

Abstract
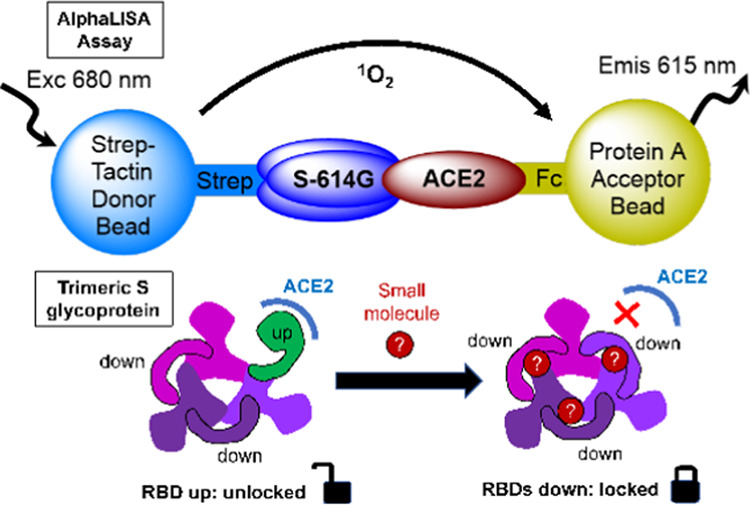
SARS-CoV-2, a coronavirus strain that started a worldwide pandemic in early 2020, attaches to human cells by binding its spike (S) glycoprotein to a host receptor protein angiotensin-converting enzyme 2 (ACE2). Blocking the interaction between the S protein and ACE2 has emerged as an important strategy for preventing viral infection. We systematically developed and optimized an AlphaLISA assay to investigate binding events between ACE2 and the ectodomain of the SARS-CoV-2 S protein (S-614G: residues 1–1208 with a D614G mutation). Using S-614G permits discovering potential allosteric inhibitors that stabilize the S protein in a conformation that impedes its access to ACE2. Over 30,000 small molecules were screened in a high-throughput format for activity against S-614G and ACE2 binding using the AlphaLISA assay. A viral entry assay was used to validate hits using lentiviral particles pseudotyped with the full-length S protein of the Wuhan-1 strain. Two compounds identified in the screen, oleic acid and suramin, blocked the attachment of S-614G to ACE2 and S protein-driven cell entry into Calu-3 and ACE2-overexpressing HEK293T cells. Oleic acid inhibits S-614G binding to ACE2 far more potently than to the receptor-binding domain (RBD, residues 319–541 of SARS-CoV-2 S), potentially indicating a noncompetitive mechanism. The results indicate that using the full-length ectodomain of the S protein can be important for identifying allosteric inhibitors of ACE2 binding. The approach reported here represents a rapidly adaptable format for discovering receptor-binding inhibitors to S-proteins of future coronavirus strains.
Keywords: SARS-CoV-2, spike protein, trimer, inhibitor, COVID-19, high-throughput
Introduction
As SARS-CoV-2 continues to impact the world, identifying new and different treatment forms and preventing infection remains vital. One successful approach implemented in FDA-approved vaccines involves targeting the cellular entry of the virus to prevent infection.1−3 The spike (S) glycoprotein facilitates coronavirus entry into host cells and projects from the virus’s surface as a transmembrane homotrimer. The S protein monomer consists of subunits S1 and S2, which mediate attachment to the cell surface and virus-cell membrane fusion, respectively.4 The S protein attaches to the host cell receptor ACE2 and is processed for viral internalization by host proteases, including TMPRSS2 and furin.5,6 The receptor-binding domain (RBD) of the S1 subunit is responsible for binding to ACE2 with a potent low-nanomolar affinity.7,8 Thus, this protein–protein interface is an important drug target for preventing SARS-CoV-2 entry into human cells.
Since the RBD directly engages ACE2, truncated forms of the S protein selectively containing this domain have been used in biochemical assays and screenings to identify interaction inhibitors. However, the S protein is dynamic and alternately presents the RBD in either an “open” or “closed” conformation, where only the open structure can bind to ACE2.9 Therefore, biochemical assays that exclude parts of the full-length S protein can potentially overlook allosteric inhibitors of ACE2 binding, including inhibitors that could function by stabilizing the closed structure. Moreover, direct competitive inhibitors targeting the RBD must be very potent to overcome the nanomolar binding affinity of the S protein to ACE2. Allosteric inhibitors of the binding interaction offer a potential way of avoiding these obstacles.
Recently, AlphaLISA and ELISA technologies have been used to detect the interaction between ACE2 and the RBD.8,10−16 AlphaLISA is a proximity-based biochemical assay platform that monitors the interaction between two analytes using a pair of functionalized beads.17 Unlike ELISA, which requires immobilization of a protein-binding partner, AlphaLISA is a homogeneous mix-and-read format amenable to high-throughput automation. We developed and optimized an AlphaLISA screen to monitor the interaction between ACE2 and the full-length ectodomain of the spike protein. We focused our screening assay on the D614G mutant S protein, which became prevalent in COVID-19 patients during our assay development.18 Our approach potentially detects binding events at allosteric sites beyond the receptor-binding motif. The S protein also contains trimeric binding interfaces and is processed by proteases at specific sites to facilitate infection and mediates membrane fusion via the S2 subunit, all of which are potential drug targets. Small molecules also have the potential to bypass the glycan shield on the S protein or target the glycans themselves (carbohydrate-binding antigens), potentially overcoming its immune evasion functionality or structural roles.19−21 Ultimately, numerous binding interactions and functionally important structural changes in the S protein can be targeted by small molecules to prevent viral infection.
We employed the AlphaLISA assay to screen a library of over 30,000 small molecules. We also used an AlphaLISA-based counter-screen and a pseudotyped viral particle cellular entry assay to validate hits.22−24 Our findings on two suspected SARS-CoV-2 antagonists, oleic acid and suramin, illustrate the potential of utilizing full-length S protein constructs instead of the shorter RBD in high-throughput screening (HTS). Our strategic and rapid assay implementation is amenable to other full-length spike protein variants and lays the foundation for addressing future similar pandemics.
Results
Optimizing the AlphaLISA Assay for the Spike Trimer
A protein–protein interaction (PPI) assay was developed to monitor the binding between ACE2 and S protein variants utilizing AlphaLISA technology (Figure 1). In designing an AlphaLISA assay, protein tags are critical since the tags must enable protein expression and purification and be compatible with the available Alpha bead pairs. A Strep-Tactin donor bead and Protein A acceptor bead were selected upon considering the efficiency of protein preparation.
Figure 1.
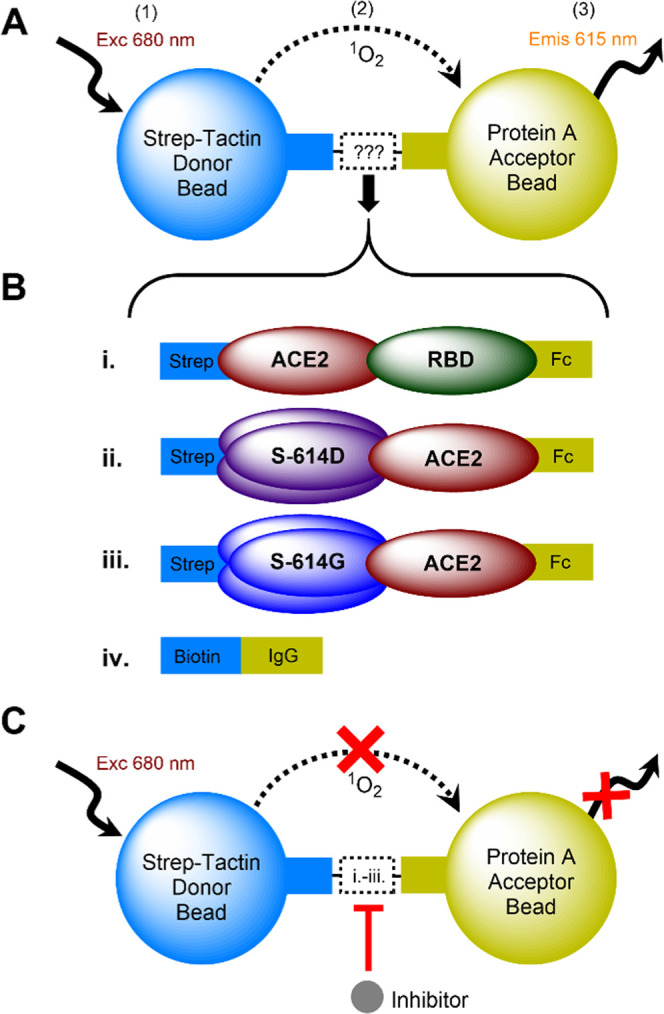
Schematic illustration of the AlphaLISA assay to monitor interactions between ACE2 and S protein variants. (A) When a Strep-Tactin donor bead and Protein A acceptor bead are brought into proximity by engagement with a tagged PPI pair, an Alpha signal can be generated by (1) excitation of the donor bead at 680 nm (exc), resulting in (2) transfer of singlet oxygen (1O2) to the acceptor bead, which then (3) emits light at 615 nm (emis). (B) PPI pairs used in our primary AlphaLISA assay include (i) ACE2-Strep/RBD-Fc, (ii) S-614D-Strep /ACE2-Fc, and (iii) S-614G-Strep/ACE2-Fc, and the counter-assay uses the dual-tagged molecule (iv) biotin-IgG. (C) Assay principle of competitive displacement where an inhibitor or a tagless protein partner for the PPIs in panel (B) will block the interaction between the two tagged proteins, reducing the Alpha signal.
As a preliminary guide, we first conducted an RBD-based assay using an approach reported by Hanson et al.16 (Figure 1). For this experiment, the RBD (residues 319–591 of SARS-CoV-2 S) was expressed and purified with a C-terminal Fc tag (RBD-Fc), while the full-length human ACE2 receptor displayed a C-terminal Strep-tag (ACE2-Strep). Figure 1A,B(i) illustrates that the Strep-Tactin-coated beads recognize Strep-tagged ACE2, and the Protein A-coated acceptor beads recognize Fc-tagged RBD. When RBD and ACE2 interact, an Alpha signal is generated. In the presence of agents that block the interaction, the Alpha signal is reduced (Figure 1C). Working concentrations of the protein partners were optimized via a cross-titration experiment. The highest sensitivity of the Alpha signal response to changes in protein binding occurred when ACE2-Strep and RBD-Fc were at 16 and 8 nM, respectively (Figure 2A,B).
Figure 2.

Optimizing protein concentrations for the AlphaLISA assay. (A) RBD-Fc/ACE2-Strep; (B) ACE2-Strep/RBD-Fc; (C) S-614D-Strep/ACE2-Fc; (D) ACE2-Fc/S-614D-Strep; (E) S-614G-Strep/ACE2-Fc; and (F) ACE2-Fc/S-614G-Strep. The first column (purple heat maps) shows the optimization of different S protein constructs. Heat maps show the fold-change in Alpha signal (relative to a background of 0 nM ACE2) for varying S protein concentrations (x-axis) at different ACE2 concentrations (y-axis). The second column (orange heat maps) shows the optimization of ACE2 constructs. Heat maps show fold-change in Alpha signal (relative to a background of 0 nM S protein) for varying ACE2 concentration (x-axis) at different S protein concentrations (y-axis). Data are shown as averages of duplicate samples.
For evaluating the PPI of the trimeric spike protein S-614D with ACE2 (Figure 1B(ii)), we expressed and purified the soluble ectodomain form of the SARS-CoV-2 S protein (residues 1–1208) of the Wuhan-1 strain. It carries the stabilizing K986P and V987P mutations, a “GSAS” substitution at the furin cleavage site (residues 682–685), and a C-terminal Strep-tag. This construct, developed by Wrapp et al.,25 will be annotated as S-614D-Strep. Similarly, the full-length human ACE2 receptor was expressed with a C-terminal Fc tag and will be annotated as ACE2-Fc. Based on the confirmation of PPI between ACE2 and RBD, optimum concentrations of S-614D-Strep and ACE2-Fc in the AlphaLISA assay were determined similarly but at a reduced bead concentration. We previously reported that an Alpha bead concentration at 4 ng/μL in an assay volume of 10 μL did not alter the assay robustness.26 Cross-titration experiments with varying concentrations (0–50 nM) of the two proteins showed a maximum fold-change in Alpha signal at 15 and 0.5 nM of ACE2-Fc and S-614D-Strep, respectively (Figure 2C,D). These results show that a 16-fold lower concentration of S-614D-Strep than RBD is required for the optimum signal. Minimizing concentrations of limited resources such as these house-prepared proteins is crucial for high-throughput assay design and cost, so these improvements indicate an additional advantage of using the full-length ectodomain of the spike protein over the RBD within the context of our custom assay conditions.
Using the optimized S-614D-Strep and ACE2-Fc concentrations, the well-to-well signal variation for samples in a 384-well plate was examined by manually filling a plate section (128 wells with negative control and 48 wells with positive control) using a multi-channel pipette. An assay mixture containing 0.5 nM S-614D-Strep, 15 nM ACE2-Fc, 4 ng/μL Alpha bead mixture, and 0.1% (v/v) dimethyl sulfoxide (DMSO) was used for the negative control. The Alpha bead mixture with 0.1% (v/v) DMSO was used for the positive control. The assay showed a Z′ factor of 0.83, S/B of 692, and a coefficient of variation (CV) of 5% (for negative controls), confirming the assay robustness at the reduced bead concentration with manual pipetting. These results indicated that the assay was ready to be adapted for high-throughput screening.
Validating the HTS Assay Using the Spike Protein 614G Variant
For our high-throughput screen, we chose to use the S protein mutant (D614G), as this point mutation became ubiquitous in variants of SARS-CoV-2 worldwide.18,27 Although the D614G mutation is not located in the predicted ACE2-binding surface of the spike protein, this mutant is associated with increased infectivity and viral replication in cells.28 The S-614G-Strep construct was prepared by mutating the D614 residue of the S-614D-Strep construct. S-614G-Strep was expressed, purified, and paired with Fc-tagged ACE2 (ACE2-Fc) for the target PPI (Figure 1B(iii)). Cross-titration experimental results were similar to those observed for the S-614D/ACE2 PPI. We selected final concentrations of 15 nM ACE2-Fc and 0.5 nM S-614G-Strep (or S-614D-Strep) (Figure 2E,F). These correspond to the lowest protein concentrations that have a minimal effect on the assay window.
Tagless ACE2 (xACE2) and tagless RBD (xRBD) were prepared as positive control competitive inhibitors that disrupt the tagged ACE2 and S proteins’ binding, causing a decrease in the Alpha signal. The S-614G-Strep/ACE2-Fc Alpha signal decreased in a dose-dependent manner with tagless counterparts, xACE2 and xRBD. The estimated IC50 values are 43 ± 5 and 28 ± 3 nM for xACE2 and xRBD, respectively (Figure 3B). Similar results were observed for S-614D-Strep/ACE2-Fc. The overall change in the Alpha signal indicates that the assay is suitable for detecting inhibitors in a small-molecule screen.
Figure 3.
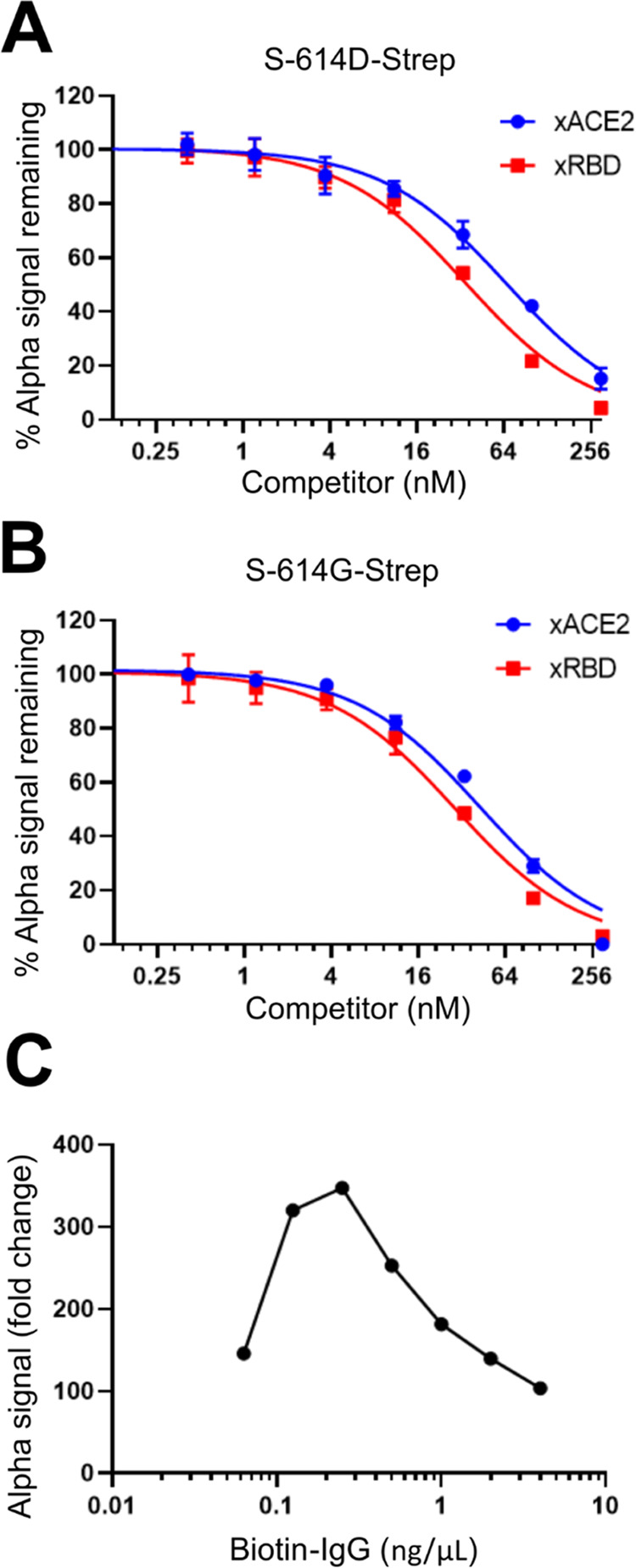
Establishing controls and counter-screen for AlphaLISA assay. Tagless ACE2 (xACE2) and tagless RBD (xRBD) were tested at varying concentrations (plotted on the Log2 scale) for a dose-dependent competitive displacement of (A) the S-614D-Strep /Fc-ACE2 PPI pair and (B) the S-614G-Strep/ACE2-Fc PPI pair. The Alpha signal is shown as a percentage of the signal for each PPI pair with no competitor present. (C) Dose-dependent Alpha signal response to various biotin-IgG concentrations (plotted on the Log10 scale) was measured for Alpha bead concentrations of 4 ng/μL and is shown as fold-change in Alpha signal relative to 0 ng/μL biotin-IgG controls. (n = 2 for A and B, n = 1 for C).
We developed a counter-screen assay to rule out small molecules that interfere with the Alpha signal or block the binding of our tagged target proteins with their corresponding Alpha beads. In this assay, we chose to use biotin-labeled IgG and the same Alpha bead pair used in our primary AlphaLISA assay. Biotin-labeled IgG is expected to bind the Strep-Tactin donor bead via the biotin tag and the Protein A acceptor beads via its Fc moiety (Figure 1B(iv)). This counter-screen can filter out molecules that disrupt the interaction between the acceptor bead and Fc tag (IgG), donor bead, and Strep-Tag (biotin) or quench singlet oxygen that interferes with Alpha signal. The AlphaLISA counter-assay was optimized using biotin-labeled IgG at a 4 ng/μL bead concentration. As shown in Figure 3C, the concentration of biotin-IgG affects the Alpha signal strength. Maximum Alpha signals were observed when [biotin-IgG] = 0.25 ng/μL. From these results, we selected final concentrations of 0.2 ng/μL biotin-IgG at 4 ng/μL of donor and acceptor beads.
Finally, we examined Alpha signal tolerances for different concentrations of the buffer additives DMSO, bovine serum albumin (BSA), and Tween-20. BSA and Tween-20 are included in HTS experiments to reduce nonspecific interactions or surface adhesion of assay components, and DMSO is the solvent used for all library compounds in our collection. The AlphaLISA assay was found to tolerate up to 1.3% (v/v) DMSO (Figure S1A), and we selected a screening concentration of 10 μM compounds with a corresponding 0.1% (v/v) DMSO well within tolerance limits. BSA concentration of 0.05 mg/mL shows a sufficient boost in Alpha signal, with a maximum signal at a threshold of approximately 0.2 mg/mL BSA (Figure S1B). The BSA concentration was selected according to reported assay buffer conditions of 0.05 mg/mL BSA in PBS, pH 7.4.16 The surfactant Tween-20 gave a maximum Alpha signal increase at approximately 0.01% (w/v), so this concentration was selected for the final screen (Figure S1C). Furthermore, the assay showed signal stability at room temperature for up to 3 h, confirming its feasibility for HTS application (Figure S1D).
High-Throughput AlphaLISA Screen of a Small-Molecule Library
A structurally diverse set of over 30,000 compounds were arrayed in 384-well assay plates and screened using our AlphaLISA assay (Figure 4A). Overall, the assay showed consistent readouts and excellent statistics throughout the entire screening, with average daily S/B, Z′, and inter-and intra-assay CV % of 389 ± 25 (Figure 4B), 0.83 ± 0.02 (Figure 4C), and 7.0 ± 1 and 2.7 ± 0.5 (Figure 4D), respectively. A total of 180 compounds showing >50% signal inhibition and passing promiscuity criteria based on other Alpha screens performed by our group were rescreened at 10 μM in duplicate. The AlphaLISA counter-screen was then conducted for the same 180 compounds. Six compounds showed reproducible activity in the AlphaLISA assay with little or no activity in counter-screen assays. These six compounds consisted of one synthetic molecule from LifeChem’s Fp3 library (F6436–2670) and five natural products (suramin, pentamycin, oleic acid, lydimycin, and s-(p-azidophenacyl) glutathione) (Figure 5A). These compounds showed more than a 100-fold greater potency in the AlphaLISA assay than in the counter AlphaLISA assay (Figure S2), with moderate IC50 values of 1.6–20.2 μM and Hill coefficients of ∼1 (Figure 5B). While lydimycin is a biotin mimetic and F6436–2670 contains furan rings that can potentially interfere with the Alpha signal, both showed limited activity in the counter-screen over the broad concentration range of 0–200 μM, which indicated that interference is unlikely. Therefore, we predict their activity at the concentrations used in our primary assay is caused by disruption of S-614G-Strep binding to ACE2-Fc.
Figure 4.

Workflow and statistics for the primary high-throughput AlphaLISA screen. (A) Illustration of compound screening workflow. Histograms of (B) signal-to-background ratio (S/B) and (C) Z′ factor for each plate are shown fitted to a normal distribution. S/B and Z′ mean values are labeled and indicated by a dashed line, with standard deviation bands shown in striped regions. (D) Average coefficients of variation (CV %) per plate are shown for each screening day. CV values are shown for positive controls (POS), negative controls (NEG), the average CV of positive and negative controls (AVG), and the root-mean-square CV of positive and negative controls (RMS) for each day of screening. The number of plates (N) screened per day is italic below the x-axis labels.
Figure 5.

Validation of hit compounds from the primary AlphaLISA screen. (A) Chemical structures of top 6 hit molecules. Suramin and oleic acid, selected for further evaluation, are shown in blue. (B) Table shows molecule names from panel (A) and IC50 values with Hill coefficients for the primary AlphaLISA assay.
Validation Studies
Of the six hit compounds from the screen, we selected suramin and oleic acid (Figure 5A shown in blue) to study in more detail since other research supports that they may be antagonists of ACE2-mediated SARS-CoV-2 cell entry.11,13,29,30 AlphaLISA assays using either S-614G-Strep or RBD-Fc were conducted using fresh stocks of the compounds to confirm the dose-dependent inhibition of ACE2 binding. As shown in Figure 6A, suramin showed similar potency against S-614G-Strep and RBD-Fc with IC50 values of approximately 2 μM. Oleic acid showed slightly weaker potency toward S-614D-Strep than suramin with an IC50 of 5.6 μM. Interestingly, it showed approximately 6-fold weaker potency against RBD-Fc (IC50 = 35.3 μM) than S-614G-Strep, with a Hill coefficient >1 (Figure 6B). These parameters for both compounds are summarized in Figure 6C.
Figure 6.

Potencies of oleic acid and suramin against S-614G-Strep and RBD-Fc were compared in the AlphaLISA assay. Dose-dependent inhibition profiles in AlphaLISA assay against both S-614G-Strep and RBD-Fc were measured in parallel with the AlphaLISA counter-assay (biotin-IgG) using freshly prepared stock solutions of suramin and oleic acid. Dose–response data for (A) suramin and (B) oleic acid fit a four-parameter variable-slope equation using nonlinear regression. IC50 values and corresponding absolute values of Hill coefficients from the nonlinear regressions are shown in panel (C).
Furthermore, we found that both compounds blocked the cellular entry of lentiviral particles pseudotyped with the full-length spike of the Wuhan-1 strain (WT) into the Calu-3 cell line with IC50s of 11.9 ± 1.3 and 25 ± 1.2 μM, respectively (Figure 7A). Lentiviral particle entry assays are a well-established and widely used model of viral infection.31 Calu-3 is a human lung epithelial cell line; it represents an excellent cellular model to characterize SARS-CoV-2 viral entry because it endogenously expresses ACE2 and the surface serine protease TMPRSS2. Most SARS-CoV-2 variants have used the human receptor ACE2 for entry and the serine protease TMPRSS2 for S protein priming.5 Monoclonal antibodies that target the spike protein32 (e.g., REGN10987, REGN10933) exhibit potent neutralization efficacy in the Calu-3 cell line. We also tested the viral entry model in HEK293T cells engineered to overexpress the human ACE2 receptor (HEK293-ACE2). In this case, we used lentiviral particles pseudotyped with SARS-COV-2 spike (D614G). According to the experimental scheme shown in Figure 7B: first, either the cells or the lentiviral particles pseudotyped with SARS-COV-2 spike (D614G) were preincubated with different concentrations of each compound for 1 h at 37 °C (for the cells) or room temperature (for the virus); second, the cells were inoculated with the virus for 1 h at 37 °C; and then, the cells were washed and treated with new media. Luminescence signals were recorded 60 h post-infection. Suramin showed a similar efficacy regardless of the treatment sequence (IC50 of 9.7 ± 1.3 and 5.9 ± 1.3 μM, for preincubation with virus and cells, respectively) (Figure 7C). In contrast, oleic acid only showed efficacy when preincubated with the virus (IC50 for virus preincubation is 17.6 ± 1.2 μM) (Figure 7D).
Figure 7.

Oleic acid and suramin inhibited the cellular entry of HIV particles pseudotyped with a full-length SARS-CoV-2 spike. Dose–response profiles of (A) suramin and (B) oleic acid inhibiting Calu-3 cells infection by HIV particles pseudotyped with SARS-CoV-2 wild-type full-length spike (S-WT). Viruses were pretreated with the indicated concentration of each compound or DMSO for 1 h at room temperature before adding to the cells. Following the experimental scheme (C), when preincubated with the virus (for 1 h at room temperature) or the cells (for 1 h at 37 °C), suramin (D) inhibits SARS-CoV-2 (S-D614G) full-length spike viral entry in HEK293-ACE2 cells. In contrast, oleic acid (E) works when preincubated with the virus rather than the cells. Viral particles or HEK293-ACE2 cells were pretreated with the indicated concentration of each compound or DMSO for 1 h. Then virus inoculation was performed for 1 h at 37 °C; cells were washed and supplied with new media. In all experiments, the luminescence signal was recorded 60 h post-infection. The entry efficiency of SARS-CoV-2 pseudoviruses and viability of infected cells preincubated with DMSO was considered 100%. Results represent the average of two different experiments using different virus preparations (n = 2 for each experiment). Both compounds had low to moderate cytotoxicity over their active concentration ranges in these cell lines.
As a control experiment, we tested HIV particles pseudotyped with the vesicular stomatitis virus (VSV) glycoprotein (VSVG) instead of the spike protein to ensure that suramin and oleic acid did not disrupt the integrity of the particles or nonspecifically interfere with viral entry. VSVG interacts with the low-density lipoprotein (LDL) receptor family instead of ACE2.33 As shown in Figure S3, viral entry is inhibited by suramin with an IC50 for virus preincubation of 94 μM, indicating approximately 9-fold selectivity of suramin for the ACE2/S protein (D614G). For oleic acid, the entry of the particles pseudotyped with VSVG was only impacted at the highest tested concentration (540 μM). For both compounds, viral entry inhibition in this control experiment coincided with the drop in cell viability, indicating that the viral entry luminescence signal cannot be uncoupled from compound cytotoxicity at high concentrations.
Discussion
To our knowledge, this is the first report of the optimization and execution of a large (>30,000 small molecules) AlphaLISA screen utilizing a trimeric spike protein construct that includes the full ectodomain and a clinically relevant point mutation (D614G) rather than using just the RBD. Our assay was highly reliable, as shown by the statistics in Figure 4. This outcome suggests that the S protein ectodomain construct25 is stably used and stored for the duration of the screening process, making it a feasible alternative to the RBD in large screening projects.
Our primary screening led to the identification of suramin and oleic acid as promising hits. Suramin is an antiparasitic drug approved for the prophylactic treatment of sleeping sickness (trypanosomiasis) and river blindness (onchocerciasis) and has diverse antiviral properties.34 Its potential for treating SARS-CoV-2 has been reported, with evidence suggesting that it can block viral entry,29 ACE2 binding,11 and viral replication via inhibition of the RNA polymerase RdRp34,35 and proteolytic processing by the 3CL protease Mpro.36 These reports corroborate our findings that suramin can block S protein binding to ACE2 and viral entry. Two recent reports using commercially available kits reached different conclusions about the potency of suramin compared to our findings. Bojadzic et al.14 found that suramin did not inhibit the ACE2/RBD interaction at concentrations up to 100 μM using cell-free ELISA-type assays. Similarly, Ao et al.11 reported around 50% inhibition of ACE2/RBD interaction at 150 μM of suramin. The two commercially available kits used in these studies include a shorter form of the RBD (residues 319–541) than the extended receptor-binding domain used in our assay (residues 319–591). The shorter form of the commercially available RBD has been reported to exhibit a higher molecular weight dimer artifact, which could be caused by a possible disulfide bond formed between Cysteine 538 of two short RBD molecules. In contrast, the longer form has an even number of cysteines that prevents the formation of intermolecular disulfide bridges. The short RBD (residues 319–541) has been known to cause consistent reproducibility problems when testing antibodies, while the longer form showed far more thermal stability than the shorter form.37 In our assays, suramin shows a strong affinity toward the longer RBD (residues 319–591) and the full-length ectodomain (residues 1–1208). Since we found similar activity profiles of suramin against both the RBD and S-614G, we can infer that in our assay, suramin inhibits ACE2 binding by interacting with the RBD region of the spike protein since it is present in both constructs. The inhibition profile of suramin in the viral entry assay (Figure 7D) could indicate several different mechanisms, including, but not limited to, blocking of viral ACE2 binding. Identifying the exact binding locations and stoichiometry of suramin on the target proteins will be part of our future studies.
Toelzer et al.30 reported that free fatty acid linoleic acid (LA) binds to three composite binding pockets on the S protein formed by adjacent RBDs, based on a 2.85 Å resolution cryo-EM structure. Linoleic acid (18:2 cis-9,12) is a very similar fatty acid to oleic acid (18:1 cis-9). Using their structure, they observed that LA binding stabilizes the “closed” S protein conformation where the RBDs are folded downward and not accessible, resulting in reduced ACE2 interaction in vitro. They also describe a similar pocket in the highly pathogenic severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), suggesting the therapeutic potential of LA for other coronaviruses. From these findings on linoleic acid, we suspected that oleic acid similarly binds the spike protein, potentially producing a closed S conformation where the RBDs have limited ability to bind ACE2. We plan to test this hypothesis in future structural studies. The lower potency of oleic acid against RBD than S-614G supports a mechanism more complex than direct binding to the ACE2/RBD interface. LA binding to the spike trimer is expected to be higher than to the RBD due to contacts with the polar headgroup of Arg-408 and Gln-409 of the adjacent RBD.30 A steep increase in percent inhibition in the AlphaLISA counter-assay at high concentrations of oleic acid is likely to be a consequence of the surfactant properties of oleic acid or a transition beyond critical micelle concentrations (Figure 6B).38 The Hill coefficient can be an indicator of cooperativity for multiple ligand binding and can therefore signify if more than one ligand is interacting with a protein. In this case, oleic acid has a Hill coefficient of 4.8 ± 0.6 (Figure 6C), which suggests it may be binding in greater than 1:1 stoichiometry on the lipophilic surfaces of RBD needed for ACE2 binding. Indeed, polyunsaturated fatty acids can align to form surfaces and are known to have antiviral properties that potentially involve disruption of the viral envelope or host cell membrane fluidity, which could alter receptor binding.13 Goc et al.13 found that polyunsaturated fatty acids can inhibit RBD binding to ACE2 and host cell protease activity against the spike protein, which supports possible spatial distortion of the transmembrane proteins involved in viral entry.
Importantly, our screen can yield different results than other AlphaLISA assays that seek inhibitors of the ACE2/RBD interaction. For example, Hanson et al. reported corilagin as an ACE2/RBD binding inhibitor with an IC50 of 5.5 μM in their AlphaLISA assay.16 In our primary screening assay using S-614G, corilagin showed approximately 16% inhibition at 10 μM and did not qualify as a hit under our criteria. Additionally, ceftazidime was recently identified by Alpha screen12 as an inhibitor of the ACE2/RBD interaction by binding to the RBD with an affinity of approximately 6 μM (from bio-layer interferometry measurements). However, this compound also did not get selected as a hit in our screening. These examples highlight the importance of working with different protein constructs in biochemical binding assays. Our results show that screening for inhibitors of the ACE2-binding interaction with the trimeric ectodomain of the S protein provides opportunities for identifying inhibitors with diverse mechanisms. This is exemplified by our findings for the small-molecule oleic acid. Oleic acid potentially weakens the binding of S protein to ACE2 allosterically, and we suggest this is caused by trapping the RBD in a closed conformation that cannot access ACE2. Furthermore, our assay can be rapidly adapted to assess mutations of the S protein located in the RBD or elsewhere on the spike protein for effects on ACE2 binding. This is made possible by advances in stable S protein expression and purification and rapid sequencing of new S protein variants. We believe our approach is valuable as SARS-CoV-2 progresses and as other future coronaviruses emerge.
Conclusions
We designed and executed a high-throughput AlphaLISA screening of over 30,000 small molecules to identify inhibitors that target the binding of the SARS-CoV-2 trimeric spike protein to the ACE2 receptor on host cells. This assay is readily adaptable to other spike protein–receptor interactions and can be used to identify molecules that indirectly target the binding interaction, offering an alternative mode of preventing viral infection.
Experimental Methods
Protein Expression, Purification, and Quality Control
Recombinant proteins were prepared according to the protocols described by Wrapp et al.25 The mammalian expression plasmid pαH expressing the WT form of the prefusion S ectodomain (noted in the manuscript as S-614D-Strep) (residues 1–1208 of 2019-nCoV S (GenBank: MN908947)) was designed to include proline substitutions at residues 986 and 987, a GSAS substitution at the furin cleavage site (residues 682–685), a C-terminal foldon trimerization motif, an HRV3C protease cleavage site, a TwinStrepTag, and an 8×HisTag.25 This plasmid was employed as a template for site-directed mutagenesis to generate the D614G mutant (noted in the manuscript as S-614G-Strep). 2019-nCoV RBD was expressed using the same plasmid backbone as the prefusion S ectodomain, pαH expressing residues 319–591 of 2019-nCoV S included a C-terminal HRV3C protease cleavage site, a monomeric Fc tag, and an 8×HisTag (noted in the manuscript as RBD-Fc). To express the full-length human ACE2 receptor, residues encoding residues 1–615 of human ACE2 were cloned in the pαH plasmid upstream of a C-terminal HRV3C protease cleavage site, a monomeric Fc tag, and an 8×HisTag (to express Fc-tagged ACE2, noted in the manuscript as ACE2-Fc) or upstream of an HRV3C protease cleavage site, a TwinStrepTag and an 8×HisTag (to express Strep-tagged ACE2, noted in the manuscript as ACE2-Strep). pαH plasmids expressing the WT form of the prefusion S ectodomain, Fc-tagged 2019-nCoV RBD, and Strep-tagged ACE2 were provided by Dr. Jason McLellan Lab (the University of Texas at Austin). Descriptions of the proteins are summarized in the Supporting Information (Table S1). Size-exclusion chromatography was used to finalize proteins, followed by SDS-PAGE to verify purified products (Figure S4). Tagless ACE2 (xACE2) and tagless RBD (xRBD) were prepared by cleavage using HRV3C protease.25
AlphaLISA Assay General Protocol
The AlphaLISA screen was designed similarly to Hanson et al.16 Assay buffer consisted of PBS, pH 7.4 (Gibco) with 0.05 mg/mL BSA and 0.01% (w/v) Tween-20. For generating the Alpha signal, Strep-tagged S protein in stable trimeric form25 (S-614D-Strep or S-614G-Strep) was paired with Strep-Tactin Alpha donor beads (PerkinElmer, #AS106D), and Fc-tagged ACE2 was paired with AlphaLISA Protein A acceptor beads (#AL101C). All assay steps were carried out at room temperature, and reaction volumes totaled 20 μL each in 384-well white OptiPlates (PerkinElmer, #6007299). After adding donor and acceptor beads to protein mixtures in assay plates, the plates were sealed with an aluminum seal, shaken at 350 rpm for 1 min, and then centrifuged at 800 rpm for 1 min. All Alpha signal measurements were collected on a Neo2 multi-mode plate reader (BioTek) equipped with an Alpha filter cube after 2 h of incubation. Given concentrations represent final assay concentrations in 20 μL reaction volumes unless otherwise noted.
AlphaLISA Assay Optimization and Validation
For the following assay optimization steps, 10 μL of 2× ACE2 and S-protein (RBD-Fc, S-614D-Strep, or S-614G-Strep) mixture was incubated with various treatments for 30 min, followed by the addition of 10 μL of 2× 1:1 donor/acceptor bead mixture (4 or 20 ng/μL of each bead). For initial comparisons to published literature,16 final concentrations of 0–64 nM RBD-Fc were cross-titrated against 0–64 nM ACE2-Strep to evaluate concentrations of each protein for the maximum Alpha signal at the bead concentration of 20 ng/μL recommended by the manufacturer’s protocol. To determine optimal protein concentrations for the screen at a 4 ng/μL bead concentration, final concentrations of 0–50 nM S-614D-Strep or S-614G-Strep were cross-titrated against 0–50 nM ACE2-Fc. The maximal fold-change determined optimal protein concentrations in the cross-titration experiments in Alpha signal relative to the background (protein of interest only). To evaluate the tolerance of the assay to different buffer additives, 0.5 nM S-614G-Strep and 15 nM ACE2-Fc were tested with 0–4% (v/v) DMSO, 0–0.2% (w/v) Tween-20, and 0–2 mg/mL BSA. The final assay buffer selected for the screening consisted of PBS, pH 7.4, with 0.05 mg/mL BSA, 0.01% (w/v) Tween-20, and a DMSO maximum of 0.1% (v/v). Protein stability at room temperature was examined by incubating a 2× mixture of S-614G-Strep and ACE2-Fc (1 and 30 nM, respectively) for 0–3 h at room temperature before the addition of 2× donor/acceptor bead mixture.
The assay principle for the inhibitor screening was validated via competitive disruption of the PPI between Alpha-bead-bound proteins using their tagless counterparts, xACE2 and xRBD. In detail, 5 μL of 4× protein mixture (60 nM ACE2-Fc and 2 nM of S-614D-Strep or S-614G-Strep protein) were equilibrated with 5 μL of 4× tagless protein (0–1200 nM of either xACE2 or xRBD) at room temperature for 30 min. Then, 10 μL 2× of donor/acceptor bead mixture (4 ng/μL assay concentrations of each bead) was mixed with the proteins and incubated for 2 h at room temperature before reading the Alpha signal. The apparent IC50 values were calculated using nonlinear regression fitting in GraphPad Prism (Version 9.3.1, GraphPad Software, LLC, San Diego, CA).
The counter-screen AlphaLISA assay was designed and carried out similarly to the primary AlphaLISA assay using biotin-labeled IgG (Jackson ImmunoResearch, #011-060-003) instead of the mixture of S-614G-Strep and ACE2-Fc. The optimum concentration of biotin-labeled IgG was determined by titration of biotin-IgG (0–4 ng/μL) at a 4 ng/μL bead concentration.
Primary AlphaLISA Screen
Molecule libraries for the screen included NIH clinical collections (Evotec, San Francisco, CA) [674 compounds], Spectrum collection (MicroSource Discovery Systems, Gaylordsville, CT) [2000 compounds], Fsp3-enriched diversity library (Life Chemical, Niagara-on-the-Lake, ON, Canada) [13,440 compounds], Natural-like compounds (Life Chemical) [1280 compounds], Diversity set (ChemDiv, San Diego, CA) [12,900 compounds], NexT Diversity set V (NCI) [1597 compounds], NexT Mechanistic set III (NCI) [821 compounds], NexT Natural products IV (NCI) [419 compounds], NexT Oncology set (NCI) [129 compounds], and Lopac library (Sigma-Aldrich, St Louis, MO) [1280 compounds].
First, 20 nL of library compounds (10 mM stocks in 100% DMSO) or 20 nL of 100% DMSO controls were dispensed to assay plates using an Access and Echo 550 liquid handler (Beckman Coulter Labcyte). For each plate, 10 μL of 2× protein mixture (1 nM S-614G-Strep and 30 nM ACE2-Fc) was dispensed to wells in plate columns 1–2 for negative controls (maximum signal) and 3–22 for sample compounds using a MultiFlo FX microplate dispenser (BioTek). Similarly, 10 μL of assay buffer was dispensed to wells in plate columns 23–24 for positive controls (minimum signal). Plates were centrifuged at 800 rpm for 2 min. After 30 min of incubation to allow binding, 10 μL of 2× donor/acceptor bead mixture (8 ng/μL each bead) was dispensed to assay plates using the MultiFlo FX. Z′ factors were calculated for each plate to assess robustness.39 Additionally, signal-to-background ratios (S/B) were calculated as the average negative control signal divided by the average positive control signal. Coefficients of variation (CV) for the Alpha signal were calculated as (standard deviation)/(mean) × 100% for positive and negative controls. Inter-assay CV (average of positive and negative control CV values divided by the number of plates) and intra-assay CV (root-mean-square of positive and negative controls divided by the number of plates) were also calculated for each screening day. Alpha signal inhibition (%) was calculated as 100 × [1 – (S – P)/(N – P)], where variables are sample Alpha signal (S) and average negative (N) and positive (P) control signals for the plate where the sample is located.
Hit Validation
A total of 180 compounds identified from the primary screen were retested in a singlet at 10 μM in the same manner as the primary screen to validate the reproducibility of their inhibition activities. Simultaneously, the compounds were also tested in a singlet at 10 μM in the AlphaLISA counter-screen to rule out compounds that interfere with the AlphaLISA assay in a nontargeted manner. This assay was performed the same as the primary screen, except for using 0.2 ng/μL final concentration of biotin-IgG instead of the S-614G-Strep/ACE2-Fc mixture. In both the primary AlphaLISA assay and counter-screen, compounds were dispensed using an Echo 550 liquid handler. Both proteins and beads were distributed manually using multi-dispense multi-channel pipettes.
For hit validation, serial dilutions of hit compounds from the screen were tested in the AlphaLISA assay and AlphaLISA counter-screen. Final concentrations included 0–200 μM compounds and either 15 nM ACE2-Fc with 0.5 nM of S-614G-Strep for the AlphaLISA assay or 0.2 ng/μL biotin-IgG for the counter-screen. After 30 min of incubating the compounds with either the S-614G-Strep/ACE2-Fc mixture or biotin-IgG, donor and acceptor Alpha bead mixture was added to each sample for a final concentration of 4 ng/μL of each bead. After 2 h of incubation, Alpha signals were read as described above. Oleic acid and suramin were tested in the AlphaLISA assay for their ability to inhibit RBD-Fc binding to ACE2-Strep. Serial dilutions of the compounds from 0–100 μM (final) were tested against final concentrations of 0.5 nM RBD-Fc with 15 nM ACE2-Strep as described above.
Viral Cellular Entry Assay Using Spike-Pseudotyped Particles
Lentiviral particles pseudotyped with the full-length SARS-CoV-2 spike (wild type and D614G mutant) were generated following previously published protocols.22−24 Briefly, HEK293T cells were cotransfected with five plasmids. The first three to express the HIV virion under CMV promotor (HDM-Hgpm2, pRC-CMV-Rev1b, and HDM-tat1b) were obtained from BEI resources with catalog numbers NR-52517, NR-52518, and NR-52519, respectively. The fourth lentiviral backbone plasmid was obtained from BEI resources with a catalog number NR-52516. It is designed to express luciferase reporter under CMV promotor, followed by an IRES and ZsGreen (pHAGE-CMV-Luc2-IRES-ZsGreen-W). And finally, the spike protein plasmid expressing a codon-optimized full-length WT SARS-COV-2 spike protein (GenBank ID NC_045512) was also provided by the BEI resources (HDM-IDTSpike-fixK, #NR-52514).22−24 The D614G mutant of this plasmid (S-D614G) was generated using the QuikChange site-directed mutagenesis (New England Biolabs, MA, following the manufacturer’s protocol). For control lentiviral particles, the pCMV-VSVG plasmid (Cell Biolabs RV-110) was used for expressing the VSVG (vesicular stomatitis virus glycoprotein) instead of the spike protein.24 Media containing the pseudovirus particles were harvested 72 h after transfection, filtered, fractionated, and stored in the −80 °C freezer. The titer of the collected virus was estimated using the Lentivirus qPCR Titer Kit (Applied Biological Materials Inc, #LV900). To perform the viral entry assays, either the target cells or the viral particles were incubated with each tested compound for 1 h at 37°C or room temperature before adding the virus to the cells. 60 h post-infection, the confluency of the cells in each well was estimated using the lncuCyte ZOOM equipment with a 10× objective, and then 25–30 μL of the Bright-Glo luciferase assay reagent (Promega, #E2610) was added to each well to quantify the luciferase signal (relative luciferase units or RLU) using the Synergy H4 plate reader. Percent (%) entry was calculated as the ratio of relative luciferase units recorded at each concentration of the tested compound to the readout in DMSO control. IC50’s (half-maximal inhibitory concentrations) were calculated using GraphPad Prism version 9.3.1 software.
The human embryonic kidney cell line, HEK293T, was purchased from ATCC (#CRL-3216), and the Calu-3 cell line was purchased from ATCC (#HTB-55). HEK293T stable cell line expressing ACE2 (HEK293-ACE2) was prepared using the lentiviral vector (pHAGE2-EF1aInt-ACE2-WT) expressing human ACE2 under an EF1a promoter (BEI resources, #NR-52512). The monoclonal cell line was selected based on the susceptibility to infection by the pseudotyped lentiviral particles; selected clones were validated using western blotting. Mycoplasma tests were performed monthly using the MycoAlert Mycoplasma Detection Kit (Lonza).
Acknowledgments
Dr. Jesse D. Bloom’s lab Fred Hutchinson Cancer Research Center, Seattle, WA, contributed the following reagents for distribution through BEI Resources, NIAID, NIH. The following reagents were obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Wuhan-Hu-1 Spike-Pseudotyped Lentiviral Kit, NR-52948 (including NR-52514, NR-52516, NR-52517, NR-51518, and NR-52519); and Vector pHAGE2 expressing human ACE2 gene (GenBank ID for human ACE2 is NM_021804) under an EF1a promoter, NR-52512. The authors also acknowledge Drs. Ashish Chougule and Anastasia Kalli of PerkinElmer for their support with the AlphaLISA technology. Modified pαH Vectors Containing the WT SARS-Related Coronavirus 2, Wuhan-Hu-1 Spike Glycoprotein Ectodomain, Fc-tagged 2019-nCoV RBD (residues 319–591), and the Strep-tagged Human Angiotensin-Converting Enzyme 2 were provided by Dr. Jason McLellan Lab (the University of Texas at Austin).
Glossary
Abbreviations
- 2019-nCoV
2019 novel coronavirus
- 3CL Mpro
3-chymotrypsin-like main protease
- ACE2
angiotensin-converting enzyme 2
- Alpha
amplified luminescent proximity homogeneous assay
- AlphaLISA
Alpha-linked immunosorbent assay
- BSA
bovine serum albumin
- CMV promoter
cytomegalovirus promoter
- COVID-19
coronavirus disease 2019
- Cryo-EM
cryogenic electron microscopy
- CV
coefficient of variation
- DMSO
dimethyl sulfoxide
- EF1a promoter
elongation factor 1 α promoter
- ELISA
enzyme-linked immunosorbent assay
- emis
emission wavelength
- exc
excitation wavelength
- Fc
fragment crystallizable region
- FDA
Food and Drug Administration
- HIV
human immunodeficiency virus
- HRV3C protease
human rhinovirus 3C protease
- HTS
high-throughput screening
- IC50
half-maximal inhibitory concentration
- IgG
immunoglobulin G
- IRES
internal ribosome entry site
- LA
linoleic acid
- LDL
low-density lipoprotein
- MERS-CoV
Middle East respiratory syndrome coronavirus
- PPI
protein–protein interaction
- qPCR
quantitative polymerase chain reaction
- RBD
receptor-binding domain
- RdRp
RNA-dependent RNA polymerase
- RNA
ribonucleic acid
- S
spike
- SARS-CoV
severe acute respiratory syndrome coronavirus
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- S/B
signal-to-background
- SDS-PAGE
sodium dodecyl sulfate- polyacrylamide gel electrophoresis
- TMPRSS2
transmembrane serine protease 2
- VSVG
vesicular stomatitis virus glycoprotein
- ZsGreen
Zoanthus sp. green fluorescence protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.2c00297.
Protein constructs; SDS-PAGE of purified proteins; dependence of AlphaLISA signal on high-throughput screening variables; IC50 curves for hit compounds; results of suramin and oleic acid tested in control lentiviral particles (PDF)
This work was supported by a Welch Foundation grant (F-1390) (KND).
The authors declare no competing financial interest.
Supplementary Material
References
- Orders M. FDA authorizes Moderna COVID-19 vaccine. Med. Lett. Drugs Ther. 2021, 63, 9–10. [PubMed] [Google Scholar]
- Tanne J. H. FDA authorizes Pfizer-BioNTech COVID-19 vaccine. Med. Lett. Drugs Ther. 2021, 63, 1–2. [PubMed] [Google Scholar]
- Mullard A. FDA authorizes first single-shot COVID-19 vaccine. Nat. Rev. Drug Discovery 2021, 20, 251–252. 10.1038/d41573-021-00046-2. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Park Y. J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Kruger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N. H.; Nitsche A.; Muller M. A.; Drosten C.; Pohlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Pohlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784 e5. 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapaillerie D.; Charlier C.; Fernandes H. S.; Sousa S. F.; Lesbats P.; Weigel P.; Favereaux A.; Guyonnet-Duperat V.; Parissi V. In Silico, In Vitro and In Cellulo Models for Monitoring SARS-CoV-2 Spike/Human ACE2 Complex, Viral Entry and Cell Fusion. Viruses 2021, 13, 365 10.3390/v13030365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juraszek J.; Rutten L.; Blokland S.; Bouchier P.; Voorzaat R.; Ritschel T.; Bakkers M. J. G.; Renault L. L. R.; Langedijk J. P. M. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 2021, 12, 244 10.1038/s41467-020-20321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna K.; Raymond W.; Jin J.; Charbit A. R.; Gitlin I.; Tang M.; Werts A. D.; Barrett E. G.; Cox J. M.; Birch S. M.; Martinelli R.; Sperber H. S.; Franz S.; Pillai S.; Healy A. M.; Duff T.; Oscarson S.; Hoffmann M.; Pohlmann S.; Simmons G.; Fahy J. V. Thiol drugs decrease SARS-CoV-2 lung injury in vivo and disrupt SARS-CoV-2 spike complex binding to ACE2 in vitro. bioRxiv 2021, 415505 10.1101/2020.12.08.415505. [DOI] [Google Scholar]
- Ao Z.; Chan M.; Ouyang M. J.; Olukitibi T. A.; Mahmoudi M.; Kobasa D.; Yao X. Identification and evaluation of the inhibitory effect of Prunella vulgaris extract on SARS-coronavirus 2 virus entry. PLoS One 2021, 16, e0251649 10.1371/journal.pone.0251649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.; Li Y.; Zhang Y.; Liu Z.; Mu X.; Gu C.; Liu J.; Li Y.; Li G.; Chen J. Ceftazidime is a potential drug to inhibit SARS-CoV-2 infection in vitro by blocking spike protein-ACE2 interaction. Signal Transduction Targeted Ther. 2021, 6, 198 10.1038/s41392-021-00619-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc A.; Niedzwiecki A.; Rath M. Polyunsaturated omega-3 fatty acids inhibit ACE2-controlled SARS-CoV-2 binding and cellular entry. Sci. Rep. 2021, 11, 5207 10.1038/s41598-021-84850-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojadzic D.; Alcazar O.; Chen J.; Chuang S. T.; Condor Capcha J. M.; Shehadeh L. A.; Buchwald P. Small-Molecule Inhibitors of the Coronavirus Spike: ACE2 Protein-Protein Interaction as Blockers of Viral Attachment and Entry for SARS-CoV-2. ACS Infect. Dis. 2021, 7, 1519–1534. 10.1021/acsinfecdis.1c00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C. W.; Chia W. N.; Qin X.; Liu P.; Chen M. I.; Tiu C.; Hu Z.; Chen V. C.; Young B. E.; Sia W. R.; Tan Y. J.; Foo R.; Yi Y.; Lye D. C.; Anderson D. E.; Wang L. F. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2-spike protein-protein interaction. Nat. Biotechnol. 2020, 38, 1073–1078. 10.1038/s41587-020-0631-z. [DOI] [PubMed] [Google Scholar]
- Hanson Q. M.; Wilson K. M.; Shen M.; Itkin Z.; Eastman R. T.; Shinn P.; Hall M. D. Targeting ACE2-RBD Interaction as a Platform for COVID-19 Therapeutics: Development and Drug-Repurposing Screen of an AlphaLISA Proximity Assay. ACS Pharmacol. Transl. Sci. 2020, 3, 1352–1360. 10.1021/acsptsci.0c00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman E. F.; Kirakossian H.; Singh S.; Wu Z. P.; Irvin B. R.; Pease J. S.; Switchenko A. C.; Irvine J. D.; Dafforn A.; Skold C. N.; et al. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 5426–5430. 10.1073/pnas.91.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B.; Fischer W. M.; Gnanakaran S.; Yoon H.; Theiler J.; Abfalterer W.; Hengartner N.; Giorgi E. E.; Bhattacharya T.; Foley B.; Hastie K. M.; Parker M. D.; Partridge D. G.; Evans C. M.; Freeman T. M.; de Silva T. I.; McDanal C.; Perez L. G.; Tang H.; Moon-Walker A.; Whelan S. P.; LaBranche C. C.; Saphire E. O.; Montefiori D. C.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; McLellan J. S.; Fadda E.; Amaro R. E. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 2007, 5, 583–597. 10.1038/nrmicro1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K. H. D.; Eguia R.; Dingens A. S.; Loes A. N.; Malone K. D.; Wolf C. R.; Chu H. Y.; Tortorici M. A.; Veesler D.; Murphy M.; Pettie D.; King N. P.; Balazs A. B.; Bloom J. D. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Nguyen A. W.; Hsieh C.-L.; Silva R.; Olaluwoye O. S.; Wilen R. E.; Kaoud T. S.; Azouz L. R.; Qerqez A. N.; Le K. C.; Bohanon A. L.; DiVenere A. M.; Liu Y.; Lee A. G.; Amengor D.; Shoemaker S. R.; Costello S. M.; Marqusee S.; Dalby K. N.; D’Arcy S.; McLellan J. S.; Maynard J. A. Identification of a conserved neutralizing epitope present on spike proteins from all highly pathogenic coronaviruses. bioRxiv 2021, 428824 10.1101/2021.01.31.428824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanmardi K.; Chou C. W.; Terrace C. I.; Annapareddy A.; Kaoud T. S.; Guo Q.; Lutgens J.; Zorkic H.; Horton A. P.; Gardner E. C.; Nguyen G.; Boutz D. R.; Goike J.; Voss W. N.; Kuo H. C.; Dalby K. N.; Gollihar J. D.; Finkelstein I. J. Rapid characterization of spike variants via mammalian cell surface display. Mol. Cell 2021, 81, 5099–5111.e8. 10.1016/j.molcel.2021.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C. L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veloria J. R.; Devkota A. K.; Cho E. J.; Dalby K. N. Development of a cost effective and robust AlphaScreen((R)) platform for application. Biotechniques 2018, 64, 181–183. 10.2144/btn-2018-2001. [DOI] [PubMed] [Google Scholar]
- Jackson C. B.; Zhang L.; Farzan M.; Choe H. Functional importance of the D614G mutation in the SARS-CoV-2 spike protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. 10.1016/j.bbrc.2020.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante J. A.; Liu Y.; Liu J.; Xia H.; Johnson B. A.; Lokugamage K. G.; Zhang X.; Muruato A. E.; Zou J.; Fontes-Garfias C. R.; Mirchandani D.; Scharton D.; Bilello J. P.; Ku Z.; An Z.; Kalveram B.; Freiberg A. N.; Menachery V. D.; Xie X.; Plante K. S.; Weaver S. C.; Shi P. Y. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado-Benvindo C.; Thaler M.; Tas A.; Ogando N. S.; Bredenbeek P. J.; Ninaber D. K.; Wang Y.; Hiemstra P. S.; Snijder E. J.; van Hemert M. J. Suramin Inhibits SARS-CoV-2 Infection in Cell Culture by Interfering with Early Steps of the Replication Cycle. Antimicrob. Agents Chemother. 2020, 64, e00900-20 10.1128/AAC.00900-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toelzer C.; Gupta K.; Yadav S. K. N.; Borucu U.; Davidson A. D.; Kavanagh Williamson M.; Shoemark D. K.; Garzoni F.; Staufer O.; Milligan R.; Capin J.; Mulholland A. J.; Spatz J.; Fitzgerald D.; Berger I.; Schaffitzel C. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt M. A. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010, 169, 365–374. 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavor E.; Choong Y. K.; Er S. Y.; Sivaraman H.; Sivaraman J. Structural Basis of SARS-CoV-2 and SARS-CoV Antibody Interactions. Trends Immunol. 2020, 41, 1006–1022. 10.1016/j.it.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein D.; Werman A.; Novick D.; Barak S.; Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7306–7311. 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey D.; Ramakumar S.; Conn G. L. Targeted Redesign of Suramin Analogs for Novel Antimicrobial Lead Development. J. Chem. Inf. Model. 2021, 61, 4442–4454. 10.1021/acs.jcim.1c00578. [DOI] [PubMed] [Google Scholar]
- Yin W.; Luan X.; Li Z.; Zhou Z.; Wang Q.; Gao M.; Wang X.; Zhou F.; Shi J.; You E.; Liu M.; Wang Q.; Jiang Y.; Jiang H.; Xiao G.; Zhang L.; Yu X.; Zhang S.; Eric Xu H. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 2021, 28, 319–325. 10.1038/s41594-021-00570-0. [DOI] [PubMed] [Google Scholar]
- Ullah A.; Ullah K. Inhibition of SARS-CoV-2 3CL M(pro) by Natural and Synthetic Inhibitors: Potential Implication for Vaccine Production Against COVID-19. Front. Mol. Biosci. 2021, 8, 640819 10.3389/fmolb.2021.640819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosi L.; Kubler E.; Weston A.; Romann P.; Panikulam S.; Dirscherl L.; Gerspach M.; Giegelmann C.; Dolce D.; Uberschlag M. E.; Melone A.; Bantleon F. I.; Villiger T. K.; Gerhold C. B. Development of a Unique Rapid Test to Detect Anti-bodies Directed Against an Extended RBD of SARS-CoV-2 Spike Protein. Chimia 2021, 75, 446–452. 10.2533/chimia.2021.446. [DOI] [PubMed] [Google Scholar]
- Serth J.; Lautwein A.; Frech M.; Wittinghofer A.; Pingoud A. The inhibition of the GTPase activating protein-Ha-ras interaction by acidic lipids is due to physical association of the C-terminal domain of the GTPase activating protein with micellar structures. EMBO J. 1991, 10, 1325–1330. 10.1002/j.1460-2075.1991.tb07651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.