1 Cardiomyocytes (A through L) and Drosophila (M though O).
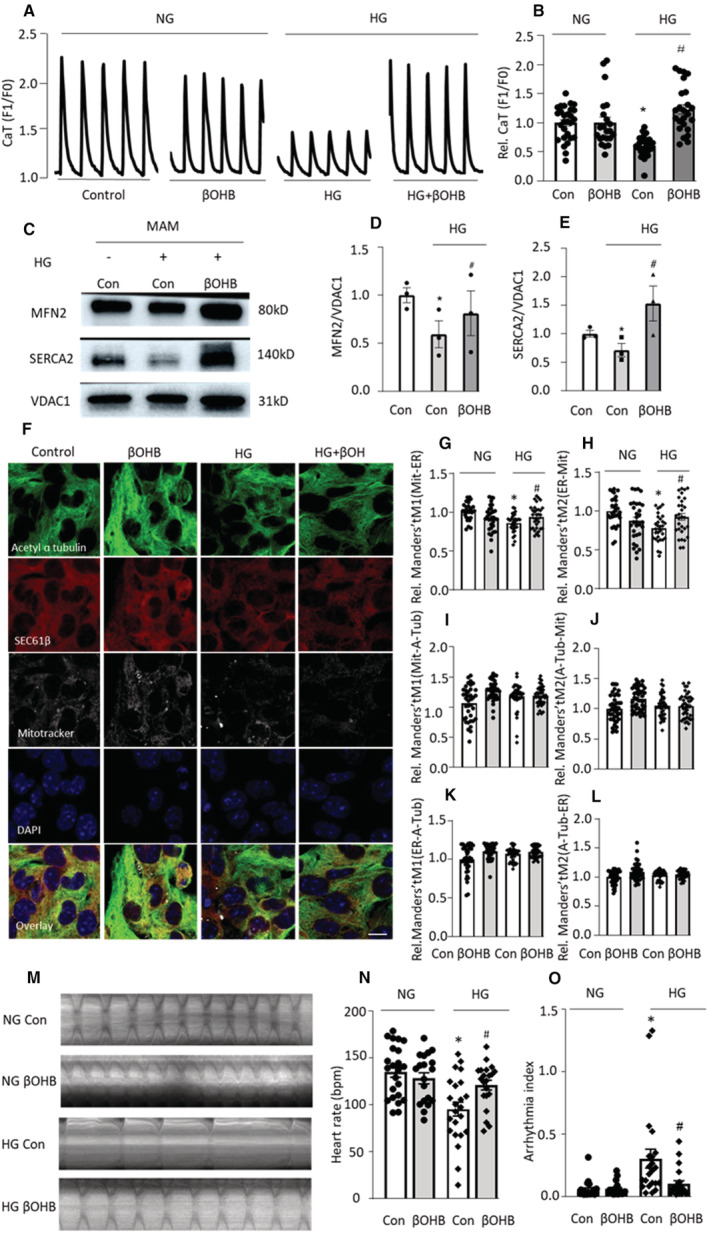
A and B, βOHB attenuated high glucose (HG)–induced loss of calcium transients (CaT) in HL‐1 cardiomyocytes. C through E, Representative Western blot images and quantified data showed that HG reduced the levels of SRMC proteins MFN2 and SERCA2, and that βOHB attenuated HG induced the reduction of SRMC proteins MFN2 and SERCA2. F, Representative immunofluorescent images showing nuclei (DAPI), mitochondria (Mitotracker deep red) and ER/SR (SEC61β) and microtubule (acetylated ɑ‐tubulin) and their overlay in HL‐1 cardiomyocyte. Scale bar = 10 μm. G and H, Quantified data of F showed HG‐induced significantly reduced co‐localization between mitochondria and ER, which was prevented by microtubule stabilizer βOHB. Manders' tM1: the fraction of mitochondria co‐localized with ER. Manders' tM2: the fraction of ER co‐localized with mitochondria. I and J, No difference was observed in the percentage of co‐localization between mitochondria and microtubule in the various conditions. Manders' tM1: the fraction of mitochondria colocalized with microtubule. Manders' tM2: the fraction of microtubule co‐localized with mitochondria. K and L, No change was found in the percentage of co‐localization between ER and microtubule in the various conditions. Manders' tM1: the fraction of ER co‐localized with microtubule. Manders' tM2: the fraction of microtubule co‐localized with ER. M through O, Drosophila prepupae were monitored to record heart wall contraction. M, Representative heart wall motions in Drosophila prepupae (during 5 s). N and O, HG reduced heart rate (bpm, beats per minute) and increased the arrhythmia index in Drosophila prepupae. βOHB attenuated HG‐induced contractile dysfunction in Drosophila prepupa. Each experiment was repeated 3 times, and for each condition 10 to 15 Drosophila prepupae were selected. NG, normal glucose. Two‐tailed Student t test was used for statistical analysis. *P<0.05 vs Control (Con) NG, # P<0.05 vs Con HG.
To further confirm the effect of HG on SRMCs and contractile dysfunction, similar experiments were conducted using Drosophila prepupae. Compared with normal diet, food with HG (300 g/L) induced contractile dysfunction with reduced heart rate and increased arrhythmia index (Figure S2D through S2F), both of which were prevented by the SRMCs stabilizer βOHB (Figure 5M through 5O).
Taken together, loss of SRMCs plays a key role in HG‐induced contractile dysfunction in HL‐1 cardiomyocytes and Drosophila, and this may thus contribute to diabetes‐related AF in patients. While both MFN2 and microtubule disruption led to the reduction of SRMCs in the tachypacing model for AF (Figure 1 and Figure 2), MFN2 reduction, but not microtubule disruption, is involved in the reduction of SRMCs in the HG models (Figure 5). Therefore, the SRMCs tether protein MFN2 is a common key player in regulation of both tachypacing (model for AF)‐ and HG‐induced contractile dysfunction in cardiomyocytes and Drosophila.
Preservation of SRMCs by Maintaining MFN2 Prevents Tachypacing/HG‐Induced Contractile Dysfunction in HL‐1 Cardiomyocytes and Drosophila
MFN2, a physical tether protein that connects SR and mitochondria, is essential for normal interorganelle Ca2+ signaling through microdomains in cardiomyocytes. 17 Both tachypacing‐ and HG‐induced reduction of MFN2 was accompanied by loss of CaT in HL‐1 cardiomyocytes (Figure 1F and 1G and Figure 5C and 5D), suggesting MFN2 as the key regulator in (diabetes‐related) AF development. To confirm this, MFN2 overexpression in HL‐1 cardiomyocytes and Drosophila was performed (Figure S3A). Indeed, MFN2 overexpression prevented tachypacing‐ and HG‐induced contractile dysfunction in HL‐1 cardiomyocytes (Figure 6A through 6D). Consistently, MFN2 overexpression prevented HG‐induced arrhythmia in Drosophila (Figure 6E through 6G). Moreover, cardiac‐specific knockdown of MFN2 alone was sufficient to cause contractile dysfunction, an increased arrhythmicity, and a decreased heart rate in Drosophila, with an effect comparable to that of tachypacing (Figure 6H through 6J). Taken together, these data strongly suggest the SRMCs tether protein MFN2 is a key player in regulating contractile function, in vitro, in HL‐1 cardiomyocytes, and in vivo, in Drosophila.
Figure 6. Preservation of SR and Mitochondria Contacts (SRMCs) via Overexpressing MFN2 Inhibits Tachypacing/HG‐Induced Contractile Dysfunction in HL‐1 Cardiomyocytes and Drosophila.
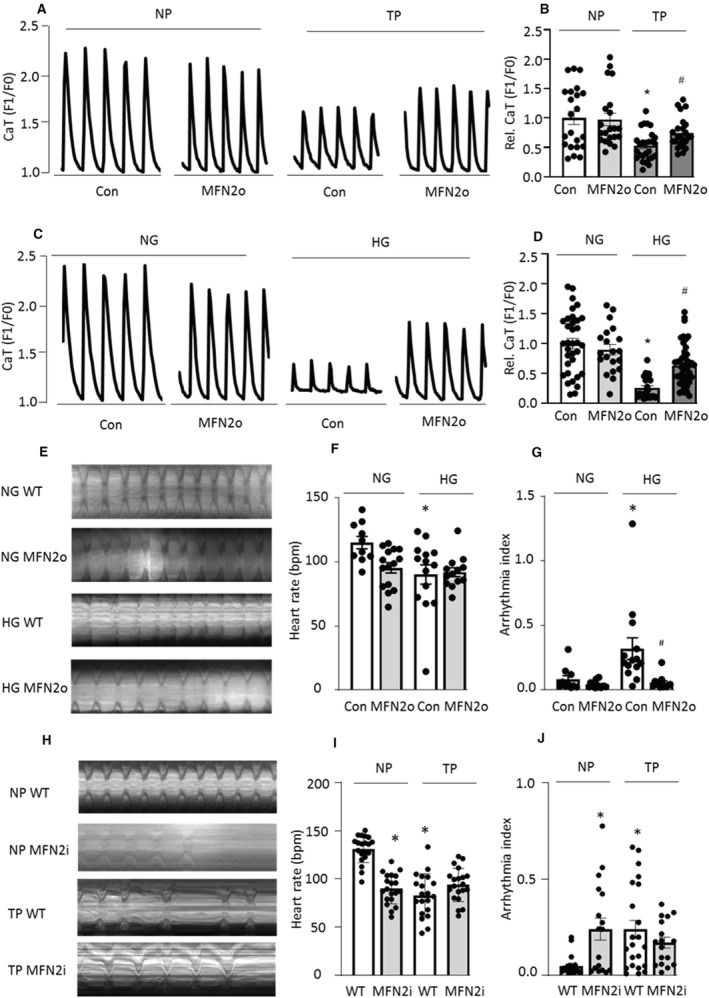
A and B, HL‐1 cardiomyocytes were transfected with MFN2‐MYC or control pcDNA3.1 plasmid for 48 h, following by 8 h tachypacing and calcium transient (CaT) measurement. MFN2 overexpression (MFN2o) inhibited tachypacing (TP)‐induced CaT loss. *P<0.05 vs pcDNA 3.1 (Con) NP, # P<0.05 vs pcDNA3.1 + TP. C and D, MFN2 overexpression inhibited high glucose (HG)–induced CaT loss in HL‐1 cardiomyocyte. *P<0.05 vs pcDNA 3.1 (Con) NG, # P<0.05 vs pcDNA3.1 + HG. N=15–20 cardiomyocytes per condition. E through G, MFN2 overexpression in Drosophila prepupae prevented HG‐induced increase in arrhythmia index, compared with HG wild‐type (WT). *P<0.05 vs normal glucose (NG) WT (Con), # P<0.05 vs HG WT. H through J, Cardiac‐specific knockdown of MFN2 (MFN2i, 40 478) in Drosophila prepupae significantly reduced heart rate and increased arrhythmia with a similar effect of tachypacing. *P<0.05 vs nonpacing (NP) WT. Each experiment was repeated 3 times, and 10 to 20 Drosophila prepupae were selected for each condition. One‐way ANOVA followed by Tukey's multiple comparisons test was used to determine statistical significance.
Loss of SRMCs Was Observed in Patient With PeAF, Which Was Aggravated by Diabetes
Finally, to investigate whether similar changes in SRMCs can be found in patients with PeAF associated with or without diabetes (Table 1), the level of SRMC protein MFN2 was determined in RAA of patients with PeAF with or without diabetes and in control SiRh patients with or without diabetes. Compared with SiRh patients, the levels of MFN2 in patients with PeAF with or without diabetes were significantly reduced (Figure 7A and 7B). Moreover, the levels of MFN2 in PeAF patients with diabetes were further decreased compared with patients with PeAF with without diabetes (Figure 7A and 7B). In addition, immunofluorescent imaging showed that the co‐localization of mitochondria with SR in RAA of all patients with PeAF was significantly lower compared with all SiRh patients (Figure 7F), although significance was not reached when compared with the separate groups with or without diabetes (Figure 7D and 7E), mainly because of the limited sample size after separation. Furthermore, male and female patients did not show a significant difference on the levels of MFN2 or SRMCs (Figure S4). Taken together, these observations show a significant reduction of SRMC tether protein MFN2 in RAA tissue samples of patients with PeAF, which is further aggravated by diabetes, and also a significant SRMCs reduction in PeAF.
Figure 7. Patients With Atrial Fibrillation (AF) Reveal Reduced Levels of SR and Mitochondria Contacts (SRMCs) Tether Protein MFN2 and Reduced SRMCs Compared With Sinus Rhythm (SiRh).
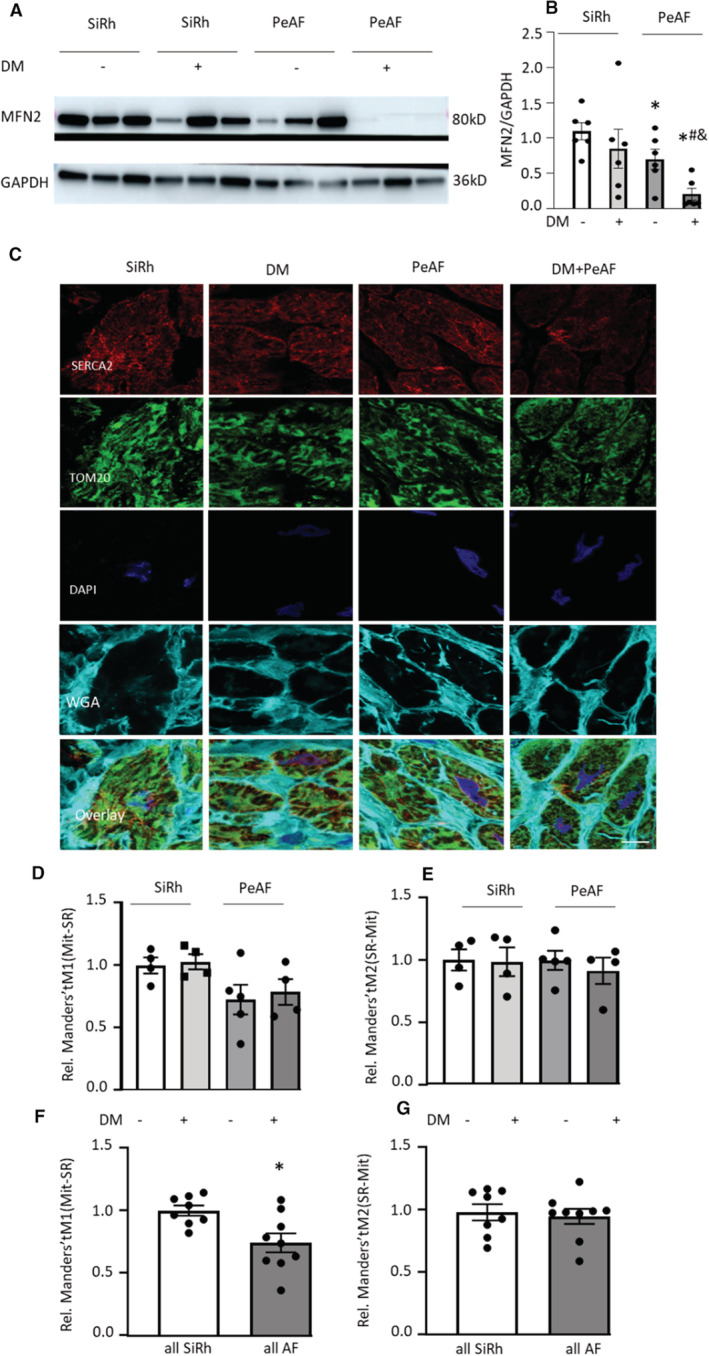
A and B, Representative Western blot images of the SRMC protein MFN2 in the right atrial appendage (RAA) of persistent AF (PeAF) with or without diabetes (DM) and sinus rhythm (SiRh) patients with or without DM. Compared with SiRh patients with DM, the levels of MFN2 in patients with PeAF with DM were significantly reduced. SiRh, N=6; SiRh+DM, N=6; PeAF, N=6 and PeAF+DM, N=6. *P<0.05 vs SR, # P<0.05 vs SiRh+DM, & P<0.05 vs PeAF. C through G, Representative immunofluorescent images showing the co‐localization between SR (SERCA2) and mitochondria (TOM20) in RAA. Scale bar = 10 μm. Compared with SiRh patients, the percentage of mitochondria colocalized with SiRh is significantly lower in patients with PeAF compared with patients with SiRh. SiRh, N=4; SiRh+DM, N=4; PeAF, N=5; and PeAF+DM, N=4. Manders' tM1: the fraction of mitochondria co‐localized with SiRh. Manders' tM2: the fraction of SiRh co‐localized with mitochondria. Statistical significance was determined using 1‐way ANOVA followed by Tukey's multiple comparisons test.
DISCUSSION
In this study, we identified a loss of SRMCs in experimental and clinical AF and showed that genetic and pharmacological preservation of SRMCs protected against tachypacing‐induced mitochondrial dysfunction and contractile dysfunction in experimental models for AF. The stability of SRMCs was regulated by both microtubules and the SR‐mitochondrial tether protein MFN2 (Figure S5). In the tachypacing model for AF, the loss of SRMCs was induced by both disruption of microtubules and reduction of MFN2. In HG condition, as a model for diabetes, the loss of SRMCs was induced by reduction of MFN2, independently of microtubule disruption (Figure S5). Pretreatment with the microtubule stabilizers tubacin, taxol, or βOHB attenuated tachypacing‐induced loss of MFN2 dose dependently (Figure S3B), and these stabilizers attenuated the reduction in SRMCs and mitochondrial dysfunction in the HL‐1 cardiomyocyte and Drosophila models for AF. Furthermore, pretreatment with βOHB prevented HG‐induced SRMC reduction and contractile dysfunction in HL‐1 cardiomyocytes and Drosophila via direct prevention of MFN2 loss. Genetic interventions support the pharmacological observations as MFN2 overexpression prevented tachypacing‐ and HG‐induced contractile dysfunction in HL‐1 cardiomyocytes and in Drosophila. Reduction of SRMCs and its tether protein MFN2 was shown consistently in RAA tissue of patients with PeAF, which was aggravated by diabetes. Together, these findings indicate that conservation of SRMCs suppresses AF promotion in both tachypaced and HG‐treated experimental model systems, and MFN2 is a shared key regulator protein in both models. In conclusion, SRMCs may play a critical role in (diabetes‐related) clinical AF development and MFN2 is a key druggable target to preserve SRMCs (Figure S5).
Key Role of SRMCs in Experimental AF and Patients With PeAF
SR, a muscle‐specialized form of the ER in cardiomyocytes, and mitochondria, are 2 key organelles involved in Ca2+ and energy (ATP) homeostasis. SR stores Ca2+ and plays a major role in excitation–contraction coupling of the heart in close cooperation with mitochondria, which play a vital role in generating ATP and buffering cytosolic Ca2+. 13 , 34 , 35 Defective cooperation between mitochondria and SR may contribute to the progression of AF. For instance, Xie et al discovered that ryanodine receptor 2, the calcium‐releasing channel located on SR, is oxidized by mitochondrial reactive oxygen species in the atria of patients with PeAF, leading to aberrant intracellular Ca2+ release (Ca2+ sparks) and thereby promoting AF development. 36 Furthermore, an increasing number of studies show that the crosstalk between SR and mitochondria via SRMCs is crucial for normal cardiomyocyte contraction and relaxation. 14 , 15 , 16 , 17 , 34 , 35 Well‐functioning SRMCs support efficient mitochondrial Ca2+ uptake and ATP production during cardiac contraction and relaxation. In particular, because of the close apposition between SR and mitochondria tethered by the SRMCs, Ca2+ released by SR during excitation–contraction coupling is able to enter mitochondria to boost the enzyme activity of the tricarboxylic acid cycle and the activity of the electron transport complexes, enhancing oxidative phosphorylation and thus ATP production in mitochondria. 17 , 34 Notably, the SRMCs are dependent on acetylated microtubules, 18 which are disrupted in experimental models for AF as well as in human AF. 12 Here, we observed that both tachypacing and HG induced a reduction in SRMCs, and consequently resulted in mitochondrial dysfunction and contractile dysfunction in HL‐1 cardiomyocytes and in Drosophila. Consistently, loss of SRMCs was observed in RAA tissue of patients with PeAF, especially those with the AF risk factor diabetes, suggesting that loss of SRMCs plays a critical role in the development of AF, especially related to diabetes‐associated AF development.
Microtubule–SRMCs Axis Underlies Tachypacing‐Induced AF Remodeling
The microtubule network is required to ensure close contact between SR and mitochondria. 18 , 37 Disruption of the microtubule network destroys the connection between microtubules and mitochondria, and consequently alters mitochondrial function. 38 Mitochondrial dysfunction reduces acetyl‐CoA levels resulting in decreased levels of acetylated microtubules, thereby promoting microtubule disruption and forming a vicious circle between microtubule disruption and mitochondrial dysfunction. 39 , 40 , 41 Previously, we found that deacetylase histone deacetylase activation induced α‐tubulin deacetylation, contributing to microtubule disruption and consequently to contractile dysfunction in experimental models for AF and in patients with AF. 12 Inhibition of deacetylase histone deacetylase, by tubacin, conserves the microtubule network and protects against AF progression in the HL‐1 cardiomyocyte and Drosophila models for AF, but the downstream molecular mechanism remained unclear. In this study, we discovered that preservation of the microtubule network prevented the downstream SRMCs reduction and mitochondrial dysfunction in tachypaced HL‐1 cardiomyocytes and Drosophila. Together, this shows that both upstream (by inhibition of HDAC6) and downstream (by targeting microtubules) conservation of the microtubule–SRMCs axis suppresses tachypacing‐induced contractile dysfunction and AF promotion in experimental models for AF.
SRMCs Underlie Diabetes‐Initiated AF Promotion
Diabetes is independently associated with new onset of AF. 5 However, the molecular mechanisms by which diabetes invokes AF are unresolved. There are indications that alterations in the interactions between SR and mitochondria may be involved in the pathophysiological process of diabetic cardiomyopathy. 35 It has been reported that reduced protein levels of ryanodine receptor 2 in streptozotocin‐induced diabetic rats contributed to a decrease in the SR Ca2+ storage and decreased rates of Ca2+ release in cardiomyocytes. 42 On the other hand, increased ER–mitochondrial contacts were observed in streptozotocin‐induced type I diabetes and reducing the contacts between ER and mitochondria inhibited apoptotic pathways in this model. 43 Therefore, whether the alterations in the structural and functional interactions between the SR/ER and mitochondria are ultimately beneficial or detrimental in clinical AF still needs to be established. In this study, we observed that HG reduced SRMCs and induced contractile dysfunction in HL‐1 cardiomyocytes, which were prevented by microtubule stabilizer βOHB or by overexpression of the SRMCs tether protein MFN2. Moreover, βOHB exposure and MFN2 overexpression inhibited HG‐induced contractile dysfunction in Drosophila prepupae. Remarkably, no changes in microtubule proteins and the contacts between microtubules and SRMCs were observed after HG exposure. This finding indicated that the effect of βOHB on preservation of SRMCs in HG‐induced contractile dysfunction in HL‐1 cardiomyocytes and Drosophila prepupae is mediated directly by MFN2, independent of the role of βOHB in stabilizing the microtubule network. We also observed aggravated reduction of MFN2 in patients with AF with diabetes. Together, these observations indicate that reduction of SRMCs may also underlie diabetes‐initiated AF promotion, and preservation of SRMCs via maintaining its tether protein MFN2 may prevent diabetes‐related AF promotion.
MFN2 Is the Key Regulator of SRMCs in AF, Especially Diabetes‐Related AF
It has been shown that cardiac‐specific ablation of MFN2 decreased SR–mitochondrial tethering by 30%, leading to a decreased mitochondrial Ca2+ uptake. 17 This established MFN2 as a key physical tether between SR and mitochondria that is required for SR–mitochondrial Ca2+ signaling in the cardiomyocytes. 17 Another study also showed that MFN2 deficiency contributes to metabolic abnormalities, such as glucose intolerance and impaired insulin signaling in liver‐specific MFN2 knockout mice, thereby modulating insulin signaling and glucose homeostasis. 44 Here, we observed that the protein level of MFN2 was reduced in both tachypacing‐ and HG‐treated HL‐1 cardiomyocytes, and that βOHB attenuated tachypacing‐ and HG‐induced reduction in MFN2 level. Moreover, MFN2 overexpression prevented tachypacing‐ and HG‐induced contractile dysfunction in HL‐1 cardiomyocytes. Interestingly, MFN2 suppression induced contractile dysfunction with a similar effect as tachypacing in Drosophila prepupae. Consistently, the RAA tissue samples of patients with PeAF with diabetes revealed an aggravated loss of MFN2. Taken together, we discovered MFN2 as a novel key regulator of SRMCs in HL‐1 cardiomyocyte and Drosophila models for AF.
Although both tachypacing and HG induced SRMCs reduction in HL‐1 cardiomyocytes, the degree of reduction of SRMCs in the HG model is much less compared with the tachypacing model. In line with this finding, tachypacing induced the reduction of both acetyl ɑ‐tubulin and MFN2, while HG treatment induced only the reduction of MFN2 without reduction of ɑ‐tubulin and acetylated ɑ‐tubulin, suggesting that microtubules and MFN2 can both stabilize SRMCs but they can function independently of each other in cardiomyocytes (Figure S5). Since MFN2 reduction is found in both conditions, MFN2 is thus a highly interesting new target for AF treatment and prevention, especially for diabetes‐related AF prevention. Accordingly, βOHB, which not only can stabilize microtubules, but also has a direct effect on preventing MFN2 reduction in both tachypacing and HG models, is a highly interesting compound to examine in the context of (diabetes)‐related AF.
Therapeutic Implications
No curative therapy exists for substrate‐driven AF. 45 , 46 In our previous study, we obtained strong indications that microtubule disruption contributes to structural remodeling and contractile dysfunction in various in vitro and in vivo models of AF. 12 The present study shows that microtubule disruption induces the loss of SRMCs and mitochondrial dysfunction, subsequently contributing to contractile dysfunction in tachypacing‐induced AF models. We also established a key role for SRMCs loss mediated by MFN2 reduction in diabetes‐related AF. Importantly, preservation of SRMCs with pharmacological and genetic approaches prevented HG‐ or tachypacing‐induced contractile dysfunction. Therefore, therapeutic interventions to stabilize SRMCs may conserve cardiac function, representing a potentially promising strategy to delay or prevent AF.
βOHB is a potential candidate to be tested in this regard, since we found that βOHB attenuated both tachypacing‐ and HG‐induced reduction of SRMCs and prevented contractile dysfunction in 2 experimental model systems for diabetes and AF, by affecting microtubules and via direct effects on MFN2. Moreover, βOHB is a metabolic intermediate that can arise during the metabolism of fatty acids and ketogenic amino acids and is enhanced by exercise, fasting, and ketogenic diets. 24 , 47 Ketogenic diets have shown effectiveness in management of type II diabetes, which further supports βOHB as a potential drug to prevent diabetes‐initiated AF promotion. 48 However, a recent study demonstrated that long exposure to ketogenic diets induced cardiac fibrosis in rats. 49 Moreover, we did not examine the effect other ketone bodies or the knockdown of mitochondrial d‐β‐hydroxybutyrate dehydrogenase, which would have been interesting in light of protective effects of βOHB especially. Therefore, more research is needed to elucidate the role of ketogenic diets in patients with AF.
Limitations and Future Directions
Our notion that SRMCs underlie contractile dysfunction is built on experiments conducted in HL‐1 cardiomyocyte and Drosophila models for AF and diabetes as well as in a limited number of right atrial samples from patients. Therefore, future research may be needed to confirm these findings in a mammalian model for AF and a more extensive analysis in patients. An additional limitation of the study is that cell shortening in single cardiomyocytes was not measured, which would have added to existing data that this correlates well with peak CaT as measured in our study.
Moreover, in the current study, we found that microtubule stabilizers (tubacin, βOHB, and taxol) could prevent MFN2 reduction induced by tachypacing and/or HG in HL‐1 cardiomyocytes. Of note, different mechanisms by which tubacin, βOHB, and taxol can stabilize microtubules have been indicated: tubacin via HADC6 inhibition, 12 taxol via directly binding to tubulin, 21 and βOHB via potential antioxidant mechanisms, 22 , 23 , 24 enforcing our conclusion that microtubule stabilizing is an effective target for AF. However, the detailed molecular mechanism by which these stabilizers regulate MFN2 expression is lacking and thus warrants further investigation. Previous research has demonstrated that the MFN2 level could be regulated at the transcriptional level as well as at the posttranslational level. At the transcriptional level, the transcription factor estrogen‐related receptor α has been shown to be a key regulator of MFN2 transcription, recruiting peroxisome proliferator‐activated receptor γ coactivator (PGC)‐1α and PGC‐1β. These 2 nuclear coactivators are potent, positive regulators of MFN2 expression in muscle cells, including the cardiac muscle. 50 At the posttranslational level, one study showed that phosphorylation of MFN2 by JNK can induce the degradation of MFN2 by the ubiquitin‐proteasome system, 51 and hence blocking this could possibly result in the reverse effect. Another study showed that the MFN2 agonists stabilized the fusion‐permissive open conformation of endogenous normal MFN2 by acting on MFN2 Ser378 phosphorylated states. 52 Therefore, compounds modulating MFN2 at transcriptional and posttranslational levels may be considered for testing their effect on SRMCs in the future. Furthermore, a recent study showed that βOHB could activate PGC‐1α and suppressed aging‐related inflammation in rat kidneys, 53 suggesting βOHB might regulate MFN2 levels by regulating PGC‐1α. However, the exact mechanism by which βOHB or taxol regulates MFN2 expression in the heart remains unknown. Therefore, future (omics) studies would be useful to delineate and dissect the exact mechanisms by which βOHB and taxol regulate MFN2.
CONCLUSIONS
In conclusion, SRMCs and its tether protein MFN2, may play a critical role in diabetes‐related clinical AF, and microtubule‐ and MFN2‐targeted preservation of SRMCs could be beneficial for AF treatment.
Sources of Funding
This work was supported by the Dutch Heart Foundation (2017T029, 2013T144, 2013T096), Dutch Heart Foundation and Dutch Research Council NWO (CVON‐STW2016‐14728), The Dutch Research Council NWO ZonMw VENI (09150161910179), The AFIP Foundation, and Medical Delta.
Disclosures
None.
Supporting information
Method S1
Figures S1–S5
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.024478
For Sources of Funding and Disclosures, see page 18.
Contributor Information
Bianca Brundel, Email: b.brundel@amsterdamumc.nl.
Deli Zhang, Email: deli.zhang@wur.nl.
REFERENCES
- 1. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener H‐C, Heidbuchel H, Hendriks J, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016; 37:2893–2962, doi: 10.1093/eurheartj/ehw210. [DOI] [PubMed] [Google Scholar]
- 2. Camm AJ, Lip GY, De Caterina R, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P, Guidelines ESCCfP . 2012 focused update of the ESC guidelines for the management of atrial fibrillation: an update of the 2010 ESC guidelines for the management of atrial fibrillation. Developed with the special contribution of the European heart rhythm association. Eur Heart J. 2012;33:2719–2747. doi: 10.1093/eurheartj/ehs253 [DOI] [PubMed] [Google Scholar]
- 3. Lippi G, Sanchis‐Gomar F, Cervellin G. Global epidemiology of atrial fibrillation: an increasing epidemic and public health challenge. Int J Stroke. 2021;16:217–221. doi: 10.1177/1747493019897870 [DOI] [PubMed] [Google Scholar]
- 4. Kotecha D, Lam CS, Van Veldhuisen DJ, Van Gelder IC, Voors AA, Rienstra M. Heart failure with preserved ejection fraction and atrial fibrillation: vicious twins. J Am Coll Cardiol. 2016;68:2217–2228. doi: 10.1016/j.jacc.2016.08.048 [DOI] [PubMed] [Google Scholar]
- 5. Goudis CA, Korantzopoulos P, Ntalas IV, Kallergis EM, Liu T, Ketikoglou DG. Diabetes mellitus and atrial fibrillation: pathophysiological mechanisms and potential upstream therapies. Int J Cardiol. 2015;184:617–622. doi: 10.1016/j.ijcard.2015.03.052 [DOI] [PubMed] [Google Scholar]
- 6. Movahed MR, Hashemzadeh M, Jamal MM. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int J Cardiol. 2005;105:315–318. doi: 10.1016/j.ijcard.2005.02.050 [DOI] [PubMed] [Google Scholar]
- 7. Huxley RR, Filion KB, Konety S, Alonso A. Meta‐analysis of cohort and case‐control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108:56–62. doi: 10.1016/j.amjcard.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du X, Ninomiya T, de Galan B, Abadir E, Chalmers J, Pillai A, Woodward M, Cooper M, Harrap S, Hamet P, et al. Risks of cardiovascular events and effects of routine blood pressure lowering among patients with type 2 diabetes and atrial fibrillation: results of the ADVANCE study. Eur Heart J. 2009;30:1128–1135. doi: 10.1093/eurheartj/ehp055 [DOI] [PubMed] [Google Scholar]
- 9. Aksnes TA, Schmieder RE, Kjeldsen SE, Ghani S, Hua TA, Julius S. Impact of new‐onset diabetes mellitus on development of atrial fibrillation and heart failure in high‐risk hypertension (from the VALUE trial). Am J Cardiol. 2008;101:634–638. doi: 10.1016/j.amjcard.2007.10.025 [DOI] [PubMed] [Google Scholar]
- 10. Li N, Brundel BJJM. Inflammasomes and proteostasis novel molecular mechanisms associated with atrial fibrillation. Circ Res. 2020;127:73–90. doi: 10.1161/CIRCRESAHA.119.316364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang D, Hu X, Henning RH, Brundel BJ. Keeping up the balance: role of HDACs in cardiac proteostasis and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2016;109:519–526. doi: 10.1093/cvr/cvv265 [DOI] [PubMed] [Google Scholar]
- 12. Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra‐Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, et al. Activation of histone deacetylase‐6 induces contractile dysfunction through derailment of α‐tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–358. doi: 10.1161/CIRCULATIONAHA.113.005300 [DOI] [PubMed] [Google Scholar]
- 13. Eisner V, Csordas G, Hajnoczky G. Interactions between sarco‐endoplasmic reticulum and mitochondria in cardiac and skeletal muscle ‐ pivotal roles in ca(2)(+) and reactive oxygen species signaling. J Cell Sci. 2013;126:2965–2978. doi: 10.1242/jcs.093609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dorn GW II, Maack C. SR and mitochondria: calcium cross‐talk between kissing cousins. J Mol Cell Cardiol. 2013;55:42–49. doi: 10.1016/j.yjmcc.2012.07.015 [DOI] [PubMed] [Google Scholar]
- 15. De la Fuente S, Sheu SS. SR‐mitochondria communication in adult cardiomyocytes: a close relationship where the ca(2+) has a lot to say. Arch Biochem Biophys. 2019;663:259–268. doi: 10.1016/j.abb.2019.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dorn GW II, Scorrano L. Two close, too close: sarcoplasmic reticulum‐mitochondrial crosstalk and cardiomyocyte fate. Circ Res. 2010;107:689–699. doi: 10.1161/CIRCRESAHA.110.225714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Y, Csordás G, Jowdy C, Schneider TG, Csordás N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, et al. Mitofusin 2‐containing mitochondrial‐reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle ca(2+) crosstalk. Circ Res. 2012;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. ER sliding dynamics and ER‐mitochondrial contacts occur on acetylated microtubules. J Cell Biol. 2010;190:363–375. doi: 10.1083/jcb.200911024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule‐driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550 [DOI] [PubMed] [Google Scholar]
- 20. Vandanmagsar B, Youm Y‐H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang CH, Horwitz SB. Taxol: the first microtubule stabilizing agent. Int J Mol Sci. 2017;18:1733. doi: 10.3390/ijms18081733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drum BM, Yuan C, Li L, Liu Q, Wordeman L, Santana LF. Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J Mol Cell Cardiol. 2016;93:32–43. doi: 10.1016/j.yjmcc.2016.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee CF, Liu CY, Hsieh RH, Wei YH. Oxidative stress‐induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann N Y Acad Sci. 2005;1042:246–254. doi: 10.1196/annals.1338.027 [DOI] [PubMed] [Google Scholar]
- 24. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brundel BJ, Shiroshita‐Takeshita A, Qi X, Yeh YH, Chartier D, van Gelder IC, Henning RH, Kampinga HH, Nattel S. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99:1394–1402. doi: 10.1161/01.RES.0000252323.83137.fe [DOI] [PubMed] [Google Scholar]
- 26. Zhang D, Hu X, Li J, Liu J, Baks‐te Bulte L, Wiersma M, NU M, van Marion DMS, Tolouee M, Hoogstra‐Berends F, et al. DNA damage‐induced PARP1 activation confers cardiomyocyte dysfunction through NAD+ depletion in experimental atrial fibrillation. Nat Commun. 2019;10:1307. doi: 10.1038/s41467-019-09014-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Musselman LP, Fink JL, Narzinski K, Ramachandran PV, Hathiramani SS, Cagan RL, Baranski TJ. A high‐sugar diet produces obesity and insulin resistance in wild‐type drosophila. Dis Model Mech. 2011;4:842–849. doi: 10.1242/dmm.007948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lanters EA, van Marion DM, Kik C, Steen H, Bogers AJ, Allessie MA, Brundel BJ, de Groot NM. HALT & REVERSE: Hsf1 activators lower cardiomyocyt damage; towards a novel approach to REVERSE atrial fibrillation. J Transl Med. 2015;13:347. doi: 10.1186/s12967-015-0714-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151 [DOI] [PubMed] [Google Scholar]
- 30. Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein‐protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manders EMM, Verbeek FJ, Aten JA. Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x [DOI] [PubMed] [Google Scholar]
- 32. Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, Berthiaume LG, Simmen T. Palmitoylated TMX and calnexin target to the mitochondria‐associated membrane. EMBO J. 2012;31:457–470. doi: 10.1038/emboj.2011.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang D, Ke L, Mackovicova K, Van Der Want JJ, Sibon OC, Tanguay RM, Morrow G, Henning RH, Kampinga HH, Brundel BJ. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for atrial fibrillation. J Mol Cell Cardiol. 2011;51:381–389. doi: 10.1016/j.yjmcc.2011.06.008 [DOI] [PubMed] [Google Scholar]
- 34. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. 2018;122:1460–1478. doi: 10.1161/CIRCRESAHA.118.310082 [DOI] [PubMed] [Google Scholar]
- 35. Li J, Zhang D, Brundel B, Wiersma M. Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells. 2019;8:1617. doi: 10.3390/cells8121617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. doi: 10.1038/srep11427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5:e10848. doi: 10.1371/journal.pone.0010848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumazawa A, Katoh H, Nonaka D, Watanabe T, Saotome M, Urushida T, Satoh H, Hayashi H. Microtubule disorganization affects the mitochondrial permeability transition pore in cardiac myocytes. Circ J. 2014;78:1206–1215. doi: 10.1253/circj.CJ-13-1298 [DOI] [PubMed] [Google Scholar]
- 39. Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56:414–424. doi: 10.1016/j.molcel.2014.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Siudeja K, Srinivasan B, Xu L, Rana A, de Jong J, Nollen EA, Jackowski S, Sanford L, Hayflick S, Sibon OC. Impaired coenzyme A metabolism affects histone and tubulin acetylation in drosophila and human cell models of pantothenate kinase associated neurodegeneration. EMBO Mol Med. 2011;3:755–766. doi: 10.1002/emmm.201100180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yaras N, Ugur M, Ozdemir S, Gurdal H, Purali N, Lacampagne A, Vassort G, Turan B. Effects of diabetes on ryanodine receptor ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54:3082–3088. doi: 10.2337/diabetes.54.11.3082 [DOI] [PubMed] [Google Scholar]
- 43. Yang F, Yu X, Li T, Wu J, Zhao Y, Liu J, Sun A, Dong S, Wu J, Zhong X, et al. Exogenous H(2)S regulates endoplasmic reticulum‐mitochondria cross‐talk to inhibit apoptotic pathways in STZ‐induced type I diabetes. Am J Physiol Endocrinol Metab. 2017;312:E190–E203. doi: 10.1152/ajpendo.00196.2016 [DOI] [PubMed] [Google Scholar]
- 44. Sebastián D, Hernández‐Alvarez MI, Segalés J, Sorianello E, Muñoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yaksh A, Kik C, Knops P, Roos‐Hesselink JW, Bogers AJ, Zijlstra F, Allessie M, de Groot NM. Atrial fibrillation: to map or not to map? Neth Heart J. 2014;22:259–266. doi: 10.1007/s12471-013-0481-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu L, Wang Z, Carmone C, Keijer J, Zhang D. Role of oxidative DNA damage and repair in atrial fibrillation and ischemic heart disease. Int J Mol Sci. 2021;22:3838. doi: 10.3390/ijms22083838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ho KL, Karwi QG, Wagg C, Zhang L, Vo K, Altamimi T, Uddin GM, Ussher JR, Lopaschuk GD. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc Res. 2021;117:1178–1187. doi: 10.1093/cvr/cvaa143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bolla AM, Caretto A, Laurenzi A, Scavini M, Piemonti L. Low‐carb and ketogenic diets in type 1 and type 2 diabetes. Nutrients. 2019;11:962. doi: 10.3390/nu11050962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu S, Tao H, Cao W, Cao L, Lin Y, Zhao SM, Xu W, Cao J, Zhao JY. Ketogenic diets inhibit mitochondrial biogenesis and induce cardiac fibrosis. Signal Transduct Target Ther. 2021;6:54. doi: 10.1038/s41392-020-00411-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zorzano A. Regulation of mitofusin‐2 expression in skeletal muscle. Appl Physiol Nutr Metab. 2009;34:433–439. doi: 10.1139/H09-049 [DOI] [PubMed] [Google Scholar]
- 51. Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM. Stress‐induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–557. doi: 10.1016/j.molcel.2012.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot‐Marie‐tooth disease type 2A. Science. 2018;360:336–341. doi: 10.1126/science.aao1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim DH, Park MH, Ha S, Bang EJ, Lee Y, Lee AK, Lee J, Yu BP, Chung HY. Anti‐inflammatory action of β‐hydroxybutyrate via modulation of PGC‐1α and FoxO1, mimicking calorie restriction. Aging (Albany NY). 2019;11:1283–1304. doi: 10.18632/aging.101838 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Method S1
Figures S1–S5
Data Availability Statement
All the data used in this study are available within the article and the supplemental material. The original data and algorithm for quantification are available from the corresponding authors upon reasonable request.