

Abstract
Purpose
Paclitaxel (PTX) has been widely utilized for the treatment of breast cancer. However, drawbacks, such as poor aqueous solubility, rapid blood clearance and severe toxicity, greatly reduce its efficacy and safety. Herein, a novel self-developed curcumin derivative (CUD) was chosen as the carrier to develop a long-acting PTX nano-delivery system (PTX-Sln@CUD) in order to improve its pharmacokinetic behavior, anti-breast cancer efficacy and safety.
Methods
PTX-Sln@CUD was prepared using solid dispersion and ultrasonic technology. Relevant physical and chemical properties, including stability and release behavior, were characterized. The clearance of PTX-Sln@CUD in vivo was studied by pharmacokinetic experiments. The anti-tumor activity of PTX-Sln@CUD was investigated in vitro and in vivo. Hemolysis experiments, acute toxicity and cumulative toxicity studies were performed in mice to determine the safety of PTX-Sln@CUD.
Results
The average particle size, PDI, Zeta potential, encapsulation efficiency and loading efficiency of the PTX-Sln@CUD were 238.5 ± 4.79 nm, 0.225 ± 0.011, −33.8 ± 1.26 mV, 94.20 ± 0.49% and 10.98 ± 0.31%, respectively. PTX-Sln@CUD was found to be stable at room temperature for half a year. The cumulative release rates of PTX-Sln@CUD at 24, 96 and 168 h were 17.98 ± 2.60, 57.09 ± 2.32 and 72.66 ± 4.16%, respectively, which were adherent to zero-order kinetics. T1/2, MRT (0-t) and AUC (0-t) of the PTX-Sln@CUD group were 4.03-fold (44.293 h), 7.78-fold (38.444 h) and 6.18-fold (14.716 mg/L*h) of the PTX group, respectively. PTX-Sln@CUD group demonstrated stronger anti-breast cancer activity than the PTX group. Importantly, the PTX-Sln@CUD group was safer compared to the PTX group both in vitro and in vivo.
Conclusion
PTX-Sln@CUD was verified as promising therapeutic nanoparticles for breast cancer and provided a novel strategy to solve the problem of low efficacy and poor safety of clinical chemotherapy drugs.
Keywords: breast cancer, long-acting, paclitaxel nanoparticles, curcumin derivative
Graphical Abstract

Introduction
Breast cancer, a malignant tumor that is predominantly associated with the female patient population, is a major disease that seriously threatens women’s health.1 Globally, the incidence and mortality of breast cancer has gained significant momentum in recent years, which has resulted in a more severe disease burden on society.2 Hence, there is an imminent need to discover and develop safe and effective therapeutic agents for the treatment of breast cancer. Paclitaxel (PTX) is a first-line chemotherapeutic agent that has exhibited therapeutic efficacy in the treatment of breast cancer, ovarian cancer, cervical cancer and lung cancer.3–6 However, issue, including poor aqueous solubility and rapid blood clearance, undermines overall clinical outcomes.7 PTX-related preparations often cause dose-limiting toxicity, which include hemolysis, myelosuppression and hypersensitivity,8–12 which not only reduces patient compliance, but also constitutes major side effects. In response to this, it is highly desirable to develop novel formulations of PTX with high aqueous solubility, improved pharmacokinetics, and favorable safety in vivo.
Distinct from conventional formulations, nanoparticles exhibit a superior tumor growth inhibition effect due to the stealthy protective effect in blood circulation and highly efficient delivery within tumors,13–16 which avoids premature drug leakage and improves the loading efficiency. In addition, the utilization of physical and chemical properties of specific carriers through nanoparticles not only alters hydrophobicity of drug molecules, but also controls loaded drugs to undergo sustained release, hence improving the safety of certain drugs with narrow therapeutic windows.17,18 Studies have reported that there is a variety of nanodrug delivery systems that can prolong the circulation of PTX by reducing recognition and elimination of the mononuclear phagocytic system (MPS), including surface modification with hydrophilic polymers (PEG)19,20 and surface-coating with albumin.21,22 These carrier-based PTX nanodrug delivery systems were experimentally chosen to be effective by modifying the pharmacokinetics and release behavior of drugs in order to solve the problems that often arise in the conventional chemotherapy of PTX, which improves therapeutic efficacy and reduces systemic toxicity.23,24 However, on the part of the carriers, there can be many issues including low drug loading capacity, high instability, their metabolites may cause additional short-term or long-term toxicity to the whole body, and others. Inevitably, this limits the development of nanodrug-delivery systems based on carriers.25,26 Therefore, the search for novel carrier materials with simple preparation methods, high therapeutic efficacy, low side effects and great biocompatibility is one of the main goals of the current research in nanodrug delivery systems. In this regard, one natural product (curcumin) and its derivatives have become great candidates for the aforementioned studies.27,28
The preclinical models have demonstrated that there is a pivotal role of curcumin in tumor progression by regulating cell proliferation, invasion, metastasis, apoptosis, epigenetic changes, and microRNA expression.29,30 Furthermore, curcumin has been shown to be extremely safe, even at relatively high doses, across various animal models and clinical studies.31 Given its significance, structural modification and derivatization of CU to develop and mine its greater application potential have gradually become a significant hotspot.32,33 Our group previously structurally modified the hydroxyl group of curcumin in order to obtain a novel target compound (Figure 1). Preliminary experiments validated that the derivative holds the potential for adjunctive drug molding. Thus, a novel long-acting nano-delivery system encapsulating paclitaxel was developed through the use of carrier material.
Figure 1.

The structure of CUD ((3S,8R,9S,10S,13R,14S,17R)-10,13-dimethyl-17-((R)-6-methylheptan-2-yl) hexadecahydro-1H-cyclopenta[a]phenanthren-3-yl (4-((1E,6E)-7-(4-hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dien-1-yl)-2-methoxyphenyl) carbonate).
In brief, in view of the defects of PTX in clinical application, a long-acting paclitaxel nanoparticle (PTX-Sln@CUD) was prepared through the use of a highly dispersing poorly soluble drug (PTX) in another carrier (CUD) using a solid dispersion technique with the expectation that it would improve its biological characteristics, release behavior, aqueous solubility, and pharmacokinetics. The physical and chemical properties of PTX-Sln@CUD were characterized by drug release and stability studies in vitro. Furthermore, sustained release of the drug from PTX-Sln@CUD was validated by in vivo pharmacokinetic behavior, which manifested by significantly prolonged retention time of PTX in vivo. This provided a basis for screening novel long-acting PTX formulations. In this way, PTX circulates continuously within the blood for a longer period of time, in which a single dose of the formulation can cause effects for a long duration of time, thereby reducing dosing frequency and improving efficacy. The anti-tumor effect of this PTX nanoparticle against breast cancer in vitro and in vivo was investigated through the use of cytotoxicity trials and the establishment of tumor models in nude mice, respectively. PTX-Sln@CUD further improves safety by combining properties of CUD, which can effectively enrich drugs in the tumor tissues, reduce drug dosage, and prevent damage to normal organs or tissues. Data from hemolytic assays, acute toxicity and cumulative toxicity determination provide important evidence for this nanoparticle that can help reduce the toxicity of PTX. We hope to achieve the development of an ultralong-circulating nano-delivery system using this strategy that can help improve the treatment efficiency of paclitaxel and reduce its toxicity. It can also further address the problem of frequent administration due to short in vivo action time of nano-preparation in clinical practice.
Materials and Methods
Materials
The following reagents, drugs and chemicals were utilized in this study. Paclitaxel (PTX; purity > 98%) was purchased from Luzhou Renkang Biotechnology Co., Ltd. The curcumin derivative (CUD; purity > 98%) was made within the laboratory. Distearoyl phosphatidylethanolamine-polyethylene glycol 2000 (DSPE-PEG2000; Lipoid GmbH, Germany), Poloxamer F68 (Shanghai Aladdin Biochemical Technology Co., Ltd.), trichloromethane (Luzhou Renkang Biotechnology Co., Ltd.) and thiazole blue (MTT; Beijing Soleboro Technology Co., Ltd.) were used in this study. All reagents were either analytical or high-performance liquid chromatography grade.
Cells
Human breast cancer (MCF-7) cells and normal hepatocytes (L02) were purchased from the Cell Bank Center of Chinese Academy of Sciences, and were cultured in DMEM high-glucose medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were cultured in a 37°C cell incubator that contained 5% carbon dioxide humidified air.
Animals
Female BALB/c mice, aged seven weeks old and weighing 20 ± 3 g, were utilized to evaluate acute and cumulative toxicity in vivo and purchased from the Chengdu Dashuo Laboratory Animal Co., Ltd. (Laboratory Animal Use License: SYXK; Sichuan; 2018-065). Female Sprague Dawley (SD) mice, weighing 220 ± 20g, were used to investigate the pharmacokinetic behavior and were purchased from the Experimental Animal Center of Southwest Medical University (Laboratory Animal License: SYXK; Sichuan; 2018-181). All mice were housed indoors with alternating light and dark cycles for 12 h. Additionally, they had free access to water and food during the adaptation period. Four-week-old female BALB/c nude mice (Laboratory Animal Production License No. SCXK; Beijing; 2016-0002) were also used to study to determine the in vivo antitumor effect. The mice were purchased from Sparford (Beijing) Biotechnology Co., Ltd. and raised in the SPF animal room of the Animal Center of Southwest Medical University. Animal procedures were approved by the Committee on the Ethics of Animal Experiments of the Southwest Medical University for Nationalities, Luzhou, People’s Republic of China (No 20180309009). All the animal studies followed the Animal Welfare Act and were compliant with the regulations of Southwest Medical University and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Methodology
Synthesis of CUD
First, 110 mg of CU was dissolved in anhydrous dichloromethane, which was then supplemented with an additional 96 μL of triethylamine (TEA). Subsequently, 135 mg of cholesteryl chloroformate was dissolved in anhydrous dichloromethane and slowly dropped to react in an ice bath for 1 h. Then, the solvent was removed using distillation under reduced pressure after completing the reaction. Finally, the desired product was obtained using an extraction method. The precipitate was retained after five ultrasonic washes with methanol. Then, the pellet was washed with isopropanol, which was followed by centrifugation to collect the supernatant. Finally, CUD was obtained using rotary evaporation of the supernatant in order to remove the solvent.
Preparation of Nanoparticles
Specified amounts of PTX (5.25 mg), CUD (4.55 mg), F68 (22.5 mg), and DSPE-PEG2000 (5.25 mg) were dissolved in 3 mL of trichloromethane in a beaker. The solvent was then evaporated at a speed of 300 r/min using a magnetic stirrer at 40 °C. In order to ensure there was no residual trichloromethane, the solvent was further removed using nitrogen, and then cured in an oven at 40°C for about 30 min. The nanoparticles were then uniformly dispersed using a probe sonicator (Ultrasonic cell disruptor; SCIENTZ-IID; China) after adding deionized water for hydration. Then, clarified PTX-SLN@CUD nano-solution was obtained using ultrasonic fragmentation at an intensity of 185 W for 10 min.34 In order to have long-term storage, 1% sucrose (w/v) was added as a lyophilization protectant to obtain a yellow loose powder through freeze-drying (Freeze-drier, LGJ-18C, China) at −68°C for about 28 h (Figure 2C).35
Figure 2.

Size and Zeta potential of optimized formulation of PTX-SLN@CUD (A) Size and PDI; (B) Zeta potential n=3 per group). (C) Imaging of PTX-SLN@CUD after freeze-drying. (D) TEM image of PTX-SLN@CUD. (E) FTIR patterns of PTX, PTX-SLN@CUD, and Blank-SLN@CUD.
Characterization of Nanoparticles
Morphology, Particle Size, Zeta Potential (ZP), Polydispersity Index (PDI), Encapsulation Efficiency (EE) and Loading Efficiency (LE)
The PTX-SLN@CUD solution was then diluted using distilled water at a ratio of 1:10 to the appropriate concentration, which was then measured three times within a sample cell (1 mL) through the use of Malvern particle size analyzer (Particle size analyzer, Zeta-sizer Nano S90, UK) at 25°C for particle size, PDI (which represents the uniformity of particle size distribution) and ZP, respectively. The light scattering angle was set at 90°. Morphology examination of the samples was conducted through the use of transmission electron microscopy (TEM) (JEM-1200EX, Japan).
Encapsulation efficiency (EE) and loading efficiency (LE) were quantified by centrifuging freshly-prepared PTX-SLN@CUD nano-solution at 3000 r/min for 10 min. The free PTX was the precipitated while the supernatant contained homogeneous PTX nanoparticles. In order to break the nanoparticles, 4 mL of methanol was added to 100 μL of the supernatant of PTX-SLN@CUD, which was followed by sonication for 5 min. Furthermore, 100 μL of uncentrifuged PTX-SLN@CUD was processed, as described above. The content of PTX was detected through HPLC in order to determine the drug content entrapped in the nano-liquid.
The encapsulation efficiency and loading efficiency formulas are as follows:
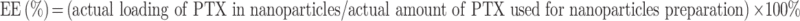 |
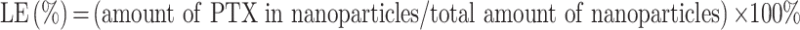 |
The chromatographic conditions are described below. The liquid chromatographic protection column was the Phenomenex C18 (4.0 mm x 3.0 mm), the liquid chromatographic column was Luna 5 μm C18 (2) 100Å (4.6 mm × 250 mm), the elution time was 10 min, the mobile phase was the acetonitrile-water phosphate (PH 4.0) = 58:42 (v/v), wavelength was 227 nm, the injection volume was 20 μL, the column temperature was 30 °C, and the flow rate was 1 mL/min.
FTIR Analysis
Using the Fourier transform infrared spectrometer (Shimadzu IRAffinity-1S, Japan), the spectra of each group of samples in the range of 4000–400 cm−1 were recorded.
In vitro Stability
An appropriate amount of nano-solution was stored at room temperature for 24 h and then collected to determine the particle size, PDI, entrapment efficiency, and loading efficiency. The stability of the sample was investigated by calculating the coefficient of variation (CV) of the four indexes compared to the original. In order to guarantee the long-term preservation of the liquid formulation of nanoparticles, the powdered solid formulation was prepared through the freeze-drying technique. The PTX-SLN@CUD lyophilized samples were kept in brown ampoules for 6 months, and then accompanied by a temperature of 25 ± 2°C, as well as a relative humidity of 40 ± 5%. The same procedure described above was then performed after dissolving the samples in deionized water at 1, 2, 3, and 6 months, respectively.
In vitro Release Investigation
The release profile of PTX-SLN@CUD was then investigated with a total volume of 200 mL of PBS phosphate buffer, containing 0.2% Tween-80, as the release medium. Due to the poor water solubility of free PTX, only a small amount of surfactant (Tween-80) was added into the release medium to ensure sink conditions.36 Next, 1 mL of PTX-SLN@CUD and free PTX at a concentration of 200 ug/mL were then separately placed into a dialysis bag (MD25 dialysis bag, 8000–14,000 Da, USA) for the release trial using a dissolution apparatus (Intelligent dissolution tester; ZRS-8G; China) at 37 °C under 120 r/min. An equivalent amount of release medium (3 mL) was withdrawn at 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48, 72, 96, 120, 144 and 168 h. It was then centrifuged at 8000 r/min for 10 min. Finally, the supernatant was subjected to HPLC detection for analysis of the cumulative release.
Three common models were used to fit the in vitro drug release kinetic models of each group of samples, and the fitting coefficient (r2) was calculated.37
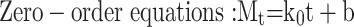 |
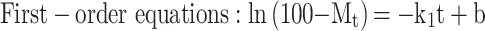 |
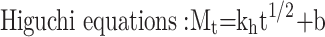 |
where Mt is the cumulative release at time t, the k0, k1, and kh are the rate constants for the zero-order, first-order, and Higuchi equations, respectively.
Pharmacokinetics
Overall, 10 SD rats were randomly divided into two groups (n=5 in each group), which were treated sequentially with 10 mg/kg of PTX and 10 mg/kg of PTX-SLN@CUD. Additionally, for each rat after drug administration, the blood was sampled at 0.08, 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48, 72, 96, and 120 h. The blood (0.3 mL) was centrifuged for 3 min (5000 rpm/min) in a sodium heparin-containing centrifuge tube, after which 100 μL of the supernatant plasma was collected for detection. The plasma was then vortexed for 3 min in 0.5 mL of extractant (methanol: ethyl acetate = 10:90, v/v), and then the supernatant was obtained after centrifugation at 8000 rpm/min for 3 min. In order to ensure adequate extraction, the remaining precipitate was added to 0.5 mL of the extractant in order to repeat the extraction. The two supernatants were then combined and blow dries with nitrogen at 35°C. Then, 200 μL of the complex solvent [phosphoric acid: acetonitrile = 75:25 (pH 3.5)] was added and then vortexed for 4 min until it was completely dissolved. After sonication for 4 min and then centrifugation at 10,000 rpm/min for 10 min, the supernatant was then collected and detected by HPLC.
The chromatographic conditions are described below. The liquid chromatographic protection column was Phenomenex C18 (4.0 mm x 3.0 mm), the liquid chromatographic column was Luna 5 μm C18 (2) 100Å (4.6 mm × 250 mm), the elution time was 10 min, the mobile phase was acetonitrile-water phosphate (PH 4.0) = 65: 35 (v/v), the wavelength was 227 nm, the injection volume was 20 μL, the column temperature was 30 °C, and the flow rate was 0.8 mL/min.
Pharmacodynamic Evaluation
In vitro Cytotoxicity Assay
The toxicity of nanoparticles on the breast cancer cell line MCF-7 and the normal hepatocyte cell line L02 was determined using thiazole blue colorimetry (MTT colorimetry). In the logarithmic growth phase, MCF-7 and L02 cells were then trypsinized in order to prepare a cell suspension. The cells were then counted and seeded onto a 96-well plate at a density of 5×103 cells/well. After the cells were cultured at 5% CO2 and 37°C for 24 h, five different concentrations (5, 10, 20, 40 and 80 ug/mL) of free PTX, PTX-SLN@CUD and blank nanoparticles were then added to the cultured cells, respectively. Each well was supplemented with 20 µL of MTT solution (5 mg/mL) after 24, 48, 72, and 96 h of continuous culture. The cultures were then terminated after 3–4 h, after which the medium was aspirated and 150 μL of dimethyl sulfoxide was added. In order to dissolve the crystals sufficiently, the mixing was done for 10 min. The absorbance value (OD) of each well was measured using a microplate reader at 490 nm wavelength. The growth inhibition rate of drugs on tumor cells was then calculated according to the formula, and then the dose-effect curve was plotted with drug concentration (ug/mL) as abscissa.
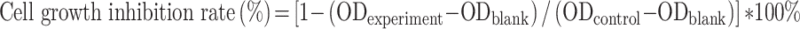 |
OD blank is the OD of the blank well that contains only medium. The OD Control is the OD of the control well. The OD experiment is the OD of the experimental well.
In vivo Antitumor Assessment
The in vivo antitumor effect of PTX nanoparticles was investigated in tumor-bearing nude mice. First, 0.1 mL of the tumor cell suspension (3 × 107 cells/mL) was inoculated into the left armpit of female BALB/tumor-bearing nude mice. Then, 30 nude mice were randomly divided into three groups, including (1) control group (normal saline), (2) 10 mg/kg PTX group, and (3) 10 mg/kg PTX-SLN@CUD group. The pharmacologic intervention was initiated when tumors grew to approximately 100 mm3 in volume. All groups were injected intraperitoneally every three days for a total of 20 days.
After drug intervention on tumor-bearing nude mice, the body weight and the tumor volume of all tumor-bearing nude mice were measured every 3 days. Furthermore, the cumulative number of deaths of tumor-bearing nude mice in each group was tallied every five days. The formula V = π/6 (AB2) was chosen in order to calculate the volume (V) of the tumor, where A and B represent the largest and shortest vertical axes, respectively. Then, the average body weight and tumor volume change curves of each group were plotted. After administration for 20 days, the nude mice were then euthanized and the tumor was removed for weighing. The tumor inhibition rate (IR) was calculated using the following formula: IR = (1-tumor weightdrug/tumor weightcontrol×100%).
Tumor tissues were dehydrated using absolute ethanol and then fixed in 4% paraformaldehyde for 24 h. Then, they were embedded by paraffin to prepare individual tumor sections. Antigen retrieval of tissue sections was performed in a high-temperature environment in 0.01 M citric acid buffer (pH 6.0) for 5 min. The sections were then incubated with the primary antibody targeted against KI67 overnight at 4 °C, followed by dropwise biotinylated secondary antibody incubation for 30 min at 37 °C. Ultimately, the sections were subjected to image acquisition using a microscopic camera system after color development with 3,30-diaminobenzidine (DAB) and hematoxylin staining. All experimental procedures were conducted in a blinded manner.
Safety Evaluation
In vitro Hemolysis
An appropriate amount of SD rat blood was continuously stirred in a clockwise manner through a glass rod for 5 min in order to destroy the fibrinogen. After adding 0.9% of the sodium chloride solution, centrifugation (1500 r/min, 10 min) was performed, and the supernatant was decanted. To obtain erythrocytes, 0.9% sodium chloride solution was added and the process of centrifugation was repeated until the supernatant exhibited no color. An erythrocyte suspension at a concentration of 2% was configured by supplementation with 0.9% sodium chloride solution. The different concentration groups of PTX-SLN@CUD (1–7), negative control (2% saline) and positive blank control (Ultrapure water) were placed in a 37 °C incubator. All groups were evaluated for hemolysis after centrifugation (1500 r/min, 10 min) at 1, 3, and 5 h, respectively. The supernatant of each sample after 5 h was supplemented into a 96 well plate. The microplate reader determined the absorbance (A) at a wavelength of 540 nm, and the hemolysis rate (%) was calculated by the following formula.38
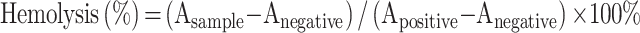 |
In vivo Acute and Cumulative Toxicity
Overall, 60 mice were randomly divided into six groups (n=10 per group) and were administered the following: (A) PTX at 40 mg/kg, (B) PTX-SLN@CUD at 40 mg/kg, (C) normal saline with the same volume as the group A or B, (D) PTX at 10 mg/kg, (E) PTX-SLN@CUD at 10 mg/kg and (F) normal saline with the same volume as the group D or E. The first three groups comprised the acute toxicity trial, and were dosed once throughout the abdominal cavity after being weighed for each mouse. Additionally, mice in groups C, D and E were to evaluate cumulative toxicity, which was injected intraperitoneally after weighing all mice. The drug was administered every three days for 60 days.
Throughout the experiment, the status of the mice, as well as the number of deaths, were observed and recorded every day. On day 60 after drug administration, the plasma and tissue (heart, liver, kidney) samples were collected from three randomly selected mice within each group for analysis and detection. The blood biochemical identification of liver and kidney function mainly manifested in the plasma samples. The following parameters were measured, including alanine transaminase (ALT), aspartate transaminase (AST), lactate dehydrogenase (LDH), urea (urea), creatinine (crea) and uric acid (UA). Tissue (heart, liver, and kidney) samples were stained using hematoxylin-eosin (HE) prior to pathological section analysis. All operating procedures were conducted according to the requirements of ethical regulations.
Statistical Analysis
All data were expressed as mean ± SEM deviation by SPSS 19.0 software. A One-way ANOVA and Tukey’s test were utilized for comparison between groups. P ≤ 0.05 was considered to be statistically significant.
Results
Characterization of CUD
CUD was characterized as follows: 1H-nuclear magnetic resonance (NMR) (400 MHz, Chloroform-d) δ 7.59 (dd, J = 15.9, 5.1 Hz, 2H), 7.21–7.01 (m, 6H), 6.98–6.88 (m, 1H), 6.51 (ddd, J = 21.3, 15.7, 3.9 Hz, 2H), 5.83 (d, J = 3.8 Hz, 1H), 5.42 (s, 1H), 4.57 (dt, J = 11.4, 5.8 Hz, 1H), 3.92 (dd, J = 17.8, 3.7 Hz, 6H), 2.48 (d, J = 14.2 Hz, 2H), 2.10–0.82 (m, 43H), 0.69 (d, J = 3.9 Hz, 3H). Electrospray ionization mass spectrometry (ESI-MS) m/z; 779.54 metal hydride [M+H]+.
For the structure of CUD, see Figure 1.
Characterization of Nanoparticles
Morphology, Particle Size, Zeta Potential (ZP), Polydispersity Index (PDI), Encapsulation Efficiency (EE) and Loading Efficiency (LE)
The data on particle size, PDI, zeta potential, loading efficiency and entrapment efficiency of PTX nanoparticles are collated in Table 1. The suitable particle size of the prepared nanoparticle (PTX-SLN@CUD) was 238.5 ± 4.79 nm. This endows the nanoparticles with a certain passive targeting ability. Since the capillary gaps in the cancer vessels exceed 400 nm, the distribution of PTX-SLN@CUD in tumor tissue increased through the EPR effect.39 The PDI is 0.225 ± 0.011, which indicates that the particles are uniformly distributed and then normalized to a single peak (Figure 2A). TEM of PTX-SLN@CUD shows the preparation was quasi-spherical in shape and uniformly dispersed without any visible signs of aggregation, which was consistent with the size distribution (Figure 2D). Zeta potential, an important indicator to evaluate the stability of nanoparticles, was quantified to be −33.8 ± 1.26 mV (Figure 2B). The surfaces of the prepared nanoparticles carry a large negative charge, which causes the electrostatic repulsion between neighboring particles to increase the distance between particles, thereby decreasing the aggregation of nanoparticles.40
Table 1.
Physicochemical Characteristics of PTX-SLN@CUD Nanoparticles
| Formulation | Size(mn) | PDI | ZP(mV) | EE(%) | DL(%) |
|---|---|---|---|---|---|
| PTX-SLN@CUD | 238.5 ± 4.79 | 0.225 ± 0.011 | −33.8 ± 1.26 | 94.20 ± 0.49 | 10.98 ± 0.31 |
Note: Each data point represents the mean ± SEM (n=3).
Abbreviations: PDI, polydispersity index; ZP, zeta potential; EE, encapsulation efficiency; LE, loading efficiency.
FTIR Analysis
FTIR (Figure 2E) showed that some of the characteristic peaks of PTX were masked in PTX-SLN@CUD, indicating that the crystal form of PTX was well encapsulated by SLN@CUD.
In vitro Stability
The coefficients of variation of various indexes of the PTX nanoparticles that were stored at room temperature for 24 h were all less than 10%, which indicated that the preparation was relatively stable (Figure 3A). For long-term preservation, PTX-SLN@CUD lyophilized powder formulation was prepared via the freeze-drying technique to improve stability. There was no significant improvement in particle size, PDI, encapsulation efficiency, and loading efficiency of the PTX-SLN@CUD lyophilized powder within 6 months, which confirmed that PTX-SLN@CUD is steady when it is stored under suitable conditions for 6 months (Figure 3B).
Figure 3.

PTX nanoparticle stability. Variation coefficients of various indicators of PTX-SLN@CUD at 24 h(A). Coefficient of variation curve of PTX-SLN@CUD lyophilized powder preparation(B), and the nanoparticles were collected at 1, 2, 3 and 6 months. Each data point represents the mean ± SEM (n=3).
Abbreviations: PDI, polydispersity index; EE, encapsulation efficiency; LE, loading efficiency.
In vitro Release Investigation
The in vitro release profile of PTX-SLN@CUD shows that the cumulative release rates at 24, 96 and 168 h were 17.98 ± 2.60, 57.09 ± 2.32 and 72.66 ± 4.16%, respectively (Figure 4A). This fits the data according to the in vitro release degree results, while the in vitro release behavior of PTX-SLN@CUD was more in accordance with the zero-order model (Table 2), and its release rate did not change with time. However, the in vitro release behavior of free PTX was similar to the first-order model. The free PTX group released over 60% of its drug content rapidly within 24 h. In brief, PTX-SLN@CUD modified by CUD carrier presented a slow-release and long circulation effect with a minimal initial burst release. The control over the initial burst release is able to significantly reduce systemic toxicity, while the sustained-release model would contribute to maintaining therapeutic drug levels within the tumor.41 Moreover, the formed nanostructured framework limited the corresponding drug to some extent, so that the nanoparticles relatively controlled and delayed the release of this drug,42 which may also be related to the excipient DSPE-PEG2000 added in the prescription.
Figure 4.

(A) Release profile of free PTX and PTX-SLN@CUD at different time points over 168 h. Each data point represents the mean ± SEM (n=3). (B) Plasma drug concentrations vs time profiles following injection administration of PTX-SLN@CUD and PTX over 120 h. Each data point represents the mean ± SEM (n=5).
Table 2.
Drug Release Kinetic Parameters and Fitting Coefficients
| Zero Order Equation | First-Order Equation | Higuchi’s Equation | ||||
|---|---|---|---|---|---|---|
| r2 | k0 | r2 | k1 | r2 | kh | |
| PTX | 0.7620 | 0.5097 | 0.9514 | 0.0711 | 0.9124 | 7.2999 |
| PTX-SLN@CUD | 0.9682 | 0.4839 | 0.9664 | 0.0068 | 0.9100 | 6.3691 |
Notes: r2, fitting coefficient; K0, K1, and Kh are the rate constants for the zero-order, first-order, and Higuchi equations, respectively.
Pharmacokinetics
The pharmacokinetic behaviors of free PTX and PTX-SLN@CUD in SD rats over 120 h were delineated through a concentration-time curve (Figure 4B). Additionally, the relevant pharmacokinetic parameters were calculated (Table 3). The blood concentration of free PTX decreased below the detection limit after an intravenous injection for 12 h, which indicated rapid clearance from the blood. However, PTX-SLN@CUD modified by CUD led to a significantly improvement in its PK profile. Significantly, higher plasma PTX concentrations were observed in rats that were subjected to PTX-SLN@CUD, compared to those exposed to free PTX, which persisted until 120 h. Hence, it can be inferred that PTX was effectively encapsulated in CUD, thereby avoiding rapid clearance from the body, with longer time required to complete the metabolism in vivo. The T1/2, MRT (0-t) and AUC (0-t) of PTX-Sln@CUD group were 4.03 times (44.293 h), 7.78 times (38.444 h) and 6.18 times (14.716 mg/L*h) compared to the PTX group, respectively (Table 3). Moreover, its clearance (CL) was 1/7 of the free PTX group. The differences in the values of these indexes validated that PTX-SLN@CUD significantly prolonged residence time of drug in the blood with an improved long-circulating effect. PTX-Sln@CUD is able to stabilize the nanoparticles in plasma or confer stealth properties, and improve bioavailability by maintaining a higher drug concentration in blood for a long time.
Table 3.
Pharmacokinetic Parameters in Rats Following Injection Administration of PTX-SLN@CUD and PTX
| Parameter | Units | 10 mg/kg | |
|---|---|---|---|
| PTX | PTX-SLN@CUD | ||
| AUC(0-t) | mg/L*h | 2.381 ± 0.276 | 14.716 ± 1.985** |
| MRT(0-t) | h | 4.94 ± 0.653 | 38.444 ± 2.983** |
| t1/2z | h | 10.991 ± 0.773 | 44.293 ± 3.012** |
| Tmax | h | 0.25 ± 0.02 | 0.61 ± 0.07** |
| Vz | L/kg | 54.467 ± 8.532 | 37.46 ± 4.82** |
| CLz | L/h/kg | 3.434 ± 0.312 | 0.443 ± 0.052** |
Note: **P < 0.01 compared with PTX.
Abbreviations: AUC, area under concentration-time curve; t1/2z, half-life time; MRT, mean residence time; Tmax, time to maximum plasma concentration; Vz, distribution volume; CLz, clearance.
Pharmacodynamic Evaluation
In vitro Cytotoxicity Assay
Next, we investigated the viability of MCF-7 cells that had been incubated with blank preparations at different concentrations for 24, 48, 72, and 96 hours at 37°C (Figure 5). As the concentration of preparation increased, the cell viability of MCF-7 cells remained above 85% at each time point, which indicated a good safety profile of the blank preparation (CUD-LN). However, both free PTX and PTX-SLN@CUD inhibited the growth rate of breast cancer cells (MCF-7), to some extent, in a concentration- and time-dependent manner. Prior to 48 h, PTX tended to be slightly better inhibited compared to PTX-SLN@CUD, but the inhibitory rate of PTX-SLN@CUD was at least 10% higher than compared to PTX at 72h and 96 h. In addition, the IC50 values of PTX-SLN@CUD against MCF-7 cells (Table 4) were also decreased by 30.5% and 35.5%, respectively, compared to PTX at 72 h and 96 h. This outcome may be affected through the sustained release effect of PTX-SLN@CUD, which also coincided with the rule of previous in vitro drug release assays of PTX-SLN@CUD. Moreover, the inhibition ratio caused by PTX-SLN@CUD was found to be significantly higher compared to the PTX group at low and medium concentrations (20 and 40 μg/mL, respectively), confirming that it has stronger cytotoxicity against MCF-7 cells.
Figure 5.

The inhibition rate (%) of PTX, CUD-LN, and PTX-SLN@CUD on MCF-7 cells was measured at 24 h (A), 48 h (B), 72 h (C) and 96 h (D), respectively ( , n=3). *P < 0.05, there was a significant difference compared with PTX.
, n=3). *P < 0.05, there was a significant difference compared with PTX.
Table 4.
IC50 Values of PTX, CUD-LN and PTX-SLN@CUD Against MCF-7 Tumor Cells and Normal Hepatocytes L02 at 24 h, 48 h, 72 h and 96 h
| Cells | Group | IC50 (μg/mL) | |||
|---|---|---|---|---|---|
| 24 h | 48 h | 72h | 96h | ||
| MCF-7 | PTX | >80 | 64.29±5.73 | 26.25±2.68 | 12.66±0.97 |
| CUD-LN | – | – | – | – | |
| PTX-SLN@CUD | >80 | 75.5±4.19 | 18.25±1.37 | 8.172±0.73 | |
| L02 | PTX | >80 | >80 | 36.86±3.18 | 17.34±0.83 |
| CUD-LN | – | – | – | – | |
| PTX-SLN@CUD | >80 | >80 | >80 | >80 | |
Note: since the inhibition of MCF-7 and L02 cells by CUD-LN at any concentration at each time point was very low (< 15%), the corresponding IC50 values could not be calculated.
Abbreviation: IC50, half maximal inhibitory concentration.
With regards to the normal hepatocyte cell line L02, the inhibitory effect of PTX was higher than that of PTX-SLN@CUD at various concentrations and time points (Figure 6). Additionally, the IC50 values of PTX-SLN@CUD on L02 cells were much higher compared to PTX at 72 h and 96 h. Free PTX at a concentration of 10 μg/mL was found to exhibit a 40.13% higher inhibitory rate on cell growth compared to PTX-SLN@CUD at 96 h, which illustrated that the safety profile of PTX-SLN@CUD was superior to that of PTX. The toxicity of the blank preparation, CUD-LN, against L02 was very mild, with no greater than 15% inhibition of L02 at different concentrations and time points.
Figure 6.

The inhibition rate (%) of PTX, CUD-LN, and PTX-SLN@CUD on L02 cells was measured at 24 h (A), 48 h (B), 72 h (C) and 96 h (D), respectively ( , n=3). *P < 0.05, there was a significant difference compared with PTX.
, n=3). *P < 0.05, there was a significant difference compared with PTX.
In vivo Antitumor Investigation
The tumors grew in different proportions after initiating drug administration to the nude mice within each group, among which the tumor volume increased more significantly in the model group, as it reached 1800 mm3 after 20 days of administration (Figure 7A). The tumor growth rate of the free PTX group and PTX-SLN@CUD group was delayed, especially in the PTX-SLN@CUD group. After 13 days of administration, tumor volume in this group of nude mice was only 750 mm3 and was subsequently maintained at 750 mm3 until the trial was completed. Meanwhile, the average weight of tumors in the PTX-SLN@CUD group was also the lightest (Figure 7F). The tumor volume was significantly suppressed in nude mice after initiating treatment with free PTX. However, tumor volume followed another upward growth trend after 13 days, reaching 1000 mm3 by 20 days. Therefore, PTX-SLN@CUD was able to effectively inhibit MCF-7 tumor growth in nude mice, and the efficacy was superior to that of free PTX. These results were also confirmed by the static tumor inhibition rate results, as the antitumor efficacy of PTX-SLN@CUD was 44.34% compared to PTX (32.93%) (P < 0.05; Figure 7E).
Figure 7.

In vivo activity of PTX-SLN@CUD. (A) Variety of tumor volume in vivo anticancer trials. (B) Changes in mouse body weight (n=6 per group). (C) After 20 days of drug administration, the mice in each group were sacrificed, and tumor tissues were collected and imaged. (D) Ki-67 immunohistochemical staining of tumors excised from animals in the control, PTX, and CU-PTX-LN groups, respectively. (E) Tumor inhibitory rates (%) of MCF-7-bearing nude mice treated with free PTX and PTX-SLN@CUD on day 20, **p < 0.01 compared with PTX. (F) Tumor weights (mg) of MCF-7-bearing nude mice treated with PTX and PTX-SLN@CUD on day 20, after which the mice were humanely sacrificed, **p < 0.01, ****p < 0.0001, administration group vs control.
After the in vivo antitumor evaluation was conducted, Ki-67 immunohistochemical staining was utilized to investigate the anti-proliferation efficacy of each experimental group. Interestingly, the weakest immunohistochemical reaction (< 20%) was observed in the PTX-SLN@CUD group (Figure 7D). In fact, H&E staining plots exhibited various degrees of necrotic areas across all treatment groups, with the exception of the saline control group. Approximately one-half of cancer cell necrosis was identified upon treatment with PTX-SLN@CUD, compared to 25% necrosis in cancer cells corresponding to free PTX, which was consistent with the above conclusions of Ki-67 IHC staining analysis.
Although the average body weight of the nude mice in each group decreased after administration, the least decrease was seen in the PTX-SLN@CUD group. There were no significant differences in the average body weight of nude mice after 20 days of drug administration compared to the beginning (Figure 7B). Free PTX led to the death of three nude mice during the test, which may be related to the combined toxicity caused by PTX, as well as the mixed solvent (ie, ethanol and hydrogenated castor oil).43 In contrast, PTX-SLN@CUD nanoparticles showed no obvious side effects, indicating that they are safer than free PTX.
Safety Evaluation
In vitro Hemolysis
Hemolysis in different PTX-SLN@CUD concentration groups as well as hemolysis rates, in which low concentration of PTX-SLN@CUD (groups 1–2) did not cause any hemolysis, either in terms of appearance or hemolysis rate (Figure 8). Similarly, the appearance of PTX-SLN@CUD within the medium and high concentration groups (groups 3–7) did not exhibit a distinct red color, and the hemolysis did not exceed more than 5%. Ethanol and hydrogenated castor oil are often chosen clinically as mixed solvents in order to improve the solubility of free PTX. However, the mixed solvents, in turn, could further aggravate the hemolytic reaction.8 PTX-SLN@CUD modified by CUD as a carrier not only helped avoid the hemolytic reaction of PTX, but also greatly improved its aqueous solubility.44
Figure 8.

The phenomenon of hemolysis of PTX-SLN@CUD with different time points ((A) 1 h; (B) 3 h; (C) 5 h). The hemolysis rates of different concentrations of PTX-SLN@CUD (D).
In vivo Acute and Cumulative Toxicity
Mouse Mortality
The conclusion of acute toxicity trials is available in Figure 9A. The 10 mice all died within 5 h after a single intraperitoneal injection of free PTX at 40 mg/kg, which deemed to be due to severe allergy and cardiotoxicity caused by free PTX and the mixed solvent (hydrogenated castor oil and absolute ethanol).45 However, only four mice died within seven days after a single intraperitoneal injection of 40 mg/kg PTX-SLN@CUD, though mice activity was decreased compared to before the injection, but the respiratory acceleration and convulsion phenomenon did not appear. The six mice that subsequently survived also gradually returned to normal, and there was no increase in dead mice between 7 and 60 days. In the cumulative toxicity assays, three mice died within seven days in the free PTX group, and all mice in this group died within 60 days (Figure 9B). Comparatively, the PTX-SLN@CUD nanoparticles group only had two numbers of mouse deaths throughout the experiment.
Figure 9.

Survival curves of mice in toxicity assays. (A) Survival curves of mice in free PTX and PTX-SLN@CUD groups over 10 days after a single 40 mg/kg dose administration. (B) Survival curves of mice in free PTX and PTX-SLN@CUD groups after continuous administration at a dose of 10 mg/kg for 2 months.
Blood Biochemical Parameters
The whole mice of the PTX acute toxicity group (group A) were weak in vital signs, and they died soon after administration. Therefore, the corresponding plasma samples were not able to be collected. Comparison of biochemical parameters among the mice in the PTX-SLN@CUD acute toxicity group compared to the control group showed that one mouse had an LDH value that was slightly below the reference range, while the CREA index of another mouse was slightly lower than the reference range without any clinical guiding significance (Table 5).46 In general, only one or two biochemical indexes were abnormal in the same group of mice, which confirmed the negligible liver and kidney injury of mice by PTX-SLN@CUD. In the cumulative toxicity assay, the relevant indicators of each mouse in the PTX group revealed that there were high values compared to the control group. Particularly, there were high levels of ALT, AST and LDH, which suggests that metabolic processes, such as normal biochemical enzymology in the liver, are affected.47 Values of UREA, CREA, and UA above the upper limit of the reference range also suggests that there was damage to the kidneys and that it affected normal physiological and biochemical processes.48 Conversely, the biochemical parameters of all mice were normal within the PTX-SLN@CUD group, which demonstrates a great safety profile.
Table 5.
Effects on Serum Biochemical Parameters of Mice After Intraperitoneal Administration in Different Experimental Groups
| Biochemical Indicators | Group B | Group C | Group D | Group E | Group F | Reference Range | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALT (U/L) | 80.4 | 69.7 | 73.5 | 51.1 | 63.8 | 47.1 | 46.8 | 107.5 | 113.2 | 83.6 | 60.7 | 79.1 | 66.4 | 74.8 | 50.8 | 33.0—98.7 |
| AST (mmol/L) | 2.68 | 6.04 | 3.55 | 5.42 | 4.82 | 6.04 | 4.42 | 8.33 | 6.06 | 4.78 | 7.94 | 6.23 | 2.69 | 5.04 | 3.43 | 2.00—7.70 |
| LDH (U/L) | 97.8 | 166.7 | 424.6 | 439.7 | 368.5 | 347.2 | 567.9 | 632.5 | 608.4 | 369.2 | 406.7 | 516.3 | 328.5 | 269.7 | 440.3 | 101.0—514.0 |
| UREA (μmol/L) | 30.9 | 49.7 | 45.8 | 58.2 | 99.2 | 73.6 | 112.1 | 93.5 | 126.0 | 48.0 | 78.8 | 56.7 | 46.2 | 57.0 | 81.6 | 22.0—97.0 |
| CREA (μmol/L) | 78.2 | 20.5 | 79.2 | 59.9 | 39.8 | 66.4 | 144.7 | 196.5 | 152.8 | 69.2 | 59.3 | 102.8 | 79.6 | 102.7 | 94.5 | 20.0—120.0 |
| UA (U/L) | 116.5 | 205.3 | 103.0 | 108.4 | 97.4 | 122.6 | 90.8 | 246.7 | 185.3 | 90.4 | 118.5 | 176.2 | 142.5 | 166.2 | 104.9 | 69.5—210.0 |
Notes: all mice in group A died after a single intraperitoneal administration, and the corresponding samples could not be collected. A Single Intraperitoneal Injection of PTX (A), PTX-SLN@CUD (B) at 40 mg/Kg and an Equal Volume of Saline (C). Intraperitoneal Injection Every Three Days for 60 Days: PTX (D), PTX-SLN@CUD (E) at 10 mg/Kg and an Equal Volume of Saline (F). (n=3).
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; CREA, creatinine; UA, uric acid.
Pathology Slides
The sliced images of the heart, liver, and kidney tissues from the acute toxicity group of PTX (group A) indicated that there was severe necrosis in the corresponding tissues (Figure 10A), which included deformation and necrosis of cardiac muscle fibers, nuclear division of hepatocytes, as well as thickening of liver fibers. However, these lesions caused after a single high-dose injection of PTX-SLN@CUD in mice were very mild, which demonstrates that its damage to heart, liver, and kidney tissues was significantly lower compared to free PTX in acute toxicity assays. Moreover, the tissues of mice in the PTX-SLN@CUD cumulative toxicity group (group E) did not reveal any apparent lesions and necrosis. Some damage to cells and muscle fibers appeared within the tissues of the heart, liver and kidney parts of mice after free PTX was continuously administered. Therefore, PTX-SLN@CUD exhibits a higher safety index, accompanied by less cumulative toxicity, compared to free PTX, which also further validated that the toxicity caused by PTX-SLN@CUD may be more tolerable than PTX during the course of long-term medication.
Figure 10.

Pathological assessments of H&E-stained organs excised from mice subjected to normal saline, PTX, and PTX-SLN@CUD. (A) The dose was 40 mg/kg and injected intraperitoneally once, (B) The dose was 10 mg/kg, injected intraperitoneally every three days for 60 days.
Discussion
Nanoparticle drug delivery system and nanomaterials, which are widely studied and applied to specific routes of administration, have become a hotspot over the years.49 For example, animal- and plant-derived polymers (natural rubber, chitosan, and cellulose, among others) have been developed for transdermal delivery and injectable delivery.50 With the development of nanotechnology, nanomaterials have also been used as nanosensors for the detection of multiple substances. Nano delivery systems developed on the basis of nanomaterials present stronger biocompatibility and tumor targeting ability, which may be beneficial to overcome many of the drawbacks of traditional delivery systems.51 Herein, a novel long-acting nanodrug delivery system that encapsulated paclitaxel was developed by directly using a curcumin derivative as a carrier material through the solid dispersion technique. Additionally, its anti-breast cancer activity and safety in preclinical in vitro and in vivo experiments were explored. This methodology was utilized to prepare the PTX-SLN@CUD nanodrug delivery system as it is simple, reliable, and expected to quantify the production.52 The resulting PTX-SLN@CUD successfully overcomes the problem of low entrapment efficiency, which is prevalent for nanomedicines, as great entrapment efficiency (94.20 ± 0.49%) is key for it to exert therapeutic effects. Considering the stability and the necessary space provided by the carrier CUD, it was able to increase its physical stability.41 In addition, zeta potential is an additional important indicator that is able to distribute cells in target organs. The surface of PTX-SLN@CUD nanoparticles carries a large negative charge, which leads to electrostatic repulsion between adjacent particles to increase the distance between particles, thereby reducing aggregation of nanoparticles. Hence, the particle size, entrapment efficiency, and loading efficiency of PTX-SLN@CUD solution were not affected at room temperature for 24 h. Lyophilization is able to effectively address long-term preservation problem, which allows the critical properties of PTX-SLN@CUD to remain stable for more than six months. In view of this, the whole drug delivery system is reliable and is conducive for further antitumor research and future medical applications.
Studies have shown that the distribution profile of nanoliposomes is similar to that in the blood in terms of tumor tissue absorption. The distribution of nanoliposomes with a particle size of 100–200 nm in tumor tissue is 4 times that of nanoliposomes larger than 300 nm or smaller than 50nm.53 Since the particle size of PTX-SLN@CUD (238.5 ± 4.79 nm) is smaller than the tumor capillary gaps (> 400 nm), giving unmodified nanoparticles a better passive targeting ability, this led to an increase in their distribution in tumor tissue. In brief, nanoparticles preferentially accumulate in solid tumors through the enhanced permeability and retention (EPR) effect. PTX-SLN@CUD passes through the blood circulation into capillaries and may also be taken up by cells in a pinocytotic manner through the inter-endothelial space into the lesion.54 This effect mediates the passive accumulation of carrier materials at the tumor site and solves the problem of suboptimal bioavailability of PTX clinically. In addition, due to the small particle size of PTX-SLN@CUD, its higher specific surface can embed hydrophobic substances to improve its solubility, which is conducive to reducing the side effects of conventional cosolvents.55 Hence, PTX-SLN@CUD significantly enhanced the anti-breast cancer efficacy and the safety in vivo.
In vitro studies are necessary in order to determine the sensitivity of cancer cells to drugs under investigation, where one significant advantage of the cell proliferation Kit (MTT) is the relative simplicity of operation. During the cytotoxicity assay, free PTX at the same concentration demonstrated a slightly better growth inhibition rate on MCF-7 cells than PTX-SLN@CUD at 24 h. However, there was no significant difference between them at 48 h. PTX-SLN@CUD exhibited a higher antitumor activity on MCF-7 cell growth inhibition rate at 72 h and 96 h. Studies have reported that most free drugs present with an initial burst release behavior in vitro, which causes excessive local drug concentrations.56 The nanostructured skeleton of PTX-SLN@CUD restricts the entrapped drug, and there is little initial release. The inhibition rate of free PTX on MCF-7 cells was found to be slightly higher compared to PTX-SLN@CUD in 24 h because the drug concentration of free PTX acting on tumor cells may be higher compared to PTX-SLN@CUD. However, controlling the initial burst release is able to significantly diminish systemic toxicity,57 while the burst release behavior of free PTX may cause drug poisoning and has suboptimal safety. The activity of PTX-SLN@CUD was significantly lower compared to free PTX in toxicity experiments against the normal hepatocyte cell line L02. CUD, as a drug carrier, is able to effectively control the rate of drug release for purposes of sustained or controlled release, which contributes to maintaining therapeutic drug levels within tumors.58 The superiority of PTX-SLN@CUD on the inhibition rate of MCF-7 cells over free PTX was evident over time (72 h and 96 h).
The slow release of the drug can help avoid rapid clearance in the blood in order to prolong the retention time of the drug within the body.59 PTX-SLN@CUD with CUD as a carrier can help stabilize the nanoparticles in plasma or impart stealth properties, as well as improve the bioavailability by maintaining a higher blood drug concentration for a longer time. Therefore, the in vivo pharmacokinetic trial observed that PTX-SLN@CUD significantly decreased the clearance of PTX after intravenous administration, which prolonged the retention time in vivo, and increased the area under the drug time curve. The residence time of this nanoparticle in vivo after administration is as long as 120 h, which is an advantage that most long-circulating nanoparticles do not have. In order to overcome the problem of short-action time of drugs in the human body, a variety of long-acting lipid nanoparticles have been developed for paclitaxel over the years.60,61 However, PTX-SLN@CUD presents a longer half-life and retention time in the body compared to the other models, and is more effective at reducing the number of administrations and dosage in order to provide smooth blood concentration.
Subsequently, a xenograft nude mice animal model bearing MCF-7 cells was established in order to further investigate the antitumor activity of PTX-SLN@CUD in vivo. PTX-SLN@CUD better inhibited tumor growth compared to free PTX at the end of the experiment (Figure 7C), and its tumor inhibition rate was greater than that free PTX (P<0.05). Tumor staining of Ki-67 antigen demonstrated that the proliferation of breast cancer cells was better inhibited by PTX-SLN@CUD, which again proves that PTX-SLN@CUD had strong anti-breast cancer activity. The in vivo pharmacodynamics were consistent with cytotoxicity results of MCF-7 cells in vitro. However, administration of free PTX for 20 days led to a significant decrease in the average body weight of nude mice and the deaths of three nude mice. PTX-SLN@CUD exhibited a good safety profile, and the average body weight of nude mice in this group was not significantly different from the beginning, and all nude mice survived. Preliminary experiments identified that the hepatotoxicity of free PTX and the severe allergic reaction caused by the mixed solvent (absolute ethanol and polyoxyethylene castor oil) led to the common occurrence of tail rot, tail docking and death in mice after tail vein injection. Hence, this could not be sustained for administration in the way of tail vein injection. PTX-SLN@CUD led to improvement in water solubility, which did not cause any hemolysis and the above phenomena in mice after the injection. Considering the consistency of other variables when comparing, a uniform intraperitoneal administration mode was adopted in the in vivo antitumor study, and subsequently in in vivo toxicity trials.
Safety is another important factor when it comes to the design of antitumor drug delivery systems. Favorable long-term safety evaluation of drug delivery systems are able to accelerate the translation of preclinical studies into clinical applications.62 Clinically, in order to improve the solubility of drugs in the PTX injection, a mixed solvent of absolute ethanol and polyoxyethylene castor oil is often chosen, but it can cause severe allergic reactions, hemolytic reactions, hepatotoxicity, and cardiotoxicity.3,45 PTX albumin nanoparticles are commonly-used PTX-loaded nanodrug delivery systems in clinic, which help avoid the toxicity of the above solvents. Although the incidence of anaphylaxis is uncommon, it still exhibits a high incidence of myelotoxicity in clinical trials with certain limitations.63,64 To the best of our knowledge, most long-acting nanodrug delivery systems have ignored the evaluation of safety in vivo when pursuing it to improve drug efficacy. However, the in vivo toxicity trial of PTX-SLN@CUD indicated that PTX-SLN@CUD at a concentration of 40 mg/kg led to very mild damage to the organs of mice after a single high-dose administration, which was much weaker than that of free PTX. Importantly, PTX-SLN@CUD at a concentration of 10 mg/kg demonstrated a good safety profile with no organ damage and related toxicity after long-term administration (Figure 10B).
By modifying the pharmacokinetics and in vitro release behavior of drugs, PTX-SLN@CUD has helped solve the problems that often arises in conventional chemotherapy with PTX formulations. This nanoparticle avoided the rapid clearance of PTX in blood, while, at the same time, enhancing the solubility and permeability of PTX. In this way, PTX-SLN@CUD is able to better access and enter tumor cells in order to improve the relative effectiveness against breast cancer, and be effectively dispersed to prevent excessive accumulation to diminish drug toxicity. PTX-SLN@CUD is a novel potential anti-neoplastic delivery system that is safe and effective for the treatment of breast cancer, which is conducive to further antitumor research and future medical applications. Herein, CUD is a potential carrier that enhances the efficacy and reduces the toxicity of PTX. However, whether CUD can be utilized as a broad carrier to improve efficacy and safety of chemotherapeutic drugs needs to be verified in combination with more experiments.
Conclusions
In conclusion, a novel structural curcumin derivative, CUD, was utilized as a carrier material in order to develop a novel long-acting paclitaxel nano-delivery system. Its efficacy and safety against breast cancer were investigated in vitro and in vivo. PTX-SLN@CUD significantly improved the in vitro release behavior of PTX, and the sustained release can contribute to maintaining therapeutic drug levels within the tumor. PTX-SLN@CUD also significantly prolongs the residence time, half-life of PTX in vivo, and increases its relative bioavailability. In this way, PTX-SLN@CUD enhanced the relative efficacy of breast cancer treatment and inhibited the proliferation of MCF-7 cells. Most importantly, hemolytic, acute toxicity, and cumulative toxicity trials demonstrated that PTX-SLN@CUD ameliorated the relative safety of PTX-based chemotherapy. Results from this present demonstrated that PTX-SLN@CUD is a relatively safe and efficacious novel strategy with great potential when it comes to improving breast cancer treatment.
Funding Statement
This study was supported by the Major R&D Plan Joint Innovation Project (No. 22ZDYF3793, 22ZDYF3798), the Youth Science and Technology Innovation Research Team (No. 2021JDTD0008) and the Basic Research fund (No. 2020YJ0336) of the Science and Technology Department of Sichuan province of China, the Science and Technology Innovation Team from Jiucheng Science and Technology Talent Cultivation Plan in Luzhou (No. 2019-1), Key Research and Development Projectors of Luzhou (No. 2021-SYF-26), the cooperation Project (No. 210027-01SZ, 200017-01SZ) of Central Nervous System Drug Key Laboratory of Sichuan Province.
Data Sharing Statement
All data generated or analyzed during this study are included in this manuscript and its Additional file.
Ethics Approval and Consent to Participate
All animal studies (including the mice euthanasia procedure) were done in compliance with the regulations and guidelines of Southwest Medical University institutional animal care and conducted according to the AAALAC and the IACUC guidelines.
Disclosure
The authors declare that they have no competing interests in this work.
References
- 1.Chong Y, Jin W. A physical mechanism and global quantification of breast cancer. PLoS One. 2016;11(7):422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev. 2016;17(S3):43–46. doi: 10.7314/APJCP.2016.17.S3.43 [DOI] [PubMed] [Google Scholar]
- 3.Abu Samaan TM, Samec M, Liskova A, Kubatka P, Büsselberg D. Paclitaxel’s mechanistic and clinical effects on breast cancer. Biomolecules. 2019;9(12):789–810. doi: 10.3390/biom9120789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Au YC, Co NN, Tsuruga T, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Della Corte L, Barra F, Foreste V, et al. Advances in paclitaxel combinations for treating cervical cancer. Expert Opin Pharmacother. 2020;21(6):663–677. doi: 10.1080/14656566.2020.1724284 [DOI] [PubMed] [Google Scholar]
- 6.Cui H, Arnst K, Miller DD, Li W. Recent advances in elucidating paclitaxel resistance mechanisms in non-small cell lung cancer and strategies to overcome drug resistance. Curr Med Chem. 2020;27(39):6573–6595. doi: 10.2174/0929867326666191016113631 [DOI] [PubMed] [Google Scholar]
- 7.Ma Y, Mou Q, Yan D, et al. Engineering small molecule nanodrugs to overcome barriers for cancer therapy. View. 2020;1:20200062. doi: 10.1002/VIW.20200062 [DOI] [Google Scholar]
- 8.Gu W, Chen J, Patra P, et al. Nanoformulated water-soluble paclitaxel to enhance drug efficacy and reduce hemolysis side effect. J Biomater Appl. 2017;32(1):66–73. doi: 10.1177/0885328217708458 [DOI] [PubMed] [Google Scholar]
- 9.Björn N, Jakobsen Falk I, Vergote I, Gréen H. ABCB1 variation affects myelosuppression, progression-free survival and overall survival in paclitaxel/carboplatin-treated ovarian cancer patients. Basic Clin Pharmacol Toxicol. 2018;123(3):277–287. doi: 10.1111/bcpt.12997 [DOI] [PubMed] [Google Scholar]
- 10.Green H, Khan MS, Ingrid JF, et al. Impact of CYP3A5*3 and CYP2C8-HapC on paclitaxel/carboplatin-induced myelosuppression in patients with ovarian cancer. J Pharm Sci. 2011;100(10):4205–4209. doi: 10.1002/jps.22680 [DOI] [PubMed] [Google Scholar]
- 11.Parisi A, Palluzzi E, Cortellini A, et al. First-line carboplatin/nab-paclitaxel in advanced ovarian cancer patients, after hypersensitivity reaction to solvent-based taxanes: a single-institution experience. Clin Transl Oncol. 2020;22(1):158–162. doi: 10.1007/s12094-019-02122-x [DOI] [PubMed] [Google Scholar]
- 12.Otani IM, Lax T, Long AA, Slawski BR, Camargo CA, Banerji A. Utility of risk stratification for paclitaxel hypersensitivity reactions. J Allergy Clin Immunol Pract. 2018;6(4):1266–1273. doi: 10.1016/j.jaip.2017.08.025 [DOI] [PubMed] [Google Scholar]
- 13.Hirpara MR, Manikkath J, Sivakumar K, et al. Long circulating PEGylated-chitosan nanoparticles of rosuvastatin calcium: development and in vitro and in vivo evaluations. Int J Biol Macromol. 2018;107(PtB):2190–2200. doi: 10.1016/j.ijbiomac.2017.10.086 [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Asghar S, Jin X, et al. Mitoxantrone-loaded chitosan/hyaluronate polyelectrolyte nanoparticles decorated with amphiphilic PEG derivates for long-circulating effect. Colloids Surf B Biointerfaces. 2018;171:468–477. doi: 10.1016/j.colsurfb.2018.07.060 [DOI] [PubMed] [Google Scholar]
- 15.Fang L, Zhang W, Wang Z, et al. Novel mitochondrial targeting charge-reversal polysaccharide hybrid shell/core nanoparticles for prolonged systemic circulation and antitumor drug delivery. Drug Deliv. 2019;26(1):1125–1139. doi: 10.1080/10717544.2019.1687614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saneja A, Kumar R, Singh A, et al. Development and evaluation of long-circulating nanoparticles loaded with betulinic acid for improved anti-tumor efficacy. Int J Pharm. 2017;531(1):153–166. doi: 10.1016/j.ijpharm.2017.08.076 [DOI] [PubMed] [Google Scholar]
- 17.Nakamae I, Morimoto T, Shima H, et al. Curcumin derivatives verify the essentiality of ros upregulation in tumor suppression. Molecules. 2019;24(22):4067. doi: 10.3390/molecules24224067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komal K, Chaudhary S, Yadav P, Parmanik R, Singh M. The therapeutic and preventive efficacy of curcumin and its derivatives in esophageal cancer. Asian Pac J Cancer Prev. 2019;20(5):1329–1337. doi: 10.31557/APJCP.2019.20.5.1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suk JS, Xu QG, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Del Rev. 2016;99:28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolate A, Baradia D, Patil S, Vhora I, Kore G, Misra A. PEG — a versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67–81. doi: 10.1016/j.jconrel.2014.06.046 [DOI] [PubMed] [Google Scholar]
- 21.Hyun H, Park J, Willis K, et al. Surface modification of polymer nanoparticles with native albumin for enhancing drug delivery to solid tumors. Biomaterials. 2018;180:206–224. doi: 10.1016/j.biomaterials.2018.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim D, Jeon H, Ahn S, Choi WI, Kim S, Jon S. An approach for half-life extension and activity preservation of an anti-diabetic peptide drug based on genetic fusion with an albumin-binding aptide. J Control Release. 2017;256:114–120. doi: 10.1016/j.jconrel.2017.04.036 [DOI] [PubMed] [Google Scholar]
- 23.Zhang W, Li C, Shen C, et al. Prodrug-based nano-drug delivery system for co-encapsulate paclitaxel and carboplatin for lung cancer treatment. Drug Deliv. 2016;23(7):2575–2580. doi: 10.3109/10717544.2015.1035466 [DOI] [PubMed] [Google Scholar]
- 24.Deng W, Qiu J, Wang S, et al. Development of biocompatible and VEGF-targeted paclitaxel nanodrugs on albumin and graphene oxide dual-carrier for photothermal-triggered drug delivery in vitro and in vivo. Int J Nanomedicine. 2018;13:439–453. doi: 10.2147/IJN.S150977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SK. Photoactivation strategies for therapeutic release in nanodelivery systems. Adv Ther. 2020;3:2000117. doi: 10.1002/adtp.202000117 [DOI] [Google Scholar]
- 26.Mishra V, Bansal KK, Verma A, et al. Solid lipid nanoparticles: emerging colloidal nano drug delivery systems. Pharmaceutics. 2018;10(4):191. doi: 10.3390/pharmaceutics10040191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farghadani R, Naidu R. Curcumin as an enhancer of therapeutic efficiency of chemotherapy drugs in breast cancer. Int J Mol Sci. 2022;23(4):2144. doi: 10.3390/ijms23042144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farghadani R, Naidu R. Curcumin: modulator of key molecular signaling pathways in hormone-independent breast cancer. Cancers. 2021;13(14):3427. doi: 10.3390/cancers13143427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mortezaee K, Salehi E, Mirtavoos-Mahyari H, et al. Mechanisms of apoptosis modulation by curcumin: implications for cancer therapy. J Cell Physiol. 2019;234(8):12537–12550. doi: 10.1002/jcp.28122 [DOI] [PubMed] [Google Scholar]
- 30.Hassan FU, Rehman MS, Khan MS, et al. Curcumin as an alternative epigenetic modulator: mechanism of action and potential effects. Front Genet. 2019;10:514. doi: 10.3389/fgene.2019.00514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soleimani V, Sahebkar A, Hosseinzadeh H. Turmeric (Curcuma longa) and its major constituent (curcumin) as nontoxic and safe substances: review. Phytother Res. 2018;32(6):985–995. doi: 10.1002/ptr.6054 [DOI] [PubMed] [Google Scholar]
- 32.Mbese Z, Khwaza V, Aderibigbe BA. Curcumin and its derivatives as potential therapeutic agents in prostate, colon and breast cancers. Molecules. 2019;24(23):4386. doi: 10.3390/molecules24234386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khorasani MY, Langari H, Sany SBT, Rezayi M, Sahebkar A. The role of curcumin and its derivatives in sensory applications. Mater Sci Eng C Mater Biol Appl. 2019;103:109792. doi: 10.1016/j.msec.2019.109792 [DOI] [PubMed] [Google Scholar]
- 34.Ramadass SK, Anantharaman NV, Subramanian S, et al. Paclitaxel/Epigallocatechin gallate coloaded liposome: a synergistic delivery to control the invasiveness of MDA-MB-231 breast cancer cells. Colloids Surf B Biointerfaces. 2015;125:65–72. doi: 10.1016/j.colsurfb.2014.11.005 [DOI] [PubMed] [Google Scholar]
- 35.Kordowska WM, Waśko A, Berecka MP, et al. Spirulina enhances the viability of Lactobacillus rhamnosus E/N after freeze-drying in a protective medium of sucrose and lactulose. Lett Appl Microbiol. 2011;53(1):79–83. doi: 10.1111/j.1472-765X.2011.03068.x [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Wei Y, Yang H, et al. A 5-fluorouracil-loaded floating gastroretentive hollow microsphere: development, pharmacokinetic in rabbits, and biodistribution in tumor-bearing mice. Drug Des Devel Ther. 2016;10:997–1008. doi: 10.2147/DDDT.S97735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei YM, Xue ZK, Wang P, et al. Formulation and pharmacokinetic evaluation of once-daily sustained-released system of nifedipine with solid dispersion and coating techniques. Arch Pharm Res. 2013;36(7):864–873. doi: 10.1007/s12272-013-0076-8 [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Lei Z, Zhong Q, et al. Enrichment of caffeic acid in peanut sprouts and evaluation of its in vitro effectiveness against oxidative stress-induced erythrocyte hemolysis. Food Chem. 2017;217:332–341. doi: 10.1016/j.foodchem.2016.07.126 [DOI] [PubMed] [Google Scholar]
- 39.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145(3):178–181. doi: 10.1016/j.jconrel.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng X, Pi C, Fu S, et al. Combination of curcumin and paclitaxel liposomes exhibits enhanced cytotoxicity towards A549/A549-T cells and unaltered pharmacokinetics. J Biomed Nanotechnol. 2020;16(8):1304–1313. doi: 10.1166/jbn.2020.2969 [DOI] [PubMed] [Google Scholar]
- 41.Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. doi: 10.1016/j.actbio.2014.08.021 [DOI] [PubMed] [Google Scholar]
- 42.Peng LH, Wei W, Shan YH, et al. Sustained release of piroxicam from solid lipid nanoparticle as an effective anti-inflammatory therapeutics in vivo. Drug Dev Ind Pharm. 2017;43(1):55–66. doi: 10.1080/03639045.2016.1220563 [DOI] [PubMed] [Google Scholar]
- 43.Selting KA, Bechtel SM, Espinosa J, et al. Evaluation of intravenous and subcutaneous administration of a novel, excipient-free, nanoparticulate formulation of paclitaxel in dogs with spontaneously occurring neoplasia. Vet Comp Oncol. 2018;16(4):650–657. doi: 10.1111/vco.12435 [DOI] [PubMed] [Google Scholar]
- 44.Shanmugam S, Im HT, Sohn YT, et al. Enhanced oral bioavailability of paclitaxel by solid dispersion granulation. Drug Dev Ind Pharm. 2015;41(11):1864–1876. doi: 10.3109/03639045.2015.1018275 [DOI] [PubMed] [Google Scholar]
- 45.Ashrafizadeh M, Zarrabi A, Hashemi F, et al. Curcumin in cancer therapy: a novel adjunct for combination chemotherapy with paclitaxel and alleviation of its adverse effects. Life Sci. 2020;256:1179–1184. doi: 10.1016/j.lfs.2020.117984 [DOI] [PubMed] [Google Scholar]
- 46.Wang F, Jiang YS, Liu F. The influence of mutant lactobacilli on serum creatinine and urea nitrogen concentrations and renal pathology in 5/6 nephrectomized rats. Ren Fail. 2016;38(9):1441–1447. doi: 10.1080/0886022X.2016.1227617 [DOI] [PubMed] [Google Scholar]
- 47.Awhin EP, Jeroh E, Anigboro AA, et al. Effect of chronic consumption of Piliostigma thonningii on activities of alanine aminotransferase and aspartate aminotransferase in serum and liver in Rattus novergicus. Pak J Biol Sci. 2013;16(24):2062–2065. doi: 10.3923/pjbs.2013.2062.2065 [DOI] [PubMed] [Google Scholar]
- 48.Zhao H, Hong J, Yu X, et al. Oxidative stress in the kidney injury of mice following exposure to lanthanides trichloride. Chemosphere. 2013;93(6):875–884. doi: 10.1016/j.chemosphere.2013.05.034 [DOI] [PubMed] [Google Scholar]
- 49.de Sousa Cunha F, Dos Santos Pereira LN, de Costa ES, de Sousa Luz RA, Nogueira Mendes A. Development of nanoparticulate systems with action in breast and ovarian cancer: nanotheragnostics. J Drug Target. 2019;27(7):732–741. doi: 10.1080/1061186X.2018.1523418 [DOI] [PubMed] [Google Scholar]
- 50.Sivasankarapillai VS, Das SS, Sabir F, et al. Progress in natural polymer engineered biomaterials for transdermal drug delivery systems. Mater Today Chem. 2021;19:100382. doi: 10.1016/j.mtchem.2020.100382 [DOI] [Google Scholar]
- 51.Pandey S, Thakur VK, Díez-Pascual AM. Amino acids, peptides, and proteins: implications for nanotechnological applications in biosensing and drug/gene delivery. Nanomaterials. 2021;11(11):3002. doi: 10.3390/nano11113002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yong Z, Wang X, Jie H, et al. Formulation, production, in vitro release and in vivo pharmacokinetics of cinnamaldehyde sub-micron emulsions. Pharm Dev Technol. 2020;16:1–10. [DOI] [PubMed] [Google Scholar]
- 53.Liu D, Mori A, Huang L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim Biophys Acta. 1992;1104(1):95–101. doi: 10.1016/0005-2736(92)90136-A [DOI] [PubMed] [Google Scholar]
- 54.Means N, Elechalawar CK, Chen WR, Bhattacharya R, Mukherjee P. Revealing macropinocytosis using nanoparticles. Mol Aspects Med. 2022;83:100993. doi: 10.1016/j.mam.2021.100993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dadwal A, Baldi A, Kumar Narang R. Nanoparticles as carriers for drug delivery in cancer. Artif Cells Nanomed Biotechnol. 2018;46(sup2):295–305. doi: 10.1080/21691401.2018.1457039 [DOI] [PubMed] [Google Scholar]
- 56.Zhao C, Liu A, Santamaria CM, et al. Polymer-tetrodotoxin conjugates to induce prolonged duration local anesthesia with minimal toxicity. Nat Commun. 2019;10(1):2566. doi: 10.1038/s41467-019-10296-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pathak S, Regmi S, Gupta B, et al. Single synchronous delivery of FK506-loaded polymeric microspheres with pancreatic islets for the successful treatment of streptozocin-induced diabetes in mice. Drug Deliv. 2017;24(1):1350–1359. doi: 10.1080/10717544.2017.1377317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li K, Liang N, Yang H, Liu H, Li S. Temozolomide encapsulated and folic acid decorated chitosan nanoparticles for lung tumor targeting: improving therapeutic efficacy both in vitro and in vivo. Oncotarget. 2017;8(67):111318–111332. doi: 10.18632/oncotarget.22791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Avnir Y, Turjeman K, Tulchinsky D, et al. Fabrication principles and their contribution to the superior in vivo therapeutic efficacy of nano-liposomes remote loaded with glucocorticoids. PLoS One. 2011;6(10):e25721. doi: 10.1371/journal.pone.0025721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Zhang P, Zou Q, et al. Co-delivery of gemcitabine and paclitaxel in crgd-modified long circulating nanoparticles with asymmetric lipid layers for breast cancer treatment. Molecules. 2018;23(11):2906. doi: 10.3390/molecules23112906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song B, Wu S, Li W, Chen D, Hu H. Folate modified long circulating nano-emulsion as a promising approach for improving the efficiency of chemotherapy drugs in cancer treatment. Pharm Res. 2020;37(12):242. doi: 10.1007/s11095-020-02811-1 [DOI] [PubMed] [Google Scholar]
- 62.Guo P, Pi C, Zhao S, et al. Oral co-delivery nanoemulsion of 5-fluorouracil and curcumin for synergistic effects against liver cancer. Expert Opin Drug Deliv. 2020;17(10):1473–1484. doi: 10.1080/17425247.2020.1796629 [DOI] [PubMed] [Google Scholar]
- 63.Herrera DA, Ashai N, Perez-Soler R, et al. Nanoparticle albumin bound-paclitaxel for treatment of advanced non-small cell lung cancer: an evaluation of the clinical evidence. Expert Opin Pharmacother. 2018;2018(10):1–8. [DOI] [PubMed] [Google Scholar]
- 64.Jiménez-López J, El-Hammadi MM, Ortiz R, et al. A novel nanoformulation of PLGA with high non-ionic surfactant content improves in vitro and in vivo PTX activity against lung cancer. Pharmacol Res. 2019;141:451–465. doi: 10.1016/j.phrs.2019.01.013 [DOI] [PubMed] [Google Scholar]