

ABSTRACT
Increased bone resorption by osteoclasts after estrogen deficiency is the main cause of postmenopausal osteoporosis. TET2 (tet methylcytosine dioxygenase 2) is a DNA demethylase that regulates cellular function and differentiation potential. Macroautophagy/autophagy maintains cellular homeostasis by recycling unnecessary and damaged organelles. This study revealed that TET2 promoted bone loss in oophorectomized (OVX) mice and that TET2 promoted osteoclast differentiation by regulating autophagy. Tet2 knockdown inhibited autophagy and osteoclast differentiation in vitro. Mechanistically, Tet2 knockdown increased BCL2 (B cell leukemia/lymphoma 2) expression and BCL2 exhibited increased binding to BECN1 and negatively regulated autophagy. Small interfering RNA specific to Bcl2 interfered with BCL2 expression in Tet2-knockdown bone marrow cells/precursors, partially reversing autophagy dysregulation and promoting osteoclast differentiation. Moreover, the LV-shTet2 lentivirus prevented bone loss in OVX mice. In summary, our findings provide evidence that TET2 promotes osteoclast differentiation by inhibiting BCL2 expression and positively regulating BECN1-dependent autophagy.
Abbreviations: ACP5/TRAP: acid phosphatase 5, tartrate resistant; ATP6V0D2: ATPase, H+ transporting, lysosomal V0 subunit D2; BCL2: B cell leukemia/lymphoma 2; BECN1: beclin 1, autophagy related; BMs: bone marrow cells; CTSK: cathepsin K; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MMP9: matrix metallopeptidase 9; OVX: oophorectomy; RUNX1: runt related transcription factor 1; SOCS3: suppressor of cytokine signaling 3; SPI1/PU.1: Spi-1 proto-oncogene; TNFSF11/RANKL: tumor necrosis factor (ligand) superfamily, member 11; TET2: tet methylcytosine dioxygenase 2.
KEYWORDS: Autophagy, BCL2, osteoclast, osteoporosis, TET2
Introduction
Estrogen deficiency affects all bone cell types and increases bone turnover, thereby accelerating bone loss [1]. Owing to acute estrogen deficiency, high expression of TNFSF11/RANKL (tumor necrosis factor (ligand) superfamily, member 11) and other osteoclast procytokines, including TNF/TNFα and IL1 (interleukin 1), released by osteoblasts and T cells, promotes osteoclast formation [2]. Subsequently, a slight increase in bone formation caused by excessive bone resorption does not maintain balanced bone reconstruction, resulting in rapid thinning of the cortex and reduction in bone trabeculae [3]. Given the important role of osteoclast overactivation in bone destruction and osteoporosis, an in-depth study of the mechanism of osteoclast activation is critical for inhibiting rapid loss of bone mass post-menopause.
Osteoclasts are large multinucleated bone-resorptive cells formed by the fusion of macrophage colony-stimulating factor and TNFSF11-stimulated mononuclear macrophage/monocyte lineage precursors [4]. TNFSF11 binds to TNFRSF11A/RANK receptor to recruit and activate TRAF6 (TNF receptor-associated factor 6), which activates MAPK (mitogen-activated protein kinase), NFKB/nuclear factor-kappa B and other signaling pathways to regulate the expression of NFATC1 (nuclear factor of activated T cells, cytoplasmic, calcineurin dependent 1) and osteoclast-associated functional proteins, thereby inducing osteoclast differentiation and maturation [5]. Osteoclasts form a unique ruffled membrane when they come into contact with bones, creating a closed extracellular microenvironment that facilitates bone matrix degradation [6].
Autophagy and autophagy-related proteins are reported to be involved in osteoclast differentiation, migration and bone resorption [5]. Autophagy can be activated to promote osteoclast differentiation during TNFSF11-stimulated differentiation [7,8]. The inhibition of chloroquine-mediated autophagy can reduce osteoclast generation in oophorectomized (OVX)-induced bone loss in mice through both the typical and atypical NFKB signaling pathways [9]. Osteoclast autophagy is significantly enhanced in OVX mice, and reduced autophagy can substantially attenuate bone loss [9–11].
TET2 (tet methylcytosine dioxygenase 2) is a DNA demethylase that oxidizes 5-methylcytosine to 5-hydroxymethylcytosine in the presence of α-ketoglutaric acid and ferrous iron [12]. TET2, a transcriptional cofactor, epigenetically regulates gene expression by maintaining DNA in an unmethylated state [13]. TET2 reportedly regulates mast cell differentiation by maintaining the expression of CEBP transcription factors [14]. TET2 demethylates the P2rx7 (purinergic receptor P2X, ligand-gated ion channel, 7) promoter to control the release of exosomes and miRNAs and maintain mesenchymal stem cell homeostasis [15]. Loss of TET2 increases the differentiation of granulocytes/monocytes from hematopoietic stem/progenitor cells [16]. Furthermore, TET2 can interact with RUNX1 (runt related transcription factor 1) and SPI1/PU.1 (Spi-1 proto-oncogene) to regulate the expression of genes crucial for osteoclast differentiation, and tet2−/− mice exhibit an osteopetrosis phenotype [17,18]. These results suggest that TET2 plays an important role in regulating the transcription of crucial genes during cell differentiation. Moreover, TET2 regulates gene expression at the post-transcriptional level. Downregulation of TET2 can reduce the demethylation of Socs3 (suppressor of cytokine signaling 3) mRNA, thus reducing the binding of Socs3 to ADAR/ADAR1 (adenosine deaminase, RNA-specific) and preventing Socs3 degradation, thereby increasing the level of Socs3 and reducing infection-induced myelopoiesis [19]. However, whether TET2 affects osteoclast autophagy and differentiation during the development of postmenopausal osteoporosis remains unclear.
In our study, increased TET2 expression was observed in the osteoclasts of OVX mice. Further in vitro experiments showed that TET2 expression increased during osteoclast differentiation and that TET2 downregulation inhibited autophagy activation and decreased osteoclast formation. It was also demonstrated that TET2 inhibition reduced osteoclast autophagy and the number of ACP5/TRAP (acid phosphatase 5, tartrate resistant)-positive cells in vivo, and inhibited bone loss in OVX mice. In conclusion, our results suggest that TET2 promotes osteoclast differentiation by regulating autophagy and provide a new basis for understanding the mechanism of osteoclastogenesis.
Results
OVX induces autophagy and upregulates TET2 expression in osteoclasts
First, an estrogen-deficient bone-loss mouse model was established using bilateral OVX. Microcomputed tomography (micro-CT) and hematoxylin and eosin (H&E) staining showed that femoral bone mass in OVX mice was significantly decreased compared with that in Sham mice 6 weeks post-operation (Figure S1A–D). ACP5 staining showed that the mean number of osteoclasts/bone surface (BS) was increased in the OVX group (Figure S1E and F), indicating that the proliferation and differentiation of osteoclast precursors increased. Furthermore, bone marrow cells/precursors (BMs) were extracted from the Sham (Sham-BMs) and OVX (OVX-BMs) groups mice, and osteoclast differentiation was induced using TNFSF11. ACP5 staining showed that OVX-BMs induced an increase in the number of ACP5-positive cells (Figure S1G and H). Bone resorption assays also revealed increased bone resorption areas in the OVX-BMs (Figure S1I and J). Western blotting showed that osteoclast functional proteins such as MMP9 (matrix metallopeptidase 9) and CTSK (cathepsin K) were highly expressed in OVX-BMs (Figure S1K and L). Additionally, the mRNA levels of osteoclast genes such as Mmp9, Ctsk, Atp6v0d2 (ATPase H+ transporting V0 subunit D2), and Acp5 were increased in OVX-BMs (Figure S1M). These results suggest that excessive osteoclast numbers and osteoclast-induced bone resorption are important contributors to OVX-induced bone loss, consistent with the results of previous studies [20,21].
To determine whether OVX-induced osteoclast activation was associated with autophagy, Sham-BMs and OVX-BMs were stimulated with TNFSF11, and autophagic activity were measured. The transmission electron microscopy (TEM) results revealed that more autophagosomes and autolysosomes were generated in OVX-BMs than in Sham-BMs (Figure 1A). Immunofluorescence staining of endogenous LC3B showed the same result, and the number of LC3B puncta, a marker of autophagosomes, was increased in OVX-BMs (Figure 1B,C). The transformation of endogenous LC3B-I to LC3B-II increases during autophagy; therefore, the LC3B-II:LC3B-I ratio was further detected by western blotting [22]. The results showed that the LC3B-II:LC3B-I ratio increased during osteoclast differentiation and was higher in OVX-BMs than in Sham-BMs (Figure 1D,E). Additionally, levels of the autophagy-related proteins ATG4B and ATG12 were higher in OVX-BMs than in Sham-BMs (Figure S1N and O). These results suggest that enhanced autophagy promotes excessive osteoclast differentiation in OVX-induced bone loss in mice.
Figure 1.
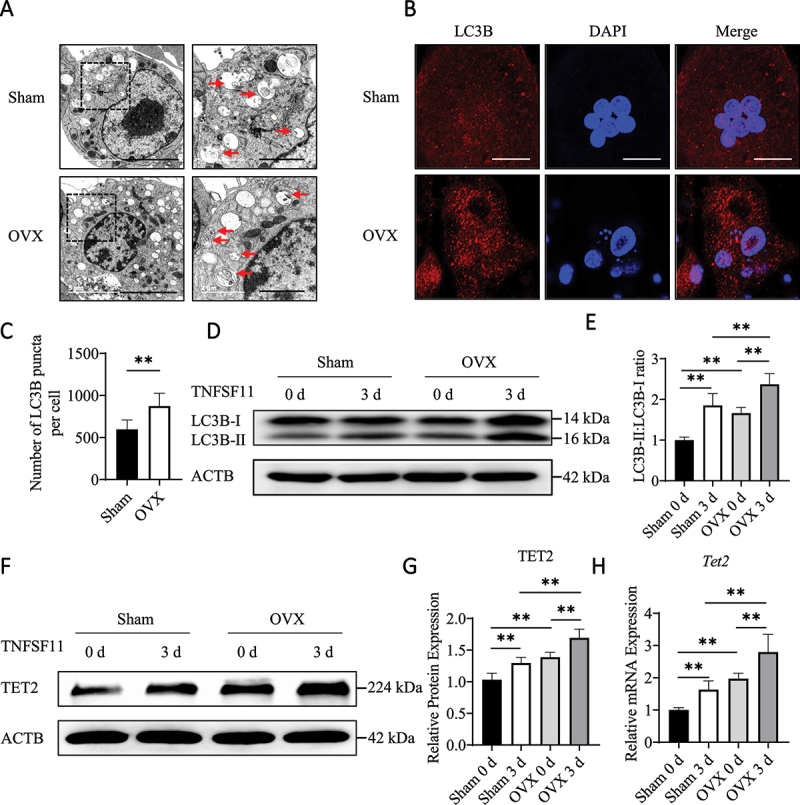
OVX induces autophagy and upregulates TET2 expression in osteoclasts. (A) After stimulation with TNFSF11/RANKL for 3 days, autophagosomes and autolysosomes in BMs from the OVX and Sham group mice were observed via TEM. Red arrows indicated autophagosomes or autolysosomes. The experiment was performed 3 times, and 15 different cells were randomly counted per experiment. Scale bars: 5 µm and 2 µm. (B) Immunofluorescence staining of LC3B in the different groups. Scale bars: 10 µm. The experiment was performed 3 times, and 15 different cells were randomly counted per experiment. (C) Quantification of the LC3B puncta per cell in the different groups. (D) Western blot analysis was performed to determine changes in the expression of autophagic molecules in BMs from OVX and Sham group mice during osteoclast differentiation. (E) The western blot results were quantified. (F) The protein levels of TET2 in BMs from OVX and Sham group mice were assessed by western blotting. (G) The western blot results were quantified. (H) The expression of Tet2 in BMs from OVX or Sham group mice during osteoclast differentiation was analyzed by RT-qPCR. The data are expressed as the mean ± SEM (* P < 0.05, ** P < 0.01, n = 3, three independent experiments were performed at an interval of 2–4 weeks, and each group was repeated three times).
To investigate whether TET2 affects osteoclast differentiation in OVX-induced bone loss, TET2 expression in Sham-BMs and OVX-BMs was measured by western blotting and real-time (RT) quantitative polymerase chain reaction (RT-qPCR). The results showed that TET2 expression was higher in OVX-BMs and further increased during osteoclast differentiation (Figure 1F–H). Immunohistochemical (IHC) staining showed more TET2-positive cells in the OVX mice (Figure S1P and Q). In addition, the western blotting and RT-qPCR results showed that TET2 expression gradually increased during the differentiation of RAW264.7 cells into osteoclasts (Figure S1R–T). These results suggest that TET2 is critical for osteoclast differentiation and OVX-induced bone destruction.
Effect of TET2 on autophagy during osteoclastogenesis
To investigate whether TET2 affects autophagy during osteoclast differentiation, LV-shTet2 lentivirus particles were used to suppress TET2 expression (Figure S2A–J), and autophagy activity was assessed. The TEM results showed that autophagosomes and autolysosomes were increased in LV-NC BMs during osteoclast differentiation, and fewer autophagosomes and autolysosomes were observed in the LV-shTet2 group than in the LV-NC group (Figure 2A). Immunofluorescence staining showed that the number of LC3B puncta was lower in LV-shTet2-treated BMs (Figure 2B,C). It was also determined that the LC3B-II:LC3B-I ratio increased in LV-NC BMs during osteoclast differentiation; however, Tet2 knockdown decreased the LC3B-II:LC3B-I ratio (Figure 2D,E). Moreover, Tet2 knockdown decreased autophagy in RAW264.7 cells (Figure S3A–D).
Figure 2.
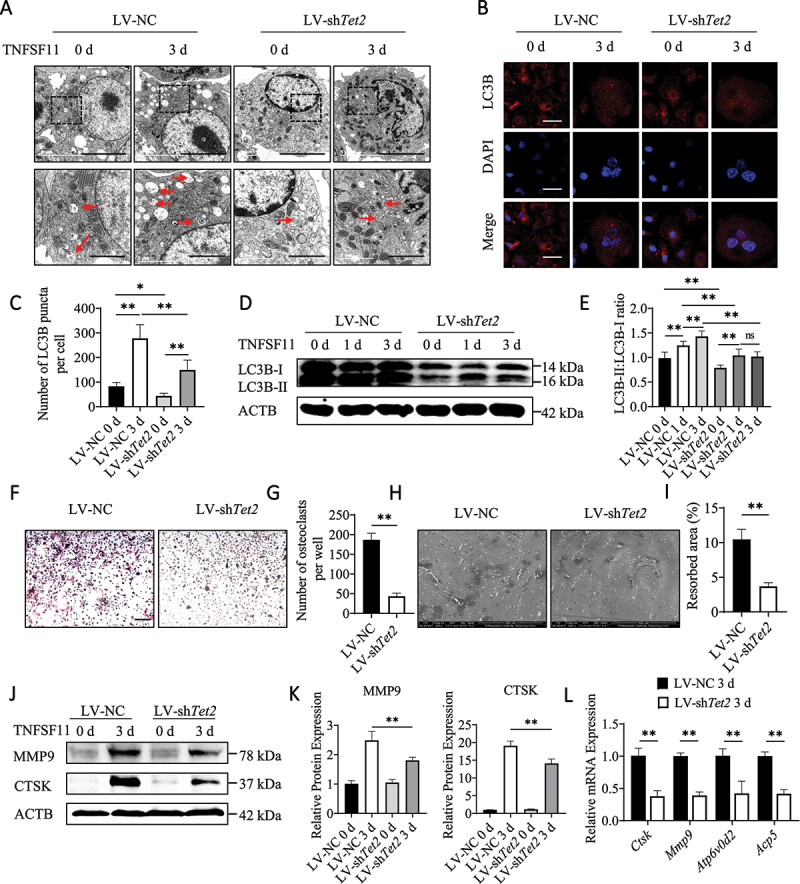
Effect of TET2 on autophagy during osteoclastogenesis. (A) Autophagosomes and autolysosomes in BMs after transfection with LV-NC or LV-shTet2 lentivirus and stimulation with TNFSF11 for 3 days were observed via TEM. Red arrows indicated autophagosomes or autolysosomes. The experiment was performed 3 times, and 15 different cells were randomly counted per experiment. Scale bars: 5 µm and 2 µm. (B) Images of LC3B puncta in the different groups. The experiment was performed 3 times, and 15 different cells were randomly counted per experiment. Scale bars: 10 µm. (C) Quantification of the LC3B puncta per cell in the different groups. (D) Western blot analysis was performed to determine changes in the expression levels of autophagic molecules in the different groups during osteoclast differentiation. (E) The western blot results were quantified. (F) ACP5 staining was used to analyze osteoclast differentiation. Scale bars: 200 µm. (G) ACP5-positive cells from 2.5 × 104 BMs per well were quantified (n = 3, three independent experiments with three replicates each). (H) Scanning electron micrographs of bone resorption pits. Scale bars: 500 µm. (I) The ratio of bone resorption pits in the entire area of a piece of a bovine bone slice was calculated. (J) The expression levels of MMP9 and CTSK in BMs after transfection with LV-NC or LV-shTet2 lentivirus and stimulation with TNFSF11 for 3 days were analyzed by western blotting. (K) The western blot results were quantified. (L) The mRNA levels of Ctsk, Mmp9, Atp6v0d2, and Acp5 were analyzed in BMs transfected with LV-NC or LV-shTet2 lentivirus after TNFSF11 stimulation for 3 days. The data are expressed as the mean ± SEM (* P < 0.05, ** P < 0.01, n = 3, three independent experiments were performed at an interval of 2–4 weeks, and each group was repeated three times).
Furthermore, we investigated the effects of TET2 inhibition on osteoclastogenesis, and ACP5 staining results showed that the formation of ACP5-positive cells was obviously reduced following Tet2 knockdown (Figure 2F,G). A bone resorption assay showed that the bone resorption area was reduced in the LV-shTet2 group (Figure 2H,I). In addition, western blotting showed that levels of the osteoclast markers MMP9 and CTSK were lower upon Tet2 knockdown (Figure 2J,K). Additionally, the mRNA levels of the osteoclast factors Ctsk, Mmp9, Atp6v0d2 and Acp5 were lower upon Tet2 knockdown (Figure 2L). Decreased osteoclastogenesis was also observed in Tet2-knockdown RAW264.7 cells (Figure S3E–K). Consistent with previous reports that decreased autophagy can inhibit osteoclast differentiation, we conclude that TET2 inhibition can decrease autophagy and impair TNFSF11-induced osteoclast formation and activation.
Tet2 knockdown alters the gene expression profiles of osteoclasts and reduces autophagic vesicle formation by increasing BCL2 expression
RNA sequencing (RNA-seq) was performed in Tet2-knockdown BMs after the cells were treated with TNFSF11 for 1 or 3 days to examine transcriptome changes, in order to explore the underlying molecular mechanisms by which TET2 regulates autophagy and osteoclast differentiation. In total, 1753 (1 day) and 1372 (3 days) genes with significantly differential expression (P < 0.05) in Tet2-knockdown BMs compared to control BMs were identified in the two groups (fold change > 1.5, and FDR < 0.05) (Figures 3A,B, S4A, and B). We observed that the expression of crucial genes in osteoclasts, such as Ctsk, Atp6v0d2, Acp5, Nfatc1, and Mmp9, was dramatically decreased in LV-shTet2 osteoclasts, and the RNA-seq results were validated by RT-qPCR (Figure S4C–G). Consistent with our previous results, Tet2 knockdown significantly inhibited functional osteoclast gene expression. Further examination revealed that the mRNA levels of functional autophagy genes, such as Ulk1 (unc-51 like kinase 1), Becn1, Atg5 (autophagy related 5), Atg7 and Atg13, did not change significantly following LV-shTet2 intervention, which was also verified by quantitative RT-qPCR (Figure S4H–L). Levels of BCL2, a negative regulator of autophagy, were significantly increased at both time points during differentiation (1 and 3 days) that were evaluated after transfection, and this finding was verified by western blotting and RT-qPCR (Figure 3C,D, and S4M). Therefore, we hypothesize that TET2 affects osteoclast autophagy through BCL2 and further affects osteoclast function.
Figure 3.
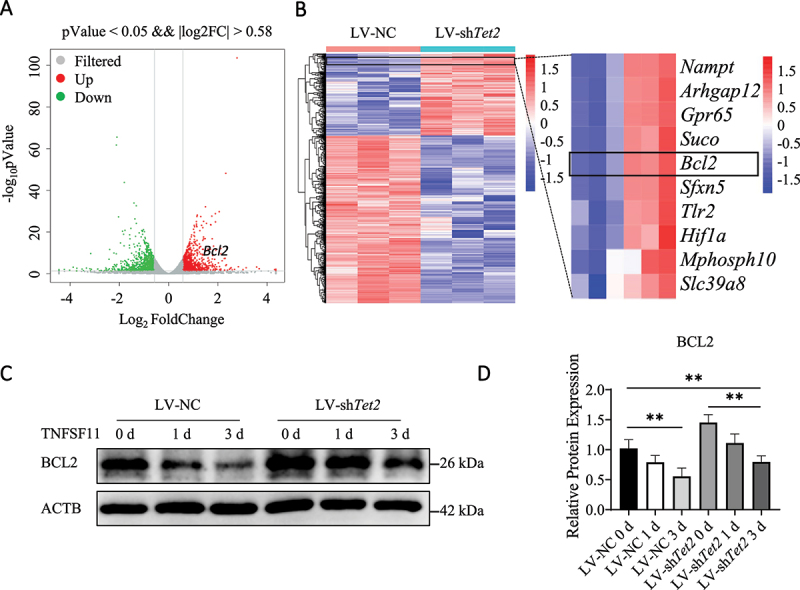
Tet2 knockdown alters the gene expression profiles of osteoclasts and reduces autophagic vesicle formation by increasing BCL2 expression. (A) Volcano plots of all differentially expressed genes (>1.5-fold) in LV-NC- and LV-shTet2-transfected BMs after stimulation with TNFSF11 for 3 days. (B) Heatmaps of all differentially expressed genes (>1.5-fold) in LV-NC- and LV-shTet2-transfected BMs after stimulation with TNFSF11 for 3 days were shown; red represented higher expression, and blue represented lower expression. (C) The expression of BCL2 in LV-NC and LV-shTet2 BMs during osteoclast differentiation was analyzed by western blot analysis. (D) The western blot results were quantified. The data are expressed as the means ± SEM (* P < 0.05, ** P < 0.01, n = 3, three biological duplications).
Interfering with BCL2 enhances autophagy and osteoclast differentiation after TET2 inhibition
To investigate the effect of BCL2 on osteoclast autophagy and differentiation, small interfering RNA (siRNA) was used to decrease BCL2 expression in osteoclasts stimulated with LV-shTet2 lentivirus particles. The knockdown efficiency of BCL2 using siRNA was confirmed by western blotting and RT-qPCR (Figure S5A–E). After Bcl2 expression was knocked down, the decrease in autophagy was partially reversed, and the numbers of autophagosomes, autolysosomes and LC3B puncta were increased (Figure 4A–C). Western blotting confirmed that the LC3B-II:LC3B-I ratio, as well as the levels of ATG4B and ATG12, were increased in the Bcl2 siRNA-treated group (Figure 4D,E, S5F and S5G). These results confirm that decreased BCL2 expression alleviates TET2 inhibition-induced autophagy dysregulation.
Figure 4.
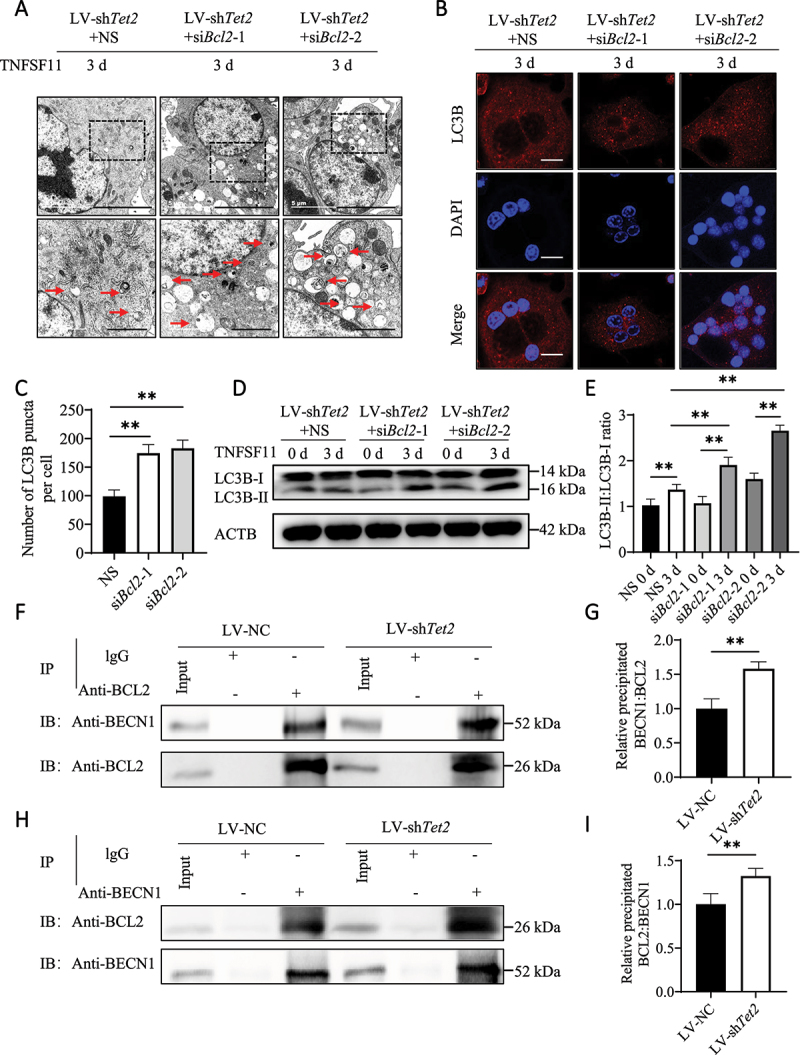
Bcl2 knockdown enhances autophagy in LV-shTet2-transfected BMs. (A) TEM was used to observe the autophagosomes and autolysosomes in osteoclasts transfected with LV-shTet2 lentivirus with or without siRNA-induced Bcl2 knockdown. Red arrows indicated autophagosomes or autolysosomes. The experiment was performed 3 times, and 15 different cells were randomly counted per experiment. Scale bars: 5 µm and 2 µm. (B) Images of LC3B puncta in the different groups. The experiment was performed 3 times, and 15 different cells were randomly counted per experiment. Scale bars: 10 µm. (C) Quantification of the LC3B puncta per cell in the different groups. (D) Western blot analysis was performed to determine changes in the expression levels of autophagic molecules in the different groups during osteoclast differentiation. (E) The western blot results were quantified. (F–I) Co-IP analysis of BCL2 and BECN1 in RAW264.7 cells was performed after transfection with LV-NC or LV-shTet2 and stimulation with TNFSF11 for 3 days. Anti-BCL2 and anti-BECN1 antibodies were used as pull-down antibodies in F and H, respectively. Cell lysates are shown as inputs, and IgG was used as an internal control. The data are expressed as the mean ± SEM (* P < 0.05, ** P < 0.01, n = 3, three independent experiments were performed at an interval of 2–4 weeks, and each group was repeated three times).
BCL2 binds to BECN1 and blocks BECN1 from promoting the localization of other autophagic proteins to the phagophore assembly site, thereby preventing autophagy. In the current study, the co-immunoprecipitation (co-IP) results showed that anti-BCL2 antibodies could bind to BECN1 in both LV-NC and LV-shTet2 RAW264.7 cells; furthermore, more BECN1 was pulled down in LV-shTet2 RAW264.7 cells (Figure 4F,G). Moreover, the anti-BECN1 antibody reciprocally bound to BCL2, and the interaction increased after Tet2 knockdown (Figure 4H,I). These results suggest that TET2 promotes BECN1-mediated osteoclast autophagy by negatively regulating BCL2 expression.
The effect of BCL2 on osteoclastogenesis was also examined. ACP5 staining revealed that the formation of ACP5-positive multinucleated cells increased after Bcl2 knockdown in LV-shTet2 BMs (Figure 5A,B). The bone resorption assay also showed that the bone resorption area increased after Bcl2 knockdown (Figure 5C,D). Additionally, the expression of functional proteins (MMP9 and CTSK) in osteoclasts was partially reversed, as shown by western blotting (Figure 5E,F). Furthermore, the mRNA levels of Mmp9, Ctsk, Acp5, and Atp6v0d2 also increased after Bcl2 knockdown (Figure 5G–J). Overall, these results suggest that Tet2 knockdown reduces osteoclast differentiation by increasing BCL2 expression and reducing BECN1-dependent autophagy.
Figure 5.
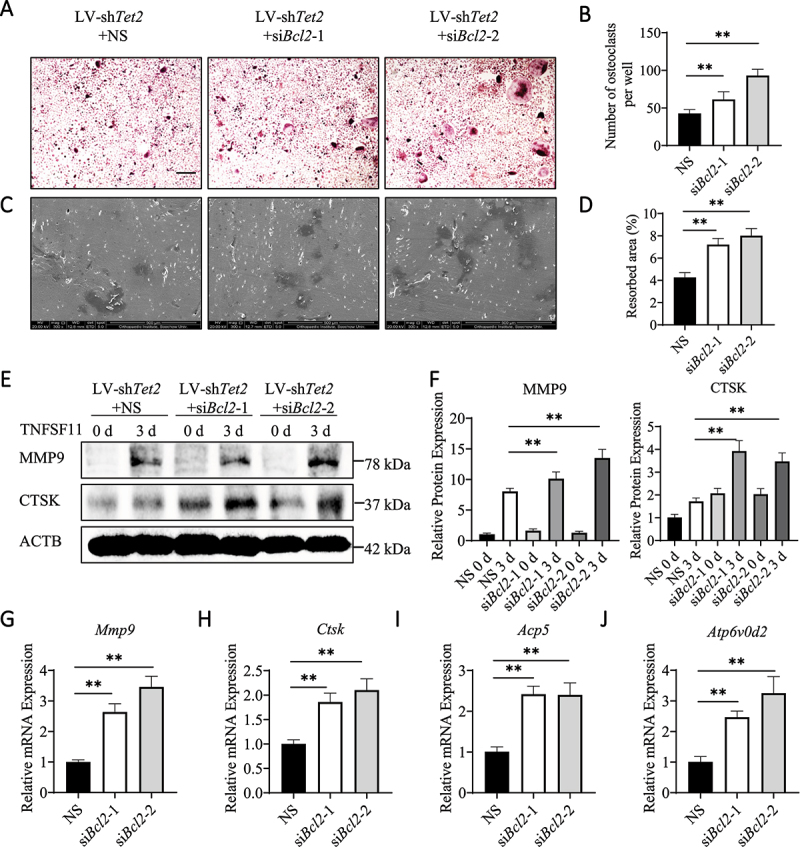
Bcl2 knockdown enhances osteoclast differentiation in LV-shTet2-transfected BMs. (A) ACP5 staining was used to analyze the differentiation of osteoclasts from BMs that were transfected with LV-shTet2 lentivirus with or without siRNA-induced Bcl2 knockdown. Scale bars: 200 µm. (B) ACP5-positive cells from 2.5 × 104 BMs per well were quantified (n = 3, three independent experiments with three replicates each). (C) Scanning electron micrographs of bone resorption pits. Scale bars: 500 µm. (D) The ratio of bone resorption pits in the entire area of a piece of the bovine bone slice was calculated (n = 3, three independent experiments with three replicates each). (E) The expression levels of MMP9 and CTSK in BMs transfected with LV-shTet2 lentivirus with or without Bcl2 knockdown under TNFSF11 stimulation for 3 days were analyzed by western blotting. (F) The western blot results were quantified. (G–J) The mRNA levels of Mmp9, Ctsk, Acp5, and Atp6v0d2 were analyzed in the different groups after TNFSF11 stimulation for 3 days. The data are expressed as the mean ± SEM (* P < 0.05, ** P < 0.01, n = 3, three independent experiments were performed at an interval of 2–4 weeks, and each group was repeated three times).
TET2 knockdown ameliorates bone loss in OVX mice
To elucidate the role of TET2 in OVX-induced bone loss, an LV-shTet2 lentivirus was used to knock down TET2 expression in the BMs of OVX mice. Western blotting showed that TET2 expression was significantly decreased in BMs isolated from OVX mice injected with LV-shTet2, and IHC staining showed that the LV-shTet2 lentivirus significantly reduced TET2 expression in OVX mice in vivo (Figure S6A–D). Micro-CT and 3D reconstruction images indicated that, consistent with the above results, the femoral bone mass in OVX mice was significantly reduced. Interestingly, the change in bone mass was partially restored following treatment with LV-shTet2 lentivirus but not LV-NC lentivirus (Figure 6A). Furthermore, quantitative analysis of bone parameters showed that, compared with those of Sham mice, the bone mineral density (BMD), bone volume per tissue volume (BV/TV), trabecular number (Tb.N), and bone volume (BV) of OVX model mice were decreased, and trabecular separation (Tb.sp) was increased. These changes were reversed after treatment with LV-shTet2, but not with LV-NC (Figure 6D–H). H&E staining was used for the histological staining of mouse femur tissue sections to further evaluate the bone tissue microstructure. Compared with that of the LV-NC-treated OVX mice, the osteoporotic phenotype of the LV-shTet2-treated OVX mice in H&E-stained tissue samples were significantly improved, which was confirmed by quantitative histomorphometrical analysis of the BS (Figure 6B,I). ACP5 staining showed that the number of osteoclasts in the OVX group was significantly reduced by LV-shTet2, but not by LV-NC (Figure 6C,J). In addition, the effect of Tet2 knockdown on osteoclast autophagy was verified in vivo. IHC analysis showed that the number of LC3B-positive cells was significantly higher in OVX mice than in Sham mice. After LV-shTet2 lentivirus injection, LC3B expression decreased in OVX mice, but not in LV-NC mice (Figure S6E and G). Moreover, compared with those in the OVX group, the number of BCL2-positive cells increased after LV-shTet2 injection; however, there was no significant change after LV-NC injection (Figure S6F and H). These results suggest that Tet2 knockdown inhibits autophagy in vivo by increasing BCL2 expression. In addition, the lentiviruses showed no organ toxicity in vivo (Figure S6I). Overall, these results suggest that Tet2 knockdown decreases osteoclast autophagy by increasing BCL2 expression and attenuating bone loss in OVX mice in vivo.
Figure 6.
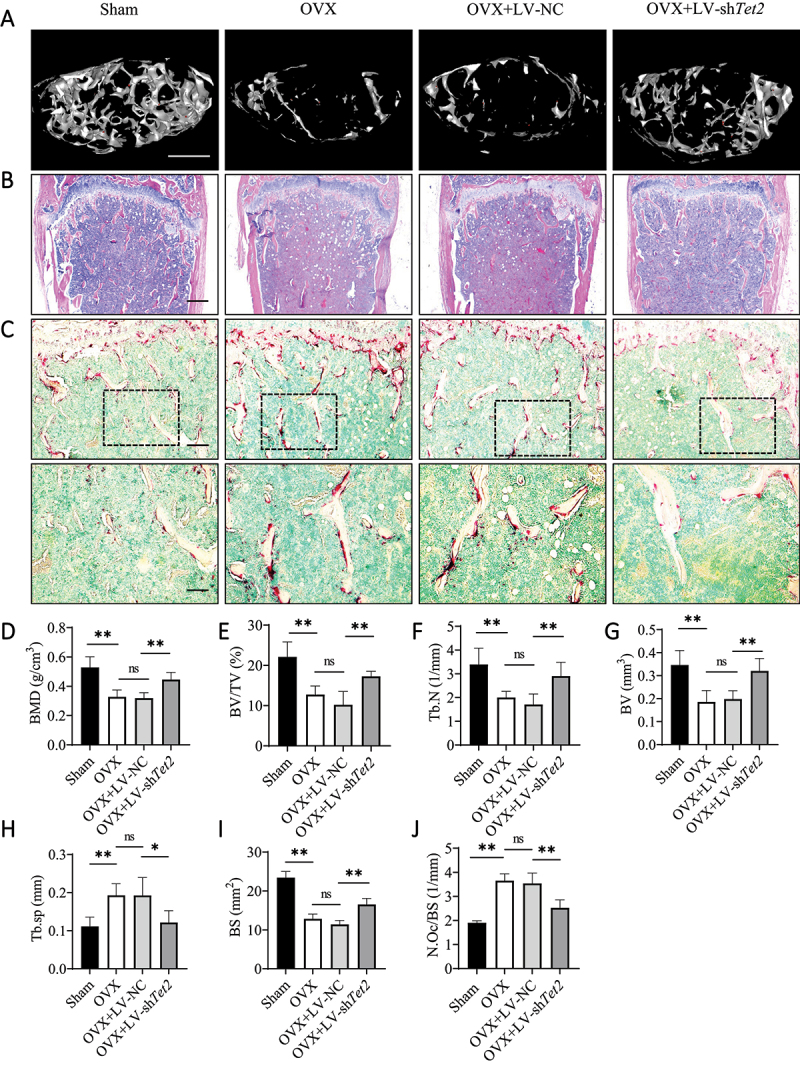
Tet2 knockdown prevents OVX-induced bone loss in vivo. (A) Representative 3D micro-CT reconstruction images of trabecular bone under the distal femur growth plate in the Sham group, OVX group, LV-NC group, and LV-shTet2 group were shown (n = 5 per group). Scale bars: 500 µm. (B) Representative H&E staining images of trabecular bone under the distal femur growth plate from the Sham group, OVX group, LV-NC group, and LV-shTet2 group were shown (n = 5 per group). Scale bars: 200 µm. (C) ACP5 staining showed the trabecular region under the distal femur growth plate from the Sham group, OVX group, LV-NC group, and LV-shTet2 group (n = 5 per group). The red stained area indicates ACP5-positive osteoclasts and methyl green stained area indicates the nuclei. Scale bars: 100 µm and 50 µm. (D-H) Quantitative analysis of bone parameters. The region of interest selected for trabecular analysis started 100 sections below the proximal end of the distal femur growth plate, and 150 slices (6 µm each) were read per sample. (D) BMD (g/cm3). (E) BV/TV (%). (F) Tb.N (1/mm). (G) BV (mm3). (H) Tb.sp (mm). (I) Quantitative analysis of histomorphometric bone parameters of BS (mm2). (J) Number of ACP5-positive osteoclasts on the trabecular bone surface (N.Oc/BS, 1/mm) in distal femora from the Sham group, OVX group, LV-NC group, and LV-shTet2 group. The data are expressed as the mean ± SEM (ns: no significance, * P < 0.05, ** P < 0.01, n = 5 biologically independent mice per group).
Discussion
In this study, TET2 is shown to promote osteoclast differentiation by promoting autophagy. In vivo, the targeted inhibition of TET2 expression attenuates the low bone mass phenotype of OVX mice. Mechanistic studies demonstrate that TET2 enhances BECN1-dependent autophagy by downregulating BCL2 expression, thereby promoting osteoclast differentiation.
TET2, a transcription cofactor, is recruited by other transcription factors to the target gene region to demethylate the target gene [23–26]. Loss of TET2 increases the differentiation of granulocytes/monocytes from hematopoietic stem/progenitor cells [16], however, the specific mechanism is unclear. However, our results showed that Tet2 knockdown reduced the TNFSF11-induced osteoclast differentiation of BMs and RAW264.7 cells. Chu et al. [18] demonstrated that TET2 interacts with RUNX1 to negatively regulate the transcription of genes that impair osteoclast differentiation. Furthermore, TET2 interacts with SPI1 to regulate the expression of genes crucial for osteoclast differentiation [17]. These results suggest that the effect of TET2 on osteoclast differentiation is partly due to the effects of the transcription factors RUNX1 and SPI1 on their target genes, which are crucial for osteoclast differentiation and function. Additionally, our results showed that TET2 regulates osteoclast differentiation by regulating autophagy.
Autophagy is an intracellular degradation pathway that improves cell survival by recycling damaged organelles and proteins [27]. Osteoclast differentiation requires turnover of organelles and intracellular proteins, and autophagy is involved in this process. The autophagy inhibitor 3-methyladenine can significantly inhibit osteoclast differentiation [7]. Through genome-wide association analysis, Zhang et al. [28] found that autophagy-related biological processes contribute to the progression of wrist osteoporosis. In our study, we found that osteoclast autophagy was significantly increased in the OVX mouse model. Autophagy activation was also found to be increased during BM differentiation into osteoclasts in vitro, and this activation was obviously upregulated in OVX-BMs compared to Sham-BMs. These results further confirm that enhanced autophagy promotes osteoclast differentiation in the OVX-induced bone loss model. We found that Tet2 knockdown decreased autophagy in BMs and RAW264.7 cells, and similar results were observed in vivo; Tet2 knockdown in the bone marrow significantly reduced LC3B expression in BMs and attenuated bone loss in OVX mice.
RNA-seq results showed that Tet2 knockdown led to increased Bcl2 expression, which was verified by RT-qPCR and western blotting. Previous studies have shown that TET2 tends to bind to genes whose promoters or coding regions are rich in CpG Islands to regulate transcription [12]. An analysis with MethPrimer software suggested that the promoter region of Bcl2 is rich in CpG Islands (Figure S6J). Therefore, we speculated that TET2 may regulate the methylation of CpG Islands in the Bcl2 promoter and inhibit its transcription. However, the specific mechanisms require further investigation. BCL2 can interact with the autophagic protein BECN1 to prevent other BECN1-dependent autophagic proteins from binding to the phagophore assembly site, thereby inhibiting the process of autophagic flux [29,30]. A co-IP assay was used to examine the binding of BCL2 to BECN1. The results showed that BCL2 could bind with BECN1 in RAW264.7 cells, and this binding was increased after Tet2 knockdown. Furthermore, we found that inhibition of BCL2 expression reversed the reduction in autophagy and osteoclastogenesis caused by Tet2 knockdown. Based on these results, we conclude that TET2 promotes osteoclast autophagy and differentiation through altering the expression of BCL2.
Bcl2 is an important osteoclast survival gene, and BCL2 overexpression in mouse hematopoietic cells can increase the number of osteoclasts [31]. BCL2 is an important regulator of apoptosis. Apoptosis is accelerated in bcl2−/− osteoclasts, but this effect can be prevented by the reintroduction of BCL2 [32]. However, though the specific mechanism by which increased BCL2 expression enhances osteoclast differentiation remains unclear, it may be related to decreased apoptosis. In our experiment, BCL2 expression was increased after the deletion of TET2; however, osteoclast differentiation was still reduced, which appears to be contrary to the above results. However, TET2 affects osteoclasts through multiple mechanisms, such as the transcriptional function of RUNX1 and SPI1 [17,18]. Our results suggest that the increased expression of BCL2 caused by Tet2 knockdown could decrease autophagy due to increased binding of BCL2 to BECN1. Inhibition of BCL2 expression partially reversed impaired osteoclast autophagy and differentiation. Yamashita et al. [33] also reported that bcl2−/− osteoclast differentiation is more rapid than bcl2+/+ osteoclast differentiation, however, the lifespan of bcl2−/− osteoclast is shorter, suggesting that BCL2 is involved in osteoclast fusion and apoptosis.
Multiple intracellular signaling pathways simultaneously regulate autophagy and apoptosis. Maintaining autophagy at an appropriate level can reduce apoptosis, whereas excess autophagy can accelerate apoptosis by removing the necessary proteins. When pressure exceeds a certain threshold or intensity, apoptotic and non-apoptotic lethal programs are activated, and caspase-mediated autophagic protein cleavage can inactivate autophagy [34]. Pattingre et al. [30] suggested that the degree to which BCL2 and BECN1 bind may regulate the transition between autophagy activation and autophagy gene-dependent cell death. Our findings confirm that TET2 regulates autophagy by negatively regulating BCL2 expression; however, whether TET2 affects osteoclast apoptosis requires further investigation.
In summary, we found that TET2 regulates BCL2 expression and affects the autophagy and differentiation of osteoclasts, and that targeted blocking TET2 reduces autophagic activity and alleviates OVX-induced bone loss. Our results reinforce the epigenetic regulation of autophagy and osteoclast differentiation by TET2, and provide a new basis for elucidating the mechanism of osteoclastogenesis.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM/high glucose; C3113-0500), alpha modification of Eagle’s medium (α-MEM; C3060-0500), and fetal bovine serum (FBS; C2820-0500) were purchased from VivaCell. Recombinant Mouse TNFSF11 (462-TEC-010/CF) and Recombinant Mouse CSF1/M-CSF (416-ML-050/CF) were purchased from R&D Systems. The antibodies used in this study included MMP9 (ABclonal, A0289), LC3B (Abclonal, A19665), TET2 (ABclonal, A1526), ATG4B (ABclonal, A5059), ATG12 (ABclonal, A19610), ACTB (actin, beta) (Abconal, AC026), CTSK (Abcam, ab187647), BECN1 (Abcam, ab207612), LC3B (Abcam, ab192890), BCL2 (Abcam, ab182858), and BCL2 (Cell Signaling Technology, 3498S). Anti-rabbit HRP-conjugated secondary antibodies (GAR0072) were purchased from Multisciences.
Cell culture
The RAW264.7 murine macrophage cell line was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM containing 10% FBS. RAW264.7 cells from passages 3 to 15 were used in this study. BMs were extracted from the femur bone marrow of female C57BL/6 mice as reported in our previous studies [35,36]. Specifically, the femurs of C57BL/6 mice were aseptically dissected, and the bone marrow was washed with phosphate buffered solution (Solarbio, P1010) using a 23-G needle several times. Then, the collected bone marrow suspension was centrifuged, and red blood cells were removed with red blood cell lysis buffer (Beyotime, C3702). After that, the single-cell suspensions were washed 3 times with phosphate buffered solution, counted, and then plated at 1 × 107 cells per 100-mm dish in α-MEM supplemented with 10% FBS and 30 ng/ml CSF1/M-CSF. Afterward, the medium was changed every other day. After 4 days of culture, the BMs had adhered to the bottom of the culture plate; the BMs were then digested, collected using trypsin (Beyotime, C0203) and counted for different experiments. Cells from one mouse were extracted from each group at a time, and each experiment was repeated three times (three biologically independent mice).
In this study, BMs in the Sham and OVX groups were isolated from the femurs of Sham and OVX mice 6 weeks after surgery. The BMs that demonstrated the effect of the lentivirus in vivo were isolated from the femurs of OVX mice (5 weeks after the injection of lentivirus), and the remaining BMs were extracted from 6- to 8-week-old female mice.
ACP5 staining
BMs (2.5 × 104 cells/well) or RAW264.7 cells (1 × 104 cells/well) were incubated overnight in 48-well plates and subsequently stimulated with 100 ng/ml TNFSF11. Three days later, the cells were stained using an ACP5 staining kit (Sigma-Aldrich, 387A-1 KT). An inverted microscope (Zeiss, Germany) was used to acquire images. Mature osteoclasts had three or more nuclei.
Bone resorption assay
BMs (1 × 104 cells/well) or RAW264.7 cells (0.8 × 104 cells/well) were incubated on bovine bone slices (JoyTech Biotechnology, 2–001-100) in 96-well plates and subsequently stimulated with 100 ng/ml TNFSF11. Seven days later, cells were removed from the bovine bone slices using a sonicator. Resorption pits on bone slices were observed using an FEI Quanta 250 scanning electron microscope (Hillsboro, USA) and quantified using ImageJ software (National Institutes of Health, USA). The pit area was normalized to the whole area of the field of bovine bone slices. Three independent experiments with three replicates each were performed.
RT-qPCR
Total cellular RNA was extracted from the different groups using TRIzol reagent (Beyotime, R0011) according to the manufacturer’s guidelines. RNA samples were quantified and qualified on a NanoDrop-2000 spectrophotometer (Thermo Fisher, USA). Then, an equal amount of RNA (0.5 µg) was mixed with dNTPs (Monad, MR05001 M) for reverse transcription. Next, real-time PCR amplification was performed in a CFX96™ thermal cycler (Bio-Rad Laboratories, USA) with a mixture containing 5 µl of qPCR Mix (Monad, MQ00401S), 0.5 µl of the corresponding primers, 2 µl of ddH2O and 2 µl of cDNA. Dissociation curves were generated to ensure the specificity of each RT-qPCR experiment. Gapdh (glyceraldehyde-3-phosphate dehydrogenase) was used as a housekeeping gene for normalization and quality control purposes. The primers utilized in this study are listed below: Acp5: 5'-TGTGGCCATCTTTATGCT-3' (forward), 5ʹ-GTCATTTCTTTGGGGCTT-3ʹ (reverse); Atp6v0d2: 5ʹ-GACCCTGTGGCACTTTTTGT-3ʹ (forward), 5ʹ-GTGTTTGAGCTTGGGGAGAA-3ʹ (reverse); Atg5: 5ʹ-AGTCAAGTGATCAACGAAATGC-3ʹ (forward), 5ʹ-TATTCCATGAGTTTCCGGTTGA-3ʹ (reverse); Atg7: 5ʹ-GTGTACGATCCCTGTAACCTAG-3ʹ (forward), 5ʹ-GATGCTATGTGTCACGTCTCTA-3ʹ (reverse); Atg13: 5ʹ-ACATCTTTTTCCACATCCCCTC-3ʹ (forward), 5ʹ-TGTTTAAGCCAGCAGTAAACAC-3ʹ (reverse); Bcl2: 5ʹ-GGTGGGGTCATGTGTGTGG-3ʹ (forward), 5ʹ-CGGTTCAGGTACTCAGTCATCC-3ʹ (reverse); Becn1: 5ʹ-CAAACAGCGTTTGTAGTTCTGA-3ʹ (forward), 5ʹ-TAATAGCTTCACTCTGATCGGG-3ʹ (reverse); Ctsk: 5ʹ-CTTCCAATACGTGCAGCAGA-3ʹ (forward), 5ʹ-TCTTCAGGGCTTTCTCGTTC-3ʹ (reverse); Gapdh: 5ʹ-GGTTGTCTCCTGCGACTTCA-3ʹ (forward), 5ʹ-TGGTCCAGGGTTTCTTACTCC-3ʹ (reverse); Mmp9: 5ʹ-CGTGTCTG GAGATTCGACTTGA-3ʹ (forward), 5ʹ-TTGGAAACTCACACGCCAGA-3ʹ (reverse); Nfatc1: 5ʹ-GAGAATCGAGATCACCTCCTAC-3ʹ (forward), 5ʹ-TTGCAGCTAGGAAGTACGTCTT-3ʹ (reverse); Tet2: 5ʹ-CTGCTGTTTGGGTCTGAAGGAAGG-3ʹ (forward), 5ʹ-GTTCTGCTGGTCTCTGTGGGAATG-3ʹ (reverse); Ulk1: 5ʹ-ACTCAGGTGCACAATTACCAG-3ʹ (forward), 5ʹ-CTTGGGGAGAAGGTGTGTAG-3ʹ (reverse). Relative mRNA expression levels were quantified using the 2−ΔΔCt method after normalization to the expression of the positive control.
Western blot analysis
Cellular proteins were extracted from the different groups with RIPA buffer (Beyotime, P0013B). Proteins (30 µg) were separated by SDS-PAGE and transferred onto nitrocellulose (NC) membranes (Beyotime, FFN03). The membranes were incubated with primary antibodies against TET2 (ABclonal, A1526; 1:1000), MMP9 (ABclonal, A0289; 1:1000), CTSK (Abcam, ab187647; 1:1000), LC3B (Abcam, ab192890; 1:2000), BECN1 (1:2000), BCL2 (1:1000), ATG4B (ABclonal, A5059; 1:1000), ACTB (actin beta) (Abconal, AC026; 1:50,000), and ATG12 (ABclonal, A19610; 1:1000) and then incubated with a secondary antibody (Multisciences;1:5000). ACTB was used as a loading control. The bands were developed using enhanced chemiluminescence (New Cell & Molecular Biotechnology, P10300) and quantified using Image Lab 3.0 (Bio-Rad, USA), and the band detections were within the linear range.
TEM
Briefly, the cells were collected in a centrifuge tube by trypsinization and centrifugation at 253 x g for 5 min and prefixed with 2.5% glutaraldehyde (12 h, 4°C). Each sample was postfixed with 1% osmium tetroxide (SPI-CHEM, 20,816–12-0; OsO4, 2 h), dehydrated sequentially with ethanol (30%, 50%, 70%, 80%, 90%, 95% and 100%) for 20 min, transferred to pure acetone (20 min), and embedded in spurr resin (SPI-CHEM, 90,529–77-4), Sections were generated on a Leica EM UC7 ultratome (Leica, Germany). After staining with uranyl acetate and alkaline lead citrate for 5 min, the sections were observed by TEM (FEI Tecnai Spirit, USA).
Lentiviral transfection
To knockdown Tet2, the lentiviruses LV-GFP-shTet2 (200902DZ) and LV-GFP-NC (H22 FZ) were purchased from GenePharma. The sequences were as follows: GGAGCTATTTGCTGAAGAATA and TTCTCCGAACGTGTCACGT. Lentiviral particles (multiplicity of infection [MOI] = 50) were used to transfect BMs and RAW264.7 cells. At 5 days postinfection, the knockdown efficiency was determined by western blot analysis and RT-qPCR. The infectious efficiency was determined by calculating the proportion of cells expressing GFP using a fluorescence microscope (Zeiss, Germany).
Immunofluorescent staining
BMs were seeded on coverslips, treated with LV-NC or LV-shTet2, and induced with TNFSF11 (100 ng/ml) in 24-well plates for 3 days. Then, 4% paraformaldehyde (New Cell & Molecular Biotechnology, N1012), 0.2% Triton X-100 (Beyotime, P0096) and QuickBlock™ blocking buffer for immunostaining (Beyotime, P0102) were used to fix, permeabilize and seal the cells, respectively, in sequence. Then, the cells were incubated with anti-LC3B (Abcam, ab192890; 1 µg/ml) antibodies, goat anti-rabbit IgG H&L (Abcam, Alexa Fluor 647; 1:500), and DAPI (Beyotime, C1006) successively. LC3B puncta were imaged using a high-resolution confocal microscope (Leica, Germany).
RNA-seq
BMs for RNA-seq were extracted from 6-week-old female C57BL/6 mice. After transfection with LV-NC or LV-shTet2 lentiviral particles, the BMs were stimulated with 100 ng/ml TNFSF11 for 1 or 3 days. Total cellular RNA was extracted from the different groups using TRIzol reagent (Beyotime, R0011). Transcriptome sequencing and analysis were conducted by OE Biotechnology (Shanghai, China). The clean reads were mapped to the Mus musculus genome (GRCm38.p6) using HISAT2 [37]. Cufflinks [38] was used to calculate the fragments per kilobase of exon per million mapped fragments (FPKM) [39] value of each gene, and htseq-Count [40] was used to calculate the read count of each gene. Differential expression analysis was performed using the DESeq [41] (2012) R package functions estimateSizeFactors and nbinomTest. A P value < 0.05 and FC > 1.5 or FC< 0.5 were set as the thresholds for significant differential expression. Volcano plots and heatmaps were used to show the expression patterns of different genes in different samples.
BCL2 knockdown by siRNA
To knockdown Bcl2, siRNAs targeting Bcl2 were purchased from GenePharma (Suzhou, China). Briefly, after transfection with LV-shTet2 lentiviral particles for five days, the cells were transfected with siRNAs using Lipofectamine RNAiMAX reagent (Invitrogen, 13,778,150). The transfection efficiency was validated by western blot analysis and RT-qPCR. The sequences of the Bcl2 siRNA oligonucleotides were as follows: siRNA1: sense, 5-GGGAGAACAGGGUAUGAUATT-3; antisense, 5-UAUCAUACCCUGUUCUCCCTT-3; siRNA2: sense, 5-GGAUGACUGAGUACCUGAATT-3; and antisense, 5-UUCAGGUACUCAGUCAUCCTT-3. The sense sequence of the negative control siRNA was 5-UUCUCCGAACGUGUCACGUTT-3, and the antisense sequence was 5-ACGUGACACGUUCGGAGAATT-3.
Co-IP
Total proteins were extracted from RAW264.7 cells transfected with LV-NC or LV-shTet2 lentiviral particles using cell lysis buffer for western blotting and IP (Beyotime, P0013). First, protein A + G magnetic beads (Beyotime, P2108) were incubated with anti-BCL2 (1:100), anti-BECN1 (1:30) or IgG antibodies (Beyotime, A7016) for 1 h on a rotating platform at room temperature. Then, the prepared protein A + G magnetic beads were incubated with 600 µg of the extracted proteins at 4°C with rotation overnight. Subsequently, the beads were separated by a magnetic shelf and washed three times with lysis buffer. The supernatant was separated on a magnetic rack for 10 seconds and analyzed by western blotting.
OVX-induced osteoporosis mouse model
The Ethics Committee of the First Affiliated Hospital of Soochow University approved all the animal experiments. In brief, (1) twenty female C57BL/6 mice (ten weeks old) were divided into two groups (n = 10 each group): the Sham group (exteriorized without resection), and the OVX group (bilateral OVX). (2) Forty female C57BL/6 mice (ten weeks old) were divided into four groups (n = 10 each group): the Sham group (exteriorized without resection), the OVX group (bilateral OVX), the LV-Tet2 group (OVX+LV-shTet2 lentivirus), and the LV-NC group (OVX+LV-NC lentivirus). One week after OVX, the lentivirus (in a volume of 25 µl) was injected into the marrow cavity through the periosteum and cortex of the femur at the epiphyseal transition once every two weeks. All animals were anesthetized with Nembutal during the operations. All animals survived and were housed at a temperature of 20–25°C and relative humidity of 40–70%. Six weeks after surgery, all of the left femur samples were extracted for micro-CT and histology experiments, the right femur was removed and immersed in saline for BM collection, and heart, liver, spleen, lung and kidney tissues were collected for lentivirus toxicity analysis.
Micro-CT imaging
Mouse femur samples were fixed for 24 h in 10% buffered formalin and then scanned with a SkyScan 1176 micro-CT (Aartselaar, Belgium) (n = 5/group). NRecon software (SkyScan micro-CT, Aartselaar, Belgium) was used to analyze the three‐dimensional (3D) histomorphometric images. SkyScan software was applied to analyze the parameters of the distal femoral metaphyseal trabecular bone, including the BV (mm3), BV/TV (%), BMD (g/cm3), Tb.sp (mm), and Tb.N (1/mm). The region of interest selected for trabecular analysis started 100 sections below the proximal end of the distal femur growth plate, and 150 slices (6 µm each) were read per sample.
Histomorphometrical and IHC staining analysis
Femur samples were fixed for 24 h in 10% buffered formalin, decalcified with 10% EDTA (Sigma-Aldrich, V900081-500 G) for 4 weeks, embedded in paraffin (Leica, 39,601,095), and sliced into 6-µm-thick sections (Leica 2135, Germany). These slices were subjected to H&E staining to observe morphological changes, and ACP5 staining (Sigma-Aldrich, 387A-1KT) and methyl green staining (Beyotime, C0115) were performed to observe osteoclasts. The slices were imaged with an Axiovert 40C optical microscope (Zeiss, Germany). The BS (mm2) and number of ACP5-positive osteoclasts/BS (N.Oc/BS, 1/mm) were used to assess differences between groups according to standard procedures [42] using Bioquant Osteo 2017 (BIOQUANT, USA). The osteoclasts on the cancellous BS were counted throughout the area of interest, and the data were normalized to the number of osteoclasts per millimeter trabecular surface perimeter (N mm−1). The region of interest that was confined under the growth plate of the distal femur per image was also selected for trabecular bone CT analysis. The levels of TET2, LC3B and BCL2 in vivo were determined by IHC staining. Briefly, the slices were dewaxed and subjected to gradient hydration and antigen retrieval, after which they were incubated with primary antibodies against LC3B (ABclonal, A19665; 1:100), BCL2 (Abcam, ab182858; 1:500), and TET2 (ABclonal, A1526; 1:100), followed by incubation with secondary and tertiary antibodies (Vector Laboratories, PK-6200). A DAB kit (Cell Signaling Technology, 8059S) was used to induce a color reaction. The region of interest that was restricted under the growth plate of the distal femur and the region containing trabecular bone were selected as the imaging area in the whole viewed area. Semiquantitative evaluation of the IHC staining results was performed as previously described [43,44].
Statistical analysis
The data are displayed as the mean ± standard error of the mean (SEM). GraphPad Prism 8.0 software was used for statistical analysis. Comparisons of data with a normal distribution between two groups were performed using Student’s t tests; otherwise, the nonparametric Mann-Whitney test was used. One-way ANOVA followed by Tukey’s test was used for comparisons of multiple groups of data that were normally distributed, and the Kruskal-Wallis test was used for comparisons of data with a nonnormal distribution. Differences with a P value <0.05 were defined as statistically significant.
Supplementary Material
Funding Statement
This work is supported by grants from the National Natural Science Foundation of China [nos. 82072425, 82072498, 82074473, 82030068, 81873991, 81873990 and 91849114], the Young Medical Talents of Jiangsu Province [no. QNRC2016751], the Natural Science Foundation of Jiangsu Province [nos. BK20180001, BK20200198, BK20191201, BE2020666, BE2021673 and BE2021650], Jiangsu Province “333 Project” research project [BRA2020129], the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Special Project of Diagnosis and Treatment Technology for Key Clinical Diseases in Suzhou [LCZX202003], the Application Fundamental Research Program of Suzhou City [SYSD2020013] and the Program for Science and Technology of Changshu Health Committee [CSWS201907].Medical and Public Health Technology Innovation and Application Project of Wuxi Science and Technology Bureau (no. N20202041)and the youth talent project of Wuxi health commission (no. Q202150)
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Eastell R, O’Neill TW, Hofbauer LC, et al. Postmenopausal osteoporosis. Nat Rev Dis Primers. 2016 Sep 29;2:16069. [DOI] [PubMed] [Google Scholar]
- [2].Weitzmann MN, Pacifici R.. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. 2006. May;116(5):1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Szulc P, Seeman E, Duboeuf F, et al. Bone fragility: failure of periosteal apposition to compensate for increased endocortical resorption in postmenopausal women. J Bone Miner Res. 2006. Dec;21(12):1856–1863. [DOI] [PubMed] [Google Scholar]
- [4].Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000 Sep 1;289(5484):1504–1508. [DOI] [PubMed] [Google Scholar]
- [5].Montaseri A, Giampietri C, Rossi M, et al. The role of autophagy in osteoclast differentiation and bone resorption function. Biomolecules. 2020 Sep 30;10(10). doi: 10.3390/biom10101398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457–484. [DOI] [PubMed] [Google Scholar]
- [7].Laha D, Deb M, Das H. KLF2 (kruppel-like factor 2 [lung]) regulates osteoclastogenesis by modulating autophagy. Autophagy. 2019. Dec;15(12):2063–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Arai A, Kim S, Goldshteyn V, et al. Beclin1 modulates bone homeostasis by regulating osteoclast and chondrocyte differentiation. J Bone Miner Res. 2019. Sep;34(9):1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xiu Y, Xu H, Zhao C, et al. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J Clin Invest. 2014. Jan;124(1):297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lin NY, Chen CW, Kagwiria R, et al. Inactivation of autophagy ameliorates glucocorticoid-induced and ovariectomy-induced bone loss. Ann Rheum Dis. 2016. Jun;75(6):1203–1210. [DOI] [PubMed] [Google Scholar]
- [11].DeSelm CJ, Miller BC, Zou W, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011 Nov 15;21(5):966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017. Sep;18(9):517–534. [DOI] [PubMed] [Google Scholar]
- [13].Wang L, Ozark PA, Smith ER, et al. TET2 coactivates gene expression through demethylation of enhancers. Sci Adv. 2018. Nov;4(11):eaau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Montagner S, Leoni C, Emming S, et al. TET2 regulates mast cell differentiation and proliferation through catalytic and non-catalytic activities. Cell Rep. 2016 May 17;15(7):1566–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang R, Yu T, Kou X, et al. Tet1 and Tet2 maintain mesenchymal stem cell homeostasis via demethylation of the P2rX7 promoter. Nat Commun. 2018 Jun 1;9(1):2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011 Jul 12;20(1):11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].de la Rica L, Rodriguez-Ubreva J, Garcia M, et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013;14(9):R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chu Y, Zhao Z, Sant DW, et al. Tet2 regulates osteoclast differentiation by interacting with Runx1 and maintaining genomic 5-Hydroxymethylcytosine (5hmC). Genomics Proteomics Bioinf. 2018. Jun;16(3):172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen Q, Zhang Q, Shi Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. 2018 Feb 1;554(7690):123–127. [DOI] [PubMed] [Google Scholar]
- [20].Yang W, Lu X, Zhang T, et al. TAZ inhibits osteoclastogenesis by attenuating TAK1/NF-kappaB signaling. Bone Res. 2021 Jul 12;9(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li J, Li X, Liu D, et al. eIF2alpha signaling regulates autophagy of osteoblasts and the development of osteoclasts in OVX mice. Cell Death Dis. 2019 Dec 4;10(12):921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010 Feb 5;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014 Dec 11;9(5):1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Y, Xiao M, Chen X, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015 Feb 19;57(4):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Okashita N, Kumaki Y, Ebi K, et al. PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development. 2014. Jan;141(2):269–280. [DOI] [PubMed] [Google Scholar]
- [26].Costa Y, Ding J, Theunissen TW, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013 Mar 21;495(7441):370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016 Jun 2;85:685–713. [DOI] [PubMed] [Google Scholar]
- [28].Zhang L, Guo YF, Liu YZ, et al. Pathway-based genome-wide association analysis identified the importance of regulation-of-autophagy pathway for ultradistal radius BMD. J Bone Miner Res. 2010. Jul;25(7):1572–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kihara A, Kabeya Y, Ohsumi Y, et al. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001. Apr;2(4):330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005 Sep 23;122(6):927–939. [DOI] [PubMed] [Google Scholar]
- [31].Bozec A, Bakiri L, Hoebertz A, et al. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature. 2008 Jul 10;454(7201):221–225. [DOI] [PubMed] [Google Scholar]
- [32].Nagase Y, Iwasawa M, Akiyama T, et al. Anti-apoptotic molecule Bcl-2 regulates the differentiation, activation, and survival of both osteoblasts and osteoclasts. J Biol Chem. 2009 Dec 25;284(52):36659–36669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yamashita J, Datta NS, Chun YH, et al. Role of Bcl2 in osteoclastogenesis and PTH anabolic actions in bone. J Bone Miner Res. 2008. May;23(5):621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014. Feb;15(2):81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li N, Li X, Zheng K, et al. Inhibition of Sirtuin 3 prevents titanium particle-induced bone resorption and osteoclastsogenesis via suppressing ERK and JNK signaling. Int J Biol Sci. 2021;17(5):1382–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang Q, Ge G, Liang X, et al. Punicalagin ameliorates wear-particle-induced inflammatory bone destruction by bi-directional regulation of osteoblastic formation and osteoclastic resorption. Biomater Sci. 2020 Sep 21;8(18):5157–5171. [DOI] [PubMed] [Google Scholar]
- [37].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015. Apr;12(4):357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010. May;28(5):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roberts A, Trapnell C, Donaghey J, et al. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015 Jan 15;31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR histomorphometry nomenclature committee. J Bone Miner Res. 2013. Jan;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li S, Liu B, Zhang L, et al. Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone. 2014. Apr;61:164–175. [DOI] [PubMed] [Google Scholar]
- [44].Da CL, Xin Y, Zhao J, et al. Significance and relationship between Yes-associated protein and survivin expression in gastric carcinoma and precancerous lesions. World J Gastroenterol. 2009 Aug 28;15(32):4055–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.